

Abstract
Background
Epidermal growth factor receptor (EGFR) inhibitors prevent cell growth and have shown benefit in the treatment of metastatic colorectal cancer, whether used as single agents or in combination with chemotherapy. Clear benefit has been shown in trials of EGFR monoclonal antibodies (EGFR MAb) but not EGFR tyrosine kinase inhibitors (EGFR TKI). However, there is ongoing debate as to which patient populations gain maximum benefit from EGFR inhibition and where they should be used in the metastatic colorectal cancer treatment paradigm to maximise efficacy and minimise toxicity.
Objectives
To determine the efficacy, safety profile, and potential harms of EGFR inhibitors in the treatment of people with metastatic colorectal cancer when given alone, in combination with chemotherapy, or with other biological agents.
The primary outcome of interest was progression‐free survival; secondary outcomes included overall survival, tumour response rate, quality of life, and adverse events.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL), the Cochrane Library, Issue 9, 2016; Ovid MEDLINE (from 1950); and Ovid Embase (from 1974) on 9 September 2016; and ClinicalTrials.gov and the World Health Organization International Clinical Trials Registry Platform (WHO ICTRP) on 14 March 2017. We also searched proceedings from the major oncology conferences ESMO, ASCO, and ASCO GI from 2012 to December 2016. We further scanned reference lists from eligible publications and contacted corresponding authors for trials for further information where needed.
Selection criteria
We included randomised controlled trials on participants with metastatic colorectal cancer comparing: 1) the combination of EGFR MAb and 'standard therapy' (whether chemotherapy or best supportive care) to standard therapy alone, 2) the combination of EGFR TKI and standard therapy to standard therapy alone, 3) the combination of EGFR inhibitor (whether MAb or TKI) and standard therapy to another EGFR inhibitor (or the same inhibitor with a different dosing regimen) and standard therapy, or 4) the combination of EGFR inhibitor (whether MAb or TKI), anti‐angiogenic therapy, and standard therapy to anti‐angiogenic therapy and standard therapy alone.
Data collection and analysis
We used standard methodological procedures defined by Cochrane. Summary statistics for the endpoints used hazard ratios (HR) with 95% confidence intervals (CI) for overall survival and progression‐free survival, and odds ratios (OR) for response rate (RR) and toxicity. Subgroup analyses were performed by Kirsten rat sarcoma viral oncogene homolog (KRAS) and neuroblastoma RAS viral (V‐Ras) oncogene homolog (NRAS) status ‐ firstly by status of KRAS exon 2 testing (mutant or wild type) and also by status of extended KRAS/NRAS testing (any mutation present or wild type).
Main results
We identified 33 randomised controlled trials for analysis (15,025 participants), including trials of both EGFR MAb and EGFR TKI. Looking across studies, significant risk of bias was present, particularly with regard to the risk of selection bias (15/33 unclear risk, 1/33 high risk), performance bias (9/33 unclear risk, 9/33 high risk), and detection bias (7/33 unclear risk, 11/33 high risk).
The addition of EGFR MAb to standard therapy in the KRAS exon 2 wild‐type population improves progression‐free survival (HR 0.70, 95% CI 0.60 to 0.82; high‐quality evidence), overall survival (HR 0.88, 95% CI 0.80 to 0.98; high‐quality evidence), and response rate (OR 2.41, 95% CI 1.70 to 3.41; high‐quality evidence). We noted evidence of significant statistical heterogeneity in all three of these analyses (progression‐free survival: I2 = 76%; overall survival: I2 = 40%; and response rate: I2 = 77%), likely due to pooling of studies investigating EGFR MAb use in different lines of therapy. Rates of overall grade 3 to 4 toxicity, diarrhoea, and rash were increased (moderate‐quality evidence for all three outcomes), but there was no evidence for increased rates of neutropenia.
For the extended RAS wild‐type population (no mutations in KRAS or NRAS), addition of EGFR MAb improved progression‐free survival (HR 0.60, 95% CI 0.48 to 0.75; moderate‐quality evidence) and overall survival (HR 0.77, 95% CI 0.67 to 0.88; high‐quality evidence). Response rate was also improved (OR 4.28, 95% CI 2.61 to 7.03; moderate‐quality evidence). We noted significant statistical heterogeneity in the progression‐free survival analysis (I2 = 61%), likely due to the pooling of studies combining EGFR MAb with chemotherapy with monotherapy studies.
We observed no evidence of a statistically significant difference when EGFR MAb was compared to bevacizumab, in progression‐free survival (HR 1.02, 95% CI 0.93 to 1.12; high quality evidence) or overall survival (HR 0.84, 95% CI 0.70 to 1.01; moderate‐quality evidence). We noted significant statistical heterogeneity in the overall survival analysis (I2 = 51%), likely due to the pooling of first‐line and second‐line studies.
The addition of EGFR TKI to standard therapy in molecularly unselected participants did not show benefit in limited data sets (meta‐analysis not performed). The addition of EGFR MAb to bevacizumab plus chemotherapy in people with KRAS exon 2 wild‐type metastatic colorectal cancer did not improve progression‐free survival (HR 1.04, 95% CI 0.83 to 1.29; very low quality evidence), overall survival (HR 1.00, 95% CI 0.69 to 1.47; low‐quality evidence), or response rate (OR 1.20, 95% CI 0.67 to 2.12; very low‐quality evidence) but increased toxicity (OR 2.57, 95% CI 1.45 to 4.57; low‐quality evidence). We noted significant between‐study heterogeneity in most analyses.
Scant information on quality of life was reported in the identified studies.
Authors' conclusions
The addition of EGFR MAb to either chemotherapy or best supportive care improves progression‐free survival (moderate‐ to high‐quality evidence), overall survival (high‐quality evidence), and tumour response rate (moderate‐ to high‐quality evidence), but may increase toxicity in people with KRAS exon 2 wild‐type or extended RAS wild‐type metastatic colorectal cancer (moderate‐quality evidence). The addition of EGFR TKI to standard therapy does not improve clinical outcomes. EGFR MAb combined with bevacizumab is of no clinical value (very low‐quality evidence). Future studies should focus on optimal sequencing and predictive biomarkers and collect quality of life data.
Plain language summary
Epidermal growth factor receptor (EGFR) inhibitors for metastatic colorectal cancer
Background
Cancer of the colon or rectum that has spread to other organs (metastatic colorectal cancer) is a commonly occurring disease that usually cannot be surgically removed. The main treatment is chemotherapy, targeted therapy (such as EGFR inhibitors, the subject of this review), or both. The epidermal growth factor receptor (EGFR) is a protein found on cells that plays a vital role in promoting cell growth. Monoclonal antibodies are molecules developed to attach to a particular protein in order to enhance or decrease action at that protein site. EGFR monoclonal antibodies (EGFR MAb), such as cetuximab and panitumumab, specifically target and block EGFR, which stops cancer cell growth. Research has shown that people with mutations (gene changes) in KRAS (a gene related to EGFR) may not benefit from these drugs ('KRAS mutant'), but those without mutations ('KRAS wild type') do benefit. Recent research also suggests that people with mutations in another related gene (NRAS) may not benefit from these drugs either – that is, patients need to have no mutation in either KRAS or NRAS (otherwise known as 'extended RAS wild type').
Another type of EGFR‐blocking drug known as tyrosine kinase inhibitor (EGFR TKI) (e.g. erlotinib and gefitinib) is effective in the treatment of lung cancer with EGFR mutations, but its benefit in colorectal cancer is unclear.
Objectives
To determine the benefit and harms of EGFR MAb and EGFR TKI in the treatment of metastatic colorectal cancer. Our primary aim was to look at whether these drugs prolonged the time before disease progression (growth of the disease, usually defined as growth more than 20% or development of a new metastasis), but we also evaluated whether the drugs prolonged survival, caused the tumour to shrink, or resulted in more side effects (particularly rash and diarrhoea).
Study investigation
We reviewed the evidence for EGFR inhibitors in people with metastatic colorectal cancer. We selected randomised studies that compared people receiving standard treatment with those who received standard treatment plus an EGFR inhibitor (both the more commonly used drug type (EGFR MAb) or the less commonly used drug type (EGFR TKI)). We searched for published studies up to September 2016 and identified 33 studies involving 15,025 participants, of which 27 studies looked at EGFR MAbs and 6 looked at EGFR TKIs.
Main results
Our main finding was that the addition of EGFR MAb drugs to standard treatment in people whose tumours were KRAS wild type reduces the risk of disease progression by 30%. The risk of death is reduced by 12% (i.e. patients live longer overall), and the chance of tumour shrinkage is increased from 31% to 46%. In people who are both KRAS and NRAS (extended RAS) wild type, the risk of disease progression is reduced by 40%; risk of death is reduced by 23%; and the rate of tumour shrinkage increases from 21% to 48%.
There was no evidence of any difference in outcome between the combination of EGFR MAb plus chemotherapy and the combination of bevacizumab (another targeted drug) plus chemotherapy.
There was no evidence that the use of EGFR TKI improved outcomes, although the number of studied participants (and trials) was too small for a formal analysis.
There was no evidence that adding EGFR MAb to both chemotherapy and bevacizumab improved outcomes, and in fact was found to increase toxicity.
Quality of the evidence
The evidence we identified was generally of moderate to high quality. Our main reason for not judging the evidence for all outcomes as high quality was that in some studies the treating doctors assessed their patients’ scans for tumour shrinkage or growth, and their knowledge of what treatment the patient received resulted in a higher risk of bias. Another reason for our judging of the evidence as lower quality was that there were differences between the studies grouped in the meta‐analyses calculations (heterogeneity).
Summary of findings
Background
Description of the condition
Bowel (colorectal) cancer is the third most common cancer worldwide. The International Agency for Research on Cancer estimated a crude colorectal cancer incidence rate of 1,361,000 in 2012 (with 694,000 deaths), with 55% of cases occurring in high‐income countries (Ferlay 2015). Although improvements in treatment, particularly over the last 10 years, have brought significant improvements in survival, metastatic colorectal cancer (mCRC) remains a major cause of morbidity and mortality.
The prognosis of untreated patients with mCRC is historically three to six months. Development of various chemotherapy agents such as 5‐fluorouracil with folinic acid, irinotecan, and oxaliplatin, used either concurrently or sequentially, increased median survival to around 20 months (Grothey 2004). The addition of new targeted therapies have provided more lines of treatment, extending median survival to around 30 months and increasing the proportion of patients who are able to proceed to curative metastatectomy (Heinemann FIRE‐3 2014), which may improve overall survival and even potentially result in long‐term cure.
Chemotherapy acts on rapidly dividing cells to block DNA replication by a variety of mechanisms, but this effect is not specific to cancer cells, and hence chemotherapy is often associated with toxicity to normal tissues. In an attempt to focus treatment effects to cancer cells, the last 15 years have seen a major effort to develop 'targeted', or biological therapies. These agents work by influencing specific cellular pathways that drive tumour growth. The main classes of targeted agents are monoclonal antibodies, which bind membrane growth factor receptors or their ligands (the proteins that bind to receptors), and small molecules (including tyrosine kinase inhibitors), which cross the cell membrane and interact with intracellular components in order to decrease processes related to cell growth or survival.
Description of the intervention
The epidermal growth factor receptor family
The epidermal growth factor receptor (EGFR) or ErbB family of receptors are cell surface receptors with tyrosine kinase activity. The family comprises of four related receptors: EGFR1 (also called EGFR, erbB1, or HER1), ErbB2 (HER2/neu), ErbB3 (HER3), and ErbB4 (HER4). They possess an extracellular ligand binding domain, a transmembrane domain, and an intracellular protein tyrosine kinase component and are overexpressed in many primary cancers. EGFR is an essential pathway in cellular growth and differentiation, with the absence of EGFR affecting development of multiple organs including the epidermis, lung, and intestine in knockout mice. It is activated by ligands such as epidermal growth factor, transforming growth factor‐a, amphiregulin, heparin‐binding epidermal growth factor, and betacellulin (Herbst 2004). Ligand binding results in dimerisation of the EGFR and activation of the intrinsic tyrosine kinase domain via autophosphorylation. (Citri 2006; Normanno 2006). Ultimately, EGFR signalling has positive downstream effects in promoting cell proliferation and increasing cell survival. Expression or upregulation of the EGFR gene occurs in up to 80% of colorectal cancers (Messa 1998; Salomon 1995), and is associated with metastatic risk (Mayer 1993). Inhibition of the EGFR signalling pathway should therefore result in interruption of this pathway and ultimately reduced cellular proliferation.
Epidermal growth factor receptor inhibitors
EGFR activation can be blocked with either monoclonal antibodies or tyrosine kinase inhibitors.
EGFR monoclonal antibodies (MAb)
Monoclonal antibodies have been extensively investigated in mCRC, and both cetuximab and panitumumab have entered routine clinical use. Cetuximab is a chimeric (IgG1) monoclonal antibody (MAb). It binds to the extracelullar domain of EGFR and therefore blocks endogenous ligand binding, which would normally have positive downstream effects on growth. It may also have immune‐mediated antitumour effects such as antibody‐dependent cell‐mediated cyotoxicity (Mendelsohn 2000). It is given as a weekly or biweekly intravenous infusion following an initial loading dose, and received US Food and Drug Administration approval for the treatment of metastatic colorectal cancer after it was shown to improve survival and reverse chemoresistance in refractory mCRC when given with irinotecan in a pivotal phase II trial. (Cunningham 2004) As well as improving survival, cetuximab maintains quality of life in mCRC patients (Jonker 2007).
Panitumumab is a humanised (IgG2) anti‐EGFR antibody which again binds to the extracellular domain of EGFR, disrupting ligand‐mediated growth signalling. It has been shown to result in clinical benefit both when added to chemotherapy and as monotherapy in mCRC in various clinical settings (Amado 2008; Douillard PRIME 2010).
The most common adverse events observed in trials of these EGFR monoclonal antibodies are skin toxicity, infusion reactions, hypomagnesemia, fatigue, abdominal pain, nausea, and diarrhoea. Serious but rare adverse events observed with these agents include pulmonary fibrosis, severe skin toxicity complicated by sepsis, and anaphylaxis or infusion reactions.
EGFR tyrosine kinase inhibitors (TKIs)
Tyrosine kinase inhibitors are small molecules derived from quinazolines that cross the cell membrane and block the intracellular tyrosine kinase domain of various receptors (e.g. EGFR, Erb2, and VEGFR). Erlotinib is a specific inhibitor of EGFR (but not other ERBb subtypes) which results in blocking of ligand‐induced EGFR receptor phosphorylation. Gefitinib inhibits EGFR in the same manner but also appears to target other pathways such as ERK 1/2 phosphorylation in mesothelioma cell lines (Favoni 2010). These drugs have been highly effective in other tumour types, particularly lung cancer harbouring mutations in the EGFR gene (Mok 2009). Consequently, there has been great interest in determining the efficacy of EGFR TKIs in mCRC.
How the intervention might work
As highlighted above, EGFR has a critical role in cell proliferation. Inhibition of EGFR function (either by targeting its extracellular or intracellular domains) should therefore decrease the amount of pro‐growth signalling, thus inhibiting cell growth and other downstream effects. EGFR inhibitors should decrease growth of colorectal cancers and may improve the efficacy of any chemotherapy with which it is partnered. However, there were some early setbacks in trials of EGFR inhibitors. The presence of an EGFR mutation in lung cancer is strongly linked to the efficacy of EGFR inhibition. This correlation was not detected in CRC, and trials showed relatively less overall patient benefit in CRC compared to lung cancer. This led to the search for another predictive biomarker and ultimately to the identification initially of KRAS, then NRAS and Harvey rat sarcoma viral oncogene homolog (HRAS) as genes that may affect the efficacy of EGFR MAbs.
The role of the RAS family (KRAS, NRAS, and HRAS) in the EGFR pathway and therapeutic implications
The RAS family of proto‐oncogenes ‐ KRAS, NRAS and HRAS ‐ encode small GTPase proteins which form an essential part of the RAS pathway, and are located downstream to EGFR. As a result, constitutively activating mutations of RAS would render the tumour cell immune to the effects of EGFR inhibition. Initially, expression of EGFR on cell surface as measured by immunohistochemistry was thought to be a marker of ability to respond to cetuximab in CRC, but no significant correlation was ultimately demonstrated (Chung 2005; Scartozzi 2004). The identification of the initial predictive biomarker for EGFR MAb came after analysis of responding participants in the large CO.17 trial of cetuximab versus best supportive care (Karapetis CO17 2008). Participants with KRAS exon 2 (codon 12 and 13) wild‐type genotypes demonstrated significantly increased benefit from EGFR inhibition compared to the KRAS unselected population; in contrast, there was minimal evidence of benefit in participants in whom a KRAS mutation was present.
KRAS mutations lead to constitutively active signal transduction and have been associated with increased risk of recurrence (Andreyev 1998), more rapid disease progression (Di Fiore 2007), and inferior survival (Lievre 2006). Activating mutations have been detected in 30% to 50% of mCRC (Amado 2008; Di Fiore 2007), and there is now broad evidence that such KRAS mutations can predict resistance to EGFR‐targeted antibodies (Bokemeyer OPUS 2009; Karapetis CO17 2008; Van Cutsem CRYSTAL 2009).
Although NRAS is found less frequently in CRC, any mutations in this gene may also result in activation of the RAS pathway with inherent resistance to EGFR inhibition for the same reasons as KRAS. More recently, retrospective analysis of multiple trials using extended RAS testing (KRAS exons 3 and 4 and NRAS exons 2 to 4) have shown that patients with extended RAS mutations (i.e. mutations not just in KRAS exon 2 but in the new areas tested) do not benefit from EGFR inhibition (Bokemeyer OPUS 2009; Douillard PRIME 2010). Extended RAS testing can therefore define a narrower patient population in whom EGFR inhibition is projected to have increased effect.
HRAS is upregulated in malignant CRC cells (Feng 2001), but HRAS mutations are rarely found in CRC; its utility as a predictive biomarker for EGFR MAb has not been formally tested to date.
Combining EGFR MAb with anti‐angiogenic agents (MAbs and TKIs)
Angiogenesis plays a vital role in tumour development, growth, progression, and metastatic potential. The 'angiogenic switch' describes the transition from pre‐malignant non‐vascular stage (when tumours can grow to around 2 to 3 mm3 but cannot form new blood vessels) into a frank malignancy capable of forming new tumour vasculature and metastasising. The vascular endothelial growth factor (VEGF) family are the principal pro‐angiogenic factors which are ligands for the VEGF receptors (VEGFR). Similar to EGFR inhibitors, anti‐angiogenic agents can be divided into monoclonal antibodies (bevacizumab, which targets circulating VEGF‐A) and tyrosine kinase inhibitors (such as sunitinib and sorafenib, which have multiple targets, but include VEGFR amongst those targets).
Bevacizumab is known to have activity in CRC, but only when combined with chemotherapy (Giantonio 2007; Hurwitz 2005). Since it was established that EGFR stimulation leads to downstream increased VEGF production, combining the blockade of both these pathways was thought to be a promising strategy that could improve antitumour effects of targeted antibodies (van Cruijsen 2005). Anti‐angiogenic TKIs such as sunitinib and sorafenib have not been shown to have a similar effect in mCRC, and consequently there has been less interest in their potential role in combination therapies.
The use of EGFR TKI
The molecular basis of action of EGFR antibodies and EGFR TKIs seemed complementary, so that it was logical to compare the effectiveness of these treatment modalities, or even to investigate a combination of both. As with all palliative cancer treatments, there were concerns about additive and excessive toxicity when combining different therapies with overlapping toxicity, even if increased efficacy was demonstrated.
Why it is important to do this review
Despite multiple positive trials, clinical trials investigating EGFR inhibitors vary widely in clinical context: monotherapy versus combination with chemotherapy, monoclonal antibodies or tyrosine kinase inhibitors, and in unselected, partially selected, or highly biomarker‐defined patient populations. The purpose of this review was to find, organise, and summarise randomised controlled trial evidence for the use of epidermal growth factor receptor inhibitors in the treatment of metastatic colorectal cancer, and to define the contexts in which EGFR inhibitor use improves clinical outcomes.
Objectives
To determine the efficacy, safety profile, and potential harms of EGFR inhibitors in the treatment of people with metastatic colorectal cancer when given alone, in combination with chemotherapy, or with other biological agents.
Methods
Criteria for considering studies for this review
Types of studies
We considered randomised controlled trials on people with metastatic colorectal cancer evaluating EGFR inhibitors. These were usally given in combination with 'standard therapy' ‐ whether chemotherapy or best supportive care, depending on clinical context. Participants had to have unresectable disease at the time of enrolment, and trials enrolling participants with resectable metastatic disease upfront were not eligible for inclusion. The intention to evaluate the participant for potential surgery at some stage after commencement of therapy (assuming they were not clearly eligible for surgery at enrolment) did not render a study ineligible. The study protocol was previously published as Herbertson 2009.
Studies were categorised by:
drug class (EGFR MAb versus EGFR TKI versus other);
clinical setting: line of therapy (first, second, or third);
therapy partner used in the trial as the comparator arm, whether chemotherapy or the combination of chemotherapy and anti‐angiogenic therapy (e.g. bevacizumab).
Eligible studies were to evaluate the following.
-
EGFR MAb
first‐line treatment with chemotherapy and an EGFR inhibitor compared to chemotherapy alone;
second‐line treatment with chemotherapy and an EGFR inhibitor compared to chemotherapy alone;
third‐line treatment (> 2 prior chemotherapy regimens) with an EGFR inhibitor alone compared to best supportive care.
-
EGFR TKI
treatment with chemotherapy and EGFR TKI compared to chemotherapy alone;
treatment with EGFR TKI compared to best supportive care.
-
Different EGFR inhibitor regimens
treatment with one EGFR inhibitor compared to treatment with another EGFR inhibitor;
treatment with one regimen of an EGFR inhibitor compared to treatment with another regimen of the same EGFR inhibitor.
-
EGFR inhibitors in combination with chemotherapy and anti‐angiogenic agents
treatment with chemotherapy and anti‐angiogenic agent compared to chemotherapy and EGFR inhibitor;
treatment with chemotherapy and anti‐angiogenic agent compared to treatment with chemotherapy, anti‐angiogenic agent, and EGFR inhibitor.
We did not exclude cross‐over studies, but we did not include these in the analysis of overall survival, as any analysis of overall survival would be hindered by all participants receiving EGFR inhibitors. Cluster randomised controlled trials were theoretically eligible for inclusion, but we considered such trials as unlikely to exist (based on ethical considerations and the review authors' clinical experience).
Types of participants
People with a histological diagnosis of colorectal carcinoma and confirmed evidence of unresectable, metastatic disease. Histological confirmation could either come from the primary site (including resection or biopsy prior to the development of metastatic disease) or a site of metastasis.
Types of interventions
Intravenous or oral EGFR inhibitors administered alone or in combination with chemotherapy or other anti‐angiogenic agents (or a combination of these treatment modalities). This includes EGFR MAb and EGFR TKI. Trials with placebo groups and trials with open control groups (no treatment or best supportive care controls) were eligible.
We defined anti‐angiogenic agents as any targeted agent (MAb or TKI) that included VEGFR as one of its targets. Bevacizumab, sorafenib, and sunitinib would therefore be included in this description, but other drugs could potentially be included as well.
Types of outcome measures
Primary outcomes
The primary endpoint was progression‐free survival, defined by time from trial enrolment to a composite of disease progression (as measured by Response Evaluation Criteria in Solid Tumours (RECIST) criteria) and death. RECIST criteria define whether a tumour is measurable on baseline imaging and require the selection of "target lesions" – index lesions for measuring disease and monitoring response. (Eisenhauer 2009). For people with RECIST measurable disease, tumour measurements on repeat imaging are separated into four categories: complete response (disappearance of all target lesions), partial response (decrease in sum of diameters of target lesions by at least 30%), progressive disease (increase in sum of diameters of target lesions by at least 20%, or appearance of a new lesion), or stable disease (none of the above). We noted discrepancies from RECIST criteria qualitatively.
Secondary outcomes
Overall survival, defined as the time from trial enrolment to death of any cause. We elected to use this measure rather than overall survival at predefined time periods (such as three or five years), as these figures are variably reported in metastatic colorectal trials (largely due to the guarded prognosis in the overall cohort). Furthermore, hazard ratios (which are used to measure overall survival) represent a summary of the difference in risk of death over the time of measurement, rather than being a point estimate.
Tumour response rate, as defined by each study. We elected to collect data according to RECIST criteria, as this is standard practice in most clinical trials. Tumour response rate is defined per clinical convention as the percentage of patients who achieve either a complete response or partial response on follow‐up imaging.
Toxicity/adverse events, as defined and graded by the Common Terminology Criteria for Adverse Events (CTCAE) guidelines (NIH 2010). These guidelines provide objective criteria to grade common adverse events from grade 1 (mild effects, often asymptomatic or minimally symptomatic) to grade 5 (death related to adverse event). We elected to measure the incidence of grade 3 to 4 toxicity overall (where available), rash, diarrhoea, and neutropenia, as these are known side effects of EGFR MAb (in the intervention arm) and chemotherapy (in either arm).
Quality of life, using validated tools. Recognising that there is no consensus on the optimal quality of life instrument in mCRC, we included all previously published quality of life scales and subscales; however, we specifically sought information with regard to the EORTC QLQ‐C30, EuroQol EQ‐5D, FACT‐C questionnaires, and the Dermatology Life Quality Index (as treatment with EGFR MAb is associated with significant incidence of rash). We were interested in the global scales as well as physical/emotional functioning (because of the potential psychological effect of rash in addition to the known physical adverse events).
Search methods for identification of studies
Electronic searches
We conducted a comprehensive literature search to identify all published and unpublished randomised controlled trials with no language or date of publication restrictions. We searched the following electronic databases on 9 September 2016:
Cochrane Central Register of Controlled Trials (CENTRAL) (the Cochrane Library, Issue 9, 2016) (Appendix 1);
Ovid MEDLINE (from 1950) (Appendix 2); and
Ovid Embase (from 1974) (Appendix 3).
We applied a sensitivity‐ and precision‐maximising search filter to the MEDLINE search strategy as recommended in Section 6.4.11 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
We also searched ClinicalTrials.gov (clinicaltrials.gov/) and the World Health Organization International Clinical Trials Registry Platform (WHO ICTRP) (www.who.int/ictrp/en/) for ongoing trials on 14 March 2017 (Appendix 4).
Searching other resources
We performed handsearches for meeting proceedings of major conferences (European Society for Medical Oncology (ESMO), American Society of Clinical Oncology (ASCO), and ASCO GI) from 2012 to March 2016 on 14‐15 January 2016 to identify any additional relevant trials.
Data collection and analysis
Selection of studies
Two review authors (DC, RW) independently assessed abstracts and (in cases of uncertainty) full‐text articles of potentially eligible studies in a blinded fashion. We included all eligible trials irrespective of whether measured outcome data were reported on. We determined eligibility based on the above criteria, resolving any disagreements by consensus with a third review author (ES).
Data extraction and management
Two review authors (DC, RW) independently extracted data from the reports of included studies, resolving any discrepancies via consensus. Data were collected in duplicate in piloted forms and stored on an online repository.
Data collected included the following.
Participant demographics and characteristics (such as gender, median age, and performance status)
Intervention: drug name, method of administration and dose, schedule of administration
Comparator: full treatment in comparator arm
Median follow‐up
Information regarding funding sources and potential conflicts of interest
Outcomes:
Progression‐free survival: hazard ratio with 95% confidence interval and P values. Observed number of events.
Overall survival: hazard ratio with 95% confidence interval and P values. Observed number of events.
Tumour response rates: complete response (incidence over total number evaluated) and partial response (incidence over total number evaluated) in both experimental and control arms, odds ratio with 95% confidence interval.
Toxicity: incidence of grade 3/4/5 toxicity (as noted above) in both arms, odds ratio with 95% confidence interval.
Quality of life.
Above outcomes by subgroups: KRAS exon 2 wild type, KRAS exon 2 mutant, extended RAS wild type, extended RAS mutant (for trials investigating EGFR MAb).
Where data from the same trial were presented in multiple publications, we extracted all of the information and reported this as a single trial whilst listing the other publications in the references.
Regarding statistical extraction, we extracted overall survival and progression‐free survival from the text of publications, conference posters, and abstracts, as well as figure legends. We obtained overall response rate and toxicity from publications and posters as above, with rates of toxicity reported in percentage form only converted to numerators by taking the denominator as the total number evaluated for safety and rounding the resultant conversion to the nearest integer. Santoro 2008 contained Kaplan‐Meier curves but no reports of hazard ratios/confidence intervals; we converted this to a hazard ratio with 95% confidence interval according to the methods outlined by Parmar 1998.
Assessment of risk of bias in included studies
Two review authors (DC, RW), independently and in a blinded fashion (to authors, journal, drug company, institutions, and results), evaluated the methods sections of included studies for quality. We used the Cochrane 'Risk of bias' tool to assess the risk of bias of included trials (Higgins 2011), using the following domains:
random sequence generation;
allocation concealment;
blinding of participants and personnel;
blinding of outcome assessment;
incomplete outcome data;
selective reporting bias; and
other bias (baseline imbalance, significant protocol deviations, inappropriate influence of funders).
We judged each domain in each study as low risk of bias, high risk of bias, or unclear risk of bias according to Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (see Appendix 5) (Higgins 2011), referring further to trial protocols in ClinicalTrials.gov where relevant for additional details. Disagreements were resolved by consensus with a third review author (ES). We then summarised the risk of bias for an outcome across studies according to the Cochrane 'Risk of bias' tool (Higgins 2011).
We presented 'Summary of findings' tables, grading each outcome according to the GRADE approach.
Measures of treatment effect
We adopted the following measures for the respective treatment effect.
Overall survival: hazard ratio, with 95% confidence interval
Progression‐free survival: hazard ratio, with 95% confidence interval
Tumour response rates: mean differences (in percentage) as well as odds ratio with 95% confidence intervals
Toxicity: mean differences (in percentage) as well as odds ratio with 95% confidence intervals
We deemed a P value of less than 0.05 as significant.
Unit of analysis issues
The unit of analysis was the individual participant. The only unit of analysis issues we encountered were from trials that had multiple intervention arms. Where possible, we utilised the summary hazard ratio (from a comparison of the combined arms versus the placebo arm) for meta‐analysis. If this was not available, we combined the hazard ratios reported for separate groups using random‐effects meta‐analysis to create one summary hazard ratio.
While we considered cluster randomised trials and cross‐over trial designs to be unlikely for trials of this therapy, they were technically eligible for inclusion. If these trials were present, we planned to seek specialised statistical advice for incorporating them into the review, however none were identified during the literature search (see below).
Dealing with missing data
Regarding trials with incompletely reported outcomes (including subgroup analyses that may have been performed but not reported), one review author (DC) contacted the lead authors of the study via email to request further information. This was successful in a minority of cases; we have detailed additional information from answered requests in the relevant sections in the Results.
Regarding missing individuals from studies, we have based analyses on the intention‐to‐treat principle to the degree permitted by published data for relevant outcome measures. For studies with dropout rates exceeding 5%, we performed best‐case/worst‐case sensitivity analyses for binary outcomes (response rate and toxicity).
Assessment of heterogeneity
We assessed clinical and methodological heterogeneity in each trial and across trials combined in meta‐analysis. Where the clinical difference between interventions tested or populations was sufficiently great to prevent meaningful synthesis, we separated the studies in terms of analysis and presented the results individually.
We defined heterogeneity as per Section 9.5.2 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). Where heterogeneity was present, we explored and commented on it as follows:
0% to 40%: might not be important;
30% to 60%: may represent moderate heterogeneity*;
50% to 90%: may represent substantial heterogeneity*;
75% to 100%: considerable heterogeneity*.
*The importance of the observed value of I2 depends on (i) magnitude and direction of effects and (ii) strength of evidence for heterogeneity (e.g. P value from the Chi2 test, or a confidence interval for I2).
Assessment of reporting biases
We investigated publication bias by visual assessment of funnel plots for the primary outcome if more than 10 studies were included.
Data synthesis
For the outcome measures of progression‐free survival and overall survival, we reported hazard ratios (HR) calculated with 95% confidence intervals (CI). We performed pooled analyses for these measures using the generic inverse‐variance method and random‐effects modelling in order to obtain a summary hazard ratio with 95% CI. We estimated the hazard ratios in the studies using a proportional hazards model. We opted to use this method because most trials reported data for overall survival and progression‐free survival as hazard ratios (with a Cox proportional hazards model) without reporting the standard deviation for each arm. In addition, the distribution of survival in each arm was not necessarily normally distributed. Consequently, we could not perform standard methods of meta‐analysis for continuous measures.
For dichotomous or categorical outcomes (tumour response rate, rates of toxicity), we reported the number of events compared to the total number of participants for each trial. We performed pooled analyses for these measures using the Mantel‐Haenszel method and random‐effects modelling with calculation of odds ratios and 95% CI. We opted for this approach as the number of observed events may be low both in response rate (e.g. with the use of EGFR MAb as monotherapy) and rates of toxicity (e.g. neutropenia with EGFR MAb monotherapy). In these situations, generic inverse‐variance methodology may give poor estimates of standard errors, and is not recommended by Cochrane (Deeks 2011). Whilst we considered the Peto odds ratio method as an alternative, it does not perform well in cases of large differences in efficacy (e.g. rash and diarrhoea with EGFR MAb) and common events (Deeks 2011). We therefore gave preference to Mantel‐Haenszel analysis for all dichotomous outcomes rather than attempting to prespecify different analysis methods for different outcomes.
Subgroup analysis and investigation of heterogeneity
We had planned to conduct the following subgroup analyses for measures of EGFR expression:
presence of KRAS mutations (given the marked correlation between KRAS mutations and lack of response to EGFR inhibitor therapy as first demonstrated in Karapetis CO17 2008);
presence of B‐Raf proto‐oncogene, serine/threonine kinase (BRAF) mutations (as BRAF and RAS mutations are mutually exclusive as demonstrated in Rajagopalan 2002);
known patient‐related prognostic factors such as age;
performance status;
number of organs involved with metastatic disease; and
the presence and grade of skin toxicity (as early trials such as Douillard PRIME 2010 and Peeters 2010 showed a potential correlation between skin toxicity and response).
However, many preplanned subgroup analyses were not possible because not all studies presented sufficient data to be stratified by these subgroups. In addition, some preplanned subgroups defined in the protocol have become less relevant with the increasing use of biomarkers (e.g. the presence of skin toxicity as a surrogate for efficacy) to define eligibility for EGFR inhibitor trials (see Differences between protocol and review). In the formal analysis, we thus included only the preplanned subgroups of KRAS exon 2 mutations (whether absent or present) and extended RAS mutations (whether absent or present).
With regard to subgroup analysis by RAS mutation status, we deemed all methods of RAS ascertainment (whether pyro‐sequencing, next‐generation sequencing, or Sanger sequencing) to be acceptable. There was no a priori barrier in terms of the rate of RAS status ascertainment in a patient population for eligibility.
We also performed preplanned subgroup analyses by line of therapy, that is first‐, second‐, or third‐line and beyond, for EGFR MAb studies.
Sensitivity analysis
We conducted sensitivity analyses to investigate the impact of excluding trials at high risk of bias from top‐level analyses. We also conducted sensitivity analyses where investigations for heterogeneity had identified one study as being the likely sole cause of heterogeneity to see whether the reported result in pooled analysis was changed.
Summary of findings
We evaluated the quality of the evidence using the GRADE approach into the following four levels (Schünemann 2009).
High: Further research is very unlikely to change our confidence in the estimate of effect.
Moderate: Further research is likely to have an impact on our confidence in the estimate of effect and may change the estimate.
Low: Further research is very likely to have an important impact on our confidence on the estimate of effect and is likely to change the effect estimate.
Very low: Any estimate of effect is very uncertain.
We have presented the quality of the evidence in 'Summary of findings' tables. The quality of the evidence can be downgraded by one (serious concern) or two levels (very serious concern) for the following reasons: risk of bias, inconsistency (unexplained heterogeneity, inconsistency of results), indirectness (indirect population, intervention, control, or outcomes) and imprecision (wide confidence intervals, single trial). The quality of the evidence can also be upgraded by one level due to a large summary effect.
We applied the GRADE approach for all outcomes, including relevant subgroups and sensitivity analyses.
Results
Description of studies
See: Characteristics of included studies; Characteristics of excluded studies; Characteristics of ongoing studies.
Results of the search
We identified 2178 references in the initial search and 1584 after de‐duplication, of which we selected 120 reports for further evaluation (Figure 1). We assessed these 120 full‐text articles for eligibility and excluded 10, leaving 110 articles. Combining these 110 articles with 105 eligible records (including some trial records) found in the handsearch, with one trial record excluded, resulted in 224 abstracts or articles (67 trials) eligible for inclusion. Of these, 28 were ongoing studies and 6 studies are awaiting classification. Therefore, 151 articles of 33 studies were included in the quantitative synthesis (meta‐analysis).
1.
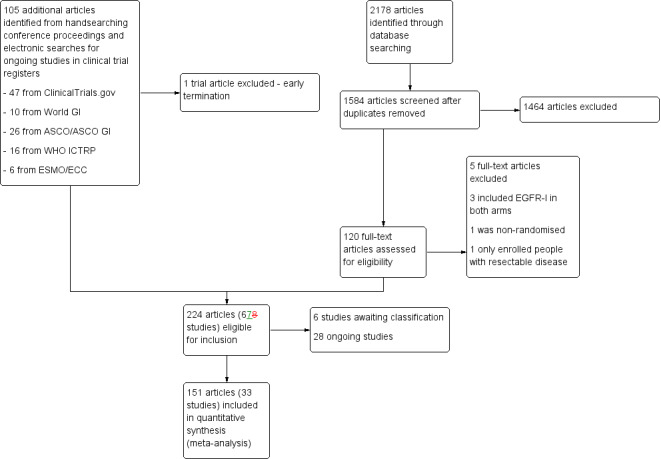
Study flow diagram.
Included studies
Altogether, we included 33 studies investigating 15,025 participants in this meta‐analysis (see Characteristics of included studies; Figure 1). We categorised the included studies as follows in order to facilitate analysis.
Twelve studies examined the effect of adding EGFR MAbs to either chemotherapy (10 studies) or best supportive care (2 studies) on progression‐free survival (PFS) in the KRAS exon 2 wild‐type (WT) setting. Ten studies compared the combination of EGFR MAb and chemotherapy (8 with cetuximab, 2 with panitumumab) to the same chemotherapy alone with KRAS status available. (Adams COIN 2011; Bokemeyer OPUS 2009; Ciardiello CAPRI‐GOIM 2016; Douillard PRIME 2010; Passardi ITACA 2015; Peeters 2010; Seymour PICCOLO 2013; Tveit NORDIC VII 2012; Van Cutsem CRYSTAL 2009; Ye 2013). Two studies (one with cetuximab and one with panitumumab) examined the effect of EGFR MAb as monotherapy (Amado 2008; Karapetis CO17 2008). In total, 7948 participants were enrolled and KRAS status was assessable in 6969 participants: 4402 were KRAS exon 2 WT and 2567 were KRAS exon 2 mutant (MT).
Three studies examined the effect of adding EGFR MAb to chemotherapy in the KRAS unselected setting; this involved 1483 KRAS unselected participants (Borner 2008; Polikoff EXPLORE 2005; Sobrero EPIC 2008).
One study involving 42 participants solely examined the effect of adding EGFR MAb to chemotherapy in the KRAS mutant setting (Siena 2013); we considered this trial in combination with the KRAS mutant cohorts of the studies in 1).
Four studies examined the effect of adding EGFR MAb to chemotherapy on progression‐free survival compared to adding another (non‐EGFR) biological agent to chemotherapy in 2189 KRAS exon 2 WT participants. All trials used bevacizumab as the second biological agent, which permitted its use as the comparator. All four trials compared the combination of chemotherapy with EGFR MAb to the combination of the same chemotherapy with bevacizumab. The chemotherapy backbone was an investigator's choice of mFOLFOX6 or FOLFIRI in Venook CALGB 80405 2014; FOLFIRI in Hecht SPIRITT 2015; mFOLFOX6 in Schwartzberg PEAK 2014; and FOLFIRI in Heinemann FIRE‐3 2014. (these, as well as subsequent chemotherapy regimens, are briefly explained in Appendix 6).
Six studies examined the effect of using one EGFR inhibitor (whether MAb or TKI) compared to another EGFR inhibitor in 1708 participants. Imgatuzumab (GA201) was compared to cetuximab in KRAS exon 2 WT participants, with FOLFIRI being the chemotherapy backbone (Bridgewater GAIN‐C 2015). Afatinib was compared to cetuximab in KRAS exon 2 WT participants in the second trial, both of which were given as monotherapy (Hickish 2014). Brodowicz 2013 compared two different regimens of cetuximab in combination with first‐line FOLFOX chemotherapy. Ma 2013 compared the combination of continuous erlotinib and CAPOX chemotherapy to intermittent erlotinib with CAPOX therapy. Price ASPECCT 2014 compared cetuximab and panitumumab as monotherapies. Finally, Wasan COIN‐B 2014 compared a strategy of intermittent mFOLFOX6 with cetuximab (with mFOLFOX6 with cetuximab ceased after 12 weeks, and assuming stable disease or better with initial treatment, re‐introduction of the same treatment on progression) with the same strategy of intermittent mFOLFOX6 with cetuximab, but with maintenance cetuximab in between these treatments.
Two studies examined the effect of adding EGFR TKI to chemotherapy on progression‐free survival in the KRAS unselected setting in 195 participants. Santoro 2008 investigated gefitinib with initiation of FOLFIRI chemotherapy, which was continued until progression. Vincent 2011 studied erlotinib plus capecitabine in people unsuitable for usual first‐line combination chemotherapy.
Six studies examined the effect of adding EGFR inhibitor (whether MAb or TKI) to a combination of chemotherapy and anti‐angiogenic agent on progression‐free survival compared to chemotherapy and anti‐angiogenic agent only in 1571 participants. (Hagman ACT2 2014; Hecht PACCE 2009; Johnsson Nordic ACT 2013; Passardi ITACA 2015; Tol CAIRO2 2008; Tournigand DREAM 2015). Two studies investigated EGFR TKI (erlotinib in Hagman ACT2 2014 and gefitinib in Tournigand DREAM 2015) added to bevacizumab in the maintenance setting commenced after stable disease or better with bevacizumab‐containing induction chemotherapy. The other three studies investigated EGFR MAb (panitumumab in Hecht PACCE 2009 and cetuximab in Passardi ITACA 2015 and Tol CAIRO2 2008) commenced at the start of first‐line chemotherapy together with bevacizumab in both arms. We note that Passardi ITACA 2015 was also mentioned in section 1) above.
The follow‐up period for included studies ranged from 9.5 to 44 months.
We identified 28 ongoing studies from searches in ClinicalTrials.gov and the WHO International Clinical Trials Registry Platform database (ICTRP). One of these studies, Ashwin 2014, has reported results in abstract form. However, as the statistics reported were sufficiently inconsistent as to prevent data extraction and incorporation into meta‐analysis at this point, we characterised it as an ongoing study.
Excluded studies
See Characteristics of excluded studies.
We excluded Cunningham BOND 2004 from the meta‐analysis as this randomised controlled trial (RCT) compared participants receiving a combination with cetuximab and irinotecan with those receiving cetuximab alone. As cetuximab was given at the same dose in both arms, the study design did not allow assessment of EGFR MAb efficacy. Similarly, we excluded Saltz BOND2 2007, which compared the combination of cetuximab, bevacizumab, and irinotecan to cetuximab and bevacizumab alone, meaning that assessment of EGFR MAb efficacy was not feasible.
We excluded Primrose NEW EPOC 2014 because enrolment was specifically restricted to people with resectable disease, and subsequent outcome was influenced by multiple factors that could not be accounted for.
We excluded Personeni 2013 because the study was initially designed as a randomised study but was subsequently amended to be a single‐arm study.
Liu 2015 compared the combination of FOLFIRI, bevacizumab, and panitumumab to FOLFIRI alone in people with KRAS mutant mCRC. We excluded this study because measures of treatment effect would incorporate use of both bevacizumab and panitumumab, thus making discernment of the contribution of EGFR MAb impractical.
NCT00950820 planned to compare the combination of CAPOX and panitumumab with CAPOX alone in people with KRAS unselected mCRC. This study was terminated after only nine participants were accrued, with no published results; we thus excluded it from analysis.
Risk of bias in included studies
See Characteristics of included studies; Figure 2.
2.
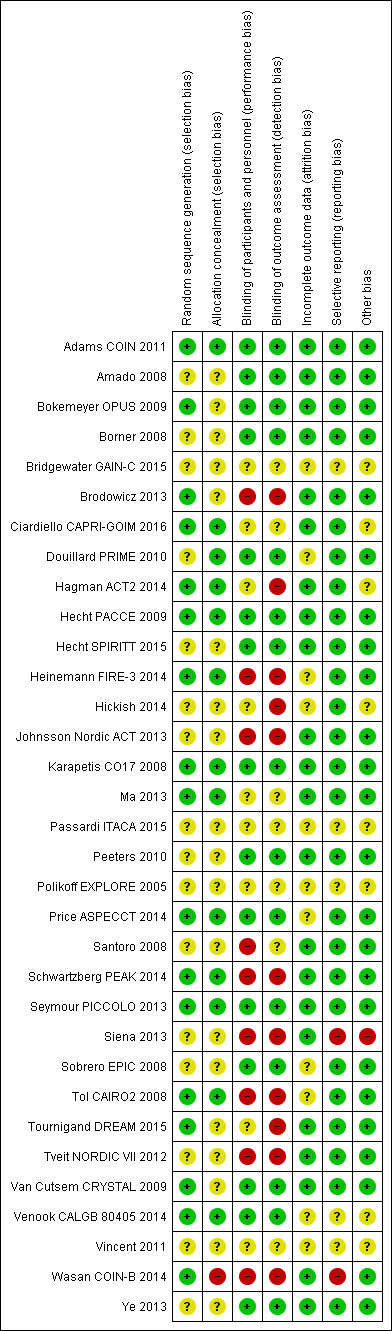
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
We assessed the included studies for risk of bias based on the domains listed in the Cochrane 'Risk of bias' tool (Appendix 5). After this assessment, we summarised the risk of bias for each outcome across the relevant studies.
Allocation
On the whole, the process of allocation was poorly described by trials, making accurate assessment of selection bias difficult.
Thirteen trials described random sequence generation or allocation concealment procedures, or both in sufficient detail to merit an assessment of low risk of bias (Adams COIN 2011; Bokemeyer OPUS 2009; Brodowicz 2013; Ciardiello CAPRI‐GOIM 2016; Heinemann FIRE‐3 2014; Karapetis CO17 2008; Ma 2013; Price ASPECCT 2014; Schwartzberg PEAK 2014; Tol CAIRO2 2008; Tournigand DREAM 2015; Van Cutsem CRYSTAL 2009; Venook CALGB 80405 2014).
Given that the remaining 20 trials reported this insufficiently, we obtained trial protocols where possible and looked for imbalanced baseline characteristics as a surrogate for well‐performed randomisation and allocation concealment. Treatment groups were well balanced in 22 studies (Adams COIN 2011; Amado 2008; Bokemeyer OPUS 2009; Borner 2008; Bridgewater GAIN‐C 2015; Hecht PACCE 2009; Heinemann FIRE‐3 2014; Hickish 2014; Johnsson Nordic ACT 2013; Karapetis CO17 2008; Peeters 2010; Price ASPECCT 2014; Santoro 2008; Schwartzberg PEAK 2014; Seymour PICCOLO 2013; Siena 2013; Sobrero EPIC 2008; Tournigand DREAM 2015; Tveit NORDIC VII 2012; Van Cutsem CRYSTAL 2009; Venook CALGB 80405 2014; Ye 2013).
We were unable to assess balancing in four studies (Hecht SPIRITT 2015; Passardi ITACA 2015; Polikoff EXPLORE 2005; Vincent 2011), as results were only available in abstract form without discussion of baseline characteristics.
Imbalanced baseline characteristics were reported in the following six studies: Brodowicz 2013 (more participants older than 65 years and more colonic primaries in fortnightly cetuximab arm); Douillard PRIME 2010 (more participants with elevated carcinoembryonic antigen, elevated lactate dehydrogenase, and three or more metastatic sites in the investigational arm of the KRAS exon 2 MT stratum); Hagman ACT2 2014 (fewer participants with rectal primaries and fewer participants with prior adjuvant treatment in the investigational arm); Ma 2013 (higher incidence of prior adjuvant 5‐fluorouracil‐based chemotherapy in intermittent erlotinib arm); Wasan COIN‐B 2014 (higher incidence in continuous cetuximab arm of age older than 75 years, performance status 2, BRAF mutations, and colon primaries compared to intermittent cetuximab arm); and Tol CAIRO2 2008 (more males in the investigational arm). Of these, we assigned Hagman ACT2 2014 high risk due to the large numerical difference in percentage of participants with rectal primaries, a known prognostic factor (19% versus 54%).
Blinding
All 33 included studies were open‐label RCTs. Outcome assessment could therefore theoretically be affected by investigators recording outcomes. Given that all trials used overall survival or outcomes related to tumour progression on imaging for their primary outcome, we have grouped together and reported the assessment of performance and detection bias below.
Nine trials selected overall survival as a primary outcome (Adams COIN 2011; Amado 2008; Karapetis CO17 2008; Peeters 2010; Polikoff EXPLORE 2005; Price ASPECCT 2014; Seymour PICCOLO 2013; Sobrero EPIC 2008; Venook CALGB 80405 2014), which would have a low risk of being affected by lack of blinding. Peeters 2010 selected co‐primary endpoints of overall survival (OS) and PFS. Of these trials, three conducted blinded or central review of imaging (Amado 2008; Peeters 2010; Seymour PICCOLO 2013), while the others did not. We note that Seymour PICCOLO 2013 only referred the imaging of one‐third of participants (not all) for central review, but the size of the study (460 participants) means that a significant number of images were double‐checked.
The other 24 trials utilised a primary endpoint related to tumour response or progression: PFS in 14, tumour response rate (TRR) in 8, and time to progression, conversion of hepatic metastases to resectability, and failure‐free survival in 1 trial each. Of these 24, 7 reported blinded assessment of progression (Bokemeyer OPUS 2009; Borner 2008; Douillard PRIME 2010; Hecht PACCE 2009; Hecht SPIRITT 2015; Van Cutsem CRYSTAL 2009; Venook CALGB 80405 2014), resulting in an assessment of low risk of detection bias. Five trials have only been reported in abstract form to date (Bridgewater GAIN‐C 2015; Ciardiello CAPRI‐GOIM 2016; Passardi ITACA 2015; Polikoff EXPLORE 2005; Vincent 2011), with resultant judgement of unclear risk of detection bias in three. Three trials did not specify whether assessment of response was blinded; protocols were not publicly available, and we await response from the corresponding authors to clarify this matter (Ma 2013; Siena 2013; Tournigand DREAM 2015). The remaining nine trials reported unblinded investigator‐performed assessment of radiology for the primary endpoint, resulting in an assessment of high risk of detection bias (Brodowicz 2013; Hagman ACT2 2014; Heinemann FIRE‐3 2014; Hickish 2014; Johnsson Nordic ACT 2013; Santoro 2008; Schwartzberg PEAK 2014; Tol CAIRO2 2008; Tveit NORDIC VII 2012; Wasan COIN‐B 2014).
Incomplete outcome data
In general, we judged the risk of attrition bias as low, being low in 22 of the 33 studies and unclear in 11 studies. Whilst several studies did not specifically report loss to follow‐up, four studies reported a high number of completed events for the primary outcome, leading to a judgement of low risk of attrition bias (Hagman ACT2 2014; Johnsson Nordic ACT 2013; Peeters 2010; Tveit NORDIC VII 2012).
Selective reporting
We judged 26 studies as being at low risk of reporting bias, 6 at unclear risk of bias, and 1 at high risk of bias. The six trials at unclear risk of bias have only been reported in abstract form to date (Bridgewater GAIN‐C 2015; Passardi ITACA 2015; Polikoff EXPLORE 2005; Venook CALGB 80405 2014; Vincent 2011). One study has planned quality of life measures but has not reported on them yet (Wasan COIN‐B 2014); we have contacted the author regarding this information. One trial was terminated early and only response rate was reported, despite initial plans to evaluate other measures such as PFS and OS, resulting in an assessment of high risk of bias (Siena 2013).
Other potential sources of bias
In terms of funding, 15 of the 33 studies were funded by pharmaceutical companies; 3 were funded by government agencies; 14 were funded by a combination of both; and 1 provided insufficient information to determine sources of funding. Given that all studies underwent peer review (either prior to full publication or by the conference's scientific committee), we consider the potential bias from funding to generally be minimal, although the need for objective evaluation of the results presented by a study funded by pharmaceutical companies is reinforced. Such funding in one trial resulted in restrictions on the principal authors being able to publish findings freely without consent of the pharmaceutical company involved; in combination with the potential for selective reporting noted in the above paragraph, this resulted in an assessment of high risk of other bias for this particular study in all endpoints (Siena 2013).
Analysis of funnel plots revealed no evidence of publication bias in the studies investigating EGFR MAb in KRAS‐assessable populations (Figure 3; Figure 4). We were unable to comment on the other funnel plots due to small number of studies (N <= 4).
3.
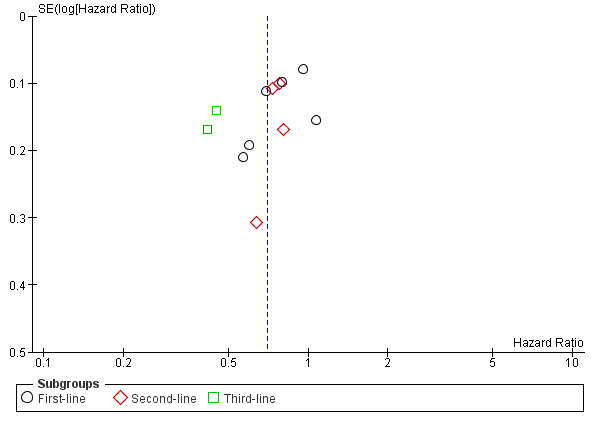
Funnel plot of comparison: 1 EGFR‐I in KRAS exon 2 WT, outcome: 1.1 Progression‐free survival.
4.
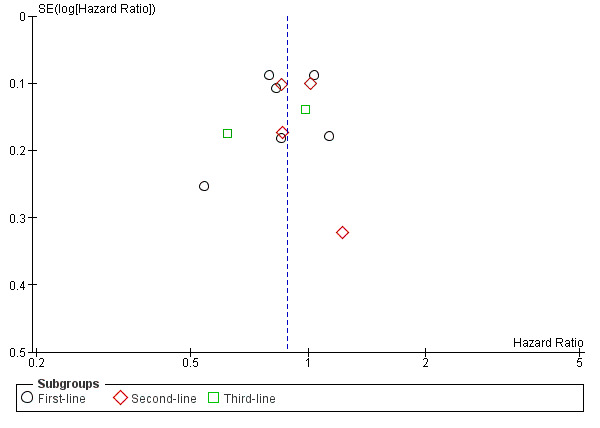
Funnel plot of comparison: 1 EGFR‐I in KRAS exon 2 WT, outcome: 1.2 Overall survival.
Summary assessments of risk of bias across studies for each outcome
Outcome 1.1, 1.3. Addition of EGFR MAb to standard therapy in KRAS exon 2 WT populations ‐ progression‐free survival and tumour response rate: unclear risk of bias
Although six studies employed blinded or central assessment of results, and two provided insufficient information to judge risk, four studies relied on unblinded investigator assessment of response (Adams COIN 2011; Karapetis CO17 2008; Tveit NORDIC VII 2012; Ye 2013). Whilst RECIST criteria are rigorous and objective, they do not completely mitigate the risk of measurement bias. We therefore judged these studies as being at unclear risk of bias with regard to PFS and TRR, resulting in an overall assessment of unclear risk of bias.
Outcome 1.2. Addition of EGFR MAb to standard therapy in KRAS exon 2 WT populations ‐ overall survival: low risk of bias
When considering the risk of bias for studies with regard to overall survival, blinding considerations are less important because the endpoint of overall survival is less amenable to performance or measurement bias. The lack of adequate blinding highlighted above is therefore less relevant, and this outcome is at less risk of bias than for PFS or TRR.
Outcome 1.4 to 1.6. Addition of EGFR MAb to standard therapy in KRAS exon 2 WT populations ‐ overall grade 3 to 4 toxicity, grade 3 to 4 diarrhoea, grade 3 to 4 rash: high risk of bias
Measurement of toxicity may be affected by inadequate blinding or open‐label trials. It is even more prone to these factors, as grading of toxicity (even with published, predefined cutoffs) remains dependent on clinical judgement. Given that all of the studies were open‐label RCTs, and that the risk of bias cannot be mitigated by factors such as central reporting as is possible for PFS, we judged these outcomes to be at high risk of bias.
Outcome 1.7. Addition of EGFR MAb to standard therapy in KRAS exon 2 WT populations ‐ grade 3 to 4 neutropenia: low risk of bias
In comparison to the toxicities in outcomes 1.4 to 1.6, neutropenia is defined by an objective laboratory test and so would not be susceptible to measurement bias. With the same rationale as for overall survival, we therefore consider this outcome to have a low risk of bias.
Outcome 2.1, 2.3. Addition of EGFR MAb to standard therapy in KRAS exon 2 MT populations ‐ progression‐free survival and tumour response rate: unclear risk of bias
Although we included only seven studies in this analysis (as compared to 12 studies in Analysis 1.1), we judged 3 of the 7 studies as having unclear risk of bias due to the reliance on an unblinded investigator to review and determine PFS and TRR. We therefore judged Analysis 2.1 as having unclear risk of bias. The removal of Adams COIN 2011 in Analysis 2.3 did not significantly change this fact, and hence the assessment of unclear risk of bias remained the same.
Outcome 2.2. Addition of EGFR MAb to standard therapy in KRAS exon 2 MT populations ‐ overall survival: low risk of bias
As noted above for outcome 1.2, lack of blinding is less likely to affect assessment of overall survival, therefore, in view of the adequate allocation concealment and low risk of attrition and reporting bias, we judged this outcome as having low risk of bias.
Outcome 2.4‐2.6. Addition of EGFR MAb to standard therapy in KRAS exon 2 MT populations ‐ overall grade 3 to 4 toxicity, grade 3 to 4 diarrhoea, grade 3 to 4 rash: high risk of bias
As noted above for outcomes 1.4 to 1.6, toxicity that is either reported by the participant or assessed by the clinician is prone to detection bias in open‐label trials. As all trials were open label in nature, we judged outcomes 2.4 to 2.6 as having high risk of bias.
Outcome 2.7. Addition of EGFR MAb to standard therapy in KRAS exon 2 MT populations ‐ grade 3 to 4 neutropenia: low risk of bias
As neutropenia is objectively defined, with the same reasoning as in outcome 1.7, we considered this outcome as being at low risk of bias.
Comparisons 3 and 4: Adding EGFR MAb to standard therapy in extended RAS WT and MT populations
As the same studies were included in Analyses 3.1 and 4.1, 3.2 and 4.2, 3.3 and 4.3, and 3.4 and 4.4, we have therefore reported the risk of bias for these outcomes together.
Outcome 3.1, 3.3, 4.1, 4.3. Addition of EGFR MAb to standard therapy in extended RAS WT and MT populations ‐ progression‐free survival, tumour response rate: low risk of bias
Of the six studies included in Analysis 3.1 (and 4.1), five employed blinded or central review of images to determine progression‐free survival and tumour response rate. One study provided insufficient information for evaluation. In view of the higher proportion of studies using these measures, we judged this outcome to have low risk of bias. All four studies included in Analysis 3.3 (and 4.3) had blinded or central review, resulting in outcome 3.3 also having low risk of bias.
Outcome 3.2, 4.2. Addition of EGFR MAb to standard therapy in extended RAS WT and MT populations ‐ overall survival: low risk of bias
The four studies included in these analyses were all well balanced with low risk of selection, attrition, or reporting bias. Furthermore, any lack of blinding would not significantly alter the measurement of OS. We therefore judged this outcome as having low risk of bias.
Outcome 3.4 to 3.7, 4.4 to 4.7. Addition of EGFR MAb to standard therapy in extended RAS WT and MT populations ‐ toxicity: not assessable
As no studies reported toxicity by extended RAS status, none were included in these analyses and hence risk of bias was not assessable.
Outcome 5.1, 5.3. EGFR MAb in KRAS unselected participants ‐ progression‐free survival, tumour response rate: unclear risk of bias
One of the two studies reported blinded assessment of progression (Borner 2008), whereas the other study did not (Sobrero EPIC 2008). As a result, we judged these outcomes as having unclear risk of bias.
Outcome 5.2. EGFR MAb in KRAS unselected participants ‐ overall survival: low risk of bias
We judged this outcome as having low risk of bias as lack of blinding is unlikely to affect overall survival in the presence of adequate allocation concealment and low attrition.
Outcome 5.4 to 5.6. EGFR MAb in KRAS unselected participants ‐ overall grade 3 to 4 toxicity, grade 3 to 4 diarrhoea, grade 3to 4 rash: high risk of bias
As above, all studies were open‐label trials, thus we judged assessment of toxicity to be at high risk of bias.
Outcome 5.7. EGFR MAb in KRAS unselected participants ‐ grade 3 to 4 neutropenia: low risk of bias
Again, the objective assessment of neutrophil count in a controlled trial setting would result in a low risk of bias for the reporting of grade 3 to 4 neutropenia.
Outcome 6.1, 6.3. Comparing EGFR MAb with chemotherapy to anti‐vascular endothelial growth factor (VEGF) MAb with chemotherapy ‐ progression‐free survival, tumour response rate: unclear risk of bias
Two of the four included studies used central or blinded assessment of response and disease progression (Hecht SPIRITT 2015; Venook CALGB 80405 2014), but the other two studies did not (Heinemann FIRE‐3 2014; Schwartzberg PEAK 2014). We therefore judged these outcomes as having unclear risk of bias.
Outcome 6.2. Comparing EGFR MAb with chemotherapy to anti‐VEGF MAb with chemotherapy ‐ overall survival: low risk of bias
All four studies had adequate allocation concealment and low attrition, with blinded assessment less relevant for this outcome. We therefore judged this outcome as having low risk of bias.
Outcome 6.4 to 6.6. Comparing EGFR MAb with chemotherapy to anti‐VEGF MAb with chemotherapy ‐ toxicity: high risk of bias
As above, all studies were open‐label trials, thus we judged assessment of toxicity to be at high risk of bias.
Outcome 7.1, 7.3. Comparing different EGFR inhibitor agents or regimens ‐ progression‐free survival, tumour response rate: high risk of bias
All of the included studies were open‐label studies that did not employ central or blinded assessment of progression. We therefore judged these outcomes to be at high risk of bias.
Outcome 7.2. Comparing different EGFR inhibitor agents or regimens ‐ overall survival: low risk of bias
Despite the open‐label nature of the trials, allocation concealment was adequate in the majority of trials, and overall survival is unlikely to be influenced by blinding. We therefore judged this outcome as having low risk of bias.
Outcome 7.4 to 7.6. Comparing different EGFR inhibitor agents or regimens ‐ overall toxicity, diarrhoea, rash: high risk of bias
As above, the non‐blinded measurement of endpoints (particularly subjective ones) in the setting of open‐label trials resulted in a judgement of high risk of bias.
Outcome 7.7. Comparing different EGFR inhibitor agents or regimens ‐ neutropenia: low risk of bias
Similar to outcome 7.2, neutropenia is judged objectively, thus we assessed this outcome as having low risk of bias.
As noted in Effects of interventions, we did not perform meta‐analysis of the six included trials due to significant between‐study heterogeneity (Bridgewater GAIN‐C 2015; Brodowicz 2013; Hickish 2014; Ma 2013; Price ASPECCT 2014; Wasan COIN‐B 2014). An assessment of the risk of bias according to each outcome was therefore not possible.
Outcome 8.1, 8.3 to 8.6. The addition of EGFR TKIs to standard therapy ‐ progression‐free survival, overall toxicity, diarrhoea, rash: high risk of bias
As both included studies were open label with no centralised or blinded review, we judged these outcomes to have high risk of bias.
Outcome 8.2, 8.7. The addition of EGFR TKIs to standard therapy ‐ overall survival, neutropenia: low risk of bias
Despite the open‐label nature of both studies, these outcomes are not affected by blinding (being objectively determined), and thus were judged to have low risk of bias.
Outcome 9.1, 9.3. Adding EGFR to the combination of bevacizumab and standard therapy ‐ progression‐free survival, tumour response rate: high risk of bias
One of the six included trials used central or blinded assessment of response and disease progression (Hecht PACCE 2009), and two studies had unclear risk of bias as results were only available in abstract form (Passardi ITACA 2015; Tournigand DREAM 2015). The remaining three studies did not utilise central or blinded assessment of disease progression (Hagman ACT2 2014; Johnsson Nordic ACT 2013; Tol CAIRO2 2008). Given the small proportion of studies with low performance bias, we judged these outcomes at having a high risk of bias
Outcome 9.2. Adding EGFR to the combination of bevacizumab and standard therapy ‐ overall survival: low risk of bias
All four studies had adequate allocation concealment and low attrition, with blinded assessment less relevant for this outcome. We therefore judged this outcome as having low risk of bias.
Outcome 9.4 to 9.6. Adding EGFR to the combination of bevacizumab and standard therapy ‐ toxicity: high risk of bias
As above, all studies were open‐label trials, thus we judged assessment of toxicity to be at high risk of bias.
Outcome 9.7. Adding EGFR to the combination of bevacizumab and standard therapy ‐ grade 3 to 4 neutropenia: no assessment made
Given that no studies reported on this outcome, it was not feasible to perform an analysis of risk of bias.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9
Summary of findings for the main comparison. EGFR MAb in KRAS exon 2 WT for metastatic colorectal cancer.
| EGFR MAb in KRAS exon 2 WT for metastatic colorectal cancer | |||||
|
Patient or population: people with metastatic colorectal cancer ‐ KRAS exon 2 WT
Intervention: EGFR MAb in addition to standard treatment Comparison: standard treatment Setting: multicentre international studies | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| Standard therapy | EGFR MAb + standard therapy | ||||
| Progression‐free survival Follow‐up: 13 to 38 months | 300 per 1000 (at 1 year)4 | 221 per 1000 (197 to 254) | HR 0.70 (0.60 to 0.82) | 4402 (12 studies) | ⊕⊕⊕⊕ high1 |
| Overall survival Follow‐up: 13 to 38 months | 400 per 1000 (at 2 years)4 | 352 per 1000 (335 to 392) | HR 0.88 (0.80 to 0.98) | 4249 (12 studies) | ⊕⊕⊕⊕ high |
| Tumour response rate Follow‐up: 13 to 38 months | Study population | OR 2.41 (1.70 to 3.41) | 4147 (12 studies) | ⊕⊕⊕⊕ high1 | |
| 331 per 1000 | 456 per 1000 (411 to 501) | ||||
| Overall grade 3 to 4 toxicity Follow‐up: 13 to 38 months | Study population | OR 2.45 (2.07 to 2.89) | 2771 (6 studies) | ⊕⊕⊕⊝
moderate2 due to risk of bias |
|
| 547 per 1000 | 747 per 1000 (714 to 777) | ||||
|
Grade 3 to 4 diarrhoea Follow‐up: 13 to 38 months |
Study population | OR 1.84 (1.47 to 2.32) | 2909 (7 studies) | ⊕⊕⊕⊝
moderate2 due to risk of bias |
|
| 95 per 1000 | 162 per 1000 (134 to 196) | ||||
|
Grade 3 to 4 rash Follow‐up: 13 to 38 months |
Study population | OR 23.42 (13.22 to 41.49) | 2909 (7 studies) | ⊕⊕⊕⊝
moderate2 due to risk of bias |
|
| 11 per 1000 | 205 per 1000 (127 to 313) | ||||
|
Grade 3 to 4 neutropenia Follow‐up: 13 to 38 months |
Study population | OR 1.22 (0.93 to 1.61) | 2666 (6 studies) | ⊕⊕⊕⊝
moderate3 due to imprecision |
|
| 256 per 1000 | 296 per 1000 (240 to 357) | ||||
| Quality of life | 4 of 5 studies showed no difference between the 2 arms or equivocal results; the last study showed significant improvement on quality of life with the addition of EGFR MAb. | 2258 (5 studies) | ⊕⊕⊕⊝
moderate2 due to risk of bias |
||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HR: hazard ratio; OR: odds ratio | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1Four of the 12 identified open‐label trials employed local (i.e. non‐centralised), non‐blinded assessment of progression‐free survival and tumour response rate, but we note the consistent findings in favour of effect in both trials with centralised and non‐centralised response assessment. In our judgement this constitutes an unclear risk of bias, and is not severe enough to merit downgrading the level of evidence for progression‐free survival and tumour response rate. We also note significant heterogeneity in these analyses, which is due more by differing degrees of benefit rather than the presence or absence of benefit itself. We therefore opted to assess the evidence for these outcomes as high quality. 2We judged the outcomes overall grade 3 to 4 toxicity, grade 3 to 4 diarrhoea, and grade 3 to 4 rash as well as quality of life as being at high risk of bias because of unblinded assessment of (somewhat) subjective symptoms in all included trials. We therefore downgraded the quality of the evidence by one level to moderate for these outcomes. 3We noted that a sufficient number of events (377 + 347 = 714) and participants were included in this analysis. However, the 95% confidence interval for the summary statistic for this outcome (0.93 to 1.61) crosses both the point of no benefit (1) and that of significant clinical harm (1.25). We therefore downgraded the quality of evidence by one level for imprecision of the estimate. 4Figures estimated based on control group of Douillard PRIME 2010.
Summary of findings 2. EGFR MAb in KRAS exon 2 MT for metastatic colorectal cancer.
| EGFR MAb in KRAS exon 2 MT for metastatic colorectal cancer | |||||
|
Patient or population: people with metastatic colorectal cancer ‐ KRAS exon 2 MT
Intervention: EGFR MAb in addition to standard treatment Comparison: standard treatment Setting: multicentre international studies | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| Standard therapy | EGFR MAb + standard therapy | ||||
| Progression‐free survival Follow‐up: 13 to 38 months | 300 per 1000 (at 1 year)7 | 307 per 1000 (272 to 348) | HR 1.03 (0.89 to 1.20) | 2567 (8 studies) | ⊕⊕⊕⊝
moderate1 due to inconsistency |
| Overall survival Follow‐up: 13 to 38 months | 300 per 1000 (at 2 years)7 | 307 per 1000 (285 to 332) | HR 1.03 (0.94 to 1.13) | 2268 (8 studies) | ⊕⊕⊕⊕ high |
| Tumour response rate Follow‐up: 13 to 38 months | Study population | OR 0.93 (0.74 to 1.16) | 1925 (8 studies) | ⊕⊕⊕⊕ high | |
| 249 per 1000 | 236 per 1000 (197 to 278) | ||||
| Overall grade 3 to 4 toxicity Follow‐up: 13 to 38 months | Study population | OR 1.63 (0.98 to 2.71) | 1635 (5 studies) | ⊕⊝⊝⊝
very low2,3,4 due to inconsistency, imprecision, and risk of bias |
|
| 545 per 1000 | 661 per 1000 (540 to 764) | ||||
|
Grade 3 to 4 diarrhoea Follow‐up: 13 to 38 months |
Study population | OR 1.45 (1.01 to 2.11) | 1635 (5 studies) | ⊕⊕⊝⊝
low4,5 due to imprecision and risk of bias |
|
| 92 per 1000 | 128 per 1000 (93 to 176) | ||||
|
Grade 3 to 4 rash Follow‐up: 13 to 38 months |
Study population | OR 32.35 (15.01 to 69.7) | 1635 (5 studies) | ⊕⊕⊕⊝
moderate4,6 due to risk of bias |
|
| 7 per 1000 | 195 per 1000 (101 to 343) | ||||
|
Grade 3 to 4 neutropenia Follow‐up: 13 to 38 months |
Study population | OR 0.7 (0.53 to 0.93) | 968 (3 studies) | ⊕⊕⊕⊕ high | |
| 383 per 1000 | 303 per 1000 (366 to 248) | ||||
| Quality of life | No data available for this outcome | ||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HR: hazard ratio; OR: odds ratio | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1Significant heterogeneity exists in this analysis (I2 = 61%). Furthermore, several included studies (e.g. Bokemeyer OPUS 2009; Johnsson Nordic ACT 2013) report confidence intervals with no overlap, adding to the likelihood of inconsistency. Given this evidence pointing towards inconsistency, we downgraded the quality of the evidence for this outcome by one level. 2Significant heterogeneity exists in this analysis (I2 = 74%). Furthermore, several included studies (e.g. Amado 2008 and Bokemeyer OPUS 2009) report confidence intervals with no overlap, adding to the likelihood of inconsistency. 3We noted that the 95% confidence interval of the effect estimate is 0.98 to 2.71, which includes both the point of no effect (1) and appreciable harm (1.25). Given this evidence for imprecision, we downgraded the quality of the evidence for this outcome by one level. 4As discussed in the Assessment of risk of bias in included studies section, we judged the outcomes overall grade 3 to 4 toxicity, grade 3 to 4 diarrhoea, and grade 3 to 4 rash as at high risk of bias due to the unblinded nature of all studies and the subjective evaluation of these measures by either unblinded participant or clinician. We therefore downgraded the quality of the evidence for these outcomes by one level each. 5There were fewer than 300 events in total for this outcome (110 + 74 = 184). Although a low number of events in a dichotomous outcome does not necessitate downgrading, we also note that the 95% confidence interval is 1.01 to 2.11 ‐ quite close to an odds ratio of 1 (zero difference) and including the point of appreciable harm (1.25). We therefore downgraded this outcome for imprecision. 6There were fewer than 300 events in total for this outcome as well (195 + 6 = 201). However, the 95% confidence interval here does not come close to the point of no difference (1) and in fact is strongly in favour of increased frequency (odds ratio 32.35, 95% confidence interval 15.01 to 69.7). Given the magnitude of this result, we feel that any small imprecision is unlikely to alter the clinical interpretation of the result; therefore, in contrast to the prior outcome, we decided not to downgrade this outcome for imprecision. 7Figures estimated based on control group of Douillard PRIME 2010.
Summary of findings 3. EGFR MAb in extended RAS WT for metastatic colorectal cancer.
| EGFR MAb in extended RAS for metastatic colorectal cancer | |||||
|
Patient or population: people with metastatic colorectal cancer ‐ extended RAS WT
Intervention: EGFR MAb in addition to standard treatment Comparison: standard treatment Setting: multicentre international studies | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| Standard therapy | EGFR MAb + standard therapy | ||||
| Progression‐free survival Follow‐up: 13 to 38 months | 300 per 1000 (at 1 year) | 193 per 1000 (157 to 235) | HR 0.60 (0.48 to 0.75) | 1237 (6 studies) | ⊕⊕⊕⊝
moderate1 due to inconsistency |
| Overall survival Follow‐up: 13 to 38 months | 400 per 1000 (at 2 years) | 325 per 1000 (290 to 362) | HR 0.77 (0.67 to 0.88) | 1053 (4 studies) | ⊕⊕⊕⊕ high |
| Tumour response rate Follow‐up: 13 to 38 months | Study population | OR 4.28 (2.61 to 7.03) | 1001 (4 studies) | ⊕⊕⊕⊝
moderate2 due to inconsistency |
|
| 213 per 1000 |
536 per 1000 (414 to 655) |
||||
| Overall grade 3 to 4 toxicity | No data available for this outcome | ||||
| Grade 3 to 4 diarrhoea | No data available for this outcome | ||||
| Grade 3 to 4 rash | No data available for this outcome | ||||
| Grade 3 to 4 neutropenia | No data available for this outcome | ||||
| Quality of life | No data available for this outcome | ||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HR: hazard ratio; OR: odds ratio | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1We noted that significant heterogeneity exists in this analysis (I2 = 61%). Furthermore, two included studies report confidence intervals with no overlap (Amado 2008; Douillard PRIME 2010), adding to the likelihood of inconsistency. Even though the differential efficacy observed between trials leading to statistical heterogeneity can be explained by the differing use of EGFR MAb ‐ either as monotherapy or in combination with chemotherapy ‐ we felt that the degree of disparity between different trials nevertheless did warrant downgrading of the quality of the evidence in this case. 2We noted that significant heterogeneity (I2 = 47%) exists in this analysis. Even though separation of clinical trials by EGFR MAb (cetuximab versus panitumumab) resolved the heterogeneity, we felt that the degree of disparity between different trial results nevertheless warranted downgrading of the quality of the evidence in this case.
Summary of findings 4. EGFR MAb in extended RAS mutation for metastatic colorectal cancer.
| EGFR MAb in extended RAS mutation for metastatic colorectal cancer | |||||
|
Patient or population: people with metastatic colorectal cancer ‐ extended RAS MT
Intervention: EGFR MAb in addition to standard treatment Comparison: standard treatment Setting: multicentre international studies | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| Standard therapy | EGFR MAb + standard therapy | ||||
| Progression‐free survival Follow‐up: 13 to 38 months | 300 per 1000 (at 1 year) | 332 per 1000 (282 to 384) | HR 1.13 (0.93 to 1.36) | 2023 (6 studies) | ⊕⊕⊕⊝
moderate1 due to inconsistency |
|
Overall survival Follow‐up: 13 to 38 months |
300 per 1000 (at 2 years) | 322 per 1000 (282 to 367) | HR 1.09 (0.93 to 1.28) | 1768 (4 studies) | ⊕⊕⊕⊝
moderate2 due to imprecision |
| Tumour response rate Follow‐up: 13 to 38 months | Study population | OR 0.76 (0.55 to 1.05) | 840 (3 studies) | ⊕⊕⊕⊝
moderate3 due to imprecision |
|
| 285 per 1000 | 233 per 1000 (180 to 295) | ||||
| Moderate | |||||
| 360 per 1000 | 299 per 1000 (236 to 371) | ||||
| Overall grade 3 to 4 toxicity | No data available for this outcome | ||||
| Grade 3 to 4 diarrhoea | No data available for this outcome | ||||
| Grade 3 to 4 rash | No data available for this outcome | ||||
| Grade 3 to 4 neutropenia | No data available for this outcome | ||||
| Quality of life | No data available for this outcome | ||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HR: hazard ratio; OR: odds ratio | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1We noted that significant heterogeneity exists (I2 = 62%) in this analysis with studies whose confidence intervals do not overlap (e.g. Peeters 2010 and Douillard PRIME 2010). Even though subgroup analysis of trials by their respective lines of therapy explained some of this heterogeneity, significant heterogeneity remained (in the subgroup of second‐line studies, I2 = 62%). We therefore downgraded the quality of the evidence by one grade for inconsistency. 2The 95% confidence interval of the effect estimate is 0.93 to 1.28, which includes both the point of no effect (1) and appreciable harm (1.25). As a result of this finding in an important outcome (where a 25% difference would certainly be clinically important), we downgraded the quality of the evidence for this outcome by one grade. 3Fewer than 300 events were observed in this analysis (113 + 115 = 228). In addition, the 95% confidence interval of the effect estimate is 0.55 to 1.05, which includes both the point of no effect (1) and significant harm (0.75). As a result of this evidence for imprecision, we downgraded the quality of the evidence for this outcome by one grade.
Summary of findings 5. EGFR inhibitors in KRAS unselected participants.
| EGFR inhibitors in KRAS unselected participants | |||||
|
Patient or population: people with metastatic colorectal cancer, not selected by KRAS status
Intervention: EGFR inhibitors in addition to standard treatment Comparison: standard treatment Setting: multicentre international studies | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| Standard therapy | EGFR inhibitor+ standard therapy | ||||
| Progression‐free survival Follow‐up: 17 months (reported by 1 study) | The majority of studies showed no change in progression‐free survival. | 1483 (2 studies) | ⊕⊕⊕⊕ high | ||
|
Overall survival Follow‐up: 17 months (reported by 1 study) |
Both studies showed no effect on overall survival. | 1382 (2 studies) | ⊕⊕⊕⊕ high | ||
| Tumour response rate Follow‐up: 17 months (reported by 1 study) | The majority of studies showed increased response rate. | 1372 (2 studies) | ⊕⊕⊕⊝
moderate1 due to imprecision |
||
|
Overall grade 3 to 4 toxicity Follow‐up: 17 months (reported by 1 study) |
The included study showed increased rate of toxicity. | 1267 (1 study) | ⊕⊕⊝⊝
low1,2 due to risk of bias and imprecision |
||
|
Grade 3 to 4 diarrhoea Follow‐up: 17 months (reported by 1 study) |
The included studies showed increased rates of diarrhoea. | 1341 (2 studies) | ⊕⊕⊝⊝
low1,2 due to risk of bias and imprecision |
||
|
Grade 3 to 4 rash Follow‐up: 17 months (reported by 1 study) |
The included studies showed increased rates of rash. | 1341 (2 studies) |
⊕⊕⊝⊝
low1,2 due to risk of bias and imprecision |
||
|
Grade 3 to 4 neutropenia Follow‐up: 17 months (reported by 1 study) |
The included studies showed a small increase or no change in rates of neutropenia. | 1341 (2 studies) |
⊕⊕⊕⊝
moderate1 due to imprecision |
||
| Quality of life | The included study reported improved quality of life in the intervention arm. | 1298 (1 study) | ⊕⊕⊕⊝
moderate2 due to risk of bias |
||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1Each of these outcomes had fewer than 400 recorded events, and given the low event rates, we downgraded the quality of the evidence for these outcomes by one grade. 2We judged overall toxicity, rash, diarrhoea, and quality of life as being at high risk of bias due to unblinded assessment of (somewhat) subjective symptoms in all included trials. We therefore downgraded the quality of the evidence by one level for each of these outcomes.
Summary of findings 6. Comparing EGFR inhibitors to another biologic agent.
| Comparing EGFR inhibitors to another biologic agent | |||||
|
Patient or population: people with metastatic colorectal cancer
Intervention: EGFR inhibitors (EGFR MAb in all identified trials) in addition to standard therapy
Comparison: another biologic agent (bevacizumab in all identified trials) in addition to standard therapy Setting: multicentre international studies | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| Bevacizumab + standard therapy | EGFR inhibitor + standard therapy | ||||
| Progression‐free survival Follow‐up: 24 to 36 months | 400 per 1000 (at 1 year)8 | 406 per 1000 (378 to 436) | HR 1.02 (0.93 to 1.12) | 2189 (4 studies) | ⊕⊕⊕⊕ high |
| Overall survival Follow‐up: 24 to 36 months | 500 per 1000 (at 2 years)8 | 441 per 1000 (384 to 503) | HR 0.84 (0.70 to 1.01) | 2189 (4 studies) | ⊕⊕⊕⊝
moderate1,2 due to imprecision |
| Tumour response rate Follow‐up: 24 to 36 months | Study population | OR 1.36 (1.15 to 1.62) | 2184 (4 studies) | ⊕⊕⊕⊝
moderate3 due to limitations in implementation |
|
| 539 per 1000 | 614 per 1000 (573 to 654) | ||||
| Overall grade 3 to 4 toxicity Follow‐up: 24 to 36 months | Study population | OR 1.37 (1.09 to 1.72) | 2133 (4 studies) | ⊕⊕⊕⊝
moderate4 due to risk of bias |
|
| 667 per 1000 | 733 per 1000 (686 to 775) | ||||
|
Grade 3 to 4 diarrhoea Follow‐up: 36 to 40 months |
Study population | OR 1.06 (0.67 to 1.67) | 1673 (2 studies) | ⊕⊝⊝⊝
very low4,5,6 due to risk of bias, inconsistency, and imprecision |
|
| 103 per 1000 | 111 per 1000 (83 to 145) | ||||
|
Grade 3 to 4 rash Follow‐up: 12 to 40 months |
Study population | OR 47.53 (14.84 to 152.19) | 1951 (3 studies) | ⊕⊕⊕⊝
moderate4,7 due to risk of bias |
|
| 2 per 1000 | 90 per 1000 (30 to 240) | ||||
| Grade 3 to 4 neutropenia | No data available for this outcome | ||||
| Quality of life | No data available for this outcome | ||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HR: hazard ratio; OR: odds ratio | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1Significant heterogeneity (I2 = 51%) was present in this analysis. However, consideration of trials by lines of therapy led to a decrease in statistical heterogeneity, which we considered to be a plausible explanation for the differential efficacy observed. We therefore did not downgrade the quality of the evidence for inconsistency for this outcome. 2The 95% confidence interval is (0.70 to 1.01), which includes both the point of no effect (1) and a point of clinically significant benefit (0.75). As a 25% difference in overall survival would certainly be clinically important, we felt that the imprecision in this estimate warranted a downgrading in the quality of the evidence. 3Significant dropout rates were noted in Heinemann FIRE‐3 2014 (28/297 participants not assessable for response in cetuximab arm, 20/295 in bevacizumab arm, for "other reasons"). On sensitivity analyses (see 6.3 Tumour response rate in the Results), best‐case/worst‐case analyses showed that the above result did not remain significant (1.26, 95% CI 0.93 to 1.71). We therefore downgraded the quality of the evidence by one level due to the limitations of implementation demonstrated by the significant dropout rate. 4We noted that we considered outcomes 6.4 to 6.6 (overall grade 3 to 4 toxicity, grade 3 to 4 diarrhoea, and grade 3 to 4 rash) as at high risk of bias due to the unblinded nature of all of the included trials and the assessment of toxicity by either unblinded clinician or participant. As a result, we downgraded each of these outcomes by one grade. 5We noted that there was significant heterogeneity in this analysis (I2 = 52%) with two studies that varied widely in their confidence intervals (the 95% confidence interval for Heinemann FIRE‐3 2014 was 0.51 to 1.34, whereas it was 0.87 to 1.98 for Venook CALGB 80405 2014). We therefore downgraded this outcome for inconsistency. 6We noted that there were fewer than 300 events in total for this outcome (93 + 85 = 178). Given that the 95% confidence interval (0.79 to 1.48) includes both the point of no effect (1) and a point of clinically significant harm (1.25), we decided to downgrade this outcome for imprecision. 7There were fewer than 300 events in total for this outcome as well (134 + 2 = 136). However, the 95% confidence interval, considered in absolute terms, differs significantly to the event rate without intervention (2 per 1000 compared to the 95% confidence interval of 30 to 240 per 1000). The imprecision noted here is unlikely to affect the clinical interpretation of this analysis (that the odds of developing rash on EGFR MAb is likely to be significantly increased). We therefore decided not to downgrade this outcome for imprecision, in contrast to the prior outcome. 8Numbers estimated based on figures provided in Heinemann FIRE‐3 2014.
Summary of findings 7. Comparing different EGFR inhibitor agents or regimens.
| Comparing different EGFR inhibitor agents or regimens | |||||
|
Patient or population: people with metastatic colorectal cancer, not selected by KRAS status
Intervention: EGFR inhibitor in combination with standard treatment Comparison: a different EGFR inhibitor (or the same one with a different regimen) in combination with standard treatment Setting: multicentre international studies | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| EGFR inhibitor + standard therapy (different agent or dose of EGFR inhibitor) | EGFR inhibitor+ standard therapy | ||||
| Progression‐free survival Follow‐up: 10 to 34 months | The majority of studies showed no difference. | 1651 (6 studies) | ⊕⊕⊕⊝
moderate1 due to risk of bias |
||
|
Overall survival Follow‐up: 10 to 34 months |
The majority of studies showed no difference. | 1482 (5 studies) | ⊕⊕⊕⊕ high | ||
| Tumour response rate Follow‐up: 10 to 34 months | The majority of studies showed no difference. | 1313 (4 studies) | ⊕⊕⊕⊝
moderate2 due to imprecision |
||
|
Overall grade 3 to 4 toxicity Follow‐up: 10 to 34 months |
The majority of studies showed no difference. | 1651 (6 studies) | ⊕⊕⊕⊝
moderate1 due to risk of bias |
||
|
Grade 3 to 4 diarrhoea Follow‐up: 10 to 34 months |
The majority of studies showed no difference. | 1651 (6 studies) | ⊕⊕⊝⊝
low1,2 due to risk of bias and imprecision |
||
|
Grade 3 to 4 rash Follow‐up: 10 to 34 months |
The majority of studies showed no difference. | 1651 (6 studies) | ⊕⊕⊝⊝
low1,2 due to risk of bias and imprecision |
||
|
Grade 3 to 4 neutropenia Follow‐up: 10 to 34 months |
The majority of studies showed no difference. | 1651 (6 studies) | ⊕⊕⊕⊝
moderate2 due to imprecision |
||
| Quality of life | No data available for this outcome | ||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1We downgraded these outcomes for high risk of bias, as noted in the 'Summary assessments of risk of bias across studies for each outcome' section in the Results. 2As these outcomes had fewer than 400 events, we downgraded the quality of the evidence by one grade.
Summary of findings 8. EGFR TKI in KRAS unselected participants.
| EGFR inhibitors in KRAS unselected participants | |||||
|
Patient or population: people with metastatic colorectal cancer, not selected by KRAS status
Intervention: EGFR TKI in addition to standard treatment Comparison: standard treatment Setting: multicentre international studies | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| Standard therapy | EGFR TKI+ standard therapy | ||||
| Progression‐free survival Follow‐up: 14.5 months (reported by 1 study) | Both studies found no difference. | 181 (2 studies) | ⊕⊕⊝⊝
low1,2 due to risk of bias and imprecision |
||
|
Overall survival Follow‐up: 14.5 months (reported by 1 study) |
The only study reporting this outcome found no difference. | 99 (2 studies) | ⊕⊕⊕⊝
moderate2 due to imprecision |
||
| Tumour response rate Follow‐up: 14.5 months (reported by 1 study) | The only study reporting this outcome found no difference. | 99 (1 study) | ⊕⊕⊝⊝
low1,2 due to risk of bias and imprecision |
||
|
Overall grade 3 to 4 toxicity Follow‐up: 14.5 months (reported by 1 study) |
The only study reporting this outcome found an increased rate of toxicity. | 99 (1 study) | ⊕⊕⊝⊝
low1,2 due to risk of bias and imprecision |
||
|
Grade 3 to 4 diarrhoea Follow‐up: 14.5 months (reported by 1 study) |
Both studies found an increased rate of diarrhoea. | 181 (2 studies) | ⊕⊕⊝⊝
low1,2 due to risk of bias and imprecision |
||
|
Grade 3 to 4 rash Follow‐up: 14.5 months (reported by 1 study) |
The only study reporting this outcome found an increased rate of rash. | 99 (1 study) | ⊕⊕⊝⊝
low1,2 due to risk of bias and imprecision |
||
|
Grade 3 to 4 neutropenia Follow‐up: 14.5 months (reported by 1 study) |
The only study reporting this outcome found an increased rate of neutropenia. | 99 (1 study) | ⊕⊕⊕⊝
moderate2 due to imprecision |
||
| Quality of life | No data available for this outcome | ||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1We judged these outcomes to be at high risk of bias due to their open‐label nature without centralised or blinded review of outcomes. We therefore downgraded the quality of the evidence by one grade. 2No outcome achieved 400 events because of the low number of included participants, thus all outcomes were downgraded one grade for imprecision.
Summary of findings 9. EGFR inhibitors added to bevacizumab for metastatic colorectal cancer.
| EGFR inhibitors added to bevacizumab for metastatic colorectal cancer | |||||
|
Patient or population: people with metastatic colorectal cancer
Intervention: EGFR inhibitors in addition to the combination of bevacizumab and standard therapy Comparison: bevacizumab and standard therapy Setting: multicentre international studies | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| Bevacizumab + standard therapy | EGFR inhibitor+ bevacizumab + standard therapy | ||||
|
Progression‐free survival Follow‐up: 23 to 37 months |
400 per 1000 (at 1 year)11 | 412 per 1000 (346 to 483) | HR 1.04 (0.83 to 1.29) | 1571 (6 studies) | ⊕⊝⊝⊝
very low1,2,3 due to risk of bias, inconsistency, and imprecision |
|
Overall survival Follow‐up: 23 to 37 months |
500 per 1000 (at 2 years)11 | 500 per 1000 (380 to 639) | HR 1.00 (0.69 to 1.47) | 1257 (5 studies) | ⊕⊕⊝⊝
low4,5 due to inconsistency and imprecision |
| Tumour response rate Follow‐up: 11 to 50 months | Study population | OR 1.2 (0.67 to 2.12) | 1310 (4 studies) | ⊕⊝⊝⊝
very low1,6,7 due to risk of bias, inconsistency, and imprecision |
|
| 387 per 1000 | 431 per 1000 (297 to 572) | ||||
|
Overall grade 3 to 4 toxicity Follow‐up: 11 to 35 months |
Study population | OR 2.57 (1.45 to 4.57) | 1831 (3 studies) | ⊕⊕⊝⊝
low1,8 due to risk of bias and inconsistency |
|
| 717 per 1000 | 867 per 1000 (786 to 921) | ||||
|
Grade 3 to 4 diarrhoea Follow‐up: 11 to 50 months |
Study population | OR 2.58 (1.44 to 4.64) | 2434 (5 studies) | ⊕⊕⊝⊝
low1,9 due to risk of bias and inconsistency |
|
| 110 per 1000 | 242 per 1000 (151 to 364) | ||||
|
Grade 3 to 4 rash Follow‐up: 11 to 50 months |
Study population | OR 67.52 (30.83 to 147.85) | 2363 (4 studies) | ⊕⊕⊕⊕ moderate1 due to risk of bias |
|
| 5 per 1000 | 257 per 1000 (136 to 431) | ||||
|
Grade 3 to 4 neutropenia Follow‐up: 11 to 37 months |
Study population | OR 0.97 (0.73 to 1.29) | 1187 (2 studies) | ⊕⊕⊕⊝
moderate10 due to imprecision |
|
| 205 per 1000 | 200 per 1000 (158 to 250) | ||||
| Quality of life | No data available for this outcome | ||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HR: hazard ratio; OR: odds ratio | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1As mentioned in the 'Summary assessments of risk of bias across studies for each outcome' section in the Results, we considered outcomes 9.1 (progression‐free survival), 9.3 (tumour response rate), 9.4 to 9.6 (overall grade 3 to 4 toxicity, grade 3 to 4 diarrhoea, grade 3 to 4 rash) as at high risk of bias largely due to the unblinded design of all included randomised controlled trials and the potential for performance bias. We therefore downgraded these outcomes by one grade for risk of bias. 2Significant heterogeneity exists in the analysis of this outcome (I2 = 66%) with studies whose confidence intervals do not overlap (e.g. Hecht PACCE 2009 and Tournigand DREAM 2015). We therefore considered there to be enough evidence to downgrade this outcome for inconsistency. 3The 95% confidence interval, 0.83 to 1.29, crosses the point of no effect (1) as well as significant harm (1.25). Given that for this outcome (progression‐free survival) a 25% change would be clinically relevant, we therefore considered the imprecision to be enough to justify downgrading of this outcome. 4The 95% confidence interval, 0.76 to 1.49, crosses the point of no effect (1) as well as significant harm (1.25). As a 25% change in overall survival would definitely be clinically relevant, we therefore considered the evidence of imprecision to be enough to justify downgrading of this outcome. 5Significant heterogeneity exists in the analysis of this outcome (I2 = 81%) with studies whose confidence intervals do not overlap (e.g. Hecht PACCE 2009 and Tournigand DREAM 2015). Although separation of trials investigating EGFR TKI in the maintenance setting and those investigating EGFR MAb on disease progression reduced the amount of heterogeneity evident, the degree of between‐study heterogeneity was such that we considered that any summary measure incorporating these disparate trials should be downgraded for inconsistency. 6We noted that the 95% confidence interval (0.67 to 2.12) includes the point of no effect (1), a point of clinically significant benefit (1.25), as well as a point of significant harm (0.75). All of these points would be clinically relevant for tumour response rate. Given that the large confidence interval spans all these points, we therefore decided to downgrade this outcome for imprecision. 7We noted that there was significant heterogeneity in this analysis (I2 = 78%). Furthermore, two of the included studies, Hecht PACCE 2009 and Tournigand DREAM 2015, reported confidence intervals with no overlap, adding to the likelihood of inconsistency. We therefore downgraded this outcome. 8We noted significant heterogeneity in this analysis (I2 = 77%). Furthermore, two studies have 95% confidence intervals that only minimally overlap (2.30 to 4.63 in Hecht PACCE 2009, compared to 1.15 to 2.32 in Tol CAIRO2 2008). We therefore considered there to be enough evidence to downgrade this outcome for inconsistency. 9We noted significant heterogeneity in this analysis (I2 = 64%). Furthermore, two studies have 95% confidence intervals that do not overlap (Tol CAIRO2 2008; Tournigand DREAM 2015). Despite clinical exploration of between‐study heterogeneity by investigating trial design and patient populations, we were unable to explain the bulk of the heterogeneity observed. We therefore considered there to be enough evidence to downgrade this outcome for inconsistency. 10There were fewer than 300 events in total for this outcome (120 + 121 = 241). In addition, the 95% confidence interval of the effect estimate (0.73 to 1.29) crosses the point of no benefit (1), significant harm (1.25), as well as significant benefit (0.75). We therefore considered there to be enough evidence to downgrade this outcome for imprecision. 11Estimated from Hecht PACCE 2009 and Tol CAIRO2 2008.
33 studies with a total of 15,250 participants were included in meta‐analyses. Findings from these analyses are summarised in Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9.
1. The addition of EGFR MAb to standard therapy in KRAS exon 2 WT populations
1.1 Progression‐free survival
See: Analysis 1.1; Figure 5.
1.1. Analysis.
Comparison 1 EGFR MAb in KRAS exon 2 WT, Outcome 1 Progression‐free survival.
5.
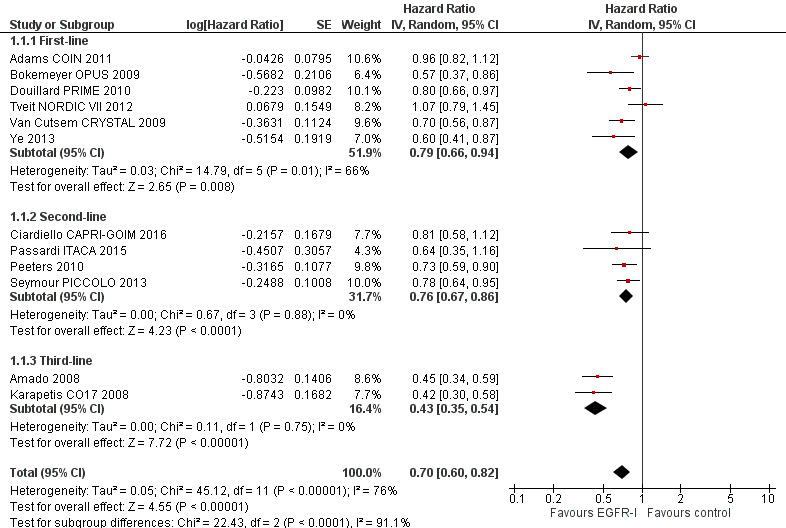
Forest plot of comparison: 1 EGFR MAb in KRAS exon 2 WT, outcome: 1.1 Progression‐free survival.
Overall, 12 RCTs (evaluating 4402 KRAS exon 2 WT participants) investigated the addition of EGFR MAb to standard therapy. Pooled analysis of these studies demonstrated that EGFR MAb reduced the risk of disease progression by 30% (hazard ratio (HR) 0.70, 95% confidence interval (CI) 0.60 to 0.82). The test for subgroup differences revealed considerable differences in effect estimates across different lines of therapy (Chi2 = 22.43, df = 2, P < 0.001, I2 = 91.1%). There was substantial heterogeneity in this analysis (Chi2 = 45.12, df = 11, P < 0.001, I2 = 76%), which may be due to pooling of studies involving differing lines of treatment and chemotherapy partners. In particular we considered that use of EGFR MAb in different lines of therapy may produce different degrees of benefit. For instance, given that the use of cytotoxic chemotherapy improves outcomes such as progression‐free survival, the incremental benefit of adding EGFR MAb in first‐ or second‐line trials (with chemotherapy) may be less than that seen with EGFR MAb (as monotherapy) in third‐line trials. We note that I2 is on the whole less when the trials are analysed by group (see below). Removal of third‐line trials lessened the heterogeneity from substantial to moderate (Chi2 = 16.61, df = 9, P = 0.06, I2 = 46%) (Amado 2008; Karapetis CO17 2008).
Pooled analysis of all first‐line trials in KRAS exon 2 WT populations (6 RCTs, 2671 participants) showed that adding EGFR MAb reduced the risk of disease progression by 21% (HR 0.79, 95% CI 0.66 to 0.94; P = 0.01; Analysis 1.1.1). The phase III trials were as follows: Van Cutsem CRYSTAL 2009 investigated the addition of cetuximab to FOLFIRI, and reported improved PFS with HR 0.70 (95% CI 0.56 to 0.87); Douillard PRIME 2010 investigated the addition of panitumumab to FOLFOX4, and reported improved PFS with HR 0.80 (95% CI 0.66 to 0.97); Adams COIN 2011 investigated the addition of cetuximab to either CAPOX or mFOLFOX6, and reported no overall PFS improvement with HR 0.96 (95% CI 0.82 to 1.12); Bokemeyer OPUS 2009 investigated the addition of cetuximab to FOLFOX4, and reported improved PFS with HR 0.57 (95% CI 0.37 to 0.86); and Tveit NORDIC VII 2012 investigated the addition of cetuximab to FLOX and reported no improvement in PFS (HR 1.09, 95% CI 0.79 to 1.45). Ye 2013 (described as a phase IV trial) investigated the addition of cetuximab to chemotherapy (either mFOLFOX6 or FOLFIRI), showing improved PFS with HR 0.60 (95% CI 0.41 to 0.87).
We noted substantial heterogeneity in the meta‐analysis (Chi2 = 14.79, df = 5, P = 0.01, I2 = 66%). This may be attributable to the pooling of studies utilising different fluoropyrimidine regimens; whilst most trials used infusional fluorouracil (5‐FU), one allowed substitution of capecitabine (Adams COIN 2011), and one utilised a bolus 5‐FU regimen, FLOX (Tveit NORDIC VII 2012). Exclusion of these two trials resulted in no important residual heterogeneity (Chi2 = 3.47, df = 3, P = 0.32, I2 = 14%). Another potential reason for the observed heterogeneity is that some studies used oxaliplatin as part of chemotherapy whilst others used irinotecan.
Pooled analysis of second‐line trials in KRAS exon 2 WT populations (4 RCTs, 1258 participants) showed that adding EGFR MAb to chemotherapy reduced the risk of disease progression by 24% (HR 0.76, 95% CI 0.67 to 0.86; P < 0.001; Analysis 1.1.2). Peeters 2010 investigated FOLFIRI plus panitumumab versus FOLFIRI alone and reported improved PFS (HR 0.73, 95% CI 0.59 to 0.90). Seymour PICCOLO 2013 investigated irinotecan plus panitumumab versus irinotecan in second‐line and subsequent settings, and reported improved PFS (HR 0.78, 95% CI 0.64 to 0.95). No important heterogeneity was present (Chi2 = 0.21, df = 1, P = 0.65, I2 = 0%). Passardi ITACA 2015 randomised participants due for first‐line therapy to physician's choice of chemotherapy with bevacizumab versus the same chemotherapy. The participants randomised to chemotherapy with bevacizumab were eligible for a subtrial (included in this analysis) investigating the combination of second‐line chemotherapy and cetuximab compared to second‐line chemotherapy alone. There were no significant differences in PFS (HR 0.64, 95% CI 0.35 to 1.16) or OS (HR 1.22, 95% CI 0.65 to 2.29), although the subtrial included only 48 participants. Ciardiello CAPRI‐GOIM 2016 commenced all participants on FOLFIRI with cetuximab, and on progression from this therapy randomised participants to FOLFOX with cetuximab or FOLFOX (the precise FOLFOX regimen was not described). Progression‐free survival did not differ significantly between the two arms (HR 0.81, 95% CI 0.58 to 1.12).
Pooled analysis of third‐line trials in KRAS exon 2 WT populations (2 RCTs, 473 participants) showed that compared to placebo, EGFR MAb reduced the risk of disease progression by 57% (HR 0.43, 95% CI 0.35 to 0.54; P < 0.001; Analysis 1.2.3). Amado 2008 compared panitumumab to best supportive care and reported improved PFS (HR 0.45, 95% CI 0.34 to 0.59). Karapetis CO17 2008 compared cetuximab to best supportive care. An improvement in PFS was observed with HR 0.42 (95% CI 0.30 to 0.58) No important heterogeneity was present (Chi2 = 0.11, df = 1, P = 0.75, I2 = 0%).
1.2 Overall survival
See: Analysis 1.2; Figure 6.
1.2. Analysis.
Comparison 1 EGFR MAb in KRAS exon 2 WT, Outcome 2 Overall survival.
6.
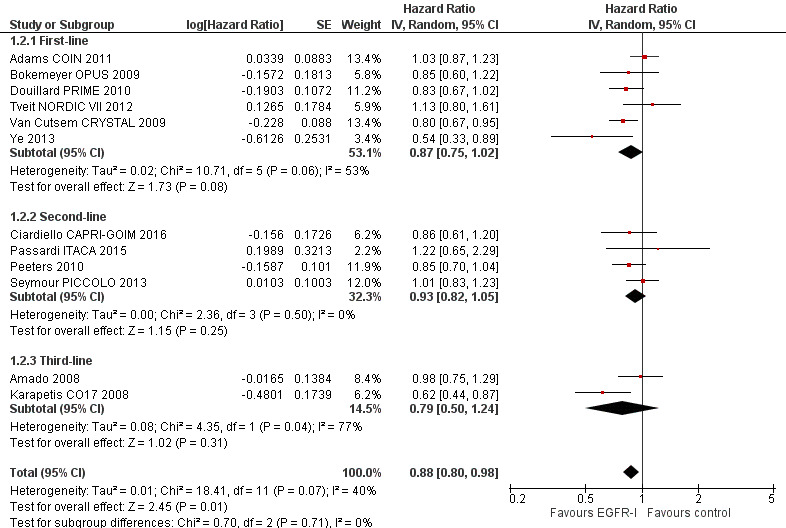
Forest plot of comparison: 1 EGFR MAb in KRAS exon 2 WT, outcome: 1.2 Overall survival.
Overall, 12 RCTs (evaluating 4402 KRAS exon 2 WT participants) investigated the addition of EGFR MAb to standard therapy in KRAS exon 2 WT populations. Pooled analysis of these trials showed that adding EGFR MAb decreased the risk of death by 12% (HR 0.88, 95% CI 0.80 to 0.98; P = 0.01). Moderate statistical heterogeneity was present (Chi2 = 18.41, df = 11, P = 0.07, I2 = 40%), again likely due to the pooling of studies investigating EGFR MAb use in different lines of treatment. Exclusion of third‐line studies resulted in ongoing moderate heterogeneity (Chi2 = 13.44, df = 9, P = 0.14, I2 = 33%). However, no important subgroup differences were present (Chi2 = 0.70, df = 2, P = 0.71, I2 = 0%).
Pooled analysis of first‐line trials (6 RCTs, 2671 participants) in KRAS exon 2 WT populations showed that adding EGFR MAb to first‐line chemotherapy did not significantly decrease the risk of death (HR 0.87, 95% CI 0.75 to 1.02; P = 0.08; Analysis 1.2.1). Moderate statistical heterogeneity was present (Chi2 = 10.71, df = 5, P = 0.06, I2 = 53%), again likely due to the pooling of studies utilising different fluoropyrimidine regimens. Exclusion of Adams COIN 2011 and Tveit NORDIC VII 2012, as per the rationale discussed above in Analysis 1.1, resulted in no residual heterogeneity (Chi2 = 2.58, df = 3, P = 0.46, I2 = 0%).
Pooled analysis of second‐line trials (4 RCTs, 1258 participants) in KRAS exon 2 WT populations showed that adding EGFR MAb to second‐line chemotherapy did not significantly decrease the risk of death (HR 0.93, 95% CI 0.82 to 1.05; P = 0.25; Analysis 1.2.2). No important heterogeneity was present (Chi2 = 2.36, df = 3, P = 0.50, I2 = 0%).
Pooled analysis of third‐line trials (2 RCTs, 473 participants) in KRAS exon 2 WT populations showed that compared to placebo, EGFR MAb did not significantly decrease the risk of death (HR 0.79, 95% CI 0.50 to 1.24; P = 0.31; Analysis 1.2.3). Substantial statistical heterogeneity was present (Chi2 = 4.35, df = 1, P = 0.04, I2 = 77%), likely attributable to the differential cross‐over in the two included studies. Karapetis CO17 2008 demonstrated significant OS benefit (HR 0.62, 95% CI 0.44 to 0.87), likely because the trial did not allow cross‐over from placebo to cetuximab on progression, with only 13 of 285 participants subsequently receiving EGFR MAb. In contrast, Amado 2008 reported no OS improvement (HR 0.98, 95% CI 0.75 to 1.29) in the context of PFS improvement. This trial allowed cross‐over, which occurred in 90 of 119 KRAS exon 2 WT participants originally in the placebo arm. Given the post hoc nature of this analysis and the high degree of heterogeneity, the results of this subgroup analysis should be interpreted with caution.
1.3 Tumour response rate
See: Analysis 1.3.
1.3. Analysis.
Comparison 1 EGFR MAb in KRAS exon 2 WT, Outcome 3 Tumour response rate.
Overall, 12 RCTs (evaluating 4147 KRAS exon 2 WT participants) investigated the addition of EGFR MAb to standard therapy. Pooled analysis of these trials showed that the addition of EGFR MAb increased the rate of response by 14.5%, from 31.1% (645/2077) to 45.6% (944/2070) with odds ratio (OR) 2.41 (95% CI 1.70 to 3.41; P < 0.001). The test for subgroup differences revealed considerable differences in effect estimates across different lines of therapy (Chi2 = 17.37, df = 2, P = 0.0002, I2 = 88.5%). Substantial statistical heterogeneity was present (Chi2 = 47.75, df = 11, P < 0.001, I2 = 77%), likely due to the differing lines of treatment investigated in the trials, which is supported by the fact that the analyses below (by line of therapy) show less heterogeneity.
Pooled analysis of first‐line trials (6 RCTs, 2447 participants) in KRAS exon 2 WT populations showed that adding EGFR MAb to first‐line chemotherapy increased the rate of response by 12.0% from 575/1243 (46.3%) to 702/1204 (58.3%) (OR 1.73, 95% CI 1.33 to 2.25; P < 0.001; Analysis 1.3.1). Moderate statistical heterogeneity was present (Chi2 = 10.86, df = 5, P = 0.10, I2 = 54%), likely due to the pooling of trials using different chemotherapy backbones.
Pooled analysis of second‐line trials (4 RCTs, 1243 participants) in KRAS exon 2 WT populations showed that adding EGFR MAb to second‐line chemotherapy increased the rate of response by 21.8% from 11.3% (70/618) to 33.1% (206/625) (OR 3.60, 95% CI 2.45 to 5.30; P < 0.001), with no important heterogeneity (Chi2 = 4.18, df = 3, P = 0.24, I2 = 28%).
Pooled analysis of third‐line trials (2 RCTs, 457 participants) in KRAS exon 2 WT populations showed that using EGFR MAb compared to placebo increased the rate of response from 0% (0/216) to 14.9% (36/241) (OR 38.44, 95% CI 5.22 to 282.91; P = 0.0003). No important heterogeneity was present (Chi2 = 0.01, df = 1, P = 0.91, I2 = 0%).
1.4 Adverse effects
See: Analysis 1.4; Analysis 1.5; Analysis 1.6; Analysis 1.7.
1.4. Analysis.
Comparison 1 EGFR MAb in KRAS exon 2 WT, Outcome 4 Grade 3/4 toxicity.
1.5. Analysis.
Comparison 1 EGFR MAb in KRAS exon 2 WT, Outcome 5 Grade 3/4 diarrhoea.
1.6. Analysis.
Comparison 1 EGFR MAb in KRAS exon 2 WT, Outcome 6 Grade 3/4 rash.
1.7. Analysis.
Comparison 1 EGFR MAb in KRAS exon 2 WT, Outcome 7 Grade 3/4 neutropenia.
Overall, six RCTs (evaluating 2771 KRAS exon 2 WT participants) reported the effects of adding EGFR MAb to standard therapy on overall grade 3 to 4 toxicity. Pooled analysis showed that the rate of overall toxicity increased by 18.4% from 54.7% (769/1405) in the control arm to 73.1% (999/1366) in the experimental arm (OR 2.45, 95% CI 2.07 to 2.89; Analysis 1.4). No important heterogeneity was present (Chi2 = 1.14, df = 5, P = 0.95, I2 = 0%).
We performed pooled analysis of the same studies for the incidence of grade 3 to 4 diarrhoea, rash, and neutropenia.
The incidence of grade 3 to 4 diarrhoea in KRAS exon 2 WT participants (7 RCTs, 2909 participants) increased by 6.5% from 9.5% (140/1473) in the control arm to 16.0% (230/1436) in the experimental arm (OR 1.84, 95% CI 1.47 to 2.32; Analysis 1.5). The incidence of grade 3 to 4 rash in KRAS exon 2 WT participants (7 RCTs, 2909 participants) increased from 1.1% (16/1473) in the control arm to 24.1% (346/1436) in the experimental arm (OR 23.42, 95% CI 13.22 to 41.49; Analysis 1.6). No important statistical heterogeneity was present for these analyses (respectively: diarrhoea: Chi2 = 1.91, df = 6, P = 0.93, I2 = 0%; rash: Chi2 = 6.82, df = 6, P = 0.34, I2 = 12%).
The incidence of grade 3 to 4 neutropenia in KRAS exon 2 WT participants (6 RCTs, 2666 participants) did not significantly increase, being 25.6% (347/1354) in the control arm versus 28.7% (377/1312) in the experimental arm (OR 1.22, 95% CI 0.93 to 1.61; Analysis 1.7). Moderate heterogeneity was present for this analysis (Chi2 = 10.39, df = 5, P = 0.06, I2 = 52%), which could be due to the use of different chemotherapy regimens, such as FOLFOX in Bokemeyer OPUS 2009 and Douillard PRIME 2010, FLOX in Tveit NORDIC VII 2012, FOLFIRI in Peeters 2010 and Van Cutsem CRYSTAL 2009, and irinotecan alone for Seymour PICCOLO 2013. Exclusion of Seymour PICCOLO 2013 (being the only trial using chemotherapy without fluoropyrimidine backbone) resulted in a decrease to no important heterogeneity (Chi2 = 4.72, df = 4, P = 0.32, I2 = 15%).
No important subgroup interactions were present in any of these four analyses (I2 = 0% for each).
1.5 Quality of life
Five included studies reported quality of life (QoL) results for the KRAS exon 2 wildtype participants (Douillard PRIME 2010; Karapetis CO17 2008; Peeters 2010; Seymour PICCOLO 2013; Van Cutsem CRYSTAL 2009), whereas a sixth study has collected this information but has not yet reported on quality of life outcomes (Adams COIN 2011). It is worth noting that a seventh study (Amado 2008) reported no sginificant differences in overall QoL results on KRAS unselected patients, but has not reported any QoL results by KRAS status. Of the five studies which have reported QoL, one study showed improved quality of life as measured on the EORTC QLQ‐C30 scale (Karapetis CO17 2008), whereas the other four studies showed neutral or equivocal results on quality of life (Douillard PRIME 2010; Peeters 2010; Seymour PICCOLO 2013; Van Cutsem CRYSTAL 2009). Van Cutsem CRYSTAL 2009 showed significant improvement in global EORTC QLQ‐C30 with the combination of irinotecan and panitumumab (as opposed to irinotecan alone) but significantly worse EORTC QLQ‐C30 symptom scores in the same arm.
2. EGFR MAb in KRAS exon 2 mutant participants
Overall, 9 studies (2609 participants) investigated the effect of adding EGFR MAb to either chemotherapy or best supportive care in KRAS exon 2 mutant populations.
2.1 Progression‐free survival
See: Analysis 2.1; Figure 7.
2.1. Analysis.
Comparison 2 EGFR MAb in KRAS exon 2 MT, Outcome 1 Progression‐free survival.
7.
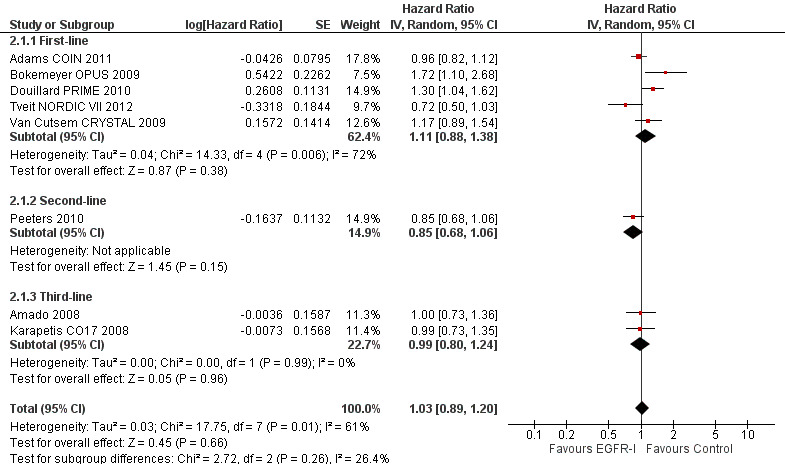
Forest plot of comparison: 2 EGFR MAb in KRAS exon 2 MT, outcome: 2.1 Progression‐free survival.
Of the above‐mentioned 9 studies, 1 study compared the combination of lenalidomide and cetuximab to lenalidomide alone (Siena 2013), but has not reported any data with regard to progression or survival, leaving 8 studies (2567 participants) for analysis of the primary outcome. No important subgroup interactions were present (Chi2 = 2.72, df = 2, P = 0.26, I2 = 26.4%). Pooled analysis of these trials showed that addition of EGFR MAb did not significantly reduce the risk of progression (HR 1.03, 95% CI 0.89 to 1.20; P = 0.66). Significant statistical heterogeneity was present (Chi2 = 17.75, df = 7, P = 0.01, I2 = 61%), likely due to pooling of studies using differing chemotherapy partners in different lines of treatment.
Pooled analysis of first‐line trials (5 RCTs, 1733 participants) in KRAS exon 2 MT populations showed that adding EGFR MAb to first‐line chemotherapy did not decrease the risk of progression (HR 1.11, 95% CI 0.88 to 1.38; P = 0.38; Analysis 2.1.1). Two trials showed significantly worse PFS with addition of EGFR MAb in this cohort (Bokemeyer OPUS 2009; Douillard PRIME 2010). Substantial statistical heterogeneity was present (Chi2 = 14.33, df = 4, P = 0.006, I2 = 72%), probably because of the utilisation of different fluoropyrimidine regimens in trials (capecitabine in some participants in Adams COIN 2011, FLOX in Tveit NORDIC VII 2012). Exclusion of these two trials resulted in no important heterogeneity (Chi2 = 2.08, df = 2, P = 0.35, I2 = 4%).
The only second‐line trial reporting PFS outcomes in KRAS exon 2 MT populations was Peeters 2010 (1 RCT, 486 participants). The risk of progression did not significantly decrease (HR 0.85, 95% CI 0.68 to 1.06; P = 0.15; Analysis 2.1.2).
Pooled analysis of third‐line trials (2 RCTs, 348 participants) showed that using EGFR MAb compared to best supportive care in KRAS exon 2 MT participants did not decrease the risk of progression (HR 0.99, 95% CI 0.80 to 1.24; P = 0.96; Analysis 2.1.3). No important heterogeneity was present (Chi2 = 0.00, df = 1, P = 0.99, I2 = 0%).
2.2 Overall survival
See: Analysis 2.2; Figure 8.
2.2. Analysis.
Comparison 2 EGFR MAb in KRAS exon 2 MT, Outcome 2 Overall survival.
8.
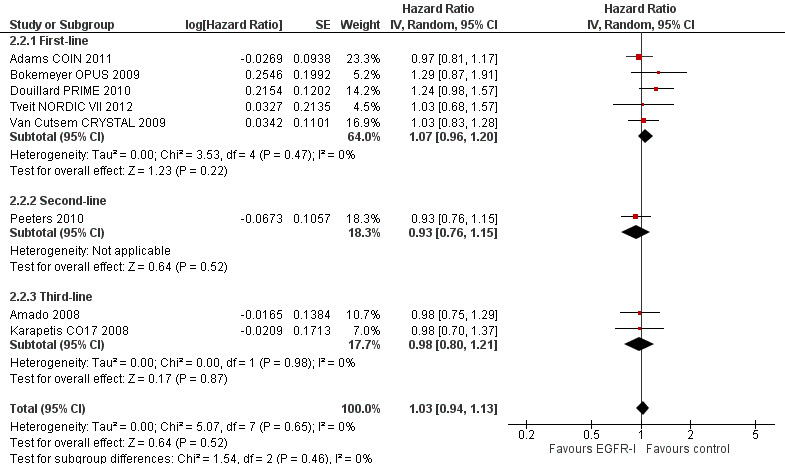
Overall, 8 trials (2567 participants) reported on the effect of adding EGFR MAb to standard therapy on overall survival in KRAS exon 2 MT populations. No important subgroup interactions were present (Chi2 = 1.54, df = 2, P = 0.46, I2 = 0%). Pooled analysis of these trials showed that adding EGFR MAb did not reduce the risk of death (HR 1.03, 95% CI 0.94 to 1.13; P = 0.52). No important heterogeneity was present (Chi2 = 5.07, df = 7, P = 0.65, I2 = 0%). Pooled analysis by line of therapy also showed no significant reduction in risk of death in the first‐line (HR 1.07, 95% CI 0.96 to 1.20; P = 0.22) and third‐line (HR 0.98, 95% CI 0.80 to 1.21; P = 0.87) settings. The one second‐line study, Peeters 2010, reported no reduction in risk of death (HR 0.93, 95% CI 0.76 to 1.15; P = 0.52). No important heterogeneity was present in these subgroup analyses (First‐line: Chi2 = 3.53, df = 4, P = 0.47, I2 = 0%; third‐line: Chi2 = 0, df = 1, P = 0.98, I2 = 0%).
2.3 Tumour response rate
See: Analysis 2.3.
2.3. Analysis.
Comparison 2 EGFR MAb in KRAS exon 2 MT, Outcome 3 Tumour response rate.
Pooled analysis of all trials (8 RCTs, 1925 participants) showed that addition of EGFR MAb in KRAS exon 2 MT populations did not increase the odds of tumour response (OR 0.93, 95% CI 0.74 to 1.16; P = 0.50). No important subgroup interactions were present (Chi2 = 1.26, df = 2, P = 0.53, I2 = 0%). No important heterogeneity was present (Chi2 = 5.45, df = 6, P = 0.49, I2 = 0%). Pooled analysis of first‐line trials (4 RCTs, 1066 participants) also demonstrated no significant increase in odds of tumour response (OR 0.90, 95% CI 0.66 to 1.22; P = 0.51; Analysis 2.3.1). No important heterogeneity was present (Chi2 = 4.11, df = 3, P = 0.25, I2 = 27%).
Only one trial with 496 participants investigated second‐line addition of EGFR MAb in KRAS exon 2 MT populations (Peeters 2010), showing response rates of 30/232 in the EGFR MAb arm and 33/237 in the control arm; hence we did not perform meta‐analysis.
Addition of EGFR MAb in the third‐line setting for KRAS exon 2 MT populations (3 RCTs, 390 participants) resulted in response rates of 6/186 in the intervention arm and 3/204 in the control arm; due to the low number of total events we did not perform meta‐analysis.
2.4 Adverse effects
See: Analysis 2.4; Analysis 2.5; Analysis 2.6; Analysis 2.7.
2.4. Analysis.
Comparison 2 EGFR MAb in KRAS exon 2 MT, Outcome 4 Overall grade 3/4 toxicity.
2.5. Analysis.
Comparison 2 EGFR MAb in KRAS exon 2 MT, Outcome 5 Grade 3/4 diarrhoea.
2.6. Analysis.
Comparison 2 EGFR MAb in KRAS exon 2 MT, Outcome 6 Grade 3/4 rash.
2.7. Analysis.
Comparison 2 EGFR MAb in KRAS exon 2 MT, Outcome 7 Grade 3/4 neutropenia.
Pooled analysis of trials, which reported the effect of adding EGFR MAb in KRAS exon 2 MT populations (5 RCTs, 1635 participants), showed that the odds of any grade 3 to 4 toxicity increased by 13.5% from 54.5% (439/806) in the control arm to 68.0% (564/829) in the experimental arm, however this decrease was not statistically significant (OR 1.63, 95% CI 0.98 to 2.71). We noted substantial subgroup differences for this outcome (Chi2 = 7.90, df = 2, P = 0.02, I2 = 74.7%). Substantial heterogeneity was present in this analysis (Chi2 = 15.28, df = 4, P = 0.004, I2 = 74%), with substantial heterogeneity remaining after analysis by line of therapy (e.g. considering first‐line trials alone the I2 statistic was 71%). Some of this heterogeneity may be due to imbalanced groups as a result of the retrospective analyses of trials and consequent between‐trial variations in clinical characteristics (such as dose intensity), which may have resulted in different odds ratios for toxicity. For instance, the Bokemeyer OPUS 2009 study (despite having adequate randomisation and stratification by other clinical factors), on retrospective analysis of KRAS status, had 59 KRAS exon 2 mutant participants in the control arm compared to 77 in the intervention arm. Duration of therapy (25 versus 21 weeks), cumulative dose of 5‐FU (median 23,755 mg/m2 versus 16,129 mg/m2), and cumulative dose of oxaliplatin (922 mg/m2 versus 765 mg/m2) were all higher in the intervention arm. Increased exposure to oxaliplatin and 5‐FU in the control arm, rather than exposure to cetuximab in the intervention arm, could therefore explain the numerically higher rate of grade 3 to 4 toxicities in this trial. In contrast, participants in the Van Cutsem CRYSTAL 2009 study had similar exposure to irinotecan and 5‐FU regardless of treatment allocation and KRAS status. Although we feel that these differences are sufficient to explain the observed heterogeneity, these post hoc observations are hypothesis‐generating, and the degree of residual heterogeneity means that this analysis should be interpreted with caution.
Pooled analysis (5 RCTs, 1635 participants) showed that addition of EGFR MAb increased the rate of grade 3 to 4 diarrhoea by 4.1% from 9.2% (74/806) of the control arm to 13.3% (110/829) in the experimental arm (OR 1.45, 95% CI 1.01 to 2.11; Analysis 2.5). No significant subgroup interactions were present (Chi2 = 0.37, df = 2, P = 0.83, I2 = 0%). No important heterogeneity was present (Chi2 = 4.95, df = 4, P = 0.29, I2 = 19%). In the same studies, addition of EGFR MAb increased the rate of grade 3 to 4 rash by 22.8% from 0.7% (6/806) in the control arm to 23.5% (195/829) in the experimental arm (OR 32.35, 95% CI 15.01 to 69.70; Analysis 2.6). No significant subgroup interactions were present (Chi2 = 0.84, df = 2, P = 0.66, I2 = 0%). No important heterogeneity was present (Chi2 = 3.15, df = 4, P = 0.53, I2 = 0%).
Pooled analysis (3 RCTs, 968 participants) showed that addition of EGFR MAb decreased the rate of grade 3 to 4 neutropenia by 8.4% from 38.3% (176/460) in the control arm to 29.9% (152/508) in the experimental arm with OR 0.70 (95% CI 0.53 to 0.93; Analysis 2.7). No important heterogeneity was present (Chi2 = 2.04, df = 2, P = 0.36, I2 = 2%). This may be potentially attributable to the toxicities from EGFR MAb use decreasing dose intensity of chemotherapy in the control arm.
3. EGFR MAb in extended RAS WT participants
In the last few years, re‐analysis of prospectively identified extended RAS populations in five pivotal studies have been published (Amado 2008; Bokemeyer OPUS 2009; Douillard PRIME 2010; Peeters 2010; Van Cutsem CRYSTAL 2009), together with one new trial reporting results by extended RAS status (Ciardiello CAPRI‐GOIM 2016). For this analysis, we excluded two studies because they compared the combination of EGFR MAb and chemotherapy to the combination of bevacizumab and chemotherapy in the control arm. (Schwartzberg PEAK 2014; Venook CALGB 80405 2014).
3.1 Progression‐free survival
See: Analysis 3.1; Figure 9.
3.1. Analysis.
Comparison 3 EGFR MAb in extended RAS WT, Outcome 1 Progression‐free survival.
9.
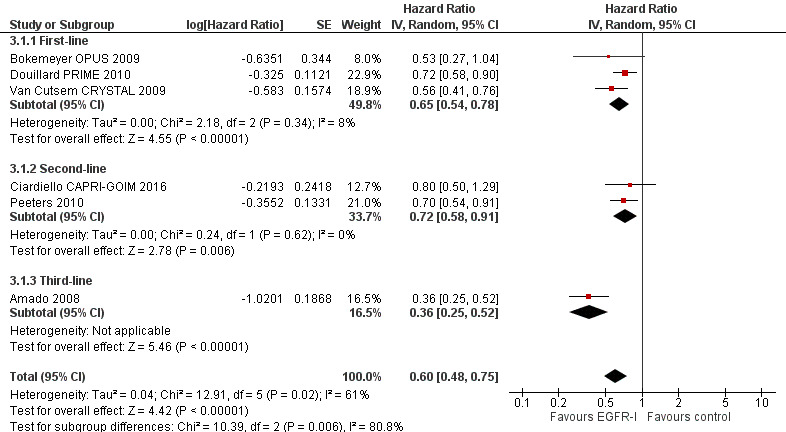
Forest plot of comparison: 3 EGFR MAb in extended RAS WT, outcome: 3.1 Progression‐free survival.
In 6 studies that included 1237 participants with mCRC and extended RAS WT genotype (Amado 2008; Bokemeyer OPUS 2009; Ciardiello CAPRI‐GOIM 2016; Douillard PRIME 2010; Peeters 2010; Van Cutsem CRYSTAL 2009), pooled analysis showed that adding EGFR MAb reduced the risk of disease progression compared to chemotherapy alone/placebo by 40% (HR 0.60, 95% CI 0.48 to 0.75; P < 0.001). Substantial subgroup differences were present (Chi2 = 10.39, df = 2, P = 0.006, I2 = 80.8%). We also found substantial heterogeneity in this analysis (Chi2 = 12.91, df = 5, P = 0.02, I2 = 61%). We noted that five of the studies investigated EGFR MAb in addition to chemotherapy, whereas the last study compared EGFR MAb alone to placebo (Amado 2008), and showed strong PFS benefit (HR 0.36, 95% CI 0.25 to 0.52). Exclusion of this study resulted in no important heterogeneity (Chi2 = 2.91, df = 4, P = 0.57, I2 = 0%).
3.2 Overall survival
See: Analysis 3.2; Figure 10.
3.2. Analysis.
Comparison 3 EGFR MAb in extended RAS WT, Outcome 2 Overall survival.
10.

Forest plot of comparison: 3 EGFR MAb in extended RAS WT, outcome: 3.2 Overall survival.
In 4 studies that included 1114 participants with mCRC and extended RAS WT genotype (Bokemeyer OPUS 2009; Douillard PRIME 2010; Peeters 2010; Van Cutsem CRYSTAL 2009), pooled analysis showed that adding EGFR MAb reduced the risk of death compared to chemotherapy alone or placebo by 23% (HR 0.77, 95% CI 0.67 to 0.88; P < 0.001). No subgroup differences (Chi2 = 0.19, df = 1, P = 0.66, I2 = 0%) nor important heterogeneity (Chi2 = 1.49, df = 3, P = 0.68, I2 = 0%) were present.
3.3 Tumour response rate
See: Analysis 3.3.
3.3. Analysis.
Comparison 3 EGFR MAb in extended RAS WT, Outcome 3 Tumour response rate.
Four RCTs reported tumour response rates in 1001 extended RAS WT participants (Amado 2008; Bokemeyer OPUS 2009; Peeters 2010; Van Cutsem CRYSTAL 2009). The addition of EGFR MAb increased the odds of tumour response compared to chemotherapy alone or placebo by 26.4% from 21.3% (108/508) to 47.7% (235/493) (OR 4.28, 95% CI 2.61 to 7.03; P < 0.001). Substantial subgroup differences (Chi2 = 5.38, df = 2, P = 0.07, I2 = 62.8%) and moderate statistical heterogeneity (Chi2 = 5.52, df = 3, P = 0.14, I2 = 46%) were present. We considered clinical differences by line of therapy (as noted above in the PFS analysis) as a potential cause of heterogeneity, but exclusion of Amado 2008 led to persistent heterogeneity (I2 = 47%). Another difference between the trials was the use of cetuximab and panitumumab. Considered separately, the two trials investigating cetuximab in the first‐line setting (I2 = 0%) had no important heterogeneity, and neither did the two trials investigating panitumumab in the second‐ and third‐line settings. However, these post hoc explanations may not fully explain the heterogeneity, and these findings should be interpreted with caution.
3.4 Adverse effects
No outcomes with regard to toxicity (overall, diarrhoea, rash, or neutropenia) were reported by any identified trial by extended RAS status.
4. EGFR MAb in extended RAS mutant participants
4.1 Progression‐free survival
See: Analysis 4.1.
4.1. Analysis.
Comparison 4 EGFR MAb in any RAS mutation, Outcome 1 Progression‐free survival.
Overall, 6 studies (2004 participants) investigated the effect of adding EGFR MAb to either chemotherapy or best supportive care in extended RAS mutant populations. Pooled analysis of all trials showed that addition of EGFR MAb did not significantly reduce the risk of progression (HR 1.13, 95% CI 0.93 to 1.36; P = 0.31). No important subgroup differences were present (Chi2 = 2.74, df = 2, P = 0.25, I2 = 27.0%). Substantial statistical heterogeneity was present (Chi2 = 13.26, df = 5, P = 0.02, I2 = 62%), potentially due to the pooling of studies involving differing lines of treatment and chemotherapy partners used. When considered by line of therapy, none of the subgroups below showed significant heterogeneity.
Pooled analysis of first‐line trials (3 RCTs, 1175 participants) in extended RAS MT populations showed that adding EGFR MAb to first‐line chemotherapy statistically increased the risk of progression by 27% (HR 1.27, 95% CI 1.08 to 1.48; P = 0.004; Analysis 4.1.1). No important heterogeneity was present (Chi2 = 2.24, df = 2, P = 0.33, I2 = 11%). This is an important finding because actual harm to participants was observed.
Pooled analysis of second‐line trials (2 RCTs, 616 participants) in extended RAS MT populations showed that adding EGFR MAb in the second‐line setting did not significantly decrease the risk of progression (HR 1.05, 95% CI 0.62 to 1.79; Analysis 4.1.2). Substantial heterogeneity was present in this analysis (Chi2 = 2.64, df = 1, P = 0.10, I2 = 62%). This was potentially due to the inclusion of different populations in the trials: Peeters 2010 enrolled participants all with KRAS genotypes, and thus their population in this analysis comprises both participants with KRAS exon 2 mutations as well as other KRAS or NRAS mutations; in contrast, Ciardiello CAPRI‐GOIM 2016 restricted enrolment to people with KRAS exon 2 WT tumours, and thus their population in this analysis would not have had KRAS exon 2 mutations, but rather mutations in other exons of KRAS or NRAS. Interpretation of this subgroup analysis should therefore be interpreted with caution.
We note that Seymour PICCOLO 2013 also provided subgroup results for KRAS mutant participants with PFS HR 0.56 (95% CI 0.13 to 2.48) and OS HR 1.73 (95% CI 0.43 to 6.58) as well as NRAS mutant participants with PFS HR 1.08 (95% CI 0.45 to 2.56) and OS HR 1.97 (95% CI 0.83 to 4.67). However, we could not incorporate these results into the same analysis as the authors did not provide the results for participants with both KRAS and NRAS mutations (i.e. the extended RAS mutant population). We note that the numbers in each subgroup were small, with 17 participants having KRAS codon 146 mutations and 29 participants having NRAS mutations.
The only third‐line trial reporting PFS outcomes in this population was Amado 2008 (1 RCT, 213 participants), which reported no significant decrease in risk of progression with HR 0.97 (95% CI 0.73 to 1.29).
4.2 Overall survival
See: Analysis 4.2.
4.2. Analysis.
Comparison 4 EGFR MAb in any RAS mutation, Outcome 2 Overall survival.
Overall, 4 trials (1768 participants) investigated the addition of EGFR MAb to standard therapy in extended RAS MT participants and reported overall survival results. Pooled analysis of these trials showed that adding EGFR MAb did not reduce the risk of death (HR 1.09, 95% CI 0.93 to 1.28; P = 0.29). We noted substantial subgroup differences (Chi2 = 4.31, df = 2, P = 0.04, I2 = 76.8%), and moderate heterogeneity was present (Chi2 = 6.22, df = 3, P = 0.10, I2 = 52%). This was possibly attributable to the pooling of studies investigating the use of EGFR MAb in differing lines of therapy; removal of Peeters 2010 (the only second‐line study) resulted in no important heterogeneity (Chi2 = 1.92, df = 2, P = 0.38, I2 = 0%).
Pooled analysis of first‐line trials (3 RCTs, 1175 participants) showed a statistically significant increase in risk of death (HR 1.16, 95% CI 1.02 to 1.33; P = 0.03). No important heterogeneity was present (Chi2 = 1.92, df = 2, P = 0.38, I2 = 0%). The one second‐line study, Peeters 2010, (574 participants) reported no reduction in risk of death (HR 0.91, 95% CI 0.76 to 1.10; P = 0.34).
4.3 Tumour response rate
See: Analysis 4.3.
4.3. Analysis.
Comparison 4 EGFR MAb in any RAS mutation, Outcome 3 Tumour response rate.
Only 3 RCTs (840 participants) reported the effect of adding EGFR MAb to standard therapy on tumour response rates in extended RAS MT populations. Adding EGFR MAb did not significantly increase the odds of tumour response (OR 0.76, 95% CI 0.55 to 1.05; P = 0.09). No important heterogeneity was present (Chi2 = 1.86, df = 2, P = 0.39, I2 = 0%). Given that only two first‐line and one third‐line studies were included, analysis by line of therapy was not performed.
4.4 Adverse effects
No outcomes with regard to toxicity (overall, diarrhoea, rash, or neutropenia) were reported by any identified trial by extended RAS status.
5. EGFR MAb in KRAS unselected participants
See: Analysis 5.1; Analysis 5.2.
5.1. Analysis.
Comparison 5 EGFR inhibitors in KRAS unselected participants, Outcome 1 Progression‐free survival.
5.2. Analysis.
Comparison 5 EGFR inhibitors in KRAS unselected participants, Outcome 2 Overall survival.
Three RCTs (evaluating 1483 participants) investigated the use of EGFR MAb in KRAS unselected participants without significant subsequent data for KRAS testing and outcomes. We did not perform meta‐analysis due to the paucity of eligible studies.
Borner 2008 investigated the addition of cetuximab to CAPOX chemotherapy in a KRAS unselected population. Time to tumour progression, the primary endpoint, was not significantly improved, being 5.8 months in the control arm and 7.2 months in the EGFR MAb arm (HR 0.96, 95% CI 0.59 to 1.56). Sobrero EPIC 2008 investigated the addition of cetuximab to second‐line irinotecan chemotherapy in a KRAS unselected population. Progression‐free survival was improved with HR 0.70 (95% CI 0.62 to 0.78). No results were available by KRAS status for analysis after communication with authors of both studies (Chan 2013 [pers comm]; Chan 2015 [pers comm]). Polikoff EXPLORE 2005 randomised participants with EGFR‐positive mCRC (without KRAS testing) to the combination of FOLFOX4 and cetuximab or FOLFOX4 alone. Response rates were 9/43 in the combination arm and 4/39 in the FOLFOX4‐only arm. Only Sobrero EPIC 2008 reported quality of life, showing an improvement in global, physical, and emotioning functioning and pain subscales of EORTC QLQ‐C30 favouring the intervention arm.
6. Comparing addition of EGFR MAb to chemotherapy with anti‐VEGF MAb and the same chemotherapy
All studies identified in this category restricted enrolment to people with KRAS exon 2 genotype mCRC.
6.1 Progression‐free survival
See: Analysis 6.1; Figure 11.
6.1. Analysis.
Comparison 6 Comparing addition of EGFR MAb to chemotherapy with anti‐VEGF MAb and the same chemotherapy, Outcome 1 Progression‐free survival.
11.
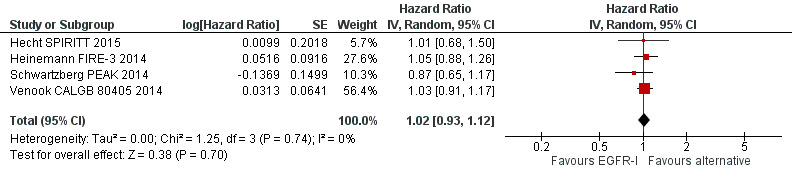
Forest plot of comparison: 6 Comparing addition of EGFR MAb to chemotherapy with anti‐VEGF MAb and the same chemotherapy, outcome: 6.1 Progression‐free survival.
Four trials (evaluating 2189 KRAS exon 2 WT participants) investigated the addition of EGFR MAb to chemotherapy compared to addition of another (non‐EGFR) biologic agent to the same chemotherapy. In all four trials the other biologic agent was bevacizumab. Venook CALGB 80405 2014 (phase III) investigated the addition of either cetuximab or bevacizumab to first‐line chemotherapy (the investigator’s choice of mFOLFOX6, FOLFIRI, or CAPOX) and showed no improvement in PFS with HR 1.03 (95% CI 0.91 to 1.17). Heinemann FIRE‐3 2014 (phase III) investigated the addition of cetuximab or bevacizumab to first‐line FOLFIRI and also showed no improvement in PFS with HR 1.05 (95% CI 0.93 to 1.12). Hecht SPIRITT 2015 (phase II) investigated the addition of panitumumab or bevacizumab to FOLFIRI in the second‐line setting and showed no improvement in PFS with HR 1.01 (95% CI 0.68 to 1.50). Schwartzberg PEAK 2014 (phase II) investigated the addition of panitumumab or bevacizumab to mFOLFOX6 in the first‐line setting and showed no improvement in PFS with HR 0.87 (95% 0.65 to 1.17).
Pooled analysis showed that use of EGFR MAb compared to bevacizumab did not reduce the risk of disease progression (HR 1.02, 95% CI 0.93 to 1.12; P = 0.74). No important heterogeneity was present (Chi2 = 1.25, df = 3, P = 0.74, I2 = 0%).
We note that two studies have subsequently published results for PFS in their extended RAS WT populations, although the trials were not powered for significance in these retrospective subgroup analyses. Schwartzberg PEAK 2014 reported significantly improved PFS with HR 0.65 (95% CI 0.44 to 0.96; P = 0.029), whereas Heinemann FIRE‐3 2014 reported no PFS benefit with HR 0.93 (95% CI 0.74 to 1.17).
6.2 Overall survival
See: Analysis 6.2; Figure 12.
6.2. Analysis.
Comparison 6 Comparing addition of EGFR MAb to chemotherapy with anti‐VEGF MAb and the same chemotherapy, Outcome 2 Overall survival.
12.

Forest plot of comparison: 6 Comparing addition of EGFR MAb to chemotherapy with anti‐VEGF MAb and the same chemotherapy, outcome: 6.2 Overall survival.
The 4 trials (2189 KRAS exon 2 WT participants) analysed in 6.1 also reported overall survival. Pooled analysis showed that compared to bevacizumab, the use of EGFR MAb did not significantly decrease the risk of death with HR 0.84 (95% CI 0.70 to 1.01; P = 0.06). Moderate heterogeneity was present in this analysis (Chi2 = 6.12, df = 3, P = 0.11, I2 = 51%), possibly due to the pooling of trials evaluating different lines of therapy. Only Schwartzberg PEAK 2014 was conducted in the second‐line setting, with the other three trials being first‐line studies. The exclusion of Schwartzberg PEAK 2014 led to no important residual heterogeneity (Chi2 = 2.78, df = 2, P = 0.25, I2 = 28%).
We note that three studies (2007 participants) have published results for OS in the extended RAS WT population. Venook CALGB 80405 2014 reported no significant difference in this population in OS with HR 0.9 (95% CI 0.7 to 1.1; P = 0.40). Heinemann FIRE‐3 2014 showed improved OS with HR 0.70 (95% CI 0.53 to 0.92). Schwartzberg PEAK 2014 reported significantly improved OS with HR 0.63 (95% CI 0.39 to 1.02). We again note that these analyses were not adequately powered to detect statistically significant differences.
6.3 Tumour response rate
See: Analysis 6.3.
6.3. Analysis.
Comparison 6 Comparing addition of EGFR MAb to chemotherapy with anti‐VEGF MAb and the same chemotherapy, Outcome 3 Tumour response rate.
Pooled analysis of the 4 trials (2184 participants) showed that the use of EGFR MAb compared to bevacizumab, both in combination with chemotherapy, improved tumour response rates by 7.2% from 53.9% (582/1080) in the bevacizumab arm to 61.1% (675/1104) in the EGFR MAb arm (OR 1.36, 95% CI 1.15 to 1.62; P = 0.0005) (Hecht SPIRITT 2015; Heinemann FIRE‐3 2014; Schwartzberg PEAK 2014; Venook CALGB 80405 2014). No important heterogeneity was present (Chi2 = 2.38, df = 3, P = 0.50, I2 = 0%). As Heinemann FIRE‐3 2014 reported dropout rates exceeding 5%, we explored the impact of including best‐ and worst‐case scenarios for participants who were lost to follow‐up on this trial. Assuming all lost participants in the EGFR MAb arm did not show tumour response, and all those in the bevacizumab arm did, the odds ratio for response rate was no longer significant (OR 1.26, 95% CI 0.93 to 1.71). Consequently, we consider the evidence for the above difference in response rates to be weak.
6.4 Adverse effects
See: Analysis 6.4; Analysis 6.5; Analysis 6.6.
6.4. Analysis.
Comparison 6 Comparing addition of EGFR MAb to chemotherapy with anti‐VEGF MAb and the same chemotherapy, Outcome 4 Overall grade 3/4 toxicity.
6.5. Analysis.
Comparison 6 Comparing addition of EGFR MAb to chemotherapy with anti‐VEGF MAb and the same chemotherapy, Outcome 5 Grade 3/4 diarrhoea.
6.6. Analysis.
Comparison 6 Comparing addition of EGFR MAb to chemotherapy with anti‐VEGF MAb and the same chemotherapy, Outcome 6 Grade 3/4 rash.
Pooled analysis of trials reporting the effect of EGFR MAb compared to bevacizumab on toxicity (4 RCTs, 2133 participants) in KRAS exon 2 WT settings showed increased odds of overall grade 3 to 4 toxicity, with incidence 72.4% (778/1074) in the EGFR MAb arm and 66.7% (706/1059) in the bevacizumab arm (net difference +5.7%) (OR 1.37, 95% CI 1.09 to 1.72; P = 0.008). No important heterogeneity was present (Chi2 = 3.80, df = 3, P = 0.28, I2 = 21%).
As only two studies (1673 participants) reported on the effect of EGFR MAb compared to bevacizumab on the rate of grade 3 to 4 diarrhoea, we did not undertake meta‐analysis. Venook CALGB 80405 2014 reported rates of 10.8% (59/547) in the cetuximab group and 8.4% (45/534) in the bevacizumab group. Heinemann FIRE‐3 2014 reported rates of 11.4% (34/297) in the EGFR MAb group and 13.6% (40/295) in the bevacizumab group.
Three trials (1951 participants) reported on the effect of EGFR MAb compared to bevacizumab on the rate of grade 3 to 4 rash. Pooled rates were 13.6% (134/983) in the EGFR MAb arm and 0.2% (2/968) in the bevacizumab arm (net increase 13.4%) (OR 47.53, 95% CI 14.84 to 152.19). No important heterogeneity was present (Chi2 = 1.04, df = 2, P = 0.60, I2 = 0%).
No trials specifically reported rates of grade 3 to 4 neutropenia. However, Heinemann FIRE‐3 2014 (592 participants) reported rates of grade 3 to 4 haematotoxicity at 73/297 for the FOLFIRI with cetuximab arm and 62/295 for the FOLFIRI with bevacizumab arm. Rates of grade 3 to 4 neutropenic infection and neutropenic fever (without infection) were 5/297 and 2/297 respectively for the FOLFIRI with cetuximab arm, and 3/295 and 1/295 respectively for the FOLFIRI with bevacizumab arm.
We did not perform analysis for toxicities specifically associated with bevacizumab and not with EGFR MAb as this was not within the scope of this review.
7. Comparing different EGFR inhibitor agents or regimens
Six trials (evaluating 1708 participants) compared the use of one EGFR inhibitor (EGFR‐I) to another EGFR‐I.
Bridgewater GAIN‐C 2015 investigated the use of imgatuzumab (GA201), a novel EGFR‐directed monoclonal antibody, in 169 participants. KRAS exon 2 WT participants were randomised to either the combination of FOLFIRI and GA201 or the combination of FOLFIRI and cetuximab, whereas KRAS exon 2 MT participants were randomised to either the combination of FOLFIRI and GA201 or FOLFIRI alone. Preliminary results from this study were reported at ASCO GI 2015.(Bridgewater GAIN‐C 2015). In KRAS exon 2 WT participants, PFS was 7.3 months for the FOLFIRI and GA201 arm versus 6.1 months for the FOLFIRI and cetuximab arm (HR 1.13, 95% CI 0.69 to 1.86). In KRAS exon 2 MT participants, PFS was 5.2 months for the FOLFIRI and GA201 arm versus 4.3 months for the FOLFIRI arm (HR 0.94, 95% CI 0.57 to 1.54). Grade 3 rash (42.5% versus 9.8% in KRAS exon 2 WT participants) and hypomagnesaemia (30.0% versus 4.9% in KRAS exon 2 WT participants) were increased in the GA201 groups, but no formal statistical comparisons were performed for these measures.
Price ASPECCT 2014 directly compared monotherapy with panitumumab to cetuximab in 1010 participants with chemotherapy‐refractory mCRC. Overall survival (HR 0.97, 95% CI 0.84 to 1.11), PFS (HR 1.00, 95% CI 0.88 to 1.14), response rate (OR 1.15, 95% CI 0.83 to 1.58), and grade 3 to 5 toxicity (49% in panitumumab arm, 47% in cetuximab arm) did not differ significantly between the two groups.
Wasan COIN‐B 2014 compared a strategy of intermittent mFOLFOX6 with cetuximab (with mFOLFOX6 with cetuximab ceased after 12 weeks and, assuming stable disease or better with initial treatment, re‐introduction of the same treatment on progression) with the same strategy of intermittent mFOLFOX6 with cetuximab, but with maintenance cetuximab in between these treatments, in 226 participants, although results have only been presented for 169 KRAS exon 2 WT participants. Failure‐free survival was defined as the time for participants to develop progressive disease after up to two re‐introductions of mFOLFOX6 with cetuximab. The median failure‐free survival of the maintenance cetuximab arm was 14.3 months compared to 12.2 months in the other arm. Median OS was 17.5 months in the maintenance cetuximab arm, compared to 16.0 months in the other arm. No formal statistical analyses were conducted.
Hickish 2014 investigated the use of afatinib in people with mCRC after progression on oxaliplatin‐ and irinotecan‐containing chemotherapies. Fifty participants with KRAS exon 2 WT tumours were randomised to afatinib (N = 36, arm 1) or cetuximab (N = 14, arm 2); 41 participants with KRAS exon 2 MT tumours were assigned to afatinib (arm 3). The primary endpoint was response rates, and confirmed response rates were 0/36 for arm 1, 2/14 for arm 2, and 0/41 for arm 3. The median PFS was 46 days for arm 1, 144.5 days for arm 2, and 41 days for afatinib arm 3, without formal statistical comparison. The median OS was 355 days for arm 1, not reached for arm 2, and 173 days for arm 3, without formal statistical comparison. Grade 3 to 4 toxicity was reported in 36% of participants in arm 1, 36% of participants in arm 2, and 32% of participants in arm 3, with diarrhoea and rash being most commonly reported.
Brodowicz 2013 compared the combination of FOLFOX4 and weekly cetuximab (400 mg/m2 loading dose, 250 mg/m2 thereafter) to the combination of FOLFOX4 and fortnightly cetuximab (500 mg/m2) in 152 participants. No differences were noted in OS (HR 0.86, 95% CI 0.56 to 1.30), PFS (HR 0.92, 95% CI 0.63 to 1.34), or response rate (53% versus 62%) (OR 1.40, 95% CI 0.74 to 2.66). Grade 3 to 5 toxicity was reported in 72% of the weekly arm and 71% of the fortnightly arm.
Similarly, Ma 2013 compared the combination of CAPOX and continuous erlotinib (100 mg daily) to the combination of CAPOX and intermittent erlotinib (150 mg on alternate days from day 2 to 14, 150 mg daily on days 15 to 21) in 60 participants. The primary endpoint, response rate, did not differ significantly between the two groups (66.7% in the intermittent arm, 56.7% in the continuous arm). No differences were noted in OS (20.7 months in the intermittent arm, 18.8 months in the continuous arm, P > 0.05, no HR or P value provided) and PFS (10.3 months in the intermittent arm, 9 months in the continuous arm, P > 0.05, no hazard ratio or exact P value provided).
We did not perform meta‐analysis due to the clinical heterogeneity between the trials with different agents under investigation, different control arms, varying schedules of administration, and different prior lines of therapy.
8. The addition of EGFR TKIs to standard therapy
8.1 Progression‐free survival
Two trials (evaluating 181 participants) investigated the addition of EGFR TKIs to standard therapy. Santoro 2008 evaluated the addition of gefitinib to FOLFIRI in the first‐line setting. Progression‐free survival was not significantly improved (HR 0.87, 95% CI 0.72 to 1.04). Vincent 2011 investigated the addition of erlotinib to low‐dose capecitabine in the first‐line setting. This trial was published in abstract form only; whilst PFS was not reported, the median time to progression was 7.9 months in the control arm versus 9.2 months in the erlotinib arm (P = 0.89). We did not perform pooled analysis due to the low number of trials.
8.2 Overall survival
Santoro 2008 showed no significant improvement in OS, with the median OS being 18.6 months in the control arm and 17.1 months in the gefitinib arm (HR 0.90, 95% CI 0.67 to 1.21). Vincent 2011 did not report overall survival in the abstract. We did not perform pooled analysis due to the low number of trials.
8.3 Tumour response rate
Only Santoro 2008 (99 participants) reported response rate. No difference was observed with response rate 47.9% (23/48) in the control arm and 45.1% (23/51) in the gefitinib arm. We did not perform pooled analysis due to the low number of trials.
8.4 Adverse effects
Only Santoro 2008 investigated the effect of adding EGFR TKI to standard therapy on rates of overall grade 3 to 4 toxicity. The incidence of overall toxicity increased from 52.1% (25/48) in the control group to 68.6% (35/51) in the EGFR TKI group.
Both Santoro 2008 and Vincent 2011 investigated the effect of adding EGFR TKI to standard therapy on rates of grade 3 to 4 diarrhoea. The pooled incidence of diarrhoea increased from 3.4% (3/88) in the control group to 31.2% (29/93) in the EGFR TKI group.
Only Santoro 2008 investigated the effect of adding EGFR TKI to standard therapy on rates of grade 3 to 4 rash and neutropenia. The incidence of rash increased from 2.1% (1/48) in the control group to 9.8% (5/51) in the EGFR TKI group. The incidence of neutropenia increased from 22.9% (11/48) in the control group to 35.3% (18/51) in the EGFR TKI group.
We did not perform pooled analysis due to the low number of trials.
9. The addition of EGFR inhibitors to the combination of standard therapy and anti‐angiogenic agent
9.1 Progression‐free survival
See: Analysis 8.1; Figure 13.
8.1. Analysis.
Comparison 8 EGFR inhibitors added to bevacizumab, Outcome 1 Progression‐free survival.
13.

Forest plot of comparison: 8 EGFR inhibitors added to bevacizumab, outcome: 8.1 Progression‐free survival.
Six trials (1571 participants) investigated the effect of the addition of EGFR‐I to bevacizumab, evaluating both EGFR MAb and EGFR TKI. Johnsson Nordic ACT 2013 (phase III) enrolled participants with KRAS unselected mCRC to receive the combination of chemotherapy (clinician's choice of CAPOX, CAPIRI, mFOLFOX6, or FOLFIRI) with bevacizumab. Those with stable disease or better after 18 weeks of this therapy were randomised to either erlotinib and bevacizumab or bevacizumab alone for maintenance therapy. Progression‐free survival was not significantly improved with HR 0.78 (95% CI 0.55 to 1.12). Hagman ACT2 2014 (phase III) similarly enrolled participants with KRAS exon 2 WT mCRC who had stable disease or better on the first‐line combination of chemotherapy with bevacizumab. Participants were randomised to either the combination of erlotinib and bevacizumab or erlotinib for maintenance therapy. We pooled participants from the above two studies for analysis, which again showed no significant PFS benefit with HR 0.93 (95% CI 0.56 to 1.56). KRAS MT participants were randomised to either bevacizumab or capecitabine maintenance treatment, however this comparison was not eligible for inclusion in the current review. Tournigand DREAM 2015 (phase III) investigated the addition of erlotinib to maintenance bevacizumab in participants who had stable disease on bevacizumab‐containing first‐line chemotherapy for six months. Progression‐free survival was not significantly improved with HR 0.79 (95% CI 0.60 to 1.06). Hecht PACCE 2009 (phase III) investigated the addition of panitumumab to combined chemotherapy and bevacizumab, stratifying analysis by type of chemotherapy (oxaliplatin‐ or irinotecan‐based). Given the evidence that KRAS WT populations alone benefit from cetuximab, only this population was included for the purposes of meta‐analysis of efficacy results. For the oxaliplatin‐based group, PFS worsened in the investigational arm with HR 1.36 (95% CI 1.04 to 1.77). For the irinotecan‐based group, PFS did not significantly worsen with HR 1.50 (95% CI 0.82 to 2.76). We combined these two subgroups of Hecht PACCE 2009 by random‐effects meta‐analysis to form hazard ratios for the overall study before pooled analysis with the other studies. Tol CAIRO2 2008 (phase III) investigated the addition of cetuximab to CAPOX with bevacizumab in the first‐line setting. Progression‐free survival in the KRAS unselected population worsened with HR 1.22 (95% CI 1.04 to 1.43). Given the evidence that KRAS WT populations alone benefit from cetuximab, we restricted analysis to the KRAS WT population, estimating PFS HR from the published survival curve according to the methods of Parmar 1998. Passardi ITACA 2015 (phase III) randomised participants due for first‐line therapy to physician's choice of chemotherapy with bevacizumab versus the same chemotherapy. The participants randomised to chemotherapy alone who were KRAS exon 2 WT were eligible for a subtrial (included here) investigating second‐line chemotherapy with both bevacizumab and cetuximab compared to second‐line chemotherapy with bevacizumab alone. This showed no significant differences in PFS (HR 1.31, 95% CI 0.72 to 2.26) or OS (HR 1.39, 95% CI 0.78 to 2.49), although only 56 participants were included.
Pooled analysis of the above trials (6 RCTs, 2012 participants) showed that adding EGFR inhibitors to combined standard therapy and bevacizumab did not significantly reduce the risk of progression (HR 1.04, 95% CI 0.83 to 1.29; P = 0.73). However, substantial heterogeneity was present in the above analysis (Chi2 = 14.50, df = 5, P = 0.01, I2 = 66%), likely due to the pooling of studies using EGFR MAb on disease progression with those investigating TKIs in the maintenance context. Removal of TKI maintenance studies resulted in no important residual heterogeneity (Chi2 = 0.70, df = 2, P = 0.71, I2 = 0%) (Hagman ACT2 2014; Johnsson Nordic ACT 2013; Tournigand DREAM 2015).
Given the different treatments investigated in the above studies, we undertook a sensitivity analysis evaluating the impact of removing the TKI maintenance studies from the analysis. Progression‐free survival worsened with HR 1.28 (95% CI 1.09 to 1.51; P = 0.003).
9.2 Overall survival
See: Analysis 8.2; Figure 14.
8.2. Analysis.
Comparison 8 EGFR inhibitors added to bevacizumab, Outcome 2 Overall survival.
14.

Forest plot of comparison: 8 EGFR inhibitors added to bevacizumab, outcome: 8.2 Overall survival.
Five trials (1257 participants) also investigated the effect of adding EGFR‐I to combined chemotherapy and bevacizumab on overall survival. Pooled analysis showed that the risk of death was not reduced with HR 1.00 (95% CI 0.69 to 1.47; P = 0.98). Significant statistical heterogeneity was present (Chi2 = 19.94, df = 4, P = 0.0005, I2 = 80%), likely due to the pooling of studies investigating both EGFR MAb and TKI in maintenance therapy, as above. We note that removal of the TKI studies (as above) resulted in no important residual heterogeneity (I2 = 0%). Although the random‐effects model incorporates some of the heterogeneity among studies in the analysis, the heterogeneity is not accounted for completely, and so these results should be interpreted with caution, especially in view of the the relatively small number of studies.
9.3 Tumour response rate
See: Analysis 8.3.
8.3. Analysis.
Comparison 8 EGFR inhibitors added to bevacizumab, Outcome 3 Tumour response rate.
Only 4 trials (1310 participants) reported response rate (Hecht PACCE 2009; Passardi ITACA 2015; Tol CAIRO2 2008; Tournigand DREAM 2015). Pooled rates were 38.7% (253/653) in the control arm and 42.6% (280/657) in the experimental arm, which were not significantly different (OR 1.20, 95% CI 0.67 to 2.12). Substantial heterogeneity was present (Chi2 = 13.48, df = 3, P = 0.004, I2 = 78%), due to the pooling of trials investigating different agents, as above. Exclusion of the only TKI trial, Tournigand DREAM 2015, still resulted in substantial heterogeneity (I2 = 72%). This remaining heterogeneity may be due to clinical differences in the above three trials. For instance, Hecht PACCE 2009 used panitumumab as the EGFR MAb, whereas the other two studies used cetuximab. The chemotherapy partner used in the three studies also differed. Hecht PACCE 2009 and Passardi ITACA 2015 used either FOLFOX (any variant in Hecht PACCE 2009, FOLFOX4 in Passardi ITACA 2015) or FOLFIRI in addition to bevacizumab, whereas Tol CAIRO2 2008 used CAPOX alone without either use of 5‐FU or irinotecan. Given the paucity of studies in these analyses and their post hoc nature, we cannot say definitively which of these factors was responsible for the heterogeneity. Although the random‐effects model incorporates some of this heterogeneity in the analysis, the heterogeneity is not accounted for completely, and so these results should be interpreted with caution.
9.4 Adverse effects
See: Analysis 8.5; Analysis 8.6.
8.5. Analysis.
Comparison 8 EGFR inhibitors added to bevacizumab, Outcome 5 Grade 3/4 diarrhoea.
8.6. Analysis.
Comparison 8 EGFR inhibitors added to bevacizumab, Outcome 6 Grade 3/4 rash.
Three RCTs (1831 participants) reported on rate of overall grade 3 to 4 toxicity. For the purposes of evaluating adverse events, we elected to include both KRAS exon 2 WT and KRAS exon 2 MT participants from Hecht PACCE 2009 and Tol CAIRO2 2008. Overall toxicity increased by 13.8% from 71.7% (653/911) in the control arm to 85.5% (787/920) in the EGFR‐I arm (OR 2.57, 95% CI 1.45 to 4.57). Substantial heterogeneity was present (Chi2 = 8.62, df = 2, P = 0.01, I2 = 77%), possibly due to the pooling of studies using EGFR MAb on disease progression and maintenance studies using EGFR TKI. We note that two of the studies investigated EGFR MAb and the third EGFR TKI; exclusion of the EGFR TKI study resulted in considerable heterogeneity (I2 = 87%). Whilst multiple clinical differences exist between Hecht PACCE 2009 and Tol CAIRO2 2008 (as discussed above), we could not identify one difference as the primary contributor of heterogeneity. Given the significant heterogeneity that could not be explained by planned subgroup analyses, these results should be interpreted with caution.
Pooled analysis (5 RCTs, 2434 participants) showed that adding EGFR‐I to standard therapy and bevacizumab increased the rate of grade 3 to 4 diarrhoea by 9.4% with incidence 11.0% (133/1210) in the control arm and 20.4% (250/1224) in the EGFR‐I arm (OR 2.58, 95% CI 1.44 to 4.64; P = 0.002). Substantial heterogeneity was present (Chi2 = 10.99, df = 4, P = 0.03, I2 = 64%), likely due to the differences in treatments investigated as described above. Exclusion of EGFR TKI studies resulted in substantial residual heterogeneity (I2 = 74%). Given the significant heterogeneity that could not be explained by planned subgroup analyses, these results should be interpreted with caution.
Pooled analysis (4 RCTs, 2363 participants) showed that adding EGFR‐I to standard therapy and bevacizumab increased the rate of grade 3 to 4 rash by 28.4% with incidence 0.5% (6/1179) in the control arm and 28.9% (342/1184) in the EGFR‐I arm (OR 67.52, 95% CI 30.83 to 147.85). No important heterogeneity was present (Chi2 = 2.00, df = 3, P = 0.57, I2 = 0%).
Two RCTs reported on rate of grade 3 to 4 neutropenia. Pooled incidence rates were 20.5% (121/589) in the control arm and 20.1% (120/598) in the EGFR‐I arm. We did not perform pooled analysis due to the low number of trials.
10. Quality of life
Only 8 of the 33 included trials reported quality of life, using various measures, as summarised in the table below. We could perform no separate analysis of quality of life in KRAS exon 2 MT participants in the trials where detrimental outcomes from adding EGFR‐I were reported.
| Study title | # Participants | Intervention | Control | QoL instrument | QoL effect |
| Seymour PICCOLO 2013 | 460 (KRAS exon 2 WT) | Irinotecan with panitumumab | Irinotecan | EORTC QLQ‐C30 Global EORTC QLQ‐C30 Symptom scores EQ‐5D Dermatology Life Quality Index |
Significantly better in intervention arm Significantly worse in intervention arm Not reported Not reported |
| Peeters 2010 | 597 (KRAS exon 2 WT) | FOLFIRI with panitumumab | FOLFIRI | EQ‐5D HSI and VAS assessments | No significant differences detected |
| Karapetis CO17 2008 | 394 (KRAS exon 2 WT) | Cetuximab | Best supportive care | EORTC QLQ‐C30 | Significantly better in intervention arm |
| Van Cutsem CRYSTAL 2009 | 351 RAS WT (1198 total) | FOLFIRI with cetuximab | FOLFIRI | EORTC QLQ‐C30, Global health status and social functioning subscales | No significant differences detected |
| Amado 2008 | 463 (KRAS unselected) | Panitumumab | Best supportive care | EQ‐5D VAS and selected questions from NCCN FACT or EORTC | No clinically meaningful differences in overall QoL were observed between the groups |
| Douillard PRIME 2010 | 456 RAS WT (1183 total) | FOLFOX4 with panitumumab | FOLFOX4 | EQ‐5D HSI and overall health rating assessments | No significant differences detected |
| Adams COIN 2011 | 1630 | mFOFLOX6 with cetuximab or CAPOX with cetuximab | mFOLFOX6 or CAPOX | EORTC QLQ‐C30, EQ‐5D, Dermatology Life Quality Index | Not reported as yet |
| Sobrero EPIC 2008 | 1298 (KRAS unselected) | Irinotecan with cetuximab | Irinotecan | EORTC QLQ‐C30 (reported in KRAS unselected population) | Improvement in global, physical, and emotional functioning, pain subscales favouring intervention arm |
Abbreviations: EQ‐5D HSI: EuroQoL 5‐Dimensions Health State Index; EQ‐5D VAS: EuroQoL 5‐Dimensions Visual Analogue Scale; NCCN FACT: National Comprehensive Cancer Network/Functional Assessment of Cancer Therapy
11. Sensitivity analysis
We performed sensitivity analyses in the above analyses as described in the Methods.
We conducted sensitivity analyses to investigate the impact of excluding trials at high risk of bias from top‐level analyses. We noted no changes in significance of the above results in any of the analyses. We have described sensitivity analyses excluding individual trials identified as being the likely sole cause of heterogeneity in the relevant sections above.
We also conducted best‐case/worst‐case analyses for binary outcomes where trials had dropout rates exceeding 5%. The only trial satisfying this criteria was Heinemann FIRE‐3 2014; the results of the analysis are described in section 6.3 above. As the endpoint of toxicity was not affected by participants lost to follow‐up, no sensitivity analysis was carried out for this endpoint.
Discussion
Summary of main results
We identified a total of 68 studies in this systematic review, of which 33 studies with a total of 15,250 participants were included in meta‐analysis.
The primary objective of this review was to examine the overall effects of EGFR‐I (primarily EGFR MAb but also EGFR TKI) in various populations. We opted to perform pair‐wise analysis for the majority of the review, except for sections where there were too few studies to allow meta‐analysis. We considered network meta‐analysis but did not think this was the appropriate choice for this review because of the different patient populations in the identified trials, as well as the use of both adjusted and unadjusted analyses in reported trials. This may be more feasible in an individual patient data meta‐analysis.
In general, the risk of bias in analyses was low to unclear. However, we considered six of the studies as at high risk of bias primarily due to the assessment of progression‐free survival in a non‐blinded manner in open‐label trials. Analysis of funnel plots revealed overall low risk of publication bias.
Outcomes that relied on assessment of imaging for determination of tumour response or progression were affected by open‐label trials with investigator review of imaging without recourse to centralised or blinded review (such as in the case of outcomes 1.1, 1.3, 2.1, and 2.3). In comparison, outcomes that did not rely on investigator assessment (overall survival and neutrophil count for determination of grade 3 to 4 neutropenia, for instance) were not affected by the open‐label randomisation of all trials, and consequently received assessments of low risk of bias (such as for outcomes 1.2 and 1.7). Significant pharmaceutical funding was present in a majority of trials (29 of 33). However, we felt that this was unlikely to affect interpretation of the trial results, except in the case of Siena 2013.
The clinical scenario with the most trials and number of participants was the addition of EGFR MAb to standard therapy in the KRAS exon 2 WT population. The likelihood of tumour progression was reduced (HR 0.70, 95% CI 0.60 to 0.82), with PFS benefit present in all lines of therapy. When trials across all lines of therapy were pooled in meta‐analysis, there was significant statistical heterogeneity (I2 = 76%). We note that the pooled HR was 0.79 for first‐line trials compared to 0.43 for third‐line trials, and this differential efficacy may explain much of the observed heterogeneity. However, there was still considerable between‐trial heterogeneity amongst the first‐line trials (I2 = 66%), which we thought was mainly due to the use of alternative 5‐FU regimens such as bolus infusion (FLOX) and one trial allowing either infusional 5‐FU or oral capecitabine (Adams COIN 2011). The issue of whether chemotherapy partner choice affects EGFR MAb efficacy is controversial, with some evidence pointing towards decreased efficacy of non‐infusional regimens (Chan 2015). Addition of EGFR MAb to standard therapy also reduced the risk of death in KRAS exon 2 WT populations (HR 0.88, 95% CI 0.80 to 0.98). Significant statistical heterogeneity was present, likely due to the grouping of studies investigating EGFR MAb in different lines of therapy. However, the reduction in risk of death was not present when considering trials grouped by individual lines of therapy. Given the PFS benefit noted across all lines of therapy, this result may be attributable to the high rate of cross‐over in many trials, which would dilute the 'real' benefit to overall survival from addition of EGFR‐I (whist not affecting PFS).
The addition of EGFR MAb to standard therapy in KRAS exon 2 WT participants increased the likelihood of tumour response with an odds ratio of 2.41. Significant heterogeneity was again present, likely attributable to varying lines of therapy where different degrees of benefit were observed (OR 1.73 in first‐line compared to OR 38.44 in third‐line settings), which was probably due to the fact that placebo was used as the control arm in third‐line trials, whereas combination chemotherapy was the control in first‐ and second‐line trials. Significant heterogeneity again remained in the first‐line subgroup of trials, which was likely due to the pooling of trials using different chemotherapy backbones. The benefits of EGFR‐I in KRAS exon 2 WT populations come at the cost of increased toxicity (OR 2.45, 95% CI 2.07 to 2.89). Considering specific toxicities, the risks of diarrhoea and rash were increased but not the risk of neutropenia.
For the above comparison, we considered the OS outcome to be at low risk of bias, PFS and response rate at unclear risk of bias, and the toxicity outcomes (not including neutropenia) as at high risk of bias with corresponding downgrading in GRADE, which was primarily due to the presence of significant measurement bias. All included studies were open label in nature, and four of the included studies relied on unblinded investigator imaging review to determine PFS and response rate. Whilst there are unambiguous objective guidelines to determine disease progression or tumour response (RECIST), this does not fully eliminate the risk of bias (e.g. in interpreting multiple new sub‐centimetre nodules in the lungs as granulomas rather than metastatic disease, or vice versa). We felt that this was even more of a concern with the assessment of toxicity. Toxicity was measured by the Common Terminology Criteria for Adverse Events (CTCAE) criteria in most of the included trials, but these criteria allow for subjective measurement of severity of patient‐reported symptoms, and hence influence the rate of grade 3 to 4 toxicity. As neutropenia is an objective laboratory measure with unambiguous cutoffs in CTCAE, we felt that this was not susceptible to measurement bias in the same manner. Significant heterogeneity was present in some analyses such as PFS, but we felt that they were less likely to affect certainty regarding the underlying effect given the magnitude of summary benefit. Correspondingly, we considered the quality of evidence for PFS, OS, and response rate to be high, and that of grade 3 to 4 overall toxicity, diarrhoea, and rash to be moderate due to risk of bias. We downgraded the outcome of grade 3 to 4 neutropenia for imprecision rather than risk of bias, which also resulted in a judgement of the evidence as moderate quality. This comparison had the largest number of participants, and generally (comparing the same outcome across different comparisons) had the highest quality of evidence.
In comparison to KRAS exon 2 WT participants, no clinical benefit was demonstrated in KRAS exon 2 mutant participants, underscoring the importance of KRAS testing as a predictive biomarker. There was no reduction in risk of progression from the addition of EGFR‐I (HR 1.03, 95% CI 0.89 to 1.20), nor reduction in risk of death (HR 1.03, 95% CI 0.94 to 1.13). Although some individual trials showed a detriment to PFS in the EGFR‐I arm in the KRAS exon 2 MT setting, no significant differences were noted on meta‐analysis. The odds of overall grade 3 to 4 toxicity were not increased in KRAS exon 2 MT populations, although we note the borderline P value (0.06) and the increase in grade 3 to 4 diarrhoea and rash. This highlights the toxicities that can result from administering EGFR MAb to this population. Although the 'Risk of bias' assessments were similar to those for comparison 1 (i.e. low risk for OS, unclear risk for PFS and response rate, and high risk for toxicity not including neutropenia), we noted that one trial, Siena 2013, mandated input from the pharmaceutical sponsor for multicentre publications, resulting in a high risk of 'other bias'. We downgraded two outcomes (PFS and overall toxicity) due to the presence of significant heterogeneity. The heterogeneity in PFS was probably attributable to the use of EGFR MAb in different lines of therapy with different chemotherapy partners, however the heterogeneity for overall toxicity had no clear clinical explanation, and was perhaps due to differential dose intensity of chemotherapy. We felt that, in the absence of clinically compelling data and the lesser number of participants who were KRAS exon 2 MT compared to KRAS exon 2 WT, it was reasonable to downgrade outcomes 2.1 and 2.3. The smaller number of participants also affected the width of confidence intervals, with overall toxicity and diarrhoea receiving further downgrading in quality due to imprecision from low event numbers and wide confidence intervals.
The publication of data in the last five years confirming extended RAS testing (KRAS exon 2, 3, 4 and NRAS exon 3, 4) as a potential predictive biomarker for EGFR MAb led us to amend the protocol to include subgroup analyses by extended RAS status. As expected, the use of EGFR MAb in extended RAS WT populations reduced the risk of disease progression to a greater degree than seen in KRAS exon 2 WT populations (HR 0.60, 95% CI 0.48 to 0.75 versus HR 0.70, 95% CI 0.60 to 0.82). Similarly, the reduction in risk of death was numerically greater in the extended RAS WT population (HR 0.77, 95% CI 0.67 to 0.88) compared to the KRAS exon 2 WT population (HR 0.88, 95% CI 0.80 to 0.98). However, due to limitations of the pair‐wise meta‐analysis methodology we cannot conclude that the benefit is statistically greater than in the KRAS exon 2 WT setting, and investigation of this important clinical question unfortunately lies outside of the current scope of this meta‐analysis.
Significant heterogeneity was present both in the PFS (I2 = 61%) and response rate analyses (I2 = 47%), which may be due to the pooling of trials investigating cetuximab and panitumumab rather than purely by line of therapy. The results for these outcomes should be interpreted with caution given the above heterogeneity, and we correspondingly downgraded the quality of the evidence by one grade to moderate for these outcomes. In contrast, participants with extended RAS mutant genotypes derived no benefit from use of EGFR MAb; in fact, use in first‐line settings increased the risk of progression (HR 1.27, 95% CI 1.08 to 1.48). We considered the evidence for all three outcomes with reported results as of moderate quality, due to heterogeneity and imprecision. Whilst the numbers of participants in these analyses were similar to those in the RAS WT analyses, the fact that the summary hazard ratios were closer to 1 (the point of no effect) meant that two of the intervals overlapped a point of no effect and a point of clinical significant harm (HR 1.25 for overall survival, 0.75 for response rate). Given that the 95% CI encompasses these two different scenarios, we downgraded the outcomes of OS and response rate due to imprecision. These data argue for the adoption of routine extended RAS testing and the restriction of EGFR MAb use to patients with this genotype.
The above results are consistent with modern‐day clinical practice. EGFR MAbs have been widely used in clinical management of mCRC, based on results from the individual positive trials which contributed to the overall positive results shown above. Assuming availability and full funding, EGFR MAbs should be used in all patients who are RAS WT and can tolerate the anticipated side effects due to the clearly demonstrated benefits to PFS, response rate, and (to a lesser extent) OS. The side effects of therapy differ from those of conventional chemotherapy; the most common 'new' effect is rash, with the rate increasing from 1% to 24% in the KRAS exon 2 WT population. Whilst these side effects should be treated seriously and may impact on patients' quality of life, rash is readily treatable with topical steroids and antibiotics (topical initially, with oral antibiotics in severe cases). In our view, the noted increase in toxicity should not be a deterrent to starting EGFR MAb in most patients, given the demonstrated clinical efficacy of these drugs and the relatively limited number of treatment options for mCRC.
We were unable to comment on the optimal line of therapy for EGFR MAb use; although proportional benefits did seem greater with third‐line use, these patients generally have a poorer PFS and OS with therapy (given that they have few chemotherapeutic options), and further research will hopefully define whether EGFR MAb should be used first‐, second‐, or third‐line, as well as its sequencing with bevacizumab (where available).
Due to the paucity of trials, meta‐analysis was not possible in the KRAS unselected population. Whilst Sobrero EPIC 2008 did report a subsequent analysis by KRAS status, we note that only 300 of 1298 participants had KRAS results available, and that OS and PFS data were incomplete (HRs only without 95% CIs). We had originally considered incorporating such KRAS unselected trials in the main analysis together with trials currently in section 1. However, we felt that this was less appropriate given the widespread adoption of KRAS testing and its effect on both clinical practice and conduct of subsequent trials. In any case, given the evidence cited above for RAS testing as a prerequisite for EGFR MAb efficacy, it seems likely that all patients commenced on EGFR MAb will have prior RAS testing. Funding for systematic RAS testing prior to EGFR MAb use is critical to optimise the use of these expensive medications.
This review also examined the overall effects of adding EGFR‐I (whether MAb or TKI) compared to adding another targeted agent. As all identified trials compared EGFR MAb with bevacizumab, bevacizumab was used as the comparator for meta‐analysis. We observed no differences in PFS (HR 1.02, 95% CI 0.93 to 1.12) or OS (HR 0.84, 95% CI 0.70 to 1.01). We noted significant heterogeneity in the OS analysis, likely due to the inclusion of a second‐line study, Hecht SPIRITT 2015, with potential differences in the patient population as a result of prior first‐line therapy and selection of a patient cohort suitable for second‐line therapy. The use of EGFR MAb compared to bevacizumab increased the odds of tumour response (OR 1.36, 95% CI 1.15 to 1.62) but also increased the odds of overall grade 3 to 4 toxicity (OR 1.37, 95% CI 1.09 to 1.72). Insufficient data were available to comment definitively on the rates of grade 3 to 4 diarrhoea and neutropenia, although the odds of rash were increased. The major source of bias was again measurement bias. Two of the four studies employed central or blinded assessment of imaging; we judged the outcomes of PFS and response rate as having unclear risk of bias, and OS as low risk of bias. Again, we felt that toxicity was at high risk of bias in the conduct of open‐label studies and downgraded this outcome accordingly. We downgraded the outcomes of OS and diarrhoea for imprecision (given that the confidence interval encompasses a point of significant difference with EGFR MAb and a point of no effect), and the outcome of diarrhoea a third time for inconsistency.
Another group of trials included in this review investigated the addition of EGFR TKIs to standard therapy. Whilst due to the small number of published trials we were unable to perform meta‐analysis, we note that neither trial showed improvements in PFS or OS. Insufficient data were available to comment on tumour response rate or toxicity. The weight of evidence is therefore much more marked for the use of EGFR MAb compared to EGFR TKI.
The final group of trials analysed investigated the addition of EGFR‐I (whether TKI or MAb) to the combination of bevacizumab and chemotherapy in people with mCRC. We restricted efficacy analysis in EGFR MAb trials to KRAS exon 2 WT participants given the evidence above supporting this in comparison 1. Unfortunately, this meant that Tol CAIRO2 2008, which provided insufficient detail for OS outcomes in the KRAS exon 2 WT cohort, could not be included in that analysis. The likelihood of tumour progression was not reduced (HR 1.04, 95% CI 0.83 to 1.29). Significant statistical heterogeneity was present, likely due to the pooling of studies investigating both MAbs and TKIs. The likelihood of death was not reduced (HR 1.00, 95% CI 0.69 to 1.47). The odds of tumour response were not increased. The addition of EGFR‐I to chemotherapy plus bevacizumab did increase the odds of grade 3 to 4 overall toxicity, diarrhoea, and rash but not neutropenia. As only one of the six trials employed blinded or central review, we judged the outcomes of PFS and response rate (in addition to overall toxicity, rash, and diarrhoea) to be at high risk of bias. In addition, we noted significant heterogeneity in the PFS, OS, response rate, overall toxicity, and diarrhoea outcomes. After investigation of multiple clinical differences, there was no compelling clinical explanation for this heterogeneity, and we further downgraded the quality of the evidence for outcomes with significant heterogeneity. Heterogeneity is to some extent expected in this analysis, which combines EGFR TKI with MAb, induction with maintenance studies, and multiple therapeutic agents. We considered not performing meta‐analysis in this comparison, but decided to proceed given the significant clinical effort invested in this topic. Although the quality of evidence is among the lowest for the comparisons in this review, we feel that there is enough evidence to show no benefit with addition of EGFR‐I to the combination of chemotherapy and bevacizumab, and potential evidence of harm. The combination of EGFR‐I, bevacizumab, and chemotherapy is therefore not supported by the current evidence, and we feel this topic does not deserve further investigation.
This meta‐analysis has provided detailed examination of the important questions regarding optimal use of EGFR‐I in people with mCRC receiving treatment in all lines of therapy. Trials varied in agents used, whether EFGR‐I were combined with chemotherapy or used as monotherapy, and whether cross‐over was allowed (which may have affected OS outcome). However, this body of work provides strong rationale for use of EFGR MAbs and provides robust and defensible outcomes of significance to patients.
Overall completeness and applicability of evidence
This review allowed complete evaluation of endpoints, some fully and some partially. In terms of efficacy endpoints, PFS and OS have been well reported with sufficient statistical information to allow meta‐analysis. On the other hand, documentation of toxicity, particularly overall grade 3 to 4 toxicity, rash, diarrhoea, and neutropenia, in relevant subgroups is less complete, despite attempts to contact study authors for more information. We have therefore evaluated these secondary outcomes less completely.
The studies identified are relevant to the aims of this review and the clinical needs of people with mCRC. As noted above, median survival for mCRC without the use of biological agents is less than 24 months, but introduction of the biological agents (particularly EGFR MAb and bevacizumab) have resulted in further incremental benefit with median survivals in recent state‐of‐the‐art sequencing trials measured at 29 to 32 months (Heinemann FIRE‐3 2014; Venook CALGB 80405 2014). Two main questions face clinicians today with unrestricted access to EGFR MAb: which populations benefit most from EGFR MAb, and which lines of therapy in which to offer EGFR MAb. With respect to the first question, the data in the current study support the use of EGFR MAb in people with extended RAS WT genotypes. In view of the moderate‐quality evidence for lack of PFS and OS benefit in RAS mutant patients, combined with the increased incidence of diarrhoea and rash noted in KRAS exon 2 MT patients, there is no clinical rationale for administering these expensive drugs in patients with a RAS mutation. This is especially the case when one considers the significant cost of EGFR MAb, which may be up to USD 6000 per month for treatment (Schrag 2015).
There were significant variations among the included trials within the broad scope of this review. Firstly, variation exists in terms of the therapeutic agents investigated in trials. The majority of trials investigated well‐known EGFR MAbs in current clinical practice (cetuximab, panitumumab) in addition to chemotherapy. However, some trials also investigated novel EGFR MAb (imgatuzumab) as well as EGFR TKI, a class of drugs that have been shown to have efficacy in metastatic non‐small cell lung cancer but not mCRC as yet. Other trials further sought to compare different EGFR agents, or different dosage regimens of agents, such as in Brodowicz 2013. Even in trials that investigated the same dose of the same EGFR MAb, there are differences (sometimes allowed in the same trial) in the chemotherapy partner, whether it be the choice of fluoropyrimidine regimen (infusional 5‐FU, bolus 5‐FU, capecitabine) or fluoropyrimidine partner (oxaliplatin or irinotecan). We have attempted to analyse the collected data by action of EGFR inhibitor (MAb versus TKI), type of trial (addition of EGFR in the intervention arm versus comparison of EGFR MAb to bevacizumab versus comparison of EGFR agent with another EGFR agent), and intervention arm (chemotherapy or best supportive care versus the combination of chemotherapy and bevacizumab).
There was also considerable between‐study variation in terms of outcome reporting. The shift towards registration of studies in the public forum and registration of planned primary and secondary outcomes in sites such as ClinicalTrials.gov have increased the transparency with which planned outcomes are reported, and thus aided in assessment of outcome reporting. Although allowing preliminary reports (such as abstracts) in the inclusion criteria increases the coverage of 'grey literature', it also increases the number of studies that have not been completely reported to date. Eight of the studies in this review were only reported in abstract form, and thus could not be assessed completely in terms of reporting bias. Two studies with published data have not reported on all of their outcomes; one has not yet published quality of life data (Wasan COIN‐B 2014), and we await information from the author regarding this data. The other, Siena 2013, was terminated early and only response rate was reported despite initial plans to evaluate other measures such as PFS and OS, leading to an assessment of high risk of bias.
Quality of life data has in general been poorly collected and reported throughout the included studies. This is obviously an important issue for both patients and clinicians, for objective QoL indices help determine whether the adverse events from EGFR MAb treatment are outweighed by the known clinical benefit. Only 8 of the 33 studies have thus far reported quality of life measures. Where reported, the results have not allowed for quantitative synthesis; P values were often reported as being more than or less than 0.05 without exact statistics either in the EQ‐5D or EORTC QLQ‐C30 subscales to allow synthesis. In addition, there is little in the way of QoL data by RAS status or other clinical subgroups (e.g. the elderly may suffer more side effects from EGFR MAb and have lower QoL from treatment). This is a missed opportunity to collect data to truly inform the patient about the anticipated effects of these treatments on their life. Some of the challenges with adopting QoL measurements in trials is the additional time and cost required, as well as the possibility in pharmaceutical‐funded trials that negative QoL impacts may delay registration of an agent. Be that as it may, careful collection and full reporting of QoL outcomes is critical if the scientific community is to move forward with patient‐centred care in mCRC, with the aim of using currently available modalities to optimise the often difficult patient journey in mCRC.
Follow‐up and surgery also differed slightly between different studies. Follow‐up ranged from 13 to 50 months where this was reported, with 11 studies not reporting median follow‐up in available publications (although a minority of these did report other forms of follow‐up). Given that many of the studies with shorter follow‐up reported a high proportion of events (either progression or death) at conclusion of follow‐up, we felt that these periods of follow‐up were reasonable to ensure accuracy of results. Patients who were eligible for surgery at enrolment to an EGFR MAb trial (e.g. the population in Primrose NEW EPOC 2014) were excluded from our review, but one study included in our review, Ye 2013, did enrol patients with liver‐limited mCRC and measured the percentage of patients who were converted to potentially resectable disease.
Whilst some EGFR‐I have entered mainstream clinical practice (cetuximab and panitumumab in particular), others have not yet progressed past the investigational setting. Even for trials investigating the same drug, different patient populations may affect the efficacy of the investigated therapy. For example, there is some evidence to suggest that the genomic profile of solid tumours change with chemotherapy (Lee 2009), and that these changes may affect the efficacy of different anticancer treatments. In addition, treatment with EGFR MAb may result in the emergence of resistant clones (Mohan 2014), diminishing the therapeutic benefit of subsequent EGFR‐directed therapy.
We noted differences in follow‐up between studies, which we have described in the 'Summary of findings' tables. Most studies utilised an intention‐to‐treat analysis model with censoring on loss to follow‐up and calculation of hazard ratios and odds ratios using a Cox proportional hazards model. This model allows for the estimation of survival percentages and comparison of different therapeutic regimens even in the context of significant participant dropout or censoring. However, the assumptions of non‐informative censoring (i.e. censoring not related to medical conditions such as progressive disease) and proportional hazards (proportional hazard functions over time between two patient strata) are present in this model, and violation of these assumptions would make the Cox model less accurate. In the context of controlled clinical trials with clearly defined exclusion and censoring criteria, we feel that the first assumption is valid. The second assumption ‐ that of proportional hazards ‐ is a standard assumption in randomised oncology trials that we feel is appropriate to this analysis.
In summary, there is vast between‐study heterogeneity in trials of EGFR inhibitors ‐ in agents investigated, dosing schedules, clinical characteristics of the patient population, risk of bias, follow‐up, and completeness of reporting.
In terms of applicability to current practice, the results of the KRAS exon 2 WT analysis are consistent with the international practice of use of EGFR MAbs in this setting, but not in patients whose tumours harbour a KRAS exon 2 mutation. The further improvement of outcomes with EGFR MAbs after extended RAS testing is the subject of evolving data. The relative efficacy of bevacizumab compared to EGFR‐I, in combination with chemotherapy, remains a controversial question with conflicting results of large phase III randomised trials. However in our meta‐analysis comparing EGFR‐I with bevacizumab, we found no significant difference in PFS or OS, although tumour response rates were higher in participants treated with cetuximab.
There is insufficient data to subject the effects of adding EGFR‐I TKI to standard therapy to meta‐analysis. However, the limited available data included in this review showed no evidence of significant improvement in PFS, OS, or tumour response rate in molecularly unselected participants, but increased toxicity including grade 3 to 4 rash and diarrhoea.
The addition of EGFR‐I to a combination of bevacizumab and chemotherapy in people with KRAS exon 2 WT mCRC did not significantly improve response rates, PFS, or OS, but did increase toxicity including grade 3 to 4 rash and diarrhoea.
We feel that the methods used were statistically sound and the results applicable to clinical practice. We restricted eligibility to studies investigating patients with histologically confirmed unresectable mCRC. Whilst sites of metastatic disease did not necessarily require histological confirmation, this was the case for all included trials. We believe that this standard is consistent with clinical practice, and maximises the applicability of this systematic review. The included studies satisfied the assumption of proportional hazards, as they all investigated chemotherapy alone (without surgery, radiotherapy, or liver‐directed therapy as study‐mandated procedures), and generally randomised participants well.
Quality of the evidence
The results included in this systematic review allow a robust conclusion regarding the primary objective addressed in the overview, that of progression‐free survival with the addition of EGFR‐I in people with KRAS exon 2 WT genotypes. Similar high‐quality evidence was available for the outcomes of overall survival and response rate, as well as the same outcomes in the extended RAS WT population.
Lower‐quality evidence was available for the outcomes of grade 3 to 4 overall toxicity, neutropenia, rash, and diarrhoea, as well as quality of life. This is disappointingly true when considering the last. Quality of life outcomes are paramount to people with a life‐limiting, incurable illness, who often undergo multiple lines of therapy. Where reported, quality of life was often not quantitatively presented, with undefined descriptions such as "no clinically meaningful differences" and lack of P values common. The reporting of quality of life needs to improve for more thorough assessments about EGFR‐I efficacy to be made.
Lower‐quality evidence was available regarding the efficacy of EGFR TKI in treatment of mCRC and the efficacy of one EGFR‐I compared to another. These questions have not been explored as thoroughly given the lack of significant differences between arms in trials conducted thus far. Nevertheless, the current available data do not provide justification for further clinical investigation of EGFR TKI in unselected people with mCRC. However, it is reasonable to further analyse the data from these or subsequent studies should they be presented in detail in the future.
Potential biases in the review process
We acknowledge that several changes were made to the protocol in consultation with the Cochrane Colorectal Cancer Group editorial board after publication of the protocol. These include:
decisions to focus on KRAS exon 2 WT populations for top‐level reporting of results (consistent with published literature and clinical use since publication of protocol);
inclusion of extended RAS analysis to the subgroup analysis.
While we believe the above changes increase the relevance of the data and reflect the change in standard of care over the last few years, this could have resulted in confirmatory bias, in that any variations in results or heterogeneity are by default attributed to KRAS/RAS status.
Our review is also biased by the decision to conduct a study‐level meta‐analysis. An individual participant data meta‐analysis would provide participant‐level information for each of the desired outcomes, offering increased accuracy and the ability to undertake more thorough subgroup analyses. In addition, data regarding randomisation processes and allocation concealment would be fully available in an ideal international collaborative process ‐ information that remains incomplete in this review despite attempts to contact authors for relevant information.
We considered the use of a network meta‐analytic approach to synthesise findings in this review, but did not find it to be appropriate given the different populations in the included trials. For instance, the populations of first‐line EGFR MAb naive patients and patients with chemo‐refractory disease with prior EGFR MAb exposure have both different prognoses and potentially different responsiveness to EGFR‐targeting agents.
Agreements and disagreements with other studies or reviews
We note that Vale and colleagues published a meta‐analysis on the efficacy of EGFR MAb in mCRC and its relation to KRAS status (Vale 2012). We agree with their conclusion that EGFR MAbs result in PFS and OS benefit used alongside infusional 5‐FU‐based regimens in KRAS exon 2 WT patients, but not in KRAS exon 2 MT patients. However, we have added to their review by including studies of EGFR TKI, evaluating EGFR‐I efficacy in extended RAS analyses, and identifying 10 additional RCTs.
We also note that a meta‐analysis was published investigating the effect of EGFR MAb on OS and PFS in mCRC patients with extended RAS genotypes that concluded that EGFR MAb in people with new RAS mutations was unlikely to provide significant benefit (Sorich 2015). We agree with these conclusions, although we again have included analyses of studies investigating EGFR TKI in our systematic review.
A systematic review considering all EGFR‐I trials together found mixed evidence for efficacy and increased toxicity in 128 identified studies (Rauw 2012). However, this review did not undertake quantitative analysis.
We note a study published by the present authors showing that the combination of EGFR MAb and oxaliplatin‐based chemotherapy regimens may show improved efficacy when infusional 5‐FU is part of the regimen (as opposed to bolus 5‐FU alone or capecitabine) (Chan 2015). However, the number of studies analysed in these subgroups was relatively small. We agree with the overall conclusion that EGFR MAbs improve PFS in mCRC.
Finally, we note that a meta‐analysis was published comparing first‐line EGFR MAb to first‐line anti‐VEGF agent in mCRC (Khattak 2015). The authors concluded that first‐line anti‐EGFR therapy improved OS and objective response rate more than first‐line anti‐VEGF therapy in both KRAS exon 2 WT and extended RAS WT populations with mCRC. We included a fourth study (Hecht SPIRITT 2015), which was performed in the second‐line setting. We agreed that EGFR MAb improves objective response rate compared to anti‐VEGF agent, but disagreed on the improvement of OS. A network meta‐analysis on the same topic, Kumachev 2014, also concluded that EGFR inhibitors improved OS (but not PFS) in direct comparison to bevacizumab, but that no significant differences were found either on indirect or network meta‐analysis. We note significant heterogeneity in these analyses, meaning that any significant difference (even if present) should be interpreted with caution. We also provided a thorough 'Risk of bias' analysis of these trials, which was not mentioned in the above papers.
Authors' conclusions
Implications for practice.
The potential benefits from epidermal growth factor receptor monoclonal antibodies (EGFR MAb) come at the cost of increased odds of toxicity, in particular overall grade 3 to 4 toxicity, rash, and diarrhoea. The lack of quality of life data makes it difficult to predict the impact of EGFR MAb on a patient's overall well‐being. As a result, the pros (improvement in efficacy parameters) and cons (increase in toxicity) of EGFR MAb therapy should be weighed with a patient when considering the optimal treatment that best suits a patient's treatment goals.
Comparing EGFR MAb to bevacizumab in combination with standard therapy in KRAS exon 2 wild‐type (WT) populations, progression‐free survival and overall survival are not improved, but tumour response rate is increased. The odds of overall grade 3 to 4 toxicity are increased with EGFR MAb compared with bevacizumab. In practice, this does not change the treatment paradigm in a country where both EGFR MAb and bevacizumab are available without restriction. In the RAS WT population, one is usually used in the first line in combination with chemotherapy, and the other is used in combination with another chemotherapy on progression (e.g. FOLFIRI with cetuximab first line, then FOLFOX with bevacizumab second line, or FOLFIRI with bevacizumab first line, then FOLFOX with cetuximab second line). The choice of line in which EGFR MAb is used, and the chemotherapy partner it is used with, remain up to clinician preference at this point given the lack of definitive evidence showing that choice of oxaliplatin or irinotecan affects EGFR MAb efficacy.
This systematic review also has important implications on the public rationing of EGFR MAb. Authorities are faced with the difficult task of balancing the provision of drugs to all who may potentially benefit with a limited budget. As evidence of EGFR MAb efficacy is limited to RAS WT populations, the evidence supports limiting provision of EGFR MAb to patients with this genotype. Given the lack of benefit with EGFR MAb in KRAS mutant (MT) and extended RAS MT populations, there is no clinical rationale for administration of these drugs in this subgroup. The cost‐benefit ratio of EGFR MAb, optimal line of therapy, and comparison of cost‐effectiveness to bevacizumab are important areas that fall outside the scope of this review.
Considering the other areas of investigation, there is no evidence that either EGFR MAb or bevacizumab is superior in combination with chemotherapy. A full discussion of sequencing of these agents for optimal benefit is again outside the bounds of this review. Nevertheless, assuming that the agents are of equal cost, there is no evidence to support restriction of EGFR MAb to a particular line of therapy or bind its provision to prior bevacizumab exposure (or lack thereof). However, it is clear that the addition of EGFR‐I to the combination of chemotherapy and bevacizumab in people with KRAS exon 2 WT metastatic colorectal cancer does not improve progression‐free survival, overall survival, or tumour response rate but does increase rates of toxicity (overall grade 3 to 4 toxicity, grade 3 to 4 diarrhoea and rash) and may cause harm. The use of EGFR MAb in addition to the combination of chemotherapy and bevacizumab is therefore not supported by the current data. Similarly, there is currently no evidence to support the use of EGFR tyrosine kinase inhibitors in metastatic colorectal cancer (whether in KRAS WT or MT populations), and their use should remain investigational at present.
Implications for research.
Basic research
The current impact of EGFR inhibitors on the genomic profile of colorectal cancer in vivo is not well understood to date. Given that multiple chemotherapeutic regimens and biological agents are available for the treatment of metastatic colorectal cancer, a more thorough understanding of these mechanisms will help clinicians understand the potential impact, or lack thereof, inherent in sequencing currently available therapies.
Given that KRAS has been thoroughly characterised as a predictive biomarker for EGFR MAb, the functional role of NRAS and HRAS needs further research.
Clinical research
An individual participant data meta‐analysis on the same topic may provide more robust trial data and allow for additional subgroup analyses.
Given that both EGFR MAb and bevacizumab individually show benefit when added to chemotherapy, but that the combination of both in addition to chemotherapy does not, future trials should not investigate the combination of EGFR MAb, bevacizumab, and chemotherapy in people with metastatic colorectal cancer without new preclinical data to suggest the efficacy of this combination.
The strength of data for the predictive role of RAS in EGFR MAb (both with regard to efficacy in the KRAS WT setting and lack of efficacy in the KRAS MT setting) means that future trials of EGFR MAb should mandate RAS testing and include RAS WT genotype as an inclusion criterion.
All future clinical trials should measure and report quality of life.
The question of optimal sequencing ‐ of EGFR MAb and bevacizumab, as well as of different chemotherapeutic regimens and the partnering of the two ‐ should continue to be investigated.
The role of additional therapies in reversing EGFR MAb resistance (e.g. the addition of vemurafenib or other B‐Raf proto‐oncogene, serine/threonine kinase (BRAF)‐targeting agents to EGFR MAb in BRAF MT patients) is incompletely defined, and deserves further investigation.
The emergence of newer data may influence future research regarding optimisation of EGFR MAb use in metastatic colorectal cancer. Recent data presented at American Society of Clinical Oncology (ASCO) 2016 regarding Venook CALGB 80405 2014 suggests that cetuximab (compared to bevacizumab) may have worse efficacy for right‐sided primaries but better efficacy in left‐sided primaries. This may be related to molecular differences between the two locations such as CpG island methylator phenotype (CIMP) and BRAF mutation status. Future research regarding the impact of these factors on the efficacy of cetuximab, as well as the molecular correlations of right‐sided and left‐sided colon cancer, should be performed to optimise benefit to patients from EGFR MAb use.
Given the lack of benefit from EGFR tyrosine kinase inhibitors in currently published trials, it is questionable as to whether future research into this area would be worthwhile.
What's new
| Date | Event | Description |
|---|---|---|
| 28 June 2017 | Amended | Included source of support and acknowledgement on NIH, US, funding for one of the authors |
History
Protocol first published: Issue 2, 2008 Review first published: Issue 6, 2017
| Date | Event | Description |
|---|---|---|
| 16 June 2009 | Amended | Protocol revised to include KRAS and BRAF mutation subgroup analysis |
| 15 June 2009 | Amended | Protocol revised to include KRAS mutation subgroup analysis |
Acknowledgements
Drs Jeremy Shapiro and Chris Karapetis contributed to the writing of the protocol for this review.
Dr Weng Ng provided the spreadsheet used to calculate hazard ratios for overall survival and progression‐free survival for Santoro 2008.
Dr Chris Pene contributed to earlier versions of the literature search.
Dr Alberto Sobrero, Dr Richard Adams, Dr Matthew Seymour, and Dr Helga Hagman provided data for Sobrero EPIC 2008, Adams COIN 2011, Seymour PICCOLO 2013, and Hagman ACT2 2014, respectively.
We would like to thank the referees and editors of the Cochrane Colorectal Cancer Group for their insightful comments regarding the manuscript.
We sincerely thank Julie Marker, Joe Levin, and Tony Hamdorf for their insightful comments and suggestions regarding the plain language summary for this review.
We acknowledge the Cochrane Colorectal Cancer Group for support in developing and carrying out the search strategies as well as substantial editorial support.
We acknowledge receiving a Core Grant (P30CA008748) at Memorial Sloan Kettering Cancer Center from the National Institutes of Health (NIH), USA, supporting the elaboration of this systematic review.
Appendices
Appendix 1. CENTRAL search strategy
CENTRAL (the Cochrane Library) Issue 9, 2016
#1 (colorect* or colon* or rect* or anal* or anus* or intestin* or bowel*) near/3 (carcinom* or neoplas* or adenocarcinom* or cancer* or tumor* or tumour* or sarcom*)
#2 MeSH descriptor: [Colorectal Neoplasms] explode all trees
#3 (#1 or #2)
#4 (epidermal growth factor or EGR or EGFR or ErbB‐1 or HER1 or Cetuximab or Panitumumab or Erlotinib or Gefitinib)
#5 MeSH descriptor: [Epidermal Growth Factor] explode all trees
#6 MeSH descriptor: [Receptor, Epidermal Growth Factor] explode all trees
#7 (#4 or #5 or #6)
#8 MeSH descriptor: [Neoplasm Metastasis] explode all trees
#9 metasta* or stage 4 or stage IV or advanced:ti,ab,kw
#10 (#8 or #9)
#11 (#3 and #7 and #10)
Appendix 2. MEDLINE search strategy
Ovid MEDLINE 1950 to 9 September 2016
1. ((colorect* or colon* or rect* or anal* or anus* or intestin* or bowel*) adj3 (carcinom* or neoplas* or adenocarcinom* or cancer* or tumor* or tumour* or sarcom*)).mp.
2. exp Colorectal Neoplasms/
3. 1 or 2
4. (epidermal growth factor or EGR or EGFR or ErbB‐1 or HER1 or Cetuximab or Panitumumab or Erlotinib or Gefitinib).mp.
5. exp Epidermal Growth Factor/
6. exp Receptor, Epidermal Growth Factor/
7. 4 or 5 or 6
8. exp Neoplasm Metastasis/
9. (metasta* or stage 4 or stage IV or advanced).mp.
10. 8 or 9
11. 3 and 7 and 10
12. randomized controlled trial.pt.
13. controlled clinical trial.pt.
14. random*.ab.
15. placebo.ab.
16. clinical trial as topic.sh.
17. trial.ti.
18. 12 or 13 or 14 or 15 or 16 or 17
19. exp animals/ not humans.sh.
20. 18 not 19
22. 11 and 20
Appendix 3. Embase search strategy
Ovid Embase 1974 to 9 September 2016
1. ((colorect* or colon* or rect* or anal* or anus* or intestin* or bowel*) adj3 (carcinom* or neoplas* or adenocarcinom* or cancer* or tumor* or tumour* or sarcom*)).m_titl.
2. exp large intestine tumor/
3. 1 or 2
4. (epidermal growth factor or EGR or EGFR or ErbB‐1 or HER1 or Cetuximab or Panitumumab or Erlotinib or Gefitinib).m_titl.
5. *epidermal growth factor/
6. *epidermal growth factor receptor/
7. *epidermal growth factor receptor antibody/
8. *epidermal growth factor receptor kinase inhibitor/
9. 4 or 5 or 6 or 7 or 8
10. exp metastasis/
11. (metasta* or stage 4 or stage IV or advanced).mp.
12. 10 or 11
13. 3 and 9 and 12
14. CROSSOVER PROCEDURE.sh.
15. DOUBLE‐BLIND PROCEDURE.sh.
16. SINGLE‐BLIND PROCEDURE.sh.
17. (crossover* or cross over*).ti,ab.
18. placebo*.ti,ab.
19. (doubl* adj blind*).ti,ab.
20. allocat*.ti,ab.
21. trial.ti.
22. RANDOMIZED CONTROLLED TRIAL.sh.
23. random*.ti,ab.
24. 14 or 15 or 16 or 17 or 18 or 19 or 20 or 21 or 22 or 23
25. (exp animal/ or exp invertebrate/ or animal.hw. or nonhuman/) not (exp human/ or human cell/ or (human or humans or man or men or wom?n).ti.)
26. 24 not 25
27. 13 and 26
Appendix 4. Other search strategies
a) ClinicalTrials.gov
Carried out on 18‐19 January 2016 and 14 March 2017
Search strategy:
"COLORECTAL" AND ("Cetuximab" OR "Panitumumab" OR "EGFR" OR "EGFR‐I")
b) ASCO/ASCO GI (2011 to 2016)
Carried out on 18‐19 January 2016 and 15 March 2017. Site searched: meetinglibrary.asco.org/
Years searched: 2011‐2016
Terms searched: 1) Colorectal, cetuximab 2) Colorectal, panitumumab 3) Colorectal, EGFR 4) Colorectal, EGFR‐I
c) ESMO (2011 to 2015)
Carried out on 18‐19 January 2016 and 15 March 2017. Site searched: annonc.oxfordjournals.org ‐ relevant supplementary sections (all abstracts in gastrointestinal tumours, colorectal)
d) World GI (2011 to 2015)
Carried out on 18‐19 January 2016 and 15 March 2017. Site searched: annonc.oxfordjournals.org ‐ relevant supplementary sections (all abstracts)
e) WHO ICTRP
Carried out on 20 January 2016 and 14 March 2017.
"Colorectal" in title and (cetuximab or panitumumab or egfr or egfr‐i) in intervention
Appendix 5. Criteria for judging risk of bias in the 'Risk of bias' assessment tool
|
RANDOM SEQUENCE GENERATION Selection bias (biased allocation to interventions) due to inadequate generation of a randomised sequence | |
| Criteria for a judgement of ‘low risk’ of bias | The investigators describe a random component in the sequence generation process such as the following.
*Minimisation may be implemented without a random element, which is considered to be equivalent to being random. |
| Criteria for a judgement of ‘high risk’ of bias | The investigators describe a non‐random component in the sequence generation process. Usually, the description would involve some systematic, non‐random approach, such as the following.
|
| Criteria for a judgement of ‘unclear risk’ of bias | Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
|
ALLOCATION CONCEALMENT Selection bias (biased allocation to interventions) due to inadequate concealment of allocations prior to assignment | |
| Criteria for a judgement of ‘low risk’ of bias | Participants and investigators enrolling participants could not foresee assignment because one of the following, or an equivalent method, was used to conceal allocation.
|
| Criteria for a judgement of ‘high risk’ of bias | Participants or investigators enrolling participants could possibly foresee assignments and thus introduce selection bias, such as allocation based on the following.
|
| Criteria for a judgement of ‘unclear risk’ of bias | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. This is usually the case if the method of concealment is not described or not described in sufficient detail to allow a definitive judgement, e.g. if the use of assignment envelopes is described, but it remains unclear whether envelopes were sequentially numbered, opaque, and sealed. |
|
BLINDING OF PARTICIPANTS AND PERSONNEL Performance bias due to knowledge of the allocated interventions by participants and personnel during the study. | |
| Criteria for a judgement of ‘low risk’ of bias | Any one of the following.
|
| Criteria for a judgement of ‘high risk’ of bias | Any one of the following.
|
| Criteria for a judgement of ‘unclear risk’ of bias | Any one of the following.
|
|
BLINDING OF OUTCOME ASSESSMENT Detection bias due to knowledge of the allocated interventions by outcome assessors | |
| Criteria for a judgement of ‘low risk’ of bias | Any one of the following.
|
| Criteria for a judgement of ‘high risk’ of bias | Any one of the following.
|
| Criteria for a judgement of ‘unclear risk’ of bias | Any one of the following.
|
|
INCOMPLETE OUTCOME DATA Attrition bias due to amount, nature, or handling of incomplete outcome data | |
| Criteria for a judgement of ‘low risk’ of bias | Any one of the following.
|
| Criteria for a judgement of ‘high risk’ of bias | Any one of the following.
|
| Criteria for a judgement of ‘unclear risk’ of bias | Any one of the following.
|
|
SELECTIVE REPORTING Reporting bias due to selective outcome reporting | |
| Criteria for a judgement of ‘low risk’ of bias | Any of the following.
|
| Criteria for a judgement of ‘high risk’ of bias | Any one of the following.
|
| Criteria for a judgement of ‘unclear risk’ of bias | Insufficient information to permit judgement of ‘low risk’ or ‘high risk’. It is likely that the majority of studies will fall into this category. |
|
OTHER BIAS Bias due to problems not covered elsewhere in the table | |
| Criteria for a judgement of ‘low risk’ of bias | The study appears to be free of other sources of bias. |
| Criteria for a judgement of ‘high risk’ of bias | There is at least one important risk of bias. For example, the study:
|
| Criteria for a judgement of ‘unclear risk’ of bias | There may be a risk of bias, but there is either:
|
Appendix 6. Glossary
CAPOX: The combination of capecitabine and oxaliplatin, given in three‐weekly cycles
CAPIRI: The combination of capecitabine and irinotecan, given in three‐weekly cycles
FOLFIRI: The combination of 5‐fluorouracil, leucovorin, and irinotecan, given in fortnightly cycles
FOLFOX4: The combination of 5‐fluorouracil, leucovorin, and oxaliplatin given in fortnightly cycles
mFOLFOX6: The combination of 5‐fluorouracil, leucovorin, and oxaliplatin given in fortnightly cycles (in a different dosing schedule to FOLFOX4)
mFOLFOX7: The combination of 5‐fluorouracil, leucovorin, and oxaliplatin given in fortnightly cycles (in a different dosing schedule to both FOLFOX4 and mFOLFOX6)
Data and analyses
Comparison 1. EGFR MAb in KRAS exon 2 WT.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Progression‐free survival | 12 | Hazard Ratio (Random, 95% CI) | 0.70 [0.60, 0.82] | |
| 1.1 First‐line | 6 | Hazard Ratio (Random, 95% CI) | 0.79 [0.66, 0.94] | |
| 1.2 Second‐line | 4 | Hazard Ratio (Random, 95% CI) | 0.76 [0.67, 0.86] | |
| 1.3 Third‐line | 2 | Hazard Ratio (Random, 95% CI) | 0.43 [0.35, 0.54] | |
| 2 Overall survival | 12 | Hazard Ratio (Random, 95% CI) | 0.88 [0.80, 0.98] | |
| 2.1 First‐line | 6 | Hazard Ratio (Random, 95% CI) | 0.87 [0.75, 1.02] | |
| 2.2 Second‐line | 4 | Hazard Ratio (Random, 95% CI) | 0.93 [0.82, 1.05] | |
| 2.3 Third‐line | 2 | Hazard Ratio (Random, 95% CI) | 0.79 [0.50, 1.24] | |
| 3 Tumour response rate | 12 | 4147 | Odds Ratio (M‐H, Random, 95% CI) | 2.41 [1.70, 3.41] |
| 3.1 First‐line | 6 | 2447 | Odds Ratio (M‐H, Random, 95% CI) | 1.73 [1.33, 2.25] |
| 3.2 Second‐line | 4 | 1243 | Odds Ratio (M‐H, Random, 95% CI) | 3.60 [2.45, 5.30] |
| 3.3 Third‐line | 2 | 457 | Odds Ratio (M‐H, Random, 95% CI) | 38.44 [5.22, 282.91] |
| 4 Grade 3/4 toxicity | 6 | 2771 | Odds Ratio (M‐H, Random, 95% CI) | 2.45 [2.07, 2.89] |
| 4.1 First‐line | 3 | 1495 | Odds Ratio (M‐H, Random, 95% CI) | 2.55 [2.01, 3.25] |
| 4.2 Second‐line | 2 | 1033 | Odds Ratio (M‐H, Random, 95% CI) | 2.42 [1.88, 3.13] |
| 4.3 Third‐line | 1 | 243 | Odds Ratio (M‐H, Random, 95% CI) | 2.08 [1.22, 3.55] |
| 5 Grade 3/4 diarrhoea | 7 | 2909 | Odds Ratio (M‐H, Random, 95% CI) | 1.84 [1.47, 2.32] |
| 5.1 First‐line | 4 | 1633 | Odds Ratio (M‐H, Random, 95% CI) | 1.95 [1.43, 2.67] |
| 5.2 Second‐line | 2 | 1033 | Odds Ratio (M‐H, Random, 95% CI) | 1.70 [1.21, 2.38] |
| 5.3 Third‐line | 1 | 243 | Odds Ratio (M‐H, Random, 95% CI) | 4.88 [0.23, 102.66] |
| 6 Grade 3/4 rash | 7 | 2909 | Odds Ratio (M‐H, Random, 95% CI) | 23.42 [13.22, 41.49] |
| 6.1 First‐line | 4 | 1633 | Odds Ratio (M‐H, Random, 95% CI) | 20.29 [5.99, 68.67] |
| 6.2 Second‐line | 2 | 1033 | Odds Ratio (M‐H, Random, 95% CI) | 24.74 [11.61, 52.72] |
| 6.3 Third‐line | 1 | 243 | Odds Ratio (M‐H, Random, 95% CI) | 80.52 [4.86, 1333.13] |
| 7 Grade 3/4 neutropenia | 6 | 2666 | Odds Ratio (M‐H, Random, 95% CI) | 1.22 [0.93, 1.61] |
| 7.1 First‐line | 4 | 1633 | Odds Ratio (M‐H, Random, 95% CI) | 1.20 [0.97, 1.49] |
| 7.2 Second‐line | 2 | 1033 | Odds Ratio (M‐H, Random, 95% CI) | 1.30 [0.49, 3.43] |
| 7.3 Third‐line | 0 | 0 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 2. EGFR MAb in KRAS exon 2 MT.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Progression‐free survival | 8 | Hazard Ratio (Random, 95% CI) | 1.03 [0.89, 1.20] | |
| 1.1 First‐line | 5 | Hazard Ratio (Random, 95% CI) | 1.11 [0.88, 1.38] | |
| 1.2 Second‐line | 1 | Hazard Ratio (Random, 95% CI) | 0.85 [0.68, 1.06] | |
| 1.3 Third‐line | 2 | Hazard Ratio (Random, 95% CI) | 0.99 [0.80, 1.24] | |
| 2 Overall survival | 8 | Hazard Ratio (Random, 95% CI) | 1.03 [0.94, 1.13] | |
| 2.1 First‐line | 5 | Hazard Ratio (Random, 95% CI) | 1.07 [0.96, 1.20] | |
| 2.2 Second‐line | 1 | Hazard Ratio (Random, 95% CI) | 0.93 [0.76, 1.15] | |
| 2.3 Third‐line | 2 | Hazard Ratio (Random, 95% CI) | 0.98 [0.80, 1.21] | |
| 3 Tumour response rate | 8 | 1925 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.74, 1.16] |
| 3.1 First‐line | 4 | 1066 | Odds Ratio (M‐H, Random, 95% CI) | 0.90 [0.66, 1.22] |
| 3.2 Second‐line | 1 | 469 | Odds Ratio (M‐H, Random, 95% CI) | 0.92 [0.54, 1.56] |
| 3.3 Third‐line | 3 | 390 | Odds Ratio (M‐H, Random, 95% CI) | 2.07 [0.50, 8.56] |
| 4 Overall grade 3/4 toxicity | 5 | 1635 | Odds Ratio (M‐H, Random, 95% CI) | 1.63 [0.98, 2.71] |
| 4.1 First‐line | 3 | 968 | Odds Ratio (M‐H, Random, 95% CI) | 1.35 [0.77, 2.38] |
| 4.2 Second‐line | 1 | 483 | Odds Ratio (M‐H, Random, 95% CI) | 1.76 [1.22, 2.53] |
| 4.3 Third‐line | 1 | 184 | Odds Ratio (M‐H, Random, 95% CI) | 81.40 [4.86, 1362.93] |
| 5 Grade 3/4 diarrhoea | 5 | 1635 | Odds Ratio (M‐H, Random, 95% CI) | 1.45 [1.01, 2.11] |
| 5.1 First‐line | 3 | 968 | Odds Ratio (M‐H, Random, 95% CI) | 1.41 [0.76, 2.62] |
| 5.2 Second‐line | 1 | 483 | Odds Ratio (M‐H, Random, 95% CI) | 1.32 [0.76, 2.29] |
| 5.3 Third‐line | 1 | 184 | Odds Ratio (M‐H, Random, 95% CI) | 3.61 [0.15, 89.81] |
| 6 Grade 3/4 rash | 5 | 1635 | Odds Ratio (M‐H, Random, 95% CI) | 32.35 [15.01, 69.70] |
| 6.1 First‐line | 3 | 968 | Odds Ratio (M‐H, Random, 95% CI) | 24.42 [8.16, 73.09] |
| 6.2 Second‐line | 1 | 483 | Odds Ratio (M‐H, Random, 95% CI) | 56.48 [13.68, 233.26] |
| 6.3 Third‐line | 1 | 184 | Odds Ratio (M‐H, Random, 95% CI) | 31.45 [1.82, 542.25] |
| 7 Grade 3/4 neutropenia | 3 | 968 | Odds Ratio (M‐H, Random, 95% CI) | 0.70 [0.53, 0.93] |
| 7.1 First‐line | 3 | 968 | Odds Ratio (M‐H, Random, 95% CI) | 0.70 [0.53, 0.93] |
| 7.2 Second‐line | 0 | 0 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 7.3 Third‐line | 0 | 0 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 3. EGFR MAb in extended RAS WT.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Progression‐free survival | 6 | Hazard Ratio (Random, 95% CI) | 0.60 [0.48, 0.75] | |
| 1.1 First‐line | 3 | Hazard Ratio (Random, 95% CI) | 0.65 [0.54, 0.78] | |
| 1.2 Second‐line | 2 | Hazard Ratio (Random, 95% CI) | 0.72 [0.58, 0.91] | |
| 1.3 Third‐line | 1 | Hazard Ratio (Random, 95% CI) | 0.36 [0.25, 0.52] | |
| 2 Overall survival | 4 | Hazard Ratio (Random, 95% CI) | 0.77 [0.67, 0.88] | |
| 2.1 First‐line | 3 | Hazard Ratio (Random, 95% CI) | 0.75 [0.64, 0.89] | |
| 2.2 Second‐line | 1 | Hazard Ratio (Random, 95% CI) | 0.81 [0.63, 1.03] | |
| 2.3 Third‐line | 0 | Hazard Ratio (Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Tumour response rate | 4 | 1001 | Odds Ratio (M‐H, Random, 95% CI) | 4.28 [2.61, 7.03] |
| 3.1 First‐line | 2 | 454 | Odds Ratio (M‐H, Random, 95% CI) | 3.18 [2.16, 4.68] |
| 3.2 Second‐line | 1 | 411 | Odds Ratio (M‐H, Random, 95% CI) | 6.08 [3.57, 10.33] |
| 3.3 Third‐line | 1 | 136 | Odds Ratio (M‐H, Random, 95% CI) | 25.81 [1.50, 445.52] |
Comparison 4. EGFR MAb in any RAS mutation.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Progression‐free survival | 6 | Hazard Ratio (Random, 95% CI) | 1.13 [0.93, 1.36] | |
| 1.1 First‐line | 3 | Hazard Ratio (Random, 95% CI) | 1.27 [1.08, 1.48] | |
| 1.2 Second‐line | 2 | Hazard Ratio (Random, 95% CI) | 1.05 [0.62, 1.79] | |
| 1.3 Third‐line | 1 | Hazard Ratio (Random, 95% CI) | 0.97 [0.73, 1.29] | |
| 2 Overall survival | 4 | Hazard Ratio (Random, 95% CI) | 1.09 [0.93, 1.28] | |
| 2.1 First‐line | 3 | Hazard Ratio (Random, 95% CI) | 1.16 [1.02, 1.33] | |
| 2.2 Second‐line | 1 | Hazard Ratio (Random, 95% CI) | 0.91 [0.76, 1.10] | |
| 2.3 Third‐line | 0 | Hazard Ratio (Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Tumour response rate | 3 | 840 | Odds Ratio (M‐H, Random, 95% CI) | 0.76 [0.55, 1.05] |
| 3.1 First‐line | 2 | 627 | Odds Ratio (M‐H, Random, 95% CI) | 0.74 [0.54, 1.03] |
| 3.2 Second‐line | 0 | 0 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.3 Third‐line | 1 | 213 | Odds Ratio (M‐H, Random, 95% CI) | 3.49 [0.14, 86.58] |
Comparison 5. EGFR inhibitors in KRAS unselected participants.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Progression‐free survival | 2 | Hazard Ratio (Fixed, 95% CI) | 0.70 [0.62, 0.78] | |
| 2 Overall survival | 2 | Hazard Ratio (Fixed, 95% CI) | 0.96 [0.84, 1.09] | |
| 3 Tumour response rate | 2 | 1372 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.70 [2.49, 5.49] |
| 4 Overall grade 3/4 toxicity | 1 | 1267 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.12 [1.69, 2.65] |
| 5 Grade 3/4 diarrhoea | 2 | 1341 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.08 [1.59, 2.71] |
| 6 Grade 3/4 rash | 2 | 1341 | Odds Ratio (M‐H, Fixed, 95% CI) | 39.89 [7.82, 203.35] |
| 7 Grade 3/4 neutropenia | 2 | 1341 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.39 [1.08, 1.78] |
5.3. Analysis.
Comparison 5 EGFR inhibitors in KRAS unselected participants, Outcome 3 Tumour response rate.
5.4. Analysis.
Comparison 5 EGFR inhibitors in KRAS unselected participants, Outcome 4 Overall grade 3/4 toxicity.
5.5. Analysis.
Comparison 5 EGFR inhibitors in KRAS unselected participants, Outcome 5 Grade 3/4 diarrhoea.
5.6. Analysis.
Comparison 5 EGFR inhibitors in KRAS unselected participants, Outcome 6 Grade 3/4 rash.
5.7. Analysis.
Comparison 5 EGFR inhibitors in KRAS unselected participants, Outcome 7 Grade 3/4 neutropenia.
Comparison 6. Comparing addition of EGFR MAb to chemotherapy with anti‐VEGF MAb and the same chemotherapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Progression‐free survival | 4 | Hazard Ratio (Random, 95% CI) | 1.02 [0.93, 1.12] | |
| 2 Overall survival | 4 | Hazard Ratio (Random, 95% CI) | 0.84 [0.70, 1.01] | |
| 3 Tumour response rate | 4 | 2184 | Odds Ratio (M‐H, Random, 95% CI) | 1.36 [1.15, 1.62] |
| 4 Overall grade 3/4 toxicity | 4 | 2133 | Odds Ratio (M‐H, Random, 95% CI) | 1.37 [1.09, 1.72] |
| 5 Grade 3/4 diarrhoea | 2 | 1673 | Odds Ratio (M‐H, Random, 95% CI) | 1.06 [0.67, 1.67] |
| 6 Grade 3/4 rash | 3 | 1951 | Odds Ratio (M‐H, Random, 95% CI) | 47.53 [14.84, 152.19] |
Comparison 7. EGFR TKI.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Progression‐free survival | 2 | Hazard Ratio (Fixed, 95% CI) | 0.87 [0.72, 1.04] | |
| 2 Overall survival | 2 | Hazard Ratio (Fixed, 95% CI) | 0.80 [0.68, 0.96] | |
| 3 Tumour response rate | 1 | 99 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.41, 1.97] |
| 4 Overall grade 3/4 toxicity | 1 | 99 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.01 [0.89, 4.56] |
| 5 Grade 3/4 diarrhoea | 2 | 181 | Odds Ratio (M‐H, Fixed, 95% CI) | 12.68 [3.71, 43.35] |
| 6 Grade 3/4 rash | 1 | 99 | Odds Ratio (M‐H, Fixed, 95% CI) | 5.11 [0.57, 45.43] |
| 7 Grade 3/4 neutropenia | 1 | 99 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.83 [0.76, 4.44] |
7.1. Analysis.
Comparison 7 EGFR TKI, Outcome 1 Progression‐free survival.
7.2. Analysis.
Comparison 7 EGFR TKI, Outcome 2 Overall survival.
7.3. Analysis.
Comparison 7 EGFR TKI, Outcome 3 Tumour response rate.
7.4. Analysis.
Comparison 7 EGFR TKI, Outcome 4 Overall grade 3/4 toxicity.
7.5. Analysis.
Comparison 7 EGFR TKI, Outcome 5 Grade 3/4 diarrhoea.
7.6. Analysis.
Comparison 7 EGFR TKI, Outcome 6 Grade 3/4 rash.
7.7. Analysis.
Comparison 7 EGFR TKI, Outcome 7 Grade 3/4 neutropenia.
Comparison 8. EGFR inhibitors added to bevacizumab.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Progression‐free survival | 6 | Hazard Ratio (Random, 95% CI) | 1.04 [0.83, 1.29] | |
| 2 Overall survival | 5 | Hazard Ratio (Random, 95% CI) | 1.00 [0.69, 1.47] | |
| 3 Tumour response rate | 4 | 1310 | Odds Ratio (M‐H, Random, 95% CI) | 1.20 [0.67, 2.12] |
| 4 Overall grade 3/4 toxicity | 3 | 1831 | Odds Ratio (M‐H, Random, 95% CI) | 2.57 [1.45, 4.57] |
| 5 Grade 3/4 diarrhoea | 5 | 2434 | Odds Ratio (M‐H, Random, 95% CI) | 2.58 [1.44, 4.64] |
| 6 Grade 3/4 rash | 4 | 2363 | Odds Ratio (M‐H, Random, 95% CI) | 67.52 [30.83, 147.85] |
| 7 Grade 3/4 neutropenia | 2 | 1187 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.73, 1.29] |
8.4. Analysis.
Comparison 8 EGFR inhibitors added to bevacizumab, Outcome 4 Overall grade 3/4 toxicity.
8.7. Analysis.
Comparison 8 EGFR inhibitors added to bevacizumab, Outcome 7 Grade 3/4 neutropenia.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Adams COIN 2011.
| Methods | Phase III open‐label RCT; n = 1630 | |
| Participants | Advanced colorectal cancer, first‐line therapy | |
| Interventions | Arm A: mFOLFOX6 or CAPOX. Arm B: mFOLFOX6 with cetuximab or CAPOX with cetuximab. Arm C: intermittent mFOLFOX6 or CAPOX. Arm C excluded from analysis given comparisons would not yield information regarding efficacy of EGFR MAb. |
|
| Outcomes | Primary outcome: OS in participants with KRAS exon 2 WT tumours. Secondary outcomes: subgroup analyses for OS for KRAS/NRAS/BRAF status, OS for all participants, PFS, ORR, toxic effects. |
|
| Notes | Sponsored by MRC. TSM and RAA have received travel, accommodation, and lecture fees from Roche and Merck Serono. Median follow‐up 21 months in control arm, 23 months in cetuximab arm | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central telephone randomisation was done by the MRC Clinical Trials Unit, using the method of minimisation with a random element. The minimisation factors were hospital, WHO performance status, chemotherapy regimen, previous adjuvant chemotherapy, liver metastases, and peritoneal metastases. Participants were randomly assigned (1:1:1). |
| Allocation concealment (selection bias) | Low risk | Centralised randomisation after faxing details to central site (COIN protocol from MRC website) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Open‐label trial (but see below) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Open‐label trial. Participant symptoms were assessed by investigators throughout treatment but primary outcome (OS) is not affected by open‐label nature. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Equal dropout: in KRAS exon 2 WT population 33/358 in arm A, 26/357 arm B. Intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | All stated outcomes reported. |
| Other bias | Low risk | No other significant bias present. |
Amado 2008.
| Methods | Phase III open‐label RCT; n = 572 | |
| Participants | Advanced colorectal cancer; prior treatment with fluorouracil, irinotecan, and oxaliplatin | |
| Interventions | Panitumumab vs best supportive care | |
| Outcomes | Primary endpoint: OS. Secondary endpoints: PFS, TRR, QoL (EORTC QLQ‐C30), safety | |
| Notes | Funded by Amgen. Median follow‐up 14.1 months for participants still alive. Amado: employment/leadership position (Amgen), stock ownership (Amgen). Chang: employment/leadership position (Amgen), stock ownership (Amgen) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available |
| Allocation concealment (selection bias) | Unclear risk | No information available |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Open label, but outcome assessment blinded (see below). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Tumour response was assessed by the investigator and by an independent central radiology review blinded to treatment and outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 91% had available KRAS tumour status results. |
| Selective reporting (reporting bias) | Low risk | All stated outcomes reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Bokemeyer OPUS 2009.
| Methods | Phase II open‐label RCT; n = 338 | |
| Participants | Nonresectable metastatic CRC | |
| Interventions | FOLFOX4 with cetuximab versus FOLFOX4 | |
| Outcomes | Primary endpoint: tumour response rate. Secondary endpoints: exploratory only | |
| Notes | Funded by Merck KGaA. Funding: Bokemeyer: consultant/advisory role (Merck Serono), honoraria (Merck Serono, Sanofi‐Aventis), research funding (Merck Serono); Koralewski: none declared. Follow‐up: 44.1 months (cetuximab arm), 31.8 months (standard arm) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation (1:1) was carried out using a stratified permuted‐block procedure, with ECOG PS (0 and 1 vs 2) as a stratification factor. |
| Allocation concealment (selection bias) | Unclear risk | No details provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Open‐label trial (but blinded review for primary outcome; see below) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The independent review committee conducted a blinded review of images and clinical data using a common set of prespecified criteria. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1/170 dropout in experimental arm, 0/168 in control arm |
| Selective reporting (reporting bias) | Low risk | All stated outcomes reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Borner 2008.
| Methods | Phase II RCT; n = 74 | |
| Participants | Advanced/metastatic unresectable adenocarcinoma of colon or rectum, no prior chemotherapy | |
| Interventions | XELOX with cetuximab versus XELOX | |
| Outcomes | Primary outcome: objective response rate. No formal statistical comparisons planned. | |
| Notes | Funded by Merck, Sanofi‐Aventis, Swiss government. Follow‐up 17.2 months | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomized" ‐ no further information given |
| Allocation concealment (selection bias) | Unclear risk | "Randomized" ‐ no further information given |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Unclear as to blinding, but blinded review of primary outcome (see below) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | An independent response review was conducted by 2 radiologists and 1 medical oncologist. The reviewers were blinded to the treatment arm and to the investigator's initial assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropout |
| Selective reporting (reporting bias) | Low risk | Declared outcomes reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Bridgewater GAIN‐C 2015.
| Methods | Phase II RCT; n = 169 | |
| Participants | People with mCRC ‐ progression post‐first‐line oxaliplatin‐containing chemotherapy | |
| Interventions | KRAS exon 2 WT patients received either FOLFIRI with cetuximab or the combination of FOLFIRI and imgatuzumab (GA201). KRAS exon 2 MT patients received either FOLFIRI with GA201 or FOLFIRI alone. | |
| Outcomes | Primary endpoint: PFS. Secondary endpoint: response rate, duration of response, clinical benefit rate, OS, safety profile, pharmacokinetics, pharmacodynamics | |
| Notes | Sponsored by Roche. Duration of follow‐up not specified. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Abstract only |
| Allocation concealment (selection bias) | Unclear risk | Abstract only |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Abstract only |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Abstract only |
| Selective reporting (reporting bias) | Unclear risk | Abstract only |
| Other bias | Unclear risk | Abstract only |
Brodowicz 2013.
| Methods | Phase II RCT; n = 152 | |
| Participants | People with KRAS exon 2 WT non‐resectable mCRC, no prior treatment | |
| Interventions | Arm A: FOLFOX4 with cetuximab weekly. Arm B: FOLFOX4 with cetuximab fortnightly | |
| Outcomes | Primary endpoint: objective response rate. Secondary endpoints: progression‐free survival, overall survival, disease control rate, and safety | |
| Notes | Sponsored by CECOG; CECOG received a medical grant/cetuximab from Merck. Follow‐up not stated. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised 1:1 stratified by study site, number of organs involved, and prior adjuvant or neoadjuvant therapy |
| Allocation concealment (selection bias) | Unclear risk | No details given. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Best overall tumour response and PFS rates were assessed by the investigator using RECIST version 1. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Equal dropout |
| Selective reporting (reporting bias) | Low risk | Declared outcomes reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Ciardiello CAPRI‐GOIM 2016.
| Methods | Randomised controlled trial | |
| Participants | 340 people with KRAS exon 2 WT mCRC | |
| Interventions | All participants received first‐line FOLFIRI with cetuximab, then proceeded on progression to FOLFOX (exact FOLFOX regimen not specified) or FOLFOX with cetuximab. | |
| Outcomes | ORR, PFS | |
| Notes | Funding: Gruppo Oncologico dell' Italia Meridionale and Merck Serono. Ciardiello: advisory boards for Merck Serono, Roche, Bayer, Lilly, Sanofi. Median follow‐up 35.3 months | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | After progression from first‐line therapy, the GOIM Clinical Trials Unit randomised the participants centrally, using the method of minimisation with a random element. |
| Allocation concealment (selection bias) | Low risk | After progression from first‐line therapy, the GOIM Clinical Trials Unit randomised the participants centrally, using the method of minimisation with a random element. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label study |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Details not available at time of review; authors being contacted for protocol |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 2/153 lost to follow‐up |
| Selective reporting (reporting bias) | Low risk | All planned endpoints reported. |
| Other bias | Unclear risk | Abstract only |
Douillard PRIME 2010.
| Methods | Phase III open‐label RCT; n = 1183 | |
| Participants | Metastatic colorectal adenocarcinoma, no prior chemotherapy | |
| Interventions | FOLFOX4 with panitumumab vs FOLFOX4 | |
| Outcomes | Primary outcome: PFS. Secondary outcome: OS | |
| Notes | Funding: Amgen. Douillard: consultant/advisory role (Amgen, Sanofi‐Aventis, Merck Serono), honoraria (Amgen, Sanofi‐Aventis). Gansert: employment/leadership position (Amgen), stock ownership (Amgen) Follow‐up 13.2 months FOLFOX4 with panitumumab, 12.5 months FOLFOX4 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned 1:1 to receive either panitumumab‐FOLFOX4 or FOLFOX4. Random assignment was stratified by geographic region (Western Europe, Canada, and Australia vs rest of the world) and ECOG PS (0 or 1 vs 2). |
| Allocation concealment (selection bias) | Low risk | No details in paper, but no significant baseline imbalance |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Open label (but blinded review of primary endpoint; see below) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Objective tumour response was evaluated by blinded central radiology review. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No dropout rate reported. Results available for 93% participants. |
| Selective reporting (reporting bias) | Low risk | All stated outcomes reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Hagman ACT2 2014.
| Methods | Phase III randomised, open‐label trial | |
| Participants | People with mCRC | |
| Interventions | All participants received first‐line XELOX/XELIRI/mFOLFOX6/FOLFIRI with bevacizumab as well as KRAS testing; those with stable disease or better after 18 weeks were randomised ‐ if KRAS exon 2 WT, to maintenance bevacizumab and erlotinib; if KRAS exon 2 MT, to bevacizumab or continuous capecitabine (500 mg twice daily). | |
| Outcomes | Primary endpoint: PFS at 3 months. Secondary endpoints: PFS, OS, toxicity | |
| Notes | Investigator‐sponsored trial, supported by Roche, Skane County Council, Sweden, Futurm, and John and Augusta Persson's Trust, Lund, Sweden. AJ: honoraria from Genentech. Median follow‐up 34.5 months | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Block randomisation was used (2 arms, 2 strata = 4 strata groups), each block 4 participants, double block size. |
| Allocation concealment (selection bias) | Low risk | Allocation concealment was ensured as randomisation procedure was conducted by a central co‐ordinated randomising service provided by the regional cancer centre in Skane/Lund, Sweden. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Whilst assessment of tumour response was undertaken using RECIST criteria, measurement of lesions was performed in an open‐label fashion by local radiologists at the participating site. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Not stated, but only 2/233 participants withdrew consent. As 184/233 participants had documented death by time of publication, we feel that there is low risk of attrition bias. |
| Selective reporting (reporting bias) | Low risk | All endpoints reported. |
| Other bias | Unclear risk | Hagman and Johnsson: No declared conflicts of interest. |
Hecht PACCE 2009.
| Methods | Phase III open‐label RCT; n = 1043 | |
| Participants | People with mCRC, no prior chemotherapy | |
| Interventions | Chemotherapy (oxaliplatin‐ or irinotecan‐based) plus bevacizumab plus panitumumab vs chemotherapy plus bevacizumab | |
| Outcomes | Primary outcome: PFS. Secondary outcomes: TRR, OS, safety | |
| Notes | Supported by Amgen; Hecht: nil declared. Amado: employment/leadership position (Amgen), stock ownership (Amgen) Median follow‐up 12.3 months oxaliplatin‐based chemotherapy; 9.0 months irinotecan‐based chemotherapy |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | No details given (but blinded central review of primary endpoint). |
| Allocation concealment (selection bias) | Low risk | No details given (but blinded central review of primary endpoint and no baseline imbalance). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Open‐label study, but blinded review of PFS as primary endpoint |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Central review censoring was based on the last available scan read centrally; local review censoring was based on the last day of participant contact or visit without known disease progression. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Composite of attrition, pregnancy, or other: 18/528 in intervention arm and 17/525 in control arm (Table A1, JCO 2008 appendix) |
| Selective reporting (reporting bias) | Low risk | All stated outcomes reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Hecht SPIRITT 2015.
| Methods | Phase III RCT; n = 182 | |
| Participants | People with KRAS exon 2 WT mCRC, prior treatment with first‐line chemotherapy containing oxaliplatin and bevacizumab | |
| Interventions | FOLFIRI with panitumumab vs FOLFIRI | |
| Outcomes | Primary outcome: PFS. Secondary outcomes: OS, TRR, safety | |
| Notes | Supported by Amgen; follow‐up not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details regarding sequence generation in publication or protocol |
| Allocation concealment (selection bias) | Unclear risk | No details regarding allocation concealment in publication or protocol |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Open‐label (but blinded assessment of endpoints ‐ see below) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessments based on blinded central radiology review per modified RECIST 1.0 |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participants lost to follow‐up |
| Selective reporting (reporting bias) | Low risk | All endpoints reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Heinemann FIRE‐3 2014.
| Methods | Phase III multicentre RCT; n = 592 | |
| Participants | People with mCRC, KRAS exon 2 WT | |
| Interventions | FOLFIRI with cetuximab vs FOLFIRI with bevacizumab | |
| Outcomes | Primary outcome: ORR. Secondary outcomes: OS, PFS, toxicity, secondary resection rate with curative intent | |
| Notes | Study funded by Merck KGaA. Heinemann: financial grants to undertake study and prepare manuscript (Merck KGaA), honoraria (Merck), financial grants to undertake clinical studies, honoraria, advisory boards (Roche, Amgen, Sanofi). Stintzing: personal fees (Merck KGaA, Roche AG, Amgen GmbH, Sanofi‐Aventis). Median follow‐up 33 months in cetuximab arm, 39 months in bevacizumab arm |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was done "using permuted blocks of randomly varying size". |
| Allocation concealment (selection bias) | Low risk | "done centrally via fax" |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Assessments based on blinded central radiology review per modified RECIST 1. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Significant but balanced rate of "not assessable for response ‐ other reasons" in intention‐to‐treat population (28/297 FOLFIRI with cetuximab arm, 20/295 FOLFIRI with bevacizumab arm) |
| Selective reporting (reporting bias) | Low risk | All findings reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Hickish 2014.
| Methods | Phase II multicentre, open‐label RCT; n = 94 | |
| Participants | People with mCRC, stratified by KRAS status | |
| Interventions | Afatinib (Arm A) vs cetuximab (Arm B) if KRAS exon 2 WT (n = 51); afatinib (single arm) if KRAS exon 2 MT (n = 43) | |
| Outcomes | Primary outcomes: response rate in arms A and B, disease control rate in arm C. Secondary outcomes: PFS, OS | |
| Notes | Funded by Boehringer Ingelheim. Hickish: no COI; Harrison: no COI Follow‐up not stated. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Open label ‐ no further details |
| Allocation concealment (selection bias) | Unclear risk | Open label ‐ no further details |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open label ‐ no further details |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Investigator‐assessed primary endpoint (response rate); no mention of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Dropout for primary endpoint 7/36 (Arm A), 5/15 (Arm B) ‐ in majority as participants "progressed rapidly after randomisation and did not have a follow‐up scan" |
| Selective reporting (reporting bias) | Low risk | All stated outcomes reported. |
| Other bias | Unclear risk | No other significant bias present; funders did not have inappropriate influence. |
Johnsson Nordic ACT 2013.
| Methods | Phase III open‐label RCT; n = 249 | |
| Participants | People with mCRC after finishing first‐line treatment with combined chemotherapy and bevacizumab | |
| Interventions | Combined erlotinib and bevacizumab vs bevacizumab | |
| Outcomes | Primary outcome: PFS. Secondary outcomes: OS, toxicity | |
| Notes | Supported by Roche Sweden, Futurum ‐ the Academy for Healthcare, Jonkoping County Council, and by the Skane Regional Council. Johnsson: presentation honoraria from Genentech. Follow‐up 36.8 months |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised 1:1 ‐ no further information |
| Allocation concealment (selection bias) | Unclear risk | No further information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No independent radiology review was done. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Primary outcome reported when 131/159 participants progressed; 6 participants (4 interventional, 2 control arms) withdrawn due to aim for curative surgery. |
| Selective reporting (reporting bias) | Low risk | All outcomes reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Karapetis CO17 2008.
| Methods | Phase III RCT; n = 572 | |
| Participants | People with mCRC, prior treatment with fluoropyrimidine, irinotecan, and oxaliplatin | |
| Interventions | Cetuximab vs best supportive care | |
| Outcomes | Primary outcome: OS. Secondary outcomes: PFS, TRR, quality of life | |
| Notes | Supported by National Cancer Institute of Canada, ImClone Systems, and Bristol‐Myers Squibb. Karapetis: consulting fees (Merck Serono), Zalcberg: research grants (Amgen, Merck Serono, Bristol‐Myers Squibb, Alphapharm) Median follow‐up 14.6 months |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomisation was performed by the NCIC CTG central office with the use of a minimisation method that dynamically balanced participants according to stratification factors." |
| Allocation concealment (selection bias) | Low risk | "Randomisation was performed by the NCIC CTG central office with the use of a minimisation method that dynamically balanced participants according to stratification factors." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Open‐label trial ‐ but OS as primary endpoint would not be affected. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assays of tissue samples for KRAS mutations were performed in a blinded fashion by members of the Department of Clinical Biomarkers–Oncology at Bristol‐Myers Squibb, Hopewell, New Jersey. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 6/285 participants in supportive care‐alone arm immediately withdrew their consent. No dropout in cetuximab group reported (although 4/287 never received cetuximab, analyses were intention‐to‐treat). |
| Selective reporting (reporting bias) | Low risk | All stated outcomes reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Ma 2013.
| Methods | Phase II RCT; n = 60 | |
| Participants | Inoperable metastatic colorectal cancer | |
| Interventions | The combination of continuous erlotinib and modified CAPOX (Arm A) versus the combination of intermittent erlotinib and modified CAPOX (Arm B) | |
| Outcomes | Primary endpoint: ORR. Secondary endpoints: OS and PFS | |
| Notes | Investigator‐initiated trial with departmental funding. Hoffmann‐La Roche (capecitabine, erlotinib) and Sanofi (oxaliplatin) provided drugs. Median follow‐up 2.8 years | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised 1:1 via central computer |
| Allocation concealment (selection bias) | Low risk | Central allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Assessed using RECIST criteria; no specification of blinding or otherwise |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participants withdrew. |
| Selective reporting (reporting bias) | Low risk | All stated outcomes reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Passardi ITACA 2015.
| Methods | Phase III randomised, multicentre study | |
| Participants | 350 | |
| Interventions | Drug: Arm A: FOLFIRI or FOLFOX4 with bevacizumab Drug: Arm B: FOLFIRI or FOLFOX4 Drug: Arm D: FOLFIRI or FOLFOX4 with cetuximab Drug: Arm F: FOLFIRI or FOLFOX4 with bevacizumab and cetuximab |
|
| Outcomes | Primary outcome measures: PFS Secondary outcome measures: ORR, OS, safety and tolerability |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Presented in abstract form only |
| Allocation concealment (selection bias) | Unclear risk | Presented in abstract form only |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Presented in abstract form only |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Presented in abstract form only |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Presented in abstract form only |
| Selective reporting (reporting bias) | Unclear risk | Presented in abstract form only |
| Other bias | Unclear risk | Presented in abstract form only |
Peeters 2010.
| Methods | Phase III open‐label RCT; n = 1186 | |
| Participants | People with mCRC, with 1 prior chemotherapy regimen | |
| Interventions | FOLFIRI with panitumumab vs FOLFIRI | |
| Outcomes | Primary outcomes: PFS, OS. Secondary outcomes: TRR, duration of response, safety | |
| Notes | Supported by Amgen. Peeters: consultancy (Amgen), honoraria (Amgen), research funding (Amgen). Gansert: employment/leadership (Amgen), stock ownership (Amgen). Median follow‐up time was 13.3 months (range 0.2 to 31.7 months) in the KRAS exon 2 WT panitumumab‐FOLFIRI arm, 10.2 months (range 0.5 to 32.9 months) in the KRAS exon 2 WT FOLFIRI arm, 10.5 months (range 0.2 to 30.1 months) in the KRAS exon 2 MT panitumumab‐FOLFIRI arm, and 9.5 months (range 0 to 31.7 months) in the KRAS exon 2 MT FOLFIRI arm. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details specified. |
| Allocation concealment (selection bias) | Unclear risk | No details specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Open label (but assessment of endpoints blinded for PFS and not affected for OS) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Tumour response was assessed by the investigator and by an independent central radiology review blinded to treatment and outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No attrition data available, but PFS occurred in 178/303 (59%) of participants in the FOLFIRI with panitumumab arm and 203/294 (69%) of participants in the FOLFIRI arm, with median follow‐up 13.3 months. |
| Selective reporting (reporting bias) | Low risk | All stated outcomes reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Polikoff EXPLORE 2005.
| Methods | Phase III randomised, open‐label, multicentre study | |
| Participants | 100 (early termination; initial plan for 1100) | |
| Interventions | FOLFOX4 with cetuximab vs FOLFOX4 | |
| Outcomes | Primary outcome: OS. Secondary outcomes: PFS, response rate, safety | |
| Notes | Sponsored by Bristol‐Myers Squibb. Langer: owns equity in and employed by Bristol‐Myers Squibb. Follow‐up not stated. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Abstract only |
| Allocation concealment (selection bias) | Unclear risk | Abstract only |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Abstract only |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Abstract only |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Abstract only |
| Selective reporting (reporting bias) | Unclear risk | Abstract only |
| Other bias | Unclear risk | Abstract only |
Price ASPECCT 2014.
| Methods | Phase III open‐label, multicentre RCT; n = 1010 | |
| Participants | People with chemotherapy‐refractory mCRC, KRAS exon 2 WT | |
| Interventions | Panitumumab vs cetuximab | |
| Outcomes | Primary endpoint: overall survival. Secondary endpoints: PFS, TRR, time to treatment failure, time to response, duration of response, safety | |
| Notes | Study funded by Amgen. Price: advisory for Amgen. Sidhu: employee/stockholder of Amgen. Median follow‐up time, defined as the time from randomisation to the last on‐study or long‐term follow‐up visit, was 41.4 weeks (22.1 to 71.6) for panitumumab and 40.5 weeks (21.3 to 68.9) for cetuximab. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Using an automated interactive voice response system (ICON Clinical Research, Dublin, Ireland), we randomly assigned patients (1:1) to either panitumumab or cetuximab treatment. Randomisation was done using a permuted block method and was stratified by geographical region (North America, western Europe, and Australia vs rest of the world) and ECOG performance status (0 or 1 vs 2)." |
| Allocation concealment (selection bias) | Low risk | Automated interactive voice response system |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and investigators were not masked to treatment assignment (open‐label treatment) ‐ but note overall survival as primary endpoint. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Participants and investigators were not masked to treatment assignment (open‐label treatment) ‐ but note overall survival as primary endpoint. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 102/1010 participants lost to follow‐up or withdrew consent |
| Selective reporting (reporting bias) | Low risk | All stated outcomes reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Santoro 2008.
| Methods | Phase II multicentre, open‐label RCT | |
| Participants | mCRC, no prior chemotherapy for metastatic disease | |
| Interventions | Arm A: the combination of FOLFIRI and gefitinib. Arm B: FOLFIRI alone | |
| Outcomes | Primary outcome: TRR. No secondary outcomes specified. | |
| Notes | Designed as a non‐comparative parallel‐group trial. Sponsored by AstraZeneca. Median follow‐up 14.5 months | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised ‐ no further details |
| Allocation concealment (selection bias) | Unclear risk | No further details |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No specification of central or blinded assessment of imaging |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis for tumour response; withdrawal prior to this evaluation 2/48 in FOLFIRI arm, 1/51 in combination arm. Subsequent withdrawal (other than progressive disease) rates 12 (25%) in FOLFIRI arm and 18 (35.3%) in combination arm. Adverse events main reason: 6 (12.5%) in FOLFIRI arm, 15 (29.4%) in combination arm |
| Selective reporting (reporting bias) | Low risk | TRR reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Schwartzberg PEAK 2014.
| Methods | Phase II open‐label, multicentre RCT; n = 285 | |
| Participants | People with mCRC, KRAS exon 2 WT, no prior treatment for metastatic disease | |
| Interventions | mFOLFOX6 with panitumumab vs mFOLFOX6 with bevacizumab | |
| Outcomes | Primary outcome: PFS. Secondary outcomes: OS, safety | |
| Notes | Sponsored by Amgen. Schwartzberg: consultant (Amgen), honoraria (Amgen). Go: employment or leadership (Amgen), stock ownership (Amgen) Additional follow‐up of participants alive 12 months after last enrolment, but median follow‐up not reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random assignment was stratified by prior adjuvant oxaliplatin therapy using permuted blocks (block size of 4). |
| Allocation concealment (selection bias) | Low risk | Randomisation through interactive voice response system (from protocol) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial (see below) |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Objective tumour response was evaluated by the investigator at each site using modified RECIST (version 1.0); no independent review was performed. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis for all randomised participants; dropout 1/142 panitumumab and 2/143 bevacizumab arm |
| Selective reporting (reporting bias) | Low risk | All outcomes reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Seymour PICCOLO 2013.
| Methods | Phase III open‐label, multicentre RCT; n = 1198 (460 participants in Arms A and B below) | |
| Participants | Advanced inoperable colorectal cancer, progressed after fluoropyrimidine chemotherapy | |
| Interventions | The combination of irinotecan and panitumumab (Arm A) vs irinotecan alone (Arm B) | |
| Outcomes | Primary outcome: OS. Secondary endpoints: PFS, TRR, QoL, toxicity | |
| Notes | Cancer Research UK ‐ independent peer review and feedback on protocols. Amgen ‐ provided panitumumab and educational grant. Follow‐up of participants still alive (n = 41): 25.4 months |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomisation was done with an automated telephonic system ... using a computer‐generated minimisation algorithm including a random element." |
| Allocation concealment (selection bias) | Low risk | Central computer‐generated randomisation after registration |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Open‐label trial but primary outcome (OS) not affected. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Primary endpoint of OS |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Equal dropout (4 withdrew in arm A, 4 in arm B) |
| Selective reporting (reporting bias) | Low risk | All secondary endpoints reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Siena 2013.
| Methods | Phase II open‐label study | |
| Participants | 42 people with KRAS exon 2 MT mCRC | |
| Interventions | Lenalidomide in combination with cetuximab (Arm 1), lenalidomide alone (Arm 2) | |
| Outcomes | Primary outcome(s): percentage of participants with dose‐limiting toxicities during first treatment cycle of safety lead‐in period, response rate. Secondary outcomes: PFS, duration of response, disease control rate, overall survival, treatment‐emergent adverse events | |
| Notes | Sponsored by Celgene. Siena: member of advisory boards for Sanofi‐Aventis, AstraZeneca, Roche, Genentech, and Amgen, and was supported by Oncologia Ca’ Granda Onlus (OCGO) Fondazione. Josep Tabernero has participated in advisory boards for Amgen, Celgene, Genentech, Merck‐Serono, Novartis, Roche, Sanofi‐Aventis, and Symphogen. Follow‐up not reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "1:1 ratio" ‐ no further information in publication |
| Allocation concealment (selection bias) | Unclear risk | "1:1 ratio" ‐ no further information in publication |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | High risk for evaluation of dose‐limiting toxicity given non‐blinding of treatment allocation |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Follow‐up until progression in all participants except possibly 1 (2.4% "other reasons" in phase IIB) |
| Selective reporting (reporting bias) | High risk | All secondary outcomes (except treatment‐emergent adverse events) not reported due to early termination. |
| Other bias | High risk | Disclosure agreement restricting rights of principal investigator to discuss or publish trial results after trial is completed as below (from ClinicalTrials.gov) Restriction Description:
|
Sobrero EPIC 2008.
| Methods | Phase III multicentre, open‐label RCT, n = 1298 | |
| Participants | 1298 people with mCRC, progression on first‐line treatment containing fluoropyrimidine and oxaliplatin, evidence of EGFR expression on histology | |
| Interventions | Arm A: combination of irinotecan with cetuximab. Arm B: irinotecan | |
| Outcomes | Primary outcome: OS. Secondary outcomes: PFS, TRR (note these were measured using WHO criteria: cutoff for objective response being 50% decrease from baseline sum of lesion diameters, cutoff for progressive disease being either 25% increase in the index‐lesion area, progression of non‐measurable lesions, or new lesions), QoL (EORTC QLQ‐C30) | |
| Notes | Sponsored by Merck KGaA, Bristol‐Myers Squibb, and ImClone Systems. Sobrero: consultant/advisory (Merck KGaA, Pfizer, Roche, Sanofi‐Aventis, Amgen), honoraria (Merck KGaA, Pfizer, Roche, Sanofi‐Aventis). Burris: consultant/advisory (Bristol‐Myers Squibb), honoraria (Bristol‐Myers Squibb). Follow‐up not reported, but results reported with 67% participants deceased. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised and stratified by study site and ECOG score (0 to 1 vs 2) ‐ no further details given |
| Allocation concealment (selection bias) | Unclear risk | No details given. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Open‐label trial (but primary outcome OS) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Open‐label trial (but primary outcome OS) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Significant dropout rate (50/648 experimental, 43/650 control), but balanced |
| Selective reporting (reporting bias) | Low risk | All findings reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Tol CAIRO2 2008.
| Methods | Phase III multicentre, open‐label RCT; n = 755 | |
| Participants | People with mCRC, previously untreated | |
| Interventions | Capecitabine, oxaliplatin, bevacizumab, and cetuximab vs capecitabine, oxaliplatin, and bevacizumab | |
| Outcomes | Primary outcome: PFS. Secondary outcomes: OS, QoL, TRR | |
| Notes | Supported by the Dutch Colorectal Cancer Group (DCCG). The DCCG received grants for data management and analysis from the Commissie Klinisch Toegepast Onderzoek of the Dutch Cancer Foundation and unrestricted scientific grants from Roche, Merck Serono, Sanofi‐Aventis, and DxS. Dr Tol reports receiving grant support from the Netherlands Organisation for Health Research and Development; and Dr Punt, grant support from the Dutch Cancer Foundation and Roche and consulting fees from Roche and Merck Serono. Median follow‐up 23 months | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation ‐ as below |
| Allocation concealment (selection bias) | Low risk | Randomisation was performed centrally by a minimisation technique with stratification according to serum lactate dehydrogenase level (normal or abnormal, according to the cutoff values of each individual centre), previous adjuvant chemotherapy (yes or no), number of affected organs (1 or more than 1), and treatment centre. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "Tumor response was assessed by the local investigators" ‐ no report of central review |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ITT analysis; no details in paper |
| Selective reporting (reporting bias) | Low risk | All stated outcomes reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Tournigand DREAM 2015.
| Methods | Phase III RCT; n = 452 | |
| Participants | People with unresectable mCRC after initial bevacizumab‐including therapy (mFOLFOX7 with bevacizumab, CAPOX with bevacizumab, or FOLFIRI with bevacizumab) | |
| Interventions | Arm A: maintenance combination of bevacizumab and erlotinib. Arm B: maintenance bevacizumab | |
| Outcomes | Primary endpoint: maintenance PFS. Secondary endpoints: PFS from inclusion, OS, safety | |
| Notes | Sponsored by GERCOR and F. Hoffmann‐La Roche. Tournigand: grants from Roche, Sanofi, Merck, Amgen, Bayer. de Gramont: personal fees for talks and participation in advisory boards from Roche and Sanofi‐Aventis Median follow‐up: 48 to 51 months |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The random allocation sequence was generated through a computer random number generator. |
| Allocation concealment (selection bias) | Unclear risk | An unblinded randomisation (1:1) was done with a minimisation technique. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Open‐label study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No statement of central or blinded review of imaging to determine PFS ‐ awaiting response from authors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Not stated, but 6/228 participants in bevacizumab arm and 4/224 participants in bevacizumab + erlotinib arm discontinued treatment due to "patient choice". In addition, long median follow‐up and balance of participants characteristics make significant attrition bias unlikely. |
| Selective reporting (reporting bias) | Low risk | All outcomes reported. |
| Other bias | Low risk | No other potential biases detected. |
Tveit NORDIC VII 2012.
| Methods | Phase III multicentre, open‐label RCT; n = 571 | |
| Participants | People with mCRC, previously untreated | |
| Interventions | Nordic FLOX (Arm A), cetuximab and FLOX (Arm B), cetuximab with intermittent FLOX (Arm C) | |
| Outcomes | Primary outcome: PFS. Secondary outcomes: OS, TRR, R0 resection rate, safety | |
| Notes | We ignored Arm C in our analysis as cannot separate effect of intermittent FLOX from that of cetuximab. Supported by Merck Serono (Darmstadt, Germany), Sanofi‐Aventis (Oslo, Norway), the Norwegian Cancer Society, and the Swedish Cancer Society (Cancerfonden). Tveit: research funding (Merck Serono); Christoffersen: nil Follow‐up not stated. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised and stratified by centre, but no further details in article or attached protocol |
| Allocation concealment (selection bias) | Unclear risk | Randomised and stratified by centre, but no further details in article or attached protocol |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Progression assessed by unblinded investigators. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 449/498 (90%) of participants had recorded progression overall: 90% of Arm A and 89% of Arm B participants; no specific details regarding dropout. |
| Selective reporting (reporting bias) | Low risk | All stated outcomes reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Van Cutsem CRYSTAL 2009.
| Methods | Phase III open‐label, multicentre study; n = 1198 | |
| Participants | People with mCRC, EGFR‐positive on histology, no prior chemotherapy | |
| Interventions | FOLFIRI with cetuximab vs FOLFIRI | |
| Outcomes | Primary outcome: PFS. Secondary outcomes: OS, TRR, safety | |
| Notes | Supported by Merck (Darmstadt). Dr Van Cutsem reports receiving consulting or advisory fees from Amgen, Merck (Darmstadt), Pfizer, Roche, and Sanofi‐Aventis; lecture fees from Amgen, Merck (Darmstadt), Roche, and Sanofi‐Aventis; and grant support from Merck (Darmstadt) and Roche; Dr Rougier, consulting or advisory fees from Merck (Darmstadt), Pfizer, Roche, and Sanofi‐Aventis and lecture fees from Merck (Darmstadt), Pfizer, and Sanofi‐Aventis. The median duration of follow‐up was 29.9 months (95% CI 29.1 to 30.5) with cetuximab plus FOLFIRI and 29.4 months (95% CI 28.8 to 30.4) with FOLFIRI alone. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Stratified permuted‐block procedure" |
| Allocation concealment (selection bias) | Unclear risk | No details in paper |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Open‐label trial (but see below) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "An independent review committee performed a preplanned, blinded, retrospective review ... to determine the day of progression and the best overall response" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 7 participants were lost to follow‐up: 2 in the cetuximab‐FOLFIRI group (n = 599), and 5 in the FOLFIRI group (n = 599). |
| Selective reporting (reporting bias) | Low risk | All stated outcomes reported. |
| Other bias | Low risk | No other significant bias present; funders did not have inappropriate influence. |
Venook CALGB 80405 2014.
| Methods | Phase III RCT; n = 1137 | |
| Participants | Metastatic colorectal cancer, no prior chemotherapy, KRAS exon 2 WT | |
| Interventions | Fluorouracil‐based chemotherapy (FOLFIRI or mFOLFOX6) with cetuximab versus fluorouracil‐based chemotherapy with bevacizumab | |
| Outcomes | Primary endpoint: OS. Secondary endpoints: PFS, toxicity, expanded RAS analysis | |
| Notes | Sponsored by Alliance for Clinical Trials in Oncology, National Cancer Institute, Southwest Oncology Group, Bristol‐Myers Squibb, Aptuit Follow‐up 40 months |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random treatment assignments for KRAS WT participants were generated according to randomly permuted blocks within strata. |
| Allocation concealment (selection bias) | Low risk | Randomised by central site (web based) after completion of registration and details |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Open‐label, but primary endpoint of OS |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Open‐label, but primary endpoint of OS |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Abstract form only |
| Selective reporting (reporting bias) | Unclear risk | Abstract form only |
| Other bias | Unclear risk | Insufficient information in presentation to determine whether other biases are likely |
Vincent 2011.
| Methods | Phase II randomised trial; n = 95 | |
| Participants | People with mCRC, opting against combination chemotherapy or not a candidate for combination chemotherapy | |
| Interventions | Arm A: combination of capecitabine (1 g/m2 twice daily, days 1 to 14 every 3 weeks) and erlotinib (150 mg daily). Arm B: capecitabine alone. Arm C: erlotinib alone | |
| Outcomes | Primary outcome: time to disease progression. Secondary outcomes: TRR, OS, safety, QoL | |
| Notes | No information about funding, potential conflicts of interest, or follow‐up available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details given, abstract only. |
| Allocation concealment (selection bias) | Unclear risk | No details given, abstract only. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details given, abstract only. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details given, abstract only. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No details given, abstract only. |
| Selective reporting (reporting bias) | Unclear risk | No details given, abstract only. |
| Other bias | Unclear risk | Insufficient information in poster to determine whether other biases are likely |
Wasan COIN‐B 2014.
| Methods | Phase II randomised, open‐label trial; n = 226 (169 KRAS exon 2 WT) | |
| Participants | People with advanced colorectal cancer, no prior chemotherapy for metastases | |
| Interventions | Intermittent mFOLFOX6 with cetuximab (ceased after 12 weeks; assuming stable disease or better, re‐introduction of mFOLFOX6 with cetuximab on progression) versus continuous mFOLFOX6 with cetuximab (same as intermittent mFOLFOX6 with cetuximab, with maintenance cetuximab in between periods of mFOLFOX6 with cetuximab) | |
| Outcomes | Primary outcome: failure‐free survival at 10 months (in participants who had completed initial 12 weeks of mFOLFOX6 with cetuximab). Main secondary outcomes: Overall survival, progression‐free survival in the interval | |
| Notes | Funded by UK Medical Research Council and Merck KGaA. Wasan: advisory boards, educational meetings (as faculty and speaker) for Merck KGaA. TM: grants, personal fees, non‐financial support from Merck KGaA unrelated to this study Follow‐up 32.8 months in intermittent cetuximab group, 34.2 months in continuous cetuximab group |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The MRC Clinical Trials Unit did the randomisation by telephone, using the method of minimisation with a random element. The minimisation factors were hospital, WHO performance status, previous adjuvant chemotherapy, liver metastases, and peritoneal metastases. Participants were randomly assigned (1:1) to intermittent chemotherapy plus intermittent cetuximab or intermittent chemotherapy plus continuous cetuximab. |
| Allocation concealment (selection bias) | High risk | Treatment allocation was not masked. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "We did not confirm responses with repeat scans nor did we do central radiological review." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 3/130 participants in primary outcome population lost to follow‐up |
| Selective reporting (reporting bias) | High risk | Data for KRAS exon 2 MT participants not yet published; quality of life data not yet published |
| Other bias | Low risk | No other significant bias present |
Ye 2013.
| Methods | Randomised controlled trial; n = 138 | |
| Participants | People with unresectable liver metastases from colorectal cancer, KRAS exon 2 WT | |
| Interventions | Arm A: the combination of chemotherapy (FOLFIRI or mFOLFOX6) and cetuximab. Arm B: the same chemotherapy alone | |
| Outcomes | Primary endpoint: rate of participants converted to resection for liver metastases. Secondary endpoints: tumour response, survival | |
| Notes | Supported by Key Projects of the Clinical Disciplines, administered by the Ministry of Health. Ye and Xu ‐ no conflicts of interest Median follow‐up 25 months |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Low risk as outcomes (blinded assessment of resectability, overall survival) not susceptible to performance bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessment of primary endpoint by > 3 liver surgeons blinded to clinical data |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low attrition: 1/70 Arm A, 2/68 Arm B |
| Selective reporting (reporting bias) | Low risk | All outcomes reported. |
| Other bias | Low risk | No other significant bias present |
CI: confidence interval COI: conflicts of interest CRC: colorectal cancer ECOG PS: Eastern Cooperative Oncology Group Performance Status EGFR: epidermal growth factor receptor ITT: intention to treat MAb: monoclonal antibodies mCRC: metastatic colorectal cancer MRC: Medical Research Council MT: mutant ORR: objective response rate OS: overall survival PFS: progression‐free survival QoL: quality of life RCT: randomised controlled trial RECIST: Response Evaluation Criteria in Solid Tumours TRR: tumour response rate WHO: World Health Organization WT: wild type
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Cunningham BOND 2004 | We excluded this randomised controlled trial as it compared participants receiving a combination of cetuximab and irinotecan with those receiving cetuximab alone. As cetuximab was given in the same dose to both arms, the study design did not allow assessment of EGFR inhibitor efficacy. |
| Liu 2015 | This trial investigated the addition of both panitumumab and bevacizumab to FOLFIRI chemotherapy, and thus did not meet the inclusion criteria for this review. The fact that both drugs were added in the investigational arm means that the effect of panitumumab cannot be accurately discerned. |
| NCT00950820 | This study, which planned to compare the combination of CAPOX and panitumumab with CAPOX alone in people with KRAS unselected metastatic colorectal cancer, was terminated after only 9 participants were accrued, with no published results and no plans to continue the trial. |
| Personeni 2013 | Study amended to single‐arm design. |
| Primrose NEW EPOC 2014 | Enrolment restricted to people with resectable disease. |
| Saltz BOND2 2007 | Similarly to Cunningham 2004, this study compared the combination of cetuximab, bevacizumab, and irinotecan to cetuximab and bevacizumab alone, meaning that assessment of EGFR inhibitor efficacy was not feasible. |
EGFR: epidermal growth factor receptor
Characteristics of studies awaiting assessment [ordered by study ID]
EVEREST 2004.
| Methods | Phase I/II multicentre randomised controlled trial |
| Participants | People with EGFR‐positive mCRC |
| Interventions | Dose‐escalated schedule of cetuximab vs standard dose of cetuximab |
| Outcomes | Main objective: to compare in skin biopsies the effects of a cetuximab dose escalation regimen with a standard cetuximab regimen on EGFR and downstream signalling pathway markers. Secondary objectives: incidence of grade 2+ skin toxicity, efficacy (not further specified), safety, tolerability, molecular markers, pharmacokinetics |
| Notes | Trial name EVEREST; listed as completed but no publicly available information to determine classification. Awaiting reply to correspondence |
Hill 2015.
| Methods | Phase II open‐label, randomised study |
| Participants | People with KRAS exon 2 WT mCRC progressing on/after oxaliplatin‐containing chemotherapy |
| Interventions | FOLFIRI plus duligotuzumab (an antibody directed against EGFR and HER3) vs FOLFIRI plus cetuximab |
| Outcomes | Primary endpoint: efficacy. Secondary endpoints: safety and tolerability |
| Notes |
Hiret 2016.
| Methods | Phase II randomised study |
| Participants | People with KRAS WT mCRC whose disease progressed on first‐line treatment with fluoropyrimidine‐based chemotherapy and bevacizumab |
| Interventions | mFOLFOX or FOLFIRI (the one not used in first‐line treatment) with bevacizumab vs mFOLFOX or FOLFIRI (the one not used in first‐line treatment) with cetuximab |
| Outcomes | Primary endpoint: PFS at 4 months. Secondary endpoints: response rate, median PFS, OS, safety, quality of life (EORTC QLQ‐C30) |
| Notes |
Kim 2016.
| Methods | Phase III randomised controlled trial |
| Participants | People with KRAS exon 2 WT mCRC, no prior administration of anti‐EGFR agents |
| Interventions | Panitumumab + best supportive care vs best supportive care |
| Outcomes | Primary endpoint: OS in people with KRAS exon 2 WT mCRC. Secondary endpoints: OS in subgroup of patients with WT RAS mCRC and PFS and safety in both WT populations. |
| Notes | Report published in BJC 2016 (PMC5104888). |
MACBETH 2016.
| Methods | Phase II randomised study |
| Participants | People with KRAS exon 2 WT mCRC |
| Interventions | Induction treatment with FOLFOXIRI and cetuximab for all participants with re‐evaluation after 8 cycles; participants deemed unsuitable for resection (with stable disease or better) are then randomised to maintenance bevacizumab or cetuximab |
| Outcomes | Primary outcome: 10‐month progression‐free rate. Secondary outcomes: best overall response rate, 10‐month resection rate, time to strategy failure, time to second progressive disease, PFS, OS, toxicity rate, overall toxicity rate |
| Notes |
TAGUS 2009.
| Methods | Phase II open‐label, randomised study |
| Participants | People with mCRC, progressing after first‐line FOLFIRI with cetuximab |
| Interventions | FOLFOX with cetuximab vs FOLFOX |
| Outcomes | Primary endpoint: response rate. Secondary endpoints: OS and time to disease progression |
| Notes | Trial name TAGUS; listed as prematurely ended but no publicly available information to determine classification. Awaiting reply to correspondence |
EGFR: epidermal growth factor receptor mCRC: metastatic colorectal cancer OS: overall survival PFS: progression‐free survival WT: wild type
Characteristics of ongoing studies [ordered by study ID]
Ashwin 2014.
| Trial name or title | Assessment of tumour response and resection rates in unresectable metastatic colorectal liver metastases following cetuximab with neoadjuvant chemotherapy |
| Methods | Prospective randomised study, n = 152 |
| Participants | Nonresectable metastatic CRC, KRAS exon 2 WT |
| Interventions | Arm A: the combination of chemotherapy (FOLFIRI or mFOLFOX6) and cetuximab. Arm B: the same chemotherapy alone |
| Outcomes | Response rate, R0 resection rate of liver metastases, perioperative morbidity, overall survival, progression‐free survival |
| Starting date | Not stated |
| Contact information | K Ashwin, Manipal Comprehensive Cancer Center, Surgical Oncology, Bangalore, India |
| Notes | Some statistics reported in the referenced abstract, but given that these are statistically inconsistent, we decided to await the final publication (see text). |
ATOM.
| Trial name or title | Achievement of Improved Survival by Molecular Targeted Chemotherapy and Liver Resection for Not Optimally Resectable Colorectal Liver Metastases (ATOM) |
| Methods | Phase II open‐label, randomised, parallel trial |
| Participants | 120 people with KRAS exon 2 WT (Protocol 1.0 to 1.2) or RAS WT (Protocol 2.0) mCRC |
| Interventions | mFOLFOX6 plus bevacizumab versus mFOLFOX6 plus cetuximab |
| Outcomes | Primary endpoint: PFS. Secondary endpoints: response rate, tumour shrinkage rate at 8 weeks, liver resection rate, R0 liver resection rate, PFS, time to treatment failure, OS, quality of life, incidence of adverse events, PFS among RAS WT subpopulation |
| Starting date | May 2013 |
| Contact information | Tasumi Shimizu +81‐3‐5684‐7767 prj‐atomdc@eps.co.jp |
| Notes | ATOM ES (NCT01834014) is an exploratory substudy of ATOM investigating predictive/prognostic biomarkers in the above cohort. |
CAIRO5.
| Trial name or title | CAIRO5 |
| Methods | Randomised, open‐label trial |
| Participants | 640 people with unresectable mCRC and liver‐only metastases |
| Interventions | Participants with RAS MT tumours will be randomised between doublet chemotherapy (FOLFOX or FOLFIRI) plus bevacizumab (schedule 1), and triple chemotherapy (FOLFOXIRI) plus bevacizumab (schedule 2). Participants with RAS WT tumours will be randomised between doublet chemotherapy (FOLFOX or FOLFIRI) plus either bevacizumab (schedule 1) or panitumumab (schedule 3). |
| Outcomes | Primary outcome: PFS. Secondary outcomes: R0/1 resection rate, median OS, response rate, toxicity (CTCAE version 4.0), rate of pathological complete response, postoperative morbidity, correlation of evaluation by panel with outcome |
| Starting date | June 2014 |
| Contact information | Dutch Colorectal Cancer Group |
| Notes |
CREPAS.
| Trial name or title | Study of medical treatment reactivity by the chemokine receptor (CXCR4) as 1st line treatment in patients with metastatic colorectal cancer (CREPAS) |
| Methods | Phase II open‐label, randomised study |
| Participants | People with KRAS exon 2 WT mCRC |
| Interventions | mFOLFOX6 with bevacizumab (Arm A), mFOLFOX6 with panitumumab (Arm B); stratified by CXCR4‐CEC >= 20 vs CXCR‐CEC < 20 (no further explanation given in trial record) |
| Outcomes | Primary endpoint: PFS. Secondary endpoints: safety profile, OS, time to treatment failure, response rate |
| Starting date | July 2012 |
| Contact information | Satoshi Matsusaka, The Cancer Institute Hospital of JFCR, Department of Gastroenterology, 3‐8‐31, Ariake, Koto, Tokyo 135‐8550 |
| Notes |
DEEPER.
| Trial name or title | A Randomized Phase II Study to Investigate the Deepness of Response of FOLFOXIRI Plus Cetuximab (Erbitux) Versus FOLFOXIRI Plus Bevacizumab as the First‐line Therapy in Metastatic Colorectal Cancer Patients With RAS Wild‐type Tumors: DEEPER |
| Methods | Phase II open‐label, randomised study |
| Participants | People with RAS WT mCRC |
| Interventions | Experimental: the combination of FOLFOXIRI and cetuximab. Comparator: the combination of FOLFOXIRI and bevacizumab |
| Outcomes | Primary outcome: best depth of response. Secondary outcomes: rate of tumour shrinkage at 8 weeks, response rate, deepness of response, OS, PFS, rate of curative resection of metastases, number of adverse events |
| Starting date | August 2015 |
| Contact information | Toshifusa Nakajima, MD, Japan Clinical Cancer Research Organization |
| Notes | JPRN‐UMIN000018412 is the adjunct biomarker study to DEEPER. |
FIRE‐4.
| Trial name or title | FIRE‐4: Randomised study of the efficacy of cetuximab rechallenge in patients with metastatic colorectal cancer (RAS wild‐type) responding to first‐line treatment with FOLFIRI plus cetuximab |
| Methods | Phase III multicentre, randomised trial |
| Participants | People with mCRC, RAS WT |
| Interventions | All participants receive first‐line FOLFIRI with cetuximab. Participants who have stable disease or better after 6 months of treatment are randomised to cetuximab rechallenge (investigational arm) or "anti‐EGFR‐free treatment" (control arm) |
| Outcomes | Primary endpoint: overall survival from randomisation to third‐line treatment (OS3). Secondary endpoints: objective response rate 1/2/3, PFS 1/2/3, overall survival from randomisation to first‐line treatment (OS1), investigation of early tumour shrinkage and depth of response during first‐line and third‐line treatment, study of molecular biomarkers for prediction of sensitivity and secondary resistance to an anti‐EGFR treatment with cetuximab (including tumour biopsies and liquid biopsies from blood samples), prospective validation of a biomarker score (AREG/EREG), prospective analysis of tumour marker evolution (CEA and CA 19‐9), recording of safety and tolerance of first‐line and third‐line treatment. |
| Starting date | March 2015 |
| Contact information | Studiensekretariat, Klinikum der Ludwig‐Maximilians‐Univ. München, Klinikum Großhadern, 0049894400 72208, Matthias.Wolff@med.uni‐muenchen.de |
| Notes |
FIRE‐4.5.
| Trial name or title | FIRE‐4.5 |
| Methods | Phase III randomised controlled trial |
| Participants | People with KRAS/NRAS WT, BRAFV600E MT mCRC |
| Interventions | Arm A: the combination of FOLFOXIRI and cetuximab. Arm B: the combination of FOLFOXIRI and bevacizumab |
| Outcomes | Primary outcome: response rate. Secondary outcomes: PFS, OS, early tumour shrinkage and depth of response, molecular biomarkers for prediction of sensitivity and secondary resistance of an anti‐EGFR treatment with cetuximab (including tumour biopsies and liquid biopsies from blood samples), analysis of tumour marker evolution (CEA and CA 19‐9), safety and tolerability of treatment |
| Starting date | September 2016 |
| Contact information | Studiensekretariat, Klinikum der Ludwig‐Maximilians‐Univ. München, Klinikum Großhadern, 0049894400 72208, Matthias.Wolff@med.uni‐muenchen.de |
| Notes |
FOCETELD.
| Trial name or title | FOCETELD |
| Methods | Phase II open‐label, randomised study |
| Participants | People with KRAS exon 2 WT mCRC, age 70 to 80, ECOG 0 to 1 |
| Interventions | FOLFIRI with cetuximab (interventional arm), FOLFIRI (control arm) |
| Outcomes | Primary endpoint: percentage of participants free of disease progression at 6 months. Secondary endpoints: safety of combination of chemotherapy with cetuximab in elderly patients; response rate and median survival in both treatment arms |
| Starting date | April 2012 |
| Contact information | Clinica di Oncologia Medica, A.O. Ospedali Riuniti di Ancona, 071.5964169, s.cascinu@univpm.it |
| Notes | Still "ongoing" on trial database as of March 2017; no further details available at time of publication. |
FOCULM.
| Trial name or title | FOLFOXIRI With or Without Cetuximab as First‐line Treatment of Patients With Non‐resectable Liver‐Only Metastatic Colorectal Cancer (FOCULM) |
| Methods | Phase II randomised controlled trial |
| Participants | 138 people with KRAS/NRAS WT mCRC |
| Interventions | Arm A: the combination of FOLFOXIRI and cetuximab. Arm B: FOLFOXIRI alone |
| Outcomes | Primary outcome: percentage of participants with curative liver treatment (complete resection/ablation) following protocol treatment. Secondary outcomes: reported adverse events, response rate, PFS, time to recurrence, quality of life (EORTC QLQ‐C30) |
| Starting date | February 2014 |
| Contact information | Yanhong Deng, MD 008613925106525 13925106525@163.com |
| Notes |
G13 study.
| Trial name or title | Randomized Phase II Study of BSC vs Cetuximab vs Irinotecan and Cetuximab in Patients with KRAS codon G13D mutant Metastatic Colorectal Cancer (G13 study) |
| Methods | Phase II open label, randomised study |
| Participants | People with KRAS G13D mutant mCRC |
| Interventions | Arm 1: cetuximab alone. Arm 2: cetuximab and irinotecan. Arm 3: best supportive care |
| Outcomes | Primary endpoint: PFS. Secondary outcomes: response rate, response rate by metastatic site, disease control rate, OS, safety |
| Starting date | September 2012 |
| Contact information | Mai Hatta, Nagoya University Graduate School of Medicine, 052‐744‐2442, m‐hatta@med.nagoya‐u.ac.jp |
| Notes | Closed to accrual as of April 2016; no results reported yet |
NCT00202787.
| Trial name or title | Open‐label, Phase II, Randomised, Pilot Study to Evaluate the Safety and Efficacy of Combination Therapy With Cetuximab and FOLFOX4 or FOLFOX4 Alone in Patients Colorectal Cancer and Initially Non‐resectable |
| Methods | Phase II open‐label, randomised trial |
| Participants | People with mCRC |
| Interventions | Experimental arm: FOLFOX4 with cetuximab. Control: FOLFOX4 |
| Outcomes | Primary endpoint: confirmed objective response rate. Secondary endpoints: safety, surgical resectability, rate of R0 resections, rate of clinical benefit, time to disease progression, time to onset of response, duration of response, time to treatment failure, OS, determination of polymorphisms of the intron 1 of the EGFR gene, TS, XRCC1, XPD, serum levels of EGFR and ATP7A and ATP7B, number of copies of EGFR gene, the levels of PTEN, EGFR, AKT, and MAPK proteins, and mutations at EGFR, PI3KCA, KRAS, and BRAF genes |
| Starting date | February 2005 |
| Contact information | Albert Abad, Spanish Cooperative Group for Gastrointestinal Tumour Therapy (TTD) |
| Notes |
NCT01442649.
| Trial name or title | Phase II, Multicentric Randomized Trial, Evaluating the Efficacy of Fluoropyrimidine‐based Standard Chemotherapy, Associated to Either Cetuximab or Bevacizumab, in KRAS Wild‐type Metastatic Colorectal Cancer Patients With Progressive Disease After Receiving First‐line Treatment With Bevacizumab |
| Methods | Phase II multicentric randomised trial |
| Participants | People with KRAS exon 2 WT mCRC with progressive disease after first‐line bevacizumab‐containing treatment |
| Interventions | Experimental: Arm A: fluoropyrimidine‐based chemotherapy and bevacizumab Experimental: Arm B: fluoropyrimidine‐based chemotherapy and cetuximab |
| Outcomes | Primary outcomes: PFS Secondary outcomes: objective response rate, OS, treatment tolerance, quality of life |
| Starting date | December 2010 |
| Contact information | Trevor Stanbury +33(1)44235567 t‐stanbury@unicancer.fr |
| Notes | Preliminary results reported at ASCO 2016; awaiting full publication. |
NCT01652482.
| Trial name or title | A Phase II, Multicenter, Open‐Label, Randomized Study Evaluating the Efficacy and Safety of MEHD7945A + FOLFIRI Versus Cetuximab + FOLFIRI in Second Line in Patients With KRAS Wildtype Metastatic Colorectal Cancer |
| Methods | Phase II multicentre, open‐label, randomised study |
| Participants | People with KRAS exon 2 WT mCRC; progressive disease on or after first‐line oxaliplatin‐containing chemotherapy |
| Interventions | Experimental arm: FOLFIRI with MEHD7945A. Control arm: FOLFIRI with cetuximab |
| Outcomes | Primary endpoint: PFS. Secondary endpoints: response rate, duration of objective response, OS, safety, pharmacokinetics of MEHD7945A in combination with FOLFIRI, incidence of anti‐MEHD7945A antibodies |
| Starting date | October 2012 |
| Contact information | Clinical Trials, Genentech Inc. |
| Notes | Listed as "completed", but no results published yet |
NCT01991873.
| Trial name or title | Maintenance Therapy With 5‐FU/FA Plus Panitumumab vs. 5‐FU/FA Alone After Prior Induction and Re‐induction After Progress for 1st‐line Treatment of Metastatic Colorectal Cancer (PanaMa) |
| Methods | Phase II multicentre, open‐label, randomised trial |
| Participants | People with RAS WT mCRC |
| Interventions | Participants with stable disease or better are randomised to maintenance chemotherapy (5‐FU/folinic acid) with panitumumab vs maintenance chemotherapy alone |
| Outcomes | Primary outcome: PFS. Secondary outcomes: time to failure of treatment strategy, PFS of re‐induction, objective response after 12 weeks of induction chemotherapy, objective best response during maintenance and re‐induction, OS, safety, health‐ and skin‐related quality of life |
| Starting date | April 2014 |
| Contact information | Tanja Trarbach, MD Praxis für interdisziplinäre Onkologie & Hämatologie Freiburg |
| Notes | Currently recruiting participants |
NCT02083653.
| Trial name or title | Open‐label, Randomized, Controlled, Multicenter Phase II Trial Investigating 2 Sym004 Doses Versus Investigator's Choice (Best Supportive Care, Capecitabine, 5‐FU) in Subjects With Metastatic Colorectal Cancer and Acquired Resistance to Anti‐EGFR Monoclonal Antibodies |
| Methods | Phase II multicentre, open‐label randomised controlled trial |
| Participants | People with mCRC; failure of or intolerance to 5‐FU, oxaliplatin, or irinotecan; acquired resistance to marketed anti‐EGFR MAbs |
| Interventions | Experimental arm A: Sym004 12 mg/kg IV weekly. Experimental arm B: Sym004 9 mg/kg IV weekly. Control arm: investigator's choice of 5‐FU, capecitabine, or BSC |
| Outcomes | Primary endpoint: OS. Secondary endpoints: response rate, PFS, time to treatment failure, relative dose intensity of Sym004, pharmacokinetic parameters, number of participants with antidrug antibodies, levels of biomarkers related to the EGFR pathway, quality of life (EORTC QLQ‐C30, QLQ‐CR29, FACT‐EGFR 18), adverse events |
| Starting date | March 2014 |
| Contact information | Ivan Horak, MD Symphogen A/S |
| Notes | Ongoing but closed to accrual; study completion date estimated to be October 2017 |
NCT02394834.
| Trial name or title | An Exploratory Study of Treatment Sensitivity and Prognostic Factors in a Efficacy and Safety Study of mFOLFOX6 + Bevacizumab Versus mFOLFOX6 + Panitumumab Therapy in Patients With Chemotherapy‐naïve Unresectable Advanced or Recurrent Colorectal Cancer |
| Methods | Phase III randomised controlled trial |
| Participants | 800 people with KRAS/NRAS WT mCRC |
| Interventions | Experimental arm: mFOLFOX6 with panitumumab. Comparator: mFOLFOX6 with bevacizumab (5 mg/kg) |
| Outcomes | Primary endpoint: OS and its correlation with each gene in baseline tumour samples. Secondary endpoints: PFS, response rate, duration of response, proportion of participants proceeding to surgical resection, proportion of participants with early tumour shrinkage, degree of maximum tumour shrinkage, evaluation of relationship of each biomarker in plasma free DNA and tumour samples from baseline or the main study, evaluation of the relationship between a change in each biomarker in plasma free DNA from baseline of the main study and efficacy, evaluation of the relationship between a change in each biomarker in plasma free DNA from baseline to discontinuation of the protocol treatment of the main study, and efficacy, evaluation of the relationship between a change in each biomarker in tumour tissue from baseline to discontinuation of the protocol treatment of the main study, and efficacy |
| Starting date | May 2015 |
| Contact information | Takeda Study Registration Call Center +1‐800‐778‐2860 (USA & EU) medicalinformation@tpna.com |
| Notes | Sponsored by Takeda. Currently recruiting participants |
PANIB.
| Trial name or title | PANIB ‐ An open‐label, randomised, controlled, multi‐center, Phase II trial comparing Panitumumab versus Bevacizumab in combination with oxaliplatin ‐ 5 FU (FOLFOX) first‐line treatment according Ras Wild Type status for patients with metastatic unresectable colorectal cancer (mCRC) |
| Methods | Phase II multicentre, open‐label, randomised trial |
| Participants | People with extended RAS WT mCRC |
| Interventions | FOLFOX with panitumumab (experimental arm), FOLFOX with bevacizumab (control arm) |
| Outcomes | Primary outcome: PFS at 1 year. Secondary endpoints: response rate, resection rate, safety profile of both combinations, OS |
| Starting date | August 2014 |
| Contact information | Prof Dr Christian Rolfo, Antwerp University Hospital, +3238213646, christian.rolfo@uza.be |
| Notes | Trial ongoing. |
PARADIGM.
| Trial name or title | PARADIGM |
| Methods | Phase III open‐label randomised controlled trial |
| Participants | People with KRAS/NRAS WT mCRC |
| Interventions | Intervention arm: mFOLFOX6 with panitumumab. Control arm: mFOLFOX6 with bevacizumab |
| Outcomes | Primary outcome: OS. Secondary outcomes: PFS, response rate, duration of response, percentage of participants treated with surgical resection after chemotherapy, percentage of participants with adverse events |
| Starting date | May 2015 |
| Contact information | Takeda Study Registration Call Center +1‐800‐778‐2860 (USA & EU) medicalinformation@tpna.com |
| Notes | Trial ongoing. |
Peeters 2012.
| Trial name or title | Colorectal Cancer (CRC) Cetuximab Elderly Frail |
| Methods | Open‐label randomised controlled trial |
| Participants | People with KRAS exon 2 WT mCRC, age >= 80 or >= 70 in combination with functional restrictions defined as limitation in at least 2 of 8 instrumental activities of daily living (IADL) |
| Interventions | 5‐FU with cetuximab (interventional arm), 5‐FU alone (control arm) |
| Outcomes | Primary outcome: PFS. Secondary outcomes: OS, response rate, change in IADL score, change in G8 geriatric assessment screening tool, change in social situation, quality of life (EORTC QLQ‐C30 and QLQ‐ELD14), occurrence of adverse events, health economy assessments, score of Elderly Minimal Dataset Comprehensive Geriatric Assessment (EMDCGA) as evaluated by G8 instrument, score of EMDCGA as evaluated by IADL questionnaire, score of EMDCGA as evaluated by social situation questionnaire |
| Starting date | April 2013 |
| Contact information | EORTC; study chairs Marc Peeters (Belgium) and Ulrich Wedding (Germany) |
| Notes | Study terminated due to poor accrual; awaiting publication of data. |
TAILOR.
| Trial name or title | An Open‐label, Randomized, Controlled, Multicenter Phase III Trial to Compare Cetuximab in Combination With FOLFOX‐4 Versus FOLFOX‐4 Alone in the First Line Treatment of Metastatic Colorectal Cancer in Chinese Subjects With RAS Wild‐type Status |
| Methods | Phase III multicentre, open‐label, randomised controlled trial |
| Participants | 503 |
| Interventions | Cetuximab with FOLFOX4 (Arm A) versus FOLFOX4 (Arm B) |
| Outcomes | Primary outcome measures: PFS, Secondary outcomes: ORR, OS, treatment failure, and rate of curative surgery for metastasis |
| Starting date | August 2010 |
| Contact information | Medical responsible Merck Serono |
| Notes | Preliminary results published at World GI 2016; awaiting full publication. |
TIME.
| Trial name or title | Randomized Phase II Study of First‐line FOLFIRI Plus Cetuximab for 8 Cycles Followed by Either Single‐agent Cetuximab as Maintenance Therapy or Observation in Patients With Wild‐type KRAS and NRAS Metastatic Colorectal Cancer |
| Methods | Phase II open‐label, randomised study |
| Participants | People with KRAS and NRAS WT mCRC |
| Interventions | All participants receive 8 cycles of FOLFIRI with cetuximab. Upon stable disease on finishing 8 cycles, participants are randomised to cetuximab maintenance or observation alone. |
| Outcomes | PFS at 6 months |
| Starting date | February 2014 |
| Contact information | Trevor Stanbury +33 (0)1 44 23 55 67 t‐stanbury@unicancer.fr |
| Notes |
UCGI 25.
| Trial name or title | Dual Targeting of EGFR With Cetuximab and Afatinib to Treat Refractory wtKRAS Metastatic Colorectal Cancer |
| Methods | Phase II multicentric randomised trial |
| Participants | 75 people with KRAS exon 2 WT mCRC |
| Interventions | Arm A: cetuximab with afatinib. Arm B: cetuximab alone |
| Outcomes | Primary outcome: disease control rate at 6 months. Secondary outcomes: response rate, PFS, OS, quality of life (EORTC QLQ‐C30, QLQ‐CR29), treatment safety |
| Starting date | October 2012 |
| Contact information | UNICANCER |
| Notes | Preliminary results presented at ASCO 2016; awaiting full publication. |
UMIN000005216.
| Trial name or title | Randomized phase II trial of FOLFIRI with either panitumumab or bevacizumab as second‐line treatment in patients with KRAS wild metastatic colorectal cancer refractory to oxaliplatin and bevacizumab with exploratory analysis to predict treatment efficacy and prognosis |
| Methods | Phase II open‐label, randomised trial |
| Participants | People with KRAS exon 2 WT mCRC |
| Interventions | Arm A: FOLFIRI with panitumumab. Arm B: FOLFIRI alone |
| Outcomes | Primary outcome: OS. Secondary outcomes: PFS, response rate, safety, translational research |
| Starting date | March 2011 |
| Contact information | Shinichiro Nakamura, West Japan Oncology Group; +81‐6‐6633‐7400, datacenter@wjog.jp |
| Notes |
UMIN000006899.
| Trial name or title | Randomized phase II study of biweekly cetuximab versus panitumumab in patients not combination of irinotecan wild‐type KRAS metastatic colorectal cancer following treatment with fluoropyrimidine and oxaliplatin chemotherapy |
| Methods | Phase II open‐label, randomised trial |
| Participants | People with KRAS exon 2 WT mCRC |
| Interventions | Panitumumab (Arm 1), cetuximab (Arm 2) |
| Outcomes | Primary outcome: OS. Secondary outcomes: response rate, time to treatment failure, PFS, safety |
| Starting date | December 2011 |
| Contact information | Koichi Taira, Machida Gastrointestinal Hospital, Department of Clinical Oncology, +81‐666491251, koichit@iris.eonet.ne.jp |
| Notes |
Venook CALGB 80203 2006.
| Trial name or title | CALGB 80203 |
| Methods | Phase 3 randomised controlled trial; n = 238 |
| Participants | People with metastatic colorectal cancer who are treatment‐naive |
| Interventions | FOLFIRI (Arm A), FOLFIRI with cetuximab (Arm B), FOLFOX (Arm C), FOLFOX with cetuximab (Arm D) |
| Outcomes | Primary endpoint: OS. Secondary endpoints: TRR, PFS, duration of response, toxicity |
| Starting date | December 2003 |
| Contact information | Alan Venook, Alan.Venook@ucsf.edu |
| Notes | Study closed early after 12 months of accrual after emergence of efficacy data for bevacizumab; awaiting efficacy results (lead author advises in private communication these may be available Q1‐2 2016). |
VISNU‐2.
| Trial name or title | Influence of BRAF and PIK3K Status on the Efficacy of 5‐Fluorouracil/Leucovorin/Oxaliplatin (FOLFIRI) Plus Bevacizumab or Cetuximab in Patients With RAS Wild‐type Metastatic Colorectal Carcinoma and < 3 Circulating Tumor Cells (CTC) (VISNU‐2) |
| Methods | Phase II open‐label, parallel assessment, randomised study |
| Participants | 240 RAS WT metastatic colorectal carcinoma and < 3 circulating tumour cells |
| Interventions | Arm A: FOLFIRI with bevacizumab. Arm B: FOLFIRI with cetuximab |
| Outcomes | Primary: PFS, Secondary: OS, RR, radical resection, CTC count basal and correlate to PFS, OS, RR; correlation of molecular status of biomarkers related to the cellular and tumoural reproduction and/or mode of action and clinical antitumour activity outcome (PFS, OS, RR) |
| Starting date | July 2012 |
| Contact information | Inmaculada Ruiz de Mena, PhD 00 34 91 378 82 75 |
| Notes | Study ongoing but closed to accrual. |
VOLFI.
| Trial name or title | An Open‐label 2:1 Randomized Phase II Study of Panitumumab Plus FOLFOXIRI or FOLFOXIRI Alone as First‐line Treatment of Patients With Non‐resectable Metastatic Colorectal Cancer and RAS Wild Type |
| Methods | Phase II randomised study |
| Participants | 93 non‐resectable mCRC and RAS WT |
| Interventions | Experimental: A (FOLFOXIRI with panitumumab) Active Comparator: B (FOLFOXIRI) |
| Outcomes | Primary: ORR. Secondary: resection rate with curative intent, disease control rate, PFS, duration of response, time to response, toxicity, quality of life |
| Starting date | April 2011 |
| Contact information | Michael Geißler, MD, PhD m.geissler@klinikum‐esslingen.de |
| Notes | Trial ongoing. |
WJOG6510G.
| Trial name or title | Randomized phase II study of panitumumab (Pmab) plus irinotecan (CPT‐11) versus cetuximab (Cmab) plus CPT‐11 in patients with KRAS wild‐type (WT) metastatic colorectal cancer (mCRC) following treatment with fluoropyrimidine, CPT‐11, and oxaliplatin (L‐OHP) chemotherapy: WJOG6510G |
| Methods | Phase II open‐label, randomised trial |
| Participants | People with KRAS exon 2 WT mCRC |
| Interventions | Irinotecan with cetuximab (Arm A), irinotecan with panitumumab (Arm B) |
| Outcomes | Primary outcome: PFS. Secondary outcomes: OS, response rate, disease control rate, safety |
| Starting date | December 2011 |
| Contact information | Shinichiro Nakamura, West Japan Oncology Group, +81‐6‐6633‐7400, datacenter@wjog.jp |
| Notes | Statistical calculations based on non‐inferiority design. Preliminary data published ASCO GI 2017; awaiting full publication. |
5‐FU: fluorouracil BSC: best supportive care CA 19‐9: carbohydrate antigen 19‐9 CEA: carcinoembryonic antigen CEC: circulating endothelial cells CRC: colorectal cancer CTC: circulating tumour cell CTCAE: Common Terminology Criteria for Adverse Events ECOG Eastern Cooperative Oncology Group IV: intravenously MAb: monoclonal antibodies mCRC: metastatic colorectal cancer MT: mutant ORR: overall response rate OS: overall survival PFS: progression‐free survival RR: response rate TRR: tumour response rate WT: wild type
Differences between protocol and review
The initial protocol specified subgroup analyses according to the degree of EGFR expression, presence of KRAS mutations, presence of BRAF mutations, known patient prognostic factors, and presence of skin toxicity. Since the protocol was written, understanding of the critical nature of KRAS in predicting efficacy of EGFR inhibition has increased. We therefore made the decision to focus on KRAS mutations.
Upon discussion with the trials review committee (on 30 March 2015), we carried out a further subgroup analysis on so‐called extended RAS mutations, testing for KRAS (exons other than exon 2) and NRAS.
We removed the outcome of cost‐effectiveness since the publication of the protocol due to the lack of published evidence in the area, with the few published studies employing different measures of cost‐effectiveness and assuming local costs, which vary considerably between countries (Graham 2014).
The introduction of EGFR inhibitors into widespread clinical use has led to robust debate regarding the optimal setting for EGFR inhibitor use ‐ whether early in the treatment paradigm with first‐line chemotherapy or reserving it as monotherapy after failure of chemotherapy. In view of this uncertainty, we decided to perform subgroup analyses by line of therapy (first‐line, second‐line, or third‐line and beyond).
Contributions of authors
DC performed the updated literature search, carried out the statistical analysis, and was responsible for writing the manuscript.
ES assisted in the literature search and major critical review of the manuscript.
RW assisted in the literature search and data extraction, as well as writing of the manuscript.
AS assisted in the literature search and data extraction, as well as writing of the manuscript.
RH developed the protocol for the review.
BL critically reviewed the manuscript.
NT critically reviewed the manuscript.
TP critically reviewed the manuscript and suggested the extended RAS amendment.
NP contributed to the writing of the protocol, advised DC in carrying out statistical analysis, contributed to development of the discussion, and co‐ordinated the project.
Sources of support
Internal sources
No relevant sources of support, Other.
External sources
-
NIH Grant supported, USA.
Core Grant (P30CA008748) at Memorial Sloan Kettering Cancer Center from the National Institutes of Health, USA.
Declarations of interest
David Lok Hang Chan has no competing interests to declare.
Eva Segelov has received travel grant support from Roche, Merck Serono, Amgen, and Sanofi‐Aventis. She has attended advisory boards for Roche, Merck Serono, and Sanofi‐Aventis.
Rachel Wong has no competing interests to declare.
Annabel Smith has no competing interests to declare.
Rebecca A Herbertson has no competing interests to declare.
Bob Li has no competing interests to declare.
Niall Tebbutt has received speaking honoraria and travel grant support from Roche, Amgen, Merck, and Sanofi‐Aventis. He has attended advisory boards for Roche, Sanofi‐Aventis, Amgen, and Merck.
Timothy Price has received speaking honoraria or travel grant support, or both from Roche, Amgen, Pfizer, and Sanofi‐Aventis. He has attended advisory boards for Roche, Merck Australia, Amgen, and Sanofi‐Aventis, and received research funding from Sanofi‐Aventis.
Nick Pavlakis has received speaking honoraria and travel grant support from Roche, Amgen, and Sanofi‐Aventis. He has attended advisory boards for Roche, Alphapharm, Amgen, and Sanofi‐Aventis, and received research funding from Sanofi‐Aventis.
Edited (no change to conclusions)
References
References to studies included in this review
Adams COIN 2011 {published data only (unpublished sought but not used)}
- Adams RA, Fisher D, Farragher S, Jasani B, Smith CG, James MD, et al. Use of epiregulin (EREG) and amphiregulin (AREG) gene expression to predict response to cetuximab (cet) in combination with oxaliplatin (Ox) and 5FU in the first‐line treatment of advanced colorectal cancer (aCRC) [COIN trial]. Journal of Clinical Oncology 2012;30(Suppl):Abstr 32. [10.1200/jco.2012.30.30_suppl.32 ] [Google Scholar]
- Adams RA, Fisher D, Farragher S, Scott A, Smith CG, James MD, et al. Epiregulin (EREG) and amphiregulin (AREG) gene expression to predict response to cetuximab therapy in combination with oxaliplatin (Ox) and 5FU in first‐line treatment of advanced colorectal cancer (aCRC) [COIN Trial]. Journal of Clinical Oncology 2012;30(Suppl):Abstr 3516. [Google Scholar]
- Adams RA, James MD, Smith CG, Wilson RH, Fisher D, Kenny SL, et al. Epidermal growth factor receptor (EGFR) as a predictive and prognostic marker in patients with advanced colorectal cancer (aCRC): The MRC COIN trial experience [COIN Trial]. Journal of Clinical Oncology Conference: 2011 Gastrointestinal Cancers Symposium 2011;29(Suppl 4):Abstr 359. [Google Scholar]
- Adams RA, Meade AM, Madi A, Fisher D, Kay E, Kenny S, et al. Toxicity associated with combination oxaliplatin plus fluoropyrimidine with or without cetuximab in the MRC COIN trial experience. British Journal of Cancer 2009;100(2):251‐8. [DOI: 10.1038/sj.bjc.6604877] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams RA, Meade AM, Seymour MT, Wilson RH, Madi A, Fisher D, et al. Addition of cetuximab to oxaliplatin‐based first‐line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet 2011;377:2013‐4. [:10.1016/S0140‐ 6736(11)60613‐2] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams RA, Meade AM, Seymour MT, Wilson RH, Madi A, Fisher D, et al. Intermittent versus continuous oxaliplatin and fluoropyrimidine combination chemotherapy for first‐line treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet Oncology 2011;12(7):642‐53. [10.1016/S1470‐ 2045(11)70102‐4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Amado 2008 {published data only}
- Amado RG, Wolf M, Peeters M, Cutsem E, Siena S, Freeman DJ, et al. Wild‐type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. Journal of Clinical Oncology 2008;26(10):1626‐34. [DOI: 10.1200/JCO.2007.14.7116] [DOI] [PubMed] [Google Scholar]
- Patterson SD, Peeters M, Siena S, Cutsem E, Humblet Y, Laethem J, et al. Comprehensive analysis of KRAS and NRAS mutations as predictive biomarkers for single agent panitumumab (pmab) response in a randomized, phase III metastatic colorectal cancer (mCRC) study (20020408). Journal of Clinical Oncology 2013;31:(Suppl; abstr 3617). [Google Scholar]
- Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, et al. Open‐label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy‐refractory metastatic colorectal cancer. Journal of Clinical Oncology 2007;25(13):1658‐64. [DOI] [PubMed] [Google Scholar]
Bokemeyer OPUS 2009 {published data only}
- Bokemeyer C, Bondarenko I, Hartmann JT, Baurd F, Schuch G, Zubel A, et al. Efficacy according to biomarker status of cetuximab plus FOLFOX‐4 as first‐line treatment for metastatic colorectal cancer. Annals of Oncology 2011;22(7):1535‐46. [DOI] [PubMed] [Google Scholar]
- Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, Braud F, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first‐line treatment of metastatic colorectal cancer. Journal of Clinical Oncology 2009;27(5):663‐71. [DOI: 10.1200/JCO.2008.20.8397] [DOI] [PubMed] [Google Scholar]
- Bokemeyer C, Boondarendo I, Hartmann JT, Braud F, Schuch G, Zubel A, et al. Overall survival of patients with KRAS wild‐type tumours treated with FOLFOX4 +/‐ cetuximab as 1st‐line treatment for metastatic colorectal cancer. European Journal of Cancer 2009;7(2):346. [Abstract number: 6079] [Google Scholar]
- Bokemeyer C, Kohne CH, Ciardiello F, Lenz HJ, Heinemann V, Klinkhardt U, et al. FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. European Journal of Cancer 2015;51(10):1243‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Borner 2008 {published and unpublished data}
- Borner M, Koeberle D, Moos R, Saletti P, Rauch D, Hess V, et al. Adding cetuximab to capecitabine plus oxaliplatin (XELOX) in first‐line treatment of metastatic colorectal cancer: a randomized phase II trial of the Swiss Group for Clinical Cancer Research SAKK. Annals of Oncology 2008;19(7):1288‐92. [DOI: 10.1093/annonc/mdn05] [DOI] [PubMed] [Google Scholar]
- Borner M, Mingrone W, Koeberle D, Moos R, Rauch D, Saletti P, et al. The impact of cetuximab on the capecitabine plus oxaliplatin (XELOX) combination in first‐line treatment of metastatic colorectal cancer (MCC): A randomized phase II trial of the Swiss Group for Clinical Cancer Research (SAKK). Journal of Clinical Oncology 2006;24(18S):3551. [DOI] [PubMed] [Google Scholar]
Bridgewater GAIN‐C 2015 {published data only (unpublished sought but not used)}
- Bridgewater JA, Cervantes A, Markman B, Siena S, Cubillo A, Carbonero RG, et al. GAIN‐(C): Efficacy and safety analysis of imgatuzumab (GA201), a novel dual‐acting monoclonal antibody (mAb) designed to enhance antibody‐dependent cellular cytotoxicity (ADCC), in combination with FOLFIRI compared to cetuximab plus FOLFIRI in second‐line KRAS exon 2 wild type (e2WT) or with FOLFIRI alone in mutated (e2MT) metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2015;33:(Suppl 3; abstr 669). [Google Scholar]
- Cervantes‐Ruiperez A, Markman B, Siena S, Pericay C, Aprile G, Bridgewater JA, et al. The GAIN‐C study (BP25438): Randomized phase II trial of RG7160 (GA201) plus FOLFIRI, compared to cetuximab plus FOLFIRI or FOLFIRI alone in second‐line KRAS wild type (WT) or mutant metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2012;30:(Suppl; abstr TPS3637). [Google Scholar]
Brodowicz 2013 {published data only}
- Brodowicz T, Ciuleanu T, Radosavljevic D, Shacham‐Shmueli E, Vrbanec D, Plate S, et al. FOLFOX4 plus cetuximab administered weekly or every second week in the first‐line treatment of patients with KRAS wild‐type metastatic colorectal cancer: a randomized phase II CECOG study. Annals of Oncology 2013;24:1769‐77. [DOI] [PubMed] [Google Scholar]
- Brodowicz T, Vrbanec D, Kaczirek K, Ciuleanu T, Knittelfelder R, Lindner E, et al. FOLFOX4 plus cetuximab administered weekly or every two weeks in first‐line treatment of patients with KRAS andNRAS wild‐type (wt) metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2014;32(Suppl 3):abstr LBA391^. [Google Scholar]
- Ciuleanu T, Nikolic V, Shmueli E, Vrbanec D, Plate S, Krmpotic ZM, et al. Cetuximab weekly (q1w) versus every two weeks (q2w) plus FOLFOX4 as first‐line therapy in patients (pts) with KRAS wild‐type (wt) metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2011;29:(Suppl; abstr 3580). [Google Scholar]
Ciardiello CAPRI‐GOIM 2016 {published data only}
- Ciardiello F, Barone C, Cartenì G, Rachiglio AM, Montesarchio V, Tonini G, et al. Clinical activity of FOLFIRI plus cetuximab according to extended gene mutation status by next‐generation sequencing: findings from the CAPRI‐GOIM trial. Annals of Oncology 2014;25:1756‐61. [DOI] [PubMed] [Google Scholar]
- Ciardiello F, Maiello E, Pisconti S, Giuliani F, Barone C, Rizzo M, et al. Optimal treatment strategy in KRAS wild type (wt) metastatic colorectal cancer (mCRC): Cetuximab plus FOLFIRI followed by FOLFOX4 with or without cetuximab ‐ the Capri trial from the Gruppo Oncologico Dell’Italia Meridionale (GOIM). Journal of Clinical Oncology 2013;31:Suppl; abstr e14565. [Google Scholar]
- Ciardiello F, Normanno N, Martinelli E, Troiani T, Cardone C, Nappi A, et al. Cetuximab beyond progression in RAS wild type (WT) metastatic colorectal cancer (mCRC): the CAPRI‐GOIM randomized phase II study of FOLFOX versus FOLFOX plus cetuximab. Annals of Oncology 2015;26 (Suppl 4):iv120‐1: Abstr LBA‐09. [Google Scholar]
- Ciardiello F, Normanno N, Martinelli E, Troiani T, Pisconti S, Cardone C, et al. Cetuximab continuation after first progression in metastatic colorectal cancer (CAPRI‐GOIM): a randomized phase II trial of FOLFOX plus cetuximab versus FOLFOX. Annals of Oncology 2016;27(6):1055‐61. [DOI] [PubMed] [Google Scholar]
Douillard PRIME 2010 {published data only}
- Douillard J, Cassidy J, Jassem J, Rivera F, Kocakova I, Rogowski W, et al. Randomized, open‐label, phase III study of panitumumab (pmab) with FOLFOX4 versus FOLFOX4 alone as first‐line treatment (tx) for metastatic colorectal cancer (mCRC): Efficacy by skin toxicity (ST). Journal of Clinical Oncology 2010;28(15 Suppl):Abstr 3528. [10.1200/jco.2010.28.15_suppl.3528 ] [Google Scholar]
- Douillard J, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, et al. Randomized phase 3 study of panitumumab (PMAB) with folfox4 compared to folfox4 alone as first line treatment (TX) for metastatic colorectal cancer (MCRC): prime trial. Journal of Clinical Oncology 2010;28(31):4697‐705. [DOI: 10.1200/JCO.2009.27.4860] [DOI] [PubMed] [Google Scholar]
- Douillard J, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel ME, et al. Randomized phase 3 study of panitumumab with FOLFOX4 compared to FOLFOX4 alone as 1st‐line treatment (tx) for metastatic colorectal cancer (mCRC). Joint ECCO 15 ‐ 34th ESMO Multidisciplinary Congress; 2009 Sep 20‐24; Berlin, Germany. 2009.
- Douillard J, Siena S, Cassidy J, Tabernero J, Burkes RL, Barugel ME, et al. Final results from PRIME: Randomized phase III study of panitumumab (pmab) with FOLFOX4 for first line metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2011;29(Suppl):Abstr 3510. [DOI] [PubMed] [Google Scholar]
- Douillard J, Siena S, Tabernero J, Burkes RL, Barugel ME, Humblet Y, et al. Overall survival (OS) analysis from PRIME: Randomized phase III study of panitumumab (pmab) with FOLFOX4 for first‐line metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2013;31(Suppl):Abstr 3620. [Google Scholar]
- Douillard JY, Siena S, Tabernero J, Burkes RL, Barugel ME, Humblet Y, et al. Final skin toxicity (ST) and patient‐reported outcomes (PRO) results from PRIME: A randomized phase III study of panitumumab (pmab) plus FOLFOX4 (CT) for first‐line metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2012;30(Suppl 4):Abstr 531. [Google Scholar]
- Oliner KS, Douillard JY, Siena S, Tabernero J, Burkes RL, Barugel ME, et al. Analysis of KRAS/NRAS and BRAF mutations in the phase III PRIME study of panitumumab (pmab) plus FOLFOX versus FOLFOX as first‐line treatment (tx) for metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2013;31(15_Suppl):3511. [Google Scholar]
- Siena S, Cassidy J, Tabernero J, Burkes RL, Barugel ME, Humblet Y, et al. Randomized phase III study of panitumumab (pmab) with FOLFOX4 compared with FOLFOX4 alone as first line treatment (tx) for metastatic colorectal cancer (mCRC): Results by Eastern Cooperative Oncology Group (ECOG) performance status (PS). Journal of Clinical Oncology 2011;29(15 Suppl):3567. [Google Scholar]
- Siena S, Douillard JY, Cassidy J, Tabernero J, Burkes R, Barugel ME, et al. Study 20050203/PRIME ‐ Effect of post‐progression anti‐epidermal growth factor receptor (EGFR) monoclonal antibody (mAb) therapy in patients with wild‐type (WT) KRAS metastatic colorectal cancer (mCRC). European Journal of Cancer 2011;47(Suppl 1):S435. [Google Scholar]
- Siena S, Douillard JY, Tabernero J, Cassidy J, Burkes R, Barugel ME, et al. Prime study: A randomised phase 3 study of panitumumab with FOLFOX4 versus FOLFOX4 alone as first‐line treatment for metastatic colorectal cancer (mCRC). Annals of Oncology 2010;21(Suppl 1):I13. [DOI] [PubMed] [Google Scholar]
- Siena S, Tabarnero J, Burkes RL, Cassidy J, Cunningham D, Barugel ME, et al. Phase III study (PRIME/20050203) of panitumumab (pmab) with FOLFOX compared with FOLFOX alone in patients (pts) with previously untreated metastatic colorectal cancer (mCRC): Pooled safety data [abstract no. 4034]. Journal of Clinical Oncology 2008;26(15s Part 1):186. [Google Scholar]
- Siena S, Tabernero J, Cunningham D, Koralewski P, Ruff P, Rother M, et al. Randomized phase III study of panitumumab (pmab) with FOLFOX4 compared to FOLFOX4 alone as first‐line treatment (tx) for metastatic colorectal cancer (mCRC): PRIME trial analysis by epidermal growth factor receptor (EGFR) tumor staining. Journal of Clinical Oncology Conference: 2010 Annual Meeting of the American Society of Clinical Oncology, ASCO 2010;28(15 Suppl):3566. [DOI: 10.1200/jco.2010.28.15_suppl.3566] [DOI] [Google Scholar]
Hagman ACT2 2014 {published and unpublished data}
- Hagman H, Frodin J, Berglund AM, Sundberg J, Vestermark LW, Albertsson M, et al. A randomized study of KRAS stratified maintenance therapy with bevacizumab, erlotinib or metronomic capecitabine after first line induction treatment of metastatic colorectal cancer: the Nordic ACT2 trial. Annals of Oncology 2014;25(Suppl 4):iv167‐209, 516P. [DOI] [PubMed] [Google Scholar]
- Hagman H, Frodin JE, Berglund A, Sundberg J, Vestermark LW, Albertsson M, et al. A randomized study of KRAS‐guided maintenance therapy with bevacizumab, erlotinib or metronomic capecitabine after first‐line induction treatment of metastatic colorectal cancer: the Nordic ACT2 trial. Annals of Oncology 2016;27(1):140‐7. [DOI] [PubMed] [Google Scholar]
Hecht PACCE 2009 {published data only}
- Hecht JR, Mitchell E, Chidiac T, Scroggin C, Hagenstad C, Spigel D, et al. A randomized phase IIIB trial of chemotherapy, bevacizumab, and panitumumab compared with chemotherapy and bevacizumab alone for metastatic colorectal cancer. Journal of Clinical Oncology 2008;27:672‐80. [DOI: 10.1200/JCO.2008.19.8135] [DOI] [PubMed] [Google Scholar]
Hecht SPIRITT 2015 {published data only}
- Cohn AL, Krishnan K, Hecht JR. SPIRITT: A multicenter, open‐label, randomized, phase II clinical trial evaluating safety and efficacy of FOLFIRI with either panitumumab or bevacizumab as second‐line treatment in patients with metastatic colorectal cancer (mCRC) with wild‐type KRAS tumors. Journal of Clinical Oncology 2010;28 (Suppl 15):30. [abstract no.TPS195] [Google Scholar]
- Hecht JR, Cohn A, Dakhil S, Saleh M, Piperdi B, Cline‐Burkhardt M, et al. SPIRITT: A randomized, multicenter, phase II study of panitumumab with FOLFIRI and bevacizumab with FOLFIRI as second‐line treatment in patients with unresectable wild type KRAS metastatic colorectal cancer. Clinical Colorectal Cancer 2015;14(2):72‐80. [DOI] [PubMed] [Google Scholar]
- Hecht JR, Cohn AL. SPIRITT: A study of second‐line treatment of metastatic colorectal cancer with FOLFIRI plus panitumumab or bevacizumab. Community Oncology 2008;5(11 (Suppl B)):1‐4. [Google Scholar]
- Hecht JR, Cohn AL, Dakhil SR, Saleh MN, Piperdi B, Cline‐Burkhardt VJM, et al. SPIRITT (study 20060141): A randomized phase II study of FOLFIRI with either panitumumab (pmab) or bevacizumab (bev) as second ‐line treatment (tx) in patients (pts) with wild‐type (WT) KRAS metastatic colorectal cancer (mCRC) [SPIRITT]. Journal of Clinical Oncology 2013;31(4 Suppl):Abstr 454. [Google Scholar]
- Hecht JR, Dakhil SR, Saleh MN, Piperdi B, Cline‐Burkhardt M, Kocs DM, et al. Pooled safety results from SPIRITT: A multicenter, open‐label, randomized, phase II study of FOLFIRI with panitumumab (pmab) or bevacizumab (bev) as second‐line treatment (tx) in patients (pts) with metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2011;29(4 Suppl):Abstr 477. [Google Scholar]
- Hecht JR, Mitchell E, Chidiac T, Scroggin C, Hagenstad C, Spigel D, et al. A randomized phase IIIB trial of chemotherapy, bevacizumab, and panitumumab compared with chemotherapy and bevacizumab alone for metastatic colorectal cancer. Journal of Clinical Oncology 2009;27(5):672‐80. [DOI: ] [DOI] [PubMed] [Google Scholar]
Heinemann FIRE‐3 2014 {published data only}
- Heinemann V, Fischer von Weikersthal L, Decker T, Kiani A, Vehling‐Kaiser U, Al‐Batran S, et al. Randomized comparison of FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first‐line treatment of KRAS wild‐type metastatic colorectal cancer: German AIO study KRK‐0306 (FIRE‐3). Journal of Clinical Oncology 2013;31(18 Suppl):LBA3506. [DOI: 10.1200/jco.2013.31.18_suppl.lba3506] [DOI] [Google Scholar]
- Heinemann V, Weikersthal LF, Decker T, Kiani A, Vehling‐Kaiser U, Al‐Batran SE, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first‐line treatment for patients with metastatic colorectal cancer (FIRE‐3): a randomised, open‐label, phase 3 trial. Lancet Oncology 2014;15(10):1065‐75. [DOI: 10.1016/S1470-2045(14)70330-4] [DOI] [PubMed] [Google Scholar]
- Stintzing S, Fischer von Weikersthal L, Decker T, Vehling‐Kaiser U, Jager E, Heintges T, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first‐line treatment for patients with metastatic colorectal cancer ‐ subgroup analysis of patients with KRAS: mutated tumours in the randomised German AIO study KRK‐0306. Annals of Oncology 2012;23(7):1693‐9. [DOI] [PubMed] [Google Scholar]
- Stintzing S, Neumann J, Jung A, Fischer von Weikersthal L, Decker T, Vehling‐Kaiser U, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first‐line treatment for patients with metastatic colorectal cancer (mCRC): Analysis of patients with KRAS‐mutated tumors in the randomized German AIO study KRK‐0306. Journal of Clinical Oncology 2011;29(15 Suppl):3575. [DOI] [PubMed] [Google Scholar]
Hickish 2014 {published and unpublished data}
- Hickish T, Cassidy J, Propper D, Chau I, Falk S, Ford H, et al. A randomised, open‐label phase II trial of afatinib versus cetuximab in patients with metastatic colorectal cancer. European Journal of Cancer 2014;50(18):3136‐44. [DOI: 10.1016/j.ejca.2014.08.008] [DOI] [PubMed] [Google Scholar]
Johnsson Nordic ACT 2013 {published data only}
- Johnsson A, Frodin J, Berglund A, Hagman H, Sundberg J, Bergstrom D, et al. A randomized phase III trial on maintenance treatment with bevacizumab (bev) alone or in combination with erlotinib (erlo) after chemotherapy and bev in metastatic colorectal cancer (mCRC). Journal of Clinical Oncology Conference: ASCO Annual Meeting 2011 2011;29(15 Suppl):3526. [DOI: 10.1200/jco.2011.29.15_suppl.3526] [DOI] [Google Scholar]
- Johnsson A, Hagman H, Frodin JE, Berglund A, Keldsen N, Fernebro E, et al. A randomized phase III trial on maintenance treatment with bevacizumab alone or in combination with erlotinib after chemotherapy and bevacizumab in metastatic colorectal cancer: the Nordic ACT Trial. Annals of Oncology 2013;24(9):2335‐41. [DOI] [PubMed] [Google Scholar]
Karapetis CO17 2008 {published data only (unpublished sought but not used)}
- Asmis TR, Powell E, Karapetis CS, Jonker DJ, Tu D, Jeffery M, et al. Comorbidity, age and overall survival in cetuximab‐treated patients with advanced colorectal cancer (ACRC) ‐ results from NCIC CTG CO.17: a phase III trial of cetuximab versus best supportive care. Annals of Oncology 2011;22(1):118‐26. [DOI: 10.1093/annonc/mdq309] [DOI] [PubMed] [Google Scholar]
- Au HJ, Karapetis CS, O'Callaghan CJ, Tu D, Moore MJ, Zalcberg JR, et al. Health‐related quality of life in patients with advanced colorectal cancer treated with cetuximab: Overall and KRAS‐specific results of the NCIC CTG and AGITG CO.17 Trial. Journal of Clinical Oncology 2009;27(11):1822‐8. [DOI: 10.1200/JCO.2008.19.6048] [DOI] [PubMed] [Google Scholar]
- Elimova E, O'Callaghan CJ, Tu D, Karapetis CS, Price TJ, Zhu L, et al. Cetuximab (CET)‐related hypersensitivity reactions (HSRs): An analysis of timing, demographics, and outcomes from the AGITG/NCIC CTG CO.17 trial. Journal of Clinical Oncology 2011;29(15 Suppl):3624. [DOI: 10.1200/jco.2011.29.15_suppl.3624] [DOI] [Google Scholar]
- Jonker DJ, Karapetis CS, O'Callaghan CJ, Marginean C, Zalcberg JR, Simes J, et al. BRAF, PIK3CA, and PTEN status and benefit from cetuximab (CET) in the treatment of advanced colorectal cancer (CRC): Results from NCIC CTG/AGITG CO.17. Journal of Clinical Oncology 2012;30(15 Suppl):3515. [Google Scholar]
- Jonker DJ, Karapetis C, Harbison C, O'Callaghan CJ, Tu D, Simes RJ, et al. High epiregulin (EREG) gene expression plus K‐ras wild‐type (WT) status as predictors of cetuximab benefit in the treatment of advanced colorectal cancer (ACRC): Results from NCIC CTG CO.17‐A phase III trial of cetuximab versus best supportive care (BSC). Journal of Clinical Oncology 2009;27(15 Suppl):4016. [Google Scholar]
- Karapetis C, Jonker D, O'Callaghan C. Cetuximab plus BSC versus BSC alone in the treatment of metastatic EGFR‐positive colorectal cancer. Signal 2005;60(1):15‐7. [Google Scholar]
- Karapetis C, Khambata‐Ford S, Jonker D, O'Callaghan C, Tu D, Vachan B, et al. KRAS mutation status is a predictive biomarker for cetuximab benefit in the treatment of advanced colorectal cancer ‐ results from NCIC CTG CO.17: A phase III trial of cetuximab versus best supportive care. Annals of Oncology 2008;19(Suppl 8):viii2. [Google Scholar]
- Karapetis CS, Khambata‐Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, et al. K‐ras mutations and benefit from cetuximab in advanced colorectal cancer. New England Journal of Medicine 2008;359(17):1757‐65. [NCT00079066] [DOI] [PubMed] [Google Scholar]
- Mittmann N, Au HJ, Tu D, O'Callaghan CJ, Isogai PK, Karapetis CS, et al. Prospective cost‐effectiveness analysis of cetuximab in metastatic colorectal cancer: Evaluation of National Cancer Institute of Canada Clinical Trials Group CO.17 Trial. Journal of the National Cancer Institute 2009;101(17):1182‐92. [DOI] [PubMed] [Google Scholar]
- O'Callaghan CJ, Tu D, Karapetis CS, Au H, Moore MJ, Tebbutt NC, et al. The relationship between the development of rash and clinical and health‐related quality of life outcomes by KRAS mutation status in patients with colorectal cancer treated with cetuximab in NCIC CTG CO.17. Journal of Clinical Oncology 2011;29(15 Suppl):3588. [Google Scholar]
- Powell ED, Asmis T, Jonker D, Tu D, Karapetis C, Jeffery M, et al. Comorbidity and overall survival (OS) in cetuximab‐treated patients with advanced colorectal cancer (ACRC) ‐ results from NCIC CTG CO.17: A phase III trial of cetuximab versus best supportive care (BSC). Journal of Clinical Oncology 2009;27(15 Suppl):4074. [DOI] [PubMed] [Google Scholar]
Ma 2013 {published data only (unpublished sought but not used)}
- Ma BB, Chan SL, Ho WM, Lau W, Mo F, Hui EP, et al. Intermittent versus continuous erlotinib with concomitant modified "XELOX" (q3W) in first‐line treatment of metastatic colorectal cancer: correlation with serum amphiregulin and transforming growth factor alpha. Cancer 2013;119(23):4145‐53. [DOI] [PubMed] [Google Scholar]
Passardi ITACA 2015 {published data only}
- Fabbri F, Passardi A, Ravaioli A, Cavanna L, Luppi G, Mucciarini C, et al. Sequential treatment strategy for metastatic colorectal cancer: A phase III study of chemotherapy (CT) with or without bevacizumab (Bev) as first‐line therapy followed by two phase III studies of CT alone or CT plus bev with or without cetuximab (Cetux) as second‐line therapy (ITACa Study IRST 153 01). Journal of Clinical Oncology Conference, ASCO Annual Meeting 2011;29(15 Suppl):Abstr e14161. [Google Scholar]
- Passardi A, Scarpi E, Fontana A, Cavanna L, Ruscelli S, Turci D, et al. Impact of second‐line cetuximab‐containing therapy in patients with KRAS wild type metastatic colorectal cancer: results from ITACa trial. Annals of Oncology 2015;26 (Suppl 4):iv83‐4: Abstr P‐283. [Google Scholar]
Peeters 2010 {published data only (unpublished sought but not used)}
- Andre T, Peeters M, Price T, Hotko Y, Cervantes A, Ducreux M, et al. Panitumumab with FOLFIRI vs FOLFIRI alone: A randomised phase 3 study for the second line treatment of patients (PTS) with metastatic colorectal cancer (mCRC). Annals of Oncology 2010;21(Suppl 1):I13. [DOI] [PubMed] [Google Scholar]
- Bennett L, Zhao Z, Barber B, Zhou X, Peeters M, Zhang J, et al. Health‐related quality of life in patients with metastatic colorectal cancer treated with panitumumab in first‐ or second‐line treatment. Journal of Clinical Oncology 2011;29(15 Suppl):Abstr e19500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliner K, Peeters M, Siena S, Cutsem E, Huang J, Humblet Y, et al. Evaluation of gene mutations beyond KRAS as predictive biomarkers of response to panitumumab in a randomized, phase III monotherapy study of metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2011;29(15 Suppl):3530. [Google Scholar]
- Patterson SD, Peeters M, Siena S, Cutsem E, Humblet Y, Laethem J, et al. Comprehensive analysis of KRAS and NRAS mutations as predictive biomarkers for single agent panitumumab (pmab) response in a randomized, phase III metastatic colorectal cancer (mCRC) study (20020408). Journal of Clinical Oncology 2013;31(15 Suppl):3617. [Google Scholar]
- Peeters M, Cervantes‐Ruiperez A, Strickland A, Ciuleanu T, Mainwaring PN, Tzekova VI, et al. Randomized phase III study of panitumumab (pmab) with FOLFIRI versus FOLFIRI alone as second‐line treatment (tx) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis by tumor epidermal growth factor receptor (EGFR) staining. Journal of Clinical Oncology 2010;28:15s. [Google Scholar]
- Peeters M, Oliner KS, Parker A, Siena S, Cutsem E, Huang J, et al. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clinical Cancer Research 2013;19(7):1902‐12. [DOI: 10.1158/1078-0432.CCR-12-1913] [DOI] [PubMed] [Google Scholar]
- Peeters M, Oliner KS, Price TJ, Cervantes A, Sobrero AF, Ducreux M, et al. Analysis of KRAS/NRAS mutations in a phase III study of panitumumab with FOLFIRI compared with FOLFIRI alone as second‐line treatment for metastatic colorectal cancer. Clinical Cancer Research 2015;21(24):5469‐79. [DOI] [PubMed] [Google Scholar]
- Peeters M, Oliner KS, Price TJ, Cervantes A, Sobrero AF, Ducreux M, et al. Analysis of KRAS/NRAS mutations in phase 3 study 20050181 of panitumumab (pmab) plus FOLFIRI versus FOLFIRI for second‐line treatment (tx) of metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2014;32:Suppl 3; abstr LBA387. [Google Scholar]
- Peeters M, Price T, Hotko Y, Cervantes A, Ducreux M, Andre T, et al. Randomized phase 3 study of panitumumab (PMAB) with FOLFIRI compared to FOLFIRI alone as second‐line treatment (TX) for metastatic colorectal cancer (MCRC): secondary endpoint results. Annals of Oncology 2010;21:15. [Google Scholar]
- Peeters M, Price T, Hotko Y, Cervantes A, Ducreux M, Andre T, et al. Randomized phase 3 study of panitumumab with FOLFIRI vs FOLFIRI alone as second‐line treatment (tx) in patients (pts) with metastatic colorectal cancer (mCRC). European Journal of Cancer, Supplement Conference: Joint ECCO 15 ‐ 34th ESMO Multidisciplinary Congress; 2009 Sept 20‐24; Berlin, Germany. 2009.
- Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second‐line treatment in patients with metastatic colorectal cancer. Journal of Clinical Oncology 2008;28(31):4706‐13. [DOI: 10.1200/JCO.2009.27.605] [DOI] [PubMed] [Google Scholar]
- Peeters M, Siena S, Cutsem E, Sobrero A, Hendlisz A, Cascinu S, et al. Association of progression‐free survival, overall survival, and patient‐reported outcomes by skin toxicity and KRAS status in patients receiving panitumumab monotherapy. Cancer 2009;115(7):1544‐54. [DOI: 10.1002/cncr.24088] [DOI] [PubMed] [Google Scholar]
- Peeters M, Wilson G, Ducreux M, Cervantes A, Andre T, Hotko Y, et al. Phase III study (2005181) of panitumumab (pmab) with FOLFIRI alone as second‐line treatment (tx) in patients (pts) with metastatic colorectal cancer (mCRC): Pooled safety results. Journal of Clinical Oncology 2008;26(15 Suppl):4064. [Google Scholar]
- Price T, Peeters M, Strickland A, Ciuleanu TE, Scheithauer W, O'Reilly S, et al. Efficacy of panitumumab plus FOLFIRI versus FOLFIRI alone in patients with wild‐type (WT) KRAS metastatic colorectal cancer (mCRC) treated with prior oxaliplatin or bevacizumab regimens: Results from 20050181. European Journal of Cancer 2011;47(Suppl 3):S431. [Google Scholar]
- Price TJ, Sobrero AF, Wilson G, Cutsem E, Aleknaviciene B, Zaniboni A, et al. Randomized, open‐label, phase III study of panitumumab (pmab) with FOLFIRI versus FOLFIRI alone as second‐line treatment (tx) in patients (pts) with metastatic colorectal cancer (mCRC): Efficacy by skin toxicity (ST). Journal of Clinical Oncology 2010;28(15 Suppl):3529. [Google Scholar]
- Sobrero AF, Peeters M, Price TJ, Hotko Y, Andres CR, Ducreux M, et al. Final results from study 181: Randomized phase III study of FOLFIRI with or without panitumumab (pmab) for the treatment of second‐line metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2012;30(4 Suppl):387. [Google Scholar]
- Sobrero AF, Peeters M, Price TJ, Hotko Y, Andres CR, Ducreux M, et al. Final results of study 20050181: A randomized phase III study of FOLFIRI with or without panitumumab (pmab) for the second‐line treatment (tx) of metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2012;30(15 Suppl):3535. [Google Scholar]
Polikoff EXPLORE 2005 {published data only (unpublished sought but not used)}
- Polikoff J, Mitchell EP, Badarinath S, Graham CD, Jennis A, Chen TT, et al. Cetuximab plus FOLFOX for colorectal cancer (EXPLORE): Preliminary efficacy analysis of a randomized phase III trial. Journal of Clinical Oncology 2005;23 (16S):3574. [Google Scholar]
Price ASPECCT 2014 {published data only}
- Price TJ, Peeters M, Kim TW, Li J, Cascinu S, Ruff P, et al. Panitumumab versus cetuximab in patients with chemotherapy‐refractory wild‐type KRAS exon 2 metastatic colorectal cancer (ASPECCT): a randomised, multicentre, open‐label, non‐inferiority phase 3 study. Lancet Oncology 2014;15(6):569‐79. [DOI] [PubMed] [Google Scholar]
Santoro 2008 {published data only (unpublished sought but not used)}
- Santoro A, Comandone A, Rimassa L, Granetti C, Lorusso V, Oliva C, et al. A phase II randomized multicenter trial of gefitinib plus FOLFIRI and FOLFIRI alone in patients with metastatic colorectal cancer.. Annals of Oncology 2008;19(11):1888‐93. [DOI] [PubMed] [Google Scholar]
Schwartzberg PEAK 2014 {published data only}
- Karthaus M, Schwartzberg L, Rivera F, Fasola G, Canon JL, Yu H, et al. Updated overall survival (OS) analysis of novel predictive KRAS/NRAS mutations beyond KRAS exon 2 in PEAK: A 1st‐line phase 2 study of FOLFOX6 plus panitumumab (pmab) or bevacizumab (bev) in metastatic colorectal cancer (mCRC). European Journal of Cancer 2013;49 (Suppl 2):Abstr 2262. [Google Scholar]
- Schwartzberg LS, Rivera F, Karthaus M, Fasola G, Canon JL, Hecht JR, et al. PEAK: a randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild‐type KRAS exon 2 metastatic colorectal cancer. Journal of Clinical Oncology 2014;32(21):2240‐7. [DOI] [PubMed] [Google Scholar]
- Schwartzberg LS, Rivera F, Karthaus M, Fasola G, Canon JL, Yu H, et al. Analysis of KRAS/NRAS mutations in PEAK: A randomized phase II study of FOLFOX6 plus panitumumab (pmab) or bevacizumab (bev) as first‐line treatment (tx) for wild‐type (WT) KRAS (exon 2) metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2013;31:Suppl; abstr 3631. [Google Scholar]
- Schwartzberg LS, Rivera F, Karthaus M, Fasola G, Canon JL, Yu H, et al. PEAK (study 20070509): A randomized phase II study of mFOLFOX6 with either panitumumab (pmab) or bevacizumab (bev) as first‐line treatment (tx) in patients (pts) with unresectable wild‑type (WT) KRAS metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2013;31:Suppl 4; abstr 446. [Google Scholar]
Seymour PICCOLO 2013 {published data only (unpublished sought but not used)}
- Seymour MT, Brown SR, Middleton G, Maughan T, Richman S, Gwyther S, et al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild‐type, fluorouracil‐resistant advanced colorectal cancer (PICCOLO): a prospectively stratified randomised trial. Lancet Oncology 2013;14:749‐59. [10.1016/ S1470‐2045(13)70163‐3] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour MT, Brown SR, Richman S, Middleton GW, Maughan T, Olivier C, et al. Addition of panitumumab to irinotecan: Results of PICCOLO, a randomized controlled trial in advanced colorectal cancer (aCRC). Journal of Clinical Oncology Conference: ASCO Annual Meeting 2011;29(15 Suppl):3523. [Google Scholar]
- Seymour MT, Brown SR, Richman S, Middleton GW, Maughan TS, Maisey N, et al. Panitumumab in combination with irinotecan for chemoresistant advanced colorectal cancer: Results of PICCOLO, a large randomised trial with prospective molecular stratification. European Journal of Cancer 2011;47(Suppl 3):S393. [Google Scholar]
Siena 2013 {published data only}
- Siena S, Cutsem E, Li M, Jungnelius U, Romano A, Beck R, et al. Phase II open‐label study to assess efficacy and safety of lenalidomide in combination with cetuximab in KRAS‐mutant metastatic colorectal cancer. PLoS ONE 2013;8(11):e62264. [PUBMED: 24244261] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sobrero EPIC 2008 {published data only (unpublished sought but not used)}
- Abubakr Y, Eng C, Wong L, Pautret V, Scheithauer W, Maurel J, et al. Cetuximab plus irinotecan for metastatic colorectal cancer (mCRC): Safety analysis of 800 patients in a randomized phase III trial (EPIC). Journal of Clinical Oncology 2006;24:3556. [Google Scholar]
- Langer C, Kopit J, Awad M, Williams K, Teegarden P, Xu L, et al. Analysis of K‐RAS mutations in patients with metastatic colorectal cancer receiving cetuximab in combination with irinotecan: results from the EPIC trial. Annals of Oncology 2009;19((Suppl 8)):133 Abstract No: 385P. [Google Scholar]
- Sobrero A, Scheithauer W, Maurel J, Mineur L, Fehrenbacher L, Kisker O, et al. Cetuximab plus irinotecan for metastatic colorectal cancer (mCRC): safety analysis of the first 400 patients in a randomized phase III trial (EPIC). Annual Meeting Proceedings of the American Society of Clinical Oncology, Abstract 2005;23(16 Suppl):266. [Google Scholar]
- Sobrero AF, Maurel J, Fehrenbacher L, Scheithauer W, Abubakr YA, Lutz MP, et al. EPIC: phase III trial of cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. Journal of Clinical Oncology 2008;26(14):2311‐9. [DOI: 10.1200/JCO.2007.13.1193] [DOI] [PubMed] [Google Scholar]
- Yang D, Bohanes P, Zhang W, Harbison C, Trifan OC, Ning Y, et al. Association of HER2 and IGF1 germline polymorphisms with outcome in metastatic colorectal cancer (mCRC) patients (pts) treated with second‐line irinotecan (IR) with or without cetuximab (CB): The EPIC experience. Journal of Clinical Oncology 2012;30(2 Suppl):3615. [Google Scholar]
Tol CAIRO2 2008 {published data only}
- Tol J, Dijkstra JR, Klomp M, Teerenstra S, Dommerholt M, Vink‐Börger ME, et al. Markers for EGFR pathway activation as predictor of outcome in metastatic colorectal cancer patients treated with or without cetuximab. European Journal of Cancer 2010;46(11):1997‐2009. [DOI] [PubMed] [Google Scholar]
- Tol J, Dijkstra JR, Vink‐Borger ME, Koopman M, Vincent AD, Krieken JHJM, et al. BRAF mutation is associated with a decreased outcome in patients (pts) with advanced colorectal cancer (ACC) treated with chemotherapy and bevacizumab with or without cetuximab. European Journal of Cancer 2009;7(2):321. [Google Scholar]
- Tol J, Koopman M, Antonini NF, Sinnige H, Valster FAA, Braun JJ, et al. Randomized phase III study of capecitabine, oxaliplatin and bevacizumab with or without cetuximab in advanced colorectal cancer (ACC), the CAIRO2 study of the Dutch Colorectal Cancer Group (DCCG). Annals of Oncology 2009;19(Suppl 8):128. [DOI] [PubMed] [Google Scholar]
- Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. New England Journal of Medicine 2009;360(6):563‐72. [DOI] [PubMed] [Google Scholar]
- Tol J, Koopman M, Rodenburg CJ, Cats A, Creemers GJ, Schrama JG, et al. A randomised phase III study on capecitabine, oxaliplatin and bevacizumab with or without cetuximab in first‐line advanced colorectal cancer, the CAIRO2 study of the Dutch Colorectal Cancer Group (DCCG). An interim analysis of toxicity. Annals of Oncology 2008;19(4):734‐8. [DOI] [PubMed] [Google Scholar]
Tournigand DREAM 2015 {published data only}
- Tournigand C, Chibaudel B, Samson B, Scheithauer W, Vernerey D, Mesange P, et al. Bevacizumab with or without erlotinib as maintenance therapy in patients with metastatic colorectal cancer (GERCOR DREAM; OPTIMOX3): a randomised, open‐label, phase 3 trial. Lancet Oncology 2015;16:1493‐505. [DOI] [PubMed] [Google Scholar]
- Tournigand C, Samson B, Scheithauer W, Lledo G, Viret F, Andre T, et al. Bevacizumab (Bev) with or without erlotinib as maintenance therapy, following induction first‐line chemotherapy plus Bev, in patients (pts) with metastatic colorectal cancer (mCRC): Efficacy and safety results of the International GERCOR DREAM phase III trial.. Journal of Clinical Oncology 2012;30:Suppl; abstr LBA3500. [Google Scholar]
Tveit NORDIC VII 2012 {published data only (unpublished sought but not used)}
- Tveit K, Guren T, Glimelius B, Pfeiffer P, Sorbye H, Pyrhonen S, et al. Randomized phase III study of 5‐fluorouracil/folinate/oxaliplatin given continuously or intermittently with or without cetuximab, as first‐line treatment of metastatic colorectal cancer: The NORDIC VII study (NCT00145314), by the Nordic Colorectal Cancer Biomodulation Group. Journal of Clinical Oncology 2011;29(4 Suppl):365. [Google Scholar]
- Tveit K, Guren T, Glimelius B, Pfeiffer P, Sorbye H, Pyrhonen S, et al. Randomized phase III study of 5‐fluorouracil/folinate/oxaliplatin given continuously or intermittently with or without cetuximab, as first‐line treatment of metastatic colorectal cancer: The Nordic VII study (NCT00145314), by the Nordic Colorectal Cancer Biomodulation Group. Annals of Oncology 2010;21(Suppl 8):viii9. [Google Scholar]
- Tveit KM, Guren T, Glimelius B, Pfeiffer P, Sorbye H, Pyrhonen S, et al. Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first‐line treatment of metastatic colorectal cancer: the NORDIC‐VII study. Journal of Clinical Oncology 2012;30(15):1755‐62. [DOI] [PubMed] [Google Scholar]
Van Cutsem CRYSTAL 2009 {published data only (unpublished sought but not used)}
- Folprecht G, Nowacki M, Lang I, Cascinu S, Shchepotin I, Maurel J, et al. Cetuximab plus FOLFIRI first line in patients (pts) with metastatic colorectal cancer (mCRC): A quality‐of‐life (QoL) analysis of the CRYSTAL trial. Journal of Clinical Oncology 2009;27(15 Suppl):4076. [Google Scholar]
- Folprecht G, Nowacki M, Lang I, Cascinu S, Shchepotin I, Maurel J, et al. Cetuximab plus FOLFIRI first line in patients (pts) with metastatic colorectal cancer (mCRC): A quality‐of‐life (QoL) analysis of the CRYSTAL trial. Journal of Clinical Oncology 2009;27(15S Part I):187 (abstract no. 4076). [Google Scholar]
- Kohne C, Stroiakovski D, Chang‐chien C, Lim R, Pinter T, Bodoky G, et al. Predictive biomarkers to improve treatment of metastatic colorectal cancer (mCRC): Outcomes with cetuximab plus FOLFIRI in the CRYSTAL trial. Journal of Clinical Oncology 2009;27(15 Suppl):4068. [Google Scholar]
- Lang I, Aleknaviciene B, Zolotukhin S, Komissarenko V, Garcia‐Alfonso P, Jurga L, et al. Impact on quality of life of cetuximab plus FOLFIRI in the first‐line treatment of patients with metastatic colorectal cancer (MCRC): results from the CRYSTAL trial. Annals of Oncology 2009;20:23‐4 (suppl 7; abstr PD‐0024). [Google Scholar]
- Lang I, Kohne CH, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, et al. Cetuximab plus FOLFIRI in 1st‐line treatment of metastatic colorectal cancer: Quality of life (QoL) analysis of patients (pts) with KRAS wild‐type (wt) tumours in the CRYSTAL trial. European Journal of Cancer, Supplement Conference: Joint ECCO 15 ‐ 34th ESMO Multidisciplinary Congress; 2009 Sept 20‐24; Berlin, Germany 2009;7(2):345. [Google Scholar]
- Lang I, Zaluski J, Changchien CR, Makhson A, Pinter T, D'Haens G, et al. Cetuximab with irinotecan in first‐line treatment of epidermal growth factor receptor (EGFR)‐expressing metastatic colorectal cancer (mCRC): Preliminary safety results (CRYSTAL). Journal of Clinical Oncology 2006;24 (18S):2555. [Google Scholar]
- Láng I, Köhne CH, Folprecht G, Rougier P, Curran D, Hitre E, et al. Quality of life analysis in patients with KRAS wild‐type metastatic colorectal cancer treated first‐line with cetuximab plus irinotecan, fluorouracil and leucovorin. European Journal of Cancer 2013;49(2):439‐48. [DOI: 10.1016/j.ejca.2012.08.023] [DOI] [PubMed] [Google Scholar]
- Custem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. New England Journal of Medicine 2009;360:1408‐17. [DOI] [PubMed] [Google Scholar]
- Wilke H, Siena S, Loos A, Berghoff K, Kohne C, Cutsem E. Premedication and the incidence and severity of infusion‐related reactions in patients treated with cetuximab: Data from the CRYSTAL and MABEL studies. Annals of Oncology 2010;21(Suppl 6):vi71. [Google Scholar]
- Wilke H, Siena S, Loos AH, Berghoff K, Kohne C, Cutsem E. Premedication and incidence of infusion‐related reactions in patients with metastatic colorectal cancer treated with cetuximab plus irinotecan‐based chemotherapy. Journal of Clinical Oncology 2010;28(15 Suppl):3561. [Google Scholar]
Venook CALGB 80405 2014 {published data only}
- Anonymous. CALGB/SWOG C80405: A phase III trial of FOLFIRI or FOLFOX with bevacizumab or cetuximab or both for untreated metastatic adenocarcinoma of the colon or rectum. Clinical Advances in Hematology & Oncology 2006;4(6):452‐3. [PubMed] [Google Scholar]
- Naughton MJ, Schrag D, Venook AP, Niedzwiecki D, Anderson RT, Lenz H‐J, et al. Quality of life (QOL) and toxicity among patients in CALGB 80405. Journal of Clinical Oncology 2013;31:Suppl 4; abstr 3611. [Google Scholar]
- Venook AP, Niedzwiecki D, Lenz HJ, Innocenti F, Mahoney MR, O'Neil BH, et al. CALGB/SWOG 80405: Phase III trial of irinotecan/5‐FU/leucovorin (FOLFIRI) or oxaliplatin/5‐FU/leucovorin (mFOLFOX6) with bevacizumab (BV) or cetuximab (CET) for patients (pts) with KRAS wild‐type (wt) untreated metastatic adenocarcinoma of the colon or rectum (MCRC). Journal of Clinical Oncology 2014;32(5s):Suppl; abstr LBA3. [Google Scholar]
Vincent 2011 {published data only}
- Vincent MD, Welch S, Soulieres D, Sanatani MS, Whiston F, Stitt L, et al. Randomized phase II trial of capecitabine (X) versus X plus erlotinib (E) in patients (pts) with metastatic colorectal cancer (mCRC): Differential impact of KRAS. Journal of Clinical Oncology 2011;29(15 Suppl):Abstr e14031. [Google Scholar]
Wasan COIN‐B 2014 {published data only}
- Wasan H, Meade AM, Adams R, Wilson R, Pugh C, Fisher D, et al. Intermittent chemotherapy plus either intermittent or continuous cetuximab for first‐line treatment of patients with KRAS wild‐type advanced colorectal cancer (COIN‐B): a randomised phase 2 trial. Lancet Oncology 2014;15(6):631‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ye 2013 {published data only (unpublished sought but not used)}
- Xu J, Ye L, Ren L, Wei Y. A randomized, controlled trial of cetuximab plus chemotherapy for patients with KRAS wild‐type unresectable colorectal liver‐limited metastases. Annals of Oncology 2012;23 (Suppl 9):ix190; Abstract 557P. [DOI] [PubMed] [Google Scholar]
- Ye LC, Liu TS, Ren L, Wei Y, Zhu DX, Zai SY, et al. A randomized controlled trial of cetuximab plus chemotherapy for patients with KRAS wild‐type unresectable colorectal liver‐limited metastases. Journal of Clinical Oncology 2013;31:1931‐8. [DOI] [PubMed] [Google Scholar]
- Ye LC, Zhong Y, Liu TS, Wei Y, Ren L, Zhu DX, et al. Impact of early tumor shrinkage on clinical outcome in KRAS wild‐type colorectal liver‐limited metastases treated with cetuximab plus chemotherapy: Lessons from a randomized controlled trial. Journal of Clinical Oncology 2013;31:Suppl; abstr 3610. [Google Scholar]
References to studies excluded from this review
Cunningham BOND 2004 {published data only}
- Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan‐refractory metastatic colorectal cancer. New England Journal of Medicine 2004;351:337‐45. [DOI] [PubMed] [Google Scholar]
Liu 2015 {published data only}
- Liu Y, Luan L, Wang X. A randomized Phase II clinical study of combining panitumumab and bevacizumab, plus irinotecan, 5‐fluorouracil, and leucovorin (FOLFIRI) compared with FOLFIRI alone as second‐line treatment for patients with metastatic colorectal cancer and KRAS mutation. Journal of OncoTargets and Therapy 2015;8:1061‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT00950820 {published data only}
- NCT00950820. Study to evaluate the effects of panitumumab if combined with chemotherapy for 2nd treatment of colorectal cancer (VOXEL). clinicaltrials.gov/ct2/show/NCT00950820 (first received 31 July 2009).
Personeni 2013 {published data only}
- Personeni N, Rimassa L, Verusio C, Barni S, Destro A, Raschioni C, et al. Prognostic factors in KRAS wild‐type (wt) metastatic colorectal cancer (mCRC) patients (pts) treated with biweekly cetuximab (C) plus irinotecan, fluorouracil, and leucovorin (FOLFIRI): A phase II study. Journal of Clinical Oncology 2013;31:Suppl; abstr e14611. [Google Scholar]
Primrose NEW EPOC 2014 {published data only}
- Primrose J, Falk S, Finch‐Jones M, Valle J, O'Reilly D, Siriwardena A, et al. Systemic chemotherapy with or without cetuximab in patients with resectable colorectal liver metastasis: the New EPOC randomised controlled trial. Lancet Oncology 2014;15(6):601‐11. [DOI] [PubMed] [Google Scholar]
Saltz BOND2 2007 {published data only}
- Saltz LB, Lenz HJ, Kindler HL, Hochster HS, Wadler S, Hoff PM, et al. Randomized phase II trial of cetuximab, bevacizumab, and irinotecan compared with cetuximab and bevacizumab alone in irinotecan‐refractory colorectal cancer: the BOND‐2 study. Journal of Clinical Oncology 2007;25(29):4557‐61. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
EVEREST 2004 {published data only}
Hill 2015 {published data only}
- Hill A, Findlay M, Burge M, Jackson C, Alfonso PG, Samuel L, et al. Randomized phase II study of duligotuzumab + FOLFIRI versus cetuximab + FOLFIRI in 2nd‐line patients with KRAS wild‐type (wt) metastatic colorectal cancer (mCRC). Cancer Research 2015;75(15 Suppl):Abstr nr CT110. [DOI: 10.1158/1538-7445.AM2015-CT110] [DOI] [Google Scholar]
Hiret 2016 {published data only}
- Hiret S, Borg C, Bertaut A, Bouche O, Adenis A, Deplanque G, et al. Bevacizumab or cetuximab plus chemotherapy after progression with bevacizumab plus chemotherapy in patients with wtKRAS metastatic colorectal cancer: A randomized phase II study (Prodige 18 –UNICANCER GI). Journal of Clinical Oncology 2016;34:Suppl; abstr 3514. [Google Scholar]
Kim 2016 {published data only}
- Kim TW, Elme A, Kusic Z, Park JO, Udrea AA, Kim SY, et al. An open label, randomized phase III trial evaluating the treatment (tx) effects of panitumumab (pmab) + best supportive care (BSC) versus BSC in chemorefractory wild‐type (WT) KRAS exon 2 metastatic colorectal cancer (mCRC) and in WT RAS mCRC. Journal of Clinical Oncology 2016;34:Suppl 4S; abstr 642. [Google Scholar]
- Kim TW, Elme A, Kusic Z, Park JO, Udrea AA, Kim SY, et al. Final results from a phase III trial evaluating panitumumab (pmab) + best supportive care (BSC) vs BSC in chemorefractory wild‐type (WT) KRAS exon 2 and WT RAS metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2016;34:Suppl; abstr 3536. [Google Scholar]
MACBETH 2016 {published data only}
- Antoniotti C, Cremolini C, Loupakis F, Bergamo F, Grande R, Tonini G, et al. Modified FOLFOXIRI (mFOLFOXIRI) plus cetuximab (cet), followed by cet or bevacizumab (bev) maintenance, in RAS/BRAF wild‐type (wt) metastatic colorectal cancer (mCRC): Results of the phase II randomized MACBETH trial by GONO. Journal of Clinical Oncology 2016;34:Suppl; abstr 3543. [Google Scholar]
- Cremolini C, Loupakis F, Salvatore L, Lonardi S, Battaglin F, Gamucci T, et al. Modified FOLFOXIRI plus cetuximab (cet) as induction treatment in unresectable metastatic colorectal cancer (mCRC) patients (pts): Preliminary results of the phase II randomized Macbeth trial by GONO group. Journal of Clinical Oncology 2014;32:5s:Abstr 3596. [Google Scholar]
TAGUS 2009 {published data only}
References to ongoing studies
Ashwin 2014 {published data only}
- Ashwin K. Assessment of tumour response and resection rates in unresectable metastatic colorectal liver metastases following cetuximab with neoadjuvant chemotherapy. European Journal of Surgical Oncology 2014;40(11):S139. [DOI] [PMC free article] [PubMed] [Google Scholar]
ATOM {published data only}
- Emi Y, Muro K, Yamanaka T, Katayose Y, Uetake H, Sugihara K, et al. The ATOM trial: A multicenter, randomized phase II study of modified FOLFOX6 plus bevacizumab and modified FOLFOX6 plus cetuximab for colorectal cancer with liver‐limited metastases. Journal of Clinical Oncology 2016;34:Suppl 4S; abstr TPS777. [Google Scholar]
CAIRO5 {published data only}
- Huiskens J, Gulik TM, Lienden KP, Engelbrecht MRW, Meijer GA, Grieken NCT, et al. Treatment strategies in colorectal cancer patients with initially unresectable liver‐only metastases: The randomized phase III CAIRO5 study of the Dutch Colorectal Cancer Group. Journal of Clinical Oncology 2015;33:Suppl; TPS3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
CREPAS {published data only}
- Study of medical treatment reactivity by the chemokine receptor (CXCR4) as 1st line treatment in patients with metastatic colorectal cancer (CREPAS). Ongoing study July 2012.
DEEPER {published data only}
- A Randomized Phase II Study to Investigate the Deepness of Response of FOLFOXIRI Plus Cetuximab (Erbitux) Versus FOLFOXIRI Plus Bevacizumab as the First‐line Therapy in Metastatic Colorectal Cancer Patients With RAS Wild‐type Tumors: DEEPER. Ongoing study August 2015.
FIRE‐4 {published data only}
- FIRE‐4: Randomised study of the efficacy of cetuximab rechallenge in patients with metastatic colorectal cancer (RAS wild‐type) responding to first‐line treatment with FOLFIRI plus cetuximab. Ongoing study March 2015.
FIRE‐4.5 {published data only}
- FIRE‐4.5. Ongoing study September 2016.
FOCETELD {published data only}
- FOCETELD. Ongoing study April 2012.
FOCULM {published data only}
- FOLFOXIRI With or Without Cetuximab as First‐line Treatment of Patients With Non‐resectable Liver‐Only Metastatic Colorectal Cancer (FOCULM). Ongoing study February 2014.
G13 study {published data only}
- Randomized Phase II Study of BSC vs Cetuximab vs Irinotecan and Cetuximab in Patients with KRAS codon G13D mutant Metastatic Colorectal Cancer (G13 study). Ongoing study September 2012.
NCT00202787 {published data only}
- Open‐label, Phase II, Randomised, Pilot Study to Evaluate the Safety and Efficacy of Combination Therapy With Cetuximab and FOLFOX4 or FOLFOX4 Alone in Patients Colorectal Cancer and Initially Non‐resectable. Ongoing study February 2005.
NCT01442649 {published data only}
- Phase II, Multicentric Randomized Trial, Evaluating the Efficacy of Fluoropyrimidine‐based Standard Chemotherapy, Associated to Either Cetuximab or Bevacizumab, in KRAS Wild‐type Metastatic Colorectal Cancer Patients With Progressive Disease After Receiving First‐line Treatment With Bevacizumab. Ongoing study December 2010.
NCT01652482 {published data only}
- A Phase II, Multicenter, Open‐Label, Randomized Study Evaluating the Efficacy and Safety of MEHD7945A + FOLFIRI Versus Cetuximab + FOLFIRI in Second Line in Patients With KRAS Wildtype Metastatic Colorectal Cancer. Ongoing study October 2012.
NCT01991873 {published data only}
- NCT01991873. Maintenance therapy with 5‐FU/FA plus panitumumab vs. 5‐FU/FA alone after prior induction and re‐induction after progress for 1st‐line treatment of metastatic colorectal cancer (PanaMa). clinicaltrials.gov/ct2/show/NCT01991873 (first received 22 October 2013).
NCT02083653 {published data only}
- Open‐label, Randomized, Controlled, Multicenter Phase II Trial Investigating 2 Sym004 Doses Versus Investigator's Choice (Best Supportive Care, Capecitabine, 5‐FU) in Subjects With Metastatic Colorectal Cancer and Acquired Resistance to Anti‐EGFR Monoclonal Antibodies. Ongoing study March 2014.
NCT02394834 {published data only}
- An Exploratory Study of Treatment Sensitivity and Prognostic Factors in a Efficacy and Safety Study of mFOLFOX6 + Bevacizumab Versus mFOLFOX6 + Panitumumab Therapy in Patients With Chemotherapy‐naïve Unresectable Advanced or Recurrent Colorectal Cancer. Ongoing study May 2015.
PANIB {published data only}
- PANIB ‐ An open‐label, randomised, controlled, multi‐center, Phase II trial comparing Panitumumab versus Bevacizumab in combination with oxaliplatin ‐ 5 FU (FOLFOX) first‐line treatment according Ras Wild Type status for patients with metastatic unresectable colorectal cancer (mCRC). Ongoing study August 2014.
PARADIGM {published data only}
- Muro K, Uetake H, Tsuchihara K, Shitara K, Yamazaki K, Oki E, et al. PARADIGM study: A multicenter, randomized, phase III study of mFOLFOX6 plus panitumumab or bevacizumab as first‐line treatment in patients with RAS (KRAS/NRAS) wild‐type metastatic colorectal cancer. Journal of Clinical Oncology 2016;34:Suppl; abstr TPS3625. [Google Scholar]
- Yoshino T, Uetake H, Tsuchihara K, Shitara K, Yamazaki K, Oki E, et al. PARADIGM study: A multicenter, randomized, phase III study of 5‐fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) plus panitumumab or bevacizumab as first‐line treatment in patients with RAS (KRAS/NRAS) wild‐type metastatic colorectal cancer. Journal of Clinical Oncology 2016;34:Suppl 4S; abstr TPS776. [Google Scholar]
Peeters 2012 {published data only}
- Colorectal Cancer (CRC) Cetuximab Elderly Frail. Ongoing study April 2013.
TAILOR {published data only}
- An Open‐label, Randomized, Controlled, Multicenter Phase III Trial to Compare Cetuximab in Combination With FOLFOX‐4 Versus FOLFOX‐4 Alone in the First Line Treatment of Metastatic Colorectal Cancer in Chinese Subjects With RAS Wild‐type Status. Ongoing study August 2010.
TIME {published data only}
- Randomized Phase II Study of First‐line FOLFIRI Plus Cetuximab for 8 Cycles Followed by Either Single‐agent Cetuximab as Maintenance Therapy or Observation in Patients With Wild‐type KRAS and NRAS Metastatic Colorectal Cancer. Ongoing study February 2014.
UCGI 25 {published data only}
- Senellart H, Samalin E, Adenis A, Malka D, Francois E, Fouchardiere C, et al. UCGI 25: A multicentric randomized phase II trial evaluating dual targeting of the epidermal growth factor (EGFR) using the combination of cetuximab and afatinib versus cetuximab alone in patients (pts) wih chemotherapy refractory wtRAS metastatic colorectal cancer (mCRC). Journal of Clinical Oncology 2014;32;5s:Abstr TPS3666. [Google Scholar]
UMIN000005216 {published data only}
- Shitara K, Yonesaka K, Denda T, Moriwaki T, Tsuda M, Takano T, et al. A randomized multicenter phase II study of FOLFIRI plus either panitumumab (Pmab) or bevacizumab (Bmab) as second‐line treatment for wild‐type KRAS exon 2 metastatic colorectal cancer (mCRC) with exploratory biomarker analysis by liquid biopsy: WJOG6210G. Journal of Clinical Oncology 2016;34:Suppl; abstr 3567. [Google Scholar]
UMIN000006899 {published data only}
- Randomized phase II study of biweekly cetuximab versus panitumumab in patients not combination of irinotecan wild‐type KRAS metastatic colorectal cancer following treatment with fluoropyrimidine and oxaliplatin chemotherapy. Ongoing study December 2011.
Venook CALGB 80203 2006 {published data only}
- Venook A, Niedzwiecki D, Hollis D, Sutherland S, Goldberg R, Alberts S, et al. Phase III study of irinotecan/5FU/LV (FOLFIRI) or oxaliplatin/5FU/LV (FOLFOX) ± cetuximab for patients (pts) with untreated metastatic adenocarcinoma of the colon or rectum (MCRC): CALGB 80203 preliminary results. Journal of Clinical Oncology 2006;24(18 Suppl):3509. [Google Scholar]
VISNU‐2 {published data only}
- Influence of BRAF and PIK3K Status on the Efficacy of 5‐Fluorouracil/Leucovorin/Oxaliplatin (FOLFIRI) Plus Bevacizumab or Cetuximab in Patients With RAS Wild‐type Metastatic Colorectal Carcinoma and < 3 Circulating Tumor Cells (CTC) (VISNU‐2). Ongoing study July 2012.
VOLFI {published data only}
- Martens UM, Wessendorf S, Riera Knorrenschild J, Buechner‐Steudel P, Florschuetz A, Atzpodien J, et al. AIO‐KRK‐0109: A randomized phase II trial of panitumumab plus FOLFOXIRI or FOLFOXIRI alone as 1st‐line treatment in RAS‐wild‐type metastatic colorectal cancer (mCRC). European Journal of Cancer 2015;51:s344‐5. [Google Scholar]
WJOG6510G {published data only}
- Sakai D, Taniguchi H, Tamura T, Sugimoto N, Esaki T, Okuda H, et al. Randomized phase II study of panitumumab (Pmab) plus irinotecan (CPT‐11) versus cetuximab (Cmab) plus CPT‐11 in patients with KRAS wild‐type (WT) metastatic colorectal cancer (mCRC) following treatment with fluoropyrimidine, CPT‐11, and oxaliplatin (L‐OHP) chemotherapy: WJOG6510G. Journal of Clinical Oncology 2014;32 (5s):TPS3654. [Google Scholar]
Additional references
Andreyev 1998
- Andreyev HJ, Norman AR, Cunningham D, Oates JR, Clarke PA. Kirsten ras mutations in patients with colorectal cancer: the multicenter "RASCAL" study. Journal of the National Cancer Institute 1998;90(9):675‐84. [DOI] [PubMed] [Google Scholar]
Chan 2013 [pers comm]
- Chan DL. Request for EPIC KRAS substudy data [personal communication]. Email to: A Sobrero 23 September 2013.
Chan 2015
- Chan DL, Pavlakis N, Shapiro J, Price TJ, Karapetis CS, Tebbutt NC, et al. Does the chemotherapy backbone impact on the efficacy of targeted agents in metastatic colorectal cancer? A systematic review and meta‐analysis of the literature. PLoS ONE 2015;10(8):e0135599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chan 2015 [pers comm]
- Chan DL. Request for SAKK XELOX+C data ‐ extended RAS [personal communication]. Email to: M Borner 10 May 2015.
Chung 2005
- Chung KY, Shia J, Kemeny NE, Shah M, Schwartz GK, Tse A, et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. Journal of Clinical Oncology 2005;23(9):1803‐10. [DOI] [PubMed] [Google Scholar]
Citri 2006
- Citri A, Yarden Y. EGF–ERBB signalling: towards the systems level. Nature Reviews Molecular Cell Biology 2006;7:505‐16. [DOI] [PubMed] [Google Scholar]
Cunningham 2004
- Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan‐refractory metastatic colorectal cancer. New England Journal of Medicine 2004;351(4):337‐45. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG. Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Di Fiore 2007
- Fiore F, Pessot F, Lamy A, Charbonnier F, Sabourin J, Paillot B, et al. KRAS mutation is highly predictive of cetuximab resistance in metastatic colorectal cancer. Journal of Clinical Oncology (Meeting Abstracts) 2007;25(18 (Suppl)):10502. [Google Scholar]
Eisenhauer 2009
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). European Journal of Cancer 2009;45(2):228‐47. [DOI] [PubMed] [Google Scholar]
Favoni 2010
- Favoni RE, Pattarozzi A, Lo Casto M, Barbieri F, Gatti M, Paleari L, et al. Gefitinib targets EGFR dimerization and ERK1/2 phosphorylation to inhibit pleural mesothelioma cell proliferation. Current Cancer Drug Targets 2010;10(2):176‐91. [DOI] [PubMed] [Google Scholar]
Feng 2001
- Feng J, Hua F, Shuo R, Chongfeng G, Huimian X, Nakajima T, et al. Upregulation of non‐mutated H‐ras and its upstream and downstream signaling proteins in colorectal cancer. Oncology Reports 2001;8(6):1409‐13. [DOI] [PubMed] [Google Scholar]
Ferlay 2015
- Ferlay J, Soerjomataram I, Dikshit R, Mathers C, Rebelo M, Parkin DM, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International Journal of Cancer 2015;136(5):E359‐86. [PUBMED: 25220842] [DOI] [PubMed] [Google Scholar]
Giantonio 2007
- Giantonio BJ, Catalano PJ, Meropol NJ, O'Dwyer PJ, Mitchell EP, Alberts SR, et al. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: Results from the Eastern Cooperative Oncology Group Study E3200. Journal of Clinical Oncology 2007;25(12):1539‐44. [DOI] [PubMed] [Google Scholar]
Graham 2014
- Graham CN, Hechmati G, Hjelmgren J, Liege F, Lanier J, Knox H, et al. Cost‐effectiveness analysis of panitumumab plus mFOLFOX6 compared with bevacizumab plus mFOLFOX6 for first‐line treatment of patients with wild‐type RAS metastatic colorectal cancer. European Journal of Cancer 2014;50(16):2791‐801. [DOI] [PubMed] [Google Scholar]
Grothey 2004
- Grothey A, Sargent D, Goldberg RM, Schmoll H. Survival of patients with advanced colorectal cancer improves with the availability of fluorouracil‐leucovorin, irinotecan, and oxaliplatin in the course of treatment. Journal of Clinical Oncology 2014;22(7):1209‐14. [DOI] [PubMed] [Google Scholar]
Herbertson 2009
- Herbertson R, Karapetis C, Price T, Tebbutt N, Pavlakis N. Epidermal growth factor receptor (EGF‐R) inhibitors for metastatic colorectal cancer. Cochrane Database of Systematic Reviews 2009, Issue 2. [DOI: 10.1002/14651858.CD007047] [DOI] [PMC free article] [PubMed] [Google Scholar]
Herbst 2004
- Herbst RS. Review of epidermal growth factor receptor biology.. International Journal of Radiation Oncology • Biology • Physics 2004;59(Suppl 2):21‐6. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Hurwitz 2005
- Hurwitz HI, Fehrenbacher L, Hainsworth JD, Heim W, Berlin J, Holmgren E, et al. Bevacizumab in combination with fluorouracil and leucovorin: an active regimen for first line metastatic colorectal cancer. Journal of Clinical Oncology 2005;23(15):3502‐8. [DOI] [PubMed] [Google Scholar]
Jonker 2007
- Jonker DJ, O'Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, et al. Cetuximab for the treatment of colorectal cancer. New England Journal of Medicine 2007;357(20):2040‐8. [DOI] [PubMed] [Google Scholar]
Khattak 2015
- Khattak MA, Martin H, Davidson A, Phillips M. Role of first‐line anti‐epidermal growth factor receptor therapy compared with anti‐vascular endothelial growth factor therapy in advanced colorectal cancer: a meta‐analysis of randomized clinical trials. Clinical Colorectal Cancer 2015;14(2):81‐90. [DOI] [PubMed] [Google Scholar]
Kumachev 2014
- Kumachev A, Yan M, Berry SR, Ko YJ, Martinez MCR, Shah K. A comparison of biologics in first‐line advanced colorectal cancer: A Bayesian network meta‐analysis of EGFR inhibitors and bevacizumab. Journal of Clinical Oncology 2014;32:Suppl 3; abstr 543. [Google Scholar]
Lee 2009
- Lee SC, Xu X, Lim YW, Iau P, Sukri N, Lim SE, et al. Chemotherapy‐induced tumor gene expression changes in human breast cancers. Pharmacogenetics and Genomics 2009;19(3):181‐92. [DOI] [PubMed] [Google Scholar]
Lievre 2006
- Lievre A, Bachet JB, Corre D, Boige V, Landi B, Emile JF, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Research 2006;66(8):3992‐5. [DOI] [PubMed] [Google Scholar]
Mayer 1993
- Mayer A, Takimoto M, Fritz E, Schellander G, Kofler K, Ludwig H. The prognostic significance of proliferating cell nuclear antigen, epidermal growth factor receptor, and mdr gene expression in colorectal cancer. Cancer 1993;71(8):2454‐60. [DOI] [PubMed] [Google Scholar]
Mendelsohn 2000
- Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene 2000;19(56):6550‐65. [DOI] [PubMed] [Google Scholar]
Messa 1998
- Messa C, Russo F, Caruso MG, Leo A. EGF, TGF‐alpha, and EGF‐R in human colorectal adenocarcinoma. Acta Oncologica 1998;37(3):285‐9. [DOI] [PubMed] [Google Scholar]
Mohan 2014
- Mohan S, Heitzer E, Ulz P, Lafer I, Lax S, Auer M, et al. Changes in colorectal carcinoma genomes under anti‐EGFR therapy identified by whole‐genome plasma DNA sequencing. PLoS Genetics 2014;10(3):e1004271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mok 2009
- Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. New England Journal of Medicine 2009;361:947‐57. [PUBMED: 19692680] [DOI] [PubMed] [Google Scholar]
NIH 2010
- National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) Version 4.03. evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010‐06‐14_QuickReference_5x7.pdf (accessed prior to 30 May 2017).
Normanno 2006
- Normanno N, Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, et al. Epidermal growth factor receptor (EGFR) signalling in cancer. Gene 2006;366(1):2‐16. [DOI] [PubMed] [Google Scholar]
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815‐34. [PUBMED: 9921604] [DOI] [PubMed] [Google Scholar]
Rajagopalan 2002
- Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch‐repair status. Nature 2002;418(6901):934. [DOI] [PubMed] [Google Scholar]
Rauw 2012
- Rauw J, Ennis M, Krzyzanowska MK, Sridhar SS. Does the addition of molecular targeted therapy to standard treatments lead to better or worse outcomes overall ‐ a systematic review of EGFR‐targeted therapies used in combination with standard treatments. Journal of Clinical Oncology 2012;30:Suppl; abstr 2572. [Google Scholar]
Salomon 1995
- Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor‐related peptides and their receptors in human malignancies. Critical Reviews in Oncology/Hematology 1995;19(3):183‐232. [DOI] [PubMed] [Google Scholar]
Scartozzi 2004
- Scartozzi M, Bearzi I, Berardi R, Mandolesi A, Fabris G, Cascinu S. Epidermal growth factor receptor (EGFR) status in primary colorectal tumors does not correlate with EGFR expression in related metastatic sites: Implications for treatment with EGFR‐targeted monoclonal antibodies. Journal of Clinical Oncology 2004;22(23):4772‐8. [DOI] [PubMed] [Google Scholar]
Schrag 2015
- Schrag D, Dueck AC, Naughton MJ, Niedzwiceki D, Earle C, Shaw JE, et al. Cost of chemotherapy for metastatic colorectal cancer with either bevacizumab or cetuximab: Economic analysis of CALGB/SWOG 80405. Journal of Clinical Oncology 2015;33(Suppl):Abstr 6504. [Google Scholar]
Schünemann 2009
- Schünemann H, Brozek J, Oxman A, editors of The GRADE Working Group. GRADE handbook for grading quality of evidence and strength of recommendations Version 3.2 [updated March 2009]. Available from guidelinedevelopment.org/handbook.
Sorich 2015
- Sorich MJ, Wiese MD, Rowland A, Kichenadasse G, McKinnon RA, Karapetis CS. Extended RAS mutations and anti‐EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta‐analysis of randomized, controlled trials.. Annals of Oncology 2015;26(1):13‐21. [DOI] [PubMed] [Google Scholar]
Vale 2012
- Vale CL, Tierney JF, Fisher D, Adams RA, Kaplan R, Maughan TS, et al. Does anti‐EGFR therapy improve outcome in advanced colorectal cancer? A systematic review and meta‐analysis. Cancer Treatment Reviews 2012;38(6):618‐25. [DOI] [PubMed] [Google Scholar]
van Cruijsen 2005
- Cruijsen H, Giaccone G, Hoekman K. Epidermal growth factor receptors and angiogenesis: Opportunities for combined anticancer strategies. International Journal of Cancer 2005;117(6):883‐8. [DOI] [PubMed] [Google Scholar]