

Abstract
Primary graft dysfunction (PGD) is a major limitation to short and long-term lung transplant survival. Recent work has shown mitochondrial damage associated molecular patterns (Mt-DAMPs) can promote solid organ injury but whether they contribute to PGD severity remains unclear. We quantitated circulating plasma Mt-DNA in 62 patients, prior to lung transplant and shortly after ICU arrival. Although all recipients released Mt-DNA, high levels were associated with severe PGD development. In a mouse orthotopic lung transplant model of PGD we detected airway cell-free damaged mitochondria and Mt-DNA in the peripheral circulation. Pharmacological inhibition or genetic deletion of the formylated peptide receptor 1 (FPR1), a chemotaxis sensor for N-formylated peptides released by damaged mitochondria, inhibited graft injury. An analysis of intragraft neutrophil trafficking patterns reveals that FPR1 enhances neutrophil transepithelial migration and retention within airways, but does not control extravasation. Using donor lungs that express a mitochondria-targeted reporter protein, we also show that FPR1-mediated neutrophil trafficking is coupled to the engulfment of damaged mitochondria, which in turn triggers reactive oxygen species (ROS)-induced pulmonary edema. Therefore, our data demonstrate an association between Mt-DAMP release and PGD development and suggest that neutrophil trafficking and effector responses to damaged mitochondria are drivers of graft damage.
Introduction
PGD is an acute multifactorial syndrome in post-transplant lung recipients that is a leading cause of morbidity and mortality (1, 2). In particular late PGD severity is associated with a higher risk of chronic rejection (3). Although the underlying pathophysiological mechanisms remain obscure, there is a general consensus PGD is worsened by ischemia-reperfusion injury (IRI) resulting from either graft retrieval, preservation or implantation (4). IRI can lead to tissue necrosis and release of DAMPs, which can be recognized by pattern recognition receptors (PRRs) that stimulate leukocyte activation and trafficking into inflamed tissue (5). Several reports have implicated the role of DAMPs in PGD. These include high mobility group box 1 (6), extracellular ATP (7) and the soluble receptor for advanced glycation end products (8).
Given their prokaryotic ancestry, Mt-DAMPs are potent activators of the innate immune response (9). Similar to bacteria, mitochondria contain hypomethylated CpG DNA motifs in their chromosome and express N-formylated peptides, which can be detected by the host with the PRRs TLR9 and FPR1, respectively (10). Both Mt-DNA and N-formylated peptides are released by damaged mitochondria (11, 12), contributing to the severity of sterile inflammation resulting from femoral fractures (12), hemorrhagic shock (13), acetaminophen-induced liver injury (14) and sepsis (15). However, whether Mt-DAMPs play a role in lung transplant-related injury is not known.
Here we show that in human lung recipients high perioperative circulating levels of Mt-DNA are associated with severe PGD. In addition, using a murine orthotopic lung transplant model that mimics PGD in humans (16, 17) we find that FPR1-mediated graft injury is coupled to neutrophil trafficking and engulfment of damaged mitochondria released by lung transplants.
Methods
Human Studies
This is a retrospective study based on prospectively collected plasma and clinical information and was approved by the Washington University School of Medicine Institutional Review Board (#201012829). Ten healthy human volunteers and 62 patients who received lung grafts at the Barnes Jewish Hospital from October 2014 to May 2017 were included in the study. Healthy volunteers were male and female subjects over 21 years of age with normal white cell counts, lacking a history of autoimmune disease and had not received a solid organ or cellular transplant. PGD grades were determined in accordance with ISHLT consensus criteria to the exclusion of other transplant associated confounders listed in this report (1). If more than 1 arterial blood gas (ABG) sample was obtained during a given day, the measurement closest to the set time point (T24, T48, or T72) was chosen for PaO2/FiO2 analysis. Patients who were on room air were graded as PGD 0 or 1 depending on the presence or absence of infiltrates consistent with pulmonary edema on chest x-ray. Patients who required ECMO support post-transplant were graded as PGD 3. All PGD data was additionally reviewed by a blinded pulmonologist to confirm accuracy. Blood samples were collected into EDTA-containing vacutainers (BD Sciences) the day before surgery and in the ICU within 6 to 12 hrs after transplantation. To obtain cell-free plasma, samples were centrifuged at 2800 x g for 10 min and immediately stored at −80°C. Written informed consent was obtained from all subjects.
Mt-DNA quantification
Real-Time PCR was performed in a BioRad CFX-Connect machine using reaction mixture containing 0.1 μL of cell-free plasma, 10 μL iQ SYBR Green Supermix (Bio-Rad), 0.5 μL of 5μM forward and reverse primers and 8.9 μL H2O. Assays were performed in triplicate under the following conditions: 1 cycle at 95°C for 3 min, then up to 40 cycles at 95°C for 10 sec and 55°C for 30 sec and then a melt curve was performed from 65°C to 95 °C (0.5 °C every 5 sec). For mice, the protocol was the same except plasma was diluted 1:5 in normal saline prior to addition the reaction mix. Primers for Human Mt-Cytochrome B (MT-CYB; forward 5′-ATGACCCCAATACGCAAAAT-3′ and reverse 5′-CGAAGTTTCATCATGCGGAG-3′), Human Mt-cytochrome C oxidase subunit III (MT-COX3: forward 5′-ATGACCCACCAATCACATGC-3′ and reverse 5′-ATCACATGGCTAGGCCGGAG-3′) and mouse MT-CYB (forward 5′-GGGTCCCTTCTAGGAGTCTGCC-3′ and reverse 5′-TTGAGGCTCCGTTTGCGTGT −3’) were synthesized by Thermofisher (Human) and Invitrogen (mouse). Mt-DNA copy number was estimated by comparison to a real-time PCR standard amplification curve generated from known amounts of purified human MT-CYB and MT-COX3 DNA.
Mouse tissue processing and assays
BAL was collected from the left bronchus and cells and pelleted at 500 x g for 20 mins to produce BALf. Blood was collected in EDTA-containing vacutainers (BD Sciences). Lung cellular fractionation was conducted by tissue mincing with forceps and digestion with RPMI with Type II collagenase (0.5 mg/ml) (Worthington Biochemical) and 5 U/mL DNAse (Sigma) and transferred to a 37°C incubator in mild agitation for 1 hour. Cell pellets were resuspended in FACS buffer (PBS with 2% FBS and 0.4% EDTA) and stained with CD45 (eBioscience, 30-F11), CD11c (BD Pharmigen, HL3), Ly6C (Biolegend, HK1.4), Ly6G (Biolegend, 1A8), Gr1 (Biolegend, RB6–8C5) and CD11b (eBioscince, M1/70). For ROS analysis BAL pellets were incubated with CellROX deep red reagent (Thermo Fisher) for 30 mins at 37 oC in DMEM complete medium (Gibco). Detection of CXCL1, CXCL2, and CXCL5 from BALf was performed using ProcartaPlex Mouse Cytokine & Chemokine Immunoassays (eBioscience) in accordance with the manufacturer`s instructions.
Mitochondria purification and analysis
Mitochondria were fractionated from BAL fluid (BALf) or lung tissue using a mouse or human Tom22+ positive isolation kit (Miltenyi Biotec) in accordance with manufacturer’s recommendations. Damaged mitochondria were prepared from lung mitochondria by 20 min 65o C incubation to induce membrane depolarization and then tittered by Bradford assay. For flow cytometric analysis mitochondrial pellets were stained with Mitotracker Green FM, Deep Red FM (0.5 μL/ml BALf) or TMRM (20 nM), both from Invitrogen. For BALf mitochondria quantification Green FM+ events were counted on a low FSC/high SSC gate as has been previously described (18).
Transmission Electron Microscopy
For ultrastructural analyses, isolated mitochondria from lung transplant recipient BALFs were viewed on a JEOL 1200 EX transmission electron microscope (JEOL USA Inc.) equipped with an AMT 8 megapixel digital camera and AMT Image Capture Engine V602 software (Advanced Microscopy Techniques).
Mice and Lung transplantation
All mice experiments were conducted with an approved IACUC Washington University protocol. C57BL/6J (B6) and B6;129S-Gt(ROSA)26Sor/J (B6 Dendra2) mice were obtained from Jackson Laboratories. LysM-EGFP on a B6 background were obtained from Klaus Ley (La Jolla Institute for Allergy and Immunology). FPR1−/− mice are on a B6 background were purchased from Taconic Biosciences and were intercrossed with LysM-EGFP to generate FPR1−/− LysM-GFP mice. Except for the B6 strain, all mice used in experiments were male, 6–10 weeks old, and bred in the same facility. Male B6 mice were purchased as needed at 6–8 weeks old and rested for 5 days before use in studies. To induce PGD, left lungs were first preserved at 4 ⁰C in low-potassium dextran glucose solution for 18 hrs, and then orthotopically transplanted using procedures previously described by our group (19). All mice received care in compliance with the “Guide for the Care and Use of Laboratory Animals” authored by the National Academy of Sciences and published by the National Institutes of Health under the title of “Principles of Laboratory Animal Care”.
Lung graft injury and histology
Wet to dry ratio was calculated by graft mass assessment before (wet) and after (dry) dehydration. BALf protein content was quantified by absorbance at 595 nm (μQuant-Biotek) using a Bradford kit Protein Assay (BioRad) and bovine serum albumin as a standard (BSA), (Sigma). For histology, lung grafts were harvested, inflation fixed in formaldehyde, embedded in paraffin, sectioned, and stained with hematoxylin and eosin. H&E slides were scored by a blinded grader using a previously described method for assessing injury in a rat lung transplant model (20). Scoring is based on 4 histological criteria, which include alveolar hemorrhage, vascular congestion, fibrin deposits and leukocyte infiltration. Each of these criteria are graded on a 4-level scale of appearance within airspaces where 0; normal (0%), 1; mild (<10%), 2; moderate (10–50%) and 3; severe (>50%). Grades for each of the injury criteria are then summed together to tabulate the lung injury score.
Neutrophil trafficking assays
Neutrophils were purified by immunomagnetic bead-mediated negative selection with biotin-labeled antibody cocktail that we have previously described (17). Labeled cells were fractionated with Streptavidin MicroBeads and LS MACS columns (Miltenyi Biotec). Isolated neutrophils (5×106/50μL) from LysM-GFP+ and LysM-GFP+ FPR1−/− mice were introduced into a B6 donor bronchus just before transplant and 90 mins after engraftment mice were euthanized for BAL and tissue FACS analysis. For transwell assays MLE 12 (murine lung epithelial cells) were grown in HITES medium according to ATCC® instructions and seeded on the underside of a gelatin-coated Transwell filters (3-μm pore polycarbonate membranes) until functional monolayers were established as confirmed by the maintenance of fluid resistance between apical and basolateral compartments. Recombinant murine CXCL1 (10 ng/ml; R&D) or N-formylated peptides (1μM; Sigma) was added into the lower chamber and 4 × 105 B6 or FPR1−/− neutrophils were placed in the upper chamber and transmigration was allowed to progress for 2 hrs at 37 °C. Transmigrating neutrophils were counted manually using a hemocytometer and quantified by FACS to prevent confusion with small numbers of detached MLE 12 cells in the bottom chamber, which was typically < 5%.
Statistical analysis
For human data, Fisher exact test or student’s t-test (with Welch’s correction when appropriate) was used to detect differences between categorical or continuous variables. The paired t test was applied to assess longitudinal differences in mean Mt-DNA levels. For mouse experiments, significant differences were evaluated with the non-parametric Mann-Whitney U test or with an unpaired Student t test if normally distributed. A one-way ANOVA or the equivalent non-parametric Kruskal-Wallis tests with post-hoc comparisons were used to analyze data with more than two groups. Statistical analyses were performed using GraphPad Prism 6.0 (GraphPad Software, Inc.) and a P value < 0.05 was considered significant.
Results
Circulating Mt-DNA levels are elevated in lung recipients with PGD
To measure Mt-DAMP release in human lung transplant recipients we developed an in situ PCR-based quantitative method for the direct assessment of cell-free circulating plasma Mt-DNA. The clinical characteristics of 62 bilateral lung transplant recipients are shown in Table 1. Mt-DNA, as measured by quantitating plasma mitochondrial cytochrome b (MT-CYB) levels, was slightly elevated above that of healthy control volunteers, but was not dependent on end-stage pulmonary disease (Fig. 1A & Fig. S1A-B). However, MT-CYB DNA levels, evaluated between 6 to 12 hours after patient arrival to the ICU, were significantly higher than prior to transplant or in healthy volunteer controls. Moreover, post-transplant increases in Mt-CYB DNA were independent of end-stage pulmonary disease, donor age, sex or the use of cardiopulmonary bypass (Fig. 1C).
Table 1.
| Total n=62 |
PGD 0 n=25 (40.3%) |
PGD 1 n=28 (45.2%) |
PGD 2 n=5 (8%) |
PGD 3 n=4 (6.5%) |
P value | |
|---|---|---|---|---|---|---|
| Donor variables | ||||||
| Age, (years)* | 31 [21–49] | 22 [18–45] | 33 [22 −48] | 47 [27 −57] | 51.5 [27 −59] | 0.1 |
| Female, n (%)† | 28 (45.1%) | 11(50%) | 14 (42.4%) | 1 (20%) | 2 (50%) | 0.6 |
| Recipient variables | ||||||
| Age (years)* | 61 [51–65] | 58 [51–64] | 61 [51–65] | 64 [51–64] | 57 [56.5–67] | 0.2 |
| Female, n (%)† | 21 (33.8%) | 3 (12%) | 12 (43%) | 2 (40%) | 4 (100%) | 0.002 |
| Afro-American, n (%)† | 5 (8%) | 0 | 2(7.1%) | 0 | 3 (75%) | 0.00001 |
| Body Mass Index (kg/m2)* | 25 [21–28] | 25.7 [22–28] | 23.6 [20–28] | 29.6 [20–30] | 24.5 [20–27] | 0.4 |
| Diagnosis: | ||||||
| COPD, n (%)† | 18 (29.1%) | 4 (16%) | 11 (39.3%) | 1 (20%) | 2 (50%) | 0.2 |
| IPF, n (%)† | 26 (41.9%) | 14 (56%) | 8 (28.6 %) | 3 (60%) | 1 (25%) | 0.1 |
| CF, n (%)† | 7 (11.3%) | 1 (4%) | 6 (21.4%) | 0 | 0 | 0.1 |
| Others, n (%)† | 11 (17.7%) | 6 (24%) | 3 (10.7%) | 1 (20)% | 1 (25%) | 0.6 |
| Surgical variables: | ||||||
| Cardio Pulmonary Bypass n (%) † |
21 | 0 | 16 (57.1%) | 1 (20%) | 4 (100%) | 0.000003 |
| Total preservation time (min)* | 351 [285–510] | 330 [282–494] | 343 [284–465] | 520 [333– 605] | 341 [284–464] | 0.3 |
Kruskal-Wallis test. Values presented as median and interquartile range
Chi square test. Values are presented as number and % of the relative column
Figure 1. Lung transplantation increase the levels of circulating Mt-DNA.

(A) Box and whiskers plot of circulating MT-CYB DNA levels obtained from lung recipients before lung transplantation and within 6 to 12 hours after ICU arrival. (B) Paired analysis plot of MT-CYB DNA levels for each patient before and after lung transplantation. (C) Box and whiskers plot of circulating Mt-CYTB DNA levels paired with qualitative variables for end-stage lung disease, gender and use of cardio pulmonary bypass (CBP). The dashed line represents the threshold limit for Mt-DNA detection (background). (**p< 0.01, *** p<0.001, ****p< 0.0001).
We next determined if high post-transplant Mt-CYTB DNA levels are associated with increased PGD severity scored at 72 hours after ICU arrival. Patients with moderate (PGD 2) or severe (PGD 3) PGD had significantly higher levels of MT-CYB when compared to patients without PGD (PGD 0) or when compared to a patient grouping with PGD 0 scores and mild PGD (PGD 1) (Fig. 2 A-B). Moreover, patients with PGD 3 had signficantly higher levels of MT-CYB relative to a patient grouping with PGD 0, 1 and 2 scores (Fig. 2C). Consistant with this observation was a similar association pattern of plasma mitochondrial cytochrome III oxidase (MT-COX3) DNA with PGD severity. (Fig. S2A-D). Taken together, these data suggest that high perioperative plasma Mt-DNA levels are associated with poorer PGD outcomes.
Figure 2. High levels of circulating Mt-DNA are associated with severe PGD in lung transplant patients.

Box and whisker plots of circulating MT-CYB levels in relation to PGD scores at 72 hours after ICU arrival as represented (A) by separate PGD grades (**p< 0.01), (B) patients grouped together with PGD 0–1 grades versus patients with PGD grades 2 or 3 (*p< 0.05) or (C) patients grouped together with PGD 0–2 grades versus patients with PGD grade 3 (*p< 0.05). Error bars represent 95% confidence intervals for (A-C).
Damaged mitochondria are released into the airway following lung transplant
Based on previous reports that Mt-DNA emanates from damaged mitochondria released by cells undergoing necrosis (11, 21), we looked for evidence of cell-free mitochondria within graft airways. To this end, we utilized a murine orthotopic transplant model where prolonged cold storage prior to implantation induces pathophysiology similar to clinical PGD (16). One day after transplantation lung grafts had the histopathological appearance of necrotic epithelial cell injury (Fig. 3A). Bronchoalveolar lavage fluid (BALf) fractionated for cell-free mitochondria using antibodies specific for Tom22, a mitochondria-specific outer membrane protein, showed significantly more evidence of cell-free mitochondria in transplanted grafts when compared to sham-operated controls (Fig. 3B). Notably, similar to humans, Mt-DNA accumulated in the circulating plasma of mice following lung transplantation (Fig. 3C). We next determined if cell-free mitochondria in the airway are derived from the donor or recipient. B6 recipients were transplanted with injured lungs that encode a mitochondria-targeted fluorescent protein Dendra2 (Fig. 3D). Approximately 70% of airway cell-free mitochondria contained Dendra2 protein indicating that most mitochondria released by transplant-mediated IRI originate from the donor lung. Additionally, cell-free mitochondria, irrespective if they were from the donor or recipient, were predominantly hypopolarized indicating ensuing or ongoing mitochondrial membrane permeabilization to release DAMPs (22) (Fig. 3F-G). In line with these observations, cell-free damaged mitochondria were also detected in the BALf of human lung transplant recipients extracted within 12 hrs after arrival to the ICU (Fig. S3A-C).
Figure 3. Cell-free damaged mitochondria are released into the airways of lung transplant recipients.
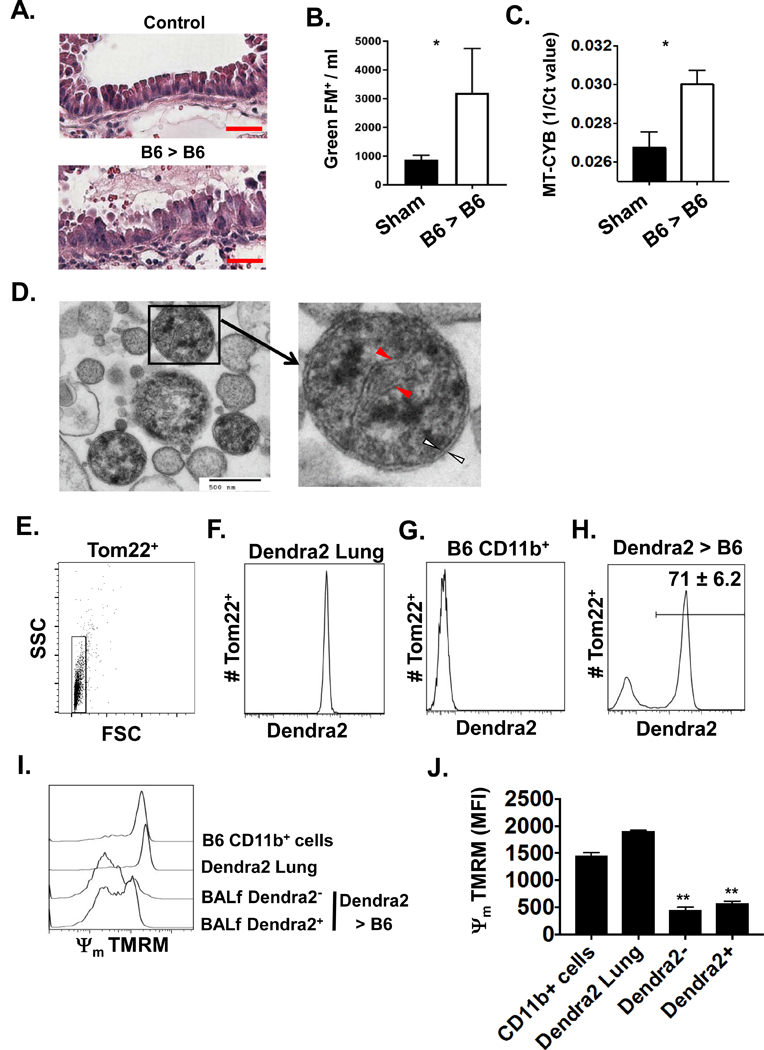
(A) Representative H&E histopathology (N=5) of bronchiolar epithelium from B6 lung grafts and the corresponding right un-transplanted lung (Control) one day after reperfusion (200X magnification, red scale bar = 50 μm). (B) Flow cytometric (FACS) quantitation of (B) BALf cell-fee mitochondria and circulating plasma (C) MT-CYB from mouse lung transplant recipients 90 mins after reperfusion. Sham surgery was used as a control. Data shown as mean Mitotracker GreenFM+ events ± SEM and mean 1/Ct ± SEM where *p <0.05 (n = 5/group). (D) Representative transmission electron micrograph of cell-free mitochondria from the BAL of B6 lung recipients (N=5). Structures resembling mitochondrial cristae (red arrows) and a mitochondrial double membrane (white arrows) at 30 × 103 X magnification where the black scale bar = 500 nM. An analysis of BALf cell-free graft- and recipient-derived mitochondria extracted from Dendra2 → B6 recipients. Tom22+ isolates were gated as in (E) to assess Dendra2 expression in (F) for control mitochondria isolated from Dendra2 lungs, (G) control mitochondria isolated from CD11b+ cells from the resting bone marrow of B6 mice and (H) BALf mitochondria isolated from Dendra2 → B6 recipients. Percent abundance of graft-derived mitochondria is shown as a mean ± SEM for 5 transplants. Measurement of inner mitochondrial membrane potential (Ψm) with the electrosensitive probe TMRM as represented by (I) histogram plot and (J) bar graph of freshly isolated mitochondria from B6 CD11b+ cells, resting Dendra2 lung tissue, cell-free mitochondria isolated from the BALf of Dendra2 > B6 lung recipients differentiated by Dendra2 expression. The result in (I) is a representative histogram of Ψm from 5 experiments and (J) is mean Ψm ± SEM where **p <0.01 (n = 5/group).
FPR1 promotes lung transplant-mediated ischemia-reperfusion injury.
Recently, FPR1 was shown to promote pulmonary injury in mouse model of acute respiratory distress syndrome (23), but whether it promotes inflammatory responses to lung transplants remains to be determined. Accordingly, we assesed lung graft injury in mouse recipients treated with Cyclosporine H (Cy-H), an FPR1 inhibitor. When compared to vehicle-treated recipients Cy-H-treated recipients had less histopathological evidence of alveolar damage (Fig. 4A-B), airway neutrophilia (Fig. 4C) and lung edema (Fig. 4D). We next determined whether FPR1 expression on infiltrating cells or graft tissue promotes tissue injury. Lung transplants were performed where the donor lung or recipient, or both, lacked FPR1. Histopathological analysis of lung grafts exhibited significantly reduced injury (Fig. 5A-C) and BAL neutrophilia (Fig. 5D) in FPR1−/− recipients irrespective of FPR1 expression in the donor graft. Additionally, lung graft edema and permeability were significantly reduced in FPR1−/− recipients (Fig. 5E). Altogether, these data show that FPR1 expression on graft-infiltrating cells is sufficient to promote graft tissue damage.
Figure 4. The FPR1 inhibitor Cyclosporine H reduces signs of lung transplant-mediated IRI.

B6 recipients were treated daily with 50μg i.p. with the FPR1 inhibitor Cyclosporine H (Cy-H) for 3 consecutive days before receiving ischemically-injured B6 lungs. One day after reperfusion grafts were analyzed for signs of acute injury by (A) H&E histopathology (100X, scale bar 200 μm; representative of n=3/group) (B) lung injury score (n=3/group), (C) airway neutrophil accumulation quantified as (left panel) total number and (right panel) percent abundance relative to CD45+ cells (n=4/group) and (D) wet to dry ratio (n=4/group). (B-C) show means ± SEM, *p<0.05, **p<0.01.
Figure 5. FPR1 expression in the recipient is sufficient to promote lung graft injury.

Lung grafts from indicated transplant combinations one-day after engraftment were assessed for signs of acute lung injury. (A) H&E stain (200X, red scale bar = 50μm, n=5/group). Red arrowheads indicate leukocyte infiltration within alveolar spaces. Histological based (B) leukocyte counts within airspaces (mean ± SEM, n=5/group) and (C) lung injury score (mean ± SEM, n=5/group). (D) Airway neutrophilia quantified by (left) total number and (right) percent abundance relative to CD45+ cells (mean ± SEM; n=5/group; *p<0.05, **p<0.01) in the BAL. (E) Lung transplant injury evaluated by (left panel) wet to dry weight ratio (mean ± SEM, n = 6/group) and (right panel) BALf protein content (mean ± SEM, n = 6/group).
FPR1 directs neutrophils to airways.
Most studies have shown that neutrophil-mediated lung injury is predominantly regulated by transendothelial migration, a distinct trafficking step that precedes transepithelial migration into airways (24). Unexpectedly, we found that neutrophil extravasation following lung transplantation was equivalent for B6 and FPR1−/− neutrophils indicating that FPR1 does not regulate neutrophil transendothelial migration (Fig. 6A). Consistent with these observations, we did not detect significant differences between the graft infiltration of B6 and FPR1−/− monocytes, which have been previously reported to promote neutrophil extravasation into transplanted lungs (25) (Fig. S4A). Moreover, chemokine concentrations of airway ELR+ CXC chemokines known to drive neutrophil chemotaxis such as CXCL1, CXLC2 and CXCL5 were all similar in B6 and FPR1−/− recipients (Fig. S4B). In light of these data, we asked if FPR1 promotes airway neutrophilia by enhancing neutrophil transepithelial migration. We analyzed neutrophil trafficking across a monolayer of mouse lung epithelial cells (Fig. 6B). N-formylated peptides elicited FPR1-dependent neutrophil transepithelial migration while both B6 and FPR1−/− responded equivalently to CXCL1. In line with these observations the number of neutrophils remaining in lung grafts after BAL extraction, which is an estimate of neutrophil accumulation within the intravascular and interstitial compartments, also did not significantly vary irrespective of FPR1 expression in the donor or recipient (Fig. 6C). Altogether, these data suggest that FPR1 promotes airway neutrophilia, but not neutrophil recruitment to other graft tissues.
Figure 6. FPR1 promotes neutrophil migration into airspaces.

(A) B6 → B6 and B6 → FPR1−/− recipients analyzed for neutrophil extravasation. PE-labeled Ly6G antibodies were injected intravenously into recipient mice 5 mins prior to euthanasia. Graft tissue was stained with Gr1-FITC antibodies allowing identification of intravascular neutrophils (Gr1-FITC+ Ly6G-PE+) and interstitial neutrophils (Gr1-FITC+ Ly6G-PE-). Results shown are (left) representative contour plots from 4 transplants per group (right) or as a histogram denoting the ratio of graft intravascular to extravascular neutrophils as mean ± SEM (n=4/group; not significant; ns). (B) Transepithelial migration of B6 and FPR1 deficient neutrophils across mouse epithelial MLE12 monolayers in response to N-formylated peptides (fMLP; 1μM) and CXCL1 (10 ng/ml). Data shown as a mean ± SEM; 2 mice pooled per group; 3 independent experiments. Statistical differences between groups were determined by 2 way ANOVA with Sidack`s multiple comparison where p* <0.05, p ****<0.0001. (C) Neutrophil accumulation in non-airway tissue compartments. Indicated transplant recipients one day after reperfusion underwent BAL extraction prior to graft digestion and flow cytometry. Neutrophils quantified by total counts (upper panel) and (lower panel) percent abundance relative to CD45+ cells (mean ± SEM; *p<0.05, **p<0.01; n=5/group). (D) Neutrophils isolated from B6 LysM-EGFP and FPR1−/− LysM-EGFP mice were introduced into the left main bronchus of ischemically-injured B6 donor lungs and immediately transplanted into B6 recipients. Lung recipients were euthanized 90 minutes after reperfusion and EGFP+ neutrophil retention within the airway (BAL), trafficking into non-airway graft tissues and peripheral blood were analyzed by FACS. Representative contour plots of EGFP+ neutrophil percent abundance for indicated tissue compartments from 5 transplants per group. (E) Histogram shows mean number of EGFP+ neutrophils retained within airways (n =5 / group; * p<0.05).
Given previous observations that neutrophils lose mobility in areas of tissue injury (26), we determined if FPR1 contributed to airway neutrophil accumulation by slowing their exit from this compartment. EGFP+ B6 and EGFP+ FPR1−/− neutrophils were introduced into the main bronchi of ischemically-injured B6 lung grafts prior transplantation. Ninety minutes after engraftment we assessed neutrophil exit from the airways into the remaining graft tissue and periphery. When compared to their wild type counterparts, more FPR1−/− neutrophils exited the airway and entered the remaining graft tissues (Fig. 6D-E). Importantly, FPR1−/− and B6 neutrophils were nearly undetectable in the peripheral blood suggesting that they had not migrated out of the graft. Taken together, these data indicate that FPR1 regulates neutrophil extravascular trafficking dynamics through promoting transepithelial migration and retention within damaged graft airways.
Neutrophil engulfment of damaged mitochondria induces ROS generation
Neutrophils play a critical role in reestablishing tissue homeostasis following sterile injuries through engulfing cellular debris. Several reports have shown that neutrophils traffic to sites of cellular injury through following a gradient of N-formylated peptides released by damaged mitochondria (27, 28). However, it remains unclear if FPR1-mediated chemotaxis results in neutrophil uptake of graft-derived mitochondria. Accordingly, B6 and FPR1−/− recipients of ischemically-injured B6 Dendra2 lungs were assessed for infiltrating neutrophils that engulfed Dendra2 protein (Fig. 7A). Significantly more airway B6 neutrophils had Dendra2 cargo when compared to FPR1−/− neutrophils. Additionally, we observed mean Dendra2 fluorescence per neutrophil was higher for B6 neutrophils indicating that the efficiency of mitochondria removal is controlled by FPR1 expression.
Figure 7. Neutrophils that engulf damaged mitochondria induce ROS-mediated pulmonary edema.

(A) BAL neutrophils from Dendra2 > B6 and Dendra2 > FPR1−/− recipients were assessed for uptake of Dendra2 containing mitochondria from lung grafts 3 hours after transplant. Left contour plots show representative percent abundance and the right panel depicts corresponding mean fluorescence intensities (MFI) for 5 transplants (**p<0.01). (B) BAL neutrophils gated on indicated Dendra2+ and Dendra2- gates, as represented in (A), were probed for cellular ROS with CellROX. Data shown are representative FACS histograms from 5 transplants per group. B6 and FPR1−/− neutrophils were co-cultured with indicated amounts of damaged mitochondria prepared from Dendra2 lungs for 20 minutes and then assessed for (C, left panel) percent engulfment, (C, right panel) engulfment per cell, and (D) ROS production. Results shown are a representative experiment (n=4/measurement; **p< 0.01, ***p< 0.001) from 3 independently conducted experiments. (E) 106 B6 and FPR1−/− neutrophils co-cultured with 25 μg/ml (Mt-DAMPs25) or 100 μg/ml (Mt-DAMPs100) of damaged mitochondria for 20 mins in the presence or absence of the ROS inhibitor DPI (10 μM), washed twice and administered down the trachea of resting B6 mice. Pulmonary edema was evaluated 18 hours later by wet to dry weight ratio (N=5/group where p** < 0.01, ****p< 0.0001). Data in (A) and (C-E) are shown with means ± SEM.
Besides driving chemotaxis, a well-described role for N-formylated peptides is the generation of ROS through FPR1 engagement (29). Airway B6 Dendra2+ neutrophils produced more ROS when compared to FPR1−/− Dendra2+ neutrophils, while Dendra2- neutrophils, irrespective of FPR1 expression, produced less ROS when compared to their Dendra2+ counterparts (Fig. 7B). To further dissect the role of FPR1 in neutrophil activation B6 and FPR1−/− neutrophils were briefly exposed to varying concentrations of damaged mitochondria prepared from Dendra2 lungs (Fig. 7C-D). Under low concentrations B6 neutrophils were more efficient at engulfing damaged mitochondria when compared to FPR1−/− neutrophils. At high concentrations, however, engulfment became comparable suggesting that FPR1 is not directly required for the uptake of damaged mitochondria. Moreover, ROS production was also comparable in B6 and FPR1−/− neutrophils that engulfed high amounts of damaged mitochondria, which could be largely reversed by treatment with diphenyleneiodonium (DPI), a NADPH oxidase inhibitor that blocks ROS production.
Because ROS generation is a major mediator of lung epithelial cell injury (30), we next determined if the presence of neutrophils carrying damaged mitochondria in the airway is sufficient to promote acute lung injury (Fig. 7E). B6 and FPR1−/− neutrophils were briefly exposed to low or high concentrations of damaged mitochondria, extensively washed, and then administered into the airways of resting B6 mice. B6 and FPR1−/− neutrophils carrying high amounts of damaged mitochondria promoted equivalent and significantly higher pulmonary edema when compared to untreated neutrophils. Notably, treatment with DPI sharply reduced pulmonary edema indicating that Mt-DAMP-mediated ROS generation from neutrophils is sufficient to induce pulmonary injury. However, under low concentration exposure, B6 neutrophils caused more pulmonary edema than FPR1−/− neutrophils underscoring a critical role for FPR1-mediated chemotaxis in potentiating damaging effector responses in lungs. Therefore, taken collectively, our data show FPR1-mediated trafficking into the airway is coupled to the uptake of Mt-DAMPs leading to ROS-mediated pulmonary injury (Fig. 8A-C).
Figure 8. Mt-DAMP release by lung transplants exacerbates neutrophil-mediated tissue injury.
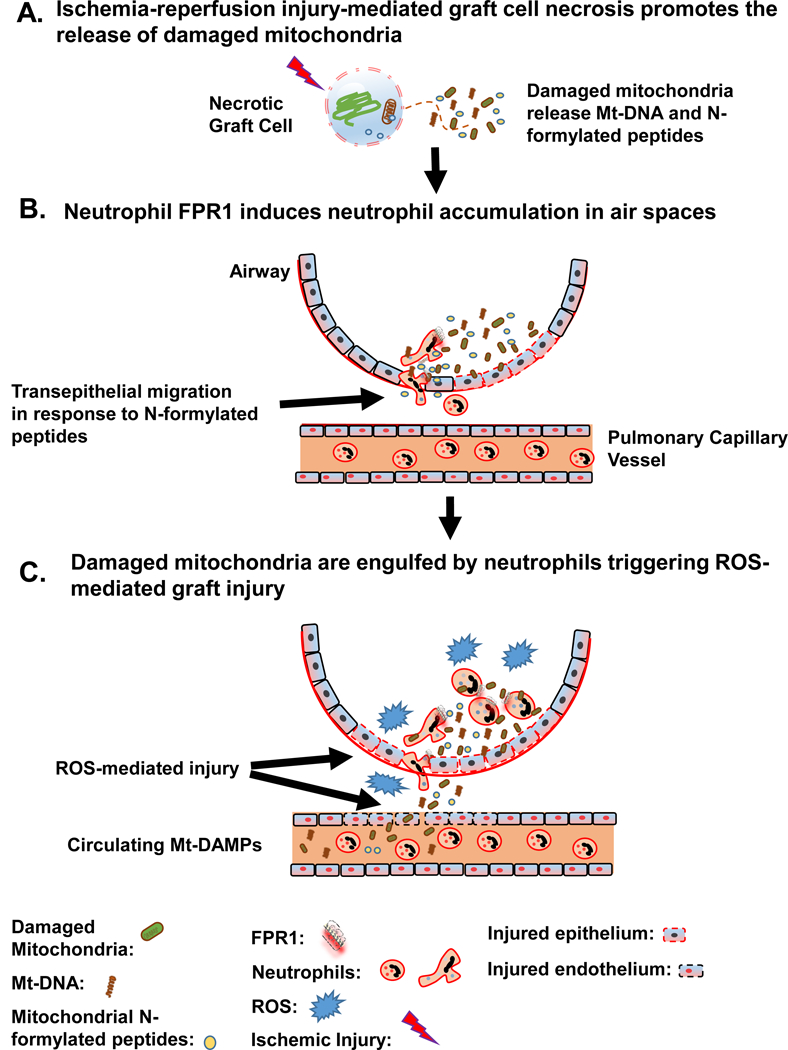
(A) Ischemia-reperfusion injury promotes necrotic cell release of damaged mitochondria, which in turn emit Mt DAMPs. (B) Neutrophil transepithelial migration into air spaces is driven by FPR1-mediated chemotaxis towards mitochondrial N-formylated peptides. (C) While in the airspaces, neutrophils engulf damaged mitochondria that leads to ROS production that compromises homeostatic barriers allowing further leakage of Mt-DAMPs, such as Mt-DNA, into the peripheral circulation.
Discussion
Here we present the first evidence that circulating Mt-DNA is associated with PGD. Notably, previously described circulating biomarkers linked to PGD development have been reported implicated in pulmonary parenchymal tissue damage including vascular endothelial growth factor (31), intercellular adhesion molecule-1 (32), IL-8 (33) and type 1 plasminogen activator inhibitor (34). However, elevated perioperative plasma Mt-DNA may be an earlier and more direct indicator of graft tissue injury. We observed that high levels of circulating Mt-DNA measured at ICU arrival were linked to PGD severity highlighting the possible pathological role of Mt-DAMP release from damaged graft tissue. Although the precise mechanisms of Mt-DAMP release are unclear several reports have implicated a role for trauma or drug-induced cellular damage (12–15). Injury in this setting may also involve known regulated cell death pathways. Recently, TNF-α-induced necroptosis was shown to induce the release of mitochondria via activation of receptor-interacting protein 1-kinase in several cell lines (35). Consistent with these reports was the necrotic appearance of airway epithelial cells in mouse lung recipients and its association with the release of graft-derived damaged mitochondria and Mt-DNA. Alveolar epithelial cell damage is commonly found in PGD patients and therefore could be a potential source of Mt-DAMPs. For example, increases in the levels of epithelial cell death markers such as M30 and M65 are related to worse lung transplant outcomes (36). The accumulation of circulating and airway soluble Receptor for Advanced Glycation End Products Soluble Receptor (RAGE), a glycoprotein highly localized to the basolateral membrane, is associated with the development of PGD (37). In contrast, other reports have linked RAGE release to vascular endothelial inflammation (38, 39) suggesting other types of cellular damage may also contribute to Mt-DAMP release following lung transplantation.
An interesting study finding was that lung recipients prior to transplant, had slightly increased Mt-DNA levels compared to healthy control volunteers. This observation suggests that pre-operative Mt DAMP levels could contribute to inflammatory responses. Although we cannot eliminate this possibility, we observed no association between end-stage lung disease and Mt-DNA levels suggesting that if there is a contribution it is uniform one with respect to pre-existing inflammation. Moreover, we observed a sharp rise in Mt-DNA levels after indicating that transplant-induced injury is a stronger trigger for the release of Mt-DAMPs.
Our studies with human lung recipients have several potential limitations. First, our study was insufficiently powered to conduct a robust multivariate analysis to determine whether elevated Mt-DNA levels are an independent risk factor for PGD. We partially addressed this constraint by demonstrating that incremental increases Mt-DNA were associated with higher grades of PGD. Secondly, we did not have access to donor-derived tissues for most of our transplants, and therefore it was not possible to differentiate donor and recipient Mt-DNA. Pollara et al. recently demonstrated that Mt-DNA levels in the donor circulation are associated to early allograft dysfunction in liver recipients suggesting the inflammatory potential of donor-derived DAMPs (40). Although in our mouse PGD model we demonstrated that most damaged mitochondria release was of donor origin, the degree to which donor-derived Mt-DAMPs contributes to human PGD-associated inflammation remains to be determined. Finally, it may be possible that blood transfusions, commonly used in the lung transplant procedure, may alter Mt-DNA measurements as recent work has shown they can accumulate in stored blood products (41).
Utilizing donor mouse lungs with the mitochondrial reporter transgene Dendra2 we detected that approximately 70% of mitochondria release was from donor cells. We also observed that remaining 30% recipient-derived mitochondria release was comparatively more damaged as evident by lower membrane potential. Interestingly, recent reports have demonstrated that damaged mitochondria can be released by neutrophils and platelets indicating that Mt-DAMPs release by recipient-derived cells are part of a coordinated response to amplify inflammation. Activated neutrophils have been demonstrated to expel their own mitochondria due to an inability to conduct mitophagy (42) or through the generation of extracellular traps (NETs) (43). Of note, NETs have been shown to accumulate in lung recipients who develop PGD and in injured mouse lung transplants (44). Platelets release mitochondria damaged by secreted phospholipase A2-IIA, which hydrolyzes mitochondrial membranes. This in turn, was shown to lead to the release of Mt DAMPs, which activates vascular endothelium resulting in the recruitment of neutrophils (45).
It is the prevailing view that neutrophil extravasation is a necessary and sufficient trafficking step to promote acute lung injury (46). Surprisingly, FPR1 did not enhance neutrophil extravasation despite exacerbating lung transplant injury. Consistent with this observation monocyte graft infiltration was FPR1-independent notwithstanding the important role these cells play in stimulating neutrophil extravasation into lung grafts (25, 47). Additionally, total neutrophil accumulation within the intravascular and interstitial graft compartments was comparable in FPR1−/− and B6 recipients. Nevertheless, FPR1 promoted neutrophil accumulation within the airway. In agreement with this observation we detected FPR1-dependent neutrophil transmigration across a mouse epithelial monolayer in response to N-formylated peptides. Recent evidence shows that infiltrating neutrophils tend to accumulate in nonperfused areas following IRI (48) and that their prolonged retention promotes tissue injury (26). These reports led us to determine if FPR1 promotes neutrophil retention within damaged airways. We observed that neutrophils administered to graft airways were retained in the airway longer if they expressed FPR1. This trafficking behavior is analogous to recently proposed chemotactic hierarchy where trafficking responses to CXC chemokines is superseded by mitochondrial N-formylated peptides within damaged tissue (27). Taken together, these data suggest that FPR1 promotes neutrophil accumulation within the airway without directly promoting recruitment from the pulmonary circulation.
FPR1 engagement by N-formylated peptides is a well-established driver of ROS generation (12, 49). Surprisingly, in B6 recipients of Dendra2+ lungs ROS generation was not directly dependent on FPR1 expression, as we could observe ROS generation in Dendra2+ FPR1−/− neutrophils. This was also evident in a comparison of wildtype and FPR1−/− neutrophil ROS activity following co-incubation with low and high concentrations of damaged mitochondria. Under low concentration exposure, where chemotaxis is required to efficiently locate damaged mitochondria, FPR1−/− neutrophils were still capable of inducing ROS-mediated pulmonary edema, although they produced less when compared to their wildtype counterparts. However, under high concentration exposure, where chemotaxis is less necessary to find damaged mitochondria, we observed comparable levels of ROS-mediated pulmonary edema. Importantly, the amount of ROS production in both cases was linked to the amount of mitochondrial engulfment irrespective FPR1 expression. Taken collectively, our data suggest other PRR-signaling pathways downstream of FPR1-mediated chemotaxis are driving ROS production. One possibility is through cytosolic DNA sensing pathways, which have been also shown to recognize Mt-DNA through multiple PRRs including the nucleotide-binding domain and leucine rich repeat (NLR) pyrin domain containing 3 (50), TLR9 (51), or cyclic GMP-AMP Synthase (43). However, it remains to be determined whether ROS generation results from the recognition of Mt-DNA or other Mt-DAMPs. In conclusion, we demonstrate that Mt-DAMPs released following lung transplantation induces FPR1-dependent inflammatory responses that exacerbate pulmonary injury. Future multicenter studies on the dynamics of Mt-DAMP release should help to confirm whether they are therapeutic targets for PGD prevention.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
This work is supported by the Barnes Jewish Foundation, the Jacqueline G and William E Maritz Chair in Immunology & Oncology, and NIH grants P01AI116501–01, R01HL113436–01A1, and R01HL121218–01.
Abbreviations
- (ABG)
Arterial blood gas
- (BSA)
Bovine serum albumin
- (BALf)
Bronchoalveolar lavage fluid
- (Cy-H)
Cyclosporine H
- (FPR1)
Formylated peptide receptor 1
- (IRI)
Ischemia-reperfusion injury
- (MT-COX3)
Mitochondrial cytochrome III oxidase
- (Mt-DAMPs)
Mitochondrial damage associated molecular patterns
- (NETs)
Neutrophil extracellular traps
- (NLR)
Nucleotide-binding domain and leucine rich repeat
- (PRRs)
Pattern recognition receptors
- (PGD)
Primary graft dysfunction
- (ROS)
Reactive oxygen species
- (RAGE)
Receptor for Advanced Glycation End Products Soluble Receptor
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Supporting Information
Additional supplemental materials may be found online in the Supporting Information section of this article.
References
- 1.Snell GI, Yusen RD, Weill D, Strueber M, Garrity E, Reed A, Pelaez A, Whelan TP, Perch M, Bag R, Budev M, Corris PA, Crespo MM, Witt C, Cantu E, and Christie JD. Report of the ISHLT Working Group on Primary Lung Graft Dysfunction, part I: Definition and grading 2014;A 2016 Consensus Group statement of the International Society for Heart and Lung Transplantation. The Journal of Heart and Lung Transplantation 36: 1097–1103. [DOI] [PubMed] [Google Scholar]
- 2.Diamond JM, Arcasoy S, Kennedy CC, Eberlein M, Singer JP, Patterson GM, Edelman JD, Dhillon G, Pena T, Kawut SM, Lee JC, Girgis R, Dark J, and Thabut G. 2017. Report of the International Society for Heart and Lung Transplantation Working Group on Primary Lung Graft Dysfunction, part II: Epidemiology, risk factors, and outcomes-A 2016 Consensus Group statement of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant 36: 1104–1113. [DOI] [PubMed] [Google Scholar]
- 3.Huang HJ, Yusen RD, Meyers BF, Walter MJ, Mohanakumar T, Patterson GA, Trulock EP, and Hachem RR. 2008. Late primary graft dysfunction after lung transplantation and bronchiolitis obliterans syndrome. Am J Transplant 8: 2454–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gelman AE, Fisher AJ, Huang HJ, Baz MA, Shaver CM, Egan TM, and Mulligan MS. Report of the ISHLT Working Group on Primary Lung Graft Dysfunction Part III: Mechanisms: A 2016 Consensus Group Statement of the International Society for Heart and Lung Transplantation. The Journal of Heart and Lung Transplantation 36: 1114–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laubach VE, and Sharma AK. 2016. Mechanisms of Lung Ischemia-Reperfusion Injury. Current opinion in organ transplantation 21: 246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weber DJ, Gracon ASA, Ripsch MS, Fisher AJ, Cheon BM, Pandya PH, Vittal R, Capitano ML, Kim Y, Allete YM, Riley AA, McCarthy BP, Territo PR, Hutchins GD, Broxmeyer HE, Sandusky GE, White FA, and Wilkes DS. 2014. The HMGB1-RAGE Axis Mediates Traumatic Brain Injury-induced Pulmonary Dysfunction in Lung Transplantation. Science translational medicine 6: 252ra124–252ra124. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Ibrahim M, Wang X, Puyo CA, Montecalvo A, Huang HJ, Hachem RR, Andreetti C, Menna C, Chen R, Krupnick AS, Kreisel D, Rendina EA, and Gelman AE. 2015. Human recombinant apyrase therapy protects against canine pulmonary ischemia-reperfusion injury. J Heart Lung Transplant 34: 247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharma AK, LaPar DJ, Stone ML, Zhao Y, Kron IL, and Laubach VE. 2013. Receptor for advanced glycation end products (RAGE) on iNKT cells mediates lung ischemia-reperfusion injury. Am J Transplant 13: 2255–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, and Hauser CJ. 2010. Circulating Mitochondrial DAMPs Cause Inflammatory Responses to Injury. Nature 464: 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakahira K, Hisata S, and Choi AMK. 2015. The Roles of Mitochondrial Damage-Associated Molecular Patterns in Diseases. Antioxidants & Redox Signaling 23: 1329–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szczesny B, Marcatti M, Ahmad A, Montalbano M, Brunyanszki A, Bibli SI, Papapetropoulos A, and Szabo C. 2018. Mitochondrial DNA damage and subsequent activation of Z-DNA binding protein 1 links oxidative stress to inflammation in epithelial cells. Sci Rep 8: 914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hauser CJ, Sursal T, Rodriguez EK, Appleton PT, Zhang Q, and Itagaki K. 2010. Mitochondrial DAMPs from femoral reamings activate neutrophils via formyl peptide receptors and P44/42 MAP Kinase. Journal of orthopaedic trauma 24: 534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simmon JD, Lee Y-L, Mulekar S, Kuck JL, Brevard SB, Gonzalez RP, Gillespie MN, and Richards WO. 2013. Elevated Levels of Plasma Mitochondrial DNA DAMPs Are Linked to Clinical Outcome in Severely Injured Human Subjects. Annals of surgery 258: 591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McGill MR, Staggs VS, Sharpe MR, Lee WM, Jaeschke H, and Acute G Liver Failure Study. 2014. Serum mitochondrial biomarkers and damage-associated molecular patterns are higher in acetaminophen overdose patients with poor outcome. Hepatology (Baltimore, Md.) 60: 1336–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Puskarich MA, Shapiro NI, Trzeciak S, Kline JA, and Jones AE. 2012. Plasma levels of mitochondrial DNA in patients presenting to the emergency department with sepsis. Shock 38: 337–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okazaki M, Krupnick AS, Kornfeld CG, Lai JM, Ritter JH, Richardson SB, Huang HJ, Das NA, Patterson GA, Gelman AE, and Kreisel D. 2007. A mouse model of orthotopic vascularized aerated lung transplantation. Am J Transplant 7: 1672–1679. [DOI] [PubMed] [Google Scholar]
- 17.Kreisel D, Sugimoto S, Tietjens J, Zhu J, Yamamoto S, Krupnick AS, Carmody RJ, and Gelman AE. 2011. Bcl3 prevents acute inflammatory lung injury in mice by restraining emergency granulopoiesis. J Clin Invest 121: 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lecoeur H, Langonne A, Baux L, Rebouillat D, Rustin P, Prevost MC, Brenner C, Edelman L, and Jacotot E. 2004. Real-time flow cytometry analysis of permeability transition in isolated mitochondria. Exp Cell Res 294: 106–117. [DOI] [PubMed] [Google Scholar]
- 19.Krupnick AS, Lin X, Li W, Okazaki M, Lai J, Sugimoto S, Richardson SB, Kornfeld CG, Garbow JR, Patterson GA, Gelman AE, and Kreisel D. 2009. Orthotopic mouse lung transplantation as experimental methodology to study transplant and tumor biology. Nat Protoc 4: 86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanou T, Ohsumi A, Kim H, Chen M, Bai X, Guan Z, Hwang D, Cypel M, Keshavjee S, and Liu M. 2018. Inhibition of regulated necrosis attenuates receptor-interacting protein kinase 1-mediated ischemia-reperfusion injury after lung transplantation. J Heart Lung Transplant 37: 1261–1270. [DOI] [PubMed] [Google Scholar]
- 21.Nerlich A, Mieth M, Letsiou E, Fatykhova D, Zscheppang K, Imai-Matsushima A, Meyer TF, Paasch L, Mitchell TJ, Tonnies M, Bauer TT, Schneider P, Neudecker J, Ruckert JC, Eggeling S, Schimek M, Witzenrath M, Suttorp N, Hippenstiel S, and Hocke AC. 2018. Pneumolysin induced mitochondrial dysfunction leads to release of mitochondrial DNA. Sci Rep 8: 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sesso A, Belizario JE, Marques MM, Higuchi ML, Schumacher RI, Colquhoun A, Ito E, and Kawakami J. 2012. Mitochondrial swelling and incipient outer membrane rupture in preapoptotic and apoptotic cells. Anat Rec (Hoboken) 295: 1647–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dorward DA, Lucas CD, Doherty MK, Chapman GB, Scholefield EJ, Conway Morris A, Felton JM, Kipari T, Humphries DC, Robb CT, Simpson AJ, Whitfield PD, Haslett C, Dhaliwal K, and Rossi AG. 2017. Novel role for endogenous mitochondrial formylated peptide-driven formyl peptide receptor 1 signalling in acute respiratory distress syndrome. Thorax 72: 928–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reutershan J, Basit A, Galkina EV, and Ley K. 2005. Sequential recruitment of neutrophils into lung and bronchoalveolar lavage fluid in LPS-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol 289: L807–815. [DOI] [PubMed] [Google Scholar]
- 25.Kreisel D, Nava RG, Li W, Zinselmeyer BH, Wang B, Lai J, Pless R, Gelman AE, Krupnick AS, and Miller MJ. 2010. In vivo two-photon imaging reveals monocyte-dependent neutrophil extravasation during pulmonary inflammation. Proc Natl Acad Sci U S A 107: 18073–18078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Devi S, Li A, Westhorpe CLV, Lo CY, Abeynaike LD, Snelgrove SL, Hall P, Ooi JD, Sobey CG, Kitching AR, and Hickey MJ. 2016. Corrigendum: Multiphoton imaging reveals a new leukocyte recruitment paradigm in the glomerulus. Nature Medicine 22: 446. [DOI] [PubMed] [Google Scholar]
- 27.Heit B, Tavener S, Raharjo E, and Kubes P. 2002. An intracellular signaling hierarchy determines direction of migration in opposing chemotactic gradients. J Cell Biol 159: 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, and Kubes P. 2010. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 330: 362–366. [DOI] [PubMed] [Google Scholar]
- 29.Dorward DA, Lucas CD, Chapman GB, Haslett C, Dhaliwal K, and Rossi AG. 2015. The role of formylated peptides and formyl peptide receptor 1 in governing neutrophil function during acute inflammation. Am J Pathol 185: 1172–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kellner M, Noonepalle S, Lu Q, Srivastava A, Zemskov E, and Black SM. 2017. ROS Signaling in the Pathogenesis of Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome (ARDS). Adv Exp Med Biol 967: 105–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krenn K, Klepetko W, Taghavi S, Lang G, Schneider B, and Aharinejad S. 2007. Recipient vascular endothelial growth factor serum levels predict primary lung graft dysfunction. Am J Transplant 7: 700–706. [DOI] [PubMed] [Google Scholar]
- 32.Covarrubias M, Ware LB, Kawut SM, De Andrade J, Milstone A, Weinacker A, Orens J, Lama V, Wille K, Bellamy S, Shah C, Demissie E, and Christie JD. 2007. Plasma intercellular adhesion molecule-1 and von Willebrand factor in primary graft dysfunction after lung transplantation. Am J Transplant 7: 2573–2578. [DOI] [PubMed] [Google Scholar]
- 33.De Perrot M, Sekine Y, Fischer S, Waddell TK, McRae K, Liu M, Wigle DA, and Keshavjee S. 2002. Interleukin-8 release during early reperfusion predicts graft function in human lung transplantation. Am J Respir Crit Care Med 165: 211–215. [DOI] [PubMed] [Google Scholar]
- 34.Christie JD, Robinson N, Ware LB, Plotnick M, De Andrade J, Lama V, Milstone A, Orens J, Weinacker A, Demissie E, Bellamy S, Kawut SM, and G. for the Lung Transplant Outcomes. 2007. Association of Protein C and Type 1 Plasminogen Activator Inhibitor with Primary Graft Dysfunction. American Journal of Respiratory and Critical Care Medicine 175: 69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maeda A, and Fadeel B. 2014. Mitochondria released by cells undergoing TNF-alpha-induced necroptosis act as danger signals. Cell Death Dis 5: e1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hashimoto K, Besla R, Zamel R, Juvet S, Kim H, Azad S, Waddell TK, Cypel M, Liu M, and Keshavjee S. 2016. Circulating Cell Death Biomarkers May Predict Survival in Human Lung Transplantation. Am J Respir Crit Care Med 194: 97–105. [DOI] [PubMed] [Google Scholar]
- 37.Christie JD, Shah CV, Kawut SM, Mangalmurti N, Lederer DJ, Sonett JR, Ahya VN, Palmer SM, Wille K, Lama V, Shah PD, Shah A, Weinacker A, Deutschman CS, Kohl BA, Demissie E, Bellamy S, and Ware LB. 2009. Plasma levels of receptor for advanced glycation end products, blood transfusion, and risk of primary graft dysfunction. Am J Respir Crit Care Med 180: 1010–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Basta G, Lazzerini G, Massaro M, Simoncini T, Tanganelli P, Fu C, Kislinger T, Stern DM, Schmidt AM, and De Caterina R. 2002. Advanced glycation end products activate endothelium through signal-transduction receptor RAGE: a mechanism for amplification of inflammatory responses. Circulation 105: 816–822. [DOI] [PubMed] [Google Scholar]
- 39.Wautier JL, Zoukourian C, Chappey O, Wautier MP, Guillausseau PJ, Cao R, Hori O, Stern D, and Schmidt AM. 1996. Receptor-mediated endothelial cell dysfunction in diabetic vasculopathy. Soluble receptor for advanced glycation end products blocks hyperpermeability in diabetic rats. The Journal of Clinical Investigation 97: 238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pollara J, Edwards RW, Lin L, Bendersky VA, and Brennan TV. 2018. Circulating mitochondria in deceased organ donors are associated with immune activation and early allograft dysfunction. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simmons JD, Lee YL, Pastukh VM, Capley G, Muscat CA, Muscat DC, Marshall ML, Brevard SB, and Gillespie MN. 2017. Potential contribution of mitochondrial DNA damage associated molecular patterns in transfusion products to the development of acute respiratory distress syndrome after multiple transfusions. J Trauma Acute Care Surg 82: 1023–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, Baisch J, Phelps K, Clayton S, Gong M, Wright T, Punaro M, Palucka K, Guiducci C, Banchereau J, and Pascual V. 2016. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med 213: 697–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, and Kaplan MJ. 2016. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 22: 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sayah DM, Mallavia B, Liu F, Ortiz-Munoz G, Caudrillier A, DerHovanessian A, Ross DJ, Lynch JP 3rd, Saggar R, Ardehali A, Ware LB, Christie JD, Belperio JA, and Looney MR. 2015. Neutrophil extracellular traps are pathogenic in primary graft dysfunction after lung transplantation. Am J Respir Crit Care Med 191: 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boudreau LH, Duchez A-C, Cloutier N, Soulet D, Martin N, Bollinger J, Paré A, Rousseau M, Naika GS, Lévesque T, Laflamme C, Marcoux G, Lambeau G, Farndale RW, Pouliot M, Hamzeh-Cognasse H, Cognasse F, Garraud O, Nigrovic PA, Guderley H, Lacroix S, Thibault L, Semple JW, Gelb MH, and Boilard E. 2014. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A(2) to promote inflammation. Blood 124: 2173–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spahn JH, Li W, Bribriesco AC, Liu J, Shen H, Ibricevic A, Pan JH, Zinselmeyer BH, Brody SL, Goldstein DR, Krupnick AS, Gelman AE, Miller MJ, and Kreisel D. 2015. DAP12 expression in lung macrophages mediates ischemia/reperfusion injury by promoting neutrophil extravasation. J Immunol 194: 4039–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng Z, Chiu S, Akbarpour M, Sun H, Reyfman PA, Anekalla KR, Abdala-Valencia H, Edgren D, Li W, Kreisel D, Korobova FV, Fernandez R, McQuattie-Pimentel A, Zhang ZJ, Perlman H, Misharin AV, Scott Budinger GR, and Bharat A. 2017. Donor pulmonary intravascular nonclassical monocytes recruit recipient neutrophils and mediate primary lung allograft dysfunction. Science Translational Medicine 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Honda M, Takeichi T, Hashimoto S, Yoshii D, Isono K, Hayashida S, Ohya Y, Yamamoto H, Sugawara Y, and Inomata Y. 2017. Intravital Imaging of Neutrophil Recruitment Reveals the Efficacy of FPR1 Blockade in Hepatic Ischemia-Reperfusion Injury. J Immunol 198: 1718–1728. [DOI] [PubMed] [Google Scholar]
- 49.Raoof M, Zhang Q, Itagaki K, and Hauser CJ. 2010. Mitochondrial peptides are potent immune activators that activate human neutrophils via FPR-1. J Trauma 68: 1328–1332; discussion 1332–1324. [DOI] [PubMed] [Google Scholar]
- 50.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, and Arditi M. 2012. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36: 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bao W, Xia H, Liang Y, Ye Y, Lu Y, Xu X, Duan A, He J, Chen Z, Wu Y, Wang X, Zheng C, Liu Z, and Shi S. 2016. Toll-like Receptor 9 Can be Activated by Endogenous Mitochondrial DNA to Induce Podocyte Apoptosis. Sci Rep 6: 22579. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.