

Abstract
The nuclear protein HMGB1 (high mobility group box 1) is secreted by monocytes-macrophages in response to inflammatory stimuli and serves as a danger-associated molecular pattern. Acetylation and phosphorylation of HMGB1 are implicated in the regulation of its nucleocytoplasmic translocation for secretion, although inflammatory stimuli are known to induce H2O2 production. Here we show that H2O2-induced oxidation of HMGB1, which results in the formation of an intramolecular disulfide bond between Cys23 and Cys45, is necessary and sufficient for its nucleocytoplasmic translocation and secretion. The oxidation is catalyzed by peroxiredoxin I (PrxI) and PrxII, which are first oxidized by H2O2 and then transfer their disulfide oxidation state to HMGB1. The disulfide form of HMGB1 showed higher affinity for nuclear exportin CRM1 compared with the reduced form. Lipopolysaccharide (LPS)–induced HMGB1 secretion was greatly attenuated in macrophages derived from PrxI or PrxII knockout mice, as was the LPS-induced increase in serum HMGB1 levels.
Keywords: HMGB1, Oxidation, Secretion, Peroxiredoxin, H2O2
Graphical abstract
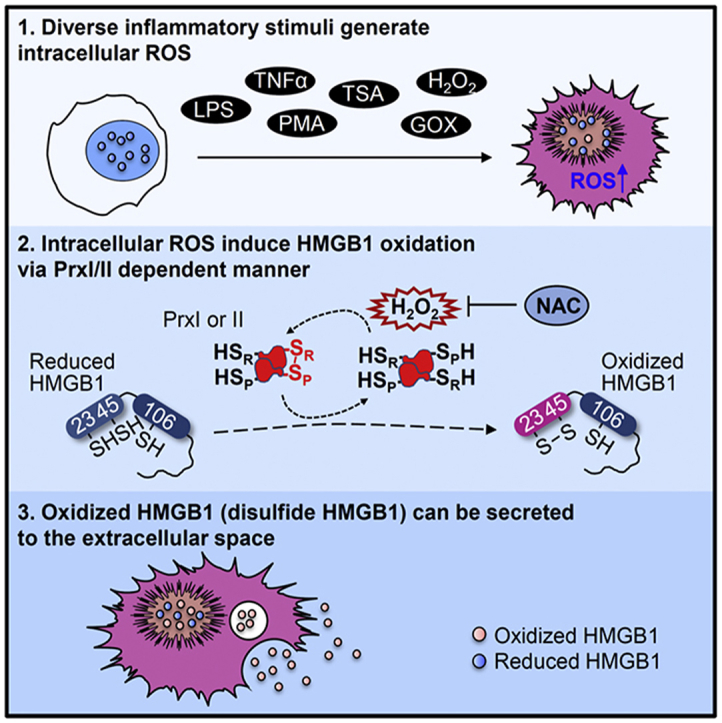
1. Introduction
HMGB1 is a nuclear protein that functions as a DNA chaperone during chromatin reorganization and transcriptional regulation [1,2] and which shuttles between the nucleus and cytoplasm of mammalian cells. Exit of HMGB1 from the nucleus occurs by both passive diffusion and an actively controlled pathway mediated by chromosome region maintenance protein 1 (CRM1), with import of the protein back into the nucleus also being actively controlled by karyopherin-α1 [3,4]. The trafficking of HMGB1 between the nucleus and cytoplasm has been thought to be regulated by protein acetylation and phosphorylation [3,4]. When HMGB1 is acetylated or phosphorylated at its nuclear localization signal (NLS) sequences in the nucleus, it is able to exit the nucleus but reimport is inhibited, resulting in its accumulation in the cytoplasm. HMGB1 is passively released from cells via necrosis, and is actively secreted from monocytes and macrophages in response to exposure of the cells to inflammatory stimuli. HMGB1 does not contain a leader sequence, with such secretion occurring through a vesicle-mediated pathway [5].
Secreted HMGB1 acts as a damage-associated molecular pattern (DAMP) molecule to signal danger to surrounding cells, triggers inflammation, and activates innate and adaptive immunity by binding to a plethora of cell surface receptors including Toll-like receptor 2 (TLR2), TLR4, TLR9, receptor for advanced glycation end products (RAGE), and C-X-C chemokine receptor 4 (CXCR4) when bound to C-X-C motif chemokine ligand 12 (CXCL12). HMGB1 also interacts with the pathogen-associated molecular pattern (PAMP) molecules LPS and lipoteichoic acid [6,7] as well as with endogenous CXCL12 [8], interleukin (IL)-1β [9], and nucleosomes [10] to augment inflammation. Furthermore, HMGB1 mediates autophagosome formation by binding to beclin-1 [11], and it inhibits polyglutamine aggregate formation in the cytoplasm through a chaperone-like function [12].
HMGB1 contains three conserved cysteine residues: Cys23, Cys45, and Cys106. The redox state of these cysteines gives rise to at least three forms of HMGB1 that are designated “reduced HMGB1” (all three cysteine residues in the thiol state), “disulfide HMGB1” (an intramolecular disulfide bond between Cys23 and Cys45; Cys106 in the thiol state), and “sulfonic HMGB1” (all three cysteines in the hyperoxidized sulfonic acid state). The specific functions of HMGB1 have recently been shown to be dependent on the redox state of its cysteines [13], with reduced HMGB1 forming a complex with CXCL12 to promote the migration of immune cells [14], disulfide HMGB1 (but not reduced HMGB1) interacting with a TLR4 adaptor protein to activate TLR4 and induce proinflammatory responses [15], and sulfonic HMGB1 manifesting neither cytokine- nor chemokine-like function [16]. HMGB1 present inside cells is predominantly in the fully reduced state, whereas the presence of disulfide HMGB1 in serum has been associated with inflammation-related pathologies [17,18]. However, fundamental issues regarding the oxidation of Cys23 and Cys45 of HMGB1—including the mechanism by which such oxidation occurs in response to inflammatory signals, the subcellular compartment where the oxidation occurs, and the effect of oxidation on HMGB1 secretion—have remained unclear.
Hydrogen peroxide (H2O2) is produced by cells in response to various extracellular stimuli and propagates intracellular signaling by oxidizing protein thiols [19]. Cells are also equipped with H2O2-eliminating enzymes such as catalase, glutathione peroxidase, and peroxiredoxin (Prx). The Prx family of peroxidases reduces H2O2 or alkyl peroxide, with the thiol of a conserved cysteine residue (the peroxidatic Cys, or CP–SH) serving as the site of oxidation by peroxides. In addition to CP–SH located in the NH2-terminal region of the molecule, most Prx enzymes contain an additional conserved cysteine (the resolving Cys, or CR–SH) in the COOH-terminal region. Mammalian cells express six isoforms of Prx (PrxI to PrxVI) that differ in their subcellular localization [20]. PrxI and PrxII are thus localized in the cytosol and nucleus. During the catalytic cycle of Prx enzymes, CP–SH is first oxidized by H2O2 to sulfenic acid (CP–SOH), which then reacts with CR–SH to form a disulfide that is subsequently reduced by an appropriate electron donor to complete the cycle.
Prxs are abundant proteins, with PrxI and PrxII together constituting a total of 0.2% to 1% of soluble protein in cultured mammalian cells [21]. In addition, Prxs possess an active-site pocket that gives rise to a high-affinity peroxide binding site [22]. This pocket is arranged so as to elicit rapid oxidation of CP–SH by H2O2, with the second-order rate constant for this oxidation being several orders of magnitude greater than that for the oxidation of thiols in well-characterized H2O2 target proteins. It was thus at first unclear how H2O2 is able to oxidize specific cysteine thiols of its target proteins in the presence of such abundant and efficient antioxidant enzymes. Two mechanisms to overcome this kinetic disadvantage with regard to oxidation by H2O2 have been proposed. One mechanism is transient localized inactivation of Prxs so as to protect H2O2 from destruction, which allows the accumulation of H2O2 in specific regions of the cell without global redox disturbance. The second mechanism is oxidation of PrxI or PrxII by H2O2 and subsequent transfer of the oxidation state to the target protein. Such an H2O2 sensor and transducer function of Prxs has been demonstrated in the oxidation of several proteins including protein disulfide isomerase (PDI) [23,24], apoptosis signal–regulating kinase 1 [25], mitogen-activated protein kinase (MAPK) [26,27], and STAT3 [28].
We now show that disulfide formation between Cys23 and Cys45 of HMGB1 is mediated by PrxI and PrxII in the nucleus and is essential for the nucleocytoplasmic translocation and secretion of HMGB1. Inflammatory stimuli such as LPS and TNFα as well as chemicals including phorbol 12-myristate 13-acetate (PMA) and trichostatin A (TSA) have been shown to induce the phosphorylation or acetylation of HMGB1 at its NLS sequences and thereby to promote its cytoplasmic translocation for secretion [3,4]. These inflammatory stimuli and chemicals also induce the intracellular production of H2O2. We show that H2O2 is first sensed by PrxI and PrxII, which then transfer the oxidant signal to HMGB1, resulting in formation of the Cys23–Cys45 disulfide bond. Importantly, whereas such disulfide formation is sufficient for HMGB1 secretion, posttranslational modification (PTM) of the NLS sequences is not essential for secretion but rather appears to function cooperatively with disulfide formation in this regard. Finally, we found that the LPS-induced increase in the circulating concentration of HMGB1 was significantly attenuated in mice lacking the PrxI or PrxII gene.
2. Materials and methods
2.1. Cell cultures
HEK293T and MEF cells were cultured in 10% FBS-Dulbecco's modified Eagle's medium (DMEM) supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM l-glutamine at 37 °C under 5% CO2. HEK293-hTLR4/MD2/CD14 (Invivogen) cells were cultured in the abovementioned culture medium supplemented with 10 μg/mL blasticidin and 50 μg/mL hygromycin B. Primary mouse BMDM cells were prepared from male C57BL/6 WT, PrxI-, and PrxII-KO mice [29,30]. Briefly, bone marrow cells were obtained by flushing bone marrow of femurs and tibias after euthanasia and then maintained in complete DMEM medium containing 20% L929 culture supernatant for 7 days [31]. BMDM cells were collected after incubation at 37 °C for 4 min with 0.25% trypsin-EDTA solution and were gently resuspended in complete DMEM.
2.2. Animals and mouse study
C57BL/6 WT, PrxI-, and PrxII-KO mice were used [29,30]. Mice were performed on age- and gender-matched randomly assigned 8- to 12-week-old male mice conducted according to procedures approved by the Institutional Animal Care and Use Committee of the Yonsei Laboratory Animal Research Center (YLARC, 2015-0275). Up to five mice per cage were housed in ventilated cages in a 12 h light and 12 h dark cycle. RT was at 22 °C, and they were fed with autoclaved standard rodent food and with drinking water at libitum.
C57BL/6 WT, PrxI- and PrxII-KO mice were used to investigate the effect of PrxI/II on HMGB1 secretion in vivo. Mice were intraperitoneally injected with 5 mg/kg LPS and then serum samples were collected at 1 and 16 h for TNF-α and HMGB1 measurements, respectively. Serum HMGB1 (IBL international) and TNF-α (R&D System) were measured according to the manufacturer's protocols.
2.3. Plasmids and in situ mutagenesis
Myc- or enhanced green fluorescent protein (EGFP)-tagged HMGB1(WT), HMGB1(C23S), and HMGB1(C45S) were inserted into the pCMV-Myc (Clontech) or pEGFP-N1 (Clontech) plasmid for mammalian cell expression. Myc/His6-tagged human PrxI and PrxII plasmids were generated using the pcDNA™3.1/Myc-His vector (Invitrogen). Myc/His6-tagged PrxI(C52S), PrxI(C173S), PrxI(C52S/173S) plasmids were generated using the QuikChange site-directed mutagenesis kit (Agilent). Streptavidin-binding peptide (SBP)-tagged PrxII(WT), PrxII(C51S), PrxII(C172S), and PrxII(C51S/172S) plasmids were kindly provided by Dr. Tobias P. Dick (German Cancer Research Center, Heidelberg, Germany) [28], and subcloned into pcDNA™3.1/Myc-His. The HMGB1(NLS1/2A)-GFP plasmid, a mutant plasmid wherein all the phosphorylation residues of six serines are changed to alanines in the NLS1 and 2 regions [4], was subcloned into pCMV-Myc. Myc-tagged HMGB1(NLS1/2R) plasmid, a mutant plasmid wherein all the acetylation residues of six lysines (K27, 28, 29 in NLS1 and K181, 182, 183 in NLS2) are changed to arginines [3], was generated using the QuikChange site-directed mutagenesis kit. Primer list for HMGB1 and PrxI/II constructs are described in Table S1.
2.4. Cell culture, transfection, and reagents
Plasmid transfections were carried out using Fugene HD reagent (Promega Corporation) or electroporation system (Invitrogen™) by MicroPorator-mini (Digital Bio). The histone deacetylase inhibitor TSA (Sigma-Aldrich) and PKC activator PMA (Sigma-Aldrich) for acetylation and phosphorylation, respectively, as well as H2O2 (Sigma-Aldrich) and glucose oxygenase (GOX) from Aspergillus niger (Sigma-Aldrich) for oxidation were used. LPS (Escherichia coli 0111:B4, 49180) (Sigma-Aldrich) and TNF-α (ATgen) were used for pro-inflammatory signaling. NAC (Enzo Life Sciences) was used as an antioxidant.
2.5. Measurement of HMGB1 secretion
To analyze the secretion of HMGB1 in the supernatants, culture media were replaced with serum-free OPTI-MEM medium (Gibco®) and treated with various stimulants. The culture supernatants were harvested and concentrated via Amicon Centricon filtration (Merck Millipore) after removing cell debris. Western blotting analysis was performed with rabbit anti-HMGB1 (Abcam, Ab18256) or mouse anti-Myc antibody (Invitrogen Life Technologies, 13-2500) as described below. Quantitative determination of HMGB1 in mouse serum or BMDM culture medium was performed using the HMGB1 ELISA Kit (IBL international) according to the manufacturer's protocol.
2.6. In vitro binding of PrxI/II proteins to HMGB1
HMGB1 and PrxI or PrxII proteins were purified. A mixture (total of 1 ml) of HMGB1 (0.5 μg/ml) and PrxI (0.4 μg/ml) or PrxII (0.4 μg/ml) was added with 0, 5, 50, and 500 μM DTT in the condition of slowly increasing amount of H2O2 to 50 μM and incubated overnight at 4 °C to observe the disulfide intermediate formation of HMGB1-PrxI/II. The mixtures were immunoprecipitated with mouse monoclonal anti-HMGB1 Ab (Abcam, Ab12029) and analysed in reducing SDS-PAGE. Immunoblots were performed with rabbit polyclonal anti-PrxI (AbFrontier, LF-PA0095), rabbit monoclonal anti-PrxII (Abcam, Ab109367) and rabbit polyclonal anti-HMGB1 (Abcam, AB18256) antibodies.
2.7. Confocal microscopy
Nucleocytoplasmic translocation of HMGB1 was observed under various stimulation conditions. HEK293T and HEK293-hTLR4/MD2/CD14 cells (InvivoGen) were transfected with EGFP-tagged HMGB1(WT), HMGB1(C23S), and HMGB1(C45S) plasmids and cultured in a LabTekTM II chamber (Nunc™) for 48 h. Cells were treated with 5 mU/mL GOX, 50 μM H2O2, 250 nM PMA, 50 ng/mL TNF-α, and 10 ng/mL TSA for 2 h. HEK293-hTLR4/MD2/CD14 cells were treated with 1 μg/mL LPS for 1 h. Cells were fixed with 4% paraformaldehyde (Biosesang) in PHEM buffer for 20 min at room temperature (RT) and washed with cold PBS. After mounting with 4′,6′-diamidino-2-phenylindole (Vector Laboratories), over 200 cells were observed to count the proportion of cytoplasmic HMGB1 (+) cells under confocal FV1000 microscopy (Olympus).
2.8. Proximity ligation assay (PLA)
The molecular interactions between HMGB1 and PrxI/II or CRM1 proteins were evaluated using a Duolink II Detection kit (Olink Bioscience). Briefly, HEK293T cells were cultured in eight well chambers (Nunc™) and transfected with HMGB1(WT), HMGB1(C23S), or HMGB1(C45S) plasmids. Cells were fixed with 4% paraformaldehyde in PHEM buffer for 20 min at RT. Cells were incubated with a blocking agent for 1 h, and rabbit polyclonal anti-PrxI and anti-PrxII were added and incubated overnight with either mouse monoclonal anti-HMGB1 or anti-Myc for endogenous or exogenous HMGB1, respectively. Mouse monoclonal anti-CRM1 (Santa Cruz Biotechnology, SC-5595) was incubated with rabbit polyclonal anti-HMGB1 to identify the HMGB1-CRM1 interaction. After three subsequent washes, PLA probes were applied and incubated for 1 h in a humidity chamber at 37 °C. Unbound PLA probes were removed and the samples were incubated in the ligation solution for 1 h. For amplification, polymerase was applied with fluorescently labeled oligonucleotides for 100 min at 37 °C. The amplified fluorescence-labeled oligonucleotide was visible as a distinct fluorescent spot and images were taken by confocal microscopy.
2.9. Mass spectrometry (LC-MS/MS)
To identify the disulfide bond formation between Cys23 and Cys45 of HMGB1 and free Cys106 of HMGB1, HEK293T cells were transfected with the Myc-HMGB1 plasmid and then stimulated with 5 mU/mL GOX for 2 h. 50 mM N-Ethyl maleimide (NEM; Sigma-Aldrich) was added to WCLs for labeling of reactive cysteine residues and the protein supernatant was applied to anti-Myc tagged-magnetic beads (Bio-Rad). The purified Myc-HMGB1 protein was treated with or without 5 mM DTT. The NEM labeled or unlabeled Myc-HMGB1 was separated on SDS-PAGE followed by Coomassie Blue staining. LC-MS/MS products were analysed after in-gel digested with trypsin at the Yonsei Proteome Research Center (Seoul, Korea).
2.10. Western blot analysis and immunoprecipitation (IP)
HEK293T or MEF cells in six-well plates were transfected with plasmids using FuGene HD or electroporated using the MicroPorator-mini. Cells were washed with phosphate buffered saline (PBS) and lysed in 1 × RIPA buffer (GenDEPOT) containing 150 mM NaCl, 1% Triton X-100, 1% deoxycholic acid sodium salt, 0.1% SDS, 50 mM Tris-HCl pH 7.5, 2 mM EDTA, and protease inhibitor cocktail (GenDEPOT). For thiol blocking, 50 mM NEM was added to inhibit the breakage or new disulfide bond formation. WCLs were centrifuged at 20,000×g for 10 min at 4 °C. Protein sample buffer (100 mM Tris-HCl pH 6.8, 2% SDS, 25% glycerol, 0.1% bromophenol blue, and with/without 5% β-mercaptoethanol) was added to the WCLs followed by heating at 94 °C for 5 min. Proteins (20 μg) were separated by molecular weight via SDS-PAGE or nonreducing SDS-PAGE and transferred to nitrocellulose membranes. Non-specific binding sites were blocked by incubating the membranes in Tris-buffered saline (TBS) supplemented with 0.1% Tween 20 and 5% w/v skim milk for 1 h. Anti-HMGB1, anti-PrxI, anti-PrxII, anti-CRM1, anti-GAPDH (AbFrontier, LF-PA0095), anti-Sp1 (Santa Cruz Biotechnology, SC-59) antibodies were used. Membranes were washed three times for 10 min with TBS-Tween 20 and probed with the appropriate horseradish peroxidase (HRP)-conjugated secondary Ab (Jackson ImmunoResearch) for 1 h. After washing three times, an enhanced chemiluminescence substrate was then used for visualization. Membranes were then stripped by submerging in EzReprobe (ATTO Corporation) at RT for 30 min.
IP was performed with 100–200 μg protein. Initially, 1 μg specific Ab was conjugated with Dynabeads® protein G (Bio-Rad) at RT for 1 h. Samples were added to 10 μL Ab conjugated-Dynabeads® Protein G at 4 °C for 18 h. The beads were washed three times with ice-cold PBS and then mixed with protein sample buffer followed by heating at 94 °C for 5 min. The proteins were separated using SDS-PAGE.
2.11. Nuclear/cytosolic fractionation
To determine the localization of the intracellular interaction between HMGB1 and PrxI or PrxII during conditions of increased intracellular H2O2 levels, HEK293T cells were transfected with Myc tagged HMGB1WT, HMGB1C23S, or HMGB1C45S with PrxI- or PrxII-Myc/His6 plasmids and then treated with 50 μM H2O2 for 30 min to induce oxidative stress. Cells were harvested by centrifugation at 600×g for 5 min at 4 °C. Nuclear/cytosolic fractionation was performed using a nuclear/cytosolic fractionation kit (BioVision Incorporated) according to the manufacturer's procedure.
2.12. Statistical analysis
Analysis of experimental data was performed with the Student's t-test using GraphPad Prism. Data represent the mean value and SEM or SD, as indicated in the individual figure legends. The difference was considered statistically significant when p < 0.05.
3. Results
3.1. HMGB1 oxidation mediated by PrxI and PrxII in the nucleus
Ligation of TLR4 by LPS elicits H2O2 production through activation of NADPH oxidase 4 (Nox4) [32]. To examine the effect of LPS on the formation of disulfide HMGB1, we stimulated mouse bone marrow–derived macrophages (BMDMs) with LPS and observed the formation of oxidized form of HMGB1 (Fig. 1A). Disulfide HMGB1 is more compact and thereby migrates faster in the electrophoretic gel compared with reduced HMGB1. HMGB1 in the nucleus of unstimulated cells existed predominantly in the fully reduced form. Stimulation with LPS, however, induced a time-dependent increase in the nuclear abundance of disulfide HMGB1. H2O2 stimulation also induced a similar oxidation effect on PrxI, and PrxII (Fig. 1A, Suppl. Fig. 1A).
Fig. 1.

HMGB1 oxidation mediated by PrxI and PrxII in the nucleus. (A) Mouse BMDMs were treated with 100 ng/mL LPS for various times, after which the nuclear fraction of cell lysates was isolated and subjected to nonreducing SDS-PAGE and immunoblotting. The intensities of bands corresponding to oxidized (Ox) forms of HMGB1 and to oxidized (dimeric) forms of PrxI and PrxII were determined. Means ± SEM (n = 4). Re, reduced form. (B) Mechanistic scheme for the formation of disulfide HMGB1 catalyzed by PrxI or PrxII. I-VI indicate the mechanistic process of reactions for HMGB1 disulfide formation by Prxs. (C, D) IP of Myc–tagged human HMGB1(WT), HMGB1(C23S) or HMGB1(C45S) as well as with those for Myc/His6-tagged human PrxI or PrxII after 50 μM H2O2 for 30 min stimulation in HEK293T cells. (E) WCLs prepared in (C) and (D) were also separated into cytosolic and nuclear fractions before IP. Sp1 and GAPDH were used as nucleus and cytoplasmic marker, respectively. (F) HEK293T cells were transfected with Myc-HMGB1(C23S) or Myc-HMGB1(C45S) and Myc/His6-tagged PrxI or PrxII, and stimulated with 50 μM H2O2 at 30 min. WCLs were subjected to 8% nonreducing SDS-PAGE. Black, blue, and red arrows show (Prx-Myc/His6)2, Myc-HMGB1-Prx-Myc/His6, Myc-HMGB1-endogenous Prx complexes, respectively. *, (Prx-Myc/His6)2-Myc-HMGB1; **, Prx-Myc/His6-protein X complexes. Endo.: endogenous. (G, H) A mixture (total of 1 ml) of HMGB1 (0.5 μg/ml) with either PrxI (0.4 μg/ml) or PrxII (0.4 μg/ml) proteins was treated with various concentrations of DTT and 50 μM H2O2. The mixtures were immunoprecipitated with anti-HMGB1 Ab and immunoblotted with anti-PrxI/II Abs. Amounts of HMGB1-PrxI/II disulfide intermediate were analysed with reducing SDS-PAGE. IgG, irrelevant Ab. (I, J) PLA assay for detection of the interaction between endogenous HMGB1 and PrxII. PLA signal (red fluorescence) were detected with antibodies HMGB1 and PrxII after1 μg/mL LPS for 1 h in MEFs or 50 μM H2O2 for 30 min in HEK293T cells. Scale bars, 10 μm. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
We examined the possibility that nuclear PrxI or PrxII might function as an H2O2 sensor and transducer for the formation of disulfide HMGB1. PrxI and PrxII are obligate homodimers arranged in a head-to-tail manner. In the presence of H2O2, the CP–SH residue (Cys52 for PrxI and Cys51 for PrxII) is oxidized to sulfenic acid (CP–SOH), which then reacts with the CR–SH residue (Cys173 for PrxI and Cys172 for PrxII) of the other subunit to form a disulfide-linked dimer. As expected, neither the higher-mobility form of HMGB1 nor dimeric PrxI/II was observed if dithiothreitol (DTT) was added to the nuclear extracts before electrophoresis (Fig. 1A).
The putative Prx-mediated oxidation of HMGB1 would likely be initiated by attack of the sulfenylated or disulfide Prx by either Cys23–SH or Cys45–SH of HMGB1, resulting in the formation of a transient intermolecular disulfide–linked intermediate containing both Prx and HMGB1 (Fig. 1B). Resolution of the intermolecular disulfide by either Cys23–SH or Cys45–SH of HMGB1, whichever does not participate in the intermolecular disulfide linkage, would then result in regeneration of reduced Prx and formation of disulfide HMGB1. To verify this possibility by detection of the disulfide-linked HMGB1-Prx complex, we transfected HEK293T cells with expression vectors for Myc epitope–tagged wild-type (WT) or Cys23-to-Ser (C23S) or Cys45-to-Ser (C45S) mutant forms of HMGB1 as well as for Myc/His6-tagged forms of PrxI or PrxII. The amount of Myc-HMGB1 that co-immunoprecipitated with Myc/His6-tagged PrxI or PrxII from cell lysates was determined by IB analysis after H2O2 stimulation. Immunoprecipitation (IP) of Myc-HMGB1(C23S) and Myc-HMGB1(C45S) with Myc/His6-tagged PrxI or PrxII was apparent in the absence of H2O2, and stimulation of the cells with H2O2 greatly increased the extent of such IP (Fig. 1C and D). Similar results were also obtained from IP of HMGB1 with PrxI/PrxII after LPS stimulation (Suppl. Fig. 1B and C). Immunoprecipitate was also detected in the PrxI(C173S)- and PrxII(C172S)-Myc/His6 with Myc-HMGB1(C23S) after H2O2 stimulation (Suppl. Fig. 1D and E). And the essential roles of the two cysteines of HMGB1 (Cys23 and Cys45) and of PrxI/II (CP and CR) in formation of the intermolecular disulfide–linked intermediates were demonstrated by analysis of HEK293T cells expressing a double mutant of either HMGB1(C23/45S) or PrxI(C52/173S) or PrxII(C51/172S). So we hypothesized that those oxidized (sulfenylated or disulfide) PrxI and PrxII recognize HMGB1 cysteine and induce its disulfide formation (Fig. 1B). Preparation of nuclear and cytosolic fractions from the whole cell lysates (WCLs) revealed that the coprecipitation was apparent with the former fraction but not the latter, where HMGB1 is mainly located (Fig. 1E). For detection of the HMGB1-PrxI or PrxII complex form, HEK293T cells transfected with PrxI or PrxII and HMGB1(C23S) or HMGB1(C45S) plasmids were stimulated with H2O2. For detection of the HMGB1-PrxI or PrxII complex form, HEK293T cells transfected with Myc/His6-tagged PrxI or PrxII and Myc-HMGB1(C23S) or Myc-HMGB1(C45S) were stimulated with H2O2. Binding complexes of His6-tagged PrxI or PrxII with HMGB1 from WCLs were eluted using imidazole, and separated by nonreducing SDS-PAGE. Coprecipitation was detected in the HMGB1-Prx intermolecular disulfide bond position; however, such complexes dissociated after DTT treatment (Suppl. Fig. 1F and G). In addition, complex formation between HMGB1 and PrxI was also analysed by mass spectrometry. High score peptide fragments of PrxI and HMGB1 from the protein band size corresponding to the HMGB1-PrxI intermolecular complex were obtained from cells expressing HMGB1(C23S) or HMGB1(C45S) with PrxI or PrxII (Table S1). In cells overexpressing Myc/His6-tagged PrxI and PrxII, amount of detected HMGB1-containing complex was notably higher. The decreased mobility of theses complexes, caused by the presence of Myc/His6-tagged PrxI and PrxII, suggests that these bands represent a disulfide complex between PrxI or PrxII and HMGB1 (Fig. 1F). The disulfide-linked complexes of HMGB1-Prx and Prx-Prx-HMGB1, which are from reactions I, II and III, IV, respectively (Fig. 1B), were directly detected through analysis of WCLs by 8% and 12% nonreducing SDS-PAGE (Fig. 1F, Suppl. Fig. 1H). Such complexes were demonstrated in wild-type cells, which were disappeared in PrxI/II double knockout (KO) HAP1 cells, after H2O2 stimulation [33]. And higher molecular weight complexes of Prx with unidentified protein (protein X) could be observed.
We also detected DTT-sensitive co-IP of purified PrxI or PrxII with HMGB1 in the presence of H2O2 in vitro (Fig. 1G and H), confirming the formation of HMGB1-Prx disulfide intermediate. The amounts of PrxI and PrxII in the nuclear fraction of HEK293T cells, Raw264.7 cells, and MEFs were similar and in the range of 60–90 pg of each protein per microgram of total nuclear protein (Suppl. Fig. 1I). These results indicated that PrxI/II-mediated generation of disulfide HMGB1 occurs in the nucleus via formation of an intermolecular disulfide as proposed in Fig. 1B. The level of HMGB1-Prx disulfide intermediate formation was decreased in PrxII-deficient (KO) MEF cells, compared to that in wild-type cells containing PrxI/II, after H2O2 stimulation (Suppl. Fig. 1J).
We performed a proximity ligation assay (PLA) to examine the close binding between endogenous HMGB1 and PrxII proteins. LPS stimulation induced a marked increase in the number of PLA signals in the nucleus, indicative of a substantial extent of complex formation in MEFs (Fig. 1I). The number of PLA signals due to the HMGB1-PrxII intermediate was also greatly increased by exposure of the cells to H2O2 in HEK293T cells (Fig. 1J). We also observed the close binding between Myc–tagged WT, C23S, or C45S forms of HMGB1 and endogenous PrxII proteins using HEK293T cells (Suppl. Fig. 1K-M). Treatment with H2O2 or GOX increased the number of PLA signals in the nucleus of cells expressing each form of HMGB1. As expected, the number of PLA signals was greater in the cells expressing HMGB1(C23S) or HMGB1(C45S) than in those expressing HMGB1(WT).
3.2. HMGB1 oxidation is decreased in PrxI or II-deficient MEF cells
We next evaluated the effect of the absence of PrxII on the formation of disulfide HMGB1 with the use of immortalized WT and PrxII-KO MEFs. The protein kinase C (PKC) activator PMA is thought to stimulate HMGB1 secretion by inducing HMGB1 phosphorylation at serine residues located in the NLS sequences [4,34]. Like LPS, PMA also induces H2O2 production by activating Nox enzymes [35]. Stimulation with LPS or PMA increased the abundance of disulfide HMGB1 in WT and PrxII KO MEFs, but this effect was markedly attenuated in the latter cells compared with the former for both stimulants (Fig. 2A and B). The amount of disulfide HMGB1 produced in PrxII KO MEFs in response to GOX or H2O2 treatment was increased greatly by ectopic expression of PrxII(WT) but was not affected by that of PrxII(C172S) (Fig. 2C and D). The CP residue (Cys51) of PrxII(C172S) is still sensitive to H2O2 and is readily oxidized to sulfenic acid (CP–SOH). Inability of PrxII (C172S) to mediate HMGB1 oxidation in PrxII KO MEFs suggests that PrxII effectively transfers its oxidation state to HMGB1 via the disulfide-linked dimer. Resolving cysteine mutant Prxs maintain lesser interaction than WT Prxs with HMGB1 and we believe the complex may have a very low reaction rate in reaction V of Fig. 1B. Similar to the results obtained with PrxI KO MEFs (Suppl. Fig. 2A and B). When we treated with cycloheximide to rule out the effect of newly synthesized cytoplasmic HMGB1 to its oxidation, there was no effect of newly synthesized HMGB1 (Suppl. Fig. 2C).
Fig. 2.

Ablation of PrxII impedes formation of disulfide HMGB1. (A, B) Immortalized WT or PrxII KO MEFs were incubated for 30 min in the absence or presence of PMA (50 or 250 nM) or LPS (0.25 or 1 μg/mL), after which WCLs were subjected to nonreducing SDS-PAGE and immunoblotting. #: nonspecific bands, M, monomer; D, dimer. (C, D) Immortalized PrxII KO MEFs that had been transfected with expression vectors for PrxII (C172S) or PrxII (WT) were incubated for 30 min in the absence or presence of GOX (0.5 or 5 mU/mL) or H2O2 (50 or 250 μM), after which WCLs were subjected to nonreducing SDS-PAGE. Means ± SEM (n = 5 for A and B, n = 3 for C and D). *P < 0.05, **P < 0.01 versus PrxII KO (A and B) or empty vector (Ev, C and D), t-test. (E) Determination of disulfide formation of HMGB1 Cys23–Cys45 and free thiol of Cys106 by LC-MS/MS analysis. WCLs of GOX exposed Myc-HMGB1 in HEK293T cells were incubated with NEM and then treated with DTT and NEM for disulfide HMGB1 analysis.
Taken together, our data indicated that (1) both PrxI and PrxII sense H2O2 and form a disulfide-linked homodimer for transduction of the H2O2 signal by transfer of their oxidation state to HMGB1, thereby generating disulfide HMGB1; (2) the transfer of oxidation state from the Prx dimer to reduced HMGB1 involves formation of a Prx-HMGB1 complex linked by an intermolecular disulfide between the CP residue of Prx and either Cys23 or Cys45 of HMGB1; and (3) the intermolecular disulfide is resolved by the cysteine residue (Cys23 or Cys45) of HMGB1 not contributing to the intermolecular linkage, resulting in the production of disulfide HMGB1. It is of note that HMGB1 oxidation occurs in both PrxI and PrxII KO MEFs, albeit a reduced rate compared with that in WT cells. This is likely because of the redundant function of PrxI and PrxII. However, it was not possible to use PrxI/II knockdown cells because they exhibited high levels of cell death in our study, which is consistent with the very short life span of PrxI/II double KO mice [36].
To determine disulfide formation of HMGB1 Cys23–Cys45 and free thiol of Cys106, Myc-tagged HMGB1 was transiently overexpressed in HEK293T cells, treated with GOX for the formation of C23-C45 linked disulfide HMGB1, and labeled the reactive cysteine residue using N-ethylmaleimide (NEM) before or after DTT treatment for LC-MS/MS analysis (Fig. 2E). Upon NEM labelling prior to DTT treatment, we identified the mass shift of the NEM-tagged C106 peptide, but not of the C23 or C45 peptide. Purified HMGB1 was also incubated with DTT for dissociation of the C23-C45 disulfide linkage, and then subjected to NEM. We detected mass shift in both NEM-tagged C106 and C23 peptides (Fig. 2E). We further calculated the degree of HMGB1 Cys23–Cys45 intramolecular disulfide formation. Fluorophore (Cy3 or Cy5)-tagged maleimide, to tag free cysteine residues, was applied without or with DTT (Suppl. Fig. 2D). HMGB1(WT) was similarly tagged with both green (Cy3) and red (Cy5) fluorescence, whereas the HMGB1(C23S) mutant was tagged only with Cy3, indicating the formation of HMGB1 Cys23–Cys45 regardless of Cys106 oxidation state. HMGB1 Cys23–Cys45 intramolecular disulfide formation could be also demonstrated using NEM and biotin-iodoacetamide (BIAM) analysis (Suppl. Fig. 2E).
3.3. Oxidation to the disulfide form is necessary for nucleocytoplasmic translocation and secretion of HMGB1
We next examined the relevance of the oxidation of HMGB1 to the disulfide form with regard to its nucleocytoplasmic translocation and secretion. HEK293-hTLR4/MD2/CD14 cells were transfected with EGFP-tagged HMGB1(WT), (C23S), or (C45S). LPS induced nucleocytoplasmic translocation of EGFP-tagged HMGB1(WT), whereas it had no such effect on HMGB1(C23S) or HMGB1(C45S), each of which forms a disulfide-linked complex with PrxI/II that cannot be further resolved to yield disulfide HMGB1 (Fig. 3A and B). IB analysis of culture supernatants revealed that LPS induced the secretion of endogenous-HMGB1 in MEFs (Fig. 3C). The secreted protein was judged to be the disulfide form on the basis of the observation that its mobility on nonreducing SDS-PAGE was decreased by the addition of DTT to the culture supernatants (Fig. 3C).
Fig. 3.

Oxidation of HMGB1 is required for nucleocytoplasmic translocation and secretion. (A, B) HEK293-hTLR4/MD2/CD14 cells expressing Myc/EGFP-tagged WT, C23S, or C45S forms of HMGB1 were incubated with 1 μg/mL LPS for 2 h, and examined for EGFP fluorescence by confocal microscopy (A). Scale bars, 10 μm. The percentage of cytoplasmic EGFP was determined (B). n > 200. (C) MEF cells were stimulated with LPS (50 or 250 ng/mL) for 24 h, after which culture supernatants were harvested, and subjected to nonreducing SDS-PAGE with/without DTT. (D-F) MEF or HEK293T cells were incubated in the presence of 250 nM PMA, 10 ng/mL TSA, 50 ng/mL TNFα, 2 mM NAC or 10 μM DPI for 24 h, after which culture supernatants were subjected to IB analysis. Means ± SEM (n = 3). *P < 0.05, **P < 0.01, t-test. NS, not significant. Dotted line is cut line of same membrane. (G-J) Oxidation effect on phosphorylation or acetylation-defective HMGB1. HMGB1 secretions of the phosphorylation-defective mutant HMGB1(NLS1/2A) and the acetylation-defective mutant HMGB1(NLS1/2R) were tested after PMA, TSA, or GOX treatments. HMGB1 knockout MEF cells (I, J) were transfected with each plasmid for 36 h and stimulated for 24 h. Culture supernatants were harvested and IB analyses were performed using an anti-Myc Ab. Means ± SEM (n = 3). *P < 0.05, **P < 0.01, t-test.
We also observed the nucleocytoplasmic transport of Myc/EGFP-HMGB1 induced by H2O2 or GOX in HEK293T cells as well as extracellular secretion of Myc-HMGB1(WT) but not of Myc-HMGB1(C23S) or Myc-HMGB1(C45S) (Suppl. Fig. 3A–D), suggesting that disulfide bond formation between Cys23 and Cys45 is also required for H2O2-induced HMGB1 translocation and secretion. Mutant protein overexpression had no effect on endogenous HMGB1 secretion and expression (Suppl. Fig. 3C–E).
Like HMGB1 phosphorylation in PMA-treated cells, acetylation of the NLS sequences of HMGB1 in cells treated with TSA promotes the nuclear transport of HMGB1 toward secretion. Whereas PKC activation by PMA results in H2O2 production through Nox activation [35], TSA-induced acetylation of multiple mitochondrial proteins increases mitochondrial H2O2 production [37]. Binding of TNFα to its cognate receptor induces both acetylation and phosphorylation of the NLS sequences of HMGB1 and thereby promotes HMGB1 secretion [3,4,38]. TNFα binding also induces H2O2 production from multiple sources including mitochondria, Nox, and xanthine oxidase [39]. Treatment of MEFs or HEK293T cells with PMA, TSA, or TNFα stimulated HMGB1 release in a manner sensitive to inhibition by the antioxidant N-acetylcysteine (NAC) (Fig. 3D and E), suggesting that oxidant production plays a key role in HMGB1 secretion. The Nox inhibitor diphenyleneiodonium (DPI) inhibited HMGB1 secretion induced by PMA but not that induced by TNFα (Fig. 3F), suggesting that H2O2 produced by Nox is required for PMA-induced HMGB1 secretion whereas non-Nox sources of H2O2 such as mitochondria and xanthine oxidase are important for HMGB1 secretion induced by TNFα. Given that H2O2 affects many signaling pathways by targeting protein cysteine thiols, we examined whether the oxidation of Cys23 and Cys45 is also required for the nuclear transport toward secretion of HMGB1 in response to PMA, TSA, or TNFα. All three stimulants triggered the nucleocytoplasmic translocation and secretion of Myc-HMGB1(WT) but not those of Myc-HMGB1(C23S) or Myc-HMGB1(C45S) (Suppl. Fig. 3F–J).
We next tested whether a phosphorylation-defective mutant (NLS1/2A, in which all six serines in the NLS1/2 sequences are replaced with alanine) of HMGB1 [4] is secreted in response to oxidative stimuli. Stimulation of HEK293T cells expressing Myc-HMGB1(WT) or (NLS1/2A) with PMA or GOX revealed that secretion of the mutant protein did occur in response to each stimulant but at a level lower than that apparent for the WT protein, with secretion of both HMGB1(WT) and HMGB1(NLS1/2A) being greatly attenuated by NAC (Fig. 3G and H). Similar results were obtained with the acetylation-defective mutant (NLS1/2R) [3], in which all six lysines in the NLS1/2 sequences are replaced with arginines (Fig. 3I and J). These results suggested that disulfide bond formation between Cys23 and Cys45 of HMGB1 is a prerequisite for its secretion whereas acetylation or phosphorylation is not sufficient.
Collectively, intramolecular disulfide bond formation between Cys23 and Cys45 of HMGB1 is required for nucleocytoplasmic transport toward secretion in response to all the stimulants tested (LPS, H2O2, PMA, TSA, and TNFα). PTM such as phosphorylation and acetylation of the NLS sequences appears not to be required for HMGB1 translocation toward secretion but rather facilitates this process. PMA thus induced the secretion of an HMGB1 mutant without NLS phosphorylation sites, but the extent of this effect was less pronounced than that apparent with the WT protein. The observation that GOX-induced HMGB1 secretion was also partially inhibited by NLS mutation is likely related to the fact that PKC isoforms are activated by H2O2 independently of diacylglycerol, the PKC activator mimicked by PMA [40].
3.4. Preferential binding of disulfide HMGB1 to CRM1
HMGB1 is sufficiently small to undergo passive diffusion through nuclear pores. Its nucleocytoplasmic translocation is also actively regulated by the nuclear exportin CRM1, to which HMGB1 binds directly [3]. We examined the affinity of HMGB1 for CRM1 with the use of HEK293T cells transfected with expression vectors for CRM1 and Myc–tagged WT, C23S, or C45S forms of HMGB1. IP revealed that CRM1 binding to HMGB1(WT) was greatly increased by exposure of the cells to H2O2 or GOX (Fig. 4A). In the treated cells, the apparent binding affinity of HMGB1 for CRM1 decreased in the rank order of WT > C23S > C45S (Fig. 4A). Similar results were obtained by PLA analysis of the proximity of the Myc–tagged forms of HMGB1 and endogenous CRM1 (Fig. 4B). Together, these results thus indicated that the affinity of disulfide HMGB1 for CRM1 is greater than that of reduced HMGB1, likely explaining the increased rate of nucleocytoplasmic translocation of the disulfide form observed in the presence of various stimulants.
Fig. 4.

Binding of disulfide HMGB1 to CRM1. (A) IP of Myc–tagged HMGB1(WT), HMGB1(C23S) or HMGB1(C45S) with CRM1 after 50 μM H2O2 or 0.5 mU/mL GOX for 2 h stimulation in HEK293T. (B) HEK293T cells expressing Myc–tagged WT, C23S, or C45S forms of HMGB1 were incubated in the absence or presence of 50 μM H2O2 or GOX (5 mU/mL) for 2 h and then subjected to a PLA assay with antibodies to Myc and to CRM1. Scale bars, 10 μm.
3.5. HMGB1 secretion from BMDMs is dependent on PrxI and PrxII
We next investigated the role of PrxI and PrxII in HMGB1 secretion with the use of PrxI- or PrxII-KO BMDMs (Suppl. Fig. 4A). HMGB1 secretion induced by LPS, PMA, TSA, TNFα, H2O2, or GOX was attenuated in PrxI- or PrxII-KO BMDMs compared with WT BMDMs (Fig. 5A–D, Suppl. Fig. 4B and C). As was the case for intramolecular disulfide formation in nonimmune cells (Fig. 2A and B), the suppression of HMGB1 secretion by PrxI or PrxII deficiency in BMDMs was partial. Partial inhibition of HMGB1 secretion induced by LPS, PMA, TSA or TNFα was also apparent in PrxI or II KO MEFs compared with WT MEFs, whereas the extent of HMGB1 secretion was restored in the mutant cells by forced expression of Prx (Fig. 5E and F).
Fig. 5.

Deficiency of PrxI or PrxII attenuates HMGB1 secretion in BMDMs and vice versa. (A-D) BMDMs derived from WT, PrxI-KO (PrxI–/–), or PrxII-KO (PrxII–/–) mice were incubated in the absence or presence of 100 ng/mL LPS, 250 nM PMA, 50 ng/mL TSA, or 50 ng/mL TNFα for 24 h, after which culture supernatants were subjected to IB analysis. Means ± SEM (n = 3). (E, F) PrxII KO MEF cells were transfected with PrxII plasmid and incubated with the indicated concentrations of LPS (ng/mL) (E) or GOX (μU/ml) (F) for 24 h, after which culture supernatants were subjected to IB analysis. WT MEF cells were used for positive control. Means ± SEM (n = 3). *P < 0.05, **P < 0.01 versus corresponding value for PrxII KO cells, t-test.
3.6. LPS-induced HMGB1 secretion is dependent on PrxI and PrxII in vivo
We examined the role of PrxI and PrxII in HMGB1 secretion in vivo. WT as well as PrxI- or PrxII-KO mice were injected intraperitoneally with a sublethal dose (5 mg/kg) of LPS, and blood samples were collected at optimal times (1 and 16 h, respectively) for the measurement of TNFα and HMGB1 concentrations in serum. LPS injection increased TNFα levels to 22.0 ± 6.7 ng/mL in WT mice, to 15.7 ± 6.1 ng/mL in PrxI-KO mice, and to 24.3 ± 6.9 ng/mL in PrxII-KO mice (Fig. 6A), values that did not differ significantly from each other. In contrast, LPS increased HMGB1 levels in PrxI- or PrxII-KO mice to 31.4 ± 13.6 and 43.6 ± 22.6 ng/mL, respectively, values that were significantly smaller than the corresponding value of 74.2 ± 14.7 ng/mL for WT mice (Fig. 6B). PrxI and PrxII-KO mice had no difference in terms of expression levels of various proteins related to influencing HMGB1 modifications (data not shown). Intramolecular oxidization of HMGB1 C23-45 is mediated by PrxI/II in the nucleus and its secretion mechanism is summarized in Fig. 7.
Fig. 6.

Serum HMGB1 levels in PrxI- or PrxII-KO mice injected with LPS. (A, B) WT, PrxI–/–, or PrxII–/– mice were injected intraperitoneally with LPS (5 mg/kg), and blood samples were collected at 1 and 16 h, and measured concentration of TNFα (A) and HMGB1 (B), respectively. Means ± SD (n = 11 per group). **P < 0.01 versus wild type, t-test. NS, not significant.
Fig. 7.

A model of PrxI/II-mediated HMGB1 oxidation and secretion. Upon receiving inflammatory stimuli such as LPS, TNFα, PMA, and TSA, cytosolic H2O2 can be generated through mitochondria and NADPH oxidase. Nucleic H2O2 can be increased by diffusion of cytosolic H2O2 or independently by nuclear Nox IV. Increasing of nuclear H2O2 leads to nuclear PrxI/II oxidation step by step, oxidized to sulfenic acid (CP-SOH) and then reacts with the CR-SH residue to disulfide-linked dimer. Sulfenic or disulfide Prx can initially attack either Cys23-SH or Cys45-SH of HMGB1, resulting in the formation of intramolecular disulfide-bond (disulfide HMGB1). Disulfide HMGB1 formation in the nucleus by PrxI/II induces nucleocytoplasmic translocation in CRM1 dependent manner and then extracellular secretion.
4. Discussion
HMGB1 passively leaks from necrotic and apoptotic cells of various types and is actively secreted from live monocytes-macrophages in response to stress signals. PTM such as acetylation or phosphorylation in the residues of HMGB1 NLS sequences has been proposed as the major mechanism for regulation of active nuclear exit of the protein. All stimulants of HMGB1 secretion tested in this study (LPS, TNFα, PMA, and TSA) are known to induce the intracellular production of H2O2 at low levels, with oxidation of various protein cysteine residues by H2O2 being an important mechanism of cell signaling [41].
Here we found that oxidation of Cys23 and Cys45 to yield disulfide HMGB1 is required for nucleocytoplasmic translocation and secretion of the protein. The C23S and C45S mutants of HMGB1, which are not able to form the intramolecular disulfide bond but in which acetylation and phosphorylation sites are intact, thus manifested almost undetectable levels of nucleocytoplasmic translocation and secretion in response to various stimulants. The dispensability of NLS phosphorylation and acetylation for HMGB1 secretion was also demonstrated by the observation that the phosphorylation- and acetylation-defective NLS1/2A and NLS1/2R mutants were still secreted in response to H2O2 stimulation. Despite the facts that oxidation of protein thiols is generally a slow process and that cellular compartments are equipped with various H2O2-eliminating enzymes, the specific oxidation of HMGB1 by H2O2 at low concentrations can be achieved in the nucleus as a result of the H2O2 sensor and transducer function of PrxI and PrxII. Such a function of PrxI and PrxII in the case of HMGB1 was demonstrated by direct detection of the reaction intermediates comprising complexes of HMGB1 and either PrxI or PrxII linked by an intermolecular disulfide between the CP residue of PrxI/II and either Cys23 or Cys45 of HMGB1, providing functional redundancy for HMGB1 oxidation. In high H2O2 concentrations, disulfide HMGB1 may proceed to the terminally oxidized form of sulfonic HMGB1, which may be localized within the band of disulfide HMGB1. However, the molecular mechanism requires further investigation. Mullen et al. reported redox independency of HMGB1 secretion, showing that HMGB1(C23S), HMGB1(C45S), and HMGB1(C23S/C45S/C106S) were secreted following TNFα treatment [42]. Although the underlying reason for this discrepancy remains unclear, it might be possibly due to cell stress imposed by the serum-free culture condition. Hopper et al. reported that HMGB1(C23S) and HMGB1(C45S) mutant forms are localized within the nuclear space after starvation stress, which is consistent with our data [43].
On the basis of our findings, we propose a model for the mechanism underlying regulated HMGB1 secretion from monocytes and macrophages. Inflammatory stimuli such as LPS and TNFα as well as chemical agents such as PMA and TSA induce H2O2 production from multiple sources including Nox and mitochondria in the cytoplasm, and the H2O2 molecules then diffuse to the nucleus. It is also likely that H2O2 produced as a result of the respiratory burst of neighboring neutrophils and macrophages can enter the cell and reach the nucleus, and that activation of Nox enzymes localized at the nuclear membrane may be a source of nuclear H2O2 [44,45]. Prx isoforms (I to VI) are abundantly present in the cytosol, mitochondria, ER, and/or peroxisome etc. In spite of high expression level of cytosolic PrxI or PrxII, PrxI/II bound to HMGB1 in the nucleus may use nuclear H2O2 for its interaction. The accumulation of H2O2 in the nucleus is first sensed by PrxI and PrxII, resulting in the rapid oxidation of CP and CR of each enzyme and the consequent formation of intersubunit disulfide bonds in each dimer. The oxidized PrxI or PrxII then transfers its oxidation state to reduced HMGB1 to generate disulfide HMGB1 with an intramolecular disulfide between Cys23 and Cys45. Disulfide HMGB1 is preferentially transported out of the nucleus as a result of its binding to the nuclear exportin CRM1 with higher affinity compared with that of reduced HMGB1—although, given that disulfide HMGB1 is more compact than the reduced form, preferential passive diffusion of disulfide HMGB1 cannot be excluded. It is also possible that, like acetylated HMGB1, disulfide HMGB1 may be less favored for nuclear import and thus accumulates in the cytosol. Disulfide HMGB1 in the cytosol is packed into lysosomes through an as yet unknown mechanism and is then secreted.
In our study, we cannot clearly explain why HMGB1 oxidation requires both PrxI and PrxII. We propose two kinds of possibility. First, HMGB1 proteins comprise ubiquitous, abundant nuclear proteins, the oxidative stage of which plays a critical role in the response to inflammatory stimuli. The concentration of PrxI and PrxII, however, is very limited in the nuclei of various cell lines (Suppl. Fig. 1H), requiring them to independently control the oxidation effect of HMGB1, thereby offering rapid and effective response to HMGB1 oxidation and inflammatory signaling. Second, thioredoxins are known to rapidly reduce disulfide HMGB1 to reduced HMGB1 [43]. Moreover, Prx has been reported to promote thioredoxin oxidation, in particular in cases of Prx-dependent Pap1 oxidation [46,47], along with PrxIV-mediated PDI oxidation in the ER [23,24]. PrxI or PrxII-mediated thioredoxin oxidation could result in the inhibition of HMGB1 reduction. In addition, depletion of cytosolic 2-Cys Prxs dramatically abolishes rapid protein thiol oxidation in an H2O2-dependent manner [33]. Therefore, the existence of the other antioxidant-dependent HMGB1 oxidation mechanisms cannot be ruled out.
Our study of HMGB1 oxidation was performed in an intracellular environment. Released HMGB1 can also be oxidized in extracellular environments such as human serum, saliva, and in cell culture medium with varying cell confluency. The reduced form of HMGB1 produced from E. coli could also be oxidized to the disulfide form within approximately ∼50 min in serum under ambient conditions in vitro, suggesting that Prx-independent HMGB1 oxidation is possible outside of cells. Considering that Prx could be secreted in an inflammatory condition [42] and our unpublished observation], both PrxI/II-dependent and independent mechanisms of HMGB1 oxidation are possible in the extracellular space. A more complete understanding of the oxidation kinetics of the reduced form of HMGB1, which is released from damaged tissue, and the oxidation mechanism of disulfide to sulfonic HMGB1 are necessary to predict the fate of HMGB1-mediated inflammation in tissue. H2O2 sensor and transducer function of PrxI and PrxII might also be needed in the cytosol to maintain HMGB1 in the disulfide form before it is packaged into lysosomes. Our results show that HMGB1 that is actively secreted in response to LPS stimulation is present in the disulfide form, not the reduced form.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
We thank Dr. Tobias P. Dick (German Cancer Research Center, Heidelberg, Germany) for Prx mutant plasmids (WT PrxII-SBP and mutants PrxII-SBP C51S, C172S, C52/172S), and Dr. In Sup Kil for discussion. This work was supported by the NRF funded by the Korean government (MEST) [2014R1A4A1008625 and 2017R1A2B3006704], the Research Center Program of Institute for Basic Science (IBS) in Korea (IBS-R026-D1), and Brain Korea 21 PLUS Project for Medical Science.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2019.101203.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Lee S.A., Kwak M.S., Kim S., Shin J.S. The role of high mobility group box 1 in innate immunity. Yonsei Med. J. 2014;55(5):1165–1176. doi: 10.3349/ymj.2014.55.5.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lotze M.T., Tracey K.J. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005;5(4):331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 3.Bonaldi T., Talamo F., Scaffidi P., Ferrera D., Porto A., Bachi A., Rubartelli A., Agresti A., Bianchi M.E. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22(20):5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Youn J.H., Shin J.S. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J. Immunol. 2006;177(11):7889–7897. doi: 10.4049/jimmunol.177.11.7889. [DOI] [PubMed] [Google Scholar]
- 5.Gardella S., Andrei C., Ferrera D., Lotti L.V., Torrisi M.R., Bianchi M.E., Rubartelli A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;3(10):995–1001. doi: 10.1093/embo-reports/kvf198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Youn J.H., Oh Y.J., Kim E.S., Choi J.E., Shin J.S. High mobility group box 1 protein binding to lipopolysaccharide facilitates transfer of lipopolysaccharide to CD14 and enhances lipopolysaccharide-mediated TNF-α production in human monocytes. J. Immunol. 2008;180(7):5067–5074. doi: 10.4049/jimmunol.180.7.5067. [DOI] [PubMed] [Google Scholar]
- 7.Kwak M.S., Lim M., Lee Y.J., Lee H.S., Kim Y.H., Youn J.H., Choi J.E., Shin J.S. HMGB1 binds to lipoteichoic acid and enhances TNF-α and IL-6 production through HMGB1-mediated transfer of lipoteichoic acid to CD14 and TLR2. J. Innate Immun. 2015;7(4):405–416. doi: 10.1159/000369972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schiraldi M., Raucci A., Munoz L.M., Livoti E., Celona B., Venereau E., Apuzzo T., De Marchis F., Pedotti M., Bachi A., Thelen M., Varani L., Mellado M., Proudfoot A., Bianchi M.E., Uguccioni M. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 2012;209(3):551–563. doi: 10.1084/jem.20111739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sha Y., Zmijewski J., Xu Z., Abraham E. HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J. Immunol. 2008;180(4):2531–2537. doi: 10.4049/jimmunol.180.4.2531. [DOI] [PubMed] [Google Scholar]
- 10.Tian J., Avalos A.M., Mao S.Y., Chen B., Senthil K., Wu H., Parroche P., Drabic S., Golenbock D., Sirois C., Hua J., An L.L., Audoly L., La Rosa G., Bierhaus A., Naworth P., Marshak-Rothstein A., Crow M.K., Fitzgerald K.A., Latz E., Kiener P.A., Coyle A.J. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007;8(5):487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 11.Tang D., Kang R., Livesey K.M., Cheh C.W., Farkas A., Loughran P., Hoppe G., Bianchi M.E., Tracey K.J., Zeh H.J., 3rd, Lotze M.T. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010;190(5):881–892. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Min H.J., Ko E.A., Wu J., Kim E.S., Kwon M.K., Kwak M.S., Choi J.E., Lee J.E., Shin J.S. Chaperone-like activity of high-mobility group box 1 protein and its role in reducing the formation of polyglutamine aggregates. J. Immunol. 2013;190(4):1797–1806. doi: 10.4049/jimmunol.1202472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang H., Lundback P., Ottosson L., Erlandsson-Harris H., Venereau E., Bianchi M.E., Al-Abed Y., Andersson U., Tracey K.J., Antoine D.J. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1) Mol. Med. 2012;18:250–259. doi: 10.2119/molmed.2011.00389. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Venereau E., Casalgrandi M., Schiraldi M., Antoine D.J., Cattaneo A., De Marchis F., Liu J., Antonelli A., Preti A., Raeli L., Shams S.S., Yang H., Varani L., Andersson U., Tracey K.J., Bachi A., Uguccioni M., Bianchi M.E. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 2012;209(9):1519–1528. doi: 10.1084/jem.20120189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang H., Hreggvidsdottir H.S., Palmblad K., Wang H., Ochani M., Li J., Lu B., Chavan S., Rosas-Ballina M., Al-Abed Y., Akira S., Bierhaus A., Erlandsson-Harris H., Andersson U., Tracey K.J. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. U. S. A. 2010;107(26):11942–11947. doi: 10.1073/pnas.1003893107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kazama H., Ricci J.E., Herndon J.M., Hoppe G., Green D.R., Ferguson T.A. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 2008;29(1):21–32. doi: 10.1016/j.immuni.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liesz A., Dalpke A., Mracsko E., Antoine D.J., Roth S., Zhou W., Yang H., Na S.Y., Akhisaroglu M., Fleming T., Eigenbrod T., Nawroth P.P., Tracey K.J., Veltkamp R. DAMP signaling is a key pathway inducing immune modulation after brain injury. J. Neurosci. 2015;35(2):583–598. doi: 10.1523/JNEUROSCI.2439-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lundback P., Stridh P., Klevenvall L., Jenkins R.E., Fischer M., Sundberg E., Andersson U., Antoine D.J., Harris H.E. Characterization of the inflammatory properties of actively released HMGB1 in juvenile idiopathic arthritis. Antioxid. Redox Signal. 2016;24(12):605–619. doi: 10.1089/ars.2014.6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rhee S.G., Woo H.A., Kang D. The role of peroxiredoxins in the transduction of H2O2 signals. Antioxid. Redox Signal. 2018;28(7):537–557. doi: 10.1089/ars.2017.7167. [DOI] [PubMed] [Google Scholar]
- 20.Wood Z.A., Schroder E., Robin Harris J., Poole L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003;28(1):32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 21.Chae H.Z., Kim H.J., Kang S.W., Rhee S.G. Characterization of three isoforms of mammalian peroxiredoxin that reduce peroxides in the presence of thioredoxin. Diabetes Res. Clin. Pract. 1999;45(2–3):101–112. doi: 10.1016/s0168-8227(99)00037-6. [DOI] [PubMed] [Google Scholar]
- 22.Rhee S.G., Kil I.S. Multiple functions and regulation of mammalian peroxiredoxins. Annu. Rev. Biochem. 2017;86:749–775. doi: 10.1146/annurev-biochem-060815-014431. [DOI] [PubMed] [Google Scholar]
- 23.Zito E., Melo E.P., Yang Y., Wahlander A., Neubert T.A., Ron D. Oxidative protein folding by an endoplasmic reticulum-localized peroxiredoxin. Mol. Cell. 2010;40(5):787–797. doi: 10.1016/j.molcel.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tavender T.J., Springate J.J., Bulleid N.J. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 2010;29(24):4185–4197. doi: 10.1038/emboj.2010.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jarvis R.M., Hughes S.M., Ledgerwood E.C. Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic. Biol. Med. 2012;53(7):1522–1530. doi: 10.1016/j.freeradbiomed.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 26.Veal E.A., Findlay V.J., Day A.M., Bozonet S.M., Evans J.M., Quinn J., Morgan B.A. A 2-Cys peroxiredoxin regulates peroxide-induced oxidation and activation of a stress-activated MAP kinase. Mol. Cell. 2004;15(1):129–139. doi: 10.1016/j.molcel.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 27.Latimer H.R., Veal E.A. Peroxiredoxins in regulation of MAPK signalling pathways; sensors and barriers to signal transduction. Mol. Cell. 2016;39(1):40–45. doi: 10.14348/molcells.2016.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sobotta M.C., Liou W., Stocker S., Talwar D., Oehler M., Ruppert T., Scharf A.N., Dick T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015;11(1):64–70. doi: 10.1038/nchembio.1695. [DOI] [PubMed] [Google Scholar]
- 29.Park H.S., Jung H.Y., Park E.Y., Kim J., Lee W.J., Bae Y.S. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-κB. J. Immunol. 2004;173(6):3589–3593. doi: 10.4049/jimmunol.173.6.3589. [DOI] [PubMed] [Google Scholar]
- 30.Stocker S., Maurer M., Ruppert T., Dick T.P. A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 2018;14(2):148–155. doi: 10.1038/nchembio.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oh Y.J., Youn J.H., Ji Y., Lee S.E., Lim K.J., Choi J.E., Shin J.S. HMGB1 is phosphorylated by classical protein kinase C and is secreted by a calcium-dependent mechanism. J. Immunol. 2009;182(9):5800–5809. doi: 10.4049/jimmunol.0801873. [DOI] [PubMed] [Google Scholar]
- 32.Karlsson A., Nixon J.B., McPhail L.C. Phorbol myristate acetate induces neutrophil NADPH-oxidase activity by two separate signal transduction pathways: dependent or independent of phosphatidylinositol 3-kinase. J. Leukoc. Biol. 2000;67(3):396–404. doi: 10.1002/jlb.67.3.396. [DOI] [PubMed] [Google Scholar]
- 33.ten Berge D., Brouwer A., Korving J., Martin J.F., Meijlink F. Prx1 and Prx2 in skeletogenesis: roles in the craniofacial region, inner ear and limbs. Development. 1998;125(19):3831–3842. doi: 10.1242/dev.125.19.3831. [DOI] [PubMed] [Google Scholar]
- 34.Sun S., Han Y., Liu J., Fang Y., Tian Y., Zhou J., Ma D., Wu P. Trichostatin A targets the mitochondrial respiratory chain, increasing mitochondrial reactive oxygen species production to trigger apoptosis in human breast cancer cells. PLoS One. 2014;9(3) doi: 10.1371/journal.pone.0091610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Willenbrock S., Braun O., Baumgart J., Lange S., Junghanss C., Heisterkamp A., Nolte I., Bullerdiek J., Murua Escobar H. TNF-α induced secretion of HMGB1 from non-immune canine mammary epithelial cells (MTH53A) Cytokine. 2012;57(2):210–220. doi: 10.1016/j.cyto.2011.11.011. [DOI] [PubMed] [Google Scholar]
- 36.Chen X., Andresen B.T., Hill M., Zhang J., Booth F., Zhang C. Role of reactive oxygen species in tumor necrosis factor-alpha induced endothelial dysfunction. Curr. Hypertens. Rev. 2008;4(4):245–255. doi: 10.2174/157340208786241336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Konishi H., Tanaka M., Takemura Y., Matsuzaki H., Ono Y., Kikkawa U., Nishizuka Y. Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc. Natl. Acad. Sci. U. S. A. 1997;94(21):11233–11237. doi: 10.1073/pnas.94.21.11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poole L.B., Nelson K.J. Discovering mechanisms of signaling-mediated cysteine oxidation. Curr. Opin. Chem. Biol. 2008;12(1):18–24. doi: 10.1016/j.cbpa.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mullen L., Hanschmann E.M., Lillig C.H., Herzenberg L.A., Ghezzi P. Cysteine oxidation targets peroxiredoxins 1 and 2 for exosomal release through a novel mechanism of redox-dependent secretion. Mol. Med. 2015;21:98–108. doi: 10.2119/molmed.2015.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoppe G., Talcott K.E., Bhattacharya S.K., Crabb J.W., Sears J.E. Molecular basis for the redox control of nuclear transport of the structural chromatin protein HMGB1. Exp. Cell Res. 2006;312(18):3526–3538. doi: 10.1016/j.yexcr.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 41.Provost C., Choufani F., Avedanian L., Bkaily G., Gobeil F., Jacques D. Nitric oxide and reactive oxygen species in the nucleus revisited. Can. J. Physiol. Pharmacol. 2010;88(3):296–304. doi: 10.1139/Y10-011. [DOI] [PubMed] [Google Scholar]
- 42.Stanicka J., Russell E.G., Woolley J.F., Cotter T.G. NADPH oxidase-generated hydrogen peroxide induces DNA damage in mutant FLT3-expressing leukemia cells. J. Biol. Chem. 2015;290(15):9348–9361. doi: 10.1074/jbc.M113.510495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown J.D., Day A.M., Taylor S.R., Tomalin L.E., Morgan B.A., Veal E.A. A peroxiredoxin promotes H2O2 signaling and oxidative stress resistance by oxidizing a thioredoxin family protein. Cell Rep. 2013;5(5):1425–1435. doi: 10.1016/j.celrep.2013.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Day A.M., Brown J.D., Taylor S.R., Rand J.D., Morgan B.A., Veal E.A. Inactivation of a peroxiredoxin by hydrogen peroxide is critical for thioredoxin-mediated repair of oxidized proteins and cell survival. Mol. Cell. 2012;45(3):398–408. doi: 10.1016/j.molcel.2011.11.027. [DOI] [PubMed] [Google Scholar]
- 45.Zandarashvili L., Sahu D., Lee K., Lee Y.S., Singh P., Rajarathnam K., Iwahara J. Real-time kinetics of high-mobility group box 1 (HMGB1) oxidation in extracellular fluids studied by in situ protein NMR spectroscopy. J. Biol. Chem. 2013;288(17):11621–11627. doi: 10.1074/jbc.M113.449942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee T.H., Kim S.U., Yu S.L., Kim S.H., Park D.S., Moon H.B., Dho S.H., Kwon K.S., Kwon H.J., Han Y.H., Jeong S., Kang S.W., Shin H.S., Lee K.K., Rhee S.G., Yu D.Y. Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood. 2003;101(12):5033–5038. doi: 10.1182/blood-2002-08-2548. [DOI] [PubMed] [Google Scholar]
- 47.Bae S.H., Sung S.H., Cho E.J., Lee S.K., Lee H.E., Woo H.A., Yu D.Y., Kil I.S., Rhee S.G. Concerted action of sulfiredoxin and peroxiredoxin I protects against alcohol-induced oxidative injury in mouse liver. Hepatology. 2011;53(3):945–953. doi: 10.1002/hep.24104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.