

Abstract
Genetic factors do not fully account for the relatively high heritability of neurodevelopmental conditions, suggesting that non-genetic heritable factors contribute to their etiology. To evaluate the potential contribution of aberrant thyroid hormone status to the epigenetic inheritance of neurological phenotypes, we examined genetically normal F2 generation descendants of mice that were developmentally overexposed to thyroid hormone due to a Dio3 mutation. Hypothalamic gene expression profiling in postnatal day 15 F2 descendants on the paternal lineage of ancestral male and female T3-overexposed mice revealed, respectively, 1089 and 1549 differentially expressed genes. A large number of them, 675 genes, were common to both sets, suggesting comparable epigenetic effects of thyroid hormone on both the male and female ancestral germ lines. Oligodendrocyte- and neuron-specific genes were strongly overrepresented among genes showing, respectively, increased and decreased expression. Altered gene expression extended to other brain regions and was associated in adulthood with decreased anxiety-like behavior, increased marble burying and reduced physical activity. The sperm of T3-overexposed male ancestors revealed significant hypomethylation of CpG islands associated with the promoters of genes involved in the early development of the central nervous system. Some of them were candidates for neurodevelopmental disorders in humans including Nrg3, Nrxn1, Gabrb3, Gabra5, Apba2, Grik3, Reln, Nsd1, Pcdh8, En1 and Elavl2. Thus, developmental levels of thyroid hormone influence the epigenetic information of the germ line, disproportionately affecting genes with critical roles in early brain development, and leading in future generations to disease-relevant alterations in postnatal brain gene expression and adult behavior.
Introduction
Epidemiological and familial studies suggest a variable but relatively large heritable component in the etiology of many neurological disorders 1–7, which may include autism spectrum disorders, schizophrenia, attention deficit and hyperactivity disorder, bipolar disorder and depression. Genome-wide association studies in recent years have increased the list of candidate genes, pointing to a complex, multigenic contribution to the onset of these conditions 8–12. However, genetic causes only account for a relatively low percentage of actual clinical cases. Although the influence of maternal pathophysiology and environmental factors are likely important modifiers of disease susceptibility 13–16, a significant proportion of the heritability of neurodevelopmental disorders remains unexplained 17–19.
Increasing evidence indicates that alterations in the epigenetic information inherited from previous generations may partly explain the “missing heritability” of neurodevelopmental conditions 20–22. Exposure to hormones, chemicals and environmental factors such as stress and diet may alter the epigenetic information of an individual’s germ line, with potential consequences for the pathophysiology of subsequent generations 23–26. However, much remains unknown about the factors that initiate or contribute to this phenomenon and how neurological phenotypes may be affected.
In this context, the role of thyroid hormones has not been sufficiently considered. Thyroid hormones are critical for normal neurological outcomes and their orchestrated action during brain development regulates neural cell proliferation, migration and differentiation, dendritic formation, axon guidance, myelination and synaptic transmission 27. Alterations in thyroid hormone status during development may occur in humans due to congenital defects 28, 29, maternal thyroid disease 29–31 and exposure to environmental chemicals 32, 33. A deficiency in thyroid hormone during development may lead to mental retardation and cretinism 34, and maternal thyroxine has been shown to be of importance to ensure appropriate levels of the principal active hormone 3,5,3’-triiodothyronine (T3) in the fetal brain before full thyroid gland function is attained 35–37. Intriguing, poorly understood observations have also linked abnormalities in thyroid hormone physiology to neurodevelopmental disorders like autism 15, 38–40, schizophrenia 41 and attention deficit and hyperactive disorder 38, 42.
Previous observations suggest that altered levels of thyroid hormone may impact brain function in later generations, presumably via intergenerational epigenetic mechanisms. Bakke et al. showed that an altered thyroid hormone state in the neonatal or adult rat results in abnormal neuroendocrine phenotypes in their offspring 43, 44. More recent work in a rat model of fetal alcohol exposure shows that developmental administration of thyroxine causes epigenetic effects in descendants, impacting behavior and the expression of genes of importance to epigenetic regulation and brain development including Dnmt1, Dio3 and Igf2 45, 46.
Thus, we investigated the neurological significance of ancestral overexposure to thyroid hormone during development. To this purpose, we used a genetic mouse model of type 3 deiodinase (DIO3) deficiency 47. DIO3 is highly expressed during development and is the principal enzyme that inactivates both thyroxine and T3 48. Here we report that genetically normal (wild type) F2-generation descendants of Dio3−/− mice exhibit alterations in their neonatal patterns of gene expression affecting the hypothalamus and other brain regions. These alterations are associated with behavioral abnormalities in adulthood. We also report an abnormal methylome in the sperm DNA of overexposed male ancestors. Our results suggest that developmental overexposure to thyroid hormone modifies the epigenetic information that is transmitted to subsequent generations, causing alterations in molecular and functional neurological traits.
Materials and Methods
Experimental Animals.
Studies on P15 neonates of both sexes were performed in mice from an outbred CD-1 genetic background. As a model of developmental overexposure to T3, we used mice genetically deficient in the type 3 deiodinase (DIO3). We have previously described that Dio3 −/− mice exhibit markedly elevated serum levels of T3 during perinatal life 47. Second generation animals from all experimental groups (Fig. 1a) were born to and raised by genetically normal females in the absence of the father. Unless otherwise stated, we used experimental mice born to first litters (see Supplementary Methods for additional details about genetic background, ancestry, litter information and mating design). Mice were euthanized by asphyxiation with CO2 and harvested tissues were frozen immediately on dry ice until further use. All experiments were approved by the Maine Medical Center Research Institute Institutional Animal Care and Use Committee (IACUC).
Fig. 1. Transgenerational effects of thyroid hormone on brain gene expression.
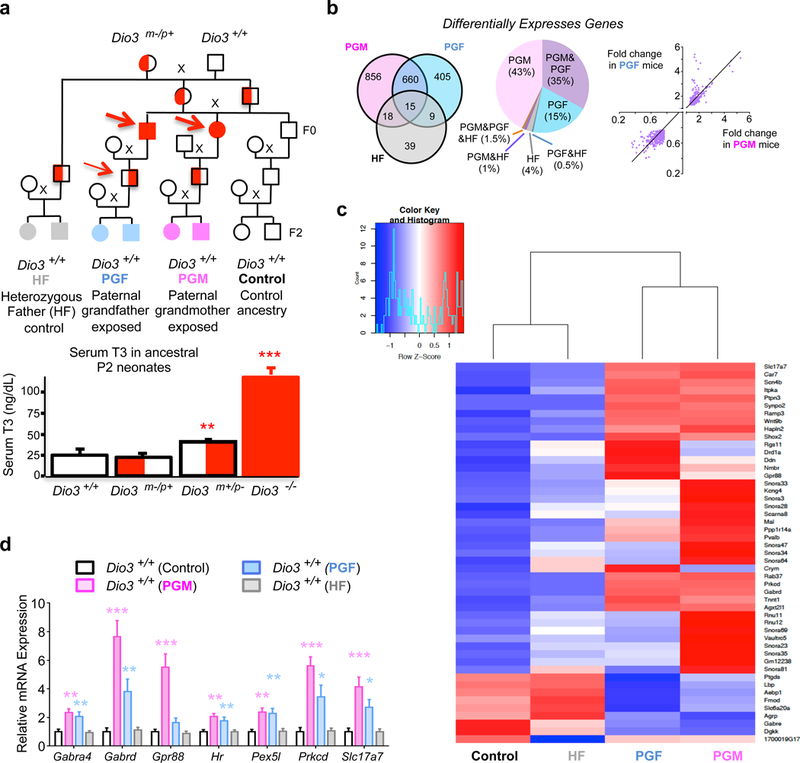
(a), Ancestry of the experimental animals studied and neonatal serum T3 in postnatal day 2 (P2) animals of either sex and of different Dio3 genotypes as previously reported. Thin and thick red arrows indicate, respectively, modest and overt overexposure to T3 during development in correspondence to the data in the graph below; (b) Distribution of differentially expressed genes in the P15 hypothalamus across experimental groups and correlation of expression levels between those common to PGF and PGF mice. (c) Clustering and heat map of 49 genes whose expression increased more than 2.5 fold and decreased more than 0.4 fold in at least one of the three experimental groups compared with the control group; (d) qPCR validation of 7 differentially expressed genes in independent samples from Control (n=11), PGF (n=11), PGM (n=10) and HF (n=9) mice. *, ** and ***, P<0.05, 0.01 and 0.001, respectively, as determined by ANOVA and Tukey’s post hoc test . PGM, paternal grandmother; PGF, paternal grandfather; HF, heterozygous father.
RNAsequencing.
Total RNA was isolated from the hypothalamus of P15 mice. For each experimental group we used three mice (n=3) of either sex (based on 80% power and an anticipated standardized effect size of 3, as we anticipated strong effects on a relatively reduced number of genes). Each mouse originated from a different litter. The RNA samples were processed for library construction and next generation sequencing by Cofactor Genomics (St. Louis, MO) (See Supplementary Methods for full details). For the purpose of the identification of differentially expressed genes, we considered only those genes (15256 genes) whose sum of expression levels between all experimental groups was higher than 10, as determined by their Fragments Per Kilobase of transcript per Million mapped reads (FPKM). A Fisher’s exact test was used to determine which genes were differentially expressed in pairwise comparisons between each of the three experimental groups and the control group. Genes showing more than a 30% change in expression and a P adjusted value lower than 0.1 were considered differentially expressed.
Representation of cell type-specific genes.
To identify any under or over-representation of genes specific to a particular brain cell type within our sets of differentially expressed genes, we took advantage of the database and webserver (http://web.stanford.edu/group/barres_lab/brain_rnaseq.html) generated by Zhang et al. 49. Using this database (see Supplementary Methods), we identified genes with the highest specificity for each brain cell type. We then implemented a Fisher’s Exact test using the R function fisher.test to test for overrepresentation of differentially expressed genes in a given list of the top set of expressed genes of a given cell type. The p-value and odds ratio for enrichment were recorded to determine statistically significant over- or under-representation.
Pathway and Disease and functional ontology analysis.
Disease and functional ontology analyses were performed on selected groups of genes using INGENUITY software (Qiagen, Valencia, CA), Functional Disease Ontology (FunDO) annotations (http://django.nubic.northwestern.edu/fundo/ and http://projects.bioinformatics.northwestern.edu/fundo), a service from Northwestern University 50–52, and the Database for Annotation, Visualization and Integrated Discovery (DAVID) (https://david.ncifcrf.gov/), a service supported by the National Institute of Allergy and Infectious Diseases 53–55.
Real time qPCR.
Real time quantitative PCR was used to validate RNA-sequencing results. This was performed following standard protocols analysis, as detailed in Supplementary Methods. Based on magnitude of the effect size observed for differentially expressed genes revealed by RNA-sequencing data, and on power analysis (80% power for an effect size of 1.5), we used samples from a minimum of 8 mice per experimental group. Animals in each experimental group represented at least three different litters.
Immunofluorescence (IF) and immunohistochemistry (IHC).
IF and IHC were performed following standard procedures (see Supplementary Methods) on testicular sections of testes fixed in Bouin’s solution.
Sperm Methylome Analysis.
Sperm DNA was isolated from 3-month old male mice following standard procedures (see Supplementary Methods). These samples were obtained from three different mice born to different mothers. We used a reduced-representation bisulfite sequencing approach to analyze methylation in sperm DNA. This was performed using the Methyl-MiniSeq service from Zymo Research (Irvine, CA). Bisulfite conversion was higher than 98%. Technical details and analytical methodology are fully described in Supplementary Methods.
Animal behavior.
All behavioral tests were performed in the same cohort of 5–6 months old mice. We aim to use a minimum of 12 mice per experimental group and sex. This number is based on acceptable standards for behavioral phenotyping and provide a 80% power to identify a significant effect size of 1.3. Animals in each group represented at least three different mothers. Tests were performed according to standard published protocols (see Supplementary Methods) with two days of separation in the following order: Elevated plus maze, dark/light box (first week), marble burying, tail suspension (second week) and physical activity (third week). Mice falling from the elevated maze were excluded from the analysis.
Statistical analysis of qPCR and behavioral data.
Data was analyzed using GraphPad Prism software. Power analysis, as indicated in the respective section, was used to determine the minimum number of biological samples or animals needed to achieve statistical significance based on the anticipated effect size. Data is represented as the mean ± SEM and exhibited normal distribution. One or two tail Student’s t test were used to compare two experimental groups, while one-way ANOVA and Tukey’s post hoc test was used for comparison between more than two experimental groups. Variances were similar between experimental groups. P<0.05 was considered significant, but more stringent levels of significance are also noted.
Results
Altered brain gene expression in the F2-generation descendants of mice overexposed to T3 during development.
As a model of developmental overexposure to T3, we utilized DIO3-deficient (Dio3−/−) mice 47. These mice exhibit consistent supraphysiological levels of T3 during fetal and neonatal life 47, 56 (Fig. 1a). We examined four groups of mice that were genetically normal (Dio3+/+) and born to genetically normal females (Fig. 1a). The control group included F2 generation mice that have only genetically normal male and female ancestry back to the F0 generation. The second group included F2-generation mice with a heterozygous father and a T3-overexposed (Dio3−/−) paternal grandmother (PGM mice). The third group included F2 generation mice with a heterozygous father and a T3-overexposed (Dio3−/−) paternal grandfather (PGF mice). The last group was an additional control group and included mice with a heterozygous father (HF mice) and no history of ancestral T3 overexposure.
All animals studied shared common ancestry previous to the F0 generation and all heterozygous animals in the ancestry tree, except one, inherited the mutated Dio3 allele from their mothers. As Dio3 is an imprinted gene preferentially expressed from the paternally inherited allele in most tissues 57, 58, although observations in rats indicate that Dio3 is preferentially expressed from the maternal allele in the adult hippocampus 59. However, circulating levels of thyroid hormones are normal in adult mice with paternal or maternal inheritance of the Dio3 mutation, and in neonates with maternal inheritance of the mutation 60, 61. Only the heterozygous male mice with paternal inheritance of the mutation (F1 generation mice that fathered the experimental PGF mice) were exposed to moderately elevated levels of thyroid hormone during development 56, 60 (Fig. 1a, thin arrow).
To determine the effects of ancestral T3 exposure on the brain developmental programs of descendants we performed RNA sequencing of total RNA isolated from the hypothalamus of postnatal day 15 (P15) animals from all experimental groups. This age was chosen as it coincides with a peak in circulating thyroid hormones and the initiation of broad thyroid hormone action in the brain 62. We identified 1549, 1089 and 81 genes that showed altered expression in the hypothalamus of PGM, PGF and HF mice, respectively (Fig. 1b, see also Supplementary Data1 and Supplementary Fig. 1). (These groups of differentially expressed genes are referred to as PGM, PGF and HF genes, respectively). We did not followed up on gene expression changes in HF mice, as remarkably, very few genes showed altered expression in HF mice and the changes were modest, suggesting that having a phenotypically normal heterozygous father has negligible effect on the brain gene expression of their wild type progeny. In contrast, most gene expression changes in the F2 generation occur as a result of a grandparent overexposed to T3.
Interestingly, 675 genes showed altered expression in both PGM and PGF mice (Fig. 1b, referred to as “common genes”). This number of “common genes” is markedly higher than predicted by chance (P=2.2 E-16), strongly suggesting a shared mechanism underlying the effects in both experimental. Furthermore, common genes showed a 100% concordance in the direction of gene expression change between PGM and PGF mice (Fig. 1b). The likelihood of this concordance is very high (P ≈ 0), strongly rejecting the null hypothesis that gene expression changes occurring in PGM and PGF mice are the result of independent mechanisms. In addition, common genes were up-regulated and down-regulated to a similar extent in PGM and PGF mice (Fig. 1b). Clustering analysis of 49 genes showing top expression differences illustrates the similarities in the gene expression profiles of PGM and PGF mice that were different from those in control and HF mice (Fig. 1d). A similar result is obtained when including all differentially expressed genes (Supplementary Fig. 2c).
RNA-sequencing results were validated using qPCR in independent samples. Expression levels of selected genes were elevated in PGM and PGF mice, but not in HF mice (Fig. 1d). Likewise, RNA-sequencing results were validated for 27 out of 32 up- and down-regulated genes in independent PGM hypothalami (Fig. 2a and 2b). The expression of many of these genes was also altered in other brain regions of PGM mice (Supplementary Fig. 2a), suggesting that gene expression programs in other brain areas are also affected.
Fig. 2. Validation and analysis of differentially expressed genes.
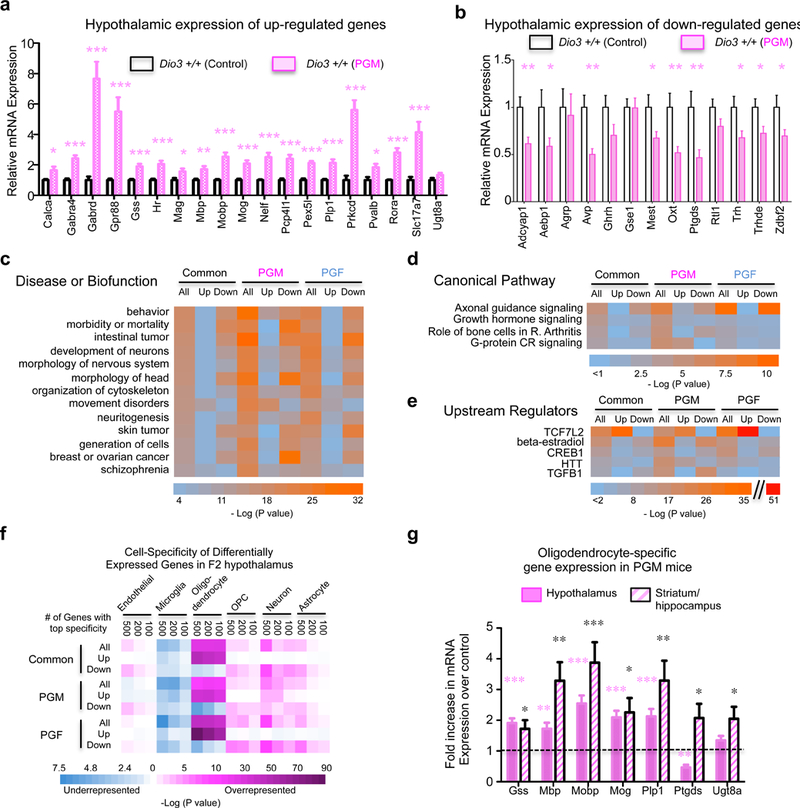
(a, b), qPCR validation in independent samples from PGM of 32 genes identified as upregulated (a) or down regulated (b) in the RNA-sequencing experiment. (c, d and e) Results summary of INGENUITY analysis of differentially expressed genes for disease, biofunction and molecular pathway; (f) Enrichment among differentially expressed genes in genes specific to particular brain cells; (g) Expression of oligodendrocyte-specific genes in the hypothalamus and striatum/hippocampus of PGM mice versus controls (dotted line). *, ** and ***, P<0.05, 0.01 and 0.001, respectively as determined by one-tail Student’s t-test (a and b) or ANOVA and Tukey’s post hoc test (g) in measurements performed on samples from 11 Control and 12 PGM mice (a and b) and in samples from 11 Control and 11 PGM mice (g).
INGENUITY analysis of differentially expressed genes in PGM and/or PGF mice identified functional enrichments in behavior, neuronal development, movement disorders, some types of cancer and schizophrenia (Fig. 2c), while the top enriched canonical pathway and upstream regulator identified were “axon guidance signaling” and TCF7L2 signaling, respectively (Fig. 2d and 2e and Supplementary Fig. 3).
We analyzed differentially expressed genes in PGM and PGF mice for enrichment in specific brain cell types using the data and tools generated by Zhang et al. 49 (see Supplemtary Methods). We observed a dramatic overrepresentation of genes specific to mature oligodendrocytes in genes upregulated in PGM and PGF mice (Fig. 2f). The upregulation of oligodendrocyte-specific genes (including Gss, Mbp, Mobp, Mog, Plp1, Ptgds and Ugt8a) was in the hypothalamus and other brain regions of PGM mice (Fig. 2g, and Supplementary Fig. 2c). In contrast, genes down-regulated in PGF and PGM mice showed enrichment in neuron-specific genes and, to a lesser extent, in astrocyte-specific genes (Fig. 2f).
Altered Adult Behavior.
We analyzed some basic behaviors in PGF mice (Supplementary Fig. 4 and 5). As these mice are F2 generation descendants through the paternal line of ancestors overtly overexposed to T3, they represent a better model to demonstrate stability in the epigenetic inheritance of any behavioral traits. Adult PGF mice of both sexes exhibited decreased anxiety-like behavior, as evidenced by increased time spent and distance traveled in the open arms of the elevated plus maze (Fig. 3a). They also showed enhanced digging behavior in the marble-burying test (Fig. 3b), but no significant depression-related behavior as assessed in the tail suspension test (Fig. 3c) Adult PGF mice also manifested decreased levels of physical activity (Fig. 5d).
Fig. 3. Behavior in adult mice descendants of exposed ancestors.
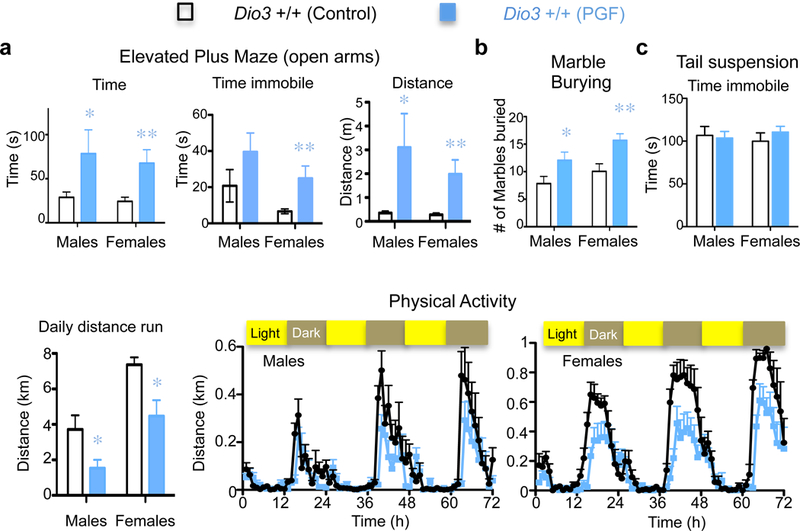
(a) Results from the elevated plus maze test; (b) Results of marble burying test; (c) Results of tail suspension test; (d) Daily distance run and physical activity. * and **, P<0.05 and 0.01 respectively as determined by ANOVA and Tukey’s post hoc test in 12 and 19 Control males and female mice, respectively, and in 12 to 18 PGF male and female mice, respectively.
Fig. 5. Descendants’ expression of genes hypomethylated in ancestors’ sperm.
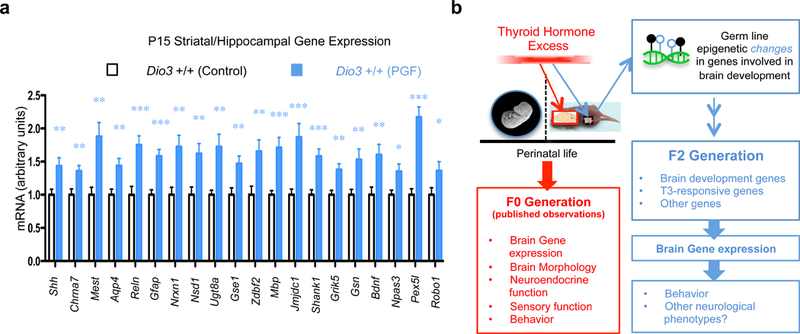
(a) Expression in the striatum/hippocampus of F2 generation descendants (PGF mice) of genes hypomethylated in ancestors germ line; (b) Diagram of findings in the current work in the context of the published neurological abnormalities of the F0 generation. *, ** and ***, P<0.05, 0.01 and 0.001, respectively as determined one-tail Student’s t test in samples from 10 Control and 11 PGF mice.
Alterations in the germ line methylome of exposed ancestors.
In view of the results above, we hypothesized that epigenetic alterations do occur in the germ line of Dio3−/− ancestors overexposed to T3 during development. Immunohistochemical analysis of Dio3−/− newborn testis revealed decreased 5-methyl-cytosine (5mC) staining (Fig. 4a), while immunofluorescence co-staining with a germ cell-specific marker (Tra98) indicated that 5mC is detected in germ cells among somatic cells at this age. Additional changes in the staining of 5mC and 5-hydroxymethyl-cytosine are observed in the testis of P1 and P11 Dio3−/− mice (Supplementary Fig. 6, 7 and 8).
Fig. 4. Epigenetic alterations in the germ line of T3-overexposed ancestors.
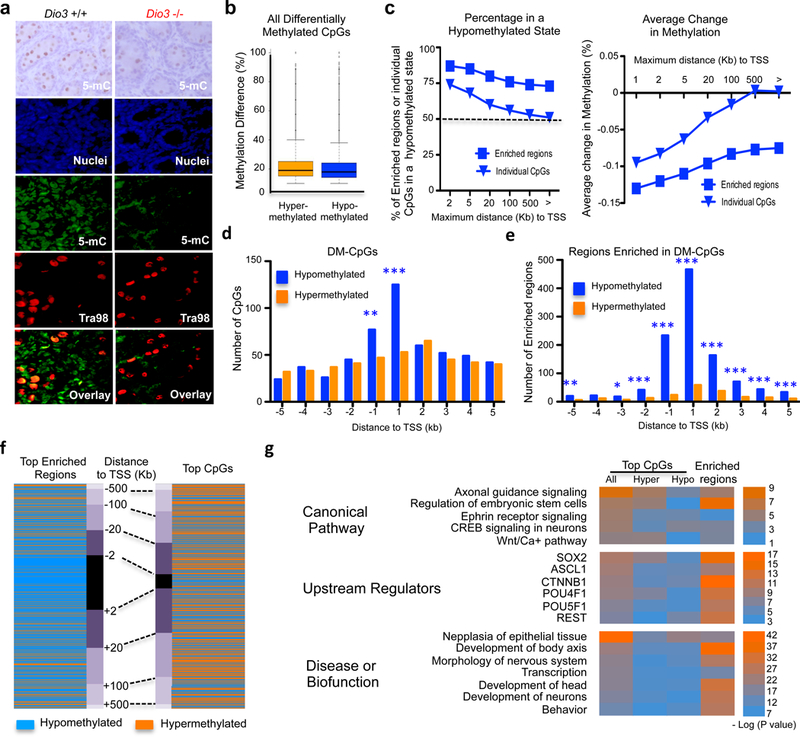
(a) 5-Methyl cytosine (5-mC) immunostaining (top), and nuclear (blue), 5-mC (green), germ cell-specific Tra98 (red), and overlay immunofluorescence in postnatal day 1 (P1) testis (see also figs. S4, S5 and S6); (b) Methylation difference in all the differentially methylated CpG sites (DM-CpGs) identified; (c) Percentage of hypomethylated sites and average change in methylation in DM-CpGs and in 1 kb genomic regions enriched in DM-CpGs, in relationship to the distance to the transcription start sites (TSSs) of genes. (d) Methylation status in DM-CpGs with top differential methylation located within 5Kb of TSSs. (e) Methylation status in DM-CpG-enriched regions with top differential methylation located within 5Kb of TSSs. (f) Methylation status comparison and distribution according to distance to TSSs between top (highest differential methylation) 1018 DM-CpGs and top (highest differential methylation average) 1042 DM-CpG-enriched regions; (g) Results highlights from INGENUITY analysis of genes associated with top individual DM-CpGs and genes associated with DM CpG islands within 5 kb of the transcription start site. *, ** and ***, indicate P<0.05, 0.01 and 0.001, respectively, as determined by χ2 test (d and e).
We then carried out a methylome analysis in DNA obtained from the sperm of adult Dio3−/− males. We identified ~110,000 differentially methylated CpG sites (DM-CpGs) (Supplementary Data 2) that showed an average methylation difference of ~20% and included a similar number of hyper- and hypomethylated DM-CpGs (Fig. 4b). We analyzed individual DM-CpGs and genomic regions 1 kb in size with a higher density of DM-CpGs (most of them CpG islands referred to as “enriched regions”)(Supplementary Data 3). We identified 3396 of these regions. Enriched regions exhibited a marked bias towards a hypomethylation state, but this bias was midler in individual DM-CpGs (Fig. 4c). Individual DM-CpGs with top methylation difference showed a hypomethylation bias only within 1kb of TSSs (Fig. 4d), but a more extensive and accentuated bias to a hypomethylation state is observed in 1326 DM-CpG-enriched regions (Fig. 4e). This is further appreciated in the Boolean heat map in Fig. 4f.
The distributions of all DM-CpGs and regions enriched in DM-CpGs were not homogeneous across the genome, but exhibited similar concentration patterns across certain chromosomal regions, including high densities of DM-CpGs in distal areas of chromosomes 4 and 15 (Supplementary Fig. 9a). Differential methylation in sex chromosomes was significantly lower than the genome’s average (Supplementary Fig. 9b).
INGENUITY analyses of 1409 genes associated with individual CpGs showing top methylation differences and 1097 did not grossly differ (Fig. 4g). They indicated enrichment in genes related to embryonic stem cells and fetal development, the development of neurons and the central nervous system, behavior, and digestive cancer. The analysis also showed common enrichment in canonical pathways associated with stem cell pluripotency and axon guidance signaling, and pointed to upstream regulators involved in cell stemness and neurogenesis, including SOX2, ASCL1, CTNNB1, POU4F1 and REST. Additional gene ontology analysis using DAVID identified enrichment in genes related to the control of transcription, embryonic development and neuronal differentiation, while disease analysis using the FunDO web tool (see Methods) determined top enrichment in genes associated with autism, cancer, schizophrenia, drug abuse and congenital abnormalities (Supplementary Fig. 9c and d). We selected 51 genes associated with top enrichment in DM-CpGs and represented their normalized tissue patterns of expression using data available from the mouse ENCODE project 63. The clustered heat map of these data illustrates a marked enrichment in genes with highly specific expression in tissues of the developing and adult central nervous system (Supplementary Fig. 10).
Descendants brain expression of genes differentially methylated in ancestors’ sperm.
Out of 597 genes (analyzed in Supplementary Fig. 9d) differentially methylated in the sperm of ancestors, we selected ~20 genes that, according to the Allen Brain Atlas, exhibit measurable expression in P15 brain tissue available from the previous RNA-sequencing experiment. All of these selected genes exhibited significantly increased expression in a striatal-hippocampal sample of P15 PGF mice (Fig. 5a). Only 9 of those genes, respectively, exhibited altered expression in the hypothalamus or cerebral cortex (Supplementary Fig. 11). Thus, at least some of the genes differentially methylated in the sperm of ancestors exhibit altered expression in the brain tissue of descendants.
Analysis of genes differentially methylated in the sperm of ancestors revealed enrichment in genes specific to neurons and oligodendrocyte precursor cells (OPCs) (Supplementary Fig. 12a), but not in oligodendrocyte-specific genes, as found for gene abnormally expressed in descendants.
Finally, we compared genes differentially methylated in the ancestor’s germ line with those differentially expressed in the F2 generation neonatal hypothalamus, and both of those sets with lists of candidate genes for schizophrenia (560 genes 3 ), autism (631 and 171 genes based on compendia by the Simmons Foundation and Peking University, respectively) and T3-regulated genes in the brain 64 (see Methods). There was a significant overlap of differentially methylated genes in ancestral sperm with autism candidate genes (P= 6.5E-07, odds ratio 6.9) and with genes down-regulated in the brain of descendants (P= 1.4E-12, odds ratio 5.2) (Supplementary Fig. 12b). There was also a significant overlap between genes differentially expressed in descendants and a dataset of genes regulated by T3 in the brain (P=2.5E-9, odds ratio 3.6).
Discussion
Using a well-characterized mouse model of developmental thyrotoxicosis 47, 65, we show that second generation wild type descendants of T3-overexposed mice exhibit an abnormal hypothalamic gene expression profile as neonates (Fig. 5b). These abnormal brain gene expression patterns are of epigenetic origin as these descendants are genetically normal and were born to genetically normal females. The abnormal gene expression profiles occur both in descendants of mice with a male or with a female overexposed ancestor in the paternal line (PGF and PGM mice). These abnormal profiles in PGF and PGM descendants largely overlap, indicating that developmental T3 excess influences the epigenetic germ line of males and females in a similar manner.
We also show that gene expression alterations in descendants do not appear to be restricted to the hypothalamus. They may affect multiple regions of the central nervous system and are associated in adulthood with behavioral abnormalities related to anxiety, repetitive behavior and physical activity. This behavioral phenotype only partly resembles that described for T3-overexposed ancestors 66, suggesting that some behavioral traits may be maintained or corrected in descendants via T3-driven intergenerational epigenetic effects .
The analysis of the sperm DNA methylome of T3-exposed male ancestors further supports a role for T3 in modifying the epigenetic information of the germ line. This process is likely initiated early in development, as indicated by the abnormal levels of DNA methylation observed in the neonatal testis, a critical period for the expansion of spermatogonia and during which high DIO3 expression 67 plays a important role protecting the testis from T3 excess and its potential deleterious effects on the chromatin landscape 68. The epigenetic effects of T3 are not surprising given its regulation of DNA methyltransferases 69 and its ability, upon receptor binding, to modify chromatin by recruiting histone modifying enzymes 68. The higher percentage of proliferating cells during development makes this period particularly susceptible to the epigenetic effects of thyroid hormone, as shown recently by aberrant epigenetic programming of the adult liver due to a loss in a neonatal hepatic peak in T3 70. It is also possible that some of the germ line epigenetic modifications observed could be secondary to other metabolic or endocrine abnormalities present in the ancestral fetus or neonate due to T3 overexposure, and not to direct T3 effects on DNA or chromatin modifying genes.
Developmental T3 excess leads in the adult sperm DNA to a similar number of hypo- and hypermethylation changes. However, we observed strong, selective hypomethylation in CpG islands associated with the promoter regions of genes that are highly specific to the central nervous system, involved in early brain development and overrepresented in neurogenic pathways regulating the proliferation and fate of neural precursor cells, including those regulated by SOX2 71, 72, ASCL1 73, CTNNB1 74 and REST 75. We show that some of these hypomethylated genes exhibit increased expression in certain brain areas of neonatal F2 generation descendants (PGF mice), suggesting a maintained hypomethylated state across generations that results in increased gene expression. Furthermore, among these group of genes are recognized candidate genes for neurodevelopmental disorders in humans including Nrxn1, Nsd1, Reln, Gfap, Mest, Bdnf and others. This raises the possibility that T3-driven epigenetic inheritance may influence susceptibility to these conditions.
Since many hypomethylated genes in the ancestral germ line are involved in early brain development, it is possible that a significant proportion of the gene expression changes observed in descendants at postnatal day 15 occurs secondary to alterations in brain cell homeostasis and development due to inherited epigenetic dysregulation of early brain genes. In addition, considering the role of thyroid hormone in brain developmental plasticity, a complementary mechanism to explain altered brain gene expression in descendants is that T3 overexposure may affect the epigenetic information of some T3 target genes 64, or of genes that control thyroid hormone action and availability in the brain. The latter may include Dio3 and Dio2, which have opposites roles in regulating T3 availability in neural tissue 76 . However, their differences in mRNA levels in descendants did not reach statistical significance in the RNA sequencing experiment. Still, this possibility is supported by the observations that thyroid hormone regulates several genes involved in myelination 27 and also influences brain cell fate favoring oligodendrocyte generation 77 , which is consistent with our observations of these processes being altered in the F2 generation. A role of inherited epigenetic alterations in Dio3 as a likely contributor to the phenotypes of descendants is supported by data indicating that the epigenetic regulation of the Dio3 gene in present and future generations is susceptible to fetal environmental factors such as alcohol exposure and thyroxine administration 45, 46.
Additional work is needed to understand how susceptibilities to particular neurodevelopmental phenotypes are influenced by ancestral overexposure to thyroid hormone and to define their patterns of inheritance. This may entail gene expression profiling in other brain regions and extended behavioral tests that may include social behavior and prepulse inhibition. Our work shows that this developmental insult may determine neurological traits in future generations via epigenetic mechanisms. This might be relevant to the “missing heritability” of neurodevelopmental conditions in humans as an abnormal thyroid hormone status in the fetus may occur largely unnoticed secondary to maternal thyroid disease or environmental exposure to chemicals that disrupt thyroid hormone action.
Supplementary Material
Acknowledgements
We are grateful to Joseph Nadeau for his critical review of the manuscript and to Amanda Drake for technical advice. This study was supported by grants MH096050 and DK095908 from the National Institute of Mental Health and National Institute of Diabetes, Digestive and Kidney Diseases, respectively. This work used the Confocal Microscopy Core, the Histopathology Core, the Physiology Core and the Molecular Phenotyping Core at Maine Medical Center Research Institute, that are supported by grants P30GM106391, P30GM103392, P20GM121301, and U54GM115516 from the National Institute of General Medical Sciences.
Footnotes
Conflict of Interest
The Authors declare that no conflict of interest exists.
References
- 1.Gupta AR, State MW. Recent advances in the genetics of autism. Biological Psychiatry 2007; 61(4): 429–437. [DOI] [PubMed] [Google Scholar]
- 2.Sullivan PF. The genetics of schizophrenia. PLoS Medicine 2005; 2(7): e212-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butler MG, McGuire AB, Masoud H, Manzardo AM. Currently recognized genes for schizophrenia: High-resolution chromosome ideogram representation. Am J Med Genet B Neuropsychiatr Genet 2016; 171b(2): 181–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sandin S, Lichtenstein P, Kuja-Halkola R, Hultman C, Larsson H, Reichenberg A. The Heritability of Autism Spectrum Disorder. Jama 2017; 318(12): 1182–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Zeeuw EL, van Beijsterveldt CEM, Hoekstra RA, Bartels M, Boomsma DI. The etiology of autistic traits in preschoolers: a population-based twin study. J Child Psychol Psychiatry 2017; 58(8): 893–901. [DOI] [PubMed] [Google Scholar]
- 6.Tick B, Bolton P, Happe F, Rutter M, Rijsdijk F. Heritability of autism spectrum disorders: a meta-analysis of twin studies. J Child Psychol Psychiatry 2016; 57(5): 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yip BHK, Bai D, Mahjani B, Klei L, Pawitan Y, Hultman CM et al. Heritable Variation, With Little or No Maternal Effect, Accounts for Recurrence Risk to Autism Spectrum Disorder in Sweden. Biol Psychiatry 2017. [DOI] [PMC free article] [PubMed]
- 8.Chen JA, Penagarikano O, Belgard TG, Swarup V, Geschwind DH. The emerging picture of autism spectrum disorder: genetics and pathology. Annu Rev Pathol 2015; 10: 111–144. [DOI] [PubMed] [Google Scholar]
- 9.Huguet G, Benabou M, Bourgeron T. The Genetics of Autism Spectrum Disorders. In: Sassone-Corsi P, Christen Y (eds). A Time for Metabolism and Hormones Springer; Copyright 2016, The Author(s). Cham (CH), 2016, pp 101–129. [PubMed] [Google Scholar]
- 10.Bourgeron T. Current knowledge on the genetics of autism and propositions for future research. C R Biol 2016; 339(7–8): 300–307. [DOI] [PubMed] [Google Scholar]
- 11.Ziats MN, Rennert OM. The Evolving Diagnostic and Genetic Landscapes of Autism Spectrum Disorder. Front Genet 2016; 7: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Rubeis S, Buxbaum JD. Genetics and genomics of autism spectrum disorder: embracing complexity. Human molecular genetics 2015; 24(R1): R24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Persico AM, Bourgeron T. Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends in Neurosciences 2006; 29(7): 349–358. [DOI] [PubMed] [Google Scholar]
- 14.Kinney DK, Munir KM, Crowley DJ, Miller AM. Prenatal stress and risk for autism. Neuroscience & Biobehavioral Reviews 2008; 32(8): 1519–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Molloy CA, Morrow AL, Meinzen-Derr J, Dawson G, Bernier R, Dunn M et al. Familial autoimmune thyroid disease as a risk factor for regression in children with Autism Spectrum Disorder: a CPEA Study. Journal of Autism & Developmental Disorders 2006; 36(3): 317–324. [DOI] [PubMed] [Google Scholar]
- 16.Dunaway KW, Islam MS, Coulson RL, Lopez SJ, Vogel Ciernia A, Chu RG et al. Cumulative Impact of Polychlorinated Biphenyl and Large Chromosomal Duplications on DNA Methylation, Chromatin, and Expression of Autism Candidate Genes. Cell reports 2016; 17(11): 3035–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vineis P, Pearce N. Missing heritability in genome-wide association study research. Nature Reviews Genetics 2010; 11(8): 589. [DOI] [PubMed] [Google Scholar]
- 18.Shen X. The curse of the missing heritability. Front Genet 2013; 4: 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ et al. Finding the missing heritability of complex diseases. [Review] [84 refs]. Nature 2009; 461(7265): 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trerotola M, Relli V, Simeone P, Alberti S. Epigenetic inheritance and the missing heritability. Hum Genomics 2015; 9: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koch L. Disease genetics: insights into missing heritability. Nat Rev Genet 2014; 15(4): 218. [DOI] [PubMed] [Google Scholar]
- 22.Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating missing heritability for disease from genome-wide association studies. American Journal of Human Genetics 88(3):294–305, 2011 Mar 11 2011; 88(3): 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan TF, Li A, Sun X, Ouyang H, Campos C, Rocha NB et al. Transgenerational Inheritance of Paternal Neurobehavioral Phenotypes: Stress, Addiction, Ageing and Metabolism. Mol Neurobiol 2015. [DOI] [PubMed]
- 24.Bohacek J, Mansuy IM. Molecular insights into transgenerational non-genetic inheritance of acquired behaviours. Nat Rev Genet 2015; 16(11): 641–652. [DOI] [PubMed] [Google Scholar]
- 25.Wei Y, Yang CR, Wei YP, Zhao ZA, Hou Y, Schatten H et al. Paternally induced transgenerational inheritance of susceptibility to diabetes in mammals. Proceedings of the National Academy of Sciences of the United States of America 2014; 111(5): 1873–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Skinner MK, Guerrero-Bosagna C, Haque M, Nilsson E, Bhandari R, McCarrey JR. Environmentally induced transgenerational epigenetic reprogramming of primordial germ cells and the subsequent germ line. PloS one 2013; 8(7): e66318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernal J. Thyroid hormones and brain development. Vitamins & Hormones 2005; 71: 95–122. [DOI] [PubMed] [Google Scholar]
- 28.Refetoff S, Weiss RE, Usala S. The syndromes of resistance to thyroid hormones. Endo Reviews 1993; 14: 348–399. [DOI] [PubMed] [Google Scholar]
- 29.Kempers MJE, van Tijn DA, van Trotsenburg ASP, de Vijlder JJM, Wiedijk BM. Central congenital hypothyroidism due to gestational hyperthyroidism: detection where prevention failed. J Clin Endocrinol Metab 2003; 88: 5851–5857. [DOI] [PubMed] [Google Scholar]
- 30.Boelaert K, Newby PR, Simmonds MJ, Holder RL, Carr-Smith JD, Heward JM et al. Prevalence and relative risk of other autoimmune diseases in subjects with autoimmune thyroid disease. Am J Med 2010; 123(2): 183.e181–189. [DOI] [PubMed] [Google Scholar]
- 31.McLachlan SM, Rapoport B. Breaking tolerance to thyroid antigens: changing concepts in thyroid autoimmunity. Endocr Rev 2014; 35(1): 59–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giera S, Bansal R, Ortiz-Toro TM, Taub DG, Zoeller RT. Individual polychlorinated biphenyl (PCB) congeners produce tissue- amd genespecific effects on thyroid hormone signaling during development. Endocrinology 2011; 152(7): 2909–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bansal R, Tighe D, Danai A, Rawn DF, Gaertner DW, Arnold DL et al. Polybrominated diphenyl ether (DE-71) interferes with thyroid hormone action independent of effects on circulating levels of thyroid hormone in male rats. Endocrinology 2014; 155(10): 4104–4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Legrand J. Effects of thyroid hormones on central nervous system development. In: Yanai J (ed). Neurobehavioral Teratology Elsevier: New York, 1984, pp 331–363. [Google Scholar]
- 35.Calvo R, Obregón MJ, Ruiz de Ona C, Escobar del Rey F, de Escobar GM. Congenital hypothyroidism, as studied in rats: crucial role of maternal thyroxine but not 3,5,3’-triiodothyronine in the protection of the fetal brain. J Clin Invest 1990; 86: 889–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crawford S, van Baar AL, Brouwers EP, Pop VJ. Neonatal effects of maternal hypothyroxinemia during early pregnancy. Pediatrics 2006; 117(1): 161–167. [DOI] [PubMed] [Google Scholar]
- 37.Reuss ML, Paneth N, Pinto-Martin JA, Lorenz JM, Susser M. The relation of transient hypothyroxinemia in preterm infants to neurologic development at two years of age. The New England journal of medicine 1996; 334(13): 821–827. [DOI] [PubMed] [Google Scholar]
- 38.Andersen SL, Laurberg P, Wu CS, Olsen J. Attention deficit hyperactivity disorder and autism spectrum disorder in children born to mothers with thyroid dysfunction: a Danish nationwide cohort study. Bjog 2014; 121(11): 1365–1374. [DOI] [PubMed] [Google Scholar]
- 39.Khan A, Harney JW, Zavacki AM, Sajdel-Sulkowska EM. Disrupted brain thyroid hormone homeostasis and altered thyroid hormone-dependent brain gene expression in autism spectrum disorders. J Physiol Pharmacol 2014; 65(2): 257–272. [PubMed] [Google Scholar]
- 40.Sadamatsu M, Kanai H, Xu X, Liu Y, Kato N. Review of animal models for autism: implication of thyroid hormone. Congenital Anomalies 2006; 46: 1–9. [DOI] [PubMed] [Google Scholar]
- 41.Gyllenberg D, Sourander A, Surcel HM, Hinkka-Yli-Salomaki S, McKeague IW, Brown AS. Hypothyroxinemia During Gestation and Offspring Schizophrenia in a National Birth Cohort. Biol Psychiatry 2016; 79(12): 962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Modesto T, Tiemeier H, Peeters RP, Jaddoe VW, Hofman A, Verhulst FC et al. Maternal Mild Thyroid Hormone Insufficiency in Early Pregnancy and Attention-Deficit/Hyperactivity Disorder Symptoms in Children. JAMA Pediatr 2015; 169(9): 838–845. [DOI] [PubMed] [Google Scholar]
- 43.Bakke JL, Lawrence NL, Robinson S, Bennett J. Observations on the untreated progeny of hypothyroid male rats. Metabolism: Clinical & Experimental 1976; 25(4): 437–444. [DOI] [PubMed] [Google Scholar]
- 44.Bakke JL, Lawrence NL, Robinson S, Bennett J. Endocrine studies in the untreated F1 and F2 progeny of rats treated neonatally with thyroxine Biol Neonate 1977; 31: 71–83. [DOI] [PubMed] [Google Scholar]
- 45.Tunc-Ozcan E, Wert SL, Lim PH, Ferreira A, Redei EE. Hippocampus-dependent memory and allele-specific gene expression in adult offspring of alcohol-consuming dams after neonatal treatment with thyroxin or metformin. Molecular psychiatry 2017. [DOI] [PMC free article] [PubMed]
- 46.Tunc-Ozcan E, Harper KM, Graf EN, Redei EE. Thyroxine administration prevents matrilineal intergenerational consequences of in utero ethanol exposure in rats. Horm Behav 2016; 82: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hernandez A, Martinez ME, Fiering S, Galton VA, St Germain D. Type 3 deiodinase is critical for the maturation and function of the thyroid axis. Journal of Clinical Investigation 2006; 116(2): 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hernandez A. Structure and function of the type 3 deiodinase gene. Thyroid : official journal of the American Thyroid Association 2005; 15(8): 865–874. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 2014; 34(36): 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Osborne JD, Flatow J, Holko M, Lin SM, Kibbe WA, Zhu LJ et al. Annotating the human genome with Disease Ontology. BMC Genomics 2009; 10 Suppl 1: S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics 2007; 23(2): 257–258. [DOI] [PubMed] [Google Scholar]
- 52.Du P, Feng G, Flatow J, Song J, Holko M, Kibbe WA et al. From disease ontology to disease-ontology lite: statistical methods to adapt a general-purpose ontology for the test of gene-ontology associations. Bioinformatics 2009; 25(12): i63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dennis G Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biology 2003; 4(5): P3. [PubMed] [Google Scholar]
- 54.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4(1): 44–57. [DOI] [PubMed] [Google Scholar]
- 55.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research 2009; 37(1): 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martinez ME, Charalambous M, Saferali A, Fiering S, Naumova AK, St Germain D et al. Genomic imprinting variations in the mouse type 3 deiodinase gene between tissues and brain regions. Mol Endocrinol 2014; 28(11): 1875–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hernandez A, Fiering S, Martinez E, Galton VA, St Germain D. The gene locus encoding iodothyronine deiodinase type 3 (Dio3) is imprinted in the fetus and expresses antisense transcripts. Endocrinology 2002; 143(11): 4483–4486. [DOI] [PubMed] [Google Scholar]
- 58.Tsai C, Lin SP, Ito M, Takagi N,ST, S. T, Ferguson-Smith AC. Genomic Imprinting contributes to thyroid hormone metabolism in the mouse embryo. Current Biology 2002; 12: 1221–1226. [DOI] [PubMed] [Google Scholar]
- 59.Sittig LJ, Herzing LBK, Shukla PK, Redei EE. Parent-of-origin allelic contributions to deiodinase-3 expression elicit localized hyperthyroid milieu in the hippocampus. Molecular psychiatry 2011; 16(8): 786–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martinez ME, Charalambous M, Saferali A, Fiering S, Naoumova A, St. Germain DL et al. Genomic Imprinting Variations in the Mouse Type 3 Deiodinase Gene Between Tissues and Brain Regions. Mol Endocrinol 2014; 28(11): 1875–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ueta Cintia B, ELO Behzad N Oskouei, Pinto Jose R, Correa Mayrin M, Simovic Gordana, Simonides Warner S, Hare Joshua M, Bianco Antonio C. Absence of myocardial thyroid hormone inactivating deiodinase results in restrictive cardiomyopathy in mice. Mol Endocrinol 2012; 26(5): 809–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barez-Lopez S, Obregon MJ, Bernal J, Guadano-Ferraz A. Thyroid Hormone Economy in the Perinatal Mouse Brain: Implications for Cerebral Cortex Development. Cerebral cortex (New York, NY : 1991) 2018; 28(5): 1783–1793. [DOI] [PubMed] [Google Scholar]
- 63.Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S et al. A map of the cis-regulatory sequences in the mouse genome. Nature 2012; 488(7409): 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chatonnet F, Flamant F, Morte B. A temporary compendium of thyroid hormone target genes in brain. Biochim Biophys Acta 2015; 1849(2): 122–129. [DOI] [PubMed] [Google Scholar]
- 65.Hernandez A, Quignodon L, Martinez ME, Flamant F, St. Germain DL. Type 3 deiodinase deficiency causes spatial an temporal alterations in brain T3 signaling that are dissociated from serum thyroid hormone levels. Endocrinology 2010; 151(11): 5550–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stohn JP, Martinez ME, Hernandez A. Decreased anxiety- and depression-like behaviors and hyperactivity in a type 3 deiodinase-deficient mouse showing brain thyrotoxicosis and peripheral hypothyroidism. Psychoneuroendocrinology 2016; 74: 46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martinez ME, Karaczyn A, Stohn JP, Donnelly WT, Croteau W, Peeters RP et al. The Type 3 Deiodinase Is a Critical Determinant of Appropriate Thyroid Hormone Action in the Developing Testis. Endocrinology 2016; 157(3): 1276–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pascual A, Aranda A. Thyroid hormone receptors, cell growth and differentiation. Biochim Biophys Acta 2013; 1830(7): 3908–3916. [DOI] [PubMed] [Google Scholar]
- 69.Kyono Y, Subramani A, Ramadoss P, Hollenberg AN, Bonett RM, Denver RJ. Liganded Thyroid Hormone Receptors Transactivate the DNA Methyltransferase 3a Gene in Mouse Neuronal Cells. Endocrinology 2016; 157(9): 3647–3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fonseca TL, Fernandes GW, McAninch EA, Bocco BM, Abdalla SM, Ribeiro MO et al. Perinatal deiodinase 2 expression in hepatocytes defines epigenetic susceptibility to liver steatosis and obesity. Proceedings of the National Academy of Sciences of the United States of America 2015; 112(45): 14018–14023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Amador-Arjona A, Cimadamore F, Huang CT, Wright R, Lewis S, Gage FH et al. SOX2 primes the epigenetic landscape in neural precursors enabling proper gene activation during hippocampal neurogenesis. Proceedings of the National Academy of Sciences of the United States of America 2015; 112(15): E1936–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lopez-Juarez A, Remaud S, Hassani Z, Jolivet P, Pierre Simons J, Sontag T et al. Thyroid hormone signaling acts as a neurogenic switch by repressing Sox2 in the adult neural stem cell niche. Cell stem cell 2012; 10(5): 531–543. [DOI] [PubMed] [Google Scholar]
- 73.Memic F, Knoflach V, Sadler R, Tegerstedt G, Sundstrom E, Guillemot F et al. Ascl1 Is Required for the Development of Specific Neuronal Subtypes in the Enteric Nervous System. The Journal of neuroscience : the official journal of the Society for Neuroscience 2016; 36(15): 4339–4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gulacsi AA, Anderson SA. Beta-catenin-mediated Wnt signaling regulates neurogenesis in the ventral telencephalon. Nat Neurosci 2008; 11(12): 1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mozzi A, Guerini FR, Forni D, Costa AS, Nemni R, Baglio F et al. REST, a master regulator of neurogenesis, evolved under strong positive selection in humans and in non human primates. Sci Rep 2017; 7(1): 9530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bianco AC. Minireview: cracking the metabolic code for thyroid hormone signaling. Endocrinology 2011; 152(9): 3306–3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Remaud S, Ortiz FC, Perret-Jeanneret M, Aigrot MS, Gothie JD, Fekete C et al. Transient hypothyroidism favors oligodendrocyte generation providing functional remyelination in the adult mouse brain. Elife 2017; 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.