

Abstract
Background
Neuropathic pain, which is caused by nerve damage, is increasing in prevalence worldwide. This may reflect improved diagnosis, or it may be due to increased incidence of diabetes‐associated neuropathy, linked to increasing levels of obesity. Other types of neuropathic pain include post‐herpetic neuralgia, trigeminal neuralgia, and neuralgia caused by chemotherapy. Antidepressant drugs are sometimes used to treat neuropathic pain; however, their analgesic efficacy is unclear. A previous Cochrane review that included all antidepressants for neuropathic pain is being replaced by new reviews of individual drugs examining chronic neuropathic pain in the first instance. Venlafaxine is a reasonably well‐tolerated antidepressant and is a serotonin reuptake inhibitor and weak noradrenaline reuptake inhibitor. Although not licensed for the treatment of chronic or neuropathic pain in most countries, it is sometimes used for this indication.
Objectives
To assess the analgesic efficacy of, and the adverse effects associated with the clinical use of, venlafaxine for chronic neuropathic pain in adults.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL) via The Cochrane Library, and MEDLINE and EMBASE via Ovid up to 14 August 2014. We reviewed the bibliographies of any randomised trials identified and review articles, contacted authors of one excluded study and searched www.clinicaltrials.gov to identify additional published or unpublished data. We also searched the meta‐Register of controlled trials (mRCT) (www.controlled‐trials.com/mrct) and the WHO International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch/) for ongoing trials but did not find any relevant trials.
Selection criteria
We included randomised, double‐blind studies of at least two weeks' duration comparing venlafaxine with either placebo or another active treatment in chronic neuropathic pain in adults. All participants were aged 18 years or over and all included studies had at least 10 participants per treatment arm. We only included studies with full journal publication.
Data collection and analysis
Three review authors independently extracted data using a standard form and assessed study quality. We intend to analyse data in three tiers of evidence as described by Hearn 2014, but did not find any first‐tier evidence (ie evidence meeting current best standards, with minimal risk of bias) or second‐tier evidence, that was considered at some risk of bias but with adequate participant numbers (at least 200 in the comparison). Third‐tier evidence is that arising from studies with small numbers of participants; studies of short duration, studies that are likely to be of limited clinical utility due to other limitations, including selection bias and attrition bias; or a combination of these.
Main results
We found six randomised, double‐blind trials of at least two weeks' duration eligible for inclusion. These trials included 460 participants with neuropathic pain, with most participants having painful diabetic neuropathy. Four studies were of cross‐over design and two were parallel trials. Only one trial was both parallel design and placebo‐controlled. Mean age of participants ranged from 48 to 59 years. In three studies (Forssell 2004, Jia 2006 and Tasmuth 2002), only mean data were reported. Comparators included placebo, imipramine, and carbamazepine and duration of treatment ranged from two to eight weeks. The risk of bias was considerable overall in the review, especially due to the small size of most studies and due to attrition bias. Four of the six studies reported some positive benefit for venlafaxine. In the largest study by Rowbotham, 2004, 56% of participants receiving venlafaxine 150 to 225 mg achieved at least a 50% reduction in pain intensity versus 34% of participants in the placebo group and the number needed to treat for an additional beneficial outcome was 4.5. However, this study was subject to significant selection bias. Known adverse effects of venlafaxine, including somnolence, dizziness, and mild gastrointestinal problems, were reported in all studies but were not particularly problematic and, overall, adverse effects were equally prominent in placebo or other active comparator groups.
Authors' conclusions
We found little compelling evidence to support the use of venlafaxine in neuropathic pain. While there was some third‐tier evidence of benefit, this arose from studies that had methodological limitations and considerable risk of bias. Placebo effects were notably strong in several studies. Given that effective drug treatments for neuropathic pain are in current use, there is no evidence to revise prescribing guidelines to promote the use of venlafaxine in neuropathic pain. Although venlafaxine was generally reasonably well tolerated, there was some evidence that it can precipitate fatigue, somnolence, nausea, and dizziness in a minority of people.
Plain language summary
Venlafaxine for neuropathic pain in adults
Background
Neuropathic pain is pain that arises from damaged nerves. It is different in nature than pain that arises from damaged tissue, such as a cut, although that type of pain is also carried along nerves. Drugs that are commonly used to treat pain, such as paracetamol, ibuprofen, or morphine, are not very good at treating neuropathic pain. However, other drugs, such as gabapentin, which are also used to prevent or treat epilepsy (fits), do appear to be of some benefit in treating neuropathic pain. There is also a great deal of interest in using antidepressant drugs to treat neuropathic pain. This does not imply that the person with neuropathic pain is depressed, but simply that these drugs may have benefits in neuropathic pain. However, while some doctors prescribe antidepressants in people with neuropathic pain, their benefits have not been confirmed in large clinical trials.
Here we reviewed clinical trials for one antidepressant drug, venlafaxine, to see if robust evidence exists that it is effective in treating neuropathic pain.
Study characteristics
In detailed searches of the medical literature, we found six trials that were suitable for inclusion in our analysis, that together included 460 adults.
Key results and quality of the evidence
All six trials were conducted in an approved statistical manner (randomised and double‐blinded); however, all had limitations that could lead to an overestimation of efficacy in treating this type of pain. Four were of very small size and five were of short duration, both of which can bias the results of chronic pain trials. Although it was not possible to combine the results of all trials to make an overall conclusion, individually they did all show some, albeit moderate, benefit for venlafaxine in treating neuropathic pain. Usually this benefit was achieved at doses of 75 to 225 mg per day. Known side effects of venlafaxine, including sleepiness, dizziness, and mild gastrointestinal problems, were reported by some studies, but were not particularly problematic.
Overall, there is currently an inadequate amount of information available to warrant any change in current prescribing practice and we cannot recommend venlafaxine as a first‐line treatment for neuropathic pain. However, it is a reasonably well‐tolerated drug and may be of some benefit in people who cannot tolerate other antidepressants or anticonvulsant drugs that are more widely prescribed to people with neuropathic pain. Larger clinical trials may provide more robust evidence for the effectiveness of venlafaxine in treating neuropathic pain.
Background
We have based the protocol for this review on a template for reviews of drugs used to relieve neuropathic pain. The aim is for all reviews to use the same methods, based on new criteria for what constitutes reliable evidence in chronic pain (Moore 2010a; Appendix 1).
Description of the condition
Neuropathic pain, unlike nociceptive pain, is caused by nerve damage, often accompanied by changes in the central nervous system (CNS) (Iannetti 2005). It is by nature chronic and may be present for months or years. The 2011 definition of neuropathic pain is "pain caused by a lesion or disease of the somatosensory system" (Jensen 2011). Many people with neuropathic pain are significantly disabled, with moderate or severe pain for many years.
In primary care in the UK, the incidences per 100,000 person‐years' observation were reported as 28 (95% confidence interval (CI) 27 to 30) for postherpetic neuralgia, 27 (95% CI 26 to 29) for trigeminal neuralgia, 0.8 (95% CI 0.6 to 1.1) for phantom limb pain, and 21 (95% CI 20 to 22) for painful diabetic neuropathy (Hall 2008). Estimates vary between studies, often because of small numbers of cases. The incidence of trigeminal neuralgia has been estimated at 4 in 100,000 per year (Katusic 1991; Rappaport 1994), while more recently a study of facial pain in The Netherlands found incidences per 100,000 person‐years of 12.6 for trigeminal neuralgia and 3.9 for postherpetic neuralgia (Koopman 2009). A systematic review of chronic pain demonstrated that some neuropathic pain conditions, such as painful diabetic neuropathy, can be more common, with prevalence rates up to 400 per 100,000 person‐years (McQuay 2007). The prevalence of neuropathic pain was reported as being 3.3% in Austria (Gustorff 2008), 6.9% in France (Bouhassira 2008), and as high as 8% in the UK (Torrance 2006). Some forms of neuropathic pain, such as diabetic neuropathy, chemotherapy‐related neuralgia, and postsurgical chronic pain (which is often neuropathic in origin) are increasing (Hall 2008). Although incidence rates for neuropathic pain have not been accurately reported in Ireland, results from the PRIME study (Prevalence, Impact and Cost of Chronic Pain) suggest a prevalence of 36% for chronic pain in the community setting (Raftery 2011). It is likely that a significant proportion of these people have pain of neuropathic origin.
Neuropathic pain is difficult to treat effectively, with only a minority of individuals experiencing a clinically relevant benefit from any one intervention. Thus, it constitutes a significant burden on healthcare systems and society at large, as well as being distressing for individual patients (Moore 2014). A multidisciplinary approach is now advocated, with pharmacological interventions being combined with physical or cognitive interventions, or both. Conventional analgesics are usually not effective, although opioids may be in some people. However, the risks of using opioids chronically to control pain tend to outweigh their benefits. Other people may derive some benefit from topical lidocaine patches or topical capsaicin. Treatment is more usually by so‐called 'unconventional' analgesics, such as antidepressants or antiepileptics.
Description of the intervention
The antidepressant agent venlafaxine is a serotonin reuptake inhibitor and weak noradrenaline reuptake inhibitor. It is used in the treatment and prevention of recurrence of major depressive disorder, as well as in the treatment of generalised anxiety disorder, social anxiety disorder, panic disorder, and agoraphobia. Although not licensed in Ireland (or the UK) for the treatment of chronic or neuropathic pain, it is commonly used for these indications. The drug is available as prolonged‐release capsules (37.5, 75, and 150 mg) suitable for once daily dosing. For the treatment and prevention of depression, the recommended starting dose for prolonged‐release venlafaxine is 75 mg given once daily. People not responding to the initial 75 mg/day dose may benefit from dose increases up to a maximum dose of 375 mg/day. Common adverse effects include nausea, dizziness, drowsiness, and dry mouth. As with other serotonergic agents, serotonin syndrome, a potentially life‐threatening condition, may occur as an adverse effect of venlafaxine treatment, particularly with concomitant use of other agents that may affect the serotonergic neurotransmitter system. Suicide‐related behaviours, mydriasis (dilated pupils), and dose‐related increases in blood pressure and heart rate have also been reported with venlafaxine.
How the intervention might work
Venlafaxine is a chimeric compound and both R‐ and S‐enantiomers, and their O‐desmethylated metabolites, are reported to mediate inhibition of serotonin and noradrenaline reuptake (Bolden‐Watson 1993; Muth 1986; Muth 1991). Its relatively clean pharmacological profile means that venlafaxine has a favourable adverse effect profile in comparison to other antidepressants used in pain management, especially the tricyclic antidepressants such as amitriptyline. The major metabolite, R‐O‐desmethylvenlafaxine, has been reported as the most potent inhibitor of both noradrenaline and serotonin reuptake (Muth 1991). In 2010, this metabolite (desvenlafaxine) was approved by the US Food and Drug Administration as a treatment for major depressive disorder (Seo 2010). Relating steady‐state concentration of venlafaxine to in vitro reuptake inhibitory concentration suggests that serotonin reuptake is maximal at low doses (less than 100 mg/day), whereas noradrenaline reuptake increases over the dose range of 100 to 375 mg/day. Inhibition of both serotonin and noradrenaline reuptake is thought to be important for the antidepressant activity of venlafaxine. It is possible that similar mechanisms underlie any analgesic activity of venlafaxine, since both noradrenaline and serotonin are primary neurotransmitters in the inhibitory descending pain pathways of the locus coeruleus and Raphe nucleus, respectively. However, direct demonstrations of the mechanism of action of venlafaxine in the treatment of neuropathic pain are lacking and it is likely that the mechanism(s) underlying its analgesic effect differ from the mechanisms underlying its antidepressant effect. For example, in animal experiments, venlafaxine‐induced analgesia was reported to be mediated via adrenergic mechanisms and via the κ and δ opioid receptors (Schreiber 1999). It is important to note that there tends to be little or no correlation between the effect of antidepressants on mood and pain in humans and that antidepressants can produce analgesia in people with and without depression, which further supports the notion that distinct mechanisms of action may underlie these effects (Onghena 1992). Furthermore, antidepressant‐induced pain relief typically emerges more rapidly and at a lower dose than the antidepressant effect, which often takes up to six weeks.
Why it is important to do this review
Venlafaxine is a reasonably well‐tolerated antidepressant and has been used as a pharmacological intervention for chronic neuropathic pain, although it is not a first‐line therapy at present. An update to an earlier Cochrane review of antidepressants for neuropathic pain reported preliminary evidence for the effectiveness of venlafaxine in relieving pain in polyneuropathy (Saarto 2007). In animal studies, venlafaxine relieved thermal hyperalgesia in rats with an induced mononeuropathy (Lang 1996). Similarly in human experimental studies, venlafaxine reduced the threshold at which repetitive electrical stimulation showed pain summation (Enggaard 2001). Taken together and considering the current lack of effective treatments for neuropathic pain with favourable risk‐benefit ratios, there is good justification for conducting a comprehensive systematic review to establish whether an evidence base exists for the clinical use of venlafaxine in neuropathic pain management. This review will be one of several updates to the Saarto 2007 review of antidepressants in neuropathic pain. It will apply more stringent criteria for validity and may include new studies that have emerged since the 2007 update by Saarto and Wiffen.
The standards used to assess evidence in chronic pain trials have changed substantially since 2008, because of an improved awareness of quality issues and the emergence of new recommendations by the Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT; Dworkin 2008), which is based on original work by Farrar 2001. Particular attention is now being paid to trial duration, withdrawals, and statistical imputation following withdrawal, all of which can substantially alter estimates of efficacy. The most important change is the move from using mean pain scores, or mean change in pain scores, to the number of people who have a large decrease in pain (by at least 50%); this level of pain relief correlates with improvements in co‐morbid symptoms, function, and quality of life. These standards are set out in the reference guide for pain studies (AUREF 2012).
This Cochrane review assessed evidence in ways that make both statistical and clinical sense, and used developing criteria for what constitutes reliable evidence in chronic pain (Moore 2010a). Trials included and analysed were required to meet a minimum of reporting quality (blinding, randomisation), validity (duration, dose and timing, diagnosis, outcomes, etc), and size (ideally at least 500 participants in a comparison in which the number needed to treat for an additional beneficial outcome (NNTB) is four or above (Moore 1998). This sets high standards and marks a departure from how reviews have been done previously.
Objectives
To assess the analgesic efficacy of, and the adverse effects associated with the clinical use of, venlafaxine for chronic neuropathic pain in adults.
Methods
Criteria for considering studies for this review
Types of studies
We included studies that were randomised controlled trials (RCTs) with double‐blind assessment of participant outcomes following two weeks of treatment or longer, though the emphasis of the review was intended to be on studies of eight weeks or longer. We required full journal publication, with the exception of online clinical trial results summaries of otherwise unpublished clinical trials and abstracts with sufficient data for analysis. We did not include short abstracts (usually meeting reports). We excluded studies that were non‐randomised, studies of experimental pain, case reports, and clinical observations.
Types of participants
All studies included adults aged 18 years and above. We considered studies if they had participants with one or more of a wide range of chronic neuropathic pain conditions including:
painful diabetic neuropathy;
postherpetic neuralgia;
trigeminal neuralgia;
phantom limb pain;
postoperative or traumatic neuropathic pain;
cancer‐related neuropathy;
chemotherapy/radiotherapy‐induced neuralgia;
atypical facial pain;
complex regional pain syndrome;
human immunodeficiency virus (HIV) neuropathy; and
spinal cord injury.
We included studies of participants with more than one type of neuropathic pain; in such cases, we planned to analyse results according to the primary condition.
Types of interventions
Venlafaxine at any dose, by any route, administered for the relief of neuropathic pain and compared with placebo or any active comparator.
Types of outcome measures
Studies used a variety of outcome measures, with the majority of studies using standard subjective scales (numerical rating scale (NRS) or visual analogue scale (VAS)) for pain intensity (VAS‐PI) or pain relief (VAS‐PR), or both. We were particularly interested in Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) definitions for moderate and substantial benefit in chronic pain studies (Dworkin 2008). These are defined as at least 30% pain relief over baseline (moderate), at least 50% pain relief over baseline (substantial), much or very much improved on Patient Global Impression of Change (PGIC) (moderate), and very much improved on PGIC (substantial). These outcomes concentrate on dichotomous outcomes where pain responses do not follow a normal (Gaussian) distribution. People with chronic pain desire high levels of pain relief, ideally more than 50%, and with pain not worse than mild (O'Brien 2010).
We planned to include a 'Summary of findings' table as set out in the author guide (AUREF 2012), but there were insufficient data.
Primary outcomes
Participant‐reported pain relief of 30% or greater.
Participant‐reported pain relief of 50% or greater.
Participant‐reported global impression of change (PGIC) much or very much improved.
Participant‐reported global impression of change (PGIC) very much improved.
Secondary outcomes
Any pain‐related outcome indicating some improvement.
Withdrawals due to lack of efficacy.
Participants experiencing any adverse effect.
Participants experiencing any serious adverse effect.
Withdrawals due to adverse effects.
Specific adverse effects, particularly somnolence and dizziness.
Search methods for identification of studies
Electronic searches
We searched the following databases with a start date of January 1990, because there are no earlier clinical trials of venlafaxine, which was developed in the late 1980s. Since the earliest clinical trials investigated its antidepressant activity, we did not miss any relevant studies by restricting the date of our searches to post‐1990.
Cochrane Central Register of Controlled Trials (CENTRAL) via The Cochrane Library 2014, Issue 8 of 12;
MEDLINE (via Ovid), January 1990 to 14 August 2014;
EMBASE (via Ovid), January 1990 to 14 August 2014.
Appendix 2 shows the search strategies. We applied no language restrictions. We tailored the searches to the individual databases.
Searching other resources
We reviewed the bibliographies of any RCTs identified and review articles, contacted authors of one excluded study, and searched www.clinicaltrials.gov to identify additional published or unpublished data. We also searched the meta‐Register of controlled trials (mRCT) (www.controlled‐trials.com/mrct) and the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch/) for ongoing trials but did not find any relevant trials.
Data collection and analysis
Selection of studies
We determined eligibility by reading the abstract of each study identified by the search. We eliminated studies that clearly did not satisfy the inclusion criteria and obtained full copies of the remaining studies. Three review authors (HCG, RMG, and MCH) read these studies independently and agreed on the study selection. We did not anonymise the studies before this assessment.
Data extraction and management
Three review authors (HCG, RMG, and MCH) independently extracted data using a standard form and checked for agreement before entry into Review Manager 5 (RevMan 2012). We included information about the pain condition and number of participants treated, drug and dosing regimen, study design (placebo or active control), study duration and follow‐up, analgesic outcome measures and results, withdrawals, and adverse effects (participants experiencing any adverse effect or serious adverse effect).
Assessment of risk of bias in included studies
We used the Oxford Quality Score as the basis for inclusion (Jadad 1996), limiting inclusion to studies that were randomised and double‐blind as a minimum. We also used the 'Risk of bias' tool to assess the likely impact on the strength of the evidence of various study characteristics relating to methodological quality (randomisation, allocation concealment, blinding, freedom from selective reporting), study validity (duration, outcome reporting, and handling of missing data), and size (Moore 2010b).
Three review authors (HCG, MCH, and RMG) independently assessed the risk of bias for each study, using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), and adapted from those used by the Cochrane Pregnancy and Childbirth Group, with any disagreements resolved by discussion. We assessed the following for each study:
Random sequence generation (checking for possible selection bias). We assessed the method used to generate the allocation sequence as: low risk of bias (any truly random process, eg random number table; computer random number generator); unclear risk of bias (method used to generate sequence not clearly stated). We planned to exclude studies that used a non‐random process (eg odd or even date of birth; hospital or clinic record number), but this did not arise.
Allocation concealment (checking for possible selection bias). The method used to conceal allocation to interventions prior to assignment determines whether intervention allocation could have been foreseen in advance of, or during, recruitment, or changed after assignment. We assessed the methods as: low risk of bias (eg telephone or central randomisation; consecutively numbered, sealed, opaque envelopes); unclear risk of bias (method not clearly stated). We excluded studies that did not conceal allocation (eg open list).
Blinding of outcome assessment (checking for possible detection bias). We assessed the methods used to blind study participants and outcome assessors from knowledge of which intervention a participant received. We assessed these methods as: low risk of bias (study stated that it was blinded and described the method used to achieve blinding, eg identical tablets; matched in appearance and smell); unclear risk of bias (study stated that it was blinded but did not provide an adequate description of how it was achieved). We excluded studies that were not, or appeared not to be, double‐blinded if we were unable to confirm double‐blinding by contact with the authors.
Incomplete outcome data (checking for possible attrition bias due to the amount, nature, and handling of incomplete outcome data). We assessed the methods used to deal with incomplete data as: low risk (less than 10% of participants did not complete the study, used 'baseline observation carried forward' analysis, or both); unclear risk of bias (used 'last observation carried forward' (LOCF) analysis); high risk of bias (used 'completer' analysis).
Size of study (checking for possible biases confounded by small size). We assessed studies as being at low risk of bias (200 or more participants per treatment arm); unclear risk of bias (50 to 199 participants per treatment arm); high risk of bias (fewer than 50 participants per treatment arm).
Measures of treatment effect
We planned to calculate the NNTB as the reciprocal of the absolute risk reduction (ARR) (McQuay 1998). For unwanted effects, the NNTB becomes the number needed to treat for an additional harmful outcome (NNTH) and is calculated in the same manner. We planned to use dichotomous data to calculate risk ratio (RR) with 95% confidence intervals (CI) using a fixed‐effect model, unless we found significant statistical heterogeneity (see Assessment of heterogeneity). We did not intend to use continuous data in analyses because it is inappropriate where there is an underlying skewed distribution.
Unit of analysis issues
We accepted randomisation at the level of individual participants only and not at the level of treatment site. We split the control treatment arm between active treatment arms in a single study if the active treatment arms were not combined for analysis. For cross‐over studies, we intended to use data from only the first period if these were available and to treat the study as a parallel study. However, if only combined data for both periods were reported, we drew attention to the possible bias associated with interpretation of this data.
Dealing with missing data
We planned to use intention‐to‐treat (ITT) analysis where the ITT population consisted of participants who were randomised, took at least one dose of the assigned study medication, and provided at least one post‐baseline assessment. We assigned missing participants to zero improvement.
Assessment of heterogeneity
We intended to deal with clinical heterogeneity by combining studies that examined similar conditions and to assess statistical heterogeneity visually (L'Abbé 1987), and with the use of the I2 statistic. However, this was not possible due to the small number of included studies.
Assessment of reporting biases
The aim of this review was to use dichotomous data of known utility (Moore 2010b). The review did not depend on what authors of the original studies chose to report or not, though clearly difficulties did arise in studies failing to report any dichotomous results. We extracted and used continuous data, which probably poorly reflect efficacy and utility, for illustrative purposes only.
We planned to assess publication bias using a method designed to detect the amount of unpublished data with a null effect required to make any result clinically irrelevant (usually taken to mean an NNTB of 10 or higher) (Moore 2008).
Data synthesis
We planned to use a fixed‐effect model for meta‐analysis, or a random‐effects model if there was significant clinical heterogeneity and it was considered appropriate to combine studies.
We analysed data for each painful condition in three tiers, according to outcome and freedom from known sources of bias.
The first tier intended to use data meeting current best standards, where studies report the outcome of at least 50% pain intensity reduction over baseline (or its equivalent), without the use of LOCF or other imputation methods for drop‐outs, report an ITT analysis, last eight or more weeks, have a parallel‐group design, and have at least 200 participants (preferably at least 400) in the comparison (Moore 1998; Moore 2010a; Moore 2012). We intended to report these top‐tier results first.
The second tier intended to use data from at least 200 participants, but where one or more of the conditions of the first tier was not met (eg reporting at least 30% pain intensity reduction, using LOCF or a completer analysis, or lasting four to eight weeks).
The third tier of evidence relates to data from fewer than 200 participants, or where there were expected to be significant problems because, for example, of very short‐duration studies of less than four weeks, where there was major heterogeneity between studies, or where there were shortcomings in allocation concealment, attrition, or incomplete outcome data. For this third tier of evidence, no data synthesis is reasonable, and may be misleading, but an indication of beneficial effects might be possible.
Subgroup analysis and investigation of heterogeneity
We planned to undertake subgroup analysis for:
dose of venlafaxine;
different painful conditions.
Sensitivity analysis
This was not possible due to a small evidence base.
Results
Description of studies
This review identified 14 clinical trials of venlafaxine in neuropathic pain, of which six met the criteria for inclusion. These six studies reported on 460 participants.
Results of the search
In total, the search identified 48 reports (after duplicates were removed) using the search criteria. However, many of these were small case reports, were of insufficient treatment duration, were open‐label studies, or involved other pain syndromes. As such, after screening titles and abstracts, only six studies met the inclusion criteria for analysis (Figure 1).
1.

Study flow diagram.
Included studies
The included studies comprised four cross‐over trials (Forssell 2004; Jia 2006; Sindrup 2003; Tasmuth 2002), and two parallel trials (Rowbotham 2004; Yucel 2005). Doses used included 25 mg twice daily and 75, 150, and 225 mg once daily. The largest study was Rowbotham 2004 with 245 participants. Three studies only reported mean data (Forssell 2004; Jia 2006; Tasmuth 2002). The earlier umbrella review did not include Jia 2006 (Saarto 2007), but overall we did not find any more recent trials. Similarly, that previous umbrella Cochrane review included Simpson 2001, but we excluded it due to small participant numbers.
Study participants were all over 18 years of age. Not all studies provided an age range, so the upper age limit was unclear but the mean age of participants taking venlafaxine ranged from 48 to 59 years and in all studies venlafaxine and control arms were age‐matched and sex‐matched appropriately.
Excluded studies
Although included in a previous Cochrane review (Saarto 2007) and another review (Lee 2010), Reuben 2004 was retracted due to falsification of data and is therefore excluded here. Durand and colleagues reported results of the EFFOX trial in which venlafaxine appeared to be effective in reducing oxiplatin‐induced neurosensory toxicity. However, the duration of treatment (10 days) was too short for inclusion in our analysis (Durand 2012). Similarly, Amr 2010 reported some benefit for venlafaxine in treating chronic postmastectomy pain, but the duration of treatment was only 10 days and so we excluded this study. Although Simpson 2001 reported the effectiveness of venlafaxine in addition to gabapentin in diabetic neuropathy, only seven participants were included in the venlafaxine arm and so we excluded this study as 10 per treatment group is the minimum we have considered.
Risk of bias in included studies
As summarised in Figure 2 and Figure 3, the risk of bias was considerable overall in this systematic review and all studies had some shortcomings in this regard.
2.
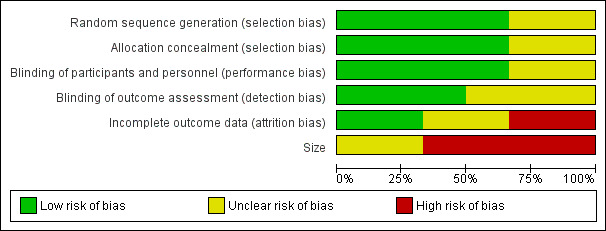
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
All studies that met the inclusion criteria were described as randomised. Selection bias was assessed by reviewing details of the randomisation of participants to study groups and the allocation concealment procedures. While four of the studies described all aspects of selection and concealment adequately and appeared to have a low risk of selection bias (Forssell 2004; Jia 2006; Sindrup 2003; Tasmuth 2002), neither Rowbotham 2004 nor Yucel 2005 described this properly, and we deemed their risk of selection bias unclear.
Blinding
Four of the included studies described the blinding procedure in detail so that it was clear that participants and investigators were unable to distinguish between active and control groups (Forssell 2004; Jia 2006; Rowbotham 2004; Sindrup 2003). These four studies were deemed to present a low risk of performance and detection bias. However, Tasmuth 2002 and Yucel 2005 did not describe how their studies were blinded, although they did mention that their studies were double‐blinded. Therefore, the risk of bias was unclear for these two studies.
Incomplete outcome data
Only two of the six included studies had a low risk of attrition bias, since for both trials more than 90% of participants completed the trial (Jia 2006; Yucel 2005). Both Tasmuth 2002 and Forssell 2004 had a greater than 10% drop‐out rate and only reported data for participants who completed trials and, therefore, we assessed them as being at high risk of bias. Rowbotham 2004 and Sindrup 2003 were of intermediate or unclear risk of attrition bias. In both studies, more than 10% of participants did not complete the trial and LOCF analysis was employed.
Selective reporting
While all studies reported the outcomes specified in their methods sections, these were not usually our preferred/primary outcome measures. Three of the included studies only reported mean data and did not provide an indication of how many participants improved according to our primary outcome measures (Forssell 2004; Jia 2006;Tasmuth 2002).
Other potential sources of bias
Although all included studies met the inclusion criterion of more than 10 participants randomised per treatment arm, lack of scale was a significant source of bias for all of them. Only Rowbotham 2004 had more than 200 participants in total enrolled on the trial, but it still had fewer than 200 per treatment arm and, therefore, we considered it, along with Jia 2006, at unclear risk of bias on the basis of size. Four of the included studies had fewer than 50 participants per treatment arm and we considered them at high risk of bias on the basis of size (Forssell 2004; Sindrup 2003; Tasmuth 2002; Yucel 2005).
Effects of interventions
First‐tier and second‐tier evidence of efficacy
We found no first‐tier or second‐tier evidence of efficacy. Of the six included studies, five had fewer than 200 participants, and the largest study employed LOCF analysis and was only of six weeks' duration (Rowbotham 2004). While small size was the primary problem with the included studies, short duration was also a major issue with only Yucel 2005 reporting on the basis of eight weeks' treatment and none of the studies was of longer than eight weeks' duration. Problems with allocation concealment, attrition, or incomplete outcome data also marred most studies and led to a significant risk of bias (Figure 2; Figure 3).
Third‐tier evidence of efficacy
Rowbotham 2004 recruited 245 adults with painful diabetic neuropathy who had experienced at least moderate neuropathic pain for at least three months and intended to treat 244 of those participants. Of these, 242 were treated and 202 completed the study. During the baseline two‐week period, their pain intensity was measured as at least 40 on the 0 to 100 VAS scale. Following randomisation to placebo, venlafaxine 75 mg, or venlafaxine 150 to 225 mg groups, their pain intensity and pain relief was measured using the VAS scales at week six. There was a significant reduction in pain intensity and a significant increase in pain relief with the higher dose of venlafaxine versus both placebo and lower dose of venlafaxine, but there were no significant differences in the frequency of adverse drug reactions (ADRs), serious ADRs, or withdrawals due to ADRs.
Importantly, the percentage of participants considered to have responded to treatment, having achieved at least a 50% reduction from baseline on the VAS‐PI assessment, was significantly greater in the venlafaxine 150 to 225 mg group (56% of participants) compared with the placebo group (34% of participants) at week six. Furthermore, the overall reductions in pain intensity scores were 27% for placebo, 32% for venlafaxine 75 mg, and 50% for venlafaxine 150 to 225 mg, with only the higher dose of venlafaxine differing significantly from placebo. The NNTB was 4.5 for the higher dose of venlafaxine. Higher‐dose venlafaxine was also significantly better than placebo at mediating pain relief (VAS‐PR) after six weeks of treatment and it was superior to both venlafaxine 75 mg and placebo on the clinician‐rated Clinical Global Impressions ‐ Improvement (CGI‐I) and Clinical Global Impressions ‐ Severity (CGI‐S) items at week six.
Sindrup 2003 conducted a placebo‐controlled study in which venlafaxine 225 mg was compared with imipramine 150 mg in 40 participants with polyneuropathy. Fifteen of the participants for whom data were presented had diabetes‐related polyneuropathy. Both venlafaxine and imipramine were effective in producing at least moderate pain relief versus placebo, but notably there was no statistically significant difference between the two drugs. At least moderate pain relief was reported in 8/30 participants taking venlafaxine compared with 2/29 participants in the placebo arm of the study. This trial was stopped early due to insufficient supply of drugs. This was the only study that reported screening participants pharmacogenetically, such that those with low metabolism of sparteine were excluded.
In one placebo‐controlled, cross‐over study of venlafaxine (dose up to 75 mg/day) in 30 participants with atypical facial pain, there was no significant reduction in pain intensity for venlafaxine, although there was a significant improvement in pain relief on the participant‐reported verbal rating scale (VRS) (Forssell 2004). The authors also reported reduced use of escape medication (nonsteroidal anti‐inflammatory drugs and paracetamol) in the period when participants were taking venlafaxine versus placebo.
Jia 2006 compared venlafaxine and carbamazepine in a randomised, parallel trial of 132 participants with painful peripheral diabetic neuropathy, with diagnosis being confirmed by abnormal nerve conduction tests. In this double‐dummy design study, venlafaxine was dosed twice daily and the total daily dose was 50 mg. There was no placebo arm in this trial and only mean data were reported. Although not stated, the twice daily dosing probably reflected the use of an immediate‐release formulation versus extended‐release formulations of venlafaxine that are designed for once‐daily dosing. Although there was a significant reduction in pain intensity in both groups at five, seven, 10, and 14 days compared with their baseline scores, venlafaxine was superior to carbamazepine at reducing pain intensity at all time points by per‐protocol analysis, with an apparent reduction from 6.8 to 2.2 in mean pain intensity after 14 days of venlafaxine, measured on an 11‐point scale. Venlafaxine was also superior to carbamazepine at improving quality of life, measured as improved sleep, routine work, and mood.
Tasmuth 2002 conducted a randomised, double‐blind cross‐over trial comparing venlafaxine and placebo in 15 women with neuropathic pain following breast cancer treatment. The treatment duration was four weeks per arm with a two‐week washout period between treatments and the dose of venlafaxine was escalated from 18.75 mg/day to a maximum of 37.5 mg (two participants) or 75 mg (11 participants). Primary outcome measures were current pain intensity during the last three days of the maximum tolerated dose (VAS‐PI and verbal rating scale ‐ pain intensity (VRS‐PI)) but these did not differ between treatment arms. Notably, there was a strong placebo effect. Using a computer program to register typical pain intensity in a diary format (VRS 0 to 7), the authors reported that 11 of the 13 participants who completed the trial had a least 50% pain relief and that this was significantly higher than pain relief reported in the placebo group. Notably, the 8‐point VRS used for current pain and the 5‐point VRS for current pain relief were non‐standard. Anxiety and depression scores were unaffected by either venlafaxine or placebo.
Yucel 2005 performed a randomised double‐blind, placebo‐controlled parallel trial with 60 participants with neuropathic pain assigned to venlafaxine 75 mg, venlafaxine 150 mg, or placebo for eight weeks. Sixteen of the 60 participants had diabetic polyneuropathy but results were not stratified according to type of neuropathic pain. A major focus of this trial was effects of venlafaxine on experimentally induced pain, which was not of interest to us. However, effects on ongoing pain intensity (VAS 0 to 10), activities of daily living, adverse effects, and global efficacy and tolerance were also reported. Although VAS‐PI scores decreased significantly from baseline in all participant groups, they did not differ between treatment arms. Since only mean data were reported, it was impossible to estimate the proportion of participants achieving moderate (or better) pain relief. In terms of global efficacy and tolerance, there was no difference between groups.
Adverse effects
All studies included in this review made some mention of adverse effects, but reporting of these was not standardised across studies and it was impossible to perform a meta‐analysis of this data. In general, venlafaxine was well tolerated and most adverse effects were minor.
Forssell 2004 asked participants about 10 adverse effects (urination difficulties, fatigue, appetite, dry mouth, constipation, sweating, nightmares, nausea, headache, and palpitations) and asked participants to self evaluate the overall severity of these adverse effects on a 100‐mm VAS. Overall, the incidence of adverse effects was similar in the venlafaxine and placebo periods but participants reported more severe dry mouth and sweating while taking venlafaxine compared with placebo.
Jia 2006 reported 29 adverse effects in the venlafaxine group, with these affecting 43.9% of participants. Common adverse effects were reported as those occurring in more than 10% of participants and these included mild gastrointestinal discomfort, dizziness, and somnolence. However, as this trial was not placebo‐controlled, it is difficult to draw a definitive conclusion. Severe gastrointestinal disturbance occurred in one participant on venlafaxine.
Rowbotham 2004 reported treatment‐emergent adverse effects in 75% of participants on placebo, 88% of participants on venlafaxine 75 mg, and 89% of participants on venlafaxine 150 to 225 mg. Nausea, dyspepsia, sweating, and somnolence were the most commonly reported and all of these were significantly more frequent in at least one of the venlafaxine groups versus placebo. The same authors reported that 10% of placebo, 9% of venlafaxine 75 mg and 12% of venlafaxine 150 to 225 mg groups had a serious adverse effect, although their precise nature was unclear. They also reported that 7/162 participants who commenced venlafaxine had clinically important electrocardiograph (ECG) changes over the course of their treatment.
Sindrup 2003 asked participants to rate adverse effects as none, slight, bothersome, or unacceptable. However, adverse‐effect ratings were not different between the venlafaxine, imipramine, and placebo arms of this study. They also reported incidence rates of specific adverse effects and suggested that venlafaxine was less well tolerated than imipramine on the basis of withdrawals due to adverse effects, and that it was associated with a higher incidence of tiredness than either placebo or imipramine.
Tasmuth 2002 reported no differences in either the number or intensity of adverse effects between venlafaxine and placebo treatment arms. Participants were asked about the same 10 adverse effects as those in the Forssell 2004 study, and they recorded both responses and spontaneous reports of the adverse effects.
Yucel 2005 mentioned nausea, vomiting, somnolence, and dizziness as occurring more commonly in the venlafaxine 150 mg group, but, since data only consisted of participant reports of none, slight, bothersome, or unacceptable adverse effects, it was unclear how many participants experienced each of these adverse effects.
Withdrawals
Forssell 2004 reported 10 drop‐outs from 30 participants who had commenced their cross‐over study. Eight participants dropped out due to adverse effects and six of these did so while taking venlafaxine (five due to nausea and one due to fatigue). They withdrew two participants due to non‐compliance and one of these drop‐outs was reported as being due to lack of efficacy.
Yucel 2005 noted five withdrawals among 60 participants who started the trial, with all being due to adverse effects. Of interest, three of the five withdrawals were in the venlafaxine 150 mg group and one was from the venlafaxine 75 mg group, consistent with the earlier comment on the higher prevalence of adverse effects associated with the higher dose of venlafaxine.
In the study by Jia 2006, there was a relatively small drop‐out rate of less than 10% in the venlafaxine group, which reduced the attrition bias compared with other studies. Six participants withdrew from 66 who commenced on venlafaxine, and four of these withdrawals were due to adverse effects (one participant with severe gastrointestinal disturbance, one participant with palpitations, and two participants with moderate gastrointestinal disturbance).
Rowbotham 2004 reported relatively high discontinuation rates of 15% with placebo, 15% with venlafaxine 75 mg, and 22% with venlafaxine 150 to 225 mg and these discontinuation rates did not differ significantly between the groups. In addition, there was no difference between venlafaxine and placebo groups in the rate of drop‐outs due to adverse effects. Overall, 14 participants taking either venlafaxine 75 mg or 150 to 225 mg withdrew due to adverse effects and 30/163 participants on venlafaxine withdrew in total. Five participants on venlafaxine were withdrawn due to inadequate efficacy, three due to protocol violations, three due to other medical events, one due to a non‐medical event, three were lost to follow‐up, and one requested to withdraw for unclear reasons.
In the cross‐over study by Sindrup 2003, there were seven reported drop‐outs from 40 participants who commenced the study and one additional participant was excluded on the basis of unacceptably high tramadol consumption. Of the seven drop‐outs, four withdrew while taking venlafaxine due to nausea, vomiting, dizziness, tiredness, or combinations thereof. The other three participants withdrew while in the non‐venlafaxine arm.
Tasmuth 2002 reported two drop‐outs from 15 participants who started the study. One was due to non‐compliance and the other due to acute nausea, sweating, and headache that started on the first day of venlafaxine 18.75 mg plus 37.5 mg dose.
Discussion
Summary of main results
Overall, it was notable that all included studies showed some positive benefit for venlafaxine in neuropathic pain. Positive outcomes included reduced use of escape medication, such as paracetamol, and reductions in pain intensity scores of less than 50%. Notably, in the largest included study, the percentage of participants considered to have responded to treatment, having achieved at least a 50% reduction in pain intensity, was significantly greater in the venlafaxine 150 to 225 mg group (56% of participants) compared with the placebo group (34% of participants) (Rowbotham 2004). However, we did not deem this to constitute strong evidence for the efficacy of this intervention, since the study was subject to significant bias (Figure 3).
Overall completeness and applicability of evidence
Since the two largest of the included studies only looked at diabetes‐associated neuropathy (Jia 2006; Rowbotham 2004), and overall more than 80% of included participants had diabetes, it is possible that any benefits shown for venlafaxine may not be applicable to other painful neuropathic conditions. Other specific types of neuropathic pain for which the efficacy of venlafaxine was assessed were atypical facial pain (Forssell 2004), and postoperative/post‐chemotherapy neuralgia in people with breast cancer (Tasmuth 2002). However, both studies were hampered by very small numbers of participants. A further confounding factor was the complex interaction between depression and pain as venlafaxine is an established and effective antidepressant drug and not all studies measured depression at baseline or accounted for the fact that relief of depression may have a positive benefit on pain scores. Applicability of these trials was further hampered by their short duration since only one trial was of six weeks' duration (Rowbotham 2004), and none was longer than this. It is thought that studies of less than six weeks' duration may not accurately predict longer‐term drug efficacy in chronic conditions, as epitomised by neuropathic pain. Similarly, short‐term use of drugs may not accurately predict ADRs that emerge with longer‐term drug dosing in chronic illness.
Quality of the evidence
By current standards, the overall reporting quality was average to poor. Although all included studies were randomised and double‐blinded, none of them provided data that met pre‐defined criteria for first‐tier or second‐tier analysis. Only two studies had more than 50 participants per treatment arm and four studies were significantly smaller than that and therefore presented a high risk of bias on the basis of size. Five of six studies were of short duration and four were of cross‐over design. Only one study was both parallel in design and placebo‐controlled. All studies used completer analysis or LOCF analyses so that only those with greater than 90% completion were not at risk of attrition bias.
Potential biases in the review process
In accordance with most Cochrane reviews completed by the Pain, Palliative and Supportive Care Review Group (PaPaS), we included only randomised double‐blind studies in an effort to limit the potential for bias. However, there were several other sources of bias.
The absence of publication bias, whereby studies showing a lack of efficacy for venlafaxine are performed but not published, cannot be proven. However, this is an inherent weakness in all systematic reviews. Nonetheless, it is considered unlikely that we have missed any published evidence, since we carried out several broad searches and we believe it is unlikely that significant amounts of data eluded us in these searches.
NNTB estimates of efficacy in chronic pain tend to increase with duration of trials, meaning that participants tend to derive less benefit as trials proceed (Moore 2010a). This means that the short duration of all included studies is particularly problematic here and that these studies are likely to have significantly over‐estimated the efficacy of venlafaxine in treating chronic pain.
Agreements and disagreements with other studies or reviews
This review does not change the conclusion drawn in an earlier review (Saarto 2007). Moreover, current treatment guidelines for neuropathic pain in Europe and the USA do not specifically recommend prescription of venlafaxine. Another review of selective serotonin re‐uptake inhibitors (SSRIs) and serotonin and noradrenaline re‐uptake inhibitors (SNRIs) in neuropathic pain concluded that there was only modest evidence for the efficacy of SSRIs overall, but that the SNRIs (ie venlafaxine and duloxetine) were effective in the treatment of painful diabetic neuropathy and polyneuropathy (Lee 2010). Notably that review, although not conducted systematically, considered several of our included studies (Forssell 2004; Rowbotham 2004; Sindrup 2003; Tasmuth 2002; Yucel 2005), however it also included data from Reuben 2004 and Simpson 2001, which we excluded due to our more stringent inclusion criteria. In their general literature reviews, not conducted along Cochrane principles, Lee 2010 and Dharmshaktu 2012 also arrived at a similar conclusion to us that while SNRIs, including venlafaxine, may have some place in treating neuropathic pain, they should probably only be considered for use in people who have not responded to tricyclic antidepressants and anticonvulsants, or if there are contraindications to these better‐established drugs.
Another issue that may have accounted for heterogeneity in the clinical findings is that there is very little standardisation of medication history and usage in chronic pain trials. Some trials preclude those people taking other analgesics, anticonvulsants, or antidepressants during or preceding the trial, while others allow the use of escape medication for analgesia as well as other concurrent medications. This is highly likely to confound study findings and makes it more difficult to compare studies directly. Use of escape medication in pain trials is considered a secondary outcome measure and two of our included studies reported that venlafaxine reduced the usage of alternative analgesics during the study period, which gives some weak indication of potential efficacy in treating neuropathic pain.
Depression and anxiety scores were also reported in some, but not all, of the reviewed studies. Due to the complex nature of pain it is considered likely that clinical improvements in anxiety and depression may be accompanied by participant‐reported improvements in pain intensity and it can be difficult to separate these complex phenomena. Due to the small number of studies reviewed here, no clear pattern emerged relating the antidepressant efficacy of venlafaxine to its analgesic efficacy; however, it is a factor that should be considered in any review of antidepressants in pain management.
Authors' conclusions
Implications for practice.
Implications for people with neuropathic pain
This review is unlikely to change clinical practice in a manner that will improve outcomes for people with neuropathic pain. Our results suggested that, for the vast majority of people with neuropathic pain, there is insufficient evidence to support the use of a venlafaxine‐based intervention in the current standard of care.
Implications for clinicians
There is little compelling evidence to support the use of venlafaxine in neuropathic pain except by experienced clinicians in exceptional circumstances.
Implications for practice for policy makers
Any evidence for venlafaxine's benefit in treating neuropathic pain is not sufficiently robust to warrant any change in policy regarding its licensed indications.
Implications for practice for funders
Although this review found some evidence to support the use of venlafaxine in treating neuropathic pain, overall this evidence was of low quality. From a pharmacoeconomic perspective, there is insufficient evidence to support a change in reimbursement practice.
Implications for research.
General
Larger, prospective well‐designed studies are required to provide more definitive conclusions on the efficacy of venlafaxine for treating neuropathic pain. While we hope these will be performed, there is no guarantee that this will occur
Design
Double‐blind, randomised, placebo‐controlled trials with over 200 participants per treatment arm and treatment period of at least eight weeks would be more conclusive if further evidence for the efficacy of venlafaxine in treating neuropathic pain is to be obtained. We believe that there is little value in the conduct of future small trials or case series.
Measurement (endpoints)
Current best standards, where studies report the outcome of at least 50% pain intensity reduction over baseline (or its equivalent), without the use of last observation carried forward (LOCF) or other imputation methods for drop‐out, should be adopted to provide first‐tier evidence in future trials. Furthermore, in future clinical studies it would be most beneficial if mean data were not reported so that proper estimates can be made of the proportion of participants benefiting from venlafaxine treatment.
Other
Further research studies on the mechanism of action of venlafaxine in treating neuropathic pain are also warranted.
Feedback
Feedback received, 5 September 2015
Summary
From: cochrane_feedback@wiley.co.uk [mailto:cochrane_feedback@wiley.co.uk]
Sent: 05 September 2015 00:09
To: Cochrane Feedback
Subject: New Feedback has been submitted for CDSR
++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
Database: Cochrane Database of Systematic Reviews
Article: Venlafaxine for neuropathic pain in adults Review Group: SYMPT
DOI: 10.1002/14651858.CD011091.pub2
++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
Date of Submission: 04‐Sep‐2015
Name: Anna Maruyama (Pharm D Student), Aaron M Tejani (PharmD) Email Address: a.maruyama@alumni.ubc.ca
Affiliation: University of British Columbia
Comment: Dear Cochrane Review Team
“Venlafaxine for neuropathic pain in adults” by Gallagher et al (2015) is a thorough and well‐done systematic review. However, a challenge for readers is deciding how to change their practice based on this review. The current conclusion under the heading “implications for clinicians” is that “venlafaxine may be of benefit in treating neuropathic pain, [but that] this evidence is not sufficiently robust to promote a change in clinical prescribing practice”. This conclusion is vague and may in fact overestimate the effects of venlafaxine, as there were limitations in the methodology in the largest trial by Rowbotham (2004) that were not mentioned or appropriately assessed in this review.
One potential source of selection bias that is not assessed by the Cochrane’s Collaboration tool for assessing risk of bias is when a trial utilizes a run‐in period prior to randomization. In Rowbotham (2004) patients were administered physical, mental and neurological exams at screening (physical and mental exams were repeated prior to randomization to reconfirm eligibility) and placebo was given daily during the two‐week baseline data collection period. Once patients were randomized, those that were unable to reduce their analgesic use to one dose per day by the first day of double‐blind treatment were excluded. Selecting patients based on the run‐in period forces differential representation of certain subpopulations relative to others (Berger 2003). For example in this study, patients that had resistant or severe pain requiring more than one dose of analgesic would have been excluded. Generalizability is impacted if this type of selection bias is not considered, as efficacy for venlafaxine is accepted for a broader, unselected population. Rowbotham’s results may not be applicable for the clinician considering venlafaxine for a patient with severe neuropathic pain, and/or patients who require other analgesics.
Blinding of participants and personnel was assessed as low risk based on Rowbotham (2004) describing a “two‐bottle system”. We disagree with this assessment, as it was not explicitly stated in the study that the capsules looked identical. The capsule size of venlafaxine increases with increasing strength, and the colour of the capsules differ for each of the strengths. Patients were also given 2 or 3 capsules at the start of week 4 when doses were titrated. Blinding of participants could have been compromised if capsules were not of the same size, colour or quantity. As well, treatment‐emergent adverse events were higher in both the venlafaxine groups (88% in the venlafaxine ER 75mg and 89% in the venlafaxine ER 150‐225mg) compared to placebo (75%). Although these differences were not considered statistically significant, it is possible that participants may have correctly anticipated which treatment they received (and personnel may have guessed what patients were receiving) and blinding may not have been maintained throughout the study. Therefore, we believe the risk of bias for blinding of participants and personnel should have been assessed as unclear. Compromised blinding would have overestimated the effects of venlafaxine.
Incomplete outcome data was assessed as intermediate or unclear risk of attrition bias for the Rowbotham (2004) study due to a loss of more than 10% of participants and because last observation carried forward (LOCF) analysis was used. The assessment of attrition bias being intermediate or unclear is ambiguous. Using LOCF could have over or underestimated the effects of venlafaxine, as it was unclear when the patients were lost to follow‐up. We have tried contacting the authors for this information, and until a response is available we suggest that attrition bias for this study is assessed as unclear only.
Although all of the included studies were considered third tier evidence, we focussed our attention to Rowbatham (2004) because it was the largest, randomized, parallel designed study included in this review. The NNT reported for this study was 4.5 (for 50% pain reduction) for the higher dose venlafaxine group (150 to 225mg). However, this result was likely an overestimation due to the unclear risk of biases for all of the categories in the Cochrane’s risk of bias tool. As well, the selection bias introduced during the run‐in period makes it difficult to know whom these results really apply to. Are the results only for patients that are early responders to venlafaxine or those that have moderate neuropathic pain requiring only one dose of analgesic per day? We cannot be sure. An overestimated benefit of venlafaxine did not come without potential harm, as treatment emergent adverse events were higher in both the venlafaxine groups compared to placebo. Before clinicians can decide what to do with the conclusions provided under “implications for clinicians” that state “venlafaxine may be of benefit in treating neuropathic pain”, they need to consider all of the above points. Therefore, we suggest revising the conclusions to: When considering venlafaxine for the treatment of neuropathic pain, it is important to recognize that certain patient populations were not included in the studies, and that the NNT is likely to be higher than reported (for 50% pain reduction) in return for potential for harm.
Sincerely,
Anna Maruyama PharmD Student and
Aaron M Tejani PharmD
References:
1) Berger VW, Rezvani A, Makarewicz VA. Direct effect on validity of response run‐in selection in clinical trials. Control Clin Trials. 2003;24:156‐166.
2) Gallagher HC, Gallagher RM, Butler M, Buggy DJ, Henman MC. Venlafaxine for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2015, Issue 8. Art. No.: CD011091. DOI: 10.1002/14651858.CD011091.pub2
3) Rowbotham MC, Goli V, Kunz NR, Lei D. Venlafaxine extended release in the treatment of painful diabetic neuropathy: a double‐blind, placebo‐controlled study. Pain. 2004;110:697‐706
Reply
Response to Maruyama and Tejani, from Professor Andrew Moore
The authors of the review have been unable to respond, so I am making a response as an editor, and as someone who is deeply involved with Cochrane neuropathic pain reviews and the methods used in them.
Far from being a challenge to how venlafaxine might form part of practice, the authors conclusions in the Abstract make it clear: “We found little compelling evidence to support the use of venlafaxine in neuropathic pain”. That is the stance that they take throughout, and make it abundantly clear that the evidence available is at best only third tier.
Defined tiers of evidence are used in these reviews because there is often little evidence from large, robust, RCTs, and yet it is often useful to examine what evidence we have. Caution is the watchword, and the essence is to imbue comments with the appropriate caution. So third tier evidence is defined in the Methods section as:
“The third tier of evidence relates to data from fewer than 200 participants, or where there were expected to be significant problems because, for example, of very short‐duration studies of less than four weeks, where there was major heterogeneity between studies, or where there were shortcomings in allocation concealment, attrition, or incomplete outcome data. For this third tier of evidence, no data synthesis is reasonable, and may be misleading, but an indication of beneficial effects might be possible.”
Maruyama and Tejani have concerns over Rowbotham’s 2004 trial. For several reasons:
1 Enrichment during run in period
That might be a cause for concern if there were any evidence that selective enrichment short of complete enrichment made a difference. It does not, as was shown in a systematic review of the subject in neuropathic pain studies, and referenced by the authors (Straube S, Derry S, McQuay HJ, Moore RA. Enriched enrollment: definition and effects of enrichment and dose in trials of pregabalin and gabapentin in neuropathic pain. A systematic review. British Journal of Clinical Pharmacology 2008;66(2):266‐75).
2 Blinding of treatments
Admittedly, the arrangements for blinding in this trial were complex. But I have read the methods section through several times, and I can find no reason to question the blinding methods described. Maruyama and Tejani surmise that there may be circumstances in which that might happen, but that is the case in virtually any trial. Rowbotham and colleagues have enormous experience in clinical trials in neuropathic pain, and it would require a considerably greater index of suspicion to reject the trial. The authors of the Cochrane review give this trial a score of unclear risk on almost all risk of bias items, so it is not as if it is given any unjustified quality score.
3 Incomplete outcome data and imputation method
This topic has been the object of some very intense study in recent years, especially the potential for overestimation of treatment effect using LOCF [last observation carried forward]. We know that excess adverse event withdrawals combined with LOCF come with particular risk, but the excess in Rowbotham is small (see Moore RA, Straube S, Eccleston C, Derry S, Aldington D, Wiffen P, et al. Estimate at your peril: imputation methods for patient withdrawal can bias efficacy outcomes in chronic pain trials using responder analyses. Pain 2012;153(2):265‐8). This is factored into second and third tier evidence, and properly referenced.
Maruyama and Tejani suggest that the conclusions be changed, based on conjecture around methodological issues that are already properly addressed in the review, and to some extent going against current best evidence. This review is properly conservative about the value of venlafaxine in the treatment of neuropathic pain throughout, and is need of no change that I can see.
4 Additional comments
Maruyama and Tejani are concerned that the current conclusion under the heading “implications for clinicians” is that “venlafaxine may be of benefit in treating neuropathic pain, [but that] this evidence is not sufficiently robust to promote a change in clinical prescribing practice” is vague.
It may be more appropriate to describe the wording as nuanced, and perhaps appropriate. Cochrane reviews are not supposed to be dogmatic about how the evidence in them is to be used, and should not cross the boundary into making recommendations. This is sensible, because medical problems are almost never simple, and complicated by individual circumstances and the settings in which evidence might be used. In chronic pain particularly, many therapies are used without much if any evidence, as here with venlafaxine. That might make their use inappropriate in primary care, but at the same time their use may not be precluded in specialist care. There are many examples of exceptional patients responding to treatments with no evidence, and even to treatments with evidence of no effect.
For venlafaxine there is too little evidence to support its use, but also too little evidence to refute it. That viewpoint comes over strongly in the Abstract conclusion and the PLS, and also in Implications for people with neuropathic pain.
I think Maruyama and Tejani have a point about the wording of Implications for clinicians, which arguably is less strong. I therefore suggest a change in language, from:
“While there is some evidence that venlafaxine may be of benefit in treating neuropathic pain, this evidence is not sufficiently robust to promote a change in clinical prescribing practice.”
To match that in the Abstract conclusions:
“There is little compelling evidence to support the use of venlafaxine in neuropathic pain except by experienced clinicians in exceptional circumstances.”
Contributors
PaPaS Feedback Editor Kate Seers and CRG editorial team.
What's new
| Date | Event | Description |
|---|---|---|
| 1 June 2017 | Review declared as stable | See Published notes. |
History
Protocol first published: Issue 4, 2014 Review first published: Issue 8, 2015
| Date | Event | Description |
|---|---|---|
| 9 February 2016 | Amended | Spelling error in the name 'Maruyama' corrected in Feedback. |
| 4 February 2016 | Feedback has been incorporated | See Feedback. |
Notes
A restricted search in May 2017 did not identify any potentially relevant studies likely to change the conclusions. Therefore, this review has now been stabilised following discussion with the authors and editors. If appropriate, we will update the review if new evidence likely to change the conclusions is published, or if standards change substantially which necessitate major revisions.
Acknowledgements
HCG was supported by a Health Research Board of Ireland Cochrane Fellowship. The review authors thank Phil Wiffen, Andrew Moore, and Anna Hobson for invaluable assistance in the preparation of this manuscript.
Cochrane Review Group funding acknowledgement: The National Institute for Health Research (NIHR) is the largest single funder of the Cochrane PaPaS Group. Disclaimer: The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the NIHR, National Health Service (NHS), or the Department of Health.
Appendices
Appendix 1. Methodological considerations for chronic pain
There have been several changes in how efficacy of conventional and unconventional treatments is assessed in chronic painful conditions. The outcomes are now better defined, particularly with new criteria for what constitutes moderate or substantial benefit (Dworkin 2008); older trials may only report participants with "any improvement". Newer trials tend to be larger, avoiding problems from the random play of chance. Newer trials also tend to be longer, up to 12 weeks, and longer trials provide a more rigorous and valid assessment of efficacy in chronic conditions. New standards have evolved for assessing efficacy in neuropathic pain, and we are now applying stricter criteria for inclusion of trials and assessment of outcomes, and are more aware of problems that may affect our overall assessment. To summarise some of the more recent insights that must be considered in this new review
Pain results tend to have a U‐shaped distribution rather than a bell‐shaped distribution. This is true in acute pain (Moore 2011a; Moore 2011b), back pain (Moore 2010b), arthritis (Moore 2010c), and fibromyalgia (Straube 2010); in all cases, mean results usually describe the experience of almost no‐one in the trial. Data expressed as means are potentially misleading, unless they can be confirmed to be suitable.
Therefore, we have to depend on dichotomous results (the person either has or does not have the outcome) usually from pain changes or participant global assessments. The Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) group has helped with their definitions of minimal, moderate, and substantial improvement (Dworkin 2008). In arthritis, trials shorter than 12 weeks, and especially those shorter than eight weeks, overestimate the effect of treatment (Moore 2009a); the effect is particularly strong for less effective analgesics, and this may also be relevant in neuropathic‐type pain.
The proportion of people with at least moderate benefit can be small, even with an effective medicine, falling from 60% with an effective medicine in arthritis, to 30% in fibromyalgia (Moore 2009b; Moore 2010c; Straube 2008; Sultan 2008). A Cochrane review of pregabalin in neuropathic pain and fibromyalgia demonstrated different response rates for different types of chronic pain (higher in diabetic neuropathy and postherpetic neuralgia and lower in central pain and fibromyalgia) (Moore 2009b). This indicates that different neuropathic pain conditions should be treated separately from one another, and that pooling should not be performed unless there are good reasons for doing so.
Presently, unpublished individual participant analyses indicate that people who get good pain relief (moderate or better) have major benefits in many other outcomes, affecting quality of life in a significant way (Moore 2010d).
Appendix 2. Search strategies
Search strategy for CENTRAL via The Cochrane Library
1. MeSH descriptor Pain explode all trees
2. MeSH descriptor Somatosensory Disorders explode all trees
3. MeSH descriptor Peripheral Nervous System Diseases explode all trees
4. ((pain* or discomfort*) and (central or complex or rheumat* or muscl* or muscul* or myofasci* or nerv* or neuralg* or n europath*)):it,ab,kw
5. ((nerv* or neur*) and (compress* or damag*)):it,ab,kw
6. (1 or 2 or 3 or 4 or 5)
7. MeSH descriptor Venlafaxine, this term only
8. (venlafaxine or Effexor):it,ab,kw
9. 7 or 8
10. 6 and 9
11. Limit 10 to CENTRAL
Search strategy for MEDLINE via Ovid
1. exp PAIN
2. exp PERIPHERAL NERVOUS SYSTEM DISORDERS/
3. exp SOMATOSENSORY DISORDERS/
4. ((pain* or discomfor*) adj10 (central or complex or rheumat* or muscl* or nerv* or neuralgia* or neuropath*)).mp
5. ((neur* or nerv*) adj6 (compress* or damag*)).mp.
6. 1 or 2 or 3 or 4 or 5
7. Venlafaxine.mp.
8. 6 and 7
9. randomized controlled trial.pt.
10. controlled clinical trial
11. randomized
12. placebo.ab.
13. drug therapy.fs.
14. randomly.ab.
15. trial.ab
16. groups.ab
17. or/9‐16
18. exp animals/ not humans.sh.
19. 17 not 18
20. 19 and 8
Search strategy for EMBASE via Ovid
1. Venlafaxine/
2. (venlafaxine or Effexor).mp
3. 1 or 2
4. exp neuralgia
5. ((pain* or discomfort*) adj10 (central or complex or rheumat* or musc* or myofasci* or nerv* or neuralg* or neuropath*)).mp.
6. ((neur* or nerv*)adj6(compress* or damag*)).mp
7. 4 or 5 or 6
8. crossover procedure/
9. double‐blind procedure/
10. randomized controlled trial/
11. (random* or crossover* or cross‐over* or cross over* or placebo* or (doubl* adj blind*) or assign* or allocat*).tw.
12. or/8‐11
13. 3 and 7 and 12
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Forssell 2004.
| Methods | Double‐blind placebo‐controlled cross‐over trial. 2 x 4‐week treatment periods with a 2‐week washout | |
| Participants | 30 adults with atypical facial pain of intensity at least 3 on an 11‐point scale. Median age 52 years (range 38‐66 years). Gender data were reported for the 18 participants for whom data were analysed (6 men and 12 women). The study was conducted in Finland | |
| Interventions | Venlafaxine 37.5‐75 mg/day vs. placebo. All participants took 37.5 mg for 2 weeks and were then allowed to regulate the dose themselves. 17 took venlafaxine 75 mg for a second 2 weeks and 1 took 37.5 mg. Paracetamol and NSAIDs were also permitted as escape medications | |
| Outcomes | Participant‐reported VAS‐PI (0‐100), VRS‐PI (8‐point scale), VRS‐PR, and VAS‐PR (0‐50) were the primary outcome measures. VAS‐PI, VAS‐PI, and VRS‐PI did not differ significantly between groups. VRS‐PR reported as significantly greater during venlafaxine period vs. placebo. Increased use of escape medication in placebo groups vs. venlafaxine but not well described. No significant difference between groups in anxiety (STAI) or depression (BDI) | |
| Notes | 10 drop‐outs including 8 due to adverse effects. 6 in venlafaxine arm (nausea 5 and fatigue 1). 2 in placebo arm (rash 1 and dizziness 1). 2 participants excluded due to non‐compliance | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed by Wyeth‐Lederle, who supplied the drug and placebo capsules 'using computer‐generated numbers' |
| Allocation concealment (selection bias) | Low risk | Participants were allocated consecutively |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Identical capsules were supplied by a pharmaceutical company (Wyeth‐Lederle) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | 'The randomization code was not opened during the trial' and along with plasma concentration data were 'kept separate from investigators carrying out the assessments until the database was closed' |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Completer analysis was employed. Only 20/30 participants completed the trial |
| Size | High risk | < 50 participants per treatment arm |
Jia 2006.
| Methods | Double‐blind, parallel, randomised trial of 2 weeks' duration comparing venlafaxine with carbamazepine. A double‐dummy design was used | |
| Participants | 132 participants with painful diabetic neuropathy, confirmed by nerve conduction testing. 80 women and 52 men were included and there was no difference in gender between the groups. The study was conducted in China | |
| Interventions | Venlafaxine 25 mg twice daily (50 mg/day) vs. carbamazepine 100 mg twice daily (200 mg/day) for 2 weeks. Participants taking other analgesic drugs in the 5 days prior to the trial period were excluded, as were those with serious diabetic complications | |
| Outcomes | Primary outcome measurement was pain intensity measured on a numeric pain intensity scale (11 points) at days 0, 2, 5, 7, and 14. However, only mean data were reported and it is unclear how many participants benefited from treatment. Frequency of pain episodes was also recorded. Interference with quality of life, activities of daily living, mood, and sleeping were also assessed. Mean pain intensity was decreased by both carbamazepine and venlafaxine over time but venlafaxine was superior to carbamazepine in relieving pain. Venlafaxine also reported to be superior in improving sleep, mood, and total quality of life | |
| Notes | 13 participants did not complete the trial (6 venlafaxine; 7 carbamazepine). 6 withdrawals were due to adverse effects (4 venlafaxine ‐ severe gastrointestinal disturbance, moderate gastrointestinal disturbance (x 2), palpitations; 2 carbamazepine). In the venlafaxine group, the 2 other participants who withdrew were lost to follow‐up. The overall incidence of adverse effects was not significantly different between groups. Included ITT analysis in the trial design and had also intended to do per‐protocol analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | 'The random sequence was generated by computer' |
| Allocation concealment (selection bias) | Low risk | The random sequence was 'sealed in opaque envelopes' |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Dummy tablets were employed |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | It is stated that 'both the researchers and the patients did not know which groups the patient was belonging to before the end of the study'. Furthermore the authors stated 'and statistician will not know, until the statistical analysis is completed' |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | < 10% of participants did not complete the study |
| Size | Unclear risk | 50‐200 participants per treatment arm |
Rowbotham 2004.
| Methods | Multicentred, randomised, double‐blind, placebo‐controlled trial of 6 weeks' duration with dose escalation over first 2 weeks. Followed by 2‐week tapering period | |
| Participants | 245 participants with painful diabetic neuropathy of at least moderate severity for ≥ 3 months. All participants had metabolically stable diabetes (type I or type II). 97 women and 145 men were included in the 242 participants in the ITT population. There was no significant difference in gender balance between the treatment groups. The study involved participants from the USA | |
| Interventions | Placebo, venlafaxine 75 mg, or venlafaxine 150‐225 mg/day for 6 weeks, followed by 2‐week tapered dose reduction | |
| Outcomes | Primary outcome measures were VAS‐PI, VAS‐PR, CGI‐S and CGI‐I, all of which were assessed by clinicians > 50% pain relief obtained by 46/82 participants (venlafaxine 150‐225 mg (derived) and by 27/80 (placebo) with NNTB of 4.5. Participant‐reported global rating of pain relief was also included but not possible to evaluate due to lack of standard deviations |
|
| Notes | 42 withdrawals in total (12/81 placebo arm; 12/81 venlafaxine 75 mg arm; 18/82 venlafaxine 150‐225 mg arm). Of these, 3/81 placebo, 6/81 venlafaxine 75 mg, and 8/82 venlafaxine 150‐225 mg were due to adverse effects. NNTH was not significantly different. Included ITT analysis in its design | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not specified |
| Allocation concealment (selection bias) | Unclear risk | Not specified |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Authors described details of a 'two‐bottle system', which was used throughout the study |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Details of double‐blinding not provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | > 10% did not complete trial. Last observation carried forward analysis was employed |
| Size | Unclear risk | 50‐200 participants per treatment arm |
Sindrup 2003.
| Methods | Double‐blind, placebo‐controlled cross‐over design with 3 x 4‐week periods and 1‐week washout | |
| Participants | 40 participants with painful polyneuropathy of at least 6 months' duration. Mean age 56 years (range 31‐69 years). 23 men and 9 women were included in the data analysis. Participants were recruited in 2 Danish hospitals | |
| Interventions | Venlafaxine 75 mg/day in week 1, 150 mg/day in week 2, 225 mg/day in weeks 3 and 4, imipramine 50 mg/day in week 1, 100 mg/day in week 2, 150 mg/day in weeks 3 and 4 vs. placebo for 5 weeks | |
| Outcomes | Participant‐reported daily rating of 4 aspects of pain were summed (pain paroxysms, constant pain, touch, and pressure‐evoked pain ‐ all on 11‐point VAS). Patient's global impression of pain relief (complete, good, moderate, slight, none). Adverse effects and use of escape medication Complete/good pain relief: 9/29 imipramine; 2/29 placebo; 7/30 venlafaxine. Complete/good/moderate pain relief: 14/29 imipramine; 2/29 placebo; 8/30 venlafaxine. Authors report NNTB of 5.2 for at least moderate pain relief with venlafaxine, but did not provide raw data or confidence intervals. Use of escape medication (paracetamol) not significantly different |
|
| Notes | 1‐week baseline observations. People with diabetes (15 participants) more likely to gain clinically relevant pain relief than people without diabetes (17 participants). 33/40 participants completed all 3 arms of study. 7 withdrew due to adverse effects (1 imipramine; 2 placebo; 4 venlafaxine). 1 lost to follow‐up | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Authors stated that 'assignment to one of six possible treatment sequences was random via a computer‐generated randomization code' and that this 'randomization plan was generated by one author who was not involved in the conduct of the trial' |
| Allocation concealment (selection bias) | Low risk | Participants were numbered consecutively. Sealed envelopes were employed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | 'Double dummy technique was employed as venlafaxine, and its corresponding placebo capsules had a different appearance than imipramine and its corresponding placebo tablets' |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The randomisation plan was generated by an author who was not involved in the conduct of the trial and double‐blinding was maintained throughout |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | > 10% of participants did not complete the trial. Last observation carried forward analysis was employed |
| Size | High risk | < 50 participants per treatment arm |
Tasmuth 2002.
| Methods | Double‐blind, placebo‐controlled cross‐over trial of 4 weeks (4 weeks' dose titration to maximum tolerated dose). 2 x 4‐week periods with 2‐week washout and no carry over effect | |
| Participants | 15 women with breast cancer of mean age 55 years (range 37 to 72), all with postoperative neuropathic pain. Baseline pain score by VRS (0‐7), median 3 (range 3 to 4) and baseline depression score by BDI (0‐63), median 10 (range 1 to 28). All were recruited in Finland | |
| Interventions | Venlafaxine dose escalation from 18.75 mg/day to 75 mg/day orally or placebo. 11 women used 75 mg/day by end of the study period. Women were not allowed to take any other medication that was significantly metabolised by the cytochrome P450 2D6 isozyme | |
| Outcomes | Participant‐reported pain relief (VRS 0‐4) and pain intensity (VRS 0‐7) were the primary outcome measures and also BDI (0‐63) Median pain relief on venlafaxine 2 (range 0 to 4) and on placebo 0 (range 0 to 4) Pain intensity on venlafaxine 1 (range 0 to 3) and on placebo 2 (range 0 to 4) BDI score on venlafaxine 7 (range 1 to 39) and on placebo 7 (range 1 to 11) |
|
| Notes | 2/15 women dropped out, 1 due to adverse effects of venlafaxine (nausea, sweating headache) and 1 due to non‐compliance. Blood sampling confirmed that 2 'poor responders' had low venlafaxine concentrations and were classified as fast hydrolysers. There was a strong placebo effect on pain | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | It is stated that the hospital pharmacy provided the venlafaxine and placebo dosage forms and 'performed the randomization using computer‐generated numbers' |
| Allocation concealment (selection bias) | Low risk | Concealment was performed centrally by the hospital pharmacy |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blinding was mentioned but details were not provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It was mentioned that the researcher who interviewed the women was not part of the clinical team. No other details were provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | > 10% of women did not complete the trial and completer analysis was employed |
| Size | High risk | < 50 participants per treatment arm |
Yucel 2005.
| Methods | Double‐blind, placebo‐controlled trial for 8 weeks | |
| Participants | 60 participants aged 33‐69 years with neuropathic pain ≥ 6 months' duration and at least 4 on 11‐point VAS‐PI. In a separate arm of this study (not considered here), participants were also subjected to experimentally induced pain. Of those that completed the study, 22 were men and 33 were women. Participants were recruited in Istanbul, Turkey | |
| Interventions | Venlafaxine 75 mg/day, venlafaxine 150 mg/day, or placebo for 8 weeks with paracetamol 500 mg 3 or 4 times daily as escape medication. Participants were not allowed to take either antidepressants or anticonvulsants | |
| Outcomes | VAS‐PI, participant satisfaction and activities of daily living, adverse effects (none, slight bothersome, or unacceptable), and global impression of change scores were all assessed. Experimental pain scores were not considered for this review. Although a reduction in VAS‐PI was seen in all groups by the end of the study, there were no inter‐group differences in either pain intensity or the use of escape medication. Daily activity scores were also not significantly different between groups while participant satisfaction was significantly higher in the venlafaxine 75 mg group compared with placebo. Global impression of improvement did not differ significantly between venlafaxine and placebo (8/16 placebo; 13/16 venlafaxine 75 mg; 1/14 venlafaxine 150 mg) | |
| Notes | 5/60 withdrew: 1 placebo; 1 venlafaxine 75 mg; 3 venlafaxine 150 mg | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Details were not provided |
| Allocation concealment (selection bias) | Unclear risk | Details were not provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blinding was mentioned but details were not provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double‐blinding was mentioned but details were not provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | < 10% of participants did not complete the trial |
| Size | High risk | < 50 participants per treatment arm |
BDI: Beck Depression Inventory; CGI‐I: Clinical Global Impressions ‐ Improvement; CGI‐S: Clinical Global Impressions ‐ Severity; ITT: intention‐to‐treat; NNTB: number needed to treat for an additional beneficial outcome; NNTH: number needed to treat for an additional harmful outcome; NSAID: nonsteroidal anti‐inflammatory drug; STAI: State‐Trait Anxiety Inventory; VAS: visual analogue scale; VAS‐PI: visual analogue scale ‐ pain intensity; VAS‐PR: visual analogue scale ‐ pain relief; VRS: verbal rating scale; VRS‐PR: verbal rating scale ‐ pain relief.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Amr 2010 | Treatment < 2 weeks' duration |
| Davis 1999 | Case report only |
| Durand 2002 | Case report only |
| Durand 2012 | Treatment < 2 weeks' duration |
| Kadiroglu 2008 | Study appeared unblinded. Authors did not respond to a query on this |
| Kiayias 2000 | Non‐randomised study with only 8 participants |
| Lithner 2000 | Case report only |
| Pernia 2000 | Case report only |
| Raskin 2006 | Venlafaxine arm only contained 7 participants |
| Rej 2014 | Open‐label study. Did not exclude neuropathic pain but most participants did not have neuropathic pain |
| Reuben 2004 | Article was retracted in 2009 due to falsification of data |
| Simpson 2001 | Venlafaxine arm was an uncontrolled study |
| Sumpton 2001 | Case report only |
Differences between protocol and review
Atypical facial pain and chemotherapy/radiotherapy‐induced neuralgia are chronic pain conditions that were included in the review, but not in the protocol.
A plan for analysis of cross‐over studies was included in the review, although this was not implemented since no data synthesis was performed.
For clarity, we added 'for adults' to the title.
Contributions of authors
HCG registered the title, wrote the protocol and review, carried out searching, identified studies for inclusion, and carried out data extraction.
RMG and MCG identified studies for inclusion, carried out data extraction, and assisted in drafting.
DJB and MB provided clinical guidance and reviewed the protocol and review. The methods section is adapted from a template protocol for antidepressants in neuropathic pain devised by the PaPaS Cochrane Review Group. All authors contributed to the final draft of the protocol and approved this published version.
HCG will be responsible for updates.
Sources of support
Internal sources
No sources of support supplied
External sources
-
Health Research Board, Ireland.
(HG is funded by a Cochrane Fellowship)
Declarations of interest
HCG has no relevant conflicts of interest. HCG was a recipient of a Health Research Board Cochrane Fellowship which allowed her protected time to complete this review. She has other aligned pharmacological research interests that are funded by L'Air Liquide and the Mater Private Hospital, but this funding does not constitute a competing interest and did not influence the outcome of this review in any manner.
RMG has no relevant conflicts of interest.
DJB has no relevant conflicts of interest.
MB has no relevant conflicts of interest.
MCH has no relevant conflicts of interest.
Stable (no update expected for reasons given in 'What's new')
References
References to studies included in this review
Forssell 2004 {published data only}
- Forssell H, Tasmuth T, Tenovuo O, Hampf G, Kalso E. Venlafaxine in the treatment of atypical facial pain: a randomized controlled trial. Journal of Orofacial Pain 2004;18(2):131‐7. [PUBMED: 15250433] [PubMed] [Google Scholar]
Jia 2006 {published data only}
- Jia H‐Y, Li Q‐F, Song D‐P, An Z‐M, Liu Y‐P, Ran X‐W, et al. Effects of venlafaxine and carbamazepine for painful peripheral diabetic neuropathy: a randomized, double‐blind and double‐dummy, controlled multi‐center trial. Chinese Journal of Evidence‐Based Medicine 2006;6(5):321‐7. [Google Scholar]
Rowbotham 2004 {published data only}
- Rowbotham MC, Goli V, Kunz NR, Lei D. Venlafaxine extended release in the treatment of painful diabetic neuropathy: a double‐blind, placebo‐controlled study. Pain 2004;110(3):697‐706. [DOI] [PubMed] [Google Scholar]
Sindrup 2003 {published data only}
- Sindrup SH, Bach FW, Madsen C, Gram LF, Jensen TS. Venlafaxine versus imipramine in painful polyneuropathy: a randomized, controlled trial. Neurology 2003;60(8):1284‐9. [DOI] [PubMed] [Google Scholar]
Tasmuth 2002 {published data only}
- Tasmuth T, Härtel B, Kalso E. Venlafaxine in neuropathic pain following treatment of breast cancer. European Journal of Pain 2002;6(1):17‐24. [DOI] [PubMed] [Google Scholar]
Yucel 2005 {published data only}
- Yucel A, Ozyalcin S, Talu GK, Kiziltan E, Yucel B, Andersen OK, et al. The effect of venlafaxine on ongoing and experimentally induced pain in neuropathic pain patients: a double blind, placebo controlled study. European Journal of Pain 2005;9(4):407‐16. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Amr 2010 {published data only}
- Amr YM, Yousef AA. Evaluation of the efficacy of the perioperative administration of venlafaxine or gabapentin on acute and chronic postmastectomy pain. Clinical Journal of Pain 2010;26(5):381‐5. [PUBMED: 20473044] [DOI] [PubMed] [Google Scholar]
Davis 1999 {published data only}
- Davis JL, Smith RL. Painful peripheral diabetic neuropathy treated with venlafaxine HCl extended release capsules. Diabetes Care 1999;22(11):1909‐10. [PUBMED: 10546032] [DOI] [PubMed] [Google Scholar]
Durand 2002 {published data only}
- Durand JP, Goldwasser F. Dramatic recovery of paclitaxel‐disabling neurosensory toxicity following treatment with venlafaxine. Anti‐Cancer Drugs 2002;13(7):777‐80. [PUBMED: 12187335] [DOI] [PubMed] [Google Scholar]
Durand 2012 {published data only}
- Durand JP, Deplanque G, Montheil V, Gornet JM, Scotte F, Mir O, et al. Efficacy of venlafaxine for the prevention and relief of oxaliplatin‐induced acute neurotoxicity: results of EFFOX, a randomized, double‐blind, placebo‐controlled phase III trial. Annals of Oncology 2012;23(1):200‐5. [DOI] [PubMed] [Google Scholar]
Kadiroglu 2008 {published data only (unpublished sought but not used)}
- Kadiroglu AK, Sit D, Kayabasi H, Kemal TA, Tasdemir N, Yilmaz ME. The effect of venlafaxine HCl on painful peripheral diabetic neuropathy in patients with type 2 diabetes mellitus. Journal of Diabetes and its Complications 2008;22(4):241‐5. [DOI] [PubMed] [Google Scholar]
Kiayias 2000 {published data only}
- Kiayias JA, Vlachou ED, Lakka‐Papadodima E. Venlafaxine HCl in the treatment of painful peripheral diabetic neuropathy. Diabetes Care 2000;23(5):699. [PUBMED: 10834432] [DOI] [PubMed] [Google Scholar]
Lithner 2000 {published data only}
- Lithner F. Venlafaxine in treatment of painful peripheral diabetic neuropathy. Diabetes Care 2000;23(11):1710‐1. [PUBMED: 11092304] [DOI] [PubMed] [Google Scholar]
Pernia 2000 {published data only}
- Pernia A, Mico JA, Calderon E, Torres LM. Venlafaxine for the treatment of neuropathic pain. Journal of Pain and Symptom Management 2000;19(6):408‐10. [PUBMED: 10991644] [DOI] [PubMed] [Google Scholar]
Raskin 2006 {published data only}
- Raskin J, Smith TR, Wong K, Pritchett YL, D'Souza D, Iyengar S, et al. Duloxetine versus routine care in the long‐term management of diabetic peripheral neuropathic pain. Journal of Palliative Medicine 2006;9(1):29‐40. [PUBMED: 16430342] [DOI] [PubMed] [Google Scholar]
Rej 2014 {published data only}
- Rej S, Dew MA, Karp JF. Treating concurrent chronic low back pain and depression with low‐dose venlafaxine: an initial identification of "easy‐to‐use" clinical predictors of early response. Pain Medicine 2014;15:1154‐62. [PUBMED: 25040462] [DOI] [PMC free article] [PubMed] [Google Scholar]
Reuben 2004 {published data only}
- Reuben SS, Makari‐Judson G, Lurie SD. Evaluation of efficacy of the perioperative administration of venlafaxine XR in the prevention of post‐mastectomy pain syndrome. Journal of Pain and Symptom Management 2004;27(2):133‐9. [PUBMED: 15157037] [DOI] [PubMed] [Google Scholar]
Simpson 2001 {published data only}
- Simpson DA. Gabapentin and venlafaxine for the treatment of painful diabetic neuropathy. Journal of Clinical Neuromuscular Disease 2001;3(2):53‐62. [DOI] [PubMed] [Google Scholar]
Sumpton 2001 {published data only}
- Sumpton JE, Moulin DE. Treatment of neuropathic pain with venlafaxine. Annals of Pharmacotherapy 2001;35(5):557‐9. [PUBMED: 11346061] [DOI] [PubMed] [Google Scholar]
Additional references
AUREF 2012
- PaPaS author and referee guidance. papas.cochrane.org/papas‐documents (accessed 22 January 2013).
Bolden‐Watson 1993
- Bolden‐Watson C, Richelson E. Blockade by newly‐developed antidepressants of biogenic amine uptake into rat brain synaptosomes. Life Sciences 1993;52(12):1023‐9. [PUBMED: 8445992] [DOI] [PubMed] [Google Scholar]
Bouhassira 2008
- Bouhassira D, Lantéri‐Minet M, Attal N, Laurent B, Touboul C. Prevalence of chronic pain with neuropathic characteristics in the general population. Pain 2008;136(3):380‐7. [DOI: 10.1016/j.pain.2007.08.013] [DOI] [PubMed] [Google Scholar]
Dharmshaktu 2012
- Dharmshaktu P, Tayal V, Singh KB. Efficacy of antidepressants as analgesics: a review. Journal of Clinical Pharmacology 2012;52:6‐17. [PUBMED: 21415285] [DOI] [PubMed] [Google Scholar]
Dworkin 2008
- Dworkin RH, Turk DC, Wyrwich KW, Beaton D, Cleeland CS, Farrar JT, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. Journal of Pain 2008;9(2):105‐21. [DOI: 10.1016/j.jpain.2007.09.005] [DOI] [PubMed] [Google Scholar]
Enggaard 2001
- Enggaard TP, Klitgaard NA, Gram LF, Arendt‐Nielsen L, Sindrup SH. Specific effect of venlafaxine on single and repetitive experimental painful stimuli in humans. Clinical Pharmacology and Therapeutics 2001;69(4):245‐51. [DOI: 10.1067/mcp.2001.114873; PUBMED: 11309553 ] [DOI] [PubMed] [Google Scholar]
Farrar 2001
- Farrar JT, Young JP Jr, LaMoreaux L, Werth JL, Poole RM. Clinical importance of changes in chronic pain intensity measured on an 11‐point numerical pain rating scale. Pain 2011;94(2):149‐58. [PUBMED: 11690728] [DOI] [PubMed] [Google Scholar]
Gustorff 2008
- Gustorff B, Dorner T, Likar R, Grisold W, Lawrence K, Schwarz F, et al. Prevalence of self‐reported neuropathic pain and impact on quality of life: a prospective representative survey. Acta Anaesthesiologica Scandinavica 2008;52(1):132‐6. [DOI: 10.1111/j.1399-6576.2007.01486.x] [DOI] [PubMed] [Google Scholar]
Hall 2008
- Hall GC, Carroll D, McQuay HJ. Primary care incidence and treatment of four neuropathic pain conditions: a descriptive study, 2002‐2005. BMC Family Practice 2008;9:26. [DOI: 10.1186/1471-2296-9-26] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hearn 2014
- Hearn L, Derry S, Phillips T, Moore RA, Wiffen PJ. Imipramine for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2014;5:Art. No: CD10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Altman DG, Sterne JAC. Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Iannetti 2005
- Iannetti GD, Zambreanu L, Wise RG, Buchanan TJ, Huggins JP, Smart TS, et al. Pharmacological modulation of pain‐related brain activity during normal and central sensitization states in humans. Proceedings of the National Academy of Sciences of the United States of America 2005;102:18195‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jadad 1996
- Jadad AR, Moore RA, Carroll D, Jenkinson C, Reynolds DJ, Gavaghan DJ, et al. Assessing the quality of reports of randomized clinical trials: is blinding necessary?. Controlled Clinical Trials 1996;17(1):1‐12. [DOI: ] [DOI] [PubMed] [Google Scholar]
Jensen 2011
- Jensen TS, Baron R, Haanpää M, Kalso E, Loeser JD, Rice AS, et al. A new definition of neuropathic pain. Pain 2011;152(10):2204‐5. [DOI: 10.1016/j.pain.2011.06.017] [DOI] [PubMed] [Google Scholar]
Katusic 1991
- Katusic S, Williams DB, Beard CM, Bergstralh EJ, Kurland LT. Epidemiology and clinical features of idiopathic trigeminal neuralgia and glossopharyngeal neuralgia: similarities and differences, Rochester, Minnesota,1945‐1984. Neuroepidemiology 1991;10:276‐81. [DOI] [PubMed] [Google Scholar]
Koopman 2009
- Koopman JS, Dieleman JP, Huygen FJ, Mos M, Martin CG, Sturkenboom MC. Incidence of facial pain in the general population. Pain 2009;147(1‐3):122‐7. [DOI: 10.1016/j.pain.2009.08.023] [DOI] [PubMed] [Google Scholar]
L'Abbé 1987
- L'Abbé KA, Detsky AS, O'Rourke K. Meta‐analysis in clinical research. Annals of Internal Medicine 1987;107:224‐33. [DOI] [PubMed] [Google Scholar]
Lang 1996
- Lang E, Hord AH, Denson D. Venlafaxine hydrochloride (Effexor) relieves thermal hyperalgesia in rats with an experimental mononeuropathy. Pain 1996;68:151‐5. [DOI: 10.1016/S0304-3959(96)03223-X; PUBMED: 9252010] [DOI] [PubMed] [Google Scholar]
Lee 2010
- Lee Y‐C, Chen P‐P. A review of SSRIs and SNRIs in neuropathic pain. Expert Opinion in Pharmacotherapy 2010;11(17):2813‐25. [PUBMED: 20642317] [DOI] [PubMed] [Google Scholar]
McQuay 1998
- McQuay H, Moore R. An Evidence‐Based Resource for Pain Relief. Oxford: Oxford University Press, 1998. [Google Scholar]
McQuay 2007
- McQuay HJ, Smith LA, Moore RA. Chronic pain. In: Stevens A, Raftery J, Mant J, Simpson S editor(s). Health Care Needs Assessment. 3rd Edition. Oxford: Radcliffe Publishing, 2007. [ISBN: 978‐1‐84619‐063‐6] [Google Scholar]
Moore 1998
- Moore RA, Gavaghan D, Tramèr MR, Collins SL, McQuay HJ. Size is everything ‐ large amounts of information are needed to overcome random effects in estimating direction and magnitude of treatment effects. Pain 1998;78(3):209‐16. [DOI: 10.1016/S0304-3959(98)00140-7] [DOI] [PubMed] [Google Scholar]
Moore 2008
- Moore RA, Barden J, Derry S, McQuay HJ. Managing potential publication bias. In: McQuay HJ, Kalso E, Moore RA editor(s). Systematic Reviews in Pain Research: Methodology Refined. Seattle: IASP Press, 2008:15‐24. [ISBN: 978–0–931092–69–5] [Google Scholar]
Moore 2009a
- Moore RA, Moore OA, Derry S, Peloso PM, Gammaitoni AR, Wang H. Responder analysis for pain relief and numbers needed to treat in a meta‐analysis of etoricoxib osteoarthritis trials: bridging a gap between clinical trials and clinical practice. Annals of the Rheumatic Diseases 2010;69(2):374‐9. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2009b
- Moore RA, Straube S, Wiffen PJ, Derry S, McQuay HJ. Pregabalin for acute and chronic pain in adults. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD007076.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2010a
- Moore RA, Eccleston C, Derry S, Wiffen P, Bell RF, Straube S, et al. "Evidence" in chronic pain ‐ establishing best practice in the reporting of systematic reviews. Pain 2010;150(3):386‐9. [DOI: ] [DOI] [PubMed] [Google Scholar]
Moore 2010b
- Moore RA, Straube S, Paine J, Phillips CJ, Derry S, McQuay HJ. Fibromyalgia: moderate and substantial pain intensity reduction predicts improvement in other outcomes and substantial quality of life gain. Pain 2010;149(2):360‐4. [DOI: 10.1016/j.pain.2010.02.039] [DOI] [PubMed] [Google Scholar]
Moore 2010c
- Moore RA, Moore OA, Derry S, Peloso PM, Gammaitoni AR, Wang H. Responder analysis for pain relief and numbers needed to treat in a meta‐analysis of etoricoxib osteoarthritis trials: bridging a gap between clinical trials and clinical practice. Annals of the Rheumatic Diseases 2010;69(2):374‐9. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2010d
- Moore RA, Smugar SS, Wang H, Peloso PM, Gammaitoni A. Numbers‐needed‐to‐treat analyses ‐ do timing, dropouts, and outcome matter? Pooled analysis of two randomized, placebo‐controlled chronic low back pain trials. Pain 2010;151(3):592‐7. [DOI: ] [DOI] [PubMed] [Google Scholar]
Moore 2011a
- Moore RA, Straube S, Paine J, Derry S, McQuay HJ. Minimum efficacy criteria for comparisons between treatments using individual patient meta‐analysis of acute pain trials: examples of etoricoxib, paracetamol, ibuprofen, and ibuprofen/paracetamol combinations after third molar extraction. Pain 2011;152(5):982‐9. [DOI: ] [DOI] [PubMed] [Google Scholar]
Moore 2011b
- Moore RA, Mhuircheartaigh RJ, Derry S, McQuay HJ. Mean analgesic consumption is inappropriate for testing analgesic efficacy in post‐operative pain: analysis and alternative suggestion. European Journal of Anaesthesiology 2011;28(6):427‐32. [DOI: 10.1097/EJA.0b013e328343c569] [DOI] [PubMed] [Google Scholar]
Moore 2012
- Moore RA, Straube S, Eccleston C, Derry S, Aldington D, Wiffen P, et al. Estimate at your peril: imputation methods for patient withdrawal can bias efficacy outcomes in chronic pain trials using responder analyses. Pain 2012;153(2):265‐8. [DOI] [PubMed] [Google Scholar]
Moore 2014
Muth 1986
- Muth EA, Haskins JT, Moyer JA, Husbands GE, Nielsen ST, Sigg EB. Antidepressant biochemical profile of the novel bicyclic compound Wy‐45,030, an ethyl cyclohexanol derivative. Biochemical Pharmacology 1986;35(24):4493‐7. [DOI: 10.1016/0006-2952(86)90769-0; PUBMED: 3790168] [DOI] [PubMed] [Google Scholar]
Muth 1991
- Muth EA, Moyer JA, Haskins JT. Biochemical, neurophysiological, and behavioral effects of Wy‐45,233 and other identified metabolites of the antidepressant venlafaxine. Drug Development Research 1991;23(2):191‐9. [DOI: 10.1002/ddr.430230210] [DOI] [Google Scholar]
O'Brien 2010
- O'Brien EM, Staud RM, Hassinger AD, McCulloch RC, Craggs JG, Atchison JW, et al. Patient‐centered perspective on treatment outcomes in chronic pain. Pain Medicine 2010;11(1):6‐15. [DOI: 10.1111/j.1526-4637.2009.00685.x] [DOI] [PubMed] [Google Scholar]
Onghena 1992
- Onghena P, Houdenhove B. Antidepressant‐induced analgesia in chronic non‐malignant pain: a meta‐analysis of 39 placebo‐controlled studies. Pain 1992;49(2):205‐19. [DOI: 10.1016/0304-3959(92)90144-Z; PUBMED: 1535121] [DOI] [PubMed] [Google Scholar]
Raftery 2011
- Raftery MN, Sarma K, Murphy AW, Harpe D, Normand C, McGuire BE. Chronic pain in the Republic of Ireland ‐ community prevalence, psychosocial profile and predictors of pain‐related disability: results from the Prevalence, Impact and Cost of Chronic Pain (PRIME) study, part 1. Pain 2011;152(5):1096‐103. [DOI: 10.1016/j.pain.2011.01.019; PUBMED: 21450402] [DOI] [PubMed] [Google Scholar]
Rappaport 1994
- Rappaport ZH, Devor M. Trigeminal neuralgia: the role of self‐sustaining discharge in the trigeminal ganglion. Pain 1994;56:127‐38. [DOI: 10.1016/0304-3959(94)90086-8] [DOI] [PubMed] [Google Scholar]
RevMan 2012 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.2. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2012.
Saarto 2007
- Saarto T, Wiffen PJ. Antidepressants for neuropathic pain. Cochrane Database of Systematic Reviews 2007, Issue 4. [DOI: 10.1002/14651858.CD005454.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schreiber 1999
- Schreiber S, Backer MM, Pick CG. The antinociceptive effect of venlafaxine in mice is mediated through opioid and adrenergic mechanisms. Neuroscience Letters 1999;273(2):85‐8. [DOI: 10.1016/S0304-3940(99)00627-8; PUBMED: 10505622] [DOI] [PubMed] [Google Scholar]
Seo 2010
- Seo HJ, Sohi MS, Patkar AA, Masand PS, Pae CU. Desvenlafaxine succinate: a newer antidepressant for the treatment of depression and somatic symptoms. Postgraduate Medicine 2010;122(1):125‐38. [DOI] [PubMed] [Google Scholar]
Straube 2008
- Straube S, Derry S, McQuay HJ, Moore RA. Enriched enrollment: definition and effects of enrichment and dose in trials of pregabalin and gabapentin in neuropathic pain. A systematic review. British Journal of Clinical Pharmacology 2008;66(2):266‐75. [DOI: 10.1111/j.1365-2125.2008.03200.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
Straube 2010
- Straube S, Derry S, Moore RA, Paine J, McQuay HJ. Pregabalin in fibromyalgia ‐ responder analysis from individual patient data. BMC Musculoskeletal Disorders 2010;11:150. [DOI: 10.1186/1471-2474-11-150] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sultan 2008
- Sultan A, Gaskell H, Derry S, Moore RA. Duloxetine for painful diabetic neuropathy and fibromyalgia pain: systematic review of randomised trials. BMC Neurology 2008;8:29. [DOI: 10.1186/1471-2377-8-29] [DOI] [PMC free article] [PubMed] [Google Scholar]
Torrance 2006
- Torrance N, Smith BH, Bennett MI, Lee AJ. The epidemiology of chronic pain of predominantly neuropathic origin. Results from a general population survey. Journal of Pain 2006;7(4):281‐9. [DOI: 10.1016/j.jpain.2005.11.008] [DOI] [PubMed] [Google Scholar]