

Abstract
eEF-2K is a potential target for treating cancer. However, potent specific inhibitors for this enzyme are lacking. Previously, we identified 2,6-diamino-4-(2-fluorophenyl)-4H-thiopyran-3,5-dicarbonitrile (DFTD) as an inhibitor of eEF-2K. In this paper, we describe its mechanism of action against eEF-2K using kinetic, mutational, and docking studies and use chemoinformatics approaches to identify a similar class of carbonitrile containing compound exhibiting the same mechanism of action. We show that DFTD behaves as a reversible covalent inhibitor of eEF-2K with a two-step mechanism of inhibition - a fast initial binding step, followed by a slower reversible inactivation step. Molecular docking suggests that a nitrile group of DFTD binds within 4.5 Å of the active site Cys-146 to form a reversible thioimidate adduct. As Cys-146 is not conserved among other related kinases, targeting this residue holds promise for the development of selective covalent inhibitors of eEF-2K.
Keywords: Cancer, covalent inhibitor, drug discovery, eEF-2 kinase, nitrile
Table of Contents
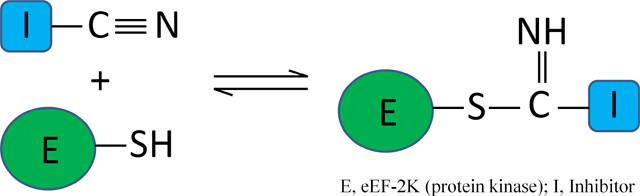
Reversible covalent kinase inhibitors. We identify a mechanism by which a non-conserved cysteine residue in the active site of eEF-2K (E) can be selectively targeted by a class of nitrile containing compounds (I), resulting in the reversible covalent inhibition of the kinase. This novel mechanism holds promise for the selective covalent drug discovery for eEF-2K.
Introduction
Elongation factor 2 kinase (eEF-2K) belongs to a sub-family of atypical protein kinases termed the alpha kinases of which there are six in humans.[1] eEF-2K plays a key role in maintaining cellular homeostasis by impeding the rate of protein synthesis, and has been implicated in enhancing tumor cell survival and proliferation. Transient inhibition of protein synthesis has been reported to induce the expression of proto-oncogenes, stimulating resting cells to enter the cell cycle.[2] Interestingly, breast cancer mitogens up-regulate the activity of eEF-2K in human breast cancer, which has been suggested to promote cell proliferation.[3] Recently, Leprivier et. al. showed that eEF-2K protects cells from nutrient deprivation and confers tumor cell adaptation to metabolic stress.[4] In addition, several other reports have suggested eEF-2K as a therapeutic target for treating cancer[5] and depression.[6]
Alpha kinases, which are Ser/Thr specific kinases, bear little sequence similarity to conventional protein kinases, however their catalytic domains exhibit similarities in structure, particularly exemplified by a common catalytic mechanism.[1, 7] Despite eEF-2K being identified as a therapeutic target, no effective approach to alter its activity in vivo has been reported to date. While A-484954 shows moderate potency in cells,[8] other attempts to inhibit eEF-2K activity have been largely unsuccessful.[9] A previous report that NH125 inhibits eEF-2K with high specificity and potency is not supported by recent data.[8, 10]
Recently, we screened recombinant eEF-2K against several collections of small molecules and identified the 2,6-diamino-4-(2-fluorophenyl)-4H-thiopyran-3,5-dicarbonitrile (DFTD) as a time-dependent inhibitor of eEF-2K. Here, we provide kinetic and modeling evidence to support a mechanism of reversible covalent targeting of Cys-146 (a non-conserved cysteine residue located at the active site of eEF-2K) by a nitrile moiety of the compound. Furthermore, we confirm the reversible covalent inhibition mechanism using 4-(3-methoxyphenyl)-2,6-dioxo-3,5-piperidinedicarbonitrile (MDP), a related class of compound containing a similarly appended nitrile group. This work provides the basis for the structure-based design of more potent covalent inhibitors of eEF-2K.
Results
Time dependent inhibition and reversibility
Recently, we reported the expression and purification of recombinant eEF-2K.[11] We determined its kinetic mechanism using a peptide substrate (Acetyl-RKKYKFNEDTERRRFL-amide)[10] and demonstrated that it undergoes rapid autophosphorylation upon incubation with calcium, calmodulin, and MgATP on Thr-348, leading to its activation.[12] Using a similar approach, a high-throughput screen was performed and DFTD (Figure 1A) was identified as an inhibitor of eEF-2K.[13]
Figure 1.

2,6-diamino-4-(2-fluorophenyl)-4H-thiopyran-3,5-dicarbonitrile
To assess the mechanism of inhibition of eEF-2K by DFTD, kinetic assays were performed, where eEF-2K was incubated with DFTD before the addition of both peptide substrate and MgATP. Under all experimental conditions, the rate of product appearance was found to be linear with time, indicating that autophosphorylation is not rate-limiting in the assays. Interestingly, it was noted that the extent of the observed inhibition, as determined by an apparent IC50 increased with the length of incubation (IC50 = 110 μM and 60 μM with 10 and 30 mintute incubations, respectively, at 100 μM ATP).
To examine time-dependent behavior, eEF-2K (100 nM) was incubated in the presence of different concentrations of DFTD for various lengths of time before initiating the reaction by diluting the enzyme inhibitor solution fifty-fold into the assay solution containing calmodulin and the substrates. Under these conditions, loosely bound inhibitor is expected to dissociate from the enzyme and so any loss in observed activity is presumed to be due to tightly bound inhibitor. The data were assessed according to a two-step model for time-dependent inhibition (Figure 2A).
Figure 2.

A) Schematic model of a two step inhibition mechanism. B) Time-dependent inhibition by DFTD. eEF-2K (100 nM) was incubated with different inhibitor concentrations (●, 500 μM; ▼, 250 μM; ▲, 125 μM; ◆, 62.5 μM; ■, 31.25 μM; and ◯, 0 μM) for different amounts of time (1, 5, 10, 20, 30, 60 and 120 min). The E+I mix was then diluted 50-fold into the assay reaction. The formation of the E•I* complex versus incubation time were fitted using KinTek Global Explorer for each inhibitor concentration. C) Reversibility of DFTD binding. eEF-2K (100 nM) was incubated with 250 μM inhibitor for 1 hour. The E+I mix was diluted 50-fold and incubated for several timepoints (0, 5, 10, 20, 30, 45, and 60 min) before starting the assay. Loss of the E•I* complex with time following dilution was fitted using KinTek Global Explorer.
According to this model, I binds rapidly to eEF-2K (step 1) to form E•I and this is followed by a second slower step (step 2), which may be reversible, to form E•I*. If we presume that E•I* is incapable of phosphorylating the peptide substrate with an appreciable rate, the concentration of E•I* may be calculated from the observed rate. Figure 2B shows the calculated concentration of E•I* after various times of incubating eEF-2K with different concentrations of DFTD. To examine whether E•I* formation is reversible, eEF-2K (100 nM) was incubated with DFTD (250 μM) for 1 hour to achieve formation of E•I*. If the formation of E•I* is reversible, the activity should be recovered according to Figure 2A. Under the conditions of the experiment, the equilibrium strongly favors the dissociation of I from E•I, thus the activity of the enzyme may be used to calculate the concentration of E•I* remaining. Figure 2C shows the loss of E•I* at different times following dilution.
To determine the kinetic parameters, the time-dependent and recovery experiments were fitted globally using KinTek Global Explorer (Figures 2B and 2C). DFTD demonstrates a fast initial binding step followed by a slower inactivation step (k2 = 0.0017 s−1). DFTD also demonstrates slow reversibility. Under the conditions of the reaction, the disappearance of E•I* is described by k-2. The best fit of the data for a first order reaction gives a value for k-2 of 0.0006 s−1, which corresponds to a half-life of approximately 19 minutes. A global fit of the data in Figures 2B and 2C furnished a Kd (= k-1/k1) of 140 μM and a slow equilibrium on the enzyme of 2.8 (=k2/k-2, stoichiometric). Thus, at saturation approximately 75% of the enzyme is covalently bound.
To determine if DFTD binds to the ATP binding site of eEF-2K, experiments were performed to determine whether incubation with 4 mM ADP (Ki ≅ 1 mM) impedes formation of E•I*. Experiments were performed in an identical manner to those described above. eEF-2K (100 nM) was incubated with DFTD (0 or 250 μM) in the presence of ADP (0 or 4 mM) for various lengths of time and then the reactions were initiated by diluting the enzyme-inhibitor-ADP solution fifty-fold into a buffer containing calmodulin and the substrates. As shown in Figure 3A, the presence of ADP significantly decreased the rate of formation of E•I*, consistent with the notion that ADP competes with DFTD for binding to the active site of eEF-2K. Together, the time dependent inactivation, reversibility, and the ADP competition assays suggested the likelyhood of covalent inhibition at the active site of the enzyme by DFTD.
Figure 3.

A) Protection with ADP against DFTD. eEF-2K was incubated in the presence of ADP (4 mM) and/or DFTD (250 μM) for different timepoints (0, 5, 10, 15, 20, and 30 min). The enzyme-inhibitor mixtures were diluted 50 fold into the assay reaction. The formation of E•I* complex in the presence (■) or absence (●) of ADP were plotted against different incubation times. Data were fitted using Equation 1, where the kobs for the formation of the E•I* complex in the presence and absence of ADP are 0.0002 ± 0.00003 and 0.0015 ± 0.0003 s−1. B) Recovery of enzyme activity against DFTD for different mutants (◆,C146A; ■, C146S; ▲, Y236F; ●, wildtype). Wildtype and mutant eEF-2K were first incubated with DFTD (1 mM) for 60 min, diluted 50 fold, and incubated for different timepoints before starting the assay. The recovery of enzyme activity upon dilution over time were fitted using Equation 1.
Docking and Mutational Studies
To gain further understanding of the mechanism of inhibition of eEF-2K by DFTD, we decided to model it into the active site of eEF-2K. The structure of eEF-2K has not been reported. However, a sequence alignment between eEF-2K and two related alpha kinases MHCK A and TRPM7, whose structures are known, reveals a 34 and 28 percent sequence similarity, respectively within the catalytic domain (Figure 4A). We constructed a homology model of eEF-2K based on these two structures according to the methods explained under Experimental Section. The crystal strucure of MHCK A and the homology model of eEF-2K are shown in Figure 4B and their alignment are shown in Figure 4C. The two structures are well-aligned, indicating good confidence in the accuracy of the predicted homology model. The eEF-2K homology model structure was further confirmed by docking ADP onto the active site, which fitted well into the active site pocket and formed H-bonds similar to that of the ADP-MHCK A complex.
Figure 4.

A) Sequence alignment of human eEF-2K with MHCK A and TRPM7. B) Crystal structure of MHCK A and homology model of eEF-2K constructed by using the crystal structures of MHCK A and TRPM7 by molecular simulation. The sequence alignments in (A) and corresponding structures in (B) are colored by sequence similarity using the BLOSUM 30 matrix and a red-white-blue color scale, with most conserved residues shown in blue. C) Superimposition of human eEF-2K homology model (orange) with the crystal structure of MHCK A (green).
When DFTD was docked to the active site, it was predicted to place a nitrile group in proximity of Cys-146 of eEF-2K. Estimates from the modeling place the sulfur atom of Cys-146 4.5 Å from the carbon atom of the nitrile (Figure 5). Nitriles are reported as inhibitors of cysteine proteases such as papain and cysteine cathepsins.[14] These enzymes contain a conserved cysteine residue which adds reversibly to the nitriles,[14] suggesting the possibility of the nitrile group of DFTD forming a covalent bond to eEF-2K through Cys-146. In addition to Cys-146, a few other nucleophilic residues (D284, K170, and Y236) were also present in the vicinity of the active site (Figure 5) that could possibly bind to DFTD. Therefore, we examined the ability of DFTD to inhibit a number of these mutants of eEF-2K.
Figure 5.

Molecular docking of ligand DFTD onto the active site of the eEF-2K homology model. A) Surface representation and B) Ribbon diagram representation. The distance from the carbon atom of the nitrile group of the compound to the possible nucleophilic residues are as follows: Cys 146, 4.5 Å; Lys 170, 3.5 Å; Asp 284, 4.9 Å; Tyr 236, 3.9 Å. Yellow represents the G-loop region, whereas the green represents the hinge region.
Mutation of D284 and K170 resulted in an inactive enzyme, and hence the time-dependent activity of these mutants could not be performed. However, the sequence alignment of eEF-2K with TRPM7 and MHCK A (Figure 4A) shows that both of these residues are conserved and are important for the catalytic activity of eEF-2K.
However, the cysteine and tyrosine mutants retained some activity allowing us to test the potential for inactivation by DFTD. If the inactivation is occurring through nucleophilic attack of the cysteine or tyrosine, then the mutation of these residues is expected to abrogate the time dependent inactivation by DFTD.
As a similar result can also be obtained due to the DFTD not binding to the mutated enzyme in the first place, we first tested the binding of DFTD to the cysteine or tyrosine mutants in a dose response assay at different concentrations of DFTD, following a 30 minute pre-incubation of the enzyme with the inhibitor. Under these conditions, DFTD inhibits C146A and C146S with IC50s of 890 and 655 μM, respectively (data not shown), suggesting that the cysteine may contribute to binding. Tyr-236 does not appear to contribute strongly to the binding of DFTD as the Tyr to Phe mutant was inhibited with an IC50 of 105 μM (data not shown), which is comparable to wild type eEF-2K. Significantly, inhibition of C146A or C146S by DFTD exhibited no time-dependence, in contrast to wildtype eEF-2K. Thus, when 1 mM of DFTD was incubated with the cysteine mutants for 1 hour and rapidly diluted, maximal activity was attained immediately (Figure 3B). In contrast, the tyrosine mutant. as well as the wild type enzyme exhibited slow recovery of activity (Figure 3B). These data are consistent with a mechanism where Cys-146 reacts with the nitrile group of DFTD to form a reversible thioimidate adduct.
Finally, to further eliminate any possibility of non-specific interaction of DFTD toward the thiols, we tested the ability of DFTD to inactivate eEF-2K in the presence of glutathione. When DFTD (250 μM) was pre-incubated in the presence of 2.5 mM glutathione for 1 hour, it had no significant effect on the inactivation or the reversibility rate (data not shown). This supports the notion that the interaction is promoted by the binding of DFTD to the active site and is selective for Cys-146 of eEF-2K.
Lastly, to confirm the selectivity of DFTD towards eEF-2K, we also performed kinase assays against TRPM7 (a related kinase) and ERK2 (an unrelated kinase) after incubating each enzyme with DFTD for 1 hour. DFTD exhibited an IC50 of 500 μM against TRPM7 and almost no inhibition (IC50 >4mM) against ERK2 (data not shown).
Derivatives identified from virtual screening show a similar mechanism
To further assess the potential for the selective targeting of cysteine by an appended nitrile group, we performed a virtual screen of the NCI diversity set using DFTD as the query compound. The virtual screen protocol is outlined in Figure 6A. As both shape and electrostatic complimentarity contribute to the biological activity of small ligands, both characteristics were assessed upon screening. 81 compounds identified from the virtual screen were further evaluated in kinase assays. Six compounds containing an appended nitrile were categorized as inhibitors of eEF-2K (Figure 6B and supplementary Figure S1) exhibiting comparable potency to DFTD with an apparent IC50 range of 25–66 μM and similar shape and electrostatic complementarity to DFTD (see Figures 6C and 6D for a comparison between DFTD and MDP).
Figure 6.

A) Virtual screening outline. B) MDP, the most potent hit identified from virtual screen (IC50 = 25 ± 0.35 μM; h = 0.93 ± 0.01) C) 3D alignment of query ligand DFTD and MDP. D) Elecrostatic maps comparison of query ligand DFTD and MDP. Positive and negative charges are shown in blue and red respectively.
An authentic sample of MDP was purchased and further assessed for time-dependent inhibition by incubating with eEF-2K for a varying length of time, to reveal a similar time-dependent inactivation (Figure 7A) to that of DFTD. Moreover with MDP, activity is recovered with an observed rate constant of 0.00056 s−1 (Figure 7B), which is almost identical to DFTD. The similarity in the kinetics of inhibition strongly supports the proposed mechanism (i.e. reversible formation of thioimidate adduct), which is summarized in Figure 8.
Figure 7.

A) Time dependent inhibition by MDP. eEF-2K was incubated with MDP (500 μM) for different amounts of time (0, 1, 5, 10, 20, 30, 45 and 60 min). The E+I mix was diluted 50 fold into the assay reaction. Formation of the E•I* complex against incubation time was fitted using Equation 1. The kobs for the formation of E•I* complex is 0.0031 ± 0.0002 s−1. B) Recovery of enzyme activity against MDP. The enzyme was first incubated with MDP (500 μM) for 60 min, then diluted 50-fold and incubated for different timepoints, before starting the assay. The loss of the E•I* complex against incubation time after dilution was fitted using Equation 2. The kobs for the loss of the E•I* complex is 0.00056 ± 0.00003 s−1.
Figure 8.

Proposed mechanism of action for DFTD. eEF-2K-S− represents a cysteine residue located in the active site of eEF-2K.
Discussion and Conclusion
Covalent inhibitors have gained significant attention from drug developers in recent years. Covalent kinase inhibitors offer several advantages over first generation reversible ATP competitive inhibitors. First, they can achieve better potency against millimolar concentrations of ATP inside the cells as the inhibition is time-dependent. Second, with two-step inhibition mechanisms, they offer better selectivity. Third, they are less affected by kinase mutations compared to reversible ATP competitive inhibitors.
Most of the covalent inhibitors developed against protein kinases target a non-conserved cysteine residue located at the active site of the kinase. Through bioinformatics analysis of the human kinome, Zhang et. al. have shown that almost 200 different protein kinases contain a cysteine residue in the vicinity of the nucleotide binding pocket.[15] However, out of all possible cysteine conformations in the kinase active site, only four positions have been targeted so far.[16] Most of the covalent inhibitors that have been developed against protein kinases such as Rsk,[17] Mek,[18] Nek2,[19] JNK,[16b] BTK,[20] EGFRs,[20] VEGFRs,[21] and FGFRs[22] are irreversible in nature. One of the major drawback of irreversible inhibitors is that they may modify off-target proteins and elucidate undesired immune response. One of the alternatives to overcome such toxicity related problems may be reversible covalent inhibitors. In a recent study, Serafimova et. al. showed that CN-NHiPr, an electron deficient cyanoacrylamide, tuned to react with cysteine thiols in a rapidly reversible manner, demonstrated similar cellular retention and better potency towards Rsk1 and Rsk2 in cellular assays compared to the known irreversible inhibitor FMK, but without the formation of an irreversible adduct.[23]
Here based on kinetic, mutational, and docking studies, we identified members of a class of dicarbonitrile containing compound as reversible, covalent inhibitors of eEF-2K. Formal elucidation of the mechanism was not possible, because its reversible nature precluded identification of the adduct using standard biophysical approaches such as mass spectrometry. However, the comparable reaction profile of MDP and DFTD, which has a similarly appended nitrile group, predicted by docking to be within 4.5 Å of Cys-146, provides strong support for the proposed mechanism where the nitrile group on these compounds target this non-conserved nucleophilic cysteine residue located at the active site of eEF-2K, forming a reversible thioimidate adduct (Figure 8). Such nitrile-thiol interactions are also widely observed in cysteine proteases such as papain and cysteine cathepsins, where the electron deficient thiol attacks a carbonyl nitrile forming a reversible thioimidate adduct, thus inactivating the enzyme.[14] Although our initial idea for the possibility of a nitrile-thiol interaction on eEF-2K came from such studies, the two mechanisms differ in a significant aspect. In the case of cysteine proteases, the active site thiol is conserved and is essential for catalysis. However, the active site cysteine in eEF-2K is not conserved among protein kinases. Therefore the mechanism is novel for protein kinases. In addition, targeting the non-conserved cysteine holds promise for the development of selective covalent inhibitors of eEF-2K.
To our knowledge, this is the first evidence of a covalent inhibitor for any atypical class of protein kinases. This is also the first time nitrile mediated reversible covalent inhibition has been identified and its mechanism extensively studied for protein kinases. Our studies show that the active site thiol in eEF-2K can be covalently targeted by small molecules. Although we demonstrated it with dicarbonitrile compounds containing nitrile as a warhead group, it opens up possibilities for several other functional groups for identifying reversible as well as irreversible covalent inhibitors for eEF-2K.
Experimental Section
Reagents and equipments:
The expression and purification of eEF-2K and the synthesis of the peptide substrate (Acetyl-RKKYKFNEDTERRRFL-amide) have been reported[11] and were followed accordingly. DFTD was purchased from Chembridge corporation (catalog no. 5212007) or made authentically. While the compound from Chembridge was used in all experiments, the authentically synthesized compound was used for confirmation. MDP was obtained from Sigma (St. Louis, MO) (catalog no. CCA002401). Structures and purity for DFTD and MDP were verified by NMR (see supplementary figures S2 and S3). Resonance forms of compound MDP were also analyzed by NMR. It was found that in DMSO the majority of the compound existed in the 4-(3-methoxyphenyl)-2,6-dioxopiperidine-3,5-dicarbonitrile form (about 86%), while the remaining (about 14%) existed in the 6-hydroxy-4-(3-methoxyphenyl)-2-oxo-1,2,3,4-tetrahydropyridine-3,5-dicarbonitrile form (see supplementary Figure S4). 81 chemical compounds identified by virtual screening and used in kinase assays were obtained from The National Cancer Institute (NCI) and used without further characterization. TRPM7[24] and ERK2[25] were prepared as previously reported.
Competent cells used for DNA amplification and protein expression were provided by Novagen (Gibbstown, NJ). Yeast extract and tryptone were purchased from US biological (Swampscott, MA). IPTG and DTT were obtained from USB (Cleveland, OH). All buffer components including HEPES, Trizma base (Tris), sodium chloride, potassium chloride, EDTA, EGTA, calcium chloride, magnesium chloride, Brij-35, Triton X-100, β-mercaptoethanol, benzamidine hydrochloride, TPCK, and PMSF were of highest quality available and were purchased from Sigma (St. Louis, MO). Ni-NTA agarose was supplied by Qiagen (Santa Clarita, CA). Amersham Biosciences (Pittsburgh, PA) provided the FPLC system and the columns for purification. P81 cellulose papers were obtained from Whatman (Piscataway, NJ). ATP was purchased from Roche (Indianapolis, IN), while ADP was from MP Biomedicals (Solon, OH). Radiolabelled [ϒ−32P]-ATP was obtained from Perkin Elmer (Waltham, MA).
Site directed mutagenesis:
The cloning of human eEF-2K (Genbank accession number NM_013302) in pET32a expression vector is already described.[11] The protein contains a thirodoxin tag followed by a His6 sequence and a TEV protease cleavage site at the N-terminus. Site directed mutagenesis was performed on eEF-2K to obtain different mutants using the following primers (mutated residues in bold): C146S, 5’ - ga gca atg agg gag agc ttc cgg acg aag – 3’ (sense) and 5’ - ctt cgt ccg gaa gct ctc cct cat tgc tc – 3’ (antisense); C146A, 5’ - a gga gca atg agg gag gcc ttc cgg acg aag aag – 3’ (sense) and 5’ - ctt ctt cgt ccg gaa ggc ctc cct cat tgc tcc t – 3’ (antisense); Y236F, 5’-c tac atc gag ggc aag ttc atc aag tac aac tcc a-3’ (sense) and 5’-t gga gtt gta ctt gat gaa ctt gcc ctc gat gta g-3’ (antisense); D284N, 5’- gga gtt ggg gat ctc tac act aac cca cag atc-3’ (sense) and 5’-gat ctg tgg gtt agt gta gag atc ccc aac tcc-3’ (antisense); K170A, 5’-tcc aac tac gtg gcg gcg cgc tac atc gag cc-3’ (sense) and 5’-gg ctc gat gta gcg cgc cgc cac gta gtt gga-3’ (antisense). PCR reactions were performed in 50 μL reaction using 1× PfuUltra™ HF reaction buffer (Agilent Technologies, Inc., Santa Clara, CA), 200 μM dNTP mix, 0.2 μM of sense and antisense primers, 10 ng of DNA template, and 1U of PfuUltra™ HF polymerase (Agilent Technologies, Inc., Santa Clara, CA). The PCR conditions included initial denaturation step (95 °C for 2 min), followed by 18 PCR cycles (95 °C denaturation step for 30 sec, 58 °C primer annealing step for 1 min, and 72 °C extension step for 4 min), followed by a final elongation step at 72 °C for 10 min. The PCR reaction was further incubated at 37 °C for 1 hour in the presence of 10 units of DpnI to degrade the methylated parent strand. 1 μL of DpnI treated sample was transformed into E. coli DH5α chemical competent cells. A single colony was isolated and amplified and the extracted plasmid DNA was verified for sequencing at the ICMB core facilities, UT-Austin, using an applied Biosystems automated DNA sequencer.
Kinase assays:
All experiments were performed in 5% DMSO to improve compound solubility since 5% DMSO did not alter enzyme activity (see supplementary Figure S5). All assays were performed at 30 °C in assay buffer (25 mM HEPES pH 7.5, 50 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 2 mM DTT, 10 mM MgCl2, 40 μg/ml BSA and 5% DMSO). For IC50 measurement with DFTD against protein variations, WT or mutant eEF-2K (2 nM for WT, C146A and Y236F; 10 nM for C146S) were preincubated with various concentrations of DFTD (0–1000 μM) in assay buffer containing 150 μM CaCl2, 1 μM calmodulin, 0.01% triton X-100 and 50 μM peptide substrate for 30 minutes. Similarly, for IC50 measurement with MDP and five additional derivatives, WT eEF-2K (2 nM) was preincubated with various concentrations of inhibitors (0–1000 μM) in assay buffer containing 150 μM CaCl2, 1 μM calmodulin, 0.01% triton X-100 and 50 μM peptide substrate for 60 minutes. After the indicated time, the reactions were started by adding 100 μM [ϒ−32P]-ATP (specific activity = 1000 cpm/pmol). The product formation was monitored over 2.5 minutes by spotting 10 μl aliquots onto P81 cellulose papers at fixed time point intervals. The papers were washed with 50 mM phosphoric acid (5 times for 10 minutes each) and then dried following acetone wash. The amounts of radiolabelled phospho-peptides were determined by counting the associated counts/min on a synctillation counter (Packard 1500) at a σ value of 2.
Time dependent inhibition experiments:
Time dependent inhibition by DFTD and MDP was tested via dilution experiments. eEF-2K at a concentration of 100 nM (50-fold higher than the assay concentration) was incubated with different concentrations of inhibitor (0–500 μM) for different amounts of time in a buffer containing 25 mM HEPES pH 7.5, 40 μg/mL BSA, 2 mM DTT, and 5% DMSO. For ADP protection experiment, a similar incubation was performed with DFTD (0 or 250 μM) in the presence or absence of 4 mM ADP. Then the incubation mixtures were diluted 50-fold into the assay buffer (25 mM HEPES pH 7.5, 50 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 2 mM DTT, 10 mM MgCl2, 40 μg/ml BSA, and 5% DMSO) containing 150 μM CaCl2, 1 μM CaM, and 50 μM peptide substrate. Immediately after the dilution, the assays were started with the addition of 100 μM [ϒ−32P]-ATP (specific activity = 1000 cpm/pmol). The product formation was monitored over 2.5 minutes by spotting 10 μl aliquots into P81 cellulose papers at fixed time point intervals. The papers were washed with 50 mM phosphoric acid (5 times for 10 minutes each) and then dried following acetone wash. The amounts of radiolabelled phospho-peptides were determined by counting the associated counts/min on a synctillation counter (Packard 1500) at a σ value of 2.
Recovery experiments:
To test the wildtype enzyme recovery after inactivation by DFTD and MDP, enzyme at a concentration 50-fold above the assay concentration was incubated for 1 hour in the presence of each inhibitor (0 or 250 μM for DFTD; 0 or 500 μM for MDP) in buffer containing 25 mM HEPES pH 7.5, 40 μg/mL BSA, 2 mM DTT, and 5% DMSO. Similarly, to test the recovery of cysteine mutants against DFTD, mutant enzymes C146A, 146S, and Y236F were incubated for 1 hour in the presence of DFTD (0 or 1000 μM). After 1 hour, the enzyme and inhibitor mixtures were diluted 50-fold into the assay buffer containing 50 μM peptide substrate. The diluted enzyme and inhibitor mixtures were incubated for various times after which the assays were then started with the addition of 150 μM calcium, 1 μM calmodulin, and 100 μM [ϒ−32P]-ATP (specific activity = 1000 cpm/pmol). The product formation was monitored over 2.5 minutes by spotting 10 μL aliquots into P81 cellulose papers at fixed time point intervals. The phosphorylated counts/min were determined similarly as mentioned above.
Homology modeling:
The amino acid sequence of human eEF-2 kinase (accession number: 21618568), was retrieved from SWISSPROT.[26] Sequences and structures of the α-kinase domain of MHCK A and TRPM7 were obtained from the RSCB protein data bank at 1.6 and 2.0 resolutions (PDB ID: 3LKM[27] and 1IA9[28]). The multiple sequence alignment was performed by using the CLUSTAL W2 server (http://www.ebi.ac.uk/Tools/msa/clustalw2/). This alignment was then manually adjusted to minimize the number of gaps and insertions. Our final alignment is similar to that published by other groups.[27–28] The final sequence and structural alignments of human eEF-2K to MHCK A and TRPM7 shows about 34% and 28% of sequence identity of the kinase domain, which suggests that the most important part of the sequence is conserved. Therefore, we conclude that this alignment is suitable for the construction of a reliable 3D model for human eEF-2K. Based on the sequence alignment and X-ray crystal structure of MHCK A and TRPM7, the three-dimentional model of the human eEF-2K was built by application of homology modeling methods implemented in the program MODELER 9V5.[29] The Modeler’s variable target function method (VTFM) and MD simulated annealing[30] were used to refine 20 initially randomized models. The final models were evaluated with PROCHECK[31] and Prosa 2003.[32] The structure and sequence were visualized by using the program VMD[33] and Jalview.[34]
Shape and Electrostatic Virtual Screening:
The compound DFTD served as the query ligand. ChemDraw 3D ultra (8.0, CambridgeSoft) was used to draw and minimize a three-dimentional conformer of DFTD. Virtual screening was performed with software from OpenEye (OpenEye Scientific Software) and the chemical database NCI diversity set (http://cactus.nci.nih.gov/download/nci/). Omega[35] was used to generate up to 20 conformers of each NCI molecule. ROCS[36] was utilized for three-dimensional shape comparisons, and the 500 molecules were ranked on the basis of the combo score function, a sum of scaled color score and shape Tanimoto score. The shape Tanimoto score is given by the following: Shape Tanimoto = VC / (VA + VB – VC), where VC is the volume in common between the two molecules, VA is the total volume of the query molecule, and VB is the total volume of the target molecule. The coefficient ranges from 0 (no shape overlap) to 1 (identical shape).
The top 1000 molecules from ROCS[36] are then reranked in EON,[37] and the top 500 are passed to next step for rescoring. In EON[37], the Poisson–Boltzmann equation is solved to calculate the electrostatic potential on the nodes of a 3D grid constructed around a molecule. The molecule has MMFF94 nuclei-centered charges assigned. The OpenEye Electrostatic Tanimoto similarity is given by: Electrostatic Tanimoto = AB / (AA + BB – AB), where A is the electrostatic potential around the query molecule and B is the electrostatic potential around the target molecule. The coefficient ranges from −1/3, when two molecules have equal but opposite potentials, and 1, when two molecules have identical potentials.
In the final rescoring stage, a composite score, which weights 50% of the shape-based and 50% of the electrostatic-based similarities, were used to rerank the 500 + 500 molecules from previous steps, resulting top 125 molecules. Among them, 81 compounds were available and tested in kinase assays.
Docking:
Docking of compound to the kinase homology model was performed using Gold5.1[38] with ChemPLP fitness function.[39] Twenty independent docking attempts were conducted for the compound using generic algorithm implemented in Gold5.1. The results of each docking were ranked according to the ChemPLP fitness score and were virtually inspected. It turns out the majority (17 out of 20) of the docked poses agree with the pose shown in Figure 5. The rest three poses, which have the lowest scores among the 20 poses, suggest a pose which has the fluorine atom pointing to the opposite direction while the pose of dicarbonitrile containing ring remains the same.
Data analysis:
Two step inhibition mechanism for DFTD were fitted globally using KinTek global explorer (KinTek Corporation, Austin, TX). The formation or the loss of product over time were fitted using Equations 1 and 2 respectively. The inhibitor dose response data were fitted using Equation 3.
| (Equation 1) |
| (Equation 2) |
| (Equation 3) |
The parameters used above are as follows: P(t), amount of product at time t; Pmax, maximum product formation; P0, amount of product at t = 0; vi, observed velocity in the presence of inhibitor; v0, observed velocity in the absence of inhibitor; IC50, inhibitor concentration required to achieve 50% inhibition; h, hill coefficient. Each data point represents the mean ± standard deviation from three replicates.
Supplementary Material
Acknowledgements
This research was supported by the grants from National Institutes of Health (GM059802, GM106137 and CA167505), Welch Foundation (F-1390 & F-1691), and CPRIT (RP110532 & RP101501).
Footnotes
Supporting information for this article is available.
References
- [1].a) Drennan D; Ryazanov AG, Prog. Biophys. Mol. Biol 2004, 85, 1–32; [DOI] [PubMed] [Google Scholar]; b) Ryazanov AG, FEBS Lett. 2002, 514, 26–29. [DOI] [PubMed] [Google Scholar]
- [2].Rosenwald IB; Setkov NA; Kazakov VN; Chen JJ; Ryazanov AG; London IM; Epifanova OI, Cell Prolif. 1995, 28, 631–644. [DOI] [PubMed] [Google Scholar]
- [3].Parmer TG; Ward MD; Yurkow EJ; Vyas VH; Kearney TJ; Hait WN, Br J Cancer 1998, 79, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Leprivier G; Remke M; Rotblat B; Dubuc A; Mateo AR; Kool M; Agnihotri S; El-Naggar A; Yu B; Prakash Somasekharan S; Faubert B; Bridon G; Tognon CE; Mathers J; Thomas R; Li A; Barokas A; Kwok B; Bowden M; Smith S; Wu X; Korshunov A; Hielscher T; Northcott PA; Galpin JD; Ahern CA; Wang Y; McCabe MG; Collins VP; Jones RG; Pollak M; Delattre O; Gleave ME; Jan E; Pfister SM; Proud CG; Derry WB; Taylor MD; Sorensen PH, Cell 2013, 153, 1064–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Cheng Y; Li H; Ren X; Niu T; Hait WN; Yang J, PLoS One 2010, 5, e9715; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tekedereli I; Alpay SN; Tavares CD; Cobanoglu ZE; Kaoud TS; Sahin I; Sood AK; Lopez-Berestein G; Dalby KN; Ozpolat B, PLoS One 2012, 7, e41171; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wu H; Zhu H; Liu DX; Niu TK; Ren X; Patel R; Hait WN; Yang JM, Cancer Res. 2009, 69, 2453–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Autry AE; Adachi M; Nosyreva E; Na ES; Los MF; Cheng PF; Kavalali ET; Monteggia LM, Nature 2011, 475, 91–95; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Monteggia LM; Gideons E; Kavalali ET, Biol. Psychiatry 2013, 73, 1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ryazanov AG; Ward MD; Mendola CE; Pavur KS; Dorovkov MV; Wiedmann M; Erdjument-Bromage H; Tempst P; Parmer TG; Prostko CR; Germino FJ; Hait WN, Proc. Natl. Acad. Sci. U. S. A 1997, 94, 4884–4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chen Z; Gopalakrishnan SM; Bui MH; Soni NB; Warrior U; Johnson EF; Donnelly JB; Glaser KB, J. Biol. Chem 2011, 286, 43951–43958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gschwendt M; Kittstein W; Marks F, FEBS Lett. 1994, 338, 85–88; [DOI] [PubMed] [Google Scholar]
- b) Cho SI; Koketsu M; Ishihara H; Matsushita M; Nairn AC; Fukazawa H; Uehara Y, Biochim. Biophys. Acta 2000, 1475, 207–215; [DOI] [PubMed] [Google Scholar]; c) Arora S; Yang JM; Kinzy TG; Utsumi R; Okamoto T; Kitayama T; Ortiz PA; Hait WN, Cancer Res. 2003, 63, 6894–6899. [PubMed] [Google Scholar]
- [10].Devkota AK; Tavares CD; Warthaka M; Abramczyk O; Marshall KD; Kaoud TS; Gorgulu K; Ozpolat B; Dalby KN, Biochemistry 2012, 51, 2100–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Abramczyk O; Tavares CD; Devkota AK; Ryazanov AG; Turk BE; Riggs AF; Ozpolat B; Dalby KN, Protein Expr. Purif 2011, 79, 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tavares CDJ; O’Brien JP; Abramczyk O; Devkota AK; Shores KS; Ferguson SB; Kaoud TS; Warthaka M; Marshall KD; Keller KM; Zhang Y; Brodbelt JS; Ozpolat B; Dalby KN, Biochemistry 2012, 51, 2232–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Devkota AK; Warthaka M; Edupuganti R; Tavares CD; Johnson WH; Ozpolat B; Cho EJ; Dalby KN, J. Biomol. Screen 2014, 19, 445–452. [DOI] [PubMed] [Google Scholar]
- [14].a) Frizler M; Stirnberg M; Sisay MT; Gutschow M, Curr. Top. Med. Chem 2010, 10, 294–322; [DOI] [PubMed] [Google Scholar]; b) Hanzlik RP; Zygmunt J; Moon JB, Biochim. Biophys. Acta 1990, 1035, 62–70; [DOI] [PubMed] [Google Scholar]; c) Liang TC; Abeles RH, Arch. Biochem. Biophys 1987, 252, 626–634; [DOI] [PubMed] [Google Scholar]; d) Otto HH; Schirmeister T, Chem. Rev 1997, 97, 133–172. [DOI] [PubMed] [Google Scholar]
- [15].Zhang J; Yang PL; Gray NS, Nat. Rev. Cancer 2009, 9, 28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Leproult E; Barluenga S; Moras D; Wurtz JM; Winssinger, J. Med. Chem 2011, 54, 1347–1355; [DOI] [PubMed] [Google Scholar]; b) Zhang T; Inesta-Vaquera F; Niepel M; Zhang J; Ficarro SB; Machleidt T; Xie T; Marto JA; Kim N; Sim T; Laughlin JD; Park H; LoGrasso PV; Patricelli M; Nomanbhoy TK; Sorger PK; Alessi DR; Gray NS, Chem. Biol 2012, 19, 140–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Cohen MS; Zhang C; Shokat KM; Taunton J, Science 2005, 308, 1318–1321; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nguyen TL, Anticancer Agents Med. Chem 2008, 8, 710–716. [DOI] [PubMed] [Google Scholar]
- [18].Schirmer A; Kennedy J; Murli S; Reid R; Santi DV, Proc. Natl. Acad. Sci. U. S. A 2006, 103, 4234–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Henise JC; Taunton J, J. Med. Chem 2011, 54, 4133–4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Singh J; Petter RC; Kluge AF, Curr. Opin. Chem. Biol 2010, 14, 475–480. [DOI] [PubMed] [Google Scholar]
- [21].Barluenga S; Jogireddy R; Koripelly GK; Winssinger N, Chembiochem 2010, 11, 1692–1699. [DOI] [PubMed] [Google Scholar]
- [22].Zhou W; Hur W; McDermott U; Dutt A; Xian W; Ficarro SB; Zhang J; Sharma SV; Brugge J; Meyerson M; Settleman J; Gray NS, Chem. Biol 2010, 17, 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Serafimova IM; Pufall MA; Krishnan S; Duda K; Cohen MS; Maglathlin RL; McFarland JM; Miller RM; Frodin M; Taunton J, Nat. Chem. Biol 2012, 8, 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kaoud TS; Yan C; Mitra S; Tseng CC; Jose J; Taliaferro JM; Tuohetahuntila M; Devkota A; Sammons R; Park J; Park H; Shi Y; Hong J; Ren P; Dalby KN, ACS Med. Chem. Lett 2012, 3, 721–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kaoud TS; Devkota AK; Harris R; Rana MS; Abramczyk O; Warthaka M; Lee S; Girvin ME; Riggs AF; Dalby KN, Biochemistry 2011, 50, 4568–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Strausberg RL; Feingold EA; Grouse LH; Derge JG; Klausner RD; Collins FS; Wagner L; Shenmen CM; Schuler GD; Altschul SF; Zeeberg B; Buetow KH; Schaefer CF; Bhat NK; Hopkins RF; Jordan H; Moore T; Max SI; Wang J; Hsieh F; Diatchenko L; Marusina K; Farmer AA; Rubin GM; Hong L; Stapleton M; Soares MB; Bonaldo MF; Casavant TL; Scheetz TE; Brownstein MJ; Usdin TB; Toshiyuki S; Carninci P; Prange C; Raha SS; Loquellano NA; Peters GJ; Abramson RD; Mullahy SJ; Bosak SA; McEwan PJ; McKernan KJ; Malek JA; Gunaratne PH; Richards S; Worley KC; Hale S; Garcia AM; Gay LJ; Hulyk SW; Villalon DK; Muzny DM; Sodergren EJ; Lu XH; Gibbs RA; Fahey J; Helton E; Ketteman M; Madan A; Rodrigues S; Sanchez A; Whiting M; Madan A; Young AC; Shevchenko Y; Bouffard GG; Blakesley RW; Touchman JW; Green ED; Dickson MC; Rodriguez AC; Grimwood J; Schmutz J; Myers RM; Butterfield YSN; Kryzywinski MI; Skalska U; Smailus DE; Schnerch A; Schein JE; Jones SJM; Marra MA; Pro MGCM, Proc. Natl. Acad. Sci. U. S. A 2002, 99, 16899–16903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ye QL; Crawley SW; Yang YD; Cote GP; Jia ZC, Sci. Signal 2010, 3, ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yamaguchi H; Matsushita M; Nairn AC; Kuriyan J, Mol. Cell 2001, 7, 1047–1057. [DOI] [PubMed] [Google Scholar]
- [29].Fiser A; Do RKG; Sali A, Protein Sci. 2000, 9, 1753–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sali A; Blundell TL, J. Mol. Biol 1993, 234, 779–815. [DOI] [PubMed] [Google Scholar]
- [31].Laskowski RA; Macarthur MW; Moss DS; Thornton JM, J. Appl. Crystallogr 1993, 26, 283–291. [Google Scholar]
- [32].Sippl MJ, Proteins 1993, 17, 355–362. [DOI] [PubMed] [Google Scholar]
- [33].Humphrey W; Dalke A; Schulten K, J. Mol. Graph 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- [34].Clamp M; Cuff J; Searle SM; Barton GJ, Bioinformatics 2004, 20, 426–427. [DOI] [PubMed] [Google Scholar]
- [35].a) Omega version 2.3.2. OpenEye Scientific Software, Santa Fe, NM: http://www.eyesopen.com; [Google Scholar]; b) Hawkins PC; Nicholls A, J. Chem. Inf. Model 2012, 52, 2919–36; [DOI] [PubMed] [Google Scholar]; c) Hawkins PC; Skillman AG; Warren GL; Ellingson BA; Stahl MT, J. Chem. Inf. Model 2010, 50, 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].a) ROCS version 3.1.2. OpenEye Scientific Software, Santa Fe, NM: http://www.eyesopen.com; [Google Scholar]; b) Hawkins PC; Skillman AG; Nicholls A, J. Med. Chem 2007, 50, 74–82. [DOI] [PubMed] [Google Scholar]
- [37].EON version 2.1.0. OpenEye Scientific Software, Santa Fe, NM: http://www.eyesopen.com. [Google Scholar]
- [38].Hartshorn MJ; Verdonk ML; Chessari G; Brewerton SC; Mooij WT; Mortenson PN; Murray CW, J. Med. Chem 2007, 50, 726–741. [DOI] [PubMed] [Google Scholar]
- [39].Korb O; Stutzle T; Exner TE, J. Chem. Inf. Model 2009, 49, 84–96. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.