

Abstract
Alterations in the coronary vascular system are likely associated with a mismatch between energy demand and energy supply and critical in triggering the cascade of events that leads to symptomatic hypertrophic cardiomyopathy. Targeting the early events, particularly vascular remodeling, may be a key approach to developing effective treatments. Improvement in our understanding of hypertrophic cardiomyopathy began with the results of early biophysical studies, proceeded to genetic analyses pinpointing the mutational origin, and now pertains to imaging of the metabolic and flow-related consequences of such mutations. Microvascular dysfunction has been an ongoing hot topic in the imaging of genetic cardiomyopathies marked by its histologically significant remodeling and has proven to be a powerful asset in determining prognosis for these patients as well as enlightening scientists on a potential pathophysiological cascade that may begin early during the developmental process. Here, we discuss questions that continue to remain on the mechanistic processes leading to microvascular dysfunction, its correlation to the morphological changes in the vessels, and its contribution to disease progression.
Keywords: Hypertrophic cardiomyopathy, Microvascular, Ischemia, Pathogenesis, Energetics
Introduction
The heart demonstrates a threshold between mechanical output and blood supply where defects in either of these interdependent variables have severe consequences on the other. Having the highest oxygen consumption per mass with the highest oxygen extraction fraction renders the heart vulnerable to energy deficits as is likely to occur in hypertrophic cardiomyopathy (HCM). Reflecting this, the capillary density of the myocardium is estimated between 3000 and 4000 capillaries/mm3. Under increased stress, coronary flow can increase many-fold (six) to meet energetic demands [30]. Compared to other organs, the heart also has the highest capillary turnover rate as determined by 3H-thymidne, particularly in the subendocardium [58]. Evolution has maximized the mechanical output and the energy requirements of the heart allowing only brief moments of overloading via sympathetic stimulation for a selective advantage in survival and competition. Endothermal birds and mammals are at one end of this evolutionary process with heavily vascularized and disproportionately compact hearts [31].
Involvement of the coronary microcirculation in cardiac disease mechanisms is becoming more evident alongside advances in tools for their analysis. The exact pathophysiology in HCM has remained elusive potentially due to the heavy focus on mechanisms intrinsic to the myocyte, the bearer of the initial mutational effect, as well as a chasm between basic and clinical research. New evidence combined with previous histological observations offers insight into how the vasculature may participate in progression into macroscopic hypertrophy and clinically symptomatic phenotypes. This review will assimilate some of the contemporary concepts and findings in the studies of genetic cardiomyopathies with how they relate to and are influenced by the coronary circulation. We end by suggesting a probable pathophysiological cascade in the progression of HCM with relevance to non-compaction cardiomyopathies (NCCMs) and restrictive cardiomyopathies (RCMs).
Epidemiology and clinical relevance
Hypertrophic cardiomyopathy has a widely heterogeneous morphology and is a cause of considerable disease burden in developed countries. It is the most common genetic cardiovascular disorder with a prevalence estimated around 1 in 500 individuals in the USA, Canada, and Europe but can be more common than originally thought [81]. Newer imaging techniques and genetic analyses indicate a carrier prevalence, or population at risk, of around 1 in 200 individuals [125]. A majority of pathogenic mutations involved reside in the thick filament, consisting mostly of missense variants in MYH7 or truncation variants of MyBPC3, followed by a variety of thin filament mutations (TNNI3, TNNT2, TPM1, ACTC1) as well as regulatory (MYL2, MYL3) sarcomeric proteins [136]. Mutations do not guarantee disease phenotype however. The incomplete penetrance and highly variable expressivity of HCM indicate that understanding factors influencing disease progression can lead to improved strategies of prevention. Current interventions are directed at symptom management after development of an overt clinical phenotype but efforts are underway to increase the proportion of genotype positive, phenotype negative individuals (HCM carriers) by focusing on prevention.
Characterization studies of 333 HCM patients by Maron et al. using cine and late gadolinium CMR showed hypertrophy to be mostly diffuse (53%), some intermediate (34%), and a few focal (12%) with particularly consistent hypertrophy at the anterior basal interventricular septum and contiguous anterior left ventricular free wall. Worth noting, the cohort consisted of older individuals (43 ± 17 years old) at varying stages of the disease excluding heart failure or surgical procedures [83]. To contrast, a study of young HCM patients who died of sudden cardiac death showed 14 of 19 had asymmetric hypertrophy of the septum [12], suggesting that besides a negligible survivor’s bias when studying older HCM patients, the morphology may change with age of onset [80] and rate of progression. Genetic studies also revealed a higher probability of the presence of known sarcomeric mutations with earlier onset of disease [120]. Alongside the phenotypic heterogeneity, disease onset can occur at any moment in an individual’s life with significant hypertrophy capable of carrying on to old age without proportionately significant complications [81]. Such features including recent SNP analyses of contributions from non-sarcomeric polymorphisms demonstrate the multi-factorial nature of the disease [3, 115, 143].
In a portion of the affected, however, HCM can progress to heart failure with preserved ejection fraction. Left ventricular outflow tract obstruction can develop if the hypertrophy becomes severe and disproportionate enough with anterior systolic motion of the anterior mitral valve leaflet, placing additional stress on the heart. This progression leads to an increased risk of complications—arrhythmias, syncope, dyspnea—and is hallmarked by severe microvascular dysfunction. As a matter of fact, coronary flow reserve, an indicator of microvascular integrity, measured by myocardial blood flow after dipyridamole infusion, was shown to be an effective predictor of adverse clinical outcomes including death [19, 23, 77]. In comparison, microvascular dysfunction in left ventricular hypertrophy of equally severe magnitude but unrelated to sarcomere mutations does not have a comparable reduction in myocardial perfusion [103].
General features: histological, genetic, and metabolic
As revealed by histological determination, HCM has the general features of cardiac remodeling: myocyte hypertrophy, interstitial fibrosis, and decreased microvascular density. Unique to HCM, however, is the extent and septal localization of myofiber disarray and the patterns of vascular remodeling. Consequently, scattered septal microinfarctions, likely a result of vascular alterations but unrelated to coronary topology, are a frequent postmortem finding in HCM [12, 122]. The vascular remodeling as closely studied in pig models by Shyu et al. includes endothelial degeneration or denudation and smooth muscle hyperplasia and intimal infiltration [128] (Fig. 1). The instigators of these histological features of vascular injury, referred to by some pathologists as fibromuscular dysplasia, are at present unknown but are presumed to be caused by any of three possibilities: compressive systolic forces, shear stress, or inflammatory mediators. Also known for its microvascular dysfunction, hypertensive heart disease (HHD) does not exhibit the same extent of remodeling of the intramyocardial arteries as does HCM. Yet when comparing autopsied hearts, or those with terminal cardiac conditions, both HCM and HHD exhibit similar percent luminal narrowing of the arteries [28, 104, 133]. The focal tendency in disease development and progression [141] is becoming increasingly more apparent with the higher resolution of contemporary imaging modalities previously unappreciated by 2D echocardiography [75, 135, 138]. This pattern in respect to myofiber disarray is a cause of false-negative results from endomyocardial biopsies performed for diagnosing HCM as the lesions can at times be located deep within the septum [59]. In some infants with HCM who died suddenly, dysplastic arteries were already present suggesting their potential early role in the disease as well as implicating ischemia as a major factor in sudden death [12, 59, 73]. Current evaluations of microvascular dysfunction would indicate otherwise though. As measured presently, the dysfunction occurs late in the disease course as a consequence of using coronary flow reserve, which sums flow diffusely throughout the myocardium supplied by the left anterior descending artery; whereas in HCM, the pathogenesis is initially localized [135]. Early endothelial dysfunction is further masked by the extensive collateral network within the heart that acts to preserve the metabolic, functional, and electrical integrity of the tissue [139]. After all, vascular remodeling has been observed in otherwise normal tissue. Most likely in HCM as in coronary artery disease, collateralization within the septum increases with chronic obstructions as may occur when sufficient number of small arteries are impacted [46]. Presence of chronic and progressive obstructions is exemplified by the occurrence of large non-specific infarctions within the hearts of HCM patients on necropsy without history of acute coronary events or atherosclerosis [82]. This partially explains why exertional ischemia usually requires severe stenosis to develop in these disorders.
Fig. 1.
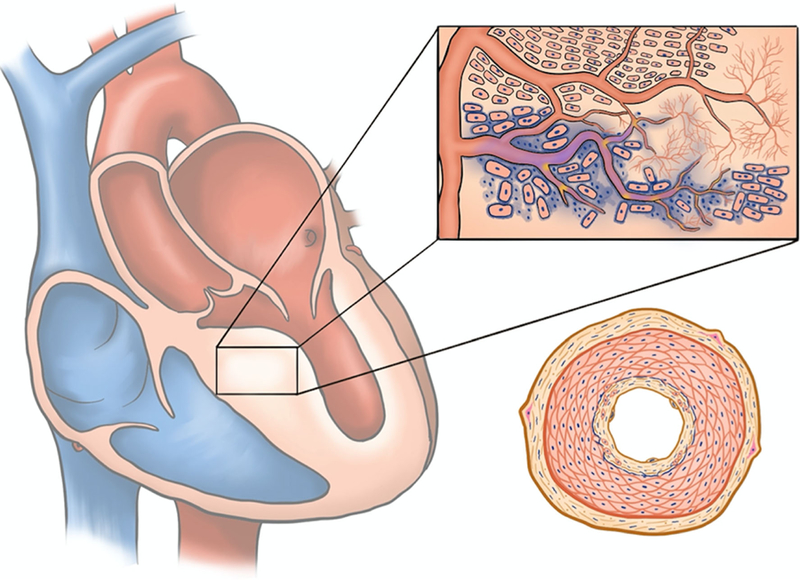
Gross and microscopic features of hypertrophic cardiomyopathy: HCM is characterized by focal lesions of replacement fibrosis within regions of gross hypertrophy as well as subendocardial fibrosis. Extensive small arterial remodeling is observed in these regions but not exclusively. The inset portrays mid-myocardial features associated with affected myocardial tissue: myofiber disarray, perivascular fibrosis (increased collagen deposition is indicated by blue as would be with Masson Trichrome staining), microvascular rarefaction, ischemic cardiac remodeling, interstitial fibrosis. Bottom right portrays a cross-section through an intra-mural artery undergoing typical changes observed during vascular injury (fibromuscular dysplasia), which include myofibroblast trans-differentiation and activation, medial hypertrophy, smooth muscle cell intimal infiltration and elaboration of extracellular matrix, and breakdown of the internal elastic membrane
Whether thick or thin filament mutations, the common feature in hypertrophic cardiomyopathy is the depletion of energy by either increased myofilament calcium sensitivity typically observed in thin filament mutations or inefficient cross-bridge cycling representative of thick filament mutations [1, 9, 57, 144]. Controversy still exists particularly in thick filament mutations as to how they affect the contractile apparatus’ functioning as disparate results have been obtained from experiments thus far [35, 69, 94]. Most likely physiological as well as pathological compensation in the form of cellular reprogramming alters critical protein content and posttranslational modifications creating variation in measurements of Ca2+ sensitivity, force generation, and cross-bridge ATPase kinetics. The distinction between thin and thick filament mutations is evident for instance by the rescue of transgenic tropomyosin E180G mice with phospholamban knockout [41] but failing to do so completely with MyBPC [123, 130] transgenics or to reverse the hypertrophy in MYH7 transgenics [38] although this approach did improve other cardiac parameters. Lack of phospholamban allows for shorter calcium transients diminishing time for cross-bridge cycling. In contrast, small molecule inhibitors that act to decrease ATPase activity of myosin rescue HCM phenotype in myosin-based mutations [48]. ATPase inhibitors in thin filament mutations have not yet been tested but direct targeting of the supposed effect of the mutation, Ca2+ sensitivity, has successfully rescued thin filament mouse models beforehand [5].
Nevertheless, the energy depletion hypothesis is supported by a diminished PCr/ATP ratio independent of the degree of hypertrophy [27] along with other relevant parameters—decreased ATP, increase ADP, decreased PCr, and increased lactate [105]. Diverging from biophysical and signaling-related models of dysfunctional relaxation, increased ADP/ATP ratio has been predicted to have significant effects on the sarcoplasmic reticulum calcium ATPase (SERCA) pump due to its high ATP consumption, but also potentially on actomyosin dissociation, both critical for effective relaxation [4, 119]. Accordingly, one of the initial prehypertrophic characteristics of HCM is diastolic dysfunction marked by an increased mitral E-to-e′ ratio [43, 52, 53, 89]. The importance of the SERCA pump in relaxation is best demonstrated by the myotonia of Brody’s disease patients whose deficient SERCA activity markedly prolongs skeletal muscle calcium transients and therefore relaxation. On the other hand, its involvement in calcium signaling pathways may in fact be behind the cellular remodeling process itself. Contemporary paradigms emphasize inappropriate Ca2+ signaling in HCM pathogenesis [50, 132].
Coronary circulation
Coronary vessels are often thought of as a separate, isolated system but there are many communications between coronary and chamber blood—arteriosinusoidal, arterioluminal, and thebesian vessels. The anastomoses may account for approximately 10% of coronary venous drainage [46] and may serve as an additional yet limited conduit for oxygenating the immediate subendocardium [112]. Potential to maintain coronary flow is dependent on upstream aortic pressure where fluctuations are met with a proportional arteriolar myogenic response to acutely autoregulate stable perfusion [78]. To complicate matters, the compressive forces of systolic contracting muscle confine a majority of left ventricular flow to diastole. Consequently, physiological retrograde flow in septal arteries and even in smaller epicardial arteries has been demonstrated [25]. Because the magnitude of systolic pressure is greatest in the subendocardium due to the summation of inward forces generated by the overlying myocardium as well as the smaller inner surface area [8, 46, 47, 51], an increased capillary blood volume (microvascular sinuses) and arteriolar density along with a compensatory hyperemia during diastole counteract the considerably decreased systolic perfusion in this layer. Similarly, adrenergic vasoconstriction in the epicardium acts to redirect more blood toward the inner layers during increased demand [32]. Evidence from positron emission tomography (PET) and cardiac magnetic resonance imaging (MRI) as well as clinical evidence from hypotensive patients suggests that this layer is indeed most susceptible to ischemia [84, 109]. It has been postulated that restoration of subendocardial perfusion by vasodilation, increased duration of diastole, or decreased oxygen consumption may be the mechanism by which verapamil ameliorates HCM-related anginal episodes [44].
Blood flow is dependent on the energy requirements of the tissue but also vice versa, and the heart exhibits a great deal of heterogeneity in metabolism and perfusion apart from the distribution generated by compressive forces [2, 114]. Bidirectional adaptability of perfusion and metabolism relates to the developmental thickening of the left ventricle even perinatally, initially accounted for by myocyte hyperplasia that promotes an environment suitable for angiogenesis [124]. Increased angiogenesis can likewise promote cellular hypertrophy [95]. Collateralization is included in this adaptive process and relates to the requirement of provocation tests with exercise, vasodilators, or inotropes to demonstrate ischemia in HCM. Along with the energy-wasting phenotype of HCM, the heterogeneity of myocardial perfusion provides an environment where foci of ischemia can develop and expand, which can be observed using contemporary imaging techniques.
Various imaging modalities have been used on human subjects to study the early functional impact of sarcomeric mutations including echocardiography, MRI, and PET. Using MRI and speckle tracking echocardiography (STE), reports of an exaggerated wringing motion, or torsion, could potentially further attenuate relaxation and diastolic flow by mechanically prolonging the total time the ventricle is in an mechanically unrelaxed state [8, 14]. Through STE, a prolonged untwist or unstrain rate was demonstrated to coincide in HCM carriers [67]. The increased torsion early in HCM mutation carriers is presumed be an indicator of impaired subendocardial contraction from deficient perfusion relative to the demand of the affected myocardium, not overt ischemia [ 121 ]. Extrapolating these findings to the transmural pressure differential across the wall of the myocardium, the initiation of diastole for adequate perfusion of the subendocardium may be postponed long after most sarcomeres have relaxed. During periods of allowed perfusion, one would expect a reactive increase in blood flow proportional to the duration of vascular compression. Such expected increases in diastolic blood flow velocity have been measured in the first septal perforator artery of symptomatic HCM patients to be 88 ± 40 cm/s, where-as normal controls were 41 ± 13 cm/s and HHD patients, 51 ± 18 cm/s [127]. An increased time to peak diastolic velocity [129] as well as a decreased diastolic perfusion acceleration and a rapid deceleration before systole [93] further support the significant relationship between diastolic dysfunction and coronary flow in the disorder. Parameters of diastolic dysfunction were found to be more substantial near the regions of greater hypertrophy as well as coronary microcirculatory abnormalities [60, 66]. Most studies of the epicardial and intramyocardial vessel flow have been done on HCM in later stages typically using Doppler echocardiography but also recently with wave intensity analysis [116]. Repeating them in early prehypertrophic phases would be more enlightening, though understandably the population of at-risk HCM individuals may be hard to come by. The general assumption is that the compressive forces may underlie changes in arterial morphology, but altered shear stress from changing flow velocities is a known inducer of vascular remodeling and endothelial dysfunction in other pathologies besides atherosclerosis [140].
Many disorders of small intramyocardial arteries other than HCM lead to diastolic dysfunction and restrictive physiology. Dysplasia (fibromuscular) of small intramyocardial arteries is seen in small/medium artery vasculitides, amyloidosis, diabetes, and Friederich ataxia. In these disorders, it is not the isolated dysplastic artery that leads to observable pathologies, but the coalescence of remodeled arteries that leads to replacement fibrosis and compensatory adaptive changes to the hypoxic/ischemic tissue [62]. Consequently, infiltrative diseases such as amyloidosis and Fabry disease can also share many of the same features of HCM depending on the pattern of involvement and in clinical practice may require further tests for proper diagnosis. Classic amyloidosis presents with systemic and diffuse interstitial deposition especially around the small vessels and if cardiac involvement occurs, concentric hypertrophy, although hypertrophy may not always be present [70, 96]. Apparently, most cases of cardiac transthyretin amyloidosis are surprisingly asymmetric with a preferential septal localization [85]. Both amyloidosis and Fabry disease have also been shown to be associated with microvascular dysfunction by measuring coronary flow reserve [76]. Other disorders causing occlusion of small coronary arterioles such as in sickle cell anemia and Churg-Strauss vasculitis have a similar tendency to lead to diastolic dysfunction [11, 98, 99].
HCM spectrum
Depending on the mutation, inadequate perfusion in HCM can occur during the fetal period. The developmental morphological characteristics of HCM carriers have been apparent to clinicians: myocardial crypts [42], mitral valve abnormalities, and false tendons [21, 102]. Sarcomeric mutations implicated in HCM have a tendency to also develop non-compaction cardiomyopathy (NCCM) and these two disorders often cooccur [13, 56, 107] with non-compaction having a predilection for the apex corresponding to the last region in the compaction process [86]. Compaction essentially follows the path of the coronary arteries: from base to apex and from the septum to lateral wall. The septum itself is formed from the fusion of trabeculae during the third week of fetal life, at that point called the inter-ampullary septum, and the process of compaction continues to some degree throughout the rest of fetal development. Early cardiomyocyte derivatives in the septum share a lineage with the vasculature and stromal tissue to a greater extent than lateral cardiac tissue probably reflecting this relationship [17]. Efforts are currently underway to study this process in detail with a focus on HCM [22].
NCCM is a disorder of diverse etiologies, many of which are related to deficient oxygen content such as coronary anomalies and congenital cardiac malformations. Here, we mainly consider the cases related to sarcomeric mutations [6]. NCCM has similar histological characteristics to HCM including arteriolar remodeling [15, 36]. Subendocardial replacement fibrosis from ischemia and microvascular dysfunction is observed even in the young [63, 65]. The primary purpose of the non-compacted, or spongy, myocardium is to increase surface area for continued expansion of the myocardial muscle with an appropriate supply of oxygenation by the chamber blood similar to the morphology seen in fish and other lower vertebrates [7, 21, 72]. It is apparently a normal stage of development prior to fusion [100, 108, 124]. Substantial evidence of the implications of the process comes from fish, whose greater amounts of compact myocardium is met with increased coronary vascular perfusion, than other fish. On one extreme are fish with no coronary vasculature and therefore only have spongy myocardium [31, 37]. Mouse models with impaired coronary development have likewise shown an increase in spongy myocardium with reduction in vascularization and may be associated with angiogenic and hypoxic signaling [24, 90]. It has been hypothesized previously that chronic hypoxia from deficient coronaries may cause greater degrees of trabeculation to compensate [124]. In addition, the susceptibility of the myocardial septum in various pathological disorders may be related to its unique embryological derivate [17, 68]. Unfortunately, attempts at creating mouse models of non-compaction using sarcomeric mutations specifically have generally failed, again probably as a result of the drastically different heart rates and coronary flows in these animals [6].
In sarcomeric RCM, adaptation of perfusion with metabolism and the compaction process itself overestimated the capability of the blood supply to match myocardial energy demand and can be related to a temporal effect of mutations on energetics—more acute or sudden. This is demonstrated by the preferential involvement of genes differentially expressed (cTnI expressed after birth and β-MHC slowly replacing α- MHC) or alternatively spliced (cTnT adult vs fetal variants) during development [10, 126]. The temporal effect of a mutation can be further potentiated by the increased rates of turn-over of troponin subunits [88]. A rather crude analysis using ClinVar variants in Table 1 demonstrates this pattern and may present potential avenues for more comprehensive statistical analyses. Other less commonly associated mutations—tropomyosin, MyBPC, or α-cardiac actin—are more difficult to explain but may involve a preferential interaction with the differentially expressed proteins during development. Mutations in α-cardiac actin have recently been shown to have an age-dependent increase in effect although the experimenters were relating this to apical HCM [142]. Likewise, restrictive phenotypes with MyBPC are related to truncations by either frameshift or splicing defects implicating allelic insufficiency in genetic pathogenesis. With increased cell size, demands for increased protein production cannot be met. Similar to NCCM, there is strong evidence of subendocardial ischemia in RCM of non-sarcomeric origin [117]. A discussion of RCM and NCCM, which are often associated with other genetic mutations with greatly variable pathogenesis involving substantial cell death and replacement, is out of the scope of this review.
Table 1.
Potential for cardiomyopathies in genes: #RCM and #NCCM represent the number of missense mutations associated with the disorder whereas %RCM and %NCCM reflect how many of those mutations occur in respect to all missense mutations that have the potential to promote metabolic inefficiency: #RCM/(total RCM, HCM, and NCCM mutations). Because of the low number of variants associated with these mutations, pathogenic and likely pathogenic were used to populate the table. Parentheses indicate variants of unknown significance. We also used conflicting interpretations of pathogenicity since the HCM spectrum is known to have clinically heterogeneous phenotypes. Not enough data for ACTC1 and TPM1 genes. Total MYH7 = 227. TNNI3 = 42, TNNT2 = 39, MYBPC = 101
| Gene | #RCM | #NCCM | %RCM | %NCCM |
|---|---|---|---|---|
| TNNI3 | 12(2) | 0(0) | 23.5% | 0 |
| MYH7 | 4(1) | 13(19) | 1.8% | 5.7% |
| TNNT2 | 19(38) | 24(40) | 48.7% | 61.5% |
| MYBPC3 | 0(0) | 15(15) | 0 | 14.9% |
Microcirculatory alterations and prehypertrophy
So far, we have discussed pathological associations of some of the larger vessels, small arteries, and proximal pre-arterioles, but microvascular changes involving capillary and distal pre-arteriolar rarefaction are also seen to occur in HCM [64]. Studies using human subendocardial biopsies have shown hypertensive heart disease to have a tendency of affecting smaller sized pre-capillary arterioles with smooth muscle hyperplasia, whereas HCM consists of a decreased density of such vessels without significant changes in medial content [92]. The rarefaction can be preferentially, though not exclusively, localized to the hypertrophied segment [91]. In general, microvascular rarefaction may occur by mechanical or molecular mechanisms [45]. Capillary rarefaction has been observed to occur when upstream blood flow is impaired, for instance [118, 134]. This phenomenon is also observed in valvular or sub-valvular aortic stenosis where coronary driving pressure is decreased [87]. Diffuse microvascular dysfunction in HCM as clinically determined occurs late in the disease course. Although some attribute the dysfunction to micro-vascular rarefaction, it likely occurs in concert with small arterial remodeling as they are codependent [23, 49, 74]. Vasomotor control has a plethora of mediators, some still unknown, that have the capacity to participate but whose function depends on the tissue and context and can compensate for each other whenever the primary mediator is suppressed. Furthermore, the acting vasoactive molecule may vary greatly among different species. Equally challenging for researchers using murine models, coronary flow rates in mice and rats barely increase with stimulation, due to their high basal flow rates and heart rates [30]. Nevertheless, models of myocardial blood flow regulation have been devised and indicate a reliance primarily on neurohormonal adrenergic stimuli, purinergic mediation from metabolites and red blood cell secretions, and endothelial H2O2 secretion acting primarily on distal arterioles [111]. Flow-mediated dilation (FMD) on the other hand is a reactionary process in larger conduit arteries primarily relying on NO release that when impaired may be substituted by prostaglandins and potentially other mediators. Direct studies of coronary FMD in HCM are lacking. Although an exact experimental approach in humans is not present, there is a convenient alternative. Peripheral endothelial dysfunction has been demonstrated to mimic coronary endothelial dysfunction in other disorders such as coronary artery disease [110]. Evidence for systemic metabolic or inflammatory states affecting endothelial function, and therefore cardiac performance, has been apparent for a while, but genetic cardiomyopathies offer a unique perspective in which expression of genetic alterations restricted to the myocardium affects the peripheral vascular function that introduces a potential for a novel interaction between the peripheral circulation and the heart.
Direct and definitive evidence for coronary microvascular dysfunction early in HCM disease course still does not exist, but there is indirect evidence from two sources of inquiry. The action of therapeutic targets related to cardiac remodeling requires early intervention suggesting a prehypertrophic remodeling process [79, 137]; mouse models demonstrate that causative processes mediating hypertrophy start early in the neonatal period and after a certain point are irreversible even with correction of the underlying genetic abnormality [20]. The necessity of understanding the mechanism of the early determination for remodeling is accentuated by the prehypertrophic effects on the peripheral vasculature in at-risk HCM individuals determined by family history. An increased response to acetylcholine induced vasodilation as determined by laser Doppler with iontophoresis was observed. It was also found that this directly correlated with the degree of diastolic dysfunction, or E/e′ ratio [33].
Given these facts, it is of interest to seek evidence for prehypertrophic processes affecting fibrotic and vascular remodeling. Current evidence in humans indicates an early remodeling process with increases in circulating cathepsin S and endostatin correlating with diastolic dysfunction [34]. Others have shown increases in collagen synthesis detected by increased PICP, an unusual finding in a pressure-overloaded system without significant hypertrophy [54]. Evidence of transcriptional changes in the prehypertrophic stage in a thick filament mouse model of HCM demonstrated upregulation of TGFB1, CTGF, and periostin (prehypertrophic) and also downregulation of SERCA2a, phospholamban, and sarcolipin—markers of fibrosis and cellular remodeling, respectively [71]. On the other hand, clinical studies, though with a limited number of biomarkers, are struggling to find markers for a prehypertrophic fibrotic process [55].
Some have chosen to focus more so on the relationship of the vessels with HCM. Polymorphisms in angiogenic genes, VEGF and HIF1-a [3], as well as renin-angiotensin-aldosterone-system genes (controlled for blood pressure) [106], showed increased susceptibility to earlier phenotypes. Whether these markers are evidence for early fibrosis, early endothelial dysfunction, or myocyte remodeling process, is still questionable and should be more extensively investigated. Although energy impairment may be the initial catalyst, the degree of hypertrophy correlates directly with stress-induced or vasodilator-induced ischemia, whereas energy impairment does not [29, 49, 61]. Ischemia can also be seen in regions of non-hypertrophied myocardium, potentially heralding extension of remodeling in those regions as well [26, 109]. Accordingly, it was previously shown using N13 PET that alterations in coronary vasodilator reserve occur in both hypertrophied and non-hypertrophied myocardium [18]. Eventually, if the heart does not decompensate toward a dilated phenotype, the microvascular dysfunction saturates and the correlation between age or duration of disease and severity of microvascular dysfunction is poor [101, 103, 131]. This new homeostatic point of a heavily remodeled yet functioning heart may be adequate for the time-being, but development of comorbidities that come with age or diet may again tip the scale to an unfavorable energetic point (Fig. 2).
Fig. 2.
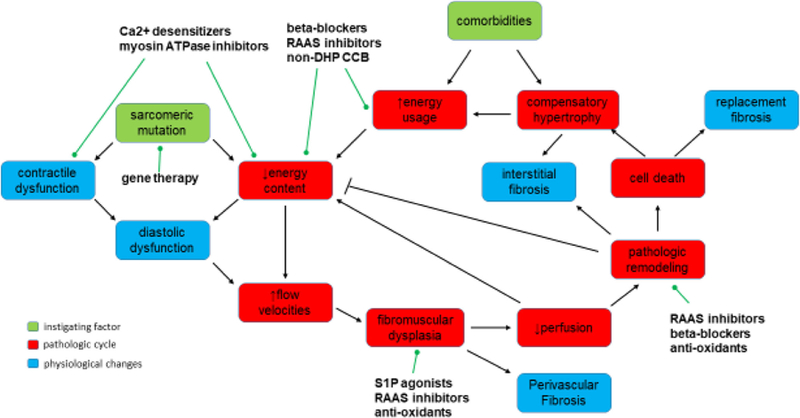
Cascade of events leading to the cyclical remodeling pattern in HCM: The initial HCM-causing mutation leads to an alteration in the contractile dynamics of the myofilaments that either directly is the cause of diastolic abnormalities or through inefficient energy consumption leads to diastolic dysfunction. Inefficient energy consumption causes arterial remodeling (fibromuscular dysplasia) likely through shear stress–related vascular injury. When remodeling is severe enough, perfusion is impaired leading to ischemic cardiac remodeling that can act to lower energy costs but when severe enough may replace cells with fibrous tissue. Loss of viable myocardium eventually leads to a compensatory hypertrophy that not only increases energy requirements but also the transmural distance required for coronary flow to traverse (not drawn). Comorbidities such as hypertension, valvular disease, or aging can increase metabolic demands of the myocardium. Possible direct targets of some current and prospective therapeutic treatments are noted. RAAS, renin-angiotensin-aldosterone-system; S1P, sphingosine-1-phosphate; non-DHP CCB, non-dihydropyridine calcium channel blockers
Summary and implications
HCM is the most common genetic cardiovascular disorder and the related sarcomeric mutations increase the energy requirements of tension [35, 81]. Resultant changes in energetics along with alterations in contractile dynamics perturb proper diastolic function. Because diastole is crucial for myocardial perfusion, the subendocardium in diastolic dysfunction is prone to ischemia with increased flow velocities required to supply this layer. Increased flow velocities with high shear stress may mediate flow-induced arterial remodeling (arteriogenesis) or a response to injury [113]. The extensive collateral network of the coronary vasculature tends to mask the effects of vascular remodeling found dispersed throughout the myocardium. It can often be found without other features of cardiac remodeling such as cellular hypertrophy [59, 84]. Therefore, contiguously altered vessels are required to create foci of vascular compromise, gradually expanding and remodeling the supplied myocardium. This in turn, through ischemia, initiates cardiac remodeling. Remodeling further changes the contractile dynamics and perfusion threshold of the myocardium causing an expansion of ischemia and remodeling. Evidence supporting these findings include induced ischemia seemingly preceding large foci of fibrosis [26, 131], the dramatic changes in diastolic flow velocities within the intramyocardial small arteries [127], and the pro-found arteriolar remodeling evident on histology [59].
Following this model, targeting the vascular remodeling process or the capillary and arteriolar density can prevent the energy perfusion mismatch that insidiously develops in HCM. Promoting endothelial function, angiogenesis, or collateralization may decrease the energy-depleted state as has been tested in hypertrophied hearts with aortic stenosis [39, 40]. Targeting arterial remodeling may be a secondary effect of many of the drugs currently used for treatment of cardiac pathologies (RAAS inhibitors, beta-blockers), and therefore a suitable alternative for preventative HCM treatments [16, 97]. We also suggest that certain cases of idiopathic hypertrophic cardiomyopathy without sarcomeric mutations may have polymorphisms increasing susceptibility to vascular remodeling which may include the angiogenic polymorphisms [3]. The knowledge gained from understanding the unique vascular pathologies occurring in HCM can then be extended to coronary small arterial modifications that occur in many cardiac disorders particularly on their way to decompensation.
Acknowledgements
We would also like to thank Jin Lim for illustrating Fig. 1.
Funding information This study received financial support from the following: NIH NHLBI PO1 HL 62426 (RJS, BMW); RO1 HL128468 (BMW, RJS), and T32 HL139439 (RJM).
Footnotes
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Adhikari AS, Kooiker KB, Sarkar SS, Liu C, Bernstein D, Spudich JA, Ruppel KM (2016) Early-onset hypertrophic cardio-myopathy mutations significantly increase the velocity, force, and actin-activated ATPase activity of human beta-cardiac myosin. Cell Rep 17(11):2857–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alders DJC, Groeneveld ABJ, de Kanter FJJ, van Beek JHGM (2004) Myocardial O2 consumption in porcine left ventricle is heterogeneously distributed in parallel to heterogeneous O2 delivery. Am J Physiol Heart Circ Physiol 287:H1353–H1361 [DOI] [PubMed] [Google Scholar]
- 3.Alkon J, Friedberg MK, Manlhiot C, Manickaraj AK, Kinnear C, McCrindle BW, Benson LN, Addonizio LJ, Colan SD, Mital S (2012) Genetic variations in hypoxia response genes influence hypertrophic cardiomyopathy phenotype. Pediatr Res 72(6):583–592 [DOI] [PubMed] [Google Scholar]
- 4.Allen DG, Orchard CH (1986) Myocardial contractile function during ischemia and hypoxia. Circulation 60(2):153–168 [DOI] [PubMed] [Google Scholar]
- 5.Alves ML, Dias FAL, Gaffin RD, Simon JN, Montminy EM, Biesiadecki BJ, Hinken AC, Warren CM, Utter MS, Davis RTR, Sakthivel S, Robbins J, Wieczorek DF, Solaro RJ, Wolska BM (2014) Desensitization of myofilaments to Ca2+ as a therapeutic target for hypertrophic cardiomyopathy with mutations in thin filament proteins. Circ Cardiovasc Genet 7(2):132–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andersson UME, Morcos PN, Adler ED (2015) Left ventricular non-compaction: current controversy and new insights. J Genet Syndr Gene Ther 06(01):1–9 [Google Scholar]
- 7.Angelini A, Melacini P, Barbero F, Thiene G (1999) Evolutionary persistence of spongy myocardium in humans. Circulation 99(18): 2475–2475 [DOI] [PubMed] [Google Scholar]
- 8.Arts MGJ, Reneman RS, Veenstra PC (1979) A model of the mechanics of the left ventricle. Ann Biomed Eng 7:299–318 [DOI] [PubMed] [Google Scholar]
- 9.Ashrafian H, McKenna WJ, Watkins H (2011) Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ Res 109(1):86–96 [DOI] [PubMed] [Google Scholar]
- 10.Bahl A, Saikia UN, Khullar M (2012) Idiopathic restrictive cardiomyopathy - perspectives from genetics studies. Is it time to redefine these disorders? Cardiogenetics 2(1):4 [Google Scholar]
- 11.Bakeera N, Jamesd J, Royb S, Wansapuraf J, Shanmukhappah SK, Lorenzi JN, Osinskae H, Backerb K, Hubyd A-C, Shresthab A, Nissa O, Fleckk R, Quinna CT, Taylord MD, Purevjave E, Aronowl BJ, Towbind JA, Malik P (2016) Sickle cell anemia mice develop a unique cardiomyopathy with restrictive physiology. PNAS 113:E5182–E5191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Basso C, Thiene G, Corrado D, Buja G, Melacini P, Nava A (2000) Hypertrophic cardiomyopathy and sudden death in the young: pathologic evidence of myocardial ischemia. Hum Pathol 31(8):988–998 [DOI] [PubMed] [Google Scholar]
- 13.Biagini E, Ragni L, Ferlito M, Pasquale F, Lofiego C, Leone O, Rocchi G, Perugini E, Zagnoni S, Branzi A, Picchio FM, Rapezzi C (2006) Different types of cardiomyopathy associated with isolated ventricular noncompaction. Am J Cardiol 98(6):821–824 [DOI] [PubMed] [Google Scholar]
- 14.Buckberg G, Mahajan A, Saleh S, Hoffman JI, Coghlan C (2008) Structure and function relationships of the helical ventricular myocardial band. J Thorac Cardiovasc Surg 136(3):578–589 589 e571–511 [DOI] [PubMed] [Google Scholar]
- 15.Burke A, Mont E, Kutys R, Virmani R (2005) Left ventricular noncompaction: a pathological study of 14 cases. Hum Pathol 36(4):403–411 [DOI] [PubMed] [Google Scholar]
- 16.Buus NH, Bottcher M, Jorgensen CG, Christensen KL, Thygesen K, Nielsen TT, Mulvany MJ (2004) Myocardial perfusion during long-term angiotensin-converting enzyme inhibition or beta-blockade in patients with essential hypertension. Hypertension 44(4):465–470 [DOI] [PubMed] [Google Scholar]
- 17.Cai CL, Martin JC, Sun Y, Cui L, Wang L, Ouyang K, Yang L, Bu L, Liang X, Zhang X, Stallcup WB, Denton CP, McCulloch A, Chen J, Evans SM (2008) A myocardial lineage derives from Tbx18 epicardial cells. Nature 454(7200):104–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Camici P, Chiriatti G, Lorenzoni R, Bellina RC, Gistri R, Italiani G, Parodi O, Salvadori PA, Nista N, Papi L, L’Abbate A (1991) Coronary vasodilation is impaired in both hypertrophied and nonhypertrophied myocardium of patients with hypertrophic cardiomyopathy: a study with nitrogen-13 ammonia and positron emission tomography. J Am Coll Cardiol 17(4):879–886 [DOI] [PubMed] [Google Scholar]
- 19.Cannon RO, Dilsizian V, O’Gara PT, Udelson JE, Schenke WH, Quyyumi A, Fananapazir L, Bonow RO (1991) Myocardial metabolic, hemodynamic, and electrocardiographic significance of reversible thallium-201 abnormalities in hypertrophic cardiomyopathy. Circulation 83(5):1660–1667 [DOI] [PubMed] [Google Scholar]
- 20.Cannon L, Yu ZY, Marciniec T, Waardenberg AJ, Iismaa SE, Nikolova-Krstevski V, Neist E, Ohanian M, Qiu MR, Rainer S, Harvey RP, Feneley MP, Graham RM, Fatkin D (2015) Irreversible triggers for hypertrophic cardiomyopathy are established in the early postnatal period. J Am Coll Cardiol 65(6):560–569 [DOI] [PubMed] [Google Scholar]
- 21.Captur G, Ho CY, Schlossarek S, Kerwin J, Mirabel M, Wilson R, Rosmini S, Obianyo C, Reant P, Bassett P, Cook AC, Lindsay S, McKenna WJ, Mills K, Elliott PM, Mohun TJ, Carrier L, Moon JC (2016) The embryological basis of subclinical hypertrophic cardiomyopathy. Sci Rep 6:27714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Captur G, Wilson R, Bennett MF, Luxan G, Nasis A, de la Pompa JL, Moon JC, Mohun TJ (2016) Morphogenesis of myocardial trabeculae in the mouse embryo. J Anat 229(2):314–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cecchi F, Olivotto I, Gistri R, Lorenzoni R, Chiriatti G, Camici PG (2003) Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. NEJM 349(11):1027–1035 [DOI] [PubMed] [Google Scholar]
- 24.Chen HI, Sharma B, Akerberg BN, Numi HJ, Kivela R, Saharinen P, Aghajanian H, McKay AS, Bogard PE, Chang AH, Jacobs AH, Epstein JA, Stankunas K, Alitalo K, Red-Horse K (2014) The sinus venosus contributes to coronary vasculature through VEGFC-stimulated angiogenesis. Development 141(23):4500–4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chilian WM, Marcus ML (1982) Phasic coronary blood flow velocity in intramural and epicardial coronary arteries. Circ Res 50: 775–781 [DOI] [PubMed] [Google Scholar]
- 26.Choudhury L, Rosen SD, Patel D, Nihoyannopoulos P, Camici PG (1997) Coronary vasodilator reserve in primary and secondary left ventricular hypertrophy: a study with positron emission tomography. Eur Heart J 18:108–116 [DOI] [PubMed] [Google Scholar]
- 27.Crilley JG, Boehm EA, Blair E, Rajagopalan B, Blamire AM, Styles P, McKenna WJ, Östman-Smith I, Clarke K, Watkins H (2003) Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J Am Coll Cardiol 41(10):1776–1782 [DOI] [PubMed] [Google Scholar]
- 28.Dai Z, Aoki T, Fukumoto Y, Shimokawa H (2012) Coronary perivascular fibrosis is associated with impairment of coronary blood flow in patients with non-ischemic heart failure. J Cardiol 60(5):416–421 [DOI] [PubMed] [Google Scholar]
- 29.Dass S, Cochlin LE, Suttie JJ, Holloway CJ, Rider OJ, Carden L, Tyler DJ, Karamitsos TD, Clarke K, Neubauer S, Watkins H (2015) Exacerbation of cardiac energetic impairment during exercise in hypertrophic cardiomyopathy: a potential mechanism for diastolic dysfunction. Eur Heart J 36(24):1547–1554 [DOI] [PubMed] [Google Scholar]
- 30.Duncker D, Bache R (2008) Regulation of coronary blood flow during exercise. Physiol Rev 88(3):1009–1086 [DOI] [PubMed] [Google Scholar]
- 31.Farrell A, Farrell N, Jourdan H, Cox G (2012) A perspective on the evolution of the coronary circulation in fishes and the transition to terrestrial life. In: Sedmera D, Wang T (eds) Ontogeny and phylogeny of the vertebrate heart Springer, New York, pp 75–102 [Google Scholar]
- 32.Feigl EO (1987) The paradox of adrenergic coronary vasoconstriction. Circulation 76(4):737–745 [DOI] [PubMed] [Google Scholar]
- 33.Fernlund E, Schlegel TT, Platonov PG, Carlson J, Carlsson M, Liuba P (2015) Peripheral microvascular function is altered in young individuals at risk for hypertrophic cardiomyopathy and correlates with myocardial diastolic function. Am J Physiol Heart Circ Physiol 308(11):H1351–H1358 [DOI] [PubMed] [Google Scholar]
- 34.Fernlund E, Gyllenhammar T, Jablonowski R, Carlsson M, Larsson A, Arnlov J, Liuba P (2017) Serum biomarkers of myocardial remodeling and coronary dysfunction in early stages of hypertrophic cardiomyopathy in the young. Pediatr Cardiol 38(4):853–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferrantini C, Belus A, Piroddi N, Scellini B, Tesi C, Poggesi C (2009) Mechanical and energetic consequences of HCM-causing mutations. J Cardiovasc Transl Res 2(4):441–451 [DOI] [PubMed] [Google Scholar]
- 36.Finsterer J, Stollberger C, Feichtinger H (2002) Histological appearance of left ventricular hypertrabeculation/noncompaction. Cardiology 98(3):162–164 [DOI] [PubMed] [Google Scholar]
- 37.Fisher SA, Burggren WW (2007) Role of hypoxia in the evolution and development of the cardiovascular system. Antioxid Redox Signal 9(9):1339–1352 [DOI] [PubMed] [Google Scholar]
- 38.Freeman K, Lerman I, Kranias EG, Bohlmeyer T, Bristow MR, Lefkowitz RJ, Iaccarino G, Koch WJ, Leinwand LA (2001) Alterations in cardiac adrenergic signaling and calcium cycling differentially affect the progression of cardiomyopathy. J Clin Investig 107(8):967–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Friehs I, Moran AM, Stamm C, Choi YH, Cowan DB, McGowan FX, del Nido PJ (2004) Promoting angiogenesis protects severely hypertrophied hearts from ischemic injury. Ann Thorac Surg 77(6):2004–2010 [DOI] [PubMed] [Google Scholar]
- 40.Friehs I, Barillas R, Vasilyev NV, Roy N, McGowan FX, del Nido PJ (2006) Vascular endothelial growth factor prevents apoptosis and preserves contractile function in hypertrophied infant heart. Circulation 114:I290–I295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaffin RD, Pena JR, Alves MS, Dias FA, Chowdhury SA, Heinrich LS, Goldspink PH, Kranias EG, Wieczorek DF, Wolska BM (2011) Long-term rescue of a familial hypertrophic cardiomyopathy caused by a mutation in the thin filament protein, tropomyosin, via modulation of a calcium cycling protein. J Mol Cell Cardiol 51(5):812–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Germans T, Wilde AA, Dijkmans PA, Chai W, Kamp O, Pinto YM, van Rossum AC (2006) Structural abnormalities of the inferoseptal left ventricular wall detected by cardiac magnetic resonance imaging in carriers of hypertrophic cardiomyopathy mutations. J Cardiovasc Magn Reson 48(12):2518–2523 [DOI] [PubMed] [Google Scholar]
- 43.Germans T, Rüssel IK, Götte MJ, Spreeuwenberg MD, Doevendans PA, Pinto YM, van der Geest RJ, van der Velden J, Wilde AA, van Rossum AC (2010) How do hypertrophic cardiomyopathy mutations affect myocardial function in carriers with normal wall thickness? Assessment with cardiovascular magnetic resonance. Journal of Cardiovascular Magnetic Resonance 12(13):1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gistri R, Cecchi F, Choudhury L, Montereggi A, Sorace O, Salvadori PA, Camici PG (1994) Effect of verapamil on absolute myocardial blood flow in hypertrophic cardiomyopathy. Am J Cardiol 74:363–368 [DOI] [PubMed] [Google Scholar]
- 45.Goligorsky MS (2010) Microvascular rarefaction. Organogenesis 6(1):1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goodwill AG, Dick GM, Kiel AM, Tune JD (2017) Regulation of coronary blood flow. Compr Physiol 7(2):321–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goto M, Flynn AE, Doucette JW, Jansen CM, Stork MM, Coggins DL, Muehrcke DD, Husseini WK, Hoffman JI (1991) Cardiac contraction affects deep myocardial vessels predominantly. Am J Physiol Heart Circ Physiol 261(5):H1417–H14129 [DOI] [PubMed] [Google Scholar]
- 48.Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, Henze M, Kawas R, Oslob JD, Rodriguez HM, Song Y, Wan W, Leinwand LA, Spudich JA, McDowell RS, Seidman JG, Seidman CE (2016) A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 351(6273):617–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gyllenhammar T, Fernlund E, Jablonowski R, Jogi J, Engblom H, Liuba P, Arheden H, Carlsson M (2014) Young patients with hypertrophic cardiomyopathy, but not subjects at risk, show decreased myocardial perfusion reserve quantified with CMR. Eur Heart J Cardiovasc Imaging 15(12):1350–1357 [DOI] [PubMed] [Google Scholar]
- 50.Heineke J, Molkentin JD (2006) Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol 7(8):589–600 [DOI] [PubMed] [Google Scholar]
- 51.Hiramatsu O, Goto M, Yada Toyotaka, Kimura A, Chiba Y, Tachibana H, Ogasawara Y, Tsujioka K, Kajiya Fumihiko (1998) In vivo observations of the intramural arterioles and venules in beating canine hearts. J Physiol 509(2):619–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ho CY (2002) Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy. Circulation 105(25):2992–2997 [DOI] [PubMed] [Google Scholar]
- 53.Ho CY, Carlsen C, Thune JJ, Havndrup O, Bundgaard H, Farrohi F, Rivero J, Cirino AL, Andersen PS, Christiansen M, Maron BJ, Orav EJ, Kober L (2009) Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2(4):314–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ho CY, Lopez B, Coelho-Filho OR, Lakdawala NK, Cirino AL, Jarolim P, Kwong R, Gonzalez A, Colan SD, Seidman JG, Diez J, Seidman CE (2010) Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N Engl J Med 363(6):552–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ho JE, Shi L, Day SM, Colan SD, Russell MW, Towbin JA, Sherrid MV, Canter CE, Jefferies JL, Murphy A, Taylor M, Mestroni L, Cirino AL, Sleeper LA, Jarolim P, Lopez B, Gonzalez A, Diez J, Orav EJ, Ho CY (2017) Biomarkers of cardiovascular stress and fibrosis in preclinical hypertrophic cardiomyopathy. Open Heart 4:e000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoedemaekers YM, Caliskan K, Majoor-Krakauer D, van de Laar I, Michels M, Witsenburg M, ten Cate FJ, Simoons ML, Dooijes D (2007) Cardiac beta-myosin heavy chain defects in two families with non-compaction cardiomyopathy: linking non-compaction to hypertrophic, restrictive, and dilated cardiomyopathies. Eur Heart J 28(22):2732–2737 [DOI] [PubMed] [Google Scholar]
- 57.Huang W, Liang J, Kazmierczak K, Muthu P, Duggal D, Farman GP, Sorensen L, Pozios I, Abraham TP, Moore JR, Borejdo J, Szczesna-Cordary D (2014) Hypertrophic cardiomyopathy associated Lys104Glu mutation in the myosin regulatory light chain causes diastolic disturbance in mice. J Mol Cell Cardiol 74:318–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hudlická O (1982) Growth of capillaries in skeletal and cardiac muscle. Circ Res 50(4):451–461 [DOI] [PubMed] [Google Scholar]
- 59.Hughes SE (2004) The pathology of hypertrophic cardiomyopathy. Histopathology 44:412–427 [DOI] [PubMed] [Google Scholar]
- 60.Inoue T, Morooka S, Hayashi T, Takayanagi K, Sakai Y, Fujito T, Fujinuma S, Takabatake Y (1991) Global and regional abnormalities of left ventricular diastolic filling in hypertrophic cardiomyopathy. Clin Cardiol 14(7):573–577 [PubMed] [Google Scholar]
- 61.Jablonowski R, Fernlund E, Aletras AH, Engblom H, Heiberg E, Liuba P, Arheden H, Carlsson M (2015) Regional stress-induced ischemia in non-fibrotic hypertrophied myocardium in young HCM patients. Pediatr Cardiol 36(8):1662–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.James TN (1977) Small arteries of the heart. Circulation 56(1):2–14 [DOI] [PubMed] [Google Scholar]
- 63.Jenni R, Wyss CA, Oechslin EN, Kaufmann PA (2002) Isolated ventricular noncompaction is associated with coronary microcirculatory dysfunction. J Am Coll Cardiol 39(3):450–454 [DOI] [PubMed] [Google Scholar]
- 64.Johansson B, Morner S, Waldenstrom A, Stal P (2008) Myocardial capillary supply is limited in hypertrophic cardiomyopathy: a morphological analysis. Int J Cardiol 126(2):252–257 [DOI] [PubMed] [Google Scholar]
- 65.Junga G, Kneifel S, Smekal AV, Steinert H, Bauersfeld U (1999) Myocardial ischaemia in children with isolated ventricular noncompaction. Eur Heart J 20:910–916 [DOI] [PubMed] [Google Scholar]
- 66.Kakoi H, Inoue T, Morooka S, Hayashi T, Takabatake Y (1996) Relationship between regional abnormality of left ventricular rapid filling and coronary microcirculation disturbance in hypertrophic cardiomyopathy. Clin Cardiol 19:379–383 [DOI] [PubMed] [Google Scholar]
- 67.Kauer F, van Dalen BM, Michels M, Schinkel AF, Vletter WB, van Slegtenhorst M, Soliman OI, Geleijnse ML (2017) Delayed and decreased LV untwist and unstrain rate in mutation carriers for hypertrophic cardiomyopathy. Eur Heart J Cardiovasc Imaging 18(4):383–389 [DOI] [PubMed] [Google Scholar]
- 68.Kaul S (1986) The interventricular septum in health and disease. Am Heart J 112(3):568–581 [DOI] [PubMed] [Google Scholar]
- 69.Kawana M, Sarkar SS, Sutton S, Ruppel KM, Spudich JA (2017) Biophysical properties of human β-cardiac myosin with converter mutations that cause hypertrophic cardiomyopathy. Sci Adv 3:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kholova I, Niessen HW (2005) Amyloid in the cardiovascular system: a review. J Clin Pathol 58(2):125–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim JB, Porreca GJ, Song L, Greenway SC, Gorham JM, Church GM, Seidman CE, Seidman JG (2007) Polony multiplex analysis of gene expression (PMAGE) in mouse hypertrophic cardiomyopathy. Science 316(5830):1482–1484 [DOI] [PubMed] [Google Scholar]
- 72.Klaassen S, Probst S, Oechslin E, Gerull B, Krings G, Schuler P, Greutmann M, Hurlimann D, Yegitbasi M, Pons L, Gramlich M, Drenckhahn JD, Heuser A, Berger F, Jenni R, Thierfelder L (2008) Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation 117(22):2893–2901 [DOI] [PubMed] [Google Scholar]
- 73.Kocovski L, Fernandes J (2015) A modern pathology approach to hypertrophic cardiomyopathy. Arch Pathol Lab Med 139:413–416 [DOI] [PubMed] [Google Scholar]
- 74.Kofflard MJ, Michels M, Krams R, Kliffen M, Geleijnse ML, Cate FJT, Serruys PW (2007) Coronary flow reserve in hypertrophic cardiomyopathy - relation with microvascular dysfunction and pathophysiological characteristics. Neth Hear J 15(6):209–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kramer CM, Reichek N, Ferrari VA, Theobald T, Dawson J, Axel L (1994) Regional heterogeneity of function in hypertrophic cardiomyopathy. Circulation 90:186–194 [DOI] [PubMed] [Google Scholar]
- 76.Kubo T, Baba Y, Hirota T, Tanioka K, Yamasaki N, Yamanaka S, Iiyama T, Kumagai N, Furuno T, Sugiura T, Kitaoka H (2015) Differentiation of infiltrative cardiomyopathy from hypertrophic cardiomyopathy using high-sensitivity cardiac troponin T: a case-control study. BMC Cardiovasc Disord 15:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lazzeroni E, Picano E, Morozzi L, Maurizio AR, Palma G, Ceriati R, Iori E, Barilli A (1997) Dipyridamole-induced ischemia as a prognostic marker of future adverse cardiac events in adult patients with hypertrophic cardiomyopathy. Circulation 96(12): 4268–4272 [DOI] [PubMed] [Google Scholar]
- 78.Le DE, Jayaweera AR, Wei K, Coggins MP, Lindner JR, Kaul S (2004) Changes in myocardial blood volume over a wide range of coronary driving pressures: role of capillaries beyond the autoregulatory range. Heart 90(10):1199–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lim D-S, Lutucuta S, Bachireddy P, Youker K, Evans A, Entman M (2001) Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation 103:789–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu D, Hu K, Nordbeck P, Ertl G, Stork S, Weidemann F (2016) Longitudinal strain bull’s eye plot patterns in patients with cardiomyopathy and concentric left ventricular hypertrophy. Eur J Med Res 21(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maron BJ (2002) Hypertrophic cardiomyopathy: a systematic review. JAMA 287(10):1308–1320 [DOI] [PubMed] [Google Scholar]
- 82.Maron BJ, Epstein SE, Roberts WC (1979) Hypertrophic cardiomyopathy and transmural myocardial infarction without significant atherosclerosis of the extramural coronary arteries. Am J Cardiol 43:1086–1102 [DOI] [PubMed] [Google Scholar]
- 83.Maron MS, Maron BJ, Harrigan C, Buros J, Gibson CM, Olivotto I, Biller L, Lesser JR, Udelson JE, Manning WJ, Appelbaum E (2009) Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J Am Coll Cardiol 54(3):220–228 [DOI] [PubMed] [Google Scholar]
- 84.Maron MS, Olivotto I, Maron BJ, Prasad SK, Cecchi F, Udelson JE, Camici PG (2009) The case for myocardial ischemia in hypertrophic cardiomyopathy. J Am Coll Cardiol 54(9):866–875 [DOI] [PubMed] [Google Scholar]
- 85.Martinez-Naharro A, Treibel TA, Abdel-Gadir A, Bulluck H, Zumbo G, Knight DS, Kotecha T, Francis R, Hutt DF, Rezk T, Rosmini S, Quarta CC, Whelan CJ, Kellman P, Gillmore JD, Moon JC, Hawkins PN, Fontana M (2017) Magnetic resonance in transthyretin cardiac amyloidosis. J Am Coll Cardiol 70(4): 466–477 [DOI] [PubMed] [Google Scholar]
- 86.McNally E, Dellefave L (2009) Sarcomere mutations in cardiogenesis and ventricular noncompaction. TCM 19(1):17–21 [DOI] [PubMed] [Google Scholar]
- 87.Michail M, Davies JE, Cameron JD, Parker KH, Brown AJ (2018) Pathophysiological coronary and microcirculatory flow alterations in aortic stenosis. Nat Rev Cardiol 15(7):420–431 [DOI] [PubMed] [Google Scholar]
- 88.Michele DE, Albayya FP, Metzger JM (1999) Thin filament protein dynamics in fully differentiated adult cardiac myocytes: toward a model of sarcomere maintenance. J Cell Biol 145(7):1483–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Michels M, Soliman OI, Kofflard MJ, Hoedemaekers YM, Dooijes D, Majoor-Krakauer D, ten Cate FJ (2009) Diastolic abnormalities as the first feature of hypertrophic cardiomyopathy in Dutch myosin-binding protein C founder mutations. JACC Cardiovasc Imaging 2(1):58–64 [DOI] [PubMed] [Google Scholar]
- 90.Montano MM, Doughman YQ, Deng H, Chaplin L, Yang J, Wang N, Zhou Q, Ward NL, Watanabe M (2008) Mutation of the HEXIM1 gene results in defects during heart and vascular development partly through downregulation of vascular endothelial growth factor. Circ Res 102(4):415–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Moon J, Cho IJ, Shim CY, Ha J-W, Jang Y, Chung N, Rim S-J (2010) Abnormal myocardial capillary density in apical hypertrophic cardiomyopathy can be assessed by myocardial contrast echocardiography. Circ J 74(10):2166–2172 [DOI] [PubMed] [Google Scholar]
- 92.Mundhenke M, Schwartzkopff B, Strauer BE (1997) Structural analysis of arteriolar and myocardial remodelling in the subendocardial region of patients with hypertensive heart disease and hypertrophic cardiomyopathy. Virchows Arch 431:265–273 [DOI] [PubMed] [Google Scholar]
- 93.N Watanabe TA, Yamaura Y, Akiyama M, Kaji S, Saito Y, Yoshida K (2003) Intramyocardial coronary flow characteristics in patients with hypertrophic cardiomyopathy: non-invasive assessment by transthoracic Doppler echocardiography. Heart 89: 657–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nag S, Sommese RF, Ujfalusi Z, Combs A, Langer S, Sutton S, Leinwand LA, Geeves MA, Ruppel KM, Spudich JA (2015) Contractility parameters of human β-cardiac myosin with the hypertrophic cardiomyopathy mutation R403Q show loss of motor function. Sci Adv 1:1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nakamura M, Sadoshima J (2018) Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol 15:387–407 [DOI] [PubMed] [Google Scholar]
- 96.Neben-Wittich MA, Wittich CM, Mueller PS, Larson DR, Gertz MA, Edwards WD (2005) Obstructive intramural coronary amyloidosis and myocardial ischemia are common in primary amyloidosis. Am J Med 118(11):1287. [DOI] [PubMed] [Google Scholar]
- 97.Neglia D, Fommei E, Varela-Carver A, Mancini M, Ghione S, Lombardi M, Pisani P, Parker H, D’Amati G, Donato L, Camici PG (2011) Perindopril and indapamide reverse coronary micro-vascular remodelling and improve flow in arterial hypertension. J Hypertens 29(2):364–372 [DOI] [PubMed] [Google Scholar]
- 98.Neumann T, Manger B, Schmid M, Kroegel C, Hansch A, Kaiser WA, Reinhardt D, Wolf G, Hein G, Mall G, Schett G, Zwerina J (2009) Cardiac involvement in Churg-Strauss syndrome: impact of endomyocarditis. Medicine (Baltimore) 88(4):236–243 [DOI] [PubMed] [Google Scholar]
- 99.Niss O, Quinn CT, Lane A, Daily J, Khoury PR, Bakeer N, Kimball TR, Towbin JA, Malik P, Taylor MD (2016) Cardiomyopathy with restrictive physiology in sickle cell disease. JACC Cardiovasc Imaging 9(3):243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Obregón R (2015) Non compaction of the ventricular myocardium: a logical reasoning J Cardiol Curr Res 2(5) [Google Scholar]
- 101.Olivotto I, Cecchi F, Gistri R, Lorenzoni R, Chiriatti G, Girolami F, Torricelli F, Camici PG (2006) Relevance of coronary micro-vascular flow impairment to long-term remodeling and systolic dysfunction in hypertrophic cardiomyopathy. J Cardiol Curr Resil 47(5):1043–1048 [DOI] [PubMed] [Google Scholar]
- 102.Olivotto I, Cecchi F, Poggesi C, Yacoub MH (2009) Developmental origins of hypertrophic cardiomyopathy phenotypes: a unifying hypothesis. Nat Rev Cardiol 6(4):317–321 [DOI] [PubMed] [Google Scholar]
- 103.Olivotto I, Girolami F, Sciagra R, Ackerman MJ, Sotgia B, Bos JM, Nistri S, Sgalambro A, Grifoni C, Torricelli F, Camici PG, Cecchi F (2011) Microvascular function is selectively impaired in patients with hypertrophic cardiomyopathy and sarcomere myofilament gene mutations. J Am Coll Cardiol 58(8):839–848 [DOI] [PubMed] [Google Scholar]
- 104.Opherk D, Mall G, Zebe H, Schwarz F, Weihe E, Manthey J, Kübler W (1984) Reduction of coronary reserve: a mechanism for angina pectoris in patients with arterial hypertension and normal coronary arteries. Circulation 69(1):1–7 [DOI] [PubMed] [Google Scholar]
- 105.Ormerod JO, Frenneaux MP, Sherrid MV (2016) Myocardial energy depletion and dynamic systolic dysfunction in hypertrophic cardiomyopathy. Nat Rev Cardiol 13(11):677–687 [DOI] [PubMed] [Google Scholar]
- 106.Ortlepp JR, Vosberg HP, Reith S, Ohme F, Mahon NG, Schröder D, Klues HG, Hanrath P, McKenna WJ (2002) Genetic polymorphisms in the renin-angiotensin-aldosterone system associated with expression of left ventricular hypertrophy in hypertrophic cardiomyopathy: a study of five polymorphic genes in a family with a disease causing mutation in the myosin binding protein C gene. Heart 87:270–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pantazis AA, Kohli SK, Elliott PM (2006) Images in cardiology. Hypertrophic cardiomyopathy and left ventricular hypertrabeculation: evidence for an overlapping phenotype. Heart 92(3):349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Patterson AJ, Zhang L (2011) Hypoxia and fetal heart development. Curr Mol Med 10(7):653–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Petersen SE, Jerosch-Herold M, Hudsmith LE, Robson MD, Francis JM, Doll HA, Selvanayagam JB, Neubauer S, Watkins H (2007) Evidence for microvascular dysfunction in hypertrophic cardiomyopathy: new insights from multiparametric magnetic resonance imaging. Circulation 115(18):2418–2425 [DOI] [PubMed] [Google Scholar]
- 110.Phillips SA, Guazzi M (2015) The vasculature in cardiovascular diseases: will the vasculature tell us what the future holds? Prog Cardiovasc Dis 57(5):407–408 [DOI] [PubMed] [Google Scholar]
- 111.Pradhan RK, Feigl EO, Gorman MW, Brengelmann GL, Beard DA (2016) Open-loop (feed-forward) and feedback control of coronary blood flow during exercise, cardiac pacing, and pressure changes. Am J Physiol Heart Circ Physiol 310(11):H1683–H1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pratt FH (1898) The nutrition of the heart through the vessels of Thebesius and the coronary veins. Am J Phys 1:86–103 [Google Scholar]
- 113.Pries AR, Secomb TW (2005) Control of blood vessel structure: insights from theoretical models. Am J Physiol Heart Circ Physiol 288(3):H1010–H1015 [DOI] [PubMed] [Google Scholar]
- 114.Pries AR, Secomb TW (2009) Origins of heterogeneity in tissue perfusion and metabolism. Cardiovasc Res 81(2):328–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rani B, Kumar A, Bahl A, Sharma R, Prasad R, Khullar M (2017) Renin-angiotensin system gene polymorphisms as potential modifiers of hypertrophic and dilated cardiomyopathy phenotypes. Mol Cell Biochem 427(1–2):1–11 [DOI] [PubMed] [Google Scholar]
- 116.Raphael CE, Cooper R, Parker KH, Collinson J, Vassiliou V, Pennell DJ, de Silva R, Hsu LY, Greve AM, Nijjer S, Broyd C, Ali A, Keegan J, Francis DP, Davies JE, Hughes AD, Arai A, Frenneaux M, Stables RH, Di Mario C, Prasad SK (2016) Mechanisms of myocardial ischemia in hypertrophic cardiomyopathy: insights from wave intensity analysis and magnetic resonance. J Am Coll Cardiol 68(15):1651–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rivenes SM, Kearney DL, Smith EOB, Towbin JA, Denfield SW (2000) Sudden death and cardiovascular collapse in children with restrictive cardiomyopathy. Circulation 102:876–882 [DOI] [PubMed] [Google Scholar]
- 118.Robbins JL, Jones WS, Duscha BD, Allen JD, Kraus WE, Regensteiner JG, Hiatt WR, Annex BH (2011) Relationship between leg muscle capillary density and peak hyperemic blood flow with endurance capacity in peripheral artery disease. J Appl Physiol 111(1):81–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Robinson P, Liu X, Sparrow A, Patel S, Zhang YH, Casadei B, Watkins H, Redwood C (2018) Hypertrophic cardiomyopathy mutations increase myofilament Ca2+ buffering, alter intracellular Ca2+ handling, and stimulate Ca2+-dependent signaling. J Biol Chem 293(27):10487–10499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rubattu S, Bozzao C, Pennacchini E, Pagannone E, Musumeci BM, Piane M, Germani A, Savio C, Francia P, Volpe M, Autore C, Chessa L (2016) A next-generation sequencing approach to identify gene mutations in early- and late-onset hypertrophic cardiomyopathy patients of an Italian cohort. Int J Mol Sci 17(8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rüssel IK, Brouwer WP, Germans T, Knaapen P, Marcus JT, van der Velden J, Götte MJ, van Rossum AC (2011) Increased left ventricular torsion in hypertrophic cardiomyopathy mutation carriers with normal wall thickness. J Cardiovasc Magn Reson 13(3): 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schwartzkopff B, Mundhenke M, Strauer BE (1998) Alterations of the architecture of subendocardial arterioles in patients with hypertrophic cardiomyopathy and impaired coronary vasodilator reserve: a possible cause for myocardial ischemia 11This study was supported by Grant SFB 242: Koronare Herzkrankheit: Prävention und Therapie akuter Komplikationen from the Deutsche Forschungsgemeinschaft, Bonn, Germany. J Am Coll Cardiol 31(5):1089–1096 [DOI] [PubMed] [Google Scholar]
- 123.Schwinger RHG, Frank KF (2003) Calcium and the failing heart: phospholamban, good guy or bad guy? Sci STKE 180(15) [DOI] [PubMed] [Google Scholar]
- 124.Sedmera D, Pexieder T, Vuillemin M, Thompson RP, Anderson RH (2000) Developmental patterning of the myocardium. Anat Rec 258:319–337 [DOI] [PubMed] [Google Scholar]
- 125.Semsarian C, Ingles J, Maron MS, Maron BJ (2015) New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 65(12):1249–1254 [DOI] [PubMed] [Google Scholar]
- 126.Sheng JJ, Jin JP (2014) Gene regulation, alternative splicing, and posttranslational modification of troponin subunits in cardiac development and adaptation: a focused review. Front Physiol 5:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sherrrid MV, Mahenthiran J, Casteneda V, Fincke R, Gasser M, Barac I, Thayaparan R, Chaudhry FA (2006) Comparison of diastolic septal perforator flow velocities in hypertrophic cardiomyopathy versus hypertensive left ventricular hypertrophy. Am J Cardiol 97(1):106–112 [DOI] [PubMed] [Google Scholar]
- 128.Shyu J-J, Cheng C-H, Erlandson RA, Lin J-H, Liu S-K (2002) Ultrastructure of intramural coronary arteries in pigs with hypertrophic cardiomyopathy. Cardiovasc Pathol 11:104–111 [DOI] [PubMed] [Google Scholar]
- 129.Solaro RJ (2010) Sarcomere control mechanisms and the dynamics of the cardiac cycle. J Biomed Biotechnol 2010:105648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Song Q, Schmidt AG, Hahn HS, Carr AN, Frank B, Pater L, Gerst M, Young K, Hoit BD, McConnell BK, Haghighi K, Seidman CE, Seidman JG, Dorn GW, Kranias EG (2003) Rescue of cardiomyocyte dysfunction by phospholamban ablation does not prevent ventricular failure in genetic hypertrophy. J Clin Investig 111(6): 859–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sotgia B, Sciagra R, Olivotto I, Casolo G, Rega L, Betti I, Pupi A, Camici PG, Cecchi F (2008) Spatial relationship between coronary microvascular dysfunction and delayed contrast enhancement in patients with hypertrophic cardiomyopathy. J Nucl Med 49(7):1090–1096 [DOI] [PubMed] [Google Scholar]
- 132.Sussman MA, HWL NG, Taigen T, Olson EN, Robbins J, Colbert MC, Gualberto A, Wieczorek DF, Molkentin JD (1998) Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science 281:1690–1693 [DOI] [PubMed] [Google Scholar]
- 133.Tanaka M, Fujiwara H, Onodera T, Wu DJ, Matsuda M, Hamashima Y, Kawai C (1987) Quantitative analysis of narrowings of intramyocardial small arteries in normal hearts, hypertensive hearts, and hearts with hypertrophic cardiomyopathy. Circulation 75(6):1130–1139 [DOI] [PubMed] [Google Scholar]
- 134.Tibirica E, Souza EG, De Lorenzo A, Oliveira GM (2015) Reduced systemic microvascular density and reactivity in individuals with early onset coronary artery disease. Microvasc Res 97: 105–108 [DOI] [PubMed] [Google Scholar]
- 135.Timmer SA, Knaapen P (2013) Coronary microvascular function, myocardial metabolism, and energetics in hypertrophic cardiomyopathy: insights from positron emission tomography. Eur Heart J Cardiovasc Imaging 14(2):95–101 [DOI] [PubMed] [Google Scholar]
- 136.Tobita T, Nomura S, Fujita T, Morita H, Asano Y, Onoue K, Ito M, Imai Y, Suzuki A, Ko T, Satoh M, Fujita K, Naito AT, Furutani Y, Toko H, Harada M, Amiya E, Hatano M, Takimoto E, Shiga T, Nakanishi T, Sakata Y, Ono M, Saito Y, Takashima S, Hagiwara N, Aburatani H, Komuro I (2018) Genetic basis of cardiomyopathy and the genotypes involved in prognosis and left ventricular reverse remodeling. Sci Rep 8(1):1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tsybouleva N, Zhang L, Chen S, Patel R, Lutucuta S, Nemoto S, DeFreitas G, Entman M, Carabello BA, Roberts R, Marian AJ (2004) Aldosterone, through novel signaling proteins, is a fundamental molecular bridge between the genetic defect and the cardiac phenotype of hypertrophic cardiomyopathy. Circulation 109(10):1284–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Valente AM, Lakdawala NK, Powell AJ, Evans SP, Cirino AL, Orav EJ, MacRae CA, Colan SD, Ho CY (2013) Comparison of echocardiographic and cardiac magnetic resonance imaging in hypertrophic cardiomyopathy sarcomere mutation carriers without left ventricular hypertrophy. Circ Cardiovasc Genet 6(3):230–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.van Lier MG, Oost E, Spaan JA, van Horssen P, van der Wal AC, vanBavel E, Siebes M, van den Wijngaard JP (2016) Transmural distribution and connectivity of coronary collaterals within the human heart. Cardiovasc Pathol 25(5):405–412 [DOI] [PubMed] [Google Scholar]
- 140.Vessieres E, Freidja ML, Loufrani L, Fassot C, Henrion D (2012) Flow (shear stress)-mediated remodeling of resistance arteries in diabetes. Vasc Pharmacol 57(5–6):173–178 [DOI] [PubMed] [Google Scholar]
- 141.Vikstrom KL, Factor SM, Leinwand LA (1996) Mice expressing mutant myosin heavy chains are a model for familial hypertrophic cardiomyopathy. Mol Med 2:556–567 [PMC free article] [PubMed] [Google Scholar]
- 142.Wang L, Bai F, Zhang Q, Song W, Messer A, Kawai M (2018) Development of apical hypertrophic cardiomyopathy with age in a transgenic mouse model carrying the cardiac actin E99K mutation. J Muscle Res Cell Motil [DOI] [PMC free article] [PubMed]
- 143.Wijnker PJM, Sequeira V, Kuster DWD, Velden JV (2018) Hypertrophic cardiomyopathy: a vicious cycle triggered by sarcomere mutations and secondary disease hits. Antioxid Redox Signal [DOI] [PMC free article] [PubMed]
- 144.Witjas-Paalberends ER, Guclu A, Germans T, Knaapen P, Harms HJ, Vermeer AM, Christiaans I, Wilde AA, Dos Remedios C, Lammertsma AA, van Rossum AC, Stienen GJ, van Slegtenhorst M, Schinkel AF, Michels M, Ho CY, Poggesi C, van der Velden J (2014) Gene-specific increase in the energetic cost of contraction in hypertrophic cardiomyopathy caused by thick filament mutations. Cardiovasc Res 103(2):248–257 [DOI] [PubMed] [Google Scholar]