

Abstract
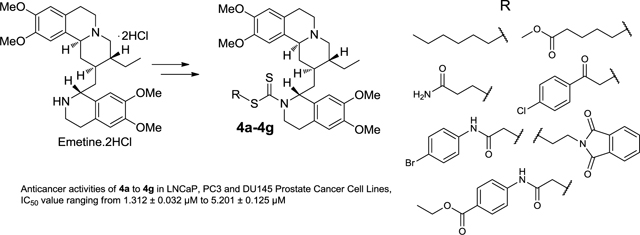
A small library of emetine dithiocarbamate ester derivatives were synthesized in 25–86% yield via derivatization of the N2’- position of emetine. Anticancer evaluation of these compounds in androgen receptor positive LNCaP and androgen receptor negative PC3 and DU145 prostate cancer cell lines revealed time dependent and dose-dependent cytotoxicity. With the exception of compound 4c, all the dithiocarbamate ester analogs in this study showed appreciable potency in all the prostate cancer cell lines (regardless of whether it is androgen receptor positive or negative) with a cytotoxicity IC50 value ranging from 1.312 ± 0.032 μM to 5.201 ± 0.125 μM by day 7 of treatment. Compared to the sodium dithiocarbamate salt 1, all the dithiocarbamate ester analogs (2 and 4a-4g) displayed lower cytotoxicity than compound 1 (PC3, IC50 = 0.087 ± 0.005 μM; DU145, IC50 = 0.079 ± 0.003 μM and LNCaP, IC50 = 0.079 ± 0.003 μM) on day 7 of treatment. Consequently, it appears that S-alkylation of compound 1 leads to a more stable dithiocarbamate ester derivative that resulted in lower anticancer activity in the prostate cancer cell lines.
Keywords: Emetine, Dithiocarbamate ester, Anti-cancer activities, Prostate cancer, Emetine derivatives
Graphical Abstract
1. Introduction
Prostate cancer is the most common non-cutaneous malignancy among American men. Patients diagnosed at stage III and IV of the disease, are usually treated with androgen-deprivation therapy (surgical or chemical castration) and virtually all patients progress to the castration resistant stage and ultimately resulting in mortality once it escapes the confines of the gland. It was estimated that more than 29,000 men in the United States would die from prostate cancer in 2014.1 Recently, several new agents have been approved for the treatment of castration resistant disease based on a few months median survival improvement over placebo. These include anti-androgen therapy abiraterone acetate and enzalutamide, second-line chemotherapy with cabazitaxel, bone-targeted denosumab, radiation therapy with radium-223 and immunotherapy vaccine, sipuleucel-T.2,3 All these agents only extend patient life by several months and there remains an urgent need to develop new therapies for castration resistant prostate cancer.3 Currently, efforts are being directed along several frontlines to develop more efficacious therapy with unique anticancer mechanisms, improved survival benefit and relatively low systemic toxicity to normal cells.3,4 Natural products continue to be valuable source of compounds with tremendous biological and medicinal importance including compounds with excellent anticancer activities. Thus, natural products remain a vital source of scaffolds and compounds for the development of useful anticancer agents. In our efforts to develop clinically useful anticancer agents based on natural products scaffold, we find emetine to be an attractive scaffold for chemical transformation.
Akinboye and Bakare recently reviewed the biological activities of emetine; this account shows the versatility of emetine as a bio-active natural product.5 Among other biological activities, it is a well known anti-cancer alkaloid from Psychotria ipecacuanha.6–8 Its anticancer activity was evaluated in several clinical studies up to Phase II for treating solid tumor, however narrow therapeutic index and dose dependent side effects stopped further studies.9 Recently, some studies reported novel mechanisms of action for the cytotoxicity of emetine. For instance, among other alkaloids screened for the inhibition of the activation of Hypoxia-inducible Factor-1 (HIF- 1) in breast cancer, emetine demonstrated an appreciable potency.6 It regulates the alternative splicing of Bcl-x Pre-mRNA, producing an up-regulation of the levels of the pro-apoptotic variant, Bcl-xS, and down-regulation of the levels of the anti-apoptotic variant Bcl-xL in different cancer cell lines.10 In a separate study on leukemia cells, emetine itself induced apoptosis, and also improved drug-induced apoptosis when combined with other chemotherapeutic agents.11 Smukste and co-workers, in a study on the use of small molecules to overcome drug resistance induced by a viral oncogene, also reported emetine and other protein synthesis inhibitors to potentiate the lethality of doxorubicin in RKO-E6 cell.12 More interestingly, emetine has been found to be a substrate of permeability glycoprotein (P-gp).13 The glycoprotein (P-gp) is a membrane protein that pumps drug molecules out of cells so that they cannot elicit their cytotoxic effects, therefore, contributing significantly to multidrug resistance (MDR). Consequently, a small library of emetine homodimers was designed and synthesized to probe the substrate binding sites of P-gp.14 One of these homodimers was reported to reverse the MDR phenotype of MCF-7/DX1 cells at a non-cytotoxic concentration when co-administered with cytotoxic agent such as doxorubicin.14 Thus, it appears that modification of emetine could lead to anticancer drug candidates with better therapeutic index.
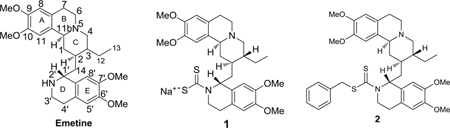
In our approach to obtain emetine derivatives without acute systemic toxicity, but still retaining anti-cancer potency, we designed the derivatization of the N2’-position of emetine into biologically interesting compounds for bioactivity studies. We initially synthesized and evaluated the anti-prostate cancer activities of a diverse library of hydrolysable analogs and prodrugs of emetine involving a variety of functional moieties. Among the compounds studied in a 7 day drug treatment, the emetine dithiocarbamate salt 1 showed considerable potency in the prostate cancer cell lines studied (PC3, IC50 = 0.087 ± 0.005 μM; LNCaP, IC50 = 0.079 ± 0.003 μM) compared to the rest of the analogs studied. Upon alkylation of 1 to form the benzyl dithiocarbamate ester 2, the cytotoxic activity was reduced (PC3, IC50 = 1.560 μM; LNCaP, IC50 = 1.970 μM).15 In order to establish the effect of alkylation of 1 on the anticancer properties of the resulting dithiocarbamate ester and establish structure activity relationship (SAR) studies that would allow us to identify a lead emetine dithiocarbamate ester derivative for further studies, we designed the synthesis and anticancer studies of a small library of emetine dithiocarbamate ester derivatives. In addition, incorporating the dithiocarbamate moiety into a library of emetine analogs is of interest to us because the presence of this moiety in a number of compounds has been associated with anti-carcinogenic, anti-mutagenic, and cancer chemo-preventive activities.16–19 In the present paper, we have carried out a time-dependent cytotoxicity study of compound 2 and seven other dithiocarbamate ester analogs in androgen receptor positive LNCaP and androgen receptor negative PC3 and DU145 prostate cancer cell lines. We herein report the synthesis, characterization and preliminary anti-prostate cancer activities of a small library of emetine dithiocarbamate ester analogs.
2. Results and Discussion
2.1. Chemistry
As we previously15 described for compound 2, the synthesis of the dithiocarbamate analogs of emetine (4a – 4g) commenced with the conversion of emetine dihydrochloride salt to the dithiocarbamate salt 1 by treating a solution of emetine dihydrochloride salt in ethanolic NaOH with carbon disulfide (Scheme 1). Compound 1 was obtained in almost quantitative yield. Subsequent reaction of 1 with different alkylating agents 3a-3g (figure 1) in acetonitrile furnished the dithiocarbamate ester analogs of emetine (4a-4g) in 25 to 86 % yield. The resulting crude product was subjected to a short flash chromatography on silica gel column using different ratios of methanol and ethyl acetate as eluent. The main impurity is believed to result from decomposition of the dithiocarbamate salt, which resulted in a more polar byproduct than the dithiocarbamate ester analog as shown by thin-layer chromatography (TLC) on silica. No attempt was made to further isolate and characterize this by-product. All compounds were characterized by infrared, 1H and 13C-nmr spectroscopy as well as by electrospray ionization mass spectrometry.
Scheme 1.
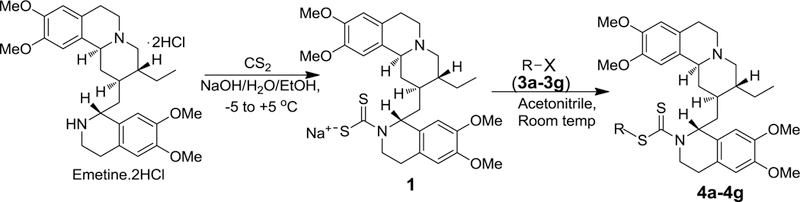
General synthetic scheme for the synthesis of dithiocarbamate ester analogs of emetine. R-X represents the various alkylating agents employed in the SN2 reactions
Fig. 1.
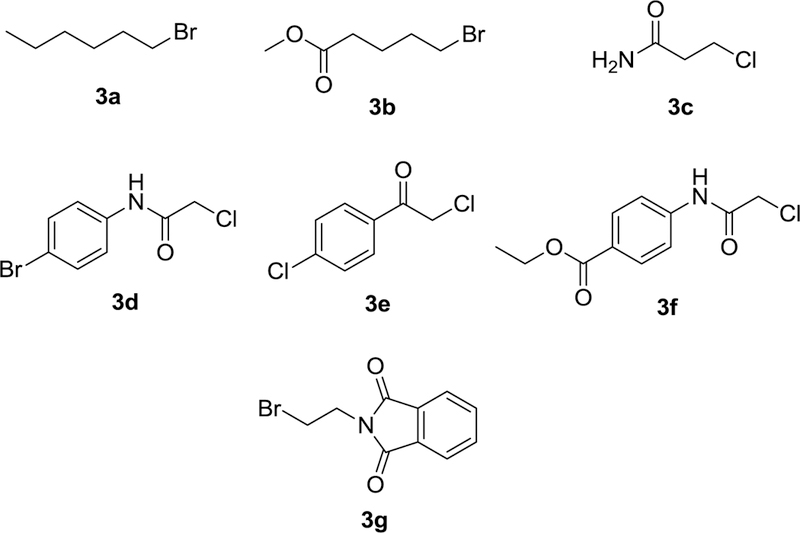
Set of alkylating agents (R-X) for the synthesis
2.2. Biology
In a seven-day exposure, we initially compared the cytotoxicity of emetine, the dithiocarbamate salt 1 and a benzyl substituted dithiocarbamate ester analog 2 of emetine in PC3 and LNCaP.15 In this study, emetine was about three folds more potent than the dithiocarbamate salt 1 while it was about 50 folds more potent than the dithiocarbamate ester analog in PC3. We established in this initial study that the dithiocarbamate salt 1 served as a prodrug of emetine and was hydrolyzed to emetine in the slightly acidic cancer cell growth medium mimicking the cancerous tumor microenvironment, whereas the dithiocarbamate ester 2 was more stable as a result of S-alkylation of the dithiocarbamyl group. More importantly, a preliminary evaluation of the safety of compound 1 in healthy mice compared to emetine suggested that the dithiocarbamate salt 1 is safer than emetine in vivo. Specifically, mice that received a single dose of emetine at 100 mg/kg body weight all died within 48 hours. On the other hand, 100 mg/kg body weight of compound 1 did not produce observable lethality. Further, when 33 mg/kg body weight of emetine or compound 1 was administered, mice that received emetine were lethargic and showed about four times weight loss compared with control; while, mice that received compound 1 appeared healthy and did not show any significant difference in weight compared with control.15 These results encouraged us to pursue further anticancer evaluation of derivatives of compound 1 similar to compound 2. We were interested in developing more stable analogs that would not necessarily act as prodrugs, but would retain anticancer activities while minimizing the toxicities associated with emetine. Such a compound would be closely related to compound 1 that we have proven to be safer than emetine in vivo. The availability of such compounds from emetine would make available clinically useful compounds that could be used in combination therapy with existing anticancer drugs, if not as a stand-alone chemotherapeutic agent. This will be beneficial for the treatment of metastatic castration-resistant prostate cancer particularly when the cancer is not responding to available treatment options as a result of drug resistance to currently used chemotherapeutic agents.
Consequently, we set out to design a small library of dithiocarbamate ester analogs of emetine with potential anticancer properties and investigate the effects of various types of “alkyl groups” on the potency of the dithiocarbamate ester analogs of emetine in the androgen receptor positive LNCaP and negative PC3 and DU145 prostate cancer cell lines. Each alkylating agent we employed is unique, but they all can be grouped into two categories: “aliphatic (non-aromatic)” which gave compounds 4a-4c and “aromatic” which gave 4d-4g. Our goal is to incorporate diversity into this small chemical library to investigate the effects of the structure and functional groups of each group on the general anticancer activity of the analogs and possibly identify a lead dithiocarbamate ester derivative. The cytotoxicity assay was done with a maximum drug concentration of 10 μM; any drug with cytotoxicity IC5o value greater than 10 μM is categorized as inactive and such values are not reported (Tables 1 to 3 and figures 2A-C).
Table 1.
Cytotoxicity of dithiocarbamate analogs of emetine in androgen receptor positive (LNCaP) and negative (PC3 and DU145) on day 3 of exposure
| Cytotoxicity IC50 (μM) on day 3 | |||
|---|---|---|---|
| Compound | LNCaP | PC3 | DU145 |
| 1 | 0.565 ± 0.012 | 0.442 ± 0.036 | 0.377 ± 0.018 |
| 2 | 1.966 ± 0.088 | 2.535 ± 0.099 | 2.741 ± 0.092 |
| 4a | 3.659 ± 0.044 | 5.978 ± 0.168 | 3.658 ± 0.053 |
| 4b | 4.785 ± 0.137 | 8.264 ± 0.006 | 3.220 ± 0.031 |
| 4c | 9.217 ± 0.709 | >10.000 | >10.000 |
| 4d | 5.280 ± 0.191 | 3.868 ± 0.277 | 5.654 ± 0.319 |
| 4e | 5.721 ± 0.092 | 3.594 ± 0.113 | 3.170 ± 0.158 |
| 4f | 4.667 ± 0.132 | 4.363 ± 0.084 | 5.157 ± 0.101 |
| 4g | 3.221 ± 0.117 | 3.207 ± 0.071 | 3.809 ± 0.051 |
| Emetine | 0.037 ± 0.001 | 0.035 ± 0.001 | 0.033 ± 0.001 |
Table 3.
Cytotoxicity of dithiocarbamate analogs of emetine in androgen receptor positive (LNCaP) and negative (PC3 and DU145) on day 7 of exposure
| Cytotoxicity IC50 (μM) on day 7 | |||
|---|---|---|---|
| Compound | LNCaP | PC3 | DU145 |
| 1 | 0.079 ± 0.003 | 0.087 ± 0.003 | 0.079 ± 0.003 |
| 2 | 1.312 ± 0.032 | 1.560 ± 0.290 | 1.980 ± 0.213 |
| 4a | 1.592 ± 0.014 | 3.027 ± 0.160 | 2.300 ± 0.067 |
| 4b | 1.763 ± 0.039 | 4.355 ± 0.091 | 2.253 ± 0.084 |
| 4c | 3.390 ± 0.339 | 5.201 ± 0.125 | >10.000 |
| 4d | 1.698 ± 0.187 | 1.507 ± 0.896 | 1.603 ± 0.093 |
| 4e | 1.951 ± 0.068 | 2.692 ± 0.145 | 2.449 ± 0.162 |
| 4f | 1.656 ± 0.564 | 1.902 ± 0.134 | 2.467 ± 0.263 |
| 4g | 2.303 ± 0.121 | 2.378 ± 0.240 | 3.050 ± 0.070 |
| Emetine | 0.037 ± 0.001 | 0.029 ± 0.003 | 0.024 ± 0.001 |
Figure 2.
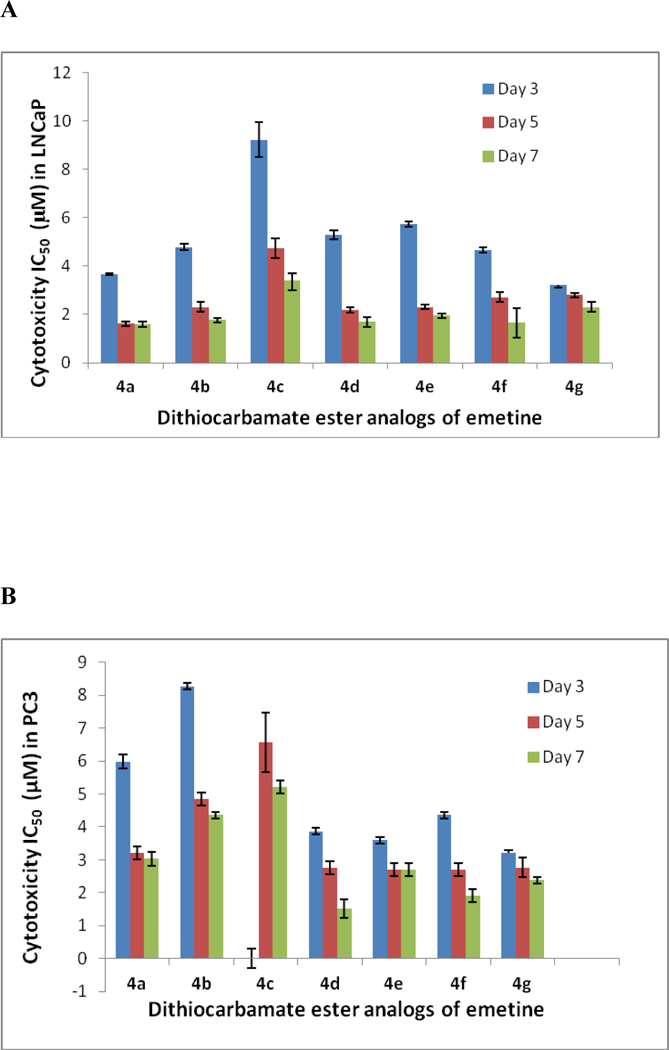
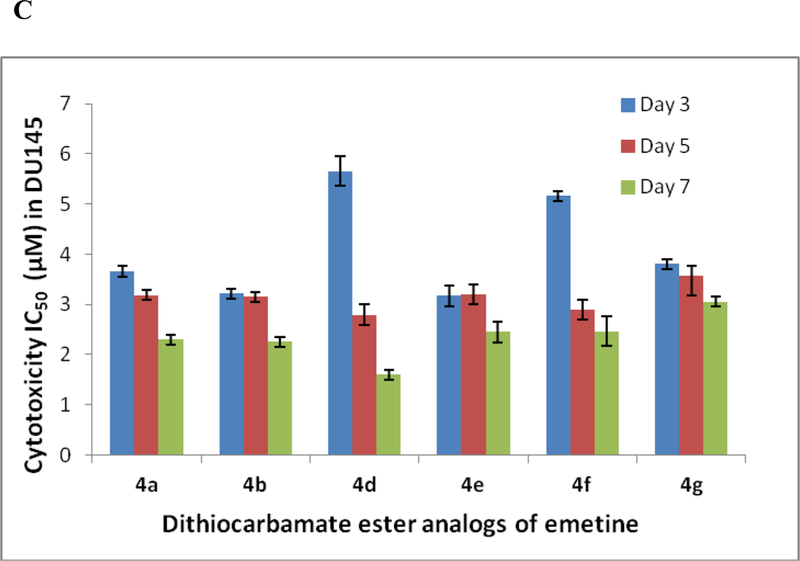
Variation of the cytotoxicity IC50 of the dithiocarbamate analogs of emetine with time over the seven-day exposure. A. Cytotoxicity IC50 values in LNCaP on the 3rd, 5th and 7th day of exposure. B. Cytotoxicity IC50 values in PC3 on the 3rd, 5th and 7th day of exposure. C. Cytotoxicity IC50 values in DU145 on the 3rd, 5th and 7th day of exposure
Looking at the effects of analogs 2 and 4a - 4g, it was observed that by day 3 all the compounds, except 4c, showed appreciable potency in all the prostate cancer cell lines (regardless of whether it is androgen receptor positive or negative) with a cytotoxicity IC50 value less than 10 μM (Table 1). Compounds 4a, 4b and 4c are designed with aliphatic (non-aromatic) alkyl group, and different functionalities at the aliphatic chain terminal. Compound 4c has an amide functional group terminal and has a shorter chain (about three carbon shorter) than 4a and 4b. As such, its relatively low potency within the first 3 days of exposure as seen in all the three cell lines, could be due to a number of factors including chain length, possible H-bonding from the terminal amide or may be due to poor cellular uptake. In both LNCaP and PC3, its potency increased as the exposure period increased to five days (LNCaP, IC50 = 4.732 μM; PC3, IC50 = 6.562 μM) and seven days (LNCaP, IC50 = 3.3 90 μM; PC3, IC50 = 5.201 μM) as shown in fig 2. Though generally more potent than 4c, compound 4b, with an ester functional group at the terminal of the aliphatic chain, is relatively less potent in the more aggressive PC3 than the other two cell lines (Tables 1, 2, 3). On the contrary, 4a without any other functionality on the hexyl chain shows consistent potency across the three cell lines with appreciable anticancer activity observed within the first three days (LNCaP, IC50 = 3.659 μM; PC3, IC50 = 5.978 μM; DU145, IC50 = 3.658 μM). Compound 4a appears to be the most potent of the three alkyl-substituted analogs showing time-dependent increase in cytotoxicity in all three cell lines by day 5 (LNCaP, IC50 = 1.613 μM; PC3, IC50 = 3.21 μM; DU145, IC50 = 3.189 μM), and day 7 (LNCaP, IC50 = 1.592 μM; PC3, IC50 = 3.027 μM; DU145, IC50 = 2.300 μM) [Tables 1, 2, 3 and Figs. 2A-C].
Table 2.
Cytotoxicity of dithiocarbamate analogs of emetine in androgen receptor positive (LNCaP) and negative (PC33 and DU145) on day 5 of exposure
| Cytotoxicity IC50 (μM) on day 5 | |||
|---|---|---|---|
| Compound | LNCaP | PC3 | DU145 |
| 1 | 0.088 ± 0.003 | 0.091 ± 0.006 | 0.080 ± 0.001 |
| 2 | 1.890 ± 0.092 | 2.021 ± 0.049 | 2.070 ± 0.103 |
| 4a | 1.613 ± 0.066 | 3.21 ± 0.117 | 3.189 ± 0.078 |
| 4b | 2.308 ± 0.174 | 4.855 ± 0.153 | 3.148 ± 0.007 |
| 4c | 4.732 ± 0.352 | 6.562 ± 0.913 | >10.000 |
| 4d | 2.189 ± 0.075 | 2.768 ± 0.146 | 2.796 ± 0.151 |
| 4e | 2.312 ± 0.023 | 2.700 ± 0.133 | 3. 197 ± 0.193 |
| 4f | 2.717 ± 0.135 | 2.706 ± 0.192 | 2.898 ± 0.137 |
| 4g | 2.801 ± 0.099 | 2.770 ± 0.060 | 3.575 ± 0.418 |
| Emetine | 0.027 ± 0.003 | 0.027 ± 0.002 | 0.025 ± 0.001 |
Compounds 2, 4d, 4e, 4f, and 4g have aromatic group in the dithiocarbamate ester substituent. Unlike compounds 4a, 4b, and 4c, each compound in this category shows almost the same level of potency across the three cell lines with compounds 2 (LNCaP, IC50 = 1.312 μM; PC3, IC50 = 1.560 μM; DU145, IC50 = 1.980 μM) and 4d (LNCaP, IC50 = 1.698 μM; PC3, IC50 = 1.507 μM; DU145, IC50 = 1.603 μM) showing the lowest IC50 values (less than 2 μM) in all three cell lines by day 7 [Tables 1, 2, 3 and Figs. 2A-C]. Further, all the analogs 2 and 4d – 4g show a gradual increase in potency between day 3 and day 7 of exposing the prostate cancer cell lines to the analogs (Tables 1 to 3). The resulting dithiocarbamate esters in this study (2, 4a – 4g) do not necessarily act as prodrugs, but rather as less toxic analogs of emetine. The slight differences in cytotoxicity observed in this study is most likely due to conformational differences resulting from the size and nature of each substituent group. As previously reported, replacement of the secondary amine hydrogen of the N-2’ position of emetine with a bulkier group would eliminate an electronic effect as a result of loss of possible H-bonding of the N-H with potential receptors at the active site. In addition, any substituted group would result in conformational changes which appear to affect the space between the tricyclic and bicyclic ring system of emetine and consequently affect the bioactivity of the resulting analog including protein synthesis inhibitory and anticancer activities.5,15,20 Hence, an appropriate substituent in this position would result in a clinically useful emetine prodrug, or an emetine derivative that does not have the same systemic toxicity as emetine. We are currently studying a diverse library of emetine analogs in this regard.
In summary, all the analogs 2 and 4a-4g consistently displayed lower cytotoxicity than compound 1, establishing the fact that once compound 1 is stabilized by S-alkylation, the cytotoxic effect of the resulting dithiocarbamate ester (2, 4a - 4g) is decreased relative to 1. In addition, for all the compounds (2, 4a-4g) there was a time dependent increase in cytotoxicity between day 3 and day 7 of the study in androgen- receptor positive LNCaP and androgen receptor negative PC3. The only exception is seen in the androgen- receptor negative DU145 where compound 4c consistently showed IC50 value of over 10 μM between days 3 to 7. The reason for this exception is not immediately apparent as the same compound 4c showed some increase in potency (IC50 of less than 10 μM by day 5 and 7) in the more aggressive androgen receptor negative PC3 prostate cancer cell line (Tables 1 to 3 and figures 2A-C). Also, it is important to note that nearly all the compounds showed appreciable potency in all the prostate cancer cell lines regardless of whether it is androgen receptor positive or androgen receptor negative. This might suggest a mechanism of action that is independent of the androgen receptor. Finally, of the eight compounds studied here, compounds 2 and 4d appear to be the most consistently potent in all three cell lines and further SAR around these two compounds could lead to more active analogs that could be clinically useful in combination therapy with existing anticancer drugs, if not as a stand-alone chemotherapeutic agent in the treatment of metastatic castration-resistant prostate cancer.
3. Experimental Section
3.1. Chemistry: general procedures
All solvents and reagents used were bought from commercial sources and used without any further purification. Melting points were determined in open capillary tubes on a Mel-Temp melting point apparatus and are uncorrected. The 1H- and 13C-NMR spectra were obtained on a Bruker Avance 400 MHz spectrometer in deuterated chloroform (CDCl3) or deuterated methanol (CD3OD). Chemical shifts are in δ units (ppm) with TMS (0.00 ppm), CHCl3 (7.27 ppm), or CH3OH (3.34 ppm) as the internal standard for 1H-NMR, and CDCl3 (77.00 ppm) or CD3OD (49.90 ppm) for 13C-NMR. The samples were analyzed with a Waters quadrupole Time-of- Flight (QTOF) mass spectrometer (micro model) equipped with both Electrospray Ionization (ESI) and Atmospheric Pressure Chemical Ionization (APCI). The instrument was operated in positive-ion (ESI+) single-stage mode. The MS was calibrated in the mass-to-charge (m/z) range = 100 −1500 using a solution of 2 μg/mL Nal and 0.05 μg/mL CsI in isopropyl alcohol:H2O 1:1 (vol:vol) introduced into the instrument by direct infusion at a flow of 25 μL/min. All the mass spectra showed signals corresponding to the molecular ion plus sodium (Na). The sodium was picked up from the conditions used to operate the instrument, as is common in this type of mass spectrometry, and was not present in the actual compounds themselves.
3.2. Synthesis of sodium salt of dithiocarbamic acid of emetine 1.
Sodium 1-(3-Ethyl-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolm-2- ylmethyl)-6,7-dimethoxy-3,4-dihydro-1H-isoquinoline-2-carbodithioate (1).
A solution of NaOH (240.0 mg) in water (1.00 ml) and ethanol (20.00ml) was added to a solution of emetine dihydrochloride hydrate (1.11 g, 2.00 mmol) in ethanol (10.0 ml) at −8°C. This was stirred at this temperature for 15min after which CS2 (0.30 mL, 4.97 mmol) was added. The resulting mixture was stirred at −5 to 1°C for 2 h and at room temperature for 30 min. The solvent was removed invacuo and the residue triturated with acetonitrile and then filtered. The filtrate was evaporated to dryness and the residue dissolved in ethyl acetate (3 mL). To this was added diethyl ether which afforded the precipitation of 1 as a white solid (950 .0mg, 82.0%). 1H NMR (400 MHz, CD3OD) δ 0.93 (3H, t, J = 7.4 Hz), 1.10–1.23 (1H, m), 1.14–1.20 (2H, m), 1.33–1.41 (2H, m), 1.66–1.72 (1H, m), 2.10 (1H, t, J = 11.0 Hz), 2.38 (1H, t, J = 12.8 Hz), 2.50 (1H, tb, J = 4.1, 11.6 Hz ), 2.63–2.70 (2H, m), 2.99–3.20 (5H, m), 3.35 (1H, s), 3.44–3.51 (1H, m) 3.80 (3H, s), 3.81 (6H, br s), 3.84 (3H, s), 5.95 (1H, dd, J = 5.9, 12.8 Hz), 6.66 (1H, s), 6.69 (1H, s), 6.72 (1H,s), 7.20 (1H,s), 7.25 (1H, dd, J = 3.6, 11.8 Hz). 13C NMR (100 MHz, CD3OD) δ 216.5, 147.4, 147.3,147.2,147.1, 133.0, 131.1, 127.0, 126.5, 112.6, 112.0, 111.0, 110.3, 65.4, 63.3, 61.6, 58.0, 56.5, 56.2, 56.1, 56.0, 55.9, 52.5, 41.7, 41.3 , 37.6 , 29.4 , 27.3 , 23.7, 11.6.
3.3. General procedure for the synthesis of the dithiocarbamate ester derivatives of emetine.
For the synthesis of each of compounds 4a-4g, the salt 1 (200 mg, 0.35 mmol) was dissolved in acetonitrile (15 mL) and to this was added a solution of alkylating agents 3a-3g (0.27 mmol.) in acetonitrile. The reaction mixtures were allowed to stir for 24 h at room temperature. Solvent was evaporated in-vacuo. Residue obtained was triturated with water (20 mL) to dissolve any inorganic substances, and then filtered under suction. The crude product was air-dried and then purified by flash chromatography on silica gel using EtOAc: MeOH (10:1) as eluent.
3.3.1. 1-(3-Ethyl-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolm-2-yl- methyl)-6,7-dimethoxy-3,4-dihydro-1H-isoquinolme-2-carbodithioic acid hexyl ester (4a).
Yield: 45.2%, mp = 78–80 °C 1H NMR (400 MHz, CDCl3) δ 0.78–0.94 (6H, m), 1.03–1.15 (1H, m), 1.16–1.32 (7H, m), 1.33–1.53 (4H, m), 1.55–1.75 (3H, m), 2.03 (1H, ds t, J = 10.0 Hz), 2.36–2.52 (2H, m), 2.60 (1H, d, J = 15.2 Hz), 2.77 (1H, d, J = 14.5 Hz), 2.90–2.98 (1H, m), 3.00–3.19 (3H, m), 3.20–3.30 (1H, m), 3.31–3.39 (2H, m), 3.61–3.72 (1H, m), 3.83 (3H, s), 3.84 (6H, s), 3.93 (3H, s), 4.68 (1H, dd, J = 4.8, 13.9 Hz), 6.55 (1H, s), 6.56 (1H, s), 6.57 (1H, s), 6.86 (1H, dd, J = 4.1, 11.5 Hz), 6.92 (1H, s). 13C NMR (100 MHz, CDCl3) δ 198.0, 148.0, 147.8, 147.3,147.2, 130.1, 126.2, 124.7, 111.6, 111.3, 111.2, 110.0, 108.8, 62.9, 61.4, 59.8, 56.3, 56.2, 56.1, 55.9, 52.4, 43.3, 41.2, 40.6, 38.9, 37.7, 37.6, 37.1, 31.4, 29.7, 28.8, 27.9, 23.8, 22.5, 14.0, 11.2. HRESIMS m/z 641.4342 ([C36H52N2O4S2 + H]+ calcd. 641.3447).
3.3.2. 5-[1-(3-Ethyl-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinoline-2- yl-methyl)-6,7-dimethoxy-3,4-dihydro-1H-isoquinoline-2-carbothioylsulfanyl]-pentanoic acid methyl ester (4b).
Yield: 86.2%, mp = 74–76 °C 1H NMR (400 MHz, CDCl3) δ 0.87 (3H, t, J = 7.4 Hz), 1.01–1.15 (1H, m), 1.15–1.31 (3H, m), 1.32–1.43 (1H, m), 1.43–1.54 (1H, m),1.54–1.67 (1H, m),1.68–1.82(4H, m), 2.03 (1H, t, J = 7.4 Hz), 2.24–2.35 (2H, m), 2.36–2.53 (2H, m), 2.59 (1H, d, J = 15.6 Hz), 2.68–2.83 (1H, m), 2.88–2.98 (1H, m), 2.98–3.26 (4H, m), 3.27–3.42 (2H, m), 3.62 (3H, s), 3.65–3.75 (1H, m), 3.81 (3H, s), 3.83 (6H, s), 3.92 (3H, s), 4.65 (1H, dd, J = 4.3, 13.6 Hz), 6.54 (1H, s), 6.55 (1H, s), 6.57 (1H, s), 6.82 (1H, dd, J = 3.7, 11.3 Hz), 6.70 (1H, s). 13C NMR (100 MHz, CDCl3) δ 197.1, 174.2, 148.5, 148.3, 147.8, 147.7, 130.5, 130.4, 128.9, 126.7, 125.1, 111.8, 110.5, 109.3, 63.3, 62.9, 60.4, 56.8, 56.6, 56.3, 52.8, 46.0, 43.9, 41.8, 41.1, 39.4, 37.3, 36.7, 34.0, 30.2, 29.7, 28.9, 28.3, 24.7, 24.3, 11.8. HRESIMS m/z 671.4057 ([C36H50N2O6S2 + H]+ calcd. 671.3189).
3.3.3. 1-(3-Ethyl-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]-isoquinoline-2- yl-methyl)-6,7-dimethoxy-3,4-dihydro-1H-isoquinoline-2-carbodithioic acid 2-carbamoyl- ethyl ester (4c).
Yield: 23.4%, mp = 107–109 °C 1H NMR (400 MHz, CDCl3) δ 0.88 (3H, t, J = 7.4 Hz), 1.05–1.13 (1H, m), 1.20–1.27 (3H, m), 1.34–1.44 (1H, m), 1.45–1.57 (1H, m), 1.58–1.69 (1H, m), 1.94–2.10 (1H, m), 2.35–2.51 (2H, m), 2.61 (1H, d, J = 15.9 Hz), 2.70 (2H, t, J =6.6 Hz), 2.78 (1H, d, J = 14.8 Hz), 2.86–3.27 (5H, m), 3.54–3.76 (3H, m), 3.83 (3H, s), 3.84 (6H, s), 3.92 (3H, s), 4.63 (1H, dd, J = 3.9, 13.9Hz ), 5.36–5.72 (2H, m), 6.55 (1H, s), 6.56 (1H, s), 6.58 (1H, s), 6.81 (1H, dd, J = 3.8, 11.2Hz ), 6.90 (1H, s). 13C NMR (100MHz, CDCl3) δ 197.1,173.2, 148.3, 147.9, 147.7, 147.3, 130.1, 129.8, 126.4, 124.6, 111.5, 111.4, 109.9, 108.9, 62.8,61.4, 59.5, 56.5, 56.4, 56.2, 56.1, 56.0, 52.4, 41.4, 40.7, 40.5, 38.8, 37.8, 35.4, 29.2, 27.1, 23.8,11.2. HRESIMS m/z 628.3776 ([C33H45N3O5S2 + H]+ calcd. 628.2879).
3.3.4. 1-(3-Ethyl-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl- methyl)-6,7-dimethoxy-3,4-dihydro-1H-isoquinoline-2-carbodithioic acid (4-bromo-phenylcarbamoyl)-methyl ester (4d).
Yield: 40.5%, mp = 136–138 °C 1H NMR (400 MHz, CDCl3) δ 0.87 (3H, t, J = 7.3 Hz), 0.99–1.29 (4H, m), 1.29–1.45 (1H, m), 1.45–1.70 (2H, m), 1.88 (1H, t, J = 10.8 Hz), 2.30 (1H, ds t, J = 9.6 Hz), 2.43 (1H, t, J = 12.8 Hz), 2.61 (1H, d, J = 15.6 Hz), 2.71–2.96 (3H, m), 2.96–3.21 (4H, m), 3.83 (3H, s), 3.86 (6H, s), 3.91 (3H, s), 4.14 (1H, d, J = 14.6 Hz), 4.37 (1H, d, J = 14.6 Hz), 4.61 (1H, br d, J = 11.09 Hz), 6.59 (2H, s), 6.60 (1H, s), 6.76 (1H, dd, J = 4.0, 11.0 Hz), 6.83 (1H, s), 7.16 (2H, d, J = 8.4 Hz), 7.26 (2H, d, J = 8.4 Hz),9.06 (1H, s). 1H NMR (400 MHz, CD3OD) δ 0.87 (3H, t, J = 7.5 Hz), 0.92–1.13 (3H, m) 1.14–1.24 (1H, m), 1.25–1.39(1H, m), 1.39–1.63 (2H, m), 2.02 (1H, t, J = 11.2 Hz), 2.26–2.42 (2H, m), 2.51–2.64 (1H, m), 2.69–2.88 (2H, m), 2.91–3.05 (4H, m), 3.13 (1H, d, J = 12.3 Hz), 3.64 (3H, s), 3.66 (3H, s), 3.71 (3H, s), 3.72 (3H, s), 3.96 (1H, d, J = 15.8 Hz), 4.32 (1H, d, J = 15.8 Hz), 4.68 (1H, dd, J = 4.8, 13.7 Hz), 6.54 (1H, s), 6.65 (1H, m), 6.66 (2H, m), 6.75 (1H, dd, J = 4.0, 11.5 Hz), 6.99 (2H, d, J = 8.8 Hz), 7.21 (2H, d, J = 8.8 Hz) 7.25 (1H, s). 13C NMR (100 MHz, CDCl3) δ 195.9, 166.8, 148.6, 148.3, 147.2, 147.4, 147.2, 137.1, 136.8, 131.8 (2C), 129.1, 127.7,126.6, 124.3, 116.8, 111.5, 111.3, 109.7, 108.6, 62.8, 61.4, 61.3, 56.2 (2C), 56.0, 55.9, 52.5,44.5, 40.7, 40.4, 38.9, 37.9, 29.7, 28.1, 27.3, 23.8, 11.2. HRESIMS m/z 768.3202 ([C38H46BrN3O5S2+H]+ calcd. 768.2140).
3.3.5. l-(3-Ethyl-9,l0-dimethoxy-l,3,4,6,7,llb-hexahydro-2H-pyrido[2,l-a]isoquinolin-2-yl-methyl)-6,7-dimethoxy-3,4-dihydro-lH-isoquinoline-2-carbodithioic acid 2-(4-chloro-phenyl)-2-oxo-ethyl ester (4e).
Yield: 75.3%, mp = 99–101 °C 1H NMR (400 MHz, CDCl3) δ 0.88 (3H, t, J = 7.4 Hz), 0.99–1.15 (1H, m), 1.16–1.28 (2H, m), 1.29–1.42 (1H, m), 1.42–1.55 (1H, m), 1.56–1.69 (1H, m), 2.03 (1H, t, J = 11.2 Hz), 2.35–2.52 (2H, m), 2.59 (1H, d, J = 15.4 Hz), 2.70–2.88 (2H, m), 2.90–3.25 (4H, m), 3.69 (3H, s), 3.83 (10H, s), 4.65–4.73 (2H, m), 4.91 (1H, d, J = 16.6 Hz), 6.54 (2H, s), 6.59 (1H, s), 6.72 (1H, dd, J = 3.9, 11.2 Hz), 6.77 (1H, s), 7.38 (2H, d, J = 8.5 Hz), 7.94 (2H, d, J = 8.5 Hz). 13C NMR (100 MHz, CDCl3) δ 195.5, 192.2, 148.1, 147.9, 147.2, 140.0, 134.5, 134.0, 130.0, 129.7, 129.0, 128.1, 126.3, 125.9, 124.5, 111.5, 111.2,109.9, 109.7, 108.7, 62.7, 61.4, 60.2, 56.2, 56.1, 56.0, 55.9, 52.4, 44.8, 44.2, 41.4, 40.6, 38.9,37.8, 29.7, 28.1, 23.8, 11.3. HRESIMS m/z 709.3492 ([C38H45ClN2O5S2+H]+ calcd. 709.2537).
3.3.6. 4-{2-[1-(3-Ethyl-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]-isoqumolin-2-ylmethyl)-6,7-dimethoxy-3,4-dihydro-1H-isoquinoline-2-carbothioylsulfanyl]-acetylamino}-benzoic acid ethyl ester (4f).
Yield: 51.4%, mp = 90–92 °C 1H NMR (400 MHz, CDCl3) δ 0.86 (3H, t, J = 7.4 Hz), 0.99–1.11 (1H, m), 1.12– 1.26 (3H, m), 1.36 (3H, t, J =7.1 Hz), 1.48–1.69 (2H, m), 1.89 (1H, t, J = 11.1 Hz), 2.30 (1H, t, J = 9.8 Hz), 2.44 (1H, t, J =12.8 Hz), 2.59 (1H, d, J = 11.8 Hz), 2.78–2.94 (3H, m), 2.94–3.20 (4H, m), 3.83 (3H, s), 3.84 (3H, s), 3.86 (3H, s), 3.87 (1H,br s), 3.91 (3H, s), 4.19 (1H, d, J = 14.6 Hz), 4.31 (2H, q, J = 7.1 Hz), 4.38 (1H, d, J = 14.6 Hz), 4.60 (1H, br d, J = 11.6 Hz), 6.58 (1H, s), 6.59 (1H, s), 6.60 (1H, s), 6.75 (1H, dd, J = 4.2, 11.0 Hz), 6.84 (1H, s), 7.44 (2H, d, J = 8.7 Hz), 7.77 (2H, d, J = 8.7 Hz), 9.27 (1H, s). 13C NMR (100 MHz, CDCl3) δ 195.9, 167.1, 166.0, 148.3, 148.1, 147.5, 147.2,141.8, 130.6, 129.7, 129.0, 126.6, 125.9, 125.8, 125.7, 124.3, 118.8, 111.5, 111.4, 109.9, 108.6,62.8, 61.4, 61.1, 60.8, 56.3, 56.2, 56.0, 55.9, 52.4, 52.0, 41.3, 40.7, 40.1, 39.0, 38.0, 29.7, 29.1,23.8, 14.4, 11.2. HRESIMS m/z 762.4441 ([C41H51N3O7S2 + H]+ calcd. 762.3247).
3.3.7. 1-(3-Ethyl-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolm-2-yl- methyl)-6,7-dimethoxy-3,4-dihydro-1H-isoquinoline-2-carbodithioic acid 2-(1,3-dioxo-1,3-dihydro-isoindol-2-yl)-ethyl ester (4g).
Yield: 27.4%, mp = 100–102 °C 1H NMR (400 MHz, CDCl3) δ 0.88 (3H, t, J = 7.4 Hz), 1.04–1.36 (4H, m), 1.37–1.52 (2H, m), 1.52–1.69 (1H, m), 1.97–2.16 (1H, m), 2.34–2.52 (2H, m), 2.60 (1H, d, J = 13.7 Hz), 2.67–2.80 (1H, m), 2.86–2.98 (1H, m), 2.98–3.27 (4H, m), 3.62–3.73 (3H, m), 3.83 (3H, s), 3.84 (6H, s), 3.95 (3H, s), 3.98–4.17, (2H, m), 4.61 (1H, dd, J = 4.8, 13.6 Hz ), 6.54 (1H, s), 6.55 (1H, s), 6.58 (1H, s ), 6.80 (1H, dd, J = 3.8, 11.4 Hz), 6.89 (1H, s), 7.62–7.69 (2H, m), 7.73–7.82 (2H, m). 13C NMR (100 MHz, CDCl3) δ 195.9, 168.0 (2C), 148.0, 147.8, 147.4, 147.2, 133.9 (2C), 133.7, 133.0, 132.0 (2C),129.8, (2C), 124.6, 123.2, 111.3, 111.2, 110.0, 108.8, 62.6, 61.3, 59.5, 56.3, 56.1, 56.0, 55.9,52.3, 43.6, 40.7, 39.0, 37.4, 36.8, 34.8, 29.7, 29.2, 28.0, 23.8, 11.3. HRESIMS m/z 730.4174 ([C40H47N3O6S2 + H]+ calcd. 730.2985).
3.4. Biology
3.4.1. Cytotoxicity Assay
3.4.1.2. General Information
The human androgen receptor positive (LNCaP) and negative (PC3 and DU145) prostate cancer cell lines were purchased from the American Type Culture Collection (Manassas, VA). All cells were grown in cell culture flasks in RPMI culture medium with phenol red (GIBCO) supplemented with only 10% fetal bovine serum, 1% L-glutamine and 1% Penicillin-Streptomycin. Cells were cultured in a humidified atmosphere of 95% air and 5% carbon dioxide at 37 °C. To sub-culture cells for experiments, cells growing as monolayer cultures were released from the tissue culture flasks by treatment with 0.05% trypson/EDTA. Cell population density was determined with aid of coulter counter (Beckman) and/or hemocytometer. For all the in-vitro experiments, cell-growth was maintained in the log phase and the cells were used while still in this logarithmic growth phase.
3.4.1.2. Cytotoxicity assay of emetine derivatives in DU145, PC3 and LNCaP cell lines.
The fast growing androgen receptor negative PC3 and DU145 were plated at a population density of 2.5 X 103 cells/well while relatively slow growing androgen receptor positive LNCaP cell lines were plated at a density of 6 X 103 cells/well into 96 well tissue culture plates. The cells were incubated for 24 h before treatment. Compounds were dissolved in 100% DMSO to give various concentrations which were serially diluted (0.01 μM to 10 μM) with complete RPMI medium to obtain a uniform DMSO concentration of 0.1% in the medium. Control cells were treated with an equivalent concentration of DMSO. Cells were exposed to the drugs for seven days and MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) cell proliferation assay was done to determine the population of viable cells was on the 3rd, 5th, and 7th, day of treatment to evaluate the cytotoxic effects of drugs and also measure if the control cells were in the logarithmic growth phase. All assays were done in six replicates and repeated at least twice.
Acknowledgment
We gratefully acknowledge grant number 5-U54-CA914–31 (Howard University/Johns Hopkins Cancer Center Partnership) and MRI grant number CHE-1126533 from the National Science Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Siegel R; Ma J; Jemal Zou, Z., A Cancer Statistics, 2014. CA-Cancer J.Clin. 2014, 64, 9–29. [DOI] [PubMed] [Google Scholar]
- 2.Acar O; Esen T; Lack NA ScientificWorldJOurnal. 2013, 20, 1–8. [Google Scholar]
- 3.Basch E; Loblaw A; Oliver TK; Cardicci M; Chen RC; Frame JN; Garrels K et al. J Clin Oncol 2014, 32, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trewartha D; Carter K Nat. Rev. Drug Discov. 2013, 12, 823–824 [DOI] [PubMed] [Google Scholar]
- 5.Akinboye ES; Bakare O The Open Nat. Prod. Journal. 2011, 4, 8–15 [Google Scholar]
- 6.Dimitrijevic S; Duncan RJ Bioactive and Compatible Polym. 1998, 13, 165–178. [Google Scholar]
- 7.Zhou Y-D; Kim Y-P; Mohammed KA; Jones DK; Muhammad I; Dunbar DC; Nagle DG J. Nat. Prod. 2005, 68, 947–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Selassie CD; Hansch C; Khwaja TA J. Med. Chem. 1990, 33, 1914–1919. [DOI] [PubMed] [Google Scholar]
- 9. (a).Siddiqui S; Firat D; Olshin S Cancer Chemother. Rep. 1973, 57, 423–428. [PubMed] [Google Scholar]; (b) Panettiere F; Coltman CA Jr. Cancer 1971, 27, 835–841. [DOI] [PubMed] [Google Scholar]; (c) Kane RC; Cohen MH; Broder LE; Bull MI; Creaven PJ; Fossieck BE Jr. Cancer Chemother Rep. 1975, 59, 1171–1172. [PubMed] [Google Scholar]; (d) Siddiqui S; Firat D; Olshin S Cancer Chemother Rep 1973, 57, 423–428. [PubMed] [Google Scholar]; (e) Moertel CG; Schutt AJ; Hahn RG; Reitemeier RJ Cancer Chemother Rep. 1974, 58, 229–232. [PubMed] [Google Scholar]; (f) Street EW. Lancer. 1972, 2, 281–282. [Google Scholar]; (g) Mastrangelo MJ; Grage TB; Bellet RE; Weiss AJ Cancer. 1973, 31, 1170–1175. [DOI] [PubMed] [Google Scholar]
- 10.Boon-Unge K; Yu Q; Zou T; Zhou A; Govitrapong P; Zhou J Chem Biol. 2007, 14, 1386–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Möller M; Herzer K; Wenger T; Herr I; Wink M Oncology Reports. 2007, 18, 737–744. [PubMed] [Google Scholar]
- 12.Smukste I; Bhalala O; Persico M; Stockwell BR Cancer Cell. 2006, 9, 133–146. [DOI] [PubMed] [Google Scholar]
- 13.Polli JW; Wring SA; Humphreys JE; Huang L; Morgan JB; Webster LO; Serabjit-Singh CS J. Pharmacol. Exp. Ther. 2001, 299, 620–628. [PubMed] [Google Scholar]
- 14.Pires MM; Hrycyna CA; Chmielewski J Biochemistry, 2006, 45, 11695–11702. [DOI] [PubMed] [Google Scholar]
- 15.Akinboye ES; Rosen MD; Denmeade SR; Kwabi-Addo B and Bakare OJ Med. Chem. 2012, 55(17), 7450–7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee B-H; Bertram B; Schmezer P; Frank N; Wiessler MJ Med. Chem. 1994, 37, 3154–3162. [DOI] [PubMed] [Google Scholar]
- 17.Mehta RG; Liu J; Constantinou A; Thomas CF; Hawthorne M; You M; Gerhuser C; Pezzuto JM; Moon RC; Moriarty RM Carcinogenesis. 1995, 16, 399–404. [DOI] [PubMed] [Google Scholar]
- 18.Gerhauser C; You M; Liu J; Moriarty RM; Hawthorne M; Mehta RG; Moon RC; Pezzuto JM Cancer Res. 1997, 57, 272–278. [PubMed] [Google Scholar]
- 19.Cao S-L; Feng Y-P; Jiang Y-Y; Liu S-Y; Ding G-Y; Li R-T Bioorg. Med. Chem. Lett. 2005, 15, 1915–1917. [DOI] [PubMed] [Google Scholar]
- 20.Troconis M; Ma W; Nichols DE; McLaughlin JJ Comput. -AidedMol. Des. 1998, 12,411–418. [DOI] [PubMed] [Google Scholar]