

Abstract
Background
This is an update of a review first published in 2003 and updated in 2012.
Ketamine is a commonly used anaesthetic agent, and in subanaesthetic doses is also given as an adjuvant to opioids for the treatment of refractory cancer pain, when opioids alone or in combination with appropriate adjuvant analgesics prove to be ineffective. Ketamine is known to have psychomimetic (including hallucinogenic), urological, and hepatic adverse effects.
Objectives
To determine the effectiveness and adverse effects of ketamine as an adjuvant to opioids for refractory cancer pain in adults.
Search methods
For this update, we searched MEDLINE (OVID) to December 2016. We searched CENTRAL (CRSO), Embase (OVID) and two clinical trial registries to January 2017.
Selection criteria
The intervention considered by this review was the addition of ketamine, given by any route of administration, in any dose, to pre‐existing opioid treatment given by any route and in any dose, compared with placebo or active control. We included studies with a group size of at least 10 participants who completed the trial.
Data collection and analysis
Two review authors independently assessed the search results and performed 'Risk of bias' assessments. We aimed to extract data on patient‐reported pain intensity, total opioid consumption over the study period; use of rescue medication; adverse events; measures of patient satisfaction/preference; function; and distress. We also assessed participant withdrawal (dropout) from trial. We assessed the quality of the evidence using GRADE (Grading of Recommendations Assessment, Development and Evaluation).
Main results
One new study (185 participants) was identified by the updated search and included in the review. We included a total of three studies in this update.
Two small studies, both with cross‐over design, with 20 and 10 participants respectively, were eligible for inclusion in the original review. One study with 20 participants examined the addition of intrathecal ketamine to intrathecal morphine, compared with intrathecal morphine alone. The second study with 10 participants examined the addition of intravenous ketamine bolus in two different doses to ongoing morphine therapy, compared with placebo. Both of these studies reported reduction in pain intensity and reduction in morphine requirements when ketamine was added to opioid for refractory cancer pain. The new study identified by the updated search had a parallel group design and 185 participants. This placebo‐controlled study examined rapid titration of subcutaneous ketamine to high dose (500 mg) in participants who were using different opioids. There were no differences between groups for patient‐reported pain intensity.
Pooling of the data from the three included trials was not appropriate because of clinical heterogeneity.
The study examining intrathecal drug administration reported no adverse events related to ketamine. In the study using intravenous bolus administration, ketamine caused hallucinations in four of 10 participants. In the rapid dose escalation/high‐dose subcutaneous ketamine study, there was almost twice the incidence of adverse events in the ketamine group, compared to the placebo group, with the most common adverse events being needle site irritation and cognitive disturbance. Two serious adverse events (bradyarrhythmia and cardiac arrest) thought to be related to ketamine were also reported in this trial.
For all three studies there was an unclear risk of bias overall. Using GRADE, we judged the quality of the evidence to be very low due to study limitations and imprecision due to the small number of participants in all comparisons.
Authors' conclusions
Current evidence is insufficient to assess the benefits and harms of ketamine as an adjuvant to opioids for the relief of refractory cancer pain. The evidence was of very low quality, meaning that it does not provide a reliable indication of the likely effect, and the likelihood that the effect will be substantially different is high. Rapid dose escalation of ketamine to high dose (500 mg) does not appear to have clinical benefit and may be associated with serious adverse events. More randomised controlled trials (RCTs) examining specific low‐dose ketamine clinical regimens in current use are needed.
Plain language summary
Adding ketamine to opioid for opioid‐resistant cancer pain
Bottom line
The benefits and harms of adding low‐dose ketamine to strong pain‐killers such as morphine for the relief of cancer pain are not yet established. High‐dose ketamine does not appear to be effective and may be associated with serious side effects.
Background
This review is an update of a review first published in 2003 and updated in 2012.
Morphine‐like drugs (opioids) are frequently prescribed for moderate and severe cancer pain, but in some cases these drugs are not effective. Ketamine, an anaesthetic agent, is used in low doses in palliative care to improve analgesia when opioids alone are ineffective.
Study characteristics
In December 2016 and January 2017, we searched for clinical trials on the addition of ketamine to morphine‐like drugs for cancer pain.
We found one new study, together with the two studies included in the original review. The three studies were very different, using different doses of ketamine, different routes of administration and different durations of treatment and it was not possible to combine the results of these studies.
Key results
The two smallest studies reported that the addition of ketamine to morphine reduced pain intensity and morphine requirements. The third study which used high doses of ketamine reported no clinical benefit of adding ketamine to different opioids. Increased doses of ketamine in some participants caused side effects such as hallucinations. The study which examined high doses of ketamine reported two serious adverse events, which may have been related to ketamine. Although two out of three studies reported reduction in pain, this could be due to chance in such small studies.
Quality of the evidence
We rated the quality of the evidence using four levels: very low, low, moderate, or high. Very low‐quality evidence means that we are very uncertain about the results. High‐quality evidence means that we are very confident in the results. The evidence from the studies was of very low quality. There were problems with the design of some studies and there were not enough data to answer some parts of our review question.
Background
This is an update of a previously published review in the Cochrane Database of Systematic Reviews (2003, Issue 1 (Bell 2003)), and updated in 2012 (Bell 2012b) on ketamine as an adjuvant to opioids for cancer pain.
Description of the condition
Studies report that moderate to severe pain is common in patients with advanced cancer (Are 2017). Cancer pain that is refractory to standard treatment occurs in 10% to 20% of these patients (Afsharimani 2015). Cancer pain is often of mixed aetiology and may have nociceptive, neuropathic and inflammatory components. Neuropathic pain which results from tumour infiltration in nerve plexi and damage of nerve tissue can be especially difficult to treat (Fallon 2013). Opioids (for example, morphine, fentanyl, hydromorphone, oxycodone, codeine) are frequently prescribed for the relief of moderate and severe cancer pain. However, not all cancer pain is sufficiently relieved by opioids alone.
Description of the intervention
The usual indication for using ketamine as an adjuvant to opioid in cancer pain is for pain which is unresponsive to opioids and adjuvant analgesics, for example in the case of refractory neuropathic pain or opioid tolerance. Clinical reports indicate that, when added to opioids, low subanaesthetic doses of ketamine may give improved analgesia (Sosnowski 1993; Fine 1999; Bell 1999). The practice of using ketamine as an adjuvant to opioids in the treatment of cancer pain that does not respond to opioids alone, or to opioids in combination with adjuvant analgesic drugs, is discussed in several pain and palliative care textbooks (Stannard 2005; Twycross 2009; Cherny 2015). Ketamine is not licensed for this purpose and this is an update of the first systematic review undertaken to establish the evidence base for this practice.
Ketamine hydrochloride has been used as a general anaesthetic agent for over 30 years, and is commonly given intravenously or intramuscularly for surgical anaesthesia (Fisher 2000). Ketamine causes dissociative anaesthesia and also has analgesic effects (Grahame‐Smith 2002); because it increases sympathetic nervous system activity, it is a useful anaesthetic for high‐risk patients who require a high degree of sympathetic activity to maintain cardiovascular function. However, the benefits are tempered by the high incidence of hallucinations and other transient psychomimetic sequelae when ketamine is used for anaesthesia in adults (BNF 2012). More recently, urological toxicity and hepatic toxicity have been described as adverse effects of ketamine (Bell 2012a).
In the 1980s ketamine was discovered to have N‐methyl‐D‐aspartate (NMDA) receptor antagonist properties and acts by blocking excitatory glutamate receptors in the central nervous system. There is an association between nociceptive activity involving the NMDA receptor and hyperalgesia/allodynia, and reduced opioid sensitivity (Dickenson 1994). The NMDA receptor plays a role in the development of opioid tolerance (Trujillo 1991; Mao 1995; Mayer 1995). Currently, there is much focus on ketamine for the treatment of major depression. A recent paper reports that ketamine metabolites exert antidepressant actions independent of NMDA receptor inhibition (Zanos 2016).
Evidence from experimental animal models, human volunteer studies and small clinical trials indicates that subanaesthetic doses of ketamine alleviate various chronic and neuropathic pain syndromes (Fisher 2000). Ketamine has anti‐inflammatory effects and may have an effect in inflammatory pain (Dale 2012; Sawynok 2014). However, the clinical use of ketamine at subanaesthetic dose levels has also been restricted by unpleasant adverse effects, typically sedation, nausea, disagreeable psychological disturbances or hallucinations (Willetts 1990).
Racemic ketamine is a mixture of two stereoisomers: R(‐) and S(+). More recently, S‐ketamine has been introduced. S(+) ketamine produces longer hypnosis than the (‐) isomer, and causes a greater rise in blood pressure and heart rate, less locomotor activity, and a shorter recovery time, and it is postulated to have twice the analgesic efficacy of racemic ketamine. S(+) ketamine is also thought to have a safer adverse effect profile (Grahame‐Smith 2002). The majority of published clinical studies in postoperative and chronic pain have used racemic ketamine. For a review on the pharmacokinetics of ketamine see Peltoniemi 2016. The oral bioavailability of ketamine is low and the drug undergoes fast cytochrome P450 (CYP) mediated N‐demethylation to norketamine. Approximately 80% of ketamine undergoes N‐demethylation to norketamine by CYP3A and CYP2B6 enzymes (Kharasch 1992; Yanagihara 2001; Hijazi 2002), with a smaller amount being metabolised to 4‐and 6‐hydroxyketamines (Woolf 1987). Compared to ketamine, norketamine is an approximately three to five times weaker NMDA receptor antagonist (Leung 1986; Ebert 1997). Being metabolised by CYP3A enzymes, ketamine may have significant interactions with opioids and other drugs. Studies in rodents indicate important interactions between ketamine and opioids. Edwards 2002 reported that in mice, distribution of ketamine into the brain was increased by low plasma concentrations of alfentanil. Recently Lilius 2015 found that ketamine co‐administration attenuates morphine tolerance and leads to increased brain concentrations of both drugs in the rat.
Ketamine has multiple routes of administration and is commonly given as an adjuvant to pre‐existing opioid treatment. A number of systematic reviews report that ketamine is effective in acute postoperative pain and reduces morphine requirements (Bell 2006; Laskowski 2011; Assouline 2016).
How the intervention might work
By blocking activity at the NMDA receptor, ketamine may reduce neuropathic‐related cancer pain. Blocking NMDA receptor activity may reduce opioid tolerance thus increasing/restoring the analgesic effect of opioid. Ketamine has anti‐inflammatory effects and may be beneficial in inflammatory cancer pain.
Why it is important to do this review
This is an update of a Cochrane review first published in 2003, and previously updated in 2012. Ketamine is routinely used in the palliative care setting for the treatment of refractory cancer pain. Earlier versions of this review found limited and heterogenous data, and there was insufficient evidence to be able to make any conclusions. In recent years the standards used to assess evidence in pain trials have changed substantially, for example there is now particular attention being paid to participant withdrawal from trials, and statistical imputation following withdrawal, which can substantially alter estimates of efficacy. The most important change is the move from using average pain scores, or average change in pain scores, to the number of people who have a large decrease in pain (by at least 50%) (PaPaS 2012). This update assesses the current evidence using the new criteria for what constitutes reliable evidence in pain trials.
Objectives
To determine the effectiveness and adverse effects of ketamine as an adjuvant to opioids for refractory cancer pain in adults.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs)
Double‐blind studies
Placebo‐ or active‐controlled trials, both with or without cross‐over, in in‐patient and out‐patient settings
We excluded studies with a group size of fewer than 10 participants who completed the study.
Types of participants
The population addressed by the review included adult patients (aged 18 or over) with cancer and pain despite being currently treated by an opioid agonist (e.g. morphine, fentanyl, oxycodone), in any dose and by any route. We excluded studies including patients who were on an established NMDA‐receptor antagonist treatment before the study began. We did not consider volunteer studies.
Types of interventions
The intervention considered by this review was the addition of ketamine, given by any route of administration, in any dose, to pre‐existing opioid treatment given by any route and in any dose.
Types of outcome measures
Primary outcomes
The primary outcome measure was patient‐reported pain intensity (e.g. visual analogue scales (VAS) and verbal rating scales).
Secondary outcomes
Secondary outcome measures were:
total opioid consumption over the study period;
rescue medication;
adverse events;
measures of patient satisfaction/ preference;
function;
distress.
Search methods for identification of studies
Electronic searches
We searched the following databases for this update:
CENTRAL (CRSO) April 2012 to January 2017;
MEDLINE (OVID) May 2012 to December 2016;
Embase (OVID) May 2012 to 2017 week 1.
Please see Appendix 1 and Appendix 2 for the searches conducted for the original review in 2003. For the searches conducted for the update in 2012 please see Appendix 3, Appendix 4 and Appendix 5. For searches conducted for the current update please see Appendix 6.
Searching other resources
We also searched two clinical trial registers (https://clinicaltrials.gov/) and the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch/) to identify additional published or unpublished data.
Language
We did not restrict searches or inclusion by language.
Data collection and analysis
Selection of studies
Two review authors (RB and EK) independently assessed the titles and abstracts from each of the electronic databases searched for relevance. We retrieved potentially relevant trial reports in full and three review authors (RB, CE, EK) assessed them for inclusion in the review.
Data extraction and management
We designed a data extraction form, and two review authors (RFB, EK) independently collected the following data items if available.
Publication details.
Patient population, number of participants, age, condition.
Description of the intervention(s) and control.
Outcomes: pain intensity, total opioid consumption, rescue medication, measures of patient satisfaction/preference, distress and function.
Adverse events (major and minor).
Quality (evaluated using the Oxford Quality Scale (Jadad 1996)).
Validity (evaluated using the Oxford Pain Validity Scale (OPVS) (Smith 2000)).
This information is recorded in the 'Characteristics of included studies' table.
Assessment of risk of bias in included studies
Two review authors (RFB, EK) independently assessed the risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) and adapted from those used by the Cochrane Pregnancy and Childbirth Group, with any disagreements resolved by discussion with a third author (CE). We completed 'Risk of bias' table for each included study using the 'Risk of bias' tool in RevMan (RevMan 2014).
We assessed the following for each included study.
Random sequence generation (checking for possible selection bias). We assessed the method used to generate the allocation sequence as: low risk of bias (any truly random process, e.g. random number table; computer random number generator); unclear risk of bias (method used to generate sequence not clearly stated). We excluded studies using a non‐random process (e.g. odd or even date of birth; hospital or clinic record number).
Allocation concealment (checking for possible selection bias). The method used to conceal allocation to interventions prior to assignment determines whether intervention allocation could have been foreseen in advance of, or during recruitment, or changed after assignment. We assessed the methods as: low risk of bias (e.g. telephone or central randomisation; consecutively numbered sealed opaque envelopes); unclear risk of bias (method not clearly stated). We excluded studies that did not conceal allocation (e.g. open list).
Blinding of participants and personnel (checking for possible performance bias). We assessed the methods used to blind study participants and personnel from knowledge of which intervention a participant received. We assessed methods as: low risk of bias (study states that it was blinded and describes the method used to achieve blinding, such as identical tablets matched in appearance and smell, or a double‐dummy technique); unclear risk of bias (study states that it was blinded but does not provide an adequate description of how it was achieved).
Blinding of outcome assessment (checking for possible detection bias). We assessed the methods used to blind study participants and outcome assessors from knowledge of which intervention a participant received. We assessed the methods as: low risk of bias (study has a clear statement that outcome assessors were unaware of treatment allocation, and ideally describes how this was achieved); unclear risk of bias (study states that outcome assessors were blind to treatment allocation but lacks a clear statement on how it was achieved).
Incomplete outcome data (checking for possible attrition bias due to the amount, nature and handling of incomplete outcome data). We assessed the methods used to deal with incomplete data as: low risk (< 10% of participants did not complete the study or used ‘baseline observation carried forward’ analysis, or both); unclear risk of bias (used 'last observation carried forward' analysis); high risk of bias (used 'completer' analysis).
Selective reporting (reporting bias). We assessed the risk of reporting bias as: low risk of bias (all intended outcomes reported); unclear risk of bias (any anomaly in reporting, such as participants contributing more than one set of data, or some outcomes not participant‐reported); high risk of bias (prespecified outcome of interest not reported).
Size of study (checking for possible biases confounded by small size). We assessed studies as being at low risk of bias (≥ 200 participants per treatment arm); unclear risk of bias (50 to 199 participants per treatment arm); high risk of bias (< 50 participants per treatment arm).
Measures of treatment effect
For dichotomous outcomes we planned to calculate the risk ratio (RR) and the corresponding 95% confidence interval (CI) and P value. We planned to calculate the number needed to treat for an additional beneficial outcome (NNTB)/number needed to treat for an additional harmful outcome (NNTH) as the reciprocal of the absolute risk difference (McQuay 1998). For continuous outcomes, we planned to calculate the mean difference (MD) and its corresponding 95% CI when means and standard deviations (SD) were available. If such information was unavailable we planned to use the methods described in Chapter 16 of the Cochrane Handbook for Systematic Reviews of Interventions to calculate standardised mean differences (SMD), from for example, F ratios, t values, Chi2 values and correlation coefficients (Higgins 2011). In cases where continuous measures were used to assess the same outcomes using different scales, we would have pooled these data using Hedges' g to estimate the SMD. When effect sizes could not be pooled, we planned to report study level effects narratively.
Unit of analysis issues
We only included studies that randomised the individual participant.
Dealing with missing data
We assessed missing data in the included studies. Where possible, we investigated and reported the reasons and numbers of those dropping out of each included study. For dichotomous outcomes, we planned to perform an intention‐to‐treat (ITT) analysis. If there was missing participant information, we recorded this and commented in the individual study's 'Risk of bias' table. Participants with missing data would be assigned to a 'zero improvement category'.
We paid particular attention to methods used for imputation of missing data due to withdrawals for adverse events and lack of efficacy. Where data were missing for substantial numbers of participants (greater than 10%), we would have rated the study as high risk of bias.
Assessment of heterogeneity
We planned to deal with clinical heterogeneity by combining studies that examined similar conditions. Statistical heterogeneity would have been assessed visually (L'Abbé 1987).
Assessment of reporting biases
We looked for the original trial protocols of the included studies and compared the results to these when they were found. When no protocol was available, we compared the reported outcomes against the Methods section of the paper to look for selective reporting of outcomes.
We planned to assess publication bias using a method designed to detect the amount of unpublished data with a null effect required to make any result for pain clinically irrelevant (usually taken to mean an NNTB of 10 or higher) (Moore 2008). In the event, there were insufficient data for statistical analysis.
Data synthesis
Quality of the evidence
We planned to combine data in a series of meta‐analyses on both primary and secondary outcomes.
We used the GRADE (Grades of Recommendation, Assessment, Development and Evaluation) system to rank the quality of the evidence using the GRADE profiler Guideline Development Tool software (GRADEpro GDT 2015), and the guidelines provided in Chapter 12.2 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
The GRADE approach uses five considerations (study limitations, consistency of effect, imprecision, indirectness and publication bias) to assess the quality of the body of evidence for each outcome. The GRADE system uses the following criteria for assigning grade of evidence:
high: we are very confident that the true effect lies close to that of the estimate of the effect;
moderate: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different;
low: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect;
very low: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect.
We decreased the grade rating by one (‐ 1) or two (‐ 2) if we identified:
serious (‐ 1) or very serious (‐ 2) limitation to study quality;
important inconsistency (‐ 1);
some (‐ 1) or major (‐ 2) uncertainty about directness;
serious (‐1) or very serious (‐2) imprecise or sparse data;
high probability of reporting bias (‐ 1).
'Summary of findings' table
We planned to include a 'Summary of findings' table as set out in the PaPaS author guide PaPaS 2012 and recommended in the Cochrane Handbook (Chapter 11, Higgins 2011) to present the main findings in a transparent and simple tabular format. However, we judged that a 'Summary of findings' table with only three very different studies would be unhelpful.
Subgroup analysis and investigation of heterogeneity
We did not plan subgroup analyses.
Sensitivity analysis
Had there been sufficient data available, we would have examined the robustness of meta‐analyses by conducting a sensitivity analysis.
Results
Description of studies
The original review included one study which compared intrathecal ketamine + intrathecal morphine with intrathecal morphine alone (Yang 1996), and one study which compared intravenous ketamine bolus with intravenous bolus of placebo as a supplement to ongoing morphine therapy (Mercadante 2000). We found one new study for this update which compared subcutaneous infusion of ketamine at three dose levels (100 mg, 300 mg, or 500 mg) with placebo in participants with ongoing treatment with opioids (Hardy 2012).
Results of the search
The updated searches of the three databases (see Electronic searches) retrieved 271 records. Our searches of the trials registers did not identify further studies. Our screening of the reference lists of the included publications did not reveal additional RCTs. We therefore had a total of 271 records.
Once duplicates had been removed, we had a total of 216 records. We excluded 215 records based on titles and abstracts. We obtained the full text of the remaining record and this study was included (Hardy 2012). For a further description of our screening process, see the study flow diagram (Figure 1).
1.
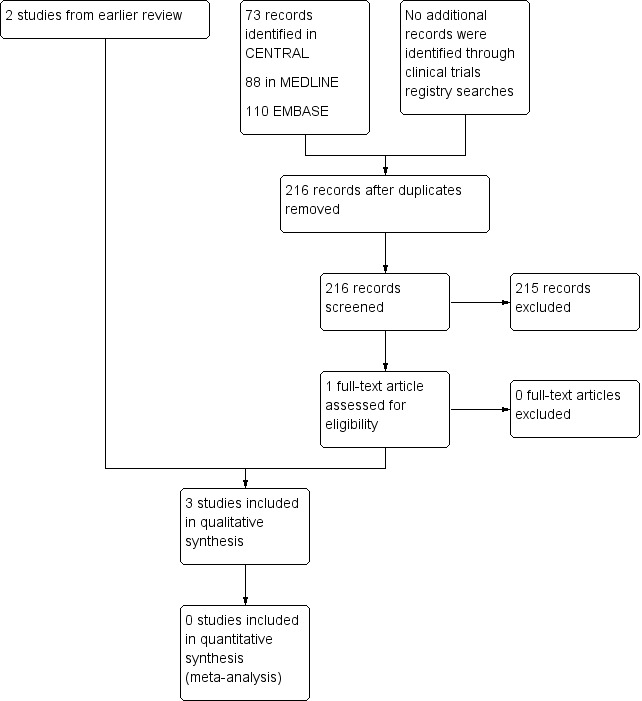
Study flow diagram.
We identified three ongoing studies and added these records to Characteristics of studies awaiting classification. Two additional trials with status 'completed' do not appear to have been published and possibly represent double registration of the same trial. These are described under Characteristics of ongoing studies.
Protocols for the studies by Yang 1996 and Mercadante 2000 were not available. The protocol for the study by Hardy 2012 was retrieved.
Included studies
Study design
Two included studies (Yang 1996; Mercadante 2000) had a cross‐over design. The trial conducted in Taiwan by Yang 1996 compared ketamine and morphine with morphine alone. The time period over which the intervention was assessed was not stated in the trial report, but there is an implication that the study was conducted over a period of days. (Attempts to contact the author to confirm the trial duration were not successful). The trial conducted in Italy by Mercadante 2000 was a placebo‐controlled trial and was conducted over a three‐hour period. The most recent trial Hardy 2012 was a multisite, dose‐escalation, double‐blind, randomised, placebo‐controlled parallel group study with a duration of five days.
Study population
Yang 1996: Twenty hospitalised participants (10 men and 10 women) aged 22 to 69 years with cancer pain of variable severity treated with opioids. The primary cancer sites were stomach, cervix, liver, lung, colon, pancreas.
Mercadante 2000: Ten participants (seven men and three women) aged 21 to 69 years who had pain unrelieved by their dose of morphine, and a Karnofsky status of 50 or more. The primary cancer sites were: bladder, rectum, lung, histiocytoma and uterus. In this study, the pain was classified as being "neuropathic" or having a "neuropathic component".
Hardy 2012: Hospitalised palliative care participants aged 18 or older, with refractory chronic nociceptive or neuropathic pain secondary to cancer or its treatment (Brief Pain Inventory (BPI) average pain score ≥ 3 despite ongoing treatment with opioids and co‐analgesics at predefined dose levels). One hundred and eighty participants were randomly assigned, two were deleted from the analysis, 93 were allocated to ketamine and 92 to placebo. Ninety‐one received ketamine and 90 received placebo. One hundred and forty‐nine were defined as having completed the trial, although only 39 participants in the ketamine arm and 35 participants in the placebo arm received either ketamine or placebo for the full five‐day period.
Intervention
Yang 1996 assessed intrathecal ketamine 1.0 mg twice daily as adjuvant to intrathecal morphine, compared with intrathecal morphine alone. The morphine dose was titrated until participants' pain relief had been stable for 48 hours, then the participants were randomly crossed over (no washout period) to morphine plus ketamine or continued on morphine (control), administered intrathecally twice a day.
Mercadante 2000 assessed two doses of ketamine (0.25 mg/kg and 0.5 mg/kg) administered intravenously as a bolus as adjuvant to ongoing morphine therapy, compared with saline. Patients were randomly assigned to receive in turn either 0.25 mg/kg or 0.5 mg/kg ketamine or saline, with a two‐day washout period between each intervention/control.
Hardy 2012 assessed either placebo (normal saline) or ketamine at three dose levels (100 mg, 300 mg, or 500 mg) as a subcutaneous infusion in a five‐day schedule, starting at the first dose level (100 mg/24 hours), as a supplement to ongoing opioid therapy. If 80% of the study drug had been delivered, and average pain improved by ≥ 2 BPI units, with no more than four doses of breakthrough medication, the dose remained the same. If not, the dose was increased to the next level.
Morphine was the only opioid participants received in the studies by Yang 1996 and Mercadante 2000. The route of administration of morphine in the study by Yang 1996 was intrathecal, while morphine was given by varied routes of administration (oral, intravenous or subcutaneous) in the trial by Mercadante 2000. The opioid was not standardised in the study by Hardy 2012 where participants used different opioids (morphine, oxycodone, hydromorphone, methadone, fentanyl, sufentanil, alfentanil) given by different routes of administration (oral, transdermal or parenteral).
It is assumed that racemic ketamine was used in all three studies.
Rescue medication
Yang 1996: In this trial a rescue dose of 5 mg morphine was administered intramuscularly as needed. Mercadante 2000 does not report the use of rescue medication. Hardy 2012 states that the participants had access to breakthrough analgesia and record the number of doses, but do not describe the rescue medication.
Outcomes
Yang 1996 measured patient‐reported pain intensity (zero to 10 numerical, 10 worst pain imaginable); pain frequency (four‐point verbal ordinal scale), group morphine dose, total titrated intrathecal morphine, total rescue medication, frequency of intrathecal titration. Mercadante 2000 measured patient‐reported pain intensity (zero to 10 numerical scale) at 30‐, 60‐, 90‐, 120‐, and 180‐minute intervals; and adverse events. Hardy 2012 defined the primary outcome as a positive response defined as a "clinically relevant improvement in pain" at the end of the ive‐day study period. A "clinically relevant improvement in pain" was defined as a reduction in BPI average pain score by ≥ 2 points from baseline in the absence of more than four breakthrough doses of analgesia over the previous 24 hours. Secondary outcomes included pain assessments at days two to five and adverse events.
See Characteristics of included studies tables.
Excluded studies
For this update we identified one eligible study (Hardy 2012), which was included. Overall, we excluded five studies. (For studies previously excluded see Characteristics of excluded studies).
Risk of bias in included studies
2.
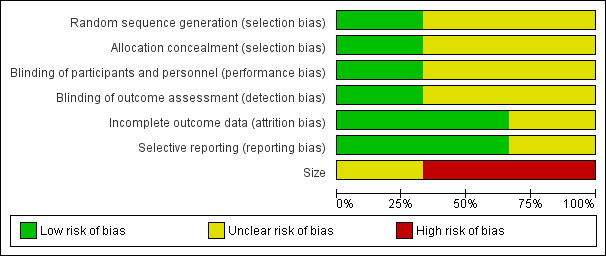
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Allocation
The studies by Yang 1996 and Mercadante 2000 stated that patients were randomised to treatment and control groups, but in neither trial was the process of randomisation described (unclear risk of bias). In the trial by Hardy 2012 each site pharmacy used randomisation tables from an independent central registry. Stratification was by pain type (neuropathic or nociceptive) and randomisation was double‐blinded, allocated by blocks of four in a 1:1 ratio for each strata by site. We judged this study to be at low risk of bias.
Blinding
Performance bias
Participants, investigators and nurses were blinded using a double‐dummy technique in Yang 1996 and the drugs were prepared in identical syringes by a person not involved in the study and administered in the same volume in Mercadante 2000. The Hardy 2012 trial is described as double‐blinded. The blinding procedure was not described in the final paper, but was described in the study protocol ("All syringes will look identical in volume and colour").
There were no specific procedures to check for performance bias in any of the three included trials. In Yang 1996, one participant in the morphine phase and no participants in the combined morphine and ketamine phase reported psychotoxicity (hallucinations), whereas in Mercadante 2000 ketamine caused hallucinations in four of 10 participants, so the participants may have been able to tell which drug they had received. The study by Hardy 2012 involved rapid dose escalation of ketamine to high doses and blinding could have been compromised due to adverse effects from ketamine. We judged the study by Yang 1996 at low risk of performance bias and the studies by Mercadante 2000 and Hardy 2012 at unclear risk of performance bias.
Detection bias
We judged the study by Yang 1996 at low risk of detection bias and the studies by Mercadante 2000 and Hardy 2012 at unclear risk of detection bias.
Incomplete outcome data
All participants were accounted for in the trials by Yang 1996 and Mercadante 2000. Hardy 2012 reported an ITT analysis, but imputed missing data using last observation carried forward (LOCF). We judged this study at unclear risk of bias.
Selective reporting
There were no problems of selective reporting detected in the trials by Mercadante 2000 and Yang 1996. In the trial by Hardy 2012, assessing "the effect of ketamine on total opioid dose" was mentioned in the protocol as a "secondary objective", but was not reported. We judged this study at unclear risk of bias.
Other potential sources of bias
Size
The cross‐over studies by Yang 1996 and Mercadante 2000 had respectively 20 and 10 participants (fewer than 50 participants per treatment arm). We judged these trials at high risk of bias. The parallel group trial by Hardy 2012 had 185 participants (between 50 and 199 participants per treatment arm) and we judged it at unclear risk of bias.
Oxford quality assessment
Quality scores derived using the Oxford quality scale (Jadad 1996) were three for both Mercadante 2000 and Yang 1996, and four for Hardy 2012 out of a possible maximum of five points.
Using the method derived by Smith 2000, the three included studies (Yang 1996; Mercadante 2000; Hardy 2012) scored 13, 12 and 12, respectively on the Oxford Pain Validity Scale, a zero to 16‐point validity scale.
Effects of interventions
It was not possible to perform a quantitative meta‐analysis because of the small number of participants in two of the trials, lack of extractable data and general heterogeneity of the data. A description of the results from the three included trials is given below. None of the trials provided data on pain relief, patient satisfaction/preference, function or distress. For all outcomes we judged the quality of the evidence to be very low. We downgraded one level for serious risk of bias and two levels for very serious imprecision due to very small number of participants in two of the comparisons and small number of participants in the third comparison.
Patient‐reported pain intensity
Ketamine 1.0 mg twice daily (intrathecal)
One study (Yang 1996) assessed adjuvant ketamine 1.0 mg administered intrathecally. The trial duration is not specified, but it was conducted over several days. Pain intensity on a numerical rating scale zero to 10 was reduced from 7.95 ± 0.25 to 2.45 ± 0.17 after adjuvant treatment with ketamine.
Ketamine 0.25 mg/kg (intravenous)
One trial (Mercadante 2000) assessed pain intensity over three hours. Mean pain intensity scores showed a reduction in pain intensity after 30 minutes compared with saline solution; after 60 minutes the analgesic effect of ketamine began to diminish but continued to have an effect for a period of three hours.
Ketamine 0.5 mg/kg (intravenous)
One trial (Mercadante 2000) assessed pain intensity over three hours. Mean pain intensity scores showed a significant reduction after 30 minutes compared with saline solution. The analgesic effect of ketamine continued throughout the three‐hour period.
Ketamine dose escalation 100 mg, 300 mg, 500 mg (subcutaneous)
One study (Hardy 2012) assessed average BPI pain score on day six, following dose escalation of ketamine subcutaneous infusion in a five‐day schedule, starting at the first dose level (100 mg/24 hours). If 80% of the study drug had been delivered, and average pain improved by ≥ 2 BPI units, with no more than four breakthrough doses, the dose remained the same. If not, the dose was increased to the next level. There was no significant difference in patient‐reported pain intensity between the placebo and ketamine arms.
Total opioid consumption
Ketamine 1.0 mg twice daily (intrathecal)
Yang 1996 reported that on the last day of the morphine phase, participants required intrathecal morphine 0.38 mg/day ± 0.04 mg/day. On the last day of the combined ketamine and morphine (K+M) phase, intrathecal morphine requirements had decreased to 0.17 mg/day ± 0.02 mg/day. The total titrated dose of intrathecal morphine, total dose of intramuscular rescue morphine during the K+M phase was less than in the morphine phase.
Ketamine 0.25 mg/kg and 0.5 mg/ kg (intravenous)
Mercadante 2000 did not provide information on this outcome.
Ketamine dose escalation 100 mg, 300 mg, 500 mg (subcutaneous)
Hardy 2012 did not provide information on this outcome.
Rescue medication
Ketamine 1.0 mg twice daily (intrathecal)
Yang 1996 reported that the total dose of rescue morphine during the K+M phase was less than the morphine phase.
Ketamine 0.25 mg/kg and 0.5 mg/kg (intravenous)
Mercadante 2000 did not report the use of rescue medication, but stated in the text that the administration of ketamine allowed for "a reduction of opioid doses".
Ketamine dose escalation 100 mg, 300 mg, 500 mg (subcutaneous)
Hardy 2012 reported that there was no significant group difference in the median number of breakthrough analgesic doses given during the study.
Adverse events
Psychomimetic adverse events
Ketamine 1.0 mg twice daily (intrathecal)
One participant in the morphine only arm of the Yang 1996 study reported hallucinations. There were none reported in the ketamine arm.
Ketamine 0.25 mg/kg and 0.5 mg/kg (intravenous)
In the study by Mercadante 2000, ketamine injection produced hallucinations in four participants: three experienced hallucinations whilst receiving 0.25 mg/kg and 0.5 mg/kg ketamine, and one further participant experienced hallucinations when receiving ketamine 0.5 mg/kg. All were treated with diazepam 1 mg. In addition, two participants experienced light flashes, a 'buzzing' feeling in the head, and sensation of insobriety. Diazepam resolved these symptoms. No significant changes in the Mini‐Mental State Examination (MMSE) were observed.
Ketamine dose escalation 100 mg, 300 mg, 500 mg (subcutaneous)
In the study by Hardy 2012, there was almost twice the incidence of adverse events in the ketamine arm compared with the placebo arm on day one and throughout the study. Psychomimetic adverse events were assessed daily using the Clinician‐Administered Dissociative States Scale (CADSS). CADSS scores were not reported but 17 cognitive disturbance events with grading worse than at baseline were recorded in the ketamine group and eight such events were recorded in the placebo group. Thirteen confusion events with grading worse than baseline were recorded in the ketamine group and nine such events were recorded in the placebo group. Psychomimetic toxicity was treated with haloperidol or midazolam at specified doses.
Other adverse events
Ketamine 1.0 mg twice daily (intrathecal)
On direct questioning, participants reported a number of adverse effects during the trial conducted by Yang 1996:
pruritis;
constipation;
urinary retention;
difficulty in urinating;
nausea and vomiting;
hallucinations;
respiratory depression.
However, these adverse events could not be attributed specifically to the study treatments as some were present prior to the commencement of the study.
Ketamine 0.25 mg/kg and 0.5 mg/kg (intravenous)
Information on the following adverse events were sought in the trial conducted by Mercadante 2000:
drowsiness;
nausea and vomiting;
dry mouth.
These adverse events were assessed on a scale from zero to three, where zero was 'not at all', and three was 'awful'. Participants treated with 0.25 mg/kg and 0.5 mg/kg ketamine reported increased drowsiness.
Ketamine dose escalation 100 mg, 300 mg, 500 mg (subcutaneous)
In the trial by Hardy 2012, adverse events were graded according to the National Institutes of Health Common Terminology Criteria for Adverse Events, version 3.0 (Cancer Therapy Evaluation Program Version 3). There was almost twice the incidence of adverse events in the ketamine arm compared with the placebo arm at the end of day one and throughout the study. The authors reported 31 episodes of injection site reactions, which were reported as nearly three times more likely than the placebo group. There were relatively few adverse events higher than grade three in severity (14 for ketamine; 16 for placebo). Seven serious adverse events were reported, two of which (bradyarrhythmia and cardiac arrest, both in participants receiving ketamine) were thought to be possibly related to the study drug.
Both the trial by Yang 1996 and the trial by Mercadante 2000 reported that the adverse events of ketamine were not serious.
Study withdrawals and dropouts
No study withdrawals or dropouts were reported in either trial by Yang 1996 or Mercadante 2000. In the study by Hardy 2012, 39 participants in the ketamine group and 55 participants in the placebo group withdrew from the trial. Sixteen participants in each group discontinued the study due to clinical deterioration, patient/ clinical request or change in therapy. Nineteen participants in the ketamine group and 37 participants in the placebo group discontinued due to treatment failure. Of these, 17 in the ketamine group and two in the placebo group discontinued due to toxicity.
Discussion
Summary of main results
There are three included studies in the current version of the review. Two small early studies Yang 1996 and Mercadante 2000 report reduction in pain intensity and reduction in morphine requirements. These two studies are of high risk of bias due to small sample size, and incomplete reporting. The new study from Hardy 2012 has unclear risk of bias due to size and incomplete reporting. Hardy 2012 reports no difference in their primary outcomes between groups. Overall, we cannot provide a reliable indication of the likely effect of ketamine, at any dose, as an adjuvant to opioids in cancer pain. Adverse events such as hallucinations and cognitive disturbance were reported for higher doses of ketamine. Two serious adverse events (bradyarrhythmia and cardiac arrest) reported in the trial examining rapid titration of ketamine to high dose were thought to be possibly related to the study drug.
There is large body of evidence demonstrating the efficacy of ketamine in acute postoperative pain. While this evidence cannot be directly extrapolated to other patient groups, it is important to note the complex pathophysiology of cancer pain and the difficulties of conducting clinical trials in palliative care. The authors of the study by Hardy 2012 should be commended for recruiting an impressive number of participants. The data from this trial could potentially provide further information on clinical questions such as whether ketamine has beneficial interactions with specific opioids, since both preclinical and clinical research have suggested that this is the case when ketamine is used as an adjuvant to morphine (Lilius 2015). To date, the fact that the participants in the Hardy 2012 trial used different opioids has not been addressed and the subgroup analyses based on type and dose of opioid have not been reported. We contacted the authors with a request for access to individual patient data, however the request was declined because such analyses were not stated in our original protocol for this review.
It is worth noting, also, that the dose escalation in the Hardy 2012 trial was very rapid, considering the pharmacokinetics of ketamine which has a short α half‐life (two to four minutes) and longer β half‐life (two to four hours) in humans (Peltoniemi 2016), and where steady state is achieved after five elimination half‐lives. The metabolite norketamine, which is also active has a much longer half‐life than ketamine, and very ill cancer patients would be likely to have a much poorer elimination than young healthy volunteers. Ketamine doses in the Hardy 2012 trial were higher than those used in the majority of ketamine regimens described in the literature. It is interesting that ketamine was found to have better effect in patients with high pain scores, however this was not mentioned in the abstract. The same finding is reported in a systematic review of 70 randomised controlled trials of intravenous ketamine for postoperative analgesia (Laskowski 2011). In clinical practice, ketamine is usually considered to be a third‐line drug which is reserved for patients with high pain intensity scores, despite adequate ongoing opioid therapy and co‐analgesics. Hardy 2012 included patients with a BPI pain intensity score of three at baseline and also patients being treated with comparatively low doses of opioid.
All three trials used pain intensity scores as the primary outcome. Percentage pain relief may be a more useful and reliable outcome measure (Dworkin 2008).
Other reports considered in the original review (2003)
Because of the paucity of data available from RCTs in the original review, we considered information presented in case studies and case series reports of ketamine for chronic cancer pain. In addition to the two RCTs included, the original review (2003) identified 32 case reports or open‐label, uncontrolled trials describing improvement of opioid analgesia with ketamine. We did not consider case studies and reports in the updated review.
Whilst the design of these studies and the issue of publication of positive outcomes preclude the inclusion of any data from these reports in this systematic review, the studies were discussed in the original review and are reported in this update in order to provide a more comprehensive review of the literature on this topic. Case reports cannot provide evidence for efficacy but may provide valuable information on adverse effects. They are, by definition, all of low quality.
The 32 reports described the use of ketamine to treat refractory cancer pain, frequently described as neuropathic pain. The total number of participants treated with ketamine in these reports was 246. The route of ketamine administration included oral, intramuscular bolus, subcutaneous bolus and infusion, intravenous bolus and infusion, epidural bolus, and intrathecal infusion. Ketamine doses ranged from 1 mg/kg/day subcutaneous infusion to 600 mg/day intravenously and 67.2 mg/day intrathecally. Treatment duration ranged from four hours to one year. Treatment was in most cases adjuvant to opioid and other drugs. Twenty‐eight reports described improved analgesia with ketamine. Where ketamine was administered as an adjuvant to opioids, the most commonly used opioid was morphine, but in some cases ketamine was given as an adjuvant to fentanyl (Ventura 1993; Bell 1999), hydromorphone (Fine 1999) or diamorphine (Garry 1996), or combinations of these. Ketamine was also used as sole analgesic in three reports (Parada 1971; Whizar‐Lugo 1987; Oshima 1990). Sixteen reports described dramatic relief of refractory cancer pain with ketamine: "complete cessation of pain" (Ventura 1993); "complete relief of pain" (Tarumi 2000); "disappearance of pain" (Parada 1971; Garry 1996); "no pain" (Fine 1999); "pain free" (Mitchell 1999),; "mostly pain free" (Lloyd‐Williams 2000); dramatic reduction in visual analogue scales (VAS) scores including VAS 100 reduced to zero (Bell 1999); average VAS score 8.3 reduced to one (Kanamaru 1990); average VAS score reduced from 5.9 +/‐ 2.0 to 0.3 +/‐ 0.8 (Ogawa 1994); VAS 7/10 reduced to 1/10 (Wood 1997); reduction of VAS 7/10 to below 2/10 (Lossignol 1999); "dramatic drop in VAS" (Lossignol 1992); "remarkable analgesia" (Fukuida 1981); "excellent analgesia" (Sosnowski 1993; Mercadante 1995).
The most commonly reported adverse events in this literature were sedation and hallucination. In general, adverse events were not reported as severe and only two studies reported patient withdrawal from treatment because of unacceptable "adverse cognitive effects" (Garry 1996), and pronounced sedation (Klahr 1997). One report described sedation which improved on tapering the opioid dose (Bell 1999). Other side effects described included evoked nystagmus (jerky eye movements) during treatment with intravenous ketamine (Lossignol 1999), and inflammation of syringe driver sites during subcutaneous treatment (Oshima 1990; Mitchell 1999). One report described generalised hyperalgesia and allodynia after abrupt termination of subcutaneous ketamine infusion (Mitchell 1999). One postmortem report described subpial vacuolar myelopathy in a participant who had received continuous intrathecal ketamine infusion (Karpinski 1997), while another described focal lymphocytic vasculitis close to the intrathecal catheter site (Stotz 1999). One report described maintenance of syringe driver sites with topical 0.1% hydrocortisone cream (Lloyd‐Williams 2000).
Overall completeness and applicability of evidence
The two small studies (30 participants) included in the original review provided insufficient data to enable any evidence‐based conclusions about the benefits and harms of adjuvant ketamine to be drawn. The larger trial by Hardy 2012 reported negative outcomes for a rapid titration, high‐dose ketamine regimen.
Quality of the evidence
The evidence from this review is limited to three very different studies which could not be combined. There are two very small studies undertaken in the 1990s of low‐dose ketamine, and one larger multi‐centre modern trial reported in 2012 on rapid titration of ketamine to high dose. Overall, the quality of the evidence base is very low and cannot provide a reliable indication of any likely effect across outcomes.
Potential biases in the review process
We are unaware of any potential biases in the review process.
Agreements and disagreements with other studies or reviews
A qualitative systematic review of ketamine for cancer pain in adults and children concluded that despite limited available data, there is evidence that ketamine may be a "viable option" for cancer pain that is poorly responsive to opioid therapy, that it appears to contribute to decreased opioid use and improved pain control (Bredlau 2013). The authors of this review specifically wanted to perform a comprehensive review of all available data. They included the same three RCTs included in our review and in addition, two trials which were excluded by our review (Lauretti 1999a; Lauretti 1999b) (see Characteristics of excluded studies). They also included six prospective, non‐randomised, uncontrolled trials and one retrospective case series of more than 10 participants.
A recent systematic review on adjuvant analgesics for cancer pain found that there is low‐grade evidence suggesting that ketamine as an adjuvant to opioid in cancer pain leads to pain reduction, but conclude that there is generally insufficient evidence on the effectiveness of NMDA receptor antagonists in cancer pain (van den Beuken‐van Everdingen 2017). This review considered our Cochrane update from 2012, the review by Bredlau 2013, and a RCT excluded from our last update (Salas 2012).
Authors' conclusions
Implications for practice.
For people with refractory cancer pain treated with opioid
The evidence base for ketamine as an adjuvant to opioids for the treatment of cancer pain is insufficient to enable us to draw any conclusions. Rapid dose escalation of ketamine to high dose (500 mg) does not appear to have clinical benefit and may be associated with serious adverse events.
For clinicians
The evidence base for ketamine as an adjuvant to opioids for the treatment of cancer pain is insufficient to enable us to draw any conclusions. Rapid dose escalation of ketamine to high dose (500 mg) does not appear to have clinical benefit and may be associated with serious adverse events.
For policy makers
The evidence base for ketamine as an adjuvant to opioids for the treatment of cancer pain is insufficient to enable us to draw any conclusions. Rapid dose escalation of ketamine to high dose (500 mg) does not appear to have clinical benefit and may be associated with serious adverse events.
For funders of the intervention
The majority of this patient group are commonly in‐patients, due to refractory symptoms. Ketamine administered to hospitalised patients is an inexpensive treatment. However, the evidence base for ketamine as an adjuvant to opioids for the treatment of cancer pain is insufficient to enable us to draw any conclusions.
Implications for research.
General implications
The amount of clinical trial evidence in this review is limited. Studies investigating specific low‐dose ketamine treatment regimens commonly used for refractory cancer pain are needed.
Design
Conducting scientifically sound trials in a population of terminally ill cancer patients is a considerable challenge, and this is perhaps reflected in the small number of published trials available for this review. It is difficult to recruit large numbers of patients from this population. Cross‐over designs, as used in the two of the three included studies, may be more appropriate than placebo‐controlled parallel group studies. Where there are large trials, planned subgroup analyses should be considered.
Measurement (endpoints)
Outcomes should be clearly defined, and trial managers should also restrict study outcomes to those that are the most clinically useful, such as which route of administration, relevant dose, co‐analgesic effects with specific opioids, and the cost to the patient in terms of adverse events.
Whether ketamine is more effective in combination with specific opioids is not known, and this is an area for further research. Rapid titration of ketamine to high dose (Hardy 2012) had no clinical benefit and was associated with adverse events. Further studies examining this specific treatment regimen are not warranted. Trials with S‐ketamine as an adjuvant to opioids might be appropriate.
Other
Opioid tolerance may also be an issue. It has been suggested that pharmacological tolerance to opioid can develop early (Laulin 2002), but it is not clear how often it is a clinical problem in cancer patients. It may be difficult in this patient population to distinguish between tolerance and disease progression, both of which require an increase in opioid dose. In patients who appear to have a problem tolerating opioids, ketamine in low dose may be a treatment option. This is a topic for future research.
More information is needed on whether the route of administration of ketamine has an impact on its effectiveness as an analgesic. If ketamine is used spinally, issues of neurotoxicity should be considered (Karpinski 1997).
Feedback
Feedback received, 20 July 2017
Summary
Name: David Currow
Email Address: david.currow@sa.gov.au
Affiliation: Flinders University
Role: Professor of Palliative & Supportive Services
Comment
The systematic review has suggested that ‘Hardy et al. Randomized, double‐blind, placebo‐controlled study to assess the efficacy and toxicity of subcutaneous ketamine in the management of cancer pain. Journal of Clinical Oncology 2012;30(29):3611‐7’ is low level evidence. Using the Cochrane Grade tool, it is not clear how this could be the conclusion given the parameters that are included in the tool. The tool is copied below and the way the design and conduct of the study was undertaken is outlined in detail for each.
Methods of sequence generation
At each centre, patients were sequentially allocated a patient number on referral to the study. This number was kept within the Patient Master Index, linking the patient name with the number allocated. Strata tables were developed for each site using random number tables, generated at an independent centre (central registry). Treatment for each patient was allocated according to a block randomisation (blocks of 4) schedule held by the central registry in a 1:1 ratio. Block randomisation ensured even allocation to each code. The central registry supplied strata tables to each site pharmacy. Stratification was according to pain type: (neuropathic or nociceptive), according to the Leeds Assessment of Neuropathic Symptoms and Signs (LANSS) scale. On notification of a participant, the pharmacist at each site consulted the strata table according to the strata determined by the LANSS scale score, and allocated the next code available according to the supplied strata table and prepared the active or inactive drug delivered in a labeled syringe. The participant ID, allocation code, dates of request, preparation, and dispensing were recorded in a log maintained by the pharmacist.
Allocation concealment
At all times, from eligibility screening to completion of the study, all study staff are unaware of the treatment allocation. Allocation was concealed from the investigator at the time of the participant inclusion in the trial.
Blinding ‐ Participants
All syringes were prepared according to the randomisation schedule. Each syringe was numbered according to the pre‐determined randomisation code and labelled as ketamine/placebo 100mg, 300mg or 500mg in normal saline according to the prescription from the investigator. All syringes looked identical in volume and colour to preserve the blinding irrespective of the contents. Study syringes were supplied to the inpatient units and stored in a locked cupboard that met state regulations for this schedule of drug.
Blinding ‐ Providers
Treatment allocation was not disclosed to study staff, treating clinicians or investigators at any stage during the study.
Blinding ‐ Outcome assessors
There were no cases of extreme emergency where knowledge of the code would have had significant consequences with respect to clinical decision making. Therefore, no unblinding occurred during the study. Adverse events including skin toxicity and psychomimetic events were documented in patients in both arms. Therefore it was not possible to differentiate clinically between arms.
Loss to follow up
No patient was lost to follow up. Those participants who had completed 24 hours at maximum dose (500 mg/24 hours) without a response were deemed to have completed the study as continuation on ineffective treatment was considered unethical. Similarly, those participants with unacceptable toxicity could complete prior to end day 5.
Failure to follow intention to treat principles in analyses. All eligible randomised participants were included in the primary ITT analysis. Four patients who were randomised, but withdrew before the commencement of study drug were deemed to have had a negative response.
Selective outcome reporting of outcomes and/or analyses
There was no difference in oral morphine equivalent doses between arms at baseline. Trial patients continued their current opioid regimen throughout the study period with breakthrough/rescue doses as below. Patients continued all adjuvant analgesics at pre‐study dose throughout the duration of the study. Patients who required the addition of a new agent or in increase in co‐analgesia during the 5 day study period had the study treatment ceased. A reduction in co‐analgesia because of toxicity was acceptable. Any changes in concomitant medication was documented in the Rescue medications. Immediate release oral or subcutaneous opioid of up to 1/6th the total daily dose of morphine or oxycodone were available q2hourly for all patients, preferably by the same route as the regular medication. For patients on transdermal fentanyl, oral /subcutaneous oxycodone or morphine at doses as recommended by the manufacturer (or parenteral/sublingual fentanyl at recommended doses) were used. There was no difference in the use of breakthrough medications between arms.
Other potential sources of bias
Size of study
A total of 150 (75 ketamine and 75 placebo) evaluable patients were required to provide approximately 85% power to detect an absolute 25% difference in response rate (30% in the placebo group vs 55% in the ketamine group), at a 2‐tailed type 1 error of 0.05. The sample size was reached.
Doses used
The regimen chosen for our study was based on the largest series reported in the literature at the time (Jackson K, Ashby M, Howell D, et al: The effectiveness and adverse event profile of “burst” ketamine in refractory cancer pain: The VCOG PM 1‐00study. J Palliat Med 14:1074‐1077, 2011).
Rapid escalation
Dose escalation only occurred if patients failed to respond after 24 hours of the previous dose as per the largest series reported in the literature at the time‐Opioid doseThe background opioid dose was 300mg OME/day (range 160‐480) and 410 (258‐700) in the ketamine and placebo arms respectively.
Last observation carried forward analysis
The primary analysis was an intention‐to‐treat (ITT) analysis. The response for participants stopping study drug before day 5 for reasons unrelated to the intervention was imputed from the last recorded assessment of pain. It is to be noted that the placebo group achieved the same response rate of analgesics but with much less toxicity.
I do not have any affiliation with or involvement in any organisation with a financial interest in the subject matter of my comment.
Reply
The authors thank Dr. Currow for his feedback regarding the recent update of the Cochrane review entitled "Ketamine as an adjuvant to opioids for cancer pain".
Dr. Currow has requested an explanation of why the paper by Hardy et al. was classified as "low level evidence".
The paper was assessed using the Cochrane Risk of bias tool. The quality of the evidence was assessed using GRADE.
Re. Risk of bias
The paper was judged to have unclear risk of bias as follows:
Detection bias and performance bias
As is evident from the Risk of bias table, we judged this trial to be at unclear risk of bias re. blinding. The detailed information on blinding that Dr. Currow provides in his email was not included in the published paper which lacks a description of the blinding procedure. We did however note that the protocol mentioned that "All syringes will look identical in volume and colour". The trial involved rapid titration of ketamine to high doses and there was a high rate of adverse events in the ketamine arm. No specific measures were taken to check for blinding. We judged that it was possible that blinding could therefore have been compromised by psychotomimetic adverse effects and skin reactions at the needle site, and a check on fidelity to blinding would have helped reduce uncertainty.
Incomplete outcome data (attrition bias)
It is unclear how missing data was imputed. The trial was therefore judged to be at unclear risk of bias due to missing outcome data.
Selective reporting
Assessing "the effect of ketamine on total opioid dose", which is a relevant outcome of interest was mentioned in the protocol as a "secondary objective", but was not reported in the final paper. We judged the study to be at unclear risk of bias due to selective reporting.
Size of study (checking for possible bias confounded by small size)( 1‐4).
We assessed studies as being at low risk of bias (200 participants or more in each treatment arm); unclear risk of bias (50‐199 participants in each treatment arm; high risk of bias (fewer than 50 participants per treatment arm). We judged the trial by Hardy et al. to be at unclear risk of bias due to size (50 ‐ 199 participants per treatment arm).
Re. GRADE assessment
For all three studies there was an unclear risk of bias overall. Using GRADE, we judged the quality of the evidence to be very low due to study limitations and imprecision due to the small number of participants in all comparisons.
We downgraded one level for serious risk of bias and two levels for very serious imprecision due to very small number of participants in two of the comparisons and small number of participants in the third comparison.
Re. the ketamine regimen used in the study by Hardy et al.
Dr. Currow writes that the ketamine regimen "was based on the largest series reported in the literature at the time" (5). The dose escalation was rapid, for example having a start dose of 100 mg/ 24 hours and tripling the dose after less than 20 hours, considering the pharmacokinetics of ketamine which has a β half‐life of two to four hours in humans (6), and where steady state is achieved after five elimination half‐lives. The metabolite norketamine, which is also active has a much longer half‐life than ketamine, and very ill cancer patients would be likely to have a much poorer elimination than young healthy volunteers.
The "burst" ketamine protocol developed by Jackson et al. appears only to have been used by that particular group, and subsequently by Hardy and colleagues. Mercadante et al. published two reports of cases treated with a "burst" ketamine regimen, but employed a dose of 100 mg daily for two days, as an adjuvant to opioid (7,8). The ketamine doses and rate of titration in the Hardy trial are higher than those generally reported and recommended in the cancer pain literature (9). The high rate of adverse events in the ketamine arm of the trial suggest that "rapid titration involving such doses of continuous subcutaneous infused ketamine is generally inadvisable" (9). The treatment regimen used in this study cannot be considered representative for ketamine regimens commonly used in palliative care, and the conclusion that "Ketamine does not have net clinical benefit when used as an adjunct to opioids and standard co‐analgesics in cancer pain" can only apply to this specific, high‐dose, rapid titration treatment regimen.
Finally, the basic opioid treatment in the Hardy study was not standardized‐ participants were treated with different opioids and different routes of administration. This lack of standardization may have influenced the results.
References
Dechartres A, Trinquart L, Boutron I, Ravaud P. Influence of trial sample size on treatment effect estimates: meta‐epidemiological study. BMJ 2013;346:f2304.
Dechartres A, Altman DG, Trinquart L, Boutron I, Ravaud P. JAMA 2014;312(6):623‐30.
Moore RA, Gavaghan D, Tramèr MR, Collins SL, McQuay HJ. Size is everything‐‐large amounts of information are needed to overcome random effects in estimating direction and magnitude of treatment effects. Pain 1998; 78(3):209‐16.
Nüesch E, Trelle S, Reichenbach S, Rutjes AW, Tschannen B, Altman DG, Egger M, Jüni P. Small study effects in meta‐analyses of osteoarthritis trials: meta‐epidemiological study. BMJ. 2010;341:c3515.
Jackson K, Ashby M, Howell D, Petersen J, Brumley D, Good P, Pisasale M, Wein S, Woodruff R.The effectiveness and adverse effects profile of "burst"ketamine in refractory cancer pain: the VCOG PM 1‐00 study. J Palliat Care. 2010;26(3):176‐83.
Peltoniemi MA, Hagelberg NM, Olkkola KT, Saari TI. Ketamine: a review of clinical pharmacokinetics and pharmacodynamics in anesthesia and pain therapy. Clin Pharmacokinet 2016;55:1059‐1077.
Mercadante S, Villari P, Ferrera P. Burst ketamine to reverse opioid tolerance in cancer pain. J Pain Symptom Manage. 2003;25(4):302‐5.
Mercadante S, Villari P, Ferrera P, Arcuri E, David F. Opioid switching and burst ketamine to improve the opioid response in patients with movement‐related pain due to bone metastases. Clin J Pain 2009;25(7):648‐9.
Quibell R, Fallon M, Mihalyo M Twycross R, Wilcock A. Ketamine. J Pain Symptom Manage 2015;50(2):268‐275.
Contributors
Response prepared by the review authors. Feedback managed by Feedback Editor Hayley Barnes, Managing Editor Anna Erskine, and Senior Editor Andrew Moore.
Feedback received, 21 February 2018
Summary
Date of Submission: 21‐Feb‐2018
Name: David Currow
Email Address: david.currow@sa.gov.au
Affiliation: Flinders University
Comment: The authors of the Cochrane review have not represented the ketamine study conducted by the Australian National Palliative Care Clinical Studies Collaborative accurately on at least two counts in their response to concerns raised by authors of the double‐blind, placebo controlled trial of ketamine for complex cancer pain. Firstly, titration occurred over 3‐5 days not 20 hours as stated in the Cochrane reviewers’ response to the concerns originally raised. This was laid out clearly in the paper, the protocol (which was delivered to this group of Cochrane reviewers in 2011) and in the Abstract. Secondly, this was a fully blinded study by all criteria required by CONSORT. The fact that there was a differential in toxicity was not known until the analysis was undertaken. Of note, this study of a dissociative anaesthetic used for complex pain in people with advanced cancer was the first randomised trial to systematically and prospectively measure dissociation. The fact that there is a differential rate of toxicities and study withdrawal was not known to the investigators until the code was broken and the analysis undertaken. As such, this was a fully double blinded study in every sense despite the assertions by the Cochrane reviewers to the contrary.
I do not have any affiliation with or involvement in any organisation with a financial interest in the subject matter of my comment.
Reply
The authors thank Dr. Currow for his additional feedback regarding the update of the Cochrane review entitled "Ketamine as an adjuvant to opioids for cancer pain". Dr. Currow states that we did not accurately represent the ketamine study by Hardy et al. “by at least two counts” in our previous response of August 2017. We reply to Dr. Currow’s specific comments below.
1. Re. titration of ketamine
“Firstly, titration occurred over 3‐5 days not 20 hours as stated in the Cochrane reviewers’ response to the concerns originally raised. This was laid out clearly in the paper, the protocol (which was delivered to this group of Cochrane reviewers in 2011) and in the Abstract.”
Reply: The dose titration was correctly recorded in the description of studies, while this discussion refers to the dose escalation. In our response to Dr. Currow we noted that “The dose escalation was rapid, for example having a start dose of 100 mg/ 24 hours and tripling the dose after less than 20 hours, considering the pharmacokinetics of ketamine which has a β half‐life of two to four hours in humans.” This is based on information in the Methods section of the paper which states that ketamine was used “at three dose levels (100, 300, or 500 mg)”, with the first dose level being “100 mg/24 hours” and that “If 80% of study drug had been delivered, and average pain improved by 2 BPI units with no more than four breakthrough doses, the dose remained the same. If not, the dose was increased to the next level.”
2. Re. study blinding
“Secondly, this was a fully blinded study by all criteria required by CONSORT. The fact that there was a differential in toxicity was not known until the analysis was undertaken….” Reply: We feel that the issue of blinding was adequately addressed in our previous response as follows: “As is evident from the Risk of bias table, we judged this trial to be at unclear risk of bias re. blinding. The detailed information on blinding that Dr. Currow provides in his email was not included in the published paper which lacks a description of the blinding procedure. We did however note that the protocol mentioned that "All syringes will look identical in volume and colour". The trial involved rapid titration of ketamine to high doses and there was a high rate of adverse events in the ketamine arm. No specific measures were taken to check for blinding. We judged that it was possible that blinding could therefore have been compromised by psychotomimetic adverse effects and skin reactions at the needle site, and a check on fidelity to blinding would have helped reduce uncertainty.”
A study published in 2009 clearly illustrates how blinding may be compromised by the adverse effects of ketamine1. The study was conducted in CRPS patients and similar to the trial by Hardy et al., involved rapid titration of ketamine to high dose, resulting in a high rate of adverse effects. Regarding blinding, the authors concluded as follows: “Patient’s and investigator’s guesses of the administered treatment were correct in 74% and 88%, respectively. This is most likely explained by ketamine’s psychomimetic side effects and suggests that in future trials an active placebo and/or the addition of an active placebo or addition of a benzodiazepine to the ketamine treatment group is warranted to preclude deblinding of treatment allocation.”
Reference
1. Sigtermans MJ et al. Ketamine produces effective and long‐term pain relief in patients with Complex Regional Pain Syndrome Type 1. Pain, 2009 Oct;145(3):304‐11. DOI: 10.1016/j.pain.2009.06.023.
Contributors
Response prepared by the review authors. Feedback managed by Feedback Editor Hayley Barnes, Managing Editor Anna Erskine, and Senior Editors Professor Philip Wiffen and Professor Andrew Moore.
What's new
| Date | Event | Description |
|---|---|---|
| 30 September 2019 | Amended | Clarification added to Declarations of interest. |
| 28 June 2017 | Review declared as stable | See Published notes. |
History
Protocol first published: Issue 4, 2001 Review first published: Issue 1, 2003
| Date | Event | Description |
|---|---|---|
| 9 November 2018 | Feedback has been incorporated | See Feedback 2. |
| 8 March 2018 | Amended | Affiliation updated. |
| 14 August 2017 | Feedback has been incorporated | See Feedback. |
| 12 April 2017 | New citation required but conclusions have not changed | We identified one new study with 185 participants (Hardy 2012). We assessed risk of bias and the quality of the evidence according to GRADE. |
| 12 April 2017 | New search has been performed | This review has been updated to include the results of a new search in December 2016/January 2017. |
| 13 May 2009 | Amended | Contact details updated. |
| 30 October 2008 | Amended | History, What's New and citation corrected |
| 4 August 2008 | Amended | Converted to new review format. |
| 19 June 2007 | New search has been performed | Review updated with revised search but no new trials were identified for this update. |
Notes
A new search within two years is not likely to identify any potentially relevant studies likely to change the conclusions. Therefore, following discussion with the authors and editors, this review has now been stabilised until 2022, at which point we will assess the review for updating. If appropriate, we will update the review before this date if new evidence likely to change the conclusions is published, or if standards change substantially which necessitate major revisions.
Acknowledgements
The updated review was supported by the Regional Centre of Excellence for Palliative Care at Haukeland University Hospital, Bergen, Norway which funds a part‐time research position for RFB.
Searches for the current update were performed by Joanne Abbott. Anna Erskine and Phil Wiffen assisted on the update.
Cochrane Review Group funding acknowledgement: this project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Pain, Palliative and Supportive Care Review Group (PaPaS). The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
We are grateful to Dr. Augusto Caraceni, Italian Ministry of Health, Rome, who provided rapid peer review for the 2017 update.
Appendices
Appendix 1. MEDLINE search strategy ‐ 2002
via OVID
KETAMINE (single term MeSH)
ketamine or inducmina or ketalar or ketanest or calypso or narkamon or keta or velonarcon or ketmin or ketava or ketalin or ketina or brevinaze or keta‐hameln
OR/1‐2
Explode NEOPLASMS
BONE MARROW TRANSPLANTATION (single term MeSH)
neoplas* or cancer* or carcinoma* or tumour* or tumor* or adenocarcinoma* or leukemi* or leukaemi* or lymphoma* or malignan* or bone‐marrow‐transplant* or "bone marrow transplant*"
Explode RADIOTHERAPY
radiotherap* or radiation or irradiat* or radiochemo* or chemotherapy*
OR/4‐8
3 AND 9
The above subject search was linked to the Cochrane Sensitive Search strategy filter for RCTs:
randomized controlled trial.pt.
controlled clinical trial.pt.
randomized controlled trials.sh.
random allocation.sh.
double blind method.sh.
single blind method.sh.
or/1‐6
(ANIMALS not HUMAN).sh.
7 not 8
clinical trial.pt.
exp clinical trials/
(clin* adj25 trial*).ti,ab.
((singl* or doubl* or trebl* or tripl*) adj25 (blind* or mask*)).ti,ab.
placebos.sh.
placebo*.ti,ab.
random*.ti,ab.
research design.sh.
or/10‐17
18 not 8
19 not 9
9 or 19
Appendix 2. Other search strategies ‐ 2002
| Database | search strategy |
| PaPaS Trials register | ((ketamine or inducmina or ketalar or ketanest or calypso or narkamon or keta or velonarcon or ketmin or ketava or ketalin or ketine or brevinaze or keta‐hameln) AND (neoplas* or cancer* or carcinoma* or tumour* or tumor* or adenocarcinoma* or leukemi* or leukaemi* or lymphoma* or malignan* or bone‐marrow‐transplant* or "bone marrow transplant*" or radiotherap* or radiation or irradiat* or radiochemo* or chemotherap*)) |
| CENTRAL | #1 KETAMINE (single term MeSH) #2 ketamine or inducmina or ketalar or ketanest or calypso or narkamon or keta or velonarcon or ketmin ketava or ketalin or ketina or brevinaze or keta‐hameln #3 #1 or #2 #4 Explode NEOPLASMS #5 BONE MARROW TRANSPLANT (single term MeSH) #6 neoplas* or cancer* or carcinoma* or tumour* or tumor* or adenocarcinoma* or leukemi* or leukaemi* or lymphoma* or malignan* or bone‐marrow‐transplant* or "bone marrow transplant*" #7 Explode RADIOTHERAPY #8 radiotherap* or radiation or irradiat* or radiochemo* or chemotherap* #9 #4 or #5 or #6 or #7 or #8 #10 #3 AND #9 |
| PubMed (for limit to Cancer subset) | #1 Search KETAMINE Field: MeSH Terms #2 Search ketamine or inducmina or ketalar or ketanest or calypso or narkamon or keta or velonarcon or ketmin ketava or ketalin or ketina or brevinaze or keta‐hameln #3 Search #1 or #2 #4 Search NEOPLASMS Field: MeSH Terms #5 Search BONE MARROW TRANSPLANTATION Field: MeSH Terms #6 Search neoplas* or cancer* or carcinoma* or tumour* or tumor* or adenocarcinoma* or leukemi* or leukaemi* or lymphoma* or malignan* or bone‐marrow‐transplant* or "bone marrow transplant*" #7 Search RADIOTHERAPY Field: MeSH Terms #8 Search radiotherap* or radiation or irradiat* or radiochemo* or chemotherap* #9 Search #4 or #5 or #6 or #7 or #8 #10 Search #3 AND #9 #11 Search (randomized controlled trial [pt] OR controlled clinical trial [pt] OR randomized controlled trials [mh] OR random allocation [mh] OR double‐blind method [mh] OR single‐blind method [mh] OR clinical trial [pt] OR clinical trials [mh] OR ("clinical trial" [tw]) OR ((singl* [tw] OR doubl* [tw] OR trebl* [tw] OR tripl* [tw]) AND (mask* [tw] OR blind* [tw])) OR (placebos [mh] OR placebo* [tw] OR random* [tw] OR research design [mh:noexp]) NOT (animals [mh] NOT humans [mh]) #12 #10 AND #11 Limit CANCER subset |
| Embase via OVID | 1 KETAMINE (single term MeSH) 2 ketamine or inducmina or ketalar or ketanest or calypso or narkamon or keta or velonarcon or ketmin or ketava or ketalin or ketina or brevinaze or keta‐hameln 3 OR/1‐2 4 Explode NEOPLASM 5 BONE MARROW TRANSPLANTATION (single term MeSH) 6 neoplas* or cancer* or carcinoma* or tumour* or tumor* or adenocarcinoma* or leukemi* or leukaemi* or lymphoma* or malignan* or bone‐marrow‐transplant* or "bone marrow transplant*" 7 Explode RADIOTHERAPY 8 radiotherap* or radiation or irradiat* or radiochemo* or chemotherapy* 9 OR/4‐8 10 3 AND 9 The above subject search was linked to the following Filter for EMBASE via OVID 1. random*.ti,ab. 2. factorial*.ti,ab. 3. (crossover* or cross over* or cross‐over*).ti,ab. 4. placebo*.ti,ab. 5. (doubl* adj blind*).ti,ab. 6. (singl* adj blind*).ti,ab. 7. assign*.ti,ab. 8. allocat*.ti,ab. 9. volunteer*.ti,ab. 10. CROSSOVER PROCEDURE.sh. 11. DOUBLE‐BLIND PROCEDURE.sh. 12. RANDOMIZED CONTROLLED TRIAL.sh. 13. SINGLE BLIND PROCEDURE.sh. 14. or/1‐13 15. ANIMAL/ or NONHUMAN/ or ANIMAL EXPERIMENT/ 16. HUMAN/ 17. 16 and 15 18. 15 not 17 19. 14 not 18 |
| THE PFIZER PRODUCT INFORMATION DATABASE (PPI) (2002‐2007) | 1 ((KETAMINE OR KETALAR) AND (CANCER* OR NEOPLASM* OR TUMOR* OR TUMOUR* OR CARCINOMA* OR MALIGNAN*)).AF 415 2 LIMIT ((KETAMINE OR KETALAR) AND (CANCER* OR NEOPLASM* OR TUMOR* OR TUMOUR* OR CARCINOMA* OR MALIGNAN*)). AF TO YR=2002‐2007 62 |
Appendix 3. CENTRAL search strategy ‐ 2012
#1 MeSH descriptor Ketamine explode all trees #2 (ketamine or inducmina or ketalar or ketanest or calypso or narkamon or keta or velonarcon or ketmin or ketava or ketalin or ketina or brevinaze or keta‐hameln):ti,ab,kw #3 (#1 OR #2) #4 MeSH descriptor Neoplasms explode all trees #5 (neoplas* or cancer* or carcinoma* or tumour* or tumor* or adenocarcinoma* or leukemi* or leukaemi* or lymphoma* or malignan*):ti,ab,kw #6 (#4 OR #5) #7 (#3 AND #6)
Appendix 4. Embase search strategy ‐ 2012
Embase <1996 to May 2012
1 exp KETAMINE/ 2 (ketamine or inducmina or ketalar or ketanest or calypso or narkamon or keta or velonarcon or ketmin or ketava or ketalin or ketina or brevinaze or keta‐hameln).tw. 3 or/1‐2 4 exp neoplasm/ 5 (neoplas* or cancer* or carcinoma* or tumour* or tumor* or adenocarcinoma* or leukemi* or leukaemi* or lymphoma* or malignan*).tw. 6 or/4‐5 7 3 and 6 8 Clinical trial/ 9 Randomized controlled trial/ 10 Randomization/ 11 Single blind procedure/ 12 Double blind procedure/ 13 Crossover procedure/ 14 Placebo/ 15 randomi?ed controlled trial*.tw. 16 RCT.tw. 17 random allocation.tw. 18 randomly allocated.tw. 19 allocated randomly.tw. 20 (allocated adj2 random).tw. 21 single blind*.tw. 22 double blind*.tw. 23 ((treble or triple) adj blind*).tw. 24 placebo*.tw. 25 Prospective study/ 26 or/8‐25 27 Case study/ 28 case report.tw. 29 abstract report/ or letter/ 30 or/27‐29 31 26 not 30 32 7 and 31 33 (2002* or 2003* or 2004* or 2005* or 2006* or 2007* or 2008* or 2009* or 2010* or 2011*).em. 34 32 and 33
Appendix 5. MEDLINE search strategy ‐ 2012
Database: Ovid MEDLINE(R) without Revisions <1996 to May 2012 1 exp Ketamine/ 2 (ketamine or inducmina or ketalar or ketanest or calypso or narkamon or keta or velonarcon or ketmin or ketava or ketalin or ketina or brevinaze or keta‐hameln).tw. 3 or/1‐2 4 exp Neoplasms/ 5 (neoplas* or cancer* or carcinoma* or tumour* or tumor* or adenocarcinoma* or leukemi* or leukaemi* or lymphoma* or malignan*).tw. 6 or/4‐5 7 3 and 6 8 randomized controlled trial.pt. 9 controlled clinical trial.pt. 10 randomized.ab. 11 placebo.ab. 12 clinical trials as topic.sh. 13 randomly.ab. 14 trial.ti. 15 or/8‐14 16 exp animals/ not humans.sh. 17 15 not 16 18 7 and 17 19 (2002* or 2003* or 2004* or 2005* or 2006* or 2007* or 2008* or 2009* or 2010* or 2011*).ed. 20 18 and 19
Appendix 6. Searches 2017
CENTRAL (CRSO)
MESH DESCRIPTOR Ketamine
(ketamine or inducmina or ketalar or ketanest or calypso or narkamon or keta or velonarcon or ketmin ketava or ketalin or ketina or brevinaze or keta‐hameln):TI,AB,KY
#1 OR #2
MESH DESCRIPTOR NEOPLASMS EXPLODE ALL TREES
MESH DESCRIPTOR BONE MARROW
MESH DESCRIPTOR Bone Marrow Transplantation
(neoplas* or cancer* or carcinoma* or tumour* or tumor* or adenocarcinoma* or leukemi* or leukaemi* or lymphoma* or malignan* or bone‐marrow‐transplant* or "bone marrow transplant*"):TI,AB,KY
MESH DESCRIPTOR RADIOTHERAPY EXPLODE ALL TREES
( radiotherap* or radiation or irradiat* or radiochemo* or chemotherap*):TI,AB,KY
#4 OR #5 OR #6 OR #7 OR #8 OR #9
#3 AND #10
MEDLINE (OVID)
1. Ketamine/
2. (ketamine or inducmina or ketalar or ketanest or calypso or narkamon or keta or velonarcon or ketmin or ketava or ketalin or ketina or brevinaze or keta‐hameln).mp. [mp=title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept word, rare disease supplementary concept word, unique identifier]
3. 1 or 2
4. exp Neoplasms/
5. Bone Marrow Transplantation/
6. (neoplas* or cancer* or carcinoma* or tumour* or tumor* or adenocarcinoma* or leukemi* or leukaemi* or lymphoma* or malignan* or bone‐marrow‐transplant* or "bone marrow transplant*").mp. [mp=title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept word, rare disease supplementary concept word, unique identifier]
7. exp Radiotherapy/
8. (radiotherap* or radiation or irradiat* or radiochemo* or chemotherapy*).mp. [mp=title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept word, rare disease supplementary concept word, unique identifier]
9. or/4‐8
10. 3 and 9
11. randomized controlled trial.pt.
12. controlled clinical trial.pt.
13. randomized.ab.
14. placebo.ab.
15. drug therapy.fs.
16. randomly.ab.
17. trial.ab.
18. groups.ab.
19. 11 or 12 or 13 or 14 or 15 or 16 or 17 or 18
20. exp animals/ not humans.sh.
21. 19 not 20
22. 10 and 21
Embase (OVID)
1. ketamine/
2. (ketamine or inducmina or ketalar or ketanest or calypso or narkamon or keta or velonarcon or ketmin or ketava or ketalin or ketina or brevinaze or keta‐hameln).tw.
3. 1 or 2
4. exp neoplasm/
5. (neoplas* or cancer* or carcinoma* or tumour* or tumor* or adenocarcinoma* or leukemi* or leukaemi* or lymphoma* or malignan*).tw.
6. or/4‐5
7. 5 or 6
8. 3 and 7
9. random$.tw.
10. factorial$.tw.
11. crossover$.tw.
12. cross over$.tw.
13. cross‐over$.tw.
14. placebo$.tw.
15. (doubl$ adj blind$).tw.
16. (singl$ adj blind$).tw.
17. assign$.tw.
18. allocat$.tw.
19. volunteer$.tw.
20. Crossover Procedure/
21. double‐blind procedure.tw.
22. Randomized Controlled Trial/
23. Single Blind Procedure/
24. or/9‐23
25. (animal/ or nonhuman/) not human/
26. 24 not 25
27. 8 and 26
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Hardy 2012.
| Methods | Randomised. Double‐blind, but blinding procedure not described. Placebo control. Parallel group Study duration 5 days |
|
| Participants | Inpatients aged 18 or older, palliative care patients with refractory chronic pain (BPI average pain score ≥3 despite ongoing treatment with opioids and co‐analgesics at predefined dose levels) secondary to cancer or its treatment N= 185, 93 randomised to ketamine group, 92 randomised to placebo Mean age in ketamine arm: 63 years. Mean age in placebo group: 64.3 |
|
| Interventions | Participants received either ketamine or placebo (normal saline) as a subcutaneous infusion in a 5‐day schedule, starting at the first dose level (100 mg/24 hrs. If 80% of the study drug had been delivered, and average pain improved by ≥ 2 BPI units, with no more than four breakthrough doses, the dose remained the same. If not, the dose was increased to the next level. | |
| Outcomes | The primary outcome was a positive response, defined as a clinically relevant improvement in pain at the end of the 5‐day study period. A clinically relevant improvement in pain was defined as a reduction in BPI average pain score by ≥ 2 points from baseline in the absence of more than four breakthrough doses of analgesia over the previous 24 hours. Secondary outcomes: pain assessments at days 2‐5. Adverse events. | |
| Notes | Data for patients who discontinued due to reasons unrelated to the intervention were imputed using LOCF. Unclear how data for those who discontinued due to adverse effects or lack of effect were handled. Quality/validity: OPVS: 12 Oxford: 4 Supported by a grant from the Palliative Care Branch, Australian Government Department of Health and Ageing |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised, process of randomisation is described and adequate. "Each site pharmacy used randomization tables from an independent central registry" |
| Allocation concealment (selection bias) | Low risk | "All non pharmacy study staff, treating clinicians,investigators and participants were unaware of treatment allocation until completion". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Described as blinded. Blinding procedure not described in final paper but was described in protocol "All syringes will look identical in volume and colour". The authors of this review update felt that blinding could have been compromised due to adverse effects from ketamine. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No specific procedure to check for detection bias. The authors of this review update felt that blinding could have been compromised due to adverse effects from ketamine |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Unclear how missing data were imputed |
| Selective reporting (reporting bias) | Unclear risk | Total opioid dose was not reported but was mentioned in the protocol |
| Size | Unclear risk | 50 ‐ 199 participants per treatment arm |
Mercadante 2000.
| Methods | Stated to be randomised but procedure not described. Blinded study: Drugs prepared in identical syringes by a person not involved in the test sessions Placebo control Cross‐over Study duration: 30 to 180 minutes |
|
| Participants | Cancer patients with neuropathic pain unrelieved by morphine N = 10 per group (cross‐over) Mean age of patients: 57 years |
|
| Interventions | Treatment 1:
Saline (IV) Treatment 2: KET bolus 0.25 mg/kg (IV) Treatment 3: KET bolus 0.5 mg/kg (IV) |
|
| Outcomes | Pain intensity
Adverse effects Results: Low‐dose KET IV + Mo (PO, SC, IV) significantly reduced pain intensity |
|
| Notes | Washout period ( "at least two days") Quality/ validity: OPVS score: 12 Oxford score: 3 Information on funding not provided |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stated to be randomised but procedure not described. |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "The drugs were prepared in identical syringes by a person not involved in the test sessions". Blinding possibly compromised due to adverse effects from ketamine. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No specific procedure to check for detection bias. Possible bias due to adverse effects from ketamine |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants accounted for |
| Selective reporting (reporting bias) | Low risk | No problems detected |
| Size | High risk | Fewer than 50 participants in total (cross‐over) |
Yang 1996.
| Methods | Stated to be randomised but procedure not described. Blinded study: double dummy Cross‐over, no washout Active control (morphine) Study duration: not defined |
|
| Participants | Hospitalised patients with terminal cancer pain, treated with opioids N = 20 per group (cross‐over) |
|
| Interventions | Treatment 1: Mo (IT) Treatment 2: KET 1.0 mg (IT) twice daily + Mo (IT) | |
| Outcomes | Pain intensity
Pain frequency
Total titrated Mo dose (IT).
Total rescue medication.
Frequency of IT titration. Results: Co‐administration of low‐dose KET reduces the amount of IT Mo required to control cancer pain |
|
| Notes | No washout period Quality/ validity: OPVS score: 13 Oxford score: 3 Supported by a grant from the National Science Council (NSC), Taipei, Taiwan |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stated to be randomised but procedure not described |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "A doubled dummy technique was used, so that the patient, investigator and nurse were unaware of the dose of morphine and ketamine" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "A doubled dummy technique was used, so that the patient, investigator and nurse were unaware of the dose of morphine and ketamine". Blinding not compromised by adverse effects from ketamine |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants accounted for |
| Selective reporting (reporting bias) | Low risk | No problems detected |
| Size | High risk | Fewer than 50 participants in total (cross‐over) |
BPI: Brief Pain Inventory IT: Intrathecal IV: Intravenous KET: Ketamine kg: Kilo LOCF:Last observation carried forward mg: Milligram Mo: Morphine N: Number of patients OPVS: Oxford Pain Validity Scale PO: Oral SC: Subcutaneous
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Currow 2011 | Poster abstract |
| Ishizuka 2007 | Dropouts leaving one group with fewer than 10 participants |
| Lauretti 1999a | Open‐label study ("pilot work"). Described as placebo‐controlled, but in fact used active control (morphine). Design flaw with fixed maximum baseline morphine dose PO and primary outcome measure: daily consumption of morphine. OPVS score: 1 Oxford Quality Scale score: 2 |
| Lauretti 1999b | Described as placebo‐controlled, but in fact used active control (morphine). Design flaw with fixed baseline dose of morphine ED, fixed maximum daily dose of morphine ED and primary outcome measure: daily consumption of morphine ED. OPVS score: 7 Oxford Quality Scale score: 3 |
| Salas 2012 | Dropouts leaving one group with fewer than 10 participants |
ED: epidural OPVS: Oxford Pain Validity Scale PO: oral
Characteristics of ongoing studies [ordered by study ID]
NCT00484484.
| Trial name or title | Ketamine associated with opioids in refractory cancer pain treatment |
| Methods | Randomised, placebo‐controlled, double‐blind parallel group study |
| Participants | 100 hospitalised adult cancer patients undergoing opioid treatment for refractory pain |
| Interventions | Ketamine versus placebo |
| Outcomes | Daily pain score on 11‐point numerical pain rating scale; patient global impression of change; daily sleep interference score;patient satisfaction; opioid consumption; adverse effects |
| Starting date | May 2007. Completed September 2009 |
| Contact information | Sylvie Rostaing‐Rigattieri, Assistance Publique‐ Hopitaux de Paris, Paris, France |
| Notes | Status 'completed'. Not published. Possibly same study as NCT01326325? |
NCT01207206.
| Trial name or title | Oral ketamine as an adjuvant to opioids for pain treatment in cancer patients |
| Methods | Randomised, placebo‐controlled, double‐blind study |
| Participants | 50 patients with unbalanced (VAS > 6) chronic cancer‐related pain despite opioid treatment |
| Interventions | Oral ketamine versus placebo |
| Outcomes | Pain reduction (VAS scores), adverse effects |
| Starting date | October 2010 |
| Contact information | Silviu Brill, Tel Aviv Sourasky Medical Center |
| Notes |
NCT01316744.
| Trial name or title | Ketamine hydrochloride and best pain management in treating cancer patients with neuropathic pain |
| Methods | Randomised, placebo‐controlled, double‐blinded, parallel group study |
| Participants | Adult cancer patients with malignant neuropathic pain |
| Interventions | Addition of ketamine hydrochloride or placebo to "best pain management" |
| Outcomes | Time to treatment failure; difference in pain scores; patient distress; adverse effects |
| Starting date | April 2009 |
| Contact information | Marie T. Fallon, Edinburgh Cancer Centre at Western General Hospital, Edinburgh, UK |
| Notes | Unknown status |
NCT01326325.
| Trial name or title | Efficacy of low analgesic doses of ketamine associated with opioids in refractory cancer pain treatment |
| Methods | Randomised, placebo‐controlled, double‐blind parallel group study |
| Participants | Adult hospitalised cancer patients with refractory pain undergoing opioid treatment |
| Interventions | IV ketamine versus placebo |
| Outcomes | Per cent reduction average daily pain intensity score; daily average pain intensity score; patient global impression of change; daily sleep interference; opioid consumption; patient satisfaction; adverse effects |
| Starting date | July 2011. Completed February 2013 |
| Contact information | Sylvie Rostaing‐Rigattieri, Center of Evaluation and Treatment of Pain, Saint‐Antoine Hospital, Paris, France |
| Notes | "Completed" study, unpublished. Possibly the same study registered as NCT00484484? |
NCT02591017.
| Trial name or title | Comparison of oral morphine versus nasal ketamine spray with chitosan in cancer pain outpatients |
| Methods | Randomised, placebo‐control double blind cross‐over study |
| Participants | 34 adult cancer outpatients with opioid base therapy because of pain, or pain breakthrough pain or extreme pain on movement |
| Interventions | 3 treatment arms investigating morphine drops, ketamine nasal spray with chitosan or morphine drops + ketamine nasal spray with chitosan |
| Outcomes | Time to onset of action; median numeric rating scale (NRS) improvement; total dose ketamine or morphine; total opioid dose increase; adverse effects |
| Starting date | February 2015 |
| Contact information | Wilhelm Ruppen, Pain Relief Unit and Anesthesiology, University Hospital Basel, Switzerland |
| Notes |
IV: intravenous VAS: visual analogue scale
Differences between protocol and review
We did not search CancerLit as we judged it redundant against current databases. We did not search the PaPaS specialised register as it is no longer updated. For this update we searched trial registries.
In this 2017 update, we excluded studies having a group size of fewer than 10 participants who completed the study, and unpublished studies.
We excluded studies that were not double‐blind. We added the secondary outcomes of total opioid consumption and rescue medication.
We also reinstated the secondary outcomes distress and function, which were omitted in previous versions of this review.
We updated the methods sections to conform to current Cochrane standards. We included 'Risk of bias' and GRADE assessments.
We reported participant withdrawal (dropouts) from the study but no longer narrate it as a secondary outcome but as a feature of the methods.
We no longer refer to pain relief as a primary outcome.
Contributions of authors
RFB: assessed the updated search results for trials for inclusion in the review, undertook quality and validity evaluation of the included studies, extracted data and wrote the update. EK: assessed the results of the updated search, undertook quality and validity evaluation of the included studies, and contributed to the writing of the update. CE: undertook quality and validity evaluation of the included studies, and contributed to the writing of the update.
Sources of support
Internal sources
-
Regional Centre of Excellence in Palliative Care, Haukeland University Hospital; Centre for Clinical Research, Haukeland University Hospital, Norway.
For the original review and all updates RF Bell has received funding for a 20% research position
External sources
-
Norwegian Research Council, Norway.
For the original review RF Bell received a grant to fund a 30% research position
Declarations of interest
RFB: none known. RFB is a retired specialist pain physician who has worked with patients having acute, chronic or cancer pain, including palliative care patients.
CE: none known. Since CE is an author as well as the PaPaS Co‐ordinating Editor at the time of writing, we acknowledge the input of Phil Wiffen who acted as Sign Off Editor for this review. CE had no input into the editorial decisions or processes for this review.
EK: none known. EK is a specialist pain physician who has worked with patients having acute, chronic or cancer pain, including palliative care patients.
Stable (no update expected for reasons given in 'What's new')
References
References to studies included in this review
Hardy 2012 {published data only}
- Hardy J, Quinn S, Fazekas B, Plummer J, Eckermann S, Agar M, et al. Randomized, double‐blind, placebo‐controlled study to assess the efficacy and toxicity of subcutaneous ketamine in the management of cancer pain. Journal of Clinical Oncology 2012;30(29):3611‐7. [DOI] [PubMed] [Google Scholar]
Mercadante 2000 {published data only}
- Mercadante S, Arcuri E, Tirelli W, Casuccio A. Analgesic effect of intravenous ketamine in cancer patients on morphine therapy: a randomized, controlled, double‐blind, crossover, double‐dose study. Journal of Pain and Symptom Management 2000;4:246‐51. [DOI] [PubMed] [Google Scholar]
Yang 1996 {published data only}
- Yang CY, Wong CS, Chang JY. Intrathecal ketamine reduces morphine requirements in patients with terminal cancer pain. Canadian Journal of Anaesthesia 1996;43(4):379‐83. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Currow 2011 {published data only}
- Currow DC, Hardy J, Sanderson C, Spruyt O, Eckermann S, Plummer J, et al. A randomised, double‐blind, placebo‐controlled, multi‐site study of subcutaneous ketamine in the management of cancer pain. European Journal of Cancer. 2011; Vol. 47:S152.
Ishizuka 2007 {published data only}
- Ishizuka P, Garcia JBS, Sakata RK, Issy AM, Mulich SL. Assessment of oral S(+) ketamine associated with morphine for the treatment of oncologic pain [Evaluación de la S(+) cetamina por vía oral asociada a la morfina en el tratamiento del dolor oncológico]. Revista Brasileira de Anestesiologia 2007;57(1):19‐31. [DOI] [PubMed] [Google Scholar]
Lauretti 1999a {published data only}
- Lauretti GR, Lima ICPR, Reis MP, Prado WA, Pereira NL. Oral ketamine and transdermal nitroglycerin as analgesic adjuvants to oral morphine therapy for cancer pain management. Anesthesiology 1999;90:1528‐33. [DOI] [PubMed] [Google Scholar]
Lauretti 1999b {published data only}
- Lauretti GR, Gomes JMA, Reis MP, Pereira NL. Low doses of epidural ketamine or neostigmine, but not midazolam, improve morphine analgesia in epidural terminal cancer pain therapy. Journal of Clinical Anaesthesia 1999;11(8):663‐8. [DOI] [PubMed] [Google Scholar]
Salas 2012 {published data only}
- Salas S, Frasca M, Planchet‐Barraud B, Burucoa B, Pascal M, Lapiana J‐M, et al. Ketamine analgesic effect by continuous intravenous infusion in refractory cancer pain: considerations about the clinical research in palliative care. Journal of Palliative Medicine 2012;15(3):287‐93. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
NCT00484484 {published data only}
- Ketamine associated with opioids in refractory cancer pain treatment. Ongoing study May 2007. Completed September 2009.
NCT01207206 {published data only}
- Oral ketamine as an adjuvant to opioids for pain treatment in cancer patients. Ongoing study October 2010.
NCT01316744 {published data only}
- Ketamine hydrochloride and best pain management in treating cancer patients with neuropathic pain. Ongoing study April 2009.
NCT01326325 {published data only}
- Efficacy of low analgesic doses of ketamine associated with opioids in refractory cancer pain treatment. Ongoing study July 2011. Completed February 2013.
NCT02591017 {published data only}
- Comparison of oral morphine versus nasal ketamine spray with chitosan in cancer pain outpatients. Ongoing study February 2015.
Additional references
Afsharimani 2015
- Afsharimani B, Kindl K, Good P, Hardy J. Pharmacological options for the treatment of refractory cancer pain‐ what is the evidence?. Supportive Care in Cancer 2015;23(5):1473‐81. [DOI] [PubMed] [Google Scholar]
Are 2017
- Are M, McIntyre A, Reddy S. Global disparities in cancer pain management and palliative care. Journal of Surgical Oncology 2017;115(5):637‐41. [DOI] [PubMed] [Google Scholar]
Assouline 2016
- Assouline B, Tramèr MR, Kreienbühl L, Elia N. Benefit and harm of adding ketamine to an opioid in a patient‐controlled analgesia device for the control of postoperative pain: systematic review and meta‐analyses of randomized controlled trials with trial sequential analyses. Pain 2016;157(12):2854‐64. [DOI] [PubMed] [Google Scholar]
Bell 1999
- Bell RF. Low‐dose subcutaneous ketamine infusion and morphine tolerance. Pain 1999;83:101‐3. [DOI] [PubMed] [Google Scholar]
Bell 2006
- Bell RF, Dahl JB, Moore RA, Kalso E. Perioperative ketamine for acute postoperative pain. Cochrane Database of Systematic Reviews 2006, Issue 1. [DOI: 10.1002/14651858.CD004603.pub2] [DOI] [PubMed] [Google Scholar]
Bell 2012a
- Bell RF. Ketamine for chronic noncancer pain: concerns regarding toxicity. Current Opinion in Supportive and Palliative Care 2012;6(2):183‐7. [DOI] [PubMed] [Google Scholar]
BNF 2012
- British Medical Association and Royal Pharmaceutical Society of Great Britain. British National Formulary. 63. London: British Medical Association and Royal Pharmaceutical Society of Great Britain, 2012 (March). [Google Scholar]
Bredlau 2013
- Bredlau AL, Thakur R, Corones DN, Dworkin RH. Ketamine for pain in adults and children with cancer: a systematic review and synthesis of the literature. Pain Medicine 2013;14(10):1505‐17. [DOI] [PubMed] [Google Scholar]
Cancer Therapy Evaluation Program Version 3
- Cancer Therapy Evaluation Program: Common Terminology Criteria for Adverse Events V3.0. http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf.
Cherny 2015
- Cherny N, Fallon M, Kaasa S, Portenoy RK, Currow DC. Oxford Textbook of Palliative Medicine. 5th Edition. Oxford: Oxford University Press, 2015. [Google Scholar]
Dale 2012
- Dale O, Somogyi AA, Li Y, Sullivan T, Shavit Y. Does intraoperative ketamine attenuate inflammatory reactivity following surgery? A systematic review and meta‐analysis. Anesthesia and Analgesia 2012;115:934‐43. [DOI] [PubMed] [Google Scholar]
Dickenson 1994
- Dickenson AH. Neurophysiology of opioid poorly responsive pain. Cancer Surveys: Palliative Medicine: Problem Areas in Pain and Symptom Management 1994;21:5‐16. [PubMed] [Google Scholar]
Dworkin 2008
- Dworkin RH, Turk DC, Wyrwich, Beaton D, Cleeland CS, Farrar JT, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. Journal of Pain 2008;9(2):105‐21. [DOI] [PubMed] [Google Scholar]
Ebert 1997
- Ebert B, Mikkelsen S, Thorkildsen C, Borgbjerg FM. Norketamine, the main metabolite of ketamine, is a non‐competitive NMDA receptor antagonist in the rat brain and spinal cord. European Journal of Pharmacology 1997;333(1):91‐104. [DOI] [PubMed] [Google Scholar]
Edwards 2002
- Edwards SR, Minto CF, Mather LE. Concurrent ketamine and alfentanil administration:pharmacokinetic considerations. British Journal of Anaesthesia 2002;88(1):94‐100. [DOI] [PubMed] [Google Scholar]
Fallon 2013
- Fallon MT. Neuropathic pain in cancer. British Journal of Anaesthesia 2013;111(1):105‐11. [DOI] [PubMed] [Google Scholar]
Fine 1999
- Fine PG. Low‐dose ketamine in the management of opioid nonresponsive terminal cancer pain. Journal of Pain and Symptom Management 1999;17:296‐300. [DOI] [PubMed] [Google Scholar]
Fisher 2000
- Fisher K, Coderre TJ, Hagen NA. Targeting the N‐Methyl‐D‐Aspartate receptor for chronic pain management: preclinical animal studies, recent clinical experience and future research directions. Journal of Pain and Symptom Management 2000;20(5):358‐73. [DOI] [PubMed] [Google Scholar]
Fukuida 1981
- Fukuida E, Gocho C, Ito K. Pain in the terminal stage of cancer. IRYO Japanese Journal of National Medical Services 1981;35(6):584‐7. [Google Scholar]
Garry 1996
- Garry AC, Simpson KH. A difficult pain problem: Use of intrathecal ketamine. Pain Clinic 1996;9(3):335‐42. [Google Scholar]
GRADEpro GDT 2015 [Computer program]
- Brozek J, Oxman A, Schünemann H. GRADEpro Guideline Development Tool [Software].. McMaster University (developed by Evidence Prime, Inc.). Available from www.gradepro.org., 2015.
Grahame‐Smith 2002
- Grahame‐Smith DG, Aronson JK. Oxford Textbook of Clinical Pharmacology and Drug herapy. 3rd Edition. Oxford: Oxford University Press, 2002. [Google Scholar]
Higgins 2011
- Higgins JPT, Altman DG, Sterne JAC. Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. [Google Scholar]
Hijazi 2002
- Hijazi Y, Boulieu R. Contribution of CYP3A4, CYP2B6, and CYP2C9 isoforms to N‐demethylation of ketamine in human liver microsomes. Drug Metabolism and Disposition 2002;30(7):853‐8. [DOI] [PubMed] [Google Scholar]
Jadad 1996
- Jadad AR, Moore RA, Carroll D, Jenkinson C, Reynolds DJ, Gavaghan DJ, et al. Assessing the quality of reports of randomized clinical trials: is blinding necessary?. Controlled Clinical Trials 1996;17:1‐12. [DOI] [PubMed] [Google Scholar]
Kanamaru 1990
- Kanamaru T, Saeki S, Katsumata N, Mizuno K, Ogawa S, Suzuki H. Ketamine infusion for control of pain in patients with advanced cancer. Masui 1990;39(10):1368‐71. [PubMed] [Google Scholar]
Karpinski 1997
- Karpinski N, Dunn J, Hansen L, Masliah E. Subpial vacuolar myelopathy after intrathecal ketamine: report of a case. Pain 1997;73(1):103‐5. [DOI] [PubMed] [Google Scholar]
Kharasch 1992
- Kharasch ED, Labroo R. Metabolism of ketamine stereoisomers by human liver microsomes. Anesthesiology 1992;77(6):1201‐7. [DOI] [PubMed] [Google Scholar]
Klahr 1997
- Klahr M, Gomas JM, Larrouture A, Vinceneux P. Treatment of neurogenic pain in adult cancer patients. A retrospective study. Presse Medicale 1997;26(20):960‐3. [PubMed] [Google Scholar]
L'Abbé 1987
- L'Abbé KA, Detsky AS, O'Rourke K. Meta‐analysis in clinical research. Annals of Internal Medicine 1987;107(2):224‐33. [DOI] [PubMed] [Google Scholar]
Laskowski 2011
- Laskowski K, Stirling A, McKay WP, Lim HJ. A systematic review of intravenous ketamine for postoperative analgesia. Canadian Journal of Anesthesia 2011;58:911‐23. [DOI] [PubMed] [Google Scholar]
Laulin 2002
- Laulin JP, Maurette P, Corcuff JB, Rivat C, Chauvin M, Simonnet G. The role of ketamine in preventing fentanyl‐induced hyperalgesia and subsequent acute morphine tolerance. Anesthesia and Analgesia 2002;94(5):1263‐9. [DOI] [PubMed] [Google Scholar]
Leung 1986
- Leung LY, Baillie TA. Comparative pharmacology in the rat of ketamine and its two principal metabolites, norketamine and (Z)‐6‐hydroxynorketamine. Journal of Medicinal Chemistry 1987;29(11):2396‐9. [DOI] [PubMed] [Google Scholar]
Lilius 2015
- Lilius TO, Jokinen V, Neuvonen MS, Niemi M, Kalso EA, Rauhala PV. Ketamine coadministration attenuates morphine tolerance and leads to increased brain concentrations of both drugs in the rat. British Journal of Pharmacology 2015;172(11):2799‐813. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lloyd‐Williams 2000
- Lloyd‐Williams M. Ketamine for cancer pain. Journal of Pain and Symptom Management 2000;19(2):79‐80. [DOI] [PubMed] [Google Scholar]
Lossignol 1992
- Lossignol D, Gil T, Rossi C, Sosnowski M, Obiols M, Vanmeerhaeghe B, et al. Cancer pain: A new concept. International Symposium on Supportive Care In Cancer; Bruges, Belgium. 8 ‐ 11 June, 1992:59.
Lossignol 1999
- Lossignol D, Obiols M, Body JJ. Ketamine and morphine in cancer pain [abstract]. 9th World Congress on Pain; Vienna, Austria. Seattle: International Association for the Study of Pain, 22 ‐ 27 August, 1999.
Mao 1995
- Mao J, Price DD, Mayer DJ. Mechanisms of hyperalgesia and morphine tolerance: a current view of their possible interactions. Pain 1995;62:259‐74. [DOI] [PubMed] [Google Scholar]
Mayer 1995
- Mayer DJ, Mao J, Price DD. The association of neuropathic pain, morphine tolerance and dependence, and the translocation of protein kinase C. NIDA‐Research Monographs 1995;147:269‐98. [PubMed] [Google Scholar]
McQuay 1998
- McQuay H, Moore R. An Evidence‐Based Resource for Pain Relief. Oxford: Oxford University Press, 1998. [Google Scholar]
Mercadante 1995
- Mercadante S, Lodi F, Sapio M, Calligara M, Serretta R. Long‐term ketamine subcutaneous continuous‐infusion in neuropathic cancer pain. Journal of Pain and Symptom Management 1995;10(7):564‐8. [DOI] [PubMed] [Google Scholar]
Mitchell 1999
- Mitchell AC. Generalized hyperalgesia and allodynia following abrupt cessation of subcutaneous ketamine infusion. Palliative Medicine 1999;13(5):427‐8. [DOI] [PubMed] [Google Scholar]
Moore 2008
- Moore RA, Barden J, Derry S, McQuay HJ. Managing potential publication bias. In: McQuay HJ, Kalso E, Moore RA editor(s). Systematic Reviews in Pain Research: Methodology Refined. Seattle: IASP Press, 2008:15‐24. [Google Scholar]
Ogawa 1994
- Ogawa S, Kanamaru T, Noda K, Saeki S, Katsumata N, Kato J, et al. Intravenous microdrip infusion of ketamine in subanaesthetic doses for intractable terminal cancer pain. Pain Clinic 1994;7(2):125‐9. [Google Scholar]
Oshima 1990
- Oshima E, Tei K, Kayazawa H, Urabe N. Continuous subcutaneous injection of ketamine for cancer pain. Canadian Journal of Anesthesia 1990;37(3):385‐6. [DOI] [PubMed] [Google Scholar]
PaPaS 2012
- Pain, Palliative and Supportive Care Group. Authoring or assessing a Cochrane Protocol, Review, or Review Update for the PaPaS Review Group (AUREF). papas.cochrane.org/papas‐documents (accessed 6 April 2017).
Parada 1971
- Parada JF. Treatment of pain in cancer with ketamine hydrochloride. 13th Congreso Argentino de Anestesiologia, 1971 vol. 1; 101‐5. Buenos Aires, Oct 1971.
Peltoniemi 2016
- Peltoniemi MA, Hagelberg NM, Olkkola KT, Saari TI. Ketamine: A review of clinical pharmacokinetics and pharmacodynamics in anaesthesia and pain therapy. Clinical Pharmacokinetics 2016;55(9):1059‐77. [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre; The Cochrane Collaboration. Review Manager. Version 5.3.. Copenhagen: The Nordic Cochrane Centre; The Cochrane Collaboration, 2014.
Sawynok 2014
- Sawynok J. Topical and peripheral ketamine as an analgesic. Anesthesia and Analgesia 2014;119(1):170‐8. [DOI] [PubMed] [Google Scholar]
Smith 2000
- Smith LA, Oldman AD, McQuay HJ, Moore RA. Teasing apart quality and validity in systematic reviews: an example from acupuncture trials in chronic neck and back pain. Pain 2000;86:119‐32. [DOI] [PubMed] [Google Scholar]
Sosnowski 1993
- Sosnowski M, Lossignol D, Fodderie L. Reversibility of opioid insensitive pain (abstract). World Congress on Pain. Seattle: IASP Publications, 1993:16.
Stannard 2005
- Stannard CF, Booth S. Churchill's Pocketbook of Pain. Second Edition. Edinburgh: Churchill Livingstone, 2005:384. [Google Scholar]
Stotz 1999
- Stotz M, Oehen HP, Gerber H. Histological findings after long‐term infusion of intrathecal ketamine for chronic pain: a case report. Journal of Pain and Symptom Management 1999;18(3):223‐8. [DOI] [PubMed] [Google Scholar]
Tarumi 2000
- Tarumi Y, Watanabe S, Bruera E, Ishitani K. High dose ketamine in the management of cancer‐related neuropathic pain. Journal of Pain and Symptom Management 2000;19(6):405‐7. [DOI] [PubMed] [Google Scholar]
Trujillo 1991
- Trujillo KA, Akil H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK‐801. Science 1991;251(4989):85‐7. [DOI] [PubMed] [Google Scholar]
Twycross 2009
- Twycross R, Wilcock A, Toller CS. Symptom Management in Advanced Cancer. Fourth. Oxford: Palliativedrugs.com Ltd, 2009. [Google Scholar]
van den Beuken‐van Everdingen 2017
- Beuken‐van Everdingen MHJ, Graeff A, Jongen JLM, Dijkstra D, Mostovaya I, Vissers C, the national guideline working group "Diagnosis treatment of cancer pain". Pharmacological treatment of pain in cancer patients: the role of adjuvant analgesics, a systematic review. Pain Practice 2017;17(3):409‐19. [DOI] [PubMed] [Google Scholar]
Ventura 1993
- Ventura GJ, Blacklock DM. Intravenous ketamine HCL for treatment of intractable neuropathic cancer pain (INCP). Proceedings of the Annual Meeting of the American Society of Clinical Oncology 1993;12:A1528. [Google Scholar]
Whizar‐Lugo 1987
- Whizar‐Lugo V, Cortez Gomez C. Epidural ketamine vs epidural morphine in severe cancer pain. Pain 1987;4 (Suppl):S142. [Google Scholar]
Willetts 1990
- Willetts J, Balster RL, Leander D. The behavioral pharmacology of NMDA receptor antagonists. Trends in Pharmacological Sciences 1990;11:423‐8. [DOI] [PubMed] [Google Scholar]
Wood 1997
- Wood T, Sloan R. Successful use of ketamine for central pain. Palliative Medicine 1997;11:57‐8. [DOI] [PubMed] [Google Scholar]
Woolf 1987
- Woolf TF, Adams JD. Biotransformation of ketamine, (Z)‐6‐hydroxyketamine, and (E)‐6‐hydroxyketamine by rat, rabbit, and human liver microsomal preparations. Xenobiotica 1987;17(7):839‐47. [DOI] [PubMed] [Google Scholar]
Yanagihara 2001
- Yanagihara Y, Kariya S, Ohtani M, Uchino K, Aoyama T, Yamamura Y, et al. Involvement of CYP2B6 in n‐demethylation of ketamine in human liver microsomes. Drug Metabolism and Disposition 2001;29(6):887‐90. [PubMed] [Google Scholar]
Zanos 2016
- Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, et al. NMDAR inhibition‐independent antidepressant actions of ketamine metabolites. Nature 2016;533(7604):481‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Bell 2003
- Bell R, Eccleston C, Kalso E. Ketamine as an adjuvant to opioids for cancer pain. Cochrane Database of Systematic Reviews 2003, Issue 1. [DOI: 10.1002/14651858.CD003351] [DOI] [PubMed] [Google Scholar]
Bell 2012b
- Bell RF, Eccleston C, Kalso EA. Ketamine as an adjuvant to opioids for cancer pain. Cochrane Database of Systematic Reviews 2012, Issue 11. [DOI: 10.1002/14651858.CD003351.pub2] [DOI] [PubMed] [Google Scholar]