

Abstract
Direct amination of sp3 C–H bonds is of broad interest in the realm of C–H functionalization because of the prevalence of nitrogen heterocycles and amines in pharmaceuticals and natural products. Here, we report a combined electrochemical/photochemical method for dehydrogenative C(sp3)–H/N–H coupling that exhibits good reactivity with both sp2 and sp3 N–H bonds. The results show how use of iodide as an electrochemical mediator, in combination with light-induced cleavage of intermediate N–I bonds, enables the electrochemical process to proceed at low electrode potentials. This approach significantly improves the functional-group compatibility of electrochemical C–H amination, for example, tolerating electron-rich aromatic groups that undergo deleterious side reactions in the presence of high electrode potentials.
Keywords: C-H Functionalization, Amination, Iodine, Electrolysis, Photochemistry
Graphical Abstract
Synergistic benefits: Visible-light irradiation is combined with an electrochemical reaction that uses iodine as a redox mediator to enable efficient intramolecular C–H amination. The combined photo/electrochemical approach allows the reactions to proceed with low anode potentials, which tolerate a wide range of functional groups.
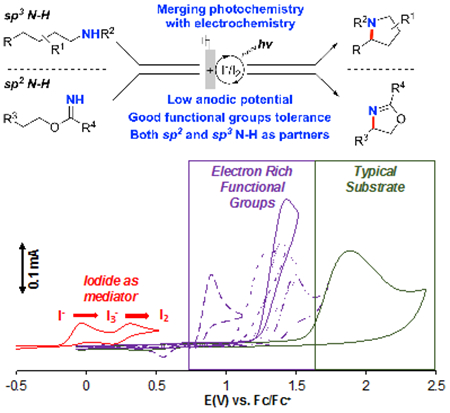
Synthetic methods for electrochemical oxidation of organic molecules are the focus of rapidly increasing interest.[1,2] Such reactions are appealing because protons are often effective as the terminal oxidant, resulting in the generation of H2 as the sole stoichiometric byproduct. Important challenges, however, must be overcome to expand the utility of these methods. For example, high electrode potentials required to initiate electron transfer from an organic molecule can lead to decomposition or oxidative side reactions of ancillary functional groups in the molecule. We have been exploring strategies to enable electrochemical oxidation reactions to proceed at lower electrode potentials.[3] Use of an electron-proton transfer mediator, such as an aminoxyl or imidoxyl species, represents one appealing strategy, as these mediators enable oxidations to proceed at electrode potentials >1 V lower than analogous non-mediated processes.[3b,c] In spite of this progress, however, additional strategies are needed. Hydrogen-atom transfer (HAT) mediators still require high electrode potentials that constrain the functional group compatibility,[3b,4] and we initiated the present study to explore possible methods to address this challenge. Herein, we show that use of iodide mediators in combination with visible-light illumination provides the basis for sp3 C–H amination at low electrode potentials.
Selective formation of C(sp3)–N bonds via C–H/N–H coupling is a transformation of longstanding interest.[5] The Hofmann–Löffler–Freytag (HLF) reaction is a pioneering example of such reactivity,[6,7] and numerous variations of the HLF reaction have emerged for C–H halogenation and nitrogen-heterocycle synthesis.[8,9,10,11] Electrochemical methods, summarized in Figure 1, have been explored as a means to bypass the use of undesirable stoichiometric oxidants or pre-generation of N–X compounds, both of which compromise the atom-economy and other appealing features of these methods. Several distinct mechanistic approaches have been investigated in these electrochemical HLF-type reactions: (i) stepwise ET-PT-ET to generate a benzyl cation, which reacts with an appended nitrogen nucleophile,[12] (ii) proton-coupled electron transfer (PCET) to generate a nitrogen-centered radical that promotes 1,5-HAT, similar to the key C–H activation step in the HLF reaction,[13] and (iii) bromide-mediated formation of an N-bromo intermediate that undergoes thermal N–Br homolysis to achieve the key 1,5-HAT step.[14,15] Each of these examples exhibits limited functional group compatibility, owing to the requirement for high anode potentials (Figure 1B). These issues could be addressed by using a mediator that undergoes regeneration at lower potentials. A number of modified-HLF reactions have been reported with stoichiometric chemical oxidants, catalytic I2, and visible light illumination,[11] raising the possibility that iodide could be used as an electrochemical mediator (Figure 1A, approach iv).[16] The I−/I3−/I2 redox couples are ~ 0.4 V lower than those for bromide[17] and 1–1.5 V lower than the electrode potentials required for the electrochemical PCET and ET-PT-ET reactions (Figure 1B).
Figure 1.
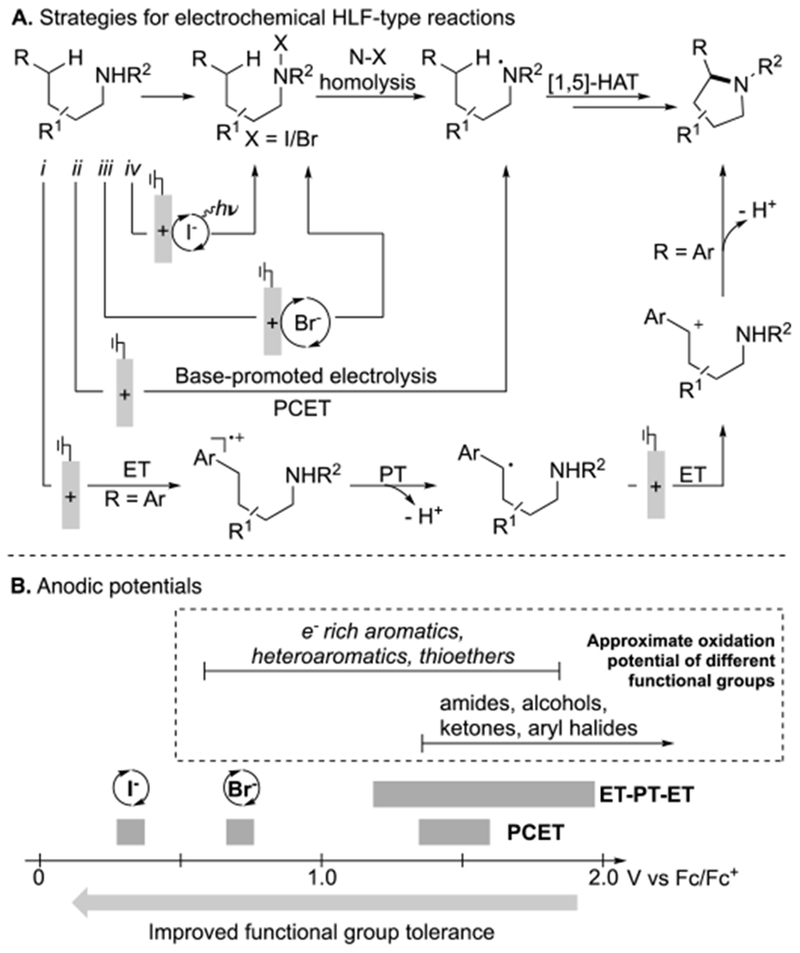
Electrochemical strategies for dehydrogenative amination of sp3 C–H bonds (A) and the associated anodic potentials required for each approach (B). ET = electron transfer, PT = proton transfer, PCET = proton-coupled electron transfer.
Our efforts were initiated by testing the three previously reported electrochemical reaction conditions with two N-alkyl sulfonamide derivatives, 1a and 1b, which differ by the presence of the electron-rich methoxynaphthyl substituent in 1b (Scheme 1). Good reactivity and product yields were observed with 1a under each of the three reported conditions. In contrast, negligible C–N bond formation was observed for 1b, and nearly complete consumption of the starting material led to a complex mixture of products in each case. With these benchmarks established, we turned our attention to electrochemical conditions with iodide as a mediator. Shono’s original report on bromide-mediated C–H amination included one example of iodide-mediated reactivity;[14] however, the N-iodo intermediates tend to undergo beta-elimination of HI to afford imine-derived products under thermal conditions. This insight, together with the use of visible-light promoted homolysis of the N–I bond in PhI(OAc)2/I2-promoted HLF-type reactions, prompted us to consider iodide-mediated electrochemical conditions illuminated by a compact fluorescent light (CFL) bulb.
Scheme 1.
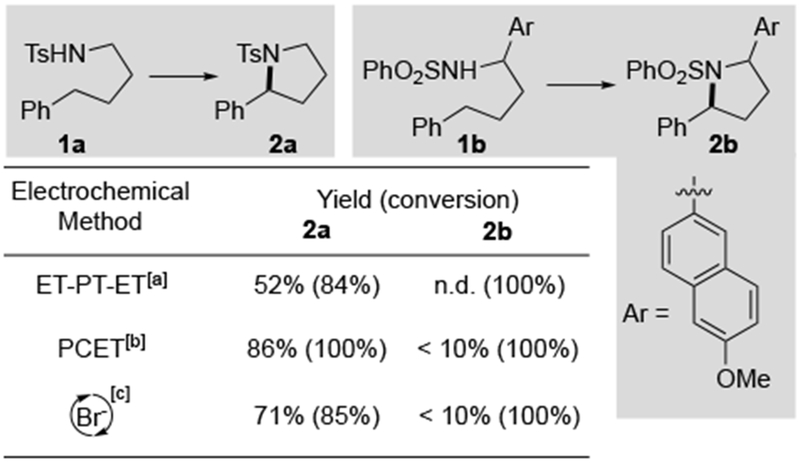
Comparison of previous electrochemical methods for sp3 C–H amination. For detail procedures, see refs. 12–14 and Supporting Information. Yields were determined by 1H NMR with m-xylene as internal standard, conversion is shown in parenthesis. [a] Conditions: 1a or 1b (0.2 mmol) and nBu4NPF6 (0.1 M) in HFIP (10 mL), 2.5 mA, RT. [b] Conditions: 1a or 1b (0.2 mmol), NaOAc (0.2 mmol) and nBu4NBF4 (0.2 mmol) in DCE/HFIP (6 mL, 2:1), 7.5 mA, RT. [c] 1a or 1b (0.4 mmol), NaOMe (0.2 mmol) and KBr (0.2 mmol) in methanol (6 mL) at 65 °C, 100 mA, DCE = 1,2-dichloroethane. HFIP = 1,1,1,3,3,3-hexafluoro-2-propanol. n.d. = not detected.
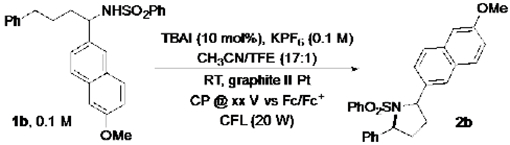
Representative data with 1b are shown in Table 1. The reactions were conducted in an undivided glass electrolysis cell with CFL illumination, using 10% tetrabutylammonium iodide (TBAI) as the mediator, 0.1 M KPF6 as supporting electrolyte, a graphite rod anode, a Pt wire cathode, and TFE as a co-solvent in CH3CN (see Supporting Information for details). An applied potential of 0.3 V vs. Fc/Fc+, which is above the I−/I3− redox wave and near the I3−/I2 mid-point potential (see Figure S1), led to only small amounts of product 2b (4%), with most of the starting material recovered unreacted (81%) (entry 1). Improved yields were observed at a higher applied potential, with a 75% yield obtained at 0.5 V (Table 1, entry 2-3). No product was generated in the absence of mediator, and much lower product yield (19%) was observed in the dark (entries 4-5).
Table 1.
Combined electrochemical/photochemical iodide-mediated process for C–H amination.[a]
 | |||
|---|---|---|---|
| Entry | Potential/V vs Fc/Fc+ | TBAI | Yield/% |
| 1 | 0.3 | 10 mol% | 4% |
| 2 | 0.4 | 10 mol% | 70% |
| 3 | 0.5 | 10 mol% | 75% (72%) |
| 4 | 0.5 | - | n.d. |
| 5[b] | 0.5 | 10 mol% | 19% |
The reaction was performed on a 0.5 mmol scale under constant potential (CP) conditions, yields were determined by 1H NMR with m-xylene as an internal standard; isolated yield shown in parenthesis.
Without irradiation.
TFE = 2,2,2-Trifluoroethanol. n.d. = not detected.
These promising results with a tosylamide substrate prompted us to consider C–H/N–H coupling with a substrate bearing an imidate nucleophile. HLF-type reactivity with this substrate class was recently demonstrated by the groups of Nagib and Chen with stoichiometric chemical oxidants (PhI(OAc)2/I3− and N-iodo- succinimide/Ag2O, respectively),[18,19] but no electrochemical precedent has been reported. The experiments were initiated with 3a, bearing an electron-rich anisole substituent. Using the conditions optimized for 1b, the desired product 4a was obtained in 54% yield with 18% recovered starting material (Table 2, entry 1). A comparable yield was obtained at lower applied potential (0.3 V vs Fc/Fc+, entry 2), while an improved result was obtained at higher potential (0.7 V; entry 3).[20] Inclusion of 1 equivalent of pyridine in the reaction mixture led to further improvement in the yield (82%, entry 4; similar beneficial effects of pyridine were not observed with 1a and 1b – see Supporting Information for details). Control experiments again revealed that no product was observed in the absence of mediator or in the dark (entries 5 and 6).
Table 2.
Combined electrochemical/photochemical iodide-mediated process for dehydrogenative amination of imidate.[a]
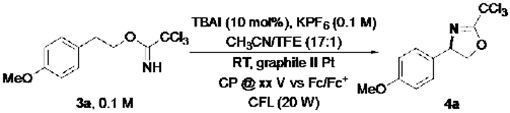 | |||
|---|---|---|---|
| Entry | Potential/V vs Fc/Fc+ | TBAI | Yield/% |
| 1 | 0.5 | 10 mol% | 54% |
| 2 | 0.3 | 10 mol% | 47% |
| 3 | 0.7 | 10 mol% | 71% |
| 4[b] | 0.7 | 10 mol% | 82% (73%) |
| 5[b] | 0.7 | - | n.d. |
| 6[b,c] | 0.7 | 10 mol% | 2% |
The reaction was performed on a 0.5 mmol scale under constant potential (CP) conditions, yields were determined by 1H NMR with m-xylene as an internal standard; isolated yield shown in parenthesis.
With 1 equiv of pyridine.
Without irradiation.
TFE = 2,2,2-Trifluoroethanol. n.d. = not detected.
A number of different N-alkyl sulfonamide and imidate substrates were then evaluated under the optimized iodine-mediated photo/electrochemical conditions to assess their ability to undergo dehydrogenative cyclization (Scheme 2). Scheme 2A shows the results with N-alkyl sulfonamide derivatives, while Scheme 2B illustrates the scope of imidates compatible with the reaction conditions. Substrates of both classes with electron-rich arenes were effectively converted to the corresponding pyrrolidine and oxazoline products under the optimized conditions (2b-2e, 2i and 4a, 4h, 4n, 4o, 4t, respectively). The oxazoline products derived from the imidate cyclization are appealing precursors to 1,2-aminoalcohol derivatives (Scheme 2C).[18a,19]
Scheme 2.
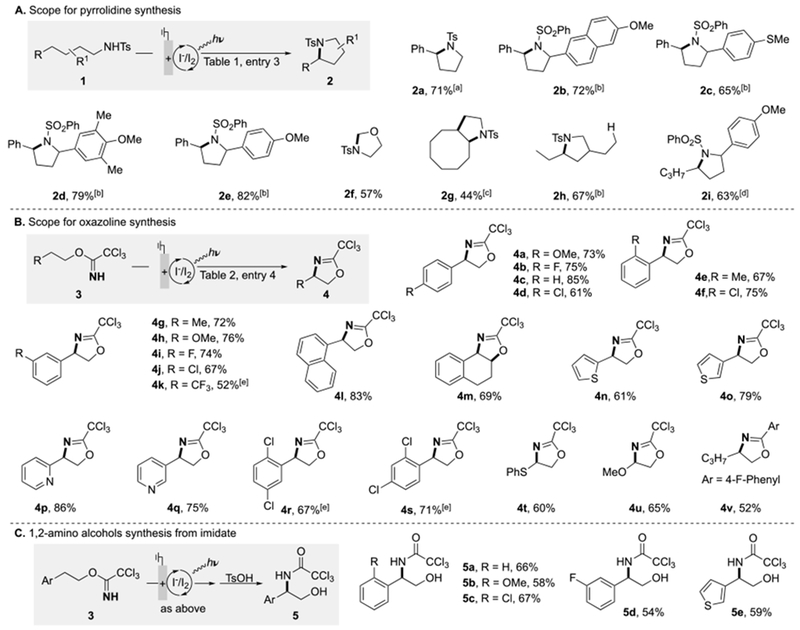
Substrate scope of iodide-mediated dehydrogenative amination. The reaction was conducted on a 0.5 mmol scale, see Supporting Information for details. All yields are isolated yield. [a] 2a has also been produced under conditions with stoichiometric chemical oxidants: PhI(OAc)2/cat. I2, 90%;11a PhI(OAc)2/I3−, 93%;10g mCPBA/cat. I2, 54%;11c [b] dr = 1:1. [c] dr = 1.8:1. [d] dr = 1.2 :1. [e] With 2,6-lutidine instead of pyridine as additive. TsOH = p-toluenesulfonic acid.
The majority of products arise from functionalization of activated C–H bonds (e.g., benzylic or adjacent to a heteroatom), but successful reactivity was also observed with unactivated aliphatic C–H bonds (2g, 2h, 2i, 4v). The generation of 2h reveals the expected preference of secondary over primary C–H functionalization.[10f] Effective substrates feature an array of aromatic substituents, including electron-rich and electron-deficient substituted arenes, in addition to thiophene and pyridine. The good functional group tolerance enabled by merging photochemistry with a low-potential mediator was further demonstrated with an intermolecular additive screening experiment for the reaction of 1a (see Table S5 in the Supporting Information for details).[21] Functional groups shown to be compatible with the reaction conditions include carbazole, (benzo)thiophene, benzofuran, benzothiazole, pyridine, and carbamate derivatives, while partial decomposition and lower yields of 2a were observed with indole, furan and aniline derivatives.
The observed reactivity is readily rationalized by an electrochemical circuit consisting of generation of I2 at the anode and hydrogen evolution at the cathode (Scheme 3A). The latter reaction contributes to the overall catalytic process by generating a Brønsted base that promotes iodination of the N–H substrate, which then initiates the non-electrochemical C–H functionalization sequence that takes place in bulk solution (Scheme 3B).[22] Photolysis contributes to homolysis of the N–I bond and enables formation of the N-centered radical that undergoes 1,5-hydrogen atom transfer (HAT) to generate an alkyl radical. Subsequent trapping of the radical with iodine generates an alkyliodide intermediate that can undergo Brønsted base-promoted nucleophilic displacement by the appended nitrogen nucleophile.
Scheme 3.
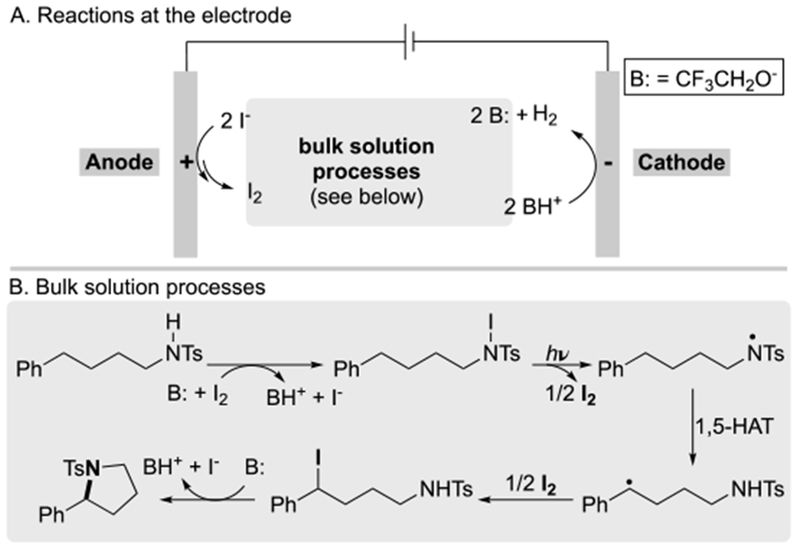
Simplified mechanism for photo/electrochemical iodide-mediated dehydrogenative C–H/N–H coupling.
Functional group tolerance is achieved by the combined use of a low-potential mediator (I−/I2) with photochemical activation of the N-iodo intermediate generated under the reaction conditions.[23] The anodic potential needed to (re)generate I2 at the anode (0.3–0.7 V vs. Fc/Fc+) is well below the redox potential of electron-rich arenes and other substituents. This feature is illustrated in Scheme 4, which shows cyclic voltammograms (CVs) of the iodide mediator, including both the I−/I3− and I3−/I2 redox couples, and several representative substrates. Comparison of the CVs for 1a and the substrates with electron-rich aromatic substituents highlights the functional-group compatibility challenges that will be encountered in reactions initiated by direct electrochemical oxidation of substrate or proton-coupled oxidation of the nitrogen nucleophile (cf. ET-PT-ET and PCET processes in Scheme 1). The ET and PCET steps associated with these reactions often require applied potentials well above that of many valuable functional groups and will initiate substrate decomposition or undesirable side reactions. For example, ET from the arene ring in 1a exhibits a peak potential of 1.9 V (Scheme 4), and the PCET-initiated reactions proceed with anodic potentials of >2 V.[24,25] While the ET-PT-ET and PCET processes will have utility for substrates that tolerate high anode potentials, the magnitude of the applied potential will intrinsically limit the scope.
Scheme 4.
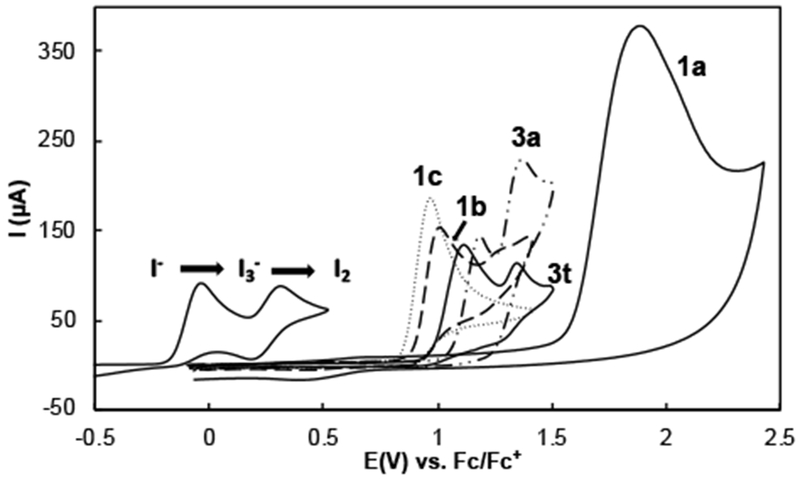
CVs of iodide and representative substrates. Conditions: 5 mM substate in acetonitrile with KPF6 (0.1 M) as supporting electrolyte, glassy carbon as working electrode (~ 7.0 mm2) and a platinum wire counter electrode, scan rate = 100 mV/s.
Overall, the results outlined herein demonstrate a novel photochemical/electrochemical strategy to enable C–H amination reactions to be conducted at lower applied potentials. In addition to the above comparisons with ET-PT-ET and PCET-based reactions, the results may be compared to other electrochemical C–H oxidation reactions that proceed via HAT mechanisms. Phthalimido-N-oxyl (PINO) is perhaps the most widely used HAT reagent in electrochemical C–H oxidation reactions.[2k,3b,4a-d] The I3−/I2 redox potential is ~0.2–0.6 V lower than the potentials needed to generate PINO from N-hydroxyphthalimide (NHPI).[26] This difference is amplified when one considers that the N-centered radicals generated in the present reactions are much stronger H-atom acceptors than PINO (estimated BDEs: NHPI(O–H) ~ 88 kcal/mol, sulfonamide and imidate (N–H) > 100 kcal/mol[27]). Thus, the combination of electrochemical and photochemical energy inputs allows more potent oxidants to be generated at lower electrode potentials, thereby enabling the C–H/N–H dehydrogenative coupling reaction to proceed with broad functional group tolerance. We anticipate that many other electrochemical redox processes will benefit from application of principles similar to those described here.
Supplementary Material
Acknowledgments
Financial support was provided from an SIOC fellowship (F.W.), and the NIH (R01 GM100143 for S.S.S.). Spectroscopic instrumentation was supported by a gift from Paul. J. Bender, the NSF (CHE- 1048642), and the NIH (1S10 OD020022-1).
Footnotes
Conflict of interest:
The authors declare no conflict of interest.
References
- [1].a) Torii S, Novel Trends in Elecroorganic Synthesis, Springer-Verlag, Tokyo, 1998; [Google Scholar]; b) Lund H, Hammerich O, Organic Electrochemistry, 5th ed., CRC Press, Boca Raton, 2015. [Google Scholar]
- [2].For representative recent reviews, see:; a) Sperry JB, Wright DL, Chem. Soc. Rev 2006, 35, 605; [DOI] [PubMed] [Google Scholar]; b) Yoshida J.-i., Kataoka K, Horcajada R, Nagaki A, Chem. Rev 2008, 108, 2265; [DOI] [PubMed] [Google Scholar]; c) Francke R, Beilstein J. Org. Chem. 2014, 10, 2858; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yan M, Kawamata Y, Baran PS, Chem. Rev 2017, 117, 13230; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Jiang Y, Xu K, Zeng C, Chem. Rev 2018, 118, 4485; [DOI] [PubMed] [Google Scholar]; f) Pletcher D, Green RA, Brown RCD, Chem. Rev 2018, 118, 4573; [DOI] [PubMed] [Google Scholar]; g) Tang S, Liu Y, Lei A, Chem 2018, 4, 27; [Google Scholar]; h) Wiebe A, Gieshoff T, Möhle S, Rodrigo E, Zirbes M, Waldvogel SR, Angew. Chem. Int. Ed 2018, 57, 5594; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Möhle S, Zirbes M, Rodrigo E, Gieshoff T, Wiebe A, Waldvogel SR, Angew. Chem. Int. Ed 2018, 57, 6018; [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Yang Q-L, Fang P, Mei T-S, Chin. J. Chem 2018, 36, 338; [Google Scholar]; k) Nutting JE, Rafiee MR, Stahl SS, Chem. Rev 2018, 118, 4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].For examples, see:; a) Badalyan A, Stahl SS, Nature 2016, 535, 406; [DOI] [PubMed] [Google Scholar]; b) Rafiee M, Wang F, Hruszkewycz DP, Stahl SS, J. Am. Chem. Soc 2018, 140, 22; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang F, Rafiee M, Stahl SS, Angew. Chem. Int. Ed 2018, 57, 6686; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lennox AJ, Goes SL, Webster MP, Koolman HF, Djuric SW, Stahl SS, J. Am. Chem. Soc 2018, 140, 11227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Masui M, Hara S, Ueshima T, Kawaguchi T, Ozaki S, Chem. Pharm. Bull 1983, 31, 4209; [Google Scholar]; b) Masui M, Kawaguchi T, Ozaki S, S. J. Chem. Soc., Chem. Commun 1985, 1484; [Google Scholar]; c) Horn EJ, Rosen BR, Chen Y, Tang J, Chen K, Eastgate MD, Baran PS, Nature 2016, 533, 77; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hruszkewycz DP, Miles KC, Thiel OR, Stahl SS, Chem. Sci 2017, 8, 1282; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kawamata Y, Yan M, Liu Z, Bao D-H, Chen J, Starr JT, Baran PS, J. Am. Chem. Soc 2017, 139, 7448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Amino Group Chemistry: From Synthesis to the Life Sciences (Eds.:Ricci A), Wiley-VCH, Weinheim, 2008, pp. 55–92; [Google Scholar]; b) Davies HML, Manning JR, Nature 2008, 451, 417; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Newhouse T, Baran PS, Angew. Chem. Int. Ed 2011, 50, 3362; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Park Y, Kim Y, Chang S, Chem. Rev 2017, 117, 9247. [DOI] [PubMed] [Google Scholar]
- [6].For reviews, see:; a) Wolff ME, Chem. Rev 1963, 63, 55; [Google Scholar]; b) Name Reactions in Heterocyclic Chemistry (Eds.: Li J-J, Corey EJ), Wiley, Hoboken, 2005; [Google Scholar]; c) Stateman LM, Nakafuku KM, Nagib DA,Synthesis 2018, 50, 1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].For early examples, see:; a) Hofmann AW, Ber. Dtsch. Chem. Ges 1879, 12, 984; [Google Scholar]; b) Hofmann AW, Ber. Dtsch. Chem. Ges 1883, 16, 558; [Google Scholar]; c) Löffler K, Freytag C, Ber. Dtsch. Chem. Ges 1909, 42, 3427; [Google Scholar]; d) Corey EJ, Hertler WR, J. Am. Chem. Soc 1960, 82, 1657; [Google Scholar]; e) Baldwin SW, Doll RJ, Tetrahedron Lett. 1979, 20, 3275. [Google Scholar]
- [8].For recent examples with pre-formed N-X species as substrates, see:; a) Togo H, Hoshina Y, Muraki T, Nakayama H, Yokoyama M, J. Org. Chem 1998, 63, 5193; [Google Scholar]; b) Reddy 1, Reddy BVS, Corey EJ, Org. Lett 2006, 8, 2819; [DOI] [PubMed] [Google Scholar]; c) Chen K, Richter JM, Baran PS, J. Am. Chem. Soc 2008, 130, 7247; [DOI] [PubMed] [Google Scholar]; d) Chen K, Baran PS, Nature 2009, 459, 824; [DOI] [PubMed] [Google Scholar]; e) Cherney EC, Lopchuk JM, Green JC, Baran PS, J. Am. Chem. Soc 2014, 136, 12592; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Qin Q, Yu S, Org. Lett 2015, 17, 1894; [DOI] [PubMed] [Google Scholar]; g) Groendyke BJ, Abusalim DI, Cook SP, J. Am. Chem. Soc 2016, 138, 12771; [DOI] [PubMed] [Google Scholar]; h) Short MA, Blackburn JM, Roizen JL, Angew. Chem. Int. Ed 2018, 57, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].For selected examples with N-H substrates, see:; a) Verma A, Patel S, Meenakshi, Kumar A, Yadav A, Kumar S, Jana S, Sharma S, Prasad CD, Kumar S, Chem. Commun 2015, 51, 1371; [DOI] [PubMed] [Google Scholar]; b) O’Broin CQ, Fernández P, Martínez C, Muñiz K, Org. Lett 2016, 18, 436; [DOI] [PubMed] [Google Scholar]; c) Long J, Cao X, Zhu L, Qiu R, Au C-T, Yin S-F, Iwasaki T, Kambe N, Org. Lett 2017, 19, 2793; [DOI] [PubMed] [Google Scholar]; d) Liu T, Myers MC, Yu J-Q, Angew. Chem. Int. Ed 2017, 56, 306; [DOI] [PubMed] [Google Scholar]; e) Sathyamoorthi S, Banerjee S, Du Bois J, Burns NZ, Zare RN, Chem. Sci 2018, 9, 100; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Becker P, Duhamel T, Martínez C, Muñiz K, Angew. Chem. Int. Ed 2018, 57, 5166. [DOI] [PubMed] [Google Scholar]
- [10].For PhI(OAc)2/I2 oxidation systems, see:; a) De Armas P, Carrau R, Concepción JI, Francisco CG, Hernández R, Suárez E, Tetrahedron Lett. 1985, 26, 2493; [Google Scholar]; b) Dorta RL, Francisco CG, Suárez E, J. Chem. Soc., Chem. Commun 1989, 1168; [Google Scholar]; c) Katohgi M, Togo H, Yamaguchi K, Masataka Y, Tetrahedron 1999, 55, 14885; [Google Scholar]; d) Francisco CG, Herrera AJ, Suárez E, J. Org. Chem 2003, 68, 1012; [DOI] [PubMed] [Google Scholar]; e) Fan R, Pu D, Wen F, Wu J, J. Org. Chem 2007, 72, 8994; [DOI] [PubMed] [Google Scholar]; f) Paz NR, Rodríguez-Sosa D, Valdés H, Marticorena R, Melián D, Copano MB, González CC, Herrera AJ, Org. Lett 2015, 17, 2370; [DOI] [PubMed] [Google Scholar]; g) Wappes EA, Fosu SC, Chopko TC, Nagib DA, Angew. Chem. Int. Ed 2016, 55, 9974. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Courtneidge JL, Lusztyk J, Pagé D, Tetrahedron Lett. 1994, 35, 1003. [Google Scholar]
- [11].For examples of I2-catalyzed HLF reactions with various oxidants, see:; a) Martínez C, Muñiz K, Angew. Chem. Int. Ed 2015, 54, 8287; [DOI] [PubMed] [Google Scholar]; b) Becker P, Duhamel T, Stein CJ, Reiher M, Muñiz K, Angew. Chem. Int. Ed 2017, 56, 8004; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Duhamel T, Stein CJ, Martínez C, Reiher M, Muñiz K, ACS Catal. 2018, 8, 3918; [Google Scholar]; d) Stateman LM, Wappes EA, Nakafuku KM, Edwardsand KM, Nagib DA, Chem. Sci 2019, DOI: 10.1039/c8sc05685d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Herold S, Bafaluy D, Muñiz K, Green Chem, 2018, 20, 3191. [Google Scholar]
- [13].Hu X, Zhang G, Bu F, Nie L, Lei A, ACS Catal. 2018, 8, 9370. [Google Scholar]
- [14].Shono T, Matsumura Y, Katoh S, Takeuchi K, Sasaki K, Kamada T, Shimizu R, J. Am. Chem. Soc 1990, 112, 2368. [Google Scholar]
- [15].See also:; Zhang S, Li L, Xue M, Zhang R, Xu K, Zeng C, Org. Lett 2018, 20, 3443. [DOI] [PubMed] [Google Scholar]
- [16].For recent review on iodide-mediated electrolysis, see:; Liu K, Song C, Lei A, Org. Biomol. Chem 2018, 16, 2375. [DOI] [PubMed] [Google Scholar]; For representative examples, see:; b) Gao W-J, Li W-C, Zeng C-C, Tian H-Y, Hu L-M, Little RD, J. Org. Chem 2014, 79, 9613; [DOI] [PubMed] [Google Scholar]; c) Chen J, Yan W-Q, Lam CM, Zeng C-C, Hu L-M, Little RD, Org. Lett 2015, 17, 986; [DOI] [PubMed] [Google Scholar]; d) Liang S, Zeng C-C, Tian H-Y, Sun B-G, Luo X-G, Ren F-Z, J. Org. Chem 2016, 81, 11565; [DOI] [PubMed] [Google Scholar]; e) Wang Q-Q, Xu K, Jiang Y-Y, Liu Y-G, Sun B-G, Zeng C-C, Org. Lett 2017, 19, 5517; [DOI] [PubMed] [Google Scholar]; f) Tang S, Gao X, Lei A, Chem. Commun 2017, 53, 3354; [DOI] [PubMed] [Google Scholar]; g) Wang H, Zhang J, Tan J, Xin L, Li Y, Zhang S, Xu K, Org. Lett 2018, 20, 2505. [DOI] [PubMed] [Google Scholar]
- [17].For CV studies of TBAI and TEABr in CH3CN with KPF6 as supporting electrolyte, see Supporting Information.
- [18].See the following and ref. 11d;; Wappes EA, Nakafuku KM, Nagib DA, J. Am. Chem. Soc 2017, 139, 10204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mou X-Q, Chen X-Y, Chen G, He G, Chem. Commun 2018, 54, 515. [DOI] [PubMed] [Google Scholar]
- [20].Although it is unclear at this stage, the higher applied potential needed to get excellent yield possibly stems from the deactivation of electrode during bulk electrolysis.
- [21].Collins KD, Glorius F, Acc. Chem. Res 2015, 48, 619. [DOI] [PubMed] [Google Scholar]
- [22].See Supporting Information for control experiments with stoichiometric iodine as oxidant in the presence of different bases.
- [23].See the Supporting Information for additive screening experiments under the present iodide-mediated photo/electrochemical conditions.
- [24].The PCET-initiated reactions in ref. 13 were conducted under constant current conditions. The >2 V value noted here was determined by monitoring the anode potential while reproducing the reported conditions.
- [25].See Supporting Information for CV studies of TsNHMe with different bases.
- [26].NHPI oxidation takes place at ~0.5–0.9 V vs. Fc/Fc+, depending on the identity of the proton acceptor. The potential is lower with stronger bases, but PINO also decomposes more rapidly under basic conditions. See refs. 2k and 3b for further discussion.
- [27].a) Luo Y-R, Handbook of Bond Dissociation Energies in Organic Compounds, CRC, Boca Raton, FL, 2002; [Google Scholar]; b) Šakić D, Zipse H, Adv. Synth. Catal 2016, 358, 3983. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.