

Abstract
Highly conserved TREX-mediated mRNA export is emerging as a key pathway in neuronal development and differentiation. TREX subunit variants cause neurodevelopmental disorders (NDDs) by interfering with mRNA export from the cell nucleus to the cytoplasm. Previously we implicated four missense variants in the X-linked THOC2 gene in intellectual disability (ID). We now report an additional six affected individuals from five unrelated families with two de novo and three maternally-inherited pathogenic or likely pathogenic variants in THOC2 extending the genotypic and phenotypic spectrum. These comprise three rare missense THOC2 variants that affect evolutionarily conserved amino acid residues and reduce protein stability and two with canonical splice-site THOC2 variants that result in C-terminally truncated THOC2 proteins. We present detailed clinical assessment and functional studies on a de novo variant in a female with an epileptic encephalopathy and discuss an additional four families with rare variants in THOC2 with supportive evidence for pathogenicity. Severe neurocognitive features, including movement and seizure disorders were observed in this cohort. Taken together our data show that even subtle alterations to the canonical molecular pathways such as mRNA export, otherwise essential for cellular life, can be compatible with life, but lead to NDDs in humans.
Keywords: XLID, THOC2, mRNA export, protein stability, partial loss-of-function variants
INTRODUCTION
Intellectual disability (ID), characterized by substantial limitations in both intellectual functioning and adaptive behaviour, affects 1-3% of the population starting before the age of 18 years and has significant impact on individuals, families and communities (Vissers, et al., 2016). Individuals with ID are more likely than members of the general population to experience poor physical and mental health, have a lower life expectancy, experience inequalities accessing health care and frequently have limited or no specific therapies for their core symptoms (Bittles, et al., 2002; Hosking, et al., 2016). Both genetic and environmental factors contribute to the development of ID (Milani, et al., 2015). Over 120 of the identified >800 ID genes are located on the X-chromosome (Chiurazzi and Pirozzi, 2016; Schwartz, 2015), and diagnosis of X-linked causes of ID remain critically important for accurate genetic counseling of families (Ropers and Hamel, 2005). Dramatic improvements in high-throughput DNA sequencing technologies and analyses software has led to identification of new ID genes and additional variants in the known ID genes (Dickinson, et al., 2016; Vissers, et al., 2016). A systematic review of clinical data suggests that ID affected individuals frequently have comorbid neurological, psychiatric and behavioural disorders (Oeseburg, et al., 2011; Vissers, et al., 2016), and disease variants in different parts of a gene can lead to a broad range of complex neurocognitive disorders (Palmer, et al., 2017; Zhu, et al., 2014). This complexity contributes to heterogeneity in clinical symptoms and indistinct boundaries between syndromic and non-syndromic forms of NDD.
In 2015, we reported genetic, molecular and protein structural data on four missense variants in an X-linked essential gene THOC2 (MIM# 300957; NM_001081550.1; c.937C>T (p.Leu313Phe), c.1313T>C (p.Leu438Pro), c.2399T>C (p.Ile800Thr) and c.3034T>C (p.Ser1012Pro), RNA not analysed) (Kumar, et al., 2015). The affected individuals had a syndromic NDD, characterized by borderline to severe ID, speech delay, short stature and adult onset truncal obesity (Kumar, et al., 2015). THOC2 encodes for the THOC2 protein - the largest subunit of the highly conserved TREX (Transcription-Export) mRNA export complex essential for exporting mRNA from the cell nucleus to the cytoplasm (Heath, et al., 2016). The TREX complex is composed of a THO sub-complex (THOC1, THOC2, THOC3, THOC5 and THOC7) and accessory proteins (UAP56, UIF, Aly, CIP29, PDIP3, ZC11A, SRRT, Chtop) (Heath, et al., 2016). The TREX complex, besides its canonical role in mRNA export in the mammalian cells, has been shown to play critical roles in gene expression, 3’ mRNA processing, stress responses, mitotic progression and genome stability as well as developmental processes such as pluripotency maintenance and hematopoiesis (Yamazaki, et al., 2010). We and others have recently demonstrated that subtle perturbations in mRNA export by gene variants or preferential cytoplasmic aggregation can lead to NDDs (Beaulieu, et al., 2013; Coe, et al., 2014; Kumar, et al., 2015), neurodegeneration (Woerner, et al., 2016) or cancer (Chinnam, et al., 2014; Hautbergue, 2017; Liu, et al., 2015; Viphakone, et al., 2015). These alterations can have tissue-specific effects as TREX subunits are shown to have tissue-specific roles; for example, mouse Thoc5 and Thoc1 deficiency interferes with the maintenance of hematopoiesis (Guria, et al., 2011; Mancini, et al., 2010) and testis development (Wang, et al., 2009). Taken together, altered TREX function can have diverse molecular and cellular consequences resulting in a range of diseases. Here we present detailed information on the clinical presentations and functional investigations on an additional eight missense and two splice THOC2 variants. These data reaffirm and extend our previous findings that THOC2 variation plays a role in complex neurodevelopmental conditions with the core clinical presentation of ID.
MATERIALS AND METHODS
Molecular and cellular studies
RNA extraction, RT-qPCR (primers listed in Supp. Table S3), cycloheximide chase, and THOC2 immunofluorescence staining were performed as reported previously (Kumar, et al., 2015). Molecular studies on the THOC2 exon35:c.4450-2A>G variant were performed using blood DNA and skin fibroblasts derived from the affected individual and his heterozygous carrier mother. Genomic DNA or cDNA (generated by reverse transcribing the fibroblast total RNAs using Superscript III reverse transcriptase; Life Technologies) was amplified with KAPA HiFi PCR Kit (Kapa Biosystems) using hTHOC2-4326F/ hTHOC2-4519-R (Supp. Table S3) at 95°C for 3 min, 35 cycles of 98°C-15sec, 59°C-15sec, 72°C-30sec, incubation at 72°C for 10 min, gel purified (MinElute Gel Extraction kit (Qiagen) and Sanger sequenced using the same primers. For the THOC2 exon28:c.3503+4A>C, blood gDNA from unaffected father, carrier mother and affected son was amplified with TaKaRa ExTaq using THOC2-F/THOC2-R primers (Supp. Table S3) at 94°C for 2 min, 40 cycles of 94°C-30sec, 60°C-30sec, 72°C-30sec, incubation at 72°C for 5 min. The cDNA was generated by reverse transcribing the white blood cell RNAs using Superscript III reverse transcriptase (Life Technologies) and amplified with TaKaRa ExTaq using THOC2-ex27F/THOC2-ex30R (Supp. Table S3) at 94°C for 2 min, 28 cycles of 94°C-30sec, 60°C-30sec, 72°C-30sec, incubation at 72°C for 5 min. The amplified products were analyzed by Sanger sequencing.
Generation of THOC2 variant expression constructs
Generation of the wild type Myc-tagged human THOC2 expression plasmid was reported earlier (Kumar, et al., 2015). Briefly, the THOC2 variants were introduced into the existing pCMV-Myc-THOC2 expression construct by overlap PCR method using the primers listed in Supp. Table S3. The variant plasmid sequences were confirmed by Sanger sequencing. Details relating generation of the THOC2 variant expression constructs are available on request.
Transient expression and Western blotting
For transient expression experiments, HEK293T and HeLa cells were transfected with expression constructs (400 ng pCMV-Myc-THOC2 plasmid and 400 ng pEGFP-C1 plasmid/transfection for stability and cycloheximide assays and 4 μg/transfection for immunofluorescence staining, IF) using Lipofectamine 3000 reagent according to manufacturer’s protocol (Life technologies). Twenty-four hours post-transfection, cells were either fixed with 4% formaldehyde for IF or collected and lysed in buffer containing 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% Triton-X 100, 1 mM EDTA, 50 mM NaF, 1× Protease inhibitor/no EDTA and 0.1 mM Na3VO4 for western blot assay as reported previously (Kumar, et al., 2015).
In silico pathogenicity prediction
We used CADD v1.3 (includes Phylop, GERP++ & PolyPhen2) (Kircher, et al., 2014), Provean (Choi and Chan, 2015) and ACMG (Richards, et al., 2015) on-line tools for in silico prediction of the pathogenicity of different variants (Table 1, Supp. Table S1).
TABLE 1.
Detailed description of the THOC2 variants with supporting molecular evidence.
| Individual | From | Method of identification | Position hg19 | NM_001081550 | Mode of inheritance | CADD | Provean score |
Provean prediction |
GERP++ | Phylop | gnomAD frequency |
Polyphen2* | Variant plasmid tested |
Reduced Protein Stability |
ACMG pathogenicity classification** |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Missense | Kumar et al 2015 AJHG | X-Chr Exome sequencing | 122799566 | c.1313T>C: p.Leu438Pro | Maternal inheritance | 28.1 | −6.08 | Deleterious | 5.7 | 1.902 | absent | D | YES | YES | LP | |
| 1 | Australia | Trio WES | 122767853 | c.2087C>T: p.Thr696Ile | De novo | 27.4 | −5.47 | Deleterious | 5.03 | 0.963 | absent | D | YES | YES | DP | |
| 2 | USA | WES | 122766890 | c.2138G>A: p.Gly713Asp | De novo | 31 | −4.69 | Deleterious | 5.73 | 2.412 | absent | D | YES | YES | DP | |
| Kumar et al 2015 AJHG | X-Chr Exome sequencing | 122765621 | c.2399T>C: p.Ile800Thr | Maternal inheritance | 23.9 | −4.01 | Deleterious | 5.97 | 2.016 | absent | P | YES | YES | LP | ||
| 3-4 | Canada/Germany/Russia Identical twins | WES | 122757079 | c.3559C>T: p.His1187Tyr | Maternal inheritance; Mother skewed (99.9:0.1%) | 23.1 | −5.07 | Deleterious | 6.07 | 2.571 | absent | P | YES | YES | LP | |
| Splice | 5 | Japan | Trio WES | 122757634 | Exon28:c.3503+4A>C | Maternal inheritance; Mother skewed 98:2% | 10.8 | N/A | N/A | 5.57 | 1.86 | absent | N/A | N/A | ND | LP |
| 6 | Canada | WES | 122747561 | Exon35:c.4450-2A>G | Maternal inheritance; Mother skewed 94:6% | 23.7 | N/A | N/A | 5.25 | 1.735 | absent | N/A | Fibroblasts of the affected male proband and carrier mother | NO | LP | |
| Missense | 7 | USA Epi4K Consortium & Epilepsy Phenome/Genome Project; Nature 501:217-221, 2013 | WES | 122778639 | c.1550A>G: p.Tyr517Cys | De novo | 26.6 | −7.87 | Deleterious | 5.84 | 1.955 | absent | D | YES | YES | DP |
Probably damaging, D; Possibly damaging, P; N/A, not applicable; ND, not determined;
De novo Pathogenic, DP; Likely Pathogenic, LP
RESULTS
Identification of THOC2 variants
We previously implicated four missense THOC2 variants in 25 individuals with ID and a range of other clinical features (Table 1, Supp. Table S1-2) (Kumar, et al., 2015). We identified an additional five THOC2 variants (three missense; de novo c.2087C>T (p.Thr696Ile), de novo c.2138G>A (p.Gly713Asp), maternally-inherited c.3559C>T (p.His1187Tyr), and two splicing-defective; maternally-inherited chrX:122747561 exon35:c.4450-2A>G and chrX:122757634 exon28:c.3503+4A>C; GenBank: NM_001081550.1) variants in a further six affected individuals, including one pair of monozygotic twins (Table 1, Figures 1–2). Whole exome (WES) or whole genome sequencing (WGS) of probands and parents was used to identify the variants that were confirmed by Sanger sequencing of the PCR amplified variant-carrying region of genomic DNA of the parents and affected individuals. The previously unreported THOC2 variants affect amino acids that are highly conserved (Supp. Figure S1), are absent in gnomAD database and are predicted to be pathogenic based on a number of in silico analyses tools (Table 1). We included in our study a de novo missense p.Tyr517Cys variant in a female with moderate-severe ID, speech problems, epileptic encephalopathy, cortical visual impairment, and gait disturbances identified using WES as part of the Epi4K Consortium & Epilepsy Phenome/Genome Project (Epi, et al., 2013) (Table 1). We have also collected further, rare and potentially pathogenic variants through international collaboration (Table1, Supp. Table S1-2, Figures 1 and 3, see Supplementary information for methods used for identifying the variants) and performed functional testing on several of these. The following three variants: c.229C>T (p.Arg77Cys), c.3034T>C (p.Ser1012Pro) and c.3781A>C (p.Asn1261His) showed no clear evidence of altered stability of variant THOC2 proteins in our cell-based assay. The reported variants have been submitted to ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/accession numbers SCV00680065-SCV000680074).
FIGURE 1.
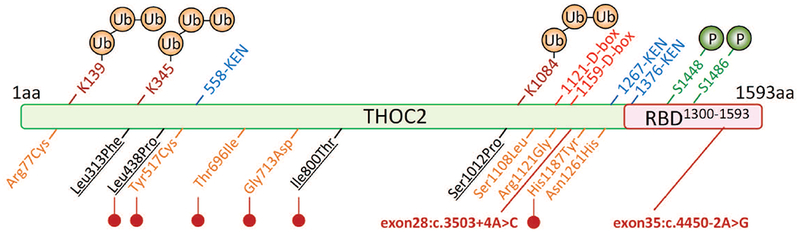
Location of variant amino acids and structural features in THOC2 protein. Ubiquitinated (K139 and K1084 (Kim, et al., 2011) and K345 (Lopitz-Otsoa, et al., 2012; Wagner, et al., 2011)), phosphorylated (S1448 and 1486 (Olsen, et al., 2006)) amino acid residues, potential RNA binding domain (RBD) and destruction box (D-box) and KEN box sequences that interact with the Anaphase Promoting Complex/Cyclosome (APC/C) for protein ubiquitination and subsequent destruction by the proteasome (Morgan, 2013) are shown. Unreported (orange) and published (black: (Kumar, et al., 2015)) missense variants effecting THOC2 protein stability are marked with red lollipops. The positions of two splice variants are shown in red.
FIGURE 2.
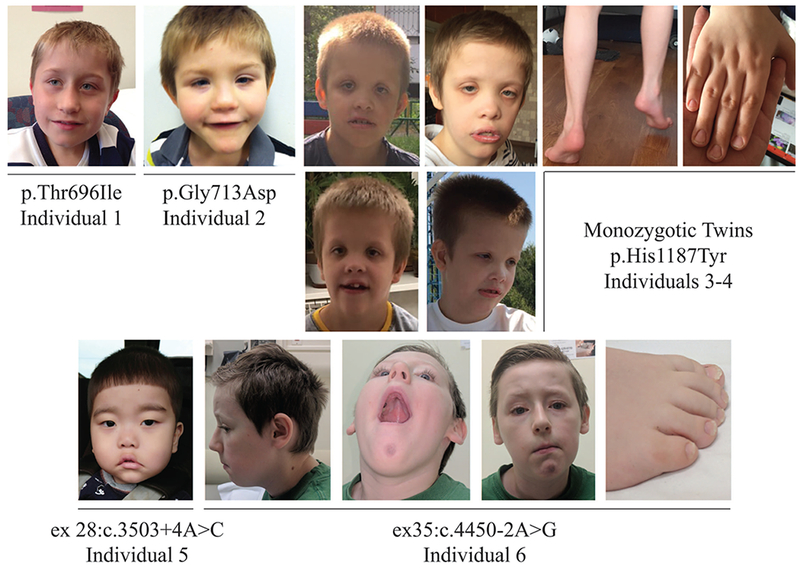
Front and side facial views of the affected individuals with THOC2 variants.
FIGURE 3.
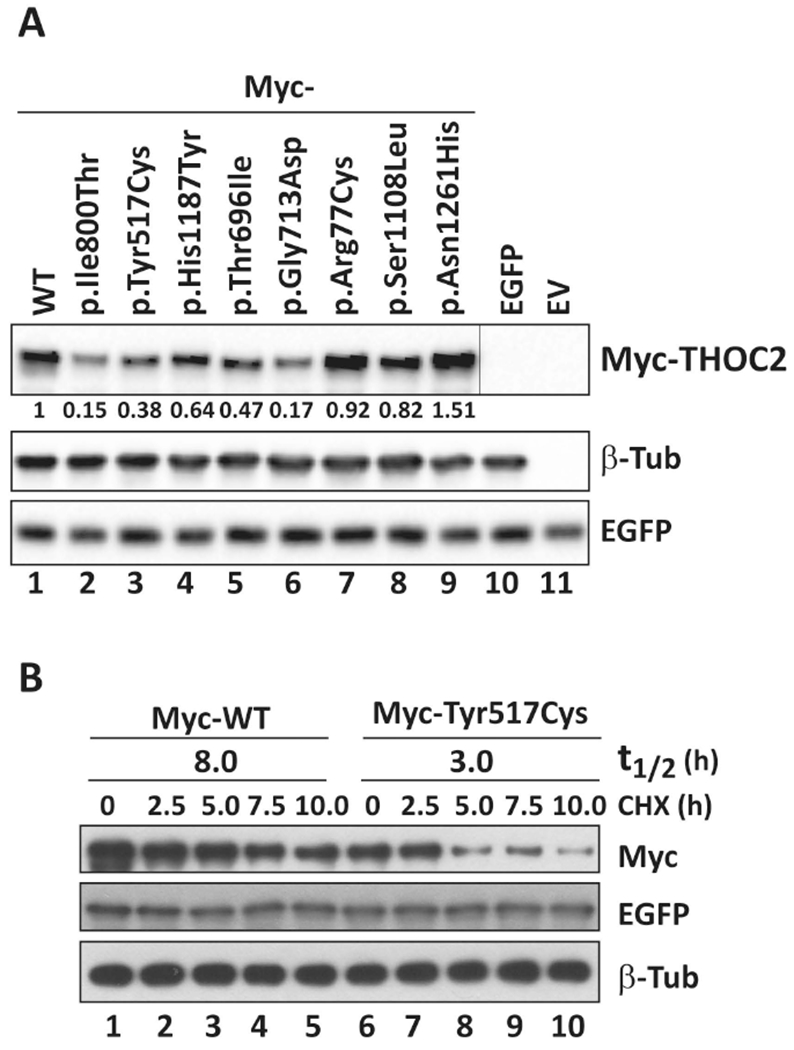
Functional testing of THOC2 missense variants. A: THOC2 variant protein stability is reduced in HEK293T cells. pCMV-Myc-THOC2 wild-type or variant expression constructs and pEGFP-C1 plasmid (transfection control) were transfected into HEK293T cells. Total protein lysates of cells 24hr post-transfection were analysed by western blotting with mouse anti-Myc (clone 9E10; Sigma), mouse anti-EGFP (clones 7.1 and 13.1; Roche) and rabbit anti-β-tubulin (loading control; Abcam) antibodies. pCMV-Myc-THOC2 p.Ile800Thr construct expressing the p.Ile800Thr protein shown to have reduced stability was used as a control (Kumar, et al., 2015). Western blot signals were quantified using ImageJ software. Averages of the Myc-THOC2 proteins normalised to the housekeeping β-tubulin signal from two independent runs are shown. B: Myc-p.Tyr517Cys THOC2 protein half-life is substantially reduced in HEK293T cells. pCMV-Myc-THOC2 or pCMV-Myc-THOC2-p.Tyr517Cys expression constructs and pEGFP-C1 plasmid (transfection control) were transfected into HEK293T cells. Next day the cells were cultured in the presence of 100 μg/ml translation inhibitor cycloheximide and harvested at the time points shown. Total protein lysates were analysed by western blotting with mouse anti-Myc, mouse anti-EGFP and rabbit anti-β-tubulin (loading control) antibodies.
Clinical presentations
The clinical features of the five previously unreported affected individuals with (likely) pathogenic THOC2 variants, aged between 3 and 12 years, and the 10 year old female with de novo p.Tyr517Cys variant are summarised in Table 2 and photographs, when available, are shown in Figure 2. Detailed clinical information is available in the supplementary data. Each individual clinical centre used local diagnostic criteria for determining degree of ID and diagnoses of co-morbidities. ID was universal and at least moderate in severity: 2/7 were non-ambulatory and 3/7 non-verbal. Behavioural problems were reported in four individuals, with one meeting diagnostic criteria for autism spectrum disorder. Additional neurological features were common. Four of the seven had infantile hypotonia and two of the seven had tremor. The monozygotic twins (individuals 3-4) had a tendency to toe walking, which was considered behavioural as it was not associated with neurological signs of lower limb spasticity. Confirmed seizure disorder was only present in the affected female (individual 7) but suspected in individual 2. Neuro-radiological studies were performed in five individuals: this was within normal limits for three individuals, with neuroanatomical differences reported in two. Individual 2 had complex neuroanatomical findings (see supplementary clinical description and Supp. Figure S2A) including changes in cortical gyral morphology, which in the inferior temporal lobes appeared finely nodular, as well as hypoplasia of the corpus callosum and reduced brainstem volume and individual 5 had mild dilatation of the lateral ventricles, mildly delayed myelination and an abnormal white matter lesion in the periventricular area close to the anterior horn (Supp. Figure S2B). Growth abnormalities were common including low birth weight (3/7), microcephaly (2/7) and short stature (2/7). Facial features are shown in Figure 2. Appropriate consent for reporting variants, clinical data and photographs of the affected individuals was obtained from their parents or legal guardians. The research has been approved by the Women’s and Children’s Health Network Human Research Ethics Committee in Adelaide, Australia.
TABLE 2.
Summary of clinical data of THOC2 variants with supporting molecular evidence.
| Individual | (likely) pathogenic | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |||
| Variant details | c.2087C>T: p.Thr696Ile | c.2138G>A: p.Gly713Asp | c.3559C>T: p.His1187Tyr Twin 1 | c.3559C>T: p.His1187Tyr Twin 2 | Exon28: c.3503+4A>C | Exon35:c.4450-2A>G | c.1550A>G: p.Tyr517Cys | ||
| Gender | Male | Male | Male | Male | Male | Male | Female | ||
| Age (years) | 12 | 5 | 7 | 7 | 3 | 10 | 10 | ||
| Perinatal features | |||||||||
| Gestation (weeks) | 36 | 37 | 37 | 37 | 37 | 41 | NA | ||
| Low birth weight (<2.5kg) | Yes | No | Yes | Yes | No | No | NA | ||
| Birth weight (g) | 2000 | 2650 | 1990 | 2420 | 3018 | 4365 | NA | ||
| Neurologic features | |||||||||
| Intellectual disability | Severe | Mod+ | Mod+ | Mod+ | Severe | Severe | Profound | ||
| Speech delay | Yes, single words, signs | Yes, non-verbal | Yes | Yes | Yes, non-verbal | Yes, nasal dysarthria | Yes, non-verbal | ||
| Hypotonia | No | Yes | NA | NA | Yes | Yes, central hypotonia | Yes | ||
| Spasticity | No | No | No | No | No | Yes- appendicular spasticity | No | ||
| Hyperkinesia | No | No | Yes | Yes | Yes | No | No | ||
| Tremor | No | Yes, intermittent | No | No | No | Yes | No | ||
| Epilepsy | No | Suspected | No | No | No | No | Yes, epileptic encephalopathy | ||
| Gait disturbances | No | Yes, gait/balance problems | Yes, toe walking* | Yes, toe walking* | Non ambulatory | Yes, ataxia/broad based gait | Non ambulatory | ||
| Behaviour problems | No | Yes | Yes | Yes | Yes, ASD | No | NR | ||
| Anxiety | No | No | No | No | No | No | NR | ||
| Depression | No | No | No | No | No | No | NR | ||
| Brain MRI/CT | MRI normal | Thin corpus callosum, low brainstem volume, variability in gyral pattern. | ND | ND | Ventricular dilatation, delayed myelination, peri-ventricular white matter lesion | MRI within normal limits | MRI normal | ||
| Growth parameters | |||||||||
| Microcephaly (≤3%) | Yes | Yes,< 1% | No | No | No | Yes,2% | No, 5% | ||
| Short stature (≤3%) | Yes | Yes | No | No | No | No | No | ||
| Overweight (BMI≥25) | No | No | No | No | No | No | No | ||
| Broad high forehead | Yes | Yes | Yes | Yes | No | No | NR | ||
| Other features | Mild joint laxity, subluxed hips, disordered sleep, feeding difficulties (g-tube dependency), laryngomalacia, micrognathia, abnormal palmar creases | Noonan facies, pes planus, hypospadias | Noonan facies, pes planus, hypospadias | Clinodactyly, nystagmus, abnormality soft palate | Cortical visual impairment | ||||
Abbreviations: %, centile; ASD, autism spectrum disorder; CT, computerised tomography scan; g-tube, gastrostomy tube; mod+, at least moderate severity;
MRI, magnetic resonance imaging; ND, not done; NA, not available; NR, not reported; NICU, neonatal intensive care unit;
VOUS, variant of uncertain significance.
Toe walking in absence of neurological signs of lower limb spasticity, therefore considered a behavioural manifestation.
THOC2 variant protein localisation and stability
Without access to affected individuals’ derived cells, we generated Myc-tagged THOC2 missense variant expression constructs to determine protein stability and localisation. The THOC2 protein stability was determined in HEK293T cells and localisation in both the HEK293T and HeLa cells. Total protein lysates of HEK293T cells ectopically-expressing the wild type or variant Myc-THOC2 proteins were western blotted for THOC2, EGFP and β-Tubulin. We used HEK293T cells expressing Myc-p.Ile800Thr THOC2 as a control for protein stability assay as this variant is shown to cause reduced protein stability (Kumar, et al., 2015). The results showed reduced stability of p.Tyr517Cys, p.Thr696Ile, p.Gly713Asp and p.His1187Tyr THOC2 compared to the wild type protein (Figure 3A). Presence of comparable levels of EGFP in the cells transfected with different expression constructs indicated that the reduced levels of THOC2 protein were not due to difference in transfection efficiency (Figure 3A). We also determined the turnover rate of Myc-p.Tyr517Cys THOC2 protein by cycloheximide chase. For this assay, the HEK293T cells transfected with pCMV-Myc-WT or pCMV-Myc-p.Tyr517Cys THOC2 and pEGFP-C1 transfection control plasmids were cultured in presence of translation inhibitor cycloheximide for different durations and western blotted for THOC2, EGFP and β-Tubulin. The results showed that p.Tyr517Cys THOC2 turnover rate was 3h compared with 8h for the wild type protein (Figure 3B). THOC2 variant proteins, similar to the wild type, were mainly localised to the nucleus in both the HEK293T and HeLa cells (Supp. Figure S3).
THOC2 splice variant: exon35:c.4450-2A>G, p.Arg1483fs52*
Sanger sequencing of amplified target region from affected son and mother’s blood genomic DNA showed that the affected boy inherited chrX:122747561 exon35:c.4450-2A>G variant from his unaffected heterozygous carrier mother (Figure 4C). A −2 A>G change in the intron-exon splicing site boundary (acceptor AG) is predicted to abolish splicing (Ohno, et al., 2018). To validate this possibility, we generated skin fibroblast cultures from the heterozygous carrier mother and the affected son. We PCR amplified their fibroblast cDNAs using primers with binding sequences located within exon 34 and 35. Amplification of a 194 bp DNA fragment from the mother indicated normal splicing but a 537 bp product from the affected son indicated retention of the intron located between these exons (Figure 4A-B). We confirmed this result by Sanger sequencing of the PCR products generated from genomic DNA that showed presence of A/G nucleotides in the carrier mother but only G (A>G) nucleotide in the affected son (Figure 4C). The cDNA sequence showed presence of normally-spliced mRNA in the mother but retention of intronic sequence upstream of the exon 35 in the affected son indicating defective splicing due to presence of −2 G variant at the intron-exon 35 junction sequence (Figure 4C). The presence of normally-spliced mRNA in the unaffected mother is consistent with X-inactivation (94% skewing) of the variant allele in her fibroblasts. We predicted that a retention of intron between exon 34-35 in the affected fibroblasts would result in loss of 110 C-terminal amino acids of the 1593 wild type THOC2 protein (that is, 1483 amino acids); however, overall the variant protein would be 58 amino acid smaller as it would now be a 1535 amino acid protein comprised of 1483 amino acids of wild type THOC2 and 52 translated from intronic sequence in the defective mRNA (Figure 4A). Consistent with our prediction, the western blot data showed presence of a slightly smaller THOC2 protein band in the affected son’s fibroblasts than his unaffected mother. Many independent western blot runs showed presence of two closely-located THOC2 bands - similar to the fruit fly THO2 (Rehwinkel, et al., 2004) - in the unaffected mother but a single highly intense band in the affected son’s fibroblasts (Figure 4D). The observed difference in levels of THOC2 protein was post-translational as we found comparable amounts of THOC2 mRNA, as assayed by real time RT-qPCR, in the mother and son (Figure. 4E). Finally, we observed no difference in THOC2 localisation in fibroblasts of the affected son and his unaffected mother (Figure 4F).
FIGURE 4.
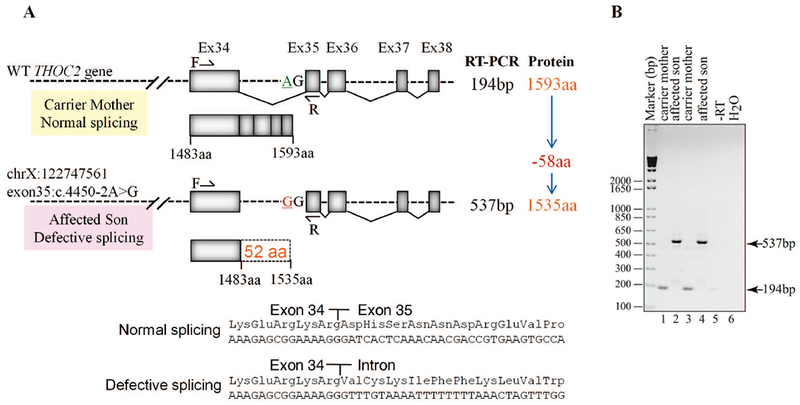
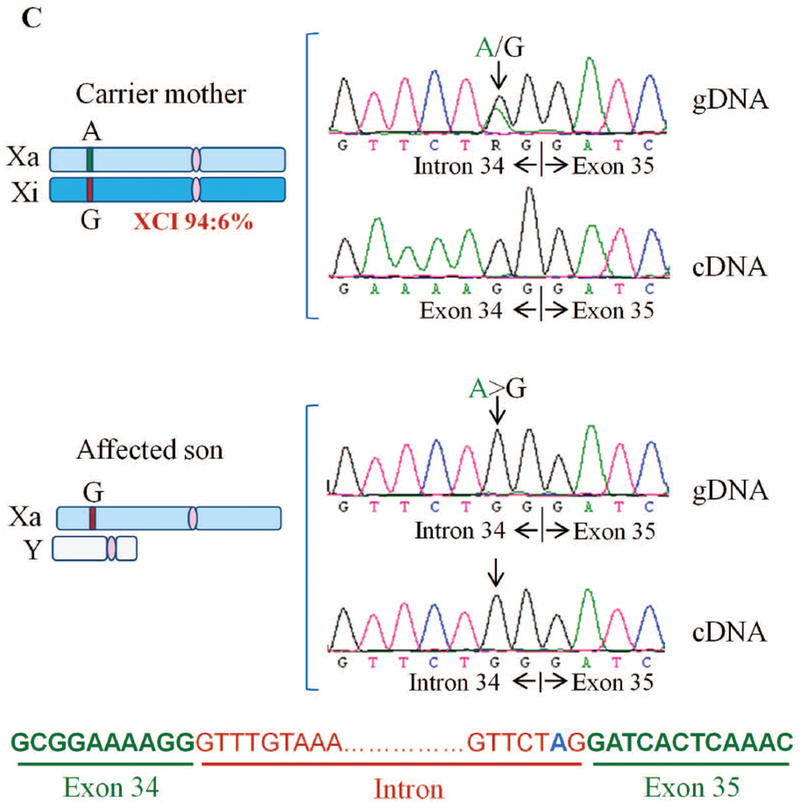
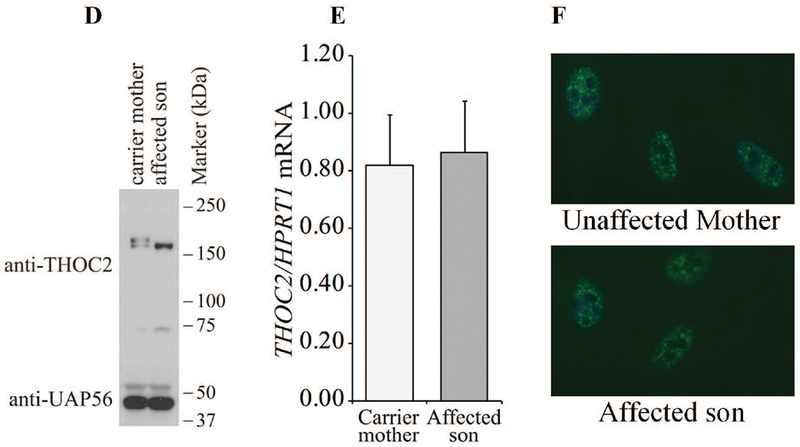
Exon35:c.4450-2A>G variant abolishes splicing of intron between exons 34-35. A: Part of the THOC2 gene showing location of the A/G nucleotide in the heterozygous carrier mother and A>G splice variant in the affected son. The C-terminal part of the 1593 amino acid wild type and 1535 amino acid (that contains 1483 normal and 52 amino acids coded by the unspliced intron) THOC2 protein in the affected boy are also shown. B: Gel showing a 194 bp RT-PCR product from the normally-spliced mRNA of the heterozygous carrier mother and a 537bp product from defective splicing of mRNA causing retention of an intron between exon 34-35 in the affected son. RT-PCR products from total RNA isolated from passage 3 (lanes 1-2) and 5 (lanes 3-4) fibroblasts. Location of the forward and reverse primers within exons 34 and 35 is shown. C: Sanger sequencing chromatograms of PCR products amplified from genomic and cDNA of the affected son and his heterozygous carrier mother using primers located within exon 34 and 35. Genomic DNA around the Exon34-Intron-Exon35 region is shown. D: Western blot showing THOC2 protein in the affected son and his carrier mother’s skin fibroblasts. TREX subunit UAP56 was used as a loading control. E: RT-qPCR showing levels of the THOC2 mRNA in the affected son and his carrier mother’s skin fibroblasts. F: Immunofluorescence detection of THOC2 in skin fibroblasts of the unaffected mother and affected son.
THOC2 splice variant: exon28:c.3503+4A>C, p.Gly1168fs7* and normal 1593 aa protein
For the second splice variant chrX:122757634 exon28:c.3503+4A>C, molecular studies were performed on the white blood cells of the unaffected father, carrier mother and the male proband. Sanger sequencing of target region amplified from the unaffected father and mother, and affected son’s genomic DNA showed that the affected son inherited the A>C change from his unaffected carrier mother who had A/C nucleotides at this position (Figure 5). The intronic nucleotide change A>C at +4 position of the 5’ exon-intron donor splicing site sequence is predicted to cause aberrant splicing (https://www.med.nagoya-u.ac.jp/neurogenetics/SD_Score/sd_score.html). To confirm this possibility, we amplified cDNA generated by reverse transcribing blood RNA of the father, mother and the affected son using primers located within exon 27 and exon 30 (Figure 5A; Supp. Table S3). Interestingly, whereas a 491 bp PCR product was observed in highly skewed carrier mother (98:2%) and normal father, 491 bp and 634 bp PCR products were detected in the affected son. A 491 bp amplified product indicated normal splicing in the mother and father, and 491 bp and 634 bp bands suggested partially-defective splicing in the affected son. Amplification of a 634 bp instead of a 994 bp fragment that would have resulted from a complete retention of intron between exon 28-29 indicated aberrant splicing event in the affected son (Figure 5). Sanger sequencing of 491 bp and 634 bp PCR products from the mother, father and son confirmed normal splicing in the mother and father and aberrant splicing in the affected son. The sequence showed retention of a 143 bp instead of complete 503 bp fragment due to activation of a cryptic splice site within the intron between exon 28-29 in the son (Figure 5). Retention of 143 bps from intron between exon 28-29 in the mRNA is predicted to result in a 1175 amino acid truncated THOC2 protein containing 1168 wild type amino acids and 7 amino acids from the translation of the intronic sequence retained in the defective mRNA. This aberrant product would be present in addition to the wild type 1593 amino acid protein from the normally-spliced mRNA in the affected son.
FIGURE 5.
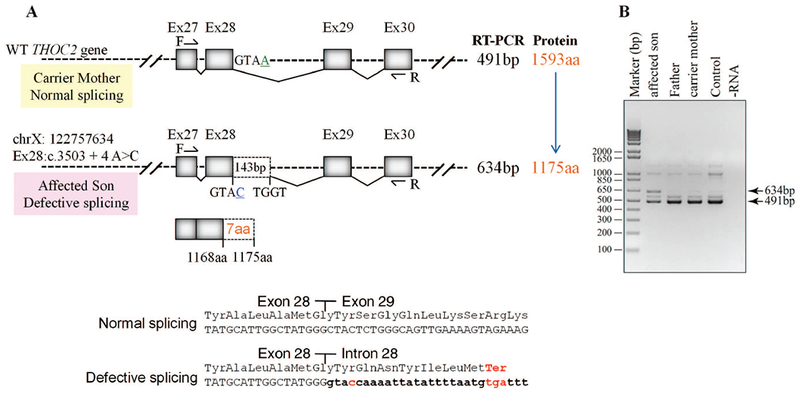
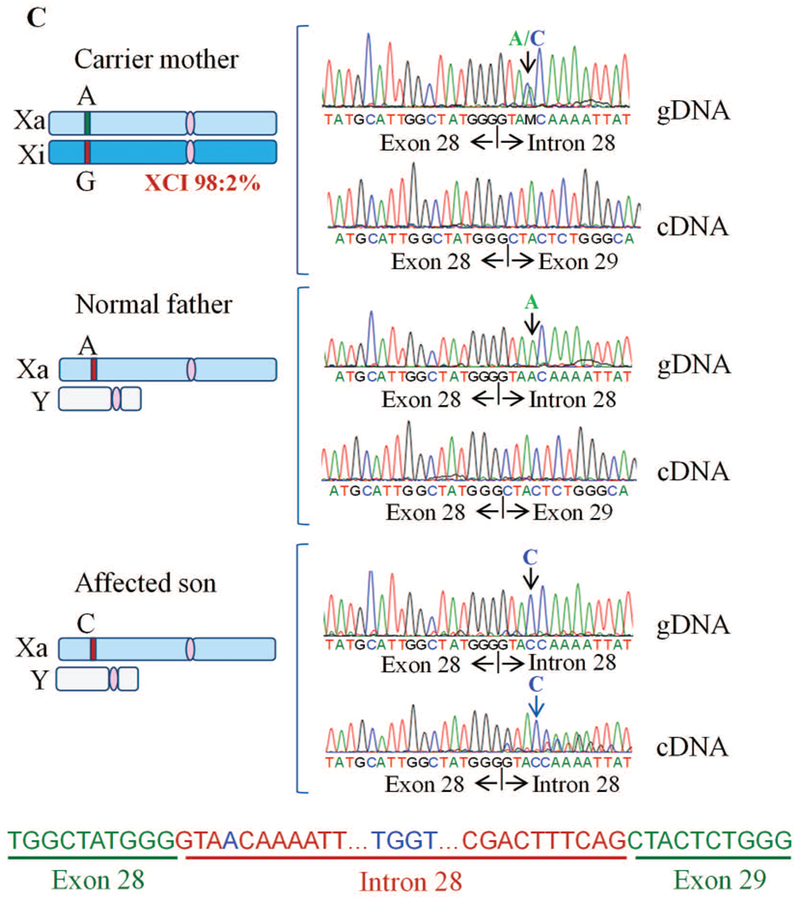
Exon28:c.3503+4A>C variant causes aberrant splicing of intron between exons 28-29 A: Part of the THOC2 gene showing location of the A/C nucleotide in the heterozygous carrier mother and A>C splice variant in the affected son. The C-terminal part of the 1593 amino acid wild type and 1175 amino acid (that contains 1168 normal and 7 amino acids coded by the unspliced intron) THOC2 protein in the affected son are also shown. B: Gel showing a 491 bp RT- PCR product from the normally-spliced heterozygous carrier mother and unaffected father, and 491 bp and 634 bp (retaining 143 bp of the 503 bp intron between exons 28-29) RT-PCR products derived from the normally and aberrantly spliced mRNAs, respectively, in the affected son. Location of the forward and reverse primers within exons 27 and 30 is shown. C: Sanger sequencing chromatograms of PCR products amplified using primers located within exons 27 and 30 from genomic and cDNA of unaffected father and mother and the affected son.
Discussion
Here we present detailed clinical information, and molecular and functional studies, on five previously unreported THOC2 variants in six affected males (two de novo variants and one maternally inherited variant in monozygotic twins) and on one affected female with a previously reported de novo p.Tyr517Cys variant. We present evidence that extends the genotypic spectrum beyond the four THOC2 missense variants that we reported previously (Kumar, et al., 2015) by including two intronic variants that affect splicing, and four missense variants that affect protein stability in a cell-based assay system. According to ACMG criteria they were classified as pathogenic or likely pathogenic (Table 1) (Richards, et al., 2015). These findings, along with the four missense variants reported earlier (Kumar, et al., 2015), add to the existing evidence that alterations in essential mRNA export pathway cause NDDs (Amos, et al., 2017; Beaulieu, et al., 2013; Kumar, et al., 2015).
We confirm that the core clinical feature of THOC2-related disorder in hemizygous males is ID, with several individuals having additional features including behavioural disorders, hypotonia, gait disturbance, tremor, low birth weight, short stature, microcephaly and variable neuroimaging findings. Although the range of neurodevelopmental features is similar, our original cohort contained males with ID in the mild or borderline range of intellectual functioning (Kumar, et al., 2015), whereas, all individuals in this cohort have ID which is at least in the moderate range. Individuals 2 and 6 had neurological signs which could be consistent with cerebellar dysfunction including tremor and a broad-based gait for individual 2 and nystagmus, tremor and an ataxic broad-based gait for individual 6, in the absence of significant cerebellar abnormalities on MRI. This is interesting given the female patient with knockdown of THOC2 function due to a de novo X;8 translocation that created a PTK2-THOC2 fusion had congenital cerebellar hypoplasia and prominent cerebellar signs with mild intellectual disability (Di Gregorio, et al., 2013). We used computerized face-matching technology to specifically evaluate the cohort to assess if a characteristic facial gestalt was evident across individuals with pathogenic or likely pathogenic variants across our original and this expanded clinical cohort (Supp. Figure S4) (Dudding-Byth, et al., 2017). Although a clearly recognizable facial gestalt was not obvious, there are some similarities. The facial gestalt spectrum associated with THOC2 pathogenic variants will continue to emerge as more individuals are reported.
As was the case in our original cohort, heterozygous mothers were clinically unaffected, and, where available, X-chromosome inactivation (XCI) was highly skewed (Table 1). In contrast individual 7, with a de novo missense variant (p.Tyr517Cys) is a female with a particularly severe neurocognitive presentation. This is consistent with other reported severely affected females with de novo variants in X linked genes (de Lange, et al., 2016; Palmer, et al., 2016; Snijders Blok, et al., 2015; Zweier, et al., 2014). Unfortunately, we did not have access to individual 7’s genomic DNA to test XCI status.
A range of protein-protein interactions are required for mRNA export (Chi, et al., 2013). Proteins with altered stability (Hirayama, et al., 2008), localization (Beaulieu, et al., 2013) (e.g., THOC6 p.Gly46Arg implicated in syndromic ID) or interaction (Chi, et al., 2013) can impact mRNA export and consequently disrupt normal cell function. We did not observe mislocalization of the THOC2 variant proteins in cultured cells and did not test alterations in their interaction with the other known or unknown TREX proteins. However, reduced levels of a number of new (p.Tyr517Cys, p.His1187Tyr, p.Thr696Ile, p.Gly713Asp) and published (p.Leu438Pro, p.Ile800Thr, (Kumar, et al., 2015) missense THOC2 variant proteins are due to impaired protein stability or reduced levels of normal mRNA due to aberrant splicing (exon28:c.3503+4A>C). We also noted increased stability of p.Asn1261His THOC2 protein. We and others have shown that THOC2 controls TREX function by maintaining the stability of THOC1, 3, 5 and 7 subunits (Chi, et al., 2013; Kumar, et al., 2015). Reduced levels of THOC2 missense variant proteins are most likely due to enhanced proteasome-mediated degradation as THOC2 is ubiquitinated (Lopitz-Otsoa, et al., 2012). THOC2 depletion has been reported to have different consequences in diverse organisms. For example, shRNA-mediated Thoc2 knockdown leads to significant increase in length of neurites in cultured rat primary hippocampal neurons (Di Gregorio, et al., 2013) although effects on neurons with persistently reduced THOC2 variant proteins in the affected individuals may be different and C. elegans thoc2 knockouts, that are completely immobile, slow-growing, sterile, have functional defects in specific sensory neurons and die prematurely (Di Gregorio, et al., 2013). D. rerio Thoc2 is essential for embryonic development (Amsterdam, et al., 2004) and in D. melanogaster S2 cells Thoc2 knockdown inhibits mRNA export and cell proliferation (Rehwinkel, et al., 2004). THOC2 depletion also results in chromosome alignment, mitotic progression and genomic stability in human HeLa cells (Yamazaki, et al., 2010). Finally, Thoc2 and Thoc5 knockdown experiments have shown their role in regulation of embryonic stem cell (ESC) self-renewal (Wang, et al., 2013).
Both the affected individuals carrying the splice-variants presented with severe neurocognitive features. The exon35:c.4450-2A>G and exon28:c.3503+4A>C THOC2 splice variants present interesting biological scenarios; the former resulting in a 1535 amino acid truncated protein that is present at higher level and the latter with both normal (albeit potentially much reduced) and a 1175 amino acid truncated THOC2 protein. We postulate that the clinical outcomes in the exon35:c.4450-2A>G individual are caused by partial loss of function due to loss of 110 amino acid C-terminal region and accumulation of the truncated THOC2 protein. However, pathogenicity in exon28:c.3503+4A>C affected individual is most likely caused by reduced levels of normal and potential dominant-negative effects of the C-terminally truncated THOC2 protein. That reduced THOC2 protein levels are associated with ID and other clinical symptoms is emerging as a frequent theme; e.g., due to reduced THOC2 protein stability caused by missense variants (see above) or aberrant splicing . Indeed reduced THOC2 levels are shown to destabilize the TREX complex in humans (Chi, et al., 2013; Kumar, et al., 2015) and removal of any THO subunit causes destabilization of other TREX components in yeast (Pena, et al., 2012).
Systematic functional analysis of the Tho2 C-terminal RNA binding region in yeast provides interesting explanation as to how the truncated THOC2 protein can perturb normal mRNA export function in human cells (Pena, et al., 2012) (Figure 6). The data showed that whereas ∆Tho2 yeast strain does not grow at 37ºC (restrictive temperature), Tho2∆1408-1597 and Tho∆1271-1597 growth is considerably reduced, suggesting that C-terminal 1271-1597 amino acids are required for cell survival at restrictive temperature (Pena, et al., 2012). If the exon28:c.3503+A>C variant caused complete splicing defect retaining intron between exon 28-29 in all mRNAs, the cells would translate only 1175 amino acids (with 1168 normal) THOC2 protein; essentially lacking the C-terminal region encompassing the RNA binding domain (RBD) that when deleted in yeast Tho∆1271-1597 strain restricts its growth at 37ºC. However, the affected boy carrying a single allele of the exon28:c.3503+A>C THOC2 variant, although with severe clinical symptoms, is alive. This could be explained by presence of reduced levels of THOC2 protein produced from translation of about 2/3rd normally-spliced mRNA in the affected white blood cells. Taken together, clinical outcomes in the affected boy may be due to perturbed mRNA export caused by reduced levels of THOC2 protein and perhaps also C-terminally truncated THOC2 protein translated from about 1/3rd aberrantly spliced mRNAs that retain a part of intron sequence between exon 28-29.
FIGURE 6.
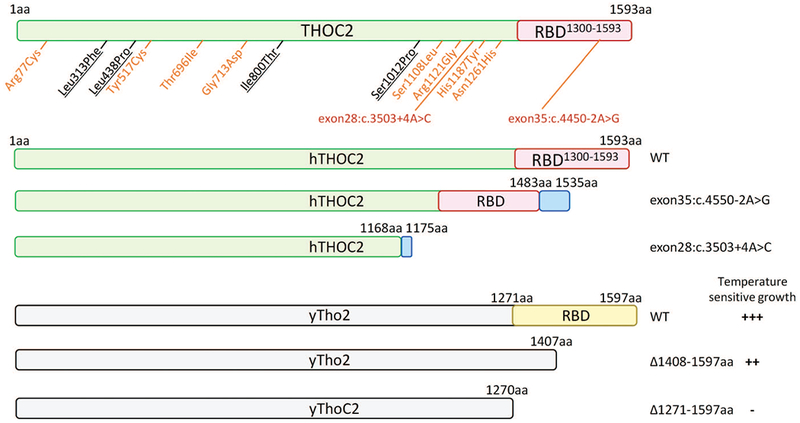
Summary of truncated human THOC2 proteins translated from aberrantly-spliced mRNAs and functional outcomes of yeast C-terminal Tho2 deletion strains (Pena, et al., 2012). Blue boxes depict the 52 and 7 amino acids coded by unspliced intron sequences of exon35:c.4550-2T>C and exon28:c.3503+4A>C variants, respectively. WT, +++ = normal, ∆1408-1597aa, ++ and ∆1271-1597aa = reduced growth at restrictive temperature.
We also identified a set of previously unreported THOC2 missense variants that, according to ACMG criteria, are variants of uncertain clinical significance (VOUS) (Supp. Table S1): namely p.Arg77Cys; p.Ser1108Leu; p.Arg1121Gly and p.Asn1261His. Nevertheless, these variants have supportive evidence pointing towards potential pathogenicity as they are rare (absent from ExAC/gnomAD databases of reference individuals) (Lek, et al., 2016), affect highly evolutionarily conserved amino acid residues, are predicted to be pathogenic by in silico analyses and are within the clinical presentations spectrum of those seen in individuals with confirmed THOC2-related ID. However, they lack supportive evidence from our existing functional assays. These variants may still have a detrimental effect on THOC2 function due to altered protein structure impacting protein-RNA and/or protein-protein interactions with known or unknown TREX subunits [E.g., (Boehringer, et al., 2017)]. The challenge of proving causality for previously unreported missense variants in NDD genes is well recognized and speaks to the need for ongoing intertwined clinical and research efforts to clarify causality of VOUS (Wright, et al., 2018). We therefore report detailed variant and clinical data (see Supp. materials, Supp. Table S1-S2 and Supp. Figure S4) with the intention of alerting researchers and clinicians to these variants, as future studies, for example identification of their recurrence in affected individuals with overlapping clinical phenotypes or pathophysiological investigations, may help clarify their clinical significance.
THOC2 is ubiquitously expressed in all human tissues (Thul, et al., 2017) and more specifically in the developing and mature human brain (Johnson, et al., 2009; Kumar, et al., 2015; Uhlen, et al., 2015) and mouse brain, with higher abundance in frontal cortex and cerebellum (Di Gregorio, et al., 2013; Kumar, et al., 2015). THOC2 is an essential mRNA export factor as its siRNA-mediated depletion results in almost complete retention of mRNAs in the cell nucleus (Chi, et al., 2013), potentially toxic to the cell. These data are consistent with the findings that THOC2 is a highly-constrained gene (Samocha, et al., 2014) and THOC (e.g. THOC1, 3, 5, 6 and 7) genes are essential for cell survival (Blomen, et al., 2015). Taken together, as THOC2 knockout cells will not survive due to complete mRNA nuclear retention, we predict that the identified THOC2 variants represent partial loss-of-function that disrupt normal mRNA export in neuronal and possibly other cell types, potentially causing variable clinical presentations.
TREX complex couples transcription and mRNA biogenesis with nuclear mRNA export, and has emerged as an essential pathway in embryogenesis, organogenesis and differentiation (Heath, et al., 2016). For example, Thoc2 and Thoc5 selectively bind and regulate export of mRNAs (e.g. Nanog, Sox2, Esrrb, and Klf4 mRNAs) involved in maintenance of pluripotency of mouse ESCs (Wang, et al., 2013) and Thoc5 in maintenance of hematopoiesis and HSP70 mRNA export (Katahira, et al., 2009; Mancini, et al., 2010). Mouse modeling shows that both Thoc1 and Thoc5 knockouts are embryonic lethal (Mancini, et al., 2010; Wang, et al., 2006). However, Thoc1 and Thoc5 expression in a range of developing and adult tissues may indicate that the two genes have a more essential role in early embryonic development compared to less stringent requirement during later stages of embryonic or adult development (Mancini, et al., 2010; Wang, et al., 2006); a functional pattern most likely followed by the THOC2 gene. Essentiality of THOC2 gene indicates that THOC2 knockout will also be lethal. However, reduced levels or perturbed functionality can lead to a range of NDD phenotypes as observed for a cohort of THOC2 variants identified by us. It is now well-established that development of brain depends on tightly regulated and complex sequence of events involving neuronal and glial cell proliferation, migration and maturation (Chiurazzi and Pirozzi, 2016). Therefore, it is not surprising that our THOC2 variant data and published work (Dickinson, et al., 2016) provides strong evidence that even subtle alterations to the canonical molecular pathways such as mRNA export, otherwise essential for cellular life, can be tolerated but at a cost of a NDD.
In summary, we present detailed clinical data on seven individuals with THOC2-associated ID caused by both missense and splice variants that meet ACMG criteria for (likely) pathogenicity. They have a core phenotype of ID, and common findings of behavioural disorders, infantile hypotonia, gait disturbance and growth impairment, similar to the affected males with THOC2-associated ID we previously reported (Kumar, et al., 2015). Other than the affected female with a de novo missense variant, heterozygote carrier females are typically unaffected. We also present data on five individuals with four previously unreported rare missense variants that show clinical overlap with our core group, but where convincing evidence for causality is still required. The significance of these variants may be clarified as additional individuals with THOC2 variants are reported. We have also ‘adopted’ THOC2 on the Human Disease Gene (HDG) Website Series (http://humandiseasegenes.nl/thoc2) in an effort to continue to explore the phenotypic-genotypic spectrum for THOC2-related ID.
Supplementary Material
Acknowledgements
We thank the individuals and families for their contribution to this study. This work was supported by NHMRC Program Grant APP1091593 and Senior Research Fellowship APP1041920 and Channel 7 Children’s Research Foundation to J.G., research grants APP19PI10/01710 from the Fondo de Investigación Sanitaria; the Spanish Ministry of Economy and Competitiveness, ‘Centro de Excelencia Severo Ochoa 2017-2021’, SEV-2016-0571; a MINECO Severo Ochoa fellowship to LD (SVP-2013-0680066); FORGE Canada and Care4Rare Canada Consortium funded by Genome Canada, the Canadian Institutes of Health Research (CIHR), the Ontario Genomics Institute, Ontario Research Fund, Génome Québec, and Children’s Hospital of Eastern Ontario Foundation; the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1 HG008900 to Daniel MacArthur and Heidi Rehm; the Undiagnosed Diseases Program-Victoria (UDP-Vic), Murdoch Children¹s Research Institute, Melbourne, Australia.
Grant sponsors: Australian NHMRC Program Grant APP1091593 and Senior Research Fellowship APP1041920; APP19PI10/01710 from the Fondo de Investigación Sanitaria; the Spanish Ministry of Economy and Competitiveness, SEV-2016-0571; a MINECO Severo Ochoa fellowship to LD (SVP-2013-0680066); FORGE Canada and Care4Rare Canada Consortium and the National Heart, Lung and Blood Institute grant UM1 HG008900 to Daniel MacArthur and Heidi Rehm.
References
- Amos JS, Huang L, Thevenon J, Kariminedjad A, Beaulieu CL, Masurel-Paulet A, Najmabadi H, Fattahi Z, Beheshtian M, Tonekaboni SH and others 2017. Autosomal recessive mutations in THOC6 cause intellectual disability: syndrome delineation requiring forward and reverse phenotyping. Clin Genet 91(1):92–99. [DOI] [PubMed] [Google Scholar]
- Amsterdam A, Nissen RM, Sun Z, Swindell EC, Farrington S, Hopkins N. 2004. Identification of 315 genes essential for early zebrafish development. Proc Natl Acad Sci U S A 101(35):12792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu CL, Huang L, Innes AM, Akimenko MA, Puffenberger EG, Schwartz C, Jerry P, Ober C, Hegele RA, McLeod DR and others 2013. Intellectual disability associated with a homozygous missense mutation in THOC6. Orphanet J Rare Dis 8(1):62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittles AH, Petterson BA, Sullivan SG, Hussain R, Glasson EJ, Montgomery PD. 2002. The influence of intellectual disability on life expectancy. J Gerontol A Biol Sci Med Sci 57(7):M470–2. [DOI] [PubMed] [Google Scholar]
- Blomen VA, Majek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, Sacco R, van Diemen FR, Olk N, Stukalov A and others 2015. Gene essentiality and synthetic lethality in haploid human cells. Science 350(6264):1092–6. [DOI] [PubMed] [Google Scholar]
- Boehringer A, Garcia-Mansfield K, Singh G, Bakkar N, Pirrotte P, Bowser R. 2017. ALS Associated Mutations in Matrin 3 Alter Protein-Protein Interactions and Impede mRNA Nuclear Export. Sci Rep 7(1):14529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi B, Wang Q, Wu G, Tan M, Wang L, Shi M, Chang X, Cheng H. 2013. Aly and THO are required for assembly of the human TREX complex and association of TREX components with the spliced mRNA. Nucleic Acids Res 41(2):1294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnam M, Wang Y, Zhang X, Gold DL, Khoury T, Nikitin AY, Foster BA, Li Y, Bshara W, Morrison CD and others 2014. The Thoc1 ribonucleoprotein and prostate cancer progression. J Natl Cancer Inst 106(11):dju306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiurazzi P, Pirozzi F. 2016. Advances in understanding - genetic basis of intellectual disability. F1000Res 5:599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Chan AP. 2015. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31(16):2745–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe BP, Witherspoon K, Rosenfeld JA, van Bon BW, Vulto-van Silfhout AT, Bosco P, Friend KL, Baker C, Buono S, Vissers LE and others 2014. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet 46(10):1063–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange IM, Helbig KL, Weckhuysen S, Moller RS, Velinov M, Dolzhanskaya N, Marsh E, Helbig I, Devinsky O, Tang S and others 2016. De novo mutations of KIAA2022 in females cause intellectual disability and intractable epilepsy. J Med Genet 53(12):850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Gregorio E, Bianchi FT, Schiavi A, Chiotto AM, Rolando M, Verdun di Cantogno L, Grosso E, Cavalieri S, Calcia A, Lacerenza D and others 2013. A de novo X;8 translocation creates a PTK2-THOC2 gene fusion with THOC2 expression knockdown in a patient with psychomotor retardation and congenital cerebellar hypoplasia. J Med Genet 50(8):543–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson ME, Flenniken AM, Ji X, Teboul L, Wong MD, White JK, Meehan TF, Weninger WJ, Westerberg H, Adissu H and others 2016. High-throughput discovery of novel developmental phenotypes. Nature 537(7621):508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudding-Byth T, Baxter A, Holliday EG, Hackett A, O’Donnell S, White SM, Attia J, Brunner H, de Vries B, Koolen D and others 2017. Computer face-matching technology using two-dimensional photographs accurately matches the facial gestalt of unrelated individuals with the same syndromic form of intellectual disability. BMC Biotechnol 17(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi KC, Epilepsy Phenome/Genome P, Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T and others 2013. De novo mutations in epileptic encephalopathies. Nature 501(7466):217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guria A, Tran DD, Ramachandran S, Koch A, El Bounkari O, Dutta P, Hauser H, Tamura T. 2011. Identification of mRNAs that are spliced but not exported to the cytoplasm in the absence of THOC5 in mouse embryo fibroblasts. RNA 17(6):1048–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hautbergue GM. 2017. RNA Nuclear Export: From Neurological Disorders to Cancer. Adv Exp Med Biol 1007:89–109. [DOI] [PubMed] [Google Scholar]
- Heath CG, Viphakone N, Wilson SA. 2016. The role of TREX in gene expression and disease. Biochem J 473(19):2911–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayama S, Yamazaki Y, Kitamura A, Oda Y, Morito D, Okawa K, Kimura H, Cyr DM, Kubota H, Nagata K. 2008. MKKS is a centrosome-shuttling protein degraded by disease-causing mutations via CHIP-mediated ubiquitination. Mol Biol Cell 19(3):899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosking FJ, Carey IM, Shah SM, Harris T, DeWilde S, Beighton C, Cook DG. 2016. Mortality Among Adults With Intellectual Disability in England: Comparisons With the General Population. Am J Public Health 106(8):1483–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MB, Kawasawa YI, Mason CE, Krsnik Z, Coppola G, Bogdanovic D, Geschwind DH, Mane SM, State MW, Sestan N. 2009. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 62(4):494–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katahira J, Inoue H, Hurt E, Yoneda Y. 2009. Adaptor Aly and co-adaptor Thoc5 function in the Tap-p15-mediated nuclear export of HSP70 mRNA. EMBO J 28(5):556–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ and others 2011. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell 44(2):325–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46(3):310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Corbett MA, van Bon BW, Woenig JA, Weir L, Douglas E, Friend KL, Gardner A, Shaw M, Jolly LA and others 2015. THOC2 Mutations Implicate mRNA-Export Pathway in X-Linked Intellectual Disability. Am J Hum Genet 97(2):302–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Yue B, Yuan C, Zhao S, Fang C, Yu Y, Yan D. 2015. Elevated expression of Thoc1 is associated with aggressive phenotype and poor prognosis in colorectal cancer. Biochem Biophys Res Commun 468(1-2):53–8. [DOI] [PubMed] [Google Scholar]
- Lopitz-Otsoa F, Rodriguez-Suarez E, Aillet F, Casado-Vela J, Lang V, Matthiesen R, Elortza F, Rodriguez MS. 2012. Integrative analysis of the ubiquitin proteome isolated using Tandem Ubiquitin Binding Entities (TUBEs). J Proteomics 75(10):2998–3014. [DOI] [PubMed] [Google Scholar]
- Mancini A, Niemann-Seyde SC, Pankow R, El Bounkari O, Klebba-Farber S, Koch A, Jaworska E, Spooncer E, Gruber AD, Whetton AD and others 2010. THOC5/FMIP, an mRNA export TREX complex protein, is essential for hematopoietic primitive cell survival in vivo. BMC Biol 8:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani D, Ronzoni L, Esposito S. 2015. Genetic Advances in Intellectual Disability. J Pediatr Genet 4(3):125–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan DO. 2013. The D box meets its match. Mol Cell 50(5):609–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeseburg B, Dijkstra GJ, Groothoff JW, Reijneveld SA, Jansen DE. 2011. Prevalence of chronic health conditions in children with intellectual disability: a systematic literature review. Intellect Dev Disabil 49(2):59–85. [DOI] [PubMed] [Google Scholar]
- Ohno K, Takeda JI, Masuda A. 2018. Rules and tools to predict the splicing effects of exonic and intronic mutations. Wiley Interdiscip Rev RNA 9(1):e1451. [DOI] [PubMed] [Google Scholar]
- Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. 2006. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127(3):635–48. [DOI] [PubMed] [Google Scholar]
- Palmer EE, Kumar R, Gordon CT, Shaw M, Hubert L, Carroll R, Rio M, Murray L, Leffler M, Dudding-Byth T and others 2017. A Recurrent De Novo Nonsense Variant in ZSWIM6 Results in Severe Intellectual Disability without Frontonasal or Limb Malformations. Am J Hum Genet 101(6):995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer EE, Stuhlmann T, Weinert S, Haan E, Van Esch H, Holvoet M, Boyle J, Leffler M, Raynaud M, Moraine C and others 2016. De novo and inherited mutations in the X-linked gene CLCN4 are associated with syndromic intellectual disability and behavior and seizure disorders in males and females. Mol Psychiatry 23(2):222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena A, Gewartowski K, Mroczek S, Cuellar J, Szykowska A, Prokop A, Czarnocki-Cieciura M, Piwowarski J, Tous C, Aguilera A and others 2012. Architecture and nucleic acids recognition mechanism of the THO complex, an mRNP assembly factor. EMBO J 31(6):1605–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehwinkel J, Herold A, Gari K, Kocher T, Rode M, Ciccarelli FL, Wilm M, Izaurralde E. 2004. Genome-wide analysis of mRNAs regulated by the THO complex in Drosophila melanogaster. Nat Struct Mol Biol 11(6):558–66. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E and others 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropers HH, Hamel BC. 2005. X-linked mental retardation. Nat Rev Genet 6(1):46–57. [DOI] [PubMed] [Google Scholar]
- Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, Kosmicki JA, Rehnstrom K, Mallick S, Kirby A and others 2014. A framework for the interpretation of de novo mutation in human disease. Nat Genet 46(9):944–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz CE. 2015. XLinked Intellectual Disability Genetics. Wiley Online Library. [Google Scholar]
- Snijders Blok L, Madsen E, Juusola J, Gilissen C, Baralle D, Reijnders MR, Venselaar H, Helsmoortel C, Cho MT, Hoischen A and others 2015. Mutations in DDX3X Are a Common Cause of Unexplained Intellectual Disability with Gender-Specific Effects on Wnt Signaling. Am J Hum Genet 97(2):343–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Bjork L, Breckels LM and others 2017. A subcellular map of the human proteome. Science 356(6340). [DOI] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A and others 2015. Proteomics. Tissue-based map of the human proteome. Science 347(6220):1260419. [DOI] [PubMed] [Google Scholar]
- Viphakone N, Cumberbatch MG, Livingstone MJ, Heath PR, Dickman MJ, Catto JW, Wilson SA. 2015. Luzp4 defines a new mRNA export pathway in cancer cells. Nucleic Acids Res 43(4):2353–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers LE, Gilissen C, Veltman JA. 2016. Genetic studies in intellectual disability and related disorders. Nat Rev Genet 17(1):9–18. [DOI] [PubMed] [Google Scholar]
- Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, Choudhary C. 2011. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics 10(10):M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Miao YL, Zheng X, Lackford B, Zhou B, Han L, Yao C, Ward JM, Burkholder A, Lipchina I and others 2013. The THO complex regulates pluripotency gene mRNA export and controls embryonic stem cell self-renewal and somatic cell reprogramming. Cell Stem Cell 13(6):676–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chang Y, Li Y, Zhang X, Goodrich DW. 2006. Thoc1/Hpr1/p84 is essential for early embryonic development in the mouse. Mol Cell Biol 26(11):4362–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chinnam M, Wang J, Wang Y, Zhang X, Marcon E, Moens P, Goodrich DW. 2009. Thoc1 deficiency compromises gene expression necessary for normal testis development in the mouse. Mol Cell Biol 29(10):2794–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woerner AC, Frottin F, Hornburg D, Feng LR, Meissner F, Patra M, Tatzelt J, Mann M, Winklhofer KF, Hartl FU and others 2016. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 351(6269):173–6. [DOI] [PubMed] [Google Scholar]
- Wright CF, McRae JF, Clayton S, Gallone G, Aitken S, FitzGerald TW, Jones P, Prigmore E, Rajan D, Lord J and others 2018. Making new genetic diagnoses with old data: iterative reanalysis and reporting from genome-wide data in 1,133 families with developmental disorders. Genet Med 10.1038/gim.2017.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Fujiwara N, Yukinaga H, Ebisuya M, Shiki T, Kurihara T, Kioka N, Kambe T, Nagao M, Nishida E and others 2010. The closely related RNA helicases, UAP56 and URH49, preferentially form distinct mRNA export machineries and coordinately regulate mitotic progression. Mol Biol Cell 21(16):2953–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Need AC, Petrovski S, Goldstein DB. 2014. One gene, many neuropsychiatric disorders: lessons from Mendelian diseases. Nat Neurosci 17(6):773–81. [DOI] [PubMed] [Google Scholar]
- Zweier C, Rittinger O, Bader I, Berland S, Cole T, Degenhardt F, Di Donato N, Graul-Neumann L, Hoyer J, Lynch SA and others 2014. Females with de novo aberrations in PHF6: clinical overlap of Borjeson-Forssman-Lehmann with Coffin-Siris syndrome. Am J Med Genet C Semin Med Genet 166C(3):290–301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.