

Abstract
Physiologically‐based pharmacokinetic models are increasingly applied for pediatric dose selection along with traditional methods such as allometry and population pharmacokinetic models. We report a retrospective evaluation of the three methods. Pediatric population pharmacokinetic models sourced from literature for a subset of eight compounds were used to predict clearances for children < 2 years when they were within the modeled age range (interpolation, N = 11) or including those outside the modeled age range (interpolation and extrapolation, N = 18). Pediatric/adult clearance ratios were evaluated with a strict performance criterion of 0.8–1.25 and with twofold criteria. For children > 2 years, 58–75% of the clinical studies (N = 10) met the strict criteria, and > 80% of the clinical studies were predicted within twofold by all three methods. For children < 2 years, physiologically‐based pharmacokinetic, allometry with age‐dependent exponents, and pediatric population pharmacokinetic models predict 54%, 82%, and 64% within twofold of the observed, respectively.
STUDY HIGHLIGHTS.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Difficulties in conducting pediatric trials lead to the suboptimal dosing of drugs approved for children. Regulatory agencies have therefore mandated pediatric study plans to be submitted and require pharmacokinetics, pharmacodynamics/efficacy, and safety studies in children. Dose selections for these pediatric studies were traditionally done with allometry and population pharmacokinetics, but they could be inadequate for children < 2 years because of organ maturation. Mechanistic physiologically‐based pharmacokinetic models may fill this gap.
WHAT QUESTION DID THIS STUDY ADDRESS?
A comparative evaluation of the methods using drugs with diverse elimination routes to address how current methods for pediatric exposure prediction can be optimally leveraged.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
All three methods are comparable for children aged > 2 years. For the children aged < 2, adult to pediatric extrapolation with physiologically‐based pharmacokinetic models is better than with simple allometry, but performance of physiologically‐based pharmacokinetic models is comparable with allometry with age‐dependent exponents. Population pharmacokinetics performs best for interpolation.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
The sparse data from physiologically‐based pharmacokinetic models or allometry with age‐dependent exponents to select starting dose and to design the confirmatory studies in children can be used for building a population pharmacokinetics model to predict exposure in different subpopulations of children < 2 years of age with better precision.
Pediatric drug development has been hampered by the cost of developing new formulation, ethical constraints to drawing blood, and other practical difficulties. The dosing of drugs approved for pediatric use were therefore often based on adult doses. To minimize the risk of treatment failures, toxicities, and other drug‐related adverse events often associated with off‐label pediatric dosing, regulatory agencies now require sponsors to submit pediatric trial plans at the end of the phase II study (following the availability of exposure‐response data in adults) and prior to the initiation of phase III studies as part of new drug approval. In the United States, the Pediatric Research Equity Act first passed in 2003, and then in 2007 with some changes, authorizes the US Food and Drug Administration (FDA) to make pediatric assessment mandatory. It is triggered by an application for a new indication, new dosage form, new dosing regimen, new route of administration, or new active ingredient of an approved drug/biologic product for certain indications. Studies for orphan indications are exempt from the Pediatric Research Equity Act. However, the Best Pharmaceuticals for Children Act authorizes the FDA to request studies of approved and/or unapproved pediatric orphan indications, paving the way for voluntary pediatric drug assessments, including clinical and nonclinical studies via a written request.1 the Pediatric Research Equity Act mandates the submission of a pediatric study plan, providing an outline of the pediatric study/studies that the sponsor plans to conduct prior to the submission of the New Drug Application or Biologic License Application as well as plans to extrapolate efficacy from adult to pediatric patients or from one pediatric age group to another. In its guidance to the industry, the FDA provides a decision tree that shows when partial or even full pediatric extrapolation may be applicable2 (Figure 1). Similar considerations apply for the submission of a Pediatric Investigation Plan by the sponsor at the end of phase II to the European Medicines Agency (EMA). Pediatric clinical trials should be designed to fill knowledge gaps. To increase the likelihood of success for a pediatric trial, guidelines from the FDA3 and the EMA4 encourage various innovative approaches.
Figure 1.
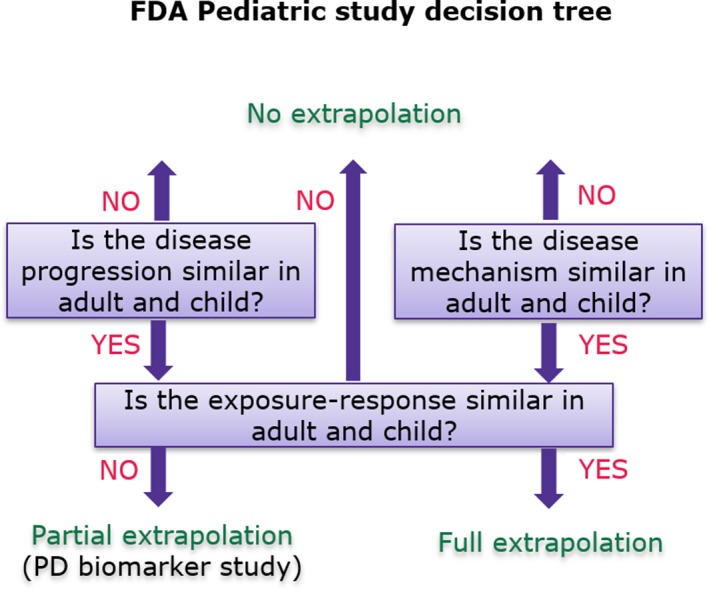
US Food and Drug Administration (FDA) decision tree for pediatric study planning. The extent of similarity in disease (with respect to pathophysiology and progression) and response to drug intervention (mode of drug action and biological pathway like a marketed drug belonging to the same therapeutic class and assessed by similar endpoints) between adults and children determines which of the three major pediatric studies should be undertaken: pharmacokinetics only, pharmacokinetics/pharmacodynamics, or pharmacokinetics/efficacy. Safety studies are required in all scenarios. PD: pharmacodynamic.
The demonstration of safety or efficacy in the dose range tested in pediatric trials requires optimal dose selection. Allometric scaling, population pharmacokinetics (popPK) modeling, or physiologically‐based pharmacokinetic (PBPK) models are some of the frequently used methods to inform pediatric dosing and study design. Plasma proteins and enzyme and transporter levels are some of the key physiological parameters that impact drug exposure in adults and children. Because growth and organ maturation in children < 2 years of age impact these key sensitive parameters, clearance (CL) may not correlate with body weight. Therefore, simple allometric methods (exponent of 0.75) are not expected to perform well for this age group. Instead, the use of body‐weight or age‐dependent exponents have been proposed.5, 6 Because maturation is completed by 2 years of age, size could be the main predictor of drug CL in a pediatric population > 2 years.7 Given the distinct physiology of children from adults (see Table S1), traditional approaches such as allometry that consider differences only related to body weight and body surface area are likely to over or underpredict drug CL in pediatric patients, especially for infants that are < 1 year.8 PopPK uses a mathematical model to describe the sparse pharmacokinetic (PK) data collected from pediatric populations in clinical studies. Variability in key model parameters are captured in equations, relating them to measurable covariates such as body weight, age, and so on. PBPK models are mechanistic mathematical models describing the disposition of drugs in an organism through the integration of multiple developmental, size, and disease differences. Both popPK and PBPK models9 can predict drug exposure across a wide range of ages and weights as well as consider differences in maturation and organ function. Pediatric PBPK models have been built for several small10, 11, 12, 13 and large molecules14 and used for pediatric trial design and dose selection involving extrapolation from adult to pediatric or from one pediatric subpopulation to another.
Several evaluations of the PBPK approach for small molecule drugs are reported in the literature.15, 16, 17, 18 Johnson et al.15 compared the performance of CL prediction by simple allometry and pediatric PBPK model incorporated in Simcyp (Certara, Sheffield, UK) for 11 drugs and demonstrated better prediction accuracy for PBPK compared with simple allometry, especially in children < 2 years old. Zhou et al.17 used 10 compounds metabolized by Cytochrome P450 (CYP)3A4 to demonstrate the value of PBPK pediatric predictions for children < 2 years of age. Although simple allometry with a constant body‐weight exponent of 0.75 performed badly for the < 2 years children,19 a recent study demonstrated that allometry with age‐dependent exponents (ADEs) predicted 87% of the 130 studies evaluated within a twofold error margin, which was comparable with PBPK.20 Evaluation of PK‐Sim (www.systems-biology.com), another widely used PBPK platform for pediatric applications, has been reported.8 Performance accuracies of PBPK and popPK have been reported to be good for renally cleared compounds6 and for valganciclovir.13 Templeton et al.16 reviewed recent pediatric clinical literature on 31 small‐molecule drugs to investigate the contribution of PBPK in understanding the impact of physiological differences in determining drug dose in children and in adults. Many of these evaluations are based on reviews of pediatric models developed by different modelers. This may introduce a modeler bias that could impact the conclusions drawn from these evaluations. Not all evaluations conformed to the learn–confirm principles practiced today. The absence of a systematic comparison of PBPK, allometry, and popPK models for pediatric dose selection, the lack of diversity in the compounds selected, and the need to reduce modeler bias prompted the initiation of this study. The objective of this work is to use a diverse set of compounds to evaluate PBPK models for pediatric dosing using CL as a surrogate for dose and compare their performance alongside traditional methods such as allometry (simple allometry and allometry with ADEs) and popPK.
Methods
Compound selection
The following criteria were applied to select compounds for the study:
Pediatric PK studies available for many age groups
The ratio of adult dose/pediatric dose > 1.25 or < 0.8
Adult intravenous (i.v.) PK available for oral drugs
The following 13 compounds met the selection criteria: amikacin, bosentan, caffeine, clindamycin, diclofenac, docetaxel, itraconazole, lorazepam, midazolam, montelukast, ropivacaine, sotalol, and theophylline. Of these compounds, 5 are in the Simcyp (midazolam, caffeine, theophylline, lorazepam, and diclofenac).
Data collection
Physicochemical properties and in vitro Absorption, Distribution, Metabolism and Elimination data were obtained from the Drug Bank (https://www.drugbank.ca) and from the literature. All data are presented in Table 1. The 13 compounds chosen for the analysis represent a broad range of physicochemical properties (lipophilicity, biopharmaceutical classification system, and compound type) and major elimination pathways (five CYP3A, three CYP1A2, one CYP2C9, one CYP2C8, one UDP‐glucuronosyltransferase (UGT)2B7, two renal). Clinical PK data for the different adult and pediatric populations were collected from the literature. A total of 59 clinical studies from the 13 compounds were available, of which 4 premature infants (< 1 month), 4 neonates (< 1 year), 19 infants (1–2 years), 11 young children (2 years up to 6 years), 13 children (9–11 years), and 6 adolescents (12–18 years) were included. The details of these studies are presented in Table S2. The plasma concentration‐time profiles were digitized using the software WebPlot Digitizer v.3.12 (https://automeris.io/WebPlotDigitizer/).
Table 1.
Summary of physicochemical and pharmacokinetic properties of compounds
| Amikacin | Bosentan | Caffeine | Clindamycin | Diclofenac | Docetaxel | Itraconazole | Lorazepam | Midazolam | Montelukast | Ropivcaine | Sotalol | Theophyline | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BCS | 3 | 3 | 1 | 1 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 3 | 1 |
| Indication | Severe sepsis | Pulmonary arterial hyper‐tension | Psychoactive agents | Infections | Infections | Cancer | Antifungal | Severe sepsis | CNS | Asthma | Visceral pain | Anti‐hypertensive | COPD |
| MW (g/mol) | 585.6 | 551.6 | 192.190 | 424.980 | 296.1 | 807.9 | 705.6 | 321.160 | 325.8 | 586.2 | 274.41 | 272.360 | 180.2 |
| log P | −7.4 | 3.7 | −0.07 | 2.160 | 4.5 | 3.2 | 5.66 | 2.4 | 3.53 | 8.98 | 2.91 | 0.370 | −0.02 |
| Compound type | Ampholyte | Acid | Base | Base | Acid | Neutral | Base | Ampholyte | Ampholyte | Acid | Base | Ampholyte | Ampholyte |
| pKas of acidic (pKa1) and basic (pKa2) centers |
pKa1 = 12.1 pKa2 = 9.8 |
pKa1 = 5.1 | pKa2 = 1.1 | pKa2 = 7.6 | pKa1 = 4.0 | — | pKa2 = 3.7 |
pKa1 = 11.5 pKa2 = 1.3 |
pKa1 = 10.95 pKa2 = 6.2 |
pKa1 = 5.7 | pKa2 = 8.07 |
pKa1 = 8.28 Pk2 = 9.72 |
pKa1 = 8.8. pKa2 = 0.99 |
| B/P | 1.00 | 0.60 | 0.98 | 0.94 | 1 | 0.68 | 0.58 | 0.642 | 0.603 | 0.65 | 0.94 | 1.02 | 0.82 |
| fu.p | 0.99 | 0.02 | 0.680 | 0.06 | 0.05 | 0.067 | 0.016 | 0.102 | 0.032 | 0.006 | 0.052 | 1 | 0.5 |
| Absorption parameters | — |
ADAM model. P eff.man = 1 × 10−4 cm/s. PSA = 145.65. HBD = 2. solubility FassiF = 20 mg/mL |
First order absorption Fa = 1. Ka = 2.03 |
— |
ADAM model. PSA = 52.16.HBD = 2.P eff.man = 10 × 10−4 cm/s Solubility FassiF = 0.805 mg/mL |
— | — | — |
ADAM model. Caco‐2 PH7.4:7.4 Passive & Active = 213 × 10−6 cm/s. P eff.man = 6.0 × 10−4 cm/s |
ADAM model. PSA = 95.72. HBD = 2. P eff.man = 0.3 × 10‐4 cm/s |
— |
First order absorption Fa = 1. Ka = 0.44 |
First order absorption fa = 1. ka = 6 |
| Main elimination pathway | Renal (100%) | Metabolism (97%), renal (3%) | Metabolism (99%), renal (1%) |
Metabolism (95%), Renal (5%) |
Metabolism (98%) renal (2%) |
Metabolism (83%), bile (4%), Renal (4%), additional (9%) | Metabolism (100%) |
Metabolism (95%) renal (5%) |
Metabolism (100%) | Metabolism (100%) |
Metabolism (99%). renal (1%) |
Renal (100%) |
Metabolism (90%) renal (10%) |
| Fractional contribution of individual CYPs/UGT | — |
CYP3A4 (60%). CYP2C9 (40%) |
CYP1A2 (98%). CYP2E1 (2%) |
CYP3A4 (88%). CYP3A5 (12%) |
CYP2C9 (100%) |
CYP3A4 (95%) CYP3A5 (5%) |
CYP3A4 (98%) CYP1A2 (2%) |
— | CYP3A4 (100%) |
CYP2C8 (72%) CYP3A4 (16%) CYP2C9 (12%) |
CYP1A2 (92%). CYP3A4 (8%) |
— |
CYP1A2, (75%) CYP2D6 (7%) CYP2E1 (10%) CYP3A4 (8%) |
| In vitro intrinsic clearance (μL/min/pmol P450) | — |
CLint CYP3A4 = 0.85 CYP2C9 = 1.07 |
CYP1A2 = 0.047 CYP1E2 = 0.000829 |
CLint CYP3A4 = 0.7 CLint CYP3A5 = 0.13 |
CLint CYP2C9 = 25.06 |
CLint CYP3A4 = 1.7 CLint CYP3A5 = 0.23 |
CYP3A4: HLM. V max = 18 pmol/min/mg. Km = 0.023 μM CLint 1A2 = 1 μL/min/mg |
CLint (UGT2B7) = 9.97 μL/min/pmol | CLint 3A4 = 3.74 |
CLint CYP2C8 = 3.68 CLint CYP3A4 = 0.13. CLint CYP2C9 = 0.18 |
CLint CYP1A2 = 2.41 CLint CYP3A4 = 0.08 |
— |
CLint CYP1A2 = 0.02 CLint CYP2D6 = 0.011 CLint CYP2E1 = 0.0022 CLint CYP3A4 = 0.00078 |
| References | Drug Bank | 23, 26, 27 | Simcyp | 12, 28 | 29, 30 | 9, 20 | Simcyp, 31 | Simcyp | Simcyp | 32, 33, 34 | 35, 36 | Drug Bank | Simcyp |
B/P, blood to plasma partition ratio; BCS, biopharmaceutical classification system; Caco‐2, Caco‐2 permeability (10−6 cm/s); CLint, in vitro intrinsic clearance; CNS, central nervous system; COPD, chronic obstructive pulmonary disease; fa, fraction available from dosage form; fu.p, fraction unbound in plasma; fuGut, unbound fraction of drug in enterocytes; HBD, number of hydrogen bond donors; ISEF, intersystem extrapolation factor for scaling of recombinant CYP in vitro kinetic data; IV, intravenous therapy; ka, first‐order absorption rate constant (L/h); Km, Michaelis‐Menten constant (μM); log P, lipophilicity; MRT, mean residence time; MW, molecular weight; P eff.man, human jejunum effective permeability (10−4 cm/s); pKa, acid/base character; PO, oral administration; PSA, polar surface area; Q Gut, a nominal flow in gut model (L/h); V max, maximum rate of metabolite formation.
Models
PBPK
Whole‐body PBPK models are in silico models that mathematically describe the mechanisms underlying drug disposition in an organism. They predict the PK profiles based on compound physicochemical properties and multiple anatomical and physiological parameters of the individual, such as organ volumes, tissue composition, and blood‐flow rates. The gastrointestinal compartment in the whole‐body PBPK model is further divided as described by the advanced compartmental absorption and transit model.21 The inclusion of gut metabolism, efflux, and influx transport in the gut compartments enables the prediction of the gut bioavailability of compounds. For the purpose of this analysis, we use the PBPK model implemented for adult and pediatric populations in Simcyp version 17.
PBPK workflow overview
Pediatric PK parameters are typically predicted using a pediatric PBPK model that has been developed using an adult PBPK model and clinical PK data. Workflows for pediatric PBPK model development with i.v. and oral adult PK data are shown in Figures 2 and 3. Intrinsic CL is obtained from the CL observed in adult PK studies (preferably i.v.) using the retrograde calculator in Simcyp.
Figure 2.
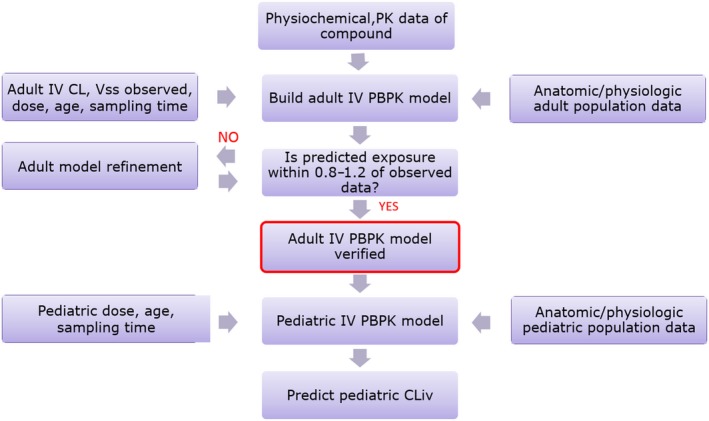
Workflow of intravenous (i.v.) pediatric physiologically‐based pharmacokinetic (PBPK) model. PK, pharmacokinetics. CL, Clearance; Vss, steady state volume of distribution; CLiv, intravenous clearance.
Figure 3.
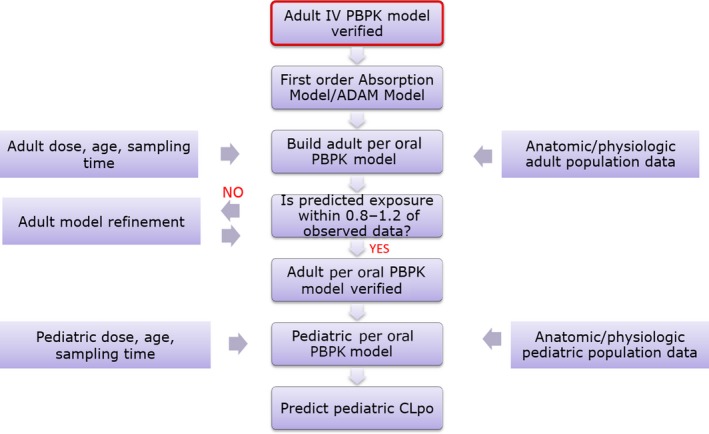
Workflow of per oral administration (PO) pediatric physiologically‐based pharmacokinetic model (PBPK). i.v., intravenous; ADAM, Advanced Dissolution, Absorption, and Metabolism model; CLpo, per oral clearance; PO, Per Oral.
Simple allometry
Pediatric CL is estimated knowing the adult CL using body size (mg/kg) according to the following equations22:
Allometry with ADEs
Allometry with ADEs used the following ADEs6, 20
| Preterm | Term | 0.25–2 years | 2–5 years | > 5 years |
| 1.2 | 1.1 | 1 | 0.9 | 0.75 |
PopPK
Pediatric popPK equations that were used in this analysis were obtained from the literature and are presented in Table 2. These equations were used to predict CL for children < 2 years when they were within the modeled age range (interpolation, N = 11) or including those outside the modeled age range (interpolation and extrapolation, N = 18). In the case of interpolation, the CL for children belonging to the age group of interest are estimated using the popPK equation derived from a base pediatric population covering that age group. In extrapolation, the age of the children for whom CL is estimated lies outside the range of age groups in the base pediatric population, described by the popPK equation.
Table 2.
Pediatric population pharmacokinetic equations sourced from the literature
| Durg | Base population | Number | Age | Equation | Reference |
|---|---|---|---|---|---|
| Amikacin | Infant to adolescent | 70 | 6 months–17 y | CL (L/h) = (5.98 × (BW/70)0.75) × 1.1 | 37 |
| Bosentan | Children | 49 |
2–12 y (40) < 2 (9) |
CL/F (L/h) = exp(0.0419 × BW) × 1.78 | 23 |
| Caffeine | Premature neonate | 75 | 22–35 w | CL (mL/h) = 5.81 × BW + 1.22 × age(w); CL × 0.757 if gestational age < 28 weeks | 38 |
| Neonate to infant | 60 | 1–100 d | CL (L/d) = 0.14 × BW + 0.0024 × age(d) | 39 | |
| Clindamycin | Premature infant to adolescent | 125 | 0–> 12 y | CL (L/h) = 13.7 × (BW/70)0.75 × (age3.1(w)/(43.63.1 + age3.1(w)) | 40 |
| Midazolam | Children to adolescent | 381 | 2–18 y | CL (L/h) = 30.7 × (BW/70)0.75 | 41 |
| Montelukast | Children to Adolescent, adult |
11 children 11 adults |
6–14 y | CL (mL/h) = 175 × BW0.635 | 42 |
| Children[Link] | 15 | 2–5 y | – | 43 | |
| Sotalol | Neonate to children | 59 | 0–12 y | CL/F (mL/min) = 60.5 + 105 × (BSA−0.7) | 44 |
| Theophylline | Neonate to young infant | 108 | 0–26 w | CL (mL/h) = 17.5 × (BW)1.28 + 1.17 × age(w) | 45 |
| Children | 84 | 1–8 y | CL (L/h) = exp (−0.229 + 0.0920*age(y)) | 46 |
BW, body weight (kg); d, days; w, weeks; y, years; BSA, Body Surface Area; exp, exponential; CL Clearance, F, oral bioavailability.
Only the result was reported in the literature. The equation was not accessible.
Comparison of model performance
To compare the predictive performance of different methods, the pediatric/adult ratios for CL and maximum plasma concentration follwing oral drug administration (Cmax) were calculated for every clinical study for both the observed and predicted data. The following two different prediction performance criteria were used: observed to predicted pediatric CL ratio in the range (i) 0.8–1.25 (similar to the bioequivalence criteria) and (ii) 0.5–2 (twofold). The percentage of clinical studies that satisfied this stringent criterion were determined for each of the three prediction methods for the 30 clinical studies in children < 2 years of age and 29 clinical studies in children > 2 years of age.
The popPK equations were available only for a subset of compounds under investigation. Therefore, the percentage of clinical studies in the < 2 and > 2 subpopulations that satisfied the performance criteria ranges were also calculated for the subset of clinical studies that had the popPK data.
Results
Plasma concentration‐time profiles from PBPK simulations corresponding to all doses in adult and pediatric subpopulations along with observed profiles are available as Figure S1. Observed and predicted (PBPK, simple allometry, and popPK models) pediatric to adult (P/A) ratios for CL and Cmax of all the studies are shown in Figure S2. Observed and predicted (PBPK, simple allometry, allometry with ADEs, and popPK methods) for CL and Cmax for all 59 pediatric studies are presented in Table S3. The predicted overobserved ratios for both CL and Cmax values from different methods are presented in Tables S4 and S5, respectively. Observed P/A ratios were > 1 for about half of the clinical studies and close to 1 for 16 studies. The percentage of predictions that met the two performance criteria are presented In Table 3. All three methods have comparable prediction performance for children > 2 years of age, with around 55% and nearly all the 29 studies meeting the stringent or twofold criteria, respectively. For children < 2 years of age, the performance of all methods is markedly poorer, especially with the stringent criteria. There were seven compounds for which the popPK model could be evaluated. The percentage of predictions that met the stringent performance criteria for a subset of the clinical studies for these seven compounds were 33%, 11%, and 28% for PBPK, simple allometry, and popPK, respectively; 67%, 33%, and 44% of the predictions met the twofold criteria. Allometry with ADEs was included for the < 2, and it was comparable with PBPK with 38% and 72% of the studies meeting the stringent and twofold criteria, respectively. For the subset of clinical studies for which popPK equations have been used only for interpolation, popPK outperformed PBPK with respect to both criteria and outperformed allometry with ADEs with respect to the twofold criteria only.
Table 3.
Performance comparison of different models evaluated
| Clearance ratio | ||||||||
|---|---|---|---|---|---|---|---|---|
| Pediatric < 2 years | Pediatric > 2 years | |||||||
| Performance criteria | Clinical studies | PBPK | Allometry exponent: 0.75 | Allometry: age‐dependent exponent | Population PK | PBPK | Allometry exponent: 0.75 | Population PK |
| 0.8–1.25 | All (< 2 years, 30; > 2 years, 29) | 37% | 17% | 30% | 52% | 69% | ||
| 0.5–2.0 | All (< 2 years, 30; > 2 years, 29) | 67% | 47% | 67% | 93% | 90% | ||
| 0.8–1.25 | Subset of studies with popPK interpolation, extrapolation (< 2 years, 18; > 2 years, 13) | 33% | 11% | 38% | 28% | 62% | 77% | 62% |
| 0.5–2.0 | Subset of studies with popPK interpolation, extrapolation (< 2 years, 18; > 2 years, 13) | 67% | 33% | 72% | 44% | 100% | 85% | 85% |
| 0.8–1.25 | Subset of studies with popPK‐only interpolation (< 2 years, 11; > 2 years, 10) | 23% | 15% | 27% | 36% | 58% | 75% | 60% |
| 0.5–2.0 | Subset of studies with popPK‐only interpolation (< 2 years, 11; > 2 years, 10) | 54% | 31% | 82% | 64% | 100% | 83% | 90% |
| Cmax ratio | ||||||||
| 0.8–1.25 | All PO studies (8, < 2 years; 14, > 2 years) | 25% | 64% | |||||
| 0.5–2.0 | All PO studies (8, < 2 years; 14, > 2 years) | 88% | 93% | |||||
The percentage values in the table refers to the percentage of clinical studies for which the predictions of pediatric to adult clearance ratio meet the performance criteria.PBPK, physiologically‐based pharmacokinetic model; PK, pharmacokinetics; PO, oral administration; popPK, population pharmacokinetics; Cmax, maximum plasma concentration follwing oral drug administration.
Discussion
A paucity of observed pediatric PK data have long been a barrier to validation efforts. With regulatory agencies mandating pediatric evaluations, more and more data are being generated. Literature reports of PBPK model evaluation for pediatric exposure predictions generally employ a bottom‐up PBPK approach,8, 15 which relies on a good recovery of the in vivo CL in the base population from in vitro intrinsic CL measured in recombinant systems. In the absence of an in vitro–in vivo correlation, a middle‐out approach may have to be employed. There are a few comparative evaluations in the literature that use a top‐down method of using adult CL to extrapolate to pediatric CL. The top‐down method used in this analysis has the advantages of not relying on in vitro–in vivo correlation, which is likely to be poor for non‐CYP pathways. Both methods assume that the elimination pathways of a drug are the same across adult and pediatric populations. To further reduce the reliance on in vitro data, most (10/13) compounds chosen for this study had one major elimination or metabolic pathway. To better characterize the elimination pathways, the selection criteria required availability of drug exposure in adults following i.v. administration. This selection criteria requirement limited the number of compounds to 13. Only 7 of these 13 compounds had oral administration (PO) data. Of these, the use of the advanced dissolution, absorption, and metabolism model was restricted to the subset of four compounds with less than complete gut bioavailability (Table S6), because formulation differences, changes in gut physiology and intestinal loss are likely to impact only these four drugs.
The volume of distribution in children younger than 2 years of age is likely to be higher in neonates and infants than in adults for hydrophilic drugs, whereas the reverse is true for lipophilic drugs. This is a result of the high total body water in neonates and infants when compared with adults (Table S1). In this analysis, 11 of the 13 compounds had volume of distribution data in both adult and pediatric populations. As might be expected, the hydrophilic compounds amikacin and caffeine (but not theophylline) had up to two‐ to threefold higher volume in neonates when compared with adults. The most lipophilic compounds (montelukast, itraconazole, and diclofenac) also showed large variations among the different populations. Contrary to expectations, there was no trend with age. These compounds generally had a high volume of distribution and high associated variability between adult and pediatric subpopulations. The volume of distribution of the remaining six drugs was comparable between adults and children. Therefore, the observed volume of distribution in adults was used as input for the pediatric simulations. This may lead to a poor simulation of the shape of the PK profile and impact the Cmax predictions in the pediatric populations for compounds that have different volumes of distribution in the pediatric subpopulations when compared with adults. However, the alternative of use of a full PBPK model is also likely to introduce uncertainty in a drug‐development setting. For drugs metabolized by the liver, the effect of volume of distribution usually becomes apparent in < 2 months. In a fit‐for‐purpose approach, CL is used as a surrogate for the pediatric dose in this analysis, assuming the volume of distribution has little impact on dose.
The P/A ratios of body‐weight–adjusted CL were calculated for pediatric studies in all 13 compounds to allow for an easy assessment of the extent of deviation of pediatric CL from adult values. Simcyp, allometry, and popPK have been evaluated with both a strict performance criterion of 0.8–1.25 as well as the more widely used twofold criteria. By virtue of the constant exponent of 0.75 in the simple allometric equation, the body‐weight–adjusted CL in children is always expected to be higher than that of adults (P/A > 1), tending toward 1 with increasing age. Simcyp and popPK can also lead to < 1 P/A ratios and do not show any trend with age, consistent with the observed. The lack of trend should be expected given that the multiple physiological factors contributing to age‐dependent CL are not always following the same direction.
The CL in neonates for amikacin and sotalol, the two renally cleared drugs in our compound set, are better predicted with PBPK when compared with allometry. Allometry overpredicts renal CL in both drugs, as may be expected. For drugs such as amikacin, whose renal CL are exclusively determined by glomerular filtration rate, the pediatric dosing of renally cleared drugs is in fact based on markers of renal excretion (creatinine CL or p‐aminohippuric acid CL)10 for the < 2 and on body surface area for > 2.
Caffeine, ropivacaine, and theophylline are predominantly cleared by CYP1A2. In caffeine, a dramatic increase in body‐weight–adjusted CL with age in the neonates could not be predicted by PBPK, popPK, or allometry. Ropivacaine and theophylline CL following i.v. dosing were well predicted for all age groups except for the < 1‐month age group. Despite the good prediction of i.v. CL of theophylline in children aged 1 to 5 years, the much higher oral CL and the consequent lower Cmax in the same age group was not predicted in PBPK, suggesting a greater role for intestinal loss in children when compared with adults.
For the three compounds (midazolam, caffeine, and theophylline) for which clinical studies in the premature were available, Simcyp predicts a 100% renal CL. Unlike the premature population, there are no dramatic shifts in the relative contribution of pathways to the overall CL of drugs in other populations.
The pediatric CL for the CYP3A substrate bosentan (PO), CYP2C9 substrate diclofenac (i.v. and PO), UGT2B7 substrate lorazepam (i.v.), and CYP2C8 substrate montelukast (i.v.) were well predicted by all methods in all subpopulations. Allometry and PBPK underpredict the CL of itraconazole (i.v.) in all pediatric subpopulations. This underprediction is more pronounced for infants as reported in the literature.23 The poor prediction of clindamycin i.v. CL in neonates and infants by all three methods is not understood. For montelukast (PO), predominantly cleared by CYP2C8, although both PBPK and allometry show little difference in oral CL with age in infants, there are dramatic changes with age observed in this population. PBPK models are most informative when supported by empirical data. For highly bound compounds, the use of protein binding measured in the different subpopulations may improve predictions.
Overall, it may be said that PBPK, allometry with ADEs, and popPK have comparable performance, consistent with what has been reported previously.6, 13, 20 However, in clinical studies where pediatric popPK models were employed for both interpolations and extrapolations, PBPK and allometry with ADEs appear to be better than popPK. However, in clinical studies where pediatric popPK models were employed for interpolations alone, pediatric popPK outperforms PBPK with respect to both criteria and allometry with ADEs with respect to the stringent criteria. It should be remembered that although the base population for the popPK approach is always children of different age groups, the base population for PBPK and allometry with ADEs in our analysis is always adult. Thus, it is fair to conclude that PBPK and allometry with ADEs are better for pediatric extrapolation from adult, whereas popPK is better when sufficient pediatric data are already available to allow for a meaningful interpolation. These results suggest that extrapolation from healthy adult using PBPK or allometry with ADEs can be used for the dose range selection and study design of pediatric studies. The margin of prediction errors can guide starting dose selection. Once the pediatric PK becomes available, popPK models can be built to optimize doses for different pediatric subpopulations.
Both PBPK and popPK models perform better than simple allometry, especially for children < 2 years of age. The lack of body‐weight correlation for the children aged < 2 years demonstrates that this subpopulation of children cannot be treated as small adults.5, 6, 20, 24, 25 It also suggests that the exponent of 0.75 in simple allometry cannot be universal. However, the use of age‐segmented5, 6, 20, 24, 25 exponents in allometry has the potential to considerably improve predictions as demonstrated in this analysis.
The tendency for better prediction performance for children aged > 2 when compared with the children aged < 2 years is also evident for Cmax predictions by PBPK. There appears to be a bias toward overprediction of Cmax for the clinical studies evaluated in this work, but with few clinical studies having Cmax data, conclusions are difficult to draw. Deviations in predicted CL from the observed in similar populations may be contributing to the discrepancy in Cmax. This is the case for bosentan, midazolam, montelukast, and theophylline, but it cannot explain the observations in caffeine and sotalol. Bosentan, midazolam, and theophylline are all CYP3A substrates. It is noteworthy that the former two drugs have a product of fraction absorbed (Fa) and gut bioavailability (Fg) Fa∙Fg < 1. A higher P/A for per oral clearance (CLpo) when compared with intravenous CL for midazolam and theophylline (two of the CYP3A substrates with both i.v. and PO) also suggests a greater role for intestinal loss (as a result of suboptimal absorption, gut metabolism, or efflux) in children. As pediatric formulations are generally designed to improve absorption in children, suboptimal absorption may be ruled out as a potential cause for intestinal loss.
Summary and conclusion
For children > 2 years of age, the performance of all methods evaluated (PBPK, allometry, and popPK) are comparable. However, for children aged < 2 years, the adult to pediatric extrapolation with PBPK is superior to that with simple allometry and comparable to allometry with ADEs. Adult to pediatric extrapolation with either PBPK or allometry with ADEs is superior to pediatric extrapolation using a pediatric popPK model. However, pediatric interpolation with pediatric popPK provides better predictions in the individual pediatric subpopulations. These results support the use of adult to pediatric extrapolation by either PBPK or allometry with ADEs to select starting dose and to design the confirmatory studies in children, the sparse data from which can be used for building a popPK model to predict exposure in different subpopulations of children < 2 years of age with better precision.
PBPK platforms generally reflect the current scientific knowledge in the biology, physiology, and pathophysiology of different pediatric age groups. Although some of the processes are well characterized, there are knowledge gaps in others (especially related to non‐CYPs and transporters) that remain to be understood. This analysis underscores the need for a better understanding of the impact of growth and maturation on drug disposition in children < 2 years of age. Filling knowledge gaps in growth and maturation trajectories, gastrointestinal fluid composition, absorptive surface area along gastrointestinal tract, development patterns for CYPs and phase II enzymes, transporter ontogeny in the gastrointestinal tract, and liver pathophysiology will allow for the full potential of PBPK models to be leveraged. The tendency for overestimating the Cmax of CYP3A substrates by PBPK highlights the need for a better understanding of gut bioavailability in children. With more data from pediatric studies expected to be generated in the coming years, a collective experience on pediatric PK will provide an opportunity for improved PBPK models in the future. Extensive and continuous evaluations of PBPK models as and when they are updated with new knowledge will serve to enhance regulatory acceptance of PBPK approach for prospective predictions of pediatric exposure.
Funding
The research was funded by Merck KGaA, Darmstadt, Germany.
Conflict of Interest
The authors declared no competing interests for this work.
Author Contributions
Q.W. and S.A.P. wrote the manuscript; S.A.P. designed the research: Q.W. performed the research; Q.W. and S.A.P. analyzed the data.
Supporting information
Table S1. Physiological differences between adult and pediatric populations.
Table S2. Clinical pharmacokinetic studies.
Table S3. Observed and predicted pharmacokinetic parameters for all 59 pediatric studies.
Table S4. Predicted to observed ratio for clearance.
Table S5. Predicted to observed ratios for maximum plasma concentration follwing oral drug administration (Cmax.)
Table S6. Rationale for selection of Advanced Dissolution, Absorption and Metabolism (ADAM) model.
Figure S1. Plasma concentration‐time profiles from physiologically‐based pharmacokinetic simulations corresponding to all doses in adult and pediatric subpopulations along with observed profiles.
Figure S2.The barcharts of pediatric over adult ratios for both clearance and maximum plasma concentration follwing oral drug administration (Cmax) values from different methods.
Supplementary Material S1. Physiologically‐based pharmacokinetic (PBPK) model simulation result adult and pediatric.
Acknowledgments
The authors acknowledge the leadership of Quantitative Pharmacology & Drug Disposition, Merck KGaA for providing the framework and technical support of this research. Christian Lüpfert kindly supported the thorough review of the physiologically‐based pharmacokinetic simulations and calculations and Faiza Rharbaoui kindly provided useful discussions.
References
- 1. Dunne, J. et al Extrapolation of adult data and other data in pediatric drug‐development programs. Pediatrics 128, 1242–1249 (2011). [DOI] [PubMed] [Google Scholar]
- 2. US Food and Drug Administration . Exposure‐response relationships—study design, data analysis, and regulatory applications, guidance for industry. <https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm072109.pdf> (2003). Accessed April 2003.
- 3. US Food and Drug Administration . General clinical pharmacology: considerations for pediatric studies for drugs and biological products, guidance for industry. <http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM425885.pdf> (2014). Accessed December 2014.
- 4. E11 International Conference on Harmonisation (ICH) . Clinical investigation of medicinal products in the paediatric population. Note for guidance on clinical investigation of medicinal products in the paediatric population (CPMP/ICH/2711/99). <http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002926.pdf>. Accessed January 2011.
- 5. Mahmood, I. Dosing in children: a critical review of the pharmacokinetic allometric scaling and modelling approaches in paediatric drug development and clinical settings. Clin. Pharmacokinet. 53, 327–346 (2014). [DOI] [PubMed] [Google Scholar]
- 6. Mahmood, I. Prediction of drug clearance in premature and mature neonates, infants, and children ≤ 2 years of age: a comparison of the predictive performance of 4 allometric models. J. Clin. Pharmacol. 56, 733–739 (2016). [DOI] [PubMed] [Google Scholar]
- 7. Anderson, B.J. & Holford, N.H.G. Tips and traps analyzing pediatric PK data. Pediatr. Anesth. 21, 222–237 (2011). [DOI] [PubMed] [Google Scholar]
- 8. Edginton, A.N. , Schmitt, W. , Voith, B. & Willmann, S. A mechanistic approach for the scaling of clearance in children. Clin. Pharmacokinet. 45, 683–704 (2006). [DOI] [PubMed] [Google Scholar]
- 9. Thai, H.‐T. , Mazuir, F. , Cartot‐Cotton, S. & Veyrat Follet, C. Optimizing pharmacokinetic bridging studies in paediatric oncology using physiologically‐based pharmacokinetic modelling: application to docetaxel. Br. J. Clin. Pharmacol. 80, 534–547 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maharaj, A.R. , Barrett, J.S. & Edginton, A.N. A workflow example of PBPK modeling to support pediatric research and development: case study with lorazepam. AAPS J. 15, 455–464 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Khalil, F. & Läer, S. Physiologically based pharmacokinetic modeling: methodology, applications, and limitations with a focus on its role in pediatric drug development. Biomed. Res. Int. 2011, e13 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hornik, C.P. , Wu, H. , Edginton, A.N. , Watt, K. , Cohen Wolkowiez, M. & Gonzalez, D. Development of a pediatric physiologically‐based pharmacokinetic model of clindamycin using opportunistic pharmacokinetic data. Clin. Pharmacokinet. 56, 1343–1353 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jorga, K. et al Bottom‐up meets top‐down: complementary physiologically based pharmacokinetic and population pharmacokinetic modeling for regulatory approval of a dosing algorithm of valganciclovir in very young children. Clin. Pharmacol. Ther. 100, 761–769 (2016). [DOI] [PubMed] [Google Scholar]
- 14. Han, K. et al Bevacizumab dosing strategy in paediatric cancer patients based on population pharmacokinetic analysis with external validation. Br. J. Clin. Pharmacol. 81, 148–160 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johnson, T.N. , Rostami‐Hodjegan, A. & Tucker, G.T. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin. Pharmacokinet. 45, 931–956 (2006). [DOI] [PubMed] [Google Scholar]
- 16. Templeton, I.E. , Jones, N.S. & Musib, L. Pediatric dose selection and utility of PBPK in determining dose. AAPS J. 20, e31 (2018). [DOI] [PubMed] [Google Scholar]
- 17. Zhou, W. et al Predictive performance of physiologically based pharmacokinetic (PBPK) modeling of drugs extensively metabolized by major cytochrome p450s in children. Clin. Pharmacol. Ther. 104, 188–200 (2018). [DOI] [PubMed] [Google Scholar]
- 18. Yellepeddi, V. , Rower, J. , Liu, X. , Kumar, S. , Rashid, J. & Sherwin, C.M.T. State‐of‐the‐art review on physiologically based pharmacokinetic modeling in pediatric drug development. Clin. Pharmacokinet. 58, 1–13 (2018). [DOI] [PubMed] [Google Scholar]
- 19. Leong, R. et al Regulatory experience with physiologically based pharmacokinetic modeling for pediatric drug trials. Clin. Pharmacol. Ther. 91, 926–931 (2012). [DOI] [PubMed] [Google Scholar]
- 20. Mahmood, I. & Tegenge, M.A. A comparative study between allometric scaling and physiologically based pharmacokinetic modeling for the prediction of drug clearance from neonates to adolescents. J. Clin. Pharmacol. 59, 189‐197 (2018). [DOI] [PubMed] [Google Scholar]
- 21. Agoram, B. , Woltosz, W.S. & Bolger, M.B. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv. Drug Deliv. Rev. 50, S41–S67 (2001). [DOI] [PubMed] [Google Scholar]
- 22. Holford, N.H.G. A size standard for pharmacokinetics. Clin. Pharmacokinet. 30, 329–332 (1996). [DOI] [PubMed] [Google Scholar]
- 23. Berger, R.M.F. et al A bosentan pharmacokinetic study to investigate dosing regimens in paediatric patients with pulmonary arterial hypertension: Future‐3. Br. J. Clin. Pharmacol. 83, 1734–1744 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou, W. et al Predictive performance of physiologically based pharmacokinetic and population pharmacokinetic modeling of renally cleared drugs in children. CPT: Pharmacomet. Syst. Pharmacol. 5, 475–483 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mahmood, I. , Staschen, C.M. & Goteti, K. Prediction of drug clearance in children: an evaluation of the predictive performance of several models. AAPS J. 16, 1334–1343 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krupa, A. , Majda, D. , Mozgawa, W. , Szlęk, J. & Jachowicz, R. Physicochemical properties of bosentan and selected pde‐5 inhibitors in the design of drugs for rare diseases. AAPS PharmSciTech. 18, 1318–1331 (2017). [DOI] [PubMed] [Google Scholar]
- 27. Dingemanse, J. & van Giersbergen, P.L.M. Clinical pharmacology of bosentan, a dual endothelin receptor antagonist. Clin. Pharmacokinet. 43, 1089–1115 (2004). [DOI] [PubMed] [Google Scholar]
- 28. Leigh, D.A. Antibacterial activity and pharmacokinetics of clindamycin. J. Antimicrob. Chemother. 7, 3–9 (1981). [DOI] [PubMed] [Google Scholar]
- 29. Bort, R. , Macé, K. , Boobis, A. , Gómez‐Lechón, M.‐J. , Pfeifer, A. & Castell, J. Hepatic metabolism of diclofenac: role of human CYP in the minor oxidative pathways. Biochem. Pharmacol. 58, 787–796 (1999). [DOI] [PubMed] [Google Scholar]
- 30. Guhmann, M. et al Design of biorelevant test setups for the prediction of diclofenac in vivo features after oral administration. Pharm. Res. 30, 1483–1501 (2013). [DOI] [PubMed] [Google Scholar]
- 31. Chen, Y. et al Best practices in design of clinical drug‐drug interaction studies with itraconazole using a mechanistic PBPK modeling approach. CPT Pharmacometrics Syst. Pharmacol. (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Okumu, A. , DiMaso, M. & Löbenberg, R. Dynamic dissolution testing to establish in vitro/in vivo correlations for montelukast sodium, a poorly soluble drug. Pharm. Res. 25, 2778–2785 (2008). [DOI] [PubMed] [Google Scholar]
- 33. Sakai, T. et al Cyp2c19 genotype and pharmacokinetics of three proton pump inhibitors in healthy subjects. Pharm. Res. 18, 721–727 (2001). [DOI] [PubMed] [Google Scholar]
- 34. Chiba, M. , Xu, X. , Nishime, J.A. , Balani, S. K. & Lin, J.H. Hepatic microsomal metabolism of montelukast, a potent leukotriene d4 receptor antagonist, in humans. Drug Metab. Dispos. 25, 1022–1031 (1997). [PubMed] [Google Scholar]
- 35. Salem, F. , Johnson, T.N. , Abduljalil, K. , Tucker, G.T. & Rostami‐Hodjegan, A. A re‐evaluation and validation of ontogeny functions for cytochrome p450 1a2 and 3a4 based on in vivo data. Clin. Pharmacokinet. 53, 625–636 (2014). [DOI] [PubMed] [Google Scholar]
- 36. Aarons, L. , Sadler, B. , Pitsiu, M. , Sjövall, J. , Henriksson, J. & Molnar, V. Population pharmacokinetic analysis of ropivacaine and its metabolite 2’, 6’‐pipecoloxylidide from pooled data in neonates, infants, and children. Br. J. Anaesth. 107, 409–424 (2011). [DOI] [PubMed] [Google Scholar]
- 37. Sherwin, C.M.T. et al Amikacin population pharmacokinetics among paediatric burn patients. Burns 40, 311–318 (2014). [DOI] [PubMed] [Google Scholar]
- 38. Falcao, A.C. et al Population pharmacokinetics of caffeine in premature neonates. Eur. J. Clin. Pharmacol. 52, 211–217 (1997). [DOI] [PubMed] [Google Scholar]
- 39. Thomson, A.H. , Kerr, S. & Wright, S. Population pharmacokinetics of caffeine in neonates and young infants. Ther. Drug Monit. 18, 245–253 (1996). [DOI] [PubMed] [Google Scholar]
- 40. Gonzalez, D. et al Use of opportunistic clinical data and a population pharmacokinetic model to support dosing of clindamycin for premature infants to adolescents. Clin. Pharmacol. Ther. 96, 429–437 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Capparelli, E.V. et al Use of pediatric and adult midazolam population pharmacokinetics to assess IM dosing and early drug exposure for status epilepticus. <https://www.pediatrictrials.org/wp-content/uploads/2014/11/Capparelli_MDZ_PAS-2016v2.pdf>.
- 42. Ramakrishnan, R. , Migoya, E. & Knorr, B. A population pharmacokinetic model for montelukast disposition in adults and children. Pharm. Res. 22, 532–540 (2005). [DOI] [PubMed] [Google Scholar]
- 43. Knorr, B. et al Montelukast dose selection in children ages 2 to 5 years: comparison of population pharmacokinetics between children and adults. Clin. Pharm. 41, 612–619 (2001). [DOI] [PubMed] [Google Scholar]
- 44. Shi, J. , Ludden, T.M. , Melikian, A.P. , Gastonguay, M.R. & Hinderling, P.H. Population pharmacokinetics and pharmacodynamics of sotalol in pediatric patients with supraventricular or ventricular tachyarrhythmia. J. Pharmacokinet. Pharmacodyn. 28, 555–575 (2001). [DOI] [PubMed] [Google Scholar]
- 45. Moore, E.S. , Faix, R.G. , Banagale, R.C. & Grasela, T.H. The population pharmacokinetics of theophylline in neonates and young infants. Pharmacokinet. Biopharmaceut. 17, 47–66 (1989). [DOI] [PubMed] [Google Scholar]
- 46. Driscoll, M.S. , Ludden, T.M. , Casto, D.T. & Littlefield, L.C. Evaluation of theophylline pharmacokinetics in a pediatric population using mixed effects models. Pharmacokinet. Biopharmaceut. 17, 141–168 (1989). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Physiological differences between adult and pediatric populations.
Table S2. Clinical pharmacokinetic studies.
Table S3. Observed and predicted pharmacokinetic parameters for all 59 pediatric studies.
Table S4. Predicted to observed ratio for clearance.
Table S5. Predicted to observed ratios for maximum plasma concentration follwing oral drug administration (Cmax.)
Table S6. Rationale for selection of Advanced Dissolution, Absorption and Metabolism (ADAM) model.
Figure S1. Plasma concentration‐time profiles from physiologically‐based pharmacokinetic simulations corresponding to all doses in adult and pediatric subpopulations along with observed profiles.
Figure S2.The barcharts of pediatric over adult ratios for both clearance and maximum plasma concentration follwing oral drug administration (Cmax) values from different methods.
Supplementary Material S1. Physiologically‐based pharmacokinetic (PBPK) model simulation result adult and pediatric.