

Abstract
Lysosomal acid lipase (LAL), encoded by the LIPA gene, hydrolyzes cholesteryl esters and triglycerides to generate free fatty acids and cholesterol in the cell. The essential role of LAL in lipid metabolism has been confirmed in mice and human with LAL deficiency. In humans, loss-of-function mutations of LIPA cause rare lysosomal disorders, Wolman Disease and Cholesteryl Ester Storage Disease, in which LAL enzyme replacement therapy has shown significant benefits in a phase 3 clinical trial. Recent studies have revealed the regulatory role of lipolytic products of lysosomal lipid hydrolysis in catabolic, anabolic and signaling pathways. In vivo studies in mice with knockout of Lipa highlight the systemic impact of Lipa deficiency on metabolic homeostasis and immune cell function. Genome-wide association studies and functional genomic studies have identified LIPA as a risk locus for coronary heart disease (CHD), but the causal variants and mechanisms remain to be determined. Future studies will continue to focus on the role of LAL in the crosstalk between lipid metabolism and cellular function in health and diseases including CHD.
Keywords: Lipids, lipase, atherosclerosis, macrophage, Lipids and cholesterol, functional genomics
INTRODUCTION
Lysosomal acid lipase (LAL), encoded by the lipase A (LIPA) gene in human, is the only known enzyme active at an acidic pH in the lysosome that hydrolyzes cholesteryl esters (CEs) and triglycerides (TGs). The critical roles of LAL in physiology and diseases are demonstrated by the phenotypes seen in mice and human with LAL deficiency.1, 2 Loss-of-function mutations of LIPA result in rare genetic disorders with massive accumulation of CEs and TGs in hepatocytes and macrophages in multiple organs causing subsequent tissue damage.3 Wolman’s disease (WD) is infant-onset and fatal due to a complete loss of LAL. Cholesteryl ester storage disease (CESD) is later-onset due to 5–10% residual LAL activity, resulting in a spectrum of clinical presentation including hyperlipidemia, hepatosplenomegaly, and premature atherosclerosis.3 In contrast to the rare loss-of-function mutations causing hyperlipidemia, common genetic variation in LIPA are specifically associated with coronary heart diseases (CHD)4–7 but not blood lipid traits8 in genome-wide association studies (GWASs). The biological mechanisms underlying the CHD association of LIPA genetic variation in the general population remain poorly understood.
In our previous review,1 we have discussed the role of LAL in intracellular and extracellular lipid catabolism, clinical studies on LAL enzyme replacement therapy for WD and CESD, and the functional genomic discoveries of LIPA as a risk locus for CHD. This review will focus on the biochemical and molecular characterization of human LAL and how the lipolytic products derived from LAL-mediated lysosomal hydrolysis contribute to catabolic, anabolic and signaling pathways both in vitro and in vivo in mice with Lipa-deficiency. We will also discuss the transcriptional regulation of LIPA in physiology and diseases, and highlight future direction.
BIOCHEMICAL AND MOLECULAR CHARACTERIZATION OF LAL
The LIPA gene is located on human chromosome 10q23.2–23.3 and consists of 10 exons spread over approximately 38 kb. LIPA has 3 transcript variants according to the RefSeq database. Variant 2 (NM_000235) lacks an internal segment in the 5’ UTR compared with variant 1 (NM_001127605). The two variants encode the same protein isoform in size of 399 amino acids (AAs), which has been experimentally validated by cDNA cloning.9 The annotated variant 3 (NM_001288979) lacks two consecutive exons in the 5’ region, which results in translation initiation at a downstream AUG and presumably a shorter protein isoform consists of 283 AAs. Previous studies on the structural and catalytic features of LAL and knowledge of other lysosomal hydrolases implicate that the signal peptide sequence and co-translational/post-translational glycosylation modification are important for LAL expression, trafficking and secretion.
LAL purified from human liver showed two silver-staining protein bands of 56 kDa and 41 kDa on SDS/PAGE (sodium dodecyl sulphate-polyacrylamide gel electrophoresis).9 Both proteins are glycosylated.9 Size-exclusion chromatography suggests that the fraction containing predominantly the 41 kDa polypeptide with only a trace amount of the 56 kDa polypeptide is enzymatically active.9 N-terminus sequencing of both 56 kDa and 41 kDa bands support that the 56 kDa band represents the proprotein of 372 AAs lacking the first 27 AAs predicted to be the signal peptide.9 The 41 kDa band has 323 AAs lacking 49 N-terminal AAs compared with the 56 kDa band.9 It has therefore been postulated that specific proteolytic processing is involved in the maturation of LAL, which removes the 49 N-terminal residues from the 372 AA proprotein and release the active lipase.9 Zschenker et al.10 further characterize the proprotein cleavage region by expressing the mutant with deletion of the 49 N-terminal residues in insect cells, which led to a complete loss of LAL activity. But the intracellular location where the cleavage occurs has not been experimentally determined. (Figure 1)
Figure 1. A schematic illustration of the expression, glycosylation modification, lysosomal targeting and secretion of human lysosomal acid lipase (LAL) based on experimental evidence and knowledge on other soluble lysosomal hydrolases.
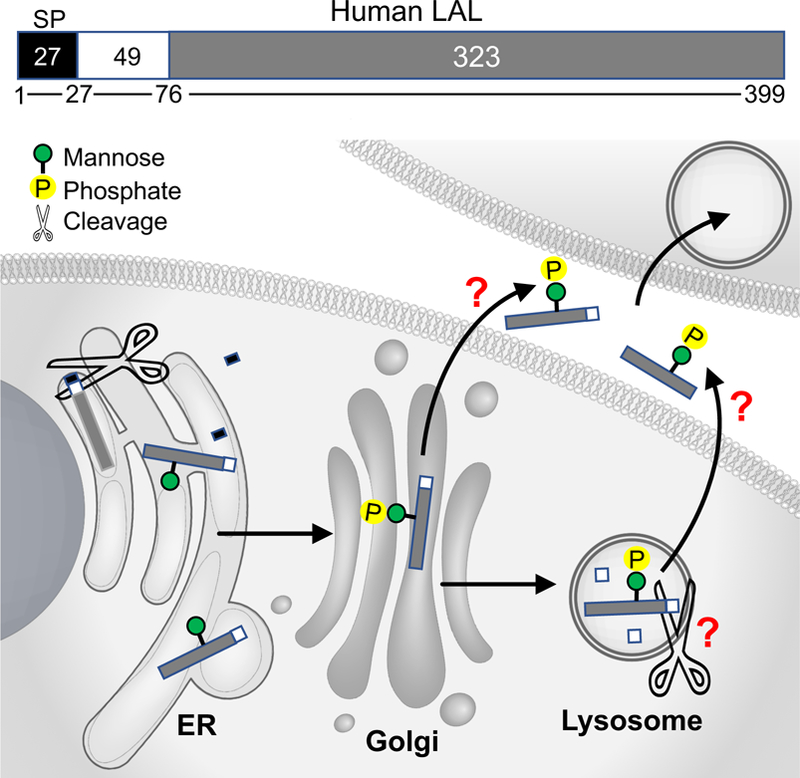
Human LAL consists of 399 amino acids (AAs).9 The first 27 AAs are predicted to be the signal peptide (SP) sequence.9 In many lysosomal proteins, the signal peptide directs their co-translational translocation to the endoplasmic reticulum (ER) for cleavage by the signal peptidase,11 although the role of signal peptide sequence in LAL has not been experimental confirmed. The proprotein of 372 AAs after signal peptide cleavage undergo co-translational/post-translational glycosylation modification in the ER,9, 48 and then transport to Golgi for the addition of mannose 6-phophate (M6P),15 which is important for lysosomal targeting of the hydrolase. It is postulated that the 372 AA proprotein undergoes cleavage of the first N-terminal 49 AAs to form the mature lipase of 323 AAs, which is enzymatically active as suggested by size-exclusion chromatography.9 Extracellular LAL with M6P modification can be taken up by other cells through M6P receptor-mediated endocytosis, and remain catalytically active.15–17 The functional significance of signal peptide sequence, the intracellular location where the proprotein cleavage occurs, and the secretory pathway of mature LAL remain to be further characterized.
Soluble lysosomal proteins are synthesized as precursor polypeptides containing an N-terminal signal peptide sequence of 20–25 AAs.11 The signal peptide sequence is important to direct their co-translational translocation into the lumen of the endoplasmic reticulum, concomitant with the cleavage of the signal peptide by signal peptidase.11 The exact cleavage site of signal peptide sequence in LAL has not been experimentally validated. Peptide sequencing of purified human LAL supports 1–27 AAs as the signal peptide as described above,9 while ab initio prediction by SignalP 4.0 suggests 1–23 AAs. A substitution of arginine for glycine (c.67G>A, p.Gly23Arg) in the signal peptide has previously been speculated to be one of the two compound heterozygous mutant alleles in a WD patient. In vitro expression of the c.67G>A mutant in insect cells caused a retained LAL activity in cell extracts, but reduced activity in the culture supernatant.12 This missense variant (c.67G>A) is, however, catalogued as a common genetic variant (rs1051339, MAF=0.12 in 1000 Genomes Project) being “likely benign” in the ClinVar Achieve, therefore unlikely to be the disease-causing variant for WD. Another missense variant, rs1051338 (c.46A>C, p.Thr16Pro), is in high linkage disequilibrium with the GWAS lead SNP (single nucleotide polymorphism) associated with CHD. The risk allele carriers of rs1051338 showed lower LAL activity and a trend toward reduced cholesterol efflux in monocyte-derived macrophages than the non-risk allele carriers, but the analysis was restricted to 4 individuals, a sample size prone to false positive and underpowered to assess the smaller effect sizes typically associated with common genetic variants.13 Characterization of these genetic variants and the signal peptide sequence in human macrophages of much larger numbers coupled to gene editing in isogenic cell lines with endogenous LIPA expression will further address the functional significance of LAL signal peptide sequence.14
N-linked oligosaccharide side chains, particularly the presence of mannose 6-phosphate (M6P) residues, are found on all soluble lysosomal enzymes.11 Deficiency in synthesizing M6P causes I-Cell disease, in which the lysosomal hydrolases fail to be sorted properly in the Golgi apparatus, therefore are secreted rather than transported to lysosomes. Indeed, decreased intracellular and increased extracellular LAL activities were found in I-Cell disease fibroblasts.15 M6P residues also mediate intercellular exchange of lysosomal enzymes. Secreted LAL in the culture medium can be taken up by fibroblast of WD patients and maintained capacity to correct CE accumulation. The uptake was abolished by adding M6P in the media or by phosphatase treatment, supporting a M6P receptor mediated endocytosis pathway.15, 16 Pretreatment of LAL with endoglycosidase H abolished its uptake by fibroblasts, suggesting that glycosylation of secreted LAL is essential for its uptake and trafficking to the lysosome.17 Indeed, intravenous delivery of recombinant human LAL purified from egg white of genetically engineered chicken was effective in WD and CESD in a Phase 3 clinical trial, indicating efficient uptake of LAL by tissues from circulation.18
To summarize, the cleavage of the signal peptide and the first 49 N-terminal AAs appear be important for the generation of the mature lipase. M6P modification is essential for LAL lysosomal targeting, secretion, and intercellular exchange. It remains to be determined whether lysosomes are responsible for propeptide cleavage, and how LAL is secreted. (Figure 1)
THE REGULATORY ROLE OF LAL-DERIVED LIPOLYTIC PRODUCTS
LAL hydrolyzes CEs and TGs and releases free cholesterol (FC) and fatty acids (FAs) that serve as important energy sources, structural components of cell membranes, and signaling molecules. LAL has recently been found to hydrolyze retinyl esters (REs) and regulate retinoid homeostasis.19 Here we will discuss how LAL couples intracellular lipid metabolism to cellular function through the regulatory role of lipolytic products. (Figure 2)
Figure 2. LAL–mediated lysosomal lipolysis links lipid metabolism to diverse cellular functions.
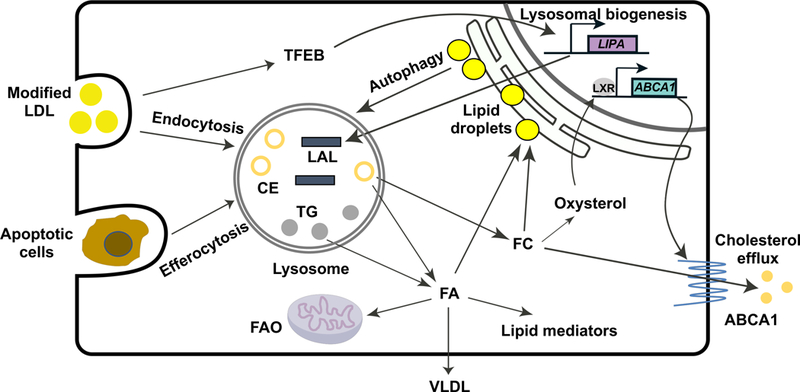
LAL hydrolyzes cholesteryl ester (CE) and triglyceride (TG) in the lysosome to release fatty acids (FAs) and free cholesterol (FC). Modified low-density lipoprotein (LDL) internalized through scavenger receptor-mediated endocytosis is an important source of CE and TG for lysosomal hydrolysis. The hydrolyzed FAs and FC can be re-esterified and form lipid droplets in the endoplasmic reticulum (ER) for storage. Lipid droplets can be delivered to the lysosome for LAL-mediated hydrolysis via autophagy to provide energy supply and maintain cellular homeostasis. The engulfed apoptotic cells by macrophages through a process called efferocytosis also deliver neutral lipids to the lysosome, and LAL is essential for maintaining the efferocytosis capacity of macrophages.29 The lipolytic products of LAL have active biological roles. Hydrolyzed FAs are substrates for fatty acid oxidation (FAO) and synthesis of very low-density lipoprotein (VLDL).32 CE-derived FAs also provide precursors for the systhesis of lipid mediators that have a broad spectrum of functional impact on inflammatory response and resolution.26 Lysosomal flux of FC is essential for the production of oxysterol that triggers the activation of liver-X-receptor (LXR),28 resulting in increased ABCA1 expression and cholesterol efflux to remove cholesterol from the cells. How is LIPA expression regulated remains incompletely understood. Under conditions of lysosomal stress induced by atherogenic lipids such as oxidized-LDL, transcription factor EB (TFEB) triggers lysosomal biogenesis, including increased expression of the LIPA gene.42 ABCA1, ATP-binding cassette transporter A1.
LAL mediates both intracellular and extracellular lipolysis in a cell type-specific mechanism. LAL hydrolyzes neutral lipids delivered to the lysosome through endocytosis of lipoproteins in many cell types. Macrophages can also form extracellular lysosomal synapse where LAL remains active and mediates catabolism of aggregated low-density lipoprotein (LDL).20 In macrophages and hepatocytes, LAL was found to hydrolyze neutral lipids stored in lipid droplets (LDs) through lysosome-dependent autophagy pathways termed lipophagy. Ablation of autophagy by deletion of critical autophagy genes led to increased lipid content in these cells, suggesting impaired LD delivery to lysosomes.21 This autophagy-mediated delivery of LDs to the lysosome can be selectively active in a certain pool of LDs. For example, in hepatic stellate cells, LAL hydrolyzed preexisting LD pools, but not newly synthesized LDs labeled by deuterated FAs.22 In adipose tissue macrophages generated by co-culturing bone marrow derived macrophage with adipose tissue explant, however, the deficiency of Atg7 did not increase the cellular lipid content, suggesting alternative pathways for the mobilization of LD pools in adipose tissue macrophages.23
LAL-mediated hydrolysis of CEs and TGs provides a source of FAs that are important metabolites for the generation of energy via fatty acid oxidation (FAO) and substrates for the biosynthesis of other lipids. FAO fueled by LAL-derived FAs was thought to be essential for interleukin-4 mediated macrophage M2 polarization,24 but the role of FAO is found to be more complex than previously envisioned. Recent studies have found that loss of Cpt1a or Cpt2, encoding the rate-limiting enzymes in FAO, in myeloid cells has little impact on the expression of M2 markers despite significantly decreased FAO.25 Thus, although knockdown of Lipa in macrophage has been shown to reduce the expression of M2 markers upon interleukin-4 treatment,24 the mechanisms must be independent of reduced FA pools. LAL-mediated lipolysis also provides source of precursors for lipid mediator synthesis. Peritoneal macrophages of mice with knockout of Lipa (lal−/−) showed an accumulation of 18:2 and 20:4 fatty acids, important precursors for lipid mediator generation, in CE fraction, but not in phospholipids or TG fractions.26 LAL inhibition by Lalistat2 in wild-type macrophages with acetylated-LDL loading increased intracellular CE concentration, reduced the release of a number of lipid mediators that have a variety of biological functions, including linoleic acid derivative compound 9-hydroxyoctadecanoic acid (9-HODE), a PPAR-γ ligand, which reduced lung inflammation in lal−/− mice when administered intranasally.27
LAL hydrolyzes CEs and release FC, important cell memberane components and precursors for other cholesterol derivatives. Increased FC flux from the lysosome inhibited sterol regulatory element binding protein (SREBP)-dependent de novo cholesterol synthesis and LDL receptor expression.2 Lysosomal release of FC also allows the production of oxysterol, potent endogenous Liver-X-Receptor agonists that activate the transcription of ABCA1 and cholesterol efflux to remove cholesterol from cells.28 Peritoneal macrophages from lal−/− mice showed reduced Abca1 expression and cholesterol efflux with lipoprotein loading.28 In addition to endocytosis of lipoproteins, the engulfment of apoptotic cells (efferocytosis) also delivers neutral lipid to macrophages for lysosomal processing. Knockdown or chemical inhibition of LIPA in human THP-1 macrophages caused a defective efferocytic response of apoptotic cells.29 Mechanistically, inhibition of LIPA led to decreased production of oxysterol upon ingestion of apoptotic cells, resulting in reduced cholesterol efflux and expression of MERTK that mediates apoptotic cell uptake.29
Thus, lysosomal lipolysis links lipid metabolism to diverse cellular functions because many metabolic derivatives of FC and FAs serve as ligands for nuclear receptors and transcription factors that have profound effects.
LAL DEFICIENCY IN MICE: SYSTEMIC EFFECTS ON METABOLIC AND IMMUNE HOMEOSTASIS
The lal−/− mice were first generated by Du et al in 1998.30 The mouse model has been widely used to study the role of Lal in multiple organ systems. The lal−/− mice on a mixed genetic background of 129Sv and CF-1 survive into adulthood, and are fertile, but die at ages of 7 to 8 months.30 The lal−/− mice show massive accumulation of TGs and CEs in liver, spleen, adrenal glands, and small intestine. These mice resemble hepatosplenomegaly, the major clinical manifestation of CESD in human, but not many of the clinical presentations in WD. The lal−/− mice provide a model to study human CESD, but more importantly, serve as a powerful tool to investigate the systemic impacts of lysosomal lipolysis on metabolic and immune homeostasis.
Deficiency in Lipa causes severe accumulation of lipids in hepatic lysosomes and progressive loss of white adipose tissue, which triggers alternative energy pathways. In liver of lal−/− mice on a C57BL/6J background, deficient CE and TG hydrolysis leads to reduced FA flux and hepatic acyl-CoA availability, and consequently, reduced very-low-density lipoprotein synthesis and plasma TG levels,31 which induced metabolic adaptations for increased systemic glucose usage. Intraperitoneal glucose tolerance tests and insulin tolerance tests revealed improved glucose tolerance and insulin sensitivity in lal−/− mice. Plasma glucose levels were reduced, together with reduced liver glucose, glycogen and glutamine concentrations, as well as increased insulin sensitivity and glucose clearance in skeletal muscle to compensate for decreased energy availability.32 The lal−/− mice also showed defective functionality of brown adipose tissue with decreased expression of uncoupling protein 1, leading to transient hypothermia in mice housed at room temperature and life threatening cold intolerance. These phenotypes in lal−/− mice highlighted the critical roles of Lipa in FA mobilization, adipocyte differentiation, and systemic metabolic homeostasis.31
Lipa deficiency causes systemic malfunction of immune cells in mice. The lal−/− mice showed neutral lipid accumulation and impaired T cell development in thymus and spleen, and reduced peripheral T cell number and maturation. These defects are also accompanied by impaired T cell function, characterized by increased apoptosis, reduced proliferation, and impaired response to T cell receptor stimulation.33 Lipa deficiency significantly increased systemic myeloid cell expansion, particularly a CD11b+Gr-1+ myeloid-derived suppressor cells (MDSCs) population that infiltrated in multiple organs including thymus, spleen, lung, liver, and small intestine.34, 35 MDSCs also directly facilitate tumor cell proliferation, growth and metastasis in lal−/− mice through mammalian target of rapamycin pathway.36 These studies provide a mechanistic basis for the role of LAL-mediated lipolysis in immune homeostasis.
Lipa deficiency-induced pathological changes can be reversed by tissue-specific human LAL reconstitution. Hepatocyte-specific expression of LAL corrects liver inflammation and tumor metastasis in lal−/− mice.37 Lung epithelial cell-specific expression of LAL reversed lung inflammation.38 Macrophages appear to play important roles in Lipa-deficiency induced systemic pathogenesis. Inducible macrophage-specific expression of LAL under the control of a c-fms promoter in lal−/− mice partially corrected adipose tissue atrophy, myeloid cell abnormality, and pathogenic phenotypes in liver, lung and intestine.39 The systemic effects of macrophage LAL reconstitution may be attributable to the presence of foamy macrophages throughout multiple organs in lal−/− mice as the major disease driver. It is also plausible that macrophage-secreted LAL can be taken up by other cell types, allowing intercellular exchange within the tissue or through circulation to exert systemic effects.
Although LAL-deficiency is disease causing, increasing LAL activity to above normal levels may not confer benefits. Epithelial cell-specific overexpression of human LAL in wild-type mice accelerated neutral lipid hydrolysis and perturbed the surfactant homeostasis, therefore disrupting lamellar body genesis and alveolar structure in the lung.40 A study by Zschenker et al. also described LAL-overexpressing transgenic mice fed with Western diet showing increased plasma very-low-density lipoprotein levels.41 Therefore, it is important to maintain a homeostasis of lysosomal neutral lipid metabolism.
TRANSCRIPTIONAL REGULATION OF LIPA IN PHYSIOLOGY AND DISEASES
Genotype-Tissue Expression (GTEx) data on the gene expression in different tissues (https://gtexportal.org/home/) suggest that LIPA is most abundantly expressed in spleen, and highly expressed in lung, intestine, and adipose tissue. The protein expression data by immunohistochemistry in Human Protein Atlas suggests that LAL is expressed in spleen red pulp macrophage and lung alveolar macrophages, supporting a macrophage-enriched expression of LAL in humans. How LIPA expression is transcriptionally regulated remains incompletely understood. Finding new molecular pathways and strategies modulating LIPA expression has therapeutic potentials in diseases involving lysosomal lipid accumulation and pathways mediated by lysosomal lipolysis.
Transcription factors involved in autophagy pathways, such as transcription factor forkhead homeobox type protein O1 (FOXO1) and transcription factor EB (TFEB), have been shown to induce LIPA expression in response to nutrient restriction.2 Under conditions of lysosomal stress induced by atherogenic lipids such as oxidized-LDL, TFEB is dephosphorylated and translocated to the nucleus and upregulated the expression of Lipa, Atp6v0d2 and Lamp1, genes involved in lysosomal biogenesis.42 TFEB overexpression augmented Lipa expression and lysosomal biogenesis, rescued the proapoptotic and proinflammatory effects of atherogenic lipid loading in mouse peritoneal macrophage,42 and reduced atherosclerosis in mice model.43
Consistent with the effects of atherogenic lipids in vitro, atherosclerosis is associated with lysosomal lipid accumulation but increased LAL levels. Several studies have reported higher LAL activity in atherosclerotic tissue and lipid-laden lysosomes isolated from atherosclerotic tissue homogenates.2 Transcriptome profilling of nonfoamy and foamy macrophage from mouse plaque showed that foamy macrophage expressed higher levels of genes involved in cholesterol and FA transport pathways, including Lipa, Abca1, and Fabp4, while nonfoamy macrophages are enriched in proinflammatory genes, such as Il1b, Il12a, Tnf, and Nlrp3.44 Collectively, these data argue that macrophages contribute quantitatively to an increase in expression and LAL activity in atherosclerotic plaque, and that lipid-loaded plaque macrophages may play a role in lipid trafficking and homeostasis in atherosclerotic plaques independent of more inflammatory subpopulations of macrophages.
Whether supplementation of LAL would be atheroprotective is an open question. Studies in LDL-receptor knockout mice suggest that supplementation of recombinant human LAL resulted in regression of lesions in coronary and aortic tissue but the mechanisms are unknown.45 The LAL treatment reduced plasma CEs and hepatic lipid levels, and therefore in this model may promote atherosclerosis regression indirectly through hepatic effects and modulation of systemic lipoproteins rather than direct actions on the lesions or lesion macrophages. Direct targeting of LAL to lesional macrophage may contribute to enhanced lipolysis and reduced lysosomal lipid accumulation in the lesion. Yet, it is uncertain at what level of substrate presentation to the lysosome does endogenous LAL become insufficient to maintain normal CE and TG hydrolysis in the lysosome, considering LAL levels are already increased in plaque macrophages. In addition, because lesional macrophages have apparent impaired lysosomal acidification42 that impedes the proper function of LAL, the effectiveness of augmented LAL levels on lysosomal lipolysis in an atherogenic environment in vivo remain very uncertain.
Human genetics and functional genomic studies provide additional insights into the role of LIPA in human CHD as highlighted in our previously published Editorial.14 Although some CESD patients develop premature atherosclerosis, hyperlipidemia and hepatomegaly are the major clinical manifestation and likely the major driver of atherosclerosis progression. Furthermore, heterozygosity of CESD mutation resulting in ~50% loss of LAL activity carries no association with CHD or myocardial infarction risk in a cohort of >27,000 individuals.46 In contrast, the risk alleles of the CHD-associated genetic variants are associated with higher LIPA expression in monocytes5 and monocyte-derived macrophages,47 supporting a potential gain-of-function effect of the LIPA common alleles in CHD. Therefore, despite the effectiveness of enzyme-replacement therapy on reducing multiple disease-related hepatic and lipid abnormalities in CESD patients,18 the long-term effects of recombinant LAL on cardiovascular events and mortality among patients with LAL deficiency and potentially in the general population should be assessed. It is also critical to determine the biological mechanisms of the CHD-associated genetic variants before increasing the activity of LAL beyond the normal cellular response may be considered as therapeutic strategies for CHD.
SUMMARY AND FUTURE DIRECTIONS
LAL has increasingly been recognized as an important regulator in a broad spectrum of pathophysiological conditions. LAL-mediated lipolysis connects intracellular lipid metabolism to cellular functions through lipolytic products that regulate catabolic, anabolic and signaling pathways. Future studies are to further characterize the biochemical and structural features of human LAL, understand the biological mechanisms underlying the genetic association between LIPA and CHD, and the potential benefits and risks in therapeutic targeting of LAL in CHD.
Supplementary Material
{kind=link}
HIGHLIGHTS.
Lysosomal acid lipase (LAL), encoded by the LIPA gene in human, is the only known enzyme active at an acidic pH in the lysosome that hydrolyzes cholesteryl esters and triglycerides.
LAL–mediated lysosomal lipolysis links lipid metabolism to diverse cellular functions through the regulatory role of lipolytic products.
In vivo studies in mice with knockout of Lipa highlight the systemic impact of Lipa deficiency on metabolic and immune homeostasis.
Genome-wide association studies and functional genomic studies have identified LIPA as a risk locus for coronary heart disease, but the causal variants and mechanisms remain to be determined.
Acknowledgments
SOURCES OF FUNDING
This work has received funding from the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH), through Grant Number UL1TR001873, and the NIH R00HL130574 (to HZ).
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Zhang H Lysosomal acid lipase and lipid metabolism: New mechanisms, new questions, and new therapies. Current opinion in lipidology 2018;29:218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dubland JA, Francis GA. Lysosomal acid lipase: At the crossroads of normal and atherogenic cholesterol metabolism. Frontiers in cell and developmental biology 2015;3:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernstein DL, Hulkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: Review of the findings in 135 reported patients with an underdiagnosed disease. Journal of hepatology 2013;58:1230–1243 [DOI] [PubMed] [Google Scholar]
- 4.Peden JF, Hopewell JC, Saleheen D, et al. A genome-wide association study in europeans and south asians identifies five new loci for coronary artery disease. Nature genetics 2011;43:339–344 [DOI] [PubMed] [Google Scholar]
- 5.Wild PS, Zeller T, Schillert A, et al. A genome-wide association study identifies lipa as a susceptibility gene for coronary artery disease. Circulation. Cardiovascular genetics 2011;4:403–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nelson CP, Goel A, Butterworth AS, et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nature genetics 2017;49:1385–1391 [DOI] [PubMed] [Google Scholar]
- 7.Consortium IKC. Large-scale gene-centric analysis identifies novel variants for coronary artery disease. PLoS genetics 2011;7:e1002260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Willer CJ, Schmidt EM, Sengupta S, et al. Discovery and refinement of loci associated with lipid levels. Nature genetics 2013;45:1274–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ameis D, Merkel M, Eckerskorn C, Greten H. Purification, characterization and molecular cloning of human hepatic lysosomal acid lipase. European journal of biochemistry / FEBS 1994;219:905–914 [DOI] [PubMed] [Google Scholar]
- 10.Zschenker O, Oezden D, Ameis D. Lysosomal acid lipase as a preproprotein. Journal of biochemistry 2004;136:65–72 [DOI] [PubMed] [Google Scholar]
- 11.Braulke T, Bonifacino JS. Sorting of lysosomal proteins. Biochimica et biophysica acta 2009;1793:605–614 [DOI] [PubMed] [Google Scholar]
- 12.Zschenker O, Jung N, Rethmeier J, Trautwein S, Hertel S, Zeigler M, Ameis D. Characterization of lysosomal acid lipase mutations in the signal peptide and mature polypeptide region causing wolman disease. Journal of lipid research 2001;42:1033–1040 [PubMed] [Google Scholar]
- 13.Morris GE, Braund PS, Moore JS, Samani NJ, Codd V, Webb TR. Coronary artery disease-associated lipa coding variant rs1051338 reduces lysosomal acid lipase levels and activity in lysosomes. Arteriosclerosis, thrombosis, and vascular biology 2017;37:1050–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H, Reilly MP. Lipa variants in genome-wide association studies of coronary artery diseases: Loss-of-function or gain-of-function? Arteriosclerosis, thrombosis, and vascular biology 2017;37:1015–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sando GN, Henke VL. Recognition and receptor-mediated endocytosis of the lysosomal acid lipase secreted by cultured human fibroblasts. Journal of lipid research 1982;23:114–123 [PubMed] [Google Scholar]
- 16.Sando GN, Ma GP, Lindsley KA, Wei YP. Intercellular transport of lysosomal acid lipase mediates lipoprotein cholesteryl ester metabolism in a human vascular endothelial cell-fibroblast coculture system. Cell regulation 1990;1:661–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sando GN, Rosenbaum LM. Human lysosomal acid lipase/cholesteryl ester hydrolase. Purification and properties of the form secreted by fibroblasts in microcarrier culture. The Journal of biological chemistry 1985;260:15186–15193 [PubMed] [Google Scholar]
- 18.Burton BK, Balwani M, Feillet F,et al. A phase 3 trial of sebelipase alfa in lysosomal acid lipase deficiency. The New England journal of medicine 2015;373:1010–1020 [DOI] [PubMed] [Google Scholar]
- 19.Grumet L, Eichmann TO, Taschler U, et al. Lysosomal acid lipase hydrolyzes retinyl ester and affects retinoid turnover. The Journal of biological chemistry 2016;291:17977–17987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh RK, Haka AS, Bhardwaj P, Zha X, Maxfield FR. Dynamic actin reorganization and vav/cdc42-dependent actin polymerization promote macrophage aggregated ldl (low-density lipoprotein) uptake and catabolism. Arteriosclerosis, thrombosis, and vascular biology 2018:Atvbaha118312087 [DOI] [PMC free article] [PubMed]
- 21.Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell metabolism 2011;13:655–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tuohetahuntila M, Molenaar MR, Spee B, Brouwers JF, Wubbolts R, Houweling M, Yan C, Du H, VanderVen BC, Vaandrager AB, Helms JB. Lysosome-mediated degradation of a distinct pool of lipid droplets during hepatic stellate cell activation. The Journal of biological chemistry 2017;292:12436–12448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grijalva A, Xu X, Ferrante AW Jr. Autophagy is dispensable for macrophage-mediated lipid homeostasis in adipose tissue. Diabetes 2016;65:967–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang SC, Everts B, Ivanova Y, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nature immunology 2014;15:846–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Divakaruni AS, Hsieh WY, Minarrieta L, et al. Etomoxir inhibits macrophage polarization by disrupting coa homeostasis. Cell metabolism 2018;28:490–503.e497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schlager S, Vujic N, Korbelius M, Duta-Mare M, Dorow J, Leopold C, Rainer S, Wegscheider M, Reicher H, Ceglarek U, Sattler W, Radovic B, Kratky D. Lysosomal lipid hydrolysis provides substrates for lipid mediator synthesis in murine macrophages. Oncotarget 2017;8:40037–40051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lian X, Yan C, Qin Y, Knox L, Li T, Du H. Neutral lipids and peroxisome proliferator-activated receptor-{gamma} control pulmonary gene expression and inflammation-triggered pathogenesis in lysosomal acid lipase knockout mice. The American journal of pathology 2005;167:813–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bowden KL, Dubland JA, Chan T, Xu YH, Grabowski GA, Du H, Francis GA. Lal (lysosomal acid lipase) promotes reverse cholesterol transport in vitro and in vivo. Arteriosclerosis, thrombosis, and vascular biology 2018;38:1191–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viaud M, Ivanov S, Vujic N, et al. Lysosomal cholesterol hydrolysis couples efferocytosis to anti-inflammatory oxysterol production. Circulation research 2018;122:1369–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Du H, Duanmu M, Witte D, Grabowski GA. Targeted disruption of the mouse lysosomal acid lipase gene: Long-term survival with massive cholesteryl ester and triglyceride storage. Human molecular genetics 1998;7:1347–1354 [DOI] [PubMed] [Google Scholar]
- 31.Du H, Heur M, Duanmu M, Grabowski GA, Hui DY, Witte DP, Mishra J. Lysosomal acid lipase-deficient mice: Depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. Journal of lipid research 2001;42:489–500 [PubMed] [Google Scholar]
- 32.Radovic B, Vujic N, Leopold C, et al. Lysosomal acid lipase regulates vldl synthesis and insulin sensitivity in mice. Diabetologia 2016;59:1743–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qu P, Du H, Wilkes DS, Yan C. Critical roles of lysosomal acid lipase in t cell development and function. The American journal of pathology 2009;174:944–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qu P, Yan C, Blum JS, Kapur R, Du H. Myeloid-specific expression of human lysosomal acid lipase corrects malformation and malfunction of myeloid-derived suppressor cells in lal−/− mice. Journal of immunology 2011;187:3854–3866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ding X, Du H, Yoder MC, Yan C. Critical role of the mtor pathway in development and function of myeloid-derived suppressor cells in lal−/− mice. The American journal of pathology 2014;184:397–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao T, Du H, Ding X, Walls K, Yan C. Activation of mtor pathway in myeloid-derived suppressor cells stimulates cancer cell proliferation and metastasis in lal(−/−) mice. Oncogene 2015;34:1938–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du H, Zhao T, Ding X, Yan C. Hepatocyte-specific expression of human lysosome acid lipase corrects liver inflammation and tumor metastasis in lal(−/−) mice. The American journal of pathology 2015;185:2379–2389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lian X, Yan C, Yang L, Xu Y, Du H. Lysosomal acid lipase deficiency causes respiratory inflammation and destruction in the lung. American journal of physiology. Lung cellular and molecular physiology 2004;286:L801–807 [DOI] [PubMed] [Google Scholar]
- 39.Yan C, Lian X, Li Y, Dai Y, White A, Qin Y, Li H, Hume DA, Du H. Macrophage-specific expression of human lysosomal acid lipase corrects inflammation and pathogenic phenotypes in lal−/− mice. The American journal of pathology 2006;169:916–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y, Qin Y, Li H, Wu R, Yan C, Du H. Lysosomal acid lipase over-expression disrupts lamellar body genesis and alveolar structure in the lung. International journal of experimental pathology 2007;88:427–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zschenker O, Illies T, Ameis D. Overexpression of lysosomal acid lipase and other proteins in atherosclerosis. Journal of biochemistry 2006;140:23–38 [DOI] [PubMed] [Google Scholar]
- 42.Emanuel R, Sergin I, Bhattacharya S, Turner JN, Epelman S, Settembre C, Diwan A, Ballabio A, Razani B. Induction of lysosomal biogenesis in atherosclerotic macrophages can rescue lipid-induced lysosomal dysfunction and downstream sequelae. Arteriosclerosis, thrombosis, and vascular biology 2014;34:1942–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sergin I, Evans TD, Zhang X, et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nature communications 2017;8:15750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim K, Shim D, Lee JS, et al. Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circulation research 2018;123:1127–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Du H, Schiavi S, Wan N, Levine M, Witte DP, Grabowski GA. Reduction of atherosclerotic plaques by lysosomal acid lipase supplementation. Arteriosclerosis, thrombosis, and vascular biology 2004;24:147–154 [DOI] [PubMed] [Google Scholar]
- 46.Stitziel NO, Fouchier SW, Sjouke B, et al. Exome sequencing and directed clinical phenotyping diagnose cholesterol ester storage disease presenting as autosomal recessive hypercholesterolemia. Arteriosclerosis, thrombosis, and vascular biology 2013;33:2909–2914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nedelec Y, Sanz J, Baharian G, et al. Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell 2016;167:657–669.e621 [DOI] [PubMed] [Google Scholar]
- 48.Zschenker O, Bahr C, Hess UF, Ameis D. Systematic mutagenesis of potential glycosylation sites of lysosomal acid lipase. Journal of biochemistry 2005;137:387–394 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.