

Abstract
Helper T cell induced plasma cells (PCs) that secrete class-switched neutralizing antibody are paramount to effective immunity. Following class switch recombination (CSR), antigen-activated B cells differentiate into extrafollicular PCs or mature in germinal centers (GCs) to produce high-affinity memory B cells and follicular PCs. Many studies focus on the core transcriptional programs that drive central PC functions of longevity and antibody secretion. However, it is becoming clear that these central programs are further subdivided across antibody isotype with separable transcriptional trajectories. Divergent functions emerge at CSR, persist through PC terminal differentiation and further assort memory PC function following antigen recall. Here, we emphasize recent work that assorts divergent isotype-specific PC function across four major modules of immune protection.
Keywords: Plasma cell, B cell memory, Follicular T helper, Isotype
Helper T cell regulated B cell immunity
Primary exposure to foreign antigen induces T follicular helper cell (TFH, see Glossary) formation which activate antigen-presenting B cells through cognate contact. Activated B cells can either differentiate into extrafollicular plasma cells (PCs) or enter a germinal center (GC) reaction. Under the direction of TFH cells, B cells undergo iterative rounds of somatic hypermutation, selectively increasing antigen affinity prior to GC exit as affinity matured memory B cells or follicular PCs [1-5]. TFH cell initiated class switch recombination (CSR) functionally diversifies the expressed B cell receptor (BCR) as each immunoglobulin (Ig) class and subclass contributes unique immune effector activity (Figure 1). In this way, antigen-driven B cell fate is imprinted by cognate TFH cell contact prior to extrafollicular PC or GC differentiation.
Figure 1: Plasma cells derive from three distinct pathways.

A schematic representation of plasma cell (PC) formation pathways is shown. Naïve B cells can be directly activated by antigen and undergo class switch recombination (CSR) without T cell help, forming unmutated PCs. Alternately, T helper (TH) cells can activate B cells through cognate contact, and direct CSR via secreted factors. Higher antigen affinity B cells are directed to form extrafollicular PCs while lower affinity B cells can enter a germinal center (GC) reaction. GC B cells undergo iterative rounds of somatic hypermutation under the direction of follicular T helper cells (TFH). B cells with highest affinity are selected for survival, exiting the GC as memory B cells and follicular PCs. T cell help and CSR likely imprint distinct transcriptional programs that can influence B cell fate and function.
Extrafollicular PCs are typically “short-lived” PCs with lifespans measured in days. After GC cycling and TFH selection, affinity matured follicular PCs exhibit unique transcriptional programs required for prolonged survival as “long-lived” PCs for years in mice and decades in humans [6-15]. Varied tissues such as bone marrow, spleen, lamina propria and the thymus present unique microenvironments that both require and contribute to PC transcriptional diversity [16-20]. These local transcriptional changes can expand PC functionality through IL-10 secretion [21, 22] and the regulation of antigen-specific TFH cells by repressing transcription factor Bcl-6 and IL-21 [23]. These secondary PC programs may further subdivide by antibody isotype exerting separable functions across different locations and target cell types.
Antigen recall responses assort and extend isotype-specific B cell immunity. The recall response progresses more swiftly as expanded compartments of memory TFH cells activate affinity-matured antigen-specific memory B cells [24], These memory B cells robustly divide and differentiate into extrafollicular memory PCs and form memory response GCs. Memory GCs produce higher affinity memory B cells and memory follicular PCs that consolidate immune protection in ways that remain poorly understood. Variations in antigen recall based on the form of antigen and/or formulation of immune challenge display altered cellular dynamics and predict divergent transcriptional programming [25]. Overall, multiple layers of cognate memory TFH cell regulation coordinate and extend protective B cell immunity according to the isotype expressed by memory B cells and their capacity for ongoing CSR and selection.
In this review, we consider the core transcriptional modules used by PCs to serve central functions of longevity and antibody secretion. We highlight how the event of CSR may be associated with additional programmatic imprinting and divergent function retained through differentiation into the terminal effector state. As with innate lymphoid cells (ILCs) and CD4+ T subsets (Figure 2), we assort B cell responses into four major modules using the isotype-specific effector functions of PC subsets [26]. Type I pro-inflammatory immunity against intracellular pathogens contrasts type II anti-inflammatory responses that restore homeostasis after extracellular pathogen challenge. Subclasses of IgG antibody are aligned with these major modules of immunity. Mucosal barrier integrity is broadly controlled by type III immunity and remains the domain of the IgM and IgA antibody classes. Finally, dampening of ongoing immunity and prevention of autoimmunity can be considered separately as “type IV” or regulatory immunity and broadly considered a secondary cellular function of long-lived PCs potentially dissociated from antibody secretion or specificity. These main immune response modules can be extended into the recall response and are discussed with regards to programming, control and origin. Finally, we consider the technological advances utilized for single cell gene and protein expression with a focus on their application in effective PC analysis. Taken together, we provide a new framework for the programming and delivery of isotype specific PC function.
Figure 2: CD4+ Helper T (TH) cell subsets have varied immune protection roles.

Naïve CD4+ T cells differentiate into TH cell subsets under the direction of environmental factors that influence the expression of specific lineage defining master transcription factors. These transcription factors induce transcriptional programming leading to unique effector functions, such as cytokine secretion for each TH cell subset.
Abbreviations: Foxp3, forkhead box protein 3; Gata3, GATA-binding factor 3; IFNγ, Interferon gamma; RORγ, retinoid-related orphan receptor gamma; Stat1, Signal transducer and activator of transcription 1; Stat3, Signal transducer and activator of transcription 3; Stat6, Signal transducer and activator of transcription 6; TGF-β, transforming growth factor-β; TH1, T-helper-1; TH2, T-helper-2; TH17, T-helper-17; TNFα, Tumor necrosis factor alpha; TREG, regulatory T.
PCs express core transcriptional programs
B cells undergo critical transcriptional changes directed by conserved master transcriptional factors that drive irreversible terminal differentiation to PCs. During B cell differentiation in murine models of immunity, DNA remodeling occurs across multiple cell division cycles where chromatin accessibility for B cell genes are reduced while the PC lineage driving prdm1 (Blimp-1) promoter region becomes more accessible [27, 28]. The transition from B cell lineage towards PC lineage can occur without the expression of Blimp-1; however, Blimp-1 is necessary for the formation of mature antibody-secreting PCs [29-32]. Moreover, increased expression of Blimp-1 can further repress B cell lineage transcriptional regulators such as Bcl-6, IRF8, Pu.1 and Pax5 with evidence in both murine and human systems [30-36].
IRF4 and Xbp-1, which are critical for PC formation and function, respectively, are upregulated during differentiation [6, 37-40]. PC formation in the murine in vivo response is deficient without IRF4, as IRF4 represses IRF8 and increases Blimp-1 and Xbp-1 expression [35, 36]. Xbp-1 controls the unfolded protein response, which increases protein production and folding capacity in PCs [6, 41]. Additionally, murine PCs increase metabolic capacity to support constitutive antibody production [8, 42, 43] and downregulate cell cycling genes such as mki67, inducing a post-mitotic quiescent state [18, 44]. Finally, increasing surface receptor expression, such as BCMA (B cell maturation antigen) permits proper PC homing and helps to promote survival niches in vivo [10, 17, 45, 46]. While all PCs express these master transcription factors, the magnitude and linked downstream protein expression may vary between PC class and subclass to influence both cellular longevity and antibody secretion.
Class Switch Recombination
Together with co-stimulatory and cognate cell contact, secreted CD4+ TH cell factors selectively direct CSR with particular cytokines directing switch to specific antibody class and subclass. Class-specific changes introduced to the BCR cytoplasmic tail alter BCR functionality and each subclass of secreted antibody elicits a varied immune response. Multiple rounds of cell division are required to complete class switching due to chromatin remodeling and heavy chain DNA excision events [47-49]; since PCs exit cell cycling, CSR must occur in activated B or memory B cells prior to PC differentiation. Thus, during CSR-driven chromatin remodeling, additional epigenetic changes are induced and further directed by antigen-specific T cell help and a multitude of TH cell derived factors.
Cytoplasmic tail functionality varies by subclass
BCR cytoplasmic tails are varied by isotype. Human and murine IgG+ BCRs possess 28 amino acid cytoplasmic tails that are mostly conserved within the IgG+ subtypes with variations within the 28 amino acid IgE+ tail. In contrast, IgA+ tails are 14 amino acids long, while IgD+ and IgM+ tails are both just 3 amino acids [50]. Deletion or truncation of murine B cell IgG+ and IgE+ cytoplasmic tails severely reduces PC formation [51-53]. Furthermore, failure to form PCs has been linked with a compromised ability to process and present antigen, thereby limiting access to T cell help [51-53]. Hence, physical variation and divergent functional impact of the BCR itself assures separable cell fate across PCs of different antibody isotype.
Artificial CSR methods deconvolute the influence of isotype-specific cytoplasmic tails from extrinsic factors such as antigen binding or TH cytokines. Prearranged antigen-specific B cells were created using cell nucleus transfer of B1-8hi NP+ memory B cells into embryonic stem cells, allowing expression of BCR without prior antigen exposure [54]. Prearranged IgG1+ B cells form GCs and PCs equivalently to prearranged antigen-specific IgM+ B cells in this mouse model [54]. In contrast, antigen experience further directed murine cell fate, given that NP (4-hydroxy-3-nitrophenyl) antigen-stimulated memory IgG1+ B cells expressed altered transcriptional programming from memory IgM+ B cells (e.g. downregulation of bach2, pax5 and bcl-6), biasing IgG1+ B cells towards the PC lineage [54]. Thus, multiple intrinsic and extrinsic influences drive B cell differentiation and PC fate towards an isotype-specific functional bias.
Antigen-independent IgG1+ B cells were also created using Cre-lox recombination to excise the constant heavy chain regions instead of AID [55]. In this murine model, prior to BCR activation, IgG1+ B cells differed minimally in transcription from IgM+ B cells. However, using a Nurr77 reporter system and following BCR signaling, IgG1+ B cells altered their transcriptional program compared to IgM+ B cells (e.g. increased c-myb and decreased CXCR5) [55]. Consequently, bone marrow PCs were significantly enriched for IgG1+ cells, while the memory compartment was enriched for IgM+ cells. NP antigen binding also induced transcriptional changes in murine bone marrow-derived IgM+ PCs, resulting in the upregulation of IL-10, Lag-3 and genes such as ccl5 and Irf8 related to cytokine production [56]. These results collectively support the notion that imprinting from CSR and its associated factors allow antigen binding to differentially induce programmatic changes, altering PC cell fate and functional potential. The precise nature and organization of these changes and their deployment in vivo remains an important feature of B cell immune protection.
Organization of B Cell immunity
Affinity can bias B cells towards the PC lineage
Multiple studies have linked higher antigen affinity of murine IgG1+ B cells with a greater propensity to form PCs, with lower affinity biased towards memory B cells [57-60]. Specifically, using a SWHEL hen egg lysosome specific B cell model in mice, higher affinity IgG1+ B cells were shown to express a more PC-like transcriptome relative to low affinity IgG1+ B cells [57]. Lower affinity SWHEL IgG1+ B cells mainly expressed a memory B cell, signature suggesting a bias toward memory B cell fate [58]. Similarly, higher affinity IgG1+ B cells can remain in stable TFH contact longer, resulting in elevated IRF4 that represses Bcl6 and Bach2 expression, thus preferentially forming PCs [59]. By contrast, lower affinity IgG1+ B cells exhibited higher Bach2 expression, thus biasing these murine B cells towards memory B cell formation [60]. Even though the IgG1+ isotype was not specifically accounted for in this study, higher affinity murine B cells produced more PCs than low affinity B cells in vitro [61]. Thus, antigen affinity provides an added layer of influence in the fate of isotype-specific B cell function.
Isotype can influence B cell differentiation, function and survival
Signaling events during CSR may initiate transcriptional changes in B cells, such as induction of master transcriptional factors, that vary by isotype. In murine B cells, our group showed that from the initiation of CSR through differentiation into memory B cells, the sustained expression of the transcription factor T-bet was critical for IgG2a+ B cell function [62]. T-bet was needed for expression of specific T-bet gene targets such as Stat1 and CXCR3, not seen in naïve IgM+ B cells. After transfer of CreERT2 Tbx21F/F B cells (conditionally deleted T-bet), IgG2a+ but not IgG1+ B cells exhibited reduced survival and consequently, compromised memory B cell and PC formation. Moreover, siRNA knockdown of RORα showed that RORα (but not T-bet) was equivalently necessary for IgA+ B cell survival [62]. In support of these findings, B cell-specific CD23-linked Cre-driven deletion of the transcriptional regulator c-myb allowed inappropriate upregulation of T-bet, which increased total IgG2a+ B cell formation [63]. In addition, T-bet driven CXCR3 elicited aberrant GC B cell differentiation into PCs, suggesting that isotype-linked master transcription factors can also influence B cell differentiation. Thus, this supports the concept that signaling events during CSR imprint unique transcriptional programming necessary for class-specific survival and function (Figure 3, Key Figure).
Key Figure, Figure 3: Plasma cell isotype defines effector function heterogeneity.
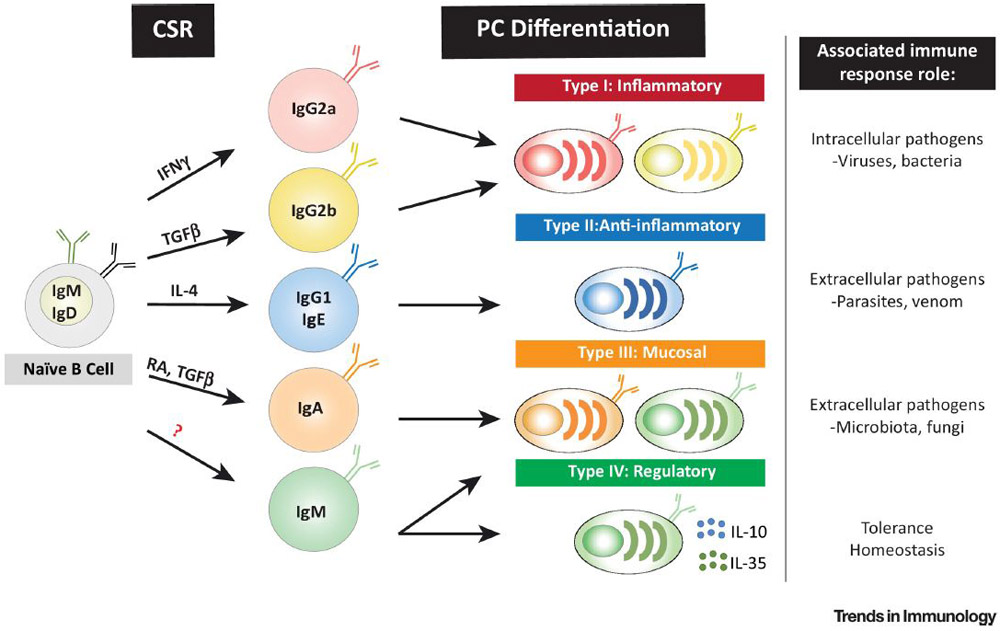
Helper T (TH) cell derived factors specifically elicit class switch recombination (CSR) in B cells to certain isotypes. Both T cell help and CSR likely imprint distinct transcriptional programs that influence B cell lineage fate and function, and which are maintained through terminal differentiation. As emphasized by terminally differentiated plasma cells, B cell isotypes participate in discrete immune responses through secreted cytokines and antibodies. B cell immunity can therefore be assorted into the four major categories of immunity used for TH and innate lymphoid cell subsets: type I inflammatory, type II anti-inflammatory, type III mucosal, and regulatory immunity (herein described as type IV). Abbreviations: IFNγ, Interferon gamma; RA, Retinoic acid; TGF-β, transforming growth factor-β
The expression of isotype specific BCR can also bias B cell differentiation fate. For example, BCR IgE+ reporter mice showed that following NP immunization, IgE+ B cells in draining lymph nodes exhibited a propensity for upregulating transcription factor Blimp-1, forming extrafollicular PCs over GC formation, suggesting an isotype-linked fate bias [64]. In contrast, a different BCR IgE+ reporter system showed that IgE+ B cells participated in GCs and exited as PCs and memory B cells [65]; however, later work proposed that these IgE+ GCs were transient, and that the resulting cells were not fully functional, as evidenced by a lack of B cells in the GC ‘light zone’, along with increased apoptosis compared to IgG1+ B cells [66]. In support of an isotype-directed fate bias, recent work revealed that heavy chain ectodomains unique to the IgE+ BCR provided the capacity to bias PC formation even before antigen encounter. Specifically, IgE+ BCRs interacted with CD19, a known modulator of BCR signaling, enhancing IRF4 expression via the PI3K-Akt signal pathway to favor PC fate, a result not seen in CD19−/− IgE+ cells [67-69]. Furthermore, other work using repertoire analysis and lineage mapping of memory B cell progeny suggested that an affinity-matured IgG1+ memory reservoir could contribute to high affinity IgE+ via secondary class switching, thus circumventing the IgE+ PC formation bias, and the dysfunctional GC output [70]. Therefore, BCR isotype expression itself, even after CSR, can be an important contributing factor to cell fate potential in B cell immunity.
IgG2a+, IgG2b+, and IgG3+ can contribute to Type I immunity
Type I immunity consists of proinflammatory responses against intracellular pathogens by ILC1 and TH1 cells; however, B cell subsets also contribute to type I immunity. Murine IgG2a+ and IgG2b+, along with their human homologs IgG1+ and IgG3+ antibodies bind the FCγRI and FCγRIV pro-inflammatory receptors with highest affinity among the IgG subclasses [71-73], [See Box 1]. Indeed, specific deletion of the murine FCγRIV receptor selectively impaired the inflammatory effector function of IgG2a+ and IgG2b+ antibodies. Specifically, in an induced melanoma model, anti-tumor IgG2a+ failed to eliminate tumors in FCγRIV−/− mice; by contrast, in intact mice these tumors were cleared by inducing antibody-dependent cellular cytotoxicity [74]. Similarly, rheumatoid arthritis development and severity was reduced in FCγRIV−/− mice, while autoantibodies induced increased inflammation in intact mice [74]. Furthermore, murine IgG2a+, IgG2b+, and IgG3+ and human IgG1+ and IgG3+ antibodies can all effectively bind and activate the pro-inflammatory complement system [75, 76]. Therefore, B cell immunity, in the form of IgG2a+, IgG2b+ and IgG3+ PCs, can support type I immune responses.
Box 1: Relating murine to human B cell isotypes.
Murine and human B cell isotypes share similar nomenclature; however, as is the case for the IgG subclasses, sharing the same name does not mean they are functionally equivalent across species. Although the comparisons are not exact, it is possible to relate murine B cell isotypes to analogous human B cell isotypes based on structure, Fc receptor binding affinity, complement activation and other functional activities. Murine IgG1+ and human IgG4+ both bind the inhibitory FCγRIIB receptor with highest affinity, block antigen crosslinking and fail to activate complement pathways [77-79]. Murine IgG3+ and human IgG2+ do not bind Fc receptors with appreciable affinity and activate complement cascades [72, 73]. Murine IgG2a+ and IgG2b+ bind activating/inflammatory Fc receptors with high affinity, as do human IgG1+ and IgG3+[72-74]. Of note, human IgA consists of 2 subclasses while murine IgA does not.
IgG1+ and IgE+ can contribute to type II immunity
IgG1+ PCs and their secreted antibodies contribute predominantly towards a type II-like immune response. For example, in a GaMD (goat anti-mouse IgD antiserum) antigen-driven renal disease model characterized by severely decreased tissue perfusion and fibrosis, IgG1+ deficient (γ1−) mice presented with increased mortality relative to wild type mice producing IgG1+ B cells; however, intravenous addition of soluble IgG1+ antibody or IgG1+ PC transfer prevented disease development equal to wild type mice [77]. Mechanistically, IgG1+ prevented the formation of pro-inflammatory IgG3+ immune complexes responsible for disease pathology through competitive binding and blockage of antigen crosslinking, a function also described for human IgG4+ antibodies [78].
Murine IgG1+ is analogous to human IgG4+ antibody: both bind the inhibitory FCγRIIB receptor with highest affinity amongst all other isotypes; they also fail to activate the pro-inflammatory complement pathway and potentially prevent other IgG+ subclasses from binding the complement subcomponent C1q [75, 76]. In addition, murine IgG1+ antibody complexes also suppress complement receptor 5a cascades, consequently preventing additional induction of inflammation [79]. Therefore, murine IgG1+ and human IgG4+ antibody can contribute to anti-inflammatory immune responses by limiting pro-inflammatory effector responses, such as via the complement pathway, as well as via inhibitory Fc receptor binding.
IgE+ is often associated with allergic responses when dysregulated, but when properly controlled, it contributes to a type II immune response. In mice, antibody binding the FCεRI was shown to be critical for IgE+ immune function, given that binding elicited the secretion of type II cytokines IL-4 and IL-13 by mast cells [80]. Following external exposure to carcinogenic DMBA (7,12-dimethylbenz[a]anthracene), FCεRI−/− mice were shown to exhibit exacerbated susceptibility to epithelial carcinogenesis based on tumor size and number compared to intact mice [81]. In this study, IgE+ antibodies in wild type mice, functioning through FCεRI binding, contributed to restoring tissue homeostasis, and limiting tumor development. Therefore, by acting through an intermediate cell such as a mast cell, IgE+ can participate in a type II immune response to reduce tissue damage and inflammation.
IgM+ and IgA+ can contribute to type III mucosal immunity
Type III immunity is associated with protection of mucosal sites from extracellular pathogens by TH17 cells and IgA+ PCs. Secreted IgA+ antibody is transported through epithelial layers; there, polyreactive IgA+ is likely induced in a T-independent manner by the metabolite retinoic acid [82]. It can then sequester commensal microbiota from gut epithelium contact, protecting microbiota from activating T cell-dependent immune responses [83]. Alternatively, pathogen-specific IgA+ binding has been shown to restrict separation of daughter cells via crosslinking, preventing pathogen growth and colonization, and thus aiding in mucosal surface protection [84]. Within the gut lamina propria, a subpopulation of IgA+ PCs has been shown to produce antimicrobial factors such as TNF-α and inducible nitric oxide synthase (iNOS); mice harboring Tnfa−/−iNOS−/− B cells are unable to form this subpopulation of IgA+ PCs, and exhibit decreased pathogen clearance as well as excessive bacterial outgrowth relative to wild type mice [85]. This subpopulation of IgA+ PCs may possess unique transcriptional programming, reflecting in part by by TNF-α and iNOS secretion, and thus enabling the multifunctional capacity of these cells in mucosal barrier defense [85].
Secreted IgM+ has also been associated with mucosal barrier defense due to its high avidity in pentameric form and presence of a J chain for epithelial transport; however, the role of IgM+ PCs extends beyond antibody secretion. In response to infection with the parasite Trypanosoma cruzi, murine splenic IgM+ PCs have been reported as the primary source of secreted IL-17, even more so than TH17 cells; indeed, selective transfer of IL-17A deficient IgM+ PCs into B cell deficient mice resulted in increased parasitemia, tissue damage and mortality after T. cruzi infection, relative to transfer of wild-type IgM+ PCs [86]. Furthermore, secretion of IL-17 was not detected from cultured or resting IgM+ B cells without exposure to T. cruzi, suggesting that antigen binding was necessary for the elicitation of this effector function [86].
IgM+ and IgA+ can contribute to regulatory responses
Within adaptive and innate immune responses, T regulatory cells (TREGs), and regulatory innate lymphoid cells (ILCREGs), respectively, both secrete factors such as IL-10 and TGFβ, which can limit ongoing immune responses or help in the prevention of autoimmunity [87]. Similarly, Regulatory B cells (BREGs) have also been identified as contributors to immune regulation, with evidence suggesting that some BREGs express the phenotypic hallmarks of PCs [21, 22, 56, 88].
Indeed, across diverse mouse models, such as Experimental Autoimmune Encephalomyelitis (EAE), Salmonella typhimurium infection and NP antigen vaccinations, secretion of IL-10 and IL-35 has been detected in a subset of IgM+ PCs [21, 22, 56, 88]. Targeted ablation of these PCs in the EAE and Salmonella models resulted in elevated inflammation based on clinical disease scoring, suggesting that these cytokines limited ongoing immune responses relative to controls [22, 88]. However, this putative regulatory activity was not thought to be limited to IgM+ PCs; specifically, in a non-alcoholic fatty liver mouse model, tissue-infiltrating IgA+ PCs secreting IL-10 were found to suppress the activity of cytotoxic CD8+ T cells by inducing their ‘exhaustion’ [89]. As a result, hepatocellular carcinoma development was reduced as non-alcoholic fatty liver carcinogenesis depends on stress and inflammation [89]. As IgM+ and IgA+ B cells have both been suggested to harbor regulatory activity, it is possible that PCs expressing other isotypes might also possess regulatory activity. Nevertheless, it remains imperative to assess the regulatory capacity of isotype-specific B cells beyond the persistent secretion of antibody.
While B cell immunity can broadly be sorted into the four main categories of immune responses, it is likely that there are additional and overlapping functional roles. Transcriptional heterogeneity derived from CSR, TH factors and even GC cycling might all contribute to the formation of B cell subsets harboring additional roles in immunity not described herein. Therefore, it will be important to continue dissecting the effector functions of immune response B cells, particularly when considering the design of more efficacious vaccine candidates that can trigger optimal protection.
Memory B cell responses
When considering the varied effector responses and differentiation potential conveyed by isotype, memory B cell-derived recall responses add another layer of complexity to the heterogeneity of B cell immunity. Memory B cells can undergo CSR, form secondary GCs where higher affinity cells are selected by memory TFH, and exit GCs as re-diversified memory B cells and PCs [24]. Why certain cells enter secondary GCs, and why others seemingly do not, remains an open question in the field [90-92]. Antigen specificity, adjuvants, the effects of CSR and memory TFH help, all likely contribute to the diversity of memory responses.
B cell isotype may be a contributing factor to the formation and persistence of the memory pool. One year after PE immunization, an increased number of memory IgM+ B cells were detected in lymph node and spleen samples via flow cytometry, relative to mutated class switched memory B cells, suggesting a reduced capacity for longevity in class switched memory B cells [93]. However, later work suggested that the reduced capacity for longevity might be due to the expression of AID during CSR and somatic hypermutation, rather than to the isotype itself. Specifically, by using Aicdacre/+R26LSL-YFP mice, where AID expression caused YFP production, YFP+ memory IgM+ B cell numbers were reported to decline at a similar rate to YFP+ class-switched memory B cells, excluding the impact of isotype in this case [55]. Moreover, in a blood-stage Plasmodium chabaudi mouse infection model, somatically-hypermutated IgM+ and IgG+ B cell memory populations were shown to decline over time, while an unmutated antigen-specific IgD+ memory population did not [94]. Additionally, murine splenic and lymph node IgM+ B cells expressing unmutated -- yet high affinity-- germline BCR against the PE antigen have been shown to form persistent B cell memory, while mutated antigen-specific IgG+ memory B cells decline over time [95]. However, not all switched memory B cells decline over time, given that NP immunized BALB/cJ and C57B/6 mouse models have exhibited antigen-specific class-switched memory B cell populations that remained stable in the spleen over time [96, 97]. Therefore, the extent to which B cell isotype affects memory B cell formation and regulation remains unclear.
The heterogeneity of recall responses is likely derived from variable imprinting during primary antigen exposures. In one study, murine IgG+ and IgM+ memory B cells participated simultaneously in antigen recall to sheep red blood cell immunization, but were found to exhibit divergent roles [98]. Memory IgG+ responded quickly to reimmunization through PC formation, while memory IgM+ supported secondary GC formation and secondary class switching [98]. Conversely, B1-8 transgenic memory B cell recall behavior to NP antigen was linked to transcriptional programming rather than to isotype [99]. In this study, programmed death ligand 2 (PD-L2) and CD80 surface expression were noted as markers of varied programming, dividing memory B cells into mixed isotype subsets that favored either PC or GC formation [99]. The division of recall response roles -- such as by isotype or transcriptional programming-- could actually be due to unique imprinting of B cells by different antigen priming, but this requires further investigation.
As previously mentioned, memory IgG+ B cells can be biased to PC formation during a recall response on the basis of transcriptional changes retained from initial antigen priming (or imprinting), while IgM+ transcriptional programs can influence memory B cell formation [54, 55]. However, in contrast to this notion, one study reported that a murine recall exposure to blood-stage Plasmodium chabaudi was dominated by high affinity IgM+ memory B cell expansion in the spleen compared to IgG+ memory B cells, resulting in early dominance of IgM+ PCs by total cell number at day 3 of recall [94]. While the IgM+ produced was of equal affinity to IgG1+, the pentameric IgM+ antibody inherently possessed higher antigen avidity and was capable of complement-mediated lysis of blood-stage Plasmodium parasites [94]. This result suggested that certain functional aspects of the IgM+ isotype may have dictated its recall dominance, but this remains to be further examined. Indeed, while transcriptional programming of memory B cell subsets is a compelling area of investigation, the extent to which Ig isotypes may contribute to transcriptional programming and B cell memory recall responses remains elusive.
Concluding remarks
As research progresses, so does the available technology for studying B cell immunology. Recent improvements in RNA sequencing technology have significantly increased throughput, resolution, and library depth by the number and coverage of detected genes. For instance, population-level RNA sequencing studies have been critical for defining core PC transcriptional programming profiles and their downstream influence on eliciting PC lineage entry [6, 7, 30]. Furthermore, transcriptome analyses have identified transcriptional variations unique to bone marrow PCs, and which have been linked to longevity [7, 8, 18]. Looking towards the future, techniques such as CyTOF (cytometry by time of flight) and single-cell RNA sequencing (scRNA-seq) are poised to provide an even greater capacity to study B cell heterogeneity at a single cell level.
CyTOF has already been utilized in studies focused on B cells, including PCs, and TFH cells. Standard flow cytometry uses fluorophore labeling; emission spectra overlap creates an upper limit of approximately 16 simultaneous markers in a four laser instrument. CyTOF uses mass spectrometry to detect heavy metal isotype markers, which have negligible signal overlap, allowing significantly more unique markers to be used together. By leveraging the extensive quantity of surface markers used concurrently, unique immune subpopulations such as GC B cells and helper T cell subsets can be defined phenotypically with greater precision [100-103]. While cells used in CyTOF are destroyed during collection and thus cannot be used for transcriptional studies, CyTOF enables proteomic analysis at a single cell level.
Single cell RNA sequencing is a useful tool for discerning heterogeneity within a defined population. By using BCR class as well as mutation number and location, single cell analysis can be used for lineage tracing and clonality studies of B cells, or secreted antibodies [24, 104, 105]. Globally-targeted scRNA seq has also been used to detect PC heterogeneity in bone marrow samples from myeloma patients [106]. Alternatively, scRNA-seq libraries can be produced using gene specific transcript priming, shown to have high resolution and low gene dropout rates, which may improve resolution for genes with biologically low expression.[107]. However, because gene-specific library formation is inherently biased and relies upon guided preselection of targets, global RNA sequencing protocols therefore retain ample utility.
Of note, isotype-linked effector function of PCs is often seen within a subset, rather than within an entire population of PCs investigated [85, 88, 89]; therefore, unique transcriptional programs may define these subsets within a total isotype population. As a result, bulk population mRNA analysis is currently insufficient to interrogate such discrete programs that may exist in only a subset of PCs within an isotype, and will require single cell level analysis for future research. PC origin, such as GC-derived follicular or extrafollicular PCs could also play a role in PC effector functionality. Thus, mutational analysis of BCR at a single cell level might reveal specific subpopulation features such as GC origin and affinity. Moving forward, the utilization of high resolution scRNA protocols will be critical for evaluating PC subsets and allow the deconvolution of transcriptional programs dictating unique effector functions.
The events of CSR, from signaling molecules to chromatin remodeling, indelibly impact B cells. CSR has been shown to induce programmatic changes such as master transcription factor expression in an isotype-specific manner, and bias B cell differentiation. Isotype-specific cytoplasmic tails that possess varied BCR signaling capacity can result in transcriptional heterogeneity after antigen binding, which may prove to be increasingly important if antigen priming is critical to imprinting memory B cell responses. With the recent emergence of studies detailing immune effector functions of B cells sorted by isotype --specifically PCs, there are critical questions that remain to be addressed (see Outstanding Questions), and it will be exciting to see the outcome of such future investigations.
Outstanding Questions:
Can we design vaccine adjuvants that promote and/or boost antigen-specific PC generation in an isotype-specific manner?
What are the isotype-specific programmatic changes that are imprinted in B cells during CSR?
Is there differential regulation of isotypes within the GC that alters the resulting functional programming of a B cell?
Do memory response-derived PCs vary transcriptionally from primary PCs?
What parallels exist between isotype specific PCs and their secondary transcriptional programs between mice and humans?
Table I:
Mouse isotypes related to human isotypes
| Mouse | IgM | IgG3 | IgG1 | IgG2a/b | IgE | IgA |
| Human | IgM | IgG2 | IgG4 | IgG1/3 | IgE | IgA1/2 |
Highlights:
Signals eliciting class switch recombination can also imprint transcriptional changes in B cells retained through terminal differentiation
B cell class can influence cell fate determination and effector function
Class-linked PC effector function appears to sort according to the four broad categories of immune protection
Single cell analysis allows increased dissection of B cell heterogeneity
Acknowledgements
The work was supported by the National Institutes of Health (AI040215 and AI121393) to M.G.M-W.).
Glossary
- BREG (Regulatory B cell):
B cell subset implicated in immunosuppressive functions, e.g. maintenance of tolerance, reduction of inflammation and induction of TREGs. BREGs can secrete cytokines such as IL-10 and IL-35.
- Class Switch Recombination (CSR):
In naïve or memory B cells, CSR is an irreversible genetic deletion event that results in the expression of different constant heavy chain DNA sequences from the originally expressed mu (IgM), to other heavy chain segments (such as gamma, alpha and epsilon), which determine the class (isotype) of the B cell receptor and secreted antibody. This process is generally induced by T cell secreted cytokines.
- Cre-Lox Recombination:
genetic tool utilizing the Cre recombinase to excise specific sections of DNA that are flanked by targeting DNA sequences, i.e. Lox sequences. Cre-driven DNA deletion can occur after a selected gene is expressed, or under the control of a specific initiating factor such as tamoxifen.
- Extrafollicular Plasma Cell:
Formed outside a GC, this subset of PCs lacks affinity increasing mutations. These PCs can form with or without T cell help. They are largely considered short-lived PCs with a lifespan measured in days.
- Follicular Plasma Cell:
A cell which has formed through GC cycling. These PCs have undergone affinity maturation and TFH cell selection. They are largely considered long-lived PCs due to transcriptional changes likely incurred during GC cycling; can have a lifespan of decades
- Germinal Center (GC):
anatomical site in lymphoid tissue that forms after antigen encounter, consisting of activated B cells, follicular T helper Cells (TFH), and follicular dendritic cells. B cells undergo somatic hypermutation and affinity maturation guided by TFH selection, resulting in mutated antigen-specific memory B cells and PCs.
- Gene dropout:
In RNA sequencing, a term used to describe when genes with low expression are not seen in sequencing libraries and are thus reported as having no expression in a given sample. Gene dropout is difficult to separate from biologically determined zero expression and thus can create false negative values for genes.
- Innate Lymphoid Cell (ILC):
An innate immunity cell subset lacking antigen specific receptors. ILC subsets elicit a variety of immune responses based on secretion of cytokines under the direction of specific master transcription factors similar to T helper cell subsets.
- TFH (T Follicular Helper) Cell:
A Cxcr5+Bcl-6+CD4+ T cell that migrates to the T cell /B cell border in the GC, engaging in cognate contact with an antigen-specific B cell. TFH can then localize to a GC reaction, supporting select GC B cells for survival based on antigen affinity.
- TREG (Regulatory T Cell):
CD4+ T cell subset defined by the expression of the transcription factor Foxp3; it plays a critical role in the prevention of autoimmunity, maintenance of tolerance and the reduction of ongoing immune responses. TREGs secrete cytokines such as IL-10 that perform immunosuppressive activities, or which directly interact with target cells through surface molecules (e.g. CTLA-4).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Vinuesa CG et al. (2016) Follicular Helper T Cells. Annu Rev Immunol 34, 335–68. [DOI] [PubMed] [Google Scholar]
- [2].Qi H (2016) T follicular helper cells in space-time. Nat Rev Immunol 16 (10), 612–25. [DOI] [PubMed] [Google Scholar]
- [3].Mesin L et al. (2016) Germinal Center B Cell Dynamics. Immunity 45 (3), 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].McHeyzer-Williams M et al. (2011) Molecular programming of B cell memory. Nat Rev Immunol 12 (1), 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dufaud CR et al. (2017) Deconstructing the germinal center, one cell at a time. Curr Opin Immunol 45, 112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tellier J et al. (2016) Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat Immunol 17 (3), 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shi W et al. (2015) Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nat Immunol 16 (6), 663–673. [DOI] [PubMed] [Google Scholar]
- [8].Lam WY et al. (2018) Metabolic and Transcriptional Modules Independently Diversify Plasma Cell Lifespan and Function. Cell Rep 24 (9), 2479–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lau D et al. (2017) Low CD21 expression defines a population of recent germinal center graduates primed for plasma cell differentiation. Sci Immunol 2 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chevrier S et al. (2014) The BTB-ZF transcription factor Zbtb20 is driven by Irf4 to promote plasma cell differentiation and longevity. J Exp Med 211 (5), 827–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bohannon C et al. (2016) Long-lived antigen-induced IgM plasma cells demonstrate somatic mutations and contribute to long-term protection. Nat Commun 7, 11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hammarlund E et al. (2016) Durability of Vaccine-Induced Immunity Against Tetanus and Diphtheria Toxins: A Cross-sectional Analysis. Clin Infect Dis 62 (9), 1111–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Amanna IJ et al. (2007) Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med 357 (19), 1903–15. [DOI] [PubMed] [Google Scholar]
- [14].Slifka MK et al. (1998) Humoral immunity due to long-lived plasma cells. Immunity 8 (3), 363–72. [DOI] [PubMed] [Google Scholar]
- [15].Benner R et al. (1974) Antibody formation in mouse bone marrow.III. Effects of route of priming and antigen dose. Immunology 27 (5), 747–60. [PMC free article] [PubMed] [Google Scholar]
- [16].Landsverk OJ et al. (2017) Antibody-secreting plasma cells persist for decades in human intestine. J Exp Med 214 (2), 309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Peperzak V et al. (2013) Mcl-1 is essential for the survival of plasma cells. Nat Immunol 14 (3), 290–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Halliley JL et al. (2015) Long-Lived Plasma Cells Are Contained within the CD19(−)CD38(hi)CD138(+) Subset in Human Bone Marrow. Immunity 43 (1), 132–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Glatman Zaretsky A et al. (2017) T Regulatory Cells Support Plasma Cell Populations in the Bone Marrow. Cell Rep 18 (8), 1906–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nuñez S et al. (2016) The human thymus perivascular space is a functional niche for viral-specific plasma cells. Sci Immunol 1 (6), eaah4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lino AC et al. (2018) LAG-3 Inhibitory Receptor Expression Identifies Immunosuppressive Natural Regulatory Plasma Cells. Immunity 49 (1), 120–133 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Matsumoto M et al. (2014) Interleukin-10-Producing Plasmablasts Exert Regulatory Function in Autoimmune Inflammation. Immunity 41 (6), 1040–1051. [DOI] [PubMed] [Google Scholar]
- [23].Pelletier N et al. (2010) Plasma cells negatively regulate the follicular helper T cell program. Nat Immunol 11 (12), 1110–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].McHeyzer-Williams LJ et al. (2015) Class-switched memory B cells remodel BCRs within secondary germinal centers. Nat Immunol 16 (3), 296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Klenovsek K et al. (2007) Protection from CMV infection in immunodeficient hosts by adoptive transfer of memory B cells. Blood 110 (9), 3472–9. [DOI] [PubMed] [Google Scholar]
- [26].O'Shea JJ and Paul WE (2010) Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 327 (5969), 1098–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Scharer CD et al. (2018) Plasma cell differentiation is controlled by multiple cell division-coupled epigenetic programs. Nat Commun 9 (1), 1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Barwick BG et al. (2016) Plasma cell differentiation is coupled to division-dependent DNA hypomethylation and gene regulation. Nat Immunol 17 (10), 1216–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Turner CA Jr. et al. (1994) Blimp-1, a novel zinc finger-containing protein that can drive the maturation of B lymphocytes into immunoglobulin-secreting cells. Cell 77 (2), 297–306. [DOI] [PubMed] [Google Scholar]
- [30].Minnich M et al. (2016) Multifunctional role of the transcription factor Blimp-1 in coordinating plasma cell differentiation. Nat Immunol 17 (3), 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kallies A et al. (2007) Initiation of plasma-cell differentiation is independent of the transcription factor Blimp-1. Immunity 26 (5), 555–566. [DOI] [PubMed] [Google Scholar]
- [32].Shapiro-Shelef M et al. (2003) Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity 19 (4), 607–620. [DOI] [PubMed] [Google Scholar]
- [33].Shaffer AL et al. (2002) Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity 17 (1), 51–62. [DOI] [PubMed] [Google Scholar]
- [34].Carotta S et al. (2014) The transcription factors IRF8 and PU.1 negatively regulate plasma cell differentiation. J Exp Med 211 (11), 2169–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ochiai K et al. (2013) Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity 38 (5), 918–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Xu HP et al. (2015) Regulation of bifurcating B cell trajectories by mutual antagonism between transcription factors IRF4 and IRF8. Nat Immunol 16 (12), 1274–1281. [DOI] [PubMed] [Google Scholar]
- [37].Reimold AM et al. (2001) Plasma cell differentiation requires the transcription factor XBP-1. Nature 412 (6844), 300–7. [DOI] [PubMed] [Google Scholar]
- [38].Klein U et al. (2006) Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol 7 (7), 773–82. [DOI] [PubMed] [Google Scholar]
- [39].Shaffer AL et al. (2004) XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 21 (1), 81–93. [DOI] [PubMed] [Google Scholar]
- [40].Sciammas R et al. (2006) Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity 25 (2), 225–36. [DOI] [PubMed] [Google Scholar]
- [41].Taubenheim N et al. (2012) High Rate of Antibody Secretion Is not Integral to Plasma Cell Differentiation as Revealed by XBP-1 Deficiency. J Immunol 189 (7), 3328–3338. [DOI] [PubMed] [Google Scholar]
- [42].Pengo N et al. (2013) Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol 14 (3), 298–305. [DOI] [PubMed] [Google Scholar]
- [43].Lam WY et al. (2016) Mitochondrial Pyruvate Import Promotes Long-Term Survival of Antibody-Secreting Plasma Cells. Immunity 45 (1), 60–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mei HE et al. (2015) A unique population of IgG-expressing plasma cells lacking CD19 is enriched in human bone marrow. Blood 125 (11), 1739–48. [DOI] [PubMed] [Google Scholar]
- [45].Wang Y and Bhattacharya D (2014) Adjuvant-specific regulation of long-term antibody responses by ZBTB20. J Exp Med 211 (5), 841–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].O'Connor BP et al. (2004) BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med 199 (1), 91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Deenick EK et al. (1999) Switching to IgG3, IgG2b, and IgA is division linked and independent, revealing a stochastic framework for describing differentiation. J Immunol 163 (9), 4707–4714. [PubMed] [Google Scholar]
- [48].Hodgkin PD et al. (1996) B cell differentiation and isotype switching is related to division cycle number. J Exp Med 184 (1), 277–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tangye SG et al. (2003) A division-linked mechanism for the rapid generation of Ig-secreting cells from human memory B cells. J Immunol 170 (1), 261–9. [DOI] [PubMed] [Google Scholar]
- [50].Reth M (1992) Antigen receptors on B lymphocytes. Annu Rev Immunol 10, 97–121. [DOI] [PubMed] [Google Scholar]
- [51].Weiser P et al. (1997) Endosomal targeting and the cytoplasmic tail of membrane immunoglobulin. Science 276 (5311), 407–409. [DOI] [PubMed] [Google Scholar]
- [52].Kaisho T et al. (1997) The roles of gamma 1 heavy chain membrane expression and cytoplasmic tail in IgG1 responses. Science 276 (5311), 412–415. [DOI] [PubMed] [Google Scholar]
- [53].Achatz G et al. (1997) Effect of transmembrane and cytoplasmic domains of IgE on the IgE response. Science 276 (5311), 409–411. [DOI] [PubMed] [Google Scholar]
- [54].Kometani K et al. (2013) Repression of the transcription factor Bach2 contributes to predisposition of IgG1 memory B cells toward plasma cell differentiation. Immunity 39 (1), 136–147. [DOI] [PubMed] [Google Scholar]
- [55].Gitlin AD et al. (2016) Independent Roles of Switching and Hypermutation in the Development and Persistence of B Lymphocyte Memory. Immunity 44 (4), 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Blanc P et al. (2016) Mature IgM-expressing plasma cells sense antigen and develop competence for cytokine production upon antigenic challenge. Nat Commun 7, 13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Krautler NJ et al. (2017) Differentiation of germinal center B cells into plasma cells is initiated by high-affinity antigen and completed by Tfh cells. J Exp Med 214 (5), 1259–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Suan D et al. (2017) CCR6 Defines Memory B Cell Precursors in Mouse and Human Germinal Centers, Revealing Light-Zone Location and Predominant Low Antigen Affinity. Immunity 47 (6), 1142–1153. [DOI] [PubMed] [Google Scholar]
- [59].Ise W et al. (2018) T Follicular Helper Cell-Germinal Center B Cell Interaction Strength Regulates Entry into Plasma Cell or Recycling Germinal Center Cell Fate. Immunity 48 (4), 702–715 [DOI] [PubMed] [Google Scholar]
- [60].Shinnakasu R et al. (2016) Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol 17 (7), 861–869. [DOI] [PubMed] [Google Scholar]
- [61].Taylor JJ et al. (2015) Humoral immunity. Apoptosis and antigen affinity limit effector cell differentiation of a single naive B cell. Science 347 (6223), 784–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wang NS et al. (2012) Divergent transcriptional programming of class-specific B cell memory by T-bet and RORalpha. Nat Immunol 13 (6), 604–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Piovesan D et al. (2017) c-Myb Regulates the T-Bet-Dependent Differentiation Program in B Cells to Coordinate Antibody Responses. Cell Rep 19 (3), 461–470. [DOI] [PubMed] [Google Scholar]
- [64].Yang ZY et al. (2012) Fluorescent In Vivo Detection Reveals that IgE(+) B Cells Are Restrained by an Intrinsic Cell Fate Predisposition. Immunity 36 (5), 857–872. [DOI] [PubMed] [Google Scholar]
- [65].Talay O et al. (2012) IgE(+) memory B cells and plasma cells generated through a germinal-center pathway. Nat Immunol 13 (4), 396–404. [DOI] [PubMed] [Google Scholar]
- [66].He JS et al. (2013) The distinctive germinal center phase of IgE(+) B lymphocytes limits their contribution to the classical memory response. J Exp Med 210 (12), 2755–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Haniuda K et al. (2016) Autonomous membrane IgE signaling prevents IgE-memory formation. Nat Immunol 17 (9), 1109–1117. [DOI] [PubMed] [Google Scholar]
- [68].Nojima T et al. (2011) In-vitro derived germinal centre B cells differentially generate memory B or plasma cells in vivo. Nat Commun 2, 465. [DOI] [PubMed] [Google Scholar]
- [69].Yang Z et al. (2016) Regulation of B cell fate by chronic activity of the IgE B cell receptor. Elife 5, e21238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].He JS et al. (2017) IgG1 memory B cells keep the memory of IgE responses. Nature Communications 8, 641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Nimmerjahn F and Ravetch JV (2005) Divergent immunoglobulin G subclass activity through selective Fc receptor binding. Science 310 (5753), 1510–1512. [DOI] [PubMed] [Google Scholar]
- [72].Dekkers G et al. (2017) Affinity of human IgG subclasses to mouse Fc gamma receptors. Mabs 9 (5), 767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Bruhns P and Jonsson F (2015) Mouse and human FcR effector functions. Immunol Rev 268 (1), 25–51. [DOI] [PubMed] [Google Scholar]
- [74].Nimmerjahn F et al. (2010) FcgammaRIV deletion reveals its central role for IgG2a and IgG2b activity in vivo. Proc Natl Acad Sci U S A 107 (45), 19396–19401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Tao MH et al. (1993) Structural features of human immunoglobulin G that determine isotype-specific differences in complement activation. J Exp Med 178 (2), 661–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Klaus GGB et al. (1979) Activation of Mouse Complement by Different Classes of Mouse Antibody. Immunology 38 (4), 687–695. [PMC free article] [PubMed] [Google Scholar]
- [77].Strait RT et al. (2015) IgG1 protects against renal disease in a mouse model of cryoglobulinaemia. Nature 517 (7535), 501–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].van der Neut Kolfschoten M et al. (2007) Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science 317 (5844), 1554–1557. [DOI] [PubMed] [Google Scholar]
- [79].Karsten CM et al. (2012) Anti-inflammatory activity of IgG1 mediated by Fc galactosylation and association of FcgammaRIIB and dectin-1. Nat Med 18 (9), 1401–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Burton OT et al. (2014) Immunoglobulin E signal inhibition during allergen ingestion leads to reversal of established food allergy and induction of regulatory T cells. Immunity 41 (1), 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Crawford G et al. (2018) Epithelial damage and tissue gammadelta T cells promote a unique tumor-protective IgE response. Nat Immunol 19 (8), 859–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Mora JR and von Andrian UH (2009) Role of retinoic acid in the imprinting of gut-homing IgA-secreting cells. Seminars in Immunology 21 (1), 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Bunker JJ et al. (2015) Innate and Adaptive Humoral Responses Coat Distinct Commensal Bacteria with Immunoglobulin A. Immunity 43 (3), 541–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Moor K et al. (2017) High-avidity IgA protects the intestine by enchaining growing bacteria. Nature 544 (7651), 498–502. [DOI] [PubMed] [Google Scholar]
- [85].Fritz JH et al. (2011) Acquisition of a multifunctional IgA+ plasma cell phenotype in the gut. Nature 481 (7380), 199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Bermejo DA et al. (2013) Trypanosoma cruzi trans-sialidase initiates a program independent of the transcription factors ROR gamma t and Ahr that leads to IL-17 production by activated B cells. Nat Immunol 14 (5), 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wang S et al. (2017) Regulatory Innate Lymphoid Cells Control Innate Intestinal Inflammation. Cell 171 (1), 201–216 e18. [DOI] [PubMed] [Google Scholar]
- [88].Shen P et al. (2014) IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 507 (7492), 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Shalapour S et al. (2017) Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 551 (7680), 340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].McHeyzer-Williams LJ et al. (2018) Do Memory B Cells Form Secondary Germinal Centers? Impact of Antibody Class and Quality of Memory T-Cell Help at Recall. Cold Spring Harb Perspect Biol 10 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Shlomchik MJ (2018) Do Memory B Cells Form Secondary Germinal Centers? Yes and No. Cold Spring Harb Perspect Biol 10 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Pape KA and Jenkins MK (2018) Do Memory B Cells Form Secondary Germinal Centers? It Depends. Cold Spring Harb Perspect Biol 10 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Pape KA et al. (2011) Different B cell populations mediate early and late memory during an endogenous immune response. Science 331 (6021), 1203–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Krishnamurty AT et al. (2016) Somatically Hypermutated Plasmodium-Specific IgM(+) Memory B Cells Are Rapid, Plastic, Early Responders upon Malaria Rechallenge. Immunity 45 (2), 402–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Pape KA et al. (2018) Naive B Cells with High-Avidity Germline-Encoded Antigen Receptors Produce Persistent IgM(+) and Transient IgG(+) Memory B Cells. Immunity 48 (6), 1135–1143 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Weisel FJ et al. (2016) A Temporal Switch in the Germinal Center Determines Differential Output of Memory B and Plasma Cells. Immunity 44 (1), 116–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Jones DD et al. (2015) Cellular Dynamics of Memory B Cell Populations: IgM(+) and IgG(+) Memory B Cells Persist Indefinitely as Quiescent Cells. Journal of Immunology 195 (10), 4753–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Dogan I et al. (2009) Multiple layers of B cell memory with different effector functions. Nat Immunol 10 (12), 1292–9. [DOI] [PubMed] [Google Scholar]
- [99].Zuccarino-Catania GV et al. (2014) CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol 15 (7), 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Nair N et al. (2016) High-dimensional immune profiling of total and rotavirus VP6-specific intestinal and circulating B cells by mass cytometry. Mucosal Immunol 9 (1), 68–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Wong MT et al. (2015) Mapping the Diversity of Follicular Helper T Cells in Human Blood and Tonsils Using High-Dimensional Mass Cytometry Analysis. Cell Rep 11 (11), 1822–1833. [DOI] [PubMed] [Google Scholar]
- [102].Good Z et al. (2018) Single-cell developmental classification of B cell precursor acute lymphoblastic leukemia at diagnosis reveals predictors of relapse. Nat Med 24 (4), 474–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Polikowsky HG et al. (2015) Cutting Edge: Redox Signaling Hypersensitivity Distinguishes Human Germinal Center B Cells. J Immunol 195 (4), 1364–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Horns F et al. (2016) Lineage tracing of human B cells reveals the in vivo landscape of human antibody class switching. Elife 5, e16578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Lee J et al. (2016) Molecular-level analysis of the serum antibody repertoire in young adults before and after seasonal influenza vaccination. Nature Medicine 22 (12), 1456–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Ledergor G et al. (2018) Single cell dissection of plasma cell heterogeneity in symptomatic and asymptomatic myeloma. Nat Med 24 (12), 1867–1876. [DOI] [PubMed] [Google Scholar]
- [107].Milpied P et al. (2018) Human germinal center transcriptional programs are de-synchronized in B cell lymphoma. Nat Immunol 19 (9), 1013–1024. [DOI] [PubMed] [Google Scholar]