

The parasitic marine dinoflagellate Amoebophrya is the first known eukaryote with aerobic mitochondria without a genome.
Abstract
Dinoflagellates are microbial eukaryotes that have exceptionally large nuclear genomes; however, their organelle genomes are small and fragmented and contain fewer genes than those of other eukaryotes. The genus Amoebophrya (Syndiniales) comprises endoparasites with high genetic diversity that can infect other dinoflagellates, such as those forming harmful algal blooms (e.g., Alexandrium). We sequenced the genome (~100 Mb) of Amoebophrya ceratii to investigate the early evolution of genomic characters in dinoflagellates. The A. ceratii genome encodes almost all essential biosynthetic pathways for self-sustaining cellular metabolism, suggesting a limited dependency on its host. Although dinoflagellates are thought to have descended from a photosynthetic ancestor, A. ceratii appears to have completely lost its plastid and nearly all genes of plastid origin. Functional mitochondria persist in all life stages of A. ceratii, but we found no evidence for the presence of a mitochondrial genome. Instead, all mitochondrial proteins appear to be lost or encoded in the A. ceratii nucleus.
INTRODUCTION
Alveolates are a highly diverse group of eukaryotes, comprising three diverse phyla—dinoflagellates, apicomplexans, and ciliates—as well as a growing number of less-studied lineages, such as colponemids, chromopodellids, and perkinsids (1, 2). Dinoflagellates include phototrophs, heterotrophs, mixotrophs, and parasites, which are characterized by chromosomes that are permanently condensed in a liquid-crystalline state throughout the cell cycle. Recently, genes encoding histone-like proteins (3) and a non-nucleosomal DNA packaging system involving unique proteins (with closest similarity to viruses) (4) have been discovered in dinoflagellates. Dinoflagellate genomes are usually 10 to 100 times larger than the human genome (5) and exhibit several unusual features whose evolutionary origins are unclear. In addition, dinoflagellate genes are typically expressed with a conserved short spliced leader (SL) sequence that is added by trans-splicing (6).
The ancestor of dinoflagellates and apicomplexans was photosynthetic (7); however, currently, only some apicomplexan relatives Chromera and Vitrella and approximately half of the known core dinoflagellates maintain photosynthesis (8). Even photosynthetic dinoflagellates have highly reduced and fragmented plastid genomes (14 genes as compared to a typical plastid genome, which contains more than 100 genes), because most plastid genes have been transferred to the nucleus (7). Dinoflagellate and apicomplexan mitochondrial genomes are even more reduced, typically harboring only three protein-coding genes and fragments of ribosomal RNA (rRNA) genes (9, 10), which represent the minimal mitochondrial genomes in aerobic species (11). However, recent examination of the respiratory chain in the photosynthetic Chromera velia showed that oxidative phosphorylation complexes I and III were lost, leaving only two protein-coding genes (coxI and coxIII) and fragments of the rRNA genes to be encoded in the mitochondrion (11).
Several species of dinoflagellates can produce potent toxins and are able to form harmful algal blooms (HABs) that have enormous impact on ecosystem functions (12). The species of the genus Alexandrium cause prominent HABs that persist for extended time periods under favorable abiotic and biotic conditions (12). Alexandrium species produce the potent neurotoxins, saxitoxin and its derivates, which are associated with paralytic shellfish poisoning (12) and have the potential to cause serious human disease and pose economic problems for fisheries.
The dynamics of HABs can be strongly affected by parasites, most commonly parasitic syndinians and perkinsids (13). Morphological features and molecular phylogenies place both lineages outside the core dinoflagellate group, together with the free-living genera Oxyrrhis and Psammosa (1). Sequencing on one deep-branching syndinian Hematodinium revealed that the parasite likely has secondarily lost the plastid organelle (14). The Amoebophryidae (Syndinea) is an exclusively endoparasitic family that comprises a large and diverse group of primarily environmental sequences, often referred to as the marine alveolate group II (MALV-II). Amoebophryidae includes a single genus, Amoebophrya (15), with seven described species that exhibit high genetic diversity (15). Amoebophrya species can infect a high proportion of blooming Alexandrium populations (13, 16), and this infection has a direct effect on HAB formation and persistence (13).
The life cycle of Amoebophrya was described more than 40 years ago and was recently examined in detail by using electron microscopy (17). The infective free-living stage, the dinospore, has two flagella (Fig. 1). The dinospore attaches to the host cell and enters its cytoplasm, losing the flagella in the process and becoming enclosed in a parasitophorous membrane. In most cases, the parasite crosses the host nuclear envelope, losing its parasitophorous membrane in the process (17). The growing parasite starts to digest its host, increases in size, and eventually forms the so-called beehive structure as a result of several consecutive mitotic divisions. The host cell wall then breaks down and releases a short-lived vermiform stage of the parasite, which divides into hundreds of infective dinospores (18). The maturation of the parasite within the host takes 2 to 3 days and is characterized by phases of differential gene expression (19).
Fig. 1. Multiprotein phylogeny of Amoebophrya isolated from three separate hosts, 15 other dinoflagellates, and 13 related eukaryotes.
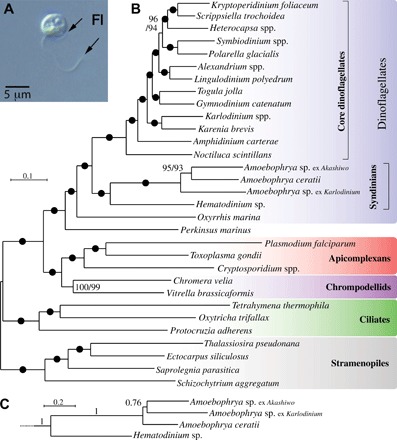
(A) Free-living stage of the parasite Amoebophrya. Fl, flagellum. (B) The best maximum likelihood tree (IQ-TREE) under the LG + G4 + I + F model with ultrafast/nonparametric bootstrap supports at branches (black circles denote 100/100 support). (C) Relationships among Amoebophrya isolates in a PhyloBayes GTR + CAT + G4 inference with posterior probabilities at branches; the rest of the tree is identical to (B) and is fully supported at all branches.
Here, we present the complete genome of Amoebophrya ceratii, a parasite of the toxin-producing species Alexandrium catenella. Examining the A. ceratii genome structure and metabolism sheds new light on the early evolution of unusual genomic characteristics in dinoflagellates and suggests that the parasite has lost its plastid organelle and its mitochondrial genome, in spite of maintaining an otherwise normal aerobic mitochondrion.
RESULTS AND DISCUSSION
Genomic characteristics and phylogenomics
Total genomic DNA (gDNA) from dinospores of A. ceratii clone AT5.2 was sequenced and assembled, and contaminant sequences were removed on the basis of identity, coverage, and GC content criteria (Materials and Methods). This resulted in 2351 A. ceratii scaffolds totaling 87.7 Mb, with an average coverage of 110-fold. This genome size is smaller than the size indicated by flow cytometry (~120 Mb; fig. S1). This size difference between assembly and flow cytometry is likely due to repetitive elements, which collapse in the assembly to small contigs. The A. ceratii genome is substantially smaller than genomes in other dinoflagellates such as Hematodinium (50 times; ~4800 Mb) (14) and Symbiodinium (15 times; ~1100 to 1500 Mb) (20–22). To the best of our knowledge, this is the smallest dinoflagellate genome reported so far. The mean GC content of A. ceratii genome was calculated to be 55.9%, which is in the range of published dinoflagellate transcriptomes (23) but relatively higher compared to Symbiodinium spp. (43.6 to 50.5%) (20–22) and Hematodinium sp. (approximately 47%) draft genomes (Table 1) (14). Gene predictions identified 19,925 protein-coding genes and 39 transfer RNAs (tRNAs). We also mapped the transcript data obtained previously (16, 19) to the scaffolds and found that 12,200 transcripts mapped to this assembly. Despite the relatively small size, by dinoflagellate standards, the genome assembly appears to be largely complete, containing 89.1% of CEGMA (core eukaryotic gene mapping approach) conserved proteins (24). This is slightly higher than the Hematodinium genome (85.9%) (14). The A. ceratii predicted proteins were clustered into 4879 families by using OrthoMCL. Of these, 499 protein clusters belonged to 12 transposon domain families (table S1), indicating a high transposon activity in the A. ceratii genome. We further searched the A. ceratii genome and found 60 general transcription factors and 46 proteins with domains corresponding to specific transcriptional regulatory factors, numbers similar to other dinoflagellates and alveolates (table S2). Although transcription factors are not abundant in dinoflagellates, they likely play an indispensable role in adapting to changing conditions, as is common in other eukaryotes. Many transcripts of dinoflagellates are trans-spliced to a 22–base pair (bp) SL sequence (6), in which individual mRNAs may be processed from larger precursors by trans-splicing and polyadenylation. The presence of such SLs in gene-coding loci indicates the potential for mRNAs to be reintegrated into the genome as intronless genes after reverse transcription (25). We examined the A. ceratii genome for SLs and traces of such reintegration events. None of the predicted gene models was associated with a full-length SL motif; however, five gene models had truncated motifs (fig. S2A and table S3A). The low frequency of SL motifs at the genomic level suggests that mRNA reintegration events are rare. Fifty-three orphan full-length SL motifs were identified across 50 scaffolds (table S3B), and another 713 truncated SL motifs with identities of 73 to 100% (table S3C) were found. In the transcriptome dataset, 70 transcripts with single SL motifs were observed (fig. S2B). Only one contig contained a second truncated SL repeat (60% identity to the consensus sequence), and no third or fourth SL repeats were identified.
Table 1. Features of A. ceratii and other dinoflagellate genomes.
CDS, coding regions. N50 measures assembly quality as a weighted median of contig length. Higher N50 values denote greater contiguity.
| A. ceratii |
Symbiodinium minutum* |
Symbiodinium kawagutii† |
Symbiodinium microadriaticum‡ |
Hematodinium sp.§ | |
| Genome | |||||
| Size (Mb) | 87.7 | ~1,500 (assembled ~616) |
~1,180 (assembled ~935.07) |
~1,100 (assembled ~808.25) |
~4,800 |
| Scaffolds (number) | 2,351 | 21,899 | 30,040 | 9,695 | 118,385 (>10 kbp) |
| GC content (%) | 55.9% | 43.6% | 43.97% | 50.5% | – |
| N50 value (kb) | 84 | 62.7 | 47.1 | 34.8 | 17.2 |
| Genes | |||||
| Predicted CDS | 19,925 | 41,925 | 36,850 | 49,109 | – |
| Average protein length (amino acids) |
643 | 1,668 | 1,041 | – | – |
| Predicted tRNAs | 39 | – | – | – | – |
| CDS with transcript data |
4,856 | 32,366 | 26,834 | 37,470 | – |
| CDS with domain annotation |
8,768 | – | – | – | – |
| Introns | |||||
| Predicted number | 51,066 | 95.3% | 61.1% | 98.2% | – |
| Median size (bases) | 184 | 499 | 893 | 504.7 | – |
| Intronless CDS | 5,464 | – | – | – | – |
A. ceratii contained 51,066 introns in 15,016 predicted genes (fig. S3). In total, 28.4% of predicted genes were intronless, more than in the Symbiodinium genomes, whereas 61.1 to 98.3% of genes have introns (Table 1). This phenomenon is observed most probably as more streamlining forces act toward maintaining the small genome size in A. ceratii, as compared to the core dinoflagellates, and their parasitic lifestyle may not favor large gene family expansions (because of their dependency on the host-prey coevolution). Mapping of RNA sequencing (RNAseq) reads (table S4) onto the A. ceratii genome revealed that genes without introns were expressed at a similar level as transcripts with introns (table S4). Our data thus suggest that gene reintegration by retroposition is rare in Amoebophrya and such events may be more common in the more complex genomes of core dinoflagellates.
The phylogenetic position of Amoebophrya with respect to other dinoflagellates has been debated, in part because only a few genes have been available for phylogenetic analysis [see (1)]. We used a concatenated set of 100 conserved nuclear proteins of three Amoebophrya species isolated from different hosts and 15 other dinoflagellates as well as 13 outgroup species to compute maximum likelihood and Bayesian phylogenies (Fig. 1). In these analyses, Amoebophrya branched before core dinoflagellates, but after Oxyrrhis marina and Perkinsus marinus, and as a specific sister group to Hematodinium (another Syndiniales parasite). This placement is in agreement with previously reported results based on concatenated ribosomal proteins (1).
Metabolic features and dependence on the host
Pathogens and parasites frequently use host resources to obtain compounds required for their own metabolism and reproduction, a relationship that often leads to losses or modifications of biosynthetic pathways in the parasite. In the A. ceratii genome, many genes encoding enzymes involved in various metabolic pathways are present in multiple copies, a common feature of dinoflagellate genomes (table S5) (26). Genes involved in amino acid biosynthesis and purine and pyrimidine biosynthesis are present in particularly high copy numbers (fig. S4 and table S5). Other prominently expanded orthologous groups include proteins involved in protein-protein or protein-carbohydrate interactions (107 proteins), carbohydrate degradation (50 proteins), and detoxification (44 proteins), which may be associated with the utilization of host-derived compounds during the infection phase of Amoebophrya (table S1).
Fatty acids are constituent building blocks for cell membranes; they act as targeting molecules to direct proteins to membranes and function as energy molecules for metabolic processes or messenger molecules, all important processes in a parasite. In photosynthetic eukaryotes, fatty acid synthesis in the plastid is carried out by a cyanobacterium-derived type II fatty acid synthase (FAS) multienzyme (27), whereas heterotrophic eukaryotes typically rely on a cytosolic multidomain type I FAS. Some apicomplexans contain both type I and type II FAS, while others have lost one or the other (27). A. ceratii has a type I FAS complex (g12138.t1) that is closely related to that of Hematodinium (14) and apicomplexans, but no plastid type II FAS enzymes were found in the A. ceratii genome (fig. S5). A. ceratii and Hematodinium also contain a type I PKS (polyketide synthase) complex (scaffold1619_size12283) (fig. S5), which is likely involved in the production of secondary metabolites and could possibly be involved in host interactions (27).
Enzymes involved in the synthesis of most amino acids were present in the A. ceratii genome (table S5), with the exception of a few individual enzymes that have likely been functionally substituted (28). This demonstrates the limited dependency of A. ceratii on its host. The shikimate pathway required for the synthesis of tyrosine, phenylalanine, and tryptophan consists of seven broadly conserved enzymes, five of which (AroB, AroA, AroK, AroD, and AroE) are fused in some eukaryotes (29). In A. ceratii, this five-domain protein is additionally fused to AroC (chorismate synthase, g6770; Fig. 2A) and all six genes are cotranscribed, a pattern not observed in any other organism to date. Moreover, the seventh enzyme of the shikimate pathway, AroG, is fused to a multifunctional tryptophan synthetase gene (g13589; Fig. 2B), which we confirmed by polymerase chain reaction (PCR) using both gDNA and complementary DNA (cDNA) templates. Gene fusions can provide a simple mechanism for concerted expression in eukaryotes. However, certain enzymes involved in converting chorismate (final product of the shikimate pathway) to tyrosine, phenylalanine, or tryptophan are not found in the A. ceratii genome (table S5). In vascular plants, approximately 20% of carbon fixed by photosynthesis is directed to the shikimate pathway, and it produces precursors not only for aromatic amino acid biosynthesis but also for various secondary metabolite pathways (30).
Fig. 2. Shikimate (g6770) and tryptophan (g13589) synthesis pathway multidomain genes.
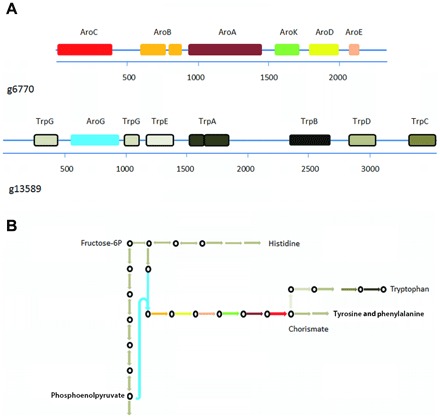
(A) Individual domains of the shikimate pathway are illustrated by colored boxes, and domains of the tryptophan pathway are represented with differently shaded gray boxes. (B) Schematic view of the biosynthetic pathway for tryptophan in A. ceratii. Circles represent intermediates that can be synthesized in A. ceratii, and arrows indicate the respective enzymatic activities. Arrows without circles indicate missing pathway components in A. ceratii. The colors for the shikimate enzymatic activities are as in (A). For simplicity, all tryptophan pathway steps are depicted in gray.
Analysis of metabolic pathways points to a loss of the plastid organelle
The ancestor of dinoflagellates and apicomplexans was an alga, and most of their current representatives are either still photosynthetic or metabolically dependent on a reduced, nonpigmented plastid (31). Plastid loss has only been shown in Cryptosporidium and Hematodinium, which have circumvented the need for plastid-derived metabolites by salvaging compounds of host origin (14). We investigated whether A. ceratii, which falls in the same lineage as Hematodinium, also lacks evidence for a relict plastid (17). We first searched for plastid metabolic genes in the A. ceratii genome (scaffolds, contigs, and gene models) by comprehensive homology searches, but we could not identify any orthologs for enzymes found in apicomplexan or dinoflagellate plastids (Materials and Methods). The synthesis of isoprenoid units is missing altogether, suggesting that A. ceratii obtains these compounds from host cells, similarly to Hematodinium. The synthesis of tetrapyrroles, fatty acids, and iron-sulfur clusters is predicted to take place in the cytosol and mitochondria (fig. S6), and in single-gene phylogenies, only one enzyme (HemD) appears to be derived from the plastidial endosymbiont (fig. S6). Unlike in typical plastids, however, the A. ceratii HemD lacks an N-terminal extension and signal and transit peptides characteristic of plastid targeting, as confirmed by the transcriptomic analysis of the 5′ gene end (Materials and Methods). This strongly indicates that HemD in A. ceratii has been relocalized to the cytosol, much like its ortholog in Hematodinium (14). Because all other enzymes for tetrapyrrole synthesis are predicted to be in the cytosol or mitochondria (fig. S6) and no other pathway necessitates plastid presence, the metabolism of A. ceratii poses no apparent barrier to plastid loss. To examine whether more endosymbiont-derived genes are present in A. ceratii, we classified all of its predicted proteins by using an automated phylogenetic pipeline (32). Proteins clustering with red algae or green plants in phylogenetic trees populated from a local database of representative eukaryotic and prokaryotic sequences were manually inspected for potential plastid functions (Materials and Methods). No putative endosymbiont-derived proteins were identified. Overall, there is no evidence for a plastid in A. ceratii, and we conclude that it has lost the organelle altogether, presumably in its common ancestor with Hematodinium.
Mitochondrial genome and function
The mitochondrial genomes of apicomplexans, P. marinus, Hematodinium, and core dinoflagellates encode genes for only three proteins: coxI, coxIII (both in complex IV), and cytb (complex III) (9). In core dinoflagellates, these genomes are also typically fragmented and transcripts sometimes undergo extensive RNA editing (10). In intermediate-branching lineages Perkinsus and Oxyrrhis, no editing has been found but the genomes are still complex and fragmented (9, 10, 14). A. ceratii is aerobic, and two mitochondria are observed in the free-living stage (Fig. 3) (17). We confirmed that A. ceratii dinospores contain respiration activity similar to other eukaryotic cells (table S6) and used a fluorescence activity assay to demonstrate that their mitochondria have an active membrane potential (fig. S7). The existence of actively respiring mitochondria is consistent with the fact that nuclear genes for mitochondrial respiration and adenosine 5′-triphosphate (ATP) synthesis are highly expressed throughout the A. ceratii infection cycle (16).
Fig. 3. Investigation of mitochondria in A. ceratii cells.
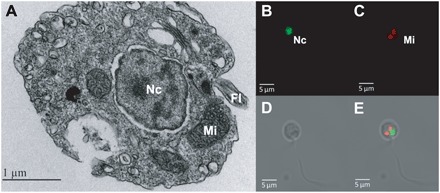
(A) Electron microscopy transmission image of A. ceratii dinospore showing the fine structure of the mitochondrion (Mi), nucleus (Nc), and flagella (Fl). Confocal microscopy images showing (B) SYTO-13–stained DNA of the nucleus (Nc), (C) mitochondria stained with MitoTracker, (D) an image of a free-swimming biflagellate dinospore cell, and (E) overlay of images.
Mitochondrial genomes are typically present in high copy numbers, and as a result, they can be visualized in the cell by using DNA stains and are typically represented with higher coverage than nuclear genomes in total DNA sequencing, as we have done here. However, we found neither to apply to A. ceratii. Using confocal laser scanning microscopy, we did not observe DNA in A. ceratii mitochondria (SYTOX DNA staining) despite the fact that nuclear DNA was clearly visible, as was A. catenella organellar DNA (fig. S7). Moreover, searches for mitochondrial genes in A. ceratii gene models and transcriptomic assemblies identified no candidates for any of the three expected genes. CoxI of A. catenella (host) was recovered, providing a positive internal control despite the very low coverage of host DNA in the assembly. Because nuclear encoded components of complex IV are present in A. ceratii, we conducted a more refined search for the missing cox genes in both the genome assembly and raw sequence reads (table S7). Two small fragments corresponding to coxI were found on scaffolds 46 and 1091, and their presence on these scaffolds was confirmed by PCR (Materials and Methods). The two scaffolds have typical characteristics of nuclear DNA: They are long (328 and 62 kbp, respectively), have a GC content comparable to the rest of the genome (55%), and encode many canonical eukaryotic genes that are functionally unrelated to mitochondria (72 and 18 genes, respectively). The coxI fragments span positions 323–370 (g15932) and 390–444 (g833) of the Pfam domain PF00115 (cytochrome c and quinol oxidase polypeptide I), which constitutes the C-terminal moiety of coxI (Fig. 4). Both are transcribed and spliced to remove a single intron, as confirmed by transcriptomic read mapping and reverse transcription PCR (RT-PCR). We found no evidence for trans-splicing, leading to a fusion of the two mRNAs. The same coxI fragments with nuclear attributes were recovered from the reference genomes of two other Amoebophrya strains, and they clustered with Plasmodium and other dinoflagellates in a phylogeny (fig. S8). No sequences encoding for coxIII, cytb, or the N-terminal part of coxI could be identified by in-depth searches of reads or assemblies from the other two Amoebophrya strains. PCR amplification of cytb from gDNA or cDNA with degenerate primers derived from conserved sites (33) likewise yielded no products. Overall, we conclude that the mitochondrial genome has therefore been lost. The mitochondrial genome has been lost many times but, to date, always in anaerobes, where most or all the respiratory complexes are lost, so although this would make A. ceratii the first described aerobic species to have completely lost the mitochondrial genome, the evolutionary rationale for this loss is not fundamentally different.
Fig. 4. CoxI fragment alignment.

Scaffold fragment, gene model, gDNA PCR amplicon sequence, and cDNA sequence with and without intron sequence. The predicted coxI domain is marked with a shaded background.
Spiked-in (in silico) reads from Physarum polycephalum mitochondrial DNA (at various sequencing depths) were recovered as fully assembled mitochondrial genomes during the assembly process, indicating that the absence of mitochondrial DNA in A. ceratii assembly was not an artifact of the assembly strategy used in this study (figs. S9 and S10). An alternative explanation for the absence of mitochondrial sequences is that it was depleted in the sequencing library, possibly due to a low GC content or possibly due to failure to lyse mitochondria. There is no evidence that either of these factors was a problem, because both low %GC DNA and Alexandrium mitochondrial sequences are present in our raw reads. Extensive editing of mitochondrial transcripts could theoretically prevent the mitochondrial genes from being detected, but edited mitochondrial transcripts are conspicuously missing from A. ceratii transcriptomes despite being highly expressed and abundant in dinoflagellate transcriptomes (because they have polyadenylated tails). A more likely reason to overlook mitochondrial genes is if they have very fast evolving sequences. This is difficult to ever rule out entirely, but one can address the likelihood by examining the existence of the protein complexes to which the missing proteins associate (complex IV for coxI and coxIII and complex III for cytb): If nuclear encoded subunits of the complex are also missing, then the mitochondrial encoded subunit has likely also been lost. We validated the presence of proteins from all five canonical complexes of the mitochondrial respiratory chain in the predicted proteome of A. ceratii. All essential components of complexes II, IV, and V were identified, but no proteins corresponding to complexes I and III were found (Fig. 5, fig. S11, and tables S7 and S8). Complex I [NADH (reduced form of nicotinamide adenine dinucleotide) dehydrogenase] has been replaced by an alternative NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) dehydrogenase in A. ceratii (g180) much like in apicomplexans and dinoflagellates (34), a transition that occurred in their common ancestor after the split with colponemids (35). The lack of complex III is more significant because it provides a direct rationale for the absence of cytb from the A. ceratii mitochondrion. Although highly unusual, a curious precedent to such loss is seen in C. velia: Electrons from the alternative NADPH dehydrogenase, complex II (succinate dehydrogenase), and presumably from other sources could be channeled directly to AOX via the lipid-soluble carrier ubiquinone (Q) (11), and that may work in a similar way in A. ceratii. Complex IV is present in A. ceratii, and it might receive electrons from alternative donors such as d-lactate:cytochrome c oxidoreductase [d-LDH (lactate dehydrogenase)] and galacto-1,4-lactone:cytochrome c oxidoreductase (G-1,4-LDH) (via cytochrome c) (Fig. 5, fig. S11, and table S8). A complete complex V is also present, which indicates that a functional proton gradient is available for ATP generation.
Fig. 5. Model of mitochondrial functions in A. ceratii based on the genome gene content.
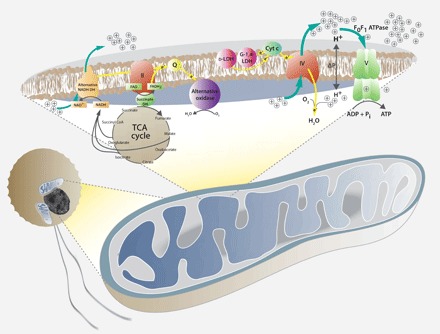
The C. velia model from (11) was taken as a template. Mitochondrial complex I has been replaced by an alternative NADH dehydrogenase (DH), which reduced the NADH from the tricarboxylic acid (TCA) cycle. Both alternative NADH dehydrogenase and succinate dehydrogenase (complex II) channel electrons through the carrier ubiquinone (Q) to the alternative oxidase (yellow arrows). Electrons may also be passed by other sources, such as d-lactate:cytochrome c oxidoreductase (d-LDH) and galacto-1,4-lactone:cytochrome c oxidoreductase (G-1,4-LDH) to cytochrome c (yellow arrows), which passes them on to complex IV (cytochrome c oxidase). Stippled yellow arrows indicate alternative pathways of electron flow as proposed in Chromera (11).
In contrast, the presence of nucleus-encoding proteins for complex IV suggests that mitochondrial coxIII and coxI should be present. CoxIII is the faster evolving of the two but is typically still easily detectable at the sequence level, so we conclude that its absence is due to outright loss, which is not unprecedented: coxIII is also absent in the related ciliate mitochondria (36). The absence of much of coxI has no precedent; however, a short sequence encoding its C terminus has been relocated to the nucleus in at least some eukaryotes (37). The two nuclear fragments of coxI in A. ceratii could therefore represent functional transfers, as observed in other fragmented cox genes: coxII has been independently fragmented and moved to the nucleus in green algae and in apicomplexans and dinoflagellates (including A. ceratii: scaffold 161, gene g4166 with three introns). It has been hypothesized that the fragmentation of coxII was crucial for its successful relocation by mitigating the difficulties of targeting a highly hydrophobic product across mitochondrial membranes (38), which is also a major rationale for the retention of coxI in the mitochondrial genome, so perhaps the nucleus-encoded fragments of coxI represent a similar process to what has taken place with coxII. Another possible explanation is that the coxI fragments are nonfunctional transfers, or nuclear mitochondrial pseudogenes (NUMTS) (39). In the absence of selection, these fragments accumulate deleterious mutations very quickly, but the A. ceratii coxI gene fragments had high GC content (suggesting that they have been in the nuclear genome for enough time to ameliorate their GC content) but still retain all aspects of a functional protein: The sequences are conserved, require no editing, are expressed, and contain one intron each that was spliced during expression. Intron gain and splicing suggest functional nuclear genes that have resided in the nucleus for some time. We therefore hypothesize that the detected coxI fragments in the A. ceratii genome are the result of a functional endosymbiotic gene transfer and that the mitochondrial genome was lost because it encoded no essential genes.
CONCLUSION
Parasite genomes are often characterized by a reduction in size, loss of genes, and loss of functions as the parasite becomes more dependent on the host. A. ceratii has retained most of the genome functionality of a free-living species, with the exception of the biosynthesis of a few amino acids and, more significantly, isoprenoids, which appears to be related to the loss of plastid organelles. The apparently minimal loss of function in this parasite is interesting because it reflects the challenges we face in inferring ecological roles from the genome of heterotrophic eukaryotes. On the basis of genomic data alone, it would be impossible to infer that A. ceratii was a parasite in the absence of other biological information about its nature.
The A. ceratii genome does, however, exhibit some intriguing unique features. The most noteworthy is the apparent transfer to the nucleus of all essential functional mitochondrial genes and the resulting loss of the mitochondrial genome. The ancestral mitochondrial genome of dinoflagellates was already highly reduced in terms of gene-coding content (10). By a combination of transferring the last essential protein moieties to the nucleus, and the loss of certain respiratory complexes, A. ceratii has eliminated the need for a mitochondrial genome, resulting in functional mitochondria with a similar electron transport system and oxidative phosphorylation as found in C. velia (Fig. 4, fig. S11, and table S8), but without a mitochondrial genome.
MATERIALS AND METHODS
Culturing and harvesting
The A. ceratii parasite strain AT5.2 (19) was isolated from Alexandrium cells sampled from the Gulf of Maine (USA) and was used to infect the A. catenella strain Alex5; RCC3037 (formerly described as Alexandrium tamarense) isolated from the North Sea coast of Scotland. A. ceratii was cultured by infecting A. catenella (Alex5; RCC3037) and then transferred to a fresh host culture every 3 to 4 days (18). Cultures were grown at 15°C in K medium, with cool-white fluorescent lamps providing photon irradiation of 150 μmol m−2 s−1 on a 14-hour/10-hour light-dark cycle.
To obtain axenic cultures, the host A. catenella strain (Alex5; RCC3037) was grown in 500 ml of K medium in four Erlenmeyer flasks supplied with a mixture of antibiotics [ampicillin (165 μg/ml), gentamicin (33.3 μg/ml), streptomycin (100 μg/ml), chloramphenicol (1 μg/ml), and ciprofloxacin (10 μg/ml)] 1 week before the experiment. The upper phase (approximately 400 ml) was transferred to a new Erlenmeyer flask, and new K medium was added, with a final volume of 500 ml. Before infecting the experimental cultures, 1 ml of each stock culture was stained with acridine orange and checked for bacterial contamination. Infection of the host culture was performed following the procedures described by Coats and Park (18). Parasite dinospores (5 μm) were harvested from infected host cultures (A. catenella > 20 μm) on day 4 by gravity filtration through a 10-μm pore size mesh. The harvested dinospores were examined microscopically to ensure the absence of host cell contamination and additionally incubated for 24 hours to allow lysis of eventual host cells as well as degradation of host environmental DNA. Dinospores were collected via gentle centrifugation at 4°C for 10 min. The supernatant was decanted, and the resulting cell pellet was ground to a fine powder with liquid nitrogen in a precooled mortar and pestle. The ground tissue was transferred immediately to a 2-ml screw-cap tube with 2 ml of Buffer G2 [with ribonuclease A (RNase A)] and 0.1 ml of the Proteinase K (Sigma-Aldrich, Germany) stock solution, according to the manufacturer’s protocol (Qiagen Genomic DNA Handbook). DNA extraction was performed using the Blood & Cell Culture DNA Kits (Qiagen, Hilden, Germany) and Qiagen Genomic-tips. DNA quality and quantity were determined using a NanoDrop ND-1000 spectrometer (PEQLAB, Erlangen, Germany) and a DNA Nano Chip assay on a 2100 Bioanalyzer device (Agilent Technologies, Böblingen, Germany).
RNA extraction
TRI Reagent–fixed and frozen dinospore cells were lysed using a Bio 101 FastPrep instrument (Thermo Savant, Illkirch, France) at maximum speed (6.5 m/s) for 2 × 45 s. Lysed cells were cooled on ice, and 200 μl of chloroform was added and vortexed for 20 s. The samples were transferred to a phase lock tube after 5 min of incubation at room temperature (Eppendorf, Hamburg, Germany) and incubated for another 5 min followed by centrifugation for 15 min at 13,000g and 4°C. The upper aqueous phase was transferred to a new tube and mixed with the same volume isopropanol, 1/10 volume of 3 M Na-acetate (pH 5.5; Ambion by Life Technologies, Carlsbad, CA, USA), and 2 μl of linear polyacrylamide (Ambion). Total RNA was precipitated for 90 min at −20°C and collected by centrifugation for 20 min at 13,000g and 4°C. The obtained pellet was washed twice, first with 1 ml of 70% ethanol (EtOH) followed by 1 ml of absolute EtOH; the RNA pellet was dried for 1 min at 37°C and resolved in 30 μl of RNase-free water (Qiagen, Hilden). RNA quality check was performed using a NanoDrop ND-1000 spectrometer (PEQLAB, Erlangen, Germany) for purity and RNA Nano Chip Assay on 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) to examine the integrity of the extracted RNA.
Sequencing
Library preparation and sequencing were performed using the Illumina NGS platform and methodology. DNA was extracted from an A. ceratii culture and used to construct paired-end (PE) and mate-pair (MP) libraries for sequencing. Approximately 5 μg of DNA was used for PE library preparation according to the manufacturer’s instructions (Illumina PE Sample Prep Kit). The library was sequenced on a single lane of HiSeq 2000 in 100-bp PE mode. Reads were extracted in FASTQ format using CASAVA v1.8.2 (supported by Illumina). Sequencing produced 127,251,149 PE reads.
Approximately 3 μg of DNA was used for MP library preparation using a Roche/Illumina hybrid protocol. First, DNA was fragmented using a HydroShear device to obtain fragments of around 3 kb in length. Circularization was performed according to the Roche PE library preparation method manual (20- and 8-kb span) using titanium linkers. Circularized fragments were nebulized and used for library preparation with TruSeq DNA Sample Prep Kit v2 (Illumina). The library was sequenced on a single lane of a HiSeq 2000 platform in 100-bp PE rapid mode. Reads were extracted in FASTQ format using bcl2fastq v1.8.3 (supported by Illumina). Sequencing produced 180,661,377 PE reads.
Assembly
Quality filtering (cutoff of Q20) and adaptor trimming (~0.3% of the total) of raw data were performed in CLC Assembly Cell (Qiagen Bioinformatics), and any reads <40 bp were discarded. All orphan (not paired) reads were also discarded. A total of 85,139,449 (PE data) and 85,139,449 (MP data) PE reads were retained after quality control and processed further.
First, PE data were assembled using the assembly pipeline in CLC Assembly Cell using standard parameters (--wordsize default=automatic; -p fb ss 250 520). To reduce the number of contigs from contaminants, we selected contigs fulfilling at least one of the following criteria: (i) coverage of 30 to 300 and a minimum length of 1 kb or (ii) contigs hit EST (expressed sequence tag) reference/database (16, 19) using BLAST with an identity threshold of 98% to account for potential sequencing errors. The resulting contigs were bridged with MP data using SSPACE2 (insert size, 2000 bp; size error, 0.65; -m 50; -k 15; -a 0.5; -x 0) forming 4630 scaffolds. From this, sequences from cocultured bacteria (e.g., Loctanella, Oceanicaulis, and Chlamydia) were identified via BLASTn and removed. We also removed the sequences originating from the Alexandrium host by matching transcript data to the genome assembly and also identified a few small apparently oomycete-derived contigs. After an initial screen of the largest contigs against the complete National Center for Biotechnology Information (NCBI) nucleotide database, genus-specific databases were set up for each identified contaminant, and these were used to rescreen the entire assembly. The coverage combined with the GC content of the contigs (GC content lower than 48%) provided a further measure of contamination. In total, these screens identified 16 Mb that were removed as contaminants, leaving 87.7 Mb (2351 scaffolds) defined as A. ceratii–specific sequences. The completeness of the genome was assessed via CEGMA using default settings (24).
Transriptomic data used in the study were obtained from two previously published studies (16, 19). Gene prediction was performed using Augustus (http://augustus.gobics.de/). A training set that contained 16 genes previously identified with BLAST was constructed. After adjusting Augustus parameters, the previously generated EST sequences were used to detect bona fide splice sites. In total, 19,925 protein-coding genes were predicted, of which 4856 had transcript support. The Augustus predictions were evaluated using the self-training prediction tool genemarkES (http://exon.gatech.edu/GeneMark/). The number of genes predicted with this tool was similar to that predicted by Augustus, and the overlap between the predicted gene sets was extensive.
Translated amino acid sequences from the predicted gene models were annotated using the Trinotate pipeline and compared with the NCBI nucleotide database using the BLAST tool, with a cutoff e value of 10−11 (http://trinotate.github.io/). Annotations were further supplemented by Kyoto Encyclopedia of Genes and Genomes mapping (https://www.genome.jp/kegg/) and were finally manually curated.
Raw and assembled sequence data were submitted to GenBank under BioProject number PRJNA306142. Contigs (data file S1), scaffolds (data file S2), gff file (data file S3), predicted protein sequences (data file S4), assembly statistics (data file S5), and the annotation table (data file S6) generated by Trinotate were uploaded as auxiliary supplementary data with the article.
Phylogeny
Predicted proteins from the A. ceratii genome were added into an alignment of 100 conserved protein sequences previously used in a multiprotein phylogeny of dinoflagellates (31). Sequences were aligned by the local pair algorithm in MAFFT v7.215 and stripped of hypervariable sites by using the -b 4 -g 0.4 settings in BMGE v1.1. Sequence orthology within single protein alignments was verified by maximum likelihood phylogenies (LG + Gamma 4 + F model) computed in RAxML v.8 (https://sco.h-its.org/exelixis/web/software/raxml/), and divergent paralogs and contaminant sequences were removed. Single protein alignments were concatenated in SCaFoS v1.25 by using the o=gclv gamma=yes l=1 m=1 setting. Chimeric sequences were created for species where overlapping sequence fragments or nonoverlapping fragments of a congruent phylogenetic position were present. The final matrix of 100 concatenated proteins contained 29,339 amino acid sites in 32 operational taxonomic units, of which 94.2% residues were present (all genes and 96% of residues were present in A. ceratii). Maximum likelihood phylogenies on the final matrix were inferred in IQ-TREE v1.0 by using the LG + I + GAMMA4 + F settings with 1000 ultrafast bootstraps (the best substitution model was determined by the implemented model tester: -m TESTONLY setting) and in RAxML by using the LG + GAMMA4 + F settings with 300 nonparametric bootstraps. Bayesian phylogeny was inferred in PhyloBayes MPI v1.5a on CIPRES Science Gateway by using GTR + CAT + GAMMA4, -dc, and maxdiff < 0.1 settings.
Metabolic analysis of plastid dependency
To determine whether A. ceratii contains a plastid organelle, we searched for plastidial and cytosolic pathways for the biosynthesis of isoprenoids, tetrapyrroles and fatty acids, and Fe-S cluster assembly. The predicted proteome of A. ceratii was searched by HMMER v3.2.1 at https://www.ebi.ac.uk/Tools/hmmer/ and BLASTP by using query sequences from Hematodinium, Oxyrrhis, apicomplexans, and chromerids. Hits at the e-value threshold of 10−5 were collected and validated for specificity by reverse BLASTP analysis against the NCBI nr database. Most tetrapyrrole biosynthesis enzymes were also host-derived (ALAS, HemB, HemC, and HemE), but one (HemD) was apparently plastid-derived. The origin of all five heme enzymes was confirmed by using single-protein phylogenies (computed in IQ-TREE as described in species phylogeny above). We then evaluated the presence of N-terminal targeting signatures within the five proteins by using HECTAR (http://webtools.sb-roscoff.fr/root?tool_id=abims_hectar), SignalP v4.1 (http://www.cbs.dtu.dk/services/SignalP/), and TargetP 1.1(http://www.cbs.dtu.dk/services/TargetP/). No signal peptide characteristic of targeting to complex plastids was identified. ALAS and ALAD had weak mitochondrial targeting signals in TargetP 1.1 (0.507 and 0.825, respectively); in ALAS, which is mitochondrion-localized in all eukaryotes, this signal was likely genuine (fig. S6). To test whether the N terminus of the plastid-derived HemD in A. ceratii was predicted correctly, transcriptomic reads (19) were mapped across the 5′ end of the gene. Four such transcriptomic reads linked the conserved region of the mature protein with the predicted methionine and a stop codon further upstream of it. This evidence confirmed that the A. ceratii HemD lacks an N-terminal extension for plastid targeting—similarly to Hematodinium and bacterial sequences but unlike all plastid-targeted HemDs—and therefore has been relocated to the cytosol. The last three enzymes of the tetrapyrrole pathway were not identified in A. ceratii, but two of them were also absent in the related genomes of Hematodinium and Symbiodinium, and they were likely replaced by new or bacteria-derived isoforms, given that the first five steps of the pathway were present and expressed in the transcriptome (all enzymes except HemE). In any case, all tetrapyrrole enzymes that were identified were predicted to localize to the cytosol and mitochondria (fig. S6), and because all other plastidial pathways were absent, A. ceratii has likely become independent of plastid-localized metabolism.
Assessment of mitochondrial activity
Dinospore cells were collected by smooth filtration over 10-μm gaze to remove host (A. catenella) cells and washed with sterile filtered seawater (150 ml) over 1-μm polycarbonate filter to remove bacterial contamination. Furthermore, cells were gently centrifuged for 15 min at 1500g using a swing-out rotor at 15°C (Eppendorf 5810, Hamburg, Germany) and suspended in 1 ml of sterile seawater. A 200-μl aliquot was used for microscopic cell quantification.
Measurements were carried out in triplicates in two parallel setups consisting of acrylic respiration chambers (Ranks Brothers, Cambridge, UK). The chambers were volume-adjusted to 400 μl and temperature-controlled to 15°C by a thermostat (Lauda, Königshofen, Germany). Respiration was measured using micro-optodes and the TX system of PreSens (PreSens GmbH, Regensburg, Germany), connected to a laptop computer running the software LabChart 7.0 for recording and analysis (ADInstruments, Castle Hill, Australia). Well-mixed purified dinospore solution (400 μl) was transferred to the chamber. The chambers were sealed airtight, and a micro-optode was inserted through the lid. Blank (culture media) respiration was recorded for 30 min. The dinospore solution was added and measured for 30 min. Blank respiration for both dinospore medium and bacterial background was conducted in the same way. In both cases, respiration was below the detection limit.
Mitochondrial staining and DNA content analysis via confocal microscopy
Dinospore cells were collected using the procedure mentioned above. Exponentially growing A. catenella cells were used for staining as a control. Cells were stained with SYTO-13 [nuclei specific, green fluorescence; Molecular Probes S7575; 5 mM in dimethyl sulfoxide (DMSO), final concentration 5, 1, or 0.5 μM] to examine the potential presence of DNA within the mitochondrion and to stain the nucleus. JC-10 (Enzo Life Sciences ENZ-52305, 1 mM stock solution in DMSO, final concentration 1 μM) staining is viable in vertebrates, invertebrates, and algae [e.g., (40)]. It was used here to visualize dinospore mitochondria and as an indicator for mitochondrial functionality. Red staining indicates more aggregate with JC-10, which is indicative of hyperpolarized electrochemical membrane potential. MitoTracker Red CMXRos (Invitrogen, M7512) was adjusted to 1 mM with DMSO and used in a final concentration of 200 nM. After 30-min incubation, the cell culture was fixed with 2% paraformaldehyde for 30 min. Cells were examined at approximately 15°C via a confocal laser scanning microscope (TCS SP5, Leica, Wetzlar, Germany) using an argon laser at an excitation of 488 nm and a HeNe laser at 633 nm. Emission was adjusted to 500 to 535 nm using SYTO and to 640 to 680 nm (633-nm excitation.) using MitoTracker. For JC-10, emissions of 505 to 540 nm and 555 to 590 nm were measured at 488-nm excitation.
Mitochondrial gene amplification
RT-PCR was conducted to test the expression of the coxI gene fragments (g833 and g15932). cDNA synthesis was performed with 1 μl of SuperScript III reverse transcriptase (200 U/μl) and 500 ng of total RNA from A. ceratii and A. catenella as a control in the presence of random hexamer primers (25 ng/μl) and anchored oligoVN(dT)20 primer (25 ng/μl) and 1 μl of RNaseOUT (40 U/μl) (Invitrogen, Darmstadt, Germany) at 42°C for 1 hour followed at 50°C for 1 hour and was finished by an inactivation cycle at 75°C for 15 min. The PCR for the target genes was conducted as described below.
gDNA and cDNA of A. ceratii dinospores (see above) were used as a PCR template to attempt to amplify genes for cytochrome oxidase 1 (coxI) and cytochrome b (cytb). A set of general cox cytb/cob primers (table S9) for dinoflagellates was used, and PCR was performed according to (33). A. catenella was used as a positive control. Additional primer sets (table S9) were designed against the identified coxI gene fragment 1 (g15932) and coxI gene fragment 2 (g833), and PCR was conducted with A. ceratii and A. catenella as negative controls. PCR was carried out in 20-μl reactions with 1 μl of 10 mM primer, 1 μl of cDNA or gDNA, 1 μl of deoxynucleoside triphosphate (dNTP) solution, 1 μl (5 U/ml) of Platinum HotMaster Taq, and 2 μl of 10× PCR buffer (5PRIME, Hamburg, Germany). The PCR cycling parameters were as follows: 94°C for 2 min and 35 cycles at 94°C for 30 s and 57°, 60°, and 68°C for 4 min, and a final extension for 10 min at 68°C. PCR products were separated on 1% agarose.
Amplification of the shikimate pathway transcripts
cDNA was generated as described above and then used as a template for PCR using primers listed in table S10. PCR was carried out in 50-μl reaction with 1 μl of 10 mM forward primer, 1 μl of 10 mM reverse primer, 1 μl of cDNA or gDNA (10 ng/μl), 1 μl of dNTP solution (10 μM), 1 μl (5 U/ml) of Phusion polymerase, and 5 μl of 5× PCR Phusion Green HF buffer. The PCR cycling parameters were as follows: 98°C for 30 s and 35 cycles at 98°C for 10 s and 52°C for 30 s and 68°C for 2 min, and a final extension for 10 min at 68°C. PCR products were separated on 1% agarose. Amplicons were used directly for Sanger sequencing.
RACE PCR for SL identification at the 5′ end
To amplify the 5′ end of a transcript and identify a potential SL, we generated cDNA and performed rapid amplification of cDNA ends (RACE) PCR. cDNA synthesis was performed with 1 ml of SuperScript III reverse transcriptase (200 U/ml) and 500 ng of total RNA from A. ceratii in the presence of random hexamer primers (50 ng/ml) and/or a special adapter ligated oligo(dT) primer (50 mM) (GCTGTCAACGATACGCTACGTAACGGCATGACAGTGTTTTTTTTTTTTTTTTTTTTTTTT) according to the manufacturer’s recommendations (Invitrogen, Darmstadt, Germany).
For 5′ RACE PCR, the SL sequence (CCGTAGCCATTTTGGCTCTTG; 21 nucleotides; temperature, 64°C) was used, as the forward and reverse primers were used for the sets for coxI gene fragments (g833 and g15932) or shikimate pathway (see tables S9 and S10), respectively. The PCR was carried out in a 50-μl reaction volume with 3 μl of 10 mM SL primer, 1 μl of 10 mM primer, 1 μl of cDNA, 1 μl of dNTP solution, 1 μl (5 U/μl) of Platinum HotMaster Taq polymerase, and 5 μl of 10× PCR buffer (5PRIME, Hamburg, Germany). A gradient touchdown PCR approach was used, and the cycling parameters were as follows: 94°C for 2 min and 5 cycles at 94°C for 30 s and 62°, 60°, and 58°C for 4 min and then 25 cycles at 94°C for 30 s and 58°, 60°, and 62°C for 4 min, with a final extension at 68°C for 10 min. PCR products were separated on 1% agarose.
Sanger sequencing of PCR fragments
Sequencing was conducted with a standard cycle sequencing chemistry (ABI 3.1, Applied Biosystems, Darmstadt, Germany). Cycle sequencing products were analyzed on an ABI 3130xl capillary sequencer (Applied Biosystems, Darmstadt, Germany), and the generated sequences were assembled and analyzed with CLC main workbench version 10.0 (www.CLCbio.com).
Supplementary Material
Acknowledgments
We thank M. Sengco for providing the parasite strain, E. Bigeard for maintaining the parasite in Roscoff, S. Lepanse (MerImage platform in Roscoff) for electron microscopic investigations, I. Goerlich for skillful and diligent assistance in the Illumina sequencing, and Nancy Kühne for support in the laboratory. Funding: Financial support was provided by the PACES Research Program of the Alfred-Wegener-Institut, Helmholtz-Zentrum für Polar- und Meeresforschung, and the China Scholarship Council (CSC). This work was also financially supported by French ANR projects HAPAR (2014-DEFI 1), the European Project Micro B3 (contract 287589), UCL Excellence Fellowship (to J.J.), and the Natural Sciences and Engineering Research Council (RGPIN-2014-03994 to P.J.K.). Findings presented in this article also formed a part of Y.L.’s thesis submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (PhD). S.Fa.’s PhD grant was supported by ANR HAPARANR-14-CE02-0007. Competing interests: The authors declare that they have no competing interests. Author contributions: Conception and design of the research were by U.J. and G.G. Gathering of data and determination, analysis, and interpretation of research data were conducted by U.J., Y.L., S.W., M.G., J.J., G.S.K., F.C.M., U.B., S.Fa., M.F., S.Fr., L.G., P.J.K., A.M., B.M.P., K.V., and G.G. The manuscript was written by U.J., G.G., and Y.L. with contributions from G.S.K., J.J., S.W., and P.J.K. All authors read and approved the final manuscript. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/4/eaav1110/DC1
Fig. S1. Flow cytometry.
Fig. S2. Alignment of 5′ ends of transcripts of A. ceratii showing SL and relict SL repeats.
Fig. S3. The distribution of intron sizes in A. ceratii genes.
Fig. S4. Expansion of gene numbers per metabolic pathway in A. ceratii.
Fig. S5. Phylogenetic analysis of prokaryotic and eukaryotic PKS and FAS.
Fig. S6. Predicted evolution of tetrapyrrole biosynthesis in A. ceratii and other dinoflagellates.
Fig. S7. Confocal microscopy images of A. ceratii and A. catenella as a control.
Fig. S8. CoxI phylogeny.
Fig. S9. Plot obtained by assembling the PE dataset including spike-in mitochondrial reads of P. polycephalum.
Fig. S10. Plot obtained by assembling the PE dataset including spike-in mitochondrial reads of P. polycephalum.
Fig. S11. Model of the mitochondrial respiratory chain in A. ceratii.
Table S1. Enriched gene families.
Table S2. Number of genes encoding transcription factors in genomes/transcriptomes of 48 genera.
Table S3. SL detected in the genome of A ceratii.
Table S4. Illumina RNAseq reads of A. ceratii at 6 and 96 hours during the infection.
Table S5. A list of enzymes involved in various metabolic pathways in the A. ceratii genome.
Table S6. Mitochondrion respiration assay.
Table S7. The search for components of the electron transport system of A. ceratii.
Table S8. Mitochondrial functions encoded in the genome of A. ceratii.
Table S9. Primers used in this study to amplify cox from A. ceratii cDNA.
Table S10. Primers used to amplify various shikimate pathway transcripts from A. ceratii cDNA.
Data file S1. Contigs of the A. ceratii AT5.2 genome.
Data file S2. Scaffolds of the A. ceratii AT5.2 genome.
Data file S3. GFF of the A. ceratii AT5.2 genome.
Data file S4. Predicted protein sequences of the A. ceratii AT5.2 genome.
Data file S5. Assembly statistics of the A. ceratii AT5.2 genome.
Data file S6. Annotation table generated via Trinotate for the A. ceratii AT5.2 genome.
REFERENCES AND NOTES
- 1.Bachvaroff T. R., Gornik S. G., Concepcion G. T., Waller R. F., Mendez G. S., Lippmeier J. C., Delwiche C. F., Dinoflagellate phylogeny revisited: Using ribosomal proteins to resolve deep branching dinoflagellate clades. Mol. Phylogenet. Evol. 70, 314–322 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keeling P. J., Burger G., Durnford D. G., Lang B. F., Lee R. W., Pearlman R. E., Roger A. J., Gray M. W., The tree of eukaryotes. Trends Ecol. Evol. 20, 670–676 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Lin S., Genomic understanding of dinoflagellates. Res. Microbiol. 162, 551–569 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Gornik S. G., Ford K. L., Mulhern T. D., Bacic A., McFadden G. I., Waller R. F., Loss of nucleosomal DNA condensation coincides with appearance of a novel nuclear protein in dinoflagellates. Curr. Biol. 22, 2303–2312 (2012). [DOI] [PubMed] [Google Scholar]
- 5.LaJeunesse T. C., Lambert G., Andersen R. A., Coffroth M. A., Galbraith D. W., Symbiodinium (Pyrrhophyta) genome sizes (DNA content) are smallest among dinoflagellates. J. Phycol. 41, 880–886 (2005). [Google Scholar]
- 6.Zhang H., Hou Y., Miranda L., Campbell D. A., Sturm N. R., Gaasterland T., Lin S., Spliced leader RNA trans-splicing in dinoflagellates. Proc. Natl. Acad. Sci. U.S.A. 104, 4618–4623 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Janouškovec J., Horák A., Oborník M., Lukeš J., Keeling P. J., A common red algal origin of the apicomplexan, dinoflagellate, and heterokont plastids. Proc. Natl. Acad. Sci. U.S.A. 107, 10949–10954 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gómez F., A quantitative review of the lifestyle, habitat and trophic diversity of dinoflagellates (Dinoflagellata, Alveolata). Syst. Biodivers. 10, 267–275 (2012). [Google Scholar]
- 9.Jackson C. J., Gornik S. G., Waller R. F., The mitochondrial genome and transcriptome of the basal dinoflagellate Hematodinium sp.: Character evolution within the highly derived mitochondrial genomes of dinoflagellates. Genome Biol. Evol. 4, 59–72 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waller R. F., Jackson C. J., Dinoflagellate mitochondrial genomes: Stretching the rules of molecular biology. Bioessays 31, 237–245 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Flegontov P., Michálek J., Janouškovec J., Lai D.-H., Jirků M., Hajdušková E., Tomčala A., Otto T. D., Keeling P. J., Pain A., Oborník M., Lukeš J., Divergent mitochondrial respiratory chains in phototrophic relatives of apicomplexan parasites. Mol. Biol. Evol. 32, 1115–1131 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Anderson D. M., Alpermann T. J., Cembella A. D., Collos Y., Masseret E., Montresor M., The globally distributed genus Alexandrium: Multifaceted roles in marine ecosystems and impacts on human health. Harmful Algae 14, 10–35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chambouvet A., Morin P., Marie D., Guillou L., Control of toxic marine dinoflagellate blooms by serial parasitic killers. Science 322, 1254–1257 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Gornik S. G., Febrimarsa , Cassin A. M., MacRae J. I., Ramaprasad A., Rchiad Z., McConville M. J., Bacic A., McFadden G. I., Pain A., Waller R. F., Endosymbiosis undone by stepwise elimination of the plastid in a parasitic dinoflagellate. Proc. Natl. Acad. Sci. U.S.A. 112, 5767–5772 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guillou L., Viprey M., Chambouvet A., Welsh R. M., Kirkham A. R., Massana R., Scanlan D. J., Worden A. Z., Widespread occurrence and genetic diversity of marine parasitoids belonging to Syndiniales (Alveolata). Environ. Microbiol. 10, 3349–3365 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Lu Y., Wohlrab S., Groth M., Glöckner G., Guillou L., John U., Transcriptomic profiling of Alexandrium fundyense during physical interaction with or exposure to chemical signals from the parasite Amoebophrya. Mol. Ecol. 25, 1294–1307 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Miller J. J., Delwiche C. F., Coats D. W., Ultrastructure of Amoebophrya sp. and its changes during the course of infection. Protist 163, 720–745 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Coats D. W., Park M. G., Parasitism of photosynthetic dinoflagellates by three strains of Amoebophrya (Dinophyta): Parasite survival, infectivity, generation time, and host specificity. J. Phycol. 38, 520–528 (2002). [Google Scholar]
- 19.Lu Y., Wohlrab S., Glöckner G., Guillou L., John U., Genomic insights into processes driving the infection of Alexandrium tamarense by the parasitoid Amoebophrya sp. Eukaryot. Cell 13, 1439–1449 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aranda M., Li Y., Liew Y. J., Baumgarten S., Simakov O., Wilson M. C., Piel J., Ashoor H., Bougouffa S., Bajic V. B., Ryu T., Ravasi T., Bayer T., Micklem G., Kim H., Bhak J., LaJeunesse T. C., Voolstra C. R., Genomes of coral dinoflagellate symbionts highlight evolutionary adaptations conducive to a symbiotic lifestyle. Sci. Rep. 6, 39734 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin S., Cheng S., Song B., Zhong X., Lin X., Li W., Li L., Zhang Y., Zhang H., Ji Z., Cai M., Zhuang Y., Shi X., Lin L., Wang L., Wang Z., Liu X., Yu S., Zeng P., Hao H., Zou Q., Chen C., Li Y., Wang Y., Xu C., Meng S., Xu X., Wang J., Yang H., Campbell D. A., Sturm N. R., Dagenais-Bellefeuille S., Morse D., The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science 350, 691–694 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Shoguchi E., Shinzato C., Kawashima T., Gyoja F., Mungpakdee S., Koyanagi R., Takeuchi T., Hisata K., Tanaka M., Fujiwara M., Hamada M., Seidi A., Fujie M., Usami T., Goto H., Yamasaki S., Arakaki N., Suzuki Y., Sugano S., Toyoda A., Kuroki Y., Fujiyama A., Medina M., Coffroth M. A., Bhattacharya D., Satoh N., Draft assembly of the Symbiodinium minutum nuclear genome reveals dinoflagellate gene structure. Curr. Biol. 23, 1399–1408 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Williams E., Place A., Bachvaroff T., Transcriptome analysis of core dinoflagellates reveals a universal bias towards “GC” rich codons. Mar. Drugs 15, E125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parra G., Bradnam K., Korf I., CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Slamovits C. H., Keeling P. J., Plastid-derived genes in the nonphotosynthetic alveolate Oxyrrhis marina. Mol. Biol. Evol. 25, 1297–1306 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Bachvaroff T. R., Place A. R., From stop to start: Tandem gene arrangement, copy number and trans-splicing sites in the dinoflagellate Amphidinium carterae. PLOS ONE 3, e2929 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kohli G. S., John U., Van Dolah F. M., Murray S. A., Evolutionary distinctiveness of fatty acid and polyketide synthesis in eukaryotes. ISME J. 10, 1877–1890 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chaudhary K., Roos D. S., Protozoan genomics for drug discovery. Nat. Biotechnol. 23, 1089–1091 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richards T. A., Dacks J. B., Campbell S. A., Blanchard J. L., Foster P. G., McLeod R., Roberts C. W., Evolutionary origins of the eukaryotic shikimate pathway: Gene fusions, horizontal gene transfer, and endosymbiotic replacements. Eukaryot. Cell 5, 1517–1531 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herrmann K. M., The shikimate pathway: Early steps in the biosynthesis of aromatic compounds. Plant Cell 7, 907–919 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Janouškovec J., Gavelis G. S., Burki F., Dinh D., Bachvaroff T. R., Gornik S. G., Bright K. J., Imanian B., Strom S. L., Delwiche C. F., Waller R. F., Fensome R. A., Leander B. S., Rohwer F. L., Saldarriaga J. F., Major transitions in dinoflagellate evolution unveiled by phylotranscriptomics. Proc. Natl. Acad. Sci. U.S.A. 114, E171–E180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moustafa A., Bhattacharya D., PhyloSort: A user-friendly phylogenetic sorting tool and its application to estimating the cyanobacterial contribution to the nuclear genome of Chlamydomonas. BMC Evol. Biol. 8, 6 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin S., Zhang H., Hou Y., Zhuang Y., Miranda L., High-level diversity of dinoflagellates in the natural environment, revealed by assessment of mitochondrial cox1 and cob genes for dinoflagellate DNA barcoding. Appl. Environ. Microbiol. 75, 1279–1290 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Danne J. C., Gornik S. G., MacRae J. I., McConville M. J., Waller R. F., Alveolate mitochondrial metabolic evolution: Dinoflagellates force reassessment of the role of parasitism as a driver of change in apicomplexans. Mol. Biol. Evol. 30, 123–139 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Janouškovec J., Tikhonenkov D. V., Mikhailov K. V., Simdyanov T. G., Aleoshin V. V., Mylnikov A. P., Keeling P. J., Colponemids represent multiple ancient alveolate lineages. Curr. Biol. 23, 2546–2552 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Smith D. G. S., Gawryluk R. M. R., Spencer D. F., Pearlman R. E., Siu K.-M., Gray M. W., Exploring the mitochondrial proteome of the ciliate protozoon Tetrahymena thermophila: Direct analysis by tandem mass spectrometry. J. Mol. Biol. 374, 837–863 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Gawryluk R. M. R., Gray M. W., An ancient fission of mitochondrial cox1. Mol. Biol. Evol. 27, 7–10 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Pérez-Martínez X., Antaramian A., Vázquez-Acevedo M., Funes S., Tolkunova E., d’Alayer J., Claros M. G., Davidson E., King M. P., González-Halphen D., Subunit II of cytochrome c oxidase in Chlamydomonad algae is a heterodimer encoded by two independent nuclear genes. J. Biol. Chem. 276, 11302–11309 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Bensasson D., Zhang D.-X., Hartl D. L., Hewitt G. M., Mitochondrial pseudogenes: Evolution’s misplaced witnesses. Trends Ecol. Evol. 16, 314–321 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Kim E. J., Park S. Y., Lee J.-Y., Park J. H. Y., Fucoidan present in brown algae induces apoptosis of human colon cancer cells. BMC Gastroenterol. 10, 96 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/4/eaav1110/DC1
Fig. S1. Flow cytometry.
Fig. S2. Alignment of 5′ ends of transcripts of A. ceratii showing SL and relict SL repeats.
Fig. S3. The distribution of intron sizes in A. ceratii genes.
Fig. S4. Expansion of gene numbers per metabolic pathway in A. ceratii.
Fig. S5. Phylogenetic analysis of prokaryotic and eukaryotic PKS and FAS.
Fig. S6. Predicted evolution of tetrapyrrole biosynthesis in A. ceratii and other dinoflagellates.
Fig. S7. Confocal microscopy images of A. ceratii and A. catenella as a control.
Fig. S8. CoxI phylogeny.
Fig. S9. Plot obtained by assembling the PE dataset including spike-in mitochondrial reads of P. polycephalum.
Fig. S10. Plot obtained by assembling the PE dataset including spike-in mitochondrial reads of P. polycephalum.
Fig. S11. Model of the mitochondrial respiratory chain in A. ceratii.
Table S1. Enriched gene families.
Table S2. Number of genes encoding transcription factors in genomes/transcriptomes of 48 genera.
Table S3. SL detected in the genome of A ceratii.
Table S4. Illumina RNAseq reads of A. ceratii at 6 and 96 hours during the infection.
Table S5. A list of enzymes involved in various metabolic pathways in the A. ceratii genome.
Table S6. Mitochondrion respiration assay.
Table S7. The search for components of the electron transport system of A. ceratii.
Table S8. Mitochondrial functions encoded in the genome of A. ceratii.
Table S9. Primers used in this study to amplify cox from A. ceratii cDNA.
Table S10. Primers used to amplify various shikimate pathway transcripts from A. ceratii cDNA.
Data file S1. Contigs of the A. ceratii AT5.2 genome.
Data file S2. Scaffolds of the A. ceratii AT5.2 genome.
Data file S3. GFF of the A. ceratii AT5.2 genome.
Data file S4. Predicted protein sequences of the A. ceratii AT5.2 genome.
Data file S5. Assembly statistics of the A. ceratii AT5.2 genome.
Data file S6. Annotation table generated via Trinotate for the A. ceratii AT5.2 genome.