

SUMMARY
A strategy for C–H functionalization of arenes and heteroarenes has been developed to allow site-selective incorporation of various anions, including Cl, Br, OMs, OTs, and OTf. This approach is enabled by in situ generation of reactive, non-symmetric iodanes by combining anions and bench-stable PhI(OAc)2. The utility of this mechanism is demonstrated via para-selective chlorination of medicinally relevant arenes, as well as site-selective C–H chlorination of heteroarenes. Spectroscopic, computational, and competition experiments describe the unique nature, reactivity, and selectivity of these transient, unsymmetrical iodanes.
eTOC Blurb
Fosu, Hambira, and colleagues describe the direct C–H functionalization of medicinally relevant arenes or heteroarenes. This strategy is enabled by transient generation of reactive, non-symmetric iodanes from anions and PhI(OAc)2. The site-selective incorporation of Cl, Br, OMs, OTs, and OTf to complex molecules, including within medicines and natural products, can be conducted by the operationally simple procedure included herein. A computational model for predicting site-selectivity is also included.
Graphical Abstract
INTRODUCTION
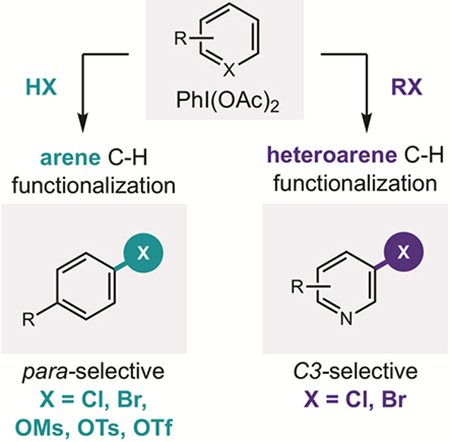
In the realm of medicinal chemistry, synthetic methods for the replacement of C–H bonds within complex molecules are of significant value.1–3 Among various approaches that enable aryl C–H functionalization,4–13 we are most interested in the suitability of hypervalent iodine-mediated chemistry14–20 to enable post-synthetic modification of medicinal agents. Since the pioneering contributions of Kita et al., λ3-iodanes have been known to substitute electron-rich arenes with certain nucleophiles (e.g. N3, CN, OAc).21–26 With the goal of expanding the scope of arenes and nucleophiles that may be coupled by this mechanism, we sought to develop a new, in situ iodane activation strategy (Figure 1).
Figure 1.

Selective C–H functionalization of arenes and heteroarenes with anions, enabled by in situ activation of an iodane
Although it is generally understood that bench-stable iodanes, like PhI(OAc)2, may be activated with Lewis (e.g. BF3, Me3Si+)21–26 or Brønsted acids (e.g. TsOH, TfOH, Ts2NH),27–30 the precise nature of this acid-activation remains unclear. Recent investigations by Shafir and co-workers revealed it entails transient formation of a non-symmetric iodane (e.g. BF3 coordination to one of the ligands of PhI(OAc)2).31 This model for increased reactivity via iodane desymmetrization may also explain the novel reactivity of Koser’s reagent, PhI(OH)OTs,27 and other hybrid iodanes.19,32–46 With this hypothesis in mind, we proposed that while unsymmetrical iodanes typically require preformation, their in situ generation may provide the dual benefits of (i) employing bench-stable precursors, and (ii) harnessing greater reactivity, including towards heteroarenes. Furthermore, we anticipated that in situ generation of non-symmetric iodanes with anions may enable the use of a wide range of nucleophiles for aryl C–H functionalization.
In designing a unified strategy for site-selective, aryl C-H functionalization via non-symmetric iodanes, we expected these reactive intermediates could be readily accessed through ligand exchange47 of bench-stable iodanes by anions (Figure 1a). As a simple and versatile source of anions, we proposed addition of acids, HX, to PhI(OAc)2 may promote ligand substitution to form PhIX2 – in analogy to acid-mediated formation of PhI(OH)OTs and Ph2IOTf.48 Moreover, we anticipated any acids stronger than AcOH should drive this acid/base equilibrium toward PhIX2 – an unstable reagent class associated with non-selective, homolytic reactivity.14–20 Essentially, however, we noted this process must proceed via an unsymmetrical iodane intermediate (I). We postulated this highly reactive species – having an elongated I-X bond and low-lying LUMO31 – could react selectively with arenes through either of two, transient-iodonium-based mechanisms (Figure 1b).49 First, anionic dissociation of X– affords cationic PhI(OAc)+, which may participate in single-electron transfer (SET) to oxidize an arene.21 The resulting radical cation is then prone to nucleophilic attack by the dissociated anion (X–). After net loss of H•, this mechanism can enable para-selective reactivity through the site-specific electrophilicity of an aryl radical cation.8, 50–51 Alternatively, AcO– dissociation from non-symmetric iodane I affords a net polarity-reversal of X– via an electrophilic PhI(X)+ iodonium species. Attack by the (hetero)arene nucleophile at its most nucleophilic position, and subsequent deprotonation would afford the X-substituted product. Thus, site-selective C–H functionalization may also be accessed by a closed-shell mechanism.
RESULTS & DISCUSSION
To test our hypothesis of the generalizability of this in situ iodane activation strategy, a series of acids was combined with N-Piv-o-toluidine and PhI(OAc)2 in DCE at 50 °C (Figure 2a). Surprisingly, several acids (spanning a range of >10 pKa units) afforded para-selective C–H functionalization by incorporation of their conjugate bases into the arene. Notably, TfOH alone facilitates iodane-mediated C–H oxytriflation to afford aryl triflate 1, without the need for stoichiometric AgOTf.52 Similarly, C–H oxy-sulfonylations are observed with TsOH and MsOH, which afford tosylate 2 and mesylate 3, also without additional promoters (e.g. BF3).53 Importantly, aqueous mineral acids, HBr and HCl, afford halogenated arenes, 4 and 5, with high efficiency (up to 88% yield) and para-selectivity (>20:1).
Figure 2.

Site-selective C–H functionalization incorporates the anions of acids, salts, and acyl halides.
With a highly, para-selective C–H functionalization observed for each of these halides and pseudo-halides, we chose to focus further studies on the most valuable of these reactions: chlorination. The post-synthetic halogenation of a medicinal candidate has two-fold utility: (i) improving biological properties by enhancing binding affinity, cellular membrane permeability, or metabolic stability,4 and (ii) enabling further synthetic manipulation. As a prototypical halide, the chloride is ubiquitous in medicines, with Cl ranking as the sixth most common element found in approved pharmaceuticals (after H, C, N, O, S),54 especially as a para-aryl substituent.55 Yet, although direct, post-synthetic modification by aryl C–H functionalization offers streamlined access to libraries of chlorinated medicinal candidates, such analogs are typically prepared by iterative synthesis with pre-halogenated arenes. In the case of chloride, the challenge arises from the traditional reliance on harsh reagents and non-selective methods for aryl chlorination, which are often not applicable to complex, drug-like molecules. An explanation is that aryl chlorination reagents either require (i) overly reactive reagents (e.g. Cl2, SO2Cl2, tBuOCl), or (ii) milder, more practically handled reagents that are sometimes not sufficiently reactive (e.g. NCS). This challenge has been addressed in recent years by the development of novel chlorination methods with reagents, such as Palau’chlor and IBA-Cl.56−57 Nonetheless, we noted that the “mild reagent vs sufficient reactivity” paradox remains unsolved in the realm of aryl C–H chlorination.58–59
Encouraged by the simple use of dilute, mineral acids (e.g. 1M HCl in water) to enable this C–H functionalization, we questioned whether other chloride reagents might also promote this ligand exchange to access this unique iodane reactivity from PhI(OAc)2 (Figure 2b). Indeed, we observed that several chlorides afford aryl chlorination, including Lewis acids (e.g. MgCl2), salts (e.g. LiCl), and acyl halides (e.g. AcCl).
Finally, cognizant of the privileged role of N-containing heterocycles in medicine,60 we focused our attention on the C–H chlorination of heteroarenes (Figure 2c). Notably, there are few ways to prepare chloro-(iso)quinolines – each analog requiring its own multi-step synthesis.61–63 With this in mind, we subjected isoquinoline to the HCl-mediated conditions. Although this acidic protocol did not afford chlorination, we observed complete protonation by 1H NMR, which likely renders the heteroarene significantly less nucleophilic. We therefore investigated other chloride reagents for aryl chlorination. While many of these chloride reagents were also ineffective (e.g. MgCl2), we found that acyl chlorides promote this reaction efficiently (up to 92% yield). We suspect these anhydrous sources of chloride anion, which may also function as desiccants, lack the acidity to deactivate the isoquinoline nucleophile. Interestingly, the observed selectivity in this case (for nucleophilic C4) is orthogonal to most heteroarene functionalizations (for electrophilic C1 or C3) via polar 64–66 or radical mechanisms.67–68
With robust conditions in hand for the selective C–H chlorination of either arenes or heteroarenes, we sought to investigate the generality and utility of this transformation (Figure 3). In the case of HCl-mediated aryl chlorination, various drug-like scaffolds are amenable to this reaction, including those containing halides, amides, esters, ketones, and imides (7–18). Notably, exclusive para-regioselectivity is observed in all cases. Additionally, this iodane-mediated reaction is chemo-selective, affording only sp2 C-H chlorination – with neither benzylic nor α-carbonyl oxidation (positions highlighted in grey), as typically observed via homolysis of PhICl2.26, 69–71 In the case of heteroarenes, aryl chlorination of both five-membered heterocycles (e.g. indoles, pyrazoles, 19–21) and six-membered heterocycles (e.g. isoquinoline, quinoline, pyrimidine, pyridine, 6, 23–30, via acyl chlorides) are also possible (up to 92%).
Figure 3. C−H Chlorination of arenes and heteroarenes via an in situ generated, non-symmetric iodane strategy.

Site-selective (hetero)aryl C–H chlorination: Arene (0.4 mmol), chloride reagent (5 equiv), PhI(OAc)2 (1.5 equiv), DCE (0.2 M), 50°C, 45 min – 24 hr. Isolated yields. No regioisomeric chlorination observed (susceptible positions highlighted with grey circles). Chloride reagents: a 1M HCl, b Bu4NCl, c PhI(OAc)2 (0.9 equiv), d 1M HCl (10 equiv), PhI(OAc)2 (2.5 equiv), e EtO2CCl, f KBr, g C6F5COCl, h AcCl, i 3:1 para:ortho, j 48% HBr, k 2M HCl in Et2O.
With our ultimate goal of medicinal application in mind, we then subjected a series of pharmaceuticals and natural products to this post-synthetic C−H chlorination strategy. Remarkably, a wide range of functionality is tolerated, including esters, amides, amines, imides, enamides, pyrroles, and dimethoxyarenes. As shown in Figure 3, the chlorinated analogs of naproxen, lidocaine, uracil, caffeine, and papaverine (31–38) were each accessed in a single-step using this protocol. Notably, the tertiary amine of lidocaine 33 remains intact, despite its oxidant-sensitivity, likely due to protection by protonation under these acidic conditions. As expected, bromination is also possible for both arenes (4) and heteroarenes (22), including within more complex drug-like scaffolds (32, 35, 38).
Finally, we sought to test this iodane-based strategy within the context of an active medicinal chemistry program. In particular, we have shown that analogs of phyllanthusmin D exhibit potent, anti-cancer activity against HT-29 human colon cancer cells.72 Although this natural product is structurally related to the DNA topoisomerase IIα inhibitor, etoposide, it appears to exhibit biological activity via a distinct mechanism of action. Notably, when non-natural sugars are affixed to the aglycone core of phyllanthusmin, especially potent analogs have been identified (IC50 < 10 nM).73–74 In order to streamline our exploration of the structure-activity relationship for substitution of the aryl core, we pursued a C−H functionalization strategy to rapidly generate a library of medicinal analogs. To this end, we subjected Ac-phyllanthusmin D to our HCl-mediated protocol. Unfortunately, the critical sugar was rapidly hydrolyzed off under these acidic conditions, affording the biologically inactive agylcone. On the other hand, employing Bu4NCl as the chloride reagent yields the C−H chlorinated derivative (39) in a single step. Similarly, LiBr affords direct C−H bromination (40). Interestingly, divergent regioselectivity is observed for these two reactions, with the smaller Cl atom substituting the more electron-rich naphthalene and the larger Br atom adding to the less-hindered, axially rotatable benzene. Importantly, in both cases, the sugar substituent is unperturbed, allowing us to access a family of potent derivatives from a single advanced intermediate.
In order to better understand the excellent regioselectivity observed in the C-H halogenations of various arenes in Figure 3, we conducted DFT experiments according to Ritter’s procedure.10 Specifically, we determined Fukui index values for several (hetero)arenes by computing a population analysis for each arene and its corresponding N-1 ionization state – and then subtracting the charge densities of the latter from the former in each case (see SI for details). As shown in Figure 4, employing this Fukui analysis correctly predicts the most reactive positions of several complex molecules from Figure 3.
Figure 4.

Computational experiments predict regioselectivity.
In particular, this analysis predicts and explains the results obtained for anilide, as well as the medicinally relevant natural products, papaverine and phyllanthusmin. Each example contains polyaromatic rings with multiple possible sites of reactivity. Yet, Fukui analysis predicts the most reactive atom in the case of papaverine (37) and the two most reactive positions for phyllanthusmin (i.e. sites of chlorination 39 and bromination 40).
On the other hand, we noted that a simple electron-density map of partial charges provides a more accurate predictor of the site-selectivity observed for heteroarene C-H functionalization. For example, the most negative position by natural population analysis correlates to the most reactive position for quinoline (25), isoquinoline (6), indole (19), and quinoxaline (26). Interestingly, for three of these four classes of heteroarene, Fukui analysis predicts alternate regioselectivity, suggesting divergent mechanisms between the arene and heteroarene reactions.
Thus, given the correct prediction of site-selectivity by these DFT calculations, we propose the mechanisms shown in Figure 1 are both viable. As shown in Figure 5, given the strong correlation of the Fukui analysis, which is dependent on radical cation character, to predict regioselectivity for the arenes, we suggest the ‘radical cation’ mechanism is likely more relevant for the electron-rich arenes. In this case, the transient, non-symmetric iodane intermediate likely promotes single-electron oxidation to afford a radical cation with electrophilicity (and nucleophilic attack) localized at the predicted para position. Alternatively, for less-electron-rich heteroarenes, the iodane likely combines directly with the heteroarene via the ‘iodonium’ mechanism. This pathway is supported by the C3 regioselectivity of the products, which are correctly (and exclusively) predicted via partial charge analysis as correlating to the most negative, or electron-dense, sites.
Figure 5.

Computationally supported divergent mechanisms explain predicted and observed regioselectivities for arenes versus heteroarenes.
The chemoselectivity observed among the various arenes in Figure 3 also led us to question the possibility of developing methods to enable reagent-based control over selectivity. To this end, we subjected a 1:1 mixture of heteroarene and arene to a series of competition experiments (Figure 6). Indeed, when heteroaryl conditions are employed (e.g. EtO2CCl), isoquinoline chlorination is exclusively observed, even in the presence of electron-rich anisoles. Yet, under acidic, aryl conditions (e.g. HCl), isoquinoline is presumably protonated, affording exclusive anisole chlorination (41).
Figure 6.

Competition experiments demonstrate chemoselectivity.
We were also interested in further probing the unique C−H chlorination of heteroarenes, especially given the importance of these motifs in medicine. To the best of our knowledge, this in situ iodane activation strategy comprises the mildest and most efficient protocol for C−H chlorination of (iso)quinolines,75–77 affording regioselective substitution of the most nucleophilic carbon (e.g. 4-Cl-isoquinoline or 3-Cl-quinoline). To test this hypothesis, we investigated the chlorination of both isoquinoline and quinoline by common aryl chlorination methods, as shown in Figure 7. Surprisingly, no chlorinated products were detected for either class of heteroarene (6 or 25) – even in the presence of state-of-the-art chlorinating reagents, Palau’chlor and IBA-Cl. Similarly, several other arenes in Figure 3 are not efficiently halogenated using alternate literature methods, such as PhICl2 (e.g. 10, 15, 33, 39; see SI).
Figure 7.

Reactivity comparison with other chlorinating reagents.
Noting a distinct difference in reactivity between our postulated, transient non-symmetric iodane and cyclic IBA-Cl in particular, we sought to investigate the precise nature of the in situ generated iodane. We first probed if a diaryliodonium pathway could promote this unprecedented heteroaryl chlorination (Figure 8a). To examine the viability of such a mechanism, we prepared 3-pyridyl-diaryliodonium and heated it in the presence of acyl chloride, both with and without PhI(OAc)2. In both cases, we did not observe any chlorination (aryl or heteroaryl) thereby precluding this type of mechanism. Similarly, Ph2I+Cl- does not afford PhCl, when heated with or without HCl. Additionally, bis-N-substituted di(hetero)aryl iodonium was prepared and subjected to various acyl chlorides (Figure 8b). In this mechanistic probe, no chloroarene was obtained, neither with nor without PhI(OAc)2, further suggesting such iodoniums are not likely intermediates in this mechanism.
Figure 8.

Mechanistic probes suggest alternate iodonium pathways are not viable.
To obtain further support for our proposed mechanism (c.f. Figure 1), we sought to spectroscopically observe the transient, non-symmetric iodane intermediate. Several representative NMR experiments from this investigation are shown in Figure 9. After identifying diagnostic, aromatic 1H NMR signals for the homo-di-substituted iodanes, PhI(OAc)2 and PhICl2, we subjected PhI(OAc)2 to varying stoichiometries of HCl and observed a new signal, which we ascribe to the non-symmetric iodane, PhI(OAc)Cl. Notably, this signal is also observed when AcCl is used as the chloride reagent. And interestingly, the same signal is observed upon combination of PhICl2 with Ac2O, which serves as an anhydrous source of acetate.
Figure 9.

1H NMR experiments indicate the intermediacy of a non-symmetric iodane intermediate.
In order to examine the role of this observed species as a possible intermediate, we then subjected isoquinoline to chlorination under various conditions (Figure 10). As expected, Willgerodt’s reagent, PhICl2, does not afford appreciable quantities of chlorination (6). However, a combination of PhICl2 and Ac2O, which also generates the unsymmetrical iodane in situ, recapitulates the reactivity of PhI(OAc)Cl that is not observed for PhICl2 alone. Additionally, a mixture of AcCl and PhI(OAc)2, which more selectively forms PhI(OAc)Cl over PhICl2, affords the desired heteroaryl chloride in high yield (86%; similar to EtO2CCl, 92%).
Figure 10.

Heteroarene chlorination by orthogonal reagents; recapitulation of reactivity supports mono-chloro-iodane hypothesis.
In summary, a site-selective hetero(aryl) C−H functionalization strategy has been developed. This practical method is enabled by in situ generation of non-symmetric iodanes from PhI(OAc)2 and the anions of acids, salts, and acyl halides. These mild conditions afford access to new types of reactivity and selectivity, as illustrated in the chemo- and regio-selective C−H halogenation of a range of medicinally relevant (hetero)arenes. Importantly, spectroscopic, computational, and experimental support for in situ generation of unsymmetrical iodanes under such conditions provides mechanistic validation to enable further development of new iodane reactivity.
Supplementary Material
Highlights.
C–H functionalization of arenes or heteroarenes with Cl, Br, OMs, OTs, and OTf anions
Transient generation of reactive, non-symmetric iodanes from anions and PhI(OAc)2
Site-selective C–H chlorination of medicinally relevant arenes or heteroarenes
Computational methods for predicting sites of reactivity with complex molecules
ACKNOWLEDGMENTS
We thank The Ohio State University, National Institutes of Health (NIH R35 GM119812 and P01 CA125066), National Science Foundation (NSF CAREER 1654656), and American Chemical Society Petroleum Research Fund for financial support. SCF is supported by an HHMI Gilliam Fellowship. CMH is supported by the Jane Chen Fellowship in Medicinal Chemistry. High performance computing resources were provided by The Ohio Supercomputer Center.
Bigger Picture
The discovery of new medicines is a time- and labor-intensive process that frequently requires over a decade to complete. A major bottleneck is the synthesis of drug candidates, wherein each complex molecule must be prepared individually via a multi-step synthesis, frequently requiring a week of effort per molecule for thousands of candidates. As an alternate strategy, direct, post-synthetic functionalization of a lead candidate could enable this diversification in a single operation. In this article, we describe a new method for direct manipulation of drug-like molecules by incorporation of motifs with either known pharmaceutical value (halides) or that permit subsequent conversion (pseudo-halides) to medicinally relevant analogs. This user-friendly strategy is enabled by combining commercial iodine reagents with salts and acids. We expect this simple method for selective, post-synthetic, incorporation of molecular diversity will streamline the discovery of new medicines.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes:
Experimental procedures and characterization for all new compounds (PDF) 1H and 13C NMR spectral data (PDF)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES AND NOTES
- (1).Cernak T, Dykstra KD, Tyagarajan S, Vachal P, and Krska SW (2016). The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev 45, 546–576. [DOI] [PubMed] [Google Scholar]
- 2).Wencel-Delord J, and Glorius F (2013). C-H bond activation enables the rapid construction and late-stage diversification of functional molecules. Nat. Chem 5, 369–375. [DOI] [PubMed] [Google Scholar]
- 3).Yamaguchi J, Yamaguchi AD, and Itami K (2012). C-H bond functionalization: emerging synthetic tools for natural products and pharmaceuticals. Angew. Chem. Int. Ed 51, 8960–9009. [DOI] [PubMed] [Google Scholar]
- 4).Zhang F, and Spring DR (2014). Arene C–H functionalisation using a removable/modifiable or a traceless directing group strategy. Chem. Soc. Rev 43, 6906–6919. [DOI] [PubMed] [Google Scholar]
- 5).Romero NA, Margrey KA, Tay NE, and Nicewicz DA (2015). Site-selective arene C-H amination via photoredox catalysis. Science 349, 1326–1330. [DOI] [PubMed] [Google Scholar]
- 6).Bag S, Patra T, Modak A, Deb A, Maity S, Dutta U, Dey A, Kancherla R, Maji A, Hazra A, et al. (2015). Remote para-C-H functionalization of arenes by a D-shaped biphenyl template-based assembly. J. Am. Chem. Soc 137, 11888–11891. [DOI] [PubMed] [Google Scholar]
- 7).Okumura S, Tang S, Saito T, Semba K, Sakaki S, and Nakao Y (2016). Para-selective alkylation of benzamides and aromatic ketones by cooperative nickel/aluminum catalysis. J. Am. Chem. Soc 138, 14699–14704. [DOI] [PubMed] [Google Scholar]
- 8).Boursalian GB, Ham WS, Mazzotti AR, and Ritter T (2016). Charge-transfer-directed radical substitution enables para-selective C-H functionalization. Nat. Chem 8, 810–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Wang P, Verma P, Xia G, Shi J, Qiao JX, Tao S, Cheng PTW, Poss MA, Farmer ME, Yeung K-S, et al. (2017). Ligand-accelerated non-directed C–H functionalization of arenes. Nature 551, 489–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Yamamoto K, Li J, Garber JAO, Rolfes JD, Boursalian GB, Borghs JC, Genicot C, Jacq J, van Gastel M, Neese F, and Ritter T (2018). Palladium-catalysed electrophilic aromatic C-H fluorination. Nature 554, 511–514. [DOI] [PubMed] [Google Scholar]
- 11).Svejstrup TD, Ruffoni A, Juliá F, Aubert VM, and Leonori D (2017). Synthesis of arylamines via aminium radicals. Angew. Chem. Int. Ed 56, 14948–14952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Imiołek M, Karunanithy G, Ng W-L, Baldwin AJ, Gouverneur V, and Davis BG (2018). Selective radical trifluoromethylation of native residues in proteins. J. Am. Chem. Soc 140, 1568–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Wang Z, Herraiz AG, del Hoyo AM, and Suero MG (2018). Generating carbyne equivalents with photoredox catalysis. Nature 554, 86–91. [DOI] [PubMed] [Google Scholar]
- 14).Moriarty RM, and Prakash O (1986). Hypervalent iodine in organic synthesis. Acc. Chem. Res 19, 244–250. [Google Scholar]
- 15).Togo H, and Katohgi M (2001). Synthetic uses of organohypervalent iodine compounds through radical pathways. Synlett 2001, 565–581. [Google Scholar]
- 16).Wirth T, ed. (2003). Hypervalent Iodine Chemistry: Modern Developments in Organic Synthesis (Springer-Verlag Berlin Heidelberg; ). [Google Scholar]
- 17).Zhdankin VV, and Stang PJ (2008). Chemistry of polyvalent iodine. Chem. Rev 108, 5299–5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Dohi T, and Kita Y (2009). Hypervalent iodine reagents as a new entrance to organocatalysts. Chem. Commun 2073–2085. [DOI] [PubMed] [Google Scholar]
- 19).Yoshimura A, and Zhdankin VV (2016). Advances in synthetic applications of hypervalent iodine compounds. Chem. Rev 116, 3328–3435. [DOI] [PubMed] [Google Scholar]
- 20).Wang X, and Studer A (2017). Iodine(III) reagents in radical chemistry. Acc. Chem. Res 50, 1712–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Kita Y, Tohma H, Hatanaka K, Takada T, Fujita S, Mitoh S, Sakurai H, and Oka S (1994). Hypervalent iodine-induced nucleophilic substitution of para-substituted phenol ethers. Generation of cation radicals as reactive intermediates. J. Am. Chem. Soc 116, 3684–3691. [Google Scholar]
- 22).Dohi T, Ito M, Morimoto K, Iwata M, and Kita Y (2008). Oxidative cross-coupling of arenes induced by single-electron transfer leading to biaryls by use of organoiodine(III) oxidants. Angew. Chem. Int. Ed 47, 1301–1304. [DOI] [PubMed] [Google Scholar]
- 23).Kita Y, Egi M, Okajima A, Ohtsubo M, Takada T, and Tohma H (1996). Hypervalent iodine(III) induced intramolecular cyclization of substituted phenol ethers bearing an alkyl azido sidechain–a novel synthesis of quinone imine ketals. Chem. Commun, 1491–1492. [Google Scholar]
- 24).Magnus P, Lacour J, Evans PA, Roe MB, and Hulme C (1996). Hypervalent iodine chemistry: new oxidation reactions using the iodosylbenzene-trimethylsilyl azide reagent combination. Direct α- and β-azido functionalization of triisopropylsilyl enol ethers. J. Am. Chem. Soc 118, 3406–3418. [Google Scholar]
- 25).Evans PA, and Brandt TA (1997). Hypervalent iodine chemistry: mechanistic investigation of the novel haloacetoxylation, halogenation, and acetoxylation reactions of 1,4-dimethoxynaphthalenes. J. Org. Chem 62, 5321–5326. [Google Scholar]
- 26).Akula R, Galligan M, and Ibrahim H (2009). Umpolung of halide reactivity: efficient (diacetoxyiodo)benzene-mediated electrophilic α-halogenation of 1,3-dicarbonyl compounds. Chem. Commun, 6991–6993. [DOI] [PubMed] [Google Scholar]
- 27).Moriarty RM, Vaid RK, and Koser GF (1990). [Hydroxy(organosulfonyloxy)iodo]arenes in organic synthesis. Synlett 1990, 365–383. [Google Scholar]
- 28).Kitamura T, Matsuyuki J, and Taniguchi H (1994). Improved preparation of diaryliodonium triflates. Synthesis 1994, 147–148. [Google Scholar]
- 29).Muñiz K (2018). Promoting intermolecular C–N bond formation under the auspices of iodine(III). Acc. Chem. Res 51, 1507–1519. [DOI] [PubMed] [Google Scholar]
- 30).Colomer I, Batchelor-McAuley C, Odell B, Donohoe TJ, and Compton RG (2016). Hydrogen bonding to hexafluoroisopropanol controls the oxidative strength of hypervalent iodine reagents. J. Am. Chem. Soc 138, 8855–8861. [DOI] [PubMed] [Google Scholar]
- 31).Izquierdo S, Essafi S, del Rosal I, Vidossich P, Pleixats R, Vallribera A, Ujaque G, Lledós A, and Shafir A (2016). Acid activation in phenyliodine dicarboxylates: direct observation, structures, and implications. J. Am. Chem. Soc 138, 12747–12750. [DOI] [PubMed] [Google Scholar]
- 32).Zhdankin VV, Krasutsky AP, Kuehl CJ, Simonsen AJ, Woodward JK, Mismash B, and Bolz JT (1996). Preparation, X-ray crystal structure, and chemistry of stable azidoiodinanes-derivatives of benziodoxole. J. Am. Chem. Soc 118, 5192–5197. [Google Scholar]
- 33).Brand JP, and Waser J (2012). Electrophilic alkynylation: the dark side of acetylene chemistry. Chem. Soc. Rev 41, 4165–4179. [DOI] [PubMed] [Google Scholar]
- 34).Charpentier J, Früh N, and Togni A (2015). Electrophilic trifluoromethylation by use of hypervalent iodine reagents. Chem. Rev 115, 650–682. [DOI] [PubMed] [Google Scholar]
- 35).Li Y, Hari DP, Vita MV, and Waser J (2016). Cyclic hypervalent iodine reagents for atom-transfer reactions: beyond trifluoromethylation. Angew. Chem. Int. Ed 55, 4436–4454. [DOI] [PubMed] [Google Scholar]
- 36).Souto JA, Zian D, and Muñiz K (2012). Iodine(III)-mediated intermolecular allylic amination under metal-free conditions. J. Am. Chem. Soc 134, 7242–7245. [DOI] [PubMed] [Google Scholar]
- 37).Lubriks D, Sokolovs I, and Suna E (2012). Indirect C–H azidation of heterocycles via copper-catalyzed regioselective fragmentation of unsymmetrical λ3 -iodanes. J. Am. Chem. Soc 134, 15436–15442. [DOI] [PubMed] [Google Scholar]
- 38).Souto JA, Becker P, Iglesias Á, and Muñiz K (2012). Metal-free iodine(III)-promoted direct intermolecular C–H amination reactions of acetylenes. J. Am. Chem. Soc 134, 15505–15511. [DOI] [PubMed] [Google Scholar]
- 39).Matcha K, and Antonchick AP (2013). Metal-free cross-dehydrogenative coupling of heterocycles with aldehydes. Angew. Chem. Int. Ed 52, 2082–2086. [DOI] [PubMed] [Google Scholar]
- 40).Moteki SA, Usui A, Zhang T, Solorio Alvarado CR, and Maruoka K (2013). Site-selective oxidation of unactivated Csp3-H bonds with hypervalent iodine(III) reagents. Angew. Chem. Int. Ed 52, 8657–8660. [DOI] [PubMed] [Google Scholar]
- 41).Huang H, Zhang G, Gong L, Zhang S, and Chen Y (2014). Visible-light-induced chemoselective deboronative alkynylation under biomolecule-compatible conditions. J. Am. Chem. Soc 136, 2280–2283. [DOI] [PubMed] [Google Scholar]
- 42).Sokolovs I, Lubriks D, and Suna E (2014). Copper-catalyzed intermolecular C–H amination of (hetero)arenes via transient unsymmetrical λ3-iodanes. J. Am. Chem. Soc 136, 6920–6928. [DOI] [PubMed] [Google Scholar]
- 43).Sharma A, and Hartwig JF (2015). Metal-catalysed azidation of tertiary C-H bonds suitable for late-stage functionalization. Nature 517, 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44).Wang X, and Studer A (2016). Regio- and stereoselective cyanotriflation of alkynes using aryl(cyano)iodonium triflates. J. Am. Chem. Soc 138, 2977–2980. [DOI] [PubMed] [Google Scholar]
- 45).Ito E, Fukushima T, Kawakami T, Murakami K, and Itami K (2017). Catalytic dehydrogenative C–H imidation of arenes enabled by photo-generated hole donation to sulfonimide. Chem 2, 383–392. [Google Scholar]
- 46).Egami H, and Sodeoka M (2014). Trifluoromethylation of alkenes with concomitant introduction of additional functional groups. Angew. Chem. Int. Ed 53, 8294–8308. [DOI] [PubMed] [Google Scholar]
- 47).Ochiai M (2003). Reactivities, properties and structures In Hypervalent Iodine Chemistry: Modern Developments in Organic Synthesis, Wirth T, ed. (Springer-Verlag Berlin Heidelberg; ), pp. 5–68. [Google Scholar]
- 48).Merritt EA, and Olofsson B (2009). Diaryliodonium salts: a journey from obscurity to fame. Angew. Chem. Int. Ed 48, 9052–9070. [DOI] [PubMed] [Google Scholar]
- 49).A third mechanism (i.e. homolytic cleavage) is also possible, albeit unlikely given the site-selectivity observed in these reactions differs from non-selective PhICl2–mediated processes.
- 50).Zhao J, and Li S (2017). Charge-transfer-induced para-selective sp2 C–H bond activation of arenes by use of a hypervalent iodine compound: a theoretical study. J. Org. Chem 82, 2984–2991. [DOI] [PubMed] [Google Scholar]
- 51).Margrey KA, McManus JB, Bonazzi S, Zecri F, and Nicewicz DA (2017). Predictive model for site-selective aryl and heteroaryl C–H functionalization via organic photoredox catalysis. J. Am. Chem. Soc 139, 11288–11299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52).Pialat A, Liégault B, and Taillefer M (2013). Oxidative para-triflation of acetanilides. Org. Lett 15, 1764–1767. [DOI] [PubMed] [Google Scholar]
- 53).Liu H, Xie Y, and Gu Y (2011). A novel method for tosyloxylation of anilides using phenyliodine bistrifluoroacetate (PIFA). Tetrahedron Lett. 52, 4324–4326. [Google Scholar]
- 54).Smith BR, Eastman CM, and Njardarson JT (2014). Beyond C, H, O, and N! Analysis of the elemental composition of U.S. FDA approved drug architectures. J. Med. Chem 57, 9764–9773. [DOI] [PubMed] [Google Scholar]
- 55).Brown DG, Gagnon MM, and Boström J (2015). Understanding our love affair with p-chlorophenyl: present day implications from historical biases of reagent selection. J. Med. Chem 58, 2390–2405. [DOI] [PubMed] [Google Scholar]
- 56).Rodriguez RA, Pan C-M, Yabe Y, Kawamata Y, Eastgate MD, and Baran PS (2014). Palau’chlor: a practical and reactive chlorinating reagent. J. Am. Chem. Soc 136, 6908–6911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57).Wang M, Zhang Y, Wang T, Wang C, Xue D, and Xiao J (2016). Story of an age-old reagent: an electrophilic chlorination of arenes and heterocycles by 1-chloro-1,2-benziodoxol-3-one. Org. Lett 18, 1976–1979. [DOI] [PubMed] [Google Scholar]
- 58).Xiong X, and Yeung Y-Y (2016). Highly ortho-selective chlorination of anilines using a secondary ammonium salt organocatalyst. Angew. Chem. Int. Ed 55, 16101–16105. [DOI] [PubMed] [Google Scholar]
- 59).Zhang L, and Hu X (2017). Room temperature C(sp2)–H oxidative chlorination via photoredox catalysis. Chem. Sci 8, 7009–7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60).Vitaku E, Smith DT, and Njardarson JT (2014). Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- 61).Yamada M, Nakamura Y, Hasegawa T, Itoh A, Kuroda S, and Shimao I (1992). Oxidative chlorination of 1,10-phenanthroline and its derivatives by phosphorus pentachloride in phosphoryl chloride. Bull. Chem. Soc. Jpn 65, 2007–2009. [Google Scholar]
- 62).Hasebe M, and Tsuchiya T (1988). Photodecarboxylative chlorination of carboxylic acids via their benzophenone oxime esters. Tetrahedron Lett. 29, 6287–6290. [Google Scholar]
- 63).Louërat F, Fort Y, and Mamane V (2009). Direct 1,4-difunctionalization of isoquinoline. Tetrahedron Lett. 50, 5716–5718. [Google Scholar]
- 64).Fier PS, and Hartwig JF (2013) Selective C-H fluorination of pyridines and diazines inspired by a classic amination reaction. Science 342, 956–960. [DOI] [PubMed] [Google Scholar]
- 65).Hilton MC, Dolewski RD, and McNally A (2016). Selective functionalization of pyridines via heterocyclic phosphonium salts. J. Am. Chem. Soc 138, 13806–13809. [DOI] [PubMed] [Google Scholar]
- 66).Iwai T, and Sawamura M (2015). Transition-metal-catalyzed site-selective C-H functionalization of quinolines beyond C2 selectivity. ACS Catal. 5, 5031–5040. [Google Scholar]
- 67).Duncton MAJ (2011). Minisci reactions: versatile CH-functionalizations for medicinal chemists. Med. Chem. Commun 2, 1135–1161. [Google Scholar]
- 68).O’Hara F, Blackmond DG, and Baran PS (2013). Radical-based regioselective C–H functionalization of electron-deficient heteroarenes: scope, tunability, and predictability. J. Am. Chem. Soc 135, 12122–12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69).Kang K, Lee S, and Kim H (2015). Radical chlorination with hypervalent iodine(III) generated by ligand exchange: revisiting palladium(II)-catalyzed directed C-H chlorination. Asian J. Org. Chem 4, 137–140. [Google Scholar]
- 70).Breslow R, Corcoran RJ, and Snider BB (1974). Remote functionalization of steroids by a radical relay mechanism. J. Am. Chem. Soc 96, 6791–6792. [DOI] [PubMed] [Google Scholar]
- 71).Cooper JC, Luo C, Kameyama R, and Van Humbeck JF (2018). Combined iron/hydroxytriazole dual catalytic system for site selective oxidation adjacent to azaheterocycles. J. Am. Chem. Soc 140, 1243–1246. [DOI] [PubMed] [Google Scholar]
- 72).Ren Y, Lantvit DD, Deng Y, Kanagasabai R, Gallucci JC, Ninh TN, Chai H-B, Soejarto DD, Fuchs JR, Yalowich JC, et al. (2014). Potent cytotoxic arylnaphthalene lignan lactones from Phyllanthus poilanei. J. Nat. Prod 77, 1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73).Woodard JL, Huntsman AC, Patel PA, Chai HB, Kanagasabai R, Karmahapatra S, Young AN, Ren Y, Cole MS, Herrera D, et al. (2018). Synthesis and antiproliferative activity of derivatives of the phyllanthusmin class of arylnaphthalene lignan lactones. Bioorganic Med. Chem 26, 2354–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74).Young AN, Herrera D, Huntsman AC, Korkmaz MA, Lantvit DD, Mazumder S, Kolli S, Coss CC, King S, Wang H, et al. (2018). Phyllanthusmin derivatives induce apoptosis and reduce tumor burden in high-grade serous ovarian cancer by late-stage autophagy inhibition. Mol. Cancer Ther 17, 2123–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75).Zucker SP, Wossidlo F, Weber M, Lentz D, and Tzschucke CC (2017). Palladiumcatalyzed directed halogenation of bipyridine N-oxides. J. Org. Chem 82, 5616–5635. [DOI] [PubMed] [Google Scholar]
- 76).Arockiam PB, Guillemard L, and Wencel-Delord J (2017). Regiodivergent visible lightinduced C–H functionalization of quinolines at C-5 and C-8 under metal-, photosensitizer- and oxidant-free conditions. Adv. Synth. Catal 359, 2571–2579. [Google Scholar]
- 77).Cruchter T, Medvedev MG, Shen X, Mietke T, Harms K, Marsch M, and Meggers E (2017). Asymmetric nucleophilic catalysis with an octahedral chiral-at-metal iridium(III) complex. ACS Catal. 7, 5151–5162. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.