

Abstract
Traumatic joint injuries produce osteoarthritic cartilage manifesting accelerated chondrocyte terminal differentiation and matrix degradation via unknown cellular and molecular mechanisms. Here we report the ability of biomechanical stress to increase expression of the calcium-sensing receptor (CaSR), a pivotal driver of chondrocyte terminal differentiation, in cultured chondrogenic cells subjected to fluid flow shear stress (FFSS) and in chondrocytes of rodent temporomandibular joint (TMJ) cartilage subjected to unilateral anterior cross-bite (UAC). In cultured ATDC5 cells or TMJ chondrocytes, FFSS induced Ca2+-loading and CaSR localization in endoplasmic reticulum (ER), casually accelerating cell differentiation that could be abrogated by emptying ER Ca2+ stores or CaSR knockdown. Likewise, acute chondrocyte-specific Casr knockout (KO) prevented the UAC-induced acceleration of chondrocyte terminal differentiation and matrix degradation in TMJ cartilage in mice. More importantly, local injections of CaSR antagonist, NPS2143, replicated the effects of Casr KO in preventing the development of osteoarthritic phenotypes in TMJ cartilage of the UAC-treated rats. Our study revealed a novel pathological action of CaSR in development of osteoarthritic cartilage due to aberrant mechanical stimuli and supports a therapeutic potential of calcilytics in preventing osteoarthritis in temporomandibular Joints by targeting the CaSR.
Keywords: calcium-sensing receptor, osteoarthritis, chondrocyte differentiation, dental malocclusion, temporomandibular joint
Introduction
Osteoarthritis (OA) is a prevalent disorder characterized by accelerated chondrocyte terminal differentiation and extracellular matrix degradation in the cartilage of movable joints (1,2). OA is often caused by acute and chronic traumatic joint injuries (3). It is generalized that alteration in chondrocyte terminal differentiation (4,5) by biomechanical stress precedes morphological and functional derangement in the affected cartilage (6). However, the mechanisms altering chondrocyte terminal differentiation remain unclear, representing a major obstacle in treating the disease.
The calcium-sensing receptor (CaSR), which is a member of family C G protein-coupled receptor (GPCR-C), senses changes in serum [Ca2+] in parathyroid glands to regulate parathyroid hormone (PTH) to control mineral and skeletal homeostasis (7,8). The CaSR is abundantly expressed in maturing and hypertrophic chondrocytes in growth plates (9) and in chondrocytes in deep layers of articular cartilage (10) with localization on cell membrane and intracellularly in Golgi and ER. In cultured chondrocytes, activation of CaSR by high extracellular [Ca2+] ([Ca2+]e) or orthosteric agonists promotes terminal differentiation and mineralizing functions of the cells (11,12). Furthermore, ablating CaSR expression specifically in chondrocytes in embryos or post-natally delays terminal cell differentiation and growth plate development, resulting in shortened skeleton and dwarfism in mice (9). Given that chondrocyte terminal differentiation is accelerated (13–15) and CaSR expression is upregulated in OA cartilage (10), we hypothesize that this receptor is involved in the induction of maladaptive chondrocyte terminal differentiation and the resulting OA phenotypes in response to mechanical assaults.
Dental malocclusion can cause OA in temporomandibular joint (TMJ) (16,17). In rodents, anterior cross-bite (UAC) imposed by dental prosthetics produces OA lesions in their TMJ cartilage as indicated by accelerated chondrocyte terminal differentiation and degradation of extracellular matrix (18–20). Herein, we first investigated the impact of biomechanical stresses on CaSR expression and terminal cell differentiation by studying the effect of fluid flow shear stress (FFSS) on isolated TMJ chondrocytes and chondrogenic ATDC5 cells. As majority of CaSR protein is localized to intracellular organelles (8,21,22) of chondrocytes, we focused on impact of Ca2+-loading and CaSR expression in endoplasmic reticulum (ER). We took genetic and/or pharmacological approaches to delineate the role of CaSR in OA development in TMJs of mice and/or rats subjected to UAC and explore the therapeutic potential of CaSR for OA treatment.
Materials and Methods
Animal Studies
Application of the unilateral anterior cross-bite (UAC) prosthesis
Male Tamoxifen (Tam)-inducible (Tam-CartCasrflox/flox) mice, which expresses Cre-ERT2 under the control of a Col-II promoter (Tam-CartCre) and Casrflox/flox alleles, were bred with female Casrflox/flox mice to produce Tam-CartCasrflox/flox and control Casrflox/flox littermates as described previously (9). Six-week-old female Tam-CartCasrflox/flox mice were injected with 5 consecutive daily doses of Tamoxifen (100 mg/kg, 06734, Sigma-Aldrich) to induce ablation of Casr genes in chondrocytes in the resulting Tam-CartCasrΔflox/Δflox (KO) mice. Female Casrflox/flox littermates were subjected to the same Tam injection schedule as “control” or “Cont”. This proof-of-principle study only focused on female mice or rat as clinical studies showed higher prevalence of TMJ OA in female vs. male patients (23). All KO and Cont mice were randomized into groups and subjected to UAC procedure as detailed previously (18,20), beginning immediately after the second Tamoxifen injection for 3 or 7 weeks before tissue harvests and analyses. Six-week-old female Sprague-Dawley rats obtained from the Animal Center of the Fourth Military Medical University (FMMU) were subjected to UAC for 2, 4, or 8 weeks before tissue analyses. No significant difference was noticed in manifestation of OA phenotype between right and left TMJs in UAC or sham group, so both joints were analyzed.
All animal procedures were approved by the Ethics Committee of School of Stomatology and carried out in accordance with the recommendations by the Committee of Care and Use of Laboratory Animals of the FMMU to minimize animal stresses. All animals were housed in pathogen-free room and fed with sterilized food and distilled water during the study. No more than four animals were housed in a single cage (measuring 50 × 40 × 25 cm) at ambient temperature of 20 ± 2°C and humidity of 55% ± 5%, with good ventilation and 12/12-hrs dark/light cycles (4 W per square meter). Sterilized wood-chip bedding was replaced every other day. Animal health status was monitored twice daily. All animals were healthy from the beginning to the end of the study. No adverse events other than TMJ pathology were observed. All animals were euthanized by a single intraperitoneal injection of overdosed pentobarbital sodium before tissue and blood harvests. This study complied with ARRIVE guidelines for preclinical animal studies.
UAC application
UAC was applied by a pair of metal prosthetics. Briefly, for rat, a small metal tube (length = 2.5 mm, inside diameter = 3 mm) was fitted onto left maxillary incisor and a larger tube (length = 4.5 mm, inside diameter = 3.5 mm) for left mandibular incisor. The end of the large tube was bended to create a 135°-angle leaning toward labial side to create a cross-bite relationship between the top and bottom incisors. Similar prosthetics was made from smaller metal tubes (inner diameter =0.61 mm, thickness = 0.3 mm) for mouse incisors.
Intra-TMJ injections in rats
Fifty microliters of NPS2143 (10−7 M in PBS, S2633, Selleckchem), Cinacalcet (10−7 M in PBS, S1260, Selleckchem) or vehicle (PBS) were injected into TMJs of anesthetized rats, beginning 4 weeks after UAC. The injections were performed by inserting a 100-μl Hamilton needle below the zygomatic arch between the eye and ear until it reached posterior mandibular ramus, which was then used as a reference point for repositioning the needle head to the outside of TMJ for final penetration and then drug delivery. Both TMJs in each animal were injected every other day for 4 weeks before tissue harvests for analyses.
Cell culture and FFSS application
Primary chondrocytes were isolated from TMJs of six-week-old female Sprague–Dawley rats (the Animal Center of the FMMU) or knee joints of six-week-old female Casrflox/flox mice as described previously (12). Briefly, cartilage deprived of subchondral bone was micro-dissected from the condyles of rat TMJ or femur head and tibial plateau of mouse knee joints, minced, and digested with 0.25% trypsin (T4674, Sigma) for 20 min to first remove outer fibrous tissues, followed by 0.2% type II collagenase (17101015, Gibco) for 2–3 hrs to release chondrocytes. Isolated chondrocytes as well as ATDC5 cells were cultured at a density of 5×103/cm3 in DMEM with 10% FCS on type I collagen-coated glass slides (Flexcell) for 48 hrs and then exposed to steady laminar flow (16 dyn/cm2) for 1 hr before protein and RNA harvests. To study the impact of calcium influx and CaSR-mediated activities on chondrocyte terminal differentiation, cells grew in DMEN with 10% FCS for 48 hrs were switched to fresh media with or without NPS2143 (10−5 M) or a FCS- and Ca2+-free medium (SH30262.01, Hyclone) before 1-hr FFSS treatment. To study the impact of ER Ca2+ stores, cells were pretreated with thapsigargin (TG) (10−6 M, ab120286, Abcam) for 2 hrs before exposure to FFSS in the FCS- and Ca2+-free medium. NPS2143 and TG were dissolved in dimethyl sulfoxide (DMSO) to make 10−1 M stocks and then diluted in culture medium to final concentrations of 10−5 M and 10−6 M, respectively. The trace amount (0.01–0.001%) of DMSO, added along with TG or NPS2143, had no acute effect on the expression of CaSR and chondrocyte differentiation markers, when compared to controls without drug (Supplemental Fig. 1). Therefore, the latter controls were presented.
To study the impact of Casr gene KO, chondrocytes cultured from the Casrflox/flox mice were pre-incubated with complexes of Cre recombinase (SCR508, Millipore) and EndoFectin™ transfection reagent (Z01010, GeneCopoeia) for 2 hrs in FCS-free medium (24) and then cultured in DMEM with 10% FCS for 24hrs before the FFSS and drug treatments described above. After FFSS stimulation for 1 hr, primary chondrocytes and chondrogenic cells were analyzed.
siRNA transfection
ATDC5 cells, cultured at a density of 5×103/cm3 in DMEM medium with 10% FCS, were transfected with CaSR-siRNA (5 nM, 5’-GAGUGCAUCAGGUAUAACUTTA GUUAUACCUGAUGCACUCTT-3’) or control siRNA (5 nM, 5’-UUCUCCGAACGUGUCACGUTTACGUGACACGUUCGGAGAATT-3’) (GenePharma) premixed with Lipofectamine 2000 (11668–027, Invitrogen) for 6 hrs. Cells were then cultured in fresh DMEM with 10% FCS for additional 24 hrs before RNA preparation for qPCR assay, or 72 hrs before protein preparations for Western blotting or FFSS experiments.
Histochemistry, Immunohistochemistry, and OARSI Score
TMJs were fixed in 4% paraformaldehyde, decalcified with 4% EDTA for 4 weeks, dehydrated in ethanol, embedded in paraffin, and cut sagittally into 5μm-thick sections. The most central sagittal sections of each joint were selected for safranin O staining (S2255, Sigma-Aldrich) and immunohistochemical detection with antibodies for CaSR (2 μg/ml, ab19347, Abcam), PTH/PTHrP receptor 1 (PPR) (2 μg/ml, ab7150, Abcam) and type X collagen (Col-X) (2 μg/ml, ab58632, Abcam). For negative controls, non-immune goat serum was substituted for the primary antibody.
To quantitate the morphological changes, images of safranin O-stained condylar cartilage was equally divided into three sections (anterior, middle and posterior). A region of interest (ROI) with a width of 200 μm for rat and 70 μm for mouse was placed at the center of each section and an OA grade (0-VI) was assigned based on the previously reported OARSI score system (25) to indicate the degree of TMJ OA progression (26). The values from three ROIs was averaged and reported for each sample.
RNA extraction and qPCR
Total RNA was extracted from condylar cartilage free of bone and fibrous tissues and cultured cells using the RNeasy Fibrous Tissue Midi Kit (75742, QIAGEN), re-purified by the RNeasy mini kit (74104, QIAGEN), reverse-transcribed into cDNA, and analyzed by an Applied Biosystems 7500 thermocycler using custom-made primers (Table.1). The amount of target cDNA, relative to GAPDH, was calculated using the formula 2−ΔCt. Each cartilage or cell sample was analyzed in triplicate and each mean value was normalized to the control group as specified.
Table.1.
Gene primers
| Genes | Forward primer | Reverse primer |
|---|---|---|
| CaSR (rat) | TTTGGAGTAGCAGCCAAAGATCAAG | ACCATCGGAATCCACGGAAG |
| Ppr (rat) | TGGATGCGGACGATGTCTTTAC | CCAGCCCTTGTCTGACTCCATTA |
| Alp (rat) | CACGTTGACTGTGGTTACTGCTGA | CCTTGTAACCAGGCCCGTTG |
| Osteocalcin (rat) | AAGGTGGTGAATAGACTCCG | AAACGGTGGTGCCATAGATG |
| Mmp13 (rat) | TGATGATGAAACCTGGACAAGCA | GAACGTCATCATCTGGGAGCA |
| Col-II(rat) | CGCCACGGTCCTACAATGTC | GTCACCTCTGGGTCCTTGTTCAC |
| Aggrecan (rat) | TGGCATTGAGGACAGCGAAG | TCCAGTGTGTAGCGTGTGGAAATAG |
| Runx2 (rat) | CATGGCCGGGAATGATGAG | TGTGAAGACCGTTATGGTCAAAGTG |
| Gapdh (rat) | GGCACAGTCAAGGCTGAGAATG | ATGGTGGTGAAGACGCCAGTA |
| CaSR (mouse) | TTTGGAGTAGCAGCCAAAGATCAAG | ACCATCGGAATCCACGGAAG |
| Ppr (mouse) | CCAGTGCTCAGCTCCGCATA | TCCTTGAGCAGCTTGTCACATTG |
| Alp (mouse) | CCCTCTTCCCACCATCTG | GGTCTCTTGGGCTTGCTG |
| Osteocalcin (mouse) | CTGCTCACTCTGCTGACC | GGACTGAGGCTCCAAGGT |
| Mmp13 (mouse) | TCCCTGGAATTGGCAACAAAG | GCATGACTCTCACAATGCGATTAC |
| Col-II(mouse) | CATCCAGGGCTCCAATGATGTA | ATGTCCATGGGTGCGATGTC |
| Aggrecan (mouse) | TTCCACCAGTGCGATGCAG | TGGTGTCCCGGATTCCGTA |
| Runx2 (mouse) | GATGAGCGACGTGAGCC | ATGGTGCGGTTGTCGTG |
| Gapdh (mouse) | TGTGTCCGTCGTGGATCTGA | TTGCTGTTGAAGTCGCAGGAG |
Extraction of endoplasmic reticulum protein
Cultured chondrocytes and ATDC5 cells were washed twice with cold PBS and collected with a fractionation buffer (20 mM HEPES, pH 7.4, 250 mM sucrose, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, and 1 mM EGTA) supplemented with proteases inhibitor PMSF (1 mM) and DTT (1 mM). Cell suspension was then passed through a 25-gage needle 10 times, incubated on ice for 20 min, and centrifuged at 800g for 5 min at 4°C to remove nuclei and unbroken cells. The nucleus-free supernatant was centrifuged at 10,000g for 15 min at 4°C to remove mitochondria and then at 100,000g for 1 hr to obtain ER fractions. The ER fraction pellets were washed and resuspended in the fractionation buffer, passed through a 25-gage needle 10 times, and centrifuged as described above. After centrifugation, ER proteins were extracted by a PBS buffer containing 10% glycerol and 0.1% SDS for immunoblotting analyses.
Immunoblotting analyses
Total and ER protein (50μg), extracted from cartilage, primary chondrocytes, or ATDC5 cells, was quantified by Bradford assay (Beyotime Biotechnology) electrophorized on SDS-PAGE gel, transferred onto a nitrocellulose membrane, and blotted with primary antibodies against CaSR (2 μg/ml), PPR (2 μg/ml), G6Pase (2 μg/ml, orb6097, Biorbyt), type X collagen (Col-II) (2 μg/ml, ab34712, Abcam), Aggrecan (2 μg/ml, sc-16492, Santa Cruz) or β-actin (1 μg/ml, ab8226, Abcam), followed by a horseradish peroxidase-conjugated secondary antibody (1 μg/ml, A0216 or A0208, Beyotime Biotechnology). Immunoreactivity was detected by a Chemiluminescent HRP Substrate kit (P90718, Millipore) and quantified by Image Lab software (version 5.1, Bio-Rad). Each protein sample was analyzed three times and the mean intensities or their ratios over β-actin were reported.
Fluorescent immunocytochemistry
For dual fluorescent detection of CaSR and G6Pase, primary chondrocytes and ATDC5 cells were cultured on coverslips, fixed in 4% paraformaldehyde, and incubated with the CaSR-antibody (2 μg/ml) and G6Pase-antibody (2 μg/ml) at 4°C overnight. After incubation with fluoro-conjugated secondary antibodies (1 μg/ml, ab6785 orab6939, Abcam) for 30 min at 37°C, cells were counter-stained with DAPI and mounted on glass slides for visualization by a confocal microscope (FV1000, OLYMPUS). For dual fluorescent staining of Ca2+ and ER, cells were incubated with ER-Tracker Red (E34250, Thermo-Fisher) for 20 min at 37°C and then with Fluo-8 (ab142773, Abcam) for 1 hr at room temperature, before DAPI staining.
Transmission electron microscopy
The dissected TMJs, primary chondrocytes, and ATDC5 cells were fixed in 2.5% glutaraldehyde in a phosphate buffer (pH 7.2) for 24 hrs, decalcified in 10% EDTA for 1 month (cartilage only), post-fixed in 1% osmium tetroxide for 1hr, dehydrated in ethanol, perfused with propylene oxide, embedded in Epon812. Ultrathin sections (40 nm thickness) were cut, stained with uranyl acetate and lead citrate, and examined by a transmission electron microscope (H-600, Hitachi).
Plasma levels of PTH detection
Blood samples were collected from caudal veins of rats one hours after NPS2143 or Cinacalcet injection on the day before and one week after the drug treatments. Blood samples were mixed with heparin sodium and centrifuged at 3,000 rpm for 5 min to separate the plasm. Plasma PTH levels were determined using the rat PTH ELISA kit (CSB-E07866r, Cusabio).
Statistics
Data presented as mean ± SD. Statistical analysis was performed using SPSS software, version 11.0. Before comparison, the normality of data distribution was tested by Shapiroe-Wilk test and Levene’s test was used to assess homogeneity of variance. Comparison between two groups was performed by 2-tails Student’s t-test. Comparison among 3 or more groups, one-way ANOVA was performed, followed by Tukey’s Post Hoc tests to evaluate the statistical significance for pairwise comparisons. The statistical significance was defined as p < 0.05.
Results
FFSS accelerates terminal differentiation of chondrocyte by stimulating Ca2+-loading and CaSR expression in ERs.
We examined the impact of biomechanical stimuli on chondrocyte terminal differentiation by culturing rat TMJ chondrocytes and subjecting them to FFSS (16 dyn/cm2) for 1hr (27). Acute FFSS induced only very modest cell death (Supplemental Fig. 2A), increased expression of terminal differentiation markers (Mmp13, Alp, Osteocalcin, and Runx2), and suppressed expression of early chondrogenic genes [Aggrecan, the α1 unit of the type II collagen (Col-II) and PTH/PTHrP receptor 1 (PPR)] (Fig. 1A), indicating accelerated terminal differentiation in these TMJ chondrocytes.
Fig. 1.
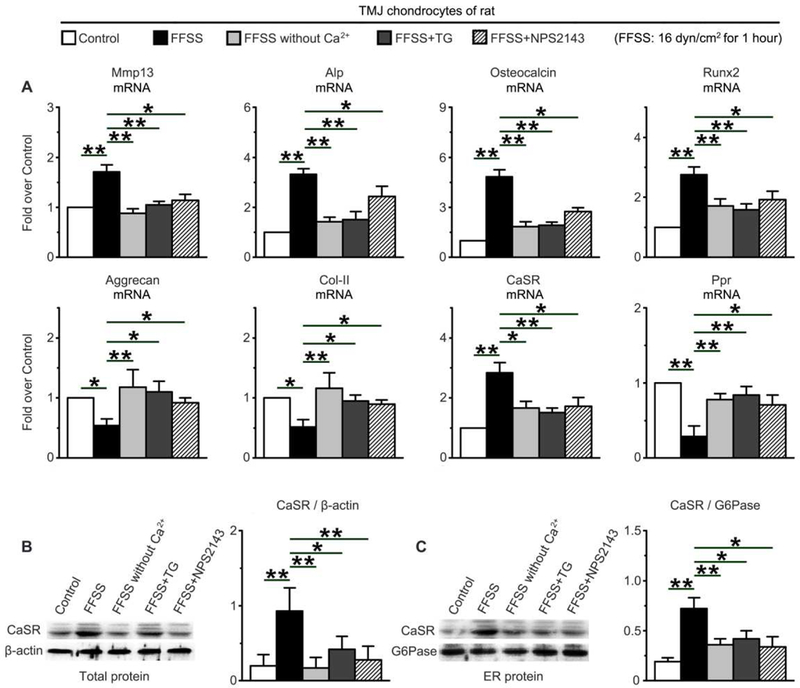
Effects of FFSS (16dyn/cm2 for 1hr) on expression of cell differentiation marker and expression and localization of CaSR protein in ERs in cultured rat TMJ chondrocytes with or without Ca2+, TG (10−6M), or NPS2143 (10−5M). (A) mRNA expression profiles by quantitative real-time PCR (qPCR) showed the ability of FFSS to upregulate the expression of terminal differentiation markers (Mmp13, ALP, Osteocalcin, and Runx2) and CaSR and downregulate the expression of early differentiation makers (Aggrecan, Col-II and PPR). These effects were abrogated by removal of extracellular Ca2+ or treatments with TG or NPS2143. The mRNA expression levels were normalized to the expression of housekeeping GAPDH and presented as fold-increase over control without FFSS treatment. (B,C) Immunoblotting of (B) total and (C) ER proteins with anti-CaSR antibody demonstrated the ability of FFSS to increase the expression of CaSR protein and its localization in ERs and the ability of Ca2+-free medium, TG, and NPS2143 to abrogate these changes. β-actin and G6Pase were used as loading controls for total and ER protein, respectively, and for normalization to calculate the expression ratios in the histograms. n=4 separate cultures. Control, cultured cells without FFSS or other treatment. Values are presented as the mean±SD. **p<0.01, *p<0.05 between groups as specified by top horizontal bars in each panel.
Previous studies have shown that Ca2+ influx is one of early cellular responses to mechanical stimulus (32,33) that increases cytoplasmic [Ca2+], refills intracellular Ca2+ stores, and subsequently alters chondrocyte functions (34,35). However, mechanisms coupling those Ca2+-mobilizing activities to chondrocyte terminal differentiation are unclear. Interestingly, along with the accelerated terminal differentiation, FFSS significantly increased CaSR mRNA (Fig. 1A) and protein expression (Fig. 1B) and its ER localization (Fig. 1C and 2A) in cultured rat TMJ chondrocytes. These cellular changes coincided with increases in ER [Ca2+] (or Ca2+ loading), as indicated by increased Flou-8 signal (green) that was co-localized with ER-tracker (red) (Fig. 2B), 1 hr after FFSS stimulation. The increased ER [Ca2+] was corroborated by profound ER swelling, a morphological indicator of Ca2+ loading (36) (Fig. 2C). The FFSS-induced CaSR expression, Ca2+ loading, and swelling of ERs required both Ca2+ entry and refilling activities, as they could be diminished by removing extracellular Ca2+ or emptying ER Ca2+ store using Thapsigargin (TG), respectively, during FFSS treatments (Fig. 2A, 2B, 2C and 1C). Interestingly, in the latter two conditions, the ability of FFSS to promote expression of terminal chondrocyte markers was also significantly attenuated (Fig. 1A), supporting a causal role for the ER CaSRs and Ca2+ loading in transducing biomechanical stimuli to down-stream cellular responses that control terminal differentiation of TMJ chondrocyte. Adding trace amount (0.01–0.001%) of DMSO (vehicle) that was included in the drug treatments did not affect the gene expression (Supplemental Fig. 1) in the cultured chondrocytes, supporting the specificity of TG.
Fig. 2.
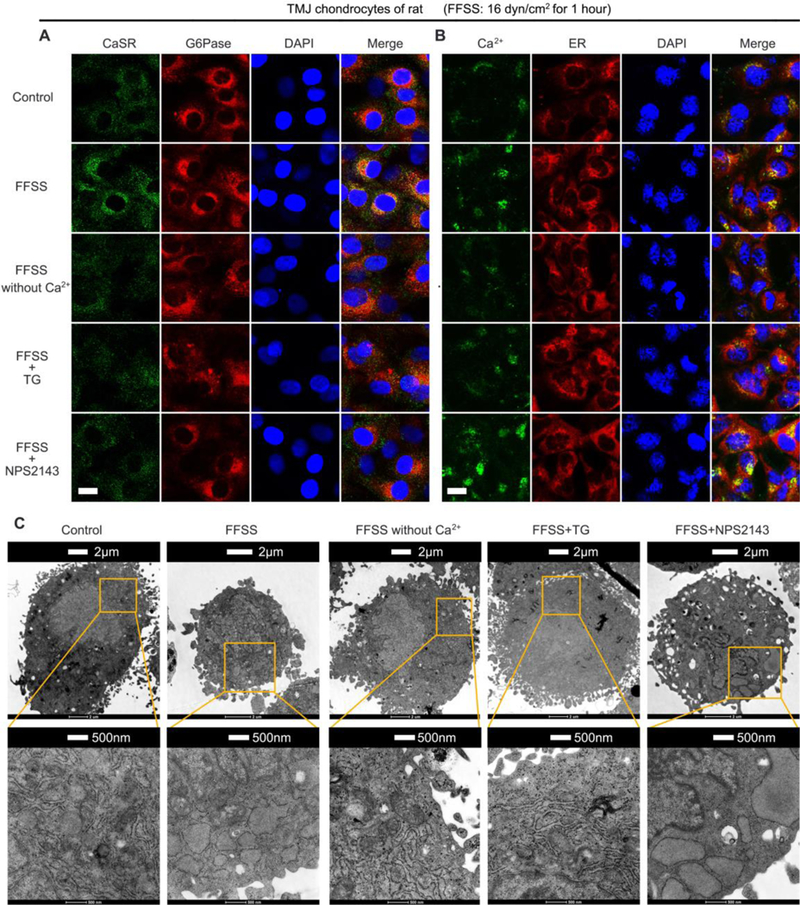
Effects of FFSS (16dyn/cm2 for 1hr) on (A) CaSR localization, (B) Ca2+ loading, and (C) organelle swelling in ERs of rat TMJ chondrocytes cultured with or without extracellular Ca2+, TG (10−6M), or NPS2143 (10−5M). (A) Dual-fluorescence staining with anti-CaSR (in green) and anti-glucose-6-phosphatase (anti-G6Pase, in red) antibodies showed the ability of FFSS to increase CaSR localization in ERs and the ability of Ca2+-free medium, TG or NPS2143 to abrogate this effect. (B) Dual-fluorescence staining with Fluo-8 (in green) and ER-tracker (in red) showed the ability of FFSS to increase [Ca2+] in ERs. This effect was abrogated by treatments of the cells with Ca2+-free medium or TG, but not with NPS2143. n=4 separate cultures. Bar=5μ,m. (C) Representative transmission electron microscopy (TEM) images showed the ability of FFSS to induce swelling of mitochondria and ERs. These morphological effects by FFSS could be abrogated by treatments Ca2+-free medium or TG, but not with NPS2143. Control, cultured cells without FFSS or other treatment.
In ER, the CaSR is assumed to situate in lipid bilayers with its Ca2+-binding domain facing the lumen of the organelle (22), enabling it to sense changes in luminal [Ca2+]. To examine whether raising ER [Ca2+] activates CaSRs to sustain its expression in ER and mediate terminal differentiation of chondrocyte, we treated rat TMJ chondrocytes with NPS2143, a hydrophobic membrane-permeable orthosteric CaSR antagonist, during FFSS treatments. Indeed, NPS2143, which did not block the ability of FFSS to increase ER [Ca2+] (Fig. 2B) or cause ER swelling (Fig. 2C), prevented an increase in CaSR expression in ERs (Fig. 2A and 1C) and attenuated the expression of terminal differentiation markers (Fig. 1A) in the FFSS-treated TMJ chondrocytes. In the absence of FFSS treatment, the removal of extracellular Ca2+ or the treatment with TG or NPS2143 did not significantly affect the expression of ER CaSR and terminal differentiation markers (Supplemental Fig. 3) and ER Ca2+-loading and swelling (Supplemental Fig. 4) in TMJ chondrocytes, albeit a trending of decrease, likely due to the relatively short period (1 hr) of the treatments.
To further establish the role of CaSR in mediating FFSS-induced chondrocyte terminal differentiation , we attempted to examine the impact of CaSR knockdown on rat TMJ chondrocytes by the siRNA approach. However, significant cell losses after siRNA transfection prevented accurate assessments of the impact of FFSS. Alternatively, we performed RNA knockdown on chondrogenic ATDC5 cells. CaSR siRNA (1 and 5 nM) suppressed both CaSR RNA and protein expression in a dose-dependent manner when compared to the cells transfected with scramble RNA (Supplemental Fig. 5). As seen in TMJ chondrocytes, FBSS promoted CaSR expression and its localization in ERs (Supplemental Fig. 6A, 6B), increased ER Ca2+-loading (Supplemental Fig. 6C) and swelling (Supplemental Fig. 7B), and altered gene-expressing profiles indicative of accelerated chondrocyte terminal differentiation (Supplemental Fig. 7A) in the control ATDC 5 cells or cell transfected with scramble RNA. In contrast, ablation of CaSR by siRNA completely or partially blocked the effects of FBSS (Supplemental Fig. 5–7), supporting a critical role of the CaSR in mediating chondrocytic responses to mechanical stimuli.
Next, we studied the effects of Casr gene KO on differentiation of chondrocytes cultured from the knee joints of Casrflox/flox mice by Cre-loxP recombination to further validate the CaSR actions in native articular chondrocytes. We were unable to perform these experiments on mouse TMJ chondrocytes due to inability to isolate sufficient amounts of cell from mouse TMJ cartilage. The cultured chondorcytes were incubated with Cre recombinase for 24 hrs to induce Casr gene excision before they were subjected to FFSS. As shown in Supplemental Fig. 8, expression of CaSR protein and RNA was reduced by >80% in the Cre-treated articular chondrocytes vs. non-treated or vehicle-treated controls. As seen in the studies of rat TMJ chondrocytes and ATDC5 cells, in the mouse articular chondrocytes treated with vehicle, FFSS increased ER [Ca2+] (Fig. 3A), enhanced expression of CaSR protein in ERs (Fig. 3B, 3C) and mRNA (Fig. 3D), caused ER swelling (Fig. 3E), and increased expression of terminal differentiation markers, but suppressed expression of early chondrogenic markers (Fig. 3D). In contrast, in cultures of Casrflox/flox chondrocytes treated with Cre, the effects of FFSS were largely abrogated (Fig. 3A-D), except ER swelling (Fig. 3E). These results together with the studies of rat TMJ chondrocytes and ATDC5 cells establish a non-redundant role for the ER CaSR in mediating chondrocyte terminal differentiation in response to FFSS-induced Ca2+ loading.
Fig. 3.
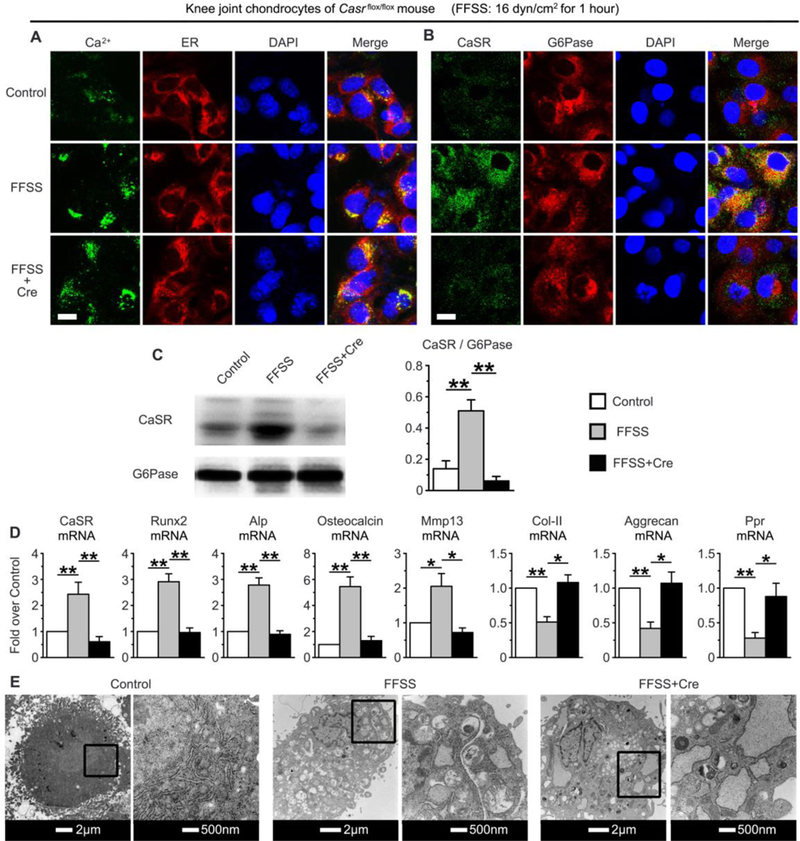
The impact of Casr gene knockout on FFSS-induced changes in (A) Ca2+ loading, (B, C) CaSR localization in ERs, (D) expression of cell differentiation markers, and (E) organelle swelling in primary chondrocytes cultured from knee joints of Casrflox/flox mice. Induction of gene KO was performed by transduction of Cre protein in chondrocyte cultures for 24 hrs before the application of FFSS. (A) Dual-fluorescence staining of Ca2+ and ERs by Fluo-8 (in green) and ER-tracker (in red) respectively, showed inability of Casr KO to prevent the FFSS-induced Ca2+ loading in ERs. (B) Dual-fluorescence staining and (C) immunoblotting of CaSR (in green) and G6Pase (in red) showed the ability of Casr KO to abrogate the FFSS-induced CaSR localization in ERs. Bar=5μm. (D) mRNA expression profiles by qPCR showed the ability of Casr KO to block the impact of FFSS on the expression of early and terminal differentiation markers. Values are presented as the mean±SD. **p<0.01, *p<0.05 between groups as specified by top horizontal bars in each panel. (E) TEM images showed the inability of Casr KO to affect the FFSS-induced swelling of mitochondria and ERs. n=4 separate cultures.
Dental malocclusion by UAC stimulated CaSR expression in ERs and terminal differentiation in chondrocytes in rat and mouse TMJ cartilage
Mandibular condylar cartilage in UAC-treated rats manifested characteristics of OA phenotypes including cartilage thinning, reduced proteoglycans content and expression of Col-II and Aggrecan mRNA and protein, and ER swelling (18,20), as well as increased OA scores by a slightly modified OARSI scoring system (26). All of these OA phenotypes worsen with time of UAC treatment in both rat (Supplemental Fig. 9) and mice (Supplemental Fig. 10). These OA phenotypes are closely associated with enhanced chondrocyte terminal differentiation as indicated by increased proportions of Col-X-positive cells in rat (Supplemental Fig. 11A) and mouse (Supplemental Fig. 11B) condylar cartilage. Interestingly, at later time point of UAC, substantial numbers of chondrocytes in superficial layer and/or “proliferation” zone became Col-X-positive (Supplemental Fig. 11), supporting accelerated or premature terminal differentiation of chondrocytes in the UAC-treatment TMJs.
To test whether the CaSR is critical for OA development, we first compared the impact of UAC on CaSR expression in TMJ cartilage. UAC increased in the numbers of CaSR-positive cells along with increased expression of CaSR protein (Fig. 4A and 4F) and mRNA (Fig. 4B) in TMJ cartilage of UAC-treated rat (Fig. 4A) and mice (Fig. 4F). The increased CaSR expression was localized to both cell membrane and ERs of chondrocytes in deep layers of cartilage (Fig. 4A and 4F). This increased expression of ER CaSR was corroborated by immunoblotting of ER protein lysates extracted from UAC vs Control cartilage (Fig. 4C). These changes in CaSR expression were accompanied by swelling of ERs and mitochondria in UAC-treated chondrocytes vs Control (Fig. 4E). In consistent with an accelerated terminal cell differentiation in the UAC-treated cartilage, we observed profound deceases in expression of PPR mRNA (Fig. 4B) and protein (Fig. 4D), which is known to delay chondrocyte terminal differentiation (37).
Fig. 4.
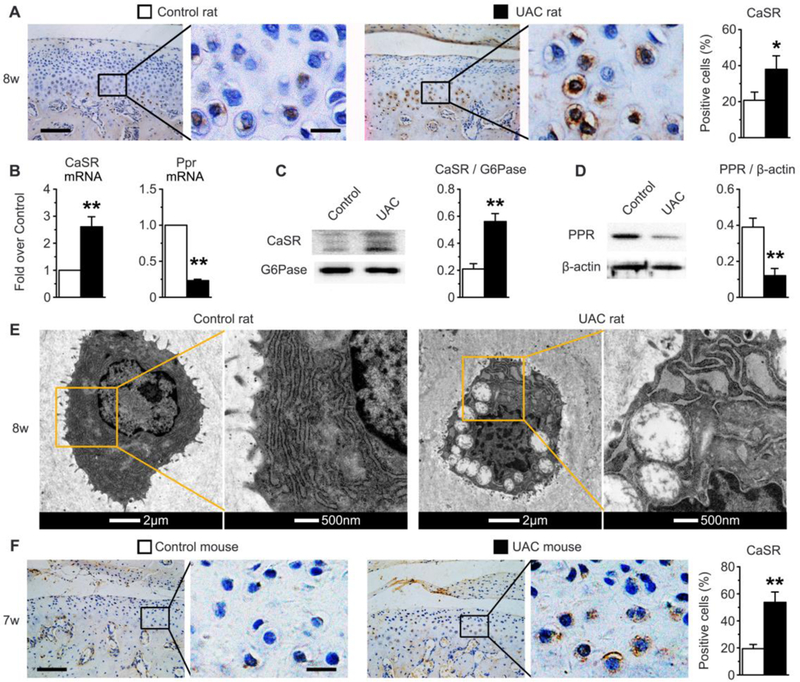
UAC induced CaSR expression, but suppressed PPR expression in condylar cartilage of rat and/or mouse TMJs. UAC was applied in rats (A-E) and in mice (F). (A) Immunohistochemical staining showed increased % of CaSR-positive chondrocytes and increased localization of the receptor in intracellular compartments, presumably ERs of the cells in TMJ cartilage of UAC vs control rat s. n=6 rats per group. (B) qPCR analyses showed increased CaSR mRNA level and reduced PPR mRNA expression in UAC vs control cartilage. Immunoblotting showed (C) increased expression of CaSR protein in the ER extracts and (D) reduced expression of total PPR protein lysates from UAC vs Control cartilage. (E) TEM images showed profound swelling of mitochondria and ERs in chondrocytes of UAC vs Control cartilage in rats. n=6 rats per group. (F) Immunohistochemical staining also showed increased % of CaSR-positive chondrocytes and increased localization of CaSR in intracellular compartments of the cells in cartilage of UAC vs control mice. n=6 mice per group. Bars in panels (A) and (F), 10μm or 20μm in digitally enlarged pictures. Values are presented as the mean±SD. **p<0.01 and *p<0.05 between UAC and Control groups.
To firmly define the role of CaSR in mediating the development of OA in mouse TMJs responding to dental malocclusion, we studied the effects of acute Casr gene KO on TMJ cartilage of Tam-CartCasrflox/flox mice. Deletion of Casr gene in chondrocytes in the latter mice was induced by five consecutive daily intraperitoneal injections of tamoxifen beginning one day before UAC treatment. Two “flox” controls (Casrflox/flox mice with or without Tam injection ), which did not render CaSR ablation, were subjected to the same UAC regimen. In both control groups, UAC produced OA morphology, characterized by decreased cartilage thickness and increased OARSI scores (Fig. 5A) along with reduced mRNA and/or protein levels of Col-II and Aggrecan and PPR (Fig. 5B), and increased expression of terminal differentiation markers (ALP, Runx2, MMP13, and Osteocalcin) in their TMJ-cartilage after UAC treatments for 3 weeks (Fig. 5B) and 7 weeks (Supplemental Fig. 12). In contrast, Casr KO in Tam-CartCasrΔflox/Δflox mice without UAC slowed down age-dependent cartilage thinning (Fig. 5A and Supplemental Fig. 12A, 12B) and loss of Col-II and Aggrecan RNA and protein (Fig. 5B, 5C; and Supplemental Fig. 12B, 12C), but increased expression of Col-X (Supplemental Fig. 13) and markers of terminal cell differentiation (Fig. 5B and Supplemental Fig. 12B), supporting a role of the CaSR in mediating age-dependent changes in articular chondrocyte differentiation. Furthermore, Casr KO significantly blocked the ability of UAC to produce OA morphology (Fig. 5A and Supplemental Fig. 12A), increase expression of Col-X (Supplemental Fig. 13) and accelerate terminal cell differentiation (Fig. 5B Supplemental Fig. 12B. These data support a non-redundant role for the CaSR in promoting both age- and mechanical stress-induced terminal cell differentiation and development of OA phenotypes in TMJs.
Fig. 5.
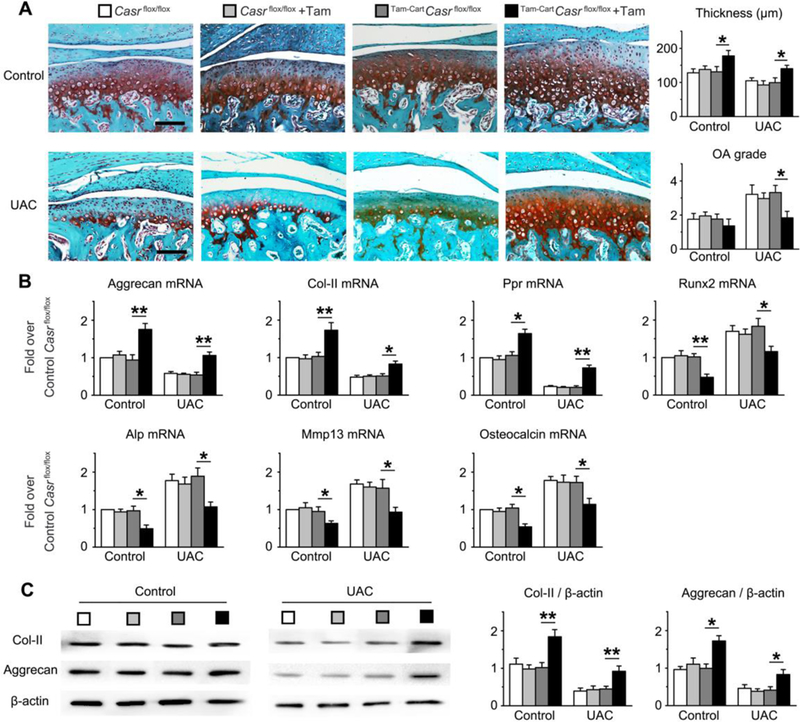
Effects of Tamoxifen (Tam)-induced Casr gene KO on chondrocyte functions in TMJ cartilage of mice without (Control) or with UAC. The Casr gene KO was induced by 5 daily intraperitoneal (IP) Tam injections in Tam-CartCasrflox/flox mice at 8 weeks of age, followed by 3 weeks of UAC. Three control groups -- Casrflox/flox, Casrflox/flox injected with Tamoxifen (Casrflox/flox+Tam), or Tam-CartCasrflox/flox without Tamoxifen injections --, which did not have their Casr genes ablated, were subjected to the same UAC regimen. (A) Safranin O staining (in red) showed retention of proteoglycan content and thickness of TMJ-cartilage and decreased OA grade in Tam-CartCasrflox/flox mice injected with Tamoxifen vs the other 3 control groups with or without UAC treatment. Bar=100μm. n=6 mice per group. (B) qPCR analyses of mRNA and (C) immunoblotting of Col-II and Aggrecan protein extracted from the TMJ-cartilage of the above mice showed inability of UAC to reduce the expression of early differentiation markers and to increase terminal differentiation makers in Tam-CartCasrflox/flox mice injected with Tamoxifen vs other 3 controls with or without UAC. Eighteen TMJ-cartilages were pooled into 3 samples (6 cartilage per sample) for qPCR and immunoblotting analysis. All values in (B) and (D) were normalized to the Casrflox/flox Control. Values are presented as the mean±SD. **p<0.01, *p<0.05 between groups as specified by top horizontal bars in each panel.
The CaSR served as a pharmaceutical target to control the progression of OA in UAC-treated TMJs.
Lastly, we tested whether chondrocytic CaSRs can serve as a pharmaceutical target to prevent OA development by assessing the effects of locally injected CaSR antagonist (NPS2143) or agonist (Cinacalcet) on OA development in rat TMJs subjected to UAC. As assessed by histological staining, OARSI score and gene expression profiling, NPS2143 injections not only increased the thickness and matrix content (Col-II and Aggrecan) (Fig. 6A and 6B), retarded chondrocyte terminal differentiation (Fig. 6B) and decreased proportions of Col-X-positive chondrocyte (Supplemental Fig. 14) in mandible condylar cartilage of control mice without UAC treatments, but also significantly blocked the ability of UAC to alter these parameters (Fig. 6C, 6D). On the other hand, injections of Cinacalcet, a CaSR agonist, promoted thinning and loss of cartilage matrix (Col-II and Aggrecan), accelerated chondrocyte terminal differentiation, and increased the proportions of Col-X-positive cells in both control (Fig. 6A, 6B and Supplemental Fig. 14) and UAC-treated (Fig. 6C, 6D and Supplemental Fig. 14) rats. Immunohistochemical staining further indicated the ability of NPS2143 to reduce the numbers of CaSR-positive chondrocytes in both control and UAC-treated cartilage (Supplemental Fig. 15), but it did not block the ability of UAC to induce ER expression of the receptor. In contract, Cinacalcet increased the numbers of CaSR-positive cells and promoted ER expression of the receptor in those cells (Supplemental Fig. 15). The lack of effect on plasma PTH levels (Supplemental Fig. 16) supports direct actions of the drugs (potential via ER CaSR) in mediating cell-autonomous responses of TMJ chondrocytes. These data indicate the pharmaceutical potential of calcilytics in slowing down OA progression in response to traumatic joint injuries by targeting CaSR in chondrocytes.
Fig. 6.
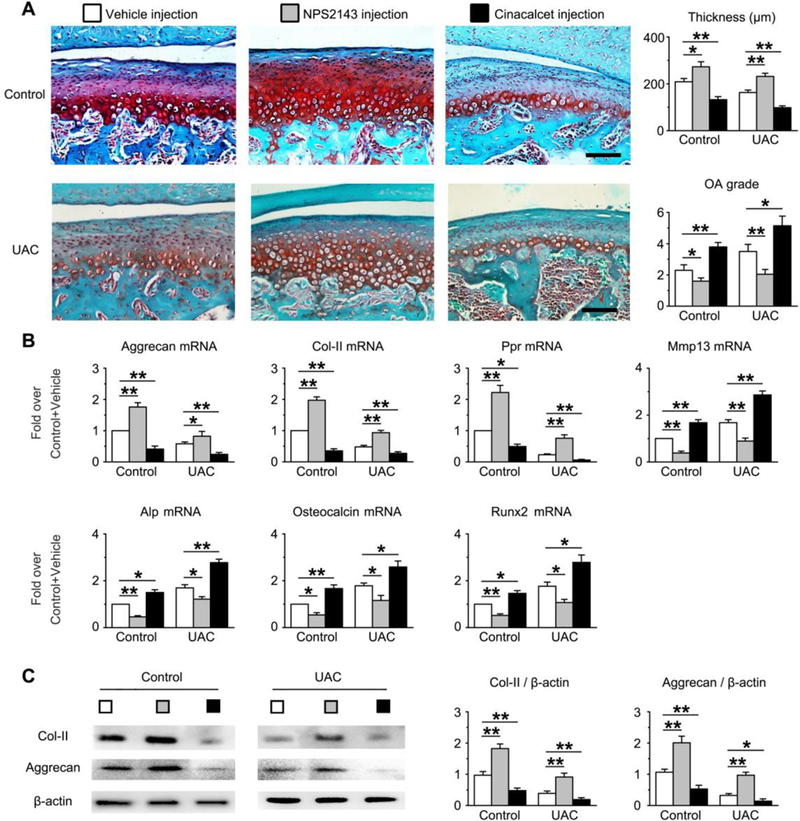
Effects of intra-synovial injections of CaSR antagonist (NPS2143) and agonist (Cinacalcet) on morphology (A), gene (B) and protein expression (C) in rat TMJ-cartilage without (Control) or with UAC stimulation. Test compounds (50 μl of 100 nM stock) and vehicle injections were performed on 10 weeks old rats every other day 2 days for 4 weeks. UAC began 4 weeks before drug injections and continued for another 4 weeks during drug treatments. (A) Safranin O staining (in red) showed the ability of NPS2143 to increase proteoglycan content and thickness of TMJ-cartilage and decreased OA grade, the ability of Cinacalcet to suppress these parameters in both control and UAC groups when compared to corresponding vehicle-injected group. Bar=100μm. n=6 rats per group. (B) qPCR analyses of mRNA and (C) immunoblotting analysis of Col-II and Aggrecan protein extracted from the TMJ-cartilage of the above mice showed the ability of NPS2143 to increase the expression of early differentiation markers and to suppress expression of terminal differentiation makers in both control and UAC groups when compared to corresponding vehicle-injected group and vice versa for effects of Cinacalcet. Six TMJ-cartilages were pooled into 3 samples (2 cartilage per sample) for qPCR and immunoblotting analysis. All values in (B) and (D) were normalized to the Control-Vehicle. Values are presented as the mean±SD. **p<0.01, *p<0.05 between groups as specified by top horizontal bars in each panel.
Discussion
Slow differentiation of articular chondrocyte into hypertrophic or terminal differentiation state retains its ability to produce abundant Col-II, proteoglycans (e.g., Aggrecan), and other matrices, which together produce a unique material property to absorb shocks and protect osseous tissues underneath. This delayed terminal cell differentiation also preserves the ability of chondrocyte to proliferate for tissue repair (38). Accelerated terminal cell differentiation as a result of cell senescence due to aging or trauma-induced biomechanical stresses lead to thinning and degradation of articular cartilage and its attaining of OA characteristics (38,39). Critical drivers of chondrocyte terminal differentiation, therefore, represent potential pharmaceutical targets to preserve articular cartilage and its functions against aging and trauma-induced joint injury. The CaSR expressed in maturing and hypertrophic chondrocytes is a critical driver of terminal differentiation which is essential for normal growth plate development and subsequent bone formation (40). Herein, our studies of cultured TMJ chondrocytes and chondrogenic ATDC5 cells support a novel signaling scheme, in which mechanical stresses induce Ca2+ influx and subsequent ER Ca2+ loading that activates ER CaSRs to alter cell differentiation program in TMJ chondrocytes (Fig.7). This signaling scheme is further supported by our genetic and pharmacological studies of normal rats and mice lacking CaSR in which the ability of UAC to increase CaSR expression in ER of articular chondrocytes and accelerate their terminal differentiation in TMJs could be reduced by reduced CaSR activity or expression.
Fig. 7.
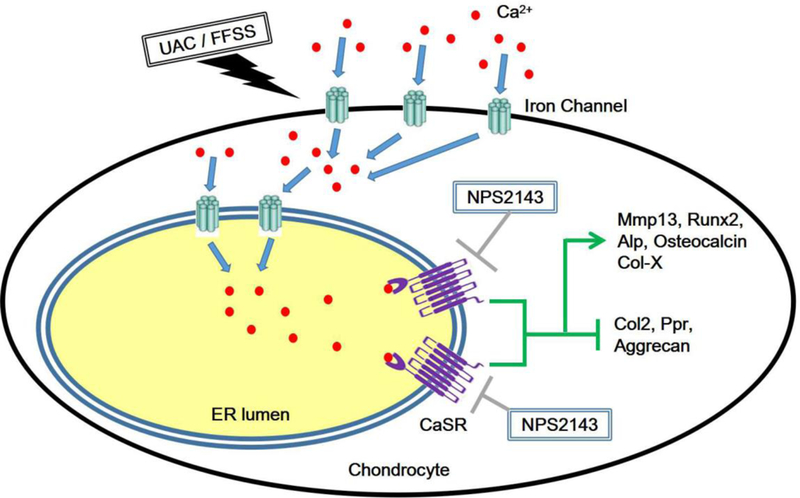
Schema for the effects of CaSR on biomechanical stress-induced chondrocyte terminal differentiation.
Our data showed reductions of Col-II and Aggrecan protein in cartilage subjected to UAC, supporting changes in homeostasis of matrix protein. These data coupled with a reduced RNA level of MMP13 in UAC-treated cartilage, support a cell-mediated increase in matrix degradation. The ability of CaSR knockdown to suppress MMP13 expression supports a role of this receptor to change matrix content potentially by altering cell differentiation program. These observations are consistent with our previous studies of cultured growth plate chondrocytes in which low extracellular [Ca2+] (0.5mM) retains high levels of Col-II and aggrecan mRNA expression, increases proteoglycan content in the surrounding matrix, and slows down terminal differentiation of the cell, while raising extracellular [Ca2+] does the opposites by activating the CaSR (12). However, our studies cannot rule out additional mechanical impact of UAC on degradation of cartilage matrix which are independent of cell differentiation.
The CaSR on the cell membrane is ascribed to sensing and responding to changes in extracellular [Ca2+] and the large amount of the receptor residing in intracellular organelles are generally described as the “immature” products trafficking to the cell membrane (8). Our study, however, for the first time, uncovers physiological actions of the CaSR in intracellular organelles, particularly in ERs. Specifically, we demonstrated the ability of FFSS-induced increases in ER Ca2+ loading and CaSR expression to accelerate chondrocyte terminal differentiation without changing the extracellular [Ca2+] in cultured chondrocytes, and furthermore the ability of CaSR mRNA knockdown or gene KO to completely abolish these effects. The role of ER CaSR in mediating chondrocyte terminal differentiation in response to biomechanical stimuli (i.e., FFSS) is further accentuated by the lack of notable increases in CaSR localization to the cell membrane of the cell. Considering that the CaSR situates in ERs or other organelles with its low-affinity Ca2+-binding domains (for mM Ca2+ concentrations) facing the lumens of the organelles, it’s plausible that raising lumen [Ca2+], which is assumed to reach a mM range in a Ca2+-loading state in response to biomechanical stresses, could activate those CaSRs directly. It has been shown that binding of CaSR to Ca2+ promote its stability in the membrane, including those in the ERs (7,8). Therefore, the increased localization of ER CaSR could be a direct result of the increased ER [Ca2+]. Our studies indicate that initial Ca2+ influx by mechanical stimuli is an essential driver of ER loading to stimulate ER CaSR expression in cultured chondrocytes. Future studies are required to assess signaling molecules associated with the ER CaSRs to determine how these receptors coupled to downstream cellular functions, including the transcriptional regulation of chondrogenic genes in chondrocytes.
Many Ca2+ channels have been reported to potentially mediate the stress-induced ER Ca2+-loading in chondrocytes (41). T-type Ca2+ channel and TRPV channel (36) are the most potential candidates and could represent other pharmaceutical targets for OA prevention and treatment. In keratinocytes, CaSRs are co-localized with PLC-1 ATP-Ca2+ transporter, and IP3-receptor in Golgi apparatus (42). PPR has been shown to activate adenylate cyclase in endosomes to increase cytosolic cAMP concentration (43,44). In the growth plate, PTHrP maintains the proliferative state of chondrocyte and prevents their terminal differentiation through the action of PPR (40). The CaSR signaling antagonizes these PTHrP/PPR actions to promote chondrocytes terminal differentiation (40). In accordance with this counteracting relationship between the CaSR and PRR, we showed profound reduction of PPR expression in FFSS-treated chondrocytes in cultures as well as articular cartilage in joints subjected to UAC. This change in PPR expression is mediated directly by the CaSR activation, as it could be enhanced by CaSR antagonist or suppressed by CaSR agonists in vitro or in vivo. The reduced PPR expression could be one of the mechanisms promoting chondrocyte terminal differentiation in FFSS- or UAC-treated chondrocytes.
NPS2143, a small hydrophobic compound that bind to the transmembrane domain of the CaSR in lipid bilayers in cell membrane or intracellular organelles (46), like ER, reproduce the effects of CaSR knockdown or knockout in promoting chondrocyte terminal differentiation. While we cannot rule out the actions of the drug on the CaSRs on the cell-surface, the ability of the drug to mimic the actions of TG and free extracellular Ca to block FFSS-induced chondrocyte terminal differentiation support its direct action on ER CaSR. In supporting this idea, Muanprasat (45) reported that NPS2143 acts through ER CaSR to suppress a Chitosan oligosaccharide-induced increase in [Ca2+]i.
CaSR antagonists, like Ronacaleret, has been developed for treating osteoporosis by increasing endogenous PTH secretion, but the results of the clinical trials were unsatisfactory for its original intent (46). These compounds are readily to be repurposed for future clinical trials in treating OA. The clinical relevance of our study is accentuated by the ability of the injectable NPS2143 to suppress the presentation of OA phenotypes in rats. The latter permits local injections, as we have demonstrated in this study, to prevent systemic actions of the drug on multiple targets, including parathyroid glands, and therefore prevent hyperparathyroidism and hypercalcemic side effects (47).
Overall, our study elucidates a novel pathological action of ER CaSR in the development of osteoarthritic cartilage due to aberrant mechanical stimuli and indicates a therapeutic potential of the clinically available calcilytics in treating OA in TMJs by targeting the CaSR. Future studies are needed to assess the roles of CaSR in OA development in other anatomical sites.
Supplementary Material
Acknowledgments
Funding:This work was supported by the National Natural Science Foundation of China grants (81700995, 81530033, 81470762, 81500875, 81371166 and 81500896), the US NIH (AR067291) and VA Merit Review (5I01BX003453) grants, the Health Science Foundation of Shanxi Province (2016D009) and the Basic Natural Science Foundation of Shanxi Province (2018JM7084 and 2017JM8131). The authors declared no conflict of interest.
Footnotes
Additional Supporting Information may be found in the online version of this article.
Competing interests: The authors declare that they have no competing interests.
This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: [10.1002/jcb.3643]
References
- 1.Guo FJ, Xiong Z, Lu X, Ye M, Han X, Jiang R 2014. ATF6 upregulates XBP1S and inhibits ER stress-mediated apoptosis in osteoarthritis cartilage. Cell Signal 26(2):332–42. [DOI] [PubMed] [Google Scholar]
- 2.Li H, Zhang XY, Wu TJ, Cheng W, Liu X, Jiang TT, Wen J, Li J, Ma QL, Hua ZC 2013. Endoplasmic reticulum stress regulates rat mandibular cartilage thinning under compressive mechanical stress. J Biol Chem 288(25): 18172–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barley RD, Adesida AB, Bagnall KM, Jomha NM 2010. Immunohistochemical characterization of reparative tissue present in human osteoarthritic tissue. Virchows Arch 456(5): 561–9. [DOI] [PubMed] [Google Scholar]
- 4.Zhong L, Huang X, Karperien M, Post JN 2016. Correlation between Gene Expression and Osteoarthritis Progression in Human. Int J Mol Sci 17(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bush JR, Beier F 2013. TGF-beta and osteoarthritis--the good and the bad. Nat Med 19(6):667–9. [DOI] [PubMed] [Google Scholar]
- 6.Papi E, Belsi A, McGregor AH 2015. A knee monitoring device and the preferences of patients living with osteoarthritis: a qualitative study. BMJ Open 5(9):e007980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Mise A, Tamma G, Ranieri M, Svelto M, Heuvel B, Levtchenko EN, Valenti G 2015. Conditionally immortalized human proximal tubular epithelial cells isolated from the urine of a healthy subject express functional calcium-sensing receptor. Am J Physiol Renal Physiol 308(11):F1200–6. [DOI] [PubMed] [Google Scholar]
- 8.Breitwieser GE 2014. Pharmacoperones and the calcium sensing receptor: exogenous and endogenous regulators. Pharmacol Res 83:30–7. [DOI] [PubMed] [Google Scholar]
- 9.Chang W, Tu C, Chen TH, Bikle D, Shoback D 2008. The extracellular calcium-sensing receptor (CaSR) is a critical modulator of skeletal development. Sci Signal 1(35):ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burton DW, Foster M, Johnson KA, Hiramoto M, Deftos LJ, Terkeltaub R 2005. Chondrocyte calcium-sensing receptor expression is up-regulated in early guinea pig knee osteoarthritis and modulates PTHrP, MMP-13, and TIMP-3 expression. Osteoarthritis Cartilage 13(5):395–404. [DOI] [PubMed] [Google Scholar]
- 11.Chang W, Tu C, Pratt S, Chen TH, Shoback D 2002. Extracellular Ca(2+)-sensing receptors modulate matrix production and mineralization in chondrogenic RCJ3.1C5.18 cells. Endocrinology 143(4):1467–74. [DOI] [PubMed] [Google Scholar]
- 12.Rodriguez L, Cheng Z, Chen TH, Tu C, Chang W 2005. Extracellular calcium and parathyroid hormone-related peptide signaling modulate the pace of growth plate chondrocyte differentiation. Endocrinology 146(11):4597–608. [DOI] [PubMed] [Google Scholar]
- 13.Goldring MB 2012. Chondrogenesis, chondrocyte differentiation, and articular cartilage metabolism in health and osteoarthritis. Ther Adv Musculoskelet Dis 4(4):269–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Filip A, Pinzano A, Bianchi A, Feve B, Jalkanen S, Gillet P, Mainard D, Lacolley P, Magdalou J, Mercier N 2016. Expression of the semicarbazide-sensitive amine oxidase in articular cartilage: its role in terminal differentiation of chondrocytes in rat and human. Osteoarthritis Cartilage 24(7):1223–34. [DOI] [PubMed] [Google Scholar]
- 15.Queirolo V, Galli D, Masselli E, Borzi RM, Martini S, Vitale F, Gobbi G, Carubbi C, Mirandola P 2016. PKCepsilon is a regulator of hypertrophic differentiation of chondrocytes in osteoarthritis. Osteoarthritis Cartilage 24(8):1451–60. [DOI] [PubMed] [Google Scholar]
- 16.Ioi H, Matsumoto R, Nishioka M, Goto TK, Nakata S, Nakasima A, Counts AL 2008. Relationship of TMJ osteoarthritis / osteoarthrosis to head posture and dentofacial morphology. Orthod Craniofac Res 11(1):8–16. [DOI] [PubMed] [Google Scholar]
- 17.Yamada K, Fukui T, Tsuruta A, Hanada K, Hosogai A, Kohno S, Hayashi T 2003. The relationship between retruded contact position and intercuspal position in patients with TMJ osteoarthritis. Cranio 21(4):240–7. [DOI] [PubMed] [Google Scholar]
- 18.Lu L, Zhang X, Zhang M, Zhang H, Liao L, Yang T, Zhang J, Xian L, Chen D, Wang M 2015. RANTES and SDF-1 Are Keys in Cell-based Therapy of TMJ Osteoarthritis. J Dent Res 94(11):1601–9. [DOI] [PubMed] [Google Scholar]
- 19.Yang H, Zhang M, Wang X, Zhang H, Zhang J, Jing L, Liao L, Wang M 2015. TNF Accelerates Death of Mandibular Condyle Chondrocytes in Rats with Biomechanical Stimulation-Induced Temporomandibular Joint Disease. PLoS One 10(11):e0141774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang M, Wang H, Zhang J, Zhang H, Yang H, Wan X, Jing L, Lu L, Liu X, Yu S, Chang W, Wang M 2016. Unilateral anterior crossbite induces aberrant mineral deposition in degenerative temporomandibular cartilage in rats. Osteoarthritis Cartilage 24(5):921–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim ES, Kim SY, Lee JY, Han JH, Sohn TS, Son HS, Moon SD 2016. Identification and functional analysis of a novel CaSR mutation in a family with familial hypocalciuric hypercalcemia. J Bone Miner Metab 34(6):662–667. [DOI] [PubMed] [Google Scholar]
- 22.Cianferotti L, Gomes AR, Fabbri S, Tanini A, Brandi ML 2015. The calcium-sensing receptor in bone metabolism: from bench to bedside and back. Osteoporos Int 26(8):2055–71. [DOI] [PubMed] [Google Scholar]
- 23.Scrivani SJ, Keith DA, Kaban LB 2008. Temporomandibular disorders. N Engl J Med 359(25):2693–705. [DOI] [PubMed] [Google Scholar]
- 24.Kadari A, Lu M, Li M, Sekaran T, Thummer RP, Guyette N, Chu V, Edenhofer F 2014. Excision of viral reprogramming cassettes by Cre protein transduction enables rapid, robust and efficient derivation of transgene-free human induced pluripotent stem cells. Stem Cell Res Ther 5(2):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pritzker KP, Gay S, Jimenez SA, Ostergaard K, Pelletier JP, Revell PA, Salter D, van den Berg WB 2006. Osteoarthritis cartilage histopathology: grading and staging. Osteoarthritis Cartilage 14(1):13–29. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, Liao L, Zhu J, Wan X, Xie M, Zhang H, Zhang M, Lu L, Yang H, Jing D, Liu X, Yu S, Lu XL, Chen C, Shan Z, Wang M 2018. Osteochondral Interface Stiffening in Mandibular Condylar Osteoarthritis. J Dent Res 97(5):563–570. [DOI] [PubMed] [Google Scholar]
- 27.Zhang J, Zhang HY, Zhang M, Qiu ZY, Wu YP, Callaway DA, Jiang JX, Lu L, Jing L, Yang T, Wang MQ 2014. Connexin43 hemichannels mediate small molecule exchange between chondrocytes and matrix in biomechanically-stimulated temporomandibular joint cartilage. Osteoarthritis Cartilage 22(6):822–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li D, Lee CW, Buckler K, Parekh A, Herring N, Paterson DJ 2012. Abnormal intracellular calcium homeostasis in sympathetic neurons from young prehypertensive rats. Hypertension 59(3):642–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang N, De Bock M, Decrock E, Bol M, Gadicherla A, Bultynck G, Leybaert L 2013. Connexin targeting peptides as inhibitors of voltage- and intracellular Ca2+-triggered Cx43 hemichannel opening. Neuropharmacology 75:506–16. [DOI] [PubMed] [Google Scholar]
- 30.Vennekens R, Voets T, Bindels RJ, Droogmans G, Nilius B 2002. Current understanding of mammalian TRP homologues. Cell Calcium 31(6):253–64. [DOI] [PubMed] [Google Scholar]
- 31.Wei H, Inan S 2013. Dual effects of neuroprotection and neurotoxicity by general anesthetics: role of intracellular calcium homeostasis. Prog Neuropsychopharmacol Biol Psychiatry 47:156–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li WE, Chen W, Ma YF, Tuo QR, Luo XJ, Zhang T, Sai WB, Liu J, Shen J, Liu ZG, Zheng YM, Wang YX, Ji G, Liu QH 2012. Methods to measure and analyze ciliary beat activity: Ca2+ influx-mediated cilia mechanosensitivity. Pflugers Arch 464(6):671–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Follonier L, Schaub S, Meister JJ, Hinz B 2008. Myofibroblast communication is controlled by intercellular mechanical coupling. J Cell Sci 121(Pt 20):3305–16. [DOI] [PubMed] [Google Scholar]
- 34.Mansfield K, Pucci B, Adams CS, Shapiro IM 2003. Induction of apoptosis in skeletal tissues: phosphate-mediated chick chondrocyte apoptosis is calcium dependent. Calcif Tissue Int 73(2):161–72. [DOI] [PubMed] [Google Scholar]
- 35.D’Andrea P, Calabrese A, Capozzi I, Grandolfo M, Tonon R, Vittur F 2000. Intercellular Ca2+ waves in mechanically stimulated articular chondrocytes. Biorheology 37(1–2):75–83. [PubMed] [Google Scholar]
- 36.Jeong SA, Kim IY, Lee AR, Yoon MJ, Cho H, Lee JS, Choi KS 2015. Ca2+ influx-mediated dilation of the endoplasmic reticulum and c-FLIPL downregulation trigger CDDO-Me-induced apoptosis in breast cancer cells. Oncotarget 6(25):21173–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim G, Schoenberger SP, Sharpe A, Kronenberg M 2006. Synergistic costimulation by both B7 molecules regulates colitis pathogenesis. Ann N Y Acad Sci 1072:233–41. [DOI] [PubMed] [Google Scholar]
- 38.Tchetina EV 2011. Developmental mechanisms in articular cartilage degradation in osteoarthritis. Arthritis 2011:683970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, Chung JW, Kim DH, Poon Y, David N, Baker DJ, van Deursen JM, Campisi J, Elisseeff JH 2017. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med 23(6):775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Santa MC, Cheng Z, Li A, Wang J, Shoback D, Tu CL, Chang W 2016. Interplay between CaSR and PTH1R signaling in skeletal development and osteoanabolism. Semin Cell Dev Biol 49:11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mobasheri A, Matta C, Uzieliene I, Budd E, Martin-Vasallo P, Bernotiene E 2018. The chondrocyte channelome: A narrative review. Joint Bone Spine. [DOI] [PubMed] [Google Scholar]
- 42.Tu CL, Chang W, Bikle DD 2007. The role of the calcium sensing receptor in regulating intracellular calcium handling in human epidermal keratinocytes. J Invest Dermatol 127(5):1074–83. [DOI] [PubMed] [Google Scholar]
- 43.Jean-Alphonse FG, Wehbi VL, Chen J, Noda M, Taboas JM, Xiao K, Vilardaga JP 2017. beta2-adrenergic receptor control of endosomal PTH receptor signaling via Gbetagamma. Nat Chem Biol 13(3):259–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vilardaga JP, Jean-Alphonse FG, Gardella TJ 2014. Endosomal generation of cAMP in GPCR signaling. Nat Chem Biol 10(9):700–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muanprasat C, Wongkrasant P, Satitsri S, Moonwiriyakit A, Pongkorpsakol P, Mattaveewong T, Pichyangkura R, Chatsudthipong V 2015. Activation of AMPK by chitosan oligosaccharide in intestinal epithelial cells: Mechanism of action and potential applications in intestinal disorders. Biochem Pharmacol 96(3):225–36. [DOI] [PubMed] [Google Scholar]
- 46.Fitzpatrick LA, Dabrowski CE, Cicconetti G, Gordon DN, Fuerst T, Engelke K, Genant HK 2012. Ronacaleret, a calcium-sensing receptor antagonist, increases trabecular but not cortical bone in postmenopausal women. J Bone Miner Res 27(2):255–62. [DOI] [PubMed] [Google Scholar]
- 47.Nemeth EF, Delmar EG, Heaton WL, Miller MA, Lambert LD, Conklin RL, Gowen M, Gleason JG, Bhatnagar PK, Fox J 2001. Calcilytic compounds: potent and selective Ca2+ receptor antagonists that stimulate secretion of parathyroid hormone. J Pharmacol Exp Ther 299(1):323–31. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.