

Abstract
Worldwide outbreaks of enterovirus D68 (EV-D68) in 2014 and 2016 have caused serious respiratory and neurological disease. To investigate diversity, spread, and evolution of EV-D68 we performed near full-length deep sequencing in fifty-four samples obtained in Sweden during the 2014 and 2016 outbreaks. In most samples, intrapatient variability was low and dominated by rare synonymous variants, but three patients showed evidence of dual infections with distinct EV-D68 variants from the same subclade. Interpatient evolution showed a very strong temporal signal, with an evolutionary rate of 0.0039 ± 0.0001 substitutions per site and year. Phylogenetic trees reconstructed from the sequences suggest that EV-D68 was introduced into Stockholm several times during the 2016 outbreak. Putative neutralization targets in the BC and DE loops of the VP1 protein were slightly more diverse within-host and tended to undergo more frequent substitution than other genomic regions. However, evolution in these loops did not appear to have been driven the emergence of the 2016 B3-subclade directly from the 2014 B1-subclade. Instead, the most recent ancestor of both clades was dated to 2009. The study provides a comprehensive description of the intra- and interpatient evolution of EV-D68, including the first report of intrapatient diversity and dual infections. The new data along with publicly available EV-D68 sequences are included in an interactive phylodynamic analysis on nextstrain.org/enterovirus/d68 to facilitate timely EV-D68 tracking in the future.
Keywords: enterovirus, intrahost evolution, whole-genome deep sequencing, interpatient evolution
1. Introduction
Enterovirus D68 (EV-D68) has been recognized as an emerging pathogen after recent worldwide outbreaks of serious respiratory and neurological disease. The virus was discovered in 1962 in California, USA in four children with pneumonia (Schieble, Fox, and Lennette 1967). Prior to 2014, EV-D68 was reported only sporadically with a total of 699 confirmed cases in Europe, Africa, and southeast Asia between 1970 and 2013 (as reviewed by Holm-Hansen, Midgley, and Fischer 2016). But in 2014, 2,287 of cases with EV-D68 infection were reported, mainly in North America and Europe, but also in southeast Asia and Chile (Holm-Hansen, Midgley, and Fischer 2016). Following the 2014 outbreak, another wave of EV-D68 infections was observed in 2016 with reports of outbreaks in several parts of the world, including Europe (Dyrdak et al., 2016; Barnadas et al. 2017; Knoester et al., 2017; Piralla et al., 2018), USA (Messacar et al., 2017; Wang et al., 2017), Argentina (Ruggieri et al., 2017), and Taiwan (Wei et al., 2018).
Even though EV-D68 was rarely reported before the recent outbreaks, a high prevalence of neutralizing antibodies have reported in samples collected before 2014 (Vogt and Crowe 2018). This indicates that infection with EV-D68 has been common also before the outbreaks in 2014 and 2016.
The main clinical presentation of EV-D68 infection is mild to severe respiratory symptoms (Holm-Hansen, Midgley, and Fischer 2016), but in rare cases EV-D68 can also cause a poliomyelitis-like disease termed acute flaccid myelitis (Dyda et al. 2018; Messacar et al. 2018). An increase of this otherwise unusual clinical presentation coincided with the outbreak of EV-D68 in 2014. In animal studies, strains from 2014 were more virulent than the prototype Fermon strain isolated in 1969 (Hixon, Clarke, and Tyler, 2017; Hixon et al., 2017; Zhang et al., 2018a).
EV-D68, as other enteroviruses, has a genome size of ∼7.5 kb and codes for a polyprotein, which is processed into four structural (VP1-4), and seven nonstructural proteins (2A–C, 3A–D). Phylogenetically EV-D68 is divided into three Clades (A–C) (Tokarz et al. 2012), and the B clade is further subdivided into three subclades or lineages (B1–B3) (Gong et al. 2016). The 2014 and 2016 outbreaks were caused by viruses belonging to the B1 and B3 subclades, respectively (Dyrdak et al., 2016; Zhang et al., 2016; Barnadas et al., 2017; Knoester et al., 2017; Wang et al., 2017; Piralla et al., 2018; Wei et al., 2018). Similar to other RNA viruses, EV-D68 has been reported to have a high substitution rate with 6.2 × 10−3 (Tokarz et al., 2012) or 5.12 × 10−3 (Ny et al., 2017) substitutions per site per year in the highly variable VP1 gene.
Mechanisms of immune protection and immune escape for EV-D68 are not well understood. Neutralizing antibodies have been shown to be protective in animal models (Hixon, Clarke, and Tyler, 2017; Hixon et al., 2017; Dai et al. 2018; Zhang et al. 2018a,b). Based on homology with the neutralizing sites NIm-IA and NIm-IB on human rhinovirus 14 (HRV-14), regions on the BC and DE loops in EV-D68 have been proposed as putative epitopes for neutralizing antibodies (Liu et al. 2015). However, also other sites in VP1 and other proteins may be of importance. Thus, amino acid variability in regions flanking the VP1 loops were reported to correlate with differences in neutralization sensitivity between the prototype Fermon strain and strains from 2014 (Zhang et al. 2015). For coxsackie virus B4, it was shown that a deletion in the BC loop reduced the neutralizing effect of CVB4-specific antisera (McPhee et al. 1994). VP2 and VP3 may also contain neutralizing epitopes as demonstrated in early poliovirus (PV) studies (Minor et al. 1986) and as recently reviewed for the enterovirus A genus (Fang and Liu 2018). Little is known about the contribution of cellular immunity to the clearance and evolution of EV-D68 (and other enteroviruses).
Here, we report deep sequencing of near full-length genomes on the Illumina next-generation sequencing platform and analyze intra- and interpatient evolution of EV-D68 during the recent outbreaks. We show that within-host diversity is typically low, but that dual infection is not uncommon, suggesting high infection incidence during the outbreaks. To facilitate exploration of the data and future analyses, we combined the whole-genome sequences reported here with publicly available genomes and implemented an interactive visualization in nextstrain available at nextstrain.org/enterovirus/d68.
2. Materials and methods
2.1 Study population and samples
For this study we used fifty-four nasopharyngeal and five other respiratory samples from two earlier studies on EV-D68 outbreaks in Stockholm in 2014 and 2016 (Dyrdak et al. 2015, 2016). The fifty-nine samples were drawn from fifty-four patients. Six patients had been sampled in 2014, and forty-eight patients (fifty-three samples) in 2016. Thirty of the patients were female. The median age was 3.6 years (range 2 months–63 years). Five patients were sampled twice during their acute illness (0, 1, 1, 2, and 7 days apart).
All, but one, samples were submitted from hospitals in Stockholm. However, we cannot exclude the possibility that a few samples might be from patients who had been transferred for care to Stockholm from other parts of Sweden.
Details about the patient demographics are given in Supplementary Table S2.
The collection dates of the samples from the outbreak in 2016 ranged between 19 August and 13 September. The samples had tested positive for EV-D68 at the Department of Clinical Microbiology at Karolinska University Hospital, by EV-D68-specific realtime-PCR and/or partial VP4/VP2 sequencing. For details see (Dyrdak et al. 2015, 2016). A few EV-D68-positive samples from the earlier studies were not included in this study because no sample material remained or because the virus levels were very low as evaluated by the cycle threshold (Ct) values in the EV-D68 realtime PCR. The Ct values for the included samples ranged from 13.9 to 33.9 (median 22.3). Details about the study samples are given in Supplementary Table S3.
The study was reviewed and approved by the Regional Ethical Review Board in Stockholm, Sweden (registration no. 2017/1317–32).
2.2 Near full-length EV-D68 amplification
Primers were designed to cover almost the entire EV-D68 genome in four overlapping fragments (F1–F4) that were ∼2,000 base pairs long (Supplementary Table S1). An alignment of published EV-D68 genomes was used to design forward and reverse primers targeting highly conserved regions of the EV-D68 genome, with similar melting temperatures, and with minimal tendency for hairpin and primer-dimer formation. Inner nested primers for fragment F1 were designed for template quantification.
RNA was manually extracted using the RNeasy Lipid Tissue Mini Kit (Qiagen Cat. No. 74804). For each nasopharyngeal sample, 200 μl was taken and mixed with 1 ml Qiazol, 200 μl chloroform and 5 μl carrier-RNA. After centrifugation and several washing steps, each aliquot was eluted with 100 μl RNase-free water and were either stored at −70°C or used directly for PCR-amplification.
Each sample was amplified in duplicate by one-step RT-PCR for each of the four overlapping fragments. The RT-PCR mixture contained: 11 μl of RNA template, 0.5 μM of forward and reverse PCR primer, 1 ng/μl random hexamers, 1 μl Superscript III RT/Platinum Taq HiFi Enzyme mix (Invitrogen, Stockholm, Sweden), 25 μl 2× reaction mix, and RNase-free water to a total volume of 50 μl. The PCR cycling profile was: ×1:30 min at 50°C, 2 min at 94°C (cDNA synthesis); ×30:15 s at 94°C, 30 s at 50°C, 90 s at 68°C (PCR-amplification); ×1:5 min at 68°C, at 4°C (final extension).
To approximately quantify the number of input EV-D68 RNA templates for sequencing, a dilution series of the RNA (1:10, 1:100, and 1:1,000) was amplified in duplicate with nested primers for fragment F1. EV-D68 RNA templates were enumerated by the Poisson distribution formula. The outer PCR is described earlier. The nested PCR mixture contained: 2.5 μl of product from the outer PCR, 0.2 μM of forward and reverse PCR primer, 1 unit of Platinum Taq HiFi (Invitrogen, Stockholm, Sweden), 5 μl 10x reaction mix, and RNase-free water to a total volume of 50 μl. The cycling profile was: ×1:2 min at 94°C, (denaturation); ×30:15 s at 94°C, 30 s at 50°C, 90 s at 68°C (PCR-amplification); ×1:6 min at 68°C, at 6 min (final extension).
Duplicates of each fragment were pooled and purified with AGENCOURT AMPure XP PCR purification kit and quantified with Qubit assays (Q32851, Life Technologies). Purified DNA from each fragment were diluted to the same concentration, pooled and sent to the Clinical Genomics Unit at Science for Life Laboratory (SciLifeLab, Stockholm, Sweden) for sequencing.
2.3 DNA library preparation and sequencing
The DNA preparations were quantified using Quant-iTTM dsDNA High-Sensitivity Assay Kit and Tecan Spark 10 M plate reader. In total 1 μl of DNA (∼0.5–2.0 ng/μl) was used in the tagmentation reaction using Nextera chemistry (Illumina) to yield fragments >150 bp. The tagmented library underwent eleven cycles of PCR with single-end indexed primers (IDT Technologies) followed by purification using Seramag beads according to the protocol. The library was quantified using Quant-iTTM dsDNA High-Sensitivity Assay Kit and Tecan Spark 10 M plate reader and pair-end (2 × 101 bp) sequenced to a depth of 100,000–1,000,000 reads per sample on a HiSeq 2500 Illumina sequencer. Base calling and demultiplexing was done using bcl2fastq v1.87, without allowing any mismatch in the index sequence.
2.4 Quality controls experiments
Quality controls were prepared from two samples with low Ct values (SWE_027_160829 and SWE_037_160829, having Ct values 14.62 and 19.27, respectively). The samples were diluted 1:100 in a pool of virus-negative nasopharyngeal samples and stored at −70°C in 200 μl aliquots. The enterovirus RNA concentration of the samples was quantified by limiting dilution in ten replicas of ten-fold dilutions, from 1:10 to 1:100,000 with an in-house enterovirus realtime-PCR (Tiveljung-Lindell et al. 2009). Based on Poisson calculations, the samples were estimated to have 4,400 and 240 EV-D68 RNA copies per μl, respectively, prior to dilution. The two samples were RNA extracted, PCR amplified and sequenced twice in two separate runs. The number of EV-D68 RNA templates in each PCR reaction was ∼48,400 and 2,640, respectively.
2.5 Read filtering, mapping, and analysis
Sequencing reads were trimmed using TrimGalore (Martin 2011; Krueger 2015) and mapped against the sequence KX675261.1 using bwa mem (Li and Durbin 2009). We used the pysam wrapper to samtools to generate a pile-up of all reads and quantify insertions relative to the reference. Custom python scripts were used to generate consensus sequences and quantify intrasample diversity from the pile-ups. All these steps were chained using the workflow engine snakemake (Köster and Rahmann 2012). The entire pipeline is available github.com/neherlab/EV-D68_analysis_Dyrdak_2019 and uses a code from a separate project github.com/neherlab/SVVC. Commit 81068ee was used to analyze the data along with commit a2cbb35 of SVVC. To analyze linkage disequilibrium, we calculated genotype frequencies p12 at pairs of positions that were covered by a read pair more than 100-fold and calculated their correlation D12 = p12 – p1p2 (aka linkage disequilibrium). This correlation was normalized such that maximal linkage corresponds to D12 = 1 and complete repulsion to D12 = −1.
The consensus sequences for successfully sequenced samples have been deposited in GenBank (accession numbers MH674111–MH674166 and MH844544).
The scripts that analyze diversity in pile-ups and generate most of the figures of this study are available at github.com/neherlab/EV-D68_analysis_Dyrdak_2019.
2.6 Phylodynamics analysis
Sequences generated in this study were combined with sequences and metadata of all EV-D68 genomes with a length of at least 6,000 bp available on Virus Pathogen Resource (ViPR) (Pickett et al. 2012) as of 2 September 2018. The combined dataset was analyzed using nextstrain (Hadfield et al. 2018) and snakemake (Köster and Rahmann 2012). The augur pipeline was run using the aligner MAFFT (Katoh et al. 2002), the tree-builder (Nguyen et al. 2015), and the phylodynamic package TreeTime (Sagulenko, Puller, and Neher 2018). The repository detailing the analysis pipeline is available at https://github.com/neherlab/enterovirus_nextstrain. Sub-genogrouping of sequences was done using the Enterovirus Genotyping Tool Version 1.0 (Kroneman et al. 2011). The resulting analysis is visualized with auspice and is available at nextstrain.org/enterovirus/d68.
Five sequences were excluded from the phylodynamic analysis because they were deemed problematic. The following three sequences are within a few mutations from other sequences sampled several years earlier or later: EV-D68/Homo_sapiens/USA/U797/2007 is identical to strain EV-D68/Homo_sapiens/USA/C7791/2014; EV_D68/Homo_sapiens/USA/O622a/2012 is very similar to strains sampled in 2003; EV_D68/Homo_sapiens/USA/MO78/2009 is very similar to strains sampled in 2014 in the USA. Given the high evolutionary rate of enteroviruses, such stasis over many years is very unlikely (we expect about thirty changes per year) and these sequences are likely dated incorrectly. The case CA/RESP/10_786 is less clear, but the sequence is several standard deviations less diverged from the root than expected given its collection date. USA/TX/2014_19267 is likely a recombinant sequence.
Mixed effects model of evolution (MEME) analysis (Murrell et al. 2012) using the DataMonkey package (Weaver et al. 2018) was used to analyze for episodic diversifying selection.
3. Results
Fifty-four of fifty-nine samples yielded a coverage of at least 100× in all four fragments and were included in further analyses. Of the excluded samples, two did not yield sequence in any fragment (Ct values 30.99 and 33.89) and three samples lacked complete sequence in one of four fragments (Ct values 20.58, 22.95, and 32.31). All five excluded samples were from 2016. Figure 1 shows the coverage plot for a representative sample. The sequencing coverage for each sample is presented in Supplementary Table S3.
Figure 1.
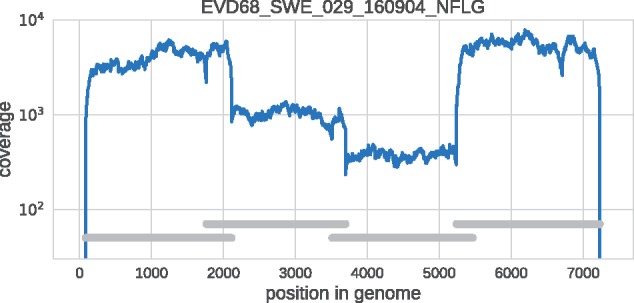
Example of coverage across the EV-D68 genome in a representative clinical sample. Note that the coverage varies somewhat in different parts of the genome due slightly different quantity of amplified DNA from the four overlapping amplicons in the material that was used for sequencing library preparation. The four overlapping are indicated by the horizontal gray bars.
3.1 Reproducibility of identification of intrasample single nucleotide variants
In addition to consensus sequences, deep population sequencing delivers information about intrasample diversity. Low genome template input, biased amplification, RT-PCR errors and sequencing errors can all potentially skew or inflate intrasample diversity estimate (Zanini et al. 2017). To control for such artifacts, we processed two high-titer patient samples in duplicate (see Section 2.4). The accuracy of the detection and quantification of intrasample single nucleotide variants (iSNVs) was evaluated by comparing results from the two runs (Fig. 2A). Minor variants down to below 1 per cent were consistently detected in both runs. Also, the estimated frequency of individual iSNVs showed good consistency between the two runs at frequencies above ∼1 per cent (Panel A), but a few iSNVs had markedly higher frequencies in one of the replicates (run 2 of sample SWE_037).
Figure 2.

Accuracy of minor variant frequencies. Panel (A) shows the consistency of minor variant frequencies across two replicate extraction, RT-PCR, and sequencing of two samples. Minor variation was relatively consistently recovered down to a frequency of about 1 per cent. Panel (B) shows the distribution of the frequencies of non-consensus calls across all sites covered in excess of 2,000-fold. At the majority of sites, no variation above 0.001 is observed. Note that this variation includes within sample variation, RT-PCR errors, and sequencing errors.
At the majority of sites, we observed background of minor variation at frequencies well below 0.1 per cent (see Fig. 2B).
Reproducibility of detection of iSNVs was also investigated in the five patients were sampled twice during their acute illness (0, 1, 1, 2, and 7 days apart). Supplementary Fig. S3 shows that frequencies of iSNVs above the 1 per cent level were largely concordant across the two consecutive samples, especially in patient SWE_037 (for whom the first sample was also sequenced twice) and less so in patient SWE_012. In patient SWE_021, with seven days between the samples, there was a tendency for an increased variation in the second sample. Unfortunately, the short time between the samples prevented us from estimating the rate of intrapatient evolution or other longitudinal aspects of intrapatient evolution of EV-D68.
Analysis of intrasample variability in all fifty-four patient samples also provided information about our ability to detect true biological variation. Figure 3A shows the distribution of sites with variation above a certain frequency cutoff among the three codon positions. The distribution was approximately even between codon sites at very low cutoffs below 10−3 and then rapidly changed around ∼0.3 per cent before plateauing at 1 per cent
Figure 3.

Within sample diversity. Panel (A) shows the distribution of the number of variable sites in coding regions among different coding positions. At very low-frequency cutoffs, variable sites are approximately equally distributed in codons. The distribution rapidly changes once the frequency cutoff exceeds 0.3 per cent, beyond which variable sites are found predominantly at third codon positions and more rarely at first and second codon positions. Panel (B) shows inverse cumulative distribution of the number of samples that have more than a certain number of sites that are variable above a level of 3 per cent. Separate distributions are shown for all sites and for first, second, and third codon position in the coding region of the genome.
Collectively, our results show that a majority of iSNVs at frequencies >1 per cent represent true biological variation, whereas variation <0.1 per cent mostly is due to amplification and sequencing errors. Between 0.1 and 1 per cent an increasing proportion of variation represents true iSNVs. The level of accuracy achieved is comparable to what we reported earlier for a similar sequencing strategy for HIV-1 (Zanini et al. 2016, 2017) and other viruses (Grubaugh et al. 2019).
3.2 Intrapatient variability
As described above intrasample variability exceeding 1 per cent likely represents true biological variation.
Approximately 60 per cent of the variable sites above this level occurred at the third codon position and around 30 and 10 per cent in the first and second codon positions, respectively (see Fig. 3A). Consequently, intrapatient variability is mostly synonymous (10, 0, and 95% of first, second, and third positions, respectively, admit synonymous mutations in our mapping reference).
Figure 3B shows that most (forty-four of fifty-four) samples had fewer than ten sites that displayed variability above a level of 3 per cent. However, three samples had more than twenty variable sites, which suggested dual infections (see below). In the remaining fifty-one samples, both synonymous and non-synonymous substitutions were mostly scattered across the genome (Fig. 4). However, there was some clustering of minor variation in first and second codon positions in the structural proteins (VP4–VP1) including putative targets for neutralizing antibodies in BC and DE loops of VP1 (Liu et al. 2015). The details about complete amino acid substitutions and minor variation in these regions are given in Supplementary Tables S4 and S5.
Figure 4.

EV-D68 intrapatient variability across the genome and codon positions. The three rows show how intrapatient variability is distributed across the EV-D68 genome for first, second, and third codon position, respectively. The panel on the left show iSNVs along the entire polypeptide, the panels on the right zoom into VP1 and highlight the BC and DE loops. This figure includes data from one sample per patient (the first sample, for patients sampled twice), but excludes the three dual infected samples (see main text).
3.3 Evidence of dual infections
As mentioned earlier, three samples (SWE_007, SWE_045, and SWE_046) drawn from three different patients showed more than twenty iSNVs. In all three samples, most minor variants were detected at a frequency around 10 per cent (Fig. 5, left). This suggested the presence of two distinct variants; a major variant constituting around 90 per cent of the virus population in the sample and a minor variant constituting around 10 per cent. To investigate the origin of these minor variants we constructed a ‘minor’ consensus sequence for each sample by exchanging the strict majority rule consensus nucleotide by the minor variant at positions where they exceeded a frequency of 1 per cent. A phylogenetic tree analysis showed that the major and minor sequences were monophyletic in all samples except the three samples with indications of dual infections. For these samples the major and minor consensus sequences occupied different positions in the tree (see Supplementary Fig. S1). To further investigate these putative dual infections we analyzed the linkage of iSNVs (Fig. 5, right). Almost all iSNVs occurred in complete linkage (LH = 1) with neighboring iSNVs, which provides strong evidence for the presence of two distinct variants, rather than rapid evolution of intrapatient variation starting from a single transmitted variant.
Figure 5.

Minor iSNVs across the EV-D68 genome for three samples with putative dual infections. These three samples had more than twenty highly covered sites with minor variants in excess of 3 per cent frequency. On the left, iSNVs are colored by codon position or as non-coding, and given opacity depending on their frequency. The majority of these variants were at third positions and had similar frequencies across the amplicons, indicated by gray bars in the lower left panel. iSNVs in three amplicons (Amplicon 1 of sample SWE_046 and Amplicons 2 and 3 of sample SWE_007), however, were found at substantially lower frequency, possibly due to primer mismatches. The three panels on the right are indexed by iSNV order on the genome and show linkage disequilibrium between iSNVs (>1%) close enough to each other that they were covered at least 100-fold by the same sequencing read. Almost all of these variants at in complete linkage (dark red). A number of iSNVs just above 1 per cent in sample SWE_045 and SWE_046 are likely variants in the background of the dominant variant. Those are in complete ‘anti-linkage’ with neighboring iSNVs are ∼10 per cent (dark blue).
3.4 Phylodynamics of EV-D68
The interpatient evolution and phylogenetic relationship of the EV-D68 variants from Sweden and 509 published EV-D68 sequences was investigated using the nextstrain platform available at https://nextstrain.org/enterovirus/d68 (Hadfield et al. 2018), which allows users to interactively explore the dataset. A screen shot is displayed in Fig. 6.
Figure 6.

Phylogenetic analysis. The left panel shows a phylogenetic tree as rendered by nextstrain with the sequences labeled by region. Those from Europe, including those from Sweden, are colored in orange. The right panel is a zoomed-in view of the boxed area on the left-hand panel colored by country and highlighting the Swedish sequences from the 2016 outbreak (orange). Swedish sequences often cluster together, but are interspersed with sequences from Canada (dark red) and the USA (red), implying multiple introductions that then spread locally.
The nextstrain phylodynamic analysis showed that all Swedish EV-D68 variants from the 2016 outbreak belonged to the B3 lineage of EV-D68, which agrees with our earlier findings based on VP2/VP4 gene sequences (Dyrdak et al. 2016). Within the B3 lineage our new Swedish sequences were interspersed with international sequences, primarily from the USA, but monophyletic clusters of Swedish sequences were also observed (Fig. 6, right). A bootstrap analysis showed that most branches were well supported (Supplementary Fig. S4).
These findings indicate that during the worldwide 2016 outbreak EV-D68 was introduced into Stockholm several times, and that these separately introduced variants thereafter spread locally.
Interestingly, the tree indicated that the 2016 B3 subclade did not evolve directly from the 2014 B1 subclade. Instead, the most recent common ancestor (MRCA) of the B1 and B3 subclades was estimated to have existed in first half of 2009 (90% CI December 2008–July 2009). Many of the proximal sequences in the B1 and B3 subclades were sampled in East Asian countries, but this should be interpreted with caution since sampling probably has been incomplete and geographically biased. A tangle-tree analysis suggested recombination within subclades, but not between clades or subclades (Supplementary Fig. S2), but also this finding should be interpreted with caution since there was limited intrasubclade variation.
3.5 Interpatient evolution
A plot of root-to-tip distances of EV-D68 sequences vs. time showed an extremely strong temporal signal (Fig. 7). We estimated an evolutionary rate of 3.8 × 10−3 ± 10−4 substitutions per site and year for the entire genome and 4.2 × 10−3 ± 4 × 10−4 for the VP1 region, which is slightly lower than previous estimates (Tokarz et al. 2012; Ny et al. 2017). Eighty-seven per cent of all substitutions were synonymous.
Figure 7.
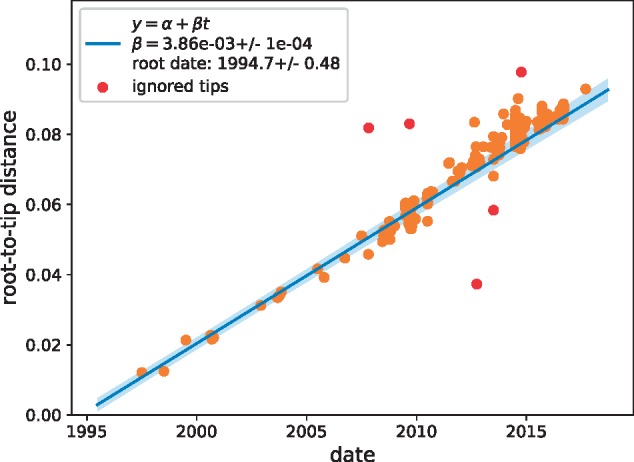
Temporal signal. A scatter plot of root-to-tip distance vs. time indicates an extremely strong temporal signal with an evolutionary rate of μ = 0.0039 ± 0.0001 substitutions per site and year.
We used nextstrain to investigate diversity across the EV-D68 genome in the sample containing the genomes reported in this study and database genomes. Ancestral sequences were reconstructed using the phylogeny in Fig. 6, and the number of transition events for each codon was counted along the tree. Figure 8 shows that amino acid substitution events were observed in all EV-D68 proteins and varied between zero and twenty-seven events per codon. The five positions with the highest number of substitution events were: codon 234 in protein VP3 (27 events), 273 in 2C (20 events), 22 in 2A (15 events), 60 in VP3 (15 events), and 277 in 2C (14 events). None of these positions have been reported to be neutralizing epitopes. Codon 277 in 2C and codon 1 in VP1 showed evidence of episodic diversifying selection as per MEME analysis (P < 0.05).
Figure 8.
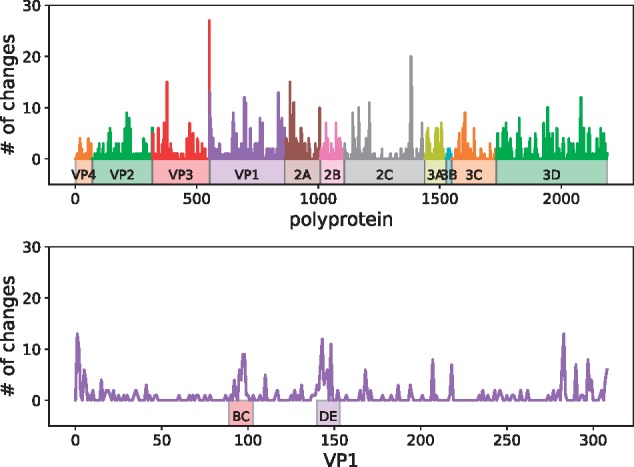
Interpatient diversity of EV-D68. Amino acid substitution events on the phylogeny in Fig. 6 left were enumerated using nextstrain. Top panel shows the number of such events for each codon along the entire coding sequence. Bottom panel shows events in VP1, where peaks were observed in the BC and DE loops, as well as both ends of the protein.
The BC and DE loops in the VP1 protein showed relatively high diversity, especially in positions 95, 97, 98, 142, 143, and 148, suggesting that immune selection may have impacted on EV-D68 evolution (see Fig. 8, bottom panel, and for details see nextstrain).
We also used nextstrain to track on which branches in the EV-D68 phylogeny amino acid substitutions had evolved and been fixated. The branches leading to the MRCA of the B1 and B3 clades had many amino acid substitutions, including substitutions at codons 90, 95, 97, 98, 145, and 148 in the BC and DE loops. In contrast, no amino acid substitutions in the loops were observed on branches separating the B1 and B3 subclades. Similarly, only very few substitutions in the loops were observed within the B1 and B3 subclades.
4. Discussion
We have performed a comprehensive investigation of the intra- and interpatient evolution of EV-D68 using new deep near full-length sequences from fifty-four Swedish patients sampled during the 2014 and 2016 outbreaks and available database sequences. The new data and previously published sequences are available on nextstrain to facilitate real-time tracking and further study of EV-D68 evolution and spread. Intrapatient variability was mostly low and dominated by rare synonymous substitutions, consistent with deep sequencing studies on influenza virus infections (Debbink et al. 2017). However, three patients showed evidence of dual infections. The phylodynamic analysis indicated EV-D68 was introduced into Stockholm several times during the 2016 outbreak and then spread locally.
Three patients showed evidence of dual infection with two different EV-D68 variants. Even though co-infections with different enteroviruses has been reported for PV and non-polio enteroviruses (Melnick et al. 1951; Parks et al. 1967; Isaacs et al. 2018), this is to our knowledge the first report of dual infections with EV-D68. Dual infections during the 2016 outbreak are not completely surprising in view of the high EV-D68 incidence during the outbreak (Dyrdak et al. 2016) and the fact that the present study showed multiple introductions of the B3 subclade into Stockholm. We also found indications of intrasubclade recombination, but not between clades or subclades. Similar findings have been reported in three recent studies (Tan et al. 2015; Ny et al. 2017; Yip et al. 2017). It is expected that recombination primarily would occur within subclades since recombination requires co-infection with two or more virus variants and EV-D68 variants that circulate during an outbreak or season tend to be closely related.
The EV-D68 genome was amplified in four overlapping amplicons that were sequenced on the Illumina HiSeq platform. Our results indicate that variation down to a frequency of 1 per cent represented ‘true’ iSNVs, which were reproducibly detected and quantified. In contrast, most variation below 0.1 per cent represented noise from cDNA synthesis, PCR and sequencing. It would be interesting to compare the performance of our sequencing protocol with the pan-enterovirus protocol recently published by (Isaacs et al. 2018). In the fifty-four samples that were sequenced to sufficient depth we observed that most nucleotides were conserved. Among iSNVs detected above the 1 per cent level, most occurred at the third codon position, that is, were usually synonymous. The finding that intrapatient variation was limited and mostly synonymous was expected since infection usually appeared to have been established by a single virion and EV-D68 is an acute infection where is limited time for the virus to diversify within the infected host.
Albeit there is a bias introduced by sampling and typing being done only at certain centras, the phylodynamic analysis showed that the splits between EV-D68 clades and subclades occurred several years ago. For instance, the MRCA of the B1 and B3 subclades was estimated to have existed in 2009. Enterovirus infections, like influenza, has a seasonal pattern. However, in contrast to influenza, which infects people of all age groups and has a ladder-like phylogenetic pattern that indicates a strong immune selection, EV-D68 infects predominantly children, and its phylogenetic pattern does not suggest continuous immune escape (Grenfell et al. 2004). In line with this, the branches leading to the B1 and B3 subclades did not have amino acid substitutions in the BC and DC loops, which are putative targets for neutralizing antibodies. However, it should be acknowledged that there is limited knowledge about targets for humoral and cellular immunity in EV-D68.
Our phylodynamic analysis suggested that the recent global outbreaks of EV-D68 might have been preceded by low-level circulation of EV-D68 in East Asia. However, this pattern should be interpreted with much caution since the number of available full-length EV-D68 sequences is low and the sequences have not been systematically collected in time or space. We hope that the nextstrain platform will allow for continuous collection and sharing of EV-D68 sequence data, which will be needed to obtain a better understanding of how new successful EV-D68 variants evolve and spread. Better knowledge about humoral and cellular immunity against EV-D68 is also needed.
Supplementary Material
Acknowledgements
We gratefully acknowledge expert sequencing service and advice by Karin Sollander, Cecilia Svensson, and Valtteri Wirta at the Clinical Genomics Unit at Science for Life Laboratory (SciLifeLab), and Lina Thebo and Eva Eriksson at the Karolinska Institute. We would also like to thank the entire nextstrain team.
Conflict of interest: None declared.
References
- Barnadas C. et al. (2017) ‘An Enhanced Enterovirus Surveillance System Allows Identification and Characterization of Rare and Emerging Respiratory Enteroviruses in Denmark, 2015-16’, Journal of Clinical Virology, 93: 40–4. [DOI] [PubMed] [Google Scholar]
- Dai W. et al. (2018) ‘A Virus-like Particle Vaccine Confers Protection against Enterovirus D68 Lethal Challenge in Mice’, Vaccine, 36: 653–9. [DOI] [PubMed] [Google Scholar]
- Debbink K. et al. (2017) ‘Vaccination Has Minimal Impact on the Intrahost Diversity of H3N2 Influenza Viruses’, PLoS Pathogens, 13: e1006194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyda A. et al. (2018) ‘The association between acute flaccid myelitis (AFM) and Enterovirus D68 (EV-D68) - what is the evidence for causation?’, Euro Surveillance, 23: doi: 10.2807/1560-7917.ES.2018.23.3.17-00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyrdak R., et al. (2016) ‘Outbreak of enterovirus D68 of the new B3 lineage in Stockholm, Sweden, August to September 2016’, Euro Surveillance, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyrdak R. et al. (2015) ‘Coexistence of Two Clades of Enterovirus D68 in Pediatric Swedish Patients in the Summer and Fall of 2014’, Infectious Diseases (London, England), 47: 734–8. [DOI] [PubMed] [Google Scholar]
- Fang C.-Y., Liu C.-C. (2018) ‘Recent development of enterovirus A vaccine candidates for the prevention of hand, foot, and mouth disease’, Expert Review of Vaccines, 17: 819--31. [DOI] [PubMed]
- Gong Y.-N. et al. (2016) ‘Molecular evolution and the global reemergence of enterovirus D68 by genome-wide analysis’, Medicine, 95: e4416. [DOI] [PMC free article] [PubMed]
- Grenfell B. T. et al. (2004) ‘Unifying the Epidemiological and Evolutionary Dynamics of Pathogens’, Science (New York, N.Y.), 303: 327–32. [DOI] [PubMed] [Google Scholar]
- Grubaugh N. D. et al. (2019) ‘An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar’, Genome Biology, 20: 8. [DOI] [PMC free article] [PubMed]
- Hadfield J. et al. (2018) ‘Nextstrain: real-time tracking of pathogen evolution’, Bioinformatics, 34: 4121–23. [DOI] [PMC free article] [PubMed]
- Hixon A. M., Clarke P., Tyler K. L. (2017) ‘Evaluating Treatment Efficacy in a Mouse Model of Enterovirus D68-Associated Paralytic Myelitis’, The Journal of Infectious Diseases, 216: 1245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hixon A. M. et al. (2017) ‘A Mouse Model of Paralytic Myelitis Caused by Enterovirus D68’, PLoS Pathogens, 13: e1006199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm-Hansen C. C., Midgley S. E., Fischer T. K. (2016) ‘Global Emergence of Enterovirus D68: A Systematic Review’, The Lancet. Infectious Diseases, 16: e64–e75. [DOI] [PubMed] [Google Scholar]
- Isaacs S. R. et al. (2018) ‘Amplification and Next Generation Sequencing of near Full-Length Human Enteroviruses for Identification and Characterisation from Clinical Samples’, Scientific Reports, 8: 11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K. et al. (2002) ‘MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform’, Nucleic Acids Research, 30: 3059–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoester M. et al. (2017) ‘Upsurge of Enterovirus D68, the Netherlands, 2016’, Emerging Infectious Diseases, 23: 140–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köster J., Rahmann S. (2012) ‘Snakemake-a Scalable Bioinformatics Workflow Engine’, Bioinformatics (Oxford, England), 28: 2520–2. [DOI] [PubMed] [Google Scholar]
- Kroneman A. et al. (2011) ‘An Automated Genotyping Tool for Enteroviruses and Noroviruses’, Journal of Clinical Virology, 51: 121–5. [DOI] [PubMed] [Google Scholar]
- Krueger F. (2015), ‘TrimGalore! A wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files’. <https://github.com/FelixKrueger/TrimGalore> accessed Aug 2018.
- Li H., Durbin R. (2009) ‘Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform’, Bioinformatics (Oxford, England), 25: 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. et al. (2015) ‘Structure and Inhibition of EV-D68, a Virus That Causes Respiratory Illness in Children’, Science (New York, N.Y.), 347: 71–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. (2011) ‘Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads’, EMBnet Journal, 17: 10–2. [Google Scholar]
- McPhee F. et al. (1994) ‘Characterization of the N-Terminal Part of the Neutralizing Antigenic Site I of Coxsackievirus B4 by Mutation Analysis of Antigen Chimeras’, Virus Research, 34: 139–51. [DOI] [PubMed] [Google Scholar]
- Melnick J. L. et al. (1951) ‘An Epidemic of Paralytic Poliomyelitis Characterized by Dual Infections with Poliomyelitis and Coxsackie Viruses’, The Journal of Experimental Medicine, 94: 471–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messacar K. et al. (2018) ‘Enterovirus D68 and acute flaccid myelitis-evaluating the evidence for causality’, The Lancet Infectious Diseases, 18: e239–47. [DOI] [PMC free article] [PubMed]
- Messacar K., et al. (2017) ‘Surveillance for Enterovirus D68 in Colorado Children Reveals Continued Circulation’, Journal of Clinical Virology, 92: 39–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor P. D. et al. (1986) ‘Antigenic Structure of Polioviruses of Serotypes 1, 2 and 3’, Journal of General Virology, 67: 1283–91. [DOI] [PubMed] [Google Scholar]
- Murrell B. et al. (2012) ‘Detecting Individual Sites Subject to Episodic Diversifying Selection’, PLoS Genetics, 8: e1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L.-T. et al. (2015) ‘IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies’, Molecular Biology and Evolution, 32: 268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ny N. T. H. et al. (2017) ‘Enterovirus D68 in Viet Nam (2009-2015)’, Wellcome Open Research, 2: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks W. P. et al. (1967) ‘Studies of Infantile Diarrhea in Karachi, Pakistan. II. Multiple Virus Isolations from Rectal Swabs’, American Journal of Epidemiology, 85: 469–78. [DOI] [PubMed] [Google Scholar]
- Pickett B. E. et al. (2012) ‘ViPR: An Open Bioinformatics Database and Analysis Resource for Virology Research’, Nucleic Acids Research, 40: D593–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piralla A. et al. (2018) ‘Enterovirus-D68 (EV-D68) in pediatric patients with respiratory infection: The circulation of a new B3 clade in Italy’, Journal of Clinical Virology, 99–100: 91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggieri V. et al. (2017) ‘Enterovirus D68 Infection in a Cluster of Children with Acute Flaccid Myelitis, Buenos Aires, Argentina, 2016’, European Journal of Paediatric Neurology, 21: 884–90. [DOI] [PubMed] [Google Scholar]
- Sagulenko P., Puller V., Neher R. A. (2018) ‘TreeTime: Maximum-Likelihood Phylodynamic Analysis’, Virus Evolution, 4: vex042.10.1093/ve/vex042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieble J. H., Fox V. L., Lennette E. H. (1967) ‘A Probable New Human Picornavirus Associated with Respiratory Diseases’, American Journal of Epidemiology, 85: 297–310. [DOI] [PubMed] [Google Scholar]
- Tan Y. et al. (2015) ‘Molecular Evolution and Intraclade Recombination of Enterovirus D68 during the 2014 Outbreak in the United States’, Journal of Virology, 90:1997–2007. [DOI] [PMC free article] [PubMed]
- Tiveljung-Lindell A. et al. (2009) ‘Development and Implementation of a Molecular Diagnostic Platform for Daily Rapid Detection of 15 Respiratory Viruses’, Journal of Medical Virology, 81: 167–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokarz R. et al. (2012) ‘Worldwide Emergence of Multiple Clades of Enterovirus 68’, The Journal of General Virology, 93: 1952–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt M. R., Crowe J. E. Jr. (2018) ‘Current Understanding of Humoral Immunity to Enterovirus D68’, Journal of the Pediatric Infectious Diseases Society, 7: S49–53. [DOI] [PubMed] [Google Scholar]
- Wang G. et al. (2017) ‘Enterovirus D68 Subclade B3 Strain Circulating and Causing an Outbreak in the United States in 2016’, Scientific Reports, 7: 1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver S. et al. (2018) ‘Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes’, Molecular Biology and Evolution, 35: 773–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H.-Y., et al. (2018) ‘Updates on the molecular epidemiology of Enterovirus D68 after installation of screening test among acute flaccid paralysis patients in Taiwan’, Journal of Microbiology, Immunology and Infection, 51: 688–91. [DOI] [PubMed]
- Yip C. C. et al. (2017) ‘First Report of a Fatal Case Associated with EV-D68 Infection in Hong Kong and Emergence of an Interclade Recombinant in China Revealed by Genome Analysis’, International Journal of Molecular Sciences, 18: 1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanini F. et al. (2017) ‘Error rates, PCR recombination, and sampling depth in HIV-1 whole genome deep sequencing’, Virus Research Deep Sequencing in Virology, 239: 106–14. [DOI] [PubMed] [Google Scholar]
- Zanini F. et al. (2016) ‘Population genomics of intrapatient HIV-1 evolution’, eLife Sciences, 4: e11282. [DOI] [PMC free article] [PubMed]
- Zhang C. et al. (2018a) ‘A Mouse Model of Enterovirus D68 Infection for Assessment of the Efficacy of Inactivated Vaccine’, Viruses, 10: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C. et al. (2018b) ‘Enterovirus D68 virus-like particles expressed in Pichia pastoris potently induce neutralizing antibody responses and confer protection against lethal viral infection in mice’, Emerging Microbes & Infections, 7: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. et al. (2016) ‘Genetic changes found in a distinct clade of Enterovirus D68 associated with paralysis during the 2014 outbreak’, Virus Evolution, 2. [DOI] [PMC free article] [PubMed]
- Zhang Y. et al. (2015) ‘Neutralization of Enterovirus D68 Isolated from the 2014 US Outbreak by Commercial Intravenous Immune Globulin Products’, Journal of Clinical Virology, 69: 172–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.