

KHK-A, a metabolic enzyme in fructose metabolism, acts as a protein kinase to phosphorylate p62 for tumor antioxidant responses.
Abstract
Cancer cells often encounter oxidative stress. However, it is unclear whether normal and cancer cells differentially respond to oxidative stress. Here, we demonstrated that under oxidative stress, hepatocellular carcinoma (HCC) cells exhibit increased antioxidative response and survival rates compared to normal hepatocytes. Oxidative stimulation induces HCC-specifically expressed fructokinase A (KHK-A) phosphorylation at S80 by 5′-adenosine monophosphate-activated protein kinase. KHK-A in turn acts as a protein kinase to phosphorylate p62 at S28, thereby blocking p62 ubiquitination and enhancing p62’s aggregation with Keap1 and Nrf2 activation. Activated Nrf2 promotes expression of genes involved in reactive oxygen species reduction, cell survival, and HCC development in mice. In addition, phosphorylation of KHK-A S80 and p62 S28 and nuclear accumulation of Nrf2 are positively correlated in human HCC specimens and with poor prognosis of patients with HCC. These findings underscore the role of the protein kinase activity of KHK-A in antioxidative stress and HCC development.
INTRODUCTION
Cancer cells exhibit altered cellular metabolism, which results in high levels of oxidative stress (1, 2). This oxidative stress is exerted by reactive oxygen species (ROS) that accumulate in response to hypoxia, metabolic defect, and endoplasmic reticulum stress (3). Nuclear factor erythroid 2–related factor 2 (Nrf2), the master transcriptional regulator of enzymes expressed in response to oxidative and electrophilic stress, is essential for cell survival and redox homeostasis (4). High activity of Nrf2 has frequently been detected in many types of human cancer and contributes to chemotherapy resistance (4–8). Nevertheless, the mechanism underlying the up-regulated Nrf2 activity is largely unclear. Under normal conditions, Kelch-like ECH-associated protein 1 (Keap1) is an adaptor between Nrf2 and the Cullin 3–Ring-box protein 1 ubiquitin ligase complex and mediates Nrf2 polyubiquitylation and proteasomal degradation (9, 10). Under oxidative stress, modifications of redox-sensitive cysteines in Keap1 disrupt its adaptor function to free Nrf2 (11, 12). In addition, the Keap1-Nrf2 complex can be regulated by p62 (also known as SQSTM1), an autophagosome cargo protein binding to other proteins for selective autophagy under multiple stress conditions (13–15). Oxidative stress promotes p62 to form a homodimer, which facilitates its oligomerization and interaction with proteins with K48-linked polyubiquitylation, including Keap1, resulting in their aggregation for autophagy-lysosomal degradation (16, 17). The p62-mediated Keap1 sequestration and degradation emancipate Nrf2 from the Keap1 inhibition, allowing for its stabilization and nuclear translocation (18–20). In the nucleus, Nrf2 binds to antioxidant response elements (AREs) or electrophile response elements on DNA and regulates the expression of genes involved in response to cellular stress, which include NADPH [reduced form of nicotinamide adenine dinucleotide phosphate (NADP+)]–generating enzymes (21) and those enzymes catalyzing glutathione biosynthesis to maintain intracellular levels of reduced glutathione (GSH) (22–24). p62 dimerization involves the formation of a K7-D69 hydrogen bond in its N-terminal Phox1 and Bem1p (PB1) domain (25), and p62 stability and function can be posttranslationally modified (26, 27). However, how p62 oligomerization is regulated, thereby activating Nrf2 transcriptional activity in cancer, remains elusive.
Here, we demonstrated that oxidative stress induced the binding of 5′-adenosine monophosphate-activated protein kinase (AMPK) to metabolic enzyme fructokinase A [also known as ketohexokinase A (KHK-A)] and KHK-A S80 phosphorylation by AMPK. Phosphorylated KHK-A acted as a protein kinase and phosphorylated p62 S28, resulting in p62 oligomerization, thereby enhancing p62 and Keap1 aggregation and nuclear translocation of Nrf2, which induced gene expression for counteracting ROS production, maintaining tumor cell survival, and promoting hepatocellular carcinoma (HCC) development.
RESULTS
KHK-A is required for oxidative stress–enhanced p62 oligomerization and Nrf2 activation in HCC cells
It was previously shown that p62 expression and oligomerization, which are necessary and sufficient for HCC induction and serve as a strong prognostic indicator of rapid HCC recurrence, are required for the activation of Nrf2 and protection of HCC-initiating cells from oxidative stress–induced cell death (28). To determine the mechanism underlying p62 oligomerization in HCC, we stimulated LO2 human normal hepatocytes and Huh7 HCC cells with hypoxia and performed mass spectrometry (MS) analyses of immunoprecipitated p62. As expected, Keap1 was shown to be a p62-associated protein in both cell lines (fig. S1A). Notably, a ~35-kDa p62-interacting protein, which was only detected in Huh7 cells upon hypoxia stimulation, was identified as KHK-A by MS analyses. This association was validated by coimmunoprecipitation analyses of Huh7 and Hep3B HCC cells stimulated with hypoxia (Fig. 1A), suggesting that KHK-A acts like other metabolic enzymes that can have nonmetabolic functions in regulating critical cellular functions (29–31).
Fig. 1. KHK-A is required for oxidative stress–enhanced p62 oligomerization and Nrf2 activation in HCC cells.
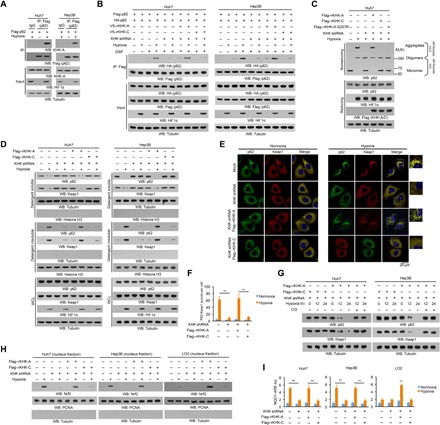
(A to D and G and H) Immunoblot analyses were performed with the indicated antibodies. (A) Huh7 and Hep3B cells expressing Flag-p62 were treated with or without hypoxia for 6 hours in the presence of 10 μM chloroquine (CQ; a lysosome inhibitor). Immunoprecipitation analyses with an anti-Flag antibody were performed. Hif 1α, hypoxia-inducible factor 1α; WB, Western blot; IP, immunoprecipitation; IgG, immunoglobulin G. (B) Huh7 and Hep3B cells with or without expressing KHK short hairpin RNA (shRNA) and with or without reconstituted expression of the indicated proteins were transfected with vectors expressing Flag-p62 and HA-p62 and treated with or without hypoxia for 6 hours in the presence of the lysosome inhibitor CQ (10 μM). After incubation with the reversible cross-linking agent DSP (0.4 mg/ml) for 2 hours, the cells were lysed in a buffer containing 1% SDS to solubilize all proteins. The lysates were subjected to immunoprecipitation analyses with an anti-Flag antibody after diluting SDS to 0.1%. (C) Huh7 cells with or without expressing KHK shRNA and with or without reconstituted expression of the indicated proteins were treated with or without hypoxia for 6 hours and lysed and analyzed by reducing (containing 2.5% β-mercaptoethanol) and nonreducing SDS–polyacrylamide gel electrophoresis (SDS-PAGE) to detect p62 aggregation. (D) Huh7 and Hep3B cells with or without expression of KHK shRNA were reconstituted with or without expression of the indicated KHK proteins. After stimulation with or without hypoxia for 6 hours in the presence of the lysosome inhibitor CQ (10 μM), the cells were lysed in a lysis buffer with 1% Triton X-100. The insoluble fraction was lysed in a lysis buffer with 1% SDS. WCL, whole-cell lysate. (E and F) Huh7 cells with or without KHK shRNA expression and with or without reconstituted expression of Flag-tagged rKHK-A or rKHK-C were stimulated with or without hypoxia for 6 hours. Immunofluorescent analyses were performed with the indicated antibodies (E). The numbers of puncta in 100 cells were counted and quantified. Data are shown as means ± SD of 100 cells per group. A two-tailed Student’s t test was used. **P < 0.01 (F). (G) Huh7 and Hep3B cells expressing KHK shRNA with or without reconstituted expression of the indicated proteins were treated with or without hypoxia and lysosome inhibitor CQ (10 μM) for the indicated periods of time. (H) The indicated cells with or without expressing KHK shRNA and with or without reconstituted expression of the indicated proteins were treated with or without hypoxia for 12 hours. The nuclear fractions were prepared. PCNA, proliferating cell nuclear antigen. (I) The indicated cells with or without expressing KHK shRNA and with or without reconstituted expression of the indicated proteins were transfected with quinone oxidoreductase 1 (NQO1)–ARE-luc and pRL-TK (Renilla luciferase control reporter vector) plasmids. Starting at 18 hours after transfection, cells were treated with or without hypoxia for 12 hours and harvested for luciferase activity analyses. The data are presented as means ± SD from triplicate samples. **P < 0.01.
Mutually exclusive splicing of the adjacent exons 3C and 3A of the KHK gene leads to alternative expression of the KHK-C and KHK-A isoforms. KHK-C, which is expressed primarily in normal hepatocytes, has much higher activity in phosphorylating fructose for fructose-1-phosphate (F1P) production than does KHK-A (32). In HCC cells, KHK-A expression replaces that of KHK-C (fig. S1B) and acts as a protein kinase, phosphorylating and activating phosphoribosyl pyrophosphate synthetase 1 (PRPS1) to promote de novo nucleic acid synthesis (32). Coimmunoprecipitation analyses revealed that hypoxia did not induce an association between p62 and KHK-C in LO2 cells (fig. S1C) or Huh7 (fig. S1D) and Hep3B (fig. S1E) HCC cells with depleted KHK and reconstituted KHK-C expression. In contrast, ectopically expressed KHK-A in KHK-depleted LO2 cells (fig. S1C) or Huh7 (fig. S1D) and Hep3B (fig. S1E) HCC cells was able to bind to p62. These results indicate that oxidative stress induces the binding of p62 to KHK-A, which is highly and specifically expressed in HCC cells.
To determine whether KHK-A regulates p62 oligomerization, we chemically cross-linked the interacting cellular proteins by treating Huh7, Hep3B, and LO2 cells with dithiobis succinimidyl propionate (DSP) and showed that hypoxic stimulation resulted in association between hemagglutinin (HA)–p62 and Flag-p62 in Huh7 and Hep3B cells (Fig. 1B), but to a much lesser extent in LO2 cells (fig. S1F). This association was dramatically reduced by KHK-A depletion in Huh7 and Hep3B cells (Fig. 1B) and enhanced by KHK-A overexpression in LO2 cells (fig. S1F). In addition, the observed reduction in KHK-A–depleted Huh7 and Hep3B cells was rescued by reconstituted expression of KHK-A but not KHK-C (Fig. 1B). Correspondingly, hypoxia reduced the amount of p62 monomer and induced p62 oligomerization in Huh7 cells (Fig. 1C), and to a much lesser extent, in LO2 cells (fig. S1G), which was detected by nonreducing gel electrophoresis analyses. The reduction of monomer and increase of oligomers were alleviated by KHK-A depletion in Huh7 cells and enhanced by reconstituted expression of KHK-A in Huh7 and LO2 cells (Fig. 1C and fig. S1G). Notably, reconstituted expression of the kinase-dead G257R mutant of KHK-A or KHK-C failed to promote p62 oligomerization (Fig. 1C and fig. S1G). These results indicate that KHK-A and its kinase activity are critical for p62 oligomerization in HCC cells.
p62 oligomerization and its interaction and aggregation with cargo proteins with K48-linked polyubiquitylation, such as Keap1, lead to autophagy-lysosomal degradation of the protein complexes (17). p62 and Keap1 aggregations were induced by hypoxic stimulation and exhibited as reduction of detergent-soluble p62 and Keap1 and increase of detergent-insoluble p62 and Keap1 in Huh7 and Hep3B cells (Fig. 1D). These aggregations were significantly inhibited by KHK-A depletion, which was rescued by reconstituted expression of KHK-A but not KHK-C (Fig. 1D). In contrast, limited p62 and Keap1 aggregations were detected in hypoxia-stimulated LO2 cells with reconstituted KHK-C expression; however, reconstituted expression of KHK-A substantially enhanced these aggregations (fig. S1H). Consistent with these results, immunofluorescence analyses showed that hypoxia stimulation resulted in aggregation of p62 and Keap1, reflected by their puncta appearances in Huh7 cells, in a manner dependent on expression of KHK-A but not KHK-C (Fig. 1, E and F). In addition, colocalization of p62 with proteins with K48-linked polyubiquitylation, but not those with K63-linked polyubiquitylation, was observed upon hypoxic stimulation (fig. S1I). As expected, hypoxia-induced degradation of both p62 and Keap1 in Huh7 and Hep3B cells was blocked by reconstituted expression of KHK-C or treatment of the cells with chloroquine (CQ), a lysosome inhibitor (Fig. 1G). In line with these findings, immunofluorescence analyses (fig. S2, A and B) and cell fractionation (Fig. 1H and fig. S2C) showed that Nrf2 was accumulated in the nucleus upon hypoxia or H2O2 stimulation, and this accumulation was dependent on expression of KHK-A but not KHK-C. Consequently, the transcriptional activity of Nrf2, measured by a luciferase reporter assay (Fig. 1I and fig. S2D), and the mRNA expression of Nrf2-regulated antioxidative genes, such as heme oxygenase-1 (HO-1), glutamate-cysteine ligase catalytic (GCLC), Glutathione S-transferase mu 1 (GSTM1), quinone oxidoreductase 1 (NQO1), UDP glucuronosyltransferase family 1 member A1 (UGT1A1), and phosphogluconate dehydrogenase (PGD) (3), measured by quantitative polymerase chain reaction (PCR) (fig. S2, E and F), were positively regulated in a KHK-A– but not KHK-C–dependent manner in Huh7 and LO2 cells.
Together, these results indicate that KHK-A, which is highly expressed in HCC cells but not in normal hepatocytes, is required for oxidative stress–enhanced p62 oligomerization and subsequent nuclear translocation and activation of Nrf2. These results also indicate that differentially expressed KHK-A enables HCC cells to have higher Nrf2 activity than normal hepatocytes.
AMPK phosphorylates KHK-A and promotes the association between KHK-A and p62
To determine the mechanism underlying the interaction between KHK-A and p62, we treated Huh7 cells with a panel of inhibitors: SP600125, SB203580, U0126, and compound C, which blocked hypoxia-induced activation of c-Jun N-terminal kinase (JNK), P38, mitogen-activated protein kinase (MAPK) kinase (MEK)/extracellular signal–regulated kinase (ERK), and AMPK, respectively (fig. S3A). We found that only AMPK inhibition blocked the interaction between KHK-A and p62 (Fig. 2A). In line with this finding, hypoxia or H2O2 treatment resulted in the binding of AMPK to KHK-A but not to KHK-C (Fig. 2B). Similarly, buthionine sulfoximine (BSO), an oxidizing agent, induced the interaction between AMPK and KHK-A (fig. S3B). This interaction was blocked by pretreatment of Huh7 cells with the AMPK inhibitor compound C before hypoxia (Fig. 2C) or H2O2 (fig. S3C) treatment. In addition, binding of KHK-A to p62 in response to AMPK activator A769662 (Fig. 2D) or hypoxia (fig. S3D) treatment was inhibited by AMPKα1/2 deficiency in embryonic fibroblasts. These results indicate that AMPK activation is required for the binding of KHK-A to p62.
Fig. 2. AMPK phosphorylates KHK-A and promotes the association between KHK-A and p62.

Immunoprecipitation or immunoblot analyses were performed with the indicated antibodies. (A) Huh7 cells transfected with Flag–KHK-A were pretreated with SB203580 (10 μM), SP600125 (25 μM), U0126 (20 μM), or compound C (5 μM) before hypoxia stimulation for 6 hours in the presence of CQ (10 μM). (B) Huh7 cells expressing Flag–KHK-C or Flag–KHK-A were cotransfected with HA-AMPKα and treated with or without hypoxia for 6 hours or H2O2 (0.5 mM) for 1 hour in the presence of the lysosome inhibitor CQ (10 μM). (C) Huh7 cells transfected with Flag–KHK-A were pretreated with or without compound C (5 μM) for 30 min before hypoxia stimulation for 6 hours in the presence of the lysosome inhibitor CQ (10 μM). ACC1, acetyl-CoA carboxylase 1. (D) WT and AMPKα1/2 double-knockout (DKO) mouse embryonic fibroblasts (MEFs) infected with lentivirus expressing Flag–KHK-A were treated with or without A769662 (0.5 mM) for 6 hours in the presence of the lysosome inhibitor CQ (10 μM). (E and F) Bacterially purified GST–KHK-A, GST–KHK-C (E), or GST–KHK-A S80A (F) was incubated with or without bacterially purified active AMPK in the presence of [γ-32P]ATP. Autoradiography and immunoblot analyses were performed. (G) Huh7 cells expressing WT Flag–KHK-A, Flag–KHK-C, or Flag–KHK-A S80A were treated with or without hypoxia for 6 hours in the presence of the lysosome inhibitor CQ (10 μM). (H) WT and AMPKα1/2 DKO MEFs were cultured with or without hypoxia for 6 hours. (I) An in vitro AMPK kinase assay was performed by mixing bacterially purified WT GST–KHK-A, GST–KHK-A S80A, or GST–KHK-C on glutathione agarose beads with or without purified active AMPK in the presence of AMP and ATP for 1 hour. The glutathione agarose beads were then washed and incubated with purified His-p62 for a GST pulldown analysis. (J) Huh7 cells expressing Flag–KHK-A, Flag–KHK-C, or Flag–KHK-A S80A were treated with or without hypoxia for 4 hours in the presence of the lysosome inhibitor CQ (10 μM). (K) Huh7 cells expressing Flag–KHK-A and HA-AMPKα were pretreated with N-acetyl-l-cysteine (NAC) (5 mM) or Trolox (100 μM) for 30 min before hypoxia stimulation for 6 hours. (L) Huh7 cells expressing Flag–KHK-A were pretreated with NAC (5 mM) or Trolox (100 μM) for 30 min before hypoxia stimulation for 6 hours in the presence of the lysosome inhibitor CQ (10 μM).
To determine whether KHK-A is a substrate of AMPK, we performed an in vitro phosphorylation assay and showed that the active recombinant AMPK protein phosphorylated purified bacteria-expressed glutathione S-transferase (GST)–KHK-A, but not KHK-C, in the presence of [γ-32P]adenosine triphosphate (ATP) (Fig. 2E). Liquid chromatography-tandem MS/MS (LC-MS/MS) analyses showed that this phosphorylation was at S80 (fig. S3E), which is conserved among species (fig. S3F) and is encoded by unique exon 3A of KHK-A (fig. S3G). S80A mutation abrogated the AMPK-dependent KHK-A phosphorylation in vitro and the recognition by a validated and specific anti–phospho–KHK-A S80 antibody (Fig. 2F and fig. S3, H and I).
Corresponding to the in vitro results, hypoxia (Fig. 2G) or A769662 treatment (fig. S3J) induced S80 phosphorylation; this phosphorylation was abrogated by the S80A mutation (Fig. 2G and fig. S3J), deficiency of AMPKα1/2 (Fig. 2H and fig. S3K), or compound C treatment (fig. S3L). These results indicate that oxidative stress results in the binding of AMPK to KHK-A and the subsequent AMPK-dependent phosphorylation of KHK-A at S80.
To determine whether KHK-A S80 phosphorylation regulates the activity of KHK-A toward fructose, we transfected Flag- or V5-tagged wild-type (WT) or S80A mutant of KHK-A into Huh7 cells and found that mutation of S80 did not affect the coimmunoprecipitation between KHK-A molecules (fig. S3M), suggesting that KHK-A S80A mutation does not affect KHK-A dimer formation. In addition, expression of KHK-A S80A, which had similar activity to WT KHK-A (fig. S3N), did not alter 13C-labeled F1P production (fig. S3O). These results suggest that KHK-A S80A mutation does not affect KHK-A dimer formation or its activity.
We next examined whether KHK-A S80 phosphorylation affects the interaction between KHK-A and p62. An in vitro AMPK phosphorylation of purified GST–KHK-A followed by GST pulldown of purified bacteria-expressed His-p62 showed that AMPK-phosphorylated WT KHK-A, but not KHK-A S80A mutant or KHK-C, was able to bind to p62 (Fig. 2I). In addition, the binding of KHK-A to p62 in Huh7 cells induced by hypoxia (Fig. 2J), H2O2 (fig. S3P), or BSO treatment (fig. S3Q) was abrogated by reconstituted expression of KHK-A S80A. Notably, treatment of Huh7 cells with the reducing agent Trolox or N-acetyl-l-cysteine (NAC), which significantly reduced hypoxia- or H2O2-induced ROS accumulation (fig. S3R), largely blocked the interaction between AMPK and KHK-A (Fig. 2K) and the binding of KHK-A to p62 (Fig. 2L). These results indicate that oxidative stress results in AMPK-dependent KHK-A S80 phosphorylation that promotes the binding of KHK-A to p62.
We previously showed that KHK-A interacted with and phosphorylated PRPS1 T225 under normoxic conditions (32). However, this interaction was disrupted by hypoxic stimulation (fig. S4A). The disrupted interaction was restored by pretreatment of Huh7 cells with compound C (fig. S4A). In addition, A769662 treatment abolished the binding of KHK-A to PRPS1 in WT mouse embryonic fibroblasts (MEFs) but not in AMPKα1/2-deficient MEFs under normoxic conditions (fig. S4B). These results indicate that AMPK activation disrupts the binding of KHK-A to PRPS1.
To determine whether AMPK-mediated KHK-A S80 phosphorylation regulates the dissociation between KHK-A and PRPS1, we performed an in vitro phosphorylation assay and showed that KHK-A S80A mutant, but not its WT counterpart, was resistant to AMPK-induced dissociation between KHK-A and PRPS1 (fig. S4C). In addition, AMPK-mediated KHK-A S80 phosphorylation inhibited WT KHK-A– but not KHK-A S80A–mediated PRPS1 phosphorylation in vitro (fig. S4D). Consistent results showed that hypoxia-inhibited binding of WT KHK-A to PRPS1 (fig. S4E) and subsequent PRPS1 T225 phosphorylation (fig. S4F) in Huh7 cells were abrogated by KHK-A S80A expression. These results indicate that hypoxia induces AMPK-mediated KHK-A S80 phosphorylation, leading to disruption of the binding of KHK-A to PRPS1 and increased binding of KHK-A to p62.
It was previously shown that AMPK oxidation reduced its activity in cardiomyocytes in response to energy stress (33). Nonreducing gel electrophoresis and immunoblotting analyses revealed that AMPK was shifted to oxidized and high–molecular weight forms with corresponding reduction of monomeric AMPK (fig. S4, G and H, left) and AMPK-mediated acetyl-CoA carboxylase 1 (ACC1) S79 phosphorylation (fig. S4I, left) upon treatment of cardiomyocytes with hypoxia or H2O2. Notably, the AMPK with high molecular weight was abolished in reducing gel electrophoresis (fig. S4, G and H, left), suggesting that hypoxia or H2O2 treatment of cardiomyocytes induces oxidation and inhibition of AMPK. In contrast, hypoxia (fig. S4G, middle and right) or H2O2 (fig. S4H, middle and right) treatment of Huh7 and Hep3B cells failed to induce AMPK oxidation and increased ACC1 S79 phosphorylation (fig. S4I, middle and right). Consistent with this finding, Huh7 and Hep3B cells had much higher levels of Trx1, an important reducing enzyme that cleaves disulfides in proteins to prevent AMPK oxidation (33), than did cardiomyocytes (fig. S4J). These results suggest that HCC cells have up-regulated Trx1 expression to counteract AMPK oxidation compared with cardiomyocytes.
KHK-A acts as a protein kinase and phosphorylates p62 at S28
KHK-A uses ATP as a phosphate donor to phosphorylate and activate PRPS1 (32). To determine whether KHK-A phosphorylates p62, we performed a phosphorylation assay by mixing AMPK-phosphorylated GST–KHK-A and purified bacteria-expressed His-p62. WT KHK-A, but not KHK-A S80A mutant (Fig. 3A), KHK-A G257R kinase-dead mutant, or KHK-C (fig. S5A), phosphorylated p62. LC-MS/MS analyses identified S28 as the phosphorylated residue (fig. S5B). p62 S28A mutation abrogated both the KHK-A–dependent p62 phosphorylation in vitro (fig. S5C), confirmed by a validated and specific anti–phospho-p62 S28 antibody (fig. S5, D and E).
Fig. 3. KHK-A acts as a protein kinase and phosphorylates p62 at S28.

Immunoblot analyses were performed with the indicated antibodies. (A) An in vitro AMPK kinase assay was performed by mixing bacterially purified WT GST–KHK-A, GST–KHK-A S80A, or GST on glutathione agarose beads with or without purified active AMPK in the presence of AMP and ATP for 1 hour. The glutathione agarose beads were then washed and incubated with purified His-p62 in the presence of [γ-32P]ATP. Autoradiography was performed. (B) Huh7 cells with or without expressing KHK shRNA and with or without reconstituted expression of the indicated proteins were treated with or without A769662 (0.5 mM) for 4 hours in the presence of the lysosome inhibitor CQ (10 μM). (C) WT and AMPKα1/2 DKO MEFs were treated with or without hypoxia for 6 hours in the presence of the lysosome inhibitor CQ (10 μM). (D) Huh7 and Hep3B cells with or without expressing KHK shRNA and with or without reconstituted expression of the indicated KHK proteins were pretreated with or without compound C (5 μM) for 30 min before hypoxia stimulation for 6 hours in the presence of the lysosome inhibitor CQ (10 μM). (E) Huh7 and Hep3B cells with or without expressing KHK shRNA and with or without reconstituted expression of the indicated proteins were treated with or without hypoxia for 6 hours in the presence of the lysosome inhibitor CQ (10 μM). (F and G) Parental Huh7 and Hep3B cells and the indicated clones of cells with knock-in of KHK-A S80A (F) or p62 S28A (G) expression were stimulated with or without hypoxia for 6 hours in the presence of the lysosome inhibitor CQ (10 μM).
We next showed that A769662 treatment induced p62 S28 phosphorylation, and this phosphorylation was alleviated by KHK-A depletion in Huh7 cells (Fig. 3B). In addition, p62 S28 phosphorylation induced by hypoxia or A769662 was inhibited by AMPKα1/2 deficiency in MEFs (Fig. 3C and fig. S5F), compound C treatment, and KHK-A depletion in Huh7 and Hep3B cells (Fig. 3D). The inhibitory effect of KHK-A depletion was abrogated by reconstituted expression of KHK-A but not KHK-C (Fig. 3, B and D). The reducing agent Trolox and NAC significantly reduced hypoxia- or H2O2-induced KHK-A S80 and p62 S28 phosphorylation (fig. S5, G and H). In addition, reconstituted expression of KHK-A G257R or KHK-A S80A in the KHK-A–depleted HCC cells (fig. S5I) inhibited p62 S28 phosphorylation induced by hypoxia (Fig. 3E). In addition, knock-in expression of KHK-A S80A (fig. S5, J and K) or p62 S28A (fig. S5, L and M) in Huh7 and Hep3B cells using CRISPR-Cas9 genome-editing knock-in technology demonstrated that expression of KHK-A S80A (Fig. 3F and fig. S5N) or p62 S28A (Fig. 3G) abolished hypoxia- or A769662-induced p62 S28 phosphorylation. These results indicate that oxidative stress results in the protein kinase activity of KHK-A–mediated phosphorylation of p62 at S28.
p62 S28 phosphorylation is required for oxidative stress–enhanced p62 oligomerization and Nrf2 activation
To determine whether p62 S28 phosphorylation regulates p62 oligomerization, we treated Huh7 cells with cross-linker DSP and showed that hypoxia-induced association between HA-p62 and Flag-p62 was reduced by knock-in of KHK-A S80A (Fig. 4A), p62 S28A, and p62 K7R (Fig. 4B), which disrupts the K7-D69 hydrogen bond in the PB1 domain. In addition, hypoxia-induced translocation of p62 from detergent-soluble to detergent-insoluble fractions was reduced by expression of KHK-A S80A and p62 S28A (fig. S6A). Correspondingly, hypoxia reduced the amount of p62 monomer and induced p62 oligomerization in Huh7 and Hep3B cells; these effects were alleviated by expression of KHK-A S80A (fig. S6B) and p62 S28A (Fig. 4C). These results indicate that KHK-A–mediated p62 S28 phosphorylation promotes p62 oligomerization.
Fig. 4. KHK-A–mediated p62 S28 phosphorylation is required for oxidative stress–enhanced p62 oligomerization and Nrf2 activation.
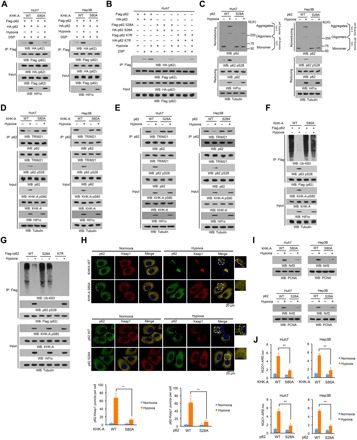
(A to G and I) Immunoprecipitation and immunoblot analyses were performed with the indicated antibodies. (A) Huh7 and Hep3B cells with or without knock-in of KHK-A S80A expression were transfected with the vectors expressing Flag-p62 and HA-p62 and treated with or without hypoxia for 6 hours in the presence of the lysosome inhibitor CQ (10 μM). After incubation with DSP (0.4 mg/ml) for 2 hours, the cells were lysed in a buffer containing 1% SDS to solubilize all proteins. The lysates were subjected to immunoprecipitation analyses with an anti-Flag antibody after diluting SDS to 0.1%. (B) Huh7 cells expressing the indicated p62 proteins were treated with or without hypoxia for 6 hours in the presence of the lysosome inhibitor CQ (10 μM). After incubation with DSP (0.4 mg/ml) for 2 hours, the cells were lysed in a buffer containing 1% SDS to solubilize all proteins. The lysates were subjected to immunoprecipitation analyses with an anti-Flag antibody after diluting SDS to 0.1%. (C) Huh7 and Hep3B cells with or without knock-in expression of p62 S28A were treated with or without hypoxia for 6 hours. The whole-cell lysates were analyzed by reducing and nonreducing SDS-PAGE to detect p62 aggregation. (D and E) Huh7 and Hep3B cells with or without knock-in expression of KHK-A S80A (D) or p62 S28A (E) were stimulated with or without hypoxia for 6 hours in the presence of the lysosome inhibitor CQ (10 μM). (F) Huh7 cells with or without knock-in of KHK-A S80A expression were treated with or without hypoxia for 6 hours in the presence of the lysosome inhibitor CQ (10 μM). (G) Endogenous p62-depleted Huh7 cells with or without reconstituted expression of WT Flag-rp62, rp62 S28A, or rp62 K7R were treated with or without hypoxia for 6 hours in the presence of the lysosome inhibitor CQ (10 μM). (H) Huh7 cells with or without knock-in of KHK-A S80A (top) or p62 S28A (bottom) expression were stimulated with or without hypoxia for 6 hours. Immunofluorescence analyses were performed with the indicated antibodies. Numbers of puncta in 100 cells were counted and quantified. Data are shown as means ± SD of 100 cells per group. A two-tailed Student’s t test was used. **P < 0.001. (I) Huh7 and Hep3B cells with or without knock-in of KHK-A S80A (top) or p62 S28A (bottom) expression were treated with or without hypoxia for 12 hours. The nuclear fractions were prepared. (J) Huh7 and Hep3B cells with or without knock-in of KHK-A S80A (top) or p62 S28A (bottom) expression were transfected with NQO1-ARE-luc and pRL-TK plasmids. After treatment with or without hypoxia for 12 hours, the cells were harvested for luciferase activity analyses. The data are presented as means ± SD from triplicate samples. **P < 0.001.
It was previously reported that proteasomal stress resulted in phosphatase and tensin homolog–induced putative kinase 1 (PINK1)–mediated p62 S28 phosphorylation and p62 oligomerization (34). Nevertheless, hypoxia-induced reduction of detergent-soluble p62 and increase of detergent-insoluble p62 (fig. S6C), aggregation (fig. S6D), and S28 phosphorylation (fig. S6E) of p62 were not affected by PINK1 depletion. However, proteasome inhibitor MG132-induced reduction of detergent-soluble p62 and increase of detergent-insoluble p62 (fig. S6F), aggregation (fig. S6G), and S28 phosphorylation of p62 (fig. S6H) were abrogated by depletion of PINK1. In contrast, depletion of KHK-A or reconstituted expression of rKHK-A or rKHK-C failed to alter MG132-induced p62 aggregation and puncta formation (fig. S6, I and J). These data suggest that p62 S28 phosphorylation and aggregation can be regulated by different upstream cellular signaling in a stress type–dependent manner.
E3 ubiquitin ligase TRIM21-mediated p62 ubiquitylation at K7 via K63 linkage blocks p62 oligomerization (26). To determine whether p62 S28 phosphorylation affects K63-linked p62 ubiquitination, we performed coimmunoprecipitation analyses and showed that hypoxia stimulation reduced the binding of TRIM21 to p62 (fig. S7A). This reduction was abrogated by replacement of KHK-A expression by KHK-C (fig. S7A) and reconstituted expression of KHK-A S80A (Fig. 4D) and p62 S28A (Fig. 4E). In addition, hypoxia stimulation inhibited K63-linked p62 ubiquitination, and this inhibition was alleviated by replacement of KHK-A expression by KHK-C (fig. S7B), knock-in of KHK-A S80A expression in Huh7 cells (Fig. 4F), and reconstituted expression of p62 S28A or p62 K7R in endogenous p62-depleted Huh7 cells (Fig. 4G). Consistent immunofluorescent results showed that S28-phosphorylated p62 was colocalized with proteins having K48-linked, but not with K63-linked, polyubiquitylation (fig. S7C). These results strongly suggest that p62 S28 phosphorylation blocks the binding of TRIM21 to p62 and subsequent ubiquitination, thereby promoting p62 oligomerization and its association with cargo proteins with K48-linked polyubiquitylation.
To determine the effect of p62 S28 phosphorylation on the Keap1-Nrf2 pathway, we performed immunofluorescence analyses and showed that hypoxia-induced puncta formation of p62 and Keap1 was blocked by expression of KHK-A S80A and p62 S28A (Fig. 4H). Consistent with this finding, expression of these mutants reduced long-term hypoxia-induced degradation of both p62 and Keap1 (fig. S7D) and nuclear translocation of Nrf2 in Huh7 and Hep3B cells, as detected by cell fractionation (Fig. 4I) and immunofluorescence analyses (fig. S7E). In addition, hypoxia-enhanced transcriptional activity of Nrf2 (Fig. 4J) and mRNA expression of Nrf2-regulated antioxidative genes (fig. S7, F and G) in Huh7 and Hep3B cells were inhibited by expression of KHK-A S80A and p62 S28A. These results indicate that KHK-A–mediated p62 S28 phosphorylation is required for oxidative stress–enhanced p62 oligomerization and subsequent nuclear translocation and activation of Nrf2.
p62 S28 phosphorylation reduces ROS production and promotes cancer cell survival
Nrf2 activation is critical for counteracting ROS production and cell survival (4). Compared with KHK-C–expressing LO2 cells, Huh7 and Hep3B cells in response to hypoxic stimulation had much reduced total cellular ROS production (Fig. 5A) and NADP+/NADPH ratios (Fig. 5B) and increased GSH/oxidized glutathione (GSSG) ratios (fig. S8A), which are indicators of reduced oxidative stress. While depletion of KHK-C in LO2 cells did not alter ROS production or NAPH+/NAPDH and GSH/GSSG ratios in response to the hypoxic condition, KHK-A depletion significantly increased these markers of oxidative stress in HCC cells, and this induction was abrogated by reconstituted expression of KHK-A but not KHK-C (Fig. 5, A and B, and fig. S8A). In addition, reconstituted expression of KHK-A in KHK-depleted LO2 cells suppressed hypoxia-induced ROS production, decreased NADP+/NADPH ratios, and increased GSH/GSSG ratios (Fig. 5, A and B, and fig. S8A).
Fig. 5. p62 S28 phosphorylation reduces ROS production and promotes cancer cell survival.
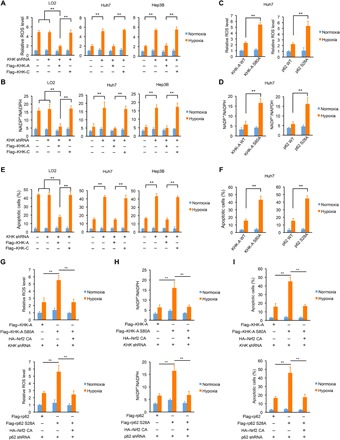
The data are presented as means ± SD from three independent experiments. **P < 0.001. A two-tailed Student’s t test was used. (A, B and E) LO2, Huh7, and Hep3B cells with or without expression of KHK shRNA were reconstituted with or without expression of the indicated KHK proteins. After treatment of the cells with or without hypoxia for 36 hours, ROS levels (A), intracellular NADP+ and NADPH levels (B), and the percentages of apoptotic cells (E) were measured. (C, D, and F) Huh7 cells with or without knock-in of KHK-A S80A (left) or p62 S28A (right) expression were treated with or without hypoxia for 36 hours. ROS levels (C), intracellular NADP+ and NADPH levels (D), and the percentages of apoptotic cells (F) were measured. (G to I) Huh7 cells with reconstituted expression of the indicated KHK-A proteins (top) or p62 proteins (bottom) were stably transfected with or without HA-tagged constitutively active Nrf2 and treated with or without hypoxia for 36 hours. ROS levels (G), intracellular NADP+ and NADPH levels (H), and the percentages of apoptotic cells (I) were measured.
Consistent with the role of p62 S28 phosphorylation in Nrf2 activation, knock-in of KHK-A S80A and p62 S28A expression in both Huh7 and Hep3B cells significantly increased total cellular ROS production (Fig. 5C and fig. S8B) and NADP+/NADPH ratios (Fig. 5D and fig. S8C) and decreased GSH/GSSG ratios (fig. S8D) under hypoxic conditions. These results indicate that HCC cells harness a specific counter-oxidative stress system, in which highly expressed KHK-A and KHK-A–mediated p62 S28 phosphorylation play an instrumental role.
We next examined cell survival under the hypoxic condition. As expected, Huh7 and Hep3B cells had much lower levels of apoptosis rates (Fig. 5E) and caspase activity, reflected by poly(adenosine diphosphate–ribose) polymerase (PARP) cleavage, in response to hypoxic stimulation than did LO2 cells (fig. S8E). Depletion of KHK-C in LO2 cells did not change the apoptosis rate, whereas reconstituted expression of KHK-A suppressed hypoxia-induced apoptosis of LO2 cells (Fig. 5E and fig. S8E). In contrast, KHK-A depletion significantly increased apoptosis of HCC cells, and this induction was inhibited by reconstituted expression of KHK-A but not KHK-C (Fig. 5E). Similarly, knock-in of KHK-A S80A and p62 S28A expression in HCC cells significantly increased apoptosis rates (Fig. 5F and fig. S8F) and PARP cleavage under hypoxic conditions (fig. S8G). Consistent results showed that reconstituted KHK-A S80A expression in Huh7 cells reduced cell proliferation (fig. S8H) and survival (fig. S8I) in response to hypoxia.
To further support the role of Nrf2 regulation by KHK-A–mediated p62 S28 phosphorylation in counteracting ROS production and promoting HCC cell survival, we expressed constitutively active Nrf2 (Nrf2 CA) mutant with the Nrf2-ECH homology 2 (Neh2) domain deletion (fig. S8J) (35). Nrf2 CA expression abrogated the enhanced effect of KHK-A S80A and p62 S28A expression on ROS production (Fig. 5G), NADP+/NADPH ratios (Fig. 5H), and apoptosis (Fig. 5I) upon hypoxic stimulation.
Oxidative stress promotes the binding of p62 to cargo proteins, including Keap1 for their autophagy-lysosomal degradation (16, 17). KHK depletion, reconstituted expression of KHK-A or KHK-C (fig. S8, K and L), or expression of KHK-A S80A (fig. S8, M and N) or p62 S28A (fig. S8, O and P) did not affect hypoxia-induced conversion of LC3B-I to LC3B-II (a phosphatidylethanolamine derivative of LC3-I) (fig. S8, K, M, and O) and GFP (green fluorescent protein)–LC3 puncta formation (fig. S8, L, N, and P), suggesting that KHK-A–mediated p62 S28 phosphorylation plays a limited role in hypoxia-induced autophagy initiation. In contrast, hypoxia-induced degradation of p62 and Keap1, which was blocked by the lysosomal inhibitor CQ, was also inhibited by reconstituted expression of KHK-C, but not KHK-A, in endogenous KHK-depleted Huh7 and Hep3B cells (Fig. 1G). In addition, this inhibition was also observed by expression of KHK-A S80A and p62 S28A in Huh7 cells (fig. S7D). These results indicate that the phosphorylation of KHK-A by AMPK and the subsequent phosphorylation of p62 by KHK-A are involved in a downstream step of autophagy and enhance the function of p62 in autophagy-dependent protein degradation.
To further support the role of the phosphorylation of KHK-A S80 and p62 S28 in counteracting oxidative stress, we expressed phosphorylation-mimicking KHK-A S80E or p62 S28E in Huh7 cells. Expression of these mutants, which promoted p62 and Keap1 aggregation (fig, S9, A and B), nuclear translocation of Nrf2 (fig, S9, C, D, E and F), and the expression of Nrf2-regulated and antioxidative genes (fig, S9, G and H), markedly reduced hypoxia-enhanced ROS accumulation (fig. S9I), NADP+/NADPH levels (fig. S9J), and apoptosis (fig. S9K). These results indicate that AMPK-mediated KHK-A S80 phosphorylation and KHK-A–mediated p62 S28 phosphorylation play an instrumental role in counteracting oxidative stress–induced cellular effects.
p62 S28 phosphorylation promotes hepatocellular tumorigenesis and is associated with the clinical aggressiveness of human HCC
To determine the effect of KHK-A–mediated p62 S28 phosphorylation in tumorigenesis, we intrahepatically (Fig. 6A) and subcutaneously (fig. S10, A to C) injected Huh7 cells into nude mice. Knock-in of KHK-A S80A or p62 S28A expression markedly inhibited tumor growth. Correspondingly, immunohistochemical (IHC) analysis revealed that expression of these mutants reduced Ki67 expression (Fig. 6B) and increased cell apoptosis (fig. S10D). In addition, KHK-A S80A expression reduced p62 S28 phosphorylation and nuclear accumulation of Nrf2 in tumor tissues (Fig. 6C), whereas p62 S28A expression also exhibited a similar inhibitory effect on nuclear amounts of Nrf2 (Fig. 6D). Notably, the inhibitory effect of KHK-A S80A (fig. S10E) and p62 S28A (fig. S10F) expression on tumor growth and Ki67 expression in tumor tissues (fig. S10, G and H) was eliminated by Nrf2 CA expression. These results indicate that KHK-A–mediated p62 S28 phosphorylation promotes HCC tumorigenesis by activating Nrf2.
Fig. 6. KHK-A–mediated p62 S28 phosphorylation promotes hepatocellular tumorigenesis and is associated with clinical aggressiveness of human HCC.
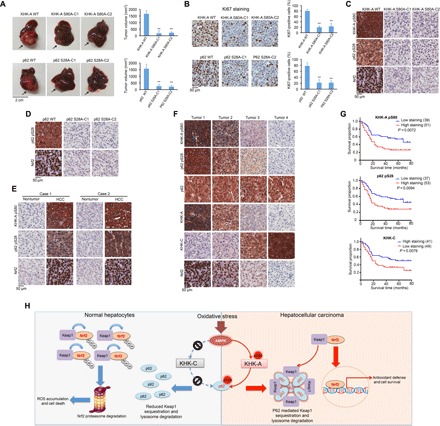
(A) Huh7 (1 × 106) cells with or without knock-in of KHK-A S80A (top) or p62 S28A (bottom) expression were intrahepatically injected into athymic nude mice (n = 7 per group). The mice were euthanized and examined for tumor growth 28 days after injection. The arrows point to the tumors. Tumor volumes were calculated (right). Data represent the means ± SD of seven mice. **P < 0.01 by a two-tailed Student’s t test. (B) IHC analyses of tumor tissues were performed with an anti-Ki67 antibody. Ki67-positive cells were quantified in 10 microscope fields. **P < 0.01 by a two-tailed Student’s t test. (C and D) IHC analyses of xenograft tumors from nude mice were performed with the indicated antibodies. (E) IHC staining of 30 human HCC and matched nontumor tissue samples was performed with the indicated antibodies. Representative photos of stains of two cases are shown. (F) IHC staining of 90 human HCC specimens was performed with the indicated antibodies. Representative images from the staining of four different specimens are shown. (G) Kaplan-Meier plots of the overall survival rates in human HCC specimens (n = 90) in groups with high (staining score, 4 to 8) and low (staining score, 0 to 3) expression of KHK-A pS80, p62 pS28, and KHK-C. The P values were calculated using the log-rank test. (H) The mechanism underlying differentiated responses of normal hepatocytes and HCC cells to oxidative stress. Oxidative stress results in AMPK-mediated phosphorylation of KHK-A S80, primarily expressed in HCC cells, but not of KHK-C, primarily expressed in normal hepatocytes. S80-phosphorylated KHK-A acts as a protein kinase and phosphorylates p62 S28, resulting in p62 oligomerization, p62 and Keap1 aggregation, and nuclear translocation and activation of Nrf2, which lead to counteracting ROS production, maintaining tumor cell survival, and promoting HCC development. In contrast, normal hepatocytes, lacking KHK-A–mediated antioxidative response, have high levels of ROS production and increased apoptosis. Ub, ubiquitin.
To determine the clinical relevance of our findings, we performed IHC analysis of 30 primary HCC samples and their adjacent normal tissues. We revealed that the levels of KHK-A S80 phosphorylation, p62 S28 phosphorylation, and nuclear Nrf2 were markedly higher in the HCC samples than in their adjacent normal tissues (Fig. 6E and fig. S10I) and were correlated with each other in 90 HCC samples (Fig. 6F and fig. S10J). Notably, p62 S28 phosphorylation levels were correlated with KHKA S80 phosphorylation levels but not with p62 expression levels, suggesting that increased p62 phosphorylation is not secondary to high levels of total p62 in the HCC tissues (Fig. 6F). In addition, we found that KHK-A and KHK-C expression levels were inversely correlated (Fig. 6F).
We next assessed the survival durations for the 90 patients with HCC, all of whom had undergone standard surgical resection of their tumors. High levels of KHK-A S80 phosphorylation and p62 S28 phosphorylation in the HCC samples were correlated with shorter patient overall survival durations, whereas high levels of KHK-C expression were correlated with better survival of patients (Fig. 6G). These results support a critical role of KHK-A–mediated p62 S28 phosphorylation in the clinical aggressiveness of human HCC.
DISCUSSION
p62, which is instrumental for HCC induction and serves as a prognostic indicator of rapid HCC recurrence after curative ablation, is required for activation of Nrf2 for counteracting oxidative stress (28). This study has revealed several key aspects of the mechanism underlying p62 oligomerization, which is essential for its functions. We demonstrated here that oxidative stress resulting from hypoxia and H2O2 stimulation induced differential responses from normal hepatocytes and HCC cells, which express KHK-C and KHK-A, respectively (32); the cancer cells exhibited much lower ROS production and NADP+/NADPH ratio and higher GSH/GSSG ratio and survival rates than their normal counterparts. Mechanistic studies showed that oxidative stress induced the binding of AMPK to KHK-A, but not KHK-C, and subsequent AMPK-mediated phosphorylation of KHK-A at S80, which is encoded by KHK-A unique exon 3A. The phosphorylated KHK-A interacted with p62. KHK-A acted as a protein kinase and phosphorylated p62 at S28 in the PB1 domain, and this phosphorylation blocked the binding of TRIM21 to p62 and promoted p62 oligomerization, which enhanced p62 aggregation with Keap1 and nuclear translocation and activation of Nrf2, thereby leading to enhanced Nrf2-regulated expression of genes that counteracted ROS production and promoted cell survival and HCC development in mice (Fig. 6H).
KHK, whose KHK-C isoform is primarily expressed in normal hepatocytes, is a metabolic enzyme that was originally characterized as a kinase that phosphorylates fructose (32). HCC-specific splicing of the adjacent exons 3C and 3A of the KHK gene leads to KHK-A expression in cancer cells. KHK-A, which loses its affinity to bind and phosphorylate fructose, gains the ability to function as a protein kinase (32). Under normoxic conditions, KHK-A–mediated PRPS1 phosphorylation promotes de novo nucleotide synthesis for HCC cell proliferation (31, 32). Upon hypoxic stress, HCC cells switch from anabolic nucleotide synthesis to antioxidative response to counteract hypoxic stress by inhibition of KHK-A–mediated PRPS1 phosphorylation and increase of KHK-A–mediated p62 phosphorylation. Thus, we here revealed an important function of KHK-A in directly phosphorylating p62 and provided a critical mechanism underlying cancer-specific antioxidative stress, which is essential for HCC development. The clinical significance of the KHK-A–mediated p62 phosphorylation is evidenced by higher levels of KHK-A S80 phosphorylation, p62 S28 phosphorylation, and nuclear accumulation of Nrf2 in HCC than in normal liver tissues; by the positive correlation among these molecular markers in human HCC specimens; and by the positive correlation of KHK-A S80 and p62 S28 phosphorylation with poor prognosis of patients with HCC. The discovery that KHK-A is a protein kinase that regulates antioxidative stress provides insight into the regulation of a redox balance in tumor cells and yields a novel and promising therapeutic target for cancer treatment.
MATERIALS AND METHODS
Materials
Rabbit polyclonal antibodies against KHK-A and KHK-C and KHK-A pS80 and p62 pS28 antibodies were obtained from Signalway Biotechnology (Pearland, TX). The antibody generation procedures were described previously (32). Normal mouse immunoglobulin G (IgG) (sc-2025), TRIM21 (sc-25351), GST (sc-138), Nrf2 (sc-13032), tubulin (sc-8035), ERK1/2 (sc-514302), pERK1/2 (Thr202/Tyr204) (sc-16982), PRPS1/2 (sc-100822), and pan–KHK-A/C (sc-377411) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit antibodies against mitogen-activated protein kinase-activated protein kinase 2 (MAPK/APK2) (#3042), MAPK/APK2 pT222 (#3316), c-Jun (#9165), c-Jun pS73 (#9164), Trx1 (#2429), AMPKα (#5831), AMPKα pT172 (#2535), ACC1 (#3676), ACC1 pS79 (#11818), LC3B (#3868), cleaved PARP (Asp214) (#9541), and anti-rabbit IgG conformation-specific secondary antibody (L27A9) (#3678) were purchased from Cell Signaling Technology (Danvers, MA). BSO (B2515), NAC (A7250), Trolox (238813), MG132 (M7449), mouse monoclonal anti-Flag (F1804), rabbit anti-Flag (F7425), and anti-His (SAB1305538) antibodies were purchased from Sigma-Aldrich (St. Louis, MO). Anti-Flag M2 agarose beads and [γ-32P]ATP were purchased from MP Biochemicals (Santa Ana, CA). Active AMPK proteins (#P47-10H) were obtained from SignalChem (Richmond, BC, Canada). A769662 was purchased from Tocris Bioscience (Avonmouth, Bristol, UK). Rabbit anti-Ki67 antibody (#AB9260) and compound C were obtained from Millipore (Billerica, MA). Anti-Keap1 (ab226997) and anti-p62 (ab56416) antibodies were obtained from Abcam (Cambridge, MA). Ub-K63 antibody (HWA4C4) was purchased from Novus (Littleton, CO). Anti–proliferating cell nuclear antigen (PCNA) (#610665) and hypoxia-inducible factor 1α (#610958) antibodies were obtained from BD Biosciences (Bedford, MA). An anti-PRPS1 T225 phosphorylation antibody was generated as previously described (32). U0126, SP600125, and SB203580 were purchased from EMD Biosciences (San Diego, CA). A fluorescein isothiocyanate (FITC)–labeled antibody against 5-bromo-2′-deoxyuridine (BrdU) (BU20A, 11-5071) was purchased from eBioscience.
DNA construction and mutagenesis
PCR-amplified human p62 was cloned into pcDNA3.1/hygro(+)-Flag, pcDNA3-HA, or pColdI (His) vector. Human AMPKα1 was cloned into pcDNA3-HA vector. The construction of Flag/V5–rKHK-A or Flag/V5–rKHK-C, GST–KHK-A or GST–KHK-C, Flag-rKHK G257R, His-PRPS1, and KHK short hairpin RNA (shRNA) was described previously (32). Nrf2 CA (with the deletion of amino acids 1 to 89) was cloned into pcDNA3.1/puro(+)-HA. PINK1 and SQSTM1 shRNA were constructed via ligation of an oligonucleotide targeting human PINK1 (5′-GCTGGAGGAGTATCTGATAGG-3′) and p62 (5′-ACTGGACCCATCTGTCTTCAA-3′) into an Xho I–/Mlu I–digested pGIPZ vector, respectively. Flag-, V5-, or GST-tagged rKHK-A S80A, Flag–rKHK-A S80E, Flag-p62 S28E, Flag-p62 S28A, His–p62 S28A, Flag–p62 K7R, and all the shRNA-resistant p62 constructs containing nonsense mutations of C1017T, T1020C, and G1023A were constructed using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). Vectors expressing GFP-LC3, HA-ubiquitin-WT, HA-ubiquitin-K48, and HA-ubiquitin-K63 were purchased from Addgene.
Cell lines and cell culture conditions
LO2, Hep3B, and Huh7 cells were obtained from the American Type Culture Collection and were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% dialyzed bovine calf serum (HyClone). AMPKα1 and AMPKα2 double-knockout MEFs (a gift from B. Viollet, Institut Cochin, Paris, France) were maintained in DMEM supplemented with 10% fetal bovine serum (FBS). No cell lines used in this study were found in the database of commonly misidentified cell lines maintained by the International Cell Line Authentication Committee and National Center for Biotechnology Information BioSample. Cell lines were authenticated by short tandem repeat profiling and were routinely tested for mycoplasma contamination. Cells were plated at a density of 4 × 105 per 60-mm dish or 1 × 105 per well of a six-well plate 18 hours before transfection. The transfection procedure was performed as previously described (36).
Immunoprecipitation and immunoblotting analysis
The extraction of proteins using a modified buffer from cultured cells was followed by immunoprecipitation and immunoblotting using corresponding antibodies, as described previously (37).
GST pulldown assay
Equal amounts of purified His-tagged protein (200 ng per sample) were incubated with GST fusion proteins (100 ng) on glutathione (GSH) agarose beads in a modified binding buffer [50 mM tris-HCl at pH 7.5, 1% Triton X-100, 150 mM NaCl, 1 mM dithiothreitol (DTT), 0.5 mM EDTA, 100 μM phenylmethylsulfonyl fluoride, 100 μM leupeptin, 1 μM aprotinin, 100 μM sodium orthovanadate, 100 μM sodium pyrophosphate, and 1 mM sodium fluoride]. The glutathione agarose beads were then washed four times with binding buffer and then subjected to immunoblotting analysis, as previously described (38).
Purification of recombinant proteins
WT His-p62, His-p62 S80A, GST–KHK-A, GST–KHK-C, GST–KHK-A S80A, and GST–KHK-A G257R were expressed in bacteria and purified, as described previously (39).
In vitro kinase assay
The kinase reactions were performed, as described previously (40). Briefly, purified active AMPK proteins (including AMPKα1/β1/γ1) were incubated with purified GST-tagged KHK-A, KHK-C, or GST–KHK-A S80A mutant (100 ng) in 25 μl of kinase buffer {50 mM tris-HCl (pH 7.5), 100 mM KCl, 50 mM MgCl2, 1 mM Na3VO4, 1 mM DTT, 5% glycerol, 0.2 mM AMP, 0.5 mM ATP, and 10 μCi [γ-32P]ATP} at 25°C for 1 hour. To test p62 phosphorylation in vitro, KHK-A or KHK-C on the glutathione agarose beads was washed in phosphate-buffered saline (PBS) five times after in vitro AMPK kinase assay and then incubated with or without bacterially purified His-p62 or His–p62 S28A in the presence of [γ-32P]ATP. The reaction was terminated by adding SDS–polyacrylamide gel electrophoresis loading buffer and heating at 100°C for 5 min.
MS analysis
In vitro KHK-A phosphorylation by AMPK and p62 phosphorylation by KHK-A were performed. The protein samples were digested and analyzed by LC-MS/MS on an Orbitrap-Elite mass spectrometer (Thermo Fisher Scientific, Waltham, MA), as described previously (41).
CRISPR-Cas9–mediated genome editing
Genomic mutations were introduced into cells using the CRISPR-Cas9 system, as described previously (41). Single-guide RNAs (sgRNAs) were designed to target the genomic area adjacent to the KHK-A S80A and p62 S28A mutation sites using the CRISPR design tool (http://crispr.mit.edu/). The annealed guide RNA oligonucleotides were inserted into a PX458 vector (Addgene, Cambridge, MA) digested with the Bbs I restriction enzyme (42). Cells were seeded at 60% confluence, followed by cotransfection of sgRNAs (0.5 μg) and single-stranded donor oligonucleotide (ssODN) (10 pmol) as a template to introduce mutations. Twenty-four hours after transfection, cells were trypsinized and diluted so that cells in single were seeded into 96-well plates. Genomic DNA was extracted from GFP-positive cells, followed by sequencing of the PCR products spanning the mutation sites. sgRNA targeting sequence for KHK-A S80A: 5′-TGACCTCCGCCGCTATTCTG-3′, ssODN sequence for KHK-A S80A: 5′-CCTCCAGTCCCCAAAACCCTTGTCGAACTGCACCCCCTTCGGGTTACTCCGCCCTAGCAGTTTTGTCCTGGATGACCTCCGaCGaTAcgCTGTGGACCTACGCTACACAGTCTTTCAGACCACAGGCTCCGTCCCCATCGCCACGGTCATCATCAACGAGGCC-3′, sgRNA targeting sequence for p62 S28A: 5′-CTGCTGCAGCCCCGAGCCTG-3′, and ssODN sequence for p62 S28A: 5′-GCGTCGCTCACCGTGAAGGCCTACCTTCTGGGCAAGGAGGACGCGGCGCGCGAGATTCGCCGCTTCAGCTTCTGCTGCgcCCCaGAaCCaGAGGCGGAAGCCGAGGCTGCGGCGGGTCCGGGACCCTGCGAGCGGCTGCTGAGCCGGGTGGCCGCCCTGTTCC-3′.
Genotyping was performed by sequencing PCR products amplified from the following primers: KHK-A forward: 5′-TTAGGACTGGGAGGGACTGA-3′, KHK-A reverse: 5′-GTGAGGGAACTGATTGAGCC-3′, p62 forward: 5′-CGACCTAGCAGCCTCCTGA-3′, and p62 reverse: 5′-CCTTGGTCACCACTCCAGTCA-3′.
Measurement of intracellular NADPH and NADP+ levels
The intracellular levels of NADPH and total NADP (which means NADPH and NADP+) were measured according to previously described methods (43, 44). Briefly, 2 × 106 cells were lysed in 400 μl of extraction buffer containing 20 mM nicotinamide, 20 mM NaHCO3, and 100 mM Na2CO3 and centrifuged for 20 min using maximum speed. The supernatant was kept on ice in the dark until use. To extract NADPH, 150 μl of the supernatant was incubated at 60°C for 30 min because heating destroys the oxidized form (NADP+) while having no effect on the reduced form (NADPH). Next, 160 μl of NADP-cycling buffer [100 mM tris-HCl (pH 8.0), 0.5 mM thiazolyl blue tetrazolium blue (MTT), 2 mM phenazine ethosulfate, and 5 mM EDTA] containing glucose-6-phosphate dehydrogenase (1.3 U) was added to a 96-well plate containing 20 μl of sample. After a 1-min incubation in the dark at 30°C, 20 μl of glucose 6-phosphate (10 mM) was added to the mixture, and the change in absorbance at 570 nm was measured every 30 s for 4 min at 30°C with a microplate reader. The concentration of NADP+ was calculated by subtracting NADPH from the total NADP.
Measurement of intracellular ROS
The fluorescence probe 2′,7′-dichlorofluorescin diacetate (DCFDA) (Abcam) was used to measure intracellular ROS following the manufacturer’s instructions. Briefly, 2 × 104 cells were seeded in a clear-bottom 96-well plate. After treatment, cells were washed with Hanks’ balanced salt solution, followed by incubation with DCFDA (25 μM)–containing HBSS for 30 min at 37°C in the dark. Cells were washed, and green fluorescence was measured with a Synergy HT microplate reader (BioTek) at 485/535 nm.
GSH/GSSG detection
GSH and GSSG concentrations were measured by a GSH/GSSG detection kit (catalog no. S0053), according to the manufacturer’s instructions (Beyotime).
ARE-luciferase assay
Three copies of the ARE (5′-GTGACAAAGCAATCCCGTGACAAAGCAATCCCGTGACAAAGCAATA-3′) were cloned into a pGL3-basic luciferase reporter plasmid. The same amounts of ARE-luciferase reporter together with KHK plasmids were transfected into cells. Each transfection included the same amount of Renilla, which was used to normalize the transfection efficiency. Twenty-four hours after transfection, cells were treated with or without hypoxia for 6 hours. The luciferase activity in cell lysates was measured with the luciferase assay system as previously described (26).
Immunofluorescence analysis
Cells were fixed and incubated with primary antibodies at a dilution of 1:100, fluorescence dye–conjugated secondary antibodies, and 4′,6-diamidino-2-phenylindole (DAPI), according to standard protocols. Immunofluorescent microscopic images of the cells were obtained and viewed with an IX81 confocal microscope system (Olympus America).
Subcellular fractionation
Nuclear fractions were isolated from cells using a nuclei isolation kit (NUC201-1KT) according to the manufacturer’s instructions (Sigma-Aldrich).
Real-time PCR
Total RNA was extracted from cells and tissue specimens using TRIzol according to the manufacturer’s instructions (Invitrogen). A total of 1 mg per sample RNA was used for complementary DNA synthesis using a TaqMan Reverse Transcription Reagents kit (Applied Biosystems). Quantitative real-time PCR (qRT-PCR) was carried out in the 7500 real-time PCR system (Applied Biosystems) using an SYBR Premix Ex Taq kit (TAKARA). The following primers were used for qRT-PCR: GSTM1, 5′-TTTGTCCTGCCCACGTTTCT-3′ and 5′-TCAAAGTCGGGAGCGTCAC-3′; HO-1, 5′-GGTCAGGTGTCCAGAGAAGG-3′ and 5′-CTTCCAGGGCCGTGTAGATA-3′; PGD, 5′-AAAGATCCGGGACAGTGCT-3′ and 5′-CACCGAGCAAAGACAGCTT-3′; GCLC, 5′-GTGGACGAGTGCAGCAAG-3′ and 5′-GTCCAGGAAATACCCCTTCC-3′; NQO1, 5′-ATCCTGCCGAGTCTGTTCTG-3′ and 5′-AGGGACTCCAAACCACTGC-3′; UGT1A1, 5′-AACAAGGAGCTCATGGCCTCC-3′ and 5′-GTTCGCAAGATTCGATGGTCG-3′; and glyceraldehyde-3-phosphate dehydrogenase, 5′-AGCCACATCGCTCAGACAC-3′ and 5′-GCCCAATACGACCAATCC-3′.
DSP cross-linking
Cells with less than 80% confluence were washed twice with ice-cold washing buffer (10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl, 0.1 mM CaCl2, and 1 mM MgCl2) and incubated in washing buffer with DSP (0.4 mg/ml) at 4°C for 2 hours. Cells were lysed in immunoprecipitation lysis buffer with 1% SDS and sonicated for 10 s. For immunoprecipitation, the lysate was diluted with immunoprecipitation lysis buffer at a ratio of 1:10 and subjected to immunoprecipitation assay.
KHK activity assay and 13C-labeled F1P detection
KHK activity was measured by a method described previously (32). We added Roche cOmplete Protease Inhibitor Cocktail and Roche PhosSTOP to the reaction mixture containing 140 mg of protein of Huh7 cell lysates and incubated the mixture at 37°C for 3 hours. Spectrophotometric absorbance measurements of the remaining fructose were detected in 96-well plates at 515 nm using a SpectraMax plate reader (Molecular Devices).
The culture media of Huh7 cells were supplied with 13C-labeled fructose for 6 hours, and then the cells were washed with PBS and whole-cell culture plates were snap-frozen on dry ice. Metabolites were extracted twice from plates with 70% ethanol (75°C). Targeted analysis 13C-labeled F1P pattern was performed by ion-pairing, reverse-phase, ultra-performance LC-MS/MS on a Thermo Quantum Ultra instrument (Thermo Fisher Scientific), as previously described (32).
Apoptosis analysis
Cells (1 × 105) were seeded into a six-well plate overnight. Cells were then fixed by direct addition of 12% formaldehyde into the culture medium. After fixation, the cells were stained with DAPI (1 μg/ml) for 5 min and washed by PBS. The cells with condensed and/or fragmented chromatin were indicative of apoptosis and counted.
Cell viability analysis
Cells (2 × 105) were plated in DMEM with 10% FBS. The viable cells were stained with trypan blue (0.5%) and counted using Beckman Coulter.
Cell proliferation assay
Cells were incubated with BrdU (50 μM) for 1 hour before harvest. Collected cells were fixed in 70% ethanol at 4°C for 1 hour, followed by incubation with 2 N HCl/0.5% Triton X-100 for 30 min and 0.1 M borate sodium for 2 min. After incubation with an anti–BrdU-FITC antibody (eBioscience) and washing five times in PBS, the BrdU incorporation rate was analyzed by flow cytometry.
Terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling assay
Mouse tumor tissues were sectioned at 5 μm thickness. Apoptotic cells were counted using the DeadEnd Colorimetric TUNEL System (Promega) according to the manufacturer’s instructions.
IHC analysis and histologic evaluation of human HCC specimens
Mouse tumor samples were fixed and prepared for staining. The samples were stained with Mayer’s hematoxylin and eosin (H&E) (BioGenex). The slides were then mounted using Universal Mount (Research Genetics).
Liquid nitrogen–frozen human HCC and adjacent matched nontumor tissue samples from 90 patients were obtained from Eastern Hepatobiliary Surgery Hospital in Shanghai, People’s Republic of China. The use of human HCC specimens and the database was approved by the Hospital’s Institutional Review Board. All tissue samples were collected in compliance with informed consent policy. Sections of paraffin-embedded human HCC tissue were stained with antibodies against Ki67, phospho–KHK-A S80, phospho-p62 S28, Nrf2, or nonspecific IgG as a negative control. The tissue sections were quantitatively scored according to the percentage of positive cells and staining intensity, as described previously (41). The following proportion scores were assigned to the sections: 0 if 0% of the tumor cells exhibited positive staining, 1 for 0 to 1%, 2 for 2 to 10%, 3 for 11 to 30%, 4 for 31 to 70%, and 5 for 71 to 100%. In addition, the staining intensity was rated on a scale of 0 to 3: 0, negative; 1, weak; 2, moderate; and 3, strong. The proportion and intensity scores were then combined to obtain a total score (range, 0 to 8), as described previously (41). Scores were compared with overall survival duration, defined as the time from date of diagnosis to death or last known follow-up examination. All patients had received standard therapies after surgery.
Animal studies
One million Huh7 cells or Huh7 cells with knock-in of KHK-A S80A or p62 S28A were collected in 20 μl of DMEM with 33% Matrigel and intrahepatically injected into livers of 6-week-old male BALB/c athymic nude mice. The injections were performed as described previously (32). Seven mice per group in each experiment were used. Animals were euthanized 28 days after injection. The liver of each mouse was dissected, fixed in 4% formaldehyde, and embedded in paraffin. Tumor formation and phenotype were determined by histologic analysis of H&E-stained sections. The tumor volume was calculated using the formula V = 1/2a2b (V, volume; a, shortest diameter; b, longest diameter). The animals were treated in accordance with relevant institutional and national guidelines and regulations. The use of the animals was approved by the Institutional Review Board at MD Anderson Cancer Center. Animals arriving in our facility were randomly put into cages with five mice each. No statistical method was used to predetermine sample size. The investigators were not blinded to allocation during experiments and outcome assessment.
Statistics and reproducibility
The mean values obtained in the control and experimental groups were analyzed for significant differences. Pairwise comparisons were performed using a two-tailed Student t test. P values less than 0.05 were considered significant.
Supplementary Material
Acknowledgments
We thank M.-C. Hung (MD Anderson Cancer Center) for the Nrf2-luciferase report plasmids. We thank L. Li (The University of Texas Health Science Center at Houston) for technical assistance and S. Patterson (MD Anderson Cancer Center) for critically reading this manuscript. The MS was supported in part by the Clinical and Translational Proteomics Service Center at the University of Texas Health Science Center. Funding: This work was supported by National Cancer Institute grants R01 CA204996 (to Z.L.), MD Anderson Cancer Center Support Grant CA016672, the Key Discipline of Zhejiang Province in Medical Technology (First Class, Category A; to J.L.), the key Science and Technology Innovation Team of Zhejiang Province in Laboratory Medicine 2010R50048 (to J.L.), and the Odyssey Fellowship from The University of Texas MD Anderson Cancer Center (to D.X.). Author contributions: Z.L., D.X., J.L., and H.W. conceived and designed the study. D.X., X.L., F.S., X.Q., Z.W., J.L., Y.X., L.D., and Y.Z. performed the experiments. Z.L. wrote the paper. All authors commented on the paper. Competing interests: Z.L. owns shares in Signalway Biotechnology (Pearland, TX), which supplied rabbit antibodies that recognize KHK-A pS80 and p62 pS28. Z.L.’s interest in this company had no bearing on its being chosen to supply these reagents. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/4/eaav4570/DC1
Fig. S1. KHK-A is required for oxidative stress–enhanced p62 oligomerization.
Fig. S2. KHK-A is required for Nrf2 activation upon oxidative stress.
Fig. S3. AMPK phosphorylates KHK-A and promotes the association between KHK-A and p62.
Fig. S4. AMPK-mediated KHK-A S80 phosphorylation inhibits the interaction between KHK-A and PRPS1 and PRPS1 T225 phosphorylation.
Fig. S5. KHK-A acts as a protein kinase and phosphorylates p62 at S28.
Fig. S6. KHK-A is required for oxidative stress–induced but not proteasomal stress−induced p62 aggregation.
Fig. S7. KHK-A–mediated p62 S28 phosphorylation is required for oxidative stress–enhanced p62 oligomerization and Nrf2 activation.
Fig. S8. KHK-A mediated p62 S28 phosphorylation reduces ROS production and promotes cancer cell survival without altering autophagy initiation.
Fig. S9. The phosphorylation-mimicking KHK-A S80E and p62 S28E mutations promote p62 oligomerization and Nrf2 activation.
Fig. S10. KHK-A–mediated p62 S28 phosphorylation promotes hepatocellular tumorigenesis and is associated with the clinical aggressiveness of human HCC.
REFERENCES AND NOTES
- 1.Cairns R. A., Harris I. S., Mak T. W., Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Holmstrom K. M., Finkel T., Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 15, 411–421 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Gorrini C., Harris I. S., Mak T. W., Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12, 931–947 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Sporn M. B., Liby K. T., NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 12, 564–571 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X.-J., Sun Z., Villeneuve N. F., Zhang S., Zhao F., Li Y., Chen W., Yi X., Zheng W., Wondrak G. T., Wong P. K., Zhang D. D., Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 29, 1235–1243 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang T., Chen N., Zhao F., Wang X.-J., Kong B., Zheng W., Zhang D. D., High levels of Nrf2 determine chemoresistance in type II endometrial cancer. Cancer Res. 70, 5486–5496 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeNicola G. M., Karreth F. A., Humpton T. J., Gopinathan A., Wei C., Frese K., Mangal D., Yu K. H., Yeo C. J., Calhoun E. S., Scrimieri F., Winter J. M., Hruban R. H., Iacobuzio-Donahue C., Kern S. E., Blair I. A., Tuveson D. A., Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inami Y., Waguri S., Sakamoto A., Kouno T., Nakada K., Hino O., Watanabe S., Ando J., Iwadate M., Yamamoto M., Lee M.-S., Tanaka K., Komatsu M., Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 193, 275–284 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaramillo M. C., Zhang D. D., The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 27, 2179–2191 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fukutomi T., Takagi K., Mizushima T., Ohuchi N., Yamamoto M., Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol. Cell. Biol. 34, 832–846 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harder B., Jiang T., Wu T., Tao S., de la Vega M. R., Tian W., Chapman E., Zhang D. D., Molecular mechanisms of Nrf2 regulation and how these influence chemical modulation for disease intervention. Biochem. Soc. Trans. 43, 680–686 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adams V., Schieber A., McCabe E. R., Hexokinase autophosphorylation: Identification of a new dual specificity protein kinase. Biochem. Med. Metab. Biol. 53, 80–86 (1994). [DOI] [PubMed] [Google Scholar]
- 13.Johansen T., Lamark T., Selective autophagy mediated by autophagic adapter proteins. Autophagy 7, 279–296 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rogov V., Dotsch V., Johansen T., Kirkin V., Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 53, 167–178 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Moscat J., Diaz-Meco M. T., p62: A versatile multitasker takes on cancer. Trends Biochem. Sci. 37, 230–236 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.White E., Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 12, 401–410 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rojo de la Vega M., Chapman E., Zhang D. D., NRF2 and the hallmarks of cancer. Cancer Cell 34, 21–43 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Komatsu M., Kurokawa H., Waguri S., Taguchi K., Kobayashi A., Ichimura Y., Sou Y.-S., Ueno I., Sakamoto A., Tong K. I., Kim M., Nishito Y., Iemura S.-i., Natsume T., Ueno T., Kominami E., Motohashi H., Tanaka K., Yamamoto M., The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 12, 213–223 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Lau A., Wang X.-J., Zhao F., Villeneuve N. F., Wu T., Jiang T., Sun Z., White E., Zhang D. D., A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 30, 3275–3285 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fan W., Tang Z., Chen D., Moughon D., Ding X., Chen S., Zhu M., Zhong Q., Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy 6, 614–621 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitsuishi Y., Taguchi K., Kawatani Y., Shibata T., Nukiwa T., Aburatani H., Yamamoto M., Motohashi H., Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22, 66–79 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Moscat J., Karin M., Diaz-Meco M. T., p62 in Cancer: Signaling adaptor beyond autophagy. Cell 167, 606–609 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katsuragi Y., Ichimura Y., Komatsu M., p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 282, 4672–4678 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Hayes J. D., Dinkova-Kostova A. T., The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 39, 199–218 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Wilson M. I., Gill D. J., Perisic O., Quinn M. T., Williams R. L., PB1 domain-mediated heterodimerization in NADPH oxidase and signaling complexes of atypical protein kinase C with Par6 and p62. Mol. Cell 12, 39–50 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Pan J. A., Sun Y., Jiang Y. P., Bott A. J., Jaber N., Dou Z., Yang B., Chen J. S., Catanzaro J. M., du C., Ding W. X., Diaz-Meco M. T., Moscat J., Ozato K., Lin R. Z., Zong W. X., TRIM21 ubiquitylates SQSTM1/p62 and suppresses protein sequestration to regulate redox homeostasis. Mol. Cell 62, 149–151 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Ichimura Y., Waguri S., Sou Y. S., Kageyama S., Hasegawa J., Ishimura R., Saito T., Yang Y., Kouno T., Fukutomi T., Hoshii T., Hirao A., Takagi K., Mizushima T., Motohashi H., Lee M. S., Yoshimori T., Tanaka K., Yamamoto M., Komatsu M., Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 51, 618–631 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Umemura A., He F., Taniguchi K., Nakagawa H., Yamachika S., Font-Burgada J., Zhong Z., Subramaniam S., Raghunandan S., Duran A., Linares J. F., Reina-Campos M., Umemura S., Valasek M. A., Seki E., Yamaguchi K., Koike K., Itoh Y., Diaz-Meco M. T., Moscat J., Karin M., p62, Upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed hcc-initiating cells. Cancer Cell 29, 935–948 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu Z., Hunter T., Metabolic kinases moonlighting as protein kinases. Trends Biochem. Sci. 43, 301–310 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X., Egervari G., Wang Y., Berger S. L., Lu Z., Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat. Rev. Mol. Cell Biol. 19, 563–578 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X., Qian X., Lu Z., Fructokinase A acts as a protein kinase to promote nucleotide synthesis. Cell Cycle 15, 2689–2690 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li X., Qian X., Peng L.-X., Jiang Y., Hawke D. H., Zheng Y., Xia Y., Lee J.-H., Cote G., Wang H., Wang L., Qian C.-N., Lu Z., A splicing switch from ketohexokinase-C to ketohexokinase-A drives hepatocellular carcinoma formation. Nat. Cell Biol. 18, 561–571 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shao D., Oka S.-i., Liu T., Zhai P., Ago T., Sciarretta S., Li H., Sadoshima J., A redox-dependent mechanism for regulation of AMPK activation by Thioredoxin1 during energy starvation. Cell Metab. 19, 232–245 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao J., Li M., Qin S., Zhang T., Jiang S., Hu Y., Deng Y., Zhang C., You D., Li H., Mu D., Zhang Z., Jiang C., Cytosolic PINK1 promotes the targeting of ubiquitinated proteins to the aggresome-autophagy pathway during proteasomal stress. Autophagy 12, 632–647 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shanmugam G., Narasimhan M., Tamowski S., Darley-Usmar V., Rajasekaran N. S., Constitutive activation of Nrf2 induces a stable reductive state in the mouse myocardium. Redox Biol. 12, 937–945 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu D., Wang Z., Zhang Y., Jiang W., Pan Y., Song B.-L., Chen Y., PAQR3 modulates cholesterol homeostasis by anchoring Scap/SREBP complex to the Golgi apparatus. Nat. Commun. 6, 8100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu Z., Liu D., Hornia A., Devonish W., Pagano M., Foster D. A., Activation of protein kinase C triggers its ubiquitination and degradation. Mol. Cell. Biol. 18, 839–845 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu D.-Q., Wang Z., Wang C.-Y., Zhang D.-Y., Wan H.-D., Zhao Z.-L., Gu J., Zhang Y.-X., Li Z.-G., Man K.-Y., Pan Y., Wang Z.-F., Ke Z.-J., Liu Z.-X., Liao L.-J., Chen Y., PAQR3 controls autophagy by integrating AMPK signaling to enhance ATG14L-associated PI3K activity. EMBO J. 35, 496–514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xia Y., Wang J., Liu T.-J., Yung W. K. A., Hunter T., Lu Z., c-Jun downregulation by HDAC3-dependent transcriptional repression promotes osmotic stress-induced cell apoptosis. Mol. Cell 25, 219–232 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qian X., Li X., Tan L., Lee J.-H., Xia Y., Cai Q., Zheng Y., Wang H., Lorenzi P. L., Lu Z., Conversion of PRPS hexamer to monomer by AMPK-mediated phosphorylation inhibits nucleotide synthesis in response to energy stress. Cancer Discov. 8, 94–107 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li X., Jiang Y., Meisenhelder J., Yang W., Hawke D. H., Zheng Y., Xia Y., Aldape K., He J., Hunter T., Wang L., Lu Z., Mitochondria-translocated PGK1 functions as a protein kinase to coordinate glycolysis and the TCA cycle in tumorigenesis. Mol. Cell 61, 705–719 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sander J. D., Joung J. K., CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 32, 347–355 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeon S.-M., Chandel N. S., Hay N., AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485, 661–665 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wagner T. C., Scott M. D., Single extraction method for the spectrophotometric quantification of oxidized and reduced pyridine nucleotides in erythrocytes. Anal. Biochem. 222, 417–426 (1994). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/4/eaav4570/DC1
Fig. S1. KHK-A is required for oxidative stress–enhanced p62 oligomerization.
Fig. S2. KHK-A is required for Nrf2 activation upon oxidative stress.
Fig. S3. AMPK phosphorylates KHK-A and promotes the association between KHK-A and p62.
Fig. S4. AMPK-mediated KHK-A S80 phosphorylation inhibits the interaction between KHK-A and PRPS1 and PRPS1 T225 phosphorylation.
Fig. S5. KHK-A acts as a protein kinase and phosphorylates p62 at S28.
Fig. S6. KHK-A is required for oxidative stress–induced but not proteasomal stress−induced p62 aggregation.
Fig. S7. KHK-A–mediated p62 S28 phosphorylation is required for oxidative stress–enhanced p62 oligomerization and Nrf2 activation.
Fig. S8. KHK-A mediated p62 S28 phosphorylation reduces ROS production and promotes cancer cell survival without altering autophagy initiation.
Fig. S9. The phosphorylation-mimicking KHK-A S80E and p62 S28E mutations promote p62 oligomerization and Nrf2 activation.
Fig. S10. KHK-A–mediated p62 S28 phosphorylation promotes hepatocellular tumorigenesis and is associated with the clinical aggressiveness of human HCC.