

Abstract
Background
Studies have reported high tumour response rates for platinum‐containing regimens in the treatment of women with metastatic breast cancer. Most of these studies were conducted prior to the 'intrinsic subtype' era, and did not specifically focus on metastatic triple‐negative breast cancers (mTNBCs).
Objectives
To identify and review the evidence from randomised trials comparing platinum‐containing chemotherapy regimens with regimens not containing platinum in the management of women with metastatic breast cancer.
Search methods
For this review update, we searched the Cochrane Breast Cancer Group's Specialised Register, CENTRAL, MEDLINE, Embase, the World Health Organization's International Clinical Trials Registry Platform and ClinicalTrials.gov on 28 May 2015. We identified further potentially relevant studies from handsearching references of previous trials, systematic reviews, and meta‐analyses. Prior to this review update, the most recent search for studies was conducted in May 2003 for the original 2004 review.
Selection criteria
Randomised trials comparing platinum‐containing chemotherapy regimens with regimens not containing platinum in women with metastatic breast cancer.
Data collection and analysis
At least two independent reviewers assessed studies for eligibility and quality, and extracted all relevant data from each study. Hazard ratios (HRs) were derived for time‐to‐event outcomes, where possible, and fixed‐effect models were used for meta‐analyses. Objective tumour response rates (OTRRs) and toxicities were analysed as binary (dichotomous) outcomes with risk ratios (RRs) used as measures of effects. Quality of life data were extracted where available. GRADE was used to rate the quality of evidence for survival and tumour response outcomes at the level of subgroups selected and unselected for mTNBC, and for toxicity outcomes based on combining data from selected and unselected populations.
Main results
This update includes 15 new eligible treatment‐comparisons from 12 studies. In total, 28 treatment‐comparisons, involving 4418 women, from 24 studies are now included in one or more meta‐analyses. Of the 28 treatment‐comparisons, 19 and 16 had published or provided extractable time‐to‐event data on overall survival (OS) or progression‐free survival/time to progression (PFS/TTP), respectively. All 28 treatment‐comparisons provided OTRR data that could be included in meta‐analyses. Most women recruited to the studies were not selected on the basis of mTNBC status.
In a subgroup of three treatment‐comparisons assessing women with mTNBC, platinum‐containing regimens may have provided a survival benefit (HR 0.75, 95% CI 0.57 to 1.00; low‐quality evidence). In women unselected for intrinsic subtypes such as mTNBC, there was little or no effect on survival (HR 1.01, 95% CI 0.92 to 1.12; high‐quality evidence). This effect was similar to the combined analysis of survival data for both populations (HR 0.98, 95% CI 0.89 to 1.07; I2 =39%, 1868 deaths, 2922 women; 19 trials). The difference in treatment effects between mTNBC women compared with unselected women was of borderline statistical significance (P = 0.05).
Data from three treatment‐comparisons with mTNBC participants showed that platinum regimens may improve PFS/TTP (HR 0.59, 95% CI 0.49 to 0.72; low‐quality evidence). Thirteen treatment‐comparisons of unselected metastatic participants showed that there was probably a small PFS/TTP benefit for platinum recipients, although the confidence interval included no difference (HR 0.92, 95% CI 0.84 to 1.01; moderate‐quality evidence). Combined analysis of data from an estimated 1772 women who progressed or died out of 2136 women selected or unselected for mTNBC indicated that platinum‐containing regimens improved PFS/TTP (HR 0.85, 95% CI 0.78 to 0.93). There was marked evidence of heterogeneity (P = 0.0004; I2 = 63%). The larger treatment benefit in mTNBC women compared with unselected women was statistically significant (P < 0.0001).
There was low‐quality evidence of better tumour response in both subgroups of women with mTNBC and unselected women (RR 1.33, 95% CI 1.13 to 1.56; RR 1.11, 95% CI 1.04 to 1.19, respectively). Combined analysis of both populations was closer to the effect in unselected women (RR 1.15, 95% CI 1.08 to 1.22; 4130 women). There was considerable evidence of heterogeneity (P < 0.0001; I2 = 64%), which may reflect between‐study differences and general difficulties in assessing response, as well as the varying potencies of the comparators.
Compared with women receiving non‐platinum regimens: rates of grade 3 and 4 nausea/vomiting were probably higher among women receiving cisplatin‐ (RR 2.65, 95% CI 2.10 to 3.34; 1731 women; moderate‐quality evidence) but the effect from carboplatin‐containing regimens was less certain (RR 0.77, 95% CI 0.47 to 1.26; 1441 women; moderate‐quality evidence); rates of grade 3 and 4 anaemia were higher among women receiving cisplatin‐ (RR 3.72, 95% CI 2.36 to 5.88; 1644 women; high‐quality evidence) and carboplatin‐containing regimens (RR 1.72, 95% CI 1.10 to 2.70; 1441 women; high‐quality evidence); rates of grade 3 and 4 hair loss (RR 1.41, 95% CI 1.26 to 1.58; 1452 women; high‐quality evidence) and leukopenia (RR 1.38, 95% CI 1.21 to 1.57; 3176 women; moderate‐quality evidence) were higher among women receiving platinum‐containing regimens (regardless of platinum agent).
Authors' conclusions
In women with metastatic breast cancer who do not have triple‐negative disease, there is high‐quality evidence of little or no survival benefit and excess toxicity from platinum‐based regimens. There is preliminary low‐quality evidence of a moderate survival benefit from platinum‐based regimens for women with mTNBC. Further randomised trials of platinum‐based regimens in this subpopulation of women with metastatic breast cancer are required.
Plain language summary
Platinum‐containing regimens for metastatic breast cancer
What is the issue? Metastatic breast cancer occurs when the cancer has spread to areas of the body beyond the breast and nearby lymph nodes. Although metastatic breast cancer is generally not curable, it is widely accepted that women with metastatic disease should receive some form of chemotherapy to help ease the severity of disease symptoms, while hopefully extending survival time. Chemotherapy containing platinum is known to be effective for treating a number of cancer types including lung, testicular, head and neck, bladder and ovarian cancers, but it also known to cause more adverse effects (such as nausea and vomiting, hair loss, anaemia, kidney damage and leukopenia (low white blood cells)) than other chemotherapy options. The two platinum agents most commonly used for treating metastatic breast cancer are carboplatin and cisplatin.
The original version of this review (2004) concluded that chemotherapy containing platinum did not increase survival time for women treated for metastatic breast cancer. Since then, however, researchers have discovered that there are a variety of subtypes of breast cancer which may respond differently to different types of chemotherapy. One of these subtypes — triple‐negative breast cancer (TNBC) — makes up approximately 12 to 17% of breast cancers and is associated with shorter survival and higher likelihood that the cancer returns. Some researchers have speculated that chemotherapy containing platinum might be more effective in treating metastatic TNBC (mTNBC) than other chemotherapy options.
Why does it matter? There are at least two reasons why it is important to update the evidence on this topic. First, it is important to assess whether our 2004 conclusions — based on 12 early studies — are representative of the 24 studies who have now published or provided results through to 2015. Second, it is important to assess whether chemotherapy containing platinum increases survival for women with mTNBC more than other chemotherapy options.
We asked whether chemotherapy treatments containing a platinum agent are more or less effective for treating women with metastatic breast cancer than chemotherapy treatments not containing a platinum agent. We also asked the same question, but with a focus on women with mTNBC.
We found 24 studies involving 4418 women. The evidence is current to May 2015. Five of the 24 studies specifically assessed women with mTNBC while the other 19 studies assessed women with metastatic breast cancer in general (mainly women without mTNBC). This review found that, compared to chemotherapy without platinum, chemotherapy with platinum did not increase survival time by any important degree for women with metastatic breast cancer in general (mainly women without mTNBC). The quality of the evidence for this was considered to be high, meaning that we are confident about the results. For women with mTNBC, however, this review found that chemotherapy containing platinum may increase survival time over chemotherapy without platinum, but the quality of the evidence for this is low at this point in time (largely due to the small number of studies that have assessed mTNBC). This review also found that chemotherapy including platinum reduced the number of breast cancer recurrences compared to chemotherapy that did not contain platinum in women with mTNBC, however these findings also currently come from low‐quality evidence. There was no difference in the number of breast cancer recurrences for women receiving platinum or non‐platinum chemotherapy for metastatic breast cancer in general. Chemotherapy with platinum was more likely to shrink tumours compared to chemotherapy without platinum, but this result needs to be considered cautiously.
Compared with women receiving chemotherapy without platinum, women receiving chemotherapy with platinum experienced higher rates of nausea/vomiting, anaemia, leukopenia and hair loss.
This means it is difficult to justify using chemotherapy containing platinum for the treatment of metastatic breast cancer that is not mTNBC, given that similarly effective but less toxic chemotherapy is commonly available. Chemotherapy containing platinum may provide a survival benefit to mTNBC participants of sufficient magnitude to justify its use, but the quality of the evidence for this is low at this point in time. Further studies are required before a more definitive conclusion can be made.
Summary of findings
Summary of findings for the main comparison. Platinum‐containing regimens for women with metastatic breast cancer unselected for triple‐negative disease.
| Platinum compared to non‐platinum chemotherapy regimens for women with metastatic breast cancer unselected for triple‐negative disease | ||||||
| Patient or population: women with metastatic breast cancer unselected for triple‐negative breast cancer (TNBC) Setting: hospital Intervention: platinum Comparison: non‐platinum chemotherapy regimens | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (treatment‐ comparisons) | Quality of the evidence (GRADE) | Comments | |
| Risk with non‐platinum chemotherapy regimens | Risk with platinum | |||||
| Overall survival ‐ trials of metastatic breast cancer participants unselected for TNBC | 1‐year risk of death | HR 1.01 (0.92 to 1.12) | 2531 (16) | ⊕⊕⊕⊕ HIGH3 | Heterogeneity: P = 0.09, I2=34% | |
| 310 per 1,000 1 | 313 per 1,000 (289 to 340)2 | |||||
| 2‐year risk of death | ||||||
| 540 per 1,000 1 | 543 per 1,000 (510 to 581)2 | |||||
| Progression‐free survival/time to progression (randomised participants) ‐ trials of metastatic breast cancer participants unselected for TNBC | 1‐year risk of progression or death | HR 0.92 (0.84 to 1.01) | 1745 (13) | ⊕⊕⊕⊝ MODERATE4 | Heterogeneity: P = 0.08, I2=38% | |
| 737 per 1,000 1 | 707 per 1,000 (674 to 740)2 | |||||
| 2‐year risk of progression or death | ||||||
| 891 per 1,000 1 | 869 per 1,000 (844 to 893)2 | |||||
| Objective tumour response rate (assessable participants) ‐ trials of metastatic breast cancer participants unselected for TNBC | 493 per 1,000 5 | 547 per 1,000 (512 to 586) | RR 1.11 (1.04 to 1.19) | 3252 (23) | ⊕⊕⊝⊝ LOW4,6 | Heterogeneity: P = 0.0002, I2=58% |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; HR: Hazard ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Estimated from the average of non‐platinum group Kaplan‐Meier probabilities from the three highest weighted non‐TNBC treatment‐comparisons for this outcome
2 Estimated as 1000*(1‐S(t)HR) where S(t) is the estimated probability of survival for non‐platinum participants and HR is the pooled hazard ratio (Davies 1998)
3 Quality of evidence for OS was not downgraded for blinding because this outcome is unlikely to be affected by non‐blinding.
4 Downgraded quality of evidence one level for 'indirectness' because this outcome is a surrogate endpoint of questionable validity for assessing the more important outcome of OS in the context of metastatic breast cancer (Burzykowski 2008)
5 Estimated from all 23 non‐TNBC treatment‐comparisons in the review
6 Downgraded quality of evidence one level for 'inconsistency' because there was substantial evidence of heterogeneity across studies (P < 0.05)
Summary of findings 2. Platinum‐containing regimens for women with metastatic triple‐negative breast cancer.
| Platinum compared to non‐platinum chemotherapy regimens for women with metastatic triple‐negative breast cancer | ||||||
| Patient or population: women with metastatic triple‐negative breast cancer (mTNBC) Setting: hospital Intervention: platinum Comparison: non‐platinum chemotherapy regimens | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (treatment‐ comparisons) | Quality of the evidence (GRADE) | Comments | |
| Risk with non‐platinum chemotherapy regimens | Risk with platinum | |||||
| Overall survival ‐ trials of mTNBC participants | 1‐year risk of death | HR 0.75 (0.57 to 1.00) | 391 (3) | ⊕⊕⊝⊝ LOW3,4,5 | Heterogeneity: P = 0.23, I2 = 32% | |
| 485 per 1,000 1 | 392 per 1,000 (315 to 485)2 | |||||
| 2‐year risk of death | ||||||
| 655 per 1,000 1 | 550 per 1,000 (455 to 655)2 | |||||
| Progression‐free survival/time to progression (randomised participants) ‐ trials of mTNBC participants | 1‐year risk of death | HR 0.59 (0.49 to 0.72) | 391 (3) | ⊕⊕⊝⊝ LOW4,6 | Heterogeneity: P = 0.07, I2 = 67% | |
| 894 per 1,000 1 | 733 per 1,000 (667 to 801)2 | |||||
| 2‐year risk of death | ||||||
| 987 per 1,000 1 | 922 per 1,000 (879 to 955)2 | |||||
| Objective tumour response rate (assessable participants) ‐ trials of mTNBC participants | 354 per 1,0007 | 470 per 1,000 (400 to 552) | RR 1.33 (1.13 to 1.56) | 878 (5) | ⊕⊕⊝⊝ LOW6,8 | Heterogeneity: P = 0.0010, I2 = 78% |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; HR: Hazard ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Estimated from the average of non‐platinum group Kaplan‐Meier probabilities from the three TNBC treatment‐comparisons contributing data for pooling on this outcome
2 Estimated as 1000*(1‐S(t)HR) where S(t) is the estimated probability of survival for non‐platinum participants and HR is the pooled hazard ratio (Davies 1998)
3 Downgraded quality of evidence one level for 'imprecision' because the confidence interval for the pooled estimate is wide and crosses or nearly crosses unity
4 Downgraded quality of evidence one level for 'suspected publication bias' because Tutt 2014 is a large study with 376 participants but has so far only reported median OS/PFS times. As a consequence, the study did not contribute to the pooled HR estimates for OS or PFS/TTP. The reported median OS/PFS times in Tutt 2014 were similar between platinum and non‐platinum regimens. Hence, it seems likely that if HRs from Tutt 2014 were able to be included in pooled HR estimates, these pooled estimates would be considerably closer to the null.
5 Quality of evidence for OS was not downgraded for blinding because this outcome is unlikely to be affected by non‐blinding.
6 Downgraded quality of evidence one level for 'indirectness' because this outcome is a surrogate endpoint of questionable validity for assessing the more important outcome of OS in the context of metastatic breast cancer (Burzykowski 2008)
7Estimated from all five TNBC treatment‐comparisons in the review
8 Downgraded quality of evidence one level for 'inconsistency' because there was substantial evidence of heterogeneity across studies (P < 0.05)
Summary of findings 3. Platinum‐containing regimens and toxicity profile.
| Platinum compared to non‐platinum chemotherapy regimens for nausea/vomiting, anaemia, hair loss and leukopenia | ||||||
| Patient or population: women with metastatic breast cancer Setting: hospital Intervention: platinum Comparison: non‐platinum chemotherapy regimens | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (treatment‐ comparisons) | Quality of the evidence (GRADE) | Comments | |
| Risk with non‐platinum chemotherapy regimens | Risk with platinum | |||||
| Nausea/vomiting* grade 3 or 4 (safety population) by type of platinum agent in platinum regimen | Carboplatin | (RR 0.77, 95% CI 0.47 to 1.26) | 1441 (7) | ⊕⊕⊕⊝ MODERATE2 | Test for carboplatin/cisplatin subgroup difference: P < 0.0001 Heterogeneity among carboplatin studies: P = 0.30, I2 = 17% Heterogeneity among cisplatin studies: P = 0.010, I2 = 32% |
|
| 80 per 1,000 1 | 62 per 1,000 (59 to 101) | |||||
| Cisplatin | (RR 2.65, 95% CI 2.10 to 3.34) | 1747 (14) | ⊕⊕⊕⊝ MODERATE3 | |||
| 80 per 1,000 1 | 210 per 1,000 (167 to 266) | |||||
| Anaemia grade 3 or 4 (safety population) by type of platinum agent in platinum regimen | Carboplatin | (RR 1.72, 95% CI 1.10 to 2.70) | 1441 (7) | ⊕⊕⊕⊕ HIGH | Test for carboplatin/cisplatin subgroup difference: P = 0.02 Heterogeneity among carboplatin studies: P = 0.67, I2 = 0% Heterogeneity among cisplatin studies: P = 0.50, I2 = 0% |
|
| 33 per 1,000 1 | 57 per 1,000 (36 to 89) | |||||
| Cisplatin | (RR 3.72, 95% CI 2.36 to 5.88) | 1644 (13) | ⊕⊕⊕⊕ HIGH |
|||
| 33 per 1,000 1 | 123 per 1,000 (78 to 194) | |||||
| Hair loss grade 3 or 4 (safety population) | Carboplatin or cisplatin | (RR 1.41, 95% CI 1.26 to 1.58) | 1452 (13) | ⊕⊕⊕⊕ HIGH | Test for carboplatin/cisplatin subgroup difference: P = 0.23 Heterogeneity: P = 0.10, I2 = 40% |
|
| 264 per 1,000 1 | 372 per 1,000 (333 to 417) | |||||
| Leukopenia** grade 3 or 4 (safety population) | Carboplatin or cisplatin | (RR 1.38, 95% CI 1.21 to 1.57) | 3176 (22) | ⊕⊕⊕⊝ MODERATE3 | Test for carboplatin/cisplatin subgroup difference: P = 0.22 Heterogeneity: P = 0.0002, I2 = 60% |
|
| 179 per 1,000 1 | 247 per 1,000 (217 to 281) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; HR: Hazard ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Estimated from all treatment‐comparisons (cisplatin and carboplatin) contributing data for pooling for this outcome
2 Downgraded quality of evidence one level for 'imprecision' because the confidence interval for the pooled estimate is wide and does not rule out 'no effect'
3 Downgraded quality of evidence one level for 'inconsistency' because there was substantial evidence of heterogeneity across studies (P < 0.05)
*data on vomiting was included if data on nausea/vomiting was reported separately
**data on neutropenia was included if data on leukopenia was not reported
Background
Description of the condition
Breast cancer is both the most common type of cancer in women and the most common cause of cancer death in women (Ferlay 2013). In 2012, there was an estimated 1.67 million estimated new cases and approximately 522,000 deaths from breast cancer worldwide, with an age standardised death rate (ASR) of 12.9 per 100,000 (Ferlay 2013). ASRs of 25 or greater were recorded that same year in Fiji (28.4), Bahamas (26.3), Nigeria (25.9), Former Yugoslav Republic (FYR) Macedonia (25.5) and Pakistan (25.2) (Ferlay 2013).
The stage of breast cancer at the time of diagnosis is an important indicator of prognosis. Once breast cancer becomes metastatic, it is not generally considered curable and most women with metastatic disease do not survive beyond five years from the time of their metastatic diagnosis (Clements 2012). Another important predictor of prognosis is the biological subtype of breast cancer. One of these subtypes — triple‐negative breast cancer (TNBC) — is characterised by a lack of expression of oestrogen and progesterone receptors (ER and PgR) and human epidermal receptor 2 (HER2). TNBC comprises approximately 12 to 17% of breast cancers and is associated with shorter survival and higher likelihood of recurrence (Foulkes 2010). The median survival time for women diagnosed with metastatic TNBC (mTNBC) is about one year from their metastatic diagnosis (Kassam 2009).
Although there is no evidence from randomised trials comparing chemotherapy with observation (no chemotherapy) in women with metastatic breast cancer, it is widely accepted that women with metastatic disease should receive some form of systemic therapy at some time during the course of their metastatic disease. Chemotherapy is considered by many to be the appropriate first treatment option for women with multiple sites of recurrence or where visceral disease is not easily treated by local modalities (Hayes 1995; Beslija 2009). Chemotherapy is also considered to be useful in women whose cancer is hormone refractory or is expected to be hormone resistant (Hortobagyi 1996).
Description of the intervention
Platinum compound, an alkylating agent, has been known to be active in metastatic breast cancer since clinical trials in the 1970s. However, it is more toxic and difficult to administer than other chemotherapy agents. The three most widely used platinum agents for treating breast cancer are cisplatin, carboplatin (both divalent complexes) and oxaliplatin (a tetravalent complex) (Sikov 2015). Cisplatin and carboplatin have demonstrated benefits in treating a number of cancer types including lung, testicular, head and neck, bladder and ovarian cancers. Oxaliplatin is often used to treat cisplatin‐ and carboplatin‐resistant tumours because it is commonly believed that cross‐resistance between oxaliplatin and cisplatin or carboplatin is incomplete (Mani 2002). More recent evidence suggests that the benefits of oxaliplatin may be due to its low toxicity and ability to be combined with other drugs rather than incomplete cross‐resistance with other platinum agents (Stordal 2007).
The use of oxaliplatin for treating breast cancers is much less common than the use of cisplatin or carboplatin, both in normal clinical practice and as an intervention in clinical trials (Sikov 2015). Cisplatin and carboplatin have been used and studied extensively as first‐line metastatic therapy in combination with other older pharmacological agents including 5‐fluorouracil and etoposide, and more recently with doxorubicin, epirubicin, vinorelbine, paclitaxel, docetaxel, cyclophosphamide, methotrexate and gemcitabine. The potential benefits of cisplatin or carboplatin as monotherapy for metastatic breast cancer, rather than as combination therapy, are rarely studied in clinical trials.
Although platinum agents have been shown to be efficacious in the treatment of a number of cancer types, their use is often associated with a variety of side effects. The known side effects of platinum agents include nausea, vomiting, myelosuppression (thrombocytopenia, leukopenia, neutropenia and anaemia), peripheral neuropathy (symptoms include tingling in fingers and toes), nephrotoxicity, ototoxicities (hearing loss and tinnitus), hypomagnesaemia and anaphylaxis. Carboplatin is reported to be more tolerable than cisplatin with less nausea and vomiting, nephrotoxicity, ototoxicity and neurotoxicity, but worse myelosuppression, especially thrombocytopenia (Sikov 2015).
How the intervention might work
The exact mechanism of action of platinum agents is not known but deoxyribonucleic acid (DNA) adducts are formed (Sikov 2015). These complexes are believed to inhibit DNA synthesis, replication and transcription by forming interstrand and intrastrand cross‐linking of DNA molecules. Interstrand cross‐links that remain intact can produce cell death, and it is this cytotoxic effect, when successful, that forms the mechanistic basis of action for cancer cell death by platinum agents (Noll 2006). For TNBC, it has been additionally hypothesised that a dysfunctional BRCA1 pathway in some TNBCs may make them more sensitive to platinum agents that selectively target cells deficient in homologous recombination DNA repair (Foulkes 2010).
Why it is important to do this review
There are a number of reasons for updating this review. First, this review was originally published in 2004 (Carrick 2004) and was based on 12 studies (13 treatment‐comparisons) identified from a May 2003 search of the literature. Although there have since been some updates to this review containing minor amendments, no new searches for relevant studies have been conducted since 2003. In the current review update, the number of included studies has increased from 12 in the original version of this review to 24 (28 treatment‐comparisons). This allows effects to be estimated with greater precision than before and provides an opportunity to conduct new subgroup analyses corresponding to emerging evidence and new hypotheses. Second, all 12 studies included in the original version of this review were conducted prior to the 'intrinsic subtype' era and thus did not specifically focus on TNBC (a term first mentioned in the medical literature in October 2006) (Foulkes 2010). Since 2009, however, five randomised control trials have published data specifically on the effects of platinum‐based regimens on survival, progression or tumour response in mTNBC participants; the inclusion of these five trials in this review update represents the first meta‐analysis of such trials. Third, some interest remains in the use of platinum agents for the treatment of women with metastatic breast cancer in general (i.e. unselected for intrinsic subtypes such as mTNBC) (Shamseddine 2012). This is notwithstanding the primary conclusion contained in the original version of this review that "...in view of the significant excess toxicity, lack of progression or survival benefit and the availability of less toxic active agents it is difficult to justify the use of platinum‐containing regimens, particularly as first‐line treatment for women with metastatic breast cancer in routine clinical practice."
Objectives
To identify and review the evidence from randomised trials comparing platinum‐containing chemotherapy regimens with regimens not containing platinum in the management of women with metastatic breast cancer.
Additional objectives of this review were to investigate whether or not women in selected subgroups of studies benefited more or less from platinum‐based chemotherapy. Some subgroup analyses included in this review update were conducted in the original version of this review, while others have been added in response to new hypotheses and the availability of new subgroups.
Methods
Criteria for considering studies for this review
Types of studies
Properly randomised controlled clinical trials (i.e. where the trial report asserts that the trial was randomised and there was no evidence to suggest otherwise).
Types of participants
Women with advanced (metastatic) breast cancer, either newly diagnosed or recurrent. Treatment‐comparisons that included both women with metastatic disease and women with loco‐regionally recurrent disease only were eligible for inclusion if it was possible to distinguish between the two groups (i.e. data were reported separately) or if women with isolated locoregional recurrence were less than 20% of the total group. There were no age restrictions. In the protocol for this review, it was proposed that treatment‐comparisons would be included if the women randomised to receive chemotherapy, were to receive it as first‐line treatment (i.e. no previous chemotherapy given except as adjuvant therapy). As few treatment‐comparisons assessing first‐line treatment were identified for inclusion in the original version of this review, those meeting the remaining eligibility criteria but which involved participants who were not first‐line naive were included. This modification of the inclusion criteria was maintained for this review update, with subgroup analysis by treatment being performed (treatment‐comparisons with first‐line therapy for > 80% of participants vs other treatment lines).
Types of interventions
Interventions were any chemotherapy regimen containing a platinum agent (see Table 4 and Table 5). Comparators were any chemotherapy regimen without a platinum agent. Endocrine therapy may also have been given to participants if it was planned to be given to both treatment groups. Studies may or may not have specified recommended treatment upon disease progression or initial treatment failure or both. This recommended treatment may have included cross‐over to the alternative treatment arm of the treatment‐comparison.
1. Platinum agents.
| Generic name | Other names |
| Carboplatin | Blastocarb, Carboplat, Carboplatin Hexal, Carboplatino, Carbosin, Carbosol, Carbotec, CBDCA, Displata, Ercar, Nealorin, Novoplatinum, Paraplat, Paraplatin AQ, Paraplatin, Paraplatine, Platinwas, Ribocarbo |
| Cisplatin | Abiplatin, Blastolem, Briplatin,CACP, CDDP, cis‐DDP, cis‐diamminedichloridoplatinum, cis‐diamminedichloro platinum (II), cis‐diamminedichloroplatinum, Cis‐dichloroammine Platinum (II), Cismaplat, Cisplatina, cis‐platinous diamine dichloride, cis‐platinum II diamine dichloride, cis‐platinum II, cis‐platinum, Cisplatyl, Citoplatino, Citosin, CPDD, Cysplatyna, DDP, DDP, Lederplatin, Metaplatin, Neoplatin, PDD, Peyrone's Chloride, Peyrone's Salt, Placis, Platamine, Platiblastin, Platiblastin‐S, Platinex, Platinol‐ AQ, Platinol, Platinol‐AQ VHA Plus, Platinol‐AQ, Platinoxan, platinum diamminodichloride, Platiran, Platistin, Platosin |
| Oxaliplatin | Ai Heng, Aiheng, diaminocyclohexane oxalatoplatinum, oxalatoplatin, oxalatoplatinum, oxaliplatine, Eloxatin, Dacotin, Dacplat, Eloxatine, 1‐OHP, L‐OHP, oxaliplatin medac |
2. Chemotherapeutic Agents (adapted from Table 1.1 in The Chemotherapy Source Book).
| Type of Agent | Action | Includes |
| Agents that damage the DNA template | by alkylation: nitrogen mustards | cyclophosphamide, melphalan, ifosfamide, chlorambucil |
| by alkylation: nitrosureas | carmustine (BCNU), lomustine (CCNU) | |
| by alkylation: other agents | thiotepa, mitomycin C | |
| by platinum coordination cross‐linking | cisplatin, carboplatin | |
| antibiotics | doxorubicin, daunorubicin, mitoxantrone, idarubicin, epirubicin, amsacrine | |
| podophyllotoxins | etoposide, teniposide | |
| by intercalation | dactinomycin, mithramycin | |
| by uncertain mechanisms | bleomycin | |
| Spindle poisons | vinca alkaloids | vincristine, vinblastine, vendesine, vinorelbine |
| taxanes | taxol, taxotere | |
| Antimetabolites | thymidylate synthase | 5‐fluorouracil |
| dihydrofolate reductase | methotrexate |
Types of outcome measures
Primary outcomes
Overall survival (OS)
Progression‐free survival/time to progression (PFS/TTP)
Secondary outcomes
Time to treatment failure (TTF)
Objective tumour response rate (OTRR)
Toxicity rates (multiple condition‐specific outcomes)
Quality of life measures (multiple outcomes)
The definitions of some outcomes varied slightly across studies included in this review. Outcomes were commonly defined as:
Overall survival (OS): time elapsed between randomisation (or study enrolment or treatment initiation) to date of death from any cause.
Progression‐free survival (PFS): time elapsed between randomisation (or study enrolment or treatment initiation) and event, with event defined as disease progression or death from any cause.
Time to progression (TTP): time elapsed between randomisation (or study enrolment or treatment initiation) and event, with event defined as disease progression (which sometimes included cause‐specific death from the study disease).
Time to treatment failure (TTF): time elapsed between randomisation (or study enrolment or treatment initiation) to treatment discontinuation for any reason, including disease progression, treatment toxicity, participant preference, or death.
Objective tumour response rate (OTRR): the proportion of participants who experienced a complete or partial tumour response (versus stable disease or no response).
Toxicity rates (multiple condition‐specific outcomes): the proportions of participants who experienced a grade 3 or 4 adverse event of nausea and vomiting, nephrotoxicity, anaemia, hair loss and leukopenia based on WHO criteria or individual protocol‐based definitions. We also investigated treatment‐related death which, for the purpose of this review, was defined as death due to the toxicity of the drug and not to disease progression. If an individual trial did not include their definition of a treatment‐related death but used the terms 'toxic death' or 'lethal toxicity', then these deaths were still included in the pooled analysis of treatment related deaths.
Quality of life measures (QoL) (multiple outcomes): various validated instruments for measuring various quality of life domains.
For the purposes of this review, PFS and TTP were analysed as the same outcome (referred to as PFS/TTP), with preference given to PFS for studies reporting both PFS and TTP extractable data.
Search methods for identification of studies
Electronic searches
For this update of the review, we searched the following databases and registries on the 28 May 2015.
(a) The Cochrane Breast Cancer Specialised Register maintained by the Cochrane Breast Cancer Group (searched 1 June 2015). Details of the search strategies used by the Cochrane Breast Cancer Group for the identification of studies and the procedure used to code references are outlined in their module (www.mrw.interscience.wiley.com/cochrane/clabout/articles/BREASTCA/frame.html). Trials coded with the key words 'advanced', 'Cisplatin', 'cisplatinum', 'carboplatin', 'carboplatinum', 'platin', 'platinum', 'platinum diamminodichloride', 'cis‐diamminedichloroplatinum', 'cis‐dichlorodiammineplatinum', 'biocisplatinum', 'dichlorodiammineplatinum', 'nsc‐119875', 'platidiam', 'platino', 'Platinol', 'cis‐diamminedichloroplatinum', 'cis‐platinum', 'cis‐diammine (cyclobutanedicarboxylato) platinum', 'cbdca', 'jm‐8', 'nsc‐241240', 'paraplatin' were extracted for consideration. (b) Cochrane Central Register of Controlled Trials (CENTRAL; 2015, Issue 4) in the Cochrane Library (searched 28 May 2015). See Appendix 1. (c) MEDLINE (via OvidSP; July 2008 to 28 May 2015). Searches were conducted from 2008 due to the Cochrane Breast Cancer Group's Specialised Register being out‐of‐date for an interim period. See Appendix 2. (d) Embase (Via Embase.com; July 2008 to 28 May 2015). Searches were conducted from 2008 due to the Cochrane Breast Cancer Group's Specialised Register being out‐of‐date for an interim period. See Appendix 3. (e) The WHO International Clinical Trials Registry Platform (ICTRP) search portal (http://apps.who.int/trialsearch/Default.aspx) for all prospectively registered and ongoing trials. See Appendix 4. (f) ClinicalTrials.gov (http://clinicaltrials.gov/ct2/home). See Appendix 5.
No restrictions were applied based on language.
Searching other resources
In addition, we searched the reference lists of other, related literature reviews.
Data collection and analysis
Selection of studies
In the original version of this review and in this review update, two reviewers applied the selection criteria (including the quality of randomisation) to each reference identified by the search strategy while masked to the study results. Any discrepancies regarding eligibility or quality were resolved by consensus or adjudication from a third reviewer. Studies that may appear to have met the eligibility criteria, but which were deemed ineligible are listed in the Characteristics of excluded studies table.
Data extraction and management
In the original version of this review and in this review update, data on the relevant outcomes were extracted by at least two reviewers, with discrepancies resolved by consensus or adjudication from another reviewer. Data were also extracted on information relating to outcome definitions, study accrual, randomisation methods, baseline characteristics of participants (e.g. age, first‐line/second‐line treatment, prior anthracyclines/anthracycline‐naïve), chemotherapy regimens (number of cycles and duration), follow‐up time and analytical methods used. Where available, multiple publications on the same study were obtained and the most complete report was assigned as the primary reference. In instances where a more recent publication was used in this review update for a study that was included in the original version of this review, the year of the reference ID was also updated. Data were entered into the Cochrane Review Manager 5.3 (RevMan 5.3) software, and RevMan 5.3 was used for most statistical analyses.
Assessment of risk of bias in included studies
In this review update, potential sources of bias for all included studies (including those in the original version of this review) were assessed using Cochrane's 'Risk of bias' assessment tool (Higgins 2011). Risk of bias for each treatment‐comparison was evaluated independently by at least two reviewers and discrepancies were resolved by consensus or adjudication from an additional reviewer. Clarification from authors was sought if the published data provided inadequate information for the review. The 'risk of bias' domains that were assessed were 'random sequence generation', 'allocation concealment', 'blinding of participants and personnel', 'blinding of outcome assessment', 'incomplete outcome data', 'selective reporting' and 'other bias'. For each included study, ratings of 'high', 'low', or 'unclear' risk of bias were assigned for each risk of bias domain following criteria outlined in the 'Risk of bias' assessment tool (Higgins 2011).
Open‐label studies are common in phase III oncology trials because it is often difficult to conceal treatments from participants, care‐providers and outcome assessors (due to differences in toxicities and treatment schedules of various treatments, for example). However, because a lack of blinding can affect risk of bias in different ways for different outcomes, we assessed 'blinding of outcome assessment' by dividing outcomes into two outcome classes: 1) OS and 2) outcomes other than OS and quality of life. This division was made because, unlike other outcomes, assessment of OS is unlikely to be affected by non‐blinding.
We also divided the 'incomplete outcome data' risk of bias domain into two outcome classes: 1) time‐to‐event outcomes and 2) binary (i.e. dichotomous) outcomes. For time‐to‐event outcomes, risk of bias was deemed low, unclear, and high risk if time‐to‐event analysis was intention‐to‐treat (ITT), modified intention‐to‐treat (mITT) or per‐protocol, respectively. For the binary outcomes (OTRRs and toxicity rates), risk of bias was deemed low, unclear, and high risk if the highest percentage of randomised participants excluded from effect estimation was less than 10%, between 10% and 15%, or more than 15%, respectively.
For 'risk of bias' domains that were divided into outcome classes, assessments were made for all studies known to be measuring the outcomes, regardless of results being reported in sufficient detail to be included in meta‐analysis or reported at all (e.g. a study might specify OS as an outcome in the study protocol but not report any results).
Measures of treatment effect
OS, PFS/TTP and TTF were analysed as time‐to‐event outcomes, for which the hazard ratio (HR) is the most appropriate measure of treatment effect. If reported, the HR and associated variance were extracted directly from the trial publication(s), and these were used to calculate observed (O) minus expected (E) numbers of events and logrank variance (V) for each treatment‐comparison using the methods described by Tierney 2007 or Parmar 1998. If not reported, O ‐ E and V were obtained indirectly from other available summary statistics or from data extracted from published Kaplan‐Meier curves using the methods described by Tierney 2007 or Parmar 1998. For studies that did not report the relevant effect estimates and required curve extraction, the numbers at risk were based on reported minimum and maximum follow‐up times. If these were not reported, minimum follow‐up was estimated as the time taken to complete treatment, and maximum follow‐up was estimated using the last event reported in the relevant time‐to‐event curve. These follow‐up estimates were recorded in the Characteristics of included studies table under 'Notes'. For the purposes of data extraction, preference was given to time‐to‐event effect estimates derived from ITT analysis, followed by mITT analysis, then per‐protocol analysis.
Pooled HRs and 95% confidence intervals (CIs) were obtained from the O ‐ E and V statistics for each treatment‐comparison using the fixed‐effect model (Yusuf 1985). The pooled HR represented the instantaneous risk of an event (such as death, disease progression or treatment failure) for women receiving platinum divided by the corresponding risk for those not receiving platinum. HRs less than 1.00 favoured the platinum‐containing regimens and values greater than 1.00 favoured non‐platinum regimens.
Toxicity rates and OTRRs were analysed as proportions using the risk ratio (RR) as the measure of treatment effect. OTRRs were most often calculated by trialists using only participants that were assessable for tumour response. These 'assessable participants' were generally defined as participants whose tumour response could be assessed according to prespecified criteria such as RECIST (Eisenhauer 2009), but this definition was sometimes extended to additionally exclude participants who had not received a specified minimum dose of chemotherapy. In this review update and in the original version of this review, we calculated OTRRs using the numbers of assessable participants in the OTRR denominators, but we also separately calculated OTRRs using the numbers of randomised participants in the denominators. In this review update, however, only the results from the assessable participants analyses were reported as there was almost no difference between the results obtained using assessable and randomised participant denominators (the latter are available on request). Toxicity rates were most often calculated by trialists using a 'safety population' of participants who received a specified minimum dose of chemotherapy. We calculated toxicity rates for each study using the population used by that study.
Pooled RRs and 95% confidence intervals were obtained through Mantel‐Haenszel fixed‐effect analysis. The pooled RR represented the cumulative risk of an event for participants receiving platinum divided by the corresponding risk for those not receiving platinum. RRs greater than 1.00 favoured platinum‐containing regimens and values less than 1.00 favoured non‐platinum regimens.
Quality of life was generally reported as a continuous outcome. Hence, if sufficient quality of life data become available for meta‐analysis in future review updates, the effect measure would most likely be the mean difference (MD) or standardised mean difference (SMD), depending on whether the same or different validated questionnaires (respectively) were employed.
Unit of analysis issues
Treatment‐comparisons were the unit of analysis in this review and corresponded to pairwise comparisons of platinum and non‐platinum regimens. Individual studies assessing more than one platinum‐based regimen or more than one non‐platinum regimen (or both) contributed more than one treatment‐comparison to the review. Consequently, there were more treatment‐comparisons in this review than there were studies.
Two studies each contained two non‐platinum regimen (control) groups for comparison against a single platinum‐based regimen (intervention) group. This was taken into account when treatment effect statistics were calculated by splitting each study into two treatment‐comparisons (Stemmler 2011 A, Stemmler 2011 B and Fountzilas 2009 A, Fountzilas 2009 B) and halving the number of participants in their intervention groups (for odd numbered group sizes, the additional participant was arbitrarily distributed to the treatment‐comparison with label ending with 'A'). Another study contained two platinum‐based regimen (intervention) groups for comparison against a single non‐platinum (control) group. This study was split into two treatment‐comparisons (Xu 2011 A, Xu 2011 B) with treatment effect statistics calculated by halving the number of participants in the control group (with additional participants again arbitrarily distributed to treatment‐comparisons with label ending with 'A'). One other study was a four‐armed trial and was also reported as two separate treatment‐comparisons (Berruti 2002 A, Berruti 2002 B). For these two treatment‐comparisons, no adjustments to participant numbers were required as their control and intervention groups were unique to each comparison. These methods for correcting for multiple intervention and/or control groups were suggested in the Cochrane Handbook (Higgins 2011).
Dealing with missing data
Attempts were made to contact a number of trial investigators for additional information. Three trialists (Cocconi, Costanza and Fountzilas) provided additional time‐to‐event data which allowed HRs to be extracted.
Assessment of heterogeneity
Heterogeneity between trial results was assessed using the Chi2 test statistic and the I2 statistic. The Chi2 test statistic assesses the amount of variation in a set of trials. Small P values for the Chi2 test statistic suggest that there is more heterogeneity present than would be expected by chance. Chi2 is not a particularly sensitive test: a cut‐off of P value less than 0.10 is often used to indicate significance, but lack of statistical significance does not mean there is no heterogeneity. I2 is the proportion of variation that is due to heterogeneity rather than chance. In conjunction with the Chi2 test, we used the I2 statistic to assess heterogeneity using the rule of thumb guide outlined in the Cochrane Handbook (Higgins 2011) (i.e. I2 between 0% to 40% might not be important; between 30% to 60% may represent moderate heterogeneity; between 50% to 90% may represent substantial heterogeneity; and between 75% to 100% considerable heterogeneity).
Assessment of reporting biases
In addition to assessing each treatment‐comparison individually for 'selective reporting' using the Cochrane Collaboration's 'Risk of bias' assessment tool (see Assessment of risk of bias in included studies above), publication bias and/or small‐study effects were assessed for the outcomes OS, PFS/TTP and OTRR by visual inspection of funnel plot asymmetry. Egger's statistical test was used to formally assess the degree of asymmetry (Egger 1997).
Data synthesis
For time‐to‐event outcomes, RevMan 5.3 was used to estimate pooled HRs and 95% CIs using fixed‐effect models of the derived or reported observed (O) and expected (E) number of events and the variance of the log‐rank statistic (V) for each trial. For binary outcomes, RevMan 5.3 was used to estimate pooled RRs and 95% CIs using the fixed‐effect Mantel‐Haenszel method.
The standard GRADE system (Guyatt 2011) was to use to rate the quality of evidence relating to the estimated treatment effects on OS, PFS/TTP, OTRR, nausea/vomiting, anaemia, hair loss and leukopenia. GRADE criteria for assessing quality of evidence include 'study design', 'risk of bias', 'inconsistency', 'indirectness', 'imprecision', 'suspected publication bias' and 'other considerations'. Assessments of these criteria and corresponding justifications are provided in three 'Summary of findings' tables largely created using GRADEproGDT (GradeproGDT). GRADE assessments were performed separately for selected subgroups related to effect estimate heterogeneity (or 'inconsistency' as labelled in the GRADE assessment criteria).
Subgroup analysis and investigation of heterogeneity
Subgroup analyses were performed to determine whether the results differed by:
mTNBC status: (a) mTNBC participants, (b) participants unselected for mTNBC;
type of regimen comparison: (a) regimen A + platinum vs regimen A, (b) regimen A + platinum vs regimen B, (c) single agent platinum vs regimen C; (note that we allowed "regimen A" to differ in dosage by small amounts between intervention and control arms)
type of platinum agent in platinum arm: (a) cisplatin, (b) carboplatin, (c) oxaliplatin;
first‐line therapy: (a) first‐line therapy for > 80% of participants, (b) second‐ or third‐line therapy for ≥ 20% of participants;
anthracycline in regimens: (a) no anthracycline in platinum or non‐platinum regimens, (b) platinum + anthracycline vs non‐platinum + anthracycline regimen, (c) platinum + anthracycline vs non‐platinum + non‐anthracycline regimens;
taxane in regimens: (a) no taxane in platinum or non‐platinum regimens, (b) platinum + taxane vs non‐platinum + taxane regimens, (c) platinum + non‐taxane vs non‐platinum + taxane regimens;
trastuzumab in regimens: (a) no trastuzumab in platinum or non‐platinum regimens, (b) platinum + trastuzumab vs non‐platinum + trastuzumab regimens
Possible subgroup differences were assessed using Chi2 tests.
Of the above seven subgroup analyses:
Subgroup analysis 2 was the only 'a priori' subgroup analysis pre‐specified in the review protocol; all other subgroup analyses were 'post hoc'.
Subgroup analyses 2, 4, 5 and 6 were conducted in the original review, although additional subgroups have been added for some of these analyses (eg. the conjugate subgroup 'second‐ or third‐line therapy for ≥ 20% of participants' has been added to the 'first‐line therapy' analysis).
In order to reduce the number of forest plots in this review update, toxicity rates were only assessed overall and by subgroup analysis 3 (as 'type of platinum agent' is known to be related to toxicity). Analyses of toxicity rates by other subgroups are available on request.
In this review update, the subgroupings of a few studies have been corrected from the original version of the review. Furthermore, the subgroup analysis 'first‐line therapy' was incorrectly labelled '100% firstline trials' in the original version of the review and the actual cut‐off that was applied was first‐line therapy for > 80% of participants.
Sensitivity analysis
Sensitivity analyses were performed to assess whether apparent inconsistencies in results relating to OS, PFS/TTP and OTRR were more likely to be due to differences in the underlying nature of these outcomes, or an artefact of the different treatment‐comparisons available for the meta‐analysis of each outcome. First, the pooled effect estimate for OTRR was recalculated restricting the meta‐analysis to the 19 treatment‐comparisons with extractable data on OS. Second, the pooled effect estimates for PFS/TTP and OS were recalculated restricting both meta‐analyses to the 12 treatment‐comparisons with extractable data on both OS and PFS/TTP.
In additional sensitivity analyses, PFS/TTP estimates were stratified according to whether the outcome was PFS or TTP. For these analyses, estimates were classified as PFS if the event of interest was defined as disease progression or death from any cause. Estimates were classified as TTP if the event of interest was defined as disease progression, which may also include cause‐specific death from breast cancer. In instances where the event of interest was ambiguously defined or not defined at all, we relied on the authors label of the outcome for classifying as PFS or TTP.
Lastly, to assess the sensitivity of our primary results to our choice of analytical method, we repeated Analysis 1.1, Analysis 1.2 and Analysis 1.3 but using random‐effects rather than fixed‐effect methods.
1.1. Analysis.
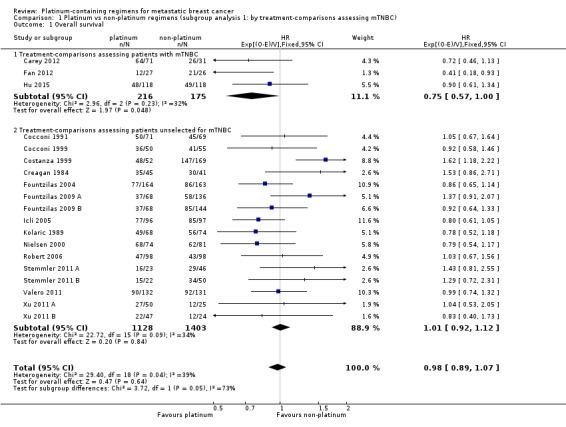
Comparison 1 Platinum vs non‐platinum regimens (subgroup analysis 1: by treatment‐comparisons assessing mTNBC), Outcome 1 Overall survival.
1.2. Analysis.
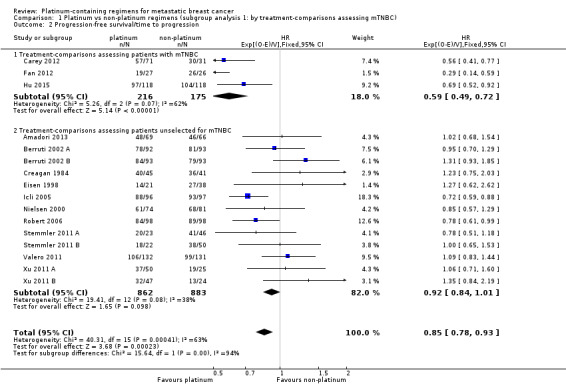
Comparison 1 Platinum vs non‐platinum regimens (subgroup analysis 1: by treatment‐comparisons assessing mTNBC), Outcome 2 Progression‐free survival/time to progression.
1.3. Analysis.
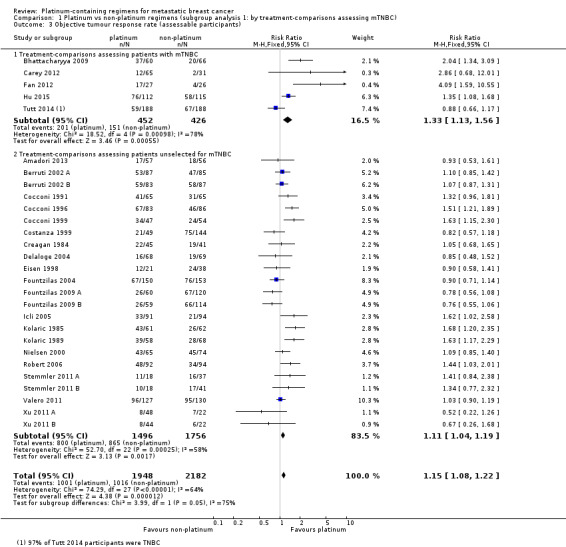
Comparison 1 Platinum vs non‐platinum regimens (subgroup analysis 1: by treatment‐comparisons assessing mTNBC), Outcome 3 Objective tumour response rate (assessable participants).
Results
Description of studies
Results of the search
For the 2016 review update, we reviewed 1644 unique records identified by the May 2015 database searches (Figure 1). Of these, 1610 could be excluded based on information in the title or abstract, and nine records from trial registries or protocol publications were considered to be potentially relevant ongoing studies yet to publish results (see Characteristics of ongoing studies). For the remaining 25 records, we retrieved full‐text articles or abstracts for further examination. Eight of the 25 articles or abstracts were excluded because they were reviews (but these were still used to search for further studies by handsearching the bibliographies) and three other full‐text articles were excluded for reasons outlined in the Characteristics of excluded studies table.
1.
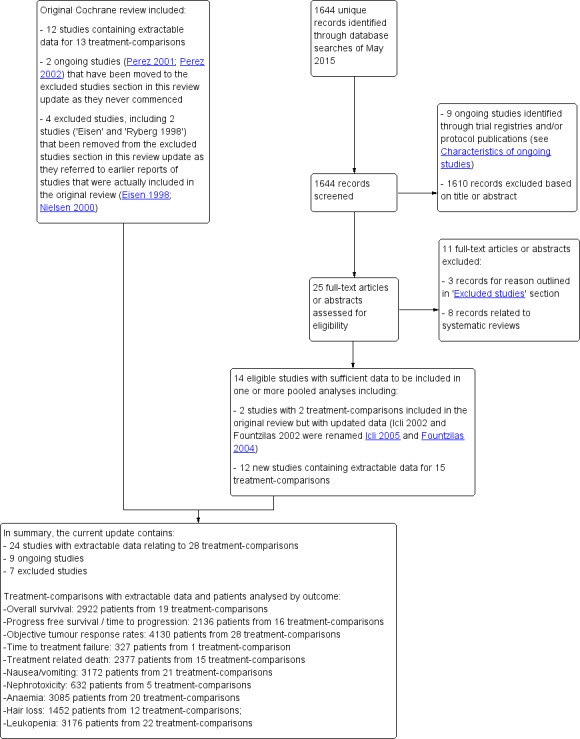
Review update 2016: study flow diagram.
Included studies
Fourteen studies containing 17 treatment‐comparisons with sufficient data to be included in one or more pooled analyses were identified by this review update. Of these 17 treatment‐comparisons, two had been included in the original review but had since published updated results (Icli 2002 and Fountzilas 2002 in the original review were renamed Icli 2005 and Fountzilas 2004 in this review update), and 15 were new to the review (Amadori 2013 ;Bhattacharyya 2009; Carey 2012; Fan 2012; Delaloge 2004; Fountzilas 2009 A; Fountzilas 2009 B; Hu 2015; Robert 2006; Stemmler 2011 A; Stemmler 2011 B; Tutt 2014; Valero 2011; Xu 2011 A; Xu 2011 B). Overall, the review now includes results from 24 studies corresponding to 28 treatment‐comparisons (see Characteristics of included studies).
Of the 28 treatment‐comparisons included in this review update (Table 6):
3. Number of treatment‐comparisons included in meta‐analyses by subgroup and three outcomes.
| Outcome | ||||
| Subgroup |
Treatment‐ comparisons N |
Overall survival n (% of N) |
Progression ‐free survival/time to progression n (% of N) |
Objective response rate n (% of N) |
| Overall: | 28 | 19 (68%) | 16 (57%) | 28 (100%) |
| Type of platinum agent in platinum arm: | ||||
| Cisplatin in platinum arm | 17 | 11 (65%) | 11 (65%) | 17 (100%) |
| Carboplatin in platinum arm | 10 | 8 (80%) | 5 (50%) | 10 (100%) |
| Oxaliplatin in platinum arm | 1 | (0%) | (0%) | 1 (100%) |
| Type of regimen comparison: | ||||
| Regimen A + platinum agent vs regimen A | 9 | 6 (67%) | 6 (67%) | 9 (100%) |
| Regimen A + platinum agent vs regimen B | 18 | 13 (72%) | 10 (56%) | 18 (100%) |
| Single agent platinum vs regimen C | 1 | (0%) | (0%) | 1 (100%) |
| First‐line therapy: | ||||
| First‐line therapy for > 80% of participants | 20 | 15 (75%) | 11 (55%) | 20 (100%) |
| Second‐ or third‐line therapy for ≥ 20% of participants | 8 | 4 (50%) | 5 (63%) | 8 (100%) |
| Participant selection for mTNBC: | ||||
| Participants with mTNBC | 5 | 3 (60%) | 3 (60%) | 5 (100%) |
| Participants unselected for mTNBC | 23 | 16 (70%) | 13 (57%) | 23 (100%) |
| Anthracycline in regimens: | ||||
| No anthracycline in platinum or non‐platinum regimens | 18 | 14 (78%) | 11 (61%) | 18 (100%) |
| Platinum + anthracycline vs non‐platinum + anthracycline regimens | 6 | 3 (50%) | 4 (67%) | 6 (100%) |
| Platinum + anthracycline vs non‐platinum + non‐anthracycline regimens | 4 | 2 (50%) | 1 (25%) | 4 (100%) |
| Taxane in regimens: | ||||
| No taxane in platinum or non‐platinum regimens | 17 | 9 (53%) | 9 (53%) | 17 (100%) |
| Platinum + taxane vs non‐platinum + taxane regimens | 6 | 6 (100%) | 3 (50%) | 6 (100%) |
| Platinum + non‐taxane vs non‐platinum + taxane regimens | 5 | 4 (80%) | 4 (80%) | 5 (100%) |
| Trastuzumab in regimens: | ||||
| No trastuzumab in platinum or non‐platinum regimens | 26 | 17 (65%) | 14 (54%) | 26 (100%) |
| Platinum + trastuzumab vs non‐platinum + trastuzumab regimens | 2 | 2 (100%) | 2 (100%) | 2 (100%) |
17 (61%) used cisplatin, 10 (36%) used carboplatin and one (4%) used oxaliplatin as the platinum agent in the intervention arm;
9 (32%) compared 'regimen A + platinum vs regimen A', 18 (64%) compared 'regimen A + platinum vs regimen B' and one (4%) compared single agent platinum vs regimen C' (Table 7);
20 (71%) had more than 80% of participants receiving first‐line therapy;
5 (18%) were designed to assess participants with mTNBC;
18 (64%) had no anthracycline in the platinum or non‐platinum regimens, six (21%) had an anthracycline in both regimens, and four (14%) had an anthracycline in the platinum regimen only;
17 (61%) had no taxane in the platinum or non‐platinum regimens; six (21%) had a taxane in both regimens and five (18%) had a taxane in the non‐platinum regimen only;
26 (93%) had no trastuzumab in the platinum or non‐platinum regimens and two (7%) had a trastuzumab in both regimens.
4. Summary of regimens included in the analysis.
| Trials ID | Arm 1 (platinum‐containing) | Arm 2 (control) | First‐line therapy for > 80% of participants | Majority participants anthracycline‐naive |
| Regimen A + platinum vs regimen A | ||||
| Berruti 2002 A | Epi + Cis (epirubicin+cisplatin) | Epi (epirubicin) | Y | Y |
| Berruti 2002 B | Epi + Cis + LND (epirubicin+cisplatin+lonidamine) | Epi + LND (epirubicin + lonidamine) | Y | Y |
| Bhattacharyya 2009 | Endoxan 50 mg per day at 10 am and methotrexate 2.5 mg twice a day at 9 am and 5 pm and with 'cisplatinum' | Endoxan 50 mg per day at 10 am and methotrexate 2.5 mg twice a day at 9 am and 5 pm | N | N |
| Carey 2012 | C + Cb (Cetuximab + carboplatin) | C (Cetuximab with carboplatin added after progression) | N | N |
| Costanza 1999 | CBDA + CAF (carboplatin + cycloheximide + doxorubicin + fluorouracil + methotrexate) | CAF (cycloheximide + doxorubicin + fluorouracil) (methotrexate substituted after total doxorubicin ‐ 540 mg) |
Y | Unclear |
| Fountzilas 2009 A | PCb (paclitaxel + carboplatin) | Pw (paclitaxel) | Y | N |
| Nielsen 2000 | Epi + Cis (epirubicin + cisplatin) | Epi (epirubicin) | Y | Y |
| Robert 2006 | TPC (trastuzumab + paclitaxel + carboplatin) | TP (trastuzumab + paclitaxel) | Y | Unclear |
| Valero 2011 | TCH (trastuzumab + docetaxel + carboplatin) | TH (trastuzumab + docetaxel) | Y | N |
| Regimen A + platinum vs regimen B | ||||
| Amadori 2013 | (pemetrexed + carboplatin) | (vinorelbine + gemcitabine) | N | N |
| Cocconi 1991 | MPEPIV(a) or MPEMi (b) (a: methotrexate, leucovorin, cisplatin, epirubicin, vincristine; b: methotrexate, leucovorin, cisplatin, etoposide, mitomycin) | CMF (cyclophosphamide, methotrexate, fluorouracil) | Y | Y |
| Cocconi 1996 | Etop + Cis (etoposide + cisplatin) | CMF (cyclophosphamide, methotrexate, fluorouracil) | Y | Unclear |
| Cocconi 1999 | PE + CMF + AL (cisplatin + etoposide + doxorubicin + cyclophosphamide, methotrexate + fluorouracil, lecovorin + allopurinol) | CMF (cyclophosphamide, methotrexate, fluorouracil) | Y | Unclear |
| Creagan 1984 | CFP + CAP (cyclophosphamide + doxorubicin + cis‐dichlordiammine + CFP) | CFP (cyclophosphamide + fluorouracil + prednisone) | Y | Y |
| Delaloge 2004 | OXA (oxaliplatin + 5‐flurouracil) | VIN (vinorelbine) | N | N |
| Eisen 1998 | EcisF (5‐flurouracil + epirubicin + cisplatin) | EcycloF (5‐flurouracil + epirubicin + cyclophosphamide) | Y | Y |
| Fan 2012 | TP (docetaxel + cisplatin) | TX (docetaxel + capecitabine) | Y | N |
| Fountzilas 2004 | Epi + Pcb (epirubicin + carboplatin) | Epi + P (epirubicin + paclitaxel) | Y | Y (54%) |
| Fountzilas 2009 B | PCb (paclitaxel + carboplatin) | GDoc (docetaxel + gemcitabine) | Y | N |
| Hu 2015 | (cisplatin + gemcitabine) | (paclitaxel + gemcitabine) | Y | N |
| Icli 2005 | Etop + Cis (etoposide + cisplatin) | P (paclitaxel) | N | N |
| Kolaric 1985 | CAP (cyclophosphamide + doxorubicin + platinum) | CMFVP (cyclophosphamide + methotrexate + 5‐fluorouracil + vincristine + prednisone) | Y | Y |
| Kolaric 1989 | CAP (cyclophosphamide + doxorubicin +platinum) | FAC (5‐flurouracil + doxorubicin + cyclophosphamide) |
Y | Y |
| Stemmler 2011 A | GemCis (gemcitabine + cisplatin) | GemVin (gemcitabine + vinorelbine) | N | N |
| Stemmler 2011 B | GemCis (gemcitabine + cisplatin) | GemCap (gemcitabine + capecitabine) | N | N |
| Xu 2011 A | GemCis (gemcitabine + cisplatin) | GemPac (gemcitabine + paclitaxel) | Y | N |
| Xu 2011 B | GemCarb (gemcitabine + carboplatin) | GemPac (gemcitabine + paclitaxel) | Y | N |
| Single agent platinum vs regimen C | ||||
| Tutt 2014 | C (carboplatin) | D (docetaxel) | N | Unclear |
Not all the studies provided sufficient information on all outcomes for inclusion in meta‐analyses. Of the 28 treatment‐comparisons:
19 (68%), 16 (57%) and 28 (100%) had sufficient data to be included in the meta‐analyses of effect estimates for OS, PFS/TTP and OTRR, respectively (Table 6 and Figure 1);
15 (54%), 21 (75%), 5 (18%), 20 (71%), 12 (43%) and 22 (79%) had sufficient data to be included in the meta‐analyses of effect estimates for treatment‐related death, nausea/vomiting, nephrotoxicity, anaemia, hair loss, and leukopenia, respectively (Table 8 and Figure 1).
5. Summary of outcomes for included trials.
| Trial ID | Extractable OS data for HR estimation1 | Median OS time2 | Extractable PFS/TTP data for HR estimation1 | Median PFS/TTP time2 | Overall response | Treatment‐related deaths | Grade III & IV Toxicity | Accrual3 |
| Regimen A + platinum vs regimen A | ||||||||
| Berruti 2002 A | NR | Y | Y | Y | Y | Y | Nausea/vomiting Nephrotoxicity Anaemia Leukopenia |
185 |
| Berruti 2002 B | NR | Y | Y | Y | Y | Y | Nausea/vomiting Nephrotoxicity Anaemia Leukopenia |
186 |
| Bhattacharyya 2009 | NR | Y | NR | Y | Y | NR | NR | 126 |
| Carey 2012 | Y | Y | Y | NR | Y | NR | Not extractable | 102 |
| Costanza 1999 | Y | Y | NR | NR | Y | Y | Nausea/vomiting Anaemia Leukopenia |
221 |
| Fountzilas 2009 A | Y | Y | NR | NR | Y | Y | Nausea/vomiting Anaemia Hair loss Leukopenia |
204 |
| Nielsen 2000 | Y | Y | Y | Y | Y | Y | Nausea/vomiting Leukopenia |
155 |
| Robert 2006 | Y | Y | Y | Y | Y | NR | Nausea/vomiting Anaemia Leukopenia |
196 |
| Valero 2011 | Y | Y | Y | Y | Y | Y | Nausea/vomiting Anaemia |
263 |
| Regimen A + platinum vs regimen B | ||||||||
| Amadori 2013 | NR | NR | Y | Y | Y | Y | Leukopenia | 135 |
| Cocconi 1991 | Y | Y | NR | Y | Y | NR | Anaemia Leukopenia |
140 |
| Cocconi 1999 | Y | Y | NR | Y | Y | NR | Nausea/vomiting Anaemia Hair loss Leukopenia |
105 |
| Cocconi 1996 | NR | Y | NR | Y | Y | DU | Not extractable | 169 |
| Creagan 1984 | Y | Y | Y | Y | Y | NR | Nausea/vomiting Hair loss Leukopenia |
86 |
| Delaloge 2004 | NR | Y | NR | Y | Y | Y | Not extractable | 137 |
| Eisen 1998 | NR | Y | Y | Y | Y | DU | Nausea/vomiting Anaemia Hair loss Leukopenia |
59 |
| Fan 2012 | Y | Y | Y | Y | Y | Y | Nausea/vomiting Anaemia Leukopenia |
53 |
| Fountzilas 2004 | Y | NR | NR | NR | Y | NR | Nausea/vomiting Anaemia Leukopenia |
327 |
| Fountzilas 2009 B | Y | Y | NR | NR | Y | Y | Nausea/vomiting Anaemia Hair loss Leukopenia |
212 |
| Hu 2015 | Y | NR | Y | Y | Y | Y | Nausea/vomiting Anaemia Hair loss Leukopenia |
236 |
| Icli 2005 | Y | Y | Y | NR | Y | Y | Nausea/vomiting Anaemia Leukopenia |
193 |
| Kolaric 1985 | NR | NR | NR | NR | Y | NR | Nausea/vomiting Nephrotoxicity Anaemia Hair loss Leukopenia |
123 |
| Kolaric 1989 | Y | Y | NR | NR | Y | Y | Nausea/vomiting Anaemia Hair loss Leukopenia |
142 |
| Stemmler 2011 A | Y | Y | Y | Y | Y | NR | Nausea/vomiting Anaemia Hair loss Leukopenia |
69 |
| Stemmler 2011 B | Y | Y | Y | Y | Y | NR | Nausea/vomiting Anaemia Hair loss Leukopenia |
72 |
| Xu 2011 A | Y | Y | Y | Y | Y | Y | Nausea/vomiting Nephrotoxicity Anaemia Hair loss Leukopenia |
75 |
| Xu 2011 B | Y | Y | Y | Y | Y | Y | Nausea/vomiting Nephrotoxicity Anaemia Hair loss Leukopenia |
71 |
| Single agent platinum vs regimen C | ||||||||
| Tutt 2014 | NR | Y | NR | Y | Y | NR | NR | 376 |
1 Sufficient data reported to estimate a HR for pooling as outlined by Parmar 1998 and Tierney 2007; this includes Kapalan‐Meier curve, HR and standard error/confidence interval or logrank statistics
2 Trials that did not explicitly report median time were classified as NR here regardless of estimable median time from Kaplan‐Meier curve
3 Accrual numbers represent the maximum numbers of participants in the trial (not study) that were included in the analyses of OS, PFS/TTP or OR (assessable participants).
*NR = not reported, DU = deaths unexplained, Y = data reported.
Three studies reported treatment effects on various quality of life domains (Fountzilas 2004; Fountzilas 2009: Fountzilas 2009 A & Fountzilas 2009 B; Amadori 2013) but these results could not be pooled in a meta‐analysis. Three studies reported TTF results (Amadori 2013; Fountzilas 2004; Xu: Xu 2011 A & Xu 2011 B), but these data were only extractable for Fountzilas 2004.
Excluded studies
In this review update, seven studies may have appeared to have met the eligibility criteria, but were deemed ineligible for reasons given in the Characteristics of excluded studies table. Of these seven excluded studies: two were registered trials that were previously listed as 'ongoing studies' in the original review but were reclassified as 'excluded' in this review update because the principal investigator had informed us that the trials never commenced (Perez 2001; Perez 2002); two were 'excluded studies' in the original review and remained so in this review update (Cartei 1996; Hogdall 1993); and three were newly identified in this review update (Crump 2008; Somlo 2015; Wang 2008). Two other studies listed as 'excluded' in the original review ('Eisen' and 'Ryberg 1998') were removed from the excluded studies section altogether in this review update as they referred to earlier reports of studies that were actually included in the original review (Eisen 1998; Nielsen 2000).
Risk of bias in included studies
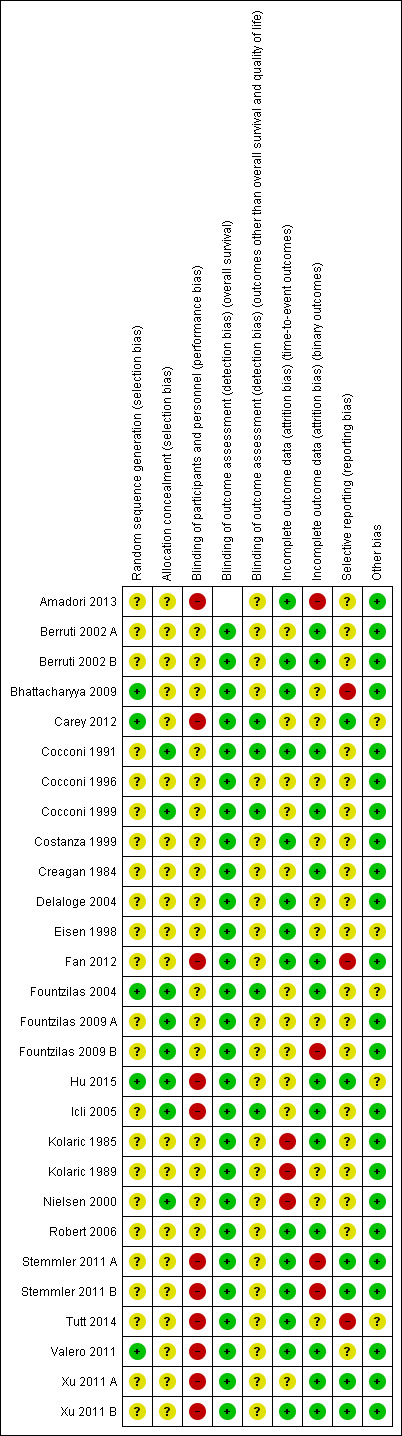
Figure 2 shows a summary of the 'Risk of bias' judgements for each 'Risk of bias' domain of the included treatment‐comparisons. Reasons for each judgement are detailed for each treatment‐comparison in the Characteristics of included studies table. For each 'risk of bias' domain, a summary of the general risk of bias for results of the included studies was as follows.
2.
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
All 24 studies, relating to 28 treatment‐comparisons, were described as randomised (this being an inclusion criterion of the review). The method of random sequence generation was described sufficiently to be judged at low risk of bias for this domain in 5 treatment‐comparisons (Bhattacharyya 2009; Carey 2012; Fountzilas 2004; Hu 2015; Valero 2011). The remaining 23 treatment‐comparisons were judged to be at unclear risk of bias for random sequence generation as the information available was insufficient to accurately assess this domain.
Eight of the 28 treatment‐comparisons described central randomisation systems and were thus judged to be at low risk of bias for treatment allocation concealment (Cocconi 1991; Cocconi 1999; Fountzilas 2004; Fountzilas 2009 A; Fountzilas 2009 B; Hu 2015; Icli 2005; Nielsen 2000). The remaining 20 treatment‐comparisons did not adequately describe methods of concealment and were thus judged as having unclear risk of bias for this domain.
Blinding
Eleven treatment‐comparisons were described as 'nonblinded', 'not blinded', 'single blind' or 'open‐label' (Amadori 2013; Carey 2012; Fan 2012; Hu 2015; Icli 2005; Stemmler 2011 A; Stemmler 2011 B; Tutt 2014; Valero 2011; Xu 2011 A; Xu 2011 B). These 11 'unblinded' treatment‐comparisons were judged to be at high risk of 'performance bias' due to the lack of blinding of participants and personnel to the treatment being administered. The remaining 17 treatment‐comparisons were judged as at unclear risk of 'performance bias' because of a lack of information needed to make a firm conclusion. It seemed highly likely, however, that a number of these 17 treatment‐comparisons would have also been 'unblinded', as open‐label studies are common in phase III oncology trials.
All 27 treatment‐comparisons known to have OS as a study outcome (including 8 not included in OS meta‐analyses but excluding Amadori 2013 which did not assess OS as an outcome) were judged to be at low risk of bias from a lack of blinding of outcome assessors, regardless of actual blinding. This is because death certification was unlikely to be affected by any lack of blinding.
For outcomes other than OS and QoL, five treatment‐comparisons were judged to be at low risk of bias from a lack of blinding of outcome assessors due to these outcomes being measured/confirmed through formal assessments including imaging, biochemical tests and/or the involvement of an independent clinical or radiological review group (Carey 2012; Cocconi 1991; Cocconi 1999; Fountzilas 2004; Icli 2005). The remaining 23 treatment‐comparisons provided insufficient detail on outcome assessments and were thus classified as having an unclear risk of bias.
Incomplete outcome data
Three treatment‐comparisons were judged to be at high risk of attrition bias for time‐to‐event outcomes because participants who did not receive a specified number of cycles of chemotherapy were excluded from time‐to‐event analyses (a form of per‐protocol analysis) (Kolaric 1985; Kolaric 1989; Nielsen 2000). Eleven treatment‐comparisons excluded randomised participants who never started treatment or who were subsequently found to have been 'ineligible' from time‐to‐event analyses (mITT analyses) (Berruti 2002 A; Carey 2012; Cocconi 1999; Cocconi 1996; Creagan 1984; Fountzilas 2004; Fountzilas 2009 A; Fountzilas 2009 B; Hu 2015; Icli 2005; Xu 2011 A). These 11 treatment‐comparisons were judged to be at unclear risk of attrition bias for time‐to‐event outcomes. The remaining 14 treatment‐comparisons were judged to be at low risk of attrition bias for time‐to‐event outcomes because all randomised participants were analysed in the groups to which they were randomised (ITT analysis).
Four treatment‐comparisons had more than 15% of participants not assessed/assessable for at least one binary outcome and were thus judged to be at high risk of attrition bias for binary outcomes (Amadori 2013; Fountzilas 2009 B; Stemmler 2011 A; Stemmler 2011 B; ). Fourteen treatment‐comparisons had less than 10% of participants not assessed/assessable for all binary outcomes and were thus judged to be at low risk of attrition bias for binary outcomes (Berruti 2002 A; Berruti 2002 B; Cocconi 1991; Cocconi 1999; Creagan 1984; Fan 2012; Fountzilas 2004; Hu 2015; Icli 2005; Kolaric 1985; Robert 2006; Valero 2011; Xu 2011 A; Xu 2011 B). The remaining 10 treatment‐comparisons were judged to be at unclear risk of attrition bias for binary outcomes (10 to 15% of participants not assessed/assessable for at least one binary outcome or it was unclear what proportion were not assessed).
Selective reporting
The assessment of risk of bias from selective reporting included crosschecking the outcomes for which there were published results against the stated outcomes reported in trial registers (WHO ICTRP and ClinicalTrials.gov) and published protocols. In our assessment of risk of bias from selective reporting, studies that began recruiting participants on or after July 1, 2005 were expected have a clinical registration or published protocol specifying the study outcome or they were deemed to be at high risk of bias from selective reporting. We chose July 1, 2005 as our cut‐off date because the International Committee of Medical Journal Editors (ICMJE) made a seminal announcement in September 2004 that clinical trials that begin recruiting on or after July 1, 2005 would not be considered for publication unless they were included on a clinical trials registry (De Angelis 2005). Studies included in this review which began recruiting participants before July 1, 2005 and which did not have a trial registration or published protocol prespecifying study outcomes, were assumed to be at unclear risk of bias from selective reporting, unless additional evidence suggested otherwise.
Six treatment‐comparisons from four studies were judged to be at low risk of bias from the selective reporting of outcomes (Carey 2012; Hu 2015; Stemmler 2011 A; Stemmler 2011 B; Xu 2011 A; Xu 2011 B). Each of these studies was included on a clinical trials registry and their prespecified outcomes either matched those in the trial reports or non‐matches were considered to be relatively minor. Three treatment‐comparisons were judged to be at high risk of bias from the selective reporting of outcomes. The first of these was Tutt 2014 which specified TTP, TTF and toxicity as outcomes in the trial registration, but did not report on these outcomes in either of two conference abstracts (future publications may report in more detail and this 'risk of bias' assessment may change). The second was Fan 2012 which did not have a trial registration or published protocol and did not report the date that participants were first enrolled. However, given that Fan 2012 was first published in December 2012 and that there were only 53 study participants, it seemed highly likely that recruitment began after July 1, 2005. Consequently, there was a high expectation of trial registration which this study failed to undertake. The third of these was Bhattacharyya 2009 in which the abstract indicated that toxicity was recorded but no results were reported. In addition, for Bhattacharyya 2009, there was no trial registration or published protocol containing prespecified outcomes. The date that participant recruitment began was not reported, but given that this was first published in September 2009, it seemed likely that recruitment began after July 1, 2005. As of April 2015, there had been no further results published other than those in the conference abstract. The remaining 19 treatment‐comparisons were judged to be at unclear risk of bias from the selective reporting of outcomes either because the trial was unregistered with recruitment starting before July 1, 2005 or the stated outcomes in the registry did not match those in the trial reports, but it was unclear if these non‐matches posed a high risk of bias.
Egger's tests for funnel plot asymmetry did not identify evidence consistent with the presence of publication bias or small‐study effects, or both, for OS (P value = 0.98; Figure 3), PFS/TTP (P value = 0.36) or OTRR (P value = 0.45).
3.
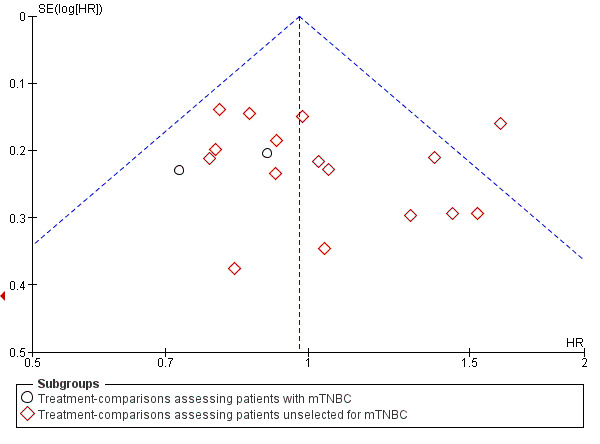
Funnel plot 1: Overall survival (OS). Assessing publication bias and/or small‐study effects. Plot includes all treatment‐comparisons with extractable data for OS. The plot does not show asymmetry (Egger's test P value = 0.98)
Other potential sources of bias
Five treatment‐comparisons were judged to be at unclear risk of 'other bias' (Carey 2012; Eisen 1998; Fountzilas 2004; Hu 2015; Tutt 2014) for various reasons outlined in the Characteristics of included studies table. The remaining 23 treatment‐comparisons were judged to be at low risk of 'other bias'.
Effects of interventions
See: Table 1; Table 2; Table 3
Refer to Table 1; Table 2; Table 3.
Overall survival (OS)
Twenty‐seven of the 28 included treatment‐comparisons assessed OS as an outcome; 19 provided sufficient OS data for pooling in meta‐analyses. From these 19 treatment‐comparisons, 2922 of 2956 randomised participants were analysed representing 99% of randomised participants in these treatment‐comparisons (and there were about 1868 deaths). There was no statistically significant difference in survival between platinum‐containing regimens or non‐platinum regimens with a HR of 0.98 (95% CI 0.89 to 1.07, P = 0.64) with some heterogeneity identified across trials (P = 0.04; I2 = 39%) (Analysis 1.1; Figure 4).
4.
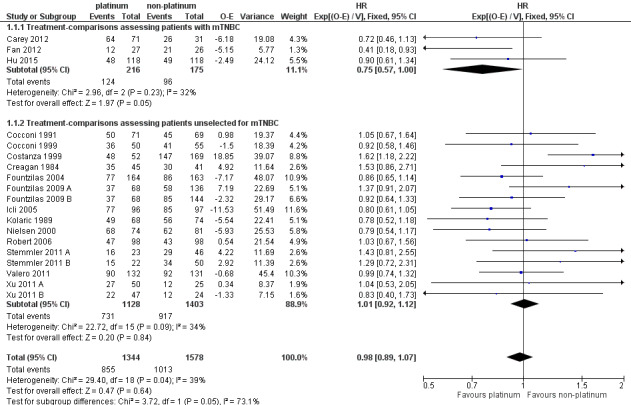
Forest plot of comparison: 1 Platinum vs non‐platinum regimens (subgroup analysis 1: by treatment‐comparisons assessing mTNBC), outcome: 1.1 Overall survival.
Subgroup analysis showed a marginally significant survival benefit in favour of platinum‐containing regimens for three treatment‐comparisons assessing women with metastatic triple‐negative breast cancer (mTNBC) only (HR 0.75, 95% CI 0.57 to 1.00; P = 0.05; low‐quality evidence) (Analysis 1.1; Figure 4). The difference between this pooled HR of 0.75 and that from 16 treatment‐comparisons assessing participants unselected for TNBC (HR 1.01, 95% CI 0.92 to 1.12; high‐quality evidence) was marginally significant (P = 0.05); heterogeneity was not large in either subgroup (P = 0.23, I2 = 32% and P = 0.09, 34% respectively). The six other subgroup analyses showed no evidence of subgroup differences (P values ranged from P = 0.09 to P = 0.86; see Analysis 2.1; Analysis 3.1; Analysis 4.1; Analysis 5.1; Analysis 6.1; Analysis 7.1).
2.1. Analysis.
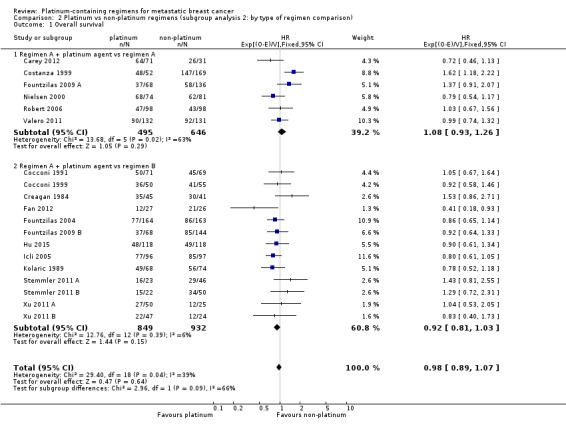
Comparison 2 Platinum vs non‐platinum regimens (subgroup analysis 2: by type of regimen comparison), Outcome 1 Overall survival.
3.1. Analysis.
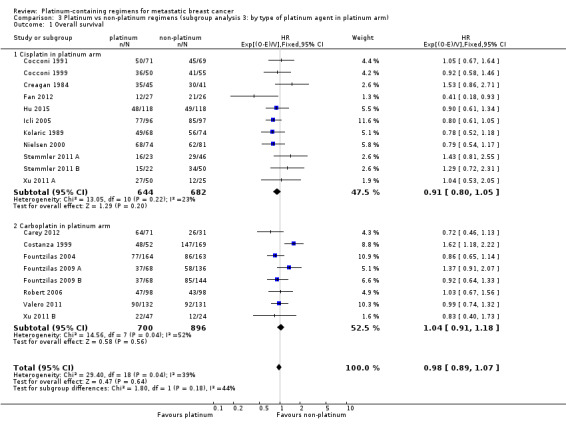
Comparison 3 Platinum vs non‐platinum regimens (subgroup analysis 3: by type of platinum agent in platinum arm), Outcome 1 Overall survival.
4.1. Analysis.
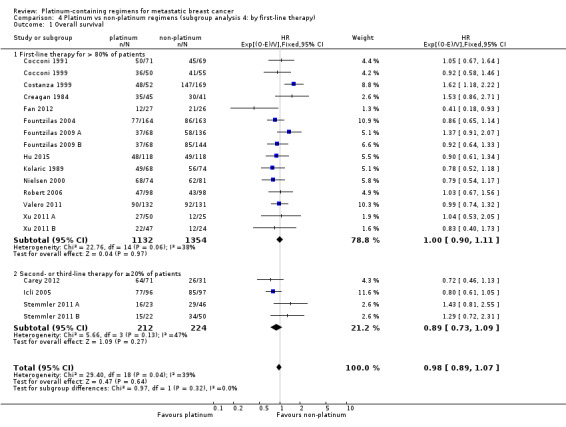
Comparison 4 Platinum vs non‐platinum regimens (subgroup analysis 4: by first‐line therapy), Outcome 1 Overall survival.
5.1. Analysis.
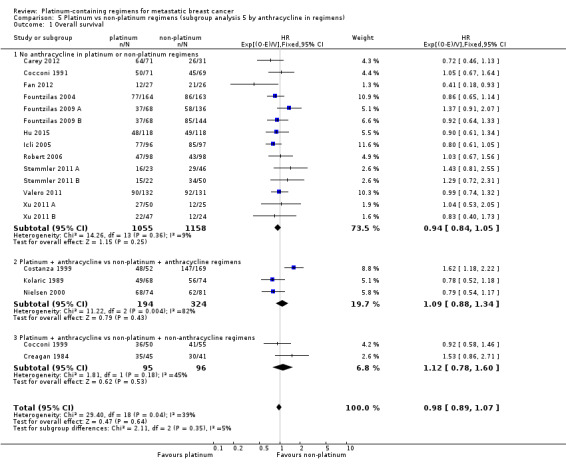
Comparison 5 Platinum vs non‐platinum regimens (subgroup analysis 5 by anthracycline in regimens), Outcome 1 Overall survival.
6.1. Analysis.
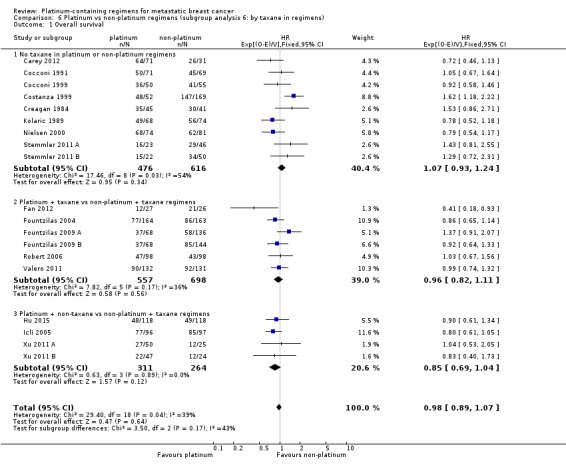
Comparison 6 Platinum vs non‐platinum regimens (subgroup analysis 6: by taxane in regimens), Outcome 1 Overall survival.
7.1. Analysis.
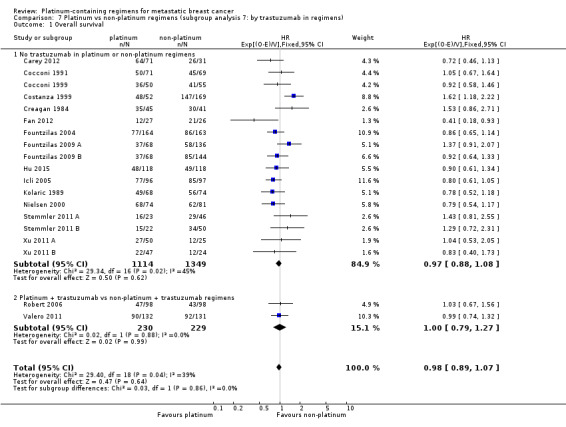
Comparison 7 Platinum vs non‐platinum regimens (subgroup analysis 7: by trastuzumab in regimens), Outcome 1 Overall survival.
Progression‐free survival/time to progression (PFS/TTP)
Twenty‐four of the 28 included treatment‐comparisons assessed PFS or TTP, or both, as an outcome; 16 provided sufficient data for pooling in meta‐analyses of the composite outcome of PFS/TTP. From these 16 treatment‐comparisons, 2136 out of 2162 (99%) randomised participants were analysed (and there were about 1772 events). There was a statistically significant difference in favour of platinum‐containing regimens (HR 0.85, 95% CI 0.78 to 0.93, P = 0.0002), although there was marked evidence of heterogeneity (P = 0.0004; I2 = 63%) (Analysis 1.2; Figure 5).
5.
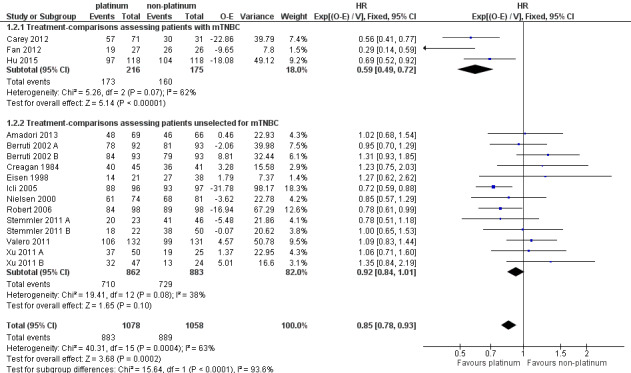
Forest plot of comparison: 1 Platinum vs non‐platinum regimens (subgroup analysis 1: by treatment‐comparisons assessing mTNBC), outcome: 1.2 Progression‐free survival/time to progression.
Significant differences in the pooled HRs of subgroups were found in three of the seven subgroup analyses involving PFS/TTP. Specifically, there were significant differences in the pooled HRs for subgroups of treatment‐comparisons:
assessing participants with mTNBC (HR 0.59, 95% CI 0.49 to 0.72; heterogeneity P = 0.07, I2 = 62%; n = 3; low‐quality evidence) and unselected for mTNBC (HR 0.92, 95% CI 0.84 to 1.01; heterogeneity P = 0.08, I2 = 38%; n = 13; moderate‐quality evidence) (P < 0.0001 for subgroup difference) (Analysis 1.2; Figure 5).
with > 80% of participants receiving first‐line therapy (HR 0.93, 95% CI 0.83 to 1.03; heterogeneity P = 0.003, I2 = 63%; n = 11) and > 20% of participants receiving second‐ or third‐line therapy (HR 0.75, 95% CI 0.65 to 0.86; heterogeneity P = 0.12, I2 = 45%; n = 5) (P = 0.01 for subgroup difference) (Analysis 4.2).
with no anthracycline in the platinum or non‐platinum regimens (HR 0.80, 95% CI 0.73 to 0.88; heterogeneity P = 0.001, I2 = 65%; n = 11), an anthracycline in both the platinum and non‐platinum regimens (HR 1.05, 95% CI 0.86 to 1.27; heterogeneity P = 0.35, I2 = 8%; n = 4) and an anthracycline in the platinum regimen only (HR 1.23, 95% CI 0.75 to 2.03; n = 1) (P = 0.02 for subgroup difference) (Analysis 5.2).
4.2. Analysis.
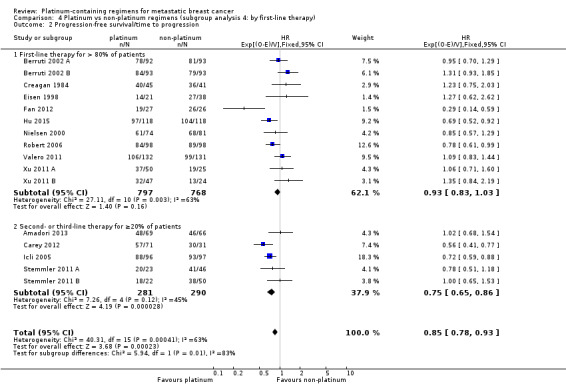
Comparison 4 Platinum vs non‐platinum regimens (subgroup analysis 4: by first‐line therapy), Outcome 2 Progression‐free survival/time to progression.
5.2. Analysis.
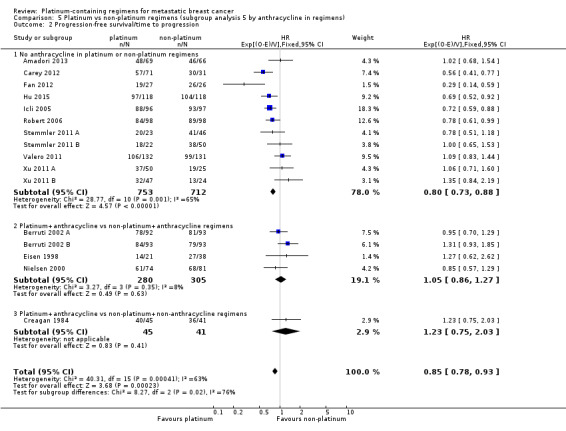
Comparison 5 Platinum vs non‐platinum regimens (subgroup analysis 5 by anthracycline in regimens), Outcome 2 Progression‐free survival/time to progression.
The four other subgroup analyses showed no evidence of subgroup differences (P values ranged from P = 0.34 to P = 0.88; see Analysis 3.2; Analysis 6.2; Analysis 7.2; Analysis 8.2).
3.2. Analysis.
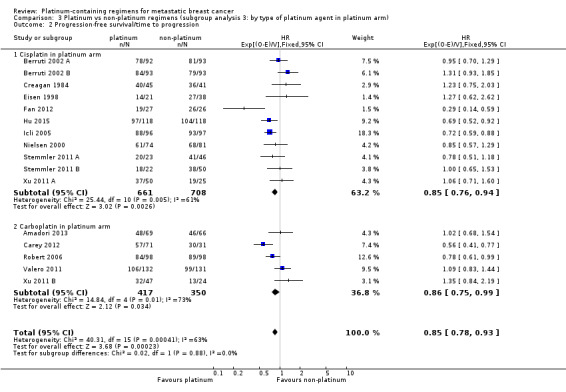
Comparison 3 Platinum vs non‐platinum regimens (subgroup analysis 3: by type of platinum agent in platinum arm), Outcome 2 Progression‐free survival/time to progression.
6.2. Analysis.
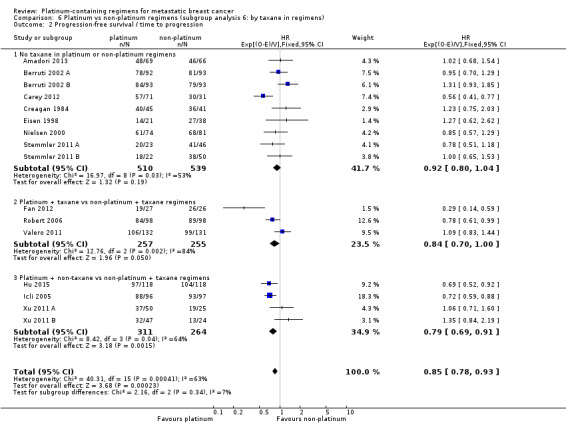
Comparison 6 Platinum vs non‐platinum regimens (subgroup analysis 6: by taxane in regimens), Outcome 2 Progression‐free survival / time to progression.
7.2. Analysis.
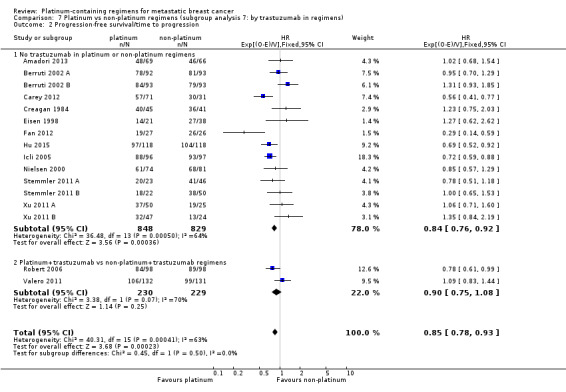
Comparison 7 Platinum vs non‐platinum regimens (subgroup analysis 7: by trastuzumab in regimens), Outcome 2 Progression‐free survival/time to progression.
8.2. Analysis.
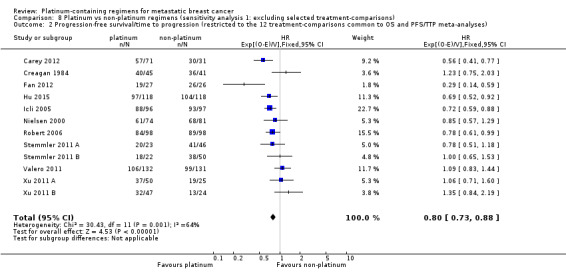
Comparison 8 Platinum vs non‐platinum regimens (sensitivity analysis 1: excluding selected treatment‐comparisons), Outcome 2 Progression‐free survival/time to progression (restricted to the 12 treatment‐comparisons common to OS and PFS/TTP meta‐analyses).
Time to treatment failure (TTF)
Three of the 28 included treatment‐comparisons assessed TTF as an outcome but only one provided sufficient data for extraction (Fountzilas 2004: HR 0.88, 95% CI 0.69 to 1.13).
Objective Tumour Response Rate (OTRR) — assessable participants
All 28 included treatment‐comparisons assessed OTRR as an outcome and provided sufficient data for extraction. From the 28 treatment‐comparisons, 4130 out of 4475 (92%) randomised participants were assessable for tumour response (and 2017 had a complete or partial response). There was a statistically significant difference in OTRRs in favour of platinum‐containing regimens (RR 1.15, 95% CI 1.08 to 1.22, P < 0.0001), but there was also considerable evidence of heterogeneity (P < 0.0001; I2 = 64%) (Analysis 1.3; Figure 6).
6.
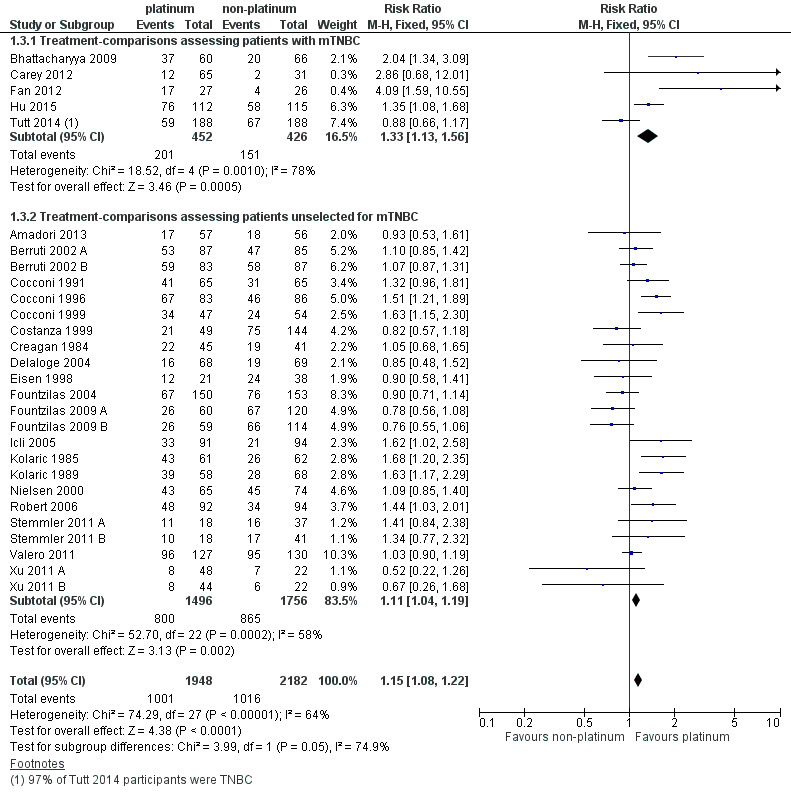
Forest plot of comparison: 1 Platinum vs non‐platinum regimens (subgroup analysis 1: by treatment‐comparisons assessing mTNBC), outcome: 1.3 Objective tumour response rate (assessable participants).
Significant differences in the pooled RRs of subgroups were found in five of the seven OTRR subgroup analyses. Specifically, there were significant differences in the pooled RRs for subgroups of treatment‐comparisons:
assessing participants with mTNBC (RR 1.33, 95% CI 1.13 to 1.56; heterogeneity P = 0.0010, I2 = 78%; n = 5; low‐quality evidence) and unselected for mTNBC (RR 1.11, 95% CI 1.04 to 1.19; heterogeneity P = 0.0002, I2 = 58%; n = 23; low‐quality evidence) (P = 0.05 for subgroup difference) (Analysis 1.3; Figure 6).
comparing 'regimen A + platinum vs regimen A' (RR 1.10, 95% CI 1.01 to 1.21; heterogeneity P = 0.009, I2 = 61%; n = 9), 'regimen A + platinum vs regimen B' (RR 1.22, 95% CI 1.12 to 1.33; heterogeneity P = 0.0002, I2 = 63%; n = 18) and 'single agent platinum vs regimen C' (RR 0.88, 95% CI 0.66 to 1.17; n = 1) (P = 0.05 for subgroup difference) (Analysis 2.3).
using cisplatin (RR 1.35, 95% CI 1.24 to 1.46; heterogeneity P = 0.006, I2 = 53%; n = 17), carboplatin (RR 0.94, 95% CI 0.86 to 1.04; heterogeneity P = 0.10, I2 = 38%; n = 10) and oxaliplatin (RR 0.85, 95% CI 0.48 to 1.52; n = 1) as the platinum agent in the platinum regimen (P < 0.0001 for subgroup difference) (Analysis 3.3).
with no anthracycline in their platinum or non‐platinum regimens (RR 1.10, 95% CI 1.02 to 1.20; heterogeneity P < 0.0001, I2 = 65%; n = 18), an anthracycline in both the platinum and non‐platinum regimens (RR 1.09, 95% CI 0.97 to 1.22; heterogeneity P = 0.12, I2 = 42%; n = 6) and an anthracycline in the platinum regimen only (RR 1.49, 95% CI 1.28 to 1.74; heterogeneity P = 0.38, I2 = 2%; n = 4) (P = 0.002 for subgroup difference) (Analysis 5.3).
with no taxane in the platinum or non‐platinum regimens (RR 1.25, 95% CI 1.15 to 1.36; heterogeneity P = 0.009, I2 = 51%; n = 17), a taxane in both platinum and non‐platinum regimens (RR 1.01, 95% CI 0.90 to 1.12; heterogeneity P = 0.002, I2 = 74%; n = 6) and a taxane in the non‐platinum regimen only (RR 1.11, 95% CI 0.94 to 1.30; heterogeneity P = 0.02, I2 = 67%; n = 5) (P = 0.007 for subgroup difference) (Analysis 6.3).
2.3. Analysis.
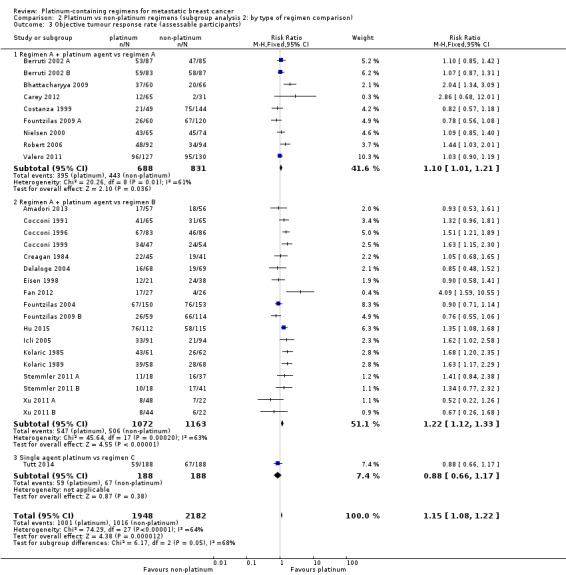
Comparison 2 Platinum vs non‐platinum regimens (subgroup analysis 2: by type of regimen comparison), Outcome 3 Objective tumour response rate (assessable participants).
3.3. Analysis.
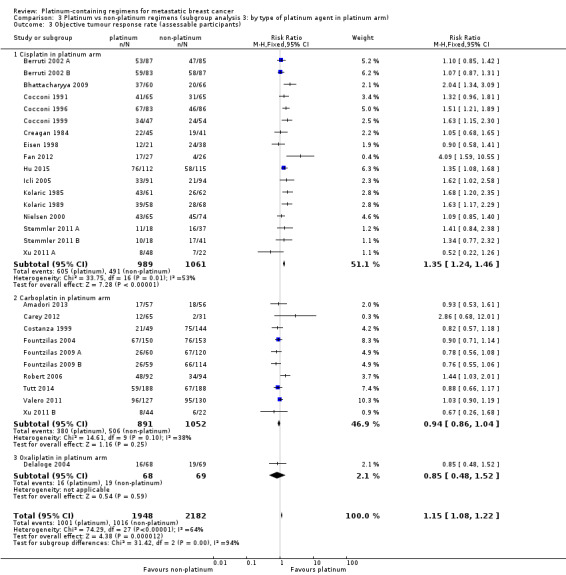
Comparison 3 Platinum vs non‐platinum regimens (subgroup analysis 3: by type of platinum agent in platinum arm), Outcome 3 Objective tumour response rate (assessable participants).
5.3. Analysis.
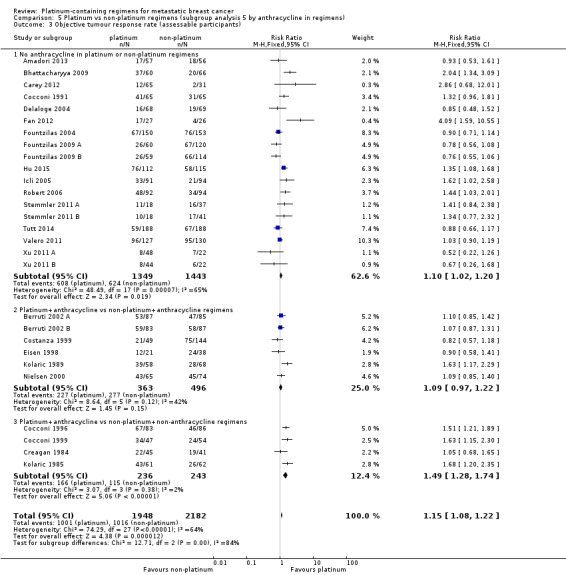
Comparison 5 Platinum vs non‐platinum regimens (subgroup analysis 5 by anthracycline in regimens), Outcome 3 Objective tumour response rate (assessable participants).
6.3. Analysis.
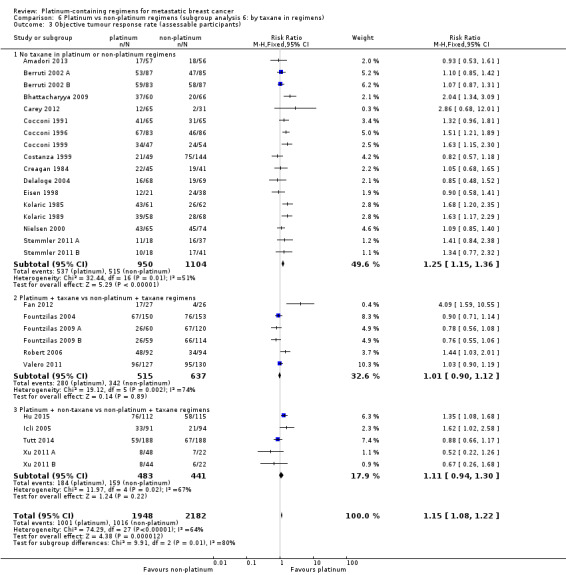
Comparison 6 Platinum vs non‐platinum regimens (subgroup analysis 6: by taxane in regimens), Outcome 3 Objective tumour response rate (assessable participants).
The two other subgroup analyses showed no evidence of subgroup differences (P = 0.55, Analysis 4.3; and P = 0.94, Analysis 7.3).
4.3. Analysis.
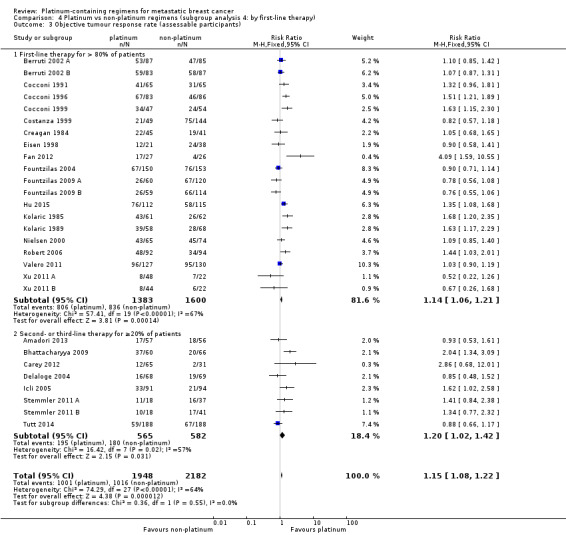
Comparison 4 Platinum vs non‐platinum regimens (subgroup analysis 4: by first‐line therapy), Outcome 3 Objective tumour response rate (assessable participants).
7.3. Analysis.
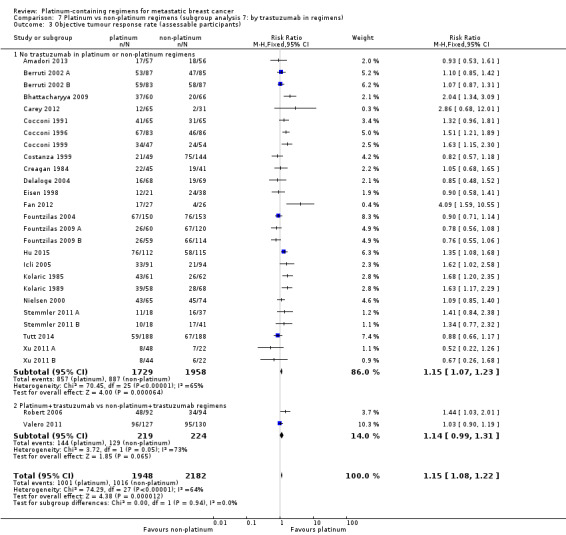
Comparison 7 Platinum vs non‐platinum regimens (subgroup analysis 7: by trastuzumab in regimens), Outcome 3 Objective tumour response rate (assessable participants).
Toxicity — safety populations
Treatment‐related death
Fifteen of the 28 included treatment‐comparisons assessed treatment‐related death and provided sufficient data for extraction. Of these 15 treatment‐comparisons, five had non‐estimable RRs due to no treatment‐related deaths and, consequently, did not contribute to the pooled estimates. For the 10 remaining treatment‐comparisons, 1688 out of 1759 (96%) randomised women were included in the safety populations, with 32 treatment‐related deaths. There was no significant difference between platinum and non‐platinum regimens in terms of treatment‐related death but the confidence interval was wide (RR 1.42, 95% CI 0.73 to 2.76; Analysis 3.4). There was no evidence of heterogeneity (P = 0.94, I2 = 0%).
3.4. Analysis.
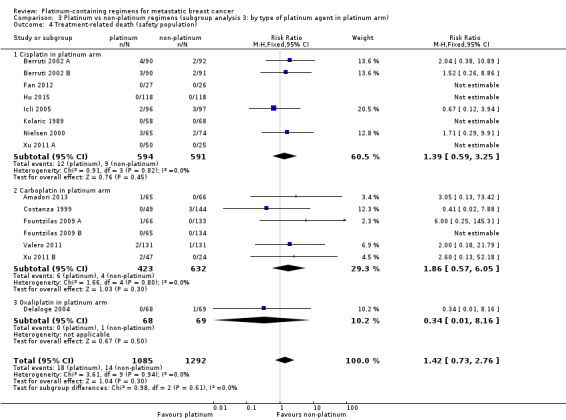
Comparison 3 Platinum vs non‐platinum regimens (subgroup analysis 3: by type of platinum agent in platinum arm), Outcome 4 Treatment‐related death (safety population).
No significant differences in pooled RRs were found according to the type of platinum agent used (P = 0.61).
Nausea/vomiting
Twenty‐one of the 28 included treatment‐comparisons assessed grade 3 and 4 nausea/vomiting with sufficient data for extraction. One treatment‐comparison had a non‐estimable RR due to there being no grade 3 or 4 cases. For the 20 remaining treatment‐comparisons, 3101 out of 3192 (97%) randomised women were included in the safety populations with 364 cases of grade 3 or 4 nausea/vomiting. Risk of grade 3 or 4 nausea/vomiting was significantly higher among women receiving platinum‐containing regimens (RR 2.07, 95% CI 1.69 to 2.54, P < 0.0001) but there was also considerable evidence of heterogeneity (P = 0.0003, I2 = 60%) (Analysis 3.5).
3.5. Analysis.
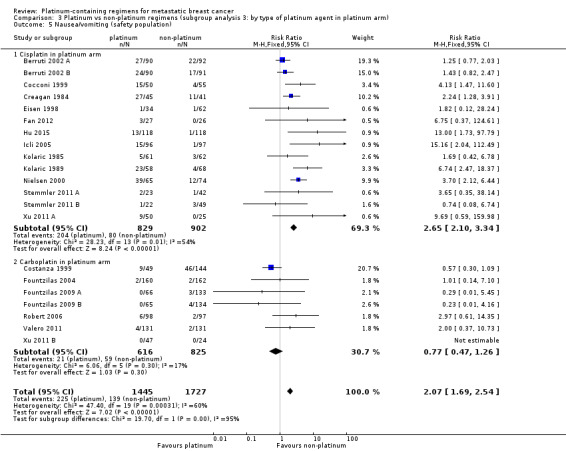
Comparison 3 Platinum vs non‐platinum regimens (subgroup analysis 3: by type of platinum agent in platinum arm), Outcome 5 Nausea/vomiting (safety population).
Subgroup analysis indicated that the increased risk of grade 3 or 4 nausea/vomiting for platinum recipients (compared to non‐platinum recipients) was primarily driven by cisplatin rather than carboplatin use. Specifically, there was a significant difference in the pooled RR for treatment‐comparisons using cisplatin (RR 2.65, 95% CI 2.10 to 3.34; n = 14; moderate‐quality evidence) rather than carboplatin (RR 0.77, 95% CI 0.47 to 1.26; n = 6; moderate‐quality evidence) in their platinum‐containing regimens (P < 0.0001 for subgroup difference). There was evidence of heterogeneity in the cisplatin (P = 0.008, I2 = 54%) but not the carboplatin (P = 0.30, I2 = 17%) subgroup.
Nephrotoxicity
Five of the 28 included treatment‐comparisons assessed grade 3 and 4 nephrotoxicity and reported sufficient data for extraction. One treatment‐comparison had a non‐estimable RR due to there being no grade 3 or 4 cases. For the four remaining treatment‐comparisons, 557 out of 571 (98%) randomised women were included in the safety populations, with 11 cases of grade 3 or 4 nephrotoxicity. Risk of grade 3 or 4 nephrotoxicity was more than three‐fold higher among women receiving platinum‐containing regimens, but this increase in risk was not significant (RR 3.06, 95% CI 0.86 to 10.84, P = 0.08) (Analysis 3.6). There was no evidence of heterogeneity (P = 0.92, I2 = 0%).
3.6. Analysis.
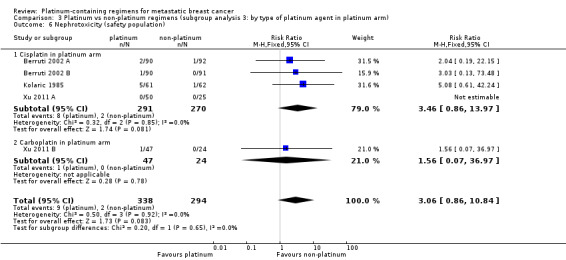
Comparison 3 Platinum vs non‐platinum regimens (subgroup analysis 3: by type of platinum agent in platinum arm), Outcome 6 Nephrotoxicity (safety population).
No significant differences in pooled RRs were found according to the type of platinum agent used (P = 0.65).
Anaemia
Twenty of the 28 included treatment‐comparisons assessed grade 3 and 4 anaemia with sufficient data for extraction. One treatment‐comparison had a non‐estimable RR due to there being no grade 3 or 4 anaemia cases. For the 19 remaining treatment‐comparisons, 3032 out of 3107 (98%) randomised women were included in the safety populations, with 176 cases of grade 3 or 4 anaemia. Risk of grade 3 or 4 anaemia was significantly higher among women receiving platinum‐containing regimens (RR 2.61, 95% CI 1.90 to 3.58, P < 0.0001) (Analysis 3.7). There was no evidence of heterogeneity (P = 0.41, I2 = 4%).
3.7. Analysis.
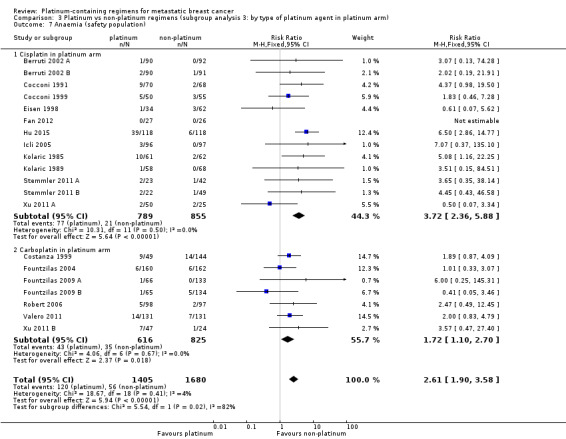
Comparison 3 Platinum vs non‐platinum regimens (subgroup analysis 3: by type of platinum agent in platinum arm), Outcome 7 Anaemia (safety population).
Subgroup analysis indicated that the increased risk of grade 3 or 4 anaemia for platinum recipients (compared to non‐platinum recipients) was worse for cisplatin recipients compared to carboplatin recipients. Specifically, there was a significant difference between the pooled RRs for treatment‐comparisons using cisplatin (RR 3.72, 95% CI 2.36 to 5.88; n =12; high‐quality evidence) rather than carboplatin (RR 1.72, 95% CI 1.10 to 2.70; n = 7; high‐quality evidence) in their platinum‐containing regimens (P = 0.02 for subgroup difference). There was no evidence of heterogeneity in the cisplatin or carboplatin subgroups (P = 0.50 and P = 0.67, respectively; I2 = 0% and I2 = 0%, respectively).
Hair Loss
Twelve of the 28 included treatment‐comparisons assessed grade 3 and 4 hair loss and reported sufficient data for extraction. Three treatment‐comparisons had a non‐estimable RR due to there being no grade 3 or 4 cases. For the nine remaining treatment‐comparisons, 1080 out of 1089 (99%) randomised women were included in the safety populations, with 469 cases of grade 3 or 4 hair loss. Risk of grade 3 or 4 hair loss was significantly higher among women receiving platinum‐containing regimens (RR 1.41, 95% CI 1.26 to 1.58, P < 0.0001; high‐quality evidence) (Analysis 3.8). There was some indication of heterogeneity (P = 0.10, I2 = 40%).
3.8. Analysis.
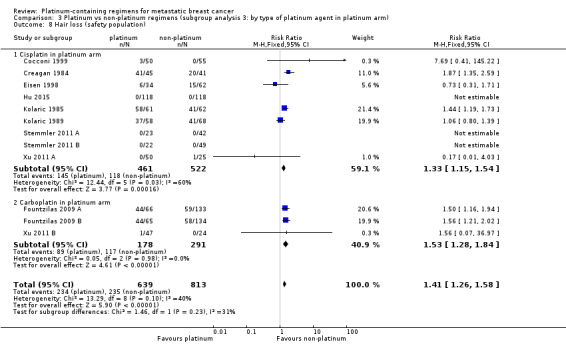
Comparison 3 Platinum vs non‐platinum regimens (subgroup analysis 3: by type of platinum agent in platinum arm), Outcome 8 Hair loss (safety population).
No significant differences in pooled RRs were found according to the type of platinum agent used (P = 0.23).
Leukopenia
Twenty‐two of the 28 included treatment‐comparisons assessed grade 3 and 4 leukopenia and reported sufficient data for extraction. One treatment‐comparison had a non‐estimable RR due to there being no grade 3 or 4 cases of leukopenia. For the 21 remaining treatment‐comparisons, 3123 out of 3222 (97%) randomised women were included in the safety populations, with 628 cases of grade 3 or 4 leukopenia. Risk of grade 3 or 4 leukopenia was significantly higher among women receiving platinum‐containing regimens (RR 1.38, 95% CI 1.21 to 1.57, P < 0.0001; moderate‐quality evidence) but with evidence of heterogeneity (P = 0.0002, I2 = 62%) (Analysis 3.9).
3.9. Analysis.
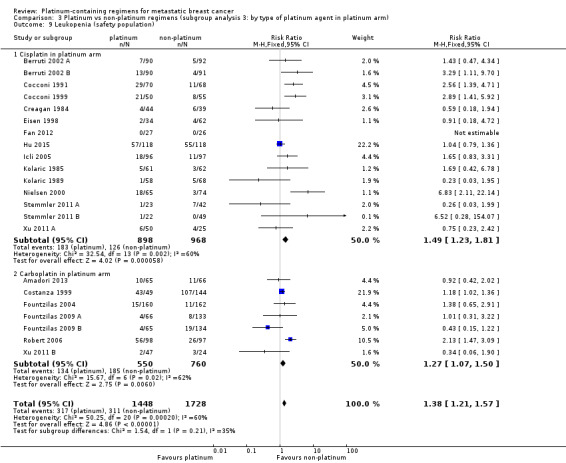
Comparison 3 Platinum vs non‐platinum regimens (subgroup analysis 3: by type of platinum agent in platinum arm), Outcome 9 Leukopenia (safety population).
No significant differences in pooled RRs were found according to the type of platinum agent used (P = 0.21).
Quality of life
Three studies reported treatment effects on quality of life; Fountzilas 2004 (EORTC QLQ‐C30), Fountzilas 2009 (Fountzilas 2009 A; Fountzilas 2009 B) (EUROQOL EQ5D and EQ VAS) and Amadori 2013 (EORTC QLQ‐C30). However, quality of life results could not be pooled in a meta‐analysis because only one quality of life domain was common to two treatment‐comparisons (the EORTC QLQ‐C30 'global' domain reported by Fountzilas 2004 and Amadori 2013) and only one reported sufficient data for extraction (Amadori 2013) .
Fountzilas 2004 found paclitaxel/carboplatin was associated with an improvement both in the emotional functioning scale and in sleep disturbance symptoms compared with paclitaxel/epirubicin, but did not find significant group differences in any other quality of life domains assessed (physical role, cognitive, social, global quality of life, fatigue, nausea and vomiting, pain, dyspnoea, appetite loss, constipation, diarrhoea, financial impact). Fountzilas 2009 (Fountzilas 2009 A; Fountzilas 2009 B) found that changes in quality across time did not differ significantly between groups for the domains reported (EQ5D index and EQ VAS score). Amadori 2013 found that pemetrexed/carboplatin participants had significantly greater deterioration in global health status scores (follow‐up minus baseline scores) than vinorelbine/gemcitabine participants, but there was no significance difference in body image scores between groups.
Sensitivity analyses
Restricting the OTRR meta‐analysis to the 19 treatment‐comparisons with extractable data on OS had little impact on the pooled RR estimate (RR = 1.15 and RR = 1.13 in the original and sensitivity analyses, respectively). Restricting the OS and PFS/TTP meta‐analyses to the 12 treatment‐comparisons with extractable data on both OS and OS and PFS/TTP lowered the pooled HR estimate from HR = 0.98 to HR = 0.92 for OS, and from 0.85 to 0.80 for PFS/TTP. This represented a 6% reduction in the HR estimates for both OS and PFS/TTP and suggested that the relative difference between OS and PFS/TTP results was unlikely to be due to the different treatment‐comparisons available for the meta‐analysis of each outcome.
Stratifying PFS/TTP estimates according to whether the outcome was PFS or TTP suggested that platinum chemotherapy was more beneficial in terms of TTP than PFS, although the difference was not statistically significant (P = 0.06). Repeating Analysis 1.1, and Analysis 1.2 using random‐effects methods did not appreciably change the point estimates for OS or PFS/TTP, but did produce slightly wider confidence intervals (Analysis 10.1; Analysis 10.2). Repeating Analysis 1.3 using a random‐effects model did not appreciably change the OTRR point estimate for participants unselected for mTNBC, but did increase the point estimate for mTNBC participants (from 1.33 to 1.60).
10.1. Analysis.
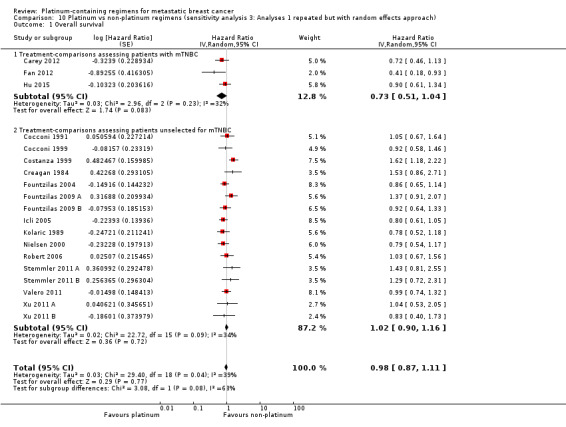
Comparison 10 Platinum vs non‐platinum regimens (sensitivity analysis 3: Analyses 1 repeated but with random effects approach), Outcome 1 Overall survival.
10.2. Analysis.
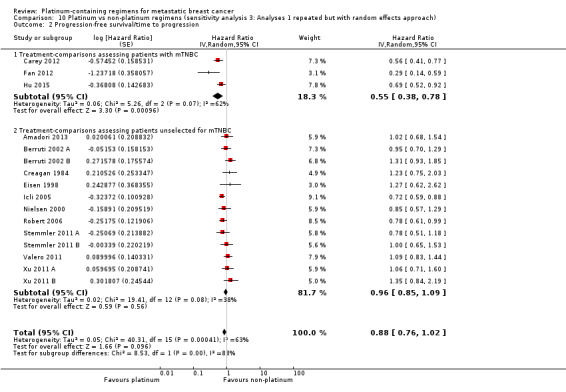
Comparison 10 Platinum vs non‐platinum regimens (sensitivity analysis 3: Analyses 1 repeated but with random effects approach), Outcome 2 Progression‐free survival/time to progression.
Discussion
Summary of main results
In this review update and consistent with the findings of the original review, our results did not show an OS benefit from platinum‐containing regimens compared to non‐platinum regimens for women with metastatic breast cancer 'in general' (i.e. without regard to subgroups). The confidence limits for the OS pooled HR estimate suggested that platinum‐containing regimens were unlikely to confer more than a 11% reduction in the risk of death and might increase the risk by up to 7%. The lack of an OS benefit for platinum‐containing regimens was somewhat at odds with the significant PFS/TTP and OTRR benefits observed in this review. Compared to women receiving non‐platinum regimens, those receiving platinum‐containing regimens had a 15% lower rate of progression or death (as measured by PFS/TTP) and a 15% higher rate of complete or partial tumour response. Sensitivity analyses indicated that the discrepancies between the OS and OTRR findings, and between the OS and PFS/TTP findings, were unlikely to be due to the different sets of treatment‐comparisons used in the various meta‐analyses of OS, PFS/TTP and OTRR. Rather, these discrepancies seemed more likely to be related to PFS/TTP and OTRR being somewhat poor surrogate outcomes for OS in metastatic breast cancer research (Burzykowski 2008), and perhaps also related to general difficulties in measuring tumour response. Given that OS is generally regarded as the outcome of choice for assessing treatment efficacy in metastatic breast cancer research (Burzykowski 2008) and given that this review now has a sufficient number of included studies to estimate the effects of treatments on OS with relatively high precision, we think that considerably more weight should be given to the OS results than the PFS/TTP or OTRR results.
In the absence of subgroup analyses, heterogeneity of effect estimates was observed for PFS/TTP, OTRR and, to a lesser degree, OS. With regards to OS and PFS/TTP, heterogeneity was explained to a large extent by the triple‐negative status of study participants. More specifically for OS, data from three treatment‐comparisons assessing mTNBC participants showed a 25% reduction in the risk of death for recipients of platinum‐containing regimens compared to recipients of non‐platinum regimens (P = 0.05). In absolute terms, this 25% risk ratio reduction corresponded to about 93 fewer deaths at one year after metastatic diagnosis for every 1000 mTNBC participants who received platinum‐containing chemotherapy, and about 105 fewer deaths at two years (Table 2). In contrast, data from 16 treatment‐comparisons, assessing participants unselected for TNBC, showed similar risks of death among platinum and non‐platinum recipients (HR = 1.01). In relation to PFS/TTP, platinum‐containing regimens reduced the risk of death or progression, or both, by about 41% for mTNBC participants (P < 0.0001) but only by about 8% for participants unselected for mTNBC (P = 0.10). The difference between the pooled effect estimates of the mTNBC and 'unselected for mTNBC' subgroups was significant for PFS/TTP (P < 0.0001) and on the margins of statistical significance for OS (P = 0.05) and OTRR (P = 0.05).
While we also found a number of other statistically significant subgroup differences for OTRR and PFS/TTP (see Effects of interventions), it is difficult to judge the importance or reliability of these findings, given that similar differences were not observed in relation to OS.
Assessments of toxicity showed that women receiving platinum‐containing regimens experienced higher rates of grade 3 and 4 nausea/vomiting, anaemia, leukopenia and hair loss than women receiving non‐platinum regimens. Subgroup analysis indicated that the higher rate of grade 3 and 4 nausea/vomiting was associated with cisplatin but not carboplatin use, and the increased risk of grade 3 and 4 anaemia was higher for cisplatin recipients than for carboplatin recipients.
Overall completeness and applicability of evidence
This review now includes data from 24 studies relating to 28 treatment‐comparisons, with publications years ranging from 1984 to 2014. Of the 28 treatment‐comparisons,19 (68%) and 16 (57%) provided sufficient time‐to‐event data to be included in OS and PFS/TTP meta‐analyses, respectively. All 28 treatment‐comparisons provided OTRR data that could be included in meta‐analyses. A majority of treatment‐comparisons had sufficient data on treatment‐related death (n =15; 54%), nausea/vomiting (n =21; 75%), anaemia (n =20; 71%) and leukopenia (n =22; 79%) for inclusion in meta‐analyses. Five (18%) and 12 (43%) treatment‐comparisons had sufficient data on nephrotoxicity and hair loss for pooling, respectively. The evidence relating to treatment effects on quality of life remained almost wholly incomplete, with only three studies reporting quality of life results that could not be pooled in meta‐analysis.
Although data for the most important outcome (OS) was extractable for 68% of treatment‐comparisons, the evidence would clearly be more complete if OS data were extractable for all 28 treatment‐comparisons. Nonetheless, it is somewhat reassuring that sensitivity analyses restricting the OTRR meta‐analysis to the 19 treatment‐comparisons with extractable OS data did not appreciably change the OTRR pooled estimate of effect (RR=1.15 and RR=1.13 in the original and sensitivity analyses, respectively). This provides some evidence that the 'overall' pooled effect estimate for OS was unlikely to have changed substantively if OS data were extractable for all 28 treatment‐comparisons. It is important to note, however, that although the 'overall' pooled estimate of effect for OS appeared robust to the missing treatment‐comparisons, this may not be the case for smaller subgroups. For example, our finding of a significant OS benefit for mTNBC participants receiving platinum‐containing regimens ― based on data from only three treatment‐comparisons ― may have been different if we were able to include OS data from Tutt 2014 in the meta‐analysis. In this relatively large treatment‐comparison of mainly mTNBC participants, median OS times were similar in the platinum and non‐platinum groups.
The evidence in this review appeared to be generally applicable to the current practice of the treatment of metastatic breast cancer for a number of reasons. First, the platinum and non‐platinum regimens used in the included trials contained most chemotherapy drugs currently used in clinical practice to treat metastatic breast cancer, including cyclophosphamide, methotrexate, fluorouracil (CMF), trastuzumab and various anthracyclines and taxanes. Second, the review included trials of women receiving first‐line treatment and women receiving treatment after failure of previous anthracycline or taxane regimens. Third, the review was well populated with trials using the two most commonly used platinum agents for treating metastatic breast cancer ― carboplatin and cisplatin. While the review had only one trial using oxaliplatin as the platinum agent, oxaliplatin is not widely used in clinical practice for treating breast cancers. Fourth, this review update is the first to use meta‐analysis to synthesise the evidence on an issue of considerable interest and importance to clinical practice; namely, whether platinum‐based chemotherapies improved survival outcomes for mTNBC participants.
Quality of the evidence
Quality of evidence ratings relating to OS, PFS/TTP and OTRR effect estimates were graded separately for the subgroups of treatment‐comparisons with women unselected for mTNBC (Table 1) and with mTNBC (Table 2). We identified no problems using the GRADE assessment criteria in relation to the OS effect estimate for the 'unselected' subgroup; hence, the quality of the evidence for this estimate was rated 'high'. For the 'mTNBC' subgroup, the quality of the evidence rating for the OS effect estimate was downgraded two levels to 'low' for 'imprecision' (because the confidence interval for the pooled estimate was wide and close to the null) and for 'suspected publication bias' (because of the absence of Tutt 2014 from the OS meta‐analysis). The quality of the evidence ratings for PFS/TTP and OTRR effect estimates ranged from 'low' to 'moderate' across the 'unselected' and 'mTNBC' subgroups for reasons including 'indirectness', 'imprecision' , 'inconsistency' and 'suspected publication bias' (see Table 1 and Table 2 for details).
Treatment effect estimates for four key toxicity outcomes were graded for quality of evidence (Table 3). Gradings were stratified by 'type of platinum agent' (cisplatin or carboplatin) for nausea/vomiting and anaemia and not stratified for hair loss and leukopenia (because hair loss and leukopenia were not significantly related to 'type of platinum agent'). The quality of the evidence ratings were 'high' for hair loss and for anaemia (both cisplatin and carboplatin subgroups), and downgraded one level to 'moderate' for nausea/vomiting (both cisplatin and carboplatin subgroups) and leukopenia due to either 'imprecision' or 'inconsistency' (see Table 3 for details).
Potential biases in the review process
Although it is possible that we may not have identified every eligible study with published results, study protocol or clinical trial registration, this seemed unlikely given our highly sensitive search strategies, including access to the Cochrane Breast Cancer Specialised Register maintained by the Cochrane Breast Cancer Group. A related but more insidious problem, however, was the risk of reporting bias arising from completed studies that never published their (largely) negative findings. While non‐publication of negative findings probably has occurred less frequently in recent years (due to increasing pressures to preregister clinical trials and publish results within reasonable time‐frames), many systematic reviews remain vulnerable to historical reporting bias. In this review, many of the included studies were conducted in an era when non‐publication of negative findings was more likely, but it seemed unlikely that this would have substantively affected our OS main finding, given that this finding was itself negative.
Agreements and disagreements with other studies or reviews
Petrelli 2016 recently published a systematic review and meta‐analysis of randomised trials to assess the efficacy and safety of therapy with platinum salts in participants with locally advanced or metastatic breast cancer. In their review, Petrelli reported overall pooled effect estimates similar to our estimates in terms of OS (HR = 0.91 vs HR = 0.98, respectively), PFS/TTP (HR = 0.84 vs HR = 0.85, respectively) and OTRR (odds ratio = 1.27 vs odds ratio = 1.33, respectively after recalculating our relative risk estimate into an odds ratio). It is important to note, however, that while the overall results of the two systematic reviews are similar, Petrelli 2016 included studies with 100% locally advanced participants whereas our review excluded studies with more than 20% of participants having isolated locoregional recurrence. In addition, our review extended the work of Petrelli through the inclusion of multiple additional subgroup analyses.
To our knowledge, there has been only one other systematic review of randomised trials comparing the effects of platinum and non‐platinum‐containing regimens among participants with mTNBC (Guan 2015). That review, however, only performed meta‐analyses of tumour response rates and not time‐to‐event outcomes. The OTRR meta‐analysis in Guan 2015 was comprised of three of the five mTNBC treatment‐comparisons included in the current review (Bhattacharyya 2009; Carey 2012; Fan 2012); the additional inclusion of Tutt 2014 and Hu 2015 in the current review resulted in a pooled OTRR estimate of effect (RR 1.33, 95% CI 1.13 to 1.56), significantly lower than that of Guan 2015 (RR 2.42, 95% CI 1.66 to 3.53).
Authors' conclusions
Implications for practice.
In view of the excess toxicity and limited survival benefits from platinum‐based regimens, it is difficult to justify their use over commonly‐available less toxic active agents as first‐line treatment for metastatic patients without mTNBC. On the other hand, this review update found preliminary low‐quality evidence of a moderate survival benefit for women with mTNBC. Consequently, although the evidence may be premature to recommend widespread use of platinum‐based regimens for mTNBC patients, some women and clinicians may consider platinum‐based regimens worth trying. While evidence from this review suggested that carboplatin was generally associated with less toxicity than cisplatin, it is not yet clear whether the possible benefits for women with mTNBC are related to the type of platinum agent.
Implications for research.
It seems unlikely that the inclusion of additional randomised trials could alter our conclusion that platinum‐containing regimens offer little or no survival benefit for women with metastatic breast cancer that is not confirmed to be mTNBC. In contrast, however, our finding of a moderate survival benefit for women with mTNBC was based on low‐quality evidence from only three trials; hence, additional randomised trials are necessary to confirm this finding. It is also worth noting that Tutt 2014 did not find an OS, PFS or OTRR advantage for women with mTNBC receiving carboplatin (vs docetaxel). However, in subgroup analysis restricted to 43 BRCA1/2 positive participants, Tutt 2014 found carboplatin was associated with significantly better OTRRs, perhaps suggesting a useful line of future research.
What's new
| Date | Event | Description |
|---|---|---|
| 23 August 2017 | Review declared as stable | The effect of platinum‐containing regimens for women with metastatic breast cancer is generally well established. The results in the 2016 review update are consistent with findings of the previous review, mainly in women without triple‐negative breast cancer. In the future, however, the scope of this review topic will be modified and likely involve an assessment of platinum‐containing regimens in women with a specific subtype of breast cancer (that is, triple‐negative breast cancer or BRCA mutation and metastatic breast cancer). Such a topic will be classified as a new review in the Cochrane Library. |
History
Protocol first published: Issue 4, 2001 Review first published: Issue 2, 2004
| Date | Event | Description |
|---|---|---|
| 28 May 2015 | New citation required and conclusions have changed | Conclusions are largely unchanged although there is now preliminary, low‐quality evidence of a survival benefit from platinum‐containing chemotherapy regimens compared to non‐platinum regimens for women with metastatic triple negative breast cancer |
| 28 May 2015 | New search has been performed | Performed searches for new studies on 28 May 2015. Twelve new studies with 15 new treatment‐comparisons were included in this review update, adding 2327 (analysed) participants since the original 2004 version of this review. Risk of bias was assessed for all domains. New subgroup analyses have been added to this review update in response to new hypotheses and available subgroups. The measure of effect for proportion (dichotomous) outcomes has been changed from odds ratio to risk ratio for ease of interpretation |
| 19 April 2012 | Amended | Additional table linked to text. |
| 4 August 2008 | Amended | Converted to new review format. |
| 25 February 2004 | New citation required and conclusions have changed | First review publication |
Acknowledgements
We would like to thank Libby Weir for her work in the identification of studies through the Cochrane Breast Cancer Group register and conducting the double data extraction in the original review, and Anna Nowak for her comments on the draft version. We also acknowledge the contribution made to the original concept for this review by Craig Henderson, Kathleen Pritchard, Martin Tattersall, Martin Stockler, Christine Brunswick, Roldano Fossati, and Alessandro Liberati.
We would also like to thank Fergus Tai and Slavica Berber for conducting the review update search strategy.
We also acknowledge Mingming Zhang and Miao Yu for their assistance with translation.
Appendices
Appendix 1. CENTRAL
MeSH descriptor: [Breast Neoplasms] explode all trees
breast near neoplasm*
breast near carcinoma*
breast near cancer*
breast near tumour*
breast near tumor*
#1 or #2 or #3 or #4 or #5 or #6
platinum or cisplatin or cisplatinum
MeSH descriptor: [Platinum] explode all trees
MeSH descriptor: [Cisplatin] explode all trees
MeSH descriptor: [Platinum Compounds] explode all trees
#8 or #9 or #10 or #11
#7 and #12
Appendix 2. MEDLINE (via Ovid SP)
| 1 | randomized controlled trial.pt. |
| 2 | controlled clinical trial.pt. |
| 3 | randomized.ab. |
| 4 | placebo.ab. |
| 5 | Clinical Trials as Topic/ |
| 6 | randomly.ab. |
| 7 | trial.ti. |
| 8 | (crossover or cross‐over).tw. |
| 9 | Pragmatic Clinical Trials as Topic/ |
| 10 | pragmatic clinical trial.pt. |
| 11 | or/1‐10 |
| 12 | exp Breast Neoplasms/ |
| 13 | advanced breast cancer$.tw,sh. |
| 14 | advanced breast neoplasm$.tw,sh. |
| 15 | advanced breast carcinoma$.tw,sh. |
| 16 | advanced breast tumor$.tw,sh. |
| 17 | advanced breast tumour$.tw,sh. |
| 18 | metastatic breast cancer$.tw,sh. |
| 19 | metastatic breast carcinoma$.tw,sh. |
| 20 | metastatic breast neoplasm$.tw,sh. |
| 21 | metastatic breast tumour$.tw,sh. |
| 22 | metastatic breast tumor$.tw,sh. |
| 23 | 12 or 13 or 14 or 15 or 16 or 17 or 18 or 19 or 20 or 21 or 22 |
| 24 | 11 and 23 |
| 25 | exp Cisplatin/ |
| 26 | exp Carboplatin/ |
| 27 | cisplatinum.tw. |
| 28 | carboplatinum.tw. |
| 29 | exp Platinum/ |
| 30 | platinum compound*.tw. |
| 31 | platinum containing regime*.tw. |
| 32 | platin*.tw. |
| 33 | platinum diamminedicholoroplatinum.tw. |
| 34 | cis‐diamminedichloroplatinum.tw. |
| 35 | cis‐dichlorodiammineoplatinum.tw. |
| 36 | biocisplatinum.tw. |
| 37 | dichlorodiammineplatinum.tw. |
| 38 | nsc‐119875.tw. |
| 39 | platidiam.tw. |
| 40 | platino.tw. |
| 41 | platinol.tw. |
| 42 | paraplatin.tw. |
| 43 | cis‐diamminedichloroplatinum.tw. |
| 44 | cis‐platinum.tw. |
| 45 | cyclobutanedicarboxylato.tw. |
| 46 | jm‐8.tw. |
| 47 | cbdca.tw. |
| 48 | nsc‐241240.tw. |
| 49 | or/25‐48 |
| 50 | 11 and 23 and 49 |
| 51 | Animals/ |
| 52 | Humans/ |
| 53 | 51 not 52 |
| 54 | 50 not 53 |
Appendix 3. Embase (via Embase.com)
random* OR factorial* OR crossover* OR cross NEXT/1 over* OR placebo* OR (doubl* AND blind*) OR (singl* AND blind*) OR assign*OR allocat* OR volunteer* OR 'crossover procedure'/exp OR 'crossover procedure' OR 'double blind procedure'/exp OR 'double blind procedure' OR 'randomized controlled trial'/exp OR 'randomized controlled trial' OR 'single blind procedure'/exp OR 'single blind procedure'
'advanced breast cancer'/exp OR 'advanced breast cancer'
'advanced breast carcinoma'
'advanced breast neoplasm'
'advanced breast tumour'
'advanced breast tumor'
'metastatic breast cancer'/exp OR 'metastatic breast cancer'
'metastatic breast carcinoma'
'metastatic breast neoplasm'
'metastatic breast tumour'
'metastatic breast tumor'
#2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11
'breast cancer'/exp OR 'breast cancer'
#12 AND #13
'cisplatin'/exp OR cisplatin
'cisplatin'/exp OR cisplatin
'cisplatinum'/exp OR cisplatinum
carboplatinum
'platinum'/exp OR platinum
'platinum containing regime'
platin*
'platinum diamminedichloroplatinum'
'cis diamminedicholoroplatinum'
'cis dichlorodiammineplatinum'/exp OR 'cis dichlorodiammineplatinum'
'biocisplatinum'/exp OR biocisplatinum
'dichlorodiammineplatinum'/exp OR 'dichlorodiammineplatinum'
'nsc 119875'/exp OR 'nsc 119875'
'platidiam'/exp OR platidiam
'platinol'/exp OR platinol
'paraplatin'/exp OR paraplatin
'cis diamminedichloroplatinum'/exp OR 'cis diamminedichloroplatinum'
'cis platinum'/exp OR 'cis platinum'
cyclobutanedicarboxylato
'cbdca'/exp OR cbdca
'jm 8'/exp OR 'jm 8'
'nsc 241240'/exp OR 'nsc 241240'
#15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 OR #30 OR #31 OR #32OR #33 OR #34 OR #35 OR #36
#1 AND #14 AND #37
#1 AND #14 AND #37
Appendix 4. WHO ICTRP Search Portal
Basic Searches:
1. Platinum‐containing regimens for metastatic breast cancer 2. Metastatic breast cancer AND platinum 3. Advanced breast cancer AND platinum
Advanced Searches:
1. Title: Platinum‐containing regimens for metastatic breast cancer Recruitment Status: ALL
2. Condition: metastatic breast cancer Intervention: platinum‐containing regime% OR platinum compound% OR platinum% OR cisplatin OR carboplatin OR platin% OR cisplatinum OR carboplatinum OR platinum diamminodichloride OR cis‐diamminedicholoroplatinum OR cis‐dichlorodiammineplatinum OR biocisplatinum OR dichlorodiammineplatinum OR nsc‐119875 OR platidiam OR platino OR platinol OR paraplatin OR cis‐diamminedichloroplatinum OR cis‐platinum OR cyclobutanedicarboxylato OR cbdca OR jn‐8 OR nsc‐241240 Recruitment Status: ALL
3. Condition: advanced breast cancer Intervention: platinum‐containing regime% OR platinum compound% OR platinum% OR cisplatin OR carboplatin OR platin% OR cisplatinum OR carboplatinum OR platinum diamminodichloride OR cis‐diamminedicholoroplatinum OR cis‐dichlorodiammineplatinum OR biocisplatinum OR dichlorodiammineplatinum OR nsc‐119875 OR platidiam OR platino OR platinol OR paraplatin OR cis‐diamminedichloroplatinum OR cis‐platinum OR cyclobutanedicarboxylato OR cbdca OR jn‐8 OR nsc‐241240 Recruitment Status: ALL
4. Condition: advanced breast cancer OR metastatic breast cancer Intervention: platinum‐containing chemotherapy regimen% OR platinum‐containing chemotherapy regimen% Recruitment Status: ALL
5. Condition: advanced breast cancer OR metastatic breast cancer Intervention: platinum AND chemotherapy% Recruitment Status: ALL
Appendix 5. ClinicalTrials.gov
Basic Searches:
1. Platinum‐containing regimens for metastatic breast cancer 2. Metastatic breast cancer AND platinum 3. Advanced breast cancer AND platinum
Advanced Searches:
1. Title: Platinum‐containing regimens for metastatic breast cancer Recruitment: All studies Study Results: All studies Study Type: All studies Gender: All studies
2. Condition: metastatic breast cancer OR advanced breast cancer Intervention: platinum‐containing regime OR platinum compound OR platinum OR cisplatin OR carboplatin OR platin OR cisplatinum OR carboplatinum OR platinum diamminodichloride OR cis‐diamminedicholoroplatinum OR cis‐dichlorodiammineplatinum OR biocisplatinum OR dichlorodiammineplatinum OR nsc‐119875 OR platidiam OR platino OR platinol OR paraplatin OR cis‐diamminedichloroplatinum OR cis‐platinum OR cyclobutanedicarboxylato OR cbdca OR jn‐8 OR nsc‐241240 Recruitment: All studies Study Results: All studies Study Type: All studies Gender: All studies
3. Condition: advanced breast cancer OR metastatic breast cancer Intervention: platinum‐containing chemotherapy regimen% OR platinum‐containing chemotherapy regimen Recruitment: All studies Study Results: All studies Study Type: All studies Gender: All studies
4. Condition: advanced breast cancer OR metastatic breast cancer Intervention: platinum AND chemotherapy Recruitment: All studies Study Results: All studies Study Type: All studies Gender: All studies
Data and analyses
Comparison 1. Platinum vs non‐platinum regimens (subgroup analysis 1: by treatment‐comparisons assessing mTNBC).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 19 | 2922 | HR (95% CI) | 0.98 [0.89, 1.07] |
| 1.1 Treatment‐comparisons assessing patients with mTNBC | 3 | 391 | HR (95% CI) | 0.75 [0.57, 1.00] |
| 1.2 Treatment‐comparisons assessing patients unselected for mTNBC | 16 | 2531 | HR (95% CI) | 1.01 [0.92, 1.12] |
| 2 Progression‐free survival/time to progression | 16 | 2136 | HR (95% CI) | 0.85 [0.78, 0.93] |
| 2.1 Treatment‐comparisons assessing patients with mTNBC | 3 | 391 | HR (95% CI) | 0.59 [0.49, 0.72] |
| 2.2 Treatment‐comparisons assessing patients unselected for mTNBC | 13 | 1745 | HR (95% CI) | 0.92 [0.84, 1.01] |
| 3 Objective tumour response rate (assessable participants) | 28 | 4130 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [1.08, 1.22] |
| 3.1 Treatment‐comparisons assessing patients with mTNBC | 5 | 878 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [1.13, 1.56] |
| 3.2 Treatment‐comparisons assessing patients unselected for mTNBC | 23 | 3252 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [1.04, 1.19] |
Comparison 2. Platinum vs non‐platinum regimens (subgroup analysis 2: by type of regimen comparison).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 19 | 2922 | HR (95% CI) | 0.98 [0.89, 1.07] |
| 1.1 Regimen A + platinum agent vs regimen A | 6 | 1141 | HR (95% CI) | 1.08 [0.93, 1.26] |
| 1.2 Regimen A + platinum agent vs regimen B | 13 | 1781 | HR (95% CI) | 0.92 [0.81, 1.03] |
| 2 Progression‐free survival / time to progression | 16 | 2136 | HR (95% CI) | 0.85 [0.78, 0.93] |
| 2.1 Regimen A+platinum agent vs regimen A | 6 | 1087 | HR (95% CI) | 0.88 [0.78, 1.00] |
| 2.2 Regimen A+platinum agent vs regimen B | 10 | 1049 | HR (95% CI) | 0.83 [0.74, 0.93] |
| 3 Objective tumour response rate (assessable participants) | 28 | 4130 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [1.08, 1.22] |
| 3.1 Regimen A + platinum agent vs regimen A | 9 | 1519 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [1.01, 1.21] |
| 3.2 Regimen A + platinum agent vs regimen B | 18 | 2235 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [1.12, 1.33] |
| 3.3 Single agent platinum vs regimen C | 1 | 376 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.66, 1.17] |
2.2. Analysis.
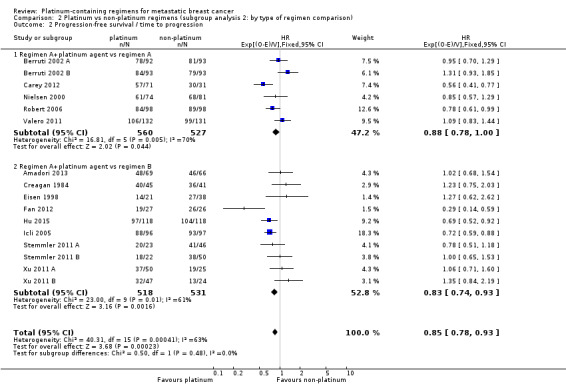
Comparison 2 Platinum vs non‐platinum regimens (subgroup analysis 2: by type of regimen comparison), Outcome 2 Progression‐free survival / time to progression.
Comparison 3. Platinum vs non‐platinum regimens (subgroup analysis 3: by type of platinum agent in platinum arm).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 19 | 2922 | HR (95% CI) | 0.98 [0.89, 1.07] |
| 1.1 Cisplatin in platinum arm | 11 | 1326 | HR (95% CI) | 0.91 [0.80, 1.05] |
| 1.2 Carboplatin in platinum arm | 8 | 1596 | HR (95% CI) | 1.04 [0.91, 1.18] |
| 2 Progression‐free survival/time to progression | 16 | 2136 | HR (95% CI) | 0.85 [0.78, 0.93] |
| 2.1 Cisplatin in platinum arm | 11 | 1369 | HR (95% CI) | 0.85 [0.76, 0.94] |
| 2.2 Carboplatin in platinum arm | 5 | 767 | HR (95% CI) | 0.86 [0.75, 0.99] |
| 3 Objective tumour response rate (assessable participants) | 28 | 4130 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [1.08, 1.22] |
| 3.1 Cisplatin in platinum arm | 17 | 2050 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.35 [1.24, 1.46] |
| 3.2 Carboplatin in platinum arm | 10 | 1943 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.86, 1.04] |
| 3.3 Oxaliplatin in platinum arm | 1 | 137 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.48, 1.52] |
| 4 Treatment‐related death (safety population) | 15 | 2377 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.42 [0.73, 2.76] |
| 4.1 Cisplatin in platinum arm | 8 | 1185 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.39 [0.59, 3.25] |
| 4.2 Carboplatin in platinum arm | 6 | 1055 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.86 [0.57, 6.05] |
| 4.3 Oxaliplatin in platinum arm | 1 | 137 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.01, 8.16] |
| 5 Nausea/vomiting (safety population) | 21 | 3172 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.07 [1.69, 2.54] |
| 5.1 Cisplatin in platinum arm | 14 | 1731 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.65 [2.10, 3.34] |
| 5.2 Carboplatin in platinum arm | 7 | 1441 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.47, 1.26] |
| 6 Nephrotoxicity (safety population) | 5 | 632 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.06 [0.86, 10.84] |
| 6.1 Cisplatin in platinum arm | 4 | 561 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.46 [0.86, 13.97] |
| 6.2 Carboplatin in platinum arm | 1 | 71 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.07, 36.97] |
| 7 Anaemia (safety population) | 20 | 3085 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.61 [1.90, 3.58] |
| 7.1 Cisplatin in platinum arm | 13 | 1644 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.72 [2.36, 5.88] |
| 7.2 Carboplatin in platinum arm | 7 | 1441 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.72 [1.10, 2.70] |
| 8 Hair loss (safety population) | 12 | 1452 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.41 [1.26, 1.58] |
| 8.1 Cisplatin in platinum arm | 9 | 983 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [1.15, 1.54] |
| 8.2 Carboplatin in platinum arm | 3 | 469 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.53 [1.28, 1.84] |
| 9 Leukopenia (safety population) | 22 | 3176 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [1.21, 1.57] |
| 9.1 Cisplatin in platinum arm | 15 | 1866 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.49 [1.23, 1.81] |
| 9.2 Carboplatin in platinum arm | 7 | 1310 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.27 [1.07, 1.50] |
Comparison 4. Platinum vs non‐platinum regimens (subgroup analysis 4: by first‐line therapy).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 19 | 2922 | HR (95% CI) | 0.98 [0.89, 1.07] |
| 1.1 First‐line therapy for > 80% of patients | 15 | 2486 | HR (95% CI) | 1.00 [0.90, 1.11] |
| 1.2 Second‐ or third‐line therapy for ≥20% of patients | 4 | 436 | HR (95% CI) | 0.89 [0.73, 1.09] |
| 2 Progression‐free survival/time to progression | 16 | 2136 | HR (95% CI) | 0.85 [0.78, 0.93] |
| 2.1 First‐line therapy for > 80% of patients | 11 | 1565 | HR (95% CI) | 0.93 [0.83, 1.03] |
| 2.2 Second‐ or third‐line therapy for ≥20% of patients | 5 | 571 | HR (95% CI) | 0.75 [0.65, 0.86] |
| 3 Objective tumour response rate (assessable participants) | 28 | 4130 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [1.08, 1.22] |
| 3.1 First‐line therapy for > 80% of patients | 20 | 2983 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [1.06, 1.21] |
| 3.2 Second‐ or third‐line therapy for ≥20% of patients | 8 | 1147 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.20 [1.02, 1.42] |
Comparison 5. Platinum vs non‐platinum regimens (subgroup analysis 5 by anthracycline in regimens).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 19 | 2922 | HR (95% CI) | 0.98 [0.89, 1.07] |
| 1.1 No anthracycline in platinum or non‐platinum regimens | 14 | 2213 | HR (95% CI) | 0.94 [0.84, 1.05] |
| 1.2 Platinum + anthracycline vs non‐platinum + anthracycline regimens | 3 | 518 | HR (95% CI) | 1.09 [0.88, 1.34] |
| 1.3 Platinum + anthracycline vs non‐platinum + non‐anthracycline regimens | 2 | 191 | HR (95% CI) | 1.12 [0.78, 1.60] |
| 2 Progression‐free survival/time to progression | 16 | 2136 | HR (95% CI) | 0.85 [0.78, 0.93] |
| 2.1 No anthracycline in platinum or non‐platinum regimens | 11 | 1465 | HR (95% CI) | 0.80 [0.73, 0.88] |
| 2.2 Platinum+anthracycline vs non‐platinum+anthracycline regimens | 4 | 585 | HR (95% CI) | 1.05 [0.86, 1.27] |
| 2.3 Platinum+anthracycline vs non‐platinum+non‐anthracycline regimens | 1 | 86 | HR (95% CI) | 1.23 [0.75, 2.03] |
| 3 Objective tumour response rate (assessable participants) | 28 | 4130 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [1.08, 1.22] |
| 3.1 No anthracycline in platinum or non‐platinum regimens | 18 | 2792 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [1.02, 1.20] |
| 3.2 Platinum+anthracycline vs non‐platinum+anthracycline regimens | 6 | 859 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.97, 1.22] |
| 3.3 Platinum+anthracycline vs non‐platinum+non‐anthracycline regimens | 4 | 479 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.49 [1.28, 1.74] |
Comparison 6. Platinum vs non‐platinum regimens (subgroup analysis 6: by taxane in regimens).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 19 | 2922 | HR (95% CI) | 0.98 [0.89, 1.07] |
| 1.1 No taxane in platinum or non‐platinum regimens | 9 | 1092 | HR (95% CI) | 1.07 [0.93, 1.24] |
| 1.2 Platinum + taxane vs non‐platinum + taxane regimens | 6 | 1255 | HR (95% CI) | 0.96 [0.82, 1.11] |
| 1.3 Platinum + non‐taxane vs non‐platinum + taxane regimens | 4 | 575 | HR (95% CI) | 0.85 [0.69, 1.04] |
| 2 Progression‐free survival / time to progression | 16 | 2136 | HR (95% CI) | 0.85 [0.78, 0.93] |
| 2.1 No taxane in platinum or non‐platinum regimens | 9 | 1049 | HR (95% CI) | 0.92 [0.80, 1.04] |
| 2.2 Platinum + taxane vs non‐platinum + taxane regimens | 3 | 512 | HR (95% CI) | 0.84 [0.70, 1.00] |
| 2.3 Platinum + non‐taxane vs non‐platinum + taxane regimens | 4 | 575 | HR (95% CI) | 0.79 [0.69, 0.91] |
| 3 Objective tumour response rate (assessable participants) | 28 | 4130 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [1.08, 1.22] |
| 3.1 No taxane in platinum or non‐platinum regimens | 17 | 2054 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [1.15, 1.36] |
| 3.2 Platinum + taxane vs non‐platinum + taxane regimens | 6 | 1152 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.90, 1.12] |
| 3.3 Platinum + non‐taxane vs non‐platinum + taxane regimens | 5 | 924 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.94, 1.30] |
Comparison 7. Platinum vs non‐platinum regimens (subgroup analysis 7: by trastuzumab in regimens).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 19 | 2922 | HR (95% CI) | 0.98 [0.89, 1.07] |
| 1.1 No trastuzumab in platinum or non‐platinum regimens | 17 | 2463 | HR (95% CI) | 0.97 [0.88, 1.08] |
| 1.2 Platinum + trastuzumab vs non‐platinum + trastuzumab regimens | 2 | 459 | HR (95% CI) | 1.00 [0.79, 1.27] |
| 2 Progression‐free survival/time to progression | 16 | 2136 | HR (95% CI) | 0.85 [0.78, 0.93] |
| 2.1 No trastuzumab in platinum or non‐platinum regimens | 14 | 1677 | HR (95% CI) | 0.84 [0.76, 0.92] |
| 2.2 Platinum+trastuzumab vs non‐platinum+trastuzumab regimens | 2 | 459 | HR (95% CI) | 0.90 [0.75, 1.08] |
| 3 Objective tumour response rate (assessable participants) | 28 | 4130 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [1.08, 1.22] |
| 3.1 No trastuzumab in platinum or non‐platinum regimens | 26 | 3687 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [1.07, 1.23] |
| 3.2 Platinum+trastuzumab vs non‐platinum+trastuzumab regimens | 2 | 443 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.99, 1.31] |
Comparison 8. Platinum vs non‐platinum regimens (sensitivity analysis 1: excluding selected treatment‐comparisons).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival (restricted to the 12 treatment‐comparisons common to OS and PFS/TTP meta‐analyses) | 12 | 1571 | HR (95% CI) | 0.92 [0.81, 1.04] |
| 2 Progression‐free survival/time to progression (restricted to the 12 treatment‐comparisons common to OS and PFS/TTP meta‐analyses) | 12 | 1571 | HR (95% CI) | 0.80 [0.73, 0.88] |
| 3 Objective tumour response rate (assessable participants‐ restricted to the 19 treatment‐comparisons in OS meta‐analyses) | 19 | 2685 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [1.05, 1.22] |
8.1. Analysis.
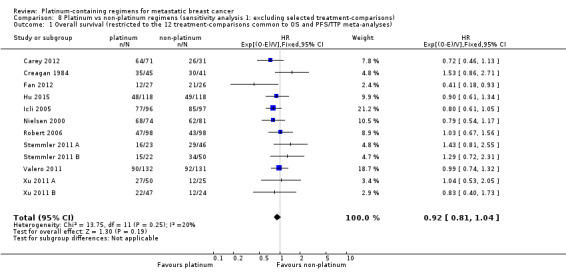
Comparison 8 Platinum vs non‐platinum regimens (sensitivity analysis 1: excluding selected treatment‐comparisons), Outcome 1 Overall survival (restricted to the 12 treatment‐comparisons common to OS and PFS/TTP meta‐analyses).
8.3. Analysis.
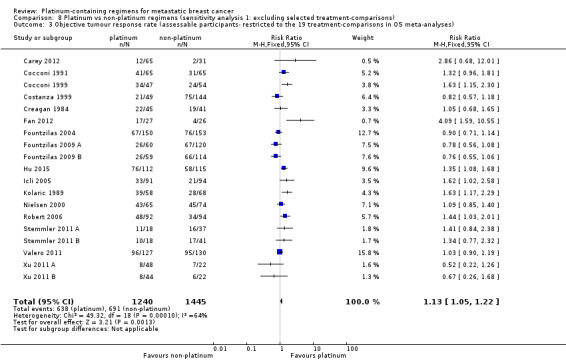
Comparison 8 Platinum vs non‐platinum regimens (sensitivity analysis 1: excluding selected treatment‐comparisons), Outcome 3 Objective tumour response rate (assessable participants‐ restricted to the 19 treatment‐comparisons in OS meta‐analyses).
Comparison 9. Platinum vs non‐platinum regimens (sensitivity analysis 2: Progression‐free survival vs. time to progression).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Progression‐free survival vs time to progression | 16 | 2136 | HR (95% CI) | 0.85 [0.78, 0.93] |
| 1.1 Progression‐free survival | 9 | 1324 | HR (95% CI) | 0.92 [0.82, 1.03] |
| 1.2 Time to progression | 7 | 812 | HR (95% CI) | 0.78 [0.69, 0.88] |
9.1. Analysis.
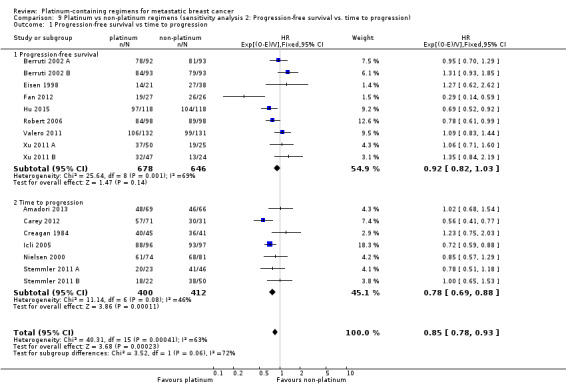
Comparison 9 Platinum vs non‐platinum regimens (sensitivity analysis 2: Progression‐free survival vs. time to progression), Outcome 1 Progression‐free survival vs time to progression.
Comparison 10. Platinum vs non‐platinum regimens (sensitivity analysis 3: Analyses 1 repeated but with random effects approach).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival | 19 | Hazard Ratio (Random, 95% CI) | 0.98 [0.87, 1.11] | |
| 1.1 Treatment‐comparisons assessing patients with mTNBC | 3 | Hazard Ratio (Random, 95% CI) | 0.73 [0.51, 1.04] | |
| 1.2 Treatment‐comparisons assessing patients unselected for mTNBC | 16 | Hazard Ratio (Random, 95% CI) | 1.02 [0.90, 1.16] | |
| 2 Progression‐free survival/time to progression | 16 | Hazard Ratio (Random, 95% CI) | 0.88 [0.76, 1.02] | |
| 2.1 Treatment‐comparisons assessing patients with mTNBC | 3 | Hazard Ratio (Random, 95% CI) | 0.55 [0.38, 0.78] | |
| 2.2 Treatment‐comparisons assessing patients unselected for mTNBC | 13 | Hazard Ratio (Random, 95% CI) | 0.96 [0.85, 1.09] | |
| 3 Objective tumour response rate (assessable participants) | 28 | 4130 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [1.05, 1.30] |
| 3.1 Treatment‐comparisons assessing patients with mTNBC | 5 | 878 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [1.04, 2.45] |
| 3.2 Treatment‐comparisons assessing patients unselected for mTNBC | 23 | 3252 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [1.01, 1.25] |
10.3. Analysis.
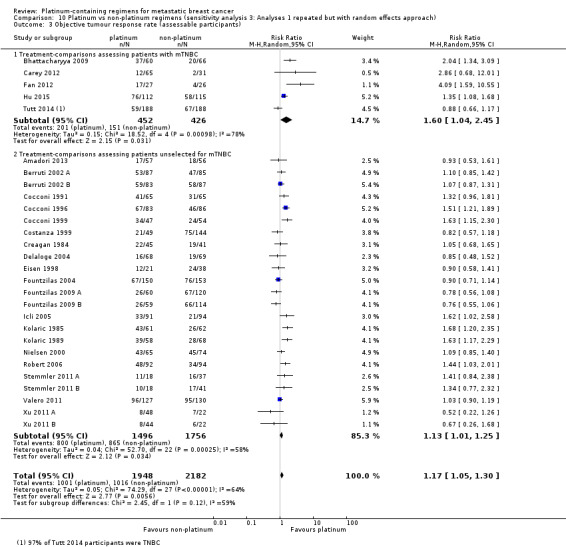
Comparison 10 Platinum vs non‐platinum regimens (sensitivity analysis 3: Analyses 1 repeated but with random effects approach), Outcome 3 Objective tumour response rate (assessable participants).
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Amadori 2013.
| Methods | Multicentre, randomised, two‑stage, open‐label, noncomparative, parallel‐group phase II study conducted between June 2006 and April 2010. | |
| Participants | 135 adult females with a histologic or cytologic diagnosis of advanced breast cancer previously treated with anthracycline and taxanes.. Median age 52 and 51.5 years in platinum and control arms, respectively. Age range 29 to 77 years. 100% advanced breast cancer. 30% 1st‐line, 70% 2nd‐line. 100% previously treated with anthracycline and taxanes. |
|
| Interventions | Pemetrexed and carboplatin vs vinorelbine and gemcitabine. ARM A: Pemetrexed 600 mg/m2 (intravenously for 10 min on day 1) and carboplatin (given over approximately 30 min beginning after the end of the pemetrexed infusion for target area under the curve (AUC) 5.0) on day 1, after pretreatment with folic acid, vitamin B12 and dexamethasone. ARM B: Vinorelbine 30 mg/m2 (given over approximately 6 to 10 min) and gemcitabine 1,200 mg/m2 (given over approximately 30 min) were administered on day 1 and day 8. |
|
| Outcomes | Response.
Time to progressive disease (TTPD; Kaplan‐Meier curve) defined as "the time from the date of study enrolment to the first documented date of progressive disease or death from study disease".
Time to treatment failure (TTTF; presented as medians in months; not extractable for hazard ratio calculation) defined as the time from date of study enrolment to the first documented date of death from any cause, progressive disease, or study treatment discontinuation due to adverse event. Grades 3 and 4 adverse events (only reported when event occurred in ≥ 10% of participants in each treatment group). QoL (EORTC questionnaires, QLQ‑C30 global health status, and QLQ‑BR23 body image). |
|
| Notes | Data for TTTF was not extractable because only event numbers and median TTFs were reported (i.e. pemetrexed and carboplatin: events/total = 60/69, median TTTF = 4.8 months; vinorelbine and gemcitabine: events/total= 58/66, median TTTF = 5.1 months). Data for some adverse event types such as anaemia were not extractable because event data were only reported for event types which occurred in ≥ 10% of participants in each treatment group. Estimated min follow‐up = 0.3 months (based on first censoring tick on TTP curve). Estimated max follow‐up = 22 months (based on last censoring tick on TTP curve). QoL: Pemetrexed/carboplatin participants had significantly greater deterioration in global health status scores (follow‐up minus baseline scores) than vinorelbine/gemcitabine participants. There was no significance difference in body image scores between groups. Median TTPD was 5.1 (95% CI: 4.1 to 8.0) months for pemetrexed/carboplatin arm and 5.6 months (95% CI: 4.2 to 7.5) months for vinorelbine/gemcitabine arm. This study was sponsored by Eli Lilly and Company, Indianapolis, IN, USA. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were "randomised either to Arm A …or to Arm B..."; no additional details were provided on how random assignment was achieved in the trial report. |
| Allocation concealment (selection bias) | Unclear risk | Method of concealment was not described. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | Open‐label study but not clear if outcome assessors were blinded to intervention. For tumour response rates, radiological assessments performed before treatment at every other cycle. No further details provided. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Low risk | All 69 and 66 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analyses (intent‐to‐treat analyses). |
| Incomplete outcome data (attrition bias) (binary outcomes) | High risk | 12 of 69 and 10 of 66 participants randomised to intervention and control groups, respectively, were not assessed/assessable for tumour response (16.3% of all randomised participants). 4 of 69 and 0 of 66 participants randomised to intervention and control groups, respectively, were not included in the safety population for evaluating toxicities (3.0% of all randomised participants). |
| Selective reporting (reporting bias) | Unclear risk | Quality of life was not included in ClinicalTrials.gov record (https://clinicaltrials.gov/ct2/show/NCT00325234) but was included in the trial report. All other outcomes in the trial report were listed in the ClinicalTrials.gov record and vice versa. |
| Other bias | Low risk | None identified. |
Berruti 2002 A.
| Methods | RCT multicentre phase III trial.
Randomisation method not reported. Accrual October 1995 to April 1999. Baseline comparability: no significant imbalance apparent or reported. |
|
| Participants | 186 women with histologically confirmed metastatic breast cancer (1 ineligible ‐185 entered trial) in the treatment‐comparison Berruti 2002 A (186 participants also for Berruti 2002 B). Median age 58. Age range 28 to 75. 100% metastatic breast cancer. 100% first‐line. All participants anthracycline‐naive. | |
| Interventions | EPI vs EPI + CDDP. ARM A: Epirubicin 60 mg/m2 on days 1 and 2 every 21 days. ARM B: EPI + CDDP: Epiubicin 60 mg/m2 IV on days 1 and 2 + platinum 30 mg/2 IV on day 1 and 2 every 21 days. |
|
| Outcomes | Overall survival measured from the date of randomisation until death (insufficient OS data reported to calculate hazard ratio for pooling). Time to progression (Kaplan‐Meier curve), defined as "the time elapsed from randomisation until disease progression or death" (this would usually be called progression‐free survival because death from any cause is treated as an event). Response. Toxicity. | |
| Notes | The trial was part of a factorial 2 x 2 design (4 arms n = 371) to answer two questions. IT IS INCLUDED IN THIS REVIEW AS TWO TRIALS: Berruti (a) and Berrutti (b). Intention‐to‐treat analyses used for overall survival and time to progression. Toxic deaths n = 6, due to either hematologic toxicity, pulmonary thromboembolism, congestive heart failure, arrhythmia, hepatorenal syndrome, or sudden death. Estimated min follow‐up = 4.5 months (based on the median number of cycles received). Estimated max follow‐up = 64 months (based on last event on the curve). Last follow‐up reported as March 2001. Median TTP: 10.8 to 12.2 months LND arms; 9.9 to 8.6 months non‐LND arms. Median survival: 28.8 CDDP arms; 29.5 non‐CDDP arms; 29.8 LND arms 27.3 non‐LND arms (insufficient OS data reported to calculate hazard ratio for pooling). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not reported in detail. |
| Allocation concealment (selection bias) | Unclear risk | Not reported ‐ stated as "randomized" only. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information in trial publication. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | No information in trial publication. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | Standard criteria used for classifying tumour response; physical examination or radiography took place. No further details provided. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Unclear risk | 92 of 93 and 93 of 93 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analysis (modified intent‐to‐treat). |
| Incomplete outcome data (attrition bias) (binary outcomes) | Low risk | 8 of 93 and 5 of 93 participants randomised to intervention and control groups, respectively, were not assessed/assessable for tumour response (7.5% of all randomised participants). 3 of 93 and 1 of 93 participants randomised to intervention and control groups, respectively, were not included in the safety population for evaluating toxicities (2.2% of all randomised participants). |
| Selective reporting (reporting bias) | Unclear risk | No trial registration or published protocol prespecifying all study outcomes. Study began recruitment before July 1, 2005 so expectation of registration or published protocol was low. |
| Other bias | Low risk | None identified. |
Berruti 2002 B.
| Methods | RCT mulitcentre phase III trial.
Randomisation method not reported. Accrual October 1995 to April 1999. Baseline comparability: no significant imbalance apparent or reported. |
|
| Participants | 186 women with histologically confirmed metastatic breast cancer. Median age 58. Age range 35 to 74. 100% metastatic breast cancer. 100% first‐line. All participants anthracycline‐naive. | |
| Interventions | EPI + LND vs EPI + CDDP + LND. ARM A: EPI + LND: Epiubicin 60 mg/m2 IV on days 1 and 2 every 21 days and Lonidamine 450 mg po every day. ARM B: EPI + CDDP + LND: Epiubicin 60 mg/m2 IV on days 1 and 2 + cisplatin 30 mg/m2 IV on days 1 and 2 every 21 days + lonidamine 450 mg po every day. LND was pursued until progression. Remaining chemotherapy was delivered up to a maximum of 6 weeks. |
|
| Outcomes | Overall survival measured from the date of randomisation until death. Time to progression (Kaplan‐Meier curve), defined as "the time elapsed from randomisation until disease progression or death" (this would usually be called progression‐free survival because death from any cause is treated as an event). Response. Toxicity. | |
| Notes | Toxic deaths n = 5, due to either hematologic, toxicity, pulmonary thromboembolism, congestive heart failure, arrhythmia, hepatorenal syndrome or sudden death. Estimated min follow‐up = 4.5 months (based on the median number of cycles received). Estimated max follow‐up = 64 months (based on last event on the curve). Last follow‐up reported as March 2001. Median TTP: 10.8 to 12.2 months LND arms; 9.9 to 8.6 months non‐LND arms. Median survival: 28.8 CDDP arms; 29.5 non‐CDDP arms; 29.8 LND arms; 27.3 non‐LND arms (insufficient OS data reported to calculate hazard ratio for pooling). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not reported in detail. |
| Allocation concealment (selection bias) | Unclear risk | Not reported ‐ stated as "randomized" only. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information in trial publication. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | No information in trial publication. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | Standard criteria used for classifying tumour response; physical examination or radiography took place. No further details provided. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Low risk | All 93 and 93 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analyses (intent‐to‐treat analyses). |
| Incomplete outcome data (attrition bias) (binary outcomes) | Low risk | 8 of 93 and 5 of 93 participants randomised to intervention and control groups, respectively, were not assessed/assessable for tumour response (8.6% of all randomised participants). 3 of 93 and 2 of 93 participants randomised to intervention and control groups, respectively, were not included in the safety population for evaluating toxicities (2.7% of all randomised participants). |
| Selective reporting (reporting bias) | Unclear risk | No trial registration or published protocol prespecifying all study outcomes. Study began recruitment before July 1, 2005 so expectation of trial registration or published protocol was low. |
| Other bias | Low risk | None identified. |
Bhattacharyya 2009.
| Methods | Randomised phase III trial. | |
| Participants | 126 mTNBC participants between age group of 38 to 72 years and who had already received anthracyclines and taxanes and had relapsed and could not afford ixabepilone and/or avastin. | |
| Interventions | 'No platinum' arm: endoxan 50 mg per day at 10 am and methotrexate 2.5 mg twice a day at 9 am and 5 pm. Platinum arm: Same as above but with 'cisplatinum'. | |
| Outcomes | Response.
Overall survival (insufficient OS data reported to calculate hazard ratio for pooling). Time to progression (insufficient TTP data reported to calculate hazard ratio for pooling). Toxicity (no results reported). |
|
| Notes | Abstract only. Median follow‐up not stated. Median TTP: Platinum arm 13 months vs 'no platinum' arm 7 months (insufficient TTP data reported to calculate hazard ratio for pooling). Median OS: Platinum arm 16 months vs 'no platinum' arm 12 months (insufficient OS data reported to calculate hazard ratio for pooling). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratified on more than one factor. Quote: "Patients were randomised to either... stratified by number of sites of metastasis and with or without visceral metastasis with or without bisphosphonates." |
| Allocation concealment (selection bias) | Unclear risk | Method of concealment was not described in the abstract. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided in the abstract. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | No information provided in the abstract. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | No information provided in the abstract. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Low risk | All 60 and 66 participants randomised to intervention and control groups, respectively, appear to have been analysed in time‐to‐event analyses (intent‐to‐treat analyses), but only median times were reported (hence no extractable time‐to‐event data). |
| Incomplete outcome data (attrition bias) (binary outcomes) | Unclear risk | All randomised participants appear to have been assessed/assessable for tumour response. This was not entirely clear though, as it was not explicitly stated and they may have simply used randomised participant denominators. |
| Selective reporting (reporting bias) | High risk | The abstract mentions that toxicity was recorded but no results were reported. In addition, there was no trial registration or published protocol containing prespecified outcomes. The date when participant recruitment began was not reported, but given that this was first published in September 2009, it seemed likely that recruitment began after July 1, 2005. As of April 2015, there has been no further results published other than those in the conference abstract. |
| Other bias | Low risk | None identified. |
Carey 2012.
| Methods | Multicentre randomised phase II study. Participants were randomised to control or platinum arms, with control participants additionally receiving platinum upon progression. |
|
| Participants | 112 women with stage IV triple‐negative metastatic breast cancer measurable by RECIST criteria and negative for ER, PR, and HER2 (0 or 1 on immunohistochemistry and/or normal gene copy number by fluorescence in situ hybridisation), of which 102 were treated and included in time‐to‐event analyses. Median age 52 and 49 years in platinum and control arms, respectively. Age range 28 to 33 years. 100% metastatic breast cancer. Of the 102 participants analysed: 55 (54%) were treated in the second‐ or third‐line setting, but not with previous EGFR inhibitor or platinum for metastatic disease; 84 (98%) had received an anthracycline; 65 (76%) had also received a taxane. |
|
| Interventions | Ce vs Ce + C. ARM 1: Cetuximab (400 mg/m2 load then 250 mg/m2 per week intravenously (IV)) alone, with carboplatin (area under the curve of 2, once per week IV) added after progression. ARM 2: Cetuximab (400 mg/m2 load then 250 mg/m2 per week intravenously and with carboplatin (area under the curve of 2, once per week IV). |
|
| Outcomes | Response.
Overall survival (Kaplan‐Meier curve).
Time to progression, defined as "treatment initiation to documented progression" (Kaplan‐Meier curve; y‐axis label typo "Progression‐free survival"). Toxicity (data not extractable because results for Arm 2 were combined with Arm 1 participants after progression). |
|
| Notes | Estimated min follow‐up = 0.25 months (based on first censoring tick on TTP curve).
Estimated max follow‐up = 38.3 months (based on last censoring tick on TTP curve). Median OS was 7.5 months (95% CI, 5.0 to 11.6) for arm one and 10.4 months (95% CI, 7.7 to 13.1) for arm 2. Study supported by Bristol‐Myers Squibb, University of North Carolina Breast Cancer Specialized Program of Research, Avon Partners‐for‐Progress awards and by National Institutes of Health. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quotes: "Patients were randomly assigned…" and "Constrained block randomizations (block size 21 plus 21) kept the imbalance between the arms to four at most". |
| Allocation concealment (selection bias) | Unclear risk | Method of concealment was not described. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Single Blind (Outcomes Assessor)" at https://clinicaltrials.gov/show/NCT00232505 implying that participants and personnel were aware of treatment allocation. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | Single Blind (Outcomes Assessor). Assessment of overall survival was unlikely to be influenced by no or incomplete blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Low risk | Cyclical evaluations including biochemical tests, CT or MRI imaging every 8 weeks, in addition to an independent evaluation of OTRR by "investigators blinded to treatment arms and not involved in the study". |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Unclear risk | 102 of 112 randomised participants were analysed in time‐to‐event analyses (modified ITT). The 10 excluded participants were excluded after enrolment but before treatment, but no information was provided on the randomised groups of these excluded participants. |
| Incomplete outcome data (attrition bias) (binary outcomes) | Unclear risk | 10 of 112 randomised participants were excluded from all analyses, with no information provided on the randomised groups of these excluded participants. In addition to these 10 excluded participants: 6 of 71 and 0 of 31 participants in the (known) intervention and control groups, respectively, were not assessed/assessable for tumour response (14.3% of all randomised participants); 6 of 71 and 0 of 31 participants in the (known) intervention and control groups, respectively, were excluded from the safety population for evaluating toxicities (14.3% of all randomised participants) (toxicity data were not extractable because results for Arm 2 were combined with Arm 1 participants after progression). |
| Selective reporting (reporting bias) | Low risk | Toxicity was not listed under 'outcomes' in ClinicalTrials.gov record (https://clinicaltrials.gov/show/NCT00232505), but it was mentioned in the 'secondary objectives' section of the record. All other outcomes in the trial report were listed in the ClinicalTrials.gov record and vice versa. |
| Other bias | Unclear risk | 26 participants in the control arm were additionally given carboplatin after progression. This may have attenuated any differences between treatment arms in overall survival. |
Cocconi 1991.
| Methods | National, multicentre, RCT, Accrual May 1985 to April 1988. Randomisation ‐ telephone call to central office. Treatment allocation by randomly permuted blocks of two. Baseline comparability: no significant imbalance apparent or reported. | |
| Participants | 140 women with histologically confirmed metastatic breast cancer. Median age 57. Age range 32 to 75. 100% metastatic breast cancer. 100% first‐line. Unclear whether the prior adjuvant therapy received by 22% of participants included anthracyclines. | |
| Interventions | CMF vs PE. ARM A: Cyclophosphamide 100 mg/m2, orally days 1 to 14; Methotrexate 40 mg/m2 IV days 1 and 8; Fluorouracil 600 mg/m2 IV days 1 and 8 repeated every 4 weeks; ARM B: Cisplatin 100 mg/m2 IV day 1 (with hydration and mannitol forced diuresis); etoposide 100 mg/m2 IV days 1, 3 and 5 repeated every 3 weeks. |
|
| Outcomes | Overall survival (curve). Time to progression (calculated from beginning of chemotherapy; insufficient TTP data reported to calculate hazard ratio for pooling). Response (calculated from beginning of chemotherapy). Toxicity. | |
| Notes | Min follow‐up: 12 months (reported).
Max follow‐up: 48 months (reported).
Median TTP: 8 m CMF, 7.7 m PE (P = 0.84) (insufficient TTP data reported to calculate hazard ratio for pooling).
Median survival: 18.7 m CMF, 19 months PE (P = 0.86).
Treatment suspended in 5 participants due to toxicity. Study supported by Associozione Italiens per la Ricerca sul Cancro and in part by Progetto Finalizzato Oncologia of Consiglio Nazionale delle Ricerche. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not reported in sufficient detail. |
| Allocation concealment (selection bias) | Low risk | Central randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information in trial publication. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | No information in trial publication. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Low risk | Tumour response rate evaluated every 3 cycles and every 3 months after suspension of treatment and were assessed "by an extramural review committee for response to treatment" (p. 666). |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Low risk | 70 of 70 and 70 of 70 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analyses (intent‐to‐treat analyses). |
| Incomplete outcome data (attrition bias) (binary outcomes) | Low risk | 5 of 70 and 5 of 70 participants randomised to intervention and control groups, respectively, were not assessed/assessable for tumour response (7.1% of all randomised participants). 0 of 70 and 2 of 70 participants randomised to intervention and control groups, respectively, were not included in the safety population for evaluating toxicities (1.4% of all randomised participants). |
| Selective reporting (reporting bias) | Unclear risk | No trial registration or published protocol prespecifying all study outcomes. Study began recruitment before July 1, 2005 so expectation of trial registration or published protocol was low. |
| Other bias | Low risk | None identified. |
Cocconi 1996.
| Methods | National multicentre RCT. Randomisation and treatment allocation methods not reported. Baseline comparability: no significant imbalance apparent or reported. | |
| Participants | 186 (183 eligible) women with metastatic breast cancer. Median age 57. Age range 32 to 75. 100% metastatic breast cancer. 100% first‐line. Unclear whether the prior adjuvant therapy received by about 45% of participants included anthracyclines. | |
| Interventions | CMF vs MPEPIV or MPEMI. ARM A: CMF: Cyclophosphamide 600 mg/m2 days 1 to 8; methotrexate 40 mg/m2 days 1 to 8; Fluorouracil 600 mg/m2 days 1 to 9; every 4 weeks. ARM B: MPEPIV: Methotrexate 100 mg/m2 days 1 to 8, + leucovorin rescue; cisplatin (P) 70 mg/m2, day 1; epirubicin 70 mg/m2 day 1 + vincristine 1.4 mg/m2 days 1 to 8; every 3 weeks. MPEMI: Methotrexate 100 mg/m2 days 1 to 8 + rescue; cisplatin 70 mg/m2 day 2; etoposide (E) 100 mg/m2 days 1 to 2; mitomycin (MI) 6 mg/m2 day 1; every 3 weeks. |
|
| Outcomes | Time to progression (insufficient TTP data reported to calculate hazard ratio for pooling). Survival (insufficient OS data reported to calculate hazard ratio for pooling). Response. Toxicity (no extractable data). | |
| Notes | Conference abstract (1996). Median TTP: 10.7 CMF, 9.5 MPEPIV or MPEMI (insufficient TTP data reported to calculate hazard ratio for pooling). Median survival: 28.7 months CMF, 31.2 MPEPIV or MPEMI (insufficient OS data reported to calculate hazard ratio for pooling). Follow‐up could not be estimated. Toxicity data was not extractable. The study reported that the platinum regimen "... was substantially more toxic than CMF, but tolerable, with no toxic deaths: the toxicities platelet, haemoglobin, vomiting, diarrhoea and mucositis were significantly more frequent in... " the platinum arm. Study "Supported by CNR Flnalyzed Project ACRO and by AIRC." |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not reported in sufficient detail. Stated as "randomised" only. |
| Allocation concealment (selection bias) | Unclear risk | Not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided in the abstract. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | No information in abstract. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | No details provided in the abstract. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Unclear risk | 91 of 93 and 92 of 93 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analysis (modified intent‐to‐treat), but only median times were reported (hence, no extractable time‐to‐event data). |
| Incomplete outcome data (attrition bias) (binary outcomes) | Unclear risk | 5 of 93 and 9 of 93 participants randomised to intervention and control groups, respectively, were not assessed/assessable for tumour response (9.1% of all randomised participants). It is not clear how many participants were included in the safety population, but toxicity data were not extractable anyway. |
| Selective reporting (reporting bias) | Unclear risk | No trial registration or published protocol prespecifying all study outcomes. Study began recruitment before July 1, 2005 so expectation of trial registration or published protocol was low. |
| Other bias | Low risk | None identified. |
Cocconi 1999.
| Methods | National, multicentre, RCT, accrual July 1988 to June 1991. Randomisation was by telephone to a central office in blocks of two. Treatment allocation not reported. Baseline comparability: no significant imbalance apparent or reported. |
|
| Participants | 109 (105 eligible) women with histologically confirmed metastatic breast cancer. Median age 53/57. Age range 26 to 72. 100% metastatic breast cancer. 100% first‐line. Unclear whether the prior adjuvant therapy received by 58% of participants included anthracyclines. | |
| Interventions | CMF vs PE + CMF + AL. ARM A: Cyclophosphamide 100 mg/m2 po days 1 to 14 + methotrexate 40 mg/m2 IV bolus days 1 and 8 + 5‐FU 6000 mg/m2 IV bolus days 1 and 8, 4 weekly cycles. ARM B: Cisplatin 100 mg/m2 i.v. infusion for 30 minutes on day 1; etoposide 100 mg/m2 i.v. infusion for 15 minutes on days 1, 3, and 5, 3 week intervals. Cyclophosphamide 100 mg/m2 po days 1 to 14 + methotrexate 40 mg/m2 IV bolus days 1 and 8 + 5‐FU 6000 mg/m2 IV bolus days 1 and 8, 4 weekly cycles. Doxorubicin 60 mg/m2 i.v. bolus on day 2; leucovorin 500 mg/m2 i.v. infusion for 2 hours on days 1 and 8; 5‐FU 600 mg/m2 i.v. bolus 1 hour after the beginning of leucovorin infusion on days 1 and 8; allopurinol, 900 mg 24 hours after each 5‐FU dose, 3 week intervals. |
|
| Outcomes | Overall survival (Kaplan‐Meier curve). Time to progression, calculated from the date of randomisation to the date of the last progression occurring during the administration of the whole program (insufficient TTP data reported to calculate hazard ratio for pooling). Response. Toxicity. | |
| Notes | The trial included 2 complex protocol treatments, rotational crossing and sequential intensification. The rotational crossing protocol was not included because of the difficulty in separating the cisplatin‐related outcomes. Intent‐to‐treat for survival, TTP and toxicity on all eligible participants.
Estimated min follow‐up: 6 months (calculated from planned months of treatment per patient).
Estimated max follow‐up: 132 months (calculated from date of randomisation to date of submission for publication).
Reported med follow‐up: 78 months.
Median TTP: 6.55 months CMF, 15 months PE + CMF + AL (P = 0.0004) (insufficient TTP data reported to calculate hazard ratio for pooling).
Median survival: 27.5 months CMF, 27.2 months PE + CMF + AL.
No toxic deaths reported. Study "Supported by Assoc. Ital. Ricerca sul Cancro (AIRC) and by PF ACRO of the Consiglio Nazionale Delle Ricerche (CNR)." |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not reported in sufficient detail. |
| Allocation concealment (selection bias) | Low risk | Central randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information in trial publication. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | No information in trial publication. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Low risk | Tumour response rate evaluated every 2 cycles and every 3 months after suspension of treatment and were assessed "by an extramural treatment response review committee". |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Unclear risk | 105 of 109 randomised participants were analysed in time‐to‐event analyses (modified ITT). The randomised groups of the 4 excluded participants were not clear. Potential bias reported as participants with progression on CMF were immediately withdrawn. |
| Incomplete outcome data (attrition bias) (binary outcomes) | Low risk | 4 of 109 randomised participants were excluded from all analyses, with no information provided on the randomised groups of these excluded participants (7.3% of all randomised participants). In addition to these 10 excluded participants: 3 of 50 and 1 of 55 participants in the (known) intervention and control groups, respectively, were not assessed/assessable for tumour response; no participants in the (known) intervention and control groups, respectively, were excluded from the safety population for evaluating toxicities (3.7% of all randomised participants). |
| Selective reporting (reporting bias) | Unclear risk | No trial registration or published protocol prespecifying all study outcomes. Study began recruitment before July 1, 2005 so expectation of trial registration or published protocol was low. |
| Other bias | Low risk | None identified. |
Costanza 1999.
| Methods | Randomised unblinded phase II study. Randomisation was 1:2 ratio of standard to experimental arm. Baseline comparability: no significant imbalance apparent or reported. | |
| Participants | 221 (193 eligible) women with histologically confirmed metastatic breast cancer. Age range < 40 to 70+. 100% metastatic breast cancer. 100% first‐line. Unclear whether the prior adjuvant therapy received by some participants included anthracyclines. | |
| Interventions | CAF vs C + CAF ARM A: CAF: cyclophosphamide 600 mg/m2 IV day 1, doxorubicin 45 mg/m2 IV day 1, fluorouracil 500 mg/m2 IV days 1 and 8, every 4 weeks. Following a total doxorubicin dose of 540 mg/m2 (including any adjuvant doxorubicin), methotrexate was substituted at 40 mg/m2 IV days 1 and 8 (30 mg/m2 for participants 60 or older). ARM B: C: Carboplatin 400 mg/m2 IV bolus escalated by 50 mg/m2 depending on day 1 nadir counts. Repeated every 28 days for up to 4 cycles followed by standard CAF. |
|
| Outcomes | Overall survival (Kaplan‐Meier, data from trialist). Response. Toxicity. | |
| Notes | Participants randomised to the phase II arm (n = 178) were randomised to CAF alone or to one of 5 phase II agents including carboplatin followed by CAF. Carboplatin data (from the published paper) only is included in the review (n = 49).
2 participants did not receive any protocol treatment, 23 were ineligible (unexplained).
Only eligible participants were included in the published analysis.
Min follow‐up: 16 months (provided by trialist).
Max follow‐up: 136 months (provided by trialist).
Median survival: 19.6 months CAF, 14.9 months C + CAF.
3 toxic deaths on CAF arm attributed to treatment‐induced sepsis. For time‐to‐event analyses, intent‐to‐treat data and numbers of participants randomised and included as eligible were provided by the trialist (ITT). Study "Supported in part by National Institutes of Health grants." |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not reported. Stated as "randomised" only. |
| Allocation concealment (selection bias) | Unclear risk | Not reported ‐ stated as "randomised" only. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information in trial publication. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | No information in trial publication. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | Blood tests and scans completed; no further details provided. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Low risk | All 52 and 169 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analyses (intent‐to‐treat analyses). |
| Incomplete outcome data (attrition bias) (binary outcomes) | Unclear risk | 3 of 52 and 25 of 169 participants randomised to intervention and control groups, respectively, were not assessed/assessable for tumour response (12.7% of all randomised participants). 3 of 52 and 25 of 169 participants randomised to intervention and control groups, respectively, were not included in the safety population for evaluating toxicities (12.7% of all randomised participants). |
| Selective reporting (reporting bias) | Unclear risk | No trial registration or published protocol prespecifying all study outcomes. Study began recruitment before July 1, 2005 so expectation of trial registration or published protocol was low. |
| Other bias | Low risk | None identified. |
Creagan 1984.
| Methods | Single centre, prospective cross‐over RCT. Randomisation by dynamic allocation. Accrual not detailed. Baseline comparability: no significant imbalance apparent or reported. | |
| Participants | 88 (86 eligible) postmenopausal women with histologically confirmed metastatic breast cancer. Median age 58. 100% metastatic breast cancer. 92% first‐line. All participants anthracycline‐naive. | |
| Interventions | CFP vs CFP + CAP ARM A: CFP: Cyclophosphamide 150 mg/m2 per day; 5‐Fluorouracil 300 mg/m2 per day IV infusion on days 1 through 5 every 5 weeks; prednisone 30 mg/d po days 1 through 14, 20 mg/d days 15 through 21; then 10 mg daily. ARM B: CAP: Cyclophosphamide 400 mg/m2 and adriamycin 40 mg/m2 on single day IV. Cis‐dichlordiammine platinum (CDDP) 40 mg/m2 in 500 mL 5% dextrose/0.5 normal saline 1 hour IV infusion for 4 cycles then cross over to CFP as above. |
|
| Outcomes | Overall survival (Kaplan‐Meier curve from time of first treatment). Time to progression (Kaplan‐Meier curve suggests breast cancer deaths and disease progression are defined events). Response. Toxicity. | |
| Notes | Abstract.
Estimated min follow‐up: 4 months (based on number of intervention cycles).
Estimated max follow‐up: 55 months OS, 44 months TTP (based on last event on curves).
Median TTP: 9.3 months CFP, 6 months CFP + CAP.
Median survival: 18.2 months CFP, 11.4 months CFP + CAP (follow‐up for survival was continued for participants with disease progression for whom the trial was terminated).
Trialists reported bias may have been introduced by the time to progression of 7 participants who refused further treatment and were censored at time off the study.
No toxic deaths reported.
Trial terminated before projected accrual due to the therapeutic trend in favour of CFP. 86 of 88 randomised participants were analysed in time‐to‐event analyses (modified ITT). Study supported in part by National Cancer Institute, National Institutes of Health. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not reported in sufficient detail. |
| Allocation concealment (selection bias) | Unclear risk | Not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information in trial publication. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | No information in trial publication. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | Standard criteria used for classifying tumour response; no further details provided. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Unclear risk | 86 of 88 randomised participants were analysed in time‐to‐event analyses (modified ITT). No information was provided on the randomised groups of the 2 excluded participants. |
| Incomplete outcome data (attrition bias) (binary outcomes) | Low risk | 2 of 88 randomised participants were excluded from all analyses, with no information provided on the randomised groups of these excluded participants. Apart from the 2 excluded participants (2.3% of all randomised participants), there were no missing data for tumour response; 1 of 45 and 2 of 41 participants in the (known) intervention and control groups, respectively, were excluded from the safety population for evaluating toxicities (5.7% of all randomised participants). |
| Selective reporting (reporting bias) | Unclear risk | No trial registration or published protocol prespecifying all study outcomes. Study began recruitment before July 1, 2005 so expectation of trial registration or published protocol was low. |
| Other bias | Low risk | None identified. |
Delaloge 2004.
| Methods | Multicentre prospective randomised controlled trial. Participant characteristics at baseline: no differences reported. |
|
| Participants | 137 women with progressing metastatic breast cancer with at least one measurable target lesion and at least one previous chemotherapy agent. | |
| Interventions | OXA + FU vs VIN + FU Arm A: OXA + FU: Oxaliplatin 130 mg/m2 2 hours IV on day 1 plus 5‐fluorouracil 750 mg/m2 daily by continuous IV infusion days 1 to 5 q 3 weeks. Arm B: VIN + FU: Vinorelbine 25 mg/m2 IV bolus plus 5‐fluorouracil 750 mg/m2 daily by continuous IV infusion days 1 to 5 q 3 weeks. |
|
| Outcomes | Overall survival (insufficient OS data reported to calculate hazard ratio for pooling). Progression‐free survival (insufficient PFS data reported to calculate hazard ratio for pooling). Response rate. Toxicity (grade‐specific data not reported). |
|
| Notes | Abstract. Follow‐up could not be estimated. Efficacy was evaluated by radiological assessment every 6 weeks; responses were confirmed at least 4 weeks later. Median PFS: 19.1 weeks OXA + FU, 22.9 weeks VIN + FU (P = 0.26) (insufficient PFS data reported to calculate hazard ratio for pooling). Median survival: 61.7 weeks OXA + FU, 71 weeks VIN + FU (P = 0.26) (insufficient OS data reported to calculate hazard ratio for pooling). The study was prematurely discontinued due to accrual difficulty related to competitive drugs introduced (Capecitabine) in the same clinical setting. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not reported. Stated as "randomised" only. |
| Allocation concealment (selection bias) | Unclear risk | Not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided in the 2 abstracts. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | No information provided in the 2 abstracts. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | Quote: "Efficacy was evaluated by radiological assessment every 6 wks; responses are confirmed at least 4 wks later" (abstract from 2004). No further details provided. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Low risk | All 68 and 69 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analyses (intent‐to‐treat analyses), but only median times were reported (hence no extractable time‐to‐event data) |
| Incomplete outcome data (attrition bias) (binary outcomes) | Unclear risk | All randomised participants appear to have been assessed/assessable for tumour response and included in the safety population for evaluating toxicities (only toxic death data extractable as grade‐specific data not reported for other conditions). This was not entirely clear though, as it was not explicitly stated and the authors may have simply used randomised participant denominators. |
| Selective reporting (reporting bias) | Unclear risk | No trial registration or published protocol prespecifying all study outcomes. Study began recruitment before July 1, 2005 so expectation of trial registration or published protocol was low. |
| Other bias | Low risk | None identified. |
Eisen 1998.
| Methods | Phase II RCT. Allocation assigned to intervention in a 2:1 randomisation. Accrual February 1994 to January 1997. Baseline comparability: no significant imbalance apparent or reported. | |
| Participants | 59 women with cytologically or histologically confirmed metastatic/advanced inoperable breast cancer. Median age 47/48. Age range 28 to 73. 61% metastatic breast cancer. 39% locally advanced. 83% first‐line. All participants anthracycline‐naive. | |
| Interventions | ECycloF vs EcisF. ARM A: ECycloF: 5‐Fluorouracil 200 mg/m2 continuous IV every 24 hours + epirubicin 60 mg/m2 by IV bolus every 3 weeks for 6 courses + Cyclophosphamide 600 mg/m2 by IV bolus every 3 weeks for 6 courses. ARM B: ECisF: 5‐Fluorouracil 200 mg/m2 IV every 24 hours + epirubicin 60 mg/m2 by IV bolus every 3 weeks for 6 courses + cisplatin 60 mg/m2 IV every 3 weeks for 6 courses. |
|
| Outcomes | Overall survival, measured from start of treatment (insufficient OS data reported to calculate hazard ratio for pooling).
Progression‐free survival, endpoints not defined (Kaplan‐Meier curve, measured from start of treatment). Time to progression, endpoints not defined (medians only). Response. Toxicity. |
|
| Notes | Metastatic and locoregional results reported separately for response.
Combined metastatic and locoregional toxicity data as published, were included in the review on the assumption that stage of disease might not influence toxicity.
2 not assessable for toxicity due to death (cause unexplained) following 1 treatment.
4 participants crossed from cisplatin to carboplatin, as per protocol, due to tinnitis (n = 2) and poor renal function (n = 2); unclear if participants had metastatic breast cancer or locally advanced.
Min follow‐up: 4 months (reported).
Max follow‐up: 32 months (reported), 23 months PFS (based on last event on curve).
Median TTP: 7 months.
Median survival: 10 m ECisF, 13 m ECycloF (insufficient OS data reported to calculate hazard ratio for pooling). All 59 randomised metastatic participants were analysed in time‐to‐event PFS analysis (ITT). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not reported. Stated as "randomised" only. |
| Allocation concealment (selection bias) | Unclear risk | Not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information in trial publication. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | No information in trial publication. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | Standard criteria used for classifying tumour response; no further details provided. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Low risk | All 21 and 38 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analyses (intent‐to‐treat analyses). |
| Incomplete outcome data (attrition bias) (binary outcomes) | Unclear risk | The study reported "Patients who received at least two cycles of chemotherapy were assessable for response... ", but they did report how many metastatic participants completed at least 2 cycles. The study calculated response rates using the numbers of randomised participants as denominators, which may or may not mean that all metastatic participants completed at least 2 cycles. Combined metastatic and locoregional toxicity data were included in this review on the assumption that stage of disease might not influence toxicity. All 96 metastatic and locoregional participants randomised to intervention and control groups, respectively, were included in the safety population for evaluating toxicities. |
| Selective reporting (reporting bias) | Unclear risk | No trial registration or published protocol prespecifying all study outcomes. Study began recruitment before July 1, 2005 so expectation of trial registration or published protocol was low. |
| Other bias | Unclear risk | None identified. |
Fan 2012.
| Methods | A prospective, open‐label, randomised phase II clinical trial carried out in the Cancer Hospital, Chinese Academy of Medical Sciences. | |
| Participants | 53 metastatic triple‐negative breast cancer (mTNBC) participants aged ≥18 years with histologically confirmed ER‐, PR‐, and HER2‐ primary breast cancer. Median age 48 and 49 years in platinum and control arms, respectively. Age range 27 to 71 years. 100% mTNBC. 100% 1st‐line. No prior treatment of advanced disease. All the participants had received anthracyclines while 66.7% of participants in the TP arm and 57.7% of participants in the TX arm received paclitaxel in the adjuvant/neoadjuvant setting.. |
|
| Interventions | TP vs TX. TP ARM: Docetaxel 75 mg/m2 plus cisplatin 75 mg/m2 IV infusion day 1. TX ARM: Docetaxel 75 mg/m2 IV infusion day 1 plus capecitabine 1000 mg/m2 bid, 2 weeks on, 1 week off. |
|
| Outcomes | Response.
Overall survival (Kaplan‐Meier curve).
Progression‐free survival, defined as "the time from the start of the treatment until disease progression or death" (Kaplan‐Meier curve). Common adverse events. |
|
| Notes | Estimated min follow‐up = 6 months (based on first event on OS curve).
Estimated max follow‐up = 42 months (based on last event on OS curve). Median PFS time: Docetaxel + cisplatin arm 10.9 months, docetaxel + capecitabine arm 4.8 months, P < 0.001. Median survival time: Docetaxel + cisplatin arm 32.8 months, docetaxel + capecitabine arm 21.5 months, P = 0.027. All 53 randomised participants were analysed in time‐to‐event analyses (ITT). Funding grant: AVON China breast cancer research grant and the National Natural Science Foundation of China. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomized…"; no additional details were provided on how random assignment was achieved in the trial report. |
| Allocation concealment (selection bias) | Unclear risk | Method of concealment was not described. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | Not clear if outcome assessors were blinded to allocated intervention. Tumour response rates evaluated by CT or MRI every two cycles; no further details provided. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Low risk | All 27 and 26 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analyses (intent‐to‐treat analyses). |
| Incomplete outcome data (attrition bias) (binary outcomes) | Low risk | All randomised participants were assessed/assessable for tumour response. All randomised participants appear to have been included in the safety population for evaluating toxicities. |
| Selective reporting (reporting bias) | High risk | No trial registration or published protocol containing prespecified outcomes could be found. The date when participant recruitment began was not reported, but given that this was first published in December 2012 and that there were only 53 participants, it seems highly likely that recruitment began after July 1, 2005. Consequently, there was a high expectation of trial registration. |
| Other bias | Low risk | Baseline characteristics similar across groups except for histological grade, where the docetaxel‐platinum arm had a greater number of grade III tumours than the docetaxel‐capecitabine arm. |
Fountzilas 2004.
| Methods | Prospective RCT. Central randomisation and stratification. Baseline comparability: no significant imbalance apparent or reported. | |
| Participants | 327 eligible women (5 excluded) with histologically confirmed metastatic breast cancer. 100% metastatic breast cancer. 100% first‐line. 54% anthracycline‐naive. | |
| Interventions | PE vs PCb. ARM A: PE: 6 cycles epirubicin 80 mg/m2 followed by paclitaxel 175 mg/m2. ARM B: PCb: 6 cycles of paclitaxel 175 mg/m2 in a 3‐hour infusion immediately followed by carboplatin 6 AUC. All cycles given every 3 weeks. |
|
| Outcomes | Overall survival (Kaplan‐Meier curve). Time to treatment failure (Kaplan‐Meier curve) calculated from the randomisation date to the date progression of the disease was documented (participants who discontinued their treatment for any reason or probably died from disease‐related causes were considered at that time, as treatment failures). Response. Toxicity. Quality of life (EORTC QLQ‐C30). | |
| Notes | Estimated min follow‐up: 3 months.
Estimated max follow‐up: 36 months TTF, 36 months OS (based on last event on curves).
Median TTF: not reported.
Median survival: not reported.
QoL: PCb was associated with an improvement both in the emotional functioning scale and in sleep disturbance symptoms compared with PE. No other QoL differences were found.
No toxic deaths reported. 327 of 332 randomised participants were analysed in time‐to‐event analyses (modified ITT) Note that 'Fountzilas 2004' was labelled 'Fountzilas 2002' in the original version of this review and its TTF estimate was incorrectly included in PFS/TTP meta‐analyses. This has been corrected in the 2016 review update. Study "Supported by a Hellenic Cooperative Oncology Group research grant (HE R‐11b/99). Dr George Fountzilas received research support from Bristol–Myers Squibb, Aventis and AstraZeneca." |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomized centrally at the HeCOG Data Office in Athens and stratified according to the history of previous adjuvant chemotherapy and risk category in a modified version of that used by Cavalli et al." |
| Allocation concealment (selection bias) | Low risk | Central randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information in trial publication. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | No information in trial publication. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Low risk | Images for tumour response were reviewed by an "independent radiological response review committee", 83% were reviewed by this committee (p. 1518). |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Unclear risk | 327 of 332 randomised participants were analysed in time‐to‐event analyses (modified ITT). No information was provided on the randomised groups of the 5 excluded participants. |
| Incomplete outcome data (attrition bias) (binary outcomes) | Low risk | 5 of 332 randomised participants were excluded from all analyses, with no information provided on the randomised groups of these excluded participants. In addition to these 5 excluded participants: 14 of 164 and 10 of 163 participants in the (known) intervention and control groups, respectively, were not assessed/assessable for tumour response (8.7% of all randomised participants); 4 of 164 and 1 of 163 participants in the (known) intervention and control groups, respectively, were excluded from the safety population for evaluating toxicities (3.0% of all randomised participants). |
| Selective reporting (reporting bias) | Unclear risk | No trial registration or published protocol prespecifying all study outcomes. Study began recruitment before July 1, 2005 so expectation of trial registration or published protocol was low. |
| Other bias | Unclear risk | None identified. |
Fountzilas 2009 A.
| Methods | Prospective RCT. Central stratified block randomisation. Baseline comparability ‐ all characteristics equal with the exception of performance status and the incidence of osseous metastases at study entry. |
|
| Participants | Total of 437 women with histologically confirmed metastatic breast cancer 'entered' the study (this included participants in both treatment‐comparisons Fountzilas 2009 A and Fountzilas 2009 B). 21 were found 'ineligible' leaving 416 'eligible' participants, of which 272 were in Fountzilas 2009 A. 100% metastatic breast cancer. 100% first‐line. | |
| Interventions | Pw vs PCb. ARM A: Pw: Paclitaxel 80 mg/m2 for 12 weeks. ARM B: PCb: Paclitaxel 175 mg/m2 + carboplatin 6 AUC for 6 (3 week) cycles. |
|
| Outcomes | Overall survival. Time to progression (insufficient TTP data reported to calculate hazard ratio for pooling). Response. Toxicity. Quality of life (EUROQOL questionnaire). | |
| Notes | The effective number of intervention participants allocated to Fountzilas 2009 A for calculating treatment effects was halved because Fountzilas 2009 A and Fountzilas 2009 B shared a common intervention group. Minimum reported follow‐up 0.01 months. Maximum reported follow‐up 56.9 months. No toxic deaths reported. Median TTP: 11.5 months PCb (0.01 to 54.6), 11.4 months Pw (0.92 to 56.9) (insufficient TTP data reported to calculate hazard ratio for pooling). Changes in QoL (EQ‐5D index and EQ VAS Score) across time did not differ significantly between groups. 21 participants in Arms A, B and C were excluded after they 'entered' the study as they were found to be ineligible. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not reported in sufficient detail. |
| Allocation concealment (selection bias) | Low risk | Centralised. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information in trial publication. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | No information in trial publication. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | Response assessed by blood and biochemistry tests, CT scans during and after treatment but "central evaluation of imaging material pertinent to tumor response was not performed in this study" and it was unclear whether or not the study was open‐label. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Unclear risk | 21 randomised participants were excluded from time‐to‐event analyses across all three treatment arms before the commencement of treatment (modified intent‐to‐treat). No information was provided on the randomised groups of the 21 excluded participants. |
| Incomplete outcome data (attrition bias) (binary outcomes) | Unclear risk | 21 randomised participants were excluded from all analyses, with no information provided on the randomised groups of these excluded participants. In addition to these 21 excluded participants: 17 of 136 and 16 of 136 participants in the (known) intervention and control groups, respectively, were not assessed/assessable for tumour response (11.8% of all randomised participants); 5 of 136 and 3 of 136 in the (known) intervention and control groups, respectively, were excluded from the safety population for evaluating toxicities (2.5% of all randomised participants). |
| Selective reporting (reporting bias) | Unclear risk | No trial registration or published protocol prespecifying all study outcomes. Study began recruitment before July 1, 2005 so expectation of trial registration or published protocol was low. |
| Other bias | Low risk | None identified. |
Fountzilas 2009 B.
| Methods | Prospective RCT. Central stratified block randomisation. Baseline comparability ‐ all characteristics equal with the exception of performance status and the incidence of osseous metastases at study entry. |
|
| Participants | Total of 437 women with histologically confirmed metastatic breast cancer 'entered' the study (this included participants in both treatment‐comparisons Fountzilas 2009 A and Fountzilas 2009 B). 21 were found 'ineligible' leaving 416 'eligible' participants, of which 280 were in Fountzilas 2009 B. 100% metastatic breast cancer. 100% first‐line. | |
| Interventions | PCb vs GDoc ARM B: PCb: Paclitaxel 175 mg/m2 + carboplatin 6 AUC for 6 (3 week) cycles. ARM C: GDoc: Gemcitabine 1000 mg/m2 + docetaxel 75 mg/m2 for 6 (3 week) cycles. |
|
| Outcomes | Overall survival. Time to progression (insufficient TTP data reported to calculate hazard ratio for pooling). Response. Toxicity. Quality of life (EUROQOL questionnaire). | |
| Notes | The effective number of intervention participants allocated to Fountzilas 2009 B for calculating treatment effects was halved because Fountzilas 2009 A and Fountzilas 2009 B shared a common intervention group. Minimum reported follow‐up 0.01 months. Maximum reported follow‐up 56.9 months. No toxic deaths reported. Median TTP: 11.5 months PCb (0.01 to 54.6), 10.4 months GDoc (0.01 to 51.4) (insufficient TTP data reported to calculate hazard ratio for pooling). Changes in QoL (EQ‐5D index and EQ VAS Score) across time did not differ significantly between groups. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not reported in sufficient detail. |
| Allocation concealment (selection bias) | Low risk | Centralised. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information in trial publication. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | No information in trial publication. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | Response assessed by blood and biochemistry tests, CT scans during and after treatment but "central evaluation of imaging material pertinent to tumor response was not performed in this study" and it was unclear whether or not the study was open‐label. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Unclear risk | 21 randomised participants were excluded from time‐to‐event analyses across all three treatment arms (modified intent‐to‐treat). No information was provided on the randomised groups of the 21 excluded participants. |
| Incomplete outcome data (attrition bias) (binary outcomes) | High risk | 21 randomised participants were excluded from all analyses, with no information provided on the randomised groups of these excluded participants. In addition to these 21 excluded participants: 17 of 136 and 30 of 144 participants in the (known) intervention and control groups, respectively, were not assessed/assessable for tumour response (18.4% of all randomised participants); 5 of 136 and 10 of 144 in the (known) intervention and control groups, respectively, were excluded from the safety population for evaluating toxicities (6.1% of all randomised participants). |
| Selective reporting (reporting bias) | Unclear risk | No trial registration or published protocol prespecifying all study outcomes. Study began recruitment before July 1, 2005 so expectation of trial registration or published protocol was low. |
| Other bias | Low risk | None identified. |
Hu 2015.
| Methods | Prospective, open‐label, multicentre, randomised, phase 3 trial at 12 institutions or hospitals in China. | |
| Participants | 240 Chinese participants (236 analysed) with breast cancer aged 18 to 70 years who had metastatic triple‐negative breast cancer (mTNBC) histologically confirmed at the primary tumour, with clinical, imaging, histological or cytological evidence of metastatic (stage IV) disease. Median age 47 and 48 years in platinum and control arms, respectively. Age interquartile range 42 to 57 and 43 to 55 years in platinum and control arms, respectively. 100% mTNBC. 100% 1st‐line. 152 (64%) of the 236 participants had received anthracyclines. 195 (83%) of the 236 participants had received taxanes. |
|
| Interventions | Cisplatin + gemcitabine vs paclitaxel + gemcitabine. Platinum ARM: Cisplatin plus gemcitabine (cisplatin 75 mg/m2 on day 1; gemcitabine 1250 mg/m2 on days 1 and 8) intravenously every 3 weeks for a maximum of eight cycles, or until disease progression or intolerable toxic effects developed. Control ARM: Paclitaxel plus gemcitabine (paclitaxel 175 mg/m2 on day 1; gemcitabine 1250 mg/m2 on days 1 and 8) intravenously every 3 weeks for a maximum of eight cycles, or until disease progression or intolerable toxic effects developed. |
|
| Outcomes | Response.
Overall survival.
Progression‐free survival, defined as "the time from the date of randomisation to progression or death from any cause". Adverse events. |
|
| Notes | 4 participants were randomised but not analysed for OS or PFS (i.e. modified ITT). An additional 9 participants were not assessable for response. Estimated min follow‐up = 3 months (based on first censoring tick on OS curve). Estimated max follow‐up = 35 months (based on last censoring tick on OS curve). Median progression‐free survival was 7.73 months (95% CI 6.16 to 9.30) in the cisplatin plus gemcitabine group and 6.47 months (5.76 to 7.18) in the paclitaxel plus gemcitabine group. Median survival time was 22.3 months in the cisplatin plus gemcitabine group and 18.6 months in the paclitaxel plus gemcitabine group; not reported in the text of the study paper but estimated from Kaplan‐Meier curve. 118 of 120 randomised metastatic participants were analysed in time‐to‐event PFS analyses (modified ITT). The study was funded by Shanghai Natural Science Foundation and gemcitabine was provided by Eli Lilly. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomisation was done centrally via a block randomisation of size eight, with no stratification factors, via an interactive web‐response system." |
| Allocation concealment (selection bias) | Low risk | Central allocation. Quote: "Randomisation was done centrally…" and "After checking the inclusion criteria, the study coordinator sent the allocated treatment back to the investigator by fax." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | The extent and/or the effectiveness of intended blinding was not clear. Quote: "Tumour response was assessed by a team of local investigators … and when needed, with independent central assessment, every two cycles until disease progression." Assessment of toxicity appeared to be unblinded. Quote: "Adverse events were recorded at each treatment visit, at each follow‐up visit, and at the end‐of‐study visit." |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Unclear risk | 118 of 120 and 118 of 120 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analysis (modified intent‐to‐treat analysis). |
| Incomplete outcome data (attrition bias) (binary outcomes) | Low risk | 8 of 120 and 5 of 120 participants randomised to intervention and control groups, respectively, were not assessed/assessable for tumour response (5.4% of all randomised participants). 2 of 120 and 2 of 120 participants randomised to intervention and control groups, respectively, were not included in the safety population for evaluating toxicities (1.7% of all randomised participants). |
| Selective reporting (reporting bias) | Low risk | Overall survival was not listed under 'outcomes' in ClinicalTrials.gov record (https://clinicaltrials.gov/ct2/show/NCT01287624) but it was mentioned in the 'purpose' section of the record. All other outcomes in the trial report were listed in the ClinicalTrials.gov record and vice versa. |
| Other bias | Unclear risk | Baseline characteristics were generally similar across groups except for ECOG performance status, number of metastatic organ sites and menopausal status. |
Icli 2005.
| Methods | Prospective randomised non‐blinded multicentre phase III study. No stratification for prognostic factors or centres. Central randomisation. Baseline comparability: no significant imbalance apparent or reported. | |
| Participants | 201 women with histologically confirmed locally advanced or metastatic breast cancer previously treated with anthracyclines (193 eligible). Median age 47/49. Age range 18 to 70. 96% metastatic breast cancer. 20% first‐line. 60% 2nd line. 20% 3rd line. All participants had previously received anthracycline therapy. | |
| Interventions | T vs VP‐16 + P. ARM A: Paclitaxel 175 mg/m2 IV, day 1 q3 weeks. ARM B: Cisplatin 70 mg/m2 IV, day 1 q3 weeks + oral etoposide (VP‐16) 50 mg bid, po, days 1 to 7 q3 weeks. |
|
| Outcomes | Overall survival (Kaplan‐Meier curve). Time to progression (Kaplan‐Meier curve) defined as "the duration between the first day of study treatment and date of progression". Response. Toxicity. | |
| Notes | Conference powerpoint slide presentation.
193 eligible:
‐ 3 died before treatment.
‐ 5 withdrew consent.
‐ 1 had an accident and did not commence treatment.
Participants crossed over after 2 cycles if disease progressed or there was no evidence of response: 47 crossed to Arm A; 37 crossed to Arm B.
185 assessable for response.
Estimated min follow‐up: 4.5 months (based on the median number of cycles received).
Estimated max follow‐up: 45 months TTP, 48 months OS (based on last events on curves).
Median TTP: 6 months Arm B, 3.9 months Arm A.
Median survival: 13 months Arm B, 10 months Arm A.
4 toxic deaths (Arm B, n = 2; Arm A, n = 3). 193 of 202 randomised metastatic participants were analysed in time‐to‐event PFS analyses (modified ITT). "Bristol Myers Squibb (Turkey) supplied limited number of paclitaxel for this trial". |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not reported other than "No stratification was carried out for prognostic factors or centers." |
| Allocation concealment (selection bias) | Low risk | Central randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "nonblinded study" |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | Non‐blinded study. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Low risk | Quote: "Responses were reviewed by two independent experts to confirm the response status blindly for treatment received" (p. 2). |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Unclear risk | 96 of 100 and 97 of 101 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analysis (modified intent‐to‐treat analysis). |
| Incomplete outcome data (attrition bias) (binary outcomes) | Low risk | 9 of 100 and 7 of 101 participants randomised to intervention and control groups, respectively, were not assessed/assessable for tumour response (8.0% of all randomised participants). 7 of 100 and 4 of 101 participants randomised to intervention and control groups, respectively, were not included in the safety population for evaluating toxicities (4.0% of all randomised participants). |
| Selective reporting (reporting bias) | Unclear risk | No trial registration or published protocol prespecifying all study outcomes. Study began recruitment before July 1, 2005 so expectation of trial registration or published protocol was low. |
| Other bias | Low risk | None identified. |
Kolaric 1985.
| Methods | Prospective phase III RCT. Randomisation, treatment, allocation methods, and accrual not detailed. Baseline comparability: no significant imbalance apparent or reported. | |
| Participants | 128 women with metastatic breast cancer. Age range 30 to 70. 100% metastatic breast cancer. 100% first‐line. All participants anthracycline‐naive. | |
| Interventions | CMFVP vs CAP. ARM A: CMFVP: Cyclophosphamide 200 mg/m2 IV days 1, 2, 3, 4, 5 + methotrexate 20 mg/m2 IV days 2, 4 + 5‐fluorouracil 500 mg/m2 IV days 1, 3, 5 + vincristine 1 mg/m2 po days 1 and 5 + prednisolone 40 mg po days 1, 2, 3, 4, 5. 3 to 4 week cycles. ARM B: CAP: cis‐platinum 30 mg/m2 IV days 1, 3, 5; + adriamycin 40 mg/m2 IV day 1 + cyclophosphamide 200 mg/m2 IV days 1, 3, 5. |
|
| Outcomes | Overall survival (insufficient OS data reported to calculate hazard ratio for pooling). Response. Toxicity. | |
| Notes | Trial analysis not intent‐to‐treat: ‐ 123 evaluable following > 2 cycles. ‐ 5 not evaluable, unexplained.. Min follow‐up: 6 months (reported). Max follow‐up: 33 months (reported). Preliminary data reported survival in favour of CAP (33%) vs CMFVP (53%), P < 0.05 (insufficient OS data reported to calculate hazard ratio for pooling). No toxic deaths reported. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not reported. Stated as "randomised" only. |
| Allocation concealment (selection bias) | Unclear risk | Not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information in trial publication. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | No information in trial publication. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | Standard criteria used to assess tumour response but no further details provided. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | High risk | 61 of 65 and 62 of 63 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analysis. Excluded participants included those who did not complete more than 2 cycles of chemotherapy (per‐protocol analysis), but only median times were reported (hence no extractable time‐to‐event data). |
| Incomplete outcome data (attrition bias) (binary outcomes) | Low risk | 4 of 65 and 1 of 63 participants randomised to intervention and control groups, respectively, were not assessed/assessable for tumour response or included in the safety population for evaluating toxicities (3.9% of all randomised participants). |
| Selective reporting (reporting bias) | Unclear risk | No trial registration or published protocol prespecifying all study outcomes. Study began recruitment before July 1, 2005 so expectation of trial registration or published protocol was low. |
| Other bias | Low risk | None identified. |
Kolaric 1989.
| Methods | Prospective RCT. Stratification prior to randomisation. Randomisation and allocation methods not detailed. Accrual June 1984 to 1986. Imbalance in prominent metastatic site (prevalence of visceral metastases 21 vs 31) was created by unevaluable participants being taken off the trial after the first cycle. | |
| Participants | 142 women with metastatic or locoregional advanced inoperable breast cancer. Median age 53. Age range 29 to 70. 58% metastatic breast cancer. 42% locally advanced non‐resectable breast cancer. 100% first‐line. All participants anthracycline‐naive. | |
| Interventions | CAP vs FAC. ARM A: FAC (n = 68): 5‐Fluorouracil 500 mg/m2 days 1 and 8; adriamycin 50 mg/m2 500 mg/m2 day 1; cyclophosphamide 500 mg/m2 day 1. ARM B: CAP (n = 58): Cyclophosphamide 200 mg/m2 IV days 1,3 and 5; adriamycin 40 mg/m2 IV day 1; platinum 30 mg/m2 IV day 1,3 and 5. |
|
| Outcomes | Overall survival (Kaplan‐Meier curve). Response. Toxicity. | |
| Notes | 126 evaluable for > 2 cycles.
16 withdrawn after 1 cycle:
‐ 1 early death due to progression.
‐ 5 further treatment refusals.
‐ 10 lost to follow‐up. Proportion of total numbers with either metastatic or locoregionally advanced disease was not reported. However, 53 reported with predominant metastatic site of soft tissue: 73 were reported with predominant site as viscera or bones; hence, an assumption made that at least 58% (73/126) were likely to have metastatic breast cancer. Min follow‐up: 1 month (reported). Max follow‐up: 30 months (reported). Median survival: 13 months CAP, 9 months FAC. No toxic deaths reported. 126 of 142 participants that had > 2 cycles were analysed in time‐to‐event OS analysis (per‐protocol analysis). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not detailed. |
| Allocation concealment (selection bias) | Unclear risk | Not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information in trial publication. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | No information in trial publication. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | Standard criteria used to assess tumour response but no further details provided. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | High risk | 58 of 68 and 68 of 74 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analysis. Excluded participants included those who did not complete more than 2 cycles of chemotherapy (per‐protocol analysis). |
| Incomplete outcome data (attrition bias) (binary outcomes) | Unclear risk | 10 of 69 and 6 of 74 participants randomised to intervention and control groups, respectively, were not assessed/assessable for tumour response or included in the safety population for evaluating toxicities (11.3% of all randomised participants). |
| Selective reporting (reporting bias) | Unclear risk | No trial registration or published protocol prespecifying all study outcomes. Study began recruitment before July 1, 2005 so expectation of trial registration or published protocol was low. |
| Other bias | Low risk | None identified. |
Nielsen 2000.
| Methods | Phase III RCT. Central randomisation registered, stratified by ECOG performance status. Accrual from July 1987 to November 1990. Baseline comparability: no significant imbalance apparent or reported. | |
| Participants | 155 women with histologically proven locally advanced or metastatic breast cancer. Median age 52/55. Age range 27 to 69. 91% metastatic breast cancer. 100% first‐line. All participants anthracycline‐naive. | |
| Interventions | EPI vs EPI + CIS. ARM A: Epirubicin 70 mg/m2 days 1 and 8 every 4 weeks. ARM B: Epirubicin 60 mg/m2 days 1 and 8 + cisplatin 100 mg/m2 day 1 every 4 weeks. |
|
| Outcomes | Overall survival (Kaplan‐Meier curve). Time to progression (Kaplan‐Meier curve, "calculated as the time from the drug administration to progression for both responders and nonresponders"). Response. Toxicity. | |
| Notes | Oopherectomy performed in premenopausal participants, n = 45 (32%). Trial claimed 'intent‐to‐treat' analysis for all randomised participants and outcomes, although: ‐ 10 were declared ineligible (EPI, n = 7; EPI + CIS, n = 3). ‐ 6 refused treatment after 1 cycle (EPI + CIS arm) and were excluded. Hence, given 139 of 155 randomised participants were analysed in time‐to‐event analyses and 6 participants who started but did not complete treatment were excluded, this represented per‐protocol analyses for time‐to‐event data. Epirubicin was continued until disease progression or to cumulative dose of 1000 mg/m2. Cisplatin was discontinued after 6 cycles. Doses of epirubicin were adjusted according to WBC and platelet counts on the day of treatment. Cisplatin was adjusted according to nephrotoxicity. Reported min follow‐up: 0.1 months. Reported max follow‐up: 77.7 months TTP and OS. Median TTP: 8.4 months EPI, 15.3 months EPI + CIS. Median survival: 15.1 months, EPI; 21.5, months EPI + CIS. 5 deaths were attributed to treatment: nephrotic syndrome: n = 1, leukaemia: n = 2, congestive cardiac failure: n = 1, and thrombocytopenia: n = 1. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not reported in sufficient detail. |
| Allocation concealment (selection bias) | Low risk | Central randomisation. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information in trial publication. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | No information in trial publication. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | "evaluation of response ... was done according to WHO criteria" (p. 460) but no further details provided. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | High risk | 65 of 74 and 74 of 81 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analysis. Excluded participants included those who did not complete more than 2 cycles of chemotherapy (per‐protocol analysis). |
| Incomplete outcome data (attrition bias) (binary outcomes) | Unclear risk | 9 of 74 and 7 of 81 participants randomised to intervention and control groups, respectively, were not assessed/assessable for tumour response or included in the safety population for evaluating toxicities (10.3% of all randomised participants). |
| Selective reporting (reporting bias) | Unclear risk | No trial registration or published protocol prespecifying all study outcomes. Study began recruitment before July 1, 2005 so expectation of trial registration or published protocol was low. |
| Other bias | Low risk | None identified. |
Robert 2006.
| Methods | Randomised, multicentre, phase III trial. Accrual between November 1998 and May 2002. Baseline characteristics reported as well balanced between study arms. |
|
| Participants | 196 women with histologically proven HER2 positive metastatic breast cancer.
Median age 55/56.
Age range 33 to 83.
100% metastatic breast cancer.
100% first‐line for metastatic breast cancer. 100% HER2 positive. |
|
| Interventions | TPa vs TPC. ARM A: TPa: Trastuzumab 4 mg/kg + paclitaxel 175mg/m2. ARM B: TPC: Trastuzumab 4 mg/kg + paclitaxel 175mg/m2 + carboplatin 6 AUC. |
|
| Outcomes | Overall survival. Progression‐free survival, defined as the interval between the date of first dose and the date of progression or death as a result of any cause. Response. Toxicity. |
|
| Notes | Minimum reported follow‐up: < 1 month. Maximum reported follow‐up: 56.8 months. No deaths due to toxicity. Randomisation procedure not stated ‐ just reported as "randomised". Stratified by IHC score (2+ or 3+). Participants with prior adjuvant or neo‐adjuvant chemotherapy: TPa, 46%; TPC, 46%. Participants were eligible provided a taxane had not been used and that cumulative doxorubicin exposure was < 360 mg/m2. Reported hazard ratios and confidence intervals were inconsistent with the Kaplan Meier curves. For example, the text stated that the difference in OS "was not statistically significant", yet the reported 95% confidence interval was 0.88 to 0.92 (not only was the confidence interval highly statistically significant, it was far too narrow, given the cohort size). Consequently, we extracted OS and TTP data from the Kaplan‐Meier curves rather than relying on the reported hazard ratios. Study "Supported by grants from Bristol‐Myers Squibb Co, Princeton, NJ, and Genentech Inc, South San Francisco, CA." |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not reported in detail. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information in trial publication. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | No information in trial publication. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | Quote: "Tumor assessments were performed by physical examination before every cycle, with imaging studies evaluating indicator lesions repeated every other cycle" (p. 2787). No further details provided on whether central assessment took place. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Low risk | All 98 and 98 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analyses (intent‐to‐treat analyses). |
| Incomplete outcome data (attrition bias) (binary outcomes) | Low risk | 6 of 98 and 4 of 98 participants randomised to intervention and control groups, respectively, were not assessed/assessable for tumour response (5.1% of all randomised participants). One randomised participant(control group) was excluded from the safety population for evaluating toxicities (0.5% of all randomised participants). |
| Selective reporting (reporting bias) | Unclear risk | No trial registration or published protocol prespecifying all study outcomes. Study began recruitment before July 1, 2005 so expectation of trial registration or published protocol was low. |
| Other bias | Low risk | None identified. |
Stemmler 2011 A.
| Methods | Randomised multicentre phase II trial. Accrual between 2003 and 2006. Groups comparable at baseline in all regards except menopausal status. |
|
| Participants | Overall, a total of 141 participants (91 in Arm A + Arm B) with histologically confirmed metastatic breast cancer. Median age: ˜ 60. 100% metastatic breast cancer. First line: ˜ 36%. Anthracycline‐naive: ˜ 43%. |
|
| Interventions | GemVin vs GemCis. ARM A: GemVin: Gemcitabine 1000 mg/m2 + vinorelbine 25 mg/m2. ARM B: GemCis: Gemcitabine 1000 mg/m2+ cisplatin 30 mg/m2. Treatment for a maximum of six (3 week) cycles. |
|
| Outcomes | Overall survival (Kaplan‐Meier). Time to progression, defined as "time from the start of therapy to first evidence of progressive disease or last follow‐up" (but TTP was also interchangeably referred to as PFS at various points in the paper) (Kaplan‐Meier). Response rate. Toxcity. Quality of life. |
|
| Notes | No reported deaths due to toxicity. Estimated min follow‐up: 1 month. Estimated max follow‐up: 47 months. Median PFS: 5.7 months, 95% CI: 3.9 to 8.2 (GemVin); 6.9 months, 95% CI: 5.8 to 8.8 (GemCis). Median OS: 17.5 months, 95% CI: 12.2 to 30.0 (GemVin); 13.0 months, 95% CI: 11.0 to 19.2 (GemCis). Randomisation procedure not stated ‐ just reported as "randomised". All randomised participants were analysed in time‐to‐event analyses (ITT). "This study was supported by Lilly GmbH Germany." |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not reported. Stated as "randomised" only. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Registered as 'open‐label' trial (https://www.clinicaltrials.gov/ct2/show/NCT00480597). |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | Open‐label study. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | Blood and biochemistry tests, and imaging took place during therapy. No details were provided on whether there was a central (independent) evaluation team for assessing tumour response rates. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Low risk | All 45 and 46 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analyses (intent‐to‐treat analyses). |
| Incomplete outcome data (attrition bias) (binary outcomes) | High risk | 10 of 45 and 9 of 46 participants randomised to intervention and control groups, respectively, were not assessed/assessable for tumour response (20.3% of all randomised participants). 0 of 45 and 4 of 46 participants randomised to intervention and control groups, respectively, were not included in the safety population for evaluating toxicities (5.8% of all randomised participants). |
| Selective reporting (reporting bias) | Low risk | All outcomes in the trial report were listed in the ClinicalTrials.gov record and vice versa (https://www.clinicaltrials.gov/ct2/show/NCT00480597). |
| Other bias | Low risk | None identified. |
Stemmler 2011 B.
| Methods | Randomised multicentre phase II trial. Accrual between 2003 and 2006. Groups comparable at baseline in all regards except menopausal status. |
|
| Participants | Overall 141 participants (95 in Arm B + Arm C) with histologically confirmed metastatic breast cancer. Median age: ˜ 60 years. 100% metastatic breast cancer. First‐line: ˜ 36%. Anthracycline‐naive: ˜ 43%. |
|
| Interventions | GemCis vs GemCap. ARM B: GemCis: Gemcitabine 1000 mg/m2 + cisplatin 30 mg/m2. ARM C: GemCap: Gemcitabine 1000 mg/m2 + capecitabine 1.300 mg/m2. Treatment for a maximum of six (3 week) cycles. |
|
| Outcomes | Overall survival (Kaplan‐Meier). Time to progression, defined as "time from the start of therapy to first evidence of progressive disease or last follow‐up" (but TTP was also interchangeably referred to as PFS at various points in the paper) (Kaplan‐Meier). Response rate. Toxcity. |
|
| Notes | No reported deaths due to toxicity. Estimated min follow‐up: 1 month. Estimated max follow‐up: 47 months. Median PFS: 6.9 months, 95% CI: 5.8 to 8.8 (GemCis); and 8.3 months, 95% CI: 4.3 to 9.6 (GemCap). Median OS: 13.0 months, 95% CI: 11.0 to 19.2 (GemCis); and 19.4 months, 95% CI: 16.6 to 22.0 (GemCap). All randomised participants were analysed in time‐to‐event analyses (ITT). "This study was supported by Lilly GmbH Germany." |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not reported. Stated as "randomised" only. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Registered as "open label" trial (https://www.clinicaltrials.gov/ct2/show/NCT00480597). |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | Open‐label study. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | Blood and biochemistry tests, and imaging took place during therapy. No details were provided on whether there was a central (independent) evaluation team for assessing tumour response rates. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Low risk | All 45 and 50 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analyses (intent‐to‐treat analyses). |
| Incomplete outcome data (attrition bias) (binary outcomes) | High risk | 10 of 45 and 9 of 50 participants randomised to intervention and control groups, respectively, were not assessed/assessable for tumour response (18.1% of all randomised participants). 0 of 45 and 1 of 50 participants randomised to intervention and control groups, respectively, were not included in the safety population for evaluating toxicities (1.4% of all randomised participants). |
| Selective reporting (reporting bias) | Low risk | All outcomes in the trial report were listed in the ClinicalTrials.gov record and vice versa (https://www.clinicaltrials.gov/ct2/show/NCT00480597). |
| Other bias | Low risk | None identified. |
Tutt 2014.
| Methods | Randomised phase III trial. | |
| Participants | 341 metastatic and 35 recurrent locally advanced triple‐negative or BRCA1/2 breast cancer. 43 BRCA + participants (29 known (i.e. 16 TNBC, 12 ER‐positive, HER2‐negative, 1 ER unknown, HER2‐negative) & 14 research test). | |
| Interventions | C vs D. C: Carboplatin (AUC 6 every 3 weeks for six cycles). D: Docetaxel (100 mg/m2 every 3 weeks for six cycles). |
|
| Outcomes | Response.
Overall survival (insufficient OS data reported to calculate hazard ratio for pooling). Progession‐free survival (insufficient PFS data reported to calculate hazard ratio for pooling). Toxicity (no results reported). |
|
| Notes | Abstracts only. Median follow‐up: 11 months. Median PFS: Carboplatin 3.1 (95% CI 2.5 to 4.2) vs docetaxel 4.5 (95% CI 4.1 to 5.2) months; absolute difference ‐0.4 (95%CI ‐1.1 to 0.3), P = 0.29 (insufficient PFS data reported to calculate hazard ratio for pooling). Median OS: Carboplatin 12.4 (95% CI 10.4 to 15.3) vs docetaxel 12.3 (95% CI 10.5 to 13.6) months; absolute difference ‐0.2 (95% CI ‐1.1 to 0.8), P = 0.31 (insufficient OS data reported to calculate hazard ratio for pooling). Subgroup analysis restricted to 43 BRCA1/2+ participants showed carboplatin was associated with significantly greater proportions of objective responses (68% vs 33%; P = 0.03). "Sponsor: Institute of Cancer Research, United Kingdom". |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomized" ; no additional details were provided on how random assignment was achieved in the abstracts or ClinicalTrials.gov record (https://clinicaltrials.gov/ct2/show/NCT00532727). |
| Allocation concealment (selection bias) | Unclear risk | Method of concealment was not described in the abstracts. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | Open‐label trial. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | Insufficient information provided in the available abstracts. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Low risk | All 188 and 188 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analyses (intent‐to‐treat analyses), but only median times were reported (hence no extractable time‐to‐event data). |
| Incomplete outcome data (attrition bias) (binary outcomes) | Unclear risk | All randomised participants were included in response rate denominators, but it was not explicitly stated that all participants were assessed/assessable. No toxicity results reported. |
| Selective reporting (reporting bias) | High risk | TTP, TTF and toxicity were specified as outcomes in ClinicalTrials.gov record (https://clinicaltrials.gov/ct2/show/NCT00532727), but no results for these outcomes were provided in the abstracts. |
| Other bias | Unclear risk | Unable to assess from the abstracts. |
Valero 2011.
| Methods | Randomised multicentre phase III trial. Accrual between 11 December 2001 and 23 March 2004. Groups comparable at baseline. |
|
| Participants | 263 participants with histologically confirmed metastatic breast cancer. Median age 52/51 years. 100% metastatic breast cancer. 100% first‐line. |
|
| Interventions | TH vs TCH ARM A: TH: Trastuzumab 2 mg/kg + docetaxel 100 mg/m2. ARM B: TCH: Trastuzumab 2 mg/kg + docetaxel 75 mg/m2 + carboplatin 6 AUC. |
|
| Outcomes | Overall survival (Kaplan‐Meier curves and unadjusted HRs). Progression‐free survival (Kaplan‐Meier curves and unadjusted HRs) defined as "the interval from the day of random assignment to the date of disease progression, diagnosis of second primary malignancy or death" (the authors used the terms TTP and PFS interchangeably). Response rate. Toxicity (frequencies for some conditions were not grade‐specific and therefore could not be used for pooling). |
|
| Notes | Estimated min follow‐up: 6 months. Estimated max follow‐up: 78 months. Median TTP: 11.1 and 10.4 months for TH and TCH arms, respectively. Median OS: 37.1 and 37.4 months for TH and TCH arms, respectively. Reported HRs were for Arm B as the reference group and were thus inverted. All 263 randomised participants were analysed in time‐to‐event analyses (ITT). Research Funding: Pfizer, Sanofi‐Aventis, Roche, GlaxoSmithKline. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned... Treatment allocation was based on a dynamic minimization procedure, stratified by center and by prior neoadjuvant chemotherapy." |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Treatment was not blinded" (p.151). |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | Open‐label study. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | Imaging for tumour response took place before, during, and after chemotherapy. No details were provided on whether there was a central (independent) evaluation team for assessing tumour response rates. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Low risk | All 132 and 131 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analyses (intent‐to‐treat analyses). |
| Incomplete outcome data (attrition bias) (binary outcomes) | Low risk | 5 of 132 and 1 of 131 participants randomised to intervention and control groups, respectively, were not assessed/assessable for tumour response (2.3% of all randomised participants). One randomised participant(control) was excluded from the safety population for evaluating toxicities (0.3% of all randomised participants). |
| Selective reporting (reporting bias) | Unclear risk | Most outcomes in the trial report were listed in the ClinicalTrials.gov record and vice versa (https://clinicaltrials.gov/ct2/show/NCT00047255). However, the ClinicalTrials.gov record indicated that pathologic, molecular, genetic and biochemical markers would also be assessed, but these were not reported on in the paper. |
| Other bias | Low risk | None identified. |
Xu 2011 A.
| Methods | Randomised multicentre phase II trial. Accrual between March 2005 and December 2007. Groups comparable at baseline except length of disease‐free interval of > 24 months at enrolment. |
|
| Participants | Total of 147 women overall (100 in Arm 1 and Arm 3) with histologically or cytologically confirmed metastatic breast cancer. Median age 48 years. 100% metastatic breast cancer. 97.3% of participants had received neoadjuvant or adjuvant anthracycline based chemotherapy. |
|
| Interventions | GemCis vs GemPac. ARM 1: GemPac: Paclitaxel 150 mg/m2 + gemcitabine 2500 mg/m2. ARM 3: GemCis: Gemcitabine 2500 mg/m2 + cisplatin 50 mg/m2. |
|
| Outcomes | Overall survival (Kaplan‐Meier). Progression‐free survival, endpoints not stated (Kaplan‐Meier). Response rate. Time to treatment failure (insufficient TTF data reported to calculate hazard ratio for pooling). Toxicity. |
|
| Notes | Estimated min follow‐up: 2 months. Estimated max follow‐up: 24 months. Median OS: GemPac = 15.5 months (10.4 to 26.7), GemCis = 20.1 months (12.4 to 21.6). Median PFS:GemPac = 4.8 months (4.2 to 7.0) , GemCis = 4.8 months (3.7 to 8.1). Median TTF: GemPac = 5.8 months (4.2 to 5.8), GemCis = not calculable. 99 of 100 randomised participants analysed in time‐to‐event analyses (modified ITT). "This study was sponsored by Eli Lilly and Company". |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not reported in detail other than stratified on one factor only. Quote: "Eligible patients were randomly assigned in a 1:1:1 ratio, stratified by country...". |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | Open‐label study. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | Qutote: "Tumor assessments were performed at baseline (within 4 weeks of enrolment) and at the end of cycles 4 and 8" (p. 205) and during follow‐up. No mention of a central (independent, blinded) adjudication team for assessing tumour response. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Unclear risk | 50 of 51 and 49 of 49 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analysis (modified intent‐to‐treat analysis). |
| Incomplete outcome data (attrition bias) (binary outcomes) | Low risk | 3 of 51 and 5 of 49 participants randomised to intervention and control groups, respectively, were not assessed/assessable for tumour response (7.9% of all randomised participants). One randomised participant(intervention) was excluded from the safety population for evaluating toxicities (1.3% of all randomised participants). |
| Selective reporting (reporting bias) | Low risk | All outcomes in the trial report were listed in the ClinicalTrials.gov record and vice versa (https://clinicaltrials.gov/ct2/show/study/NCT00191854). |
| Other bias | Low risk | None identified. |
Xu 2011 B.
| Methods | Randomised multicentre phase II trial. Accrual between March 2005 and December 2007. Groups comparable at baseline except length of disease‐free interval of > 24 months at enrolment. |
|
| Participants | Total of 147 women overall (96 in Arm 1 and Arm 2) with histologically or cytologically confirmed metastatic breast cancer. Median age 48 years. 100% metastatic breast cancer. 97.3% of participants had received neoadjuvant or adjuvant anthracycline‐based chemotherapy. |
|
| Interventions | GemCarb vs GemPac. ARM 1: GemPac: Paclitaxel 150 mg/m2 + gemcitabine 2500 mg/m2. ARM 2: GemCarb: Gemcitabine 2500 mg/m2 + carboplatin 2.5 AUC. |
|
| Outcomes | Overall survival (Kaplan‐Meier). Progression‐free survival, endpoints not stated (Kaplan‐Meier). Response rate. Time to treatment failure (insufficient TTF data reported to calculate hazard ratio for pooling). Toxicity. |
|
| Notes | Estimated min follow‐up: 2 months. Estimated max follow‐up: 24 months. Median OS: GemPac = 15.5 months (10.4 to 26.7), GemCarb = 22.8 months (13.3 – not calculable). Median PFS: GemPac = 4.8 months (4.2 to 7.0), GemCarb = 4.3 months (3.7 to 5.9). Median TTF: GemPac = 5.8 months (4.2 to 5.8), GemCarb = 4.3 months (3.7 – not calculable). All 96 randomised participants analysed in time‐to‐event analyses (ITT). "This study was sponsored by Eli Lilly and Company". |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not reported in detail other than stratified on one factor only. Quote: "Eligible patients were randomly assigned in a 1:1:1 ratio, stratified by country...". |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. |
| Blinding of outcome assessment (detection bias) (overall survival) | Low risk | Open‐label study. Unlikely that assessment of overall survival would be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) (outcomes other than overall survival and quality of life) | Unclear risk | Quote: "Tumor assessments were performed at baseline (within 4 weeks of enrolment) and at the end of cycles 4 and 8" (p. 205) and during follow‐up. No mention of a central (independent, blinded) adjudication team for assessing tumour response. |
| Incomplete outcome data (attrition bias) (time‐to‐event outcomes) | Low risk | All 47 and 49 participants randomised to intervention and control groups, respectively, were analysed in time‐to‐event analyses (intent‐to‐treat analyses). |
| Incomplete outcome data (attrition bias) (binary outcomes) | Low risk | 3 of 47 and 5 of 49 participants randomised to intervention and control groups, respectively, were not assessed/assessable for tumour response (7.0% of all randomised participants). No participants were excluded from the safety population for evaluating toxicities. |
| Selective reporting (reporting bias) | Low risk | All outcomes in the trial report were listed in the ClinicalTrials.gov record and vice versa (https://clinicaltrials.gov/ct2/show/study/NCT00191854). |
| Other bias | Low risk | None identified. |
5‐FU:5‐fluorouracil AL: doxorubicin, leucovorin, 5‐fluorouracil, allopurinol AUC: Area under the curve bid: Twice a day C: Carboplatin CAF: Cyclophosphamide, doxorubicin, fluorouracil CAP: Cyclophosphamide, adriamycin, Cis‐dichlordiammine platinum CDDP: Cis‐dichlordiammine platinum Ce: Cetuximab CFP: Cyclophosphamide, 5‐Fluorouracil, prednisone CI: Confidence interval CIS: Cisplatin CMF: Cyclophosphamide, methotrexate, fluorouracil CMFVP: Cyclophosphamide, methotrexate, 5‐fluorouracil, vincristine, prednisolone CT: X‐ray image made using computerized axial tomography D: Docetaxel EcisF: 5‐Fluorouracil, epirubicin, cisplatin ECOG: Eastern Cooperative Oncology Group EcycloF: 5‐Fluorouracil, epirubicin, cyclophosphamide EGFR: Epidermal growth factor receptor EORTC: European Organisation for Research and Treatment of Cancer EPI: Epirubicin EQ‐5D: EuroQol five dimensions questionnaire EQ VAS: EuroQol visual analogue scale ER: oestrogen receptor EUROQOL: The EuroQOL research Group FAC: 5‐Fluorouracil, adriamycin, cyclophosphamide FU: 5‐fluorouracil GemCap: Gemcitabine, capecitabine GemCarb: Gemcitabine, carboplatin GemCis: Gemcitabine, cisplatin GemPac: Gemcitabine, paclitaxel GemVin: Gemcitabine, vinorelbine GDoc: Gemcitabine, docetaxel HeCOG: Hellenic Cooperative Oncology Group HER2: Human epidermal growth factor receptor 2 HR: Hazard ratio IHC: Immunohistochemistry ITT: Intention‐to‐treat IV: Intravenous LND: Lonidamine Max: Maximum Min: Minimum MPEMI: Methotrexate + rescue, cisplatin, etoposide, mitomycin MPEPIV: Methotrexate+ leucovorin rescue; cisplatin, epirubicin, vincristine MRI: Magnetic resonance imaging mTNBC: metastatic triple‐negative breast cancer OS: overall survival OXA: Oxaliplatin P: Cisplatin p.: Page PCb: Paclitaxel, carboplatin PE: Cisplatin, etoposide PFS: progression‐free survival po: by mouth PR: Progesterone receptor Pw: Paclitaxel q: every QLQ‐BR23: 23 item quality of life questionnaire QLQ‐C30: 30 item quality of life questionnaire QoL: quality of life RECIST: Response Evaluation Criteria In Solid Tumors T: Paclitaxel TCH: Trastuzumab 2 mg/kg + docetaxel 75 mg/m2 + carboplatin TH: Trastuzumab, docetaxel TP: Docetaxel, cisplatin TPa: Trastuzumab, paclitaxel TPC: Trastuzumab, carboplatin TTF: Time to treatment failure TTP: Time to progression TTPD: Time to progressive disease TTTF: Time to treatment failure TX: Docetaxel, capecitabine VIN: Vinorelbine VP‐16:Oral etoposide WBC: White blood cell
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Cartei 1996 | Conference abstract only with insufficient data. Attempts to contact authors were unsuccessful. This study was listed as an 'excluded study' in the original version of this review. |
| Crump 2008 | Included 38% participants with locoregional disease. Attempts to contact authors were unsuccessful. |
| Hogdall 1993 | Trial reported only the serum tetranectin levels in relation to survival and response.This study was listed as an 'excluded study' in the original version of this review. |
| Perez 2001 | Trial was registered but never started. Hence, this study has been moved from the 'ongoing studies' section in the original version of this review to the 'excluded studies' of this review update. |
| Perez 2002 | Trial was registered but never started. Hence, this study has been moved from the 'ongoing studies' section in the original version of this review to the 'excluded studies' of this review update. |
| Somlo 2015 | Participants not randomised. |
| Wang 2008 | > 20% participants with locally advanced disease only. Data were not reported separately. |
Characteristics of ongoing studies [ordered by study ID]
BRCA.
| Trial name or title | Carboplatin or Docetaxel in Treating Women With Metastatic Genetic Breast Cancer (BRCA Trial). |
| Methods | A randomised phase II pilot trial. |
| Participants | Participants with metastatic genetic breast cancer. |
| Interventions | Carboplatin vs docetaxel. |
| Outcomes | Response. Time to progression. Toxicity. |
| Starting date | January 2005. Estimated primary completion date: 2009. |
| Contact information | Andrew Tutt, King's College London, email: andrew.tutt@kcl.ac.uk. |
| Notes | Emails sent to the principal investigator in 2015/16 requesting a progress report on the study were not answered. |
NCT00201760.
| Trial name or title | Gemcitabine/Trastuzumab and Gemcitabine/Cisplatin/Trastuzumab in Patients With Metastatic Breast Cancer. |
| Methods | Randomised phase II study. |
| Participants | Participants with metastatic breast cancer. |
| Interventions | Gemcitabine/trastuzumab vs gemcitabine/cisplatin/trastuzumab. |
| Outcomes | Disease‐free progression. Response. Side effects. |
| Starting date | February 2005. Estimated study completion date: December 2012. |
| Contact information | Kari Kendra, MD, email: Kari.Kendra@osumc.edu. |
| Notes | Emails sent to the principal investigator in 2015 requesting a progress report on the study were not answered. Sponsor: Eli Lilly and Company. |
NCT00717951.
| Trial name or title | A Randomised,Multi‐Center Study of Docetaxol Plus Capecitabine or Cisplatin in Anthracycline‐Pretreated Patients With Advanced Breast Cancer. |
| Methods | Randomised, phase 2, multicentre study. |
| Participants | Participants with advanced breast cancer. |
| Interventions | Docetaxel + capecitabine vs docetaxel + cisplatin. |
| Outcomes | Response. Time to progression. Time to treatment failure. 2 year progression‐free survival. Safety. QoL. |
| Starting date | May 2008. Estimated study completion date: May 2010. |
| Contact information | Jiang Zefei, Ph.D, emails: jiangzf@hotmail.com; jiangzefei@medmail.com.cn. |
| Notes | Emails sent to the principal investigator in 2015 requesting a progress report on the study were not answered. |
NCT01506609.
| Trial name or title | The Study Evaluating Efficacy And Tolerability Of Veliparib in Combination With Temozolomide or In Combination With Carboplatin and Paclitaxel Versus Placebo in Subjects With BRCA1 and BRCA2 Mutation and Metastatic Breast Cancer. |
| Methods | Randomised, phase 2 study. |
| Participants | Women with BRCA1 or BRCA2 mutation and metastatic breast cancer. |
| Interventions | Veliparib with temozolomide vs veliparib with carboplatin and paclitaxel vs placebo with carboplatin and paclitaxel. |
| Outcomes | Progression‐free survival. Overall survival. Clinical benefit. Objective response. Chemotherapy‐induced peripheral neuropathy. |
| Starting date | Study start date: January 2012. Estimated study completion date: May 2016. |
| Contact information | Stacie P. Shepherd, MD, AbbVie. |
| Notes | Sponsors and Collaborators: AbbVie (prior sponsor, Abbott). |
NCT01898117.
| Trial name or title | Biomarker Discovery Randomized Phase IIb Trial With Carboplatin‐cyclophosphamide Versus Paclitaxel With or Without Bevacizumab as First‐line Treatment in Advanced Triple Negative Breast Cancer. |
| Methods | Randomised phase IIb trial. |
| Participants | Participants with advanced triple‐negative breast cancer. |
| Interventions | Carboplatin‐cyclophosphamide vs paclitaxel with or without bevacizumab. |
| Outcomes | Progression‐free survival. Overall survival. Toxicity. |
| Starting date | July 2013. Estimated primary completion date: December 2019. |
| Contact information | Sabine C Linn, Prof, MD, email; s.linn@nki.nl. |
| Notes |
NCT02207335.
| Trial name or title | Trial of Gemcitabine_Capecitabine Versus Gemcitabine_Carboplatin in Breast Cancer. |
| Methods | A multicentre randomised phase Ⅲ clinical trial. |
| Participants | Participants with triple‐negative recurrent or metastatic breast cancer. |
| Interventions | Gemcitabine + capecitabine vs gemcitabine + carboplatin. |
| Outcomes | Response (RECIST 1.1). |
| Starting date | December 2013. Estimated study completion date: December 2016. |
| Contact information | Zhongsheng Tong, Master, email: 18622221181@163.com. |
| Notes |
NCT02207361.
| Trial name or title | Paclitaxel in Combination With Carboplatin Versus Paclitaxel Plus Epirubicin in Metastatic Breast Cancer. |
| Methods | Randomised prospective clinical trial. |
| Participants | Participants with metastatic breast cancer. |
| Interventions | Paclitaxel + carboplatin vs paclitaxel + epirubicin. |
| Outcomes | Response (RECIST 1.1). |
| Starting date | December 2013. Estimated study completion date: December 2016. |
| Contact information | Zhongsheng Tong, Master, email: 18622221181@163.com. |
| Notes |
TnAcity.
| Trial name or title | TnAcity: A phase 2/3 randomised study of weekly nab‐paclitaxel in combination with either gemcitabine or carboplatin vs gemcitabine/carboplatin as first‐line treatment for triple‐negative metastatic breast cancer. |
| Methods | Phase 2/3 randomised study. |
| Participants | Women with ER‐, PR‐, and HER2 negative (triple‐negative) metastatic breast cancer. |
| Interventions | Carboplatin plus gemcitabine vs nab‐paclitaxel plus carboplatin OR gemcitabine. |
| Outcomes | Progression‐free survival. Overall survival. Response. Duration of response. Safety. |
| Starting date | Date of registration: 12/06/2013. |
| Contact information | |
| Notes | Sponsored by Abraxis BioScience, LLC, a wholly‐owned subsidiary of Celgene Corporation. |
BRCA: Breast cancer susceptibility gene ER: Oestrogen receptor HER2: Human epidermal growth factor receptor 2 PR: Progesterone receptor QoL: Quality of life RECIST:Response Evaluation Criteria In Solid Tumors
Differences between protocol and review
The protocol for this review proposed that trials would be included if study participants were to receive first‐line treatment. As few trials assessing first‐line treatment were identified for inclusion in the original version of this review, those meeting the remaining eligibility criteria but which involved participants who were not first‐line naive were included. This modification of the inclusion criteria was maintained for this review update with subgroup analysis by treatment line being performed (treatment‐comparisons with first‐line therapy for > 80% of participants vs second‐ or third‐line therapy for ≥ 20% of participants).
Six of the seven subgroup analyses in this review were not prespecified in the review protocol and were added to the review in response to new hypotheses and available subgroups. Three of the six 'post hoc' subgroup analyses were added in this review update and the other three were included in the original version of the review.
Contributions of authors
Original review
DG designed the review and wrote the protocol. JS and NW provided clinical input and commented on the drafts of the review. SC undertook the review including assessing trial eligibility, data extraction, analyses and writing the review. All other authors commented on the draft review.
2016 review update
SE, NW, MW, JM and HW assessed trial eligibility. SE, MW, JM and HW performed data extraction and 'risk of bias' assessments. SE, JM and HW entered the data. SE performed the statistical analysis and wrote the update. NW provided clinical input. All authors commented on and contributed to the writing of the update.
Sources of support
Internal sources
NHMRC Clinical Trials Centre, Australia.
External sources
U.S. Army Medical Research Acquisition Activity, USA.
Declarations of interest
SE: none; MW: none; JM: none; HW: none; DO'C: none; SC: none; DG: none; NW: none.
Stable (no update expected for reasons given in 'What's new')
References
References to studies included in this review
Amadori 2013 {published data only}
- Amadori D, Carrasco E, Roesel S, Labianca R, Uziely B, Soldatenkova V, et al. A randomized phase II non‐comparative study of pemetrexed‑carboplatin and gemcitabine‑vinorelbine in anthracycline‐ and taxane‐pretreated advanced breast cancer patients. International Journal of Oncology 2013;42(5):1778‐85. [DOI] [PubMed] [Google Scholar]
- Amadori D, Torre I, Carrasco EM, Roesel S, Labianca R, Moreau‐Donnet V, et al. A randomized phase II study of pemetrexed‐carboplatin and gemcitabine‐vinorelbine in patients with anthracycline‐ and taxane‐pretreated advanced breast cancer. Journal of Clinical Oncology 2011;29(15 Suppl):1051‐1051. [Google Scholar]
- NCT00325234. Pemetrexed‐carboplatin and gemcitabine‐vinorelbine in advanced breast cancer. clinicaltrials.gov/ct2/show/results/NCT00325234 (accessed 13 April 2016).
Berruti 2002 A {published data only}
- Berruti A, Bitossi R, Gorzegno G, Bottini A, Alquati P, Matteis A, et al. Time to progression in metastatic breast cancer patients treated with epirubicin is not improved by the addition of either cisplatin or lonidamine: final results of a phase III study with a factorial design. Journal of Clinical Oncology 2002;20(20):4150‐9. [DOI] [PubMed] [Google Scholar]
Berruti 2002 B {published data only}
- Berruti A, Bitossi R, Gorzegno G, Bottini A, Alquati P, Matteis A, et al. Time to progression in metastatic breast cancer patients treated with epirubicin is not improved by the addition of either cisplatin or lonidamine: final results of a phase III study with a factorial design. Journal of Clinical Oncology 2002;20(20):4150‐9. [DOI] [PubMed] [Google Scholar]
Bhattacharyya 2009 {published data only}
- Bhattacharyya GS, Basu S, Agarwal V, Malhotra H, Pareekh PM, Babu KG, et al. 41LBA Single institute phase II study of weekly cisplatinum and metronomic dosing of endoxan and methotrexate in second line metastatic breast cancer triple‐negative. European Journal of Cancer Supplements. 2009; Vol. 7, issue 3:18‐19.
Carey 2012 {published data only}
- Carey LA, Rugo HS, Marcom PK, Mayer EL, Esteva FJ, Ma CX, et al. TBCRC 001: randomized phase II study of cetuximab in combination with carboplatin in stage IV triple‐negative breast cancer. Journal of Clinical Oncology 2012;30(21):2615‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT00232505. Cetuximab with or without carboplatin in treating women with estrogen receptor‐negative, progesterone receptor‐negative metastatic breast cancer. clinicaltrials.gov/show/NCT00232505 (accessed 13 April 2016).
Cocconi 1991 {published data only}
- Cocconi G, Bisagni G, Bacchi M, Boni C, Bartolucci R, Ceci G, et al. Cisplatin and etoposide as first‐line chemotherapy for metastatic breast carcinoma: a prospective randomized trial of the Italian Oncology Group for Clinical Research. Journal of Clinical Oncology 1991;9(4):664‐9. [DOI] [PubMed] [Google Scholar]
Cocconi 1996 {published data only}
- Cocconi G, Bella M, Bartolucci R, Basurto C, Colozza M, Indelli M. Continuous CMF compared to a short chemotherapy using cisplatin‐containing combinations in metastatic breast carcinoma. A prospective randomized study. Annals of Oncology 1996;7(Suppl 5):22. [Google Scholar]
Cocconi 1999 {published data only}
- Cocconi G, Bisagni G, Bella M, Acito L, Anastasi P, Carpi A, et al. Comparison of CMF (cyclophosphamide, methotrexate, and 5‐fluorouracil) with a rotational crossing and a sequential intensification regimen in advanced breast cancer: a prospective randomized study. American Journal of Clinical Oncology 1999;22(6):593‐600. [DOI] [PubMed] [Google Scholar]
Costanza 1999 {published data only}
- Costanza ME, Weiss RB, Henderson IC, Norton L, Berry DA, Cirrincione C, et al. Safety and efficacy of using a single agent or a phase II agent before instituting standard combination chemotherapy in previously untreated metastatic breast cancer patients: report of a randomized study‐‐Cancer and Leukemia Group B 8642. Journal of Clinical Oncology 1999;17(5):1397‐406. [DOI] [PubMed] [Google Scholar]
Creagan 1984 {published data only}
- Creagan ET, Green SJ, Ahmann DL, Ingle JN, Edmonson JH, Marschke RF‐J. A phase III clinical trial comparing the combination cyclophosphamide, adriamycin, cisplatin with cyclophosphamide, 5‐ fluorouracil, prednisone in patients with advanced breast cancer. Journal of Clinical Oncology 1984;2(11):1260‐5. [DOI] [PubMed] [Google Scholar]
Delaloge 2004 {published data only}
- Delaloge S, Tubiana‐Hulin M, Wardley A, Mastro L, Santoro A, Zambelli A, et al. A multistep randomized phase II/III trial comparing Oxaliplatin (OXA)+5 fluorouracil (FU) to vinorelbine (VIN) + FU (FUN) after taxane (T)/anthracycline (A) failure in advanced/metastatic breast cancer (MBC) patients (pts): Final results. Journal of Clinical Oncology. 2004; Vol. 22 Supplement:14S.
Eisen 1998 {published data only}
- Eisen T, Smith IE, Boer R, Ellis PA. A randomised phase II trial of infusional 5‐FU and epirubicin with cyclophosphamide versus cisplatin in advanced breast cancer. Breast Cancer Research & Treatment 1997;46(1):94. [Google Scholar]
- Eisen T, Smith IE, Johnston S, Ellis PA, Prendiville J, Seymour MT, et al. Randomized phase II trial of infusional fluorouracil, epirubicin, and cyclophosphamide versus infusional fluorouracil, epirubicin, and cisplatin in patients with advanced breast cancer. Journal of Clinical Oncology 1998;16(4):1350‐7. [DOI] [PubMed] [Google Scholar]
Fan 2012 {published data only}
- Fan Y, Xu BH, Yuan P, Ma F, Wang JY, Ding XY, et al. Docetaxel–cisplatin might be superior to docetaxel–capecitabine in the first‐line treatment of metastatic triple‐negative breast cancer. Annals of Oncology 2013;24(5):1219‐25. [DOI] [PubMed] [Google Scholar]
Fountzilas 2004 {published data only}
- Fountzilas G, Kalofonos HP, Bafaloukos D, Papakostas P, Kosmidis P, Gogas H, et al. Paclitaxel and epirubicin versus paclitaxel and carboplatin as first‐line chemotherapy in patients with advanced breast cancer: A phase III study conducted by the Hellenic Cooperative Oncology Group. Annals of Oncology 2002;13(Suppl 5):46‐69. [DOI] [PubMed] [Google Scholar]
- Fountzilas G, Kalofonos HP, Dafni U, Papadimitriou C, Bafaloukos D, Papakostas P, et al. Paclitaxel and epirubicin versus paclitaxel and carboplatin as first‐line chemotherapy in patients with advanced breast cancer: a phase III study conducted by the Hellenic Cooperative Oncology Group. Annals of Oncology 2004;15(10):1517‐26. [DOI] [PubMed] [Google Scholar]
Fountzilas 2009 A {published data only}
- Fountzilas G, Dafni U, Dimopoulos MA, Koutras A, Skarlos D, Papakostas P, et al. A randomized phase III study comparing three anthracycline‐free taxane‐based regimens, as first‐line chemotherapy, in metastatic breast cancer. A Hellenic Cooperative Oncology Group Study. Breast Cancer Research and Treatment 2009;11:87‐99. [DOI] [PubMed] [Google Scholar]
Fountzilas 2009 B {published data only}
- Fountzilas G, Dafni U, Dimopoulos MA, Koutras A, Skarlos D, Papakostas P, et al. A randomized phase III study comparing three anthracycline‐free taxane‐based regimens, as first‐line chemotherapy, in metastatic breast cancer. A Hellenic Cooperative Oncology Group Study. Breast Cancer Research and Treatment 2009;11:87‐99. [DOI] [PubMed] [Google Scholar]
Hu 2015 {published data only}
- Hu XC, Zhang J, Xu BH, Cai L, Ragaz J, Wang ZH, et al. Cisplatin plus gemcitabine versus paclitaxel plus gemcitabine as first‐line therapy for metastatic triple‐negative breast cancer (CBCSG006): a randomised, open‐label, multicentre, phase 3 trial. Lancet Oncology 2015;16(4):436‐46. [DOI] [PubMed] [Google Scholar]
- NCT01287624. Gemcitabine plus cisplatin versus gemcitabine plus paclitaxel in triple negative breast cancer (TNBC). clinicaltrials.gov/ct2/show/NCT01287624 (accessed 13 April 2016).
Icli 2005 {published data only}
- Icli F, Akbulut H, Uner A, Bulent Y, Altinbas M, Baltali E, et al. Paclitaxel (T) vs cisplatin + VP‐16 (EP) in metastatic breast cancer patients treated with anthracyclines: A phase III randomized study, Turkish Oncology Group. Annals of Oncology 2002; Vol. 13:47.
- Icli F, Akbulut H, Uner A, Yalcin B, Baltali E, Altinbas M, et al. Cisplatin plus oral etoposide (EoP) combination is more effective than paclitaxel in patients with advanced breast cancer pretreated with anthracyclines: a randomised phase III trial of Turkish Oncology Group. British Journal of Cancer 2005;92:639‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kolaric 1985 {published data only}
- Kolaric K, Vukas D, Roth A, Potrebica V, Cervek J, Cerar O. Cyclophosphamide, adriamycin and platinum (CAP) combination chemotherapy, a new effective approach in the treatment of disseminated breast cancer. Preliminary report. Tumori 1985;71:159‐65. [DOI] [PubMed] [Google Scholar]
Kolaric 1989 {published data only}
- Kolaric K, Vukas D, Potrebica V. Combination of cyclophosphamide, adriamycin and platinum (CAP) versus 5‐fluorouracil, adriamycin and cyclophosphamide (FAC) as primary treatment in metastatic breast cancer: results of a prospective randomized study. Tumori 1989;75:132‐6. [DOI] [PubMed] [Google Scholar]
Nielsen 2000 {published data only}
- Nielsen D, Dombernowsky P, Larsen SK, Hansen OP, Skovsgaard T. Epirubicin or epirubicin and cisplatin as first‐line therapy in advanced breast cancer. A phase III study. Cancer Chemotherapy & Pharmacology 2000;46(6):459‐66. [DOI] [PubMed] [Google Scholar]
- Ryberg M, Nielsen D, Skovsgaard T, Hansen J, Jensen BV, Dombernowsky P. Epirubicin cardiotoxicity: An analysis of 469 patients with metastatic breast cancer. Journal of Clinical Oncology 1998;16(11):3502‐8. [DOI] [PubMed] [Google Scholar]
Robert 2006 {published data only}
- Robert N, Leyland‐Jones B, Asmar L, Belt R, Ilegbodu D, Loesch D, et al. Randomized phase III study of trastuzumab, paclitaxel, and carboplatin compared with trastuzumab and paclitaxel in women with HER‐2‐overexpressing metastatic breast cancer. Journal of Clinical Oncology 2006;24(18):2786‐92. [DOI] [PubMed] [Google Scholar]
Stemmler 2011 A {published data only}
- NCT00480597. Gemcitabine/vinorelbine versus gemcitabine/cisplatin versus gemcitabine/capecitabine in metastatic breast cancer. clinicaltrials.gov/ct2/show/NCT00480597 (accessed 13 April 2016).
- Stemmler HJ, DiGioia D, Freier W, Tessen HW, Gitsch G, Jonat W, et al. Randomised phase II trial of gemcitabine plus vinorelbine vs gemcitabine plus cisplatin vs gemcitabine plus capecitabine in patients with pretreated metastatic breast cancer. British Journal of Cancer 2011;104(7):1071‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Stemmler 2011 B {published data only}
- NCT00480597. Gemcitabine/vinorelbine versus gemcitabine/cisplatin versus gemcitabine/capecitabine in metastatic breast cancer. clinicaltrials.gov/ct2/show/NCT00480597 (accessed 13 April 2016).
- Stemmler HJ, DiGioia D, Freier W, Tessen HW, Gitsch G, Jonat W, et al. Randomised phase II trial of gemcitabine plus vinorelbine vs gemcitabine plus cisplatin vs gemcitabine plus capecitabine in patients with pretreated metastatic breast cancer. British Journal of Cancer 2011;104(7):1071‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tutt 2014 {published data only}
- NCT00532727. Triple negative breast cancer trial (TNT). clinicaltrials.gov/show/NCT00532727 (accessed 13 April 2016).
- Tutt A, Ellis P, Kilburn L, Gilett C, Pinder S, Abraham J, et al. The TNT trial: A randomized phase III trial of carboplatin (C) compared with docetaxel (D) for patients with metastatic or recurrent locally advanced triple negative or BRCA1/2 breast cancer (CRUK/07/012). Cancer Research 2015;75(9 Supplement):S3‐01. [Google Scholar]
- Tutt A, Ellis P, Kilburn LS. The TNT trial: A randomized phase III trial of carboplatin compared with docetaxel for patients with metastatic or recurrent locally advanced trip‐negative or BRCA1/2 breast cancer. Thirty‐Seventh Annual CTRC‐AACR San Antonio Breast Cancer Symposium; December 9‐13, 2014; San Antonio, TX. 2014; Vol. 75:S3‐1.
Valero 2011 {published data only}
- Forbes JF, Pienkowski T, Valero V, Eiermann W, Minckwitz G, Martin M, et al. BCIRG 007: Randomized phase III trial of trastuzumab plus docetaxel with or without carboplatin first‐line in HER2 positive metastatic breast cancer (MBC). Journal of Clinical Oncology 2006;24(Supplement):18S. [Google Scholar]
- NCT00047255. Docetaxel and trastuzumab with or without carboplatin in treating women with HER2‐positive breast cancer. clinicaltrials.gov/ct2/show/NCT00047255 (accessed 13 April 2016).
- Valero V, Forbes J, Pegram MD, Pienkowski T, Eiermann W, Minckwitz G, et al. Multicenter phase III randomized trial comparing docetaxel and trastuzumab with docetaxel, carboplatin, and trastuzumab as first‐line chemotherapy for patients with HER2‐gene‐amplified metastatic breast cancer (BCIRG 007 Study): Two highly active therapeutic regimens. Journal of Clinical Oncology 2011;29(2):149‐56. [DOI] [PubMed] [Google Scholar]
Xu 2011 A {published data only}
- NCT00191854. Gemcitabine combinations in metastatic breast cancer (MBC), 1st Line. clinicaltrials.gov/ct2/show/results/NCT00191854 (accessed 13 April 2016).
- Xu B, Jiang Z, Kim SB, Yu S, Feng J, Malzyner A, et al. Biweekly gemcitabine‐paclitaxel, gemcitabine‐carboplatin, or gemcitabine‐cisplatin as first‐line treatment in metastatic breast cancer after anthracycline failure: a phase II randomized selection trial. Breast Cancer 2011;18(3):203‐12. [DOI] [PubMed] [Google Scholar]
Xu 2011 B {published data only}
- NCT00191854. Gemcitabine combinations in metastatic breast cancer (MBC), 1st Line. clinicaltrials.gov/ct2/show/results/NCT00191854 (accessed 13 April 2016).
- Xu B, Jiang Z, Kim SB, Yu S, Feng J, Malzyner A, et al. Biweekly gemcitabine‐paclitaxel, gemcitabine‐carboplatin, or gemcitabine‐cisplatin as first‐line treatment in metastatic breast cancer after anthracycline failure: a phase II randomized selection trial. Breast Cancer 2011;18(3):203‐12. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Cartei 1996 {published data only}
- Cartei, MPP. Epirubicin versus platin and etoposide: A cross over study in breast cancer. Tumori. 1996; Vol. 82, issue Supplement:125.
Crump 2008 {published data only}
- Crump M, Gluck S, Tu D, Stewart D, Levine M, Kirkbride P, et al. Randomized trial of high‐dose chemotherapy with autologous peripheral‐blood stem‐cell support compared with standard‐dose chemotherapy in women with metastatic breast cancer: NCIC MA.16. Journal of Clinical Oncology 2008;26(1):37‐43. [DOI] [PubMed] [Google Scholar]
Hogdall 1993 {published data only}
- Hogdall CK, Soletormos G, Nielsen D, Norgaard PB, Dombernowsky P, Clemmensen I. Prognostic value of serum tetranectin in patients with metastatic breast cancer. Acta Oncologica 1993;32:631‐6. [DOI] [PubMed] [Google Scholar]
Perez 2001 {published data only}
- NCT00025688. A randomized, phase II trial of weekly taxol (paclitaxel) versus weekly taxol plus paraplatin (carboplatin) as first‐line chemotherapy in patients age 65 years or older with metastatic breast cancer. clinicaltrials.gov/ct2/show/study/NCT00025688 (accessed April 12 2016).
Perez 2002 {published data only}
- Perez EA. Phase III randomized study of Paclitaxel, Carboplatin, and Trastuzumab (Herceptin) as first‐line chemotherapy in women with overexpressed HER‐2, metastatic breast cancer. www.cancer.gov/clinical trials 2002.
Somlo 2015 {published and unpublished data}
- Somlo G, Frankel PH, Luu TH, Ma CX, Arun B, Garcia AA, et al. Efficacy of the PARP inhibitor (PI) ABT‐888 (veliparib [vel]) either with carboplatin (carb) or as a single agent followed by post‐progression therapy in combination with carb in patients (pts) with BRCA1‐or BRCA2‐(BRCA)‐associated metastatic breast cancer (MBC). Journal of Clinical Oncology 2015;33(15 Suppl):520. [Google Scholar]
Wang 2008 {published data only}
- Wang Y, Wu Q, Su F, Zhou L, Ye Z, Yang J, et al. Phase II study of docetaxel plus epirubicin versus docetaxel plus cisplatin as first‐line chemotherapy for metastatic breast cancer. Chinese Journal of Oncology 2008;30(7):541‐4. [PubMed] [Google Scholar]
References to ongoing studies
BRCA {unpublished data only}
- Triple negative trial: a randomised phase III trial of carboplatin compared to docetaxel for patients with metastatic or recurrent locally advanced ER‐, PR‐ and HER2‐breast cancer. clinicaltrials.gov/show/NCT00532727 (accessed 12 April 2016).
NCT00201760 {unpublished data only}
- NCT00201760. A randomized phase II study of gemcitabine/ trastuzumab and gemcitabine/ cisplatin/ trastuzumab in patients with metastatic breast cancer. clinicaltrials.gov/show/NCT00201760 (accessed 12 April 2016).
NCT00717951 {unpublished data only}
- NCT00717951. A randomised, multi‐center study of docetaxol plus capecitabine or cisplatin in anthracycline‐pretreated patients with advanced breast cancer. clinicaltrials.gov/show/NCT00717951 (accessed April 12 2016).
NCT01506609 {unpublished data only}
- NCT01506609. A randomized, phase 2 study of the efficacy and tolerability of veliparib in combination with temozolomide or veliparib in combination with carboplatin and paclitaxel versus placebo plus carboplatin and paclitaxel in subjects with BRCA1 or BRCA2 mutation and metastatic breast cancer. clinicaltrials.gov/show/NCT01506609 (accessed 12 April 2016).
NCT01898117 {unpublished data only}
- NCT01898117. Triple‐B study;carboplatin‐cyclophosphamide versus paclitaxel with or without bevacizumab as first‐line treatment in advanced triple negative breast cancer (Triple‐B). clinicaltrials.gov/show/NCT01898117 (accessed 12 April 2016).
NCT02207335 {unpublished data only}
- NCT02207335. A multicenter randomized phase III clinical trial of gemcitabine in combination with capecitabine versus gemcitabine plus carboplatin as first‐line treatment in triple‐negative recurrent or metastatic breast cancer. clinicaltrials.gov/ct2/show/NCT02207335 (accessed 12 April 2016).
NCT02207361 {unpublished data only}
- NCT02207361. A randomized prospective clinical trial of paclitaxel in combination with carboplatin versus paclitaxel plus epirubicin as first‐line treatment in metastatic breast cancer. clinicaltrials.gov/ct2/show/NCT02207361 (accessed 12 April 2016).
TnAcity {unpublished data only}
- Yardley DA, Brufsky A, Conte P, Cortes J, Glück S, Nabholtz J‐MA, et al. TnAcity: A phase 2/3 randomized study of weekly nab‐paclitaxel in combination with either gemcitabine or carboplatin vs gemcitabine/carboplatin as first‐line treatment for triple‐negative metastatic breast cancer. Cancer Research 2013;73(24 Supplement):OT3. [Google Scholar]
Additional references
Beslija 2009
- Beslija S, Bonneterre J, Burstein HJ, Cocquyt V, Gnant M, Heinemann V, et al. Third consensus on medical treatment of metastatic breast cancer. Annals of Oncology 2009;20(11):1771‐85. [DOI] [PubMed] [Google Scholar]
Burzykowski 2008
- Burzykowski T, Buyse M, Piccart‐Gebhart MJ, Sledge G, Carmichael J, Lück HJ, et al. Evaluation of tumor response, disease control, progression‐free survival, and time to progression as potential surrogate end points in metastatic breast cancer. Journal of Clinical Oncology 2008;26(12):1987‐92. [DOI] [PubMed] [Google Scholar]
Carrick 2004
- Carrick S, Ghersi D, Wilcken N, Simes J. Platinum containing regimens for metastatic breast cancer. Cochrane Database of Systematic Reviews 2004, Issue 3. [DOI: 10.1002/14651858.CD003374.pub3] [DOI] [PubMed] [Google Scholar]
Clements 2012
- Clements MS, Roder DM, Yu XQ, Egger S, O'Connell DL. Estimating prevalence of distant metastatic breast cancer: a means of filling a data gap. Cancer Causes & Control 2012;23(10):1625‐34. [DOI] [PubMed] [Google Scholar]
Davies 1998
- Davies HT. Interpreting measures of treatment effect. Hospital Medicine 1998;59(6):499‐501. [PubMed] [Google Scholar]
De Angelis 2005
- Angelis CD, Drazen JM, Frizelle FA, Haug C, Hoey J, Horton R, et al. Is this clinical trial fully registered?—A statement from the International Committee of Medical Journal Editors. New England Journal of Medicine 2005;352(23):2436‐8. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Smith GD, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315(7109):629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Eisenhauer 2009
- Eisenhauer E, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). European Journal of Cancer 2009;45(2):228‐47. [DOI] [PubMed] [Google Scholar]
Ferlay 2013
- Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, et al. Cancer incidence and mortality worldwide. GLOBOCAN 2012 : IARC Cancerbase No. 11. globocan.iarc.fr/Default.aspx (accessed prior to 25 May 2017).
Foulkes 2010
- Foulkes WD, Smith IE, Reis‐Filho JS. Triple‐negative breast cancer. NEJM 2010;363(20):1938‐48. [DOI] [PubMed] [Google Scholar]
GradeproGDT
- GRADEproGDT: GRADEpro Guideline Development Tool [Software]. McMaster University, 2015 (developed by Evidence Prime, Inc.). Available from www.gradepro.org.
Guan 2015
- Guan X, Ma F, Fan Y, Zhu W, Hong R, Xu B. Platinum‐based chemotherapy in triple‐negative breast cancer: a systematic review and meta‐analysis of randomized‐controlled trials. Anti‐cancer Drugs 2015;26(8):894‐901. [DOI] [PubMed] [Google Scholar]
Guyatt 2011
- Guyatt GH, Oxman AD, Sultan S, Glasziou P, Akl EA, Alonso‐Coello P, et al. the GRADE Working Group. GRADE guidelines: 9. Rating up the quality of evidence. Journal of Clinical Epidemiology 2011;64(12):1311‐6. [DOI] [PubMed] [Google Scholar]
Hayes 1995
- Hayes DF, Henderson IC, Shapiro CL. Treatment of metastatic breast cancer: present and future prospects. Seminars in Oncology 1995;22(Suppl 5):5‐21. [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Hortobagyi 1996
- Hortobagyi GN, Piccart‐Gebhart MJ. Current management of advanced breast cancer. Seminars in Oncology 1996;23(Suppl 11):1‐5. [PubMed] [Google Scholar]
Kassam 2009
- Kassam F, Enright K, Dent R, Dranitsaris G, Myers J, Flynn C, et al. Survival outcomes for patients with metastatic triple‐negative breast cancer: implications for clinical practice and trial design. Clinical Breast Cancer 2009;9(1):29‐33. [DOI] [PubMed] [Google Scholar]
Mani 2002
- Mani S, Graham MA, Bregman DB, Ivy P, Chaney SG. Oxaliplatin: a review of evolving concepts. Cancer Investigation 2002;20(2):246‐63. [DOI] [PubMed] [Google Scholar]
Noll 2006
- Noll DM, Mason TM, Miller PS. Formation and repair of interstrand cross‐links in DNA. Chemical Reviews 2006;106(2):277‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
Parmar 1998
- Parmar MKB, Torri V, Stewart L. Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17:2815‐34. [DOI] [PubMed] [Google Scholar]
Petrelli 2016
- Petrelli F, Barni S, Bregni G, Braud F, Cosimo S. Platinum salts in advanced breast cancer: a systematic review and meta‐analysis of randomized clinical trials. Breast Cancer Research and Treatment 2016;160(3):425‐37. [DOI] [PubMed] [Google Scholar]
RevMan 5.3 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Shamseddine 2012
- Shamseddine AI, Farhat FS. Platinum‐based compounds for the treatment of metastatic breast cancer. Chemotherapy 2012;57(6):468‐87. [DOI] [PubMed] [Google Scholar]
Sikov 2015
- Sikov WM. Assessing the role of platinum agents in aggressive breast cancers. Current Oncology Reports 2015;17(2):1‐10. [DOI] [PubMed] [Google Scholar]
Stordal 2007
- Stordal B, Pavlakis N, Davey R. Oxaliplatin for the treatment of cisplatin‐resistant cancer: a systematic review. Cancer Treatment Reviews 2007;33(4):347‐57. [DOI] [PubMed] [Google Scholar]
Tierney 2007
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time‐to‐event data into meta‐analysis. Trials 2007;8(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yusuf 1985
- Yusuf S, Peto R, Lewis J, Collins R, Sleight P. Beta blockade during and after myocardial infarction: an overview of the randomized trials. Progress in Cardiovascular Diseases. 1985;5:335‐71. [DOI] [PubMed] [Google Scholar]