

Abstract
Neurocysticercosis is a parasitic brain disease caused by the larval form (Cysticercus cellulosae) of Taenia solium and is the leading cause of preventable epilepsy worldwide. However, the pathophysiology and relation to the wide range of clinical features remains poorly understood. Axonal swelling is emerging as an important early pathological finding in multiple neurodegenerative diseases and as a cause of brain injury, but has not been well described in neurocysticercosis. Histological analysis was performed on human, rat and porcine NCC brain specimens to identify axonal pathology. Rat infection was successfully carried out via two routes of inoculation: direct intracranial injection and oral feeding. Extensive axonal swellings, in the form of spheroids, were observed in both humans and rats and to a lesser extent in pigs with NCC. Spheroids demonstrated increased immunoreactivity to amyloid precursor protein and neurofilament indicating probable impairment of axonal transport. These novel findings demonstrate that spheroids are present in NCC which is conserved across species. Not only is this an important contribution toward understanding the pathogenesis of NCC, but it also provides a model to analyze the association of spheroids with specific clinical features and to investigate the reversibility of spheroid formation with antihelminthic treatment.
Keywords: APP, neurocysticercosis, neurofilament, spheroids, T. solium oncospheres
Introduction
Neurocysticercosis (NCC) is the leading preventable cause of epilepsy worldwide 27. It remains a significant cause of morbidity in areas endemic to Taenia solium, but it is also a growing problem in developed nations because of immigration 28, 35. NCC occurs when humans ingest fecal matter containing eggs of the T. solium tapeworm. These then hatch and oncospheres migrate, invading the central nervous system (CNS) and developing into cysticerci, where they can live in the CNS for years before the onset of clinical symptoms. Seizures are the predominant clinical feature of NCC but the spectrum of disease is wide‐ranging, from asymptomatic cases to life‐threatening intracranial hypertension 11, 17.
Despite being a serious public health concern in endemic countries, the pathogenesis of NCC and correlation with clinical evolution remain poorly understood. Human studies to assess NCC pathogenesis are limited because of the slow progression of the disease, the limited availability of post‐mortem infected brain tissue and the selective bias associated with biopsied specimens. Previous human post‐mortem studies have shown alterations at the interface between parasite and host tissue including: inflammatory changes, perilesional edema 12, disrupted myelin sheaths 29, necrotic brain tissue 17 and β‐amyloid fibrils 19. However, pathological changes occurring beyond the zone of gliosis are not well defined in either human studies or animal models.
Axonal swelling is emerging as an important finding in multiple neurodegenerative diseases and various mechanisms of brain injury 7. Interestingly, in a study of NCC naturally infected pigs in 2009, Sikasunge et al reported an increase in the neurofilament protein (NFP) expression in both neurons and axons, as well as enlarged axon fibers with increased NFP expression surrounding the area of fibrosis around the cysticercus 36. However, to our knowledge, the extent of axonal swelling in NCC and conservation across species has not previously been reported.
Despite the fact that experimental animal models have been used to describe histopathological changes in NCC, few of them have used T. solium and none of them have described axonal pathology 2, 6, 20, 22, 37. Studies in pigs, the natural intermediate host, have been hindered because of the variable success in experimental infection of pigs, high logistical costs and the lack of commercially available immunohistochemical antibodies. Recently, our group reported a rat model for NCC obtained by the intracranial injection of T. solium onchospheres into the rat brain 41. In this model, rats developed mature viable cysts, which appear similar in morphology to those present in both porcine and human NCC and develop seizures, a frequent clinical manifestation of human NCC.
The rat model for NCC was used in our study to define inflammatory changes around the cyst and to identify changes in the brain tissue within and beyond the area of gliosis. Immunohistochemistry (IHC) was carried out to identify and characterize axonal pathology in the rat model, as well as in pig and human NCC brain specimens. Furthermore, in order to demonstrate that the observed changes are not an artifact of intracranial inoculation and to mimic the natural route of the infection, rats were also infected via the oral route with activated oncospheres.
Materials and Methods
Rat model
Outbred Holtzman rats of 6–14 days of life were obtained from the animal facility of the Universidad Peruana Cayetano Heredia, Lima, Peru. Animals were kept under 12‐hour light/dark cycles with food ad libitum during the entire period of the study. Infection with T. solium oncospheres was performed by either intracranial injection as previously reported 41 or by oral inoculation. All procedures were conducted under strict compliance with the Animal Welfare Committee of the Universidad Peruana Cayetano Heredia in Lima, Peru (ICAC codes. 61242).
Oncosphere preparation
Oncospheres were processed according to Verastegui 40. Briefly, T. solium eggs were obtained from gravid proglottids by disruption with tweezers, followed by three rounds of centrifugation at 2500 rpm and washing in distilled water. Eggs were immersed in 0.75% sodium hypochlorite solution and washed in RPMI medium to obtain oncospheres. Oncospheres were activated by the addition of artificial intestinal fluid (AIF) for 45 min at 37°C and washed three times in the RPMI medium. Oncosphere activation was verified using microscopy; those moving their hooks and body were considered activated 40.
Intracranial route of infection
Briefly, 70 pup rats, 10 to 14 days old, were infected by intracranial injection at 2 mm depth with 500 activated T. solium oncospheres suspended in 100 µL of sterile physiologic saline solution (0.9%). Ten age‐matched sham rats received 100‐µL sterile physiological saline solution. The intracranial route of infection delivered oncospheres to the space between the brain parenchyma and the skull. This method avoids the penetration of the needle into the brain parenchymal tissue 41.
Oral route of infection
The oral route of infection by gavage was performed on 30 pup rats at days 6–10 of life with 20,000 activated T. solium oncospheres suspended in 50 µL of sterile physiological saline solution (0.9%). Ten age‐matched sham rats were fed 50 µL of the saline solution (0.9%). The oral route of infection simulates the natural process of the neurocysticercosis infection but requires a larger number of oncospheres to establish the infection than does the intracranial route.
Human and porcine samples
Four anonymized human brain tissue samples corresponding to four patients with a post‐mortem diagnosis of neurocysticercosis were collected from the Department of Pathology, Layton Aging & Alzheimer’s Disease Center, Oregon Health & Science University. Necropsies had been performed under appropriate consent from the relatives. Brain samples from five pigs naturally infected with T. solium cysticercosis were donated by Dr. Armando Gonzalez, from the Cysticercosis Working Group in Peru, and stored in formalin. Histological processing of the human and porcine samples was performed at the Department of Pathology, Layton Aging & Alzheimer’s Disease Center, Oregon Health & Science University. The samples were processed according to standard histological techniques, and the human samples were stained for amyloid precursor protein (APP), while pigs were stained for APP and medium and heavy chain neurofilament proteins.
Rat histology
All rats were euthanized between four and five months post‐infection for histological and immunohistochemical analyses. Animals were first anaesthetized with ketamine (100 mg/kg; Ket‐A‐100, Agrovet Market SA, Peru) and xylazine (10 mg/kg; Dormi‐Xyl‐2, Agrovet Market SA, Peru). Next, rats were perfused through direct cardiac puncture with 300 mL of phosphate buffered saline (PBS) and fixed with 200 mL of 4% paraformaldehyde/PBS (PFA/PBS). The brains were removed and immediately post‐fixed in PFA/PBS overnight. The brains were embedded in paraffin and the entire brain was cut into 3‐ and 6‐µm sections, only in the coronal plane. The sections to be analyzed were randomly selected from the sections with a visible scolex on H&E staining. The scolex tends to be at the center of the cyst, so was used as a reference point to increase the comparability of the sections 41. Tissue sections of 3 µm were deparaffinized and rehydrated for standard Hematoxylin & Eosin (H&E) staining to observe the histopathological features of the rat tissue and the characteristics of the brain cysticercus. Intracranially and orally infected rats had 3‐µm sections stained with Masson’s trichrome (Chromaview kit) to visualize the fibrosis around the parasite and Nissl (Polysciences Inc, USA) in order to visualize cellular alteration.
Immunohistochemistry
Immunohistochemistry was performed at the Infectious Diseases Research Laboratory, Universidad Peruana Cayetano Heredia, Lima, Peru. Consecutive 3‐µm sections from both routes of infection and their respective sham controls (10–15 congruent and comparable sections) were processed with 14 different primary antibodies (Supplementary file I, Table S1). To identify axonal swellings, stains were performed for phosphorylated heavy chain of neurofilament (p‐NFP200kD) and their variants, super oxide dismutase I (SOD1) and ubiquitin. Additionally, GFAP and Iba‐1 antibodies were used to identify astrocytes and microglia, respectively, and NeuN for neuron identification.
In addition, because of the lack of sufficient sections, the following stains were run on only 15 intracranially infected and 4 orally infected sections with established cysticerci: non‐phosphorylated neurofilament (np‐NFP200kD), pan neurofilament (pan‐NFP200kD), heat shock protein 10 (Hsp70), Tar DNA binding protein 43 (TDP43) and Fused‐in‐Sarcoma/Translocated‐in‐Sarcoma protein (FUS/TLS).
Briefly, the brain sections were deparaffinized and antigen retrieval was performed using citrate buffer pH 6 at 95°C for 20 minutes. Next, endogenous peroxidase activity was blocked with 3% H2O2/PBS and nonspecific antibody binding was blocked with 10% normal goat serum in PBS‐Triton X‐100 (PBS‐T). Primary antibodies were diluted in normal goat or rabbit serum in 0.1M PBS‐T and incubated with the tissue sections overnight at 4°C. Secondary antibodies were added according to their respective protocols (Supplementary file, Table S1). Antigen‐antibody reactions were then detected using the chromogenic substrate 3′3′‐diaminobenzidine (DAB; 50 µL/1–3 mL, DAKO, USA). Finally, the tissue sections were dehydrated, counterstained with hematoxylin and mounted. All images were obtained with an Axiocam light microscope (Zeiss, Germany), and digitalized and processed with blue ZEN software (Zeiss, Germany) and Adobe Photoshop Elements CS6.
One infected and one noninfected rat sample was sent to the Department of Pathology, Layton Aging & Alzheimer’s Disease Center, Oregon Health & Science University and immunohistochemistry was performed for APP.
Immunofluorescence
Confocal imaging was performed on 3‐µm sections for each marker. The sections were deparaffinized and rehydrated as performed for light immunohistochemistry. Antigen retrieval was performed using citrate buffer pH 6 at 95°C for 20 minutes and then 10% donkey and 10% horse serum diluted in PBS were added to the sections to block nonspecific reactions. The sections were incubated overnight with both primary antibodies (rabbit anti‐GFAP, mouse anti‐GFAP, goat anti‐Iba‐1, mouse anti‐rat p‐NFP200kD, mouse anti‐rat np‐NFP200kD, rabbit anti‐NeuN, mouse anti‐NeuN. Mouse anti‐PD41, rabbit anti‐SOD1, rabbit anti‐Hsp70; supplementary file I, Table S1). Subsequently after washing, the appropriate secondary antibody (Alexa 488 donkey anti‐mouse, Alexa 488 donkey anti‐rabbit and Alexa 594 goat anti‐rabbit, Life Technology, USA) was diluted in 10% normal goat or 10% normal donkey serum in PBS‐T pH 7.4 and added to each section. Finally, the sections were mounted using ProLong Gold Antifade Mountant with DAPI (Life Technologies, USA). Digital images were obtained using Confocal Microscopy LSM880 (Zeiss, Germany) through Airy scanning fast mode processing (Zen black, Zeiss).
Neuropathological assessment of the lesion surrounding the cysticercus
The anatomical location of the cysticerci from both intracranial and oral routes of infection was classified in accordance with previous human NCC reports 1, 11, and in agreement with the Paxinos Rat Brain Atlas. The locations of the cysticerci were defined as follows: parenchymal cysticerci were those lodged exclusively in the gray or white matter of the brain without invading the ventricles or subarachnoid space; ventricular cysticerci were defined as those located inside the ventricles; meningeal cysticerci were those that partially invaded or compressed the cortical brain tissue and were easily observed on the exterior surface of the brain.
Measurement of the cysticercus size and ventricular dilation was performed using light microscopy by H&E staining. All measurements performed for the diameter of the cyst and the extension of spheroids or inflammation were taken from one coronal section per cyst for each stain. A semiquantitative analysis of the infiltrating inflammatory cells and the pyknotic nuclei was performed by manual evaluation of the entire area surrounding the cyst in the coronal section, under a light microscope at 40× objective. Infiltrating cells were identified as plasmocytes, macrophages, lymphocytes and heterophils (eosinophils and neutrophils). Spongy changes and perivascular cuffing were quantified as the percentage of the surrounding tissue where the lesion was observed. The thickness of the fibrotic collagen layer was calculated by averaging the width of the lesion at 4–6 different points randomly selected around the cysticerci.
To quantify the area of immunoreactivity to GFAP, Iba‐1 and CD68, images were analyzed using Image J v1.49 (National Institute of Health, MD, USA). First, stacks of images were white‐balanced to normalize the brightness/contrast of each image. Next, a color deconvolution function was applied to separate the DAB‐stained tissue from the hematoxylin‐stained background. The resulting images were then converted to gray scale (8 bit) and the area of immunoreactivity was measured in mm2. Finally, the immunoreactivity of the infected brains was obtained by dividing the area of reactivity by the total area analyzed and normalizing against their respective sham groups.
Axonal spheroids were defined as an accumulation or deposition immunoreactive to either APP, p‐NFP200kD, np‐NFP200kD, pan NFP200kD, SOD‐1, Hsp70, ubiquitin or TDP43 or FUS/TLS occurring in a well‐defined oval body measuring 10–50 µm. The total number of spheroids in one plane (coronal) was counted manually from rats infected by the oral and intracranial routes of infection. The areas of analysis included the cortex, the hippocampus, caudate‐putamen, and the thalamus. The perpendicular distance between the spheroids and the fibrotic scar was calculated for all spheroids.
Statistical analysis
Statistical analysis was performed using the software package Stata 12 (Stata Corp. College Station, TX, USA). Quantitative data and semiquantitative approaches were presented as mean ± SEM to describe the area of immunoreactivity, inflammation and spheroids. To compare the immunoreactivity, spheroids, and type of infiltrating cells between the parenchymal and meningeal cysticerci from both routes of infection, the Mann‐Whitney U and Kruskal‐Wallis tests were used. A Kernel density plot was used to analyze the extension of spheroids beyond the fibrotic scar. Significance was set at P‐values of less than 0.05.
Results
T. solium cysticerci in orally and intracranially infected rats
The rate of infection for intracranially infected rats was 71% (50/70) and for orally infected rats it was 16.7% (5/30). Only brains containing one or two cysts were used for analysis, as when three or more cysts were present it was difficult to analyze the pathological response to an individual cyst. For the intracranial route of infection, 27 rats developed: 11 parenchymal, 16 meningeal and 5 ventricle cysticerci in total. For rats infected by the oral route, 5 out of the 30 rats developed: 3 parenchymal and 4 meningeal cysticerci. No ventricular cysticerci were found in rats infected by the oral route (Supplementary file, Table S2). All T. solium cysticerci found contained translucent vesicular fluid, indicating that the vesicular or viable stage was present. These cysts were morphologically indistinguishable from those in natural human and porcine infections 11.
Parenchymal cysticerci from the intracranially infected group were lodged specifically in the primary motor and somatosensory cortices, the hippocampus, the thalamus and caudate‐putamen. Parenchymal cysticerci from the orally infected group were found in the primary motor cortex and adjacent to the hippocampus. Meningeal cysticerci from the intracranially infected group were found abutting the motor and visual cortexes, olfactory bulb, entorhinal cortex, superior colliculus and dorsal cortex of the inferior colliculus. Similarly, meningeal cysticerci from the orally infected group were found adjacent to the sensorimotor cortex, primary motor cortex and olfactory bulb. In the intracranial group, ventricular cysts had an average 2.9‐fold increase in size of the ipsilateral ventricle compared to the contralateral side, and a 4.8‐fold increase in the size of the lateral ventricles compared to sham rats. The diameters of the cysticerci of both routes of infection were not significantly different (Table 1). Mass effect because of the relatively large size of the cysticercus in the rat brains of both routes of infection was evidenced by abnormal cortical neuronal orientation and displacement of the brain anatomy in areas adjacent to the cysticerci.
Table 1.
NCC neuroinflammatory characteristics
| Sham | Parenchyma | P‐value* | Meninges | P‐value† | Ventricle | ||||
|---|---|---|---|---|---|---|---|---|---|
| Intracranial route | Oral route | Intracranial route | Oral route | Intracranial route | |||||
| Cyst dimensions and area | Cysticercus Diameter | – | 2705 ± 177 | 2003 ± 223 | 0.08 | 2350 ± 196 | 1921 ± 609 | 0.39 | 2036 ± 206 |
| Cyst area as % of brain area | – | 10.50 ± 1.4 | 7.65 ± 0.97 | 0.33 | 8.08 ± 1.04 | 7.25 ± 0.86 | 0.72 | 6.52 ± 1.60 | |
| Histopathological changes | Spongy changes | 0 | 34.09 ± 5.99 | 18.33 ± 7.26 | 0.10 | 18.75 ± 5.05 | 6.25 ± 1.25 | 0.50 | 11.00 ± 9.80 |
| Perivascular cuffing | 0 | 9.55 ± 0.81 | 5.00 ± 0.00 | 0.01 | 5.94 ± 0.94 | 5.00 ± 0.00 | 0.50 | 5.00 ± 1.58 | |
| Frequency of cells with nuclear pyknosis | 0 | 91% | 100% | 0.60 | 100% | 100% | nd | 80% | |
| Thickness of collagen layer | 0 | 28.14 ± 3.89 | 26.02 ± 9.68 | 0.80 | 30.33 ± 5.31 | 28.14 ± 8.52 | 0.80 | 12.41 ± 7.26 | |
| Inflammatory cell infiltrate | Macrophages | 0 | 19.45 ± 3.48 | 22.00 ± 11.02 | 0.50 | 13.88 ± 1.58 | 18.00 ± 8.67 | 0.20 | 31.20 ± 10.42 |
| Heterophils | 0 | 12.55 ± 2.08 | 6.67 ± 0.88 | 0.03 | 9.75 ± 0.87 | 6.50 ± 0.50 | 0.02 | 15.60 ± 4.88 | |
| Plasmocytes | 0 | 60.45 ± 6.33 | 33.67 ± 1.76 | 0.01 | 50.88 ± 3.97 | 31.25 ± 0.48 | 0.02 | 58.80 ± 13.60 | |
| Lymphocytes | 0 | 38.27 ± 6.45 | 21.33 ± 3.76 | 0.18 | 26.81 ± 2.47 | 20.50 ± 1.89 | 0.39 | 39.40 ± 10.23 | |
NCC neuroinflammatory characteristics. Dimensions and Cyst area, histopathological alterations and inflammatory cell infiltrate were analyzed in the tissue surrounding the cysticercus by H&E staining and are presented as mean ± SEM. Sham groups from either intracranial and oral routes of infection did not show any degree of inflammatory cell infiltrate in the brain tissue. Cysticercus diameter (µm) is the mean of two measurements. % cystcicercus area is the percentage area occupied by the cyst as a proportion of the total brain area per H&E stained section. Spongy changes were characterized by fluid‐filled spaces of variable size in the tissue surrounding the cysticercus. Perivascular cuffing refers to a variable cell infiltrate around small brain vessels in the tissue surrounding the cysticercus. Pyknotic cells were characterized by nuclear shrinking. The thickness of the collagen layer was obtained by the mean of 4–6 measurements of the collagen layer surrounding the cysticerci. Heterophils refer to either eosinophils or neutrophils.
P‐value represents individual P‐values of the two‐sample test of ranges (Mann‐Whitney) between each variable for parenchymal cysticerci by intracranial vs. oral route.
P‐value represents individual P‐values of the two‐sample test of ranges (Mann‐Whitney) between each variable for meningeal cysticerci by intracranial vs. oral route.
Cellular pathology in rat NCC
Sham rats, that received intracranial injection of physiological saline only, showed no spongiform lesions, vasculitis or alteration in the brain architecture in H&E staining (Figure 1, Table 1). In the infected rats, viable cysticerci with a scolex containing the suckers and hooklets were surrounded first by an initial layer of fibrotic tissue between 20 and 30 µm. Numerous macrophages, lymphocytes, heterophils, plasma cells, perivascular infiltrates and cells with nuclear pyknosis were found within the area of gliosis up to 300 µm from the border of the cysticerci, with plasma cells being the most abundant (Figure 1, Table 1). Spongy changes were observed up to 600 µm from the border of the cysticerci, extending beyond the zone of gliosis and inflammation. Sixty‐five and seventy‐five percent of meningeal cysticerci from intracranial and oral routes of infection, respectively, had an associated increase in inflammatory cells and signs of vasculitis in the subarachnoid space. Comparing both routes of infection: increased perivascular cuffing as well as a significantly higher number of heterophils and plasma cells adjacent to cysticerci was found in rats infected via the intracranial route of infection compared to the oral route of infection (Table 1).
Figure 1.
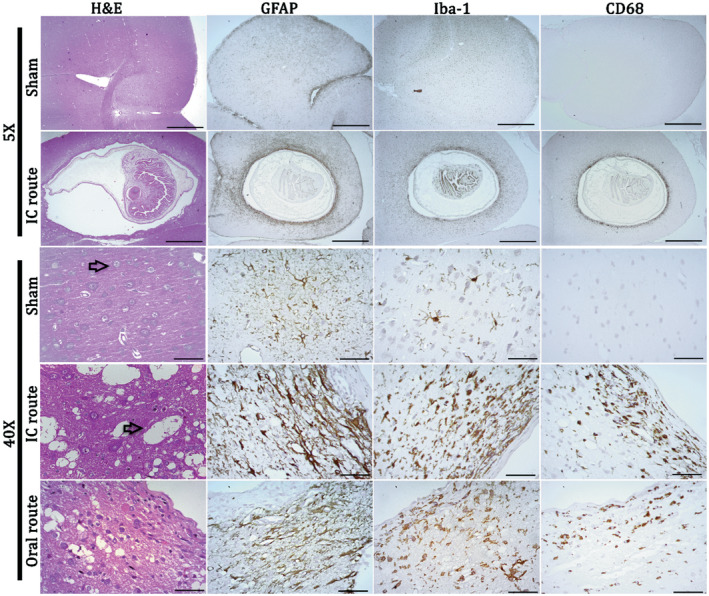
Images showing pathologic changes surrounding T. solium cysticercosis in rat brain. In H & E images rats have well‐preserved tissue and neurons (indicated by an arrow in sham brain). Spongy changes are observed in rats infected by both intracranial and oral feeding (arrow in IC route), and show an accumulation of inflammatory cells adjacent to the collagen layer in both routes of infection. There is an intense reaction by reactive astrocytes (fibrillary gliosis), activated microglia and phagocytes in the brain tissue surrounding cysticerci of both routes of infection. Astrocytes are restricted to areas beyond the fibrotic layer while macrophage cells invade the layer of fibrosis. Inflammatory cells and gliosis were observed up to 300 μm away from the cysticercus. Scale bars: for 10× objective = 1 mm and for 40× = 50 μm.
Reactive astrocytes, activated microglia and phagocytes were present in infected brains only for both routes of infection
In the sham brains of both routes of infection, astrocytes and microglia were in a resting state as characterized by their star‐like shape and highly branched processes with a small nucleus. In the infected brain, reactive astrocytes and activated microglia were found up to 300 µm from the fibrotic capsule (Table 1). Astrocytes surrounding the cysticerci were markedly different from those in sham brains, with thickened processes and a rounder shape (Figure 1, Table 2). Microglia in the area immediately surrounding the cysticerci took on a more amoeboid shape with fewer branched processes, staining more intensely with Iba‐1 when compared to sham sections, while microglia outside the area of definitive inflammation closely resembled the microglia in sham sections (Figure 1). Phagocytes, which include microglia in a late activated stage and invading macrophages (CD68 positive cells), were characterized by strong cytoplasmic staining and were clustered in the area of astrogliosis surrounding the fibrotic layer (Table 2).
Table 2.
Spheroid, astrocyte, microglia and phagocyte evaluation in rNCC
| Parenchyma | P‐value* | Meninges | P‐value† | Ventricle | ||||
|---|---|---|---|---|---|---|---|---|
| Intracranial route | Oral route | Intracranial route | Oral route | Intracranial route | ||||
| Glial cells | GFAP | 25.60 ± 5.67 | 12.77 ± 4.20 | 0.24 | 15.68 ± 3.12 | 34.32 ± 10.45 | 0.06 | 12.62 ± 4.33 |
| Iba‐1 | 5.24 ± 1.57 | 5.33 ± 2.04 | 0.58 | 2.20 ± 0.55 | 0.92 ± 0.53 | 0.34 | 3.74 ± 1.13 | |
| CD68 | 2.98 ± 0.58 | 2.13 ± 1.65 | 0.48 | 1.10 ± 0.50 | 0.41 ± 0.23 | 0.63 | 2.65 ± 1.68 | |
| Frequency (%) of spheroids | p‐NFP200kD | 64% | 100% | 0.51 | 88% | 75% | 0.51 | 80% |
| SOD1 | 82% | 100% | 1.00 | 94% | 100% | 1.00 | 40% | |
| Ubiquitin | 72% | 75% | 1.00 | 100% | 100% | – | 60% | |
| Mean no. of spheroids per cysticercus | p‐NFP200kD | 110.63 ± 23.90 | 45.50 ± 25.60 | 0.10 | 35.30 ± 10.70 | 38.00 ± 21.00 | 0.96 | 32.00 ± 14.00 |
| np‐NFP200kD | 16.60 ± 10.30 | 33.30 ± 20.53 | 0.29 | 8.00 ± 3.99 | 3.00 ± 0.00 | 0.58 | – | |
| pan NFP200kD | 44.50 ± 17.67 | 66.60 ± 39.89 | 0.72 | 14.00 ± 7.46 | 12.30 ± 7.50 | 0.84 | 2.00 ± 2.00 | |
| SOD1 | 150.00 ± 22.40 | 144.00 ± 67.10 | 0.58 | 64.40 ± 28.30 | 69.70 ± 18.80 | 0.39 | 92.40 ± 40.60 | |
| Ubiquitin | 97.10 ± 19.90 | 129.00 ± 64.90 | 0.07 | 53.90 ± 15.80 | 39.00 ± 18.20 | 0.82 | 53.70 ± 32.30 | |
| Hsp70 | 36.00 ± 23.40 | 22.00± | 0.76 | 20.00 ± 9.96 | 6.66 ± 4.05 | 0.84 | – | |
Neuronal lesion evaluation in rNCC. For glial cells and phagocytes, the histological area analyzed was up to 300 µm from the collagen layer and is presented as the mean area of immunoreactivity ± SEM. For the frequency (%) of spheroids and spheroid number, the histological area analyzed was up to 1500 µm from the collagen layer. The frequency (%) of spheroids shows the proportion of cysticerci that are associated with any number of spheroids; statistics performed using Fisher exact test. Spheroid number is the mean number of spheroids per cysticerci and is presented as mean ± SEM. Sham groups from either intracranial or oral routes of infection did not show any structure resembling spheroids. For np‐NFP200kD we only performed IHC for 3 ventricular, 6 parenchymal and 13 meningeal cysticerci, and for Hsp70 only 1 ventricular, 3 parenchymal and 9 meningeal cysticerci were analyzed.
P‐value represents individual P‐values of the two‐sample test of ranges (Mann‐Whitney) between each variable for parenchymal cysticerci comparing intracranial vs. oral route.
P‐value represents individual P‐values of the two‐sample test of ranges (Mann‐Whitney) between each variable for meningeal cysticerci comparing intracranial vs. oral route.
Axonal swelling is found in areas surrounding the cysticerci in orally and intracranially infected rats
Axonal swelling in the form of spheroids was identified by staining areas for any of the following proteins: neurofilament proteins [phosphorylated (pNFp 200KDa) and non‐phosphorylated (NFp 200KDa)], ubiquitin, SOD1 and Hsp70 (Figure 2). Spheroids did not stain for either TDP43 or FUS/TLS (data not shown). APP staining was only performed on one rat brain specimen in an external laboratory as detailed above, which showed axonal swellings with APP immunoreactivity. No significant difference was found for the number of spheroids per cysticerci between the oral and intracranial routes of infection, when compared for each marker by location (Table 2). In sham rats and infected rat brains that did not develop cysticerci, no spheroids were seen.
Figure 2.
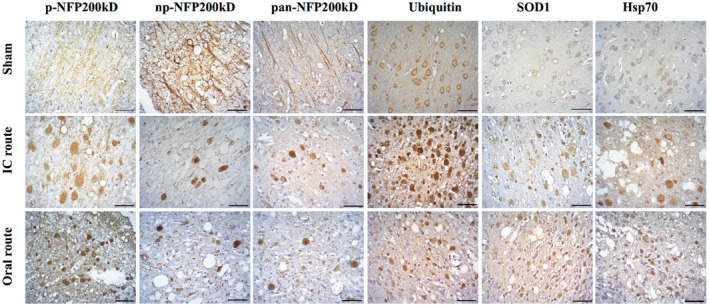
Images showing rat axonal swellings (spheroids) surrounding T. solium cysticercus. Spheroids were detected using IHC to neurofilament (phosphorylated (p‐NFp), non‐phosphorylated (np‐NFp) or pan), ubiquitin, SOD1 or Hsp70 antibodies. No spheroids were found in sham animals. The images show spheroids located in cortical areas adjacent to the cysticercus for the intracranial (IC) and oral routes of infection but were also found in the hippocampus (data not shown). Spheroids were more clearly observed using NFp than with np‐NFp or pan‐NFp antibody. Ubiquitin and SOD1 in sham sections were restricted to the cytoplasm as expected. In both IC and oral routes of infection, ubiquitin and SOD1 spheroids were scattered in the cortex and associated with spongy changes. Hsp70 spheroids were fewer compared to other markers. Scale bars = 50 μm.
Spheroids had diffuse morphologies but were similar in shape and size in the brains infected by the oral and the intracranial routes of infection, with an average diameter of 20 µm (Figure S1), although in some cases p‐NFP200kD and SOD1 swellings reached a diameter of 50 µm. As measured in the coronal plane, spheroids were more abundant closer to the cyst within the area of gliosis, the majority observed within 100–500 µm, and became sparser further away from the cyst, extending as far as 1600 µm away from the fibrotic layer. SOD1 was more widely dispersed than other spheroid markers (Figure 3). While SOD1 and ubiquitin were observed as far as 1600 µm away from the fibrotic lesion, Hsp70 and p‐NFp only extended to 1200 µm (Figure 3). Spheroids were not uniformly distributed around the cyst. Spheroids associated with parenchymal cysticerci were more abundant (Table 2).
Figure 3.
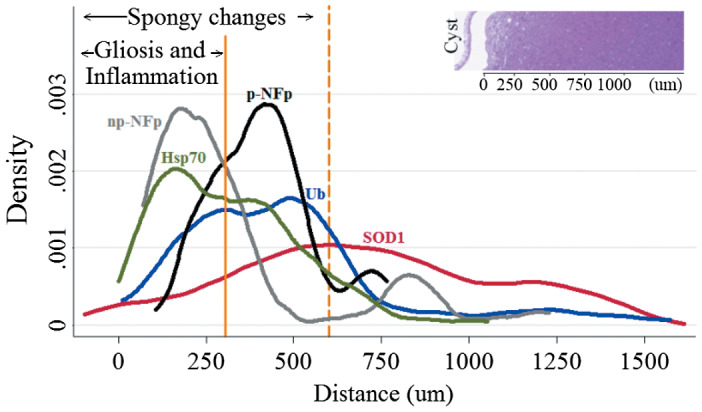
Spheroid distribution in intracranially infected rats. The graph shows a kernel density plot of the different spheroid markers. Distance represents the extension of the different markers from the fibrotic layer of the cysticerci. Most spheroids are found in conjunction with spongy changes (600 um) and gliosis but extend more than 1500 um into the tissue that appears to be otherwise normal.
Distribution of different spheroid markers, neurons and glial cells
Interestingly, SOD1, HSP70 and ubiquitin spheroids frequently merged on immunofluorescence images (Figure 4). In general, SOD1 and neurofilament proteins did not merge (Figure 4). In addition, immunofluorescence for spheroid markers (np‐NFp, pNFp pan NFp, SOD1, ubiquitin, Hsp70) and glial cells (GFAP and IBA‐1) did not merge (Figures S2 and S3). Neuron nuclei (NeuN) were observed in close proximity to p‐NFp, np‐NFp and pan‐NFp (Figure S3) and in some cases, NeuN was observed in proximity to SOD1 and ubiquitin (Figure S3).
Figure 4.
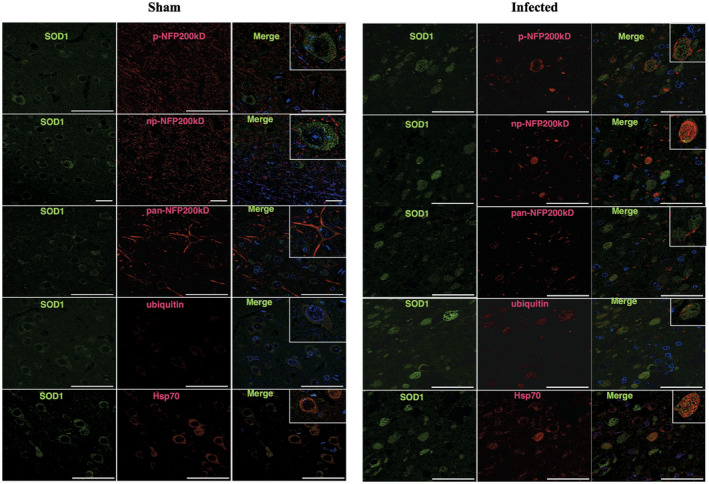
Spheroids shown by confocal imaging. Sham sections showed the normal distribution of SOD1, neurofilament forms, ubiquitin and Hsp70, with no spheroids seen. Infected brains (intracranial route) show most neurofilament protein (p‐NFP200, np‐NFP200KDa and pan‐NFP200KDa) forms do not overlap with SOD1. However, as is shown in SOD1 and p‐NFP200KDa immunofluorescence, SOD1 spheroids can be surrounded by or can coexist with NFp. SOD1 and ubiquitin double labeling shows that spheroids share a similar distribution as well as SOD1 and Hsp70 spheroids.
Axonal swelling is a common feature present in human, pig and rat neurocysticercosis
In order to demonstrate that axonal swelling was not restricted to the rat model of cysticercosis, four pathological specimens were analyzed from four human cases and five specimens from five pig cases of neurocysticercosis. Tissue regions adjacent to the cyst from human cases typically were involved in inflammatory and reactive changes accompanied by axonal destruction, and all those cysts were invariably degenerated. However, the more distal tissue showed extensive staining for APP demonstrating the presence of axonal spheroids in three out of the four analyzed human specimens. (Figure 5). In addition, three out of the five brain specimens from five naturally infected pigs showed axonal swelling in close proximity to the cyst, although to a much lesser extent than did human and rat specimens; these spheroids stained only for medium and heavy chain neurofilament protein and not for APP. Cysts presented in pigs were viable with no calcifications.
Figure 5.
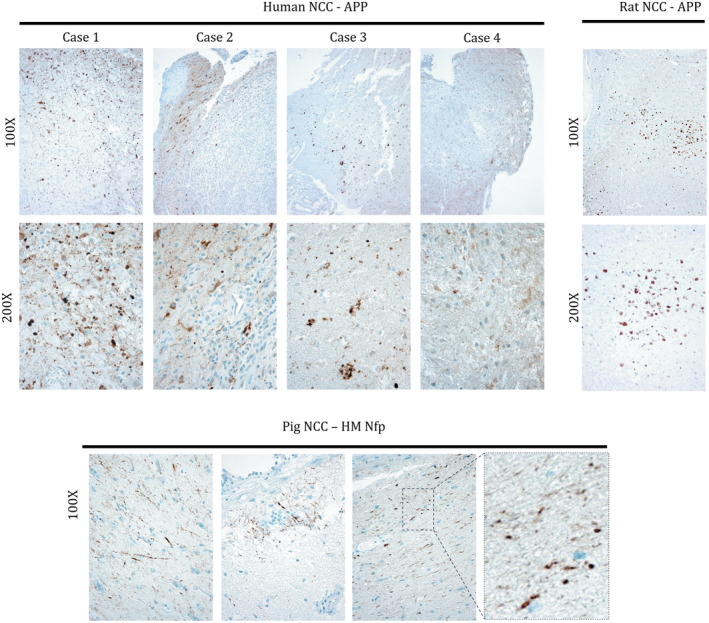
Spheroids found in human, rat and pig NCC. Amyloid precursor protein (APP) staining shows that spheroids are present in human and rat NCC. Cases 1–3 demonstrate axonal swellings while in case 4 axonal swellings are less conspicuous among inflammatory changes. Pig samples do not show evidence of spheroids positive for APP (not shown) but axonal swellings were detected with high/medium‐MW neurofilament (HM NfP) IHC.
Discussion
To our knowledge, these are the first data demonstrating the presence of axonal swelling, evidenced by the formation of spheroids, not only in a rat model of NCC, but also in human NCC brain tissues. These spheroids were not confined to the area of gliosis but extended into the otherwise normal tissue. In addition, this is the first study showing rats can simulate the oral route of infection via ingestion of T. solium oncopheres with subsequent development of viable intracranial cysts. Importantly, the route of infection did not affect the appearance of axonal spheroids indicating that this finding is not an artifact of intracranial inoculation.
Spheroids are focal swellings of an axon often filled with organelles and disrupted cytoskeleton, 7. Although axonal enlargement with increased staining for NFP was described by Sikasunge et al in naturally infected pigs, the quantity, size and distribution were not characterized 36. Here, for the first time, we have demonstrated the presence of spheroids in humans, pigs and a rat model of neurocysticercosis and have characterized the histopathology and immunoreactivity of spheroids in rats. Although conserved across these species, axonal spheroids appeared more prominently in rats and humans compared to pigs.
Axonal swellings, which encompass spheroids and smaller varicosities, are emerging as an important early finding in Alzheimer’s disease 38, amyotrophic lateral sclerosis 25, metabolic encephalopathies, multiple sclerosis and other demyelinating diseases 24. It is also more commonly reported in traumatic, ischaemic and toxic brain injuries 10, 21, 23, 33, 39, thus representing a common pathway of Wallerian degeneration and neurodegenerative disease. Histological findings in our rat model revealed similar reactivity to that found in various aforementioned neurodegenerative diseases, supporting the likelihood of shared common pathways in axonal degeneration of NCC with these diseases. Axonal degeneration has been shown to precede neuronal cell death in various diseases. Furthermore, new longitudinal imaging methods have shown that axonal spheroids are present on unbroken axons, raising the possibility of reversibility of this stage of the disease process 5. Although it has been possible to slow this process in animal models 7, the reversibility of this stage of axonal degeneration is currently not known.
Increasing evidence shows that impairment of axonal transport leads to axonal degeneration in various diseases 24, 25, 38. Amyloid precursor protein (APP) is transported rapidly along the axon and its levels are usually undetectable; however, with impairment of axonal transport it can accumulate in axonal spheroids serving as a marker of disrupted transport 7. Our findings show accumulation of APP in human and rat samples, indicating that the impairment of axonal transport may also contribute to axonal pathology in NCC. Furthermore, our results show an accumulation of both phosphorylated (p‐NFP) and non‐phosphorylated (np‐NFP) neurofilaments within axonal spheroids. These structural elements have been shown to accumulate secondary to aberrant phosphorylation and disruption of slow axonal transport 9, 18, 26, 42. Interestingly, increased levels of neurofilaments have been found in the CSF of patients with multiple neurodegenerative diseases and are thought to correspond to axonal damage 15, 30, 43. Non‐useful proteins are marked by ubiquitin for degradation by the ubiquitin protease system (UPS) 3. Our finding of increased ubiquitin immunoreactivity suggests that there is an increase in proteins recognized as abnormal within the spheroids. However, this could simply reflect the accumulation of proteins because of the impairment of axonal transport. The frequent co‐localization of SOD1 and ubiquitin could indicate ubiquitination of SOD1 and hence an abnormality of this protein within axonal spheroids of NCC. Further studies would be needed to confirm this hypothesis.
Many diseases with axonal swellings have increased frequency of seizures, most prominent in Seitelberger’s disease 13, 34, infantile neuroaxonal dystrophy 32, cerebral malaria 16, 23, traumatic brain injury 33 and Hallervorden‐Spatz Syndrome 14 but also seen with increased frequency in Alzheimer’s disease 4. The principal clinical feature of NCC is seizures; however, the trigger for seizures in NCC remains unclear. A recent study in humans compared magnetic resonance imaging (MRI) of the brain between those with NCC and seizures and asymptomatic individuals with NCC and found there was no correlation between cyst stage or burden and seizure development. Furthermore, asymptomatic individuals had a similar proportion of degenerating cysts with perilesional edema and contrast enhancement, a finding commonly implicated with acute seizure development in NCC 31. Although likely multifactorial, it is possible that axonal spheroid formation may contribute to seizure development. Further studies would need to correlate the degree of spheroid formation with seizure development, before investigating causality.
Various mechanisms are known to lead to axonal degeneration, including direct physical trauma to the axon, oxidative stress and inflammation 7, 8. In our rat model, because of the relatively large size of the cysticerci in the rat brain, physical compression of the axon likely contributes to the impairment of axonal transport and axonal spheroid formation. However, we have demonstrated a significant inflammatory response surrounding the cysticerci which could also be contributing to the axonal pathology. The finding of spheroids at a distance from the cysticerci in an otherwise normal tissue could be explained by the mass effect of the cysticerci in rat brains, or it could be a downstream effect on the distal axon because of the damage more proximally. In humans, the cyst is relatively smaller compared to the human brain, so a mass effect alone is unlikely to explain the finding of spheroids at a distance from the cyst. It is plausible that the intense immune reaction in the vicinity of the cyst provokes a broader zone of axonal degeneration.
Humans develop NCC after accidental ingestion of T. solium eggs. In this study, for the first time, we have infected rats orally with T. solium oncospheres obtained from eggs, with the successful development of cysticerci in their brains. However, this technique required a high number of pre‐activated oncospheres to produce infection, with a lower rate of infection compared to the intracranial route, limiting the use of this technique. Histopathological findings were similar between the oral and intracranial routes of infection, indicating that spheroid formation is not an artifact of the intracranial route of infection. However, differences in inflammatory infiltrate cells were observed; with increased perivascular cuffing, increased heterophils and plasmocytes were seen in the intracranial compared to the oral route of infection.
This study has shown that axonal spheroids are a prominent finding in NCC in humans, pigs and our rat model, and share common histopathological features to axonal pathology seen in many neurodegenerative diseases and Wallerian degeneration. Future studies should focus on using this rat model to analyze the relationship of spheroid formation to specific clinical signs, such as seizures, as well as to test for the reversibility of spheroid formation, using known, effective antihelminthic treatments.
Conflict of Interest
The authors declare that they have no conflict of interest.
Author Contributions
All authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Conceptualization, AJ MM, RP CO, MR V, RL W and RH G; Methodology, AJ MM, RP CO, DG D, G C and MR V; Investigation, AJ MM, RP CO, DG D, G C, JD M and J M; Formal Analysis, AJ MM and RP CO; Resources: MR V, HH G and RH G; Writing—Original Draft, AJ MM, RP CO and ES C; Writing—Review and Editing, AJ MM, RP CO, ES C, C G, J A, C S, A G, HH G, RL W, RH G and MR V; Visualization, AJ MM and RP CO; Supervision, RH G and MR V; Project Administration, MR V; Funding Acquisition, RH G, HH G and MR V. All authors reviewed the manuscript.
Supporting information
Table S1. List of antibodies used in this study.
Table S2. Distribution of cysts.
Figure S1. Higher magnification images of spheroids stained with the different antibodies.
Figure S2. Confocal images of double labeling with GFAP and the different spheroids markers (NFP, SOD1, ubiquitin and Hsp70). Astrogliosis (GFAP) is seen in the tissue surrounding the T. solium parasite (P). Most spheroids are present outside of the area of gliosis. No spheroid markers overlapped with GFAP.
Figure S3. Confocal images of double labeling between Iba‐1 and the different spheroids markers (p‐NFP200kD, SOD1, ubiquitin and Hsp70). Normal tissue shows microglial cells in their normal resting state. A dense Iba‐1‐positive reaction is seen surrounding T. solium parasite (P). None of the spheroid markers overlay with Iba‐1.
Figure S4. Confocal images of double labeling with NeuN and the different spheroid markers (NFP, SOD1, ubiquitin and Hsp70). In sham tissue heavy neurofilament protein is present in its normal form distributed along the length of the axon of most neurons, close to the soma (NeuN marker). In infected tissue, spheroids surrounding the cysticercus are not in close proximity to the soma (NeuN marker). Ubiquitin, SOD1 and Hsp70 were abundant in the tissue close to the cysticercus and formed spheroids which appear close to the soma of some neurons. P represents the location of the parasite (cysticercus).
Acknowledgments
We are grateful to Dr. Diana Rivas, Head of the Department of Neuropathology of the Instituto Nacional de Ciencias Neurologicas in Lima, Peru; Carla Cangalaya, Casey Krebs, Homero Céliz, Ana Delgado, Cesar Quispe for their technical support in the rat model for neurocysticercosis as part of the Cysticercosis working group in Peru and, Dr. Armando Gonzalez for his help in obtaining samples for histological processing and Dr. Richard lerner.
We would like to thank Michelle Beam, MPH, Oregon Health and Science University, for her help and advice in processing of human and porcine samples.
This study was funded by Innovate Peru 135, PNP‐PNIC‐2015; Fondecyt‐Convenio, No.118‐2015; National Institutes of Health P30 AG008017; and National Institutes of Health D43 TW001140.
[Correction added on 20 March 2020, after first online publication: The middle name of one of the authors' names was changed after publication.]
References
- 1. Alvarez JI, Colegial CH, Castaño CA, Trujillo J, Teale JM, Restrepo BI (2002) The human nervous tissue in proximity to granulomatous lesions induced by Taenia solium metacestodes displays an active response. J Neuroimmunol 127:139–144. [DOI] [PubMed] [Google Scholar]
- 2. Arora N, Tripathi S, Kumar P, Mondal P, Mishra A, Prasad A (2017) Recent advancements and new perspectives in animal models for Neurocysticercosis immunopathogenesis. Parasite Immunol 39:e12439. [DOI] [PubMed] [Google Scholar]
- 3. Atkin G, Paulson H (2014) Ubiquitin pathways in neurodegenerative disease, Front Mol Neurosci 7:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Born HA (2015) Seizures in Alzheimer’s disease. Neuroscience 286:251–263. [DOI] [PubMed] [Google Scholar]
- 5. Burke RE, O’Malley K (2013) Axon degeneration in Parkinson’s disease. Exp Neurol 246:72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cardona AE, Restrepo BI, Jaramillo JM, Teale JM. 1999. Development of an animal model for neurocysticercosis: immune response in the central nervous system is characterized by a predominance of gamma delta T cells. J Immunol Baltim Md 1950 162:995–1002. [PubMed] [Google Scholar]
- 7. Coleman M (2005) Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci 6:889–898. [DOI] [PubMed] [Google Scholar]
- 8. Coleman MP, Perry VH (2002) Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci 25:532–537. [DOI] [PubMed] [Google Scholar]
- 9. Dale JM, Garcia ML (2012) Neurofilament phosphorylation during development and disease: which came first, the phosphorylation or the accumulation? J Amino Acids 2012:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dolinak D, Smith C, Graham DI (2000) Hypoglycaemia is a cause of axonal injury. Neuropathol Appl Neurobiol 26:448–453. [DOI] [PubMed] [Google Scholar]
- 11. Garcia HH, Nash TE, Del Brutto OH (2014) Clinical symptoms, diagnosis, and treatment of neurocysticercosis. Lancet Neurol 13:1202–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gonzales I, Rivera JT, Garcia HH, for The Cysticercosis Working Group in Peru (2016) Pathogenesis of Taenia solium taeniasis and cysticercosis. Parasite Immunol 38:136–146. [DOI] [PubMed] [Google Scholar]
- 13. Gordon N (2007) Infantile neuroaxonal dystrophy (Seitelberger’s disease). Dev Med Child Neurol 44:849–851. [DOI] [PubMed] [Google Scholar]
- 14. Gothwal S, Nayan S (2016) Hallervorden‐Spatz syndrome with seizures. Basic Clin Neurosci J 7:165–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Håkansson I, Tisell A, Cassel P, Blennow K, Zetterberg H, Lundberg P et al. (2017) Neurofilament light chain in cerebrospinal fluid and prediction of disease activity in clinically isolated syndrome and relapsing‐remitting multiple sclerosis. Eur J Neurol 24:703–712. [DOI] [PubMed] [Google Scholar]
- 16. Idro R, Marsh K, John CC, Newton CRJ (2010) Cerebral malaria: mechanisms of brain injury and strategies for improved neurocognitive outcome. Pediatr Res 68:267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ignacio Madrazo N, Olhagaray B, Becerra M, Sandoval MA, Raúl SL (1983) Acute cysticercosis encephalitis: description of a histologically confirmed case. Neurosurgery 13:593–595. [DOI] [PubMed] [Google Scholar]
- 18. Kumar S, Yin X, Trapp BD, Hoh JH, Paulaitis ME (2002) Relating interactions between neurofilaments to the structure of axonal neurofilament distributions through polymer brush models. Biophys J 82:2360–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lino‐Junior RDS, Faleiros ACG, Vinaud MC, Oliveira FAD, Guimarães JV, Reis MAD et al (2007) Anatomopathological aspects of neurocysticercosis in autopsied patients. Arq Neuropsiquiatr 65:87–91. [DOI] [PubMed] [Google Scholar]
- 20. Liu YJ, Li QZ, Hao YH (2002) Oncospheres of Taenia solium develop into cysticerci in normal mice. J Vet Med Ser B 49:371–372. [DOI] [PubMed] [Google Scholar]
- 21. Lynch DS, Jaunmuktane Z, Sheerin U‐M, Phadke R, Brandner S, Milonas I et al (2016) Hereditary leukoencephalopathy with axonal spheroids: a spectrum of phenotypes from CNS vasculitis to parkinsonism in an adult onset leukodystrophy series. J Neurol Neurosurg Psychiatry 87:512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Matos‐Silva H, Reciputti BP, de Paula EC, Oliveira AL, Moura VBL, Vinaud MC et al (2012) Experimental encephalitis caused by Taenia crassiceps cysticerci in mice. Arq Neuropsiquiatr 70:287–292. [DOI] [PubMed] [Google Scholar]
- 23. Medana IM, Day NP, Hien TT, Mai NTH, Bethell D, Phu NH et al (2002) Axonal injury in cerebral malaria. Am J Pathol 160:655–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Medana IM, Esiri MM (2003) Axonal damage: a key predictor of outcome in human CNS diseases. Brain J Neurol 126(Pt 3):515–530. [DOI] [PubMed] [Google Scholar]
- 25. Millecamps S, Julien J‐P (2013) Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci 14:161–176. [DOI] [PubMed] [Google Scholar]
- 26. Mizusawa H, Matsumoto S, Yen SH, Hirano A, Rojas‐Corona RR, Donnenfeld H (1989) Focal accumulation of phosphorylated neurofilaments within anterior horn cell in familial amyotrophic lateral sclerosis. Acta Neuropathol (Berl) 79:37–43. [DOI] [PubMed] [Google Scholar]
- 27. Nash TE, Mahanty S, Garcia HH (2013) Neurocysticercosis—more than a neglected disease. PLoS Negl Trop Dis 7:e1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ong S, Talan DA, Moran GJ, Mower W, Newdow M, Tsang VC et al (2002) Neurocysticercosis in radiographically imaged seizure patients in U.S. emergency departments. Emerg Infect Dis 8:608–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peeters D, Ceuterick C, De Jonghe P, Martin JJ (1980) Cerebral cysticercosis. Light‐ and electron microscopy report on one case. Ann Soc Belg Med Trop 60:183–194. [PubMed] [Google Scholar]
- 30. Poesen K, De Schaepdryver M, Stubendorff B, Gille B, Muckova P, Wendler S et al (2017) Neurofilament markers for ALS correlate with extent of upper and lower motor neuron disease. Neurology 88:2302–2309. [DOI] [PubMed] [Google Scholar]
- 31. Prasad A, Gupta RK, Pradhan S, Tripathi M, Pandey CM, Prasad KN (2008) What triggers seizures in neurocysticercosis? A MRI‐based study in pig farming community from a district of North India. Parasitol Int 57:166–171. [DOI] [PubMed] [Google Scholar]
- 32. Riku Y, Ikeuchi T, Yoshino H, Mimuro M, Mano K, Goto Y et al (2013) Extensive aggregation of α‐synuclein and tau in juvenile‐onset neuroaxonal dystrophy: an autopsied individual with a novel mutation in the PLA2G6 gene‐splicing site. Acta Neuropathol Commun 1:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sahuquillo J, Vilalta J, Lamarca J, Rubio E, Rodriguez‐Pazos M, Salva JA (1989) Diffuse axonal injury after severe head trauma. A clinico‐pathological study. Acta Neurochir (Wien) 101:149–158. [DOI] [PubMed] [Google Scholar]
- 34. Scheithauer BW, Forno LS, Dorfman LJ, Kane CA (1978) Neuroaxonal dystrophy (Seitelberger’s disease) with late onset, protracted course and myoclonic epilepsy. J Neurol Sci 36:247–258. [DOI] [PubMed] [Google Scholar]
- 35. Serpa JA, Clinton White A (2012) Neurocysticercosis in the United States. Pathog Glob Health 106:256–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sikasunge CS, Johansen MV, Phiri IK, Willingham AL, Leifsson PS (2009) The immune response in Taenia solium neurocysticercosis in pigs is associated with astrogliosis, axonal degeneration and altered blood–brain barrier permeability. Vet Parasitol 160:242–250. [DOI] [PubMed] [Google Scholar]
- 37. Singh AK, Prasad KN, Prasad A, Tripathi M, Gupta RK, Husain N (2013) Immune responses to viable and degenerative metacestodes of Taenia solium in naturally infected swine. Int J Parasitol 43:1101–1107. [DOI] [PubMed] [Google Scholar]
- 38. Stokin GB (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 307:1282–1288. [DOI] [PubMed] [Google Scholar]
- 39. Takahashi T, Yagishita S, Amano N, Yamaoka K, Kamei T (1997) Amyotrophic lateral sclerosis with numerous axonal spheroids in the corticospinal tract and massive degeneration of the cortex. Acta Neuropathol (Berl) 94:294–299. [DOI] [PubMed] [Google Scholar]
- 40. Verastegui M, Gilman RH, Arana Y, Barber D, Velasquez J, Farfán M et al (2007) Taenia solium oncosphere adhesion to intestinal epithelial and chinese hamster ovary cells in vitro. Infect Immun 75:5158–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Verastegui MR, Mejia A, Clark T, Gavidia CM, Mamani J, Ccopa F et al (2015) Novel rat model for neurocysticercosis using Taenia solium . Am J Pathol 185:2259–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yuan A, Rao MV, Veeranna Nixon RA (2012) Neurofilaments at a glance. J Cell Sci 125:3257–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zetterberg H, Skillbäck T, Mattsson N, Trojanowski JQ, Portelius E, Shaw LM et al (2016) Association of cerebrospinal fluid neurofilament light concentration with Alzheimer disease progression. JAMA Neurol 73:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of antibodies used in this study.
Table S2. Distribution of cysts.
Figure S1. Higher magnification images of spheroids stained with the different antibodies.
Figure S2. Confocal images of double labeling with GFAP and the different spheroids markers (NFP, SOD1, ubiquitin and Hsp70). Astrogliosis (GFAP) is seen in the tissue surrounding the T. solium parasite (P). Most spheroids are present outside of the area of gliosis. No spheroid markers overlapped with GFAP.
Figure S3. Confocal images of double labeling between Iba‐1 and the different spheroids markers (p‐NFP200kD, SOD1, ubiquitin and Hsp70). Normal tissue shows microglial cells in their normal resting state. A dense Iba‐1‐positive reaction is seen surrounding T. solium parasite (P). None of the spheroid markers overlay with Iba‐1.
Figure S4. Confocal images of double labeling with NeuN and the different spheroid markers (NFP, SOD1, ubiquitin and Hsp70). In sham tissue heavy neurofilament protein is present in its normal form distributed along the length of the axon of most neurons, close to the soma (NeuN marker). In infected tissue, spheroids surrounding the cysticercus are not in close proximity to the soma (NeuN marker). Ubiquitin, SOD1 and Hsp70 were abundant in the tissue close to the cysticercus and formed spheroids which appear close to the soma of some neurons. P represents the location of the parasite (cysticercus).