

Abstract
Background
The projected rise in the incidence of type 2 diabetes mellitus (T2DM) could develop into a substantial health problem worldwide. Whether dipeptidyl‐peptidase (DPP)‐4 inhibitors or glucagon‐like peptide (GLP)‐1 analogues are able to prevent or delay T2DM and its associated complications in people at risk for the development of T2DM is unknown.
Objectives
To assess the effects of DPP‐4 inhibitors and GLP‐1 analogues on the prevention or delay of T2DM and its associated complications in people with impaired glucose tolerance, impaired fasting blood glucose, moderately elevated glycosylated haemoglobin A1c (HbA1c) or any combination of these.
Search methods
We searched the Cochrane Central Register of Controlled Trials; MEDLINE; PubMed; Embase; ClinicalTrials.gov; the World Health Organization (WHO) International Clinical Trials Registry Platform; and the reference lists of systematic reviews, articles and health technology assessment reports. We asked investigators of the included trials for information about additional trials. The date of the last search of all databases was January 2017.
Selection criteria
We included randomised controlled trials (RCTs) with a duration of 12 weeks or more comparing DPP‐4 inhibitors and GLP‐1 analogues with any pharmacological glucose‐lowering intervention, behaviour‐changing intervention, placebo or no intervention in people with impaired fasting glucose, impaired glucose tolerance, moderately elevated HbA1c or combinations of these.
Data collection and analysis
Two review authors read all abstracts and full‐text articles and records, assessed quality and extracted outcome data independently. One review author extracted data which were checked by a second review author. We resolved discrepancies by consensus or the involvement of a third review author. For meta‐analyses, we planned to use a random‐effects model with investigation of risk ratios (RRs) for dichotomous outcomes and mean differences (MDs) for continuous outcomes, using 95% confidence intervals (CIs) for effect estimates. We assessed the overall quality of the evidence using the GRADE instrument.
Main results
We included seven completed RCTs; about 98 participants were randomised to a DPP‐4 inhibitor as monotherapy and 1620 participants were randomised to a GLP‐1 analogue as monotherapy. Two trials investigated a DPP‐4 inhibitor and five trials investigated a GLP‐1 analogue. A total of 924 participants with data on allocation to control groups were randomised to a comparator group; 889 participants were randomised to placebo and 33 participants to metformin monotherapy. One RCT of liraglutide contributed 85% of all participants. The duration of the intervention varied from 12 weeks to 160 weeks. We judged none of the included trials at low risk of bias for all 'Risk of bias' domains and did not perform meta‐analyses because there were not enough trials.
One trial comparing the DPP‐4 inhibitor vildagliptin with placebo reported no deaths (very low‐quality evidence). The incidence of T2DM by means of WHO diagnostic criteria in this trial was 3/90 participants randomised to vildagliptin versus 1/89 participants randomised to placebo (very low‐quality evidence). Also, 1/90 participants on vildagliptin versus 2/89 participants on placebo experienced a serious adverse event (very low‐quality evidence). One out of 90 participants experienced congestive heart failure in the vildagliptin group versus none in the placebo group (very low‐quality evidence). There were no data on non‐fatal myocardial infarction, stroke, health‐related quality of life or socioeconomic effects reported.
All‐cause and cardiovascular mortality following treatment with GLP‐1 analogues were rarely reported; one trial of exenatide reported that no participant died. Another trial of liraglutide 3.0 mg showed that 2/1501 in the liraglutide group versus 2/747 in the placebo group died after 160 weeks of treatment (very low‐quality evidence).
The incidence of T2DM following treatment with liraglutide 3.0 mg compared to placebo after 160 weeks was 26/1472 (1.8%) participants randomised to liraglutide versus 46/738 (6.2%) participants randomised to placebo (very low‐quality evidence). The trial established the risk for (diagnosis of) T2DM as HbA1c 5.7% to 6.4% (6.5% or greater), fasting plasma glucose 5.6 mmol/L or greater to 6.9 mmol/L or less (7.0 mmol/L or greater) or two‐hour post‐load plasma glucose 7.8 mmol/L or greater to 11.0 mmol/L (11.1 mmol/L). Altogether, 70/1472 (66%) participants regressed from intermediate hyperglycaemia to normoglycaemia compared with 268/738 (36%) participants in the placebo group. The incidence of T2DM after the 12‐week off‐treatment extension period (i.e. after 172 weeks) showed that five additional participants were diagnosed T2DM in the liraglutide group, compared with one participant in the placebo group. After 12‐week treatment cessation, 740/1472 (50%) participants in the liraglutide group compared with 263/738 (36%) participants in the placebo group had normoglycaemia.
One trial used exenatide and 2/17 participants randomised to exenatide versus 1/16 participants randomised to placebo developed T2DM (very low‐quality evidence). This trial did not provide a definition of T2DM. One trial reported serious adverse events in 230/1524 (15.1%) participants in the liraglutide 3.0 mg arm versus 96/755 (12.7%) participants in the placebo arm (very low quality evidence). There were no serious adverse events in the trial using exenatide. Non‐fatal myocardial infarction was reported in 1/1524 participants in the liraglutide arm and in 0/55 participants in the placebo arm at 172 weeks (very low‐quality evidence). One trial reported congestive heart failure in 1/1524 participants in the liraglutide arm and in 1/755 participants in the placebo arm (very low‐quality evidence). Participants receiving liraglutide compared with placebo had a small mean improvement in the physical component of the 36‐item Short Form scale showing a difference of 0.87 points (95% CI 0.17 to 1.58; P = 0.02; 1 trial; 1791 participants; very low‐quality evidence). No trial evaluating GLP‐1‐analogues reported data on stroke, microvascular complications or socioeconomic effects.
Authors' conclusions
There is no firm evidence that DPP‐4 inhibitors or GLP‐1 analogues compared mainly with placebo substantially influence the risk of T2DM and especially its associated complications in people at increased risk for the development of T2DM. Most trials did not investigate patient‐important outcomes.
Plain language summary
Dipeptidyl‐peptidase (DPP)‐4 inhibitors and glucagon‐like peptide (GLP)‐1 analogues for prevention or delay of type 2 diabetes mellitus
Review question
Are the glucose‐lowering medicines, DPP‐4 inhibitors (e.g. linagliptin or vildagliptin) and GLP‐1 analogues (e.g. exenatide or liraglutide) able to prevent or delay the development of type 2 diabetes and its associated complications in people at risk for the development of type 2 diabetes?
Background
DPP‐4 inhibitors and GLP‐1 analogues are widely used to treat people with type 2 diabetes. People with moderately elevated blood glucose are said to be at an increased risk for developing type 2 diabetes (often referred to as 'prediabetes'). It is currently not known whether DPP‐4 inhibitors or GLP‐1 analogues should be prescribed for people with raised blood glucose levels who do not have type 2 diabetes. We wanted to find out whether these medicines could prevent or delay type 2 diabetes in people at increased risk. We also wanted to know the effects on patient‐important outcomes such as complications of diabetes (e.g. kidney and eye disease, heart attacks, strokes), death from any cause, health‐related quality of life (a measure of a person's satisfaction with their life and health) and side effects of the medicines.
Study characteristics
Participants had to have blood glucose levels higher than considered normal, but below the glucose levels that are used to diagnose type 2 diabetes mellitus. We found seven randomised controlled trials (clinical studies where people are randomly put into one of two or more treatment groups) with 2702 participants. The duration of the treatments varied from 12 weeks to 160 weeks. One study investigating liraglutide dominated the evidence (2285/2702 participants). The participants in this study were overweight or obese.
This evidence is up to date as of January 2017.
Key results
DPP‐4 inhibitors did not reduce the risk of developing type 2 diabetes compared with placebo (a dummy medicine). In the big study investigating the GLP‐1‐analogue liraglutide, given in a dose used for obese people (3.0 mg), the development of type 2 diabetes was delayed: 26/1472 (1.8%) participants randomised to liraglutide compared with 46/738 (6.2%) participants randomised to placebo developed type 2 diabetes after 160 weeks. On the other side, 970/1472 (66%) participants randomised to liraglutide compared with 268/738 (36%) participants randomised to placebo switched back to normal glucose levels. This study was extended for another 12 weeks without treatment and five additional participants developed diabetes in the liraglutide group, compared with one participant in the placebo group. After the 12 weeks without treatment, 740/1472 (50%) participants in the liraglutide group compared with 263/738 (36%) participants in the placebo group had glucose levels considered as normal. This means that to keep chances high to prevent type 2 diabetes in people at risk one probably needs to continuously take this drug. Of note, serious adverse events (e.g. defined as hospitalisation or a hazard putting the participant at risk, such as an interaction with another medicine) happened more often following liraglutide treatment (230/1524 (15%) participants in the liraglutide group and 96/755 (13%) participants in the placebo group) and it is unclear whether taking this drug is safe in the long term.
We detected neither an advantage nor a disadvantage of DPP‐4 inhibitors or GLP‐1 analogues in relation to non‐fatal heart attacks, non‐fatal strokes or heart failure. Our included studies did not report on other complications of diabetes such as kidney or eye disease. The effects on health‐related quality of life were inconclusive. In the included studies, very few participants died and there was no apparent relation to treatment.
Future studies should investigate more patient‐important outcomes like complications of diabetes and especially the side effects of the medications, because we do not know for sure whether 'prediabetes' is just a condition arbitrarily defined by a laboratory measurement, is in fact a real risk factor for type 2 diabetes mellitus and whether treatment of this condition translates into better patient‐important outcomes.
Quality of the evidence
All included trials had deficiencies in the way they were conducted or how key items were reported. For the individual comparisons, the number of participants was small, resulting in a high risk of random errors (play of chance).
Summary of findings
Background
Description of the condition
'Prediabetes', 'borderline diabetes', the 'prediabetic stage', 'high risk of diabetes', 'dysglycaemia' or 'intermediate hyperglycaemia' are often characterised by various measurements of elevated blood glucose concentrations, such as isolated impaired fasting glucose (IFG), isolated impaired glucose tolerance (IGT), isolated elevated glycosylated haemoglobin A1c (HbA1c) or combinations thereof (WHO/IDF 2006). These elevated blood glucose levels that indicate hyperglycaemia are too high to be considered normal, but are below the diagnostic threshold for type 2 diabetes mellitus (T2DM). Therefore, due to the continuous glycaemic spectrum from the normal to the diabetic stage, a sound evidence base is needed to define glycaemic thresholds for people at high risk of T2DM. The different terms used to describe various stages of hyperglycaemia may cause people to have different emotional reactions. For example, the term 'prediabetes' may imply (at least for lay people) that the disease diabetes is unavoidable, whereas (high) risk of diabetes has the positive connotation of possibly being able to avoid the disease altogether. In addition to the disputable construct of intermediate health states termed 'prediseases' (Viera 2011), many people may associate the label 'prediabetes' with dire consequences. Alternatively, any diagnosis of 'prediabetes' may be an opportunity to review, for example, eating habits and physical activity levels, thus enabling affected people to actively change their way of life.
The American Diabetes Association (ADA) and the World Health Organization (WHO) established the most commonly used criteria to define people who are at a high risk of developing T2DM. IGT was the first glycaemic measurement used by the US National Diabetes Data Group to define the prediabetes stage (NDDG 1979). It is based on the measurement of plasma glucose two hours after ingestion of glucose 75 g. The dysglycaemic range is defined as plasma glucose concentrations between 7.8 mmol/L and 11.1 mmol/L (140 mg/dL and 200 mg/dL) two hours after the glucose load. Studies indicate that IGT is caused by insulin resistance and defective insulin secretion (Abdul‐Ghani 2006; Jensen 2002). In 1997, the ADA and later the WHO introduced the IFG concept to define 'prediabetes' and intermediate hyperglycaemia (ADA 1997; WHO 1999). The initial definition of IFG was fasting blood glucose concentrations between 6.1 mmol/L and 6.9 mmol/L (110 mg/dL and 125 mg/dL). Later, the ADA reduced the lower threshold for defining IFG to 5.6 mmol/L (100 mg/dL) (ADA 2003). However, the WHO did not endorse this lower cut‐off point for IFG to define 'prediabetes' (WHO/IDF 2006). IFG seems to be associated with β‐cell dysfunction (impaired insulin secretion) and an increase in the hepatic glucose output (DeFronzo 1989). More recently, HbA1c has been introduced to identify people at high risk of developing T2DM. In 2009, the International Expert Committee (IEC) suggested certain HbA1c ranges to identify people at a high risk of T2DM. People with HbA1c measurements between 6.0% and 6.4% fulfilled this criterion (IEC 2009). Shortly afterwards, the ADA redefined this HbA1c level as 5.7% to 6.4% to identify people at a high risk of developing T2DM (ADA 2010), a decision not endorsed by WHO, IEC or other organisations. Unlike IFG and IGT, HbA1c reflects longer‐term glycaemic control, that is how a person's blood glucose concentrations have been during the preceding two to three months (Inzucchi 2012).
In 2010, the International Diabetes Federation (IDF) estimated the prevalence of IGT to be 343 million people, and this is predicted to increase to 642 million people by 2040 (IDF 2015). Studies have shown poor correlations between HbA1c and IFG/IGT (Gosmanov 2014; Selvin 2011). Notably, the various glycaemic tests do not seem to identify the same people, as there is an imperfect overlap among the glycaemic modalities available to define intermediate hyperglycaemia (Gosmanov 2014; Selvin 2011). The risk of progression from people at risk to T2DM depends on the diagnostic criteria used to identify the risk. Some people with intermediate risk will never develop T2DM, and some people will return to normoglycaemia. IGT is often accepted as the best glycaemic variable for risk to predict progression to T2DM. However, studies indicate that less than half of the people defined as 'prediabetic' by means of IGT will develop T2DM in the following 10 years (Morris 2013). IFG and HbA1c are thought to predict a different risk spectrum for developing T2DM (Cheng 2006; Morris 2013). Most importantly, dysglycaemia is commonly an asymptomatic condition, and naturally often remains 'undiagnosed' (CDC 2015).
It has not been clarified whether any particular intervention, especially glucose‐lowering drugs, should be recommended for people at risk for T2DM (Yudkin 2014). Trials have indicated that the progression to T2DM is reduced, or possibly only delayed, with behavioural interventions (increased physical activity, dietary changes, or both) (Diabetes Prevention Program 2009; Knowler 2002; Tuomilehto 2001). One meta‐analysis of 22 trials with interventions that changed behaviour in people at high risk of T2DM concluded that the effect of these interventions on longer‐term diabetes prevention is unclear (Dunkley 2014). Therefore, more research is needed to establish optimal strategies for reducing T2DM with behavioural approaches (Dunkley 2014).
International diabetes associations and clinicians do not generally accept the prescription of pharmacological glucose‐lowering interventions for the prevention of T2DM. Several groups of pharmacological glucose‐lowering interventions have been investigated for people at risk of T2DM. Some findings indicate that the progression to T2DM is reduced or may only be delayed (Knowler 2002; Diabetes Prevention Program 2009). However, the ADA recommends metformin for people at risk of T2DM, especially for those with a body mass index (BMI) over 35 kg/m², aged less than 60 years, women with prior gestational diabetes mellitus, people with rising HbA1c despite lifestyle intervention, or a combination of these (ADA 2017).
Description of the intervention
Sitagliptin was the first dipeptidyl‐peptidase (DPP)‐4 inhibitor approved as a glucose‐lowering intervention for people with T2DM (FDA 2006). Since then, several other types of DPP‐4 inhibitors have been approved for T2DM, such as alogliptin, anagliptin, linagliptin, saxagliptin, teneligliptin and vildagliptin. The DPP‐4 inhibitors are administered orally.
Exenatide was the first glucagon‐like peptide 1 (GLP‐1) analogue approved as a glucose‐lowering intervention for people with T2DM (FDA 2005). Since then, several other types of GLP‐1 analogues have been approved. The GLP‐1 analogues can be categorised as either short‐acting (e.g. exenatide) or long‐acting (e.g. liraglutide). Currently available GLP‐1 analogues are administered subcutaneously. Peroral GLP‐1 analogues are in the pipeline (NCT02161588).
For people with T2DM, GLP‐1 analogues and DPP‐4 inhibitors can be prescribed as monotherapy or in combination with existing glucose‐lowering interventions (ADA 2014). Currently, the GLP‐1 analogues or DPP‐4 inhibitors are not recommended for people with intermediate hyperglycaemia (ADA 2014). However, it has been shown that people with IGT have alterations in circulating incretin hormones (Rask 2004).
Adverse effects of the intervention
The most common adverse effects of GLP‐1 analogues are gastrointestinal disturbances and nausea. Nausea and vomiting associated with the GLP‐1 analogues are dose‐dependent and appear to decrease over time (Reid 2014). For DPP‐4 inhibitors, the most common adverse effects reported are gastrointestinal disturbances (Reid 2014).
Both the GLP‐1 analogues and the DPP‐4 inhibitors have been suggested to increase risk of pancreatitis and potentially pancreatic cancer (Nagel 2015). The incidence of acute pancreatitis has been low in large‐scale randomised controlled trials (RCTs), and was slightly increased compared with placebo (Scirica 2013; Zannad 2015). The US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have made comprehensive evaluations of the safety of the incretin‐based interventions based on post‐marketing reports of pancreatitis and pancreatic cancer in people with T2DM. These post‐marketing data have not been convincing regarding these adverse effects (Egan 2014). Nevertheless, current opinion holds that DPP‐4 inhibitors should not be prescribed to people at risk of or having existing pancreatitis (De Heer 2014). Besides, the DPP‐4 inhibitors have been reported to increase the risk of heart failure; however, this is still not clarified (Elgendy 2017; Rehman 2017). However, one large‐scale trial investigating the DPP‐4 inhibitor, saxagliptin, was associated with an increased risk of heart failure leading to hospitalisation (hazard ratio (HR) 1.27, 95% confidence interval (CI) 1.07 to 1.51) (Scirica 2013).
Results of systematic reviews are conflicting and adverse effects could vary with the type of DPP‐4 inhibitor used (Elgendy 2017).
An increased risk of thyroid cell neoplasm has been observed in rodents exposed to GLP‐1 analogues. However, this increased risk has not been observed in people (Reid 2014).
How the intervention might work
A two‐ to three‐fold greater increase in plasma insulin is observed when glucose is administered orally compared with an intravenous application (Nauck 1986). This phenomenon is called the incretin effect and accounts for approximately 70% to 80% of total insulin release after orally administered glucose (Nauck 1986). The incretin effect is mediated mainly by GLP‐1 and glucose‐dependent insulinotropic polypeptide (GIP) (Nauck 1986). GLP‐1 is a 30‐amino acid polypeptide produced in the intestinal L cell upon processing of the precursor proglucagon (Bell 2013). GLP‐1 and GIP exert powerful and strictly glucose‐dependent insulinotropic activity via specific GLP‐1 and GIP receptors in the plasma membrane of pancreatic β‐cells (Holst 2007). In addition, GLP‐1 reduces glucagon secretion, an effect that might be as clinically important as the insulinotropic effect of GLP‐1 (Holst 2007; Orskov 1988). Furthermore, GLP‐1 reduces food intake (most likely via activation of GLP‐1 receptors in the central nervous system) and delays gastric emptying, whereby postprandial glucose excursions and food intake are reduced (Holst 2007). Lastly, in animal models GLP‐1 promotes β‐cell growth and inhibits β‐cell apoptosis (Holst 2007; Holz 1993). During the progression from normal glucose tolerance to insulin resistance and eventually to T2DM, some studies have shown that plasma GLP‐1 concentrations decline (Rask 2004; Toft‐Nielsen 2001). However, one meta‐analysis suggested that people with T2DM do not exhibit reduced GLP‐1 secretion in response to an oral glucose tolerance test (OGTT) or meal test (Calanna 2013).
Native GLP‐1 and GIP are rapidly inactivated (half‐life: GLP‐1: 2 minutes; GIP: 5 to 7 minutes) by the enzyme DPP‐4, expressed in many tissues (e.g. kidney and intestine) (Creutzfeldt 1979). DPP‐4 inhibitors exert competitive inhibition of DPP‐4 and thereby increase the concentrations of active (endogenous) GLP‐1 and GIP (Holst 2007). GLP‐1 analogues are resistant to DPP‐4 degradation (Holst 2007).
Why it is important to do this review
There has been an increased focus on the prevention or delay of T2DM with non‐pharmacological interventions and glucose‐lowering medications. Currently, several trials are being conducted to clarify whether the progression from an at‐risk status to T2DM can be stopped or postponed with glucose‐lowering agents (ClinicalTrials.gov). However, a more important issue for people with dysglycaemia is whether these interventions reduce the risk of death or complications ‐ especially cardiovascular disease ‐ related to T2DM.
Objectives
To assess the effects of DPP‐4 inhibitors and GLP‐1 analogues on the prevention or delay of T2DM and its associated complications in people with impaired glucose tolerance, impaired fasting blood glucose, moderately elevated glycosylated haemoglobin A1c (HbA1c) or any combination of these.
Methods
Criteria for considering studies for this review
Types of studies
We included RCTs.
Types of participants
We included non‐diabetic people with increased risk of T2DM.
Diagnostic criteria for people at risk of type 2 diabetes mellitus development
To be consistent with changes to the classification of, and diagnostic criteria for, intermediate hyperglycaemia (IFG, IGT and elevated HbA1c) over the years, the diagnosis should be established using the standard criteria valid at the trial start (e.g. ADA 1997; ADA 2010; NDDG 1979; WHO 1999). Ideally, the diagnostic criteria should have been described. If necessary, we used the trial authors' definition of risk but we contacted trial authors for additional information. Differences in the glycaemic measurements used to define risk may introduce substantial heterogeneity. Therefore, we planned to subject the diagnostic criteria to a subgroup analysis.
Types of interventions
We planned to investigate the following comparisons of DPP‐4 inhibitors or GLP‐1 analogues versus all other pharmacological glucose‐lowering interventions, behaviour‐changing interventions, placebo or no intervention.
Intervention
DPP‐4 inhibitors as monotherapy.
DPP‐4 inhibitors as a part of a combination therapy.
GLP‐1 analogues as monotherapy.
GLP‐1 analogues as a part of a combination therapy.
Comparator
Any pharmacological glucose‐lowering intervention (e.g. acarbose, metformin, sulphonylurea) compared with DPP‐4 inhibitors as monotherapy or GLP‐1 analogues as monotherapy.
Any pharmacological glucose‐lowering agent (e.g. acarbose, metformin, sulphonylurea) compared with DPP‐4 inhibitors as a part of a combination therapy or GLP‐1 analogues as a part of a combination therapy if this glucose‐lowering agent was the same in both the intervention and comparator groups (e.g. DPP‐4 inhibitor plus metformin versus metformin).
Behaviour‐changing interventions (e.g. diet, exercise, diet and exercise) compared with DPP‐4 inhibitors as monotherapy or GLP‐1 analogues as monotherapy.
Placebo compared with DPP‐4 inhibitors as monotherapy or GLP‐1 analogues as monotherapy.
No intervention compared with DPP‐4 inhibitors as monotherapy or GLP‐1 analogues as monotherapy.
Other concomitant interventions (e.g. educational programmes or additional pharmacotherapy) had to be the same in both the intervention and comparator groups to establish fair comparisons.
Minimum duration of intervention
We included trials with a duration of the intervention of 12 weeks or more.
Exclusion criteria
We excluded trials of people diagnosed with the 'metabolic syndrome' because this is a special population which is not representative of people with only intermediate hyperglycaemia. Also, the composite of risk indicators such as elevated blood lipids, insulin resistance, obesity and hypertension which is termed 'metabolic syndrome' is of doubtful clinical usefulness and uncertain distinct disease entity. However, should we identify trials investigating participants with any definition of the metabolic syndrome, we summarised some basic trial information in an additional table.
We excluded trials evaluating participants with intermediate hyperglycaemia in combination with another condition (e.g. cystic fibrosis).
We excluded trials evaluating participants with intermediate hyperglycaemia because of other medical interventions (e.g. glucocorticoids).
We tried to include trials explicitly describing that a portion of the included participants had intermediate hyperglycaemia. We contacted the investigators to obtain separate data on the group with intermediate hyperglycaemia and include these in the meta‐analyses.
We included trials in obese people and participants with previous gestational diabetes, if trial investigators described that the participants had intermediate hyperglycaemia.
We included a trial even if it did not report one or more of our primary or secondary outcome measures in the publication. If a trial did not report any of our primary or secondary outcomes, we included this trial and contacted the corresponding trial author for supplementary data. We listed information about trials with a duration of the intervention shorter than 12 weeks in Appendix 1.
Some trials evaluated GLP‐1 analogues in doses recommended for achieving a weight‐reducing effect rather than a glucose‐lowering effect. We included such trials and performed subgroup analyses according to the dose of GLP‐1 analogues.
Types of outcome measures
Primary outcomes
All‐cause mortality.
Incidence of T2DM.
Serious adverse events.
Secondary outcomes
Cardiovascular mortality.
Non‐fatal myocardial infarction.
Non‐fatal stroke.
Congestive heart failure.
Amputation of lower extremity.
Blindness or severe vision loss.
End‐stage renal disease.
Non‐serious adverse events.
Hypoglycaemia.
Health‐related quality of life (HRQoL).
Time to progression to T2DM.
Measures of blood glucose control.
Socioeconomic effects.
Method and timing of outcome measurement
All‐cause mortality: defined as death from any cause. Measured at any time of the intervention and during follow‐up.
Incidence of T2DM and time to progression to T2DM: defined according to diagnostic criteria valid at the time the diagnosis was established using the standard criteria valid at the time of the trial commencing (e.g. ADA 2008; WHO 1998). If necessary, we used the trial authors' definition of T2DM. Measured at the end of the intervention and the end of follow‐up. We also investigated regression from intermediate hyperglycaemia back to normoglycaemia.
Serious adverse events: defined according to the International Conference on Harmonization Guidelines as any event that led to death, was life‐threatening, required hospitalisation or prolongation of existing hospitalisation, resulted in persistent or significant disability, or any important medical event which may have had jeopardised the person or required intervention to prevent it (ICH 1997), or as reported in trials. Measured at any time of the intervention and during follow‐up.
Cardiovascular mortality, non‐fatal myocardial infarction, non‐fatal stroke, congestive heart failure, amputation of lower extremity, blindness or severe vision loss, hypoglycaemia (mild, moderate, severe/serious): defined as reported in trials. Measured at the end of the intervention and at the end of follow‐up.
End‐stage renal disease: defined as dialysis, renal transplantation or death due to renal disease. Measured at the end of the intervention and at the end of follow‐up.
Non‐serious adverse events: defined as the number of participants with any untoward medical occurrence not necessarily having a causal relationship with the intervention. Measured at any time of the intervention and during follow‐up.
HRQoL: defined as mental and physical HRQoL as separate domains and combined, evaluated by a validated instrument such as the 36‐item Short‐Form (SF‐36). Measured at the end of the intervention and at the end of follow‐up.
Measures of blood glucose control: fasting blood glucose, blood glucose two hours after ingestion of glucose 75 g and HbA1c measurements. Measured at the end of the intervention and at the end of follow‐up.
Socioeconomic effects: for example, costs of the intervention, absence from work and medication consumption. Measured at the end of the intervention and at the end of follow‐up.
Specification of key prognostic variables
Age.
Gender.
Equity issues (access to health care, social determinants).
Ethnicity.
Hypertension.
Cardiovascular disease.
Obesity.
Previous gestational diabetes.
'Summary of findings' table
We present a 'Summary of findings' table to report the following outcomes, listed according to priority.
All‐cause mortality.
Incidence of T2DM.
Serious adverse events.
Cardiovascular mortality.
Non‐fatal myocardial infarction/stroke and congestive heart failure.
HRQoL.
Socioeconomic effects.
Search methods for identification of studies
Electronic searches
We searched the following sources from inception of each database to the specified date, and placed no restrictions on the language of publication.
Cochrane Central Register of Controlled Trials (26 January 2017).
Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations, Ovid MEDLINE(R) Daily and Ovid MEDLINE(R) 1946 to 26 January 2017).
PubMed (subsets not available on Ovid) (12 February 2016).
Embase (1974 to 2017 week 4, 26 January 2017).
ClinicalTrials.gov (26 January 2017).
WHO International Clinical Trials Registry Platform (ICTRP) Search Portal (apps.who.int/trialsearch/) (26 January 2017).
We continuously used a MEDLINE (via OvidSP) email alert service established by the Cochrane Metabolic and Endocrine Disorders (CMED) Group to identify newly published trials using the same search strategy as described for MEDLINE (for details on search strategies, see Appendix 2).
We obtained evaluations of all relevant non‐English articles.
Searching other resources
We searched the reference lists of retrieved included trials, systematic reviews, meta‐analyses and health technology assessment reports for other potentially eligible trials or ancillary publications. In addition, we contacted authors of included trials to identify any additional information on the retrieved trials and if further trials existed, that we may have missed.
As none of the existing DPP‐4 inhibitors or GLP‐1 analogues is approved in glucose‐lowering doses for intermediate hyperglycaemia, we did not search databases of the regulatory agencies (EMA, US FDA). However, GLP‐1 analogues in higher doses are approved as a weight‐reducing intervention in obese people with intermediate hyperglycaemia. Therefore, we searched EMA and FDA for GLP‐1 analogues approved as a weight‐reducing intervention.
Data collection and analysis
Selection of studies
Two review authors (BH and DS) independently scanned the abstract, title, or both, of every record we retrieved in the literature searches, to determine which trials should be assessed further. We investigated the full text of all potentially relevant articles. We resolved discrepancies through consensus or by recourse to a third review author (BR). We prepared a flow diagram of the number of trials identified and excluded at each stage in accordance with the PRISMA flow diagram of trial selection (Liberati 2009).
Data extraction and management
For trials that fulfilled the inclusion criteria, two review authors (BH and DS) independently extracted outcome data and assessed the risk of bias. Key characteristics of participants and interventions were extracted by one review author (BH) and controlled by another (DS). We reported data on efficacy outcomes and adverse events using standard data extraction sheets from the CMED Group. We resolved any disagreements by discussion or, if required, by consultation with a third review author (BR) (for details, see Characteristics of included studies table; Table 3; Appendix 3; Appendix 4; Appendix 5; Appendix 6; Appendix 7; Appendix 8; Appendix 9; Appendix 10; Appendix 11; Appendix 12; Appendix 13; Appendix 14; Appendix 15; Appendix 17; Appendix 18).
1. Overview of trial populations.
| Trial (design) | Intervention and comparator | Description of power and sample size calculation and handling of missing data | Screened/eligible (n) | Randomised (n) | ITT (n) | Analysed (n) | Finishing trial (n) | Randomised finishing trial (%) | Follow‐up (extended follow‐up)a |
|
Ariel 2014 (parallel RCT) |
I: liraglutide 1.8 mg, once daily | "Targeted sample size of 30 subjects in each group provided 90% power to detect a 20% difference (2.2 mmol/L) in SSPG concentration. With 24 subjects per group, there was 82% power to detect a 20% difference. A difference of 20% was chosen because this is the degree of difference in SSPG concentration seen with modest weight loss of 7%." "Only subjects who had end‐of‐study testing were included in the analyses." |
161 | 35 | 24 | 24 | 24 | 68.6 | 14 weeks |
| C: placebo, once daily | 33 | 27 | 27 | 27 | 81.8 | ||||
| total: | 68 | 51 | 51 | 51 | 75 | ||||
|
Kelly 2012 (parallel RCT) |
I: exenatide 10 μg twice daily | ‐ | ‐ | 25 | 25 | 25 | 25 | 100 | 3 months |
| C: metformin 1000 mg twice daily | 25 | 25 | 25 | 25 | 100 | ||||
| total: | 50 | 50 | 50 | 50 | 100 | ||||
|
Martinez‐Abundis 2015 (parallel RCT) |
I: linagliptin 5 mg + placebo in the evening | ‐ | ‐ | 8 | 8 | 8 | 8 | 100 | 90 days |
| C: metformin 500 mg twice daily | 8 | 8 | 8 | 8 | 100 | ||||
| total: | 16 | 16 | 16 | 16 | 100 | ||||
|
McLaughlin 2011 (parallel RCT) |
I: exenatide 10 μg twice daily | ‐ | ‐ | 32 | 32 | 32 | ‐ | ‐ | 30 weeks (1 year) |
| C: placebo | 34 | 34 | 34 | ‐ | ‐ | ||||
| total: | 66e | 66 | 66 | ‐ | ‐ | ||||
|
Rosenstock 2008 (parallel RCT) |
I: vildagliptin 50 mg once daily | ITT | 956 | 90 | 89 | 89 | 84 | 93.3 | 12 weeks |
| C: placebo | 89 | 89 | 89 | 84 | 94.4 | ||||
| total: | 179 | 178 | 178 | 168 | 93.9 | ||||
|
Rosenstock 2010 (parallel RCT) |
I: exenatide 10 μg twice daily | Includes participants that received at least 1 dose of study drug | 322b | ‐ | 17d | 17d | 17d | ‐ | 24 weeks |
| C: placebo | ‐ | 16d | 16d | 16d | ‐ | ||||
| total: | 38c | 33d | 33d | 33d | 86.8 | ||||
|
SCALE (parallel RCT) |
I: liraglutide 3.0 mg, once daily | "The power for the primary endpoint weight change is calculated based on a two sided t‐test with a significance level of 5%. The power with regard to the co‐primary dichotomous endpoints proportion of subjects with a weight loss of at least 5% or more than 10%, respectively, is calculated based on a two‐sided chi‐square test." "The large number of randomised subjects also provides sufficient power for the fourth primary endpoint new onset of diabetes among subjects with pre‐diabetes. The endpoint new onset of diabetes will be analysed using methods for analysis of interval censored failure time data. A conservative estimate of the power may be calculated as if the endpoint diabetes yes/no among completers during the 160 weeks is analysed by use of a two‐sided Chisquare test with a significance level of 5%. It is assumed that the annual conversion rate of subjects with pre‐diabetes to diabetes equals 7% among placebo treated subjects, whereas it is 60% lower, 2.1%, among liraglutide treated subjects. After 160 weeks of treatment with liraglutide/placebo, the percentage of subjects with diabetes is therefore equal to 1‐(1‐0.07)3 = 20% among liraglutide placebo treated subjects and 1‐(1‐0.021)3 = 6% among liraglutide treated subjects. It is assumed that the drop‐out during the 160 weeks may be as large as 65% in both groups. The power for conversion rates for placebo of 5, 7 and 9% and conversion rates 60 and 70% lower in the liraglutide group may be seen in Table 18‐1." From the numbers in Table 18‐1 following numbers are specified: "Based on these figures it is apparent that a sample size of 2400 liraglutide treated subjects and 1200 liraglutide placebo treated subjects will provide sufficient power also for the fourth primary endpoint onset of diabetes" "Missing values were imputed with the use of the last‐observation‐carried‐forward method for measurements made after baseline." |
4992b | Period week 0‐56: 1528 Period week 0‐172: 1505 |
1472f | 1472f | Period week 0‐56: 1110 Period week 0‐172: 783g |
Period week 0‐56: 72.6 Period week 0‐172: 52.0 |
160 weeks (172 weeks) |
| Period week 0‐56: 757 Period week 0‐172: 749 |
738f | 738f | Period week 0‐56: 505 Period week 0‐172: 327g |
Period week 0‐56: 66.7 Period week 0‐172: 43.7 |
|||||
| C: placebo | |||||||||
| total: | 2285 | 2210 | 2210 | 1110 | 48.6 | ||||
| Grand total | All interventionsh | ‐ | 1718 | 924 | ‐ | ||||
| All comparatorsh | 946 | 471 | |||||||
| All interventions and comparatorsi | 2702 | 1428j | |||||||
‐ denotes not reported.
aFollow‐up under randomised conditions until end of trial or if not available, duration of intervention; extended follow‐up refers to follow‐up of participants once the original study was terminated as specified in the power calculation. bTotal number of screened. cArticle stated that 38 participants out of the total randomised population had impaired fasting glucose or impaired glucose tolerance, or both. dNumber retrieved from abstract. eOne of the abstracts reports that 68 participants were randomised and another abstract reports 66 participants. fThe SCALE had different number of participants included in the analyses depending on outcome. A full analyses set is reported in the table and was defined as: "All randomised subjects exposed to at least one dose of the trial product and with at least one post baseline assessment of any efficacy endpoint will be included. Subjects in the FAS will be analysed according to randomised treatment." Beside safety analysis set was defined as: "All randomised subjects who have been exposed to at least one dose of trial product. Subjects in the safety analysis set will be analysed "as treated" ". The number of participants started in the trial according to clinicaltrial.gov varied from the period of week 0 to week 56 and from week 0 to 172 due to misclassification of screened participants. gIn the publication reporting long‐term data, it was stated that 791 (53%) participants in the liraglutide group and 337 (15%) participants in the placebo group completed the trial (Le Roux 2017). hNot all trials described the number of participants randomised to each intervention/comparator group. iOne trial did not report the number of randomised participants per intervention group. Therefore, numbers do not add up accurately. jNot all trials reported the number of participants finishing the trial.
C: comparator; I: intervention; ITT: intention to treat; n: number of participants; N/A: not applicable; RCT: randomised controlled trial; SCALE: Satiety and Clinical Adiposity ‐ Liraglutide Evidence in Nondiabetic and Diabetic Individuals; SSPG: steady‐state plasma glucose.
We provided information about potentially relevant ongoing trials including trial identifier in the Characteristics of ongoing studies table and in Appendix 7 'Matrix of trial endpoint (publications and trial documents)'. For each included trial, we tried to retrieve the protocol. If not available from the search of the databases, reference screening or Internet searches, we asked authors to provide a copy of the protocol. Predefined outcomes were entered in a 'Matrix of trial endpoint (publications and trial documents)' (see Appendix 7).
We emailed all authors of the included trials to enquire whether they were willing to answer questions regarding their trials. We presented the results of this survey in 'Survey of trial investigators providing information on included trials' (see Appendix 14). We sought relevant missing information on the trial from the primary author(s) of the articles, if possible.
Dealing with duplicate and companion publications
In the event of duplicate publications, companion documents or multiple reports of a primary trial, we maximised the information yield by collating all available data and used the most complete data set aggregated across all known publications. Duplicate publications, companion documents or multiple reports of a primary trial would be listed as secondary references under the primary reference of the included, ongoing or excluded trial.
Data from clinical trial registers
If data of included trials were available as study results in clinical trial registers such as ClinicalTrials.gov or similar sources, we made full use of this information and extracted data. If there was also a full publication of the trial, we collated and critically appraised all available data.
Assessment of risk of bias in included studies
Two review authors (BH and DS) independently assessed the risk of bias of each included trial. We resolved any disagreements by consensus, or by consultation with a third review author (BR). If adequate information was not available from the trial publication, trial protocol, or both we contacted trial authors for missing data on 'Risk of bias' items.
We used the Cochrane 'Risk of bias' assessment tool (Higgins 2011a; Higgins 2011b), and judged 'Risk of bias' criteria as 'low', 'high' or 'unclear' risk and evaluated individual bias items as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a), where any of the specified criteria for a judgement on 'low', unclear' or 'high' risk of bias justified the associated categorisation.
Random sequence generation (selection bias due to inadequate generation of a randomised sequence) ‐ assessment at trial level
We described for each included trial the method used to generate the allocation sequence in sufficient detail to allow an assessment of whether it should produce comparable groups.
Low risk of bias: sequence generation was achieved using computer random number generation or a random number table. Drawing of lots, tossing a coin, shuffling cards or envelopes, and throwing dice were adequate if performed by an independent person not otherwise involved in the trial. Use of the minimisation technique was considered as equivalent to being random.
Unclear risk of bias: insufficient information about the sequence generation process.
High risk of bias: the sequence generation method was non‐random (e.g. sequence generated by odd or even date of birth; sequence generated by some rule based on date (or day) of admission; sequence generated by some rule based on hospital or clinic record number; allocation by judgement of the clinician; allocation by preference of the participant; allocation based on the results of a laboratory test or a series of tests; allocation by availability of the intervention). Such trials were excluded.
Allocation concealment (selection bias due to inadequate concealment of allocations prior to assignment) ‐ assessment at trial level
We described for each included trial the method used to conceal allocation to interventions prior to assignment and assessed whether intervention allocation could have been foreseen in advance of, or during, recruitment, or changed after assignment.
Low risk of bias: central allocation (including telephone, interactive voice‐recorder, web‐based and pharmacy‐controlled randomisation); sequentially numbered drug containers of identical appearance; sequentially numbered, opaque, sealed envelopes.
Unclear risk of bias: insufficient information about the allocation concealment.
High risk of bias: used an open random allocation schedule (e.g. a list of random numbers); assignment envelopes were used without appropriate safeguards; alternation or rotation; date of birth; case record number; any other explicitly unconcealed procedure. Such trials were excluded.
We also evaluated trial baseline data to incorporate assessment of baseline imbalance into the 'Risk of bias' judgement for selection bias (Corbett 2014; Egbewale 2014; Riley 2013). Chance imbalances may also affect judgements on the risk of attrition bias. In the case of unadjusted analyses, we distinguished between trials we rated as at low risk of bias on the basis of both randomisation methods and baseline similarity, and trials rated as at low risk of bias on the basis of baseline similarity alone (Corbett 2014). We reclassified judgements of unclear, low or high risk of selection bias as specified in 'Selection bias decisions' (Appendix 18).
Blinding of participants and study personnel (performance bias due to knowledge of the allocated interventions by participants and personnel during the trial) ‐ assessment at outcome level
We evaluated the risk of performance bias separately for each outcome (Hróbjartsson 2013). We noted whether outcomes were self‐reported, investigator‐assessed or adjudicated outcome measures (see below).
Low risk of bias: blinding of participants and key study personnel ensured, and unlikely that the blinding could have been broken; no blinding or incomplete blinding, but the review authors judged that the outcome was not likely to be influenced by lack of blinding.
Unclear risk of bias: insufficient information about the blinding of participants and study personnel; the trial did not address this outcome.
High risk of bias: no blinding or incomplete blinding, and the outcome was likely to be influenced by lack of blinding; blinding of trial participants and key personnel attempted, but likely that the blinding could have been broken, and the outcome was likely to be influenced by lack of blinding.
Blinding of outcome assessment (detection bias due to knowledge of the allocated interventions by outcome assessment) ‐ assessment at outcome level
We evaluated the risk of detection bias separately for each outcome (Hróbjartsson 2013). We noted whether outcomes were self‐reported, investigator‐assessed or adjudicated outcome measures (see below).
Low risk of bias: blinding of outcome assessment ensured, and unlikely that the blinding could have been broken; no blinding of outcome assessment, but the review authors judged that the outcome measurement was not likely to be influenced by lack of blinding.
Unclear risk of bias: insufficient information about the blinding of outcome assessors; the trial did not address this outcome.
High risk of bias: no blinding of outcome assessment, and the outcome measurement was likely to be influenced by lack of blinding; blinding of outcome assessment, but likely that the blinding could have been broken, and the outcome measurement was likely to be influenced by lack of blinding.
Incomplete outcome data (attrition bias due to amount, nature or handling of incomplete outcome data) ‐ assessment at outcome level
We described for each included trial, and for each outcome, the completeness of data including attrition and exclusions from the analysis. We stated whether attrition and exclusions were reported and the number included in the analysis at each stage (compared with the number of randomised participants per intervention/comparator groups), if reasons for attrition or exclusion were reported, and whether missing data were balanced across groups or were related to outcomes. We considered the implications of missing outcome data per outcome such as high dropout rates (e.g. above 15%) or disparate attrition rates (e.g. difference of 10% or more between trial arms).
Low risk of bias: no missing outcome data; reasons for missing outcome data unlikely to be related to true outcome (for survival data, censoring unlikely to be introducing bias); missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups; for dichotomous outcome data, the proportion of missing outcomes compared with observed event risk not enough to have a clinically relevant impact on the intervention effect estimate; for continuous outcome data, plausible effect size (mean difference (MD) or standardised mean difference (SMD)) among missing outcomes not enough to have a clinically relevant impact on observed effect size; appropriate methods, such as multiple imputation, were used to handle missing data.
Unclear risk of bias: insufficient information to assess whether missing data in combination with the method used to handle missing data were likely to induce bias; the trial did not address this outcome.
High risk of bias: reason for missing outcome data likely to be related to true outcome, with either imbalance in numbers or reasons for missing data across intervention groups; for dichotomous outcome data, the proportion of missing outcomes compared with observed event risk enough to induce clinically relevant bias in intervention effect estimate; for continuous outcome data, plausible effect size (MD or SMD) among missing outcomes enough to induce clinically relevant bias in observed effect size; 'as‐treated' or similar analysis done with substantial departure of the intervention received from that assigned at randomisation; potentially inappropriate application of simple imputation.
Selective reporting (reporting bias due to selective outcome reporting) ‐ assessment at trial level
We assessed outcome reporting bias by integrating the results of the appendix 'Matrix of trial endpoints (publications and trial documents' (Appendix 7) (Boutron 2014; Mathieu 2009), with those of the appendix 'High risk of outcome reporting bias according to ORBIT classification' (Appendix 8) (Kirkham 2010). This analysis formed the basis for the judgement of selective reporting.
Low risk of bias: the trial protocol was available and all the trial's prespecified (primary and secondary) outcomes that were of interest in the review were reported in the prespecified way; the study protocol was not available but it was clear that the published reports include all expected outcomes (ORBIT classification).
Unclear risk of bias: insufficient information about selective reporting.
High risk of bias: not all the trial's prespecified primary outcomes have been reported; one or more primary outcomes was reported using measurements, analysis methods or subsets of the data (e.g. subscales) that were not prespecified; one or more reported primary outcomes were not prespecified (unless clear justification for their reporting was provided, such as an unexpected adverse effect); one or more outcomes of interest in the review were reported incompletely so that they could not be entered in a meta‐analysis; the trial report failed to include results for a key outcome that would be expected to have been reported for such a trial (ORBIT classification).
Other bias (bias due to problems not covered elsewhere) ‐ assessment at trial level
Other risk of bias reflected other circumstances that may threaten the validity of the trials.
Low risk of bias: the trial appeared to be free of other sources of bias.
Unclear risk of bias: insufficient information to assess whether an important risk of bias existed; insufficient rationale or evidence that an identified problem introduced bias.
High risk of bias: had a potential source of bias related to the specific trial design used; has been claimed to have been fraudulent; had some other serious problem.
We established a 'Risk of bias' graph and a 'Risk of bias' summary figure.
We distinguished between self‐reported, investigator‐assessed and adjudicated outcome measures.
We defined the following outcomes as self‐reported.
Non‐serious adverse events.
Hypoglycaemia, if reported by participants.
HRQoL.
Measures of blood glucose control, if measured by trial participants.
We defined the following outcomes as investigator‐assessed.
All‐cause mortality.
Incidence of T2DM.
Serious adverse events.
Cardiovascular mortality.
Non‐fatal myocardial infarction.
Non‐fatal stroke.
Congestive heart failure.
Amputation of lower extremity.
Blindness or severe vision loss.
End‐stage renal disease.
Hypoglycaemia, if measured by trial personnel.
Time to progression to T2DM.
Blood glucose control, if measured by trial personnel.
Socioeconomic effects.
Summary assessment of risk of bias
Risk of bias for a trial across outcomes: some risk of bias domains, such as selection bias (sequence generation and allocation sequence concealment), affected the risk of bias across all outcome measures in a trial. Otherwise, we did not perform a summary assessment of the risk of bias across all outcomes for a trial. In case of high risk of selection bias, we excluded the trial.
Risk of bias for an outcome within a trial and across domains: we assessed the risk of bias for an outcome measure by including all entries relevant to that outcome (i.e. both trial‐level entries and outcome‐specific entries). 'Low' risk of bias was defined as low risk of bias for all key domains, 'unclear' risk of bias as unclear risk of bias for one or more key domains and 'high' risk as high risk of bias for one or more key domains.
Risk of bias for an outcome across trials and across domains: these were our main summary assessments that were incorporated in our judgements about the quality of evidence in the 'Summary of findings' tables. 'Low' risk of bias was defined as most information coming from trials at low risk of bias, 'unclear' risk of bias as most information coming from trials at low or unclear risk of bias and 'high' risk of bias as sufficient proportion of information coming from trials at high risk of bias.
Measures of treatment effect
When at least two trials were available for a comparison of a given outcome, we expressed dichotomous data as risk ratio (RR) with 95% CIs and with Trial Sequential Analysis (TSA)‐adjusted 95% CIs if the diversity‐adjusted required information size was not reached. We expressed continuous data reported on the same scale as MD with 95% CIs and with TSA‐adjusted 95% CIs if the diversity‐adjusted required information size was not reached. For trials addressing the same outcome but using different outcome measure scales, we used SMD with 95% CI. We planned to calculate time‐to‐event data as HR with 95% CI with the generic inverse variance method. Unadjusted HRs would have been preferred, as adjustment may differ among the included trials. For outcomes meta‐analysed as SMD and the generic inverse variance method, we are presently unable to conduct TSA and adjust the 95% CIs.
The scales measuring HRQoL may go in different directions. Some scales increase in values with improved HRQoL, whereas other scales decrease in values with improved HRQoL. To adjust for the different directions of the scales, we planned to multiply the scales that reported better HRQoL with decreasing values by ‐1.
Unit of analysis issues
We took into account the level at which randomisation occurred, such as cross‐over trials, cluster‐randomised trials and multiple observations for the same outcome. If more than one comparison from the same trial was eligible for inclusion in the same meta‐analysis, we would have either combined groups to create a single pair‐wise comparison or appropriately reduced the sample size so that the same participants do not contribute multiply (splitting the 'shared' group into two or more groups). While the latter approach offers some solution to adjusting the precision of the comparison, it does not account for correlation arising from the same set of participants being in multiple comparisons (Higgins 2011a).
We planned to reanalyse cluster‐randomised trials that did not appropriately adjust for potential clustering of participants within clusters in their analyses. The variance of the intervention effects was planned to be inflated by a design effect (DEFF). Calculation of a DEFF involves estimation of an intra‐cluster correlation (ICC). We planned to obtain estimates of ICCs through contact with authors, or impute using estimates from other included studies that report ICCs, or using external estimates from empirical research (e.g. Bell 2013). We planned to examine the impact of clustering using sensitivity analyses.
Dealing with missing data
We tried to obtain missing data from trial authors and carefully evaluate important numerical data such as screened, randomly assigned participants as well as intention‐to‐treat (ITT), and as‐treated and per‐protocol populations.
We investigated attrition rates (e.g. dropouts, losses to follow‐up, withdrawals), and we critically appraised issues concerning missing data and imputation methods (e.g. last observation carried forward (LOCF)).
We converted standard errors and CIs to standard deviations (SD) (Higgins 2011a). When there were no differences in means and SDs from baseline, we used the end‐of follow‐up values (Higgins 2011a). Where means and SDs for outcomes were not reported and we did not receive the needed information from trial authors, we calculated the SDs from standard errors, if possible. Otherwise, we would have imputed the values by assuming the SDs of the missing outcome to be the mean of the SDs from the trials that reported this information.
We planned to investigate the impact of imputation on meta‐analyses by performing sensitivity analyses.
Assessment of heterogeneity
In the event of substantial clinical or methodological heterogeneity, we planned not to report trial results as the pooled effect estimate in a meta‐analysis.
We investigated heterogeneity (inconsistency) by visually inspecting the forest plots and by using a standard Chi² test with a significance level of 0.1. In view of the low power of this test, we also considered the I² statistic, which quantifies inconsistency across trials to assess the impact of heterogeneity on the meta‐analysis (Higgins 2002; Higgins 2003); an I² statistic of 75% or greater indicated a considerable level of heterogeneity (Higgins 2011a).
Assessment of reporting biases
If we included 10 or more trials investigating a particular outcome, we planned to use funnel plots to assess small‐trial effects. Several explanations may account for funnel plot asymmetry, including true heterogeneity of effect with respect to trial size, poor methodological design (and hence bias of small trials) and publication bias. Therefore, we planned to interpret results carefully (Sterne 2011).
Data synthesis
Unless good evidence showed homogeneous effects across trials, we would primarily summarise low risk of bias data using a random‐effects model (Wood 2008). We interpreted random‐effects meta‐analyses with consideration to the whole distribution of effects, ideally by presenting a prediction interval (Higgins 2009). A prediction interval specifies a predicted range for the true treatment effect in an individual trial (Riley 2011). In addition, we performed statistical analyses according to the statistical guidelines presented in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a).
Trial Sequential Analyses
In a single trial, sparse data and interim analyses increase the risk of type I and type II errors. To avoid type I errors, group sequential monitoring boundaries are applied to decide whether a trial could be terminated early because of a sufficiently small P value (i.e. the cumulative Z‐curve crosses the monitoring boundaries) (Lan 1983). Likewise, before reaching the planned sample size of a trial, the trial may be stopped due to futility if the cumulative Z‐score crosses the futility monitoring boundaries (Higgins 2011a). Sequential monitoring boundaries for benefit, harm or futility can be applied to meta‐analyses as well (termed trial sequential monitoring boundaries) (Higgins 2011c; Wetterslev 2008). In TSA, the addition of each trial in a cumulative meta‐analysis is regarded as an interim meta‐analysis and helps to clarify whether significance or futility is reached, or whether additional trials are needed (Wetterslev 2008).
TSA combines a calculation of the diversity‐adjusted required information size (cumulated meta‐analysis sample size to detect or reject a specific relative intervention effect) for meta‐analysis with the threshold of data associated with statistics. We performed TSA on all outcomes (Brok 2009; Pogue 1997; Wetterslev 2008).
The idea in TSA is that if the cumulative Z‐curve crosses the boundary for benefit or harm before a diversity‐adjusted required information size is reached, a sufficient level of evidence for the anticipated intervention effect has been reached with the assumed type I error and no further trials may be needed. If the cumulative Z‐curve crosses the boundary for futility before a diversity‐adjusted required information size is reached, the assumed intervention effect can be rejected with the assumed type II error and no further trials may be needed. If the Z‐curve does not cross any boundary, then there is insufficient evidence to reach a conclusion. To construct the trial sequential monitoring boundaries, the required information size is needed and is calculated as the least number of participants needed in a well‐powered single trial and subsequently adjusted for diversity among the included trials in the meta‐analysis (Brok 2009; Wetterslev 2008). We applied TSA as it decreases the risk of type I and II errors due to sparse data and multiple updating in a cumulative meta‐analysis, and it provides us with important information to estimate the risks of imprecision when the required information size is not reached. Additionally, TSA provides important information regarding the need for additional trials and the required information size of such trials (Wetterslev 2008).
We applied trial sequential monitoring boundaries according to an estimated clinically important effect. We based the required information size on an a priori effect corresponding to a 10% relative risk reduction (RRR) for beneficial effects of the interventions and a 30% relative risk increase for harmful effects of the interventions.
TSA for continuous outcomes was performed with MDs, by using trials applying the same scale to calculate the required sample size. For continuous outcomes, we tested the evidence for the achieved differences in cumulative meta‐analyses.
For adjustment of heterogeneity of the required information size we used the diversity (D²) estimated in the meta‐analyses of included trials. When diversity was zero in a meta‐analysis, we performed a sensitivity analysis using an assumed diversity of 20%.
Quality of evidence
We presented the overall quality of the evidence for each outcome according to the GRADE approach, which takes into account issues relating not only to internal validity (risk of bias, inconsistency, imprecision, publication bias) but also to external validity, such as directness of results. Two review authors (BH and DS) independently rated the quality of evidence for each outcome. We presented a summary of the evidence in Table 1; Table 2. This provides key information about the best estimate of the magnitude of the effect, in relative terms and as absolute differences, for each relevant comparison of alternative management strategies, the numbers of participants and trials addressing each important outcome, and rates the overall confidence in effect estimates for each outcome. We created the 'Summary of findings' tables on the basis of methods described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a) by means of the table editor in Review Manager 5 (RevMan 2014), and included two appendices (Appendix 17; Appendix 18) providing checklists as guides to the consistency and reproducibility of GRADE assessments (Meader 2014) to help with the standardisation of the 'Summary of findings' tables. Alternatively, we would have used the GRADEpro GDT software (GRADEpro GDT 2015) and presented evidence profile tables as an appendix. We presented results for the outcomes as described in the Types of outcome measures section. If meta‐analysis was not possible, we presented the results in a narrative format in the 'Summary of findings' tables. We justified all decisions to downgrade the quality using footnotes, and we made comments to aid the reader's understanding of the review where necessary.
Summary of findings for the main comparison. DPP‐4 inhibitors for prevention or delay of type 2 diabetes mellitus and its associated complications in people at risk for the development of type 2 diabetes mellitus.
| DPP‐4 inhibitors for prevention or delay of type 2 diabetes mellitus and its associated complications in people at risk for the development of type 2 diabetes mellitus | ||||||
|
Population: people at risk for development of T2DM Settings: outpatients Intervention: DPP‐4 inhibitors Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trial(s)) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | DPP‐4 inhibitors | |||||
|
All‐cause mortality Follow‐up: 12 weeks |
See comment | See comment | See comment | 179 (1) | ⊕⊝⊝⊝ Very lowa | 1 trial on vildagliptin reported that none of the participants died (Rosenstock 2008) |
|
Incidence of T2DM Definition: WHO criteria Follow‐up: 12 weeks |
See comment | See comment | See comment | 179 (1) | ⊕⊝⊝⊝ Very lowa | 1 trial reported that 3/90 in the vildagliptin group vs 1/89 in the placebo group developed T2DM (Rosenstock 2008) |
|
Serious adverse events Follow‐up: 12 weeks |
See comment | See comment | See comment | 179 (1) | ⊕⊝⊝⊝ Very lowa | 1 trial reported that 1/90 in the vildagliptin group vs 2/89 in the placebo group experienced a serious adverse event (Rosenstock 2008) |
|
Cardiovascular mortality Follow‐up: 12 weeks |
See comment | See comment | See comment | 179 (1) | ⊕⊝⊝⊝ Very lowa | 1 trial reported that none of the participants died (Rosenstock 2008) |
|
(1) Non‐fatal myocardial infarction (2) Non‐fatal stroke (3) Congestive heart failure Follow‐up: 12 weeks |
See comment | See comment | See comment | (1) ‐ (2) ‐ (3) 179 (1) |
⊕⊝⊝⊝ Very lowa | (1) + (2): not reported (3): 1 trial on vildagliptin reported 1/90 in the vildagliptin group vs 0/89 in the placebo group experienced heart failure (Rosenstock 2008) |
| Health‐related quality of life | See comment | See comment | See comment | See comment | See comment | Not reported |
| Socioeconomic effects | See comment | See comment | See comment | See comment | See comment | Not reported |
| *The basis for the assumed risk (e.g. the median control group risk across trials) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; DPP‐4: dipeptidyl‐peptidase‐4; RR: risk ratio; T2DM: type 2 diabetes mellitus; WHO: World Health Organization. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
*Assumed risk was derived from the event rates in the comparator groups.
aDowngraded by three levels because of indirectness, imprecision (very sparse data) and risk of publication bias (see Appendix 15).
Summary of findings 2. GLP‐1 analogues for prevention or delay of type 2 diabetes mellitus and its associated complications in people at risk for the development of type 2 diabetes mellitus.
| GLP‐1 analogues for prevention or delay of type 2 diabetes mellitus and its associated complications in people at risk for the development of type 2 diabetes mellitus | ||||||
|
Population: people at risk for development of T2DM Settings: outpatients Intervention: GLP‐1 analogues Comparison: placebo | ||||||
| Outcomes | Placebo | GLP‐1 analogues | Relative effect (95% CI) | No of participants (trial(s)) | Quality of the evidence (GRADE) | Comments |
|
All‐cause mortality Follow‐up: up to 172 weeks |
See comment | See comment | See comment | 2281 (2) | ⊕⊝⊝⊝ Very lowa | 1 trial reported that 2/1501 in the liraglutide group vs 2/747 in the placebo group died after 160 weeks of intervention. Liraglutide used in doses approved for weight‐reducing purposes (3.0 mg) (SCALE). 1 trial using exenatide reported that none of the participants died (Rosenstock 2010) |
|
Incidence of T2DM Definition/description: 1 trial established the diagnosis of T2DM defined as HbA1c ≥ 6.5%, or fasting plasma glucose ≥ 7.0 mmol/L, or 2‐hour plasma glucose post‐challenge (oral glucose tolerance test) ≥ 11.1 mmol/L (SCALE). Other trial did not report how the diagnosis of T2DM was established. Follow‐up: up to 172 weeks |
See comment | See comment | See comment | 2243 (2) | ⊕⊝⊝⊝ Very lowa | At 160 weeks, 26/1472 (1.8%) participants in the liraglutide group vs 46/738 (6.2%) participants in the placebo group developed T2DM (SCALE). The incidence of T2DM after the 12‐week off‐treatment extension period (i.e. after 172 weeks) showed that 5 additional participants were diagnosed with T2DM in the liraglutide group, compared with 1 participant in the placebo group. Liraglutide used in doses approved for weight‐reducing purposes (3.0 mg). 1 trial reported that 2/17 in the exenatide group vs1/16 in the placebo group developed T2DM (Rosenstock 2010) |
|
Serious adverse events Follow‐up: up to 172 weeks |
See comment | See comment | See comment | 2312 (2) | ⊕⊕⊝⊝ Lowb | 1 trial on liraglutide reported that 227/1501 (15.1%) participants in the liraglutide 3.0 mg group vs 96/747 (12.7%) participants in the placebo group experienced a serious adverse event after 160 weeks (SCALE). 1 trial on exenatide reported that none of the participants experienced a serious adverse event (Rosenstock 2010) |
|
Cardiovascular mortality Follow‐up: up to 172 weeks |
See comment | See comment | See comment | 2281 (2) | ⊕⊝⊝⊝ Very lowa | 1 trial reported that none of the participants died (Rosenstock 2010). 1 trial reported that 1/1501 participants in the liraglutide group died from cardiac arrest; no participant in the placebo group died of cardiovascular reasons (SCALE). |
|
(1) Non‐fatal myocardial infarction (2) Non‐fatal stroke (3) Congestive heart failure Follow‐up: up to 172 weeks |
See comment | See comment | See comment | (1) 2279 (1) (2) See comments (3) 2279 (1) |
⊕⊝⊝⊝ Very lowa | (1) 1 trial reported 1/1524 participants in the liraglutide 3.0 mg group vs 0/755 participants in the placebo group (SCALE) (2) Not reported (3) 1 trial reported 1/1524 in the liraglutide 3.0 mg group vs 1/755 participants in the placebo group (SCALE) |
|
Health‐related quality of life SF‐36 scale: total score 0‐100, 8 subscales. Higher values mean better health‐related quality of life. Follow‐up: 160 weeks |
See comment | See comment | See comment | 1791 (1) | ⊕⊝⊝⊝ Very lowa | Physical functioning component score had mean difference of 0.87 in favour of liraglutide (SCALE) |
| Socioeconomic effects | See comment | See comment | See comment | See comment | See comment | Not reported |
| *The basis for the assumed risk (e.g. the median control group risk across trials) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; GLP‐1: glucagon‐like peptide‐1; HbA1c: glycosylated haemoglobin A1c; RR: risk ratio; SF‐36: 36‐item Short Form health survey; T2DM: type 2 diabetes mellitus. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aDowngraded by three levels because of indirectness, imprecision (sparse data) and risk of attrition and publication bias (see Appendix 16). bDowngraded by two levels because of imprecision (sparse data) and risk of attrition and publication bias (see Appendix 16).
Subgroup analysis and investigation of heterogeneity
We expected the following characteristics to introduce clinical heterogeneity, and planned to carry out the following subgroup analyses with investigation of interactions.
Type of DPP‐4 inhibitor or GLP‐1 analogue.
Trials with long duration (two years or greater) versus trials with short duration (less than two years).
Diagnostic criteria (IFG, IGT, HbA1c).
Age, depending on data.
Gender.
Ethnicity, depending on data.
Comorbid conditions, such as hypertension, obesity, or both.
Participants with previous gestational diabetes mellitus.
GLP‐1 analogues dose (up to the recommended dose for a glucose‐lowering effect in people with T2DM versus higher doses).
Sensitivity analysis
We planned to perform sensitivity analyses to explore the influence of the certain factors (when applicable) on effect sizes by restricting analysis to the following.
Published trials.
Taking into account risk of bias, as specified in the Assessment of risk of bias in included studies section.
Very long or large trials, to establish the extent to which they dominated the results.
Trials using the following filters: diagnostic criteria, imputation, language of publication, source of funding (industry versus other) or country.
Results
Description of studies
For a detailed description of trials, see Table 3; and the Characteristics of included studies, Characteristics of excluded studies, Characteristics of studies awaiting classification and Characteristics of ongoing studies tables.
Results of the search
The initial search of the databases identified 2074 records after duplicates were removed. The applied MEDLINE (via OvidSP) email alert service established by the CMED Group to identify newly published trials using the same search strategy as described for MEDLINE (for details on search strategies, see Appendix 2) did not identify any additional references. We excluded most of the references on the basis of their titles and abstracts because they clearly did not meet the inclusion criteria (Figure 1). We evaluated 164 references further. After screening the full texts, seven RCTs published in 50 records met our inclusion criteria. One trial published in three references did not report any of the primary or secondary outcomes of this review (McLaughlin 2011). The trial was only published in abstracts. The investigators were asked for additional data, but stated they would not provide additional data before the trial was published in full (McLaughlin 2011) (see Appendix 14).
1.
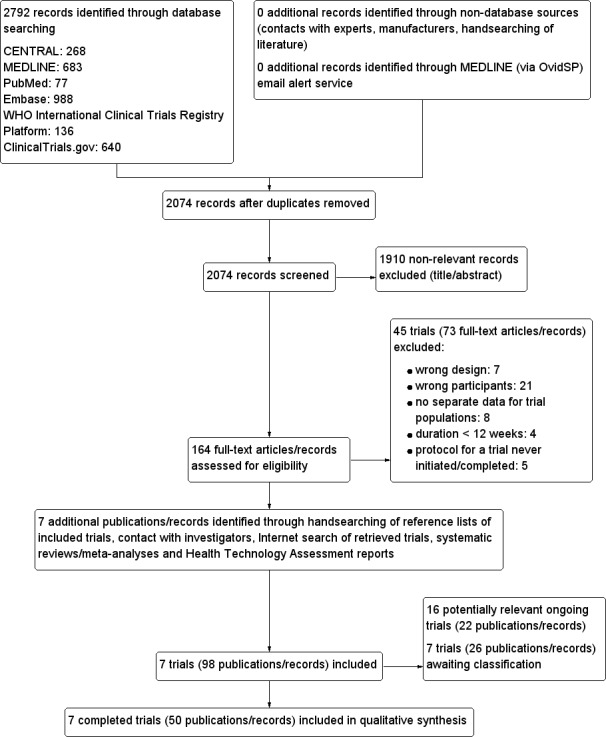
Study flow diagram.
We excluded 73 references after full‐text evaluation.
We identified two applications for approval of liraglutide 3.0 mg in people with obesity when searching the US FDA and the EMA websites (EMA/143005/2015; FDA 2014). As some of these people might have had intermediate hyperglycaemia and therefore fulfilled the inclusion criteria, these applications were screened for trials and data to be included. We screened the statistical and medical reviews in the FDA application, but did not identify additional trials or data with relevance for this review (EMA/143005/2015; FDA 2014). We did not find a health technology assessment report for DPP‐4 inhibitors or GLP‐1 analogues in people at increased risk for the development of T2DM. Furthermore, no systematic review or meta‐analysis focusing on the DPP‐4 inhibitors or GLP‐1 analogues in people at increased risk for the development of T2DM could be identified. Four systematic reviews published in five records in people at increased risk for the development of T2DM did not include any trials using a DPP‐4 inhibitor or GLP‐1 analogue as a comparator (Anderson 2005; Bhardwaj 2010; Hopper 2011; Phung 2012; Van de Laar 2006). We evaluated all these systematic reviews but did not identify additional trials.
Through Internet searches of retrieved trials, we retrieved seven references with additional information on four trials (Astrup 2009; Rosenstock 2008; SCALE; SCALE‐SLEEP).
We sent all corresponding trial authors of the included trials a reference list and a request for information on additional trials of relevance. All the investigators of the included trials replied, except one (Kelly 2012). Two of the investigators provided information that we could not retrieve from the publications (Martinez‐Abundis 2015; SCALE) (see Appendix 14). Another trial could be included in the quantitative analyses as the investigators provided information on some of the outcomes of interest for this review. These data were not presented in the published abstract (Martinez‐Abundis 2015).
Studies awaiting classification
We classified seven trials in 26 references as studies awaiting classification (see Characteristics of studies awaiting classification table). Three of the trials awaiting classification were performed by Novo Nordisk and we requested separate information on the people with intermediate hyperglycaemia (Astrup 2009; SCALE‐SLEEP; SCALE 2013). Novo Nordisk has replied that we would get access to trial data. Once available, these data will be used in updates of this review. One completed trial included participants both with intermediate hyperglycaemia and T2DM (Santilli 2015). The investigators replied to our request and will provide separate data on the people with intermediate hyperglycaemia when all data are published (Santilli 2015). Three trials were listed in ClinicalTrials.gov as completed in September 2014 (NCT01521312), December 2014 (NCT01960205), and May 2016 (NCT02294084). The corresponding investigators were contacted for each trial. Two of the investigators replied that data were not yet available (NCT01960205; NCT02294084). One investigator did not reply (NCT01521312) (see Appendix 14).
Ongoing trials
We found 16 ongoing RCTs in 22 references (EudraCT 2013‐000418‐39; Naidoo 2016; NCT01234649; NCT01336322; NCT01548651; NCT01779362; NCT01795248; NCT01856907; NCT02104739; NCT02140983; NCT02488057; NCT02576288; NCT02847403; NCT02969798; NCT03004612; UMIN000008620) (see Characteristics of ongoing studies table). When insufficient information was reported in the available protocol documents, we asked investigators for additional information (see Appendix 14). We estimate the ongoing trials to include 5921 participants. Three of the ongoing trials have predefined the assessment of one or more of the primary outcomes included in our review (EudraCT 2013‐000418‐39; Naidoo 2016; NCT01795248). Seven ongoing trials did not predefine the primary outcomes of our review; however, one or more of our secondary outcomes were planned to be assessed (NCT01234649; NCT01779362; NCT01856907; NCT02488057; NCT02969798; NCT03004612; UMIN000008620). Six other trials did not predefine any outcomes of relevance to our review (NCT01336322; NCT01548651; NCT02104739; NCT02140983; NCT02576288; NCT02847403).
Six ongoing RCTs, estimated to include 1115 participants have a GLP‐1 analogue as the intervention (NCT01234649; NCT01779362; NCT01795248; NCT02104739; NCT02140983; NCT02488057). The largest of the ongoing RCTs plans to include around 3000 participants allocated to a DPP‐4 inhibitor, metformin or placebo. The intervention period has been estimated to be three years and the total follow‐up period will be five years (EudraCT 2013‐000418‐39). The trial is estimated to be completed in 2018 (EudraCT 2013‐000418‐39).
Future updates will include all ongoing trials, if possible.
Included studies
A detailed description of the characteristics of included trials is presented elsewhere (see Characteristics of included studies table; Table 3; and Appendix 3; Appendix 4; Appendix 5; Appendix 6; Appendix 7; Appendix 8; Appendix 9; Appendix 10; Appendix 11; Appendix 12; Appendix 13; Appendix 14). The following is a succinct overview.
Overview of trial populations
Four of the trials reported the total number of participants screened (Ariel 2014; Rosenstock 2008; Rosenstock 2010; SCALE). For trials not only including participants with intermediate hyperglycaemia, the total number of screened was reported (Rosenstock 2010; SCALE). Three trials did not report the number of participants with intermediate hyperglycaemia randomised to each intervention group upon trial initiation (BEGAMI 2013; McLaughlin 2011; Rosenstock 2010). However, one trial provided the number of randomised participants with intermediate hyperglycaemia on request (BEGAMI 2013). About 123 participants were randomised to a DPP‐4 inhibitor (Martinez‐Abundis 2015; Rosenstock 2008), and 1620 participants were randomised to a GLP‐1 analogue (Ariel 2014; Kelly 2012; McLaughlin 2011; Rosenstock 2010; SCALE). All included trials used the intervention as monotherapy. About 946 participants were randomised to a comparator group; five trials used placebo as the comparator (Ariel 2014; McLaughlin 2011; Rosenstock 2008; Rosenstock 2010; SCALE), and two trials used metformin as a comparator (Kelly 2012; Martinez‐Abundis 2015). One trial randomised the participants to a GLP‐1 analogue using a dose approved for weight reduction (liraglutide 3.0 mg) (SCALE).
Two trials provided information about sample size calculation (Ariel 2014; SCALE).
The proportion of participants finishing the trial varied from 49% to 100% (Kelly 2012; Martinez‐Abundis 2015; SCALE).
One of the included trials included participants with intermediate hyperglycaemia and T2DM (Rosenstock 2010). One trial included participants with intermediate hyperglycaemic and normoglycaemia but had prespecified and reported the data on the participants with intermediate hyperglycaemia separately (SCALE).
Trial design
All seven included trials were parallel RCTs. Six trials performed blinding of the participants and investigators (Ariel 2014; Martinez‐Abundis 2015; McLaughlin 2011; Rosenstock 2008; Rosenstock 2010; SCALE). In addition, one trial had a double‐blind intervention period for the first year (pharmaceutical company, investigators and participants were blinded), and a single‐blind period for the remaining two years (pharmaceutical company was unblinded, whereas investigators and participants remained blinded) (SCALE). One trial had an open‐label design (Kelly 2012). One trial had a run‐in period where for one week a single‐blind placebo was given (Rosenstock 2010). The duration of the intervention in the included trials varied from 12 to 160 weeks (Kelly 2012; Martinez‐Abundis 2015; Rosenstock 2008; SCALE). One trial had a duration of the intervention of two years or more (SCALE). Two trials had an extended follow‐up period after the intervention period had stopped (McLaughlin 2011; SCALE). One trial had an intervention period for 30 weeks, but followed the participants for one year (McLaughlin 2011). Another trial followed the participants 12 weeks after the end of the intervention period (i.e. 172 weeks) (SCALE).
The number of participants varied from 16 (Martinez‐Abundis 2015) to 2285 (SCALE). One trial contributed 85% of the total number of all randomised participants (SCALE). Four trials were multicentre trials (Kelly 2012; Rosenstock 2008; Rosenstock 2010; SCALE), one trial was single‐centre trial (Ariel 2014), and two trials did not provide a centre description (Martinez‐Abundis 2015; McLaughlin 2011). All trials were performed in outpatient settings.
Five of the included trials stated that they had received grants from a pharmaceutical company (Ariel 2014; Kelly 2012; Rosenstock 2008; Rosenstock 2010; SCALE), and two trials explicitly acknowledged individuals employed by a pharmaceutical company for their contribution to the trials (Rosenstock 2008; SCALE). Two trials did not report the funding source (Martinez‐Abundis 2015; McLaughlin 2011).
Participants
Three trials reported the ethnicity of the participants; two trials included mainly white people (Ariel 2014; Rosenstock 2008) and another trial only white people (Kelly 2012). Only one trial included participants from low‐income countries (SCALE).
One trial did not report the gender of the participants (Martinez‐Abundis 2015). For the remaining trials authors provided gender information, and both men and women were included in these trials. The age of the included participants varied from 46.1 to 67 years (see Appendix 5).
Six trials reported fasting glucose values at baseline, which varied from 5.5 mmol/L to 6.2 mmol/L (Rosenstock 2008; SCALE). Two trials did not report any glycaemic baseline value (McLaughlin 2011; Rosenstock 2010). Four trials reported 2‐hour glucose values after a glucose‐load at baseline which varied from 7.4 mmol/L to 9.3 mmol/L (Martinez‐Abundis 2015; SCALE). HbA1c values were reported at baseline in three trials and varied from 5.7% to 6.2% (Martinez‐Abundis 2015; SCALE).
All trials reported BMI at baseline. All trials reported a baseline mean BMI above 30 kg/m². Three trials had participants with a mean BMI above 35 kg/m² at baseline (Kelly 2012; Rosenstock 2010; SCALE). One trial reported the number of participants with previous cardiovascular diseases at baseline (SCALE). Most trials excluded participants with other endocrine conditions, or hepatic or kidney disease.
The diagnosis applied in the included trials for identifying intermediate hyperglycaemia varied. Two trials included participants with intermediate hyperglycaemia according to the criteria by ADA regarding elevated fasting glucose (5.6 mmol/L to 6.9 mmol/L) or elevated two‐hour glucose concentrations (7.8 mmol/L to 11.0 mmol/L) after an OGTT (Ariel 2014; McLaughlin 2011). HbA1c was not applied as a criterion for intermediate hyperglycaemia in these trials (Ariel 2014; McLaughlin 2011). Two trials used all the glycaemic cut‐off points recommended by ADA, including HbA1c (5.7% to 6.4%) to diagnose intermediate hyperglycaemia (Kelly 2012; SCALE). One trial used the diagnostic criteria for IGT as recommended by the WHO (fasting plasma glucose below 7.0 mmol/L and elevated two‐hour glucose concentration after an OGTT (7.8 mmol/L to 11.0 mmol/L)) (Rosenstock 2008). One trial used the diagnostic criteria for both IFG and IGT as recommended by the WHO (Rosenstock 2010). One trial just reported that participants with IGT were included, with no further details specified (Martinez‐Abundis 2015).
One trial had explicitly defined withdrawal criteria for the participants (see Characteristics of included studies table) (SCALE). One of the applied withdrawal criteria was not tolerating the dose of the study drug (SCALE). Another trial explicitly stated that intolerability of the maximum dose led to dose‐reduction (Ariel 2014). In this trial, one participant received liraglutide 1.2 mg and another participant received liraglutide 0.6 mg due to intolerance to the originally planned dose of 1.8 mg (Ariel 2014).
Interventions
All the participants of the included trials were treatment naive regarding pharmacological glucose‐lowering interventions. Two trials used a DPP‐4 inhibitor in the intervention arm (Martinez‐Abundis 2015; Rosenstock 2008); one used linagliptin 5 mg/day (Martinez‐Abundis 2015), and one used vildagliptin 50 mg/day (Rosenstock 2008). Five trials used GLP‐1 analogues in the intervention arms (Ariel 2014; Kelly 2012; McLaughlin 2011; Rosenstock 2010; SCALE). Two trials used liraglutide in the intervention arms (Ariel 2014; SCALE). One trial used liraglutide 1.8 mg/day (Ariel 2014), whereas the other trial used liraglutide 3.0 mg/day (SCALE). Three trials used exenatide in doses of 10 μg twice daily (Kelly 2012; McLaughlin 2011; Rosenstock 2010).
Five of the included trials had placebo as comparator (Ariel 2014; McLaughlin 2011; Rosenstock 2008; Rosenstock 2010; SCALE). Two trials used metformin as comparator (Kelly 2012; Martinez‐Abundis 2015); one trial used metformin 500 mg twice daily (Martinez‐Abundis 2015); the other trial used metformin 1000 mg twice daily (Kelly 2012) (see Appendix 3). In two trials, the participants did not take the study drug on the day glycaemic tests were performed (Kelly 2012; Rosenstock 2010). In two trials, the participants received the study drug after the glycaemic test had been performed (Kelly 2012; Rosenstock 2010). In one trial, the study drug was withheld on the days where the OGTTs were performed (every sixth month) (SCALE). However, the participants were allowed to take the study medication on the days where FPG was measured (except if OGTT was planned to be measured at the same visit) (SCALE). Three trials did not specify if the study drug was withheld the days of measuring glycaemic variables (Martinez‐Abundis 2015; McLaughlin 2011; Rosenstock 2008). In one trial, it was explicitly stated that the study drug was not given before the test meal at baseline but was given 15 minutes before the test meal at week 12. However, it was not described if the study drug was provided before the measurement of OGTT and fasting glucose (Rosenstock 2008).
Outcomes
Six trials had specified primary outcomes (Ariel 2014; Kelly 2012; McLaughlin 2011; Rosenstock 2008; Rosenstock 2010; SCALE). Five trials had not defined the secondary outcomes (Ariel 2014; McLaughlin 2011; Rosenstock 2008; Rosenstock 2010; SCALE). One trial did not specify primary or secondary outcomes (Martinez‐Abundis 2015). Six trials were registered at ClinicalTrials.gov (Ariel 2014; Kelly 2012; McLaughlin 2011; Rosenstock 2008; Rosenstock 2010; SCALE). The documented changes at ClinicalTrials.gov varied from 0 to 20 (see Appendix 7).
Four trials reported one or more of the primary outcomes of relevance for this review (Kelly 2012; Rosenstock 2008; Rosenstock 2010; SCALE). Three of them assessed the incidence of T2DM as an outcome (Rosenstock 2008; Rosenstock 2010; SCALE). The trials reporting data on T2DM applied different definitions. One trial did not report how the incidence of T2DM was defined, but defined T2DM as an exclusion criterion (FPG 7.0 mmol/L or greater or two‐hour post‐challenge plasma glucose (after an OGTT with glucose 75 g) 11.1 mmol/L or greater) (Rosenstock 2008). One trial used FPG 7.0 mmol/L or greater or two‐hour post‐challenge plasma glucose (after an OGTT with glucose 75 g) 11.1 mmol/L or greater and HbA1c values of 6.5% or more (SCALE). One trial did not report how T2DM was defined (Rosenstock 2010).
The reporting of adverse events was lacking in most trials. Four trials reported non‐serious adverse events experienced during the trial (Ariel 2014; Kelly 2012; Rosenstock 2008; SCALE) (see Appendix 11; Appendix 12; Appendix 13).
One trial reported data on HRQoL (SCALE) (see Appendix 17).
Six trials reported one or more glycaemic variables predefined to be assessed in our review (Ariel 2014; Kelly 2012; Martinez‐Abundis 2015; Rosenstock 2008; Rosenstock 2010; SCALE). One trial reported glycaemic variables in a format that made them unsuitable for meta‐analysis (McLaughlin 2011).
None of the included trials reported microvascular outcomes or socioeconomic effects.
Source of data
We contacted all trial authors or investigators through email (see Appendix 14). One trial could only be included when the investigators provided additional information on the trial (Martinez‐Abundis 2015). When important information was lacking on ongoing studies and excluded studies, we contacted investigators for clarification (see Appendix 14).
Excluded studies
We excluded 73 articles or records (45 trials) after full‐text evaluation (Figure 1). These references are listed in Characteristics of excluded studies table and some are detailed in Appendix 1.
We excluded seven trials published in 11 references due to the trial design (Aoki 2014; Armato 2012; EudraCT 2013‐001240‐64; Gonzalez‐Ortiz 2015; Koska 2015; UMIN000006197; Utzschneider 2008); three trials did not allocate the participants to DPP‐4 inhibitors or GLP‐1 analogues by randomisation (Armato 2012; UMIN000006197; Utzschneider 2008); one trial compared two different types of GLP‐1 analogues with each other (Gonzalez‐Ortiz 2015); and three trials were excluded as they used the study drug intravenously (Aoki 2014; EudraCT 2013‐001240‐64; Koska 2015).
We excluded 21 trials in 28 references as they did not include participants of relevance for this review (Acosta 2015; ACTRN12615001029583; BEGAMI 2013; Best 2015; Cui 2016; EudraCT 2011‐005980‐26; Gudipaty 2014; Larsen 2014; NCT00101712; NCT00198146; NCT00721552; NCT00886626; NCT01054118; NCT01346254; NCT01845259; NCT01970462; NCT02016846; NCT02022007; NCT02446834; UMIN000014249; Werzowa 2013). One of these trials included participants with acute coronary disease and intermediate hyperglycaemia (BEGAMI 2013). Participants were randomised to sitagliptin 100 mg/day or placebo for 12 weeks. The trial reported that none participants died; 1/24 participants in the sitagliptin group versus 4/23 participants in the placebo group developed T2DM according to the criteria for the WHO (FPG 7.0 mmol/L or greater (126 mg/dL or greater) or two‐hour plasma glucose 11.1 mmol/L or greater (200 mg/dL or greater)).
We excluded eight trials published in 15 records as it was not possible to obtain separate data on the participants of interest for our review, neither from the publication nor through correspondence with the investigators (Daniele 2015; Dushay 2012; Ishikawa 2014; NCT00845182; NCT01018602; NCT01122641; NCT01472640; Tsuchiya 2011). We contacted trial authors but did not receive additional data (see Appendix 14).
Six records representing five trials were never initiated or completed (NCT00845559; NCT00961363; NCT01006018; NCT01038648; NCT02284230).
We excluded four trials reported in 13 references because of a trial duration of less than 12 weeks (Almeda‐Valdes 2012; Bock 2010; Kaku 2015; Schwartz 2010) (Appendix 1). We screened five systematic reviews investigating people at increased risk for the development of T2DM for additional data (Anderson 2005; Bhardwaj 2010; Hopper 2011; Phung 2012; Van de Laar 2006).
Risk of bias in included studies
For details on the risk of bias of the included trials, see the Characteristics of included studies table.
For an overview of review authors' judgements about each risk of bias item for individual trials and across all trials, see Figure 2 and Figure 3.
2.
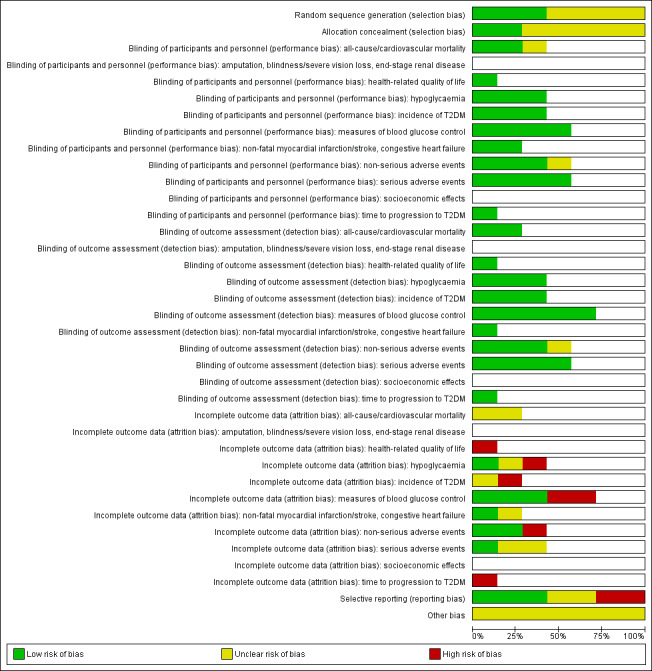
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included trials (blank cells indicate that the particular outcome was not measured in some trials).
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included trial (blank cells indicate that the particular outcome was not measured in some trials).
None of the included trials reported on microvascular outcomes or socioeconomic effects.
Allocation
We judged two trials at low risk of selection bias regarding the method of randomisation and allocation concealment (Ariel 2014; SCALE). One trial only reported the method of randomisation but not how allocation concealment was achieved (Martinez‐Abundis 2015). The remaining trials only reported that the participants were randomised but did not provide any further description (Kelly 2012; McLaughlin 2011; Rosenstock 2008; Rosenstock 2010). Therefore, these trials were judged at unclear risk of bias regarding randomisation and allocation concealment.
We evaluated trial baseline data for our predefined prognostic baseline variables. Only one trial reported all the prognostic baseline variables of interest, which all were balanced between the intervention groups (SCALE). The remaining trials only reported some of our predefined key prognostic variables of interest (Ariel 2014; Kelly 2012; Martinez‐Abundis 2015; McLaughlin 2011; Rosenstock 2008; Rosenstock 2010). None of the trials reporting one or more key prognostic variables showed important differences between the intervention groups (see Appendix 4; Appendix 5; Appendix 6).
Blinding
Six trials explicitly reported blinding of participants and investigators (Ariel 2014; Martinez‐Abundis 2015; McLaughlin 2011; Rosenstock 2008; Rosenstock 2010; SCALE). The blinding of participants and investigators was ensured by using placebo injections or tablets. In addition, one trial had a double‐blind intervention period for the first year (pharmaceutical company, investigators and participants were blinded), and a single‐blind period for the remaining two years (pharmaceutical company was unblinded, whereas investigators and participants remained blinded) (SCALE). One trial described a blinded outcome committee evaluating mortality, acute coronary syndromes, cerebrovascular events (stroke or transient ischaemic attacks), heart failure requiring hospitalisation, stent thrombosis, coronary revascularisations, pancreatitis or acute severe, persistent abdominal pain leading to a suspicion of pancreatitis, neoplasms, thyroid disorders requiring thyroidectomy, serious adverse events and severe hypoglycaemia (SCALE). None of the remaining trials reported that a blinded outcome committee was instituted to assess any of the reported outcomes.
Where measured, all primary outcomes of this review were investigator assessed and we judged these at low risk of performance and detection bias. The trials reporting blood glucose measurements were all performed by the investigators and we judged these outcomes measures at low risk of performance and detection bias.
When reported, non‐serious adverse events and mild hypoglycaemia were partly or exclusively self‐reported. Blinding of participants and investigators was ensured in three trials reporting these outcomes (Martinez‐Abundis 2015; Rosenstock 2008; SCALE). One trial with an open‐label design reported that no participants experienced a non‐serious adverse event (Kelly 2012). Overall, the risk of performance bias and detection bias was judged as low or unclear for our secondary outcomes.
Incomplete outcome data
Six trials reported the complete number of participants randomised and finishing the trial (Ariel 2014; Kelly 2012; Martinez‐Abundis 2015; McLaughlin 2011; Rosenstock 2008; SCALE). The percentage of randomised participants completing the trials varied from 49% to 100%. One trial did not describe how many participants were originally randomised but reported the number analysed (Rosenstock 2010).
Two trials stated that all randomised participants completed the trial and were included in the analyses (Kelly 2012; Martinez‐Abundis 2015). One trial reported the number of missing participants, which were balanced across the intervention groups and the number and reasons were unlikely to introduce clinically relevant bias (Rosenstock 2008).
One trial reported five participants were missing in each intervention group (Rosenstock 2008). Detailed reason were stated (vildagliptin group: adverse events (three participants); withdrew consent (one); protocol violation (one); lost to follow‐up (one); placebo group: adverse events (two); withdrew consent (one); protocol violation (two); lost to follow‐up (none) (Rosenstock 2008).
We judged two trials to have high risk of incomplete outcome data for the outcomes reported with relevance for our review (Ariel 2014; SCALE). In one trial, 31% in the liraglutide group and 18% in the placebo group dropped out during the trial. Eight of the dropouts were due to adverse events in the liraglutide group versus no dropouts happened because of adverse events in the placebo group (Ariel 2014). In one trial, only about half of the participants completed the trial (SCALE). Missing data were imputed with the LOCF. We judged this as high risk of attrition bias due to the large proportion of missing data and the method of imputation (SCALE). One trial was judged at unclear risk of bias regarding incomplete outcome data for the reported outcomes with relevance to our review (Rosenstock 2010). A total of 38 participants were randomised and 33 participants were analysed, which presumably received at least one dose of the study drug. The number of participants with IFG or IGT completing the visits, as well as reasons for the dropping out, were not reported. The method of imputation of missing data was not explained (Rosenstock 2010).
Selective reporting
All included trials, except one, had a published protocol (Martinez‐Abundis 2015). We judged two of the included trials at high risk of reporting bias on one or more of the outcomes of relevance to our review (Kelly 2012; McLaughlin 2011). Two trials had unclear risk of reporting bias (Martinez‐Abundis 2015; Rosenstock 2010). Three trials were at low risk of selective outcome reporting bias (Ariel 2014; Rosenstock 2008; SCALE). For more details, see Appendix 7 and Appendix 8.
Other potential sources of bias
Five of the included trials stated that they had received support from a pharmaceutical company (Ariel 2014; Kelly 2012; Rosenstock 2008; Rosenstock 2010; SCALE). Two trials did not report the funding source (Martinez‐Abundis 2015; McLaughlin 2011). It is known that trials receiving funding or provision of free drug or devices from a pharmaceutical company lead to more favourable results and conclusions than trials sponsored by other sources (Lundh 2017). Therefore, all eight trials were judged at unclear risk of bias in the 'other sources' bias‐domain.
Effects of interventions
See Table 1 and Table 2 for details.
Baseline characteristics
For details of baseline characteristics, see Appendix 4; Appendix 5; Appendix 6.
DPP‐4 inhibitors as monotherapy versus any pharmacological glucose‐lowering intervention (e.g. acarbose, metformin, sulphonylurea)
One trial compared a DPP‐4 inhibitor as monotherapy (linagliptin 5 mg in the morning plus a placebo tablet in the evening) with another pharmacological glucose‐lowering intervention (metformin 500 mg twice daily) (Martinez‐Abundis 2015). The corresponding investigator provided additional information regarding outcomes of relevance for this review (internal correspondence).
There were eight participants allocated to each group. None of the participants progressed to T2DM or experienced a serious adverse event. None of the participants experienced mild hypoglycaemia and severe hypoglycaemia was not assessed. One participant in the linagliptin group experienced a non‐serious adverse event versus four participants in the metformin group (Analysis 1.1).
1.1. Analysis.
Comparison 1 Dipeptidyl‐peptidase (DPP)‐4 inhibitors versus metformin, Outcome 1 Non‐serious adverse events.
The trial evaluated FPG, glucose concentrations two hours after an OGTT and HbA1c. The mean FPG concentration at the end of the intervention in the linagliptin plus placebo group was 5.5 (SD 0.6) mmol/L versus 5.7 (SD 0.7 ) mmol/L in the metformin group (MD ‐0.20 mmol/L, 95% CI ‐0.84 to 0.44; Analysis 1.2). Two‐hour post‐OGTT glucose concentrations were 9.0 (SD 0.9) mmol/L with linagliptin plus placebo versus 9.5 (SD 1.0) mmol/L with metformin (MD ‐0.50 mmol/L, 95% CI ‐1.43 to 0.43; Analysis 1.3). Mean HbA1c was 6.1% (SD 0.6%) linagliptin plus placebo versus 6.2% (SD 0.4%) with metformin (MD ‐0.10%, 95% CI ‐0.60% to 0.40%; Analysis 1.4).
1.2. Analysis.

Comparison 1 Dipeptidyl‐peptidase (DPP)‐4 inhibitors versus metformin, Outcome 2 Fasting blood glucose.
1.3. Analysis.

Comparison 1 Dipeptidyl‐peptidase (DPP)‐4 inhibitors versus metformin, Outcome 3 2‐hour glucose.
1.4. Analysis.

Comparison 1 Dipeptidyl‐peptidase (DPP)‐4 inhibitors versus metformin, Outcome 4 Haemoglobin A1c.
DPP‐4 inhibitors as monotherapy versus behaviour‐changing interventions (e.g. diet, exercise, diet plus exercise)
We identified no trials comparing DPP‐4 inhibitors as monotherapy with behaviour‐changing interventions.
DPP‐4 inhibitors as monotherapy versus placebo
One trial compared a DPP‐4 inhibitor (vildagliptin) as monotherapy with placebo only in people with IGT (Rosenstock 2008). The trial had a duration of intervention of 12 weeks.
Primary outcomes
All‐cause mortality
The trial reported that none of the participants died (very low‐quality evidence).
Incidence of type 2 diabetes mellitus
The trial defined T2DM as FPG 7.0 mmol/L or greater (126 mg/dL or greater) or two‐hour plasma glucose 11.1 mmol/L or greater (200 mg/dL or greater). Three out of 90 in the vildagliptin group versus 1/89 in the placebo group reported T2DM during the trial (Analysis 2.1) (very low‐quality evidence).
2.1. Analysis.

Comparison 2 DDP‐4 inhibitors versus placebo, Outcome 1 Incidence of type 2 diabetes mellitus.
Serious adverse events
A total of 1/90 participants in the vildagliptin group versus 2/89 participants in the placebo group had a serious adverse event (RR 0.49, 95% CI 0.05 to 5.36; Analysis 2.2) (very‐low quality evidence).
2.2. Analysis.

Comparison 2 DDP‐4 inhibitors versus placebo, Outcome 2 Serious adverse events.
Secondary outcomes
Cardiovascular mortality
The trial reported that none of the participants died (very low‐quality evidence).
Non‐fatal myocardial infarction
The trial did not report data on non‐fatal myocardial infarction (very low‐quality evidence).
Non‐fatal stroke
The trial did not report data on non‐fatal stroke (very low‐quality evidence).
Congestive heart failure
One out of 90 participants in the vildagliptin group compared with 0/89 participants in the placebo group experienced heart failure (Analysis 2.3) (very low‐quality evidence).
2.3. Analysis.

Comparison 2 DDP‐4 inhibitors versus placebo, Outcome 3 Congestive heart failure.
Amputation of lower extremity
The trial did not report data on amputation of lower extremity.
Blindness or severe vision loss
The trial did not report data on blindness or severe vision loss.
End‐stage renal disease
The trial did not report data on end‐stage renal disease.
Non‐serious adverse events
A total of 49/90 participants in the vildagliptin group versus 44/89 participants in the placebo group experienced non‐serious adverse events (RR 1.10, 95% CI 0.83 to 1.46; Analysis 2.4).
2.4. Analysis.

Comparison 2 DDP‐4 inhibitors versus placebo, Outcome 4 Non‐serious adverse events.
Hypoglycaemia
None of the participants in the trial experienced either mild or severe hypoglycaemic episodes.
Health‐related quality of life
The trial did not report data on HRQoL (very low‐quality evidence).
Time to progression to type 2 diabetes mellitus
The trial did not report data on time to progression to T2DM.
Measures of blood glucose control
The trial did not describe whether the trial drug was used the day of measuring glycaemic variables (Rosenstock 2008).
Fasting plasma glucose
The trial reported FPG concentrations as adjusted mean change from baseline. However, these adjustments were not specified. The MD of FPG for vildagliptin compared with placebo was ‐0.03 mmol/L (95% CI ‐0.21 to 0.15;179 participants; Analysis 2.5).
2.5. Analysis.
Comparison 2 DDP‐4 inhibitors versus placebo, Outcome 5 Fasting glucose.
Two‐hour plasma glucose concentrations (oral glucose tolerance test)
The trial reported glucose values two hours after an OGTT as adjusted mean change from baseline, but these adjustments were not specified (Rosenstock 2008). The MD of glucose values after an OGTT for vildagliptin compared with placebo was ‐0.30 mmol/L (95% CI ‐0.57 to ‐0.03; 179 participants; Analysis 2.6).
2.6. Analysis.
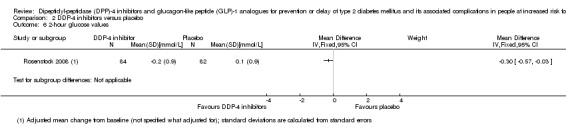
Comparison 2 DDP‐4 inhibitors versus placebo, Outcome 6 2‐hour glucose values.
Glycosylated haemoglobin A1c
The trial reported HbA1c as an adjusted mean change from baseline, but the adjustments were not further specified (Rosenstock 2008). The MD in HbA1c for vildagliptin compared with placebo was ‐0.15% (95% CI ‐0.24% to ‐0.06%; 179 participants; Analysis 2.7).
2.7. Analysis.

Comparison 2 DDP‐4 inhibitors versus placebo, Outcome 7 Haemoglobin A1c.
Socioeconomic effects
The trial did not report data on socioeconomic effects (very low‐quality evidence).
DPP‐4 inhibitors as monotherapy versus no intervention
We identified no trials comparing DPP‐4 inhibitors as monotherapy with no intervention.
DPP‐4 inhibitors as a part of a combination therapy versus any other pharmacological glucose‐lowering agent (e.g. acarbose, metformin, sulphonylurea)
We identified no trials comparing DPP‐4 inhibitors as a part of a combination therapy with any other pharmacological glucose‐lowering agent.
GLP‐1 analogues as monotherapy versus any other pharmacological glucose‐lowering intervention (e.g. acarbose, metformin, sulphonylurea)
One trial compared a GLP‐1 analogue as monotherapy (exenatide 5 μg twice daily for the first month followed by 10 μg twice daily for the remaining two months) with another glucose‐lowering intervention (metformin initiated at 500 mg twice daily for the first month, and thereafter uptitrated to 1000 mg twice daily for the remaining two months) (Kelly 2012).
Primary outcomes
All‐cause mortality
None of the participants died.
Incidence of type 2 diabetes mellitus
The trial did not report data on the incidence of T2DM.
Serious adverse events
None of the participants experienced serious adverse events.
Secondary outcomes
Cardiovascular mortality
None of the participants died.
Non‐fatal myocardial infarction
The trial did not report data on non‐fatal myocardial infarction.
Non‐fatal stroke
The trial did not report data on non‐fatal stroke.
Congestive heart failure
The trial did not report data on congestive heart.
Amputation of lower extremity
The trial did not report data on amputation of the lower extremity.
Blindness or severe vision loss
The trial did not report data on blindness or severe vision loss.
End‐stage renal disease
The trial did not report data on end‐stage renal disease.
Non‐serious adverse events
None of the participants reported non‐serious adverse events. The threshold above which other adverse events were reported was 5%. Consequently, non‐serious adverse events may have been under‐reported due to low number of total events. Therefore, this trial was judged at high risk of selective outcome reporting bias regarding non‐serious adverse events.
Hypoglycaemia
None of the participants experienced hypoglycaemic events.
Health‐related quality of life
The trial did not report data on HRQoL.
Time to progression to type 2 diabetes mellitus
The trial did not report data on time to progression to T2DM.
Measures of blood glucose control
Glycaemic testing was performed in the morning after fasting. The trial drug was withheld on these mornings. The only glycaemic variable reported in the trial was change in fasting glucose from baseline. The change from baseline was ‐0.2 (SD 0.6) mmol/L in 25 participants allocated to exenatide versus ‐0.2 (SD 0.5) mmol/L in 25 participants allocated to metformin (MD 0.00 mmol/L, 95% CI ‐0.31 to 0.31; Analysis 3.1).
3.1. Analysis.
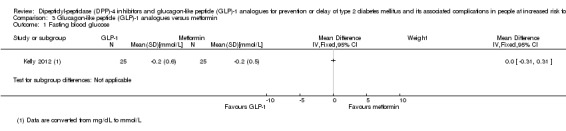
Comparison 3 Glucagon‐like peptide (GLP)‐1 analogues versus metformin, Outcome 1 Fasting blood glucose.
Socioeconomic effects
The trial did not report data on socioeconomic effects.
GLP‐1 analogues as monotherapy versus behaviour‐changing interventions (e.g. diet, exercise, diet and exercise)
We identified no trials comparing GLP‐1 analogues as monotherapy with behaviour‐changing interventions.
GLP‐1 analogues as monotherapy versus placebo
Four trials compared a GLP‐1 analogue as monotherapy with placebo (Ariel 2014; McLaughlin 2011; Rosenstock 2010; SCALE). One trial reported no outcomes of relevance to this review (McLaughlin 2011). Two trials used the GLP‐1 analogue exenatide (McLaughlin 2011; Rosenstock 2010); the other two trials used liraglutide (Ariel 2014; SCALE). In both the exenatide trials, exenatide was uptitrated to 10 μg twice daily (McLaughlin 2011; Rosenstock 2010). One of the trials randomising the participants to liraglutide used a dose of 1.8 mg (Ariel 2014); the other trial used 3.0 mg (SCALE). One of the trials included participants with and without intermediate hyperglycaemia (Rosenstock 2010). However, it was possible to retrieve data for some outcomes on the participants with IGT or IFG at baseline (38/152 participants) (Rosenstock 2010).
Primary outcomes
All‐cause mortality
One trial reported that no participants died (Rosenstock 2010). Another trial reported that four participants died; two in the liraglutide group (due to cardiac arrest and metastatic cholangiocarcinoma) and two in the placebo group (pulmonary failure and cancer (primary tumour unknown) (SCALE) (very low‐quality evidence).
Incidence of type 2 diabetes mellitus
Two trials comparing GLP‐1 analogues as monotherapy with placebo reported data on the incidence of T2DM (Rosenstock 2010; SCALE). In the Satiety and Clinical Adiposity ‐ Liraglutide Evidence in Nondiabetic and Diabetic Individuals (SCALE) trial, participants were not supposed to take the study medication in the morning on the OGTT days. The OGTT was performed about every sixth month and FPG and HbA1c were measured every third month. The participants were allowed to take the trial medication on the days when FPG was measured (except if the OGTT was planned at the same visit). The diagnosis of T2DM was defined as HbA1c 6.5% or greater, FPG 7.0 mmol/L or greater or two‐hour plasma glucose 11.1 mmol/L or greater (OGTT). The T2DM incidence was reported after 56 weeks of the intervention and after 160 weeks of the intervention period with an extended follow‐up period of 12 weeks after the intervention was stopped (i.e. 172 weeks). At 56 weeks, four participants in the liraglutide group and 13 participants in the placebo group developed T2DM (SCALE). At 160 weeks, 26 participants in the liraglutide group and 46 participants in the placebo group developed T2DM (SCALE). After the extension period (i.e. after 172 weeks), five additional participants were diagnosed with T2DM in the liraglutide group compared with one participant in the placebo group. Missing data were imputed with LOCF (SCALE) (very low‐quality evidence).
A post‐hoc analysis was performed to address the lack of follow‐up information for withdrawn participants at week 172, and assumed that 1% of those withdrawn in the liraglutide group had undiagnosed T2DM at withdrawal, whereas none of those in the placebo group did (HR 0.34, 95% CI 0.22 to 0.53; P < 0.0001).
Of the 1468 participants in the liraglutide group, several glycaemic indices at screening were associated with the later development of T2DM after 160 weeks of the intervention.
173 with IFG: one (0.6%) progressed to T2DM.
158 with IGT: two (1.3%) progressed to T2DM.
376 with elevated HbA1c: one (0.3%) progressed to T2DM.
103 with IFG plus IGT: none (0%) progressed to T2DM.
262 with IFG plus elevated HbA1c: nine (3.4%) progressed to T2DM.
161 with IGT plus elevated HbA1c: three (1.9%) progressed to T2DM.
235 with IFG plus IGT plus elevated HbA1c: 10 (3.8%) progressed to T2DM.
Of the 736 participants in the placebo group, several glycaemic indices at screening were associated with the later development of T2DM after 160 weeks of the intervention.
90 with IFG: four (4.4%) progressed to T2DM.
94 with IGT: one (1.1%) progressed to T2DM.
187 with elevated HbA1c: five (2.7%) progressed to T2DM.
46 with IFG plus IGT: two (4.3%) progressed to T2DM.
128 with IFG plus elevated HbA1c: 11 (8.6%) progressed to T2DM.
75 with IGT plus elevated HbA1c: four (5.3%) progressed to T2DM.
116 with IFG plus IGT plus elevated HbA1c: 18 (15.5%) progressed to T2DM.
The numbers above was presented in an appendix of the publication of the long‐term data from the SCALE trial (Le Roux 2017). However, the article included 1472 participants in the liraglutide group and 738 in the placebo group (Le Roux 2017). The reason for the discrepancy in number of participants included in the analysis as well in the number of participants developing T2DM (45 instead of 46 participants) was not stated. The corresponding author of the article was contacted twice, but no reply was provided (Appendix 14).
In the SCALE trial after 160 weeks of the intervention, 970/1472 (66%) participants had regressed from intermediate hyperglycaemia to normoglycaemia compared with 268/738 (36%) participants in the placebo group. After 12‐week treatment cessation, 740/1472 (50%) participants in the liraglutide group compared with 263/738 (36%) participants in the placebo group had normoglycaemia.
The other trial reporting T2DM incidence stated that the trial drug was withheld before the OGTT (Rosenstock 2010). The trial had a final follow‐up visit four weeks after trial completion. There was no definition of the T2DM diagnosis provided (Rosenstock 2010).
The RR for the incidence of T2DM differed for the analysed GLP‐1 analogues: exenatide therapy compared with placebo was associated with an RR of 1.88 (95% CI 0.19 to 18.80; 2/17 participants in the GLP‐1 analogue group versus 1/16 participants in the placebo group; Analysis 4.1). Liraglutide treatment compared with placebo was associated with an RR of 0.28 (95% CI 0.18 to 0.45; 26/1472 (1.8%) participants in the liraglutide group versus 46/738 (6.2%) participants in the placebo group; Analysis 4.1).
4.1. Analysis.

Comparison 4 Glucagon‐like peptide (GLP)‐1 analogues versus placebo, Outcome 1 Incidence of type 2 diabetes mellitus.
TSA showed that 0.36% of the diversity‐adjusted information size was accrued to detect or reject a 10% RRR. Diversity was 93%. As only a minor fraction of the diversity‐adjusted required information size was accrued, the TSA‐adjusted 95% CI could not be calculated.
Serious adverse events
Two trials comparing GLP‐1 analogues as monotherapy with placebo reported data on serious adverse events (Rosenstock 2010; SCALE). One of the trials reported that none of the participants experienced a serious adverse event (Rosenstock 2010). The other trial reported two different numbers for serious adverse events in 227/1501 participants (15%) in the liraglutide arm versus 96/747 participants (13%) in the placebo arm after 160 weeks (RR 1.18, 95% CI 0.94 to 1.47; Analysis 4.2; very low‐quality evidence). In ClinicalTrials.gov, serious adverse events were reported in 230/1524 (14.9%) participants the liraglutide arm versus 96/755 (12.7%) participants in the placebo arm after 172 weeks (i.e. including 12 weeks after the intervention had stopped) (SCALE).
4.2. Analysis.
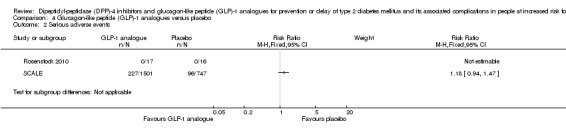
Comparison 4 Glucagon‐like peptide (GLP)‐1 analogues versus placebo, Outcome 2 Serious adverse events.
Secondary outcomes
Cardiovascular mortality
One trial reported that no participants died (Rosenstock 2010). Another trial reported that 1/1501 participants died from cardiac arrest in the liraglutide group and none of the participants in the placebo group died of cardiovascular reasons (SCALE) (very low‐quality evidence).
Non‐fatal myocardial infarction
One trial reported data on myocardial infarction (SCALE). Under the serious adverse events, 1/1524 participants in the liraglutide arm and 0/755 participants in the placebo arm experienced a myocardial infarction at 172 weeks (very low‐quality evidence).
Non‐fatal stroke
None of the trials reported data on non‐fatal stroke (very low‐quality evidence).
Congestive heart failure
One trial reported data on heart failure (SCALE). Under serious adverse events, 1/1524 participants in the liraglutide arm and 1/755 participants in the placebo arm experienced congestive heart failure at 172 weeks (very low‐quality evidence).
Amputation of lower extremity
None of the trials reported data on amputation of lower extremity.
Blindness or severe vision loss
None of the trials reported data on blindness or severe vision loss.
End‐stage renal disease
None of the trials reported data on end‐stage renal disease loss.
Non‐serious adverse events
One trial reported data on non‐serious adverse events (SCALE). A total of 1342/1524 (88.1%) participants in the liraglutide arm versus 586/755 (77.6%) participants in the placebo arm experienced a non‐serious adverse event at 172 weeks (RR 1.13, 95% CI 1.09 to 1.18 in favour of placebo; Analysis 4.3). Gastrointestinal disorders were the most common adverse effects in the liraglutide group; 41% of the participants in the liraglutide group reported nausea; 25% diarrhoea and 22% constipation; compared with 17% nausea; 14% diarrhoea and 11% constipation in the placebo group. Gastrointestinal adverse events were also the most common cause of withdrawal (118/1501 (8%) participants in the liraglutide group versus 11747 (2%) participants in the placebo group) (SCALE).
4.3. Analysis.

Comparison 4 Glucagon‐like peptide (GLP)‐1 analogues versus placebo, Outcome 3 Non‐serious adverse events.
Hypoglycaemia
Two trials reported data on the incidence of hypoglycaemic episodes (Rosenstock 2010; SCALE). One of the trials reported that no participant experienced mild hypoglycaemia (Rosenstock 2010). The SCALE trial reported hypoglycaemia as a non‐serious adverse event after 172 weeks in 295/1524 (19.4%) participants in the liraglutide group and in 35/755 (4.6%) participants in the placebo group (RR 4.18, 95% CI 2.97 to 5.86 in favour of placebo; Analysis 4.4). More individuals with liraglutide compared with placebo had biochemical hypoglycaemia adverse events during fasting plasma glucose visits (3.6% versus 0.8%).
4.4. Analysis.

Comparison 4 Glucagon‐like peptide (GLP)‐1 analogues versus placebo, Outcome 4 Mild hypoglycaemia.
One of the trials reported that none of the participants experienced severe hypoglycaemia (Rosenstock 2010). No severe hypoglycaemic events were defined as serious or requiring third party assistance in the other trial (SCALE).
Health‐related quality of life
One trial reported HRQoL using the 36‐item Short Form (SF‐36) (see Appendix 17) (SCALE). Changes in scores were reported from baseline to week 160. Participants on liraglutide 3.0 mg had greater mean improvements in the physical functioning component score (PCS) (MD 0.87 points, 95% CI 0.17 to 1.58; P = 0.02). There was no substantial difference for the mental component score (MCS) between the interventions (MD 0.88, 95% CI ‐0.09 to 1.63; P = 0.08; very low‐quality evidence). In this cohort, a one point lower score on the PCS is approximately associated with an RR for mortality of 1.09 (Bjorner 2013).
The total number of randomised participants was 1203 in the liraglutide group versus 588 in the placebo group; the number of participants completing the 160 weeks' follow‐up was 586 in the liraglutide group and 244 in the placebo group (SCALE). HRQoL was only evaluated in participants from countries with validated translations of SF‐36 (14/29 countries). The method of imputation for missing data was LOCF.
Time to progression to type 2 diabetes mellitus
The mean time from randomisation to the diagnosis of T2DM was 99 (SD 47) weeks for the 26 participants who developed T2DM during the trial in the liraglutide group versus 87 (47) weeks for the 46 participants who developed T2DM during the trial in the placebo group. The time to onset of diabetes over 160 weeks among all participants was 2.7 times longer with liraglutide than with placebo (95% CI 1.9 to 3.9; P < 0.0001), corresponding with an HR of 0.21 (95% CI 0.13 to 0.34) (SCALE). None of the predefined sensitivity analyses (e.g. completer population, excluding potentially unblinded participants) showed any influence on the time of onset of T2DM. No sensitivity analyses were performed according to diagnostic criteria (IFG, IGT, HbA1c), age, gender, ethnicity or BMI.
Measures of blood glucose control
Two trials reported data on glycaemic variables that could be used in our review (Ariel 2014; SCALE). In the SCALE trial, the participants were not supposed to take the trial medication in the morning on the OGTT days. OGTT was performed every sixth month. FPG and HbA1c were measured every third month. The participants were allowed to take the trial medication on the days when FPG was measured (except if OGTT was planned to be measured at the same visit) (SCALE). In the other trial, the medication was given after glycaemic testing (Ariel 2014). In the SCALE trial, the mean changes in glycaemic variables were reported between baseline and week 56 and week 160.
In one trial (published as an abstract only), it was stated that FPG and two‐hour blood glucose concentrations were measured, but data were reported in a format unsuitable for meta‐analyses. Useable data might be available in updates (McLaughlin 2011).
Fasting plasma glucose
Two trials reported data on fasting plasma glucose concentrations (Ariel 2014; SCALE). One of the trials reported end of follow‐up concentrations of fasting plasma glucose (Ariel 2014). The other trial reported change from baseline after 56 and 160 weeks of intervention (SCALE). At 56 weeks, the change in FPG from baseline was ‐0.46 mmol/L in 1495 participants in the liraglutide group and ‐0.006 mmol/L in 746 participants in the placebo group (glucose values were converted from mg/dL to mmol/L by multiplying by 0.0555; numbers were read from a figure in the primary publication). The change in FPG from baseline to week 160 was ‐0.37 (SD 0.68) mmol/L in 1472 participants in the liraglutide group and 0.05 (SD 0.62) mmol/L in 738 participants in the placebo group (estimated MD ‐0.42 mmol/L, 95% CI ‐0.48 to ‐0.36; Analysis 4.5). After 12 weeks of treatment cessation, the effect of liraglutide on fasting plasma glucose was reduced (a change in FPG in the liraglutide group of ‐0.08 (SD 0.66) mmol/L in 783 participants versus a change in FPG in the placebo group of 0.06 (0.67) mmol/L in 326 participants (estimated treatment difference ‐0.13 mmol/L, 95% CI ‐0.21 to ‐0.05).
4.5. Analysis.
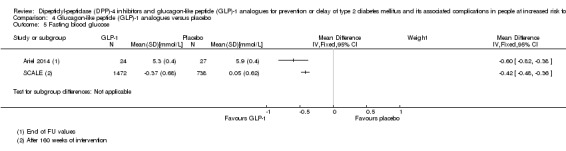
Comparison 4 Glucagon‐like peptide (GLP)‐1 analogues versus placebo, Outcome 5 Fasting blood glucose.
TSA could not be performed as the required information size was exceeded already when analysing one trial.
Two‐hour plasma glucose concentrations (oral glucose tolerance test)
One trial reported data on two‐hour plasma glucose concentrations (SCALE). Glucose values two hours after an OGTT could be read from a figure in the primary publication. In week 56, two‐hour plasma glucose concentrations were estimated to be 5.4 mmol/L in the 1495 participants of the liraglutide group versus 7.1 mmol/L in the 746 participants of the placebo group. Data after 160 weeks were presented at the European Association for the Study of Diabetes meeting in 2016; two‐hour glucose concentration was 7.4 (SD 1.8) mmol/L in the 1505 participants of the liraglutide group versus 7.4 (SD 1.7) mmol/L in the 749 participants of the placebo group. In the publication reporting data after 160 weeks of follow‐up, the two‐hour glucose concentration was lowered by ‐1.6 (SD 2.1) mmol/L in the 1472 participants of the liraglutide group versus ‐0.2 (2.2) mmol/L in the 738 participants of the placebo group (MD ‐1.40, 95% CI ‐1.59 to ‐1.21 in favour of liraglutide; Analysis 4.6). By week 172, the effects on two‐hour glucose levels were comparable with placebo levels (SCALE).
4.6. Analysis.

Comparison 4 Glucagon‐like peptide (GLP)‐1 analogues versus placebo, Outcome 6 2‐hour glucose.
Glycosylated haemoglobin A1c
One trial reported data on HbA1c (SCALE). The mean change from baseline in HbA1c was ‐0.32% (SD 0.34%) in the 1495 participants of the liraglutide group and ‐0.07% (SD 0.34%) in the 746 participants of the placebo group (MD ‐0.25%, 95% CI ‐0.28 to ‐0.22 in favour of liraglutide; Analysis 4.7). Estimated treatment difference for liraglutide versus placebo was ‐0.25% (95% CI ‐0.28% to ‐0.23%; P < 0.001). At the EASD meeting 2016, HbA1c at week 160 was reported as 5.8% (SD 0.3%) in the 1505 participants of the liraglutide group and 5.7% (SD 0.3%) in the 749 participants of the placebo group (SCALE). In the publication reporting data after 160 weeks of follow‐up, the HbA1c was lowered with ‐0.35% (SD 0.32%) in the 1472 participants of the liraglutide group versus ‐0.14% (0.34%) in the 738 participants of the placebo group after. After 12 weeks of treatment cessation, the effect of liraglutide on fasting plasma glucose were no longer evident (SCALE).
4.7. Analysis.

Comparison 4 Glucagon‐like peptide (GLP)‐1 analogues versus placebo, Outcome 7 Haemoglobin A1c.
Socioeconomic effects
None of the trials reported data on socioeconomic effects.
GLP‐1 analogues as monotherapy versus no intervention
We identified no trials comparing GLP‐1 analogues as monotherapy with no intervention.
GLP‐1 analogues as a part of a combination therapy versus any pharmacological glucose‐lowering agent (e.g. acarbose, metformin, sulphonylurea)
We identified no trials comparing GLP‐1 analogues as a part of a combination therapy with any pharmacological glucose‐lowering agent.
Subgroup analyses
We did not perform subgroups analyses because there were insufficient trials to estimate effects in various subgroups.
Sensitivity analyses
We did not perform sensitivity analyses because there were insufficient trials to explore the influence of our predefined factors on effect sizes.
Assessment of reporting bias
We did not draw funnel plots due to limited number of trials (seven).
Discussion
Summary of main results
This Cochrane Review is the first systematic review investigating the effects of DPP‐4 inhibitors and GLP‐1 analogues compared with other pharmacological glucose‐lowering interventions, placebo, diet and exercise, or no intervention in people at increased risk for developing T2DM. We included seven trials with 2702 participants. We judged all trials to have unclear or high risk of bias in one or more 'Risk of bias' domains. The amount of evidence on patient‐important outcomes was limited. Use of DPP‐4 inhibitors neither revealed an advantage nor a disadvantage for the incidence of T2DM. Use of liraglutide has the potential to delay the onset of T2DM but the evidence base is weak. For incidence of T2DM as well as for all‐cause mortality and cardiovascular mortality, we judged the quality of evidence as very low.
Overall completeness and applicability of evidence
We conducted an extensive search for trials, including publications in all languages, and tried to obtain additional data on all trials. However, no additional data were provided. We looked for additional trials and cross‐checked our data with the data from other meta‐analyses and Cochrane Reviews of relevance (Anderson 2005; Bhardwaj 2010; Hopper 2011; Phung 2012; Van de Laar 2006).
The diagnosis of intermediate hyperglycaemia was mostly established by the criteria from the WHO and ADA. Only one trial did not specify how the diagnosis of intermediate hyperglycaemia was established (Martinez‐Abundis 2015). The included trials used different types of DPP‐4 inhibitors (Martinez‐Abundis 2015; Rosenstock 2008), as well as GLP‐1 analogues (Ariel 2014; Kelly 2012; McLaughlin 2011; Rosenstock 2010; SCALE). Moreover, one of the included trials used the GLP‐1 analogue in doses approved for weight reduction (SCALE).
A potential selection bias exists as more healthy and motivated people may participate in a clinical trial. However, one Cochrane systematic review observed that clinical outcomes in people participating in RCTs are comparable to outcomes in comparable individuals outside RCTs (Vist 2008).
Quality of the evidence
For all trials, we contacted one or more authors to obtain supplemental information on baseline data, bias domains and outcomes. In addition, authors were asked to confirm risk of bias assessments, extracted outcome data and other issues if unclear in the publication. Two trial investigators (29%) either just confirmed a question or provided additional data that could be implemented for the risk of bias assessment or the meta‐analyses of outcomes (Martinez‐Abundis 2015; SCALE). We excluded eight trials because they did not provide separate data on participants with intermediate hyperglycaemia (Daniele 2015; Dushay 2012; Ishikawa 2014; NCT01018602; NCT01122641; NCT01472640; NCT00845182; Tsuchiya 2011). We contacted all the investigators of these trials, but due to lack of additional information, these could not be included. Three trials including participants with intermediate hyperglycaemia as a part of the study population might be included in the updates of this review (Astrup 2009; SCALE‐SLEEP; SCALE 2013). Application to the sponsor has been send to request additional data. We made a concerted effort to obtain additional data from all trial authors. If we were unable to retrieve contact information for the corresponding author, we attempted to contact one of the coauthors. For all trials, we identified contact information for one or more authors.
None of the seven included trials in our review was classified as having low risk of bias in all 'Risk of bias' domains. In general, the description of randomisation and allocation in the included studies was insufficient. Three trials had insufficient reporting of one or more outcomes of relevance to our review and were, therefore, classified as having high risk of bias for selective outcome reporting bias. We were able to assess one or more of our predefined outcomes in six of the seven included trials. The largest of the included trials imputed missing data with LOCF (SCALE), which is not an optimal imputation method (Siddiqui 2009). Therefore, all outcomes reported from this trial were judged at high risk of incomplete outcome data.
For the DPP‐4 inhibitors and the GLP‐1 analogues, we judged the quality of evidence to be low or very low because of very limited data and various risk of bias.
Most trials had received free drugs or financial funding from the pharmaceutical industry. It is known that trials receiving funding or provision of free drugs or devices from a pharmaceutical company lead to more favourable results and conclusions compared to trials sponsored by other sources (Lundh 2017).
Potential biases in the review process
We were unable to draw funnel plots to assess small‐study bias due to lack of data. If more data had been available and more meta‐analyses could have been performed, we would have investigated heterogeneity and the potential reasons for it.
We were dealing with a substantial heterogeneous group of trials. Our meta‐analyses, when performed, were limited by the inability to use individual participant data to assess whether distinct clinical characteristics may have influenced the effect estimates of the interventions. We would have explored heterogeneity using sensitivity analyses for our patient‐important outcomes, if possible. However, data were limited, so no subgroup analyses or sensitivity analyses were performed. Many of the included trials were not designed or powered to detect our predefined patient‐important outcomes.
Some trials required the participants to take the study drug on the days the glycaemic variables were measured, whereas others did not. This may have influenced the glucose measurements in these trials, as well as the incidence of T2DM (which is based on glycaemic measurements) making it difficult to reliably compare incidence rates.
Most of the included trials had a relatively small number of participants and the information sizes in the meta‐analyses were equally small. This increases the risk of unrealistic estimates of the intervention effects due to bias (systematic errors) and chance (random errors) (Wetterslev 2008; Wood 2008). We have attempted to clarify systematic errors. We contacted all trial authors for clarification if one of the bias domains was not adequately reported. To reduce the risk of random errors, we conducted TSA on all predefined outcomes, whenever possible.
Several trials were published in more than one publication, which for some trials made it difficult to separate the primary publication from companion papers (for details, see Included studies).
We excluded trials in participants with IGT due to other conditions (e.g. cystic fibrosis or glucocorticoid treatment).
We included trials with a minimum duration of 12 weeks to detect clinically relevant differences for the predefined outcomes. We identified four trials with a duration of less than 12 weeks (Almeda‐Valdes 2012; Bock 2010; Kaku 2015; Schwartz 2010). Unfortunately, the reporting of long‐term data in the included trials was poor.
Two review authors carried out data extraction. However, the review authors extracting the data were not blinded as to which trial they were extracting data from.
Agreements and disagreements with other studies or reviews
Several RCTs have assessed the effects of different pharmacological glucose‐lowering interventions for the prevention of T2DM (DeFronzo 2011; Diabetes Prevention Program 2009; SCALE). A pharmacological approach to the prevention or delay of T2DM is appealing to both the clinician and the pharmaceutical industry. However, although a reduction in, or delay of, the incidence of T2DM is important, the major public health impact of prevention trials will be determined by the extent to which prevention or delay of T2DM will translate into a reduction in diabetes‐specific macrovascular and microvascular complications.
Given the intertwined relationship between obesity and T2DM, one well‐established approach to reduce the risk of T2DM is weight loss. Weight loss with liraglutide is dose‐dependent up to 3.0 mg once daily. In the SCALE trial, participants lost a mean of 6.5 (SD 7.3) kg of body weight versus 2.0 (SD 7.3) kg in the placebo group after 160 weeks of intervention. Liraglutide reduced the incidence or delayed the onset of T2DM (SCALE). Twelve weeks after the end of intervention, a mean weight regain occurred in the liraglutide group, so the total loss of mean weight was 5.6 (SD 9.2) kg in the liraglutide group versus 2.2 (SD 8.4) kg in the placebo group. The people progressing to T2DM during the study period had a median weight gain of 0.3% in the liraglutide group and 1.7% in the placebo group (SCALE). The number of participants progressing from intermediate hyperglycaemia to T2DM was low. The effect on reducing the risk of T2DM with liraglutide seemed to depend on how intermediate hyperglycaemia was measured. It appeared more pronounced in people with IFG, intermediate elevated HbA1c levels, or both. However, the number of participants with different subtypes of intermediate hyperglycaemia was low. The subtype of intermediate hyperglycaemia with the lowest incidence of T2DM in the placebo group was people with IGT only. Furthermore, 12 weeks after the end of intervention, five participants in the liraglutide group compared with one participant in the placebo group developed T2DM. After 160 weeks of the intervention, 970/1472 (66%) participants had regressed from intermediate hyperglycaemia to normoglycaemia compared with 268/738 (36%) in the placebo group. Furthermore, after 12‐week treatment cessation, 740/1472 (50%) participants in the liraglutide group compared with 263/738 (36%) participants in the placebo group had normoglycaemia. Even though the duration of post‐treatment follow‐up was short, the data did not indicate long‐term beneficial effects on the incidence of T2DM. Also, the glycaemic improvements in HbA1c and fasting plasma glucose were no longer evident. Whether liraglutide is more effective in the longer term (e.g. 10 years) or whether other GLP‐1 analogues are more effective than other interventions (e.g. metformin, diet and exercise) recommended by the ADA to prevent the progression to T2DM and its associated complications remains to be confirmed (ADA 2015).
Several ongoing trials investigating the effects of a DPP‐4 inhibitors or a GLP‐1 analogues in people at increased risk for the development of T2DM exist. This reflects the interest from the scientific community and the pharmaceutical companies as several of the DPP‐4 inhibitors and GLP‐1 analogues still are patent‐registered.
Authors' conclusions
Implications for practice.
There is no firm evidence whether dipeptidyl‐peptidase (DPP)‐4 inhibitors or glucagon‐like peptide (GLP)‐1 analogues compared with other pharmacological glucose‐lowering interventions, placebo, behaviour‐changing interventions or no intervention substantially influence the risk of type 2 diabetes mellitus and its associated complications. Data on patient‐important outcomes such as mortality, and macrovascular and microvascular complications are sparse.
Implications for research.
It remains to be clarified whether there are any substantial beneficial or harmful effects of DPP‐4 inhibitors or GLP‐1 analogues in people with intermediate hyperglycaemia if given for a prolonged period of time. Several ongoing trials are investigating this topic. Future randomised controlled clinical trials should focus on patient‐important outcomes.
Notes
Portions of the background and methods sections, the appendices, additional tables and figures 1 to 3 of this review are based on a standard template established by the Cochrane Metabolic and Endocrine Disorders Group.
Acknowledgements
Dr Rickels, Dr Chuang, Dr Elkind‐Hirsch, Dr Tuomilehto, Dr Ramachandran, Dr Gutierrez, Dr Vilsboll, Dr Watson, Dr Villagomez, Dr Much, Dr Santilli, Dr Gabriel and Dr Zhou for replying our requests. Dr Martínez‐Abundis and Dr Hage for providing unpublished outcome data with relevance to our review.
Appendices
Appendix 1. Trials with a duration of less than 12 weeks
| Trial | Intervention and comparator | Design | Duration of intervention | Randomised (n) | Description of participants | Outcomes reported | Outcomes reported with interest of this review | Stated purpose of study |
| Almeda‐Valdes 2012 | I: sitagliptin 100 mg/day, PO | Assume single centre, double blind (blinded to participants and investigators), parallel | 2 weeks | 13 | Intermediate hyperglycaemia not defined, only described as clinical and biochemical diagnosis of prediabetic reactive hypoglycaemia. Mainly non‐obese women | Area under curve in early and late insulin secretion phase, symptoms of reactive hypoglycaemia | None | Quote: "The purpose of this study is to determine whether sitagliptin is effective in the treatment of reactive hypoglycemia by dysinsulinism." |
| C: placebo, PO | 15 | |||||||
| Bock 2010 | I: sitagliptin 100 mg/day, PO | Single centre, double blind (blinded to participants and investigators), parallel | 8 weeks | 11 | Impaired fasting glucose, mainly obese women | Fasting and postprandial glucose, insulin and C‐peptide, glucagon‐like peptide, glucose‐dependent insulinotropic polypeptide, glucagon and endogenous glucose production. Glucose disappearance, systemic meal appearance, insulin action, insulin secretion | Postprandial glucose, fasting glucose | Quote: "The current experiments tested this hypothesis by measuring insulin secretion and action and fasting and postprandial glucose turnover before and after 8 weeks of therapy with a DPP‐4 inhibitor." |
| C: placebo, PO | 11 | |||||||
| Kaku 2015 | I1: sitagliptin 25 mg/day, PO | Multicentre, double blind (blinded to participants and investigators), parallel | 8 weeks | 82 | Japanese people, IGT | Glucose, glucagon and insulin area under the curve 0‐2 hour during meal tolerance tests and oral glucose tolerance test, HbA1c, fasting glucose, 2‐hour glucose, adverse events, hypoglycaemia, DDP‐4 activity, reversion to normoglycaemia, electrocardiogram changes | HbA1c, fasting glucose, 2‐hour glucose, serious adverse events, adverse events, hypoglycaemia | Quote: "To evaluate the efficacy and tolerability of sitagliptin in subjects with impaired glucose tolerance (IGT)." |
| I2: sitagliptin 50 mg/day, PO | 77 | |||||||
| C: placebo, PO | 83 | |||||||
| Schwartz 2010 | I: exenatide 10 μg/day, subcutaneous (administered prior to a high‐calorie, fat‐enriched breakfast meal) | Assumed single centre, double blind (blinded to participants and investigators), cross‐over | Participants studied twice within 1‐3 weeks. Only single injection exenatide and placebo | 35 | Mainly men, 20 participants had IGT and 15 had recent‐onset diet controlled T2DM. The authors provided separate data for the participants with IGT | Concentrations of triglycerides, apolipoproteins B‐48 and CIII, non‐esterified fatty acids, remnant lipoprotein cholesterol and triglycerides in serum or plasma, endothelial function, measured prior to the injection and 8 h postprandially | None | Quote: "In the present study, we tested the effects of a single acute injection of exenatide to determine what direct benefits (in the absence of changes in satiety, weight loss and other chronic effects) this agent may have on increments in triglycerides, apolipoproteins, and cholesterol‐and triglyceride‐rich remnant particles, following a standardized fat‐enriched meal challenge." |
| C: placebo, subcutaneous (administered prior to a high‐calorie, fat‐enriched break fast meal) | ||||||||
| C: control; DPP‐4: dipeptidyl‐peptidase‐4; HbA1c: glycosylated haemoglobin A1c; I: intervention; IGT: impaired glucose tolerance; n: number of participants; PO: per os (orally); T2DM: type 2 diabetes mellitus. | ||||||||
Appendix 2. Search strategies
| Cochrane Central Register of Controlled Trials (Cochrane Register of Studies Online) |
| 1. MESH DESCRIPTOR Prediabetic state 2. MESH DESCRIPTOR Glucose Intolerance 3. (prediabet* or pre diabet*):TI,AB,KY 4. (intermediate hyperglyc?emi*):TI,AB,KY 5. ((impaired fasting ADJ2 glucose) or IFG or impaired FPG):TI,AB,KY 6. glucose intolerance:TI,AB,KY 7. ((impaired glucose ADJ (tolerance or metabolism)) or IGT):TI,AB,KY 8. ((risk or progress* or prevent* or inciden* or conversion or develop* or delay*) ADJ4 (diabetes or T2D* or NIDDM or "type 2" or "type II")):TI,AB,KY 9. #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 10. MESH DESCRIPTOR Dipeptidyl‐Peptidase IV Inhibitors 11. gliptin*:TI,AB,KY 12. ((dipeptidyl peptidase or dipeptidylpeptidase or dpp) ADJ ("4" or IV) ADJ inhibitor?):TI,AB,KY 13. (alogliptin or SYR 322 or SYR322):TI,AB,KY 14. anagliptin:TI,AB,KY 15. bisegliptin:TI,AB,KY 16. (carmegliptin or R1579 or RO4876904):TI,AB,KY 17. (denagliptin or GW 823093 or GW823093):TI,AB,KY 18. (dutogliptin or PHX1149):TI,AB,KY 19. (evogliptin or DA 1229):TI,AB,KY 20. (gemigliptin or LC15 0444):TI,AB,KY 21. (gosogliptin or PF 00734200 or PF 734200):TI,AB,KY 22. (linagliptin or BI 1356 or BS 1356):TI,AB,KY 23. (melogliptin or GRC 8200):TI,AB,KY 24. (omarigliptin or MK 3102):TI,AB,KY 25. (sitagliptin or MK 0431):TI,AB,KY 26. (saxagliptin or BMS 477118):TI,AB,KY 27. (teneligliptin):TI,AB,KY 28. (trelagliptin or SYR 472):TI,AB,KY 29. (vildagliptin or LAF 237 or LAF237):TI,AB,KY 30. MESH DESCRIPTOR Glucagon‐Like Peptide 1 EXPLODE ALL TREES WITH QUALIFIERS AA,AG 31. ((glucagon like peptide* or GLP 1 or GLP1) ADJ3 (analog* or agonist*)):TI,AB,KY 32. (exenatide or AC 2993 or ITCA 650):TI,AB,KY 33. (liraglutide or NN 2211 or NN2211 or NNC 90 1170 or NNC90 1170):TI,AB,KY 34. (albiglutide or GSK 716155):TI,AB,KY 35. (elsiglutide):TI,AB,KY 36. (lixisenatide or AVE 0010):TI,AB,KY 37. (dulaglutide or LY2189265 or LY 2189265):TI,AB,KY 38. (taspoglutide or BIM 51077 or BIM51077 or ITM 077 or ITM077 or R 1583 or R1583 or RO 5073031 or RO5073031):TI,AB,KY 39. (semaglutide or NN 9535 or NN9535):TI,AB,KY 40. (teduglutide or ALX 0600 or ALX0600):TI,AB,KY 41. #9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 OR #30 OR #31 OR #32 OR #33 OR #34 OR #35 OR #36 OR #37 OR #38 OR #39 OR #40 42. #9 AND #41 |
| MEDLINE (OvidSP) |
|
Block 1: Prediabetes 1. Prediabetic state/ 2. Glucose Intolerance/ 3. (prediabet* or pre diabet*).tw. 4. intermediate hyperglyc?emi*.tw. 5. ((impaired fasting adj2 glucose) or IFG or impaired FPG).tw. 6. glucose intolerance.tw. 7. ((impaired glucose adj (tolerance or metabolism)) or IGT).tw. 8. ((risk or progress* or prevent* or inciden* or conversion or develop* or delay*) adj4 (diabetes or T2D* or NIDDM or "type 2" or "type II")).tw. 9. or/1‐8 Block 2: DPP‐4 inhibitors/GLP‐1 analogues 10. Dipeptidyl‐Peptidase IV Inhibitors/ 11. gliptin*.tw. 12. ((dipeptidyl peptidase or dipeptidylpeptidase or dpp) adj ("4" or IV) adj inhibitor?).tw. 13. (alogliptin or SYR 322 or SYR322).tw. 14. anagliptin.tw. 15. bisegliptin.tw. 16. (carmegliptin or R1579 or RO4876904).tw. 17. (denagliptin or GW 823093 or GW823093).tw. 18. (dutogliptin or PHX1149).tw. 19. (evogliptin or DA 1229).tw. 20. (gemigliptin or LC15 0444).tw. 21. (gosogliptin or PF 00734200 or PF 734200).tw. 22. (linagliptin or BI 1356 or BS 1356).tw. 23. (melogliptin or GRC 8200).tw. 24. (omarigliptin or MK 3102).tw. 25. (sitagliptin or MK 0431).tw. 26. (saxagliptin or BMS 477118).tw. 27. (teneligliptin).tw. 28. (trelagliptin or SYR 472).tw. 29. (vildagliptin or LAF 237 or LAF237).tw. 30. exp Glucagon‐Like Peptide 1/aa,ag [Analogs & Derivatives, Agonists] 31. ((glucagon like peptide* or GLP 1 or GLP1) adj3 (analog* or agonist*)).tw. 32. (exenatide or AC 2993 or ITCA 650).tw. 33. (liraglutide or NN 2211 or NN2211 or NNC 90 1170 or NNC90 1170).tw. 34. (albiglutide or GSK 716155).tw. 35. (elsiglutide).tw. 36. (lixisenatide or AVE 0010).tw. 37. (dulaglutide or LY2189265 or LY 2189265).tw. 38. (taspoglutide or BIM 51077 or BIM51077 or ITM 077 or ITM077 or R 1583 or R1583 or RO 5073031 or RO5073031).tw. 39. (semaglutide or NN 9535 or NN9535).tw. 40. (teduglutide or ALX 0600 or ALX0600).tw. 41. or/10‐40 Block 1 AND block 2 AND filters 42. 9 and 41 [43‐53: Cochrane Handbook 2008 RCT filter ‐ sensitivity max. version] 43. randomized controlled trial.pt. 44. controlled clinical trial.pt. 45. randomi?ed.ab. 46. placebo.ab. 47. drug therapy.fs. 48. randomly.ab. 49. trial.ab. 50. groups.ab. 51. or/43‐50 52. exp animals/ not humans/ 53. 51 not 52 54. 42 and 53 [55:Wong 2006a– systematic reviews filter – SensSpec version] 55. meta analysis.mp,pt. or review.pt. or search*.tw. 56. 42 and 55 57. 54 or 56 |
| Embase (OvidSP) |
|
Block 1: Prediabetes 1. impaired glucose tolerance/ 2. (prediabet* or pre diabet*).tw. 3. intermediate hyperglyc?emi*.tw. 4. ((impaired fasting adj2 glucose) or IFG or impaired FPG).tw. 5. glucose intolerance.tw. 6. ((impaired glucose adj (tolerance or metabolism)) or IGT).tw. 7. ((risk or progress* or prevent* or inciden* or conversion or develop* or delay*) adj4 (diabetes or T2D* or NIDDM or "type 2" or "type II")).tw. 8. or/1‐7 Block 2: DPP‐4 inhibitors/GLP‐1 analogues 9. gliptin*.tw. 10. ((dipeptidyl peptidase or dipeptidylpeptidase or dpp) adj ("4" or IV) adj inhibitor?).tw. 11. (alogliptin or SYR 322 or SYR322).tw. 12. anagliptin.tw. 13. bisegliptin.tw. 14. (carmegliptin or R1579 or RO4876904).tw. 15. (denagliptin or GW 823093 or GW823093).tw. 16. (dutogliptin or PHX1149).tw. 17. (evogliptin or DA 1229).tw. 18. (gemigliptin or LC15 0444).tw. 19. (gosogliptin or PF 00734200 or PF 734200).tw. 20. (linagliptin or BI 1356 or BS 1356).tw. 21. (melogliptin or GRC 8200).tw. 22. (omarigliptin or MK 3102).tw. 23. (sitagliptin or MK 0431).tw. 24. (saxagliptin or BMS 477118).tw. 25. (teneligliptin).tw. 26. (trelagliptin or SYR 472).tw. 27. (vildagliptin or LAF 237 or LAF237).tw. 28. ((glucagon like peptide* or GLP 1 or GLP1) adj3 (analog* or agonist*)).tw. 29. (exenatide or AC 2993 or ITCA 650).tw. 30. (liraglutide or NN 2211 or NN2211 or NNC 90 1170 or NNC90 1170).tw. 31. (albiglutide or GSK 716155).tw. 32. (elsiglutide).tw. 33. (lixisenatide or AVE 0010).tw. 34. (dulaglutide or LY2189265 or LY 2189265).tw. 35. (taspoglutide or BIM 51077 or BIM51077 or ITM 077 or ITM077 or R 1583 or R1583 or RO 5073031 or RO5073031).tw. 36. (semaglutide or NN 9535 or NN9535).tw. 37. (teduglutide or ALX 0600 or ALX0600).tw. 38. or/9‐37 Block 1 AND block 2 AND filter 39. 8 and 38 [40:Wong 2006b"sound treatment studies" filter – BS version] 40. random*.tw. or clinical trial*.mp. or exp health care quality/ 41. 39 and 40 |
| PubMed (subsets not available on Ovid) |
| #1 ((prediabet*[tiab] OR pre diabet*[tiab] OR hyperglyc*[tiab] OR ("impaired fasting"[tiab] AND glucose[tiab]) OR IFG[tiab] OR "impaired FPG"[tiab] OR "glucose intolerance"[tiab] OR ("impaired glucose"[tiab] AND (tolerance[tiab] OR metabolism[tiab])) OR IGT[tiab] OR ((risk[tiab] OR progress*[tiab] OR prevent*[tiab] OR inciden*[tiab] OR conversion[tiab] OR develop*[tiab] OR delay*[tiab]) AND (diabetes[tiab] OR T2D*[tiab] OR NIDDM[tiab] OR "type 2"[tiab] OR "type II"[tiab])))) #2 (gliptin*[tiab] OR "dipeptidyl peptidase 4"[tiab] OR "DPP 4"[tiab] OR DPP4[tiab] OR "dipeptidyl peptidase IV"[tiab] OR "DPP IV"[tiab] OR alogliptin[tiab] OR "SYR 322"[tiab] OR SYR322[tiab] OR anagliptin[tiab] OR bisegliptin[tiab] OR carmegliptin[tiab] OR R1579[tiab] OR RO4876904[tiab] OR denagliptin[tiab] OR "GW 823093"[tiab] OR GW823093[tiab] OR dutogliptin[tiab] OR PHX1149[tiab] OR evogliptin[tiab] OR "DA 1229"[tiab] OR gemigliptin[tiab] OR "LC15 0444"[tiab] OR gosogliptin[tiab] OR "PF 00734200"[tiab] OR "PF 734200"[tiab] OR linagliptin[tiab] OR "BI 1356"[tiab] OR "BS 1356"[tiab] OR melogliptin[tiab] OR "GRC 8200"[tiab] OR omarigliptin[tiab] OR "MK 3102"[tiab] OR sitagliptin[tiab] OR "MK 0431"[tiab] OR saxagliptin[tiab] OR "BMS 477118"[tiab] OR teneligliptin[tiab] OR trelagliptin[tiab] OR "SYR 472"[tiab] OR vildagliptin[tiab] OR "LAF 237"[tiab] OR LAF237[tiab] OR "glucagon like peptide"[tiab] OR "GLP 1"[tiab] OR GLP1[tiab] OR exenatide[tiab] OR "AC 2993"[tiab] OR "ITCA 650"[tiab] OR liraglutide[tiab] OR "NN 2211"[tiab] OR NN2211[tiab] OR "NNC 90 1170"[tiab] OR "NNC90 1170"[tiab] OR albiglutide[tiab] OR "GSK 716155"[tiab] OR elsiglutide[tiab] OR lixisenatide[tiab] OR "AVE 0010"[tiab] OR dulaglutide[tiab] OR LY2189265[tiab] OR "LY 2189265"[tiab] OR taspoglutide[tiab] OR "BIM 51077"[tiab] OR BIM51077[tiab] OR "ITM 077"[tiab] OR ITM077[tiab] OR "R 1583"[tiab] OR R1583[tiab] OR "RO 5073031"[tiab] OR RO5073031[tiab] OR semaglutide[tiab] OR "NN 9535"[tiab] OR NN9535[tiab] OR teduglutide[tiab] OR "ALX 0600"[tiab] OR ALX0600[tiab]) #3 #1 AND #2 #4 publisher[sb] #5 #3 AND #4 #6 (random*[tiab] OR placebo[tiab] OR trial[tiab] OR groups[tiab]) OR (meta analysis[tiab] OR review[tiab] OR search*[tiab]) #7 #5 AND #6 |
| ClinicalTrials.gov (Expert search) |
| (prediabetes OR prediabetic OR "pre diabetes" OR "pre diabetic" OR hyperglycemia OR hyperglycaemia OR hyperglycemic OR hyperglycaemic OR "impaired glucose tolerance" OR "impaired fasting glucose" OR "glucose intolerance" OR IGT OR IFG OR ((diabetes OR "type 2" OR "type II" OR T2D OR T2DM) AND (risk OR progress OR progression OR progressed OR incident OR incidence OR conversion OR developed OR development OR develop OR delay OR delayed OR prevention OR prevent OR prevented))) AND (gliptin OR "dipeptidyl peptidase 4" OR "DPP 4" OR DPP4 OR "dipeptidyl peptidase IV" OR "DPP IV" OR alogliptin OR "SYR 322" OR SYR322 OR anagliptin OR bisegliptin OR carmegliptin OR R1579 OR RO4876904 OR denagliptin OR "GW 823093" OR GW823093 OR dutogliptin OR PHX1149 OR evogliptin OR "DA 1229" OR gemigliptin OR "LC15 0444" OR gosogliptin OR "PF 00734200" OR "PF 734200" OR linagliptin OR "BI 1356" OR "BS 1356" OR melogliptin OR "GRC 8200" OR omarigliptin OR "MK 3102" OR sitagliptin OR "MK 0431" OR saxagliptin OR "BMS 477118" OR teneligliptin OR trelagliptin OR "SYR 472" OR vildagliptin OR "LAF 237" OR LAF237 OR "glucagon like peptide" OR "GLP 1" OR GLP1 OR exenatide OR "AC 2993" OR "ITCA 650" OR liraglutide OR "NN 2211" OR NN2211 OR "NNC 90 1170" OR "NNC90 1170" OR albiglutide OR "GSK 716155" OR elsiglutide OR lixisenatide OR "AVE 0010" OR dulaglutide OR LY2189265 OR "LY 2189265" OR taspoglutide OR "BIM 51077" OR BIM51077 OR "ITM 077" OR ITM077 OR "R 1583" OR R1583 OR "RO 5073031" OR RO5073031 OR semaglutide OR "NN 9535" OR NN9535 OR teduglutide OR "ALX 0600" OR ALX0600) AND INFLECT EXACT "Interventional" [STUDY‐TYPES] |
| ICTRP Search Portal (Standard search) |
| [string to be run in six parts] 1. prediabetes AND alogliptin OR pre diabetes AND alogliptin OR impaired glucose tolerance AND alogliptin OR impaired fasting glucose AND alogliptin OR glucose intolerance AND alogliptin OR diabetes AND risk AND alogliptin OR diabetes AND prevent* AND alogliptin OR prediabetes AND anagliptin OR pre diabetes AND anagliptin OR impaired glucose tolerance AND anagliptin OR impaired fasting glucose AND anagliptin OR glucose intolerance AND anagliptin OR diabetes AND risk AND anagliptin OR diabetes AND prevent* AND anagliptin OR prediabetes AND bisegliptin OR pre diabetes AND bisegliptin OR impaired glucose tolerance AND bisegliptin OR impaired fasting glucose AND bisegliptin OR glucose intolerance AND bisegliptin OR diabetes AND risk AND bisegliptin OR diabetes AND prevent* AND bisegliptin OR prediabetes AND carmegliptin OR pre diabetes AND carmegliptin OR impaired glucose tolerance AND carmegliptin OR impaired fasting glucose AND carmegliptin OR glucose intolerance AND carmegliptin OR diabetes AND risk AND carmegliptin OR diabetes AND prevent* AND carmegliptin OR prediabetes AND denagliptin OR pre diabetes AND denagliptin OR impaired glucose tolerance AND denagliptin OR impaired fasting glucose AND denagliptin OR glucose intolerance AND denagliptin OR diabetes AND risk AND denagliptin OR diabetes AND prevent* AND denagliptin 2. prediabetes AND dutogliptin OR pre diabetes AND dutogliptin OR impaired glucose tolerance AND dutogliptin OR impaired fasting glucose AND dutogliptin OR glucose intolerance AND dutogliptin OR diabetes AND risk AND dutogliptin OR diabetes AND prevent* AND dutogliptin OR prediabetes AND evogliptin OR pre diabetes AND evogliptin OR impaired glucose tolerance AND evogliptin OR impaired fasting glucose AND evogliptin OR glucose intolerance AND evogliptin OR diabetes AND risk AND evogliptin OR diabetes AND prevent* AND evogliptin OR prediabetes AND gemigliptin OR pre diabetes AND gemigliptin OR impaired glucose tolerance AND gemigliptin OR impaired fasting glucose AND gemigliptin OR glucose intolerance AND gemigliptin OR diabetes AND risk AND gemigliptin OR diabetes AND prevent* AND gemigliptin OR prediabetes AND gosogliptin OR pre diabetes AND gosogliptin OR impaired glucose tolerance AND gosogliptin OR impaired fasting glucose AND gosogliptin OR glucose intolerance AND gosogliptin OR diabetes AND risk AND gosogliptin OR diabetes AND prevent* AND gosogliptin OR prediabetes AND linagliptin OR pre diabetes AND linagliptin OR impaired glucose tolerance AND linagliptin OR impaired fasting glucose AND linagliptin OR glucose intolerance AND linagliptin OR diabetes AND risk AND linagliptin OR diabetes AND prevent* AND linagliptin 3. prediabetes AND melogliptin OR pre diabetes AND melogliptin OR impaired glucose tolerance AND melogliptin OR impaired fasting glucose AND melogliptin OR glucose intolerance AND melogliptin OR diabetes AND risk AND melogliptin OR diabetes AND prevent* AND melogliptin OR prediabetes AND omarigliptin OR pre diabetes AND omarigliptin OR impaired glucose tolerance AND omarigliptin OR impaired fasting glucose AND omarigliptin OR glucose intolerance AND omarigliptin OR diabetes AND risk AND omarigliptin OR diabetes AND prevent* AND omarigliptin OR prediabetes AND sitagliptin OR pre diabetes AND sitagliptin OR impaired glucose tolerance AND sitagliptin OR impaired fasting glucose AND sitagliptin OR glucose intolerance AND sitagliptin OR diabetes AND risk AND sitagliptin OR diabetes AND prevent* AND sitagliptin OR prediabetes AND saxagliptin OR pre diabetes AND saxagliptin OR impaired glucose tolerance AND saxagliptin OR impaired fasting glucose AND saxagliptin OR glucose intolerance AND saxagliptin OR diabetes AND risk AND saxagliptin OR diabetes AND prevent* AND saxagliptin OR prediabetes AND teneligliptin OR pre diabetes AND teneligliptin OR impaired glucose tolerance AND teneligliptin OR impaired fasting glucose AND teneligliptin OR glucose intolerance AND teneligliptin OR diabetes AND risk AND teneligliptin OR diabetes AND prevent* AND teneligliptin 4. prediabetes AND trelagliptin OR pre diabetes AND trelagliptin OR impaired glucose tolerance AND trelagliptin OR impaired fasting glucose AND trelagliptin OR glucose intolerance AND trelagliptin OR diabetes AND risk AND trelagliptin OR diabetes AND prevent* AND trelagliptin OR prediabetes AND vildagliptin OR pre diabetes AND vildagliptin OR impaired glucose tolerance AND vildagliptin OR impaired fasting glucose AND vildagliptin OR glucose intolerance AND vildagliptin OR diabetes AND risk AND vildagliptin OR diabetes AND prevent* AND vildagliptin OR prediabetes AND GLP OR pre diabetes AND GLP OR impaired glucose tolerance AND GLP OR impaired fasting glucose AND GLP OR glucose intolerance AND GLP OR diabetes AND risk AND GLP OR diabetes AND prevent* AND GLP OR prediabetes AND exenatide OR pre diabetes AND exenatide OR impaired glucose tolerance AND exenatide OR impaired fasting glucose AND exenatide OR glucose intolerance AND exenatide OR diabetes AND risk AND exenatide OR diabetes AND prevent* AND exenatide OR prediabetes AND liraglutide OR pre diabetes AND liraglutide OR impaired glucose tolerance AND liraglutide OR impaired fasting glucose AND liraglutide OR glucose intolerance AND liraglutide OR diabetes AND risk AND liraglutide OR diabetes AND prevent* AND liraglutide 5. prediabetes AND albiglutide OR pre diabetes AND albiglutide OR impaired glucose tolerance AND albiglutide OR impaired fasting glucose AND albiglutide OR glucose intolerance AND albiglutide OR diabetes AND risk AND albiglutide OR diabetes AND prevent* AND albiglutide OR prediabetes AND elsiglutide OR pre diabetes AND elsiglutide OR impaired glucose tolerance AND elsiglutide OR impaired fasting glucose AND elsiglutide OR glucose intolerance AND elsiglutide OR diabetes AND risk AND elsiglutide OR diabetes AND prevent* AND elsiglutide OR prediabetes AND lixisenatide OR pre diabetes AND lixisenatide OR impaired glucose tolerance AND lixisenatide OR impaired fasting glucose AND lixisenatide OR glucose intolerance AND lixisenatide OR diabetes AND risk AND lixisenatide OR diabetes AND prevent* AND lixisenatide OR prediabetes AND dulaglutide OR pre diabetes AND dulaglutide OR impaired glucose tolerance AND dulaglutide OR impaired fasting glucose AND dulaglutide OR glucose intolerance AND dulaglutide OR diabetes AND risk AND dulaglutide OR diabetes AND prevent* AND dulaglutide OR prediabetes AND taspoglutide OR pre diabetes AND taspoglutide OR impaired glucose tolerance AND taspoglutide OR impaired fasting glucose AND taspoglutide OR glucose intolerance AND taspoglutide OR diabetes AND risk AND taspoglutide OR diabetes AND prevent* AND taspoglutide 6. prediabetes AND semaglutide OR pre diabetes AND semaglutide OR impaired glucose tolerance AND semaglutide OR impaired fasting glucose AND semaglutide OR glucose intolerance AND semaglutide OR diabetes AND risk AND semaglutide OR diabetes AND prevent* AND semaglutide OR prediabetes AND teduglutide OR pre diabetes AND teduglutide OR impaired glucose tolerance AND teduglutide OR impaired fasting glucose AND teduglutide OR glucose intolerance AND teduglutide OR diabetes AND risk AND teduglutide OR diabetes AND prevent* AND teduglutide |
Appendix 3. Description of interventions
| Trial | Intervention (route, frequency, total dose/day) | Intervention appropriate as used in a clinical practice settinga (description) | Comparator (route, frequency, total dose/day) | Comparator appropriate as used in a clinical practice settinga (description) |
| Ariel 2014 | Liraglutide 1.8 mg, subcutaneous. Starting dose 0.6 mg; then dose titrated by 0.6 mg weekly to maximum of 1.8 mg.
Dose decreased by 0.6 mg if intolerable adverse effects present. Participants instructed to decrease caloric intake by 500 Kcal/day and advised to consume a diet containing 43% carbohydrate, 42% fat (< 7% saturated fat) and 15% protein. Participants were regularly informed about importance of both dietary compliance and maintaining baseline levels of activity |
Yes | Placebo, subcutaneous. Uptitrated. Participants instructed to decrease caloric intake by 500 Kcal/day and advised to consume a diet containing 43% carbohydrate, 42% fat (< 7% saturated fat) and 15% protein. Participants regularly informed about importance of both dietary compliance and maintaining baseline levels of activity |
Yes |
| Kelly 2012 | Exenatide initiated at 5 μg, twice daily for 1 month and uptitrated to 10 μg twice daily for remaining 2 months, subcutaneous | Yes | Metformin initiated at 500 mg, twice daily for 1 month and uptitrated to 1000 mg, twice daily for remaining 2 months, PO | Yes |
| Martinez‐Abundis 2015 | Linagliptin 5 mg + placebo in evening, PO | Yes | Metformin 500 mg twice daily, PO | Yes |
| McLaughlin 2011 | Exenatide 5 μg twice daily for 4 weeks followed by exenatide 10 μg twice daily for 26 weeks. During first 16 weeks, participants advised to consume a hypocaloric diet | Yes | Placebo. During first 16 weeks, participants advised to consume a hypocaloric diet | Yes |
| Rosenstock 2008 | Vildagliptin 50 mg once daily, PO | Yes | Placebo once daily, PO | Yes |
| Rosenstock 2010 | Exenatide 5 μg twice daily for 4 weeks + lifestyle modification, followed by exenatide 10 μg twice daily for 20 weeks + lifestyle modification, subcutaneous | Yes | Placebo (volume equivalent to exenatide injection) twice daily for 24 weeks + lifestyle modification, subcutaneous | Yes |
| SCALE | Liraglutide starting dose 0.6 mg with weekly 0.6 mg increments to 3.0 mg, once daily subcutaneously for initial 56 weeks then continued treatment to 160 weeks, followed by an off‐drug, observational follow‐up period of 12 weeks. Total duration of this treatment arm from randomisation to follow‐up 172 weeks. All participants received standardised counselling on 'lifestyle' modification approximately monthly |
Yes | Placebo once daily subcutaneously for initial 56 weeks, then continued treatment to 160 weeks, followed by an off‐drug, observational follow‐up period of 12 weeks. Total duration of this treatment arm from randomisation to follow‐up was 172 weeks. All participants received standardised counselling on 'lifestyle' modification approximately monthly |
Yes |
| ‐ denotes not reported. aThe term 'clinical practice setting' refers to the specification of the intervention/comparator as used in the course of a standard medical treatment (such as dose, dose escalation, dosing scheme, provision for contraindications and other important features). PO: per os (orally); SCALE: Satiety and Clinical Adiposity ‐ Liraglutide Evidence in nondiabetic and Diabetic Individuals. | ||||
Appendix 4. Baseline characteristics (I)
| Trial | Intervention and comparator | Duration of intervention/duration of follow‐up | Description of participants (diagnostic criteria) | Trial period (year to year) | Country | Setting | Ethnic groups (%) |
| Ariel 2014a | I: liraglutide 1.8 mg, subcutaneous | 14 weeks/14 weeks |
IFG or IGT, or both (ADA: FPG 5.6‐6.9 mmol/L or 2 h OGTT (7.8‐11.0 mmol/L). However, in 1 publication elevated 2‐h glucose was defined as 7.8‐10.5 mmol/L after a OGTT 75‐g glucose)) |
2009‐2013 | US | Outpatients | White: 75 |
| C: placebo, subcutaneous | White: 63 | ||||||
| Kelly 2012 | I: exenatide 10 μg twice daily | 3 months/3 months |
IGT or IFG or intermediate elevated HbA1c and abdominal obesity (ADA: IFG: FPG ≥ 100 mg/dL (5.6 mmol/L), HbA1c ≥ 5.7%; IGT: 2‐h OGTT > 140 mg/dL (7.8 mmol/L)) |
‐ | US | Outpatients | White: 100 |
| C: metformin 1000 mg twice daily | White: 100 | ||||||
| Martinez‐Abundis 2015 | I: linagliptin 5 mg + placebo | 90 days/90 days | IGT, being overweight or obese (not reported) | ‐ | ‐ | Outpatients | ‐ |
| C: metformin 500 mg twice daily | ‐ | ||||||
| McLaughlin 2011 | I: exenatide 10 μg twice daily | 30 weeks/52 weeks |
IFG or IGT, or both, being overweight or obese (ADA: FPG ≥ 100 mg/dL to ≤ 125 mg/dL (5.6 mmol/L to 6.9 mmol/L); 2‐hr OGTT ≥ 140 mg/dL to ≤ 199 mg/dL (7.8 mmol/L to 11.0 mmol/L) |
2007‐2013 | US | Outpatients | ‐ |
| C: placebo | ‐ | ||||||
| Rosenstock 2008 | I: vildagliptin 50 mg once daily | 12 weeks/12 weeks | About 15% of the participants had isolated IGT, 75% had IGT + IFG (WHO: IGT: FPG < 126 mg/dL (7.0 mmol/L) and 2‐h OGTT ≥ 140 mg/dL (7.8 mmol/L) to < 200 mg/dL (11.1 mmol/L)) |
2005‐2006 | US, Spain, Finland, UK, Sweden and Germany | Outpatients | White: 90 Hispanic or Latino: 4.4 Black: 3.3 All other: 2.2 |
| C: placebo | White: 89.9 Hispanic or Latino: 7.9 Black: 1.1 All other: 1.1 | ||||||
| Rosenstock 2010 | I: exenatide 10 μg twice daily | 24 weeks/24 weeks | Obese people with IFG or IGT, or both (only a fraction of the included participants in the trial) (WHO: IFG: FPG 6.1‐6.9 mmol/L and 2‐h postprandial glucose < 7.8 mmol/L; IGT: FPG < 7 mmol/L and 2‐h postprandial glucose ≥ 7.8 to < 11.1 mmol/L) |
2007 | US, Puerto Rico | Outpatients | ‐ |
| C: placebo | ‐ | ||||||
| SCALE | I: liraglutide 3.0 mg once daily | 160 weeks/172 weeks | Being overweight or obese with intermediate hyperglycaemia (ADA: HbA1c 5.7‐6.4% both inclusive or FPG ≥ 100 mg/dL (5.6 mmol/L) to ≤ 125 mg/dL (6.9 mmol/L), or 2‐h OGTT FPG ≥ 140 mg/dL (7.8 mmol/L) to ≤ 199 mg/dL (11.0 mmol/L)), or a combinationb |
1 June 2011 through to 18 March 2013 | 27 countries: Germany, Spain, France, Italy, Belgium, Austria, Switzerland, Hungary, Poland, Serbia, UK, Norway, Finland, Denmark, Netherlands, Ireland, Turkey, Israel, Brazil, Mexico, India, Russia, Hong Kong, South Africa, US, Canada, Australia | Outpatients | White: 83.5 Black or African‐American: 9.7 Asian: 5.0 American Indian or Alaska Native: 0.3 Native Hawaiian or other Pacific Islander: < 0.1 Other: 1.4 |
| C: placebo | White: 83.9
Black or African‐American: 9.5
Asian: 5.2
American Indian or Alaska Native: 0.3 Native Hawaiian or other Pacific Islander: 0.1 Other: 1.1 |
||||||
| ‐ denotes not reported aBaseline characteristics only reported for the participants completing the trial; the trial reported separate baseline data for the participants who did not complete the trial. bOf the 1468 participants in the liraglutide group; 173 had IFG; 158 had IGT; 376 had intermediate elevated HbA1c; 103 had IFG + IGT; 262 had IFG + intermediate elevated HbA1c; 161 had IGT + intermediate elevated HbA1c; 235 had IFG + IGT + intermediate elevated HbA1c at baseline. Of the 736 in the placebo group; 90 had IFG; 94 had IGT; 187 had intermediate elevated HbA1c; 46 had IFG + IGT; 128 had IFG + intermediate elevated HbA1c; 75 had IGT+ intermediate elevated HbA1c; 116 had IFG + IGT + intermediate elevated HbA1c at baseline. ADA: American Diabetes Association; C: comparator; FPG: fasting plasma glucose; h: hour; HbA1c: glycosylated haemoglobin A1c; I: intervention; IFG: impaired fasting glucose; IGT: impaired glucose tolerance; OGTT: oral glucose tolerance test; SCALE: Satiety and Clinical Adiposity ‐ Liraglutide Evidence in Nondiabetic and Diabetic Individuals; WHO: World Health Organization. | |||||||
Appendix 5. Baseline characteristics (II)
| Trial | Intervention and comparator | Sex (female %) | Age (mean/range years (SD)) | Systolic/diastolic blood pressure (mean mmHg (SD)) | Access to health care, social determinants |
| Ariel 2014a | I: liraglutide 1.8 mg, subcutaneous | 67 | 58 (7) | ‐ | ‐ |
| C: placebo, subcutaneous | 63 | 58 (8) | ‐ | ‐ | |
| Kelly 2012 | I: exenatide 10 μg twice daily | 80 | 58.7 (10.0) | 130.6 (17.1)/74.3 (9.3) | ‐ |
| C: metformin 1000 mg twice daily | 72 | 58.4 (10.1) | 125.8 (12.3)/75.6 (10.0) | ‐ | |
| Martinez‐Abundis 2015 | I: linagliptin 5 mg + placebo | ‐ | 49.3 (5.7) | 121.8 (17.7)/79.5 (9.2) | ‐ |
| C: metformin 500 mg twice daily | ‐ | 51.9 (6.4) | 120.6 (12.9)/79.9 (8.0) | ‐ | |
| McLaughlin 2011 | I: exenatide 10 μg twice daily | ‐c | ‐c | ‐ | ‐ |
| C: placebo | ‐c | ‐c | ‐ | ‐ | |
| Rosenstock 2008 | I: vildagliptin 50 mg once daily | 52.2 | 57.1 (10.7) | ‐ | ‐ |
| C: placebo | 57.3 | 59.8 (11.5) | ‐ | ‐ | |
| Rosenstock 2010 | I: exenatide 10 μg twice daily | 79.4d | 47.0 (11.0)d | ‐ | ‐ |
| C: placebo | 84.8d | 45.2 (12.7)d | ‐ | ‐ | |
| SCALE | I: liraglutide 3.0 mg once daily | 75.7 | 47.4 (11.8) | 124.8 (12.9)/79.4 (8.4) | ‐ |
| C: placebo | 76.8 | 47.2 (11.8) | 125.0 (12.8)/79.9 (8.3) | ‐ | |
| ‐ denotes not reported. aBaseline characteristics only reported for the participants completing the trial; the trial reported separate baseline data for the participants that did not complete the trial. bPresented as median (quartile 1‐3). cReported in abstract that there was no significant difference between groups. dOnly reported for all participants and not for the 38 participants with impaired fasting glucose/impaired glucose tolerance. C: comparator; I: intervention; SCALE: Satiety and Clinical Adiposity ‐ Liraglutide Evidence in Nondiabetic and Diabetic Individuals; SD: standard deviation. | |||||
Appendix 6. Baseline characteristics (III)
| Trial | Intervention and comparator | Fasting plasma glucose (mean mmol/L (SD)) | 2‐hour plasma glucose (mean mmol/L (SD)) | HbA1c (%) | BMI (mean kg/m² (SD)) | Comedications/cointerventions (n (%)) | Comorbidities (n (%)) |
| Ariel 2014a | I: liraglutide 1.8 mg, subcutaneous | 5.9 (0.4) | 7.9 (1.8) | ‐ | 31.9 (2.7) | ‐ | ‐ |
| C: placebo, subcutaneous | 6.1 (0.4) | 7.8 (1.7) | ‐ | 31.9 (3.5) | ‐ | ‐ | |
| Kelly 2012 | I: exenatide 10 μg twice daily | 5.7 (0.6)d | ‐ | ‐ | 35.3 (5.5) | ‐ | ‐ |
| C: metformin 1000 mg twice daily | 5.7 (0.5)d | ‐ | ‐ | 35.8 (7.0) | ‐ | ‐ | |
| Martinez‐Abundis 2015 | I: linagliptin 5 mg + placebo | 5.5 (0.6) | 9.0 (0.9) | 6.1 (0.6) | 31.1 (3.6) | ‐ | ‐ |
| C: metformin 500 mg twice daily | 5.7 (0.6) | 9.5 (1.0) | 6.2 (0.4) | 31.0 (2.4) | ‐ | ‐ | |
| McLaughlin 2011 | I: exenatide 10 μg twice daily | ‐ | ‐ | ‐ | 33 | ‐ | ‐ |
| C: placebo | ‐ | ‐ | ‐ | 33 | ‐ | ‐ | |
| Rosenstock 2008 | I: vildagliptin 50 mg once daily | 6.2 (0.7) | 9.1 (0.9) | 5.9 (0.5) | 31.7 (4.8) | ‐ | ‐ |
| C: placebo | 6.1 (0.7) | 9.2 (0.9) | 5.9 (0.4) | 30.9 (5.3) | ‐ | ‐ | |
| Rosenstock 2010 | I: exenatide 10 μg twice daily | ‐ | ‐ | ‐ | 39.6 (7.0)e | ‐ | ‐ |
| C: placebo | ‐ | ‐ | ‐ | ‐ | ‐ | ||
| SCALE | I: liraglutide 3.0 mg once daily | 5.5 (0.6) | 7.4 (1.8) | 5.8 (0.3) | 38.8 (6.4) | Antihypertensives: 581 (38.9) Lipid‐lowering drugs: 292 (19.5) | Cardiovascular disease: 150 (9.8) Dyslipidaemia: 504 (33) Hypertension: 639 (41.8) Dyslipidaemia and hypertension: 318 (20.8) |
| C: placebo | 5.5 (0.5) | 7.4 (1.7) | 5.7 (0.3) | 39.0 (6.3) | Antihypertensives: 293 (39.3) Lipid‐lowering drugs: 133 (17.8) |
Cardiovascular disease: 76 (10.1) Dyslipidaemia: 246 (32.5) Hypertension: 316 (41.7) Dyslipidaemia and hypertension: 155 (20.5) | |
| ‐ denotes not reported. aBaseline characteristics only reported for participants completing trial; trial reported separate baseline data for participants who did not complete trial. bPresented as median (quartile 1; quartile 3). cHbA1c converted from mmol/mol to percentage. dGlucose values converted from mg/dL to mmol/L. eReported as mean BMI for both group and reported no statistical significant difference between groups. Only reported for all participants included in the trial (participants with normal glucose tolerance, impaired fasting glucose and impaired glucose tolerance). BMI: body mass index; C: comparator; HbA1c: glycosylated haemoglobin A1c; I: intervention; n: number; SCALE: Satiety and Clinical Adiposity ‐ Liraglutide Evidence in Nondiabetic and Diabetic Individuals; SD: standard deviation. | |||||||
Appendix 7. Matrix of trial endpoints (publications and trial documents)
| Trial | Endpoints quoted in trial document(s) (ClinicalTrials.gov, FDA/EMA document, manufacturer's website, published design paper)a | Trial results available in trial register | Endpoints quoted in publication(s)b,c | Endpoints quoted in abstract of publication(s)b,c |
| Ariel 2014 |
Source: NCT01784965 Primary outcome measure: magnitude of weight loss |
No | Primary outcome measure: weight loss | Primary outcome measure: weight loss |
| Secondary outcome measures: change in glucose‐stimulated insulin secretion; change in insulin resistance | Secondary outcome measures: glucose‐stimulated insulin secretion, insulin resistance | Secondary outcome measures: glucose‐stimulated insulin secretion, insulin resistance | ||
| Other outcome measures: ‐ | Other outcome measures: dropouts (total and due to adverse effects), systolic blood pressure, fasting blood glucose, triglyceride and cholesterol profiles concentration, reversion to normoglycaemia, inflammatory markers, change in pulse, metabolic syndrome components | Other outcome measures: fasting plasma glucose, lipid and cholesterol concentrations, dropouts, blood pressure, inflammatory markers | ||
| History of changes: no documented changes | ||||
| Kelly 2012 |
Source:
NCT00546728 Primary outcome measure: change in reactive hyperaemic index |
Yes | Primary outcome measure: change in reactive hyperaemic index | Primary outcome measure: change in reactive hyperaemic index |
| Secondary outcome measures: ‐ | Secondary outcome measures: ‐ | Secondary outcome measures: ‐ | ||
| Other outcome measures: ‐ | Other outcome measures: inflammatory markers, blood pressure, cholesterol and triglyceride levels, body fat, BMI, insulin resistance, fasting blood glucose | Other outcome measures: inflammatory markers, triglycerides | ||
| History of changes: 7 documented changes; last change 7 November 2013 | ||||
| Martinez‐Abundis 2015 | Source: N/T | Primary outcome measures: ‐ | Primary outcome measures: glycaemic variability evaluated with mean amplitude of glycaemic excursions and area under curve | |
| Secondary outcome measures: ‐ | Secondary outcome measures: ‐ | |||
| Other outcome measures: ‐ | Other outcome measure: cholesterol | |||
| McLaughlin 2011 |
Source:
NCT02084654 Primary outcome measures: first‐phase insulin response |
No | Primary outcome measures: ‐ | Primary outcome measure: first‐phase insulin response |
| Secondary outcome measures: reversal of prediabetes measured by fasting, 2‐hour plasma glucose during OGTT | Secondary outcome measures: ‐ | Secondary outcome measures: reversal of prediabetes | ||
| Other outcome measures: insulin‐mediated glucose uptake | Other outcome measures: ‐ | Other outcome measures: body weight, fasting blood glucose, 2‐hour blood glucose | ||
| History of changes: zero documented changes | ||||
| Rosenstock 2008 |
Source: NCT00237250 Primary outcome measure: change in area under 0‐ to 2‐hour prandial glucose curve at 12 weeks |
No | Primary outcome measure: "The primary efficacy variable was the change from baseline to endpoint (week 12 or last available post baseline value) in the prandial plasma glucose AUC0‐2 h [area under curve]" | Primary outcome measure(s): in abstract, not specified as primary or secondary outcomes. Outcomes quoted in abstract: intact GLP‐1, GIP, glucagon, postprandial insulin levels and postprandial glucose excursions area under the curve. β‐cell function, adverse events, hypoglycaemia |
| Secondary outcome measures: adverse event profile after 12 weeks of treatment; change in ratio for postprandial insulin area under the curve and postprandial glucose area under the curve (0‐2 hours) after 12 weeks of treatment; change in homeostatic model assessment B at 12 weeks; change in fasting insulin at 12 weeks; change in fasting proinsulin/insulin ratio at 12 weeks | Secondary outcome measures: not explicitly described as secondary outcomes in publication, but stated the following in 'Data analysis' section: "Insulin secretory rate (ISR) was estimated by deconvolution of C‐peptide levels and expressed per square meter of body surface area. The total and incremental (Δ) areas under the curve (AUC) for GLP‐1, GIP, glucose, insulin, glucagon, C‐peptide, and ISR were calculated with the trapezoidal method for the 0‐ to 2‐h postmeal time interval. Insulin secretion relative to glucose (ISR AUC0‐2h/glucose AUC0‐2h) was calculated as a measure of β‐cell function. In addition, homeostasis model assessment of insulin resistance (HOMA‐IR) and the meal‐derived insulin sensitivity index (ISI) were calculated." | Secondary outcome measures: in the abstract outcomes were not specified as primary or secondary outcomes, see above | ||
| Other outcome measure(s): none | Other outcome measures:‐ | Other outcome measures:‐ | ||
| History of changes: 8 documented changes; last change 4 May 2012 | ||||
| Rosenstock 2010 |
Source: NCT00500370 Primary outcome measure: change in body weight from baseline after 24 weeks of treatment |
Yes | Primary outcome measure: change in body weight | Primary outcome measure: change in body weight |
| Secondary outcome measures: change in BMI; waist‐to‐hip ratio; percentage of exenatide‐ and placebo‐treated participants experiencing ≥ 5% weight loss after 24 weeks of treatment; change in total cholesterol; change in HDL cholesterol; change in LDL cholesterol; ratio of triglycerides; change in fasting serum glucose; change in serum glucose area under the curve following OGTT; ratio of HOMA‐B at week 24 to HOMA‐B at week 0; ratio of HOMA‐S at week 24 to HOMA‐S at week 0; number of participants in each treatment group who demonstrated overt signs of diabetes mellitus diagnosis by week 24; number of participants in each treatment group who demonstrated normalisation of IFG or IGT (or both) by week 24; change in HbA1c from baseline following 24 weeks of treatment; change in high sensitivity C‐reactive protein following 24 weeks of treatment | Secondary outcome measure(s): percentage of exenatide‐ and placebo‐treated participants experiencing ≥ 5% weight loss; progression to T2DM; HbA1c; lipid concentration | Secondary outcome measure(s): percentage of participants in each treatment group that demonstrate normalisation of IFG or IGT (or both) | ||
| Other outcome measures: ‐ | Other outcome measures: withdrawal rate, adverse events, mortality, serious adverse events, hypoglycaemia, calorie intake, blood pressure | Other outcome measure: calorie intake | ||
| History of changes: 11 documented changes; last change 6 April 2015 | ||||
| SCALE |
Source:
NCT01272219 and protocol (appendix) to main publication Primary outcome measures: change from baseline in fasting body weight after 56 weeks of treatment (main treatment period); proportion of participants losing ≥ 5% of baseline fasting body weight after 56 weeks of treatment (main treatment period); proportion of participants losing > 10% of baseline fasting body weight after 56 weeks of treatment (main treatment period); proportion of participants with onset of T2DM at week 160 (main + extension treatment period) among participants with prediabetes at baseline |
Yes | Primary outcome measures: change from baseline in fasting body weight after 56 weeks of treatment; proportion of participants losing ≥ 5% of baseline fasting body weight after 56 weeks of treatment; proportion of participants losing > 10% of baseline fasting body weight after 56 weeks of treatment; proportion of participants with onset of T2DM at week 160 (main + extension treatment period) among participants with prediabetes at baseline | Primary outcome measures: change from baseline in fasting body weight after 56 weeks of treatment; proportion of participants losing ≥ 5% of baseline fasting body weight after 56 weeks of treatment; proportion of participants losing > 10% of baseline fasting body weight after 56 weeks of treatment; proportion of participants with onset of T2DM at week 160 (main + extension treatment period) among participants with prediabetes at baseline |
|
Secondary outcome measures: according to NCT01272219: change from baseline in waist circumference after 56 weeks of treatment; change from baseline in waist circumference (participants with prediabetes at baseline) after 160 weeks of treatment (main + extension treatment period); prediabetes status after 56 weeks of treatment in participants with prediabetes; prediabetes status in participants with prediabetes at baseline after 160 weeks of treatment; mean change from baseline in fasting body weight (participants with prediabetes at baseline) after 160 weeks of treatment (main + extension treatment period); proportion of participants losing ≥ 5% and proportion of participants losing > 10% of baseline fasting body weight (participants with prediabetes at baseline) after 160 weeks of treatment (main + extension treatment period); change from week 56 in fasting body weight (re‐randomised participants with no prediabetes) from week 56 to week 68; change from baseline in fasting body weight (re‐randomised participants with no prediabetes) Secondary outcomes according to protocol (appendix) to main publication: change from baseline to week 56 in: glucose‐related parameters (HbA1c, fasting plasma glucose, fasting insulin, C‐peptide, endpoints from 2‐hour OGTT parameters; proportion of participants with T2DM; prediabetes status; HOMA parameters;urinary‐albumin‐to‐creatinine ratio; systolic and diastolic blood pressure and pulse pressure; cardiovascular biomarkers; Impact Of Weight On Quality Of Life‐Lite Questionnaire, 36‐item Short Form Health Survey and Treatment Related Impact measure ‐ Weight (TRIm‐Weight); proportion of participants with change in concomitant medication from baseline to week 56 in: antihypotensive drugs; lipid‐lowering agent and oral glucose‐lowering drugs Outcomes written initalic differ from NCT01272219 and protocol (appendix) to main publication |
Secondary outcome measures: change from baseline to week 56 in systolic and diastolic blood pressure; change from baseline to week 56 in glucose‐related parameters; change from baseline to week 56 in cardiovascular biomarkers; change from baseline to week 56 in health‐related quality of life; proportion of participants with T2DM; adverse effects and adverse events; prediabetes status; proportion of participants with change in concomitant medication from baseline to week 56 in: antihypotensive drugs; lipid‐lowering agent and oral glucose‐lowering drugs. After 160 weeks of intervention: prediabetes status in participants with prediabetes at baseline after 160 weeks of treatment; mean change from baseline in fasting body weight (participants with prediabetes at baseline) after 160 weeks of treatment; proportion of participants losing ≥ 5% and proportion of participants losing > 10% of baseline fasting body weight (participants with prediabetes at baseline) after 160 weeks of treatment; glucose‐related parameters (HbA1c, fasting plasma glucose, fasting insulin, C‐peptide, endpoints from 2‐hour OGTT parameters) mortality, serious adverse events |
Secondary outcome measures: adverse events, serious adverse events, incidence of T2DM | ||
| Other outcome measures: ‐ | Other outcome measures: ‐ | Other outcome measures: ‐ | ||
| History of changes: 20 documented changes; last change 3 May 2016 | ||||
| ‐ denotes not reported. aTrial document(s) referred to all available information from published design papers and sources other than regular publications (e.g. FDA/EMA documents, manufacturer's websites, trial registers). bPublication(s) referred to trial information published in scientific journals (primary reference, duplicate publications, companion documents or multiple reports of a primary trial). cOther outcome measures referred to all outcomes not specified as primary or secondary outcome measures. BMI: body mass index; EMA: European Medicines Agency; FDA: Food and Drug Administration (US); GIP: glucose‐dependent insulinotropic polypeptide; GLP‐1: glucagon‐like peptide‐1; HbA1c: glycosylated haemoglobin A1c; HDL: high‐density lipoprotein; HOMA: Homeostatic model assessment; HS‐CRP: high‐sensitivity C‐reactive protein; IFG: impaired fasting glucose; IGT: impaired glucose tolerance; LDL: low‐density lipoprotein; N/A: not applicable; N/T: no trial document available; OGTT: oral glucose tolerance test; SCALE: Satiety and Clinical Adiposity ‐ Liraglutide Evidence in Nondiabetic and Diabetic Individuals; T2DM: type 2 diabetes mellitus. | ||||
Appendix 8. High risk of outcome reporting bias according to ORBIT classification
| Trial | Outcome | High risk of bias (category A)a | High risk of bias (category D)b | High risk of bias (category E)c | High risk of bias (category G)d |
| Ariel 2014 | Fasting blood glucose | No | No | No | No |
| 2‐hour blood glucose | No | No | No | No | |
| Kelly 2012 | Serious adverse events | No | No | No | No |
| Non‐serious adverse events | No | Yes | No | No | |
| Fasting glucose | No | No | No | No | |
| Martinez‐Abundis 2015 | Fasting glucose | No | No | No | No |
| 2‐hour glucose | No | No | No | No | |
| HbA1c | No | No | No | No | |
| Non‐serious adverse events | No | No | No | No | |
| Hypoglycaemia | No | No | No | No | |
| McLaughlin 2011 | Fasting glucose | Yes | No | No | No |
| 2‐hour glucose | Yes | No | No | No | |
| Rosenstock 2008 | Incidence of T2DM | No | No | No | No |
| Serious adverse events | No | No | No | No | |
| Adverse events | No | No | No | No | |
| Hypoglycaemia | No | No | No | No | |
| Fasting glucose | No | No | No | No | |
| 2‐hour glucose | No | No | No | No | |
| HbA1c | No | No | No | No | |
| Rosenstock 2010e | All‐cause mortality | No | No | No | No |
| Incidence of T2DM | No | No | No | No | |
| Serious adverse events | No | No | No | No | |
| Hypoglycaemia | No | No | No | No | |
| SCALE | All‐cause mortality | No | No | No | No |
| Incidence of T2DM (specifically assessed for the participants with intermediate hyperglycaemia at baseline) ‐ end of intervention period | No | No | No | No | |
| Serious adverse events (specifically assessed for the participants with intermediate hyperglycaemia at baseline) | No | No | No | No | |
| Health‐related quality of life (specifically assessed for the participants with intermediate hyperglycaemia at baseline) | No | No | No | No | |
| Adverse events (specifically assessed for the participants with intermediate hyperglycaemia at baseline) | No | No | No | No | |
| Fasting glucose (specifically assessed for the participants with intermediate hyperglycaemia at baseline) | No | No | No | No | |
| 2‐hour glucose (specifically assessed for the participants with intermediate hyperglycaemia at baseline) | No | No | No | No | |
| HbA1c (specifically assessed for the participants with intermediate hyperglycaemia at baseline) | No | No | No | No | |
|
aClear that outcome was measured and analysed; trial report stated that outcome was analysed but reported only that result was not significant.
(Classification 'A', table 2, Kirkham 2010).
bClear that outcome was measured and analysed; trial report stated that outcome was analysed but reported no results.
( Classification 'D', table 2, Kirkham 2010).
cClear that outcome was measured but was not necessarily analysed; judgement says likely to have been analysed but not reported because of non‐significant results.
(Classification 'E', table 2, Kirkham 2010).
dUnclear whether outcome was measured; not mentioned, but clinical judgement says likely to have been measured and analysed but not reported on the basis of non‐significant results.
(Classification 'G', table 2, Kirkham 2010). eOnly a fraction of the participants had impaired glucose tolerance or impaired fasting glucose (or both). Several of the predefined outcomes of interest of this review were only reported for all participants. HbA1c: glycosylated haemoglobin A1c; ORBIT: Outcome Reporting Bias In Trials; SCALE: Satiety and Clinical Adiposity ‐ Liraglutide Evidence in Nondiabetic and Diabetic Individuals; T2DM: type 2 diabetes mellitus. | |||||
Appendix 9. Definition of endpoint measurementa (I)
| Trial | All‐cause mortality | Incidence of T2DM | Serious adverse events | Cardiovascular mortality | Non‐fatal myocardial infarction | Non‐fatal stroke | Congestive heart failure | Amputation of lower extremity |
| Ariel 2014 | N/I | N/I | N/I | N/I | N/I | N/I | N/I | N/I |
| Kelly 2012 | N/I | N/I | SAE | N/I | N/I | N/I | N/I | N/I |
| Martinez‐Abundis 2015 | N/I | N/I | N/I | N/I | N/I | N/I | N/I | N/I |
| McLaughlin 2011 | N/I | N/I | N/I | N/I | N/I | N/I | N/I | N/I |
| Rosenstock 2008 | Deaths (IO) |
Reported as adverse events. Assume definition was identical with the definition of T2DM in the exclusion criteria: FPG ≥ 126 mg/dL (7.0 mmol/L) at visit 1 (week ‐4); 2‐hour post‐challenge plasma glucose (after OGTT 75‐g glucose) ≥ 200 mg/dL (11.1 mmol/L) (IO) | "All adverse events were recorded and assessed as to their severity and possible relationship to the study medication as judged by the investigator" (IO) | N/I | N/I | N/I | Heart failure (IO) | N/I |
| Rosenstock 2010 | Deaths (IO) | T2DM (IO) | SAE (IO) | N/I | N/I | N/I | N/I | N/I |
| SCALE | Deaths (IO/AO) |
Quote: "Furthermore, patients were diagnosed with type 2 diabetes based on the following criteria:
Measurements of FPG and 2‐hour glucose values after an OGTT had to be confirmed in repeated assessments (IO) |
Quote: "A SAE is an experience that at any dose results in any of the following:
* "life‐threatening" in the definition of SAE refers to an event in which the participant was at risk of death at the time of the event. It does not refer to an event which hypothetically might have caused death if it were more severe. ** "hospitalisation" is the definition of a participant admitted to a hospital/inpatient (irrespective of the duration of physical stay), or not admitted to a hospital/not inpatient, but stays at the hospital for treatment or observation for > 24 hours. Medical judgement must always be exercised, and when in doubt, the hospital contact should be regarded as a hospitalisation. Hospitalisations for administrative, trial related and social purposes do not constitute AEs and should therefore neither be reported as AEs or SAEs. Likewise, hospital admissions for surgical procedures planned prior to trial inclusion are not considered AEs or SAEs. *** "disability/incapacity" means that following the event the participant or clinical investigation participant has significant, persistent or permanent change, impairment, damage or disruption in his body function or structure, physical activity or quality of life (or both). ****"important medical events" means events which may jeopardise the participant or require intervention to prevent a seriousness criterion. It can be adverse events which suggest a significant hazard or puts the participants or clinical investigation participants at risk, such as drug interaction, contraindications or precautions, occurrence of malignancies or development of drug dependency or drug abuse (IO/AO) |
"Cardiovascular death, includes
1 participant died from cardiac arrest (IO/AO) |
Acute coronary syndrome (myocardial infarction or hospitalisation for unstable angina) (IO/AO) | Cerebrovascular event (stroke or transient ischaemic attack) (IO/AO) | Heart failure requiring hospitalisation (IO/AO) |
N/I |
|
aIn addition to definition of endpoint measurement, description who measured the outcome (AO: adjudicated outcome measurement; IO: investigator‐assessed outcome measurement; SO: self‐reported outcome measurement). AE: adverse event; AO: adjudicated outcome measurement; FPG: fasting plasma glucose; HbA1c: glycosylated haemoglobin A1c; IO: investigator‐assessed outcome measurement; N/I: not investigated; OGTT: oral glucose tolerance test; SAE: serious adverse event; SCALE: Satiety and Clinical Adiposity ‐ Liraglutide Evidence in Nondiabetic and Diabetic Individuals; T2DM: type 2 diabetes mellitus. | ||||||||
Appendix 10. Definition of endpoint measurementa (II)
| Trial | Blindness or severe vision loss | End‐stage renal disease | Non‐serious adverse events | Hypoglycaemic events | Health‐related quality of life | Time to progression to T2DM | Measures of blood glucose control | Socioeconomic effects |
| Ariel 2014 | N/I | N/I | N/I | N/I | N/I | N/I | Fasting blood glucose (IO) | N/I |
| Kelly 2012 | N/I | N/I | Total, other (not including serious) adverse events | N/I | N/I | N/I | Fasting blood glucose (IO) | N/I |
| Martinez‐Abundis 2015 | N/I | N/I | "Non‐serious adverse events" (SO/IO) | N/I | N/I | N/I | Fasting blood glucose, 2‐hour glucose, HbA1c (IO) | N/I |
| McLaughlin 2011 | N/I | N/I | N/I | N/I | N/I | N/I | Fasting blood glucose, 2‐hour glucose (IO) | N/I |
| Rosenstock 2008 | N/I | N/I | "All adverse events were recorded and assessed as to their severity and possible relationship to the study medication as judged by the investigator" (SO/IO) | "Hypoglycemia was defined as symptoms suggestive of low blood glucose confirmed by self‐monitored blood glucose measurement 3.1 mmol/L plasma glucose equivalent" (SO) | N/I | N/I | Fasting blood glucose, 2‐hour glucose, HbA1c (IO) | N/I |
| Rosenstock 2010 | N/I | N/I | Adverse events (SO/IO) | Hypoglycaemia | N/I | N/I | HbA1c (IO) | N/I |
| SCALE | N/I | N/I | "Any untoward medical occurrence in a subject or clinical investigation subject administered a pharmaceutical product, and which does not necessarily have to have a causal relationship with this treatment" (SO/IO) | "Severe hypoglycaemia: an episode requiring assistance of another person to actively administer carbohydrate, glucagon, or other resuscitative actions. Documented symptomatic hypoglycaemia: an episode during which typical symptoms of hypoglycaemia are accompanied by a measured plasma glucose concentration ≤ 3.9 mmol/L (70 mg/dL). Asymptomatic hypoglycaemia: an episode not accompanied by typical symptoms of hypoglycaemia, but with a measured plasma glucose concentration ≤ 3.9 mmol/L (70 mg/dL). Probable symptomatic hypoglycaemia: an episode during which symptoms of hypoglycaemia are not accompanied by a plasma glucose determination (but that was presumably caused by a plasma glucose concentration ≤ 3.9 mmol/L (70 mg/dL)). Relative hypoglycaemia: an episode during which the person with diabetes reports any of the typical symptoms of hypoglycaemia, and interprets those as indicative of hypoglycaemia, but with a measured plasma glucose concentration > 3.9 mmol/L (70 mg/dL)" And "Minor hypoglycaemic episode were defined as:
|
N/I | N/I | N/I | N/I |
|
aIn addition to definition of endpoint measurement, description who measured the outcome (AO: adjudicated outcome measurement; IO: investigator‐assessed outcome measurement; SO: self‐reported outcome measurement) HbA1c: glycosylated haemoglobin A1c; IO: investigator‐assessed outcome measurement; N/I: not investigated; SCALE: Satiety and Clinical Adiposity ‐ Liraglutide Evidence in Nondiabetic and Diabetic Individuals; SO: self‐reported outcome measurement; T2DM: type 2 diabetes mellitus. | ||||||||
Appendix 11. Adverse events (I)
| Trial | Intervention and comparator | Participants included in analysis (n) | Deaths (n) | Deaths (% participants) | Participants with ≥ 1 adverse event (n) | Participants with ≥ 1 adverse event (%) | Participants with ≥ 1 severe/serious adverse event (n) | Participants with ≥ 1 severe/serious adverse event (%) |
| Ariel 2014 | I: liraglutide 1.8 mg, subcutaneous | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| C: placebo, subcutaneous | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Kelly 2012 | I: exenatide 10 μg twice daily |
25 | 0 | 0 | 0 | 0 | 0 | 0 |
| C: metformin 1000 mg twice daily | 25 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Martinez‐Abundis 2015 | I: linagliptin 5 mg + placebo | 8 | ‐ | ‐ | 1 | 12.5 | 0 | 0 |
| C1: metformin 500 mg twice daily | 8 | ‐ | ‐ | 4 | 50 | 0 | 0 | |
| McLaughlin 2011 | I1: exenatide 10 μg twice daily |
‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| C: placebo | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Rosenstock 2008 | I: vildagliptin 50 mg | 90 | 0 | 0 | 49 | 54.4 | 1 | 1.1 |
| C: placebo | 89 | 0 | 0 | 44 | 49.4 | 2 | 2.2 | |
| Rosenstock 2010 | I: exenatide 10 μg twice daily |
17a | 0 | 0 | ‐b | ‐b | 0 | 0 |
| C: placebo | 16a | 0 | 0 | ‐b | ‐b | 0 | 0 | |
| SCALE | I: liraglutide 3.0 mg once daily | 1524 | ‐ | ‐ | ‐ | ‐ | 230 | 15.1 |
| C: placebo | 755 | ‐ | ‐ | ‐ | ‐ | 96 | 12.7 | |
| ‐ denotes not reported. aUnknown if all randomised participants were included in the analyses of adverse events or only the 33 participants included in the remaining analyses. bOnly reported for all the included participants and not separately for the fraction of participants with impaired glucose tolerance or impaired fasting glucose (or both). C: comparator; I: intervention; n: number of participants; SCALE: Satiety and Clinical Adiposity ‐ Liraglutide Evidence in Nondiabetic and Diabetic Individuals. | ||||||||
Appendix 12. Adverse events (II)
| Trial | Intervention and comparator | Participants included in analysis (n) | Participants discontinuing trial due to an adverse event (n) | Participants discontinuing trial due to an adverse event (%) | Participants with ≥ 1 hospitalisation (n) | Participants with ≥ 1 hospitalisation (%) | Participants with ≥ 1 outpatient treatment (n) | Participants with ≥ 1 outpatient treatment (%) |
| Ariel 2014 | I: liraglutide 1.8 mg, subcutaneous | 35 | 8 | 22.9 | ‐ | ‐ | ‐ | ‐ |
| C: placebo, subcutaneous | 33 | 0 | 0 | ‐ | ‐ | ‐ | ‐ | |
| Kelly 2012 | I: exenatide 10 μg twice daily |
25 | 0 | 0 | ‐ | ‐ | ‐ | ‐ |
| C: metformin 1000 mg twice daily | 25 | 0 | 0 | ‐ | ‐ | ‐ | ‐ | |
| Martinez‐Abundis 2015 | I: linagliptin 5 mg + placebo | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| C: metformin 500 mg twice daily | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| McLaughlin 2011 | I: exenatide 10 μg twice daily |
‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| C: placebo | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Rosenstock 2008 | I: vildagliptin 50 mg | 90 | 3 | 3.3 | ‐ | ‐ | ‐ | ‐ |
| C: placebo | 89 | 2 | 2.2 | ‐ | ‐ | ‐ | ‐ | |
| Rosenstock 2010 | I: exenatide 10 μg twice daily |
‐a | ‐a | ‐a | ‐a | ‐a | ‐a | ‐a |
| C: placebo | ‐a | ‐a | ‐a | ‐a | ‐a | ‐a | ‐a | |
| SCALE | I: liraglutide 3.0 mg | 1524 | 191b | 12.5 | ‐ | ‐ | ‐ | ‐ |
| C: placebo | 755 | 43c | 5.7 | ‐ | ‐ | ‐ | ‐ | |
| ‐ denotes not reported. aOnly reported for all the included participants and not separately for the fraction of participants with impaired glucose tolerance or impaired fasting glucose (or both). bFrom the main + extension period (weeks 0‐172). Number of participants discontinuing trial due to an adverse event in the main period (56 weeks) were 152/1528 in the liraglutide group and 29/757 in the placebo group. C: comparator; I: intervention; SCALE: Satiety and Clinical Adiposity ‐ Liraglutide Evidence in Nondiabetic and Diabetic Individuals. | ||||||||
Appendix 13. Adverse events (III)
| Trial | Intervention and comparator | Participants included in analysis (n) | Participants with a specific adverse event (description) | Participants with at least one specific adverse events (n) | Participants with at least one specific adverse event (%) |
| Ariel 2014 | I: liraglutide 1.8 mg, subcutaneous | 24 | Non‐serious adverse events for completers (1) nausea (2) vomiting (3) diarrhoea (4) constipation (5) abdominal pain (6) ≥ 1 gastrointestinal symptom (7) headache (8) injection‐site irritation |
(1) 16 (2) 3 (3) 6 (4) 8 (5) 3 (6) 19 (7) 2 (8) 7 | (1) 67 (2) 13 (3) 25 (4) 33 (5) 13 (6) 79 (7) 8 (8) 29 |
| C: placebo, subcutaneous | 27 | Non‐serious adverse events for completers (1) nausea (2) vomiting (3) diarrhoea (4) constipation (5) abdominal pain (6) ≥ 1 gastrointestinal symptom (7) headache (8) injection‐site irritation |
(1) 7 (2) 1 (3) 6 (4) 3 (5) 6 (6) 12 (7) 6 (8) 3 | (1) 26 (2) 4 (3) 23 (4) 11 (5) 22 (6) 46 (7) 22 (8) 11 | |
| Kelly 2012 | I: exenatide 10 μg twice daily |
25 | ‐ | ‐ | ‐ |
| C: metformin 1000 mg twice daily | 25 | ‐ | ‐ | ‐ | |
| Martinez‐Abundis 2015 | I: linagliptin 5 mg + placebo | 8 | ‐ | ‐ | ‐ |
| C: metformin 500 mg twice daily | 8 | ‐ | ‐ | ‐ | |
| McLaughlin 2011 | I: exenatide 10 μg twice daily |
‐ | ‐ | ‐ | ‐ |
| C: placebo | ‐ | ‐ | ‐ | ‐ | |
| Rosenstock 2008 | I: vildagliptin 50 mg | 90 | Reported adverse events occurring in ≥ 2% of participants (1) headache (2) dizziness (3) influenza (4) nasopharyngitis (5) back pain (6) diabetes mellitus non‐insulin‐dependent (7) arthralgia (8) diarrhoea (9) generalised oedema (10) tooth infection (11) tremor (12) urinary tract infection (13) asthenia (14) joint swelling (15) pharyngolaryngeal pain (16) upper respiratory tract infection (17) hunger (18) hyperhidrosis |
(1) 4 (2) 4 (3) 4 (4) 3 (5) 3 (6) 3 (7) 2 (8) 2 (9) 2 (10) 2 (11) 2 (12) 2 (13) 1 (14) 1 (15) 1 (16) 1 (17) 0 (18) 0 | (1) 4.4 (2) 4.4 (3) 4.4 (4) 3.3 (5) 3.3 (6) 3.3 (7) 2.2 (8) 2.2 (9) 2.2 (10) 2.2 (11) 2.2 (12) 2.2 (13) 1.1 (14) 1.1 (15) 1.1 (16) 1.1 (17) 0.0 (18) 0.0 |
| C: placebo | 89 | Reported adverse events occurring in ≥ 2% of participants (1) headache (2) dizziness (3) influenza (4) nasopharyngitis (5) back pain (6) diabetes mellitus non‐insulin‐dependent (7) arthralgia (8) diarrhoea (9) generalised oedema (10) tooth infection (11) tremor (12) urinary tract infection (13) asthenia (14) joint swelling (15) pharyngolaryngeal pain (16) upper respiratory tract infection (17) hunger (18) hyperhidrosis |
(1) 4 (2) 2 (3) 2 (4) 5 (5) 2 (6) 1 (7) 1 (8) 1 (9) 0 (10) 0 (11) 2 (12) 3 (13) 2 (14) 2 (15) 2 (16) 4 (17) 2 (18) 2 | (1) 4.5 (2) 2.2 (3) 2.2 (4) 5.6 (5) 2.2 (6) 1.1 (7) 1.1 (8) 1.1 (9) 0.0 (10) 0.0 (11) 2.2 (12) 3.4 (13) 2.2 (14) 2.2 (15) 2.2 (16) 4.5 (17) 2.2 (18) 2.2 | |
| Rosenstock 2010 | I: exenatide 10 μg twice daily |
‐a | ‐a | ‐a | ‐a |
| C: placebo | ‐a | ‐a | ‐a | ‐a | |
| SCALE | I: liraglutide 3.0 mg | 1524 | Non‐serious adverse events. Threshold above which adverse events were reported was 5 (1) abdominal pain (2) abdominal pain upper (3) constipation (4) diarrhoea (5) dyspepsia (6) eructation (7) flatulence (8) gastro‐oesophageal reflux disease (9) nausea (10) vomiting (11) fatigue (12) injection site hematoma (13) oedema peripheral (14) bronchitis (15) gastroenteritis (16) influenza (17) nasopharyngitis (18) sinusitis (19) upper respiratory tract infection (20) urinary tract infection (21) lipase increased (22) decreased appetite (23) hypoglycaemia (24) arthralgia (25) back pain (26) pain in extremity (27) dizziness (28) headache (29) cough (30) oropharyngeal pain (31) hypertension |
(1) 112 (2) 110 (3) 333 (4) 388 (5) 156 (6) 86 (7) 81 (8) 101 (9) 622 (10) 300 (11) 154 (12) 91 (13) 53 (14) 114 (15) 138 (16) 181 (17) 404 (18) 129 (19) 236 (20) 123 (21) 145 (22) 169 (23) 295 (24) 186 (25) 198 (26) 108 (27) 149 (28) 273 (29) 112 (30) 74 (31) 74 | (1) 7.4 (2) 7.2 (3) 21.9 (4) 25.5 (5) 10.2 (6) 5.6 (7) 5.3 (8) 6.6 (9) 40.8 (10) 20.0 (11) 10.1 (12) 6.0 (13) 3.5 (14) 7.5 (15) 9.1 (16) 11.9 (17) 26.5 (18) 8.5 (19) 15.5 (20) 8.1 (21) 9.5 (22) 11.1 (23) 19.4 (24) 12.2 (25) 13.0 (26) 7.1 (27) 9.8 (28) 17.9 (29) 7.4 (30) 4.9 (31) 4.9 |
| C: placebo | 755 | Non‐serious adverse events. Threshold above which adverse events were reported was 5 (1) abdominal pain (2) abdominal pain upper (3) constipation (4) diarrhoea (5) dyspepsia (6) eructation (7) flatulence (8) gastro‐oesophageal reflux disease (9) nausea (10) vomiting (11) fatigue (12) injection site hematoma (13) oedema peripheral (14) bronchitis (15) gastroenteritis (16) influenza (17) nasopharyngitis (18) sinusitis (19) upper respiratory tract infection (20) urinary tract infection (21) lipase increased (22) decreased appetite (23) hypoglycaemia (24) arthralgia (25) back pain (26) pain in extremity (27) dizziness (28) headache (29) cough (30) oropharyngeal pain (31) hypertension |
(1) 37 (2) 40 (3) 85 (4) 107 (5) 156 (6) 4 (7) 22 (8) 18 (9) 128 (10) 42 (11) 58 (12) 61 (13) 47 (14) 62 (15) 46 (16) 80 (17) 210 (18) 65 (19) 120 (20) 43 (21) 24 (22) 27 (23) 35 (24) 97 (25) 118 (26) 54 (27) 55 (28) 124 (29) 60 (30) 44 (31) 48 | (1) 4.9 (2) 5.3 (3) 11.3 (4) 14.2 (5) 10.2 (6) 0.5 (7) 2.9 (8) 2.4 (9) 17.0 (10) 5.6 (11) 7.7 (12) 8.1 (13) 6.2 (14) 8.2 (15) 6.1 (16) 10.6 (17) 27.8 (18) 8.6 (19) 15.9 (20) 5.7 (21) 3.2 (22) 3.6 (23) 4.6 (24) 12.9 (25) 15.6 (26) 7.2 (27) 7.3 (28) 16.4 (29) 8.0 (30) 5.8 (31) 6.4 | |
| ‐ denotes not reported. aOnly reported for all the included participants and not separately for the fraction of participants with impaired glucose tolerance or impaired fasting glucose (or both). C: comparator; I: intervention; n: number of participants; SCALE: Satiety and Clinical Adiposity ‐ Liraglutide Evidence in Nondiabetic and Diabetic Individuals. | |||||
Appendix 14. Survey of trial investigators providing information on trials
| Trial | Date trial author contacted | Date trial author replied | Date trial author was asked for additional information (short summary) | Date trial author provided data (short summary) |
| Almeda‐Valdes 2012 | 29 March 2016 | 30 March 2016 | 30 March 2016 Asked if the trial was published anywhere |
5 April 2016 Trial only published as a conference abstract (Almeda‐Valdes 2012) |
| Aoki 2014 | 1 April 16 and 6 April 2016 | 8 April 2016 | 8 April 2016 Questions regarding method and outcomes reported |
11 April 2016 Did not provide information |
| Ariel 2014 | 29 August 2016 | 29 August 2016 | 29 August 2016 Extraction sheet and list of included trials were provided to get additional information on outcomes of interest for review. |
29 August 2016 Authors could not provide any additional information about outcomes |
| Astrup 2009 | 6 April, 29 June and 1 July 2016 | Immediate auto reply; also, an employee sent us an email | 6 April, 29 June and 1 July 2016 Extraction sheet and list of included trials were provided to get additional information on participants with intermediate hyperglycaemia |
19 September 2016 Novo Nordisk received all required information for data sharing. Novo Nordisk has approved access to raw trial data, which are not yet available |
| BEGAMI 2013 | 1 July 2016 | 6 July 2016 Would try to find data as soon as possible |
Extraction sheet and list of included trials were provided to get additional information on participants with intermediate hyperglycaemia | 7 July 2016 Provided separate outcome data on participants with intermediate hyperglycaemia |
| Daniele 2015 | 8 April 2016 | 9 April 2016 | 9 April 2016 Question regarding duration of intervention: ClinicalTrials.gov reported 12 weeks but publication reported 26 days. Requested separate data on participants with IGT |
9 April 2016 Replied that trial was not suitable for the meta‐analysis |
| Dushay 2012 | 27 March, 7 April and 29 June 2016 | 7 April and 14 April 2016 | Asked for separate data on the participants with IGT/IFG | 7 April and 14 April 2016 Replied they would try to find the separate data on people with IGT/IFG but did not provide these data |
| EudraCT 2013‐000418‐39 | 20 March and 7 July 2016 | 24 March 2016 with an auto reply; again 7 July 2016 | 20 March 2016 Asked for information about who was blinded, the intervention, trial start and completion data |
The author's reply referred to a web page which gave an error when following the link. The information was provided by another email to 1 of the investigators |
| Gudipaty 2014 | 27 March 2016 | 29 March 2016 | 3 April 2016 Asked for separate data on the participants with IGT/IFG |
6 April 2016 Clarified that all participants had a history of type 2 diabetes mellitus |
| Ishikawa 2014 | 3 July and 12 August 2016 | No reply | Extraction sheet and list of included trials were provided to get additional information on participants with intermediate hyperglycaemia | N/A |
| Kelly 2012 | 30 August 2016 | No reply | Extraction sheet and list of included trials were provided to get additional information on participants | N/A |
| McLaughlin 2011 | 30 March 2016, 7 July 2016 and 15 February 2017 | 30 March 2016 | Extraction sheet and list of included trials were provided to get additional information | 30 March 2016 Replied that complete trial was not yet published. No reply when contacted again in July 2016 and February 2017 |
| Martinez‐Abundis 2015 | 30 March 2016 | 30 March 2016 | 30 March 2016 Asked if trial was published as full text, if trial protocol was available and if additional data could be provided |
9 April 2016 Author replied that trial was only published as an abstract, but provided unpublished data on some of the outcomes relevant for our review |
| NCT01018602 | 3 July 2016 | 6 July 2016 When trial is unblinded, investigators will provide data |
Extraction sheet and list of included trials were provided to get additional information on participants with intermediate hyperglycaemia | 6 July 2016 When trial is unblinded, investigators will provide data |
| NCT01234649 | 31 March 2016 | 31 March 2016 | Asked about duration of intervention | 31 March 2016 Clarified duration of intervention period |
| NCT01960205 | 30 March and 15 April 2016 | 19 April 2016 | 30 March and 15 April 2016 Asked for data on trial. Trial completed December 2014, but no data published |
19 April 2016 Unable to publish data yet, so no additional data could be provided |
| NCT01122641 | 17 and 23 March 2016 | No reply | Asked how many participants had IGT, IFG or intermediate elevated HbA1c at baseline | N/A |
| NCT01336322 | 30 March 2016 | 31 March 2016 | Asked if trial was published as completion data registered on ClinicalTrials.gov was exceeded | 31 March 2016 Trial still ongoing. Was delayed due to study drug supply |
| NCT01521312 | 24 March and 15 April 2016 | No reply | Asked for trial data | N/A Trial completed September 2014 according to ClinicalTrials.gov, but no data are published. Authors asked if the trial was published, or if they could provide additional data |
| NCT01038648 | 23 March 2016 | 23 March 2016 | According to ClinicalTrials.gov, trial was stopped prior to enrolment. Asked for reason | 23 March 2016 Trial not approved by Indian authorities |
| NCT02104739 | 30 March 2016 | 30 March 2016 | Asked regarding duration of intervention | 31 March 2016 Clarified the duration of the intervention period |
| NCT00961363 | 30 March 2016 | 30 March 2016 | According to ClinicalTrials.gov trial is fulfilling inclusion criteria and was completed in 2011 | 31 March 2016 Trial terminated early due difficulties in recruiting participants with IGT |
| NCT00845559 | 24 March 16 | No reply | Asked for reason for withdrawal of trial before enrolment | N/A |
| NCT02140983 | 30 March and 7 April 2016 | 7 April 2016 | 30 March and 7 April 2016 Asked if it was possible to get data for this review, as the trial was completed February 2016 according to ClinicalTrials.gov |
7 April 2016 Trial was not yet completed, but would be completed in the end of 2016. The reason for the delay was that the investigators had to wait for the drug supply |
| NCT01006018 | 23 March 2016 | 24 March 2016 | Asked why the trial was stopped prematurely | 24 March 2016 Trial was stopped before recruitment due to difficulties of GLP‐1 supplies |
| NCT02488057 | 25 March 2016 | 25 March 2016 | Asked for diagnostic criteria for intermediate hyperglycaemia | 26 March 2016 Authors replied that the diagnostic criteria applied were from the American Diabetes Association |
| NCT01472640 | 1 July 2016 | 4 July 2016 | Extraction sheet and list of included trials were provided to get additional information on participants with intermediate hyperglycaemia | 4 July 2016 Not possible to get separate data on the people with IGT before the main publication is published |
| PIO‐EX 2009 | 30 March and 8 April 2016 | No reply | Authors asked for separate data on participants with IGT. Trial completed in 2010 | N/A |
| Rosenstock 2008 | 8 April and 4 July 2016 | 22 March 2016 | Extraction sheet and list of included trials were provided to get information on participants with intermediate hyperglycaemia | The pharmaceutical company was contacted, and it was required that the request was send through a link. It was done, but no reply was received. The contact person from the pharmaceutical company was therefore contacted again in July 2016, but no reply was given |
| Rosenstock 2010 | 6 April 2016, 7 July 2016 and 16 January 2017 | 6 April 2016 | Extraction sheet and list of included trials were provided to get information on participants with intermediate hyperglycaemia | Trial authors replied that a pharmaceutical company should be contacted. The company was contacted, and it was required that the request was send through a link. It was done, but no reply was received. The contact person from the pharmaceutical company was therefore contacted again in July 2016 and February 2017, but no reply was given |
| Santilli 2015 | 5 April and 1 July 2016 | 11 April and 7 July 2016 | 5 April 2016 Extraction sheet and list of included trials were provided to get additional information |
11 April and 7 July 2016 Trial not yet published. When it is published, they will provide data |
| SCALE | 15 September 2016, 22 March 2017 and 23 March 2017 | 16 September 2016, 22 March 2017 and 23 March 2017: no reply | Novo Nordisk: asked for additional information regarding outcomes for the participants with intermediate hyperglycaemia at baseline. Corresponding author: asked about the application of study medication the days of drug testing. 22 and 23 March 2017: investigator asked for the reason of the discrepancy in the number with diabetes in the text of the publication compared with the appendix (Lancet 2017) |
16 September 2016 Primary investigator replied that participants were not supposed to take the study medication on the days of measuring oral glucose tolerance test. No reply regarding fasting plasma glucose. 19 September 2016 Novo Nordisk received all required information for data sharing. Novo Nordisk has approved access to raw trial data, but data are not yet available |
| SCALE‐SLEEP | 8 April and 1 July 2016 | Immediate auto reply; also, an employee sent us an email | 8 April and 1 July 2016 Extraction sheet and list of included trials were provided to get additional information on participants with intermediate hyperglycaemia |
19 September 2016 Novo Nordisk received all required information for data sharing. Novo Nordisk has approved access to raw trial data, but data are not yet available |
| SCALE‐TM 2013 | 7 April and 1 July 2016 | No reply | N/A | 19 September 2016 Novo Nordisk received all required information for data sharing. Novo Nordisk has approved access to raw trial data, but data are not yet available |
| Tsuchiya 2011 | 6 and 14 April 2016 | No reply | N/A | N/A |
| UMIN000008620 | 7 July 2016 | No reply | Asked about completion date | N/A |
| GLP‐1: glucagon‐like peptide‐1; HbA1c: glycosylated haemoglobin A1c; IFG: impaired fasting glucose; IGT: impaired glucose tolerance; N/A: not applicable. | ||||
Appendix 15. Checklist to aid consistency and reproducibility of GRADE assessments: dipeptidyl‐peptidase‐4 inhibitors
| (1) All‐cause mortality | (2) Incidence of T2DM | (3) Serious adverse events | (4) Cardiovascular mortality | (5) Non‐fatal myocardial infarction/stroke and congestive heart failure | (6) Health‐related quality of life | (7) Socioeconomic effects | ||
| Trial limitations (risk of bias)a | Was random sequence generation used (i.e. no potential for selection bias)? | Yes | Yes | Yes | Yes | Yes | N/A | N/A |
| Was allocation concealment used (i.e. no potential for selection bias)? | Yes | Yes | Yes | Yes | Yes | |||
| Was there blinding of participants and personnel (i.e. no potential for performance bias) or outcome not likely to be influenced by lack of blinding? | Yes | Yes | Yes | Yes | Yes | |||
| Was there blinding of outcome assessment (i.e. no potential for detection bias) or was outcome measurement not likely to be influenced by lack of blinding? | Yes | Yes | Yes | Yes | Yes | |||
| Was an objective outcome used? | Yes | Yes | Yes | Yes | Yes | |||
| Were more than 80% of participants enrolled in trials included in the analysis (i.e. no potential reporting bias)?e | Yes | Yes | Yes | Yes | Yes | |||
| Were data reported consistently for the outcome of interest (i.e. no potential selective reporting)? | Yes | Yes | Yes | Yes | Yes | |||
| No other biases reported (i.e. no potential of other bias)? | Unclear | Unclear | Unclear | Unclear | Unclear | |||
| Did the trials end up as scheduled (i.e. not stopped early)? | Yes | Yes | Yes | Yes | Yes | |||
| Inconsistencyb | Point estimates did not vary widely? | N/A | N/A | N/A | N/A | N/A | ||
| To what extent did confidence intervals overlap (substantial: all confidence intervals overlap at least one of the included studies point estimate; some: confidence intervals overlap but not all overlap at least one point estimate; no: at least one outlier: where the confidence interval of some of the studies do not overlap with those of most included studies)? | N/A | N/A | N/A | N/A | N/A | |||
| Was the direction of effect consistent? | N/A | N/A | N/A | N/A | N/A | |||
| What was the magnitude of statistical heterogeneity (as measured by I²) ‐ low (I²<40%), moderate (I² 40%‐60%), high I²>60%)? | N/A | N/A | N/A | N/A | N/A | |||
| Was the test for heterogeneity statistically significant (P < 0.1)? | N/A | N/A | N/A | N/A | N/A | |||
| Indirectness | Were the populations in included studies applicable to the decision context? | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | ||
| Were the interventions in the included studies applicable to the decision context? | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | |||
| Was the included outcome not a surrogate outcome? | Yes | No (↓) | Yes | Yes | Yes | |||
| Was the outcome timeframe sufficient? | Insufficient (↓) | Insufficient (↓) | Insufficient (↓) | Insufficient (↓) | Insufficient (↓) | |||
| Were the conclusions based on direct comparisons? | Yes | Yes | Yes | Yes | Yes | |||
| Imprecisionc | Was the confidence interval for the pooled estimate not consistent with benefit and harm? | N/A | N/A | N/A | N/A | N/A | ||
| What is the magnitude of the median sample size (high: >300 participants, intermediate: 100‐300 participants, low: <100 participants)?e | Low (↓) | Intermediate | Intermediate | Low (↓) | Low (↓) | |||
| What was the magnitude of the number of included studies (large: >10 studies, moderate: 5‐10 studies, small: <5 studies)?e | Small (↓) | Small (↓) | Small (↓) | Small (↓) | Small (↓) | |||
| Was the outcome a common event (e.g. occurs more than 1/100)? | Yes | Yes | Yes | Yes | Yes | |||
| Publication biasd | Was a comprehensive search conducted? | Yes | Yes | Yes | Yes | Yes | ||
| Was grey literature searched? | Yes | Yes | Yes | Yes | Yes | |||
| Were no restrictions applied to study selection on the basis of language? | Yes | Yes | Yes | Yes | Yes | |||
| There was no industry influence on studies included in the review? | No (↓) | No (↓) | No (↓) | No (↓) | No (↓) | |||
| There was no evidence of funnel plot asymmetry? | N/A | N/A | N/A | N/A | N/A | |||
| There was no discrepancy in findings between published and unpublished trials? | N/A | N/A | N/A | N/A | N/A | |||
|
aQuestions on risk of bias are answered in relation to the majority of the aggregated evidence in the meta‐analysis rather than to individual trials.
bQuestions on inconsistency are primarily based on visual assessment of forest plots and the statistical quantification of heterogeneity based on I². cWhen judging the width of the CI it is recommended to use a clinical decision threshold to assess whether the imprecision is clinically meaningful. dQuestions address comprehensiveness of the search strategy, industry influence, funnel plot asymmetry and discrepancies between published and unpublished trials. eDepends on the context of the systematic review area. (↓): key item for potential downgrading the quality of the evidence (GRADE) as shown in the footnotes of the 'Summary of finding' table(s). CI: confidence interval; N/A: not applicable; T2DM: type 2 diabetes mellitus. | ||||||||
Appendix 16. Checklist to aid consistency and reproducibility of GRADE assessments: glucagon‐like peptide‐1 analogues
| Glucagon‐like peptide‐1 analogues compared with placebo | ||||||||
| (1) All‐cause mortality | (2) Incidence of T2DM | (3) Serious adverse events | (4) Cardiovascular mortality | (5) Non‐fatal myocardial infarction/stroke and congestive heart failure | (6) Health‐related quality of life | (7) Socioeconomic effects | ||
| Trial limitations (risk of bias)a | Was random sequence generation used (i.e. no potential for selection bias)? | Unclear | Yes | Yes | Unclear | Yes | Yes | N/A |
| Was allocation concealment used (i.e. no potential for selection bias)? | Unclear | Yes | Yes | Unclear | Yes | Yes | ||
| Was there blinding of participants and personnel (i.e. no potential for performance bias) or outcome not likely to be influenced by lack of blinding? | Yes | Yes | Yes | Yes | Yes | Yes | ||
| Was there blinding of outcome assessment (i.e. no potential for detection bias) or was outcome measurement not likely to be influenced by lack of blinding? | Yes | Yes | Yes | Yes | Yes | Yes | ||
| Was an objective outcome used? | Yes | Yes | Yes | Yes | Yes | Yes | ||
| Were more than 80% of participants enrolled in trials included in the analysis (i.e. no potential reporting bias)?e | Unclear | No (↓) | Yes | Unclear | Unclear | No (↓) | ||
| Were data reported consistently for the outcome of interest (i.e. no potential selective reporting)? | Yes | Yes | Unclear | Yes | Unclear | Unclear | ||
| No other biases reported (i.e. no potential of other bias)? | No (↓) | No (↓) | No (↓) | No (↓) | No (↓) | |||
| Did the trials end up as scheduled (i.e. not stopped early)? | Yes | Yes | Yes | Yes | Yes | Yes | ||
| Inconsistencyb | Point estimates did not vary widely? | N/A | N/A | N/A | N/A | N/A | N/A | |
| To what extent did confidence intervals overlap (substantial: all confidence intervals overlap at least one of the included studies point estimate; some: confidence intervals overlap but not all overlap at least one point estimate; no: at least one outlier: where the confidence interval of some of the studies do not overlap with those of most included studies)? | N/A | N/A | N/A | N/A | N/A | N/A | ||
| Was the direction of effect consistent? | N/A | N/A | N/A | N/A | N/A | N/A | ||
| What was the magnitude of statistical heterogeneity (as measured by I²) ‐ low (I²<40%), moderate (I² 40%‐60%), high I²>60%)? | N/A | N/A | N/A | N/A | N/A | N/A | ||
| Was the test for heterogeneity statistically significant (P < 0.1)? | N/A | N/A | N/A | N/A | N/A | N/A | ||
| Indirectness | Were the populations in included studies applicable to the decision context? | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | |
| Were the interventions in the included studies applicable to the decision context? | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | ||
| Was the included outcome not a surrogate outcome? | Yes | No (↓) | Yes | Yes | Yes | Yes | ||
| Was the outcome timeframe sufficient? | Sufficient | Sufficient | Sufficient | Sufficient | Sufficient | Sufficient | ||
| Were the conclusions based on direct comparisons? | Yes | Yes | Yes | Yes | Yes | Yes | ||
| Imprecisionc | Was the confidence interval for the pooled estimate not consistent with benefit and harm? | N/A | No (↓) | N/A | N/A | N/A | N/A | |
| What is the magnitude of the median sample size (high: >300 participants, intermediate: 100‐300 participants, low: <100 participants)?e | Low (↓) | High | High | Low (↓) | High | High | ||
| What was the magnitude of the number of included studies (large: >10 studies, moderate: 5‐10 studies, small: <5 studies)?e | Small (↓) | Moderate | Moderate | Small (↓) | Moderate | Moderate | ||
| Was the outcome a common event (e.g. occurs more than 1/100)? | Yes | Yes | Yes | Yes | Yes | N/A | ||
| Publication biasd | Was a comprehensive search conducted? | Yes | Yes | Yes | Yes | Yes | Yes | |
| Was grey literature searched? | Yes | Yes | Yes | Yes | Yes | Yes | ||
| Were no restrictions applied to study selection on the basis of language? | Yes | Yes | Yes | Yes | Yes | Yes | ||
| There was no industry influence on studies included in the review? | No (↓) | No (↓) | No (↓) | No (↓) | No (↓) | No (↓) | ||
| There was no evidence of funnel plot asymmetry? | N/A | N/A | N/A | N/A | N/A | N/A | ||
| There was no discrepancy in findings between published and unpublished trials? | N/A | N/A | N/A | N/A | N/A | N/A | ||
|
aQuestions on risk of bias are answered in relation to the majority of the aggregated evidence in the meta‐analysis rather than to individual trials.
bQuestions on inconsistency are primarily based on visual assessment of forest plots and the statistical quantification of heterogeneity based on I². cWhen judging the width of the CI it is recommended to use a clinical decision threshold to assess whether the imprecision is clinically meaningful. dQuestions address comprehensiveness of the search strategy, industry influence, funnel plot asymmetry and discrepancies between published and unpublished trials. eDepends on the context of the systematic review area. (↓): key item for potential downgrading the quality of the evidence (GRADE) as shown in the footnotes of the 'Summary of finding' table(s). CI: confidence interval; GLP‐1: glucagon‐like peptide‐1; N/A: not applicable; T2DM: type 2 diabetes mellitus. | ||||||||
Appendix 17. Health‐related quality of life: instruments
| Instrument | Dimensions (subscales) (number of items) | Validated instrument | Answer options | Scores |
Minimum score Maximum score |
Weighting of scores | Direction of scales | Minimal important difference | |
| SF‐36 (G) Employed in: SCALE |
Physical Functioning (PF) (10), Role‐Physical (RP) (4), Bodily Pain (BP) (2), General Health (GH) (5), Vitality (VT) (4) Social Functioning (SF) (2) Role‐Emotional (RE) (3), Mental Health (MH) (5) | Yes | Likert‐scale | Scores for dimensions
Physical Component Summary (PCS) Mental Component Summary (MCS) |
Minimum scores: 0 Maximum scores: 100 |
No | Higher values mean better assessment | PCS: 2‐3 points MCS: 3 points Dimensions: PF/BT/VT: 2 points, if score < 40; 3 points, if score ≥ 40 RP: 2 points SF/MH: 3 points RE: 4 points |
|
| G: generic; SF‐36: 36‐item Short Form health survey. | |||||||||
Appendix 18. Selection bias decisions
| Selection bias decisions for trials reporting unadjusted analyses ‐ comparison of results obtained using method details alone with results using method details and trial baseline informationa | |||
| Reported randomisation and allocation concealment methods | Risk of bias judgement using methods reporting | Information gained from study characteristics data | Risk of bias using baseline information and methods reporting |
| Unclear methods | Unclear risk | Baseline imbalances present for important prognostic variable(s) | High risk |
| Groups appear similar at baseline for all important prognostic variables | Low risk | ||
| Limited or no baseline details | Unclear risk | ||
| Would generate a truly random sample, with robust allocation concealment | Low risk | Baseline imbalances present for important prognostic variable(s) | Unclear riskc |
| Groups appear similar at baseline for all important prognostic variables | Low risk | ||
| Limited baseline details, showing balance in some important prognostic variablesb | Low risk | ||
| No baseline details | Unclear risk | ||
| Sequence is not truly random, or allocation concealment is inadequate | High risk | Baseline imbalances present for important prognostic variable(s) | High risk |
| Groups appear similar at baseline for all important prognostic variables | Low risk | ||
| Limited baseline details, showing balance in some important prognostic variablesb | Unclear risk | ||
| No baseline details | High risk | ||
| aTaken from Corbett 2014; judgements highlighted in bold indicate situations in which the addition of baseline assessments would change the judgement about risk of selection bias, compared with using methods reporting alone. bDetails for the remaining important prognostic variables not reported. cImbalance identified which appears likely to be due to chance. | |||
Data and analyses
Comparison 1. Dipeptidyl‐peptidase (DPP)‐4 inhibitors versus metformin.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Non‐serious adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2 Fasting blood glucose | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3 2‐hour glucose | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4 Haemoglobin A1c | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only |
Comparison 2. DDP‐4 inhibitors versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Incidence of type 2 diabetes mellitus | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2 Serious adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3 Congestive heart failure | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4 Non‐serious adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5 Fasting glucose | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6 2‐hour glucose values | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 7 Haemoglobin A1c | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only |
Comparison 3. Glucagon‐like peptide (GLP)‐1 analogues versus metformin.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Fasting blood glucose | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only |
Comparison 4. Glucagon‐like peptide (GLP)‐1 analogues versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Incidence of type 2 diabetes mellitus | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2 Serious adverse events | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3 Non‐serious adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4 Mild hypoglycaemia | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5 Fasting blood glucose | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6 2‐hour glucose | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 7 Haemoglobin A1c | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Ariel 2014.
| Methods |
Design: parallel RCT Randomisation ratio: 1:1 Number of study centres: 1 |
|
| Participants |
Inclusion criteria: aged 40‐70 years; IFG or IGT; BMI 27.0‐37.0 kg/m²; stable weight (< 5% reported change) in the previous 3 months Exclusion criteria: T2DM; use of medications that can affect carbohydrate metabolism or promote weight loss; gallstones; history of pancreatitis; medullary carcinoma; family history of medullary carcinoma or multiple endocrine neoplasia type 2; and known cardiac, liver or kidney disease Diagnostic criteria: criteria for intermediate hyperglycaemia as defined by ADA: intermediate hyperglycaemia defined as elevated fasting glucose (5.6‐6.9 mmol/L) or elevated 2‐h glucose (7.8‐11.0 mmol/L) concentration after OGTT with 75‐g glucose, or both; 1 of the publications defined intermediate hyperglycaemia as elevated fasting glucose (5.6‐6.9 mmol/L) or elevated 2‐h glucose (7.8‐10.5 mmol/L) concentration after OGTT with 75‐g glucose, or both |
|
| Interventions |
Intervention: liraglutide 1.8 mg subcutaneous Comparator: placebo subcutaneous Run‐in period: none Study drug administration free period before glucose testing during trial: fasting glucose measured on the days of the insulin suppression test. On these days, the trial medication was given after the testing Extension period: none |
|
| Outcomes | Composite outcome measures reported: no | |
| Study details | Trial terminated early: no | |
| Publication details |
Language of publication: English Funding: commercial funding (liraglutide and matching placebo were provided by Novo Nordisk); non‐commercial funding (ADA) Publication status: peer‐reviewed journal/full article |
|
| Stated aim for study | Quote: "To evaluate the effects of 14 weeks of liraglutide plus modest caloric restriction on lipid/lipoprotein metabolism in overweight/obese persons with prediabetes." | |
| Notes | The participants visited a dietitian weekly for the first 4 weeks, and then every 2 weeks. They were advised to eat a moderate‐carbohydrate diet (43% carbohydrate, 42% fat and 15% protein) and to decrease total caloric intake by 500 kcal/day. Individualised meal plan was provided | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk |
Quote: "This was a double‐blind, randomised, placebo controlled, parallel‐group study. Participants were randomised to receive either liraglutide (n =35) or matching placebo (n =33) by block randomisation by sex and BMI (<31 vs ≥31 kg/m²) via a computerised randomisation system." Comment: method of random sequence generation adequately described |
| Allocation concealment (selection bias) | Low risk |
Quote: "This was a double‐blind, randomised, placebo controlled, parallel‐group study. Participants were randomised to receive either liraglutide (n =35) or matching placebo (n =33) by block randomisation by sex and BMI (<31 vs ≥31 kg/m²) via a computerised randomisation system." Comment: method of allocation concealment adequately described |
| Blinding of participants and personnel (performance bias) measures of blood glucose control | Low risk |
Quote: "We conducted a double‐blind ..." Comment: investigator‐assessed |
| Blinding of outcome assessment (detection bias) measures of blood glucose control | Low risk |
Quote: "We conducted a double‐blind ..." Comment: investigator‐assessed |
| Incomplete outcome data (attrition bias) measures of blood glucose control | High risk |
Quote: "Only subjects who had end‐of‐study testing were included in the analyses." Comment: 31% (n = 11) in liraglutide group and 18% (n = 6) in placebo group dropped out during the trial and were not included in the final analysis of fasting blood glucose |
| Selective reporting (reporting bias) | Low risk | Comment: no selective outcome reporting bias |
| Other bias | Unclear risk | Quote: "funding was received by a pharmaceutical company." |
Kelly 2012.
| Methods |
Design: parallel RCT Randomisation ratio: 1:1 Number of study centres: 2 |
|
| Participants |
Inclusion criteria: aged ≥ 18 years; IGT or IFG or moderately elevated HbA1c; abdominal obesity: waist circumference > 102 cm (men) and > 88 cm (women); stable cardiovascular medication regimen (or other medications known to affect endothelial function) at least 1 month prior to enrolment and throughout the study Exclusion criteria: T2DM; current use of glycaemic control medications within 1 month of randomisation; fasting glucose > 126 mg/dL (7.0 mmol/L); current use of weight loss medication; previous weight loss surgery; history of severe gastrointestinal disease; standard clinical contraindications to exenatide or metformin; unstable angina; heart failure; stroke or coronary artery bypass graft within 3 months of screening; women who were currently pregnant or planning to become pregnant; breastfeeding women; clinically significant liver disease; creatinine > 1.5 mg/dL; hepatic function > 3 times upper limit of normal; mentally incompetent and cannot sign a patient informed consent form Diagnostic criteria: criteria for intermediate hyperglycaemia as defined by ADA: IGT: 2‐h OGTT plasma glucose > 140 mg/dL (7.8 mmol/L); IFG: fasting glucose ≥ 100 mg/dL (5.6 mmol/L); moderately elevated HbA1c ≥ 5.7% |
|
| Interventions |
Intervention: exenatide 10 μg twice daily Comparator: metformin 1000 mg twice daily Run‐in period: none Study drug administration free period before glucose testing during trial: all testing was performed in the morning after fasting. The study drugs were withheld on these mornings Extension period: none |
|
| Outcomes | Composite outcome measures reported: no | |
| Study details | Trial terminated early: no | |
| Publication details |
Language of publication: English Funding: commercial funding (funding was provided by an investigator‐initiated grant from Amylin Pharmaceuticals and Eli Lilly and Company) Publication status: peer‐reviewed journal/full article |
|
| Stated aim for study | Quote: "The purpose of this study is to compare the effects of exenatide versus metformin on vascular health with chronic (3‐month) therapy and during a 2‐hour period following a meal in patients with pre‐diabetes. It is predicted that exenatide will improve vascular health to a greater degree compared to metformin." | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote: "We performed a 3‐month, randomized (1:1), open‐label, head‐to‐head (exenatide vs. metformin) clinical trial." Comment: method of random sequence generation not adequately described |
| Allocation concealment (selection bias) | Unclear risk |
Quote: "We performed a 3‐month, randomized (1:1), open‐label, head‐to‐head (exenatide vs. metformin) clinical trial." Comment: method of allocation concealment not adequately described |
| Blinding of participants and personnel (performance bias) measures of blood glucose control | Low risk |
Quote: "We performed a randomized, open‐label ..." Comment: investigator‐assessed outcome measurement, outcome unlikely to be influenced by lack of blinding |
| Blinding of participants and personnel (performance bias) non‐serious adverse events | Unclear risk |
Quote: "We performed a randomized, open‐label ..." Comment: self‐reported and investigator‐assessed outcome measurement. Trial observed no non‐serious adverse events |
| Blinding of participants and personnel (performance bias) serious adverse events | Low risk |
Quote: "We performed a randomized, open‐label ..." Comment: investigator‐assessed outcome measurement, outcome unlikely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) measures of blood glucose control | Low risk |
Quote: "We performed a randomized, open‐label ..." Comment: investigator‐assessed outcome measurement, outcome unlikely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) non‐serious adverse events | Unclear risk |
Quote: "We performed a randomized, open‐label ..." Comment: self‐reported and investigator‐assessed outcome measurement. Trial observed no non‐serious adverse events. |
| Blinding of outcome assessment (detection bias) serious adverse events | Low risk |
Quote: "We performed a randomized, open‐label ..." Comment: investigator‐assessed outcome measurement, outcome unlikely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) measures of blood glucose control | Low risk | Comment: all randomised participants completed study and were included in analysis |
| Incomplete outcome data (attrition bias) non‐serious adverse events | Low risk | Comment: all randomised participants completed study and were included in analysis |
| Selective reporting (reporting bias) | High risk |
Quote from ClinicalTrials.gov: "Threshold above which other adverse events are reported 5." Comment: quote referred to non‐serious adverse events, meaning that adverse events with a lower frequency were not reported |
| Other bias | Unclear risk | Quote: funding received from a pharmaceutical company |
Martinez‐Abundis 2015.
| Methods |
Design: parallel RCT Randomisation ratio: 1:1 Number of study centres: NR, presumably 1 |
|
| Participants |
Inclusion criteria: IGT with overweight or obesity, no other inclusion criteria specified Exclusion criteria: not specified Diagnostic criteria: IGT, not further specified |
|
| Interventions |
Intervention: linagliptin 5 mg + placebo. Comparator: metformin 500 mg twice daily Run‐in period: NR Study drug administration free period before glucose testing during trial: NR Extension period: none |
|
| Outcomes | Composite outcome measures reported: no | |
| Study details | Trial terminated early: no | |
| Publication details |
Language of publication: English Funding: NR Publication status: abstract |
|
| Stated aim for study | Quote: "The aim of this study was to assess the effect of linagliptin on glycemic control and GV in patients with IGT." | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk |
Quote from internal correspondence: "Yes, we used a table of random numbers for randomisation." Comment: method of random sequence generation judged sufficient |
| Allocation concealment (selection bias) | Unclear risk | Comment: method for allocation concealment NR |
| Blinding of participants and personnel (performance bias) measures of blood glucose control | Low risk |
Quote: "A randomized, double blind clinical trial with parallel groups ..." Comment: measured by investigator at baseline and end of follow‐up. Blinding ensured by placebo tablet |
| Blinding of participants and personnel (performance bias) non‐serious adverse events | Low risk |
Quote: "A randomized, double blind clinical trial with parallel groups ..." Comment: unclear how non‐serious adverse events were adjudicated. Blinding ensured by placebo tablet |
| Blinding of outcome assessment (detection bias) measures of blood glucose control | Low risk |
Quote: "A randomized, double blind clinical trial with parallel groups ..." Comment: measured by investigator at baseline and end of follow‐up. Blinding ensured by placebo tablet |
| Blinding of outcome assessment (detection bias) non‐serious adverse events | Low risk |
Quote: "A randomized, double blind clinical trial with parallel groups ..." Comment: unclear how non‐serious adverse events were adjudicated. Blinding ensured by placebo tablet |
| Incomplete outcome data (attrition bias) measures of blood glucose control | Low risk | Comment: according to data provided by investigators all participants were included in analysis |
| Selective reporting (reporting bias) | Unclear risk | Comment: insufficient information about selective reporting. No trial protocol available |
| Other bias | Unclear risk | Comment: funding source NR |
McLaughlin 2011.
| Methods |
Design: parallel RCT, superiority design, controlled clinical trial Randomisation ratio: 1:1 Number of study centres: NR |
|
| Participants |
Inclusion criteria: aged 30‐70 years; healthy men and women, BMI 27‐37 kg/m²; FPG ≥ 100 mg/dL and ≤ 125 mg/dL or a 2‐h post OGTT ≥ 140 mg/dL or ≤ 199 mg/dL Exclusion criteria: diabetes; active cardiac, kidney, liver, pulmonary or other major organ diseases that are cause for exclusion; other exclusionary criteria include: use of corticosteroids, diet medications or antipsychotic medications, history of eating disorder, history of bariatric surgery, active malignancy, recent weight change > 2%, inability to attend follow‐up visits, excessive alcohol use, investigator's discretion that it is not in person's best interest. Diagnostic criteria: criteria for intermediate hyperglycaemia as defined by ADA: FPG ≥ 100 mg/dL and ≤ 125 mg/dL or a 2‐h post OGTT ≥ 140 mg/dL or ≤ 199 mg/dL |
|
| Interventions |
Intervention: exenatide 10 μg twice daily Comparator: placebo Run‐in period: none Study drug administration free period before glucose testing during trial: NR Extension period: none |
|
| Outcomes | Composite outcome measures reported: no | |
| Study details | Trial terminated early: no | |
| Publication details |
Language of publication: English Funding: NR Publication status: abstract |
|
| Stated aim for study | Quote: "We sought to evaluate persistence of weight and metabolic benefits of exenatide in moderately‐obese prediabetics one year after discontinuing the drug." | |
| Notes | Contacted investigator Dr Tracey, but the trial is not yet published as an article | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote: "In a double blinded, randomized controlled design ..." Comment: method of random sequence generation not adequately described |
| Allocation concealment (selection bias) | Unclear risk |
Quote: "In a double blinded, randomized controlled design ..." Comment: method of allocation concealment not adequately described |
| Selective reporting (reporting bias) | High risk | Comment: stated in abstract that fasting blood glucose and 2‐h blood glucose were measured, but data not reported in a way that they could be included in review. Useable data might be available in updates |
| Other bias | Unclear risk | Comment: funding source NR |
Rosenstock 2008.
| Methods |
Design: parallel RCT Randomisation ratio: 1:1 Number of study centres: 28 |
|
| Participants |
Inclusion criteria: aged 18‐80 years with IGT as defined as FPG < 126 mg/dL (7.0 mmol/L) and 2‐h post‐challenge plasma glucose (after OGTT 75‐g glucose) 140 mg/dL (7.8 mmol/L) to < 200 mg/dL (< 11.1 mmol/L) and BMI 23‐45 kg/m². Agreement to maintain prior diet and exercise habits during the full course of trial. Women with childbearing potential required to use medically approved contraception Exclusion criteria: pregnant or lactating women; diabetes (defined as any of: FPG ≥ 126 mg/dL (7.0 mmol/L) at visit 1 (week ‐4); 2‐h post‐challenge plasma glucose (after OGTT 75‐g glucose) ≥ 200 mg/dL (11.1 mmol/L) at visit 1 (week ‐4); diabetes diagnosed by physician and confirmed by other clinical data, other than gestational diabetes; use of insulin or any oral antidiabetic agents prior to visit 1 (week ‐4), other than during pregnancy); acute infections that may have affected blood glucose control within 4 weeks prior to visit 1 (week ‐4); history of torsades de pointes, sustained and clinically relevant ventricular tachycardia or ventricular fibrillation; percutaneous coronary intervention within the past 3 months; any of the following within the past 6 months: myocardial infarction, coronary artery bypass surgery, unstable angina or stroke; congestive heart failure NYHA class III or IV; second‐degree AV block (Mobitz 1 and 2), third degree AV block; prolonged QTc; malignancy including leukaemia and lymphoma (not including basal cell skin cancer) within last 5 years; liver disease; acromegaly or treatment with growth hormone or similar drugs; concurrent medical condition that may interfere with interpretation of efficacy and safety data during trial; donation of ≥ 1 unit (500 mL) blood, significant blood loss ≥ 1 unit of blood within past 2 weeks, or a blood transfusion within past 8 weeks; chronic insulin treatment (> 4 weeks of treatment in the absence of an intercurrent illness) within past 6 months; chronic oral or parenteral corticosteroid treatment (> 7 consecutive days of treatment) within 8 weeks prior to visit 1 (week ‐4); treatment with class Ia, Ib and Ic, or III anti‐arrhythmic drugs; thyroid hormone replacement allowed if dosage had been stable for ≥ 3 months; use of other investigational drugs at visit 1 (week ‐4), or within 30 days or 5 half‐lives of visit 1 (week ‐4), whichever longer, unless local health authority guidelines mandate a longer period; treatment with any drug with a known and frequent toxicity to a major organ system within the past 3 months; any of the following significant laboratory abnormalities alanine aminotransferase, aspartate aminotransferase > 3 times the upper limit of the normal range, direct bilirubin > 1.3 times the upper limit of the normal range at visit 2 (week ‐2), serum creatinine levels ≥ 2.5 mg/dL (220 μmol/L) at visit 2 (week ‐2), thyroid‐stimulating hormone outside normal range at visit 2 (week ‐2), clinically significant laboratory abnormalities confirmed by repeat measurement at visit 2 (week ‐2), fasting triglycerides > 700 mg/dL (> 7.9 mmol/L) at visit 2 (week ‐2); history of active substance abuse (including alcohol) within the past 2 years; potentially unreliable people, and people judged by investigator to be unsuitable for trial Diagnostic criteria: criteria for IGT as defined by WHO: FPG < 126 mg/dL (7.0 mmol/L) and 2‐h post‐challenge plasma glucose (after a OGTT 75‐g glucose) ≥ 140 mg/dL (7.8 mmol/L) to < 200 mg/dL (11.1 mmol/L) |
|
| Interventions |
Intervention: vildagliptin 50 mg once daily Comparator: placebo Run‐in period: none Study drug administration free period before glucose testing during trial: study drug not given before test meal at baseline but was given 15 minutes before test meal at week 12; not described if study drug provided before measurement of OGTT and fasting glucose Extension period: none |
|
| Outcomes | Composite outcome measures reported: no | |
| Study details | Trial terminated early: no | |
| Publication details |
Language of publication: English Funding: commercial funding (Novartis Pharmaceuticals Corporation) Publication status: peer‐reviewed journal. |
|
| Stated aim for study | Quote: "This study was conducted to determine the effects of vildagliptin on incretin hormone levels, islet function, and postprandial glucose control in subjects with impaired glucose tolerance (IGT)." | |
| Notes | Each potential relevant participant attended a prescreening visit (week ‐4) where an OGTT was performed. Possible participants with confirmed IGT then attended main screening visit (week ‐2) where all the inclusion/exclusion criteria were assessed. Eligible participants randomised at visit 3 (baseline, day 1) and completed 2 further visits over 12 weeks of treatment with vildagliptin or placebo | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote: "... double‐blind, randomized, placebo‐controlled, parallel‐group study conducted ..." Comment: method of random sequence generation not adequately described |
| Allocation concealment (selection bias) | Unclear risk |
Quote: "... double‐blind, randomized, placebo‐controlled, parallel‐group study conducted ..." Comment: method of allocation concealment not adequately described |
| Blinding of participants and personnel (performance bias) hypoglycaemia | Low risk | Comment: self‐reported outcome measurement. Blinding of participants ensured using placebo tablet |
| Blinding of participants and personnel (performance bias) incidence of T2DM | Low risk | Comment: listed as adverse events. Investigator assessed outcome measurement. Blinding ensured using placebo tablet |
| Blinding of participants and personnel (performance bias) non‐fatal myocardial infarction/stroke, congestive heart failure | Low risk | Comment: investigator assessed outcome measurement. Blinding ensured using placebo tablet |
| Blinding of participants and personnel (performance bias) non‐serious adverse events | Low risk | Comment: investigator assessed outcome measurement. Blinding ensured using placebo tablet |
| Blinding of participants and personnel (performance bias) serious adverse events | Low risk | Comment: investigator assessed outcome measurement. Blinding ensured using placebo tablet |
| Blinding of outcome assessment (detection bias) hypoglycaemia | Low risk | Comment: self‐reported outcome measurement. Blinding of participants ensured by placebo tablet |
| Blinding of outcome assessment (detection bias) incidence of T2DM | Low risk | Comment: listed as adverse events; investigator assessed outcome measurement. Blinding ensured by placebo tablet |
| Blinding of outcome assessment (detection bias) measures of blood glucose control | Low risk | Comment: measured by investigator at baseline and end of follow‐up. Blinding ensured by placebo tablet |
| Blinding of outcome assessment (detection bias) non‐serious adverse events | Low risk | Comment: adjudicated by investigator at baseline and end of follow‐up. Blinding ensured by placebo tablet |
| Blinding of outcome assessment (detection bias) serious adverse events | Low risk | Comment: adjudicated by investigator at baseline and end of follow‐up. Blinding ensured by placebo tablet |
| Incomplete outcome data (attrition bias) hypoglycaemia | Low risk | Comment: 5 participants missing in each group. Reasons for vildagliptin group: adverse events (n = 3); withdrew consent (n = 1); protocol violation (n = 1); lost to follow‐up (n = 0). For placebo group: adverse events (n = 2); withdrew consent (n = 1); protocol violation (n = 2); lost to follow‐up (n = 0); missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups; proportion of missing outcomes not enough to have a clinically relevant impact on intervention effect estimate |
| Incomplete outcome data (attrition bias) measures of blood glucose control | Low risk | Comment: 5 participants missing in each group. Reasons for vildagliptin group: adverse events (n = 3); withdrew consent (n = 1); protocol violation (n = 1); lost to follow‐up (n = 0). For placebo group: adverse events (n = 2); withdrew consent (n = 1); protocol violation (n = 2); lost to follow‐up (n = 0); missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups; proportion of missing outcomes not enough to have a clinically relevant impact on intervention effect estimate |
| Incomplete outcome data (attrition bias) non‐fatal myocardial infarction/stroke, congestive heart failure | Low risk | Comment: 5 participants missing in each group. Reasons for vildagliptin group: adverse events (n = 3); withdrew consent (n = 1); protocol violation (n = 1); lost to follow‐up (n = 0). For placebo group: adverse events (n = 2); withdrew consent (n = 1); protocol violation (n = 2); lost to follow‐up (n = 0); missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups; proportion of missing outcomes not enough to have a clinically relevant impact on intervention effect estimate |
| Incomplete outcome data (attrition bias) non‐serious adverse events | Low risk | Comment: 5 participants missing in each group. Reasons for vildagliptin group: adverse events (n = 3); withdrew consent (n = 1); protocol violation (n = 1); lost to follow‐up (n = 0). For placebo group: adverse events (n = 2); withdrew consent (n = 1); protocol violation (n = 2); lost to follow‐up (n = 0); missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups; proportion of missing outcomes not enough to have a clinically relevant impact on intervention effect estimate |
| Incomplete outcome data (attrition bias) serious adverse events | Low risk | Comment: 5 participants missing in each group. Reason for vildagliptin group: adverse events (n = 3); withdrew consent (n = 1); protocol violation (n = 1); lost to follow‐up (n = 0). For placebo group: adverse events (n = 2); withdrew consent (n = 1); protocol violation (n = 2); lost to follow‐up (n = 0); missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups; proportion of missing outcomes not enough to have a clinically relevant impact on the intervention effect estimate |
| Selective reporting (reporting bias) | Low risk | Comment: trial protocol available and all the trial's prespecified outcomes that were of interest in review were reported in prespecified way |
| Other bias | Unclear risk | Comment: funding received by a pharmaceutical company |
Rosenstock 2010.
| Methods |
Design: parallel RCT Randomisation ratio: 1:1 Number of study centres: 16 (identified through ClinicalTrials.gov) |
|
| Participants |
Inclusion criteria: BMI ≥ 30 kg/m²; aged ≥ 18 years Exclusion criteria: participated in this study previously, or any other study using exenatide or GLP‐1 analogues; participated in an interventional medical, surgical or pharmaceutical study (a study in which an experimental, drug, medical or surgical treatment was given) within 30 days of study start (this criterion included drugs that had not received regulatory approval for any indication at time of study entry); diagnosis of diabetes mellitus (other than gestational diabetes), or previous use of antidiabetic medications for > 3 months; change in prescribed lipid‐lowering or blood pressure agents within 4 weeks of screening; used drugs for weight loss (e.g. orlistat, sibutramine, phenylpropanolamine, rimonabant, low‐dose orlistat or other similar non‐prescription weight loss remedies or medications) within 3 months of screening; actively participating in, or have participated in a formal weight loss programme within the last 3 months; have a history of chronic use of drugs that directly affect gastrointestinal motility, including, but not limited to, metoclopramide and chronic macrolide antibiotics; treated with any antidiabetic medications within 3 months of screening; receiving chronic (lasting longer than 2 weeks) systemic glucocorticoid therapy or have received such therapy within the 4 weeks immediately prior to study start; have had bariatric surgery; have had an organ transplant Diagnostic criteria: criteria for intermediate hyperglycaemia as defined by WHO; IGT (fasting glucose < 7 mmol/L and 2‐h postprandial glucose ≥ 7.8 to < 11.1 mmol/L); IFG (fasting glucose 6.1‐6.9 mmol/L and 2‐h postprandial glucose < 7.8 mmol/L) |
|
| Interventions |
Intervention: exenatide 10 μg twice daily Comparator: placebo Run‐in period: prior to randomisation, a 1‐week single‐blind placebo lead‐in period was performed Study drug administration free period before glucose testing during trial: study drug was withheld before OGTT assessments. Follow‐up visit conducted 4 weeks after study completion Extension period: none |
|
| Outcomes | Composite outcome measures reported: no | |
| Study details | Trial terminated early: no | |
| Publication details |
Language of publication: English Funding: commercial funding (Eli Lilly and Company and Amylin Pharmaceuticals) Publication status: peer‐reviewed journal. |
|
| Stated aim for study | Quote: "To assess the effects of exenatide on body weight and glucose tolerance in nondiabetic obese subjects with normal or impaired glucose tolerance (IGT) or impaired fasting glucose (IFG)." | |
| Notes | 38/152 participants had IFG or IGT at baseline | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote: "... were randomized to receive exenatide ..." Comment: method of random sequence generation not adequately described. No separate baseline variables available for participants with IFG or IGT, or both |
| Allocation concealment (selection bias) | Unclear risk |
Quote: "... were randomized to receive exenatide ..." Comment: method of allocation concealment not adequately described. No separate baseline variables available for participants with IFG or IGT, or both |
| Blinding of participants and personnel (performance bias) all‐cause/cardiovascular mortality | Low risk |
Quote: "No deaths, serious adverse events, or hypoglycemia were observed during the study." Blinding was ensured by placebo injection Comment: investigator‐assessed outcome measurement |
| Blinding of participants and personnel (performance bias) hypoglycaemia | Low risk |
Quote: "No deaths, serious adverse events, or hypoglycemia were observed during the study." Blinding was ensured by placebo injection Comment: investigator‐assessed outcome measurement/self‐reported (mild hypoglycaemia) |
| Blinding of participants and personnel (performance bias) incidence of T2DM | Low risk |
Quote: "Five participants (three exenatide, two placebo) developed type 2 diabetes during the study, three of which (two exenatide, one placebo) had IGT or IFG at baseline." Blinding was ensured by placebo injection Comment: investigator‐assessed outcome measurement |
| Blinding of participants and personnel (performance bias) serious adverse events | Low risk |
Quote: "No deaths, serious adverse events, or hypoglycemia were observed during the study." Blinding was ensured by placebo injection Comment: investigator‐assessed outcome measurement/self‐reported (mild hypoglycaemia) |
| Blinding of outcome assessment (detection bias) all‐cause/cardiovascular mortality | Low risk |
Quote: "No deaths, serious adverse events, or hypoglycemia were observed during the study." Blinding was ensured by placebo injection Comment: investigator‐assessed outcome measurement |
| Blinding of outcome assessment (detection bias) hypoglycaemia | Low risk |
Quote: "No deaths, serious adverse events, or hypoglycemia were observed during the study." Blinding was ensured by placebo injection Comment: investigator‐assessed outcome measurement/self‐reported (mild hypoglycaemia) |
| Blinding of outcome assessment (detection bias) incidence of T2DM | Low risk |
Quote: "Five participants (three exenatide, two placebo) developed type 2 diabetes during the study, three of which (two exenatide, one placebo) had IGT or IFG at baseline." Blinding was ensured by placebo injection Comment: investigator‐assessed outcome measurement |
| Blinding of outcome assessment (detection bias) serious adverse events | Low risk |
Quote: "No deaths, serious adverse events, or hypoglycemia were observed during the study." Blinding was ensured by placebo injection Comment: investigator‐assessed outcome measurement |
| Incomplete outcome data (attrition bias) all‐cause/cardiovascular mortality | Unclear risk | Comment: 163 participants randomised. 152 participants included in analyses, 96 completed final follow‐up visit. Balanced across groups. 38 randomised with IFG or IGT, 33 included in analyses of abstract in publication, and presumably received at least 1 dose of study drug. Unknown how many participants with IFG or IGT completed final visit. Method of imputation of missing data not explained |
| Incomplete outcome data (attrition bias) hypoglycaemia | Unclear risk | Comment: 163 participants randomised. 152 participants included in analyses, 96 completed final follow‐up visit. Balanced across groups. 38 randomised with IFG or IGT, 33 included in analyses of abstract in publication, and presumably received at least 1 dose of study drug. Unknown how many participants with IFG or IGT completed final visit. Method of imputation of missing data not explained. |
| Incomplete outcome data (attrition bias) incidence of T2DM | Unclear risk | Comment: 163 participants randomised. 152 participants included in analyses, 96 completed final follow‐up visit. Balanced across groups. 38 randomised with IFG or IGT, 33 included in analyses of abstract in publication, and presumably received at least 1 dose of study drug. Unknown how many participants with IFG or IGT completed final visit. Method of imputation of missing data not explained |
| Incomplete outcome data (attrition bias) serious adverse events | Unclear risk | Comment: 163 participants randomised. 152 participants included in analyses, 96 completed final follow‐up visit. Balanced across groups. 38 randomised with IFG or IGT, 33 included in analyses of abstract in publication, and presumably received at least 1 dose of study drug. Unknown how many participants with IFG or IGT completed final visit. Method of imputation of missing data not explained |
| Selective reporting (reporting bias) | Unclear risk | Comment: subgroup of participants with IFG or IGT, or both not prespecified in available protocol, except for normalisation of glycaemic levels (NCT00500370). Several outcomes of interest for this review reported for all participants but not separately reported for participants with IFG or IGT, or both |
| Other bias | Unclear risk | Comment: participants with IFG or IGT, or both were only a subgroup of participants; funding received from a pharmaceutical company |
SCALE.
| Methods |
Design: parallel RCT Randomisation ratio: 2:1 Number of study centres: 191 |
|
| Participants |
Inclusion criteria: aged ≥ 18 years; informed consent obtained before any trial‐related activity took place; obesity (BMI ≥ 30.0 kg/m²) or overweight (BMI ≥ 27.0 kg/m²) with treated or untreated comorbid dyslipidaemia (LDL ≥ 160 mg/dL, or triglycerides ≥ 150 mg/dL, or HDL < 40 mg/dL for men and < 50 mg/dL for women) or hypertension (systolic blood pressure ≥ 140 mmHg or diastolic blood pressure ≥ 90 mmHg), or both; stable body weight (< 5 kg self‐reported change during the previous 3 months); preceding failed dietary effort Exclusion criteria: diagnosis of T1DM or T2DM per the judgement of investigator; HbA1c ≥ 6.5% or FPG ≥ 126 mg/dL (7.0 mmol/L) or 2‐h post‐challenge plasma glucose ≥ 200 mg/dL (11.1 mmol/L) (at screening); previous treatment with glucagon‐like peptide‐1 receptor agonists (including liraglutide or exenatide) within last 3 months; untreated or uncontrolled hypothyroidism/hyperthyroidism defined as thyroid‐stimulating hormone > 6 mIU/L or < 0.4 mIU/L; screening calcitonin ≥ 50 ng/L; family or personal history of multiple endocrine neoplasia type 2 or familial medullary thyroid carcinoma; personal history of non‐familial medullary thyroid carcinoma; history of chronic pancreatitis or idiopathic acute pancreatitis; obesity induced by other endocrinological disorders (e.g. Cushing's syndrome); current or history of treatment with medications that may cause significant weight gain, within 3 months prior to screening, including systemic corticosteroids (except for a short course of treatment, i.e. 7‐10 days), tricyclic antidepressants, atypical antipsychotic and mood stabilisers (e.g. imipramine, amitriptyline, mirtazepine, paroxetine, phenelzine, chlorpromazine, thioridazine, clozapine, olanzapine, valproic acid and its derivatives, and lithium); diet attempts using herbal supplements or non‐prescription medications within 3 months before screening; current participation (or within the last 3 months) in an organised weight reduction programme or currently using or used within 3 months before screening: pramlintide, sibutramine, orlistat, zonisamide, topiramate, phentermine or metformin (either by prescription or as part of a clinical trial); participation in a clinical trial within the last 3 months prior to screening; simultaneous participation in any other clinical trial of an investigational drug; previous surgical treatment for obesity (excluding liposuction if performed > 1 year before trial entry); history of major depressive disorder within the last 2 years; history of other severe psychiatric disorders(e.g. schizophrenia, bipolar disorder; Patient Health Questionnaire (PHQ‐9) score ≥ 15); any lifetime history of a suicidal attempt; history of any suicidal behaviour in month prior to randomisation; any suicidal ideation of type 4 or 5 on the Columbian Suicidality Severity Rating Scale (C‐SSRS) in month prior to randomisation; surgery scheduled for the trial duration period, except for minor surgical procedures, at the discretion of investigator; uncontrolled treated/untreated hypertension (systolic blood pressure ≥ 160 mmHg or diastolic blood pressure ≥ 100 mmHg, or both) (if white‐coat hypertension suspected at screening, a repeated measurement prior to other trial‐related activities allowed); cancer (past or present, except basal cell skin cancer or squamous cell skin cancer), which in investigator's opinion could interfere with the results of the trial; known or suspected hypersensitivity to trial product or related products; previous participation in the randomised phase of this trial. Re‐screening allowed once within limit of recruitment period; known or suspected abuse of alcohol or narcotics; language barrier, mental incapacity, unwillingness or inability to understand and be able to complete the mental health questionnaire in the provided language; people from the same household participating in the trial; women of child‐bearing potential who were pregnant, breastfeeding or intend to become pregnant or were not using adequate contraception (adequate contraceptive measures as required by local law or practice). US: abstinence and the following methods: diaphragm with spermicide, condom with spermicide (by male partner), intrauterine device, sponge, spermicide, Norplant, Depo‐Provera or oral contraceptives. Germany: adequate contraceptive measures are implants, injectables, combined oral contraceptives, hormonal intrauterine device, sexual abstinence or vasectomised partner. UK: adequate contraceptive measures are defined as sterilisation, intrauterine device, oral contraceptives, consistent use of barrier methods, male sterilisation or true abstinence; receipt of any investigational drug within 4 weeks prior to screening for this trial (Brazil: the receipt of any investigational drug within 1 year prior to screening for this trial, unless there was direct benefit to the person at investigator's discretion; France: abnormality of the thyroid identified during the physical examination at screening) Diagnostic criteria: criteria for intermediate hyperglycaemia as defined by ADA:
OGTT done at screening for diagnosis of intermediate hyperglycaemia. Diagnosis confirmed by second repeated measurement |
|
| Interventions |
Intervention: liraglutide 3.0 mg once daily Comparator: placebo Run‐in period: none Study drug administration free period before glucose testing during trial: protocol stated that trial participants were not supposed to inject liraglutide/liraglutide placebo in morning on days where OGTT measurements were made. OGTT performed every sixth month. FPG and HbA1c measured every third month. Participants allowed to take study medication on days where FPG was measured (except if OGTT was planned to be measured at same visit). This was not written clearly in protocol, but confirmed through internal correspondence with corresponding author of main publication Extension period: yes; intervention period 160 weeks, and followed by an off‐drug, observational follow‐up period of 12 weeks |
|
| Outcomes | Composite outcome measures reported: no | |
| Study details | Trial terminated early: no | |
| Publication details |
Language of publication: English Funding: commercial funding (pharmaceutical company) Publication status: peer‐reviewed journal |
|
| Stated aim for study | Quote: "The aim of this clinical trial is to evaluate the potential of liraglutide to induce and maintain weight loss over 56 weeks in obese subjects or overweight subjects with co‐morbidities. Furthermore, the aim is to investigate the long term potential of liraglutide to delay the onset of type 2 diabetes in subjects diagnosed with pre‐diabetes at baseline." | |
| Notes | Participants stratified based on prediabetes status and BMI at screening. Prespecified subgroup analyses performed in participants with or without prediabetes at enrolment (all endpoints) and in those with different baseline BMI categories (body weight and HbA1c endpoints). Trial had predefined withdrawal criteria: "6.6 Withdrawal criteria. The subject may be withdrawn from the trial at the discretion of investigator or Novo Nordisk due to a safety concern or if judged non‐compliant with trial procedures. A subject must be withdrawn if the following applies: 1. The subject may withdraw from the trial at will at any time 2. If the target treatment dose of the randomised trial product is not tolerated by the subjects 3. Pregnancy or intention of becoming pregnant 4. Subjects who develop diabetes during the trial will not be withdrawn but should receive the best standard of care at the discretion of the Investigator. If the Investigator determines that insulin, GLP‐1 receptor agonist (e.g., Byetta® or Victoza®), or DPP‐4 inhibitor is the best treatment option, the subject must be withdrawn. The medication prescribed by the Investigator will not be provided by Novo Nordisk 5. If the investigator suspects acute pancreatitis, all suspected drugs should be discontinued until confirmatory tests have been conducted and appropriate treatment should be initiated. Subjects that are diagnosed with acute pancreatitis (as a minimum 2 of 3: characteristic abdominal pain, amylase and/or lipase >3x UNR [upper normal range] or characteristic findings on CT [computer tomography]/MRI [magnetic resonance imaging]), must be withdrawn from the trial 6. A subject should be referred to a Mental Health Professional (MHP) if he/she has: • a PHQ‐9 [Patient Health Questionnaire] score ≥ 10, OR, • any suicidal behaviour, OR, • any suicidal ideation of type 4 (active suicidal ideation with some intent to act, without specific plan) or type 5 (active suicidal ideation with specific plan and intent) on any C‐SSRS assessment ‐ A referral to a Mental Health Professional (MHP) should also be made if in the opinion of the Investigator it is necessary for the safety of the subject. If a subject's psychiatric disorder can be adequately treated with psycho‐ and/or pharmacotherapy, then the subject, at the discretion of the Investigator (in agreement with the MHP), may be continued in the trial on randomised therapy, otherwise, the subject must be withdrawn. In case of withdrawal, the End of Trial form must be filled in and in the IV/WRS the Withdrawal session must be completed. If possible, the subject should be called in for a final visit. Procedures according to Visit 17 should be performed for all subjects who discontinue the trial prematurely." |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was performed with the use of a telephone or Web‐based system provided by the sponsor." |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomization was performed with the use of a telephone or Web‐based system provided by the sponsor." |
| Blinding of participants and personnel (performance bias) all‐cause/cardiovascular mortality | Low risk |
Quote: "Treatment allocation for subjects with pre‐diabetes will be double‐blind for the first year (Novo Nordisk, Investigator and subject are blinded), and single blind for the remaining 2 years (i.e. Novo Nordisk is unblinded, whereas investigator and subjects remain blinded)." Comment: adjudicated outcome measurement |
| Blinding of participants and personnel (performance bias) health‐related quality of life | Low risk |
Quote: "Treatment allocation for subjects with pre‐diabetes will be double‐blind for the first year (Novo Nordisk, Investigator and subject are blinded), and single blind for the remaining 2 years (i.e. Novo Nordisk is unblinded, whereas investigator and subjects remain blinded)." Comment: self‐reported outcome measurement |
| Blinding of participants and personnel (performance bias) hypoglycaemia | Low risk |
Quote: "Treatment allocation for subjects with pre‐diabetes will be double‐blind for the first year (Novo Nordisk, Investigator and subject are blinded), and single blind for the remaining 2 years (i.e. Novo Nordisk is unblinded, whereas investigator and subjects remain blinded)." Comment: self‐reported and investigator‐assessed outcome measurement |
| Blinding of participants and personnel (performance bias) incidence of T2DM | Low risk |
Quote: "Treatment allocation for subjects with pre‐diabetes will be double‐blind for the first year (Novo Nordisk, Investigator and subject are blinded), and single blind for the remaining 2 years (i.e. Novo Nordisk is unblinded, whereas investigator and subjects remain blinded)." Comment: investigator‐assessed outcome measurement |
| Blinding of participants and personnel (performance bias) measures of blood glucose control | Low risk |
Quote: "Treatment allocation for subjects with pre‐diabetes will be double‐blind for the first year (Novo Nordisk, Investigator and subject are blinded), and single blind for the remaining 2 years (i.e. Novo Nordisk is unblinded, whereas investigator and subjects remain blinded)." Comment: investigator‐assessed outcome measurement |
| Blinding of participants and personnel (performance bias) non‐fatal myocardial infarction/stroke, congestive heart failure | Low risk |
Quote: "Treatment allocation for subjects with pre‐diabetes will be double‐blind for the first year (Novo Nordisk, Investigator and subject are blinded), and single blind for the remaining 2 years (i.e. Novo Nordisk is unblinded, whereas investigator and subjects remain blinded)." Comment: investigator‐assessed outcome measurement/adjudicated outcome measurement |
| Blinding of participants and personnel (performance bias) non‐serious adverse events | Low risk |
Quote: "Treatment allocation for subjects with pre‐diabetes will be double‐blind for the first year (Novo Nordisk, Investigator and subject are blinded), and single blind for the remaining 2 years (i.e. Novo Nordisk is unblinded, whereas investigator and subjects remain blinded)." Comment: self‐reported and investigator‐assessed outcome measurement |
| Blinding of participants and personnel (performance bias) serious adverse events | Low risk |
Quote: "Treatment allocation for subjects with pre‐diabetes will be double‐blind for the first year (Novo Nordisk, Investigator and subject are blinded), and single blind for the remaining 2 years (i.e. Novo Nordisk is unblinded, whereas investigator and subjects remain blinded)." Comment: investigator‐assessed outcome measurement |
| Blinding of participants and personnel (performance bias) time to progression to T2DM | Low risk |
Quote: "Treatment allocation for subjects with pre‐diabetes will be double‐blind for the first year (Novo Nordisk, Investigator and subject are blinded), and single blind for the remaining 2 years (i.e. Novo Nordisk is unblinded, whereas investigator and subjects remain blinded)." Comment: investigator‐assessed outcome measurement |
| Blinding of outcome assessment (detection bias) all‐cause/cardiovascular mortality | Low risk |
Quote: "Treatment allocation for subjects with pre‐diabetes will be double‐blind for the first year (Novo Nordisk, Investigator and subject are blinded), and single blind for the remaining 2 years (i.e. Novo Nordisk is unblinded, whereas investigator and subjects remain blinded)." Comment: adjudicated outcome measurement |
| Blinding of outcome assessment (detection bias) health‐related quality of life | Low risk |
Quote: "Treatment allocation for subjects with pre‐diabetes will be double‐blind for the first year (Novo Nordisk, Investigator and subject are blinded), and single blind for the remaining 2 years (i.e. Novo Nordisk is unblinded, whereas investigator and subjects remain blinded)." Comment: self‐reported outcome measurement |
| Blinding of outcome assessment (detection bias) hypoglycaemia | Low risk |
Quote: "Treatment allocation for subjects with pre‐diabetes will be double‐blind for the first year (Novo Nordisk, Investigator and subject are blinded), and single blind for the remaining 2 years (i.e. Novo Nordisk is unblinded, whereas investigator and subjects remain blinded)." Comment: self‐reported and investigator‐assessed outcome measurement |
| Blinding of outcome assessment (detection bias) incidence of T2DM | Low risk |
Quote: "Treatment allocation for subjects with pre‐diabetes will be double‐blind for the first year (Novo Nordisk, Investigator and subject are blinded), and single blind for the remaining 2 years (i.e. Novo Nordisk is unblinded, whereas investigator and subjects remain blinded)." Comment: investigator‐assessed outcome measurement |
| Blinding of outcome assessment (detection bias) measures of blood glucose control | Low risk |
Quote: "Treatment allocation for subjects with pre‐diabetes will be double‐blind for the first year (Novo Nordisk, Investigator and subject are blinded), and single blind for the remaining 2 years (i.e. Novo Nordisk is unblinded, whereas investigator and subjects remain blinded)." Comment: investigator‐assessed outcome measurement |
| Blinding of outcome assessment (detection bias) non‐fatal myocardial infarction/stroke, congestive heart failure | Low risk |
Quote: "Treatment allocation for subjects with pre‐diabetes will be double‐blind for the first year (Novo Nordisk, Investigator and subject are blinded), and single blind for the remaining 2 years (i.e. Novo Nordisk is unblinded, whereas investigator and subjects remain blinded)." Comment: investigator‐assessed outcome measurement/adjudicated outcome measurement |
| Blinding of outcome assessment (detection bias) non‐serious adverse events | Low risk |
Quote: "Treatment allocation for subjects with pre‐diabetes will be double‐blind for the first year (Novo Nordisk, Investigator and subject are blinded), and single blind for the remaining 2 years (i.e. Novo Nordisk is unblinded, whereas investigator and subjects remain blinded)." Comment: self‐reported and investigator‐assessed outcome measurement |
| Blinding of outcome assessment (detection bias) serious adverse events | Low risk |
Quote: "Some types of AEs [adverse events] were also evaluated via a blinded adjudication process by an independent, external adjudication committee of medical experts (those marked 'Adjudicated' below). Based on predefined diagnostic criteria, the adjudication committee could either confirm or not confirm the AE classification/diagnosis." Comment: deaths, cardiovascular events, pancreatitis/suspicion of pancreatitis and neoplasms were evaluated by an independent adjudication committee |
| Blinding of outcome assessment (detection bias) time to progression to T2DM | Low risk |
Quote: "Treatment allocation for subjects with pre‐diabetes will be double‐blind for the first year (Novo Nordisk, Investigator and subject are blinded), and single blind for the remaining 2 years (i.e. Novo Nordisk is unblinded, whereas investigator and subjects remain blinded)." Comment: investigator‐assessed outcome measurement |
| Incomplete outcome data (attrition bias) all‐cause/cardiovascular mortality | Unclear risk | Comment: not described how missing data were handled |
| Incomplete outcome data (attrition bias) health‐related quality of life | High risk | Comment: only about half of participants completed assessment of health‐related quality of life, this proportion of missingness is high enough to indicate relevant bias. Missing data were imputed with last observation carried forward method |
| Incomplete outcome data (attrition bias) hypoglycaemia | High risk |
Quote: "Missing values were imputed with the use of the last observation carried forward method for measurements made after baseline." Comment: only about half of participants completed the trial, this proportion of missingness was high enough to indicate relevant bias |
| Incomplete outcome data (attrition bias) incidence of T2DM | High risk |
Quote: "Missing values were imputed with the use of the last observation carried forward method for measurements made after baseline" and "Data from all pre‐diabetic subjects in the FAS will be analysed using a Weibull model. Pre‐diabetic subjects who incorrectly entered the re‐randomised treatment period will be censored at the date corresponding to visit 17 unless they had onset of type 2 diabetes prior to this date. The model will include treatment, gender and BMI stratification factor as fixed factors and baseline FPG will be included as a covariate." Comment: only about half of participants completed the trial, this proportion of missingness was high enough to indicate relevant bias |
| Incomplete outcome data (attrition bias) measures of blood glucose control | High risk |
Quote: "Missing values were imputed with the use of the last observation carried forward method for measurements made after baseline." Comment: only about half of the participants completed the trial, this proportion of missingness was high enough to indicate relevant bias |
| Incomplete outcome data (attrition bias) non‐fatal myocardial infarction/stroke, congestive heart failure | Unclear risk | Comment: not described how missing data were handled |
| Incomplete outcome data (attrition bias) non‐serious adverse events | High risk |
Quote: "Missing values were imputed with the use of the last observation carried forward method for measurements made after baseline." Comment: only about half of participants completed the trial, this proportion of missingness was high enough to indicate relevant bias. |
| Incomplete outcome data (attrition bias) serious adverse events | Unclear risk | Comment: not described how missing data were handled. |
| Incomplete outcome data (attrition bias) time to progression to T2DM | High risk | Comment: 1128 completed 160 weeks (52.6% on liraglutide, 45% on placebo). Proportion of missingness was high enough to indicate clinical relevant bias. |
| Selective reporting (reporting bias) | Low risk | Comment: trial protocol was available and all the trial's prespecified (primary and secondary) outcomes that are of interest of the review were reported in the prespecified way. |
| Other bias | Unclear risk | Comment: trial conducted by Novo Nordisk. |
Note: where the judgement was 'unclear' with a blank 'Support for judgement', the trial did not report that particular outcome.
ACS: acute coronary syndrome; ADA: American Diabetes Association; AV: atrioventricular; BEGAMI: beta‐cell function in glucose abnormalities and acute myocardial Infarction; BMI: body mass index; FPG: fasting plasma glucose; GAD: glutamic acid decarboxylase; GITS: glipizide gastrointestinal therapeutic system; GV: glycaemic variability; h: hour; HbA1c: glycosylated haemoglobin A1c; HDL: high‐density lipoprotein; IFG: impaired fasting glucose; IGT: impaired glucose tolerance; LDL: low‐density lipoprotein; n: number of participants; NR: not reported; NYHA: New York Heart Association; OGTT: oral glucose tolerance test; RCT: randomised controlled trial; SCALE: Satiety and Clinical Adiposity ‐ Liraglutide Evidence in Nondiabetic and Diabetic Individuals; T1DM: type 1 diabetes mellitus; T2DM: type 2 diabetes mellitus; WHO: World Health Organization.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Acosta 2015 | Included obese participants, but did not state intermediate hyperglycaemia as an inclusion criterion. |
| ACTRN12615001029583 | Protocol for a randomised trial including participants with cystic fibrosis‐related diabetes or IGT. |
| Almeda‐Valdes 2012 | Duration of intervention < 12 weeks. |
| Aoki 2014 | Protocol of trial described it as a non‐randomised trial, but both full‐text articles stated that participants were randomised. Authors did not respond to this question. Not possible to obtain separate data for the participants with IGT. Single administration of interventions applied, and duration < 12 weeks. |
| Armato 2012 | Not a randomised clinical trial. |
| BEGAMI 2013 | Included participants with intermediate hyperglycaemia and acute coronary syndrome. |
| Best 2015 | Participants were HIV positive. |
| Bock 2010 | Duration of intervention < 12 weeks. |
| Cui 2016 | Participants had hepatic steatosis. |
| Daniele 2015 | Duration of intervention 26 days according to publication, stated as 12 weeks on ClinicalTrials.gov. Trial included both participants with IGT and T2DM. Authors did not provide separate data. |
| Dushay 2012 | Not possible to obtain separate data on the participants with prediabetes. Authors contacted. |
| EudraCT 2011‐005980‐26 | Protocol for a trial including participants with intermediate hyperglycaemia and stroke. |
| EudraCT 2013‐001240‐64 | Protocol for a trial where exenatide was administered intravenously. |
| Gonzalez‐Ortiz 2015 | Trial compared 2 different types of GLP‐1 analogues with each other. |
| Gudipaty 2014 | All participants had a history of T2DM. |
| Ishikawa 2014 | Not possible to obtain separate data on the participants with IGT. Authors contacted twice, but no replies. |
| Kaku 2015 | Duration of intervention < 12 weeks. |
| Koska 2015 | Exenatide administered intravenously, duration of intervention < 12 weeks. |
| Larsen 2014 | Protocol for a randomised clinical trial including participants on antipsychotic medicine and intermediate hyperglycaemia. |
| NCT00101712 | Protocol for a randomised clinical trial including participants with T2DM. |
| NCT00198146 | Protocol for a trial including participants with T2DM. |
| NCT00721552 | Protocol for trial including glucocorticoid‐induced impairment of glucose metabolism. |
| NCT00845182 | Authors did not reply to our request for separate data on the participants with IGT. |
| NCT00845559 | Protocol for a trial fulfilling the inclusion criteria, but stopped prior to enrolment. |
| NCT00886626 | Protocol for a randomised clinical trial including obese children. |
| NCT00961363 | Protocol for a trial fulfilling the inclusion criteria. However, the trial was terminated early due to difficulties in recruiting participants. |
| NCT01006018 | Protocol for a trial that according to the primary investigator never was initiated. According to ClinicalTrials.gov: "Unanticipated delays due to sterilization/stabilization testing of GLP‐1." |
| NCT01018602 | Not possible to obtain separate data on the people with intermediate hyperglycaemia. |
| NCT01038648 | Protocol for a trial that was never initiated. |
| NCT01054118 | Included participants with T2DM. Cross‐over trial with a duration of intervention of 28 days for each intervention period. |
| NCT01122641 | Trial protocol for a completed trial including obese participants with high FinRisk score. Principal investigator approached twice to request clarification if there are separate data on people with IFG, IGT or intermediate elevated HbA1c but without any reply. |
| NCT01346254 | Included participants with prediabetes after kidney transplantation. |
| NCT01472640 | Not possible to obtain separate data on the participants with IGT. |
| NCT01845259 | Protocol for a randomised clinical trial including participants on antipsychotic medicine and intermediate hyperglycaemia. |
| NCT01970462 | Protocol for a trial including participants with stress hyperglycaemia or mild diabetes following cardiac surgery. |
| NCT02016846 | Protocol for trial including glucocorticoid‐induced impairment of glucose metabolism. |
| NCT02022007 | Protocol for trial including participants with polycystic ovary syndrome and IFG, IGT, or both. |
| NCT02284230 | Included participants with IGT or IFG (or both) and kidney failure. Not completed due to inability to recruit participants. |
| NCT02446834 | Protocol for trial including participants with polycystic ovary syndrome and IGT. |
| Schwartz 2010 | Duration < 12 weeks. Included participants with T2DM and IGT. |
| Tsuchiya 2011 | Not possible to obtain separate data for participants with intermediate hyperglycaemia. Authors approached twice through email, but no reply. |
| UMIN000006197 | Protocol for a non‐randomised clinical trial. |
| UMIN000014249 | Protocol for non‐randomised clinical study including participants with IGT or diabetes due to partial pancreatectomy. |
| Utzschneider 2008 | Not a randomised clinical trial. |
| Werzowa 2013 | Included participants with IGT after renal transplantation. |
GLP‐1: glucagon‐like peptide‐1; HbA1c: glycosylated haemoglobin A1c; IFG: impaired fasting glucose; IGT: impaired glucose tolerance; T2DM: type 2 diabetes mellitus.
Characteristics of studies awaiting assessment [ordered by study ID]
Astrup 2009.
| Methods |
Allocation: randomised clinical trial Intervention model: parallel assignment Masking: double blind for liraglutide and placebo (participants and investigators), open label for orlistat |
| Participants |
Condition: normoglycaemia and intermediate hyperglycaemia Enrolment: total number of participants 564. From baseline table, it is stated that about one‐third had intermediate hyperglycaemia Inclusion criteria: men and women aged 18‐65 years, body mass index 30‐40 kg/m², stable body weight (< 5% reported change during the previous 3 months) and fasting plasma glucose < 7 mmol/L Exclusion criteria: key criteria included known T1DM or T2DM, obesity induced by drug treatment, use of approved weight‐lowering pharmacotherapy or participation in a clinical weight control study within the previous 3 months, previous surgical obesity treatment and major medical conditions |
| Interventions |
Intervention (1): liraglutide 1.2 mg, subcutaneous Intervention (2): liraglutide 1.8 mg, subcutaneous Intervention (3): liraglutide 2.4 mg, subcutaneous Intervention (4): liraglutide 3.0 mg, subcutaneous Comparator (1): orlistat, PO Comparator (2): placebo, subcutaneous Duration of intervention: 20 weeks of intervention followed by an 84‐week open‐label extension period |
| Outcomes |
Primary outcomes: intervention period: mean change from baseline in body weight at week 20. Interim analysis (weeks 21‐52): efficacy of liraglutide to induce weight loss. Extension period (weeks 21‐104): long‐term safety and tolerability of liraglutide Secondary outcomes: intervention period: secondary efficacy endpoints included change in waist circumference, systolic and diastolic blood pressure, prevalence of metabolic syndrome, prediabetes status, fasting lipids (total cholesterol, LDL cholesterol, HDL cholesterol, very‐low‐density lipoprotein cholesterol and triglycerides), cardiovascular biomarkers (highly sensitive C‐reactive protein, plasminogen activator inhibitor‐1, fibrinogen and adiponectin), glucose metabolism parameters (fasting plasma glucose, fasting insulin and HbA1c) and homoeostasis model assessment of β‐cell function and insulin resistance; 2‐h glucose, insulin and C‐peptide concentrations during OGTT; patient‐reported outcome scores with Impact of Weight on Quality of Life ‐ Lite Interim analysis (weeks 21‐52): to compare weight‐lowering effect of liraglutide to orlistat; to investigate long‐term efficacy of liraglutide to induce and maintain weight loss; to investigate effects induced by liraglutide on: body composition as assessed by waist circumference; cardiovascular risk factors as assessed by systolic and diastolic blood pressures and fasting lipid profile; glucose metabolism, including β‐cell function, as assessed by prediabetes status; presence of the metabolic syndrome as assessed using criteria introduced by the Adult Treatment Panel III under the National Cholesterol Education Program. Extension period (weeks 21‐104): to summarise long‐term efficacy of liraglutide to induce and maintain weight loss; to summarise effects induced by liraglutide on: waist circumference; cardiovascular risk markers as assessed by blood pressure, lipids, cardiovascular biomarkers, metabolic syndrome status and glucose metabolism; patient‐reported quality of life; prediabetes status |
| Study details | Trial terminated early: no |
| Publication details |
Language of publication: English Funding: commercial funding Publication status: peer‐reviewed journal/full article for complete study population |
| Stated aim of study | Quote: "The purpose of the 20‐week trial is to investigate the efficacy of liraglutide to induce body weight loss and the purpose of the extension is to evaluate the long term safety and tolerability of liraglutide." |
| Notes | Application sent to Novo Nordisk to request separate data on participants with intermediate hyperglycaemia. Novo Nordisk has approved access to raw trial data, but data are not yet available |
NCT01521312.
| Methods |
Allocation: randomised Intervention model: parallel assignment Masking: double blind (participants and investigators) |
| Participants |
Condition: impaired glucose tolerance Enrolment: 24 Inclusion criteria: social security affiliation; people without tutorship that can freely agree to participate to study; aged 18‐70 years; impaired glucose tolerance diagnosed during the previous month Exclusion criteria: pregnancy; breastfeeding; diabetes; no contraception; BMI > 45 kg/m²; arterial blood pressure > 160/110 mmHg; creatinine clearance < 60 mL/minute; severe hepatocellular insufficiency; chronic respiratory disease; anaemia (haemoglobin < 10 g/dL); peripheral arterial occlusive disease; heart failure; cardiac arrhythmia |
| Interventions |
Intervention: sitagliptin 5 mg, PO Comparator: placebo pill, PO Duration of intervention: 11‐14 weeks |
| Outcomes |
Primary outcomes: vago‐sympathetic activity; arterial stiffness; endothelial function; OGTT Secondary outcomes: NR |
| Study details | Trial terminated early: NR |
| Publication details |
Language of publication: not published Funding: NR Publication status: not published |
| Stated aim of study |
Quote: "Glucose ACCES study will explore the acute and long term (12‐week treatment) effects of saxagliptin in patients with impaired glucose tolerance during fasting and after a standardised breakfast. The investigations will be performed on:
|
| Notes | Trial registered in ClinicalTrials.gov as completed in September 2014. However, it is not published. Authors asked for publication status and data, but no reply provided. As it can take time to obtain data published, the trial is characterised as awaiting classification |
NCT01960205.
| Methods |
Allocation: randomised Intervention model: parallel assignment Masking: open label |
| Participants |
Condition: obese with glucose intolerance. Enrolment: 80 Inclusion criteria: newly diagnosed insulin resistance or glucose intolerance; age 20‐70 years; BMI ≥ 28 kg/m² or > 25 kg/m² beside waist line ≥ 80 cm (women); ≥ 90 cm (men) Exclusion criteria: under diabetes mellitus treatment; allergy to DPP‐4 inhibitors; active heart failure; unwilling or unable to sign inform consents |
| Interventions |
Intervention (1): saxagliptin 5 mg PO, once daily + lifestyle intervention Intervention (2): saxagliptin 2.5 mg PO, once daily + lifestyle intervention Comparator (1): lifestyle intervention Comparator (2): metformin 500 mg PO, 3 times daily + lifestyle intervention Duration of intervention: 6 months |
| Outcomes |
Primary outcome: OGTT (change of blood sugar from baseline at 6 months) Secondary outcomes: NR Other outcomes: NR |
| Study details | Trial terminated early: unknown |
| Publication details |
Language of publication: not published Funding: NR Publication status: not published |
| Stated aim of study | Quote: "The purpose of the study is to examine the effect of saxagliptin in the newly diagnosed people with pre‐diabetes and obesity besides lifestyle intervention, there to evaluate DPP 4 inhibitors of reversing pre‐diabetes curative effect to normal blood sugar, and observe its influences on the targets of obesity related metabolic abnormalities, to explore new ways for intervention on populations with pre‐diabetes and obesity." |
| Notes | Trial registered as completed in December 2014, investigators were contacted and no data available yet |
NCT02294084.
| Methods |
Allocation: randomised Intervention model: parallel assignment Masking: double blind (participants, investigator) |
| Participants |
Condition: impaired glucose tolerance Enrolment: 30 Inclusion criteria: men, white, born in the Netherlands, age 35‐50 years, BMI > 25 and < 30 kg/m², plasma glucose levels 2 h after OGTT between 7.8 and 11 mM (e.g. impaired glucose tolerance) Exclusion criteria: T2DM (determined on basis of OGTT) defined by American Diabetes Association criteria; BMI > 30 kg/m² or < 25 kg/m²; plasma glucose levels 2 h after OGTT < 7.8 or > 11.1 mM; use of medication known to influence glucose or lipid metabolism (or both) or BAT activity (e.g. beta‐blockers); any significant chronic disease renal, hepatic or endocrine disease; smoking; participation in an intensive weight‐loss programme or vigorous exercise programme during last year before start of the study; difficulties to insert an intravenous catheter; recent participation in other research projects (within the last 3 months) |
| Interventions |
Intervention: sitagliptin 100 mg/day, PO Comparator: placebo, PO Duration of intervention: 12 weeks |
| Outcomes |
Primary outcomes: effect of sitagliptin treatment on BAT activity in overweight, prediabetic people, BAT volume and activity measured by cold‐induced 18F‐FDG PET‐CT scans (after 12 weeks of treatment); BAT volume and activity measured by cold‐induced 18F‐FDG PET‐CT scans Secondary outcomes: energy expenditure (indirect calorimetrie) (after 12 weeks of treatment), muscle glucose metabolism, expression or activation (or both) of biomarkers for insulin signalling and glucose and lipid metabolism in skeletal muscle biopsies (after 12 weeks of treatment), fat mass (measured via DEXA scan) (after 12 weeks of treatment); glucose metabolism (serum glucose, insulin and HbA1c); Insulin secretion (determined by OGTT and C‐peptine, glucose and insulin area under the curve) (after 12 weeks of treatment); plasma lipid levels (total cholesterol, HDL cholesterol, LDL cholesterol, triglycerides and free fatty acids in plasma) (after 12 weeks of treatment) Other outcomes: none |
| Study details | Trial terminated early: unknown |
| Publication details |
Language of publication: not published Funding: NR Publication status: not published |
| Stated aim of study | Quote: "The investigators hypothesize that STG [sitagliptin] enhances BAT activation, thereby increasing energy expenditure and combustion of TG‐derived fatty acids, resulting in lowering of plasma TG levels and body weight." |
| Notes | According to ClinicalTrials.gov, the trial should be completed in May 2016. However, no data available yet |
Santilli 2015.
| Methods |
Allocation: randomised Intervention model: parallel assignment Masking: open label |
| Participants |
Condition: impaired glucose tolerance, impaired fasting glucose or newly diagnosed T2DM Enrolment: 29 Inclusion criteria: metformin‐treated obese participants with impaired glucose tolerance, impaired fasting glucose or newly diagnosed T2DM Exclusion criteria: NR |
| Interventions |
Intervention(s): liraglutide, subcutaneous Comparator (s): placebo Duration of intervention: 4 months |
| Outcomes |
Primary outcomes: not specified, assume: change in subcutaneous fat and visceral adipose tissue Secondary outcomes: not specified, assume: degree of non‐alcoholic fatty liver disease, weight loss, insulin sensitivity, β‐cell performance, C‐reactive protein, leptin, glycaemic measures |
| Study details | Trial terminated early: no |
| Publication details |
Language of publication: English Funding: NR Publication status: abstract |
| Stated aim of study | Quote: "... we hypothesized that this class of drugs may exert additional cardiometabolic actions on top of those anticipated for lifestyle intervention‐mediated weight loss." |
| Notes | Separate data for participants with intermediate hyperglycaemia are not yet available, but will be provided from the authors when the full article is published |
SCALE 2013.
| Methods |
Allocation: randomised Intervention model: parallel Masking: double blind (participants and investigators) |
| Participants |
Condition: normoglycaemia and intermediate hyperglycaemia Enrolment: in total 422 participants; abstract stated that 224 participants had impaired fasting glucose at baseline Inclusion criteria: men and women age ≥ 18 years, with stable body weight and BMI ≥ 30 kg/m² or ≥ 27 kg/m² with comorbidities of treated or untreated dyslipidaemia or treated or untreated hypertension (or both) Exclusion criteria: main criteria: diagnosis of T1DM or T2DM; fasting plasma glucose ≥ 7 mmol/L at run‐in (week 12); treatment with glucagon‐like peptide‐1 receptor agonists or medications causing significant weight gain/loss; bariatric surgery; history of idiopathic acute or chronic pancreatitis; history of major depressive disorder or other severe psychiatric disorders; or clinically significant active cardiovascular disease |
| Interventions |
Intervention: liraglutide 3.0 mg, subcutaneous Comparator: placebo, subcutaneous Duration of intervention: 12‐week run‐in period followed by a 56‐week main trial period and a 12‐week follow‐up period |
| Outcomes |
Primary outcomes: change in body weight; percentage of maintained run‐in fasting weight loss; percentage of who lost ≥ 5% of fasting body weight Secondary outcomes: percentage who lost > 10% of fasting body weight; percentage with weight regain ≥ 5%; percentage with weight regain ≥ 10%; percentage with weight regain > 50% of fasting run‐in weight loss maintained; percentage with weight regain > 75% of fasting run‐in weight loss maintained; change from baseline in fasting weight; change from baseline in fasting weight for participants completing the main trial period and entering the follow‐up period; change from baseline in blood pressure; change from baseline in pulse; change from baseline in fasting lipid profile and cardiovascular biomarkers; metabolic syndrome status; waist circumference; BMI; glycaemic parameters; β‐cell function; insulin resistance; insulin; concomitant medication; Binge Eating Scale |
| Study details | Trial terminated early: no |
| Publication details |
Language of publication: English Funding: commercial funding Publication status: peer‐reviewed journal/full article |
| Stated aim of study | Quote: "The present trial provides the first evaluation of liraglutide for maintenance of prior weight loss achieved by treatment with a LCD in obese/overweight individuals without T2D [type 2 diabetes]." |
| Notes | To qualify for randomisation, participants had to lose ≥ 5% of initial body weight during a variable‐length (4‐12 weeks) low calorie diet run‐in period. Application sent to Novo Nordisk to request separate data on the participants with intermediate hyperglycaemia. Novo Nordisk has approved access to raw trial data, but data are not yet available |
SCALE‐SLEEP.
| Methods |
Allocation: randomised Intervention model: parallel Masking: double blind (participants and investigators) |
| Participants |
Condition: normoglycaemia and intermediate hyperglycaemia Enrolment: in total 359 participants; the abstract stated that 63.2% of participants had intermediate hyperglycaemia at baseline Inclusion criteria: informed consent; BMI ≥ 30 kg/m²; stable body weight (< 5% self‐reported change during the previous 3 months); diagnosis of moderate or severe obstructive sleep apnoea; unwilling or unable to use continuous positive airway pressure (or other positive airway pressure) treatment. No continuous positive airway pressure (or other positive airway pressure) treatment for at least 4 weeks prior to screening; ability and willingness to comply with all protocol procedures, e.g. correct handling of trial product, compliance to visit schedule and dietary advice and complete trial related questionnaires Exclusion criteria: treatment with glucagon‐like peptide‐1 receptor agonists, DPP‐4 inhibitors or insulin within the last 3 months prior to screening; diagnosis of T1DM or T2DM per judgement of investigator; HbA1c ≥ 6.5%; significant craniofacial abnormalities that may cause obstructive sleep apnoea; respiratory and neuromuscular diseases that could interfere with the results of the trial in the opinion of investigator; use of central stimulants, hypnotics, mirtazapine, opioids or trazodone within the previous 3 months prior to screening; obesity induced by drug treatment; treatment with pramlintide, sibutramine, orlistat, zonisamide, topiramate or phentermine within the last 3 month prior to screening; previous surgical treatment for obesity; screening calcitonin ≥ 50 ng/L; familial or personal history of multiple endocrine neoplasia type 2 or familial medullary thyroid carcinoma; personal history of non‐familial medullary thyroid carcinoma; history of chronic pancreatitis or idiopathic acute pancreatitis; history of major depressive disorder or suicide attempts; systolic blood pressure ≥ 160 mmHg or diastolic blood pressure ≥ 100 mmHg (or both) |
| Interventions |
Intervention: liraglutide 3.0 mg, subcutaneous Comparator: placebo, subcutaneous Duration of intervention: 32 weeks |
| Outcomes |
Primary outcome: change from baseline in apnoea‐hypopnoea index Secondary outcomes: change from baseline in body weight; glycaemic measures |
| Study details | Trial terminated early: no |
| Publication details |
Language of publication: English Funding: commercial funding Publication status: peer‐reviewed journal/full article |
| Stated aim of study | Quote: "The aim of the trial is to investigate the effect of liraglutide in obese subjects with sleep apnoea." |
| Notes | Application sent to Novo Nordisk to request separate data on the participants with intermediate hyperglycaemia. Novo Nordisk has approved access to raw trial data, but data are not yet available |
18F‐FDG PET‐CT: 18‐fluorodeoxyglucose positron emission tomography computed tomography; BAT: brown adipose tissue; BMI: body mass index; DEXA: dual‐energy x‐ray absorptiometry; DPP‐4: dipeptidyl‐peptidase‐4; h: hour; HbA1c: glycosylated haemoglobin A1c; HDL: high‐density lipoprotein; LDL: low‐density lipoprotein; OGTT: oral glucose tolerance test; PO: per os (orally); NR: not reported; T1DM: type 1 diabetes mellitus; T2DM: type 2 diabetes mellitus.
Characteristics of ongoing studies [ordered by study ID]
EudraCT 2013‐000418‐39.
| Trial name or title | Acronym: ePREDICE |
| Methods |
Type of trial: efficacy trial Allocation: randomised Intervention model: parallel assignment Masking: double blind Primary purpose: not specified in protocol |
| Participants |
Condition: IGT or IFG, or both Enrolment: 3000 Inclusion criteria: age 45‐74 years; IFG (FPG 6.1‐6.9 mmol/L and 2‐h PG < 7.8 mmol/L) or IGT (FPG < 7.0 mmol/L and 2‐h PG ≥ 7.8 to < 11.1 mmol/L) or both conditions; informed consent given Exclusion criteria: T1DM; known or unknown T2DM (including screen‐detected T2DM) with or without pharmacological treatment; use of a GLP‐1 receptor agonist (exenatide or other) or pramlintide or any DPP‐4 inhibitor or metformin within the 3 months prior to enrolment; use of insulin or long‐acting insulin analogue within 3 months prior to enrolment; any previous cardiovascular or cerebrovascular clinically documented event or revascularisation procedure; clinical evidence of macrovascular complications (overt clinical cardiovascular disease) at enrolment, including angina (stable or unstable) and evidence of previous myocardial infarction in baseline electrocardiogram; current renal replacement therapy; previous diagnosis of liver cirrhosis or chronic hepatitis, or an elevation of liver enzymes (aspartate aminotransferase and or alanine aminotransferase) > 3 times normal ranges; previous diagnosis of chronic heart failure (NYHA class III or higher); prior solid organ transplant or awaiting solid organ transplant; malignant neoplasm requiring chemotherapy, surgery, radiation or palliative therapy in the previous 5 years. Participants with intraepithelial squamous cell carcinoma of the skin (Bowen's disease) treated with topical 5‐fluorouracil and people with basal cell skin cancer allowed to enter trial; any acute condition or exacerbation of chronic condition that would, in investigator's opinion, interfere with the initial trial visit schedule and procedures; known or suspected hypersensitivity to trial products or related products; known use of non‐prescribed narcotics or illicit drugs; simultaneous participation in any other clinical trial of an investigational agent; women of childbearing potential who are pregnant (all fertile women will be tested for before randomisation), breastfeeding or intend to become pregnant; presence of cataract that impedes the retinal evaluation of both eyes; other previously diagnosed retinal diseases; any diseases that would prevent the measurement of primary endpoints; dementia, mental disorder or evident cognitive impairment unable to give informed consent; end‐stage or metastatic cancer; institutionalisation; renal function impairment: GFR < 60 mL/minute/1.73 m².; contraindication to any of the study drugs (metformin or linagliptin). This includes: alanine aminotransferase > 3 times the upper limit of normal, history of cirrhosis or hepatitis, suspected renal artery stenosis, recent gastrointestinal bleeding (within last year), pregnant, breastfeeding or a female of childbearing potential not on reliable contraception and also any circumstance where ongoing medication might lead to potential adverse drug interaction with components of the trial medications; any other reason, medical condition, ongoing medication or significant disability that would prevent the participant complying with trial consent, treatment and follow‐up procedures or potentially jeopardise her/his medical care |
| Interventions |
Intervention: 2 tablets of linagliptin 5 mg + diet and physical activity Comparator (1): 2 tablets of metformin 850 mg/day + diet and physical activity Comparator (2): 2 tablets of linagliptin 2.5 mg + metformin 850 mg plus diet and physical activity Comparator (3): 2 tablets of placebo + diet and physical activity Duration of intervention: at least 3 years, and additional follow‐up to 5 years |
| Outcomes |
Primary outcome: a combined continuous variable, "the microvascular complication índex" (MCI), composed of linear combination of ETDRS score, the level of urinary albumin to creatinine ratio, and sudomotor test (SUDOSCAN) score, measured during the 36th and 60th month visits. From email correspondence: primary purpose: prevention of complications of hyperglycaemia/ prevention of progression to diabetes Secondary outcomes: retinopathy score at last visit defined as 2 steps' progression on ETDRS scale between baseline and visits at months 36 and 60; 1 SD increase in level of urinary albumin to creatinine ratio between baseline and visits at months 36 and 60; 1 SD decrease change in level of hands and feet conductance in SUDOSCAN between baseline and visits at months 36 and 60; change in microvascular endothelial function measured by EndoPAT method (in a subset); change in the Non‐Alcoholic Fatty Liver Index (in a subset); change in biomarkers of microvascular damage, endothelial function, per‐oxidation, inflammation and metabolomics (in a subset); change in the insulin secretion and β‐cell function; change in self‐perceived quality of life; change in symptoms of peripheral neuropathy; change in neuropsychological parameters: cognitive function, anxiety and depressive symptoms and indices; changes in obstructive sleep apnoea indices as measured by Somnomedics (in a subset); changes in ambulatory blood pressure monitoring (in a subset); change in the mean common carotid intimae‐media thickness (in a subset); incidence of major cardiovascular events, defined as an expanded composite of total coronary events, total stroke events, revascularisation procedures (coronary artery bypass graft, percutaneous coronary angioplasty and peripheral revascularisation), hospitalisation for heart failure, TIA and cardiovascular or cerebrovascular death. Secondary outcomes will be evaluated at 36 and 60 months Other outcome: none |
| Starting date |
Trial start date: 2015 Trial completion date: 2018 |
| Contact information | Responsible party/principal investigator: Prof Jaakko Tuomilehto; Prof Rafael Gabriel (co‐principal investigators) |
| Study identifier | EudraCT‐number: 2013‐000418‐39 |
| Official title | Early Prevention of Diabetes Complications in People with Hyperglycaemia in Europe |
| Stated purpose of study | Quote: "To assess the effect of treatment with linagliptin, metformin or the combination of linagliptin with metformin, plus lifestyle intervention (diet and physical activity), compared to lifestyle intervention alone, for at least 3 years, and up to 5 years, on different microvascular parameters (retinal, renal and neurological), as defined by the primary and secondary endpoints, in adults with non diabetic hyperglycaemia (IGT, IFG or IFG plus IGT)." |
| Notes | Multinational trial with 15 clinical centres from 12 countries: Australia, Austria, Bulgaria, Germany, Greece, Italy, Lithuania, Poland, Serbia, Spain, Switzerland and Turkey. Clarified though e‐mail correspondence that the trial is double‐blind, trial start date and trial completion date |
Naidoo 2016.
| Trial name or title | Acronym: SiMePreD |
| Methods |
Type of trial: efficacy trial Allocation: randomised Intervention model: parallel assignment Masking: double blind Primary purpose: efficacy |
| Participants |
Condition: IGT, IFG, or both Enrolment: 820 Inclusion criteria: informed consent; IGT (2‐h postprandial glucose 7.8‐11.0 mmol/L); IFG glucose (fasting glucose 5.6‐6.9 mmol/L); age 18‐65 years; no history of liver disease; negative pregnancy test. Exclusion criteria: impaired liver function tests; cardiac failure or history of congestive heart failure in the close family; medication that may affect insulin resistance (e.g. oral hypoglycaemic agents, thiazide diuretics); contraindications to exercise; pregnancy; planning to move residence within the next 5‐10 years; history of hypersensitivity reaction to sitagliptin, such as anaphylaxis or angio‐oedema |
| Interventions |
Intervention: sitagliptin 25 mg/day + metformin extended release 500 mg/day Comparator: placebo + metformin extended release 500 mg/day Duration of intervention: 5 years |
| Outcomes |
Primary outcomes: number of participants progressing from prediabetes to T2DM, number of cardiovascular events and number of deaths Secondary outcomes: lipo grams, urea and electrolytes, liver function tests, full blood count, fasting blood glucose, fasting blood insulin, weight, blood pressure. Other anthropometric parameters Other outcomes: adverse effects |
| Starting date |
Trial start date: not stated Trial completion date: not stated |
| Contact information | Responsible party/principal investigator: not defined, presumably the corresponding author of the article: N Poobalan, Johannesburg, South Africa |
| Study identifier | Study has not started |
| Official title | Sitagliptin and Metformin in PreDiabetes (SiMePreD) Study |
| Stated purpose of study | Quote: "The aim of this study is to determine the effect of sitagliptin and metformin on progression from prediabetes to type 2 DM." |
| Notes | Study currently searching for funding. Study has not started |
NCT01234649.
| Trial name or title | Effects of Intervention with the Glucagon‐like Peptide 1 (GLP‐1) Analog Liraglutide Plus Metformin Versus Metformin Monotherapy in Overweight/Obese Women with Metabolic Defects and Recent History of Gestational Diabetes Mellitus (GDM) |
| Methods |
Type of trial: efficacy trial Allocation: randomised Intervention model: parallel assignment Masking: double blind (participant, carer, investigator) Primary purpose: prevention |
| Participants |
Condition: obese with previous GDM and IFG, IGT, or both with or without β‐cell dysfunction postpartum requiring pharmacological intervention Enrolment: 150 Inclusion criteria: women age 18‐45 years who experienced GDM within 52 weeks of index pregnancy; actual BMI > 25 kg/ m²; written consent for participation in the trial; women completed lactation; dysglycaemia (IFG, IGT, or both) or β‐cell dysfunction postpartum requiring pharmacological intervention (except T1DM or T2DM), or both Exclusion criteria: personal or family history of medullary thyroid carcinoma or in people with multiple endocrine neoplasia syndrome type 2; history of pancreatitis; significant cardiovascular, cerebrovascular, renal or hepatobiliary diseases (viral hepatitis, toxic hepatic damage, jaundice of unknown aetiology); serum liver enzymes (aspartate aminotransferase or alanine aminotransferase (or both) levels) exceeding more than twice normal laboratory values; uncontrolled hypertension (systolic blood pressure > 150 mmHg or diastolic blood pressure > 90 mmHg, or both); fasting serum triglycerides ≥ 800 mg/dL at screening. Lipid‐lowering medications must have been maintained at the same dose for 3 months prior to enrolment; haematological profiles considered to be clinically significant; cholestasis during past pregnancy; presence of contradictions for GLP‐1 receptor agonist or metformin administration such as allergy or hypersensitivity; current use of metformin, thiazolidinediones, DPP‐4 inhibitors or GLP‐1 receptor agonist medications; use of drugs known to exacerbate glucose tolerance; use of prescription or non‐prescription weight‐loss drugs; diabetes postpartum or history of diabetes or prior use of medications to treat diabetes other than GDM; creatinine clearance < 60 mL/minute; history or currently undergoing chemotherapy or radiotherapy for cancer; pregnancy planned during the coming 2 years; currently breastfeeding; any condition that, in the opinion of investigator, would place the woman at increased risk or otherwise make the women unsuitable for participation in trial |
| Interventions |
Intervention: liraglutide, subcutaneous (titrated up to 1.8 mg) + metformin, PO (titrated up to 1000 mg twice daily) Comparator: placebo, subcutaneous plus metformin, PO (titrated up to 1000 mg twice daily) Duration of intervention: 84 weeks at full dose (8‐12 weeks for up titrate to full dose) |
| Outcomes |
Primary outcomes: index of insulin secretion in relation to insulin resistance will be calculated (change in index from baseline at 32‐36 weeks, 56 ‐60 weeks and study end (80‐84 weeks)). β‐cell compensatory capacity will be evaluated by insulin sensitivity‐secretion index defined as the product of composite insulin sensitivity index and first‐phase insulin release index (insulinogenic index) Secondary outcomes: insulin resistance ‐ baseline (HOMA‐IR) and composite insulin sensitivity index), and pancreatic β‐cell function (corrected insulin response (CIRglupeak) and insulinogenic index/HOMA‐IR (change in indexes from baseline at 32‐36 weeks, 56‐60 weeks, and trial end (80‐84 weeks)). Indexes of insulin sensitivity and secretion using the serum glucose and insulin concentrations obtained in the fasting state and during the 2‐h glucose tolerance test with insulin levels will be computed by several measures previously validated in women. Cardiometabolic risk measures (change in measures (lipids, liver enzymes, blood pressure) from baseline at 32‐36 weeks, 56‐60 weeks and study end (80‐84 weeks). Lipids, liver enzymes, blood pressure. Anthropometric measurements (change in measures of total and central adiposity from baseline at 32‐36 weeks, 56‐60 weeks, and study end (80‐84 weeks)). BMI, absolute body weight, waist circumference, waist:hip ratio. Development of dysglycaemia (changes in glucose tolerance will be evaluated at baseline, 32‐36 weeks, 56‐60 weeks and study end (80‐84 weeks)). Change in glycaemic status from baseline (at 32‐36 weeks, 56‐60 weeks and study end (80‐84 weeks). Dysglycaemia will be defined as IFG, IGT, combined IFG/IGT and diabetic according to the American Diabetes Association. Women diagnosed with diabetes will be withdrawn and referred to a specialised physician Other outcomes: none specified |
| Starting date |
Trial start date: January 2011 Trial completion date: October 2017 |
| Contact information | Responsible party/principal investigator: Karen E Elkind‐Hirsch, PhD and Martha Paterson, MD, Woman's Hospital, Louisiana |
| Study identifier | NCT number: NCT01234649 |
| Official title | Effects of Intervention with the Glucagon‐like Peptide 1 (GLP‐1) Analog Liraglutide Plus Metformin Versus Metformin Monotherapy in Overweight/Obese Women with Metabolic Defects and Recent History of Gestational Diabetes Mellitus (GDM) |
| Stated purpose of study | Quote: "This study will examine if the addition of liraglutide to metformin therapy is more effective than metformin alone in improving insulin sensitivity and normalizing insulin secretion in at‐risk overweight/obese women with prior GDM." |
| Notes | Investigator clarified the intervention period through email correspondence |
NCT01336322.
| Trial name or title | Acronym: SITA‐previousGDM |
| Methods |
Type of trial: efficacy trial Allocation: randomised Intervention model: parallel assignment Masking: double blind (participants, investigator) Primary purpose: treatment |
| Participants |
Condition: IFG or IGT in women with previous GDM Enrolment: 45 Inclusion criteria: women age 18‐45 years; Caucasian race; history of GDM (in screening) during pregnancy, defined according to Carpenter and Coustan criteria; women of childbearing potential must use effective contraceptive measures for at last 1 month prior to entry into study and should continue to use some contraceptive method during overall trial period; written informed consent obtained. IFG and IGT not listed in inclusion criteria, but in the text it is stated: "Women with IFG or IGT will be recruited and undergo to a hyperglycaemic clamp with arginin bolus at the end of the test." Exclusion criteria: diagnosed with type 1 insulin‐dependent diabetes; diagnosis of diabetes in OGTT 75‐g glucose performed at entry; BMI ≤ 18 or ≥ 50 kg/m²; chronic impaired renal function; impaired liver function as shown by transaminase levels ≥ twice above upper normal range; history of hypersensitivity to metformin; pregnant or breastfeeding women, or women planning to become pregnant during trial; failure to use adequate contraception (women of current reproductive only); mental condition rending the person unable to understand the nature, scope and possible consequences of trial; any clinically significant major organ system disease; underlying concomitant illness requiring a long‐term use of drugs potentially acting on glucose metabolism (e.g. corticosteroids, diuretics, beta‐adrenergic drugs or others); treatment or likelihood of requiring treatment during the study period with drugs not permitted by the clinical trial protocol; history of drug or alcohol abuse within the last 2 years or current addiction to substances of abuse; any disease or condition that, in the opinion of investigator, may interfere with the completion of the trial; unlikely to comply with protocol |
| Interventions |
Intervention (1): sitagliptin 100 mg, PO, once daily Intervention (2): sitagliptin 100 mg, PO, once daily + metformin 850 mg, PO, twice daily Comparator: metformin 850 mg, PO, twice daily Duration of intervention: 4 months |
| Outcomes |
Primary outcome: β‐cell function (at 4 months) Secondary outcomes: insulin resistance (at 4 months); glucose control (at 4 months) Other outcomes: none |
| Starting date |
Trial start date: May 2011 Trial completion date: according to ClinicalTrials.gov December 2012, but principal investigator informed us through email that trial is still ongoing |
| Contact information | Responsible party/principal investigator: Stefano Del Prato, MD, University of Pisa |
| Study identifier | NCT number: NCT01336322 |
| Official title | Effects of Treatment with Metformin and/or Sitagliptin on Beta‐cell Function and Insulin Resistance in Women with Previous Gestational Diabetes |
| Stated purpose of study | Quote: "The goal of the present research is to compare the effects of treatment with metformin and sitagliptin, alone or in association, in women with previous gestational diabetes to evaluate the impact of the two drugs on beta‐cell function." |
| Notes | Through correspondence it was clarified that the trial was extended due to delay in study drug supply |
NCT01548651.
| Trial name or title | Effect of Saxagliptin Treatment on Myocardial Fat Content, Left Ventricular Function, and Monocyte Inflammation in Patients with Impaired Glucose Tolerance |
| Methods |
Type of trial: not reported in available protocol, but assume efficacy Allocation: randomised Intervention model: parallel assignment Masking: double blind (participant, carer, investigator, outcomes assessor) Primary purpose: treatment |
| Participants |
Condition: IGT Enrolment: estimated 40 Inclusion criteria: men and women with IGT (i.e. FPG ≤ 125 mg/dL, 2‐h post OGTT 75‐g glucose 140‐199 mg/dL, HbA1c < 6.5% as per American Diabetes Association criteria; age 30‐70 years; using an acceptable method of contraception to avoid pregnancy throughout the study in such a manner that the risk of pregnancy is minimised; BMI 30‐35 kg/m² and stable body weight Exclusion criteria: must not be on anti‐diabetes therapy for treatment of IGT and must have a FPG concentration ≤ 125 mg/dL; T1DM or T2DM (FPG > 125 mg/dL); must not be on or have received metformin, thiazolidinediones, sulphonylureas, DPP‐4 inhibitor or exenatide/liraglutide treatment for IGT at any time; must not be receiving any of the following: thiazide or furosemide diuretics, beta‐blockers or other chronic medications such as hormone replacement therapy with known adverse effects on glucose tolerance levels; people taking systemic glucocorticoids excluded; history of clinically significant heart disease, peripheral vascular disease or pulmonary disease; must not have clinically significant liver disease (aspartate aminotransferase < 2.5 times upper limit of normal, alanine transaminase < 2.5 times upper limit of normal, alkaline phosphatase < 2.5 times upper limit of normal), kidney disease (serum creatinine < 1.5 mg/dL in men and 1.4 mg/dL in women) or significant anaemia (hematocrit < 34 vol%); history of any serious hypersensitivity reaction to saxagliptin or a DPP‐4 inhibitor; concomitant treatment with systemic cytochrome P450 3A4 inducers; pregnant or breastfeeding |
| Interventions |
Intervention: saxagliptin 5 mg/day, PO Comparator: placebo daily Duration of intervention: 6 months |
| Outcomes |
Primary outcomes: myocardial and hepatic fat content (%) (at 6 months); % change in hepatic fat and myocardial fat from baseline as measured by magnetic resonance imaging and spectroscopy Secondary outcomes: left ventricular ejection fraction (%) (at 6 months); % change in left ventricular ejection fraction from baseline as measured by magnetic resonance imaging; monocyte inflammatory protein NFkappaB (%) (at 6 months); % change in monocyte inflammatory proteins NFkappaB from baseline Other outcomes: none |
| Starting date |
Trial start date: February 2012 Trial completion date: December 2016 (final data collection date for primary outcome measure) |
| Contact information | Responsible party/principal investigator: Mandeep Bajaj, MD, Baylor College of Medicine |
| Study identifier | NCT number: NCT01548651 |
| Official title | Effect of Saxagliptin Treatment on Myocardial Fat Content, Left Ventricular Function, and Monocyte Inflammation in Patients with Impaired Glucose Tolerance |
| Stated purpose of study | Quote: "The purpose of the study is to examine the effect of saxagliptin, an anti‐diabetes medication, on hepatic and myocardial fat content and monocyte inflammation in patients with Impaired Glucose Tolerance (IGT)." |
| Notes | ‐ |
NCT01779362.
| Trial name or title | Acronym: RISE Adult |
| Methods |
Type of trial: efficacy trial Allocation: randomised Intervention model: parallel assignment Masking: double blind (participants, carer, investigator, outcomes assessor) Primary purpose: treatment |
| Participants |
Condition: prediabetes and early T2DM Enrolment: 255 Inclusion criteria: FPG 95‐125 mg/dL + 2‐h glucose ≥ 140 mg/dL on OGTT 75‐g glucose + HbA1c ≤ 7.0%. No upper limit for the 2‐h glucose on OGTT; age 20‐65 years; BMI ≥ 25 kg/m² but ≤ 50 kg/m²; self‐reported diabetes < 1 year in duration; drug naive (no prior to oral glucose lowering agent(s), insulin or other injectable glucose lowering agents) Exclusion criteria: underlying disease likely to limit life span or increase risk of intervention (or both) or an underlying condition that is likely to limit ability to participate in outcomes assessment; underlying disease that affects glucose metabolism other than T2DM; taking medications that affect glucose metabolism, or has an underlying condition that is likely to require such medications; active infections; renal disease (serum creatinine > 1.4 mg/dL for men; > 1.3 mg/dL for women) or serum potassium abnormality (< 3.4 mmol/L or > 5.5 mmol/L); anaemia (haemoglobin <11 g/dL for women, < 12 g/dL for men) or known coagulopathy; cardiovascular disease, including uncontrolled hypertension; intolerant to administration of intravenous fluids required during clamp studies; history of conditions that may be precipitated or exacerbated by a study drug (pancreatitis, serum alanine transaminase more than 3 times the upper limit of normal, excessive alcohol intake, suboptimally treated thyroid disease, medullary carcinoma of the thyroid or multiple endocrine neoplasia‐2 (in participant or a family history), hypertriglyceridaemia (> 400 mg/dL despite treatment); conditions or behaviours likely to affect the conduct of the RISE Study; unable or unwilling to give informed consent; unable to adequately communicate with clinic staff; another household member is a participant or staff member in RISE; current, recent or anticipated participation in another intervention research project that would interfere with any of the interventions/outcomes in RISE; weight loss > 5% in past 3 months for any reason other than postpartum weight loss; taking weight loss drugs or using preparations taken for intended weight loss; likely to move away from participating clinics in next 2 years; women of childbearing potential unwilling to use adequate contraception; current (or anticipated) pregnancy and lactation; major psychiatric disorder that, in the opinion of clinic staff, would impede the conduct of RISE; additional conditions may serve as criteria for exclusion at the discretion of the local site |
| Interventions |
Intervention: liraglutide + open‐label metformin. Liraglutide titrated to maximum dose tolerated (up to 1.8 mg/day) after which metformin titrated to maximum dose tolerated (up to 2000 mg/day) Comparator: metformin alone. Metformin titrated to maximum dose tolerated (up to 2000 mg/day). Participants randomised to metformin‐alone arm will be blinded to intervention Duration of intervention: participants will have 12‐months of active therapy and 3‐months of washout |
| Outcomes |
Primary outcome: β‐cell function measured by hyperglycaemic clamp techniques (at 3‐months after a medication washout). Participants will have 12‐months of active therapy and 3 months of washout after which the primary outcome will be assessed Secondary outcome: hyperglycaemic clamp and OGTT measures of β‐cell function and glucose tolerance (at 3‐months after a medication washout). Measures derived from the hyperglycaemic clamp that are not specified as primary outcomes and measures derived from the OGTT Other outcomes: hyperglycaemic clamp and OGTT measures of β‐cell function and glucose tolerance (at after 12 months of active treatment); measures derived from the hyperglycaemic clamp and the OGTT related to treatment effect at end of 12‐month active intervention period compared to pretreatment baseline |
| Starting date |
Trial start date: April 2013 Trial completion date: March 2019 |
| Contact information | Responsible party/principal investigator(s): David Ehrmann, MD, Kieren Mather, MD, Steven Kahn, MB, ChB |
| Study identifier | NCT number: NCT01779362 |
| Official title | Restoring Insulin Secretion Adult Medication Study |
| Stated purpose of study | Quote: "The primary clinical question RISE will address is: Are improvements in β‐cell function following 12 months of active treatment maintained for 3 months following the withdrawal of therapy? Secondary outcomes will assess durability of glucose tolerance following withdrawal of therapy, and whether biomarkers obtained in the fasting state predict parameters of β‐cell function, insulin sensitivity and glucose tolerance and the response to an intervention." |
| Notes | Other treatment arms in the trial are: insulin glargine followed by metformin; placebo |
NCT01795248.
| Trial name or title | Acronym: GDM‐TREAT |
| Methods |
Type of trial: efficacy trial Allocation: randomised Intervention model: parallel assignment Masking: double blind (participants, carer, investigator) in 1 year, thereafter open‐label Primary purpose: prevention |
| Participants |
Condition: NGT, IFG, IGT or a combination Enrolment: 100 Inclusion criteria: informed oral and written consent; previous diagnosis of GDM according to current Danish guidelines (mainly PG concentration at 120 minutes after OGTT 75‐g glucose ≥ 9.0 mM) during pregnancy within last 5 years; age > 18 years; BMI 25‐45 kg/m²; NGT, IFG, IGT or a combination, safe contraception and negative pregnancy test Exclusion criteria: diabetes, HbA1c ≥ 6.5%, previous pancreatitis or previous neoplasia, pregnant or breastfeeding, anaemia (haemoglobin < 7 mM), women planning to become pregnant within the next 5 years, women using other contraception than IUD or oral contraceptives (women who do not use safe contraception will be offered an IUD), women treated with statins, corticosteroids or other hormone therapy (except oestrogens and gestagens); ongoing abuse of alcohol or narcotics; impaired hepatic function (liver transaminases > 3 times upper normal limit); impaired renal function (serum creatinine > 120 μM or albuminuria (or both)), uncontrolled hypertension (systolic blood pressure > 180 mmHg, diastolic blood pressure > 100 mmHg); any condition that investigator feels would interfere with trial participation, receiving any investigational drug within the last 3 months |
| Interventions |
Intervention: liraglutide 1.8 mg, subcutaneous, once daily Comparator: placebo, subcutaneous, once daily. Placebo only given in first year, thereafter no intervention Duration of intervention: 5 years |
| Outcomes |
Primary outcomes: change in glucose tolerance (from baseline to 52, 53, 260 and 261 weeks), changes in glucose measured by area under the curve for the PG excursion following 4‐h OGTT 75‐g glucose. Secondary outcomes: deterioration in glycaemic status (from baseline to 52, 53, 260 and 261 weeks), % in each treatment arm with NGT at inclusion who develop IFG or IGT (or both) or T2DM; or with IFG or IGT who develop combined IFG/IGT; or with combined IFG/IGT who develop T2DM Other outcomes: changes in HbA1c (from baseline to 52 and 260 weeks), from normoglycaemic to prediabetic or T2DM and from prediabetic to T2DM, changes in anthropometric measurements (from baseline to 52 and 260 weeks), changes in BMI, absolute body weight, and waist:hip ratio, changes in β‐cell secretory responses (from baseline to 52, 53, 260 and 261 weeks), changes in area under the curve during OGTT and isoglycaemic intravenous glucose infusion, homeostatic model assessment and proinsulin ratio, changes in insulin sensitivity assessed by homeostatic model assessment and proinsulin ratio and Matsuda insulin sensitivity index (from baseline to 52, 53, 260 and 261 weeks), changes in incretin hormone secretion (baseline to 52, 53, 260 and 261 weeks), measured as fasting plasma concentrations and plasma responses of GLP‐1, GLP2, and glucose‐dependent insulinotropic polypeptide and plasma glucagon during OGTT, changes in incretin effect (from baseline to 52, 53, 260 and 261 weeks), insulin and C‐peptide responses after OGTT vs isoglycaemic intravenous glucose infusion, changes in cardiometabolic risk measures (from baseline to 52 and 260 weeks), changes in subjective appetite (from baseline to 52, 53, 260 and 261 weeks), quality of life (from baseline to 52 and 260 weeks) |
| Starting date |
Trial start date: July 2012 Trial completion date: August 2019 (final data collection date for primary outcome measure) |
| Contact information | Responsible party/principal investigator: Tina Vilsbøll, MD, DMSc, University Hospital Gentofte, Denmark |
| Study identifier |
NCT number: NCT01795248 EudraCT number: 2012‐001371‐37 |
| Official title | The Impact of Liraglutide on Glucose Tolerance and the Risk of Type 2 Diabetes in Women with Previous Gestational Diabetes Mellitus |
| Stated purpose of study | Quote: "It is well‐known that women with previous gestational diabetes mellitus are in risk of developing type 2 diabetes later in life; approximately half of the women develop overt type 2 diabetes within the first 10 years after pregnancy. Knowing this, we want to examine the effect of the type 2 diabetes medicine, liraglutide (Victoza), in women with previous gestational diabetes with the aim of reducing the risk of developing type 2 diabetes." |
| Notes | 15 healthy women without previous GDM will make up a baseline control group. There has been correspondence with the principal investigator, Tina Vilsbøll. So far 2 out 3 of the included participants have IFT or IGT (or both). The first year, the trial is double blind |
NCT01856907.
| Trial name or title | A Randomized Pilot Study Evaluating Combination Dipeptidyl Peptidase‐4 Inhibitor Sitagliptin Plus Metformin Compared to Metformin Monotherapy and Placebo on Metabolic Abnormalities in Women with a Recent History of GDM |
| Methods |
Type of trial: efficacy trial Allocation: randomised Intervention model: parallel assignment Masking: single blind (investigator) Primary purpose: treatment |
| Participants |
Condition: IFG, IGT, or both postpartum Enrolment: 36 Inclusion criteria: women age 18‐42 years who experienced GDM during recent (within 12 months) pregnancy with prediabetic hyperglycaemia determined by an OGTT 75‐g glucose postpartum; IFG, IGT, or both postpartum; written consent for participation in trial Exclusion criteria: cholestasis during the past pregnancy; any hepatic diseases in the past (viral hepatitis, toxic hepatic damage, jaundice of unknown aetiology); serum aspartate transaminase or alanine aminotransferase level exceeding more than twice normal laboratory values; presence of hypersensitivity to sitagliptin or other DPP‐4 inhibitor; current use of metformin, thiazolidinediones, GLP‐1 receptor agonists, DPP‐4 inhibitors or weight loss medications (prescription or non‐prescription); prior use of medication to treat diabetes except GDM; use of drugs known to exacerbate glucose tolerance; history of diabetes or prior use of medications to treat diabetes except GDM; creatinine clearance < 60 mL/minute; pregnancy planned during the coming 2 years; currently lactating; not willing to use adequate contraception during trial period (unless sterilised) |
| Interventions |
Intervention: sitagliptin 5 mg + metformin 1000 mg, PO, twice daily Comparator (1): placebo tablet, PO, twice daily Comparator (2): metformin, PO, twice daily Duration of intervention: 16 weeks (inclusive up titration period) |
| Outcomes |
Primary outcomes: β‐cell compensatory function (change from baseline to 16 weeks); surrogate measures of insulin sensitivity and secretion (change from baseline to 16 weeks); fasting and 2‐h glucose levels after glucose load (change from baseline to 16 weeks) Secondary outcome: cardiometabolic risk factors (change from baseline to 16 weeks) Other outcome: liver enzymes (change from baseline to 16 weeks) |
| Starting date |
Trial start date: September 2013 Trial completion date: February 2017 |
| Contact information | Responsible party/principal investigator: Karen Elkind‐Hirsch, PhD, and Martha Paterson, MD, Woman's Hospital, Louisiana |
| Study identifier | NCT number: NCT01856907 |
| Official title | A Randomized Pilot Study Evaluating Combination Dipeptidyl Peptidase‐4 Inhibitor Sitagliptin Plus Metformin Compared to Metformin Monotherapy and Placebo on Metabolic Abnormalities in Women With a Recent History of GDM |
| Stated purpose of study | Quote: "This study will examine if combination sitagliptin (a DPP‐4 inhibitor)‐plus metformin is more effective than metformin alone or placebo in improving metabolic parameters, specifically the impact on β‐cell function, in prior GDM women with glucose abnormalities." |
| Notes | ‐ |
NCT02104739.
| Trial name or title | Comparative Effects of Antidiabetic Medications on Postprandial Hyperlipidemia, Free Fatty Acid Signaling, and Endothelial Dysfunction in Individuals with Prediabetes |
| Methods |
Type of trial: efficacy trial Allocation: randomised Intervention model: cross‐over assignment Masking: double blind (participant, carer, investigator, outcomes assessor) Primary purpose: treatment |
| Participants |
Condition: obese with intermediate hyperglycaemia Enrolment: 40 Inclusion criteria: men and women; age 30‐70 years of age inclusive; diagnosis of prediabetes defined as either IFG (fasting glucose 100‐125 mg/dL), IGT (2‐h postprandial blood glucose of 140‐199 mg/dL after 75‐g oral glucose challenge), HbA1c 5.7‐6.4%, or a combination of these; participants are allowed, but not required, to be on statins, ACE‐inhibitors, beta‐blockers, angiotensin‐receptor blockers, thiazide diuretics, loop diuretics, or a combination of these, at doses stable ≥ 3 months; BMI 30‐35 kg/m² (± 1 kg/m²); body weight stable (± 4‐5 pounds) over the prior 3 months; women of childbearing age using acceptable contraception (barrier methods, abstinence or surgical sterilisation) for duration of trial; must have: hematocrit ≥ 34 vol%, serum creatinine < 1.5 mg/dL in men and 1.4 mg/dL in women, aspartate aminotransferase < 2.5 times upper limit of normal, alanine aminotransferase < 2.5 times upper limit of normal, alkaline phosphatase < 2.5 times upper limit of normal Exclusion criteria: history of T1DM or T2DM; history of diabetic ketoacidosis or hyperosmolar non‐ketotic coma; pregnant or breastfeeding; receiving lipid‐lowering medications other than statins within the last 3 months; receiving metformin, DPP‐IV inhibitors, GLP‐1 agonists, thiazolidinediones, insulin, sulphonylureas, acarbose, sodium‐glucose co‐transporter‐2 inhibitors, corticosteroids or immunosuppressive therapy within the last 3 months and cannot take them for duration of study; receiving non‐steroidal anti‐inflammatory drugs or antioxidant vitamins within the last 1 week, and cannot take them for duration of study; receiving hormone replacement therapy; diabetic gastroparesis; current tobacco use; active malignancy; history of urinary bladder cancer; dietary restrictions precluding a high‐fat meal; history of clinically significant heart disease (NYHA III or IV; more than non‐specific ST‐T wave changes on the electrocardiogram), peripheral vascular disease (history of claudication) or pulmonary disease (dyspnoea on exertion of 1 flight of stairs or less; abnormal breath sounds on auscultation); history of any serious hypersensitivity reaction to study medications; prisoners or people who are involuntarily incarcerated; people who are compulsorily detained for treatment of either a psychiatric or physical (e.g. infectious disease) illness; known allergic reactions to study medications or test meal; unwilling or unable to provide informed consent; people determined by investigator(s) to not be appropriate candidates for the trial |
| Interventions |
Intervention (1): saxagliptin 5 mg PO, once daily Intervention (2): exenatide 10 μg subcutaneous, once daily Comparator (1): pioglitazone 45 mg PO, once daily Comparator (2): placebo tablets and placebo (normal saline) injections All drugs taken immediately before a high‐fat meal Duration of intervention: participants receive 1 dose (1 day for each intervention). Minimum 10‐day washout between interventions |
| Outcomes |
Primary outcome: free fatty acids (6 h after ingestion of meal) Secondary outcome: triglycerides (6 h after ingestion of meal) Other outcome: forearm blood flow (6 h after meal) |
| Starting date |
Trial start date: March 2014 Trial completion date: March 2018 |
| Contact information | Responsible party/principal investigator: Absalaon D Gutierrez, MD, University of Texas Health Science Center at Houston, Dept of Medicine |
| Study identifier | NCT number: NCT02104739 |
| Official title | Comparative Effects of Antidiabetic Medications on Postprandial Hyperlipidemia, Free Fatty Acid Signaling, and Endothelial Dysfunction in Individuals with Prediabetes |
| Stated purpose of study | Quote: "This project addresses cardiovascular disease risk in patients with prediabetes." |
| Notes | Duration of intervention is less than required for inclusion in this review, but the trial will be listed in 'Supplementary table' when completed. Investigators planning an extension period |
NCT02140983.
| Trial name or title | Acronym: LGT |
| Methods |
Type of trial: efficacy trial Allocation: randomised Intervention model: parallel assignment Masking: double blind (participants, carer, outcome assessors) Primary purpose: not specified |
| Participants |
Condition: IFG 100‐125 mg/dL or impaired 2‐h glucose concentration 140‐199 mg/dL and < 200 mg/dL on OGTT 75‐g glucose, or both Enrolment: 80 Inclusion criteria: men and women age 50‐70 years, BMI 27‐37 kg/m² and ≥ 12 years of education; medically stable (i.e. no uncontrolled or poorly controlled medical illnesses); cognitively intact as defined by MMSE score > 27 and will have adequate visual and auditory acuity to allow for cognitive testing; metabolic function determined as IFG 100‐125 mg/dL or impaired 2‐h glucose concentration 140‐199 mg/dL and < 200 mg/dL (or both) on 75‐g oral glucose challenge; half of participants will have a family history of dementia Exclusion criteria: diagnosis of possible or probable Alzheimer's dementia, mild cognitive impairment or any other dementia; evidence of cognitive decline by MMSE score < 27 or self‐reported significant decline in memory within the past year (per the Memory Function Questionnaire); history of T1DM or T2DM, or FPG > 126 mg/dL; history of significant cardiovascular disease or myocardial infarction; unstable cerebrovascular or pulmonary disease, gallstones, pancreatitis or cancer, multiple endocrine neoplasia untreated hypothyroidism, unstable or untreated hypertension, anaemia as determined by hematocrit < 30%; abnormal renal clearance as determined by serum creatinine 1.5 mg/dL, hepatic dysfunction as determined by alanine aminotransferase > 2 times the upper limit of normal; presence of medications known to affect insulin action or insulin secretion; premature birth (which may affect magnetic resonance imaging findings), history of neurological disorder (ischaemic attacks, carotid bruits or lacunes upon magnetic resonance imaging scan), or evidence of neurological or other physical illness that could produce cognitive deterioration; use of any drug that may significantly affect the OGTT results or cognitive testing results; drug or alcohol abuse or dependence within the past 6 months; positive urine toxicology screen for illicit substances at eligibility screening; history of mental illness, with the exception of past mood disorder, or evidence of acute depression as determined by a 17‐item Hamilton Depression Rating Scale score ≥ 8; participants with history of mood disorder must be in remission for at least 6 months prior to study entry |
| Interventions |
Intervention: liraglutide up to 1.8 mg/day, subcutaneous Comparator: placebo, subcutaneous Duration of intervention: 90 days |
| Outcomes |
Primary outcomes: cognitive outcomes (change from baseline to 3 months); the following battery of tests will take approximately 90 minutes (at both baseline and 12‐week follow‐up) to complete and will be administered during the afternoon to avoid diurnal effects: Auditory Consonant Trigrams; Benton Visual Retention Test 5th Edition, Boston Naming Test, Buschke‐Fuld Selective Reminding Test, DKEFS, Color‐Word subtest, DKEFS Tower Test, DKEFS Trail Making Test, DKEFS Verbal Fluency subtest, Purdue Pegboard, Rey‐Osterrieth Complex Figure Test, Taylor Complex Figure Task, Wechsler Abbreviated Scale of Intelligence, and the Wechsler Adult Intelligence Scale‐3rd Edition; OGTT (change from baseline to 3 months) Secondary outcomes: none reported Other outcomes: none reported |
| Starting date |
Trial start date: August 2013 Trial completion date: according to ClinicalTrials.gov in February 2016; however, investigator has replied that trial will be completed at end of 2016 as they had to wait for drug supply (internal communication) |
| Contact information | Responsible party/principal investigator: Natalie Rasgon, Professor, Stanford University |
| Study identifier | NCT number: NCT02140983 |
| Official title | Effects of Liraglutide on Hippocampal Structure and Function in Aging Adults with Prediabetes |
| Stated purpose of study | Quote: "The purpose of this study is to evaluate the effects of liraglutide on the memory and attention of people with insulin resistance." |
| Notes | It was clarified through correspondence that the trial is still ongoing |
NCT02488057.
| Trial name or title | Improving Beta Cell Function in Mexican American Women with Prediabetes |
| Methods |
Type of trial: efficacy trial Allocation: randomised Intervention model: parallel assignment Masking: open label Primary purpose: prevention |
| Participants |
Condition: IGT Enrolment: 450 Inclusion criteria: Mexican‐American women aged 18‐40 years, BMI 31‐42 kg/m², willingness to complete protocol Exclusion criteria: pregnant, ≥ 30 minutes of moderate‐to‐vigorous activity > 3 times per week, cardiovascular disease, physical limitations that might be aggravated by moderate physical activity, planning to move in next 12‐24 months, diabetic |
| Interventions |
Intervention: weight loss + liraglutide 0.6 mg injection daily for 1 week, increased to 1.2 mg for 1 week and then 3.0 mg for the next 10 weeks Comparator: diet‐induced weight loss Duration of intervention: 12 weeks |
| Outcomes |
Primary outcomes : Secondary outcomes: waist circumference; fasting glucose; triglycerides; high‐density lipoprotein cholesterol; blood pressure; highly sensitive C‐reactive protein; presence of genetic polymorphisms Other outcomes: none |
| Starting date |
Trial start date: July 2015 Trial completion date: September 2020 |
| Contact information | Responsible party/principal investigator: Willa A Hsueh, MD, Ohio State University |
| Study identifier | NCT number: NCT02488057 |
| Official title | Improving Beta Cell Function in Mexican American Women with Prediabetes |
| Stated purpose of study | Quote: "This study will examine the benefits of weight loss alone or in combination with a GLP1 receptor agonist, liraglutide, on beta cell function in young adult Mexican American (MA) women with prediabetes." |
| Notes | Through correspondence we were informed that the diagnostic criterion for prediabetes was IGT defined by the American Diabetes Association |
NCT02576288.
| Trial name or title | Acronym: SAVORO |
| Methods |
Type of trial: safety/efficacy trial Allocation: randomised (3:1) Intervention model: parallel assignment Masking: double blind (participants, carer, investigator, outcomes assessor) Primary purpose: treatment |
| Participants |
Condition: FPG 100‐125 mg/dL, HbA1c 5.7‐6.4% or HOMA‐IR ≥ 3.0 and abdominal obesity Enrolment: 40 Inclusion criteria: age 18‐40 years; stable weight (no change > 3% in prior 6 months); waist circumference ≥ 102 cm for men, ≥ 88 cm for women; FPG 100‐125 mg/dL, HbA1c 5.7‐6.4% or HOMA‐IR* ≥ 3.0. Exclusion criteria: regular use of non‐steroidal anti‐inflammatory drug; unwilling to stop non‐steroidal anti‐inflammatory drug; receiving statin or other prescription anti‐inflammatory drugs; diabetes or clinically evident cardiovascular disease; smoking daily or consuming > 200 g alcohol/day |
| Interventions |
Intervention: sitagliptin 100 mg, PO, once daily Comparator: placebo, PO, once daily Duration of intervention: 28 days |
| Outcomes |
Primary outcome: ultrasound quantification of change in brachial artery flow‐mediated dilation and carotid stiffness (elasticity and dispensability) (immediately before and after 28 days of study therapy) Secondary outcome: deep subcutaneous adipose tissue inflammation (immediately before and after 28 days of study therapy) Other outcomes: systemic markers of inflammation/atherogenic mediators and insulin resistance (immediately before and after 28 days of study therapy) |
| Starting date |
Trial start date: January 2016 Trial completion date: July 2017 |
| Contact information | Responsible party/principal investigator: Fred Sattler, MD, University of Southern California |
| Study identifier | NCT number: NCT02576288 |
| Official title | Effects of Sitagliptin on Arterial Vasoreactivity and Proatherogenic Mediators in Obesity |
| Stated purpose of study | Quote: "The investigators will evaluate a novel approach using a dipeptidyl peptidase 4 inhibitor (DPP4i) sitagliptin, which blocks signal transduction for monocyte/macrophage activation." |
| Notes | Duration of intervention is less than required for inclusion in this review, but the trial will be listed in 'Supplementary table' when completed |
NCT02847403.
| Trial name or title | Acronym: DRINN |
| Methods |
Type of trial: efficacy trial Allocation: randomised Intervention model: parallel assignment Masking: open label Primary purpose: treatment |
| Participants |
Condition: dysglycaemia/prediabetes defined as FPG 100‐125 mg/dL or 2‐h PG 140‐199 mg/dL after a OGTT 75‐g glucose or HbA1c 5.7‐6.4% or a combination of these Enrolment: 40 Inclusion criteria: dysglycaemia/prediabetes defined as FPG 100‐125 mg/dL or 2‐h PG 140‐199 mg/dL after a OGTT 75‐g glucose or HbA1c 5.7‐6.4%, or a combination of these; mild cognitive impairment; age 50‐80 years; stable medication for past 3 months; white Exclusion criteria: age < 50 or > 80 years; incapability to give informed consent; T2DM; clinically significant liver or kidney dysfunction; endocrinological diseases other than well‐controlled hypothyroidism, personal or family history of medullary thyroid carcinoma or multiple endocrine neoplasia syndrome, severe gastrointestinal diseases (i.e. gastroparesis, dumping syndromes), current or history of chronic or acute pancreatitis; any contraindication to use of exenatide as per the 'Summary of product characteristics'; known abuse of alcohol or drugs; ferromagnetic prosthesis, pacemaker or other metals incorporated in the body; significant neurological disease other than mild cognitive impairment (i.e. Parkinson's disease, multiple system atrophy, normal pressure hydrocephalus, progressive supranuclear palsy, subarachnoid haemorrhage, brain neoplasms, Huntington's disease, epilepsy or head trauma); BMI ≤ 22 kg/m² in people age ≥ 70 years; magnetic resonance imaging/computer tomography showing unambiguous aetiological evidence of cerebrovascular disease with regard to mild cognitive impairment; severe sensory defects; current presence of clinically significant psychiatric disorder; warfarin treatment, clinically significant systemic condition; history of cancer within the last 5 years; known allergy to exenatide or any of other components |
| Interventions |
Intervention: long‐acting exenatide 2 mg, subcutaneously once‐weekly Comparator: not clear whether placebo is provided or no intervention Duration of intervention: 32 weeks |
| Outcomes |
Primary outcome: improvement of Alzheimer's Disease Assessment Scale ‐ cognitive at 16 and 32 weeks compared to baseline Secondary outcomes: at 16 and 32 weeks compared to baseline: improvement of MMSE test, MMSE quality test, Phonemic verbal fluency test, Semantic verbal fluency test, Geriatric Depression Scale test, Clinical Dementia Rating Scale test, Activities of Daily Living test, Neuropsychiatric Inventory test and Instrumental Activities of Daily Living; changes in structural and functional connectivity of neural networks as assessed by functional magnetic resonance imaging Other outcomes: not stated |
| Starting date |
Trial start date: February 2016 Trial completion date: July 2018 |
| Contact information | Responsible party/principal investigator: Alessandra Dei Cas, MD, Azienda Ospedaliero‐Universitaria di Parma, Italy |
| Study identifier | NCT number: NCT02847403 |
| Official title | Long‐acting Exenatide and Cognitive Decline in Dysglycemic Patients (DRINN) |
| Stated purpose of study | Quote: "The overall objective of the study is to assess the potential effects of the long‐acting GLP‐1 analogue exenatide in preventing/slowing the progression of cognitive dysfunction and related biomarkers in dysglycaemic/prediabetic patients with mild cognitive impairment (MCI)." |
| Notes | ‐ |
NCT02969798.
| Trial name or title | Pre‐diabetes in Subject with Impaired Fasting Glucose (IFG) and Impaired Glucose Tolerance (IGT) |
| Methods |
Type of trial: efficacy trial Allocation: randomised Intervention model: crossover assignment Masking: open label Primary purpose: treatment |
| Participants |
Condition: IGT and IFG NGT participants will serve as controls and will be matched in age, gender, ethnicity and BMI to IGT and IFG participants Enrolment: 700 Inclusion criteria: age 18‐65 years; FPG < 100 mg/dL and 2‐h PG < 140 mg/dL; BMI 24‐40 kg/m²; stable body weight (± 4 pounds) over the preceding 3 months; no evidence of major organ system disease as determined by physical examination, history and screening laboratory data; women of childbearing potential with a negative pregnancy test at screening and treatment visits, using contraception for the duration of participation in the study (i.e. until follow‐up 7‐14 days after last dose) (oral contraceptive, injectable progesterone, subdermal implant, spermicidal foam/gel/film/cream/suppository, diaphragm with spermicide, copper or hormonal‐containing IUD, vasectomised male partner > 6 month predosing); signed and dated informed consent document indicating that participant has been informed of all pertinent aspects of study; willing and able to comply with scheduled visits, treatment, laboratory tests and study procedures Exclusion criteria: recent (i.e. within 3 months prior to screening) evidence or medical history of unstable concurrent disease such as: documented evidence or history of clinically significant haematological, endocrine, pulmonary, gastrointestinal, cardiovascular, hepatic, psychiatric, immunological or clinically significant neurological disease; family history of diabetes in a first‐degree relative; BMI < 24 or > 40 kg/m²; unstable body weight (change ± 4 pounds over the preceding 3 months); participating in an excessively heavy exercise programme; feeding/sleeping schedule different from a daytime feeding/night‐time sleeping schedule; receiving medications known to alter glucose metabolism (with the exception of metformin or pioglitazone, or both) or which effect brain neurosynaptic function; evidence of major organ system disease as determined by physical examination, history and screening laboratory data; pregnant or unwilling to use contraception during study; blood donation of approximately 1 pint (500 mL) within 8 weeks prior to screening; other severe acute or chronic medical or psychiatric condition or laboratory abnormality that may increase risk associated with study participation or investigational product administration or may interfere with the interpretation of study results and, in judgement of investigator, would make participant inappropriate for entry into study; people haematuria; evidence or prior history of heart failure; family history of pancreatic, bladder and breast cancer; history of pancreatitis; estimated GFR < 60 ± 5 mL/minute/1.73 m²; elevated serum creatinine (> 1.5 mg/dL for men/1.4 mg/dL for women); history of orthostatic hypotension (> 15/10 mmHg); liver enzymes > 3‐fold above upper normal limit; history of hypersensitivity to pioglitazone, dapagliflozin or saxagliptin. |
| Interventions |
Intervention: saxagliptin 5 mg/day Comparator (1): dapagliflozin 100 mg/day Comparator (2): pioglitazone 30 mg/day Comparator (3): metformin 200 mg/day The trial will randomise participants exclusively with IGT to 1 treatment group; participants exclusively with IFG to 1 treatment group and participants with IGT plus IFG to 1 treatment group Duration of intervention: 24 months |
| Outcomes |
Primary outcomes: β‐cell function, insulin sensitivity and glucose tolerance status in people with isolated IGT; β‐cell function, insulin sensitivity and glucose tolerance status in people with isolated IFG; β‐cell function, insulin sensitivity and glucose tolerance status in people with IGT plus IFG Secondary outcomes: not stated Other outcomes: not stated |
| Starting date |
Trial start date: January 2014 Trial completion date: January 2017 |
| Contact information | Responsible party/principal investigator: Ralph A DeFronzo, The University of Texas Health Science Center at San Antonio |
| Study identifier | NCT number: NCT02969798 |
| Official title | Preservation of Beta Cell Function in Pre‐diabetes in Subject with Impaired Fasting Glucose (IFG) and Impaired Glucose Tolerance (IGT). |
| Stated purpose of study | Quote: "Impaired glucose tolerance (IGT) and impaired fasting glucose (IFG) have distinct pathophysiologic etiologies. Therefore, therapeutic interventions designed to correct the specific underlying pathogenic abnormalities in IGT and IFG will be required to optimally prevent the progressive beta cell failure and development of overt type 2 diabetes." |
| Notes | There is a control arm with participants with NGT ‐ these will not be included in updates of our review |
NCT03004612.
| Trial name or title | Acronym: PRELLIM |
| Methods |
Type of trial: efficacy trial Allocation: randomised Intervention model: parallel assignment Masking: double blind Primary purpose: prevention |
| Participants |
Condition: IGT and IFG Enrolment: 75 Inclusion criteria: coexistence of IFG (fasting glucose 100‐125 mg/dL) and IGT (glucose 140‐199 mg/dL at 2‐h OGTT Exclusion criteria: T2DM; actual treatment or during the last 3 months with metformin, pioglitazone or another antidiabetic drug, including insulin; serum creatinine > 1.6 mg/dL; hypertriglyceridaemia very high (> 500 mg/dL); pregnant women; altered arterial hypertension (systolic > 180 mmHg or diastolic > 105 mmHg); excessive alcohol intake; medications or medical conditions that affect glucose homeostasis |
| Interventions |
Intervention: linagliptin 2.5 mg/day + metformin 850 mg/day every 12 h Comparator: metformin 850 mg every 12 h Duration of intervention: 12 months |
| Outcomes |
Primary outcome: change from basal fasting and post 2‐h OGTT glucose levels at 6 and 12 months Secondary outcomes: change from basal pancreatic β‐cell function at 12 months; change from basal insulin sensitivity at 6 and 12 months; change from basal weight at 6 and 12 months; change from basal lipid profile at 6 and 12 months Other outcomes: not stated |
| Starting date |
Trial start date: December 2015 Trial completion date: August 2018 |
| Contact information | Responsible party/principal investigator: Rodolfo Guardado‐Mendoza, Universidad de Guanajuato, Mexico |
| Study identifier | NCT number: NCT03004612 |
| Official title | Effect of Linagliptin + Metformin vs Metformin Alone in Patients with Prediabetes (PRELLIM) |
| Stated purpose of study | Quote: "The goal of this clinical trial is to evaluate the effect of linagliptin + metformin vs metformin alone on physiopathological parameters, such as glucose metabolism, insulin resistance, insulin secretion and pancreatic beta cell function in patients with impaired fasting glucose plus impaired glucose tolerance, during 12 months." |
| Notes | The study is currently recruiting participants |
UMIN000008620.
| Trial name or title | Acronym: VOGUE‐KOBE |
| Methods |
Type of trial: efficacy trial Allocation: randomised Intervention model: parallel assignment Masking: open label Primary purpose: efficacy |
| Participants |
Condition: people with coronary artery disease and IGT Enrolment: 50 Inclusion criteria: undergoing percutaneous coronary intervention, untreated IGT and 2‐h plasma/serum glucose level: 140‐199 mg/dL in 75‐g OGTT, LDL cholesterol < 100 mg/dL in people not taking statins; LDL‐cholesterol < 120 mg/dL in people taking statins; age 20‐80 years; written consent for participation in study Exclusion criteria: severe T1DM or T2DM liver dysfunction; severe renal dysfunction; severe heart failure (NYHA Stage III or more severe); malignancies or other diseases with poor prognosis; pregnant, lactating and possibly pregnant women and women planning to become pregnant; history of hypersensitivity to investigational drugs; judged as ineligible by clinical investigators |
| Interventions |
Intervention: vildagliptin 50 mg/day Comparator: diet + exercise Duration of intervention: 6 months |
| Outcomes |
Primary outcomes: change in coronary plaque character analysed by coronary angiography, intravascular ultrasound and optical coherence tomography; daily glucose profile analysed by 24‐h continuous glucose monitoring system before and after 6 months Secondary outcomes: changes in the intima media thickness value measured by carotid arterial echography; changes in HbA1c and OGTT 75‐g glucose (glucose and insulin levels after glucose load) Other outcomes: not stated |
| Starting date |
Trial start date: July 2012 Trial completion date: not stated |
| Contact information | Responsible party/principal investigator: Toshiro Shinke, Kobe University Graduate School of Medicine, Japan |
| Study identifier | UMIN number: 000008620 |
| Official title | The Impact of Vildagliptin on Daily Glucose Profile and Coronary PlaqUE Character in Impaired Glucose Tolerance Patients with Coronary Artery Disease: VOGUE‐KOBE |
| Stated purpose of study | Quote: "Comparison of vildagliptin versus conventional treatment without DDP‐4 inhibitor on daily glucose profile analyzed by 24‐hour continuous glucose monitoring system and coronary plaque character using coronary imaging devices in IGT patients with coronary artery disease." |
| Notes | Investigators asked for completion date |
ACE: angiotensin‐converting enzyme; BMI: body mass index; DPP‐4: dipeptidyl‐peptidase‐4; DKEFS: Delis Kaplan Executive Function System; ETDRS: Early Treatment Diabetic Retinopathy Study Scale; FPG: fasting plasma glucose; GDM: gestational diabetes mellitus; GFR: glomerular filtration rate; GLP‐1; glucagon‐like peptide‐1; h: hour; HbA1c: glycosylated haemoglobin A1c; HOMA‐IR: homeostatic model assessment insulin resistance; IFG: impaired fasting glucose; IGT: impaired glucose tolerance; IUD: intrauterine device; LDL: low‐density lipoprotein; MMSE: Mini Mental State Examination; NGT: normal glucose tolerance; NYHA: New York Heart Association; OGTT: oral glucose tolerance test; PG: plasma glucose; PO: per os (orally); SD: standard deviation; T1DM: type 1 diabetes mellitus; T2DM: type 2 diabetes mellitus; TIA: transient ischaemic attack.
Differences between protocol and review
We also investigated regression from intermediate hyperglycaemia back to normoglycaemia because this is part of the overall transition phases between intermediate hyperglycaemia and development of type 2 diabetes mellitus.
Contributions of authors
All review authors read and approved the final review draft.
BH: protocol and review draft, search strategy development, acquisition of trial reports, trial selection, data extraction of all trials, data analysis, contact to trial authors, data interpretation, GRADE assessment and writing of drafts.
DS: trial selection, data extraction, data interpretation, GRADE assessment and review of drafts.
MIM: search strategy development and review of drafts.
BR: protocol and review draft, search strategy development, data interpretation and review of drafts.
Sources of support
Internal sources
No sources of support supplied
External sources
-
World Health Organization, Other.
This review is part of a series of reviews on interventions for prevention or delay of type 2 diabetes mellitus and its associated complications in pople at increased risk for the development of type 2 diabetes mellitus which is funded by the WHO. (Hemmingsen 2016a; Hemmingsen 2016c)
Declarations of interest
BH: this review is part of a series of reviews on interventions for the prevention or delay of type 2 diabetes mellitus and its associated complications in people at increased risk for the development of type 2 diabetes mellitus, which is funded by the World Health Organization (Hemmingsen 2016a; Hemmingsen 2016b; Hemmingsen 2016c).
DS: none known.
MIM: none known.
BR: none known.
New
References
References to studies included in this review
Ariel 2014 {published data only}
- Ariel D, Kim SH, Abbasi F, Lamendola CA, Liu A, Reaven GM. Effect of liraglutide administration and a calorie‐restricted diet on lipoprotein profile in overweight/obese persons with prediabetes. Nutrition, Metabolism, and Cardiovascular diseases 2014;24(12):1317‐22. [PUBMED: 25280957] [DOI] [PubMed] [Google Scholar]
- Kim S, Abbasi F, Lamendola C, Liu A, Ariel D, Schaaf P, et al. Liraglutide for overweight, older individuals with prediabetes. Diabetes 2013;62(0):A260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Abbasi F, Lamendola C, Liu A, Ariel D, Schaaf P, et al. Benefits of liraglutide treatment in overweight and obese older individuals with prediabetes. Diabetes Care 2013;36(10):3276‐82. [PUBMED: 23835684] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Liu A, Ariel D, Abbasi F, Lamendola C, Grove K, et al. Pancreatic beta cell function following liraglutide‐augmented weight loss in individuals with prediabetes: analysis of a randomised, placebo‐controlled study. Diabetologia 2014;57(3):455‐62. [PUBMED: 24326527] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01784965. Liraglutide and a calorie restricted diet augments weight loss and decreases risk of type 2 diabetes and CVD. clinicaltrials.gov/ct2/show/NCT01784965 Date first received: 4 February 2013.
Kelly 2012 {published data only}
- Kelly AS, Bergenstal RM, Gonzalez‐Campoy JM, Katz H, Bank AJ. Effects of exenatide vs. metformin on endothelial function in obese patients with pre‐diabetes. Diabetes 2012;61(0):A112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly AS, Bergenstal RM, Gonzalez‐Campoy JM, Katz H, Bank AJ. Effects of exenatide vs. metformin on endothelial function in obese patients with pre‐diabetes: a randomized trial. Cardiovascular Diabetology 2012;11:64. [PUBMED: 22681705] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT00546728. The vascular effects of exenatide versus metformin in patients with pre‐diabetes. clinicaltrials.gov/ct2/show/NCT00546728 Date first received: 17 October 2007.
Martinez‐Abundis 2015 {published and unpublished data}
McLaughlin 2011 {published data only}
- McLaughlin T. Exenatide/diet vs diet alone for treatment of prediabetes. Diabetes 2011;60(Suppl 1):34‐LB. [Google Scholar]
- NCT02084654. Exenatide and weight loss for diabetes prevention. clinicaltrials.gov/ct2/show/NCT02084654 Date first received: 2 December 2013.
- Tam JK, McLaughlin TL, Lamendola CA, Lerner KD, Peck M, Liu LF. Effect of exenatide on weight and metabolic profile in obese insulin‐resistant individuals and results after drug discontinuation. Endocrine Reviews 2013;34(3):SUN‐854. [Google Scholar]
Rosenstock 2008 {published data only}
- CLAF237A2357. www.clinicaltrialsregister.eu/ctr‐search/rest/download/result/attachment/2005‐002778‐30/1/17121 (accessed 9 May 2017).
- EudraCT 2005‐002778‐30. A multicenter, randomized, double‐blind, parallel‐group, placebo‐controlled study to assess the efficacy and safety of 12 week treatment with vildagliptin (LAF237) 50 mg QD in subjects with impaired glucose tolerance (IGT). www.clinicaltrialsregister.eu/ctr‐search/trial/2005‐002778‐30/DE Date first registered: 11 August 2005.
- NCT00237250. Efficacy and safety of vildagliptin in subjects with impaired glucose tolerance. www.clinicaltrials.gov/ct2/show/NCT00237250 Date first received: 9 October 2005.
- Rosenstock J, Foley JE, Rendell M, Landin‐Olsson M, Holst JJ, Deacon CF, et al. Effects of the dipeptidyl peptidase‐IV inhibitor vildagliptin on incretin hormones, islet function, and postprandial glycemia in subjects with impaired glucose tolerance. Diabetes Care 2008;31(1):30‐5. [PUBMED: 17947341] [DOI] [PubMed] [Google Scholar]
Rosenstock 2010 {published data only}
- NCT00500370A. Pilot study of effects of exenatide on body weight in non‐diabetic, obese patients. clinicaltrials.gov/ct2/show/NCT00500370 Date first received: 10 July 2007.
- Rosenstock J, Klaff LJ, Schwartz S, Northrup J, Holcombe JH, Wilhelm K, et al. Effects of exenatide and lifestyle modification on body weight and glucose tolerance in obese subjects with and without pre‐diabetes. Diabetes Care 2010;33(6):1173‐5. [PUBMED: 20332357] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautmann M, Northrup J, Cao D, Glass L, Holcombe J. Exenatide was associated with body weight reduction and normalisation of impaired glucose tolerance in non‐diabetic, obese patients. Journal of Diabetes 2009;1(0):A196‐7. [Google Scholar]
SCALE {published data only}
- Astrup A, Fujioka K, Violante Ortiz R, Jensen CB, Lillerre SK, O'Neil P. Efficacy and safety of liraglutide 3.0 Mg in adult overweight and obese weight loss responders without diabetes: results of the 56‐week randomised, controlled, scale obesity and prediabetes trial. Obesity Facts 2015;8(0):42. [Google Scholar]
- Bjorner JB, Brett JH, Meincke HH, Kolotkin RL. The effect of liraglutide 3.0 mg for weight management on HRQoL, as measured by SF‐36: 3‐year data. Diabetologia 2016; Vol. 59:S330.
- Bjorner JB, Fujioka K, Lay A, Brett J, Kolotkin R. Liraglutide 3.0 mg improves health utilities in weight management compared with placebo. Diabetes 2015;64(0):A565‐6. [Google Scholar]
- Bluher M, Hermansen K, Greenway F, Fujioka K, Donsmark M, Jensen CB, et al. Early weight loss with liraglutide 3.0 mg is good predictor of clinically meaningful weight loss after 56 weeks. Diabetologia 2015;58(1):S310. [Google Scholar]
- EudraCT 2008‐001049‐24. Effect of liraglutide on body weight in non‐diabetic obese subjects or overweight subjects with comorbidities. www.clinicaltrialsregister.eu/ctr‐search/trial/2008‐001049‐24/IE Date first registered: 15 September 2008.
- Fujioka K, Wilding JPH, Astrup A, Greenway F, Halpern A, Krempf M, et al. Efficacy and safety of liraglutide 3.0 mg for weight management in overweight/obese adults: the SCALE obesity and prediabetes trial. Diabetologia 2014;57(1):S368‐9. [Google Scholar]
- Gaal LV, Pi‐Sunyer X, Hemmingsson JU, Claudius B, Svendsen CB, Bays H. Liraglutide 3.0 mg: weight‐loss dependent and independent effects. Obesity Facts 2015;8(0):41‐2. [Google Scholar]
- Kolotkin RL, Fujioka K, Wolden ML, Brett JH, Bjorner JB. Improvements in health‐related quality of life with liraglutide 3.0 mg compared with placebo in weight management. Clinical Obesity 2016;6(4):233‐42. [PUBMED: 27198973] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolotkin RL, Meincke HJ, Manning LS, Bjorner JB. Impact of weight loss on patient‐reported outcomes in the SCALE obesity and prediabetes trial of liraglutide 3.0 mg as adjunct to a diet and exercise (D&E) programme. Value in Health 2015;18(7):A674‐5. [PUBMED: 26533777] [Google Scholar]
- Krempf M, Astrup A, Fujioka K, Greenway F, Halpern A, Lau DC, et al. Additional analyses of the weight‐lowering efficacy of liraglutide 3.0 Mg in overweight and obese adults: the scale obesity and prediabetes randomised trial. Obesity Facts 2015;8(0):210. [Google Scholar]
- Krempf M, Astrup A, Roux C, Fujioka K, Greenway F, Halpern A, et al. Liraglutide 3.0 mg reduces body weight and improves cardiometabolic risk factors in overweight/obese adults: the SCALE obesity and prediabetes randomised trial. Diabetologia 2014;57(1):S368. [Google Scholar]
- Lau DC, Davies M, Pi‐Sunyer FX, Wilding J, Manning LS, Jensen CB, et al. Hypoglycemia reported with liraglutide 3.0 mg in overweight and obese adults with and without prediabetes: results of the randomized, double‐blind, placebo‐controlled 56‐week scale obesity and prediabetes trial. Endocrine Society's 97th Annual Meeting and Expo; 2015 Mar 5‐8; San Diego (CA) 2015:OR07‐2. [Google Scholar]
- Lau DC, Fujioka K, Astrup A, Greenway F, Halpern A, Krempf M, et al. Liraglutide 3.0 Mg reduces body weight and improves HRQoL in overweight or obese adults without diabetes: SCALE obesity and prediabetes randomized, double‐blind, placebo controlled, 56‐week trial. Obesity Facts 2015;8(0):185. [Google Scholar]
- Lau DC, Greenway F, Fujioka K, Astrup A, Halpern A, Krempf M, et al. Additional analyses of the weight‐lowering efficacy of liraglutide 3.0 mg in adults with overweight and obesity: the SCALE obesity and prediabetes randomized trial. Canadian Journal of Diabetes 2015;39(0):S48. [Google Scholar]
- Lau DC, Krempf M, Astrup A, Roux CW, Fujioka K, Greenway F, et al. Liraglutide 3.0 mg reduces body weight and improves cardiometabolic risk factors in adults with overweight/obesity: the SCALE obesity and prediabetes randomised trial. Canadian Journal of Diabetes 2015;39(0):S48‐9. [Google Scholar]
- Lau DCW, Wilding JPH, Astrup A, Fujioka K, Greenway F, Halpern A, et al. Liraglutide 3.0 mg improves insulin secretion and action in overweight/ obese adults without type 2 diabetes: the SCALE obesity and prediabetes trial. Diabetologia 2014;57(1):S358. [Google Scholar]
- Roux C, Lau DCW, Astrup A, Fujioka K, Greenway F, Halpern A, et al. Safety and tolerability of liraglutide 3.0 mg in overweight and obese adults: the SCALE obesity and prediabetes randomised trial. Diabetologia 2014;57(1):S338. [Google Scholar]
- Roux C, Lau DCW, Fujioka K, Caterson ID, Cancino AP, Jensen CB, et al. The impact of gastrointestinal adverse events on weight loss with liraglutide 3.0 mg as adjunct to a diet and exercise programme. Diabetologia 2015;58(1):S311. [Google Scholar]
- Roux CW, Astrup A, Fujioka K, Greenway F, Lau DC, Gaal L, et al. 3 years of liraglutide versus placebo for type 2 diabetes risk reduction and weight management in individuals with prediabetes: a randomised, double‐blind trial. Lancet 2017;389(10077):1399‐409. [PUBMED: 28237263] [DOI] [PubMed] [Google Scholar]
- Liutkus JF, Fujioka K, Astrup A, Greenway F, Halpern A, Krempf M, et al. Liraglutide 3.0 mg reduces body weight and improves health‐related quality of life (HRQoL) in overweight or obese adults without diabetes: the SCALE obesity and prediabetes randomized, double‐blind, placebo‐controlled, 56‐week trial. Canadian Journal of Diabetes 2015;39(0):S36. [Google Scholar]
- Madsbad S, Lieberman G, Skjoth TV, Lilleore SK, Fronzo RA. Comparable efficacy and safety of liraglutide 3.0 mg for weight management across baseline BMI subgroups: results from the SCALE obesity and prediabetes trial. Diabetologia 2016; Vol. 59, issue 0:540‐1.
- NCT01272219. Effect of liraglutide on body weight in non‐diabetic obese subjects or overweight subjects with co‐morbidities: SCALE™ ‐ obesity and pre‐diabetes. clinicaltrials.gov/ct2/show/NCT01272219 Date first received: 6 January 2011.
- NN8022‐1839. Effect of liraglutide on body weight in non‐diabetic obese subjects or overweight subjects with co‐morbidities: SCALE™ ‐ obesity and pre‐diabetes. www.novonordisk‐trials.com/Website/pdf/registry/nn80221839.pdf (assessed 22 September 2016).
- Pi‐Sunyer X, Astrup A, Fujioka K, Greenway F, Halpern A, Krempf M, et al. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. New England Journal of Medicine 2015;373(1):11‐22. [DOI: 10.1056/NEJMoa1411892] [DOI] [PubMed] [Google Scholar]
- Pi‐Sunyer X, Fujioka K, Roux C, Astrup A, Greenway F, Halpern A, et al. Liraglutide 3.0 mg reduces the prevalence of prediabetes and delays onset of type 2 diabetes in overweight/obese adults: the SCALE obesity and prediabetes trial. Diabetologia 2014;57(1):S37. [Google Scholar]
- Proietto J, Roux CW, Pi‐Sunyer X, Astrup A, Fujioka K, Greenway F, et al. Efficacy and safety of liraglutide 3.0 mg for weight management in overweight and obese adults: the SCALE obesity and prediabetes, a randomised, double‐blind and placebo‐controlled trial. Obesity Research and Clinical Practice 2014;8(0):117. [Google Scholar]
- Wilding J, Astrup A, Fujioka K, Greenway F, Krempf M, Lau D, et al. In overweight and obese adults, liraglutide 3.0mg reduces the prevalence of prediabetes and delays the onset of type 2 diabetes: SCALE obesity and pre‐diabetes trial. Diabetic Medicine 2015;32(0):71. [Google Scholar]
- Wilding J, Lau D, Astrup A, Fujioka K, Greenway F, Krempf M, et al. Safety and tolerability of liraglutide 3.0mg following 56 weeks of randomised treatment in overweight and obese adults: SCALE obesity and pre‐diabetes trial. Diabetic Medicine 2015;32:70. [Google Scholar]
- Wilding JP, Overgaard RV, Jacobsen LV, Jensen CB, Roux CW. Exposure‐response analyses of liraglutide 3.0 mg for weight management. Diabetes, Obesity & Metabolism 2016;18(5):491‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilding JPH, Bluher M, Hermansen K, Greenway F, Fujioka K, Claudius B, et al. Early weight loss responders to liraglutide 3.0 mg in the scale obesity and prediabetes trial achieve greater reversal of prediabetes than placebo. Diabetes 2015;64:A568. [Google Scholar]
- Wittert G, Roux CW, Astrup A, Fujioka K, Greenway F, Halpern A, et al. Liraglutide 3.0mg improves body weight and cardiometabolic risk factors in overweight or obese adults without diabetes: the SCALE obesity and prediabetes randomised, double‐blind, placebo‐controlled 56‐week trial. Obesity Research and Clinical Practice 2014;8:118. [Google Scholar]
References to studies excluded from this review
Acosta 2015 {published data only}
- Acosta A, Camilleri M, Burton D, O'Neill J, Eckert D, Carlson P, et al. Exenatide in obesity with accelerated gastric emptying: a randomized, pharmacodynamics study. Physiological Reports 2015;3(11):e12610. [DOI: 10.14814/phy2.12610] [DOI] [PMC free article] [PubMed] [Google Scholar]
ACTRN12615001029583 {published data only}
- ACTRN12615001029583. Does exenatide improve post prandial glycaemic control in young people with cystic fibrosis related diabetes or impaired glucose tolerance?. www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=369121 Date first registered: 1 October 2015.
Almeda‐Valdes 2012 {published data only}
- Almeda‐Valdes P, Cuevas‐Ramos D, Brito‐Cordova G, Gomez‐Perez FJ. Treatment with sitagliptin phosphate in patients with symptomatic reactive hypoglycemia. A randomized, placebo‐controlled, double‐blinded, clinical trial. 94th Annual Meeting and Expo; 2012 23‐26 Jun; Houston (TX) 2012:SUN‐209.
- NCT00847080. Treatment with sitagliptin for reactive hypoglycemia secondary to dysinsulinism. clinicaltrials.gov/ct2/show/NCT00847080 Date first received: 18 February 2009.
Aoki 2014 {published data only}
- Aoki K, Kamiyama H, Masuda K, Kamiko K, Noguchi Y, Tajima K, et al. Effects of miglitol, vildagliptin, or their combination on serum insulin and peptide YY levels and plasma glucose, cholecystokinin, ghrelin, and obestatin levels. Endocrine Journal 2014;61(3):249‐56. [PUBMED: 24389993] [DOI] [PubMed] [Google Scholar]
- Aoki K, Masuda K, Miyazaki T, Togashi Y, Terauchi Y. Effects of miglitol, sitagliptin or their combination on plasma glucose, insulin and incretin levels in non‐diabetic men. Endocrine Journal 2010;57(8):667‐72. [PUBMED: 20519806] [DOI] [PubMed] [Google Scholar]
- UMIN000010481. Effect of miglitol and vildagliptin on gastrointestinal hormone. upload.umin.ac.jp/cgi‐open‐bin/ctr/ctr.cgi?function=brows&action=brows&type=summary&recptno=R000012252&language=E Date first registered: 13 April 2013.
Armato 2012 {published data only}
- Armato J, DeFronzo RA, Abdul‐Ghani M, Ruby R. Successful treatment of prediabetes in clinical practice: targeting insulin resistance and beta‐cell dysfunction. Endocrine Practice 2012;18(3):342‐50. [PUBMED: 22068250] [DOI] [PubMed] [Google Scholar]
BEGAMI 2013 {published and unpublished data}
- EUCTR2007‐003963‐31‐SE. BEGAMI. www.clinicaltrialsregister.eu/ctr‐search/trial/2007‐003963‐31/SE Date first registered: 29 August 2007.
- Hage C, Brismar K, Efendic S, Lundman P, Ryden L, Mellbin L. Sitagliptin improves beta‐cell function in patients with acute coronary syndromes and newly diagnosed glucose abnormalities ‐ the BEGAMI study. Journal of Internal Medicine 2013;273(4):410‐21. [PUBMED: 23331339] [DOI] [PubMed] [Google Scholar]
- Hage C, Brismar K, Lundman P, Mellbin L, Ryden L. The DPP‐4 inhibitor sitagliptin and endothelial function in patients with acute coronary syndromes and newly detected glucose perturbations. European Heart Journal 2013;34(0):793. [DOI] [PubMed] [Google Scholar]
- Hage C, Brismar K, Lundman P, Norhammar A, Ryden L, Mellbin L. The DPP‐4 inhibitor sitagliptin and endothelial function in patients with acute coronary syndromes and newly detected glucose perturbations: a report from the BEGAMI study. Diabetes & Vascular Disease Research 2014;11(4):290‐3. [PUBMED: 24845072] [DOI] [PubMed] [Google Scholar]
- NCT00627744. Beta‐cell function in glucose abnormalities and acute myocardial infarction (BEGAMI). clinicaltrials.gov/ct2/show/NCT00627744 Date first received: 22 February 2008.
Best 2015 {published data only}
- Best C, Struthers H, Laciny E, Royal M, Reeds DN, Yarasheski KE. Sitagliptin reduces inflammation and chronic immune cell activation in HIV+ adults with impaired glucose tolerance. Journal of Clinical Endocrinology and Metabolism 2015;100(7):2621‐9. [PUBMED: 25938633] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bock 2010 {published data only}
- Bock G, Dalla Man C, Micheletto F, Basu R, Giesler PD, Laugen J, et al. The effect of DPP‐4 inhibition with sitagliptin on incretin secretion and on fasting and postprandial glucose turnover in subjects with impaired fasting glucose. Clinical Endocrinology 2010;73(2):189‐96. [DOI: 10.1111/j.1365-2265.2009.03764.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheletto F, Dalla Man C, Vella A, Cobelli C. Estimation of GLP‐1 induced potentiation of insulin secretion from an oral model. Diabetes 2011;60(0):A447. [Google Scholar]
- NCT00364377. Incretins in impaired fasting glucose. clinicaltrials.gov/ct2/show/NCT00364377 Date first received: 14 August 2006.
- Nelson RH, Miles JM, Vella A. Sitagliptin effects on triglyceride and free fatty acid metabolism. Diabetologia 2009;52:S313. [Google Scholar]
Cui 2016 {published data only}
- Cui J, Philo L, Nguyen P, Hofflich H, Hernandez C, Bettencourt R, et al. Sitagliptin vs. placebo for non‐alcoholic fatty liver disease: a randomized controlled trial. Journal of Hepatology 2016;65(2):369‐76. [PUBMED: 27151177] [DOI] [PMC free article] [PubMed] [Google Scholar]
Daniele 2015 {published data only}
- Daniele G, Iozzo P, Molina‐Carrion M, Lancaster J, Ciociaro D, Cersosimo E, et al. Exenatide regulates cerebral glucose metabolism in brain areas associated with glucose homeostasis and reward system. Diabetes 2015;64(10):3406‐12. [PUBMED: 26116695] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniele G, Iozzo P, Molina‐Wilkins M, Lancaster J, Ciociaro D, Triplitt C, et al. Effect of exenatide on postprandial cerebral and liver glucose metabolism: a double‐blind randomised clinical trial. Diabetologia 2014;57(1):S37‐8. [Google Scholar]
- Daniele G, Molina‐Wilkins M, Lancaster J, Ciociaro D, Triplitt C, Cersosimo E, et al. Exenatide affects the distribution of cerebral postprandial glucose uptake: a double‐blind, randomized clinical trial. Diabetes 2014;63:A85. [Google Scholar]
- NCT01588418. Effect of exenatide on brain, adipose tissue, pancreas, and liver function. clinicaltrials.gov/ct2/show/NCT01588418 Date first received: 19 March 2012.
Dushay 2012 {published data only}
- Dushay J, Gao C, Gopalakrishnan GS, Crawley M, Mitten EK, Wilker E, et al. Short‐term exenatide treatment leads to significant weight loss in a subset of obese women without diabetes. Diabetes Care 2012;35(1):4‐11. [DOI: 10.2337/dc11-0931] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT00456885. The effect of exenatide on weight and hunger in obese, healthy women. clinicaltrials.gov/ct2/show/NCT00456885 Date first received: 4 April 2007.
EudraCT 2011‐005980‐26 {published data only}
- EudraCT 2011‐005980‐26. Metformin and sitagliptin in patients with impaired glucose tolerance and a recent TIA or minor ischemic stroke ‐ a multicenter, randomized, open‐label phase II trial. www.clinicaltrialsregister.eu/ctr‐search/trial/2011‐005980‐26/NL Date first registered: 13 February 2012.
- Osei E, Fonville S, Zandbergen AA, Brouwers PJ, Mulder LJ, Lingsma HF, et al. Metformin and sitAgliptin in patients with impAired glucose tolerance and a recent TIA or minor ischemic Stroke (MAAS): study protocol for a randomized controlled trial. Trials 2015;16:332. [PUBMED: 26242578] [DOI] [PMC free article] [PubMed] [Google Scholar]
EudraCT 2013‐001240‐64 {published and unpublished data}
- EudraCT 2013‐001240‐64. Effect of intact GLP‐1 (7‐36) and GLP‐1 metabolite (9‐36) on coronary microvascular function in adults with prediabetes. www.clinicaltrialsregister.eu/ctr‐search/search?query=2013‐001240‐64 (accessed 5 April 2016).
Gonzalez‐Ortiz 2015 {published data only}
- Gonzalez‐Ortiz M, Perez‐Rubio KG, Martinezabundis E. Lixisenatide vs. exenatide on metabolic control, insulin secretion, and insulin sensitivity in impaired glucose tolerance patients. Diabetes 2015; Vol. 64:A297.
Gudipaty 2014 {published data only}
- Effect of exenatide, sitagliptin or glimepiride on functional β‐cell mass. clinicaltrials.gov/ct2/show/NCT00775684 Date first received: 17 October 2008.
- Gudipaty L, Fuller C, Rosenfeld N, Rickels MR. No effect of exenatide or sitagliptin therapy on beta‐cell secretory capacity in early type 2 diabetes. Diabetes 2013; Vol. 62:A288. [DOI] [PMC free article] [PubMed]
- Gudipaty L, Rosenfeld NK, Fuller CS, Gallop R, Schutta MH, Rickels MR. Effect of exenatide, sitagliptin, or glimepiride on beta‐cell secretory capacity in early type 2 diabetes. Diabetes Care 2014;37(9):2451‐8. [PUBMED: 24969577] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ishikawa 2014 {published data only}
- Ishikawa S, Shimano M, Watarai M, Koyasu M, Uchikawa T, Ishii H, et al. Impact of sitagliptin on carotid intima‐media thickness in patients with coronary artery disease and impaired glucose tolerance or mild diabetes mellitus. American Journal of Cardiology 2014;114(3):384‐8. [PUBMED: 24929624] [DOI] [PubMed] [Google Scholar]
Kaku 2015 {published data only}
- Kaku K, Kadowaki T, Terauchi Y, Okamoto T, Sato A, Okuyama K, et al. Sitagliptin improves glycaemic excursion after a meal or after an oral glucose load in Japanese subjects with impaired glucose tolerance. Diabetes, Obesity & Metabolism 2015;17(11):1033‐41. [PUBMED: 26094974] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01405911. Dose response finding study of MK‐0431/ONO‐5435 in Japanese subjects with impaired glucose tolerance (MK‐0431‐105). clinicaltrials.gov/ct2/show/NCT01405911 Date first received: 28 July 2011.
Koska 2015 {published data only}
- Koska J, Sands M, Burciu C, D'Souza KM, Raravikar K, Liu J, et al. Exenatide protects against glucose‐ and lipid‐induced endothelial dysfunction: evidence for direct vasodilation effect of GLP‐1 receptor agonists in humans. Diabetes 2015;64(7):2624‐35. [PUBMED: 25720388] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01181986. The study of exenatide action on vessel function in type 2 diabetes and prediabetes. clinicaltrials.gov/ct2/show/NCT01181986 Date first received: 13 August 2010.
Larsen 2014 {published data only}
- Larsen JR, Vedtofte L, Holst JJ, Oturai P, Kjaer A, Correll CU, et al. Does a GLP‐1 receptor agonist change glucose tolerance in patients treated with antipsychotic medications? Design of a randomised, double‐blinded, placebo‐controlled clinical trial. BMJ Open 2014;4(3):e004227. [PUBMED: 24667381] [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT00101712 {published data only}
- NCT00101712. Efficacy and safety of vildagliptin compared to placebo in patients with type 2 diabetes and mild hyperglycemia. clinicaltrials.gov/ct2/show/NCT00101712 Date first received: 12 January 2005.
NCT00198146 {published data only}
- NCT00198146. Prevention of Diabetes Progression Trial (PDPT). clinicaltrials.gov/ct2/show/NCT00198146 Date first received: 12 September 2005.
NCT00721552 {published data only}
- NCT00721552. Sitagliptin prophylaxis for glucocorticoid‐induced impairment of glucose metabolism in males with the metabolic syndrome (SPHINX). clinicaltrials.gov/ct2/show/NCT00721552 Date first received: 22 July 2008.
NCT00845182 {published data only}
- NCT00845182. Effect of pioglitazone and exenatide on body weight and beta cell function (PIO‐EX). clinicaltrials.gov/ct2/show/study/NCT00845182 Date first received: 17 February 2009.
NCT00845559 {published data only}
- NCT00845559. The effects of exenatide on post‐meal sugar peaks and vascular health in obese/pre‐diabetic young adults. clinicaltrials.gov/ct2/show/NCT00845559 Date first received: 16 February 2009.
NCT00886626 {published data only}
- NCT00886626. GLP‐1 therapy for weight loss and improved glucose tolerance in obese children. clinicaltrials.gov/ct2/show/NCT00886626 Date first received: 22 April 2009.
NCT00961363 {published and unpublished data}
- NCT00961363. Effect of sitagliptin in impaired glucose tolerance. clinicaltrials.gov/ct2/show/NCT00961363 Date first received: 11 August 2009.
NCT01006018 {published data only}
- NCT01006018. DPP‐4 Inhibition and TZD for DM Prevention (DInT DM). clinicaltrials.gov/ct2/show/NCT01006018 Date first received: 27 October 2009.
NCT01018602 {published data only}
- NCT01018602. PINGUIN (Postpartum Intervention in Women with Gestational Diabetes Using Insulin). clinicaltrials.gov/ct2/show/NCT01018602 Date first received: 20 November 2009.
NCT01038648 {published and unpublished data}
- NCT01038648. Sitagliptin in Prevention of Type 2 Diabetes Mellitus (SITAGLIPTIN). clinicaltrials.gov/ct2/show/NCT01038648 Date first received: 22 December 2009.
NCT01054118 {published data only}
- NCT01054118. A study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of single doses of JNJ‐38431055, sitagliptin, and co‐administration of JNJ‐38431055 and sitagliptin. clinicaltrials.gov/ct2/show/NCT01054118 Date first received: 15 January 2010.
NCT01122641 {published data only}
- NCT01122641. The vascular effects of vildagliptin in insulin resistant individuals. clinicaltrials.gov/ct2/show/NCT01122641 Date first received: 12 May 2010.
NCT01346254 {published data only}
- NCT01346254. Glucose control in pre‐diabetic renal transplant patients (GCPD). clinicaltrials.gov/ct2/show/NCT01346254 Date first received: 29 April 2011.
NCT01472640 {published data only}
- NCT01472640. The effect of liraglutide on left ventricular function in chronic heart failure patients with and without type 2 diabetes mellitus. clinicaltrials.gov/ct2/show/NCT01472640 Date first received: 11 November 2011.
NCT01845259 {published data only}
- NCT01845259. Does a GLP‐1 receptor agonist change glucose tolerance in antipsychotic‐treated patients? (GREAT). clinicaltrials.gov/ct2/show/NCT01845259 Date first received: 14 April 2013.
NCT01970462 {published data only}
- NCT01970462. Use of sitagliptin for stress hyperglycemia or mild diabetes following cardiac surgery. clinicaltrials.gov/ct2/show/NCT01970462 Date first received: 22 October 2013.
NCT02016846 {published data only}
- NCT02016846. Liraglutide efficacy on glucocorticoid induced hyperglycemia in patients high risk for diabetes. clinicaltrials.gov/ct2/show/NCT02016846 Date first received: 15 December 2013.
NCT02022007 {published data only}
- NCT02022007. Saxagliptin + metformin compared to saxagliptin or metformin monotherapy in PCOS women Wwith impaired glucose homeostasis (BMS‐AZPCOS). clinicaltrials.gov/ct2/show/NCT02022007 Date first received: 17 December 2013. [NCT02022007]
NCT02284230 {published data only}
- 2014‐001778‐32/DK. The LiRA2 study. www.clinicaltrialsregister.eu/ctr‐search/trial/2014‐001778‐32/DK Date first registered: 17 June 2014.
- NCT02284230. The effect of liraglutide in patients with prediabetes and kidney failure (LiRA2). clinicaltrials.gov/ct2/show/NCT02284230 Date first received: 3 November 2014.
NCT02446834 {published data only}
- NCT02446834. Research of intensive lifestyle Intervention for PCOS patients with IGT. clinicaltrials.gov/ct2/show/NCT02446834 Date first received: 6 May 2015.
Schwartz 2010 {published data only}
- Koska J, Sands M, Burciu C, D'Souza KM, Raravikar K, Liu J, et al. Exenatide protects against glucose‐ and lipid‐induced endothelial dysfunction: evidence for direct vasodilation effect of GLP‐1 receptor agonists in humans. Diabetes 2015;64(7):2624‐35. [PUBMED: 25720388] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koska J, Schwartz EA, Mullin MP, Schwenke DC, Reaven PD. Improvement of postprandial endothelial function after a single dose of exenatide in individuals with impaired glucose tolerance and recent‐onset type 2 diabetes. Diabetes Care 2010;33(5):1028‐30. [PUBMED: 20200309] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullin MP, Koska J, Schwartz EA, Schwenke DC, Reaven PD. Acute administration of exenatide improves endothelial function following a high‐fat meal. Diabetes 2009;58:P‐640. [Google Scholar]
- Schwartz EA, Koska J, Mullin MP, Schwenke DC, Reaven PD. Effects of acute exenatide administration on postprandial lipids and lipoproteins in individuals with impaired glucose metabolism. Diabetes 2009; Vol. 58:375‐OR.
- Schwartz EA, Koska J, Mullin MP, Syoufi I, Schwenke DC, Reaven PD. Exenatide suppresses postprandial elevations in lipids and lipoproteins in individuals with impaired glucose tolerance and recent onset type 2 diabetes mellitus. Atherosclerosis 2010;212(1):217‐22. [PUBMED: 20557887] [DOI] [PubMed] [Google Scholar]
Tsuchiya 2011 {published data only}
- Aramaki M. Alogliptin, but not voglibose, ameliorates dyslipidemia and LDL particle size in patients with impaired glucose tolerance or Type 2 diabetes. Clinical Lipidology 2013;8(5):533‐40. [Google Scholar]
- Tsuchiya M. Alogliptin ameliorates dyslipidemia and LDL‐size in patients with IGT or type 2 diabetes: comparing alogliptin and voglibose. Diabetes 2011;60:A280. [Google Scholar]
- Tsuchiya M. Alogliptin decreases liver fat content in patients with IGT or type Q2 DM: comparing alogliptin and voglibose. Diabetes 2011;60:A595. [Google Scholar]
- Tsuchiya M, Maegawa H. DPP4 inhibition ameliorates intramuscular fat content in patients with IGT or type 2 DM: comparing alogliptin and voglibose. Diabetes 2013;62:A297. [Google Scholar]
UMIN000006197 {published data only}
- UMIN000006197. Therapeutic efficacy of sitagliptin in patients with nonalcoholic fatty liver disease (NAFLD) in impaired glucose tolerance (IGT): a pilot study. upload.umin.ac.jp/cgi‐open‐bin/ctr/ctr.cgi?function=brows&action=brows&recptno=R000007311&type=summary&language=E Date first registered: 1 September 2011.
UMIN000014249 {published data only}
- UMIN000014249. Efficacy and safety of combination therapy with GLP‐1 analog lixisenatide and long‐acting insulin glargine in patients with glucose intolerance or diabetes caused by hyposecretion of endogenous insulin who underwent partial pancreatectomy. upload.umin.ac.jp/cgi‐open‐bin/ctr/ctr.cgi?function=brows&action=brows&type=summary&recptno=R000016598&language=E Date first registered: 12 June 2014.
Utzschneider 2008 {published data only}
- NCT00312130. A study to evaluate the effects of vildagliptin on the insulin response to glucose in subjects with pre‐diabetes. clinicaltrials.gov/ct2/show/NCT00312130 Date first received: 6 April 2006.
- Utzschneider KM, Tong J, Montgomery B, Udayasankar J, Gerchman F, Marcovina SM, et al. The dipeptidyl peptidase‐4 inhibitor vildagliptin improves beta‐cell function and insulin sensitivity in subjects with impaired fasting glucose. Diabetes Care 2008;31(1):108‐13. [PUBMED: 17909087] [DOI] [PubMed] [Google Scholar]
Werzowa 2013 {published data only}
- Werzowa J, Hecking M, Haidinger M, Lechner F, Doller D, Pacini G, et al. Vildagliptin and pioglitazone in patients with impaired glucose tolerance after kidney transplantation: a randomized, placebo‐controlled clinical trial. Transplantation 2013;95(3):456‐62. [PUBMED: 23380864] [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Astrup 2009 {published data only}
- Astrup A, Carraro R, Finer N, Harper A, Kunesova M, Lean ME, et al. Safety, tolerability and sustained weight loss over 2 years with the once‐daily human GLP‐1 analog, liraglutide. International Journal of Obesity (2005) 2012;36(6):843‐54. [PUBMED: 21844879] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astrup A, Rossner S, Gaal L, Rissanen A, Niskanen L, Al Hakim M, et al. Effects of liraglutide in the treatment of obesity: a randomised, double‐blind, placebo‐controlled study. Lancet 2009;374(9701):1606‐16. [PUBMED: 19853906] [DOI] [PubMed] [Google Scholar]
- EudraCT 2006‐004481‐13. Effect of liraglutide on body weight in obese subjects without diabetes. A 20‐week randomised, double‐blind, placebo‐controlled, six armed parallel group, multi‐centre, multinational trial with an open label orlistat comparator arm with an 84 week extension period. www.clinicaltrialsregister.eu/ctr‐search/trial/2006‐004481‐13/DK Date first received: 3 October 2006.
- Finer N, Astrup A, Carraro R, Hartvig H, Kunesova M, Lean ME, et al. Liraglutide once daily reduces the prevalence of prediabetes and metabolic syndrome in obese non‐diabetic adults over two years. Journal of Diabetes 2011;3:30. [Google Scholar]
- Finer N, Hakim MA, Astrup A, Harper A, Lean M, Niskanen L, et al. Liraglutide, a once‐daily human GLP‐1 analog, reverses indices of prediabetes in obese subjects: a randomized placebo‐controlled 20‐week trial. Diabetes 2009;58. [Google Scholar]
- Finer N, Hakim MA, Astrup A, Harper A, Lean M, Niskanen, L, et al. Liraglutide, a once‐daily human GLP‐1 analogue, reduces the prevalence of prediabetes in obese subjects over 20 weeks: a randomized placebo‐controlled trial. Diabetologia 2009;52(0):S11‐2. [Google Scholar]
- Lean ME, Carraro R, Finer N, Hartvig H, Lindegaard ML, Rossner S, et al. Tolerability of nausea and vomiting and associations with weight loss in a randomized trial of liraglutide in obese, non‐diabetic adults. International Journal of Obesity 2014;38(5):689‐97. [PUBMED: 23942319] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT00422058. The effect of liraglutide on body weight in obese subjects without diabetes. clinicaltrials.gov/ct2/show/NCT00422058 Date first received: 12 January 2007.
- NN8022‐1807. The effect of liraglutide on body weight in obese subjects without diabetes. www.novonordisk‐trials.com/Website/pdf/registry/nn80221807.pdf (assessed 3 July 2016).
- Niskanen L, Astrup A, Hakim MA, Carraro R, Finer N, Hartvig H, et al. Liraglutide once daily reduces the prevalence of prediabetes and metabolic syndrome in obese non‐diabetic adults: a 2‐year randomized trial. Obesity 2010;18:S56. [Google Scholar]
- Synopsis NN8022‐1807. Effect of liraglutide on body weight in obese subjects without diabetes. A 20‐week randomised, double‐blind, placebo‐controlled, six armed parallel group, multi‐centre, multinational trial with an open label orlistat comparator arm with an 84 week extension period. novonordisk‐trials.com/website/pdf/registry/bin_20110928‐123116‐617.pdf (assessed 3 July 2016).
NCT01521312 {published data only}
- NCT01521312. Acute and chronic effects of saxagliptin (ACCES). clinicaltrials.gov/ct2/show/NCT01521312 Date first received: 22 November 2011.
NCT01960205 {published and unpublished data}
- NCT01960205. Effect of saxagliptin on pre‐diabetes mellitus and obesity. clinicaltrials.gov/ct2/show/NCT01960205 Date first received: 7 October 2013.
NCT02294084 {published data only}
- EudraCT: 2014‐003532‐39. The effect of the diabetes medication sitagliptin on brown fat and whole‐body metabolism in men with overweight and impaired glucose tolerance (or 'pre‐diabetes'). www.clinicaltrialsregister.eu/ctr‐search/trial/2014‐003532‐39/NL Date first registered: 15 October 2014.
- NCT02294084. Sitagliptin and brown adipose tissue (Sita01). clinicaltrials.gov/ct2/show/NCT02294084 Date first received: 17 November 2014.
Santilli 2015 {published data only}
- Santilli F, Guagnano M, Tartaro A, Simeone PG, Liani R, Tripaldi R, et al. Effects of liraglutide vs lifestyle changes on subcutaneous and visceral fat, liver steatosis, insulin sensitivity and beta cell function after comparable weight loss. Diabetologia 2015;58(1):S375. [Google Scholar]
- Santilli F, Guagnano MT, Tartaro A, Angelucci E, Simeone PG, Laronga G, et al. Liraglutide is more effective than lifestyle changes in modulating subcutaneous and visceral fat distribution, liver steatosis, insulin sensitivity and beta‐cell function after comparable weight loss. European Heart Journal 2015;36(0):473. [Google Scholar]
- Santilli F, Tartaro A, Guagnano MT, Simeone PG, Laronga G, Sborgia C, et al. Effects of liraglutide or lifestyle counseling on subcutaneous and visceral fat distribution, liver fat content, insulin sensitivity, and beta‐cell performance. Diabetes 2015;64:A287. [Google Scholar]
SCALE 2013 {published data only}
- Hollander P, Aronne L, Klein S, Niswender K, Jensen CB, Woo V, et al. Diet‐induced weight loss and subsequent addition of liraglutide 3.0 mg reduces impaired fasting glucose in overweight/obese adults in the SCALEtm maintenance 56‐week randomised trial. Journal of Diabetes 2013;5:124. [Google Scholar]
- Klein S, Aronne L, Hollander P, Niswender K, Bjorn Jensen C, Lindegaard M, et al. Effect of diet‐induced weight loss and subsequent addition of liraglutide 3.0 MG on impaired fasting glucose in overweight/obese adults in the SCALEtm maintenance 56‐week phase 3 randomised trial. Obesity Facts 2012;5:206. [Google Scholar]
- Klein S, Aronne LJ, Hollander P, Niswender K, Woo V, Hale PM, et al. Effect of liraglutide on cardiovascular (CV) risk factors in adults without diabetes after diet‐induced weight loss: the SCALE 56‐week randomized study. Obesity (Silver Spring, Md.) 2011; Vol. 19:S177.
- NCT00781937. Comparison of liraglutide versus placebo in weight loss maintenance in obese subjects: SCALE ‐ maintenance. clinicaltrials.gov/show/NCT00781937 Date first received: 28 October 2008.
- Wadden TA, Hollander P, Klein S, Niswender K, Woo V, Hale PM, et al. NN8022‐1923 Investigators. Weight maintenance and additional weight loss with liraglutide after low‐calorie‐diet‐induced weight loss: the SCALE maintenance randomized study. International Journal of Obesity 2013;37(11):1443‐51. [DOI: 10.1038/ijo.2013.120] [DOI] [PubMed] [Google Scholar]
SCALE‐SLEEP {published data only}
- Blackman A, Foster GD, Zammit G, Rosenberg R, Aronne L, Wadden T, et al. Effect of liraglutide 3.0 mg in individuals with obesity and moderate or severe obstructive sleep apnea: the SCALE sleep apnea randomized clinical trial. International Journal of Obesity 2016;40(8):1310‐9. [PUBMED: 27005405] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01557166. Effect of liraglutide in obese subjects with moderate or severe obstructive sleep apnoea: SCALE™ ‐ sleep apnoea. clinicaltrials.gov/ct2/show/NCT01272232 Date first received: 6 January 2011.
- NN8022‐3970. Effect of liraglutide in obese subjects with moderate or severe obstructive sleep apnoea: SCALE™ ‐ sleep apnoea. novonordisk‐trials.com/Website/pdf/registry/nn80223970.pdf (accessed 30 September 2016).
References to ongoing studies
EudraCT 2013‐000418‐39 {published and unpublished data}
- Unpublished protocol. Provided by the investigators.
- EUCTR2013‐000418‐39‐AT. Early prevention of diabetes complications in people with hyperglycaemia in Europe ‐ e‐PREDICE. apps.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2013‐000418‐39‐AT (accessed 8 April 2016).
- EudraCT 2013‐000418‐39. Early prevention of diabetes complications in people with hyperglycaemia in Europe. www.clinicaltrialsregister.eu/ctr‐search/search?query=2013‐000418‐39 (accessed 17 March 2016).
- ePRECIDE. The project. www.epredice.eu/en/the‐project (accessed 17 March 2016).
Naidoo 2016 {published data only}
- Naidoo P, Wing J, Rambiritch V. Effect of sitagliptin and metformin on prediabetes progression to type 2 diabetes ‐ a randomized, double‐blind, double‐arm, multicenter clinical trial: protocol for the Sitagliptin and Metformin in PreDiabetes (SiMePreD) study. JMIR Research Protocols 2016;5(3):e145. [PUBMED: 27491324] [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT01234649 {published and unpublished data}
- NCT01234649. Combined liraglutide and metformin therapy in women with previous gestational diabetes mellitus (GDM). clinicaltrials.gov/ct2/show/NCT01234649 Date first received: 3 November 2010.
NCT01336322 {published and unpublished data}
- NCT01336322. Metformin and sitagliptin in women with previous gestational diabetes. clinicaltrials.gov/ct2/show/NCT01336322 Date first received: 13 April 2011.
NCT01548651 {published data only}
- NCT01548651. Effect of saxagliptin treatment on myocardial fat content, and monocyte inflammation. clinicaltrials.gov/ct2/show/NCT01548651 Date first received: 6 February 2012.
NCT01779362 {published data only}
- NCT01779362. RISE adult medication study (RISE Adult). clinicaltrials.gov/ct2/show/NCT01779362 Date first received: 28 January 2013.
- RISE Consortium. Restoring Insulin Secretion (RISE): design of studies of beta‐cell preservation in prediabetes and early type 2 diabetes across the life span. Diabetes Care 2014;37(3):780‐8. [PUBMED: 24194506] [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT01795248 {published and unpublished data}
- EudraCT 2012‐001371‐37. GDM‐TREAT. www.clinicaltrialsregister.eu/ctr‐search/trial/2012‐001371‐37/DK Date first registered: 9 July 2012.
- Foghsgaard S, Vedtofte L, Mathiesen ER, Svare JA, Gluud LL, Holst JJ, et al. The effect of a glucagon‐like peptide‐1 receptor agonist on glucose tolerance in women with previous gestational diabetes mellitus: protocol for an investigator‐initiated, randomised, placebo‐controlled, double‐blinded, parallel intervention trial. BMJ Open 2013;3(10):e003834. [PUBMED: 24176797] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01795248. The impact of liraglutide on glucose tolerance and the risk of type 2 diabetes in women with previous pregnancy‐induced diabetes. clinicaltrials.gov/ct2/show/NCT01795248 Date first received: 18 February 2013.
NCT01856907 {published and unpublished data}
- NCT01856907. Sitagliptin + metformin compared to metformin monotherapy and placebo in women with a recent GDM. clinicaltrials.gov/ct2/show/NCT01856907 Date first received: 14 May 2013.
NCT02104739 {published and unpublished data}
- NCT02104739. Effects of antidiabetic medications on the postprandial state in prediabetes. clinicaltrials.gov/ct2/show/NCT02104739 Date first received: 1 April 2014.
NCT02140983 {published and unpublished data}
- NCT02140983. Effects of liraglutide on hippocampal structure and function in aging adults with prediabetes (LGT). clinicaltrials.gov/ct2/show/NCT02140983 Date first received: 27 January 2014.
NCT02488057 {published and unpublished data}
- NCT02488057. Improving beta cell function in Mexican American women with prediabetes. clinicaltrials.gov/ct2/show/NCT02488057 Date first received: 11 June 2015.
NCT02576288 {published data only}
- NCT02576288. Sitagliptin effects on arterial vasculature and inflammation in obesity (SAVORO). clinicaltrials.gov/ct2/show/NCT02576288 Date first received: 12 October 2015.
NCT02847403 {published data only}
- NCT02847403. Long‐acting exenatide and cognitive decline in dysglycemic patients (DRINN). clinicaltrials.gov/show/NCT02847403 Date first received: 21 July 2016.
NCT02969798 {published data only}
- NCT02969798. Pre‐diabetes in subject with impaired fasting glucose (IFG) and impaired glucose tolerance (IGT). clinicaltrials.gov/show/NCT02969798 Date first received: 16 March 2016.
NCT03004612 {published data only}
- NCT03004612. Effect of linagliptin + metformin vs metformin alone in patients with prediabetes (PRELLIM). clinicaltrials.gov/show/NCT03004612 Date first received: 21 December 2016.
UMIN000008620 {published data only}
- UMIN000008620. The impact of DPP‐4 inhibitor on daily glucose profile and coronary plaque character in impaired glucose tolerance patients with coronary artery disease. upload.umin.ac.jp/cgi‐open‐bin/ctr/ctr.cgi?function=brows&action=brows&recptno=R000010058&type=summary&language=E Date first received: 6 August 2012.
Additional references
Abdul‐Ghani 2006
- Abdul‐Ghani MA, Jenkinson CP, Richardson DK, Tripathy D, DeFronzo RA. Insulin secretion and action in subjects with impaired fasting glucose and impaired glucose tolerance: results from the Veterans Administration Genetic Epidemiology Study. Diabetes 2006;55(5):1430‐5. [PUBMED: 16644701] [DOI] [PubMed] [Google Scholar]
ADA 1997
- The Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Report of the expert committee on the diagnosis and classification of diabetes mellitus. Diabetes Care 1997;20(7):1183‐97. [DOI] [PubMed] [Google Scholar]
ADA 2003
- Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Report of the expert committee on the diagnosis and classification of diabetes mellitus. Diabetes Care 2003;26(Suppl 1):S5‐20. [DOI] [PubMed] [Google Scholar]
ADA 2008
- American Diabetes Association. Standards of medical care in diabetes ‐ 2008. Diabetes Care 2008;31(Suppl 1):S12‐54. [PUBMED: 18165335] [DOI] [PubMed] [Google Scholar]
ADA 2010
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2010;33(Suppl 1):S62‐9. [PUBMED: 20042775] [DOI] [PMC free article] [PubMed] [Google Scholar]
ADA 2014
- American Diabetes Association. Standards of medical care in diabetes ‐ 2014. Diabetes Care 2014;37 Suppl 1:S14‐80. [PUBMED: 24357209] [DOI] [PubMed] [Google Scholar]
ADA 2015
- American Diabetes Association. Standards of medical care in diabetes ‐ 2015. Diabetes Care 2015;38(Suppl 1):S1‐93. [PUBMED: 24357209] [PubMed] [Google Scholar]
ADA 2017
- American Diabetes Association. Standards of medical care in diabetes ‐ 2017. Diabetes Care 2017;40(Suppl 1):S4‐S5. [PubMed] [Google Scholar]
Anderson 2005
- Anderson DC Jr. Pharmacologic prevention or delay of type 2 diabetes mellitus. Annals of Pharmacotherapy 2005;39(1):102‐9. [PUBMED: 15562143] [DOI] [PubMed] [Google Scholar]
Bell 2013
- Bell ML, McKenzie JE. Designing psycho‐oncology randomised trials and cluster randomised trials: variance components and intra‐cluster correlation of commonly used psychosocial measures. Psycho‐oncology 2013;22:1738‐47. [DOI] [PubMed] [Google Scholar]
Bhardwaj 2010
- Bhardwaj A, Agarwal V, Phung OJ, Baker WL, Tongbram V, Coleman CI. Can oral hypoglycemic agents restore normoglycemia in individuals with insulin resistance?. 70th Scientific Sessions of the American Diabetes Association; 2010 25‐29 June; Orlando (FL). 2010.
Bjorner 2013
- Bjorner JB, Lyng WM, Gundgaard J, Miller KA. Benchmarks for interpretation of score differences on the SF‐36 health survey for patients with diabetes. Value in Health 2013;16(6):993‐1000. [DOI] [PubMed] [Google Scholar]
Boutron 2014
- Boutron I, Altman DG, Hopewell S, Vera‐Badillo F, Tannock I, Ravaud P. Impact of spin in the abstracts of articles reporting results of randomized controlled trials in the field of cancer: the SPIIN randomized controlled trial. Journal of Clinical Oncology 2014;32:4120‐6. [DOI] [PubMed] [Google Scholar]
Brok 2009
- Brok J, Thorlund K, Wetterslev J, Gluud C. Apparently conclusive meta‐analyses may be inconclusive ‐ trial sequential analysis adjustment of random error risk due to repetitive testing of accumulating data in apparently conclusive neonatal meta‐analyses. International Journal of Epidemiology 2009;38(1):287‐98. [PUBMED: 18824466] [DOI] [PubMed] [Google Scholar]
Calanna 2013
- Calanna S, Christensen M, Holst JJ, Laferrere B, Gluud LL, Vilsboll T, et al. Secretion of glucagon‐like peptide‐1 in patients with type 2 diabetes mellitus: systematic review and meta‐analyses of clinical studies. Diabetologia 2013;56(5):965‐72. [PUBMED: 23377698] [DOI] [PMC free article] [PubMed] [Google Scholar]
CDC 2015
- Centers for Disease Control and Prevention. 2014 National Diabetes Statistics Report. www.cdc.gov/diabetes/data/statistics/2014statisticsreport.html (accessed 15 October 2015).
Cheng 2006
- Cheng C, Kushner H, Falkner BE. The utility of fasting glucose for detection of prediabetes. Metabolism: Clinical and Experimental 2006;55(4):434‐8. [PUBMED: 16546472] [DOI] [PubMed] [Google Scholar]
ClinicalTrials.gov
- Search terms: prediabetes drugs. clinicaltrials.gov/ct2/results?term=prediabetes+drugs&Search=Search (accessed 12 February 2016).
Corbett 2014
- Corbett MS, Higgins JP, Woolacott NF. Assessing baseline imbalance in randomised trials: implications for the Cochrane risk of bias tool. Research Synthesis Methods 2014;5:79‐85. [DOI] [PubMed] [Google Scholar]
Creutzfeldt 1979
- Creutzfeldt W. The incretin concept today. Diabetologia 1979;16(2):75‐85. [PUBMED: 32119] [DOI] [PubMed] [Google Scholar]
De Heer 2014
- Heer J, Göke B. Are incretin mimetics and enhancers linked to pancreatitis and malignant transformations in pancreas?. Expert Opinion on Drug Safety 2014;13(11):1469‐81. [DOI] [PubMed] [Google Scholar]
DeFronzo 1989
- DeFronzo RA, Ferrannini E, Simonson DC. Fasting hyperglycemia in non‐insulin‐dependent diabetes mellitus: contributions of excessive hepatic glucose production and impaired tissue glucose uptake. Metabolism: Clinical and Experimental 1989;38(4):387‐95. [PUBMED: 2657323] [DOI] [PubMed] [Google Scholar]
DeFronzo 2011
- DeFronzo RA, Tripathy D, Schwenke DC, Banerji M, Bray GA, Buchanan TA, et al. Pioglitazone for diabetes prevention in impaired glucose tolerance. New England Journal of Medicine 2011;364(12):1104‐15. [PUBMED: 21428766] [DOI] [PubMed] [Google Scholar]
Diabetes Prevention Program 2009
- Diabetes Prevention Program Research Group, Knowler WC, Fowler SE, Hamman RF, Christophi CA, Hoffman HJ, et al. 10‐year follow‐up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet 2009;374(9702):1677‐86. [DOI: 10.1016/S0140-6736(09)61457-4; PUBMED: 19878986] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dunkley 2014
- Dunkley AJ, Bodicoat DH, Greaves CJ, Russell C, Yates T, Davies MJ, et al. Diabetes prevention in the real world: effectiveness of pragmatic lifestyle interventions for the prevention of type 2 diabetes and of the impact of adherence to guideline recommendations: a systematic review and meta‐analysis. Diabetes Care 2014;37(4):922‐33. [PUBMED: 24652723] [DOI] [PubMed] [Google Scholar]
Egan 2014
- Egan AG, Blind E, Dunder K, Graeff PA, Hummer BT, Bourcier T, et al. Pancreatic safety of incretin‐based drugs ‐ FDA and EMA assessment. New England Journal of Medicine 2014;370(9):794‐7. [PUBMED: 24571751] [DOI] [PubMed] [Google Scholar]
Egbewale 2014
- Egbewale BE, Lewis M, Sim J. Bias, precision and statistical power of analysis of covariance in the analysis of randomized trials with baseline imbalance: a simulation study. BMC Medical Research Methodology 2014;14:49. [DOI: 10.1186/1471-2288-14-49] [DOI] [PMC free article] [PubMed] [Google Scholar]
Elgendy 2017
- Elgendy IY, Mahmoud AN, Barakat AF, Elgendy AY, Saad M, Abuzaid A, et al. Cardiovascular safety of dipeptidyl‐peptidase IV inhibitors: a meta‐analysis of placebo‐controlled randomized trials. American Journal of Cardiovascular Drugs 2017;17(2):143‐55. [PUBMED: 27873238] [DOI] [PubMed] [Google Scholar]
EMA/143005/2015
- European Medicines Agency. Assessment Report. Saxenda. International non‐proprietary name: liraglutide. www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/003780/WC500185788.pdf (accessed 21 September 2016).
FDA 2005
- US Food, Drug Administration. Drug approval package: Byetta (exenatide) injection. www.accessdata.fda.gov/drugsatfda_docs/nda/2005/021773_ByettaTOC.cfm (accessed 26 February 2016).
FDA 2006
- US Food, Drug Administration. FDA approves new treatment for diabetes. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2006/ucm108770 (assessed 26 February 2016).
FDA 2014
- US Food, Drug Administration. Saxenda Injection (Liraglutide [rDNA origin]). www.accessdata.fda.gov/drugsatfda_docs/nda/2014/206321Orig1s000TOC.cfm (assessed 22 September 2016).
Gosmanov 2014
- Gosmanov AR, Wan J. Low positive predictive value of hemoglobin A1c for diagnosis of prediabetes in clinical practice. American Journal of the Medical Sciences 2014;348(3):191‐4. [PUBMED: 24556928] [DOI] [PubMed] [Google Scholar]
GRADEpro GDT 2015 [Computer program]
- GRADE Working Group, McMaster University. GRADEpro GDT. Hamilton (ON): GRADE Working Group, McMaster University, 2015.
Hemmingsen 2016a
- Hemmingsen B, Krogh J, Metzendorf MI, Richter B. Sodium‐glucose cotransporter (SGLT) 2 inhibitors for prevention or delay of type 2 diabetes mellitus and its associated complications in people at risk for the development of type 2 diabetes mellitus. Cochrane Database of Systematic Reviews 2016, Issue 4. [DOI: 10.1002/14651858.CD012106.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hemmingsen 2016b
- Hemmingsen B, Krogh J, Metzendorf MI, Richter B. Dipeptidyl‐peptidase (DPP)‐4 inhibitors or glucagon‐like peptide (GLP)‐1 analogues for prevention or delay of type 2 diabetes mellitus and its associated complications in persons at increased risk for the development of type 2 diabetes mellitus. Cochrane Database of Systematic Reviews 2016, Issue 5. [DOI: 10.1002/14651858.CD012204] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hemmingsen 2016c
- Hemmingsen B, Sonne DP, Metzendorf MI, Richter B. Insulin secretagogues for prevention or delay of type 2 diabetes mellitus and its associated complications in persons at increased risk for the development of type 2 diabetes mellitus. Cochrane Database of Systematic Reviews 2016, Issue 10. [DOI: 10.1002/14651858.CD012151.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2002
- Higgins JPT, Thompson SG. Quantifying heterogeneity in a meta‐analysis. Statistics in Medicine 2002;21:1539‐58. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analysis. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2009
- Higgins JPT, Thompson SG, Spiegelhalter DJ. A re‐evaluation of random‐effects meta‐analysis. Journal of the Royal Statistical Society. Series A, (Statistics in Society) 2009;172(1):137‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JPT, Altman DG, Gøtzsche PC, Jüni P, Moher D, Oxman AD, et al. The Cochrane Collaboration's tool for assessing risk of bias in randomised trials. BMJ 2011;343:d5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011c
- Higgins JP, Whitehead A, Simmonds M. Sequential methods for random‐effects meta‐analysis. Statistics in Medicine 2011;30(9):903‐21. [PUBMED: 21190240] [DOI] [PMC free article] [PubMed] [Google Scholar]
Holst 2007
- Holst JJ. The physiology of glucagon‐like peptide 1. Physiological Reviews 2007;87(4):1409‐39. [PUBMED: 17928588] [DOI] [PubMed] [Google Scholar]
Holz 1993
- Holz GG 4th, Kuhtreiber WM, Habener JF. Pancreatic beta‐cells are rendered glucose‐competent by the insulinotropic hormone glucagon‐like peptide‐1(7‐37). Nature 1993;361(6410):362‐5. [PUBMED: 8381211] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hopper 2011
- Hopper I, Billah B, Skiba M, Krum H. Prevention of diabetes and reduction in major cardiovascular events in studies of subjects with prediabetes: meta‐analysis of randomised controlled clinical trials. European Journal of Cardiovascular Prevention and Rehabilitation 2011;18(6):813‐23. [PUBMED: 21878448] [DOI] [PubMed] [Google Scholar]
Hróbjartsson 2013
- Hróbjartsson A, Thomsen AS, Emanuelsson F, Tendal B, Hilden J, Boutron I, et al. Observer bias in randomized clinical trials with measurement scale outcomes: a systematic review of trials with both blinded and nonblinded assessors. Canadian Medical Association Journal 2013;185(4):E201‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
ICH 1997
- International Conference on Harmonisation Expert Working Group. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline. Guideline for Good Clinical Practice 1997 CFR & ICH Guidelines. Philadelphia (PA): Barnett International/PAREXEL, 1997. [Google Scholar]
IDF 2015
- International Diabetes Federation. IDF Diabetes Atlas. 7th Edition. Brussels: International Diabetes Federation, 2015. [Google Scholar]
IEC 2009
- International Expert Committee. International Expert Committee report on the role of the A1C assay in the diagnosis of diabetes. Diabetes Care 2009;32(7):1327‐34. [PUBMED: 19502545] [DOI] [PMC free article] [PubMed] [Google Scholar]
Inzucchi 2012
- Inzucchi SE. Clinical practice. Diagnosis of diabetes. New England Journal of Medicine 2012;367(6):542‐50. [PUBMED: 22873534] [DOI] [PubMed] [Google Scholar]
Jensen 2002
- Jensen CC, Cnop M, Hull RL, Fujimoto WY, Kahn SE. Beta‐cell function is a major contributor to oral glucose tolerance in high‐risk relatives of four ethnic groups in the U.S. Diabetes 2002;51(7):2170‐8. [PUBMED: 12086947] [DOI] [PubMed] [Google Scholar]
Kirkham 2010
- Kirkham JJ, Dwan KM, Altman DG, Gamble C, Dodd S, Smyth R, et al. The impact of outcome reporting bias in randomised controlled trials on a cohort of systematic reviews. BMJ 2010;340:c365. [DOI: 10.1136/bmj.c365] [DOI] [PubMed] [Google Scholar]
Knowler 2002
- Knowler WC, Barrett‐Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. New England Journal of Medicine 2002;346(6):393‐403. [PUBMED: 11832527] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lan 1983
- Lan KKG, DeMets DL. Discrete sequential boundaries for clinical trials. Biometrika 1983;70:659‐63. [Google Scholar]
Le Roux 2017
- Lle Roux CW, Astrup A, Fujioka K, Greenway F, Lau DC, Gaal L, et al. 3 years of liraglutide versus placebo for type 2 diabetes risk reduction and weight management in individuals with prediabetes: a randomised, double‐blind trial. Lancet 2017;389(10077):1399‐409. [PUBMED: 28237263] [DOI] [PubMed] [Google Scholar]
Liberati 2009
- Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gøtzsche PC, Ioannidis JPA, et al. The PRISMA statement for reporting systematic and meta‐analyses of studies that evaluate interventions: explanation and elaboration. PLoS Medicine 2009;6(7):1‐28. [DOI: 10.1371/journal.pmed.1000100] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lundh 2017
- Lundh A, Lexchin J, Mintzes B, Schroll JB, Bero L. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2017, Issue 2. [DOI: 10.1002/14651858.MR000033.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mathieu 2009
- Mathieu S, Boutron I, Moher D, Altman DG, Ravaud P. Comparison of registered and published primary outcomes in randomized controlled trials. JAMA 2009;302:977‐84. [DOI] [PubMed] [Google Scholar]
Meader 2014
- Meader N, King K, Llewellyn A, Norman G, Brown J, Rodgers M, et al. A checklist designed to aid consistency and reproducibility of GRADE assessments: development and pilot validation. Systematic Reviews 2014;3:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Morris 2013
- Morris DH, Khunti K, Achana F, Srinivasan B, Gray LJ, Davies MJ, et al. Progression rates from HbA1c 6.0‐6.4% and other prediabetes definitions to type 2 diabetes: a meta‐analysis. Diabetologia 2013;56(7):1489‐93. [PUBMED: 23584433] [DOI] [PubMed] [Google Scholar]
Nagel 2015
Nauck 1986
- Nauck MA, Homberger E, Siegel EG, Allen RC, Eaton RP, Ebert R, et al. Incretin effects of increasing glucose loads in man calculated from venous insulin and C‐peptide responses. Journal of Clinical Endocrinology and Metabolism 1986;63(2):492‐8. [PUBMED: 3522621] [DOI] [PubMed] [Google Scholar]
NCT02161588
- NCT02161588. A trial to assess the pharmacokinetics, pharmacodynamics, safety and tolerability of oral semaglutide in healthy male Japanese and Caucasian subjects. clinicaltrials.gov/ct2/show/NCT02161588 Date first received: 10 June 2014.
NDDG 1979
- National Diabetes Data Group. Classification and diagnosis of diabetes mellitus and other categories of glucose intolerance. National Diabetes Data Group. Diabetes 1979;28(12):1039‐57. [PUBMED: 510803] [DOI] [PubMed] [Google Scholar]
Orskov 1988
- Orskov C, Holst JJ, Nielsen OV. Effect of truncated glucagon‐like peptide‐1 [proglucagon‐(78‐107) amide] on endocrine secretion from pig pancreas, antrum, and nonantral stomach. Endocrinology 1988;123(4):2009‐13. [PUBMED: 2901341] [DOI] [PubMed] [Google Scholar]
Phung 2012
- Phung OJ, Baker WL, Tongbram V, Bhardwaj A, Coleman CI. Oral antidiabetic drugs and regression from prediabetes to normoglycemia: a meta‐analysis. Annals of Pharmacotherapy 2012;46(4):469‐76. [PUBMED: 22474136] [DOI] [PubMed] [Google Scholar]
Pogue 1997
- Pogue JM, Yusuf S. Cumulating evidence from randomized trials: utilizing sequential monitoring boundaries for cumulative meta‐analysis. Controlled Clinical Trials 1997;18(6):580‐93; discussion 661‐6. [PUBMED: 9408720] [DOI] [PubMed] [Google Scholar]
Rask 2004
- Rask E, Olsson T, Soderberg S, Holst JJ, Tura A, Pacini G, et al. Insulin secretion and incretin hormones after oral glucose in non‐obese subjects with impaired glucose tolerance. Metabolism: Clinical and Experimental 2004;53(5):624‐31. [PUBMED: 15131768] [DOI] [PubMed] [Google Scholar]
Rehman 2017
- Rehman MB, Tudrej BV, Soustre J, Buisson M, Archambault P, Pouchain D, et al. Efficacy and safety of DPP‐4 inhibitors in patients with type 2 diabetes: Meta‐analysis of placebo‐controlled randomized clinical trials. Diabetes & Metabolism 2017;43(1):48‐58. [DOI] [PubMed] [Google Scholar]
Reid 2014
- Reid TS. Practical use of glucagon‐like peptide‐1 receptor agonist therapy in primary care. Clinical Diabetes 2014;31(4):148‐57. [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Riley 2011
- Riley RD, Higgins JP, Deeks JJ. Interpretation of random effects meta‐analyses. BMJ 2011;342:d549. [DOI] [PubMed] [Google Scholar]
Riley 2013
- Riley RD, Kauser I, Bland M, Thijs L, Staessen JA, Wang J, et al. Meta‐analysis of randomised trials with a continuous outcome according to baseline imbalance and availability of individual participant data. Statistics in Medicine 2013;32(16):2747‐66. [DOI] [PubMed] [Google Scholar]
Scirica 2013
- Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. New England Journal of Medicine 2013;369(14):1317‐26. [DOI: ] [DOI] [PubMed] [Google Scholar]
Selvin 2011
- Selvin E, Steffes MW, Ballantyne CM, Hoogeveen RC, Coresh J, Brancati FL. Racial differences in glycemic markers: a cross‐sectional analysis of community‐based data. Annals of Internal Medicine 2011;154(5):303‐9. [PUBMED: 21357907] [DOI] [PMC free article] [PubMed] [Google Scholar]
Siddiqui 2009
- Siddiqui O, Hung HM, O'Neill R. MMRM vs. LOCF: a comprehensive comparison based on simulation study and 25 NDA datasets. Journal of Biopharmaceutical Statistics 2009;19(2):227‐46. [DOI] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JA, Sutton AJ, Ioannidis JP, Terrin N, Jones DR, Lau J, et al. Recommendations for examining and interpreting funnel plot asymmetry in meta‐analyses of randomised controlled trials. BMJ 2011;343:d4002. [DOI] [PubMed] [Google Scholar]
Toft‐Nielsen 2001
- Toft‐Nielsen MB, Damholt MB, Madsbad S, Hilsted LM, Hughes TE, Michelsen BK, et al. Determinants of the impaired secretion of glucagon‐like peptide‐1 in type 2 diabetic patients. Journal of Clinical Endocrinology and Metabolism 2001;86(8):3717‐23. [PUBMED: 11502801] [DOI] [PubMed] [Google Scholar]
Tuomilehto 2001
- Tuomilehto J, Lindstrom J, Eriksson JG, Valle TT, Hamalainen H, Ilanne‐Parikka P, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. New England Journal of Medicine 2001;344(18):1343‐50. [PUBMED: 11333990] [DOI] [PubMed] [Google Scholar]
Van de Laar 2006
- Laar FA, Lucassen PL, Akkermans RP, Lisdonk EH, Grauw WJ. Alpha‐glucosidase inhibitors for people with impaired glucose tolerance or impaired fasting blood glucose. Cochrane Database of Systematic Reviews 2006, Issue 4. [DOI: 10.1002/14651858.CD005061.pub2] [DOI] [PubMed] [Google Scholar]
Viera 2011
- Viera AJ. Predisease: when does it make sense?. Epidemiologic Reviews 2011;33:122‐34. [DOI] [PubMed] [Google Scholar]
Vist 2008
- Vist GE, Bryant D, Somerville L, Birminghem T, Oxman AD. Outcomes of patients who participate in randomized controlled trials compared to similar patients receiving similar interventions who do not participate. Cochrane Database of Systematic Reviews 2008, Issue 3. [DOI: 10.1002/14651858.MR000009.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wetterslev 2008
- Wetterslev J, Thorlund K, Brok J, Gluud C. Trial sequential analysis may establish when firm evidence is reached in cumulative meta‐analysis. Journal of Clinical Epidemiology 2008;61(1):64‐75. [PUBMED: 18083463] [DOI] [PubMed] [Google Scholar]
WHO 1998
- Alberti KM, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part I: diagnosis and classification of diabetes mellitus. Provisional report of a WHO consultation. Diabetic Medicine 1998;15(7):539‐53. [DOI] [PubMed] [Google Scholar]
WHO 1999
- World Health Organization. Definition, diagnosis and classification of diabetes mellitus and its complications: report of a WHO consultation. Part 1. Diagnosis and classification of diabetes mellitus. Geneva: WHO, 1999. [Google Scholar]
WHO/IDF 2006
- World Health Organization/International Diabetes Federation. Definition and diagnosis of diabetes mellitus and intermediate hyperglycaemia: report of a WHO/IDF consultation. www.who.int/diabetes/publications/Definition%20and%20diagnosis%20of%20diabetes_new.pdf 2006; Vol. (assessed 7 May 2017).
Wong 2006a
- Wong SS, Wilczynski NL, Haynes RB. Comparison of top‐performing search strategies for detecting clinically sound treatment studies and systematic reviews in MEDLINE and EMBASE. Journal of the Medical Library Association 2006;94(4):451‐5. [PMC free article] [PubMed] [Google Scholar]
Wong 2006b
- Wong SSL, Wilczynski NL, Haynes RB. Developing optimal search strategies for detecting clinically sound treatment studies in EMBASE. Journal of the Medical Library Association 2006;94(1):41‐7. [PMC free article] [PubMed] [Google Scholar]
Wood 2008
- Wood L, Egger M, Gluud LL, Schulz KF, Jüni P, Altman DG, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta‐epidemiological study. BMJ 2008;336(7644):601‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yudkin 2014
- Yudkin JS, Montori VM. The epidemic of pre‐diabetes: the medicine and the politics. BMJ 2014;349:g4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zannad 2015
- Zannad F, Cannon CP, Cushman WC, Bakris GL, Menon V, Perez AT, et al. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double‐blind trial. Lancet 2015;385(9982):2067‐76. [PUBMED: 25765696] [DOI] [PubMed] [Google Scholar]