

 Berberine is a bioactive alkaloid used in Chinese medicine and has numerous positive effects on biological systems.
Berberine is a bioactive alkaloid used in Chinese medicine and has numerous positive effects on biological systems.
Abstract
Berberine is a bioactive alkaloid used in Chinese medicine and has numerous positive effects on biological systems. This paper describes the facile and highly efficient synthesis of some carbohydrate modified berberine derivatives, and conjugation of the carbohydrate moiety with berberine was finished by “click” chemistry. The cytotoxicity and anti-diabetic measurements of all berberine derivatives were accomplished on HepG2 cell lines, and the results indicated that most of the derivatives exhibit higher anti-diabetic activity than berberine. The mannose modified berberine derivative has significantly lower cytotoxicity than berberine, and the induced IC50 value of this derivative is nearly 1.5 times that of berberine. Furthermore, this mannose modified berberine derivative exhibits high anti-diabetic activity at both high and low drug concentrations, thereby indicating its potential application for the development of novel anti-diabetic drugs.
Introduction
Berberine (Fig. 1), an active isoquinoline alkaloid extracted from Chinese herb Coptis chinensis,1 has been widely used for the treatment of gastrointestinal infection and diarrhea in China.2,3 Recent studies revealed that berberine has various pharmacological activities such as anti-microbial,4 anti-diabetic,5 anti-hyperlipidemic,6 anti-tumor,7 anti-inflammatory,8 and anti-parasitic.9 As an anti-diabetic agent, berberine obviously reduces the blood glucose in type 2 diabetic patients and has been optionally used by physicians in China for several years.10,11 It has been proved that berberine can inhibit the activity of α-glucosidase, which hydrolyzes the oligosaccharides and polysaccharides to release free glucose, thereby leading to the inefficient absorption rates of glucose, as shown in Fig. 2.12 Furthermore, berberine can activate AMP-activated protein kinase (AMPK), an important fuel gauge protein that monitors the energy levels in mammalian cells, which might partially contribute to glucose consumption.13,14 There are some other proposed mechanisms that can explain the anti-diabetic activity of berberine, such as mitochondrial function inhibition,15 inhibition of PPARγ and C/EBPα,16,17 down regulation of the PEPCK and G6Pase gene expression,18etc.
Fig. 1. Chemical structure of berberine.
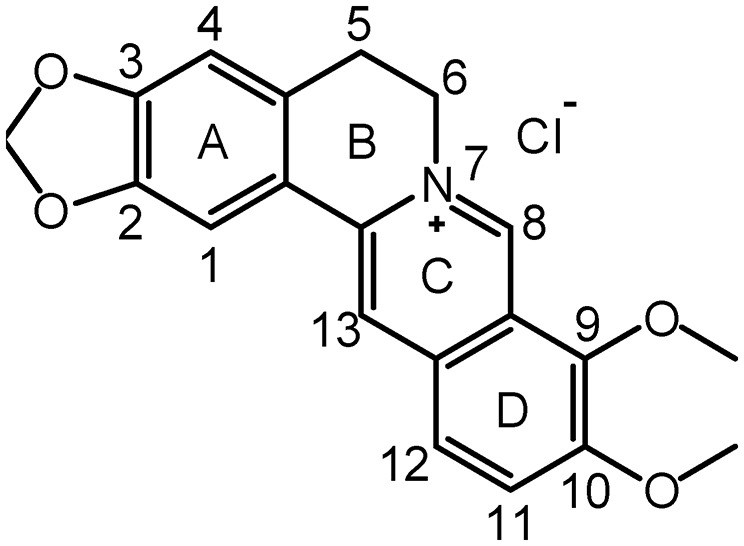
Fig. 2. Proposed anti-diabetic mechanisms of berberine.

Therefore, berberine is a promising medicine for the treatment of type 2 diabetes. But the problem is that oral administration of berberine usually gives extremely low bioavailability (<5%),10 while increasing the oral dose and parenteral drug delivery usually causes severe side effects. Consequently, several novel strategies have been developed for improving the bioavailability of berberine, which alternatively enhance its anti-diabetic activity. The most important strategy is the structural modification of berberine to provide novel fully-synthetic anti-diabetic drug candidates.19 For example, the 9-O-hydrophilic and hydrophobic modification of berberine can both improve its bioavailability and enhance the anti-diabetic activity,19,20 and some C-8,13-substituted berberine derivatives have significant glucose-lowering properties,4,21 and the 9-OH or 10-OH pseudoberberines also exhibited high anti-diabetic activity.22 But, there is no berberine derivative that showed potential application as an anti-diabetic drug, and many of these derivatives are unstable under mild conditions which are easily degraded into berberine and related analogs in vitro. Consequently, some stable berberine derivatives should be designed and synthesized to develop effective anti-diabetic drug candidates.
Previous research revealed that the presence of a carbohydrate moiety in drugs had the ability to improve their bio-availability and receptor-binding affinity, thus resulting in significant drug activity enhancement.23,24 Studies on metabolites of berberine in rats revealed that 9-O-glucosyl-berberine (Fig. 3) existed as one of the major metabolites.25,26 Therefore, synthesis of 9-O-hydrophilic berberine derivatives to improve its water solubility might be a new idea to increase its bio-availability.19,20 This conjecture was confirmed by Chen and co-workers who had synthesized 9-O-glucosyl-berberine (Fig. 3) and proved that hydrophilic modification of berberine could obviously improve its bio-availability.19,20 However, this derivative showed poor stability under acidic conditions and at high temperature (>45 °C).19,20 In order to improve the stability of 9-O-glucosyl-berberine and simultaneously enhance its anti-diabetic activity, we now focused on the preparation of some carbohydrate modified berberine derivatives 1–5 (Fig. 4). The N-linked glucose, galactose, mannose, ribose, and rhamnose, which have different contents of hydroxyl groups and water solubilities, were chosen as carbohydrate moieties. The modification was mainly on the C-9 position of berberine, because the key intermediate 9-OH pseudoberberine (6, Scheme 1) could be easily available through a pyrolysis strategy. Furthermore, the highly efficient “click” chemistry was used to accomplish the conjugation of monosaccharides with berberine, which could form a triazole27 linker to give stable glucosyl berberines. Compared with O-glycosides, N-glycosides are stable enough in the presence of glycoside hydrolases under physiological conditions. The synthesized derivatives were subjected to the anti-diabetic evaluation in HepG2 cell lines, and their cytotoxicity activity was determined through the cell counting kit-8 (CCK-8) assay.
Fig. 3. 9-O-Glucosyl-berberine.
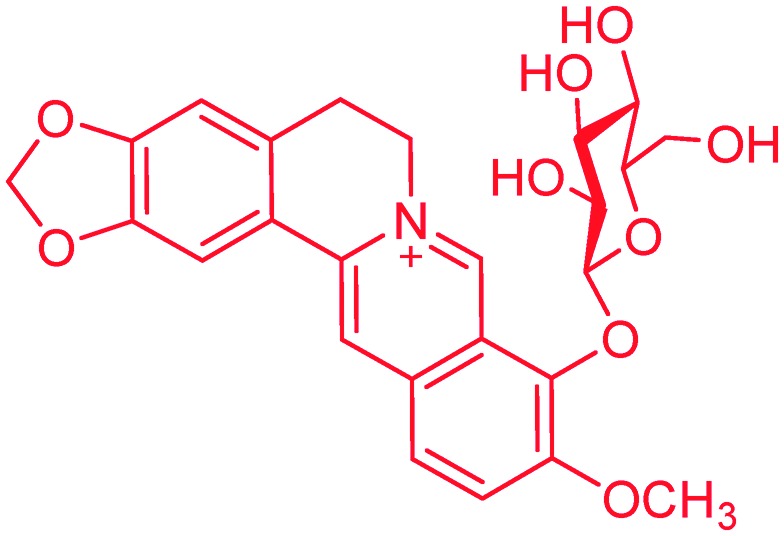
Fig. 4. Carbohydrate modified berberine derivatives 1–5.

Scheme 1. Synthesis of berberine derivatives 1 and 2.

Results and discussion
Synthesis of berberine derivatives
The synthesis routes of glucose and galactose modified berberine derivatives 1 and 2 are outlined in Scheme 1. The commercially available berberine chloride was heated at 185–195 °C under high vacuum (20–30 mm Hg) to produce berberrubine 6.28 Without any purification, crude 6 was reacted with propargyl bromide 7 in DMF to give 8 with an alkynyl group. In parallel, treatment of pentaacetyl β-d-glucose 9 with HBr (33% in AcOH) afforded glycosyl bromide 11, which was immediately converted into compound 13 in the presence of NaN3 and DMF with 69% overall yield. Due to the neighboring group participation effect29–33 of the 2-O-acetyl group in 11, this reaction was highly stereoselective and gave β-linked azido sugar 13 as the only product. Thereafter, the acetyl groups in 13 were removed with CH3ONa, and the resultant 15 was subjected to the Cu-catalyzed “click” reaction with alkylated berberine derivative 8 to afford the desired compound 1 in 56% total yield. Using the same protocol, we synthesized the galactose modified berberine derivative 2 from 10 in four steps with a high overall yield (50%). Therefore, this synthetic route was highly efficient and could be applied for the preparation of various carbohydrate modified berberine derivatives.
Using this highly efficient synthetic strategy, we focused on the synthesis of a mannose modified berberine derivative 3 as shown in Scheme 2. Our initial work was the synthesis of azido sugar 19α, through treatment of d-mannose pentaacetate 17 with HBr followed by reaction of the resultant glucosyl bromide 18 with NaN3 in DMF. This transformation generated an α, β mixture of 19 with only 29% total yield. As an alternative, we employed a new strategy based on Carrington's method,34,35 which could directly afford the azido sugar 19α under mild conditions. Therefore, treatment of 17 with TMSN3 under the promotion of SnCl4 in CH2Cl2 produced 19α with absolute α-selectivity and 88% isolated yield. The high α-stereoselectivity of this reaction was also caused by the neighboring participation effect of the 2-O-acetyl group in 17. Subsequent deacetylation of 19α using CH3ONa followed by the Cu-catalyzed “click” reaction of 20 with 8 under the same condition as described previously, facilitated the target derivative 3 in 52% overall yield. This simplified and highly efficient strategy was further applied for the synthesis of rhamnopyranose modified berberine derivative 4 (in three steps and 35% total yield) and ribofuranose modified berberine derivative 5 (in three steps and 44% total yield).
Scheme 2. Synthesis of berberine derivatives 3–5.

CCK-8 assay for cytotoxicity
It has been proved that berberine and its derivatives have the ability to up-regulate the reactive oxygen species (ROS), hence depressing the proliferation of HepG2 cells.36 This feature can be applied for the cytotoxicity assessment of berberine derivatives. Therefore, all the synthesized berberine derivatives were tested for their cytotoxicity through the standard CCK-8 assay37 at different concentrations, and their half maximal inhibitory concentration (IC50) was calculated (Fig. 5 and Table 1). The viability of HepG2 cells obviously decreased with the concentration increasing from 0.2 to 625 μg mL–1, which indicated that all the synthesized berberine derivatives exerted cytotoxicity in a dose-dependent manner. The glucose, galactose and mannose modified berberine derivatives 1–3 exhibited lower cytotoxicity than berberine, in particular compound 3 with an IC50 value of 72.19 μg mL–1, which was nearly 1.5 times that of berberine (Fig. 5A and Table 1). Meanwhile, the rhamnose and ribose modified berberine derivatives 4 and 5 showed slightly higher cytotoxicity than berberine (Fig. 5B and Table 1).
Fig. 5. The effect of (A) glucose, galactose and mannose modified berberine derivatives (1, 2 and 3) and (B) rhamnose and ribose modified berberine derivatives (4 and 5) on the HepG2 cell viability. Data is expressed as means ± SD; n = 3.

Table 1. The half maximal inhibitory concentrations (IC50) of berberine and its derivatives on HepG2 cells.
| Compounds | R = | IC50 (μg mL–1) |
| 1 |
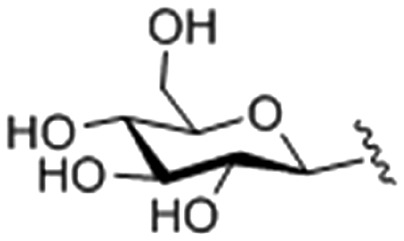
|
62.94 ± 0.032 |
| 2 |
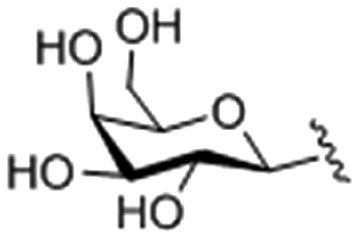
|
59.29 ± 0.014 |
| 3 |
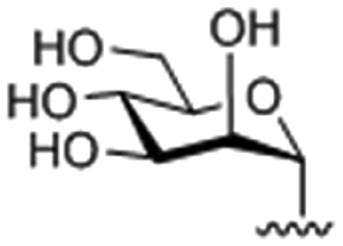
|
72.19 ± 0.051 |
| 4 |
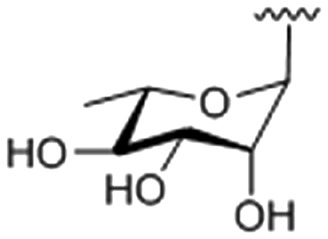
|
49.27 ± 0.013 |
| 5 |
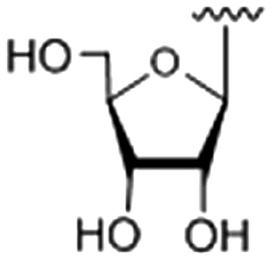
|
44.02 ± 0.017 |
| Berberine | — | 53.91 ± 0.014 |
Anti-diabetic investigation on HepG2 cell lines
The HepG2 cell line is derived from human hepatocellular carcinoma and exhibits a similar phenotype to a hepatocyte.4 The insulin receptors on the surface of HepG2 cells can significantly decrease under the stimulation of a high level of insulin, which then results in low glucose consumption (GC).38,39 Consequently, the HepG2 cell line has been widely used as a model for in vitro anti-diabetic investigation. In order to screen the anti-diabetic activity of the synthesized berberine derivatives 1–5, we determined the GC values of HepG2 cells19 after treatment with different concentrations (0.2, 1 and 5 μg mL–1) of berberine and its derivatives (Fig. 6). All the GC values of berberine and its derivatives on HepG2 cell lines were higher than that of the negative control group (0.5% DMSO), which indicated that berberine and its derivatives had different degrees of anti-diabetic activity. Compared with berberine, the carbohydrate modified derivatives showed higher anti-diabetic activity, in particular the mannose and ribose modified berberine derivatives 3 and 5 (Table 2). These two compounds showed the highest anti-diabetic activity when the drug concentration was 5 μg mL–1, and induced an increase of GC values on HepG2 cell lines by 10.81% and 14.83%, respectively. More importantly, compounds 3 and 5 still showed high anti-diabetic activity at concentrations of 0.2 and 1 μg mL–1, suggesting their low effective concentrations. The galactose modified berberine derivative 2 had similar activity to berberine at high concentrations (1 and 5 μg mL–1), but with poor activity at low concentration (0.2 μg mL–1). The rhamnose modified berberine derivative 4 showed relatively poor anti-diabetic activity both at high and low concentrations. Furthermore, modification of berberine with glucose (compound 1) exhibited similar anti-diabetic activity at both high and low concentrations.
Fig. 6. The effect of glucose, galactose, mannose, rhamnose and ribose modified berberine derivatives (1, 2, 3, 4 and 5) on glucose consumption of HepG2 cells after 48 h of treatment. Data are expressed as means ± SD; n = 6.

Table 2. Data of glucose consumption activity in vitro for all compounds with a concentration of 5 μg mL–1 (means ± SD, n = 8).
| Compound | GC (Mm) | Percentage increase in GC (%) compared with berberine |
| 0.5 % DMSO | 3.41 ± 0.033 | — |
| Berberine | 4.72 ± 0.071 | — |
| 1 | 4.83 ± 0.053 | 2.23 |
| 2 | 4.71 ± 0.033 | 0.21 |
| 3 | 5.23 ± 0.025 | 10.81 |
| 4 | 4.55 ± 0.023 | –3.60 |
| 5 | 5.42 ± 0.028 | 14.83 |
Stability under acidic conditions
Berberine derivative 3, with low cytotoxicity and high anti-diabetic activity, was further subjected to stability investigation under acidic conditions. Both berberine and compound 3 were dissolved in the mixed solution of acetonitrile and water (1/1, v/v, 0.5 mg mL–1), in which its pH value was adjusted to 1.5 using phosphoric acid. The solution was then analyzed by HPLC with the absorption wavelength at 320 nm (Shim-pack C8 250 mm × 4.6 mm/5 μm column). The elution was 50% acetonitrile in 0.05 mol L–1 of aqueous KH2PO4, and the flow rate was 1 mL min–1. The results (Fig. 7) indicated that the content of berberine declined by 1.8% after 5 hours, while that of derivative 3 only declined by 0.3%. Moreover, derivative 3 showed higher aqueous solubility (9 mg mL–1) than berberine (1.75 mg mL–1). The improvement on acid stability and aqueous solubility of berberine derivatives has been proved critical for enhancing their bio-availability.4,21
Fig. 7. Stability of berberine and compound 3 under acidic conditions.

Conclusions
In conclusion, some novel carbohydrate modified berberine derivatives were designed and synthesized. Synthesis of azido sugars was carried out using Carrington's method, which could directly convert the acetylated sugar into azido sugar with high efficiency and stereoselectivity. Attachment of the azido sugars to the alkynylated berberine derivative 8 was finished by “click” chemistry, and all the target compounds were achieved with high total yields. Therefore, this strategy could be applied for the convenient synthesis of more complex carbohydrate modified berberine derivatives. All the synthesized compounds were subjected to the cytotoxicity measurement through the CCK-8 assay on HepG2 cell lines, and the glucose, galactose and mannose modified berberine derivatives 1–3 exhibited lower cytotoxicity than berberine. Furthermore, their anti-diabetic activity was investigated on the HepG2 cell line. The results indicated that the mannose and ribose modified berberine derivatives 3 and 5 showed higher anti-diabetic activity than berberine both at high and low concentrations. Consequently, the mannose modified berberine derivative 3 with low cytotoxicity, high anti-diabetic activity on the HepG2 cell lines and high stability under acidic conditions could be a potential candidate for further research. To further investigate the structure–activity relationship, it is interesting to prepare more berberine derivatives with hydrophilic carbohydrate moieties, such as the oligosaccharide-based berberine conjugates, and this work is now under processing by our group.
Experimental procedures
General procedures
Berberine chloride was purchased from Tokyo Chemical Industry (TCI, B0450). HepG2 cell lines were acquired from Shanghai Gefan Biotechnology Co., Ltd (GF589). CCK-8 was obtained from Dōjindo Laboratories (Dojindo). DMEM cell culture was bought from GE Healthcare Life Sciences (SH30243.01). All the other reagents were commercially available and used without further purification unless otherwise noted. Thin layer chromatography (TLC) was performed with silica gel HF254 plates and detection was conducted by charring with 30% (v/v) H2SO4 in methanol or using a UV detector. The 1H NMR and 13C NMR spectra were recorded on a Bruker AV 400 spectrometer, and chemical shifts (δ) are given in ppm downfield from internal TMS as an internal standard. The high-resolution mass spectra (HRMS) were recorded on an Agilent QTOF 6520 using the electrospray ionization (ESI) technique to introduce the sample.
9-O-(Propargyl) berberine chloride (8)
Berberine chloride (5 g, 13.48 mmol) was heated at 190 °C under high vacuum (20–30 mmHg) for 30 min to give berberrubine 6 as a purple solid. The crude solid was dissolved in 50 mL of DMF, and propargyl bromide (7, 4.73 g, 40.44 mmol) was added. The reaction mixture was heated to 80 °C and stirred for 6 h, and the color of the mixture changed from purple to yellow. Then, the reaction mixture was cooled to room temperature and stirred for another 2 h, the mixture was filtered, and the filter cake was washed with cold methanol and dried to give 9-O-(propargyl) berberine chloride 8 (3.83 g, 72%) as a yellow solid. 1H NMR (400 MHz, DMSO-d6): δ 9.88 (s, 1H, CH N), 8.97 (s, 1H, ArH),8.23 (d, J = 9.2 Hz, 1H, ArH), 8.04 (d, J = 9.2 Hz, 1H, ArH), 7.80 (s, 1H, ArH), 7.10 (s, 1H, ArH), 6.18 (s, 2H, –OCH2O–), 5.10 (d, J = 2.4 Hz, 2H, –CH2C CH), 4.96 (t, J = 6.4 Hz, 2H, CH2), 4.08 (s, 3H, OCH3), 3.62–3.61 (m, 1H, –CH2C CH), 3.21 (t, J = 6.0 Hz, 2H, CH2); 13C NMR (100 MHz, DMSO-d6): δ 151.18, 150.36, 148.17, 145.79, 141.18, 138.11, 133.40, 131.20, 127.03, 124.73, 122.56, 120.87, 120.77, 108.90, 105.94, 102.58, 80.26, 79.24, 61.42, 57.63, 55.78, 26.81; ESI-TOF HRMS (m/z): calcd for C22H18NO4+, [M – Cl]+, 360.1230; found, 360.1238.
2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl azide (13)
To the solution of 1,2,3,4,6-penta-O-acetyl-d-glucopyranose 9 (5.0 g, 12.82 mmol) in dry CH2Cl2 (30 mL) was added hydrogen bromide (33% in acetic acid, 15 mL) at 0 °C. The reaction mixture was allowed to warm to room temperature, stirred for 2 h, and then poured into 150 mL of ice water. After which, the mixture was extracted with 150 mL of CH2Cl2, and the organic layer was washed with cold saturated NaHCO3 solution (50 mL × 2) followed by cold water (50 mL). The organic phase was dried with anhydrous Na2SO4 and the solvent was evaporated to give 11 (2,3,4,6-tetra-O-acetyl-α-d-glucopyranosyl bromide) as a colorless oil. Next, the crude oil was dissolved in 30 mL of DMF, and a solution of NaN3 (2.5 g, 38.46 mmol) in 5 mL of water was added at 0 °C. The reaction mixture was slowly warmed to room temperature and stirred for 3 h. After TLC (petroleum ether/ethyl acetate, 2/1, v/v) showed the conversion of the starting material, the reaction mixture was diluted with 150 mL of CH2Cl2 and washed with water (100 mL × 3). The organic layer was dried and evaporated, and the desired 13 (3.30 g, 69%) was obtained as white crystals after recrystallization from ethanol. 1H NMR (400 MHz, CDCl3): δ 5.22 (t, J = 9.6 Hz, 1H, H-3), 5.11 (t, J = 9.6 Hz, 1H, H-2), 4.96 (t, J = 9.2 Hz, 1H, H-4), 4.64 (d, J = 8.8 Hz, 1H, H-1), 4.27 (dd, J = 4.8, 12.4 Hz, 1H, H-6), 4.18 (dd, J = 2.0, 12.4 Hz, 1H, H-6′), 3.80 (dq, J = 2.4, 10.0 Hz, 1H, H-5), 2.11 (s, 3H, CH3CO), 2.08 (s, 3H, CH3CO), 2.04 (s, 3H, CH3CO), 2.01 (s, 3H, CH3CO); 13C NMR (100 MHz, CDCl3): δ 170.57, 170.09, 169.28, 169.18, 87.93, 74.06, 72.63, 70.67, 67.92, 61.68, 20.70, 20.56, 20.54; ESI-TOF HRMS (m/z): calcd for C14H19N3O9Na, [M + Na]+, 396.1014; found, 396.1007; calcd for C14H23N4O9, [M + NH4]+, 391.1460; found, 391.1453.
9-O-(1-(β-d-glucopyranosyl)-4-methylene-1H-1,2,3-triazole)berberine chloride (1)
To the solution of 2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl azide 13 (200 mg, 0.536 mmol) in methanol (5 mL) was added CH3ONa (1 M in methanol) until the pH value reached 10.0. The reaction mixture was stirred at room temperature for 2 h, and TLC (CH2Cl2/methanol, 5/1, v/v) showed the conversion of the starting material. To this mixture was added 9-O-(propargyl) berberine chloride (8, 176 mg, 0.447 mmol) followed by CuSO4·5H2O (67 mg, 0.268 mmol) and sodium ascorbate (53 mg, 0.268 mmol). The reaction mixture was then heated to 60 °C and stirred for 5 h. After TLC (CH2Cl2/methanol/water, 10/10/1, v/v) showed the conversion of the starting material, the solvent was evaporated, and the resultant residue was subjected to column chromatography to give the desired 1 (150 mg, 56%) as a yellow solid. 1H NMR (400 MHz, DMSO-d6): δ 9.59 (s, 1H, CH N), 8.92 (s, 1H, ArH), 8.61 (s, 1H, triazole-H), 8.22 (d, J = 9.2 Hz, 1H, ArH), 8.04 (d, J = 9.2 Hz, 1H, ArH), 7.78 (s, 1H, ArH), 7.09 (s, 1H, ArH), 6.18 (s, 2H, –OCH2O–), 5.54 (d, J = 9.2 Hz, 1H, glucose-H1), 5.50–5.43 (m, 2H, CH2), 5.38 (s, 2H, CH2), 5.20 (s, 1H, OH), 4.89 (t, J = 15.2 Hz, 2H, CH2), 4.62 (s, 1H, OH), 4.11 (s, 3H, OCH3), 3.76 (t, J = 8.8 Hz, 1H), 3.71–3.67 (m, 1H), 3.45–3.41 (m, 2H), 3.25–3.17 (m, 4H); 13C NMR (100 MHz, DMSO-d6): δ 151.40, 150.32, 148.14, 145.54, 142.89, 142.06, 137.94, 133.38, 131.17, 127.02, 124.97, 124.53, 122.42, 120.85, 120.74, 108.90, 105.91, 102.56, 87.95, 80.42, 77.34, 72.60, 70.12, 66.97, 61.22, 57.56, 55.88, 26.82; ESI-TOF HRMS (m/z): calcd for C28H29N4O9+, [M – Cl]+, 565.1929; found, 565.1936.
2,3,4,6-Tetra-O-acetyl-β-d-galactopyranosyl azide (14)
Compound 14 was achieved from 10 (5.0 g, 12.82 mmol) as a white solid (3.68 g, 77%), using the same procedure for the synthesis of 13. 1H NMR (400 MHz, CDCl3): δ 5.42 (d, J = 3.2 Hz, 1H, H-4), 5.17 (t, J = 9.2 Hz, 1H, H-2), 5.05 (dd, J = 3.6, 10.4 Hz, 1H, H-3), 4.59 (d, J = 8.8 Hz, 1H, H-1), 4.21–4.13 (m, 2H, 2 × H-6), 4.02 (t, J = 6.4 Hz, 1H, H-5), 2.17 (s, 3H, CH3CO), 2.10 (s, 3H, CH3CO), 2.07 (s, 3H, CH3CO), 1.99 (s, 3H, CH3CO); 13C NMR (100 MHz, CDCl3): δ 170.34, 170.09, 169.97, 169.34, 88.33, 72.91, 70.75, 68.10, 66.87, 61.23, 20.67, 20.66, 20.62, 20.52; ESI-TOF HRMS (m/z): calcd for C14H19N3O9Na, [M + Na]+, 396.1014; found, 396.1012; calcd for C14H23N4O9, [M + NH4]+, 391.1460; found, 391.1455.
9-O-(1-(β-d-galactopyranosyl)-4-methylene-1H-1,2,3-triazole)berberine chloride (2)
Compound 2 was achieved from 14 (200 mg, 0.536 mmol) and 8 (176 mg, 0.447 mmol) as a yellow solid (174 mg, 65%), using the same procedure for the synthesis of 1. 1H NMR (400 MHz, DMSO-d6): δ 9.62 (s, 1H, CH N), 8.88 (s, 1H, ArH), 8.66 (s, 1H, triazole-H), 8.20 (d, J = 8.4 Hz, 1H, ArH), 8.02 (d, J = 8.8 Hz, 1H, ArH), 7.75 (s, 1H, ArH), 7.07 (s, 1H, ArH), 6.17 (s, 2H, –OCH2O–), 5.52–5.26 (m, 4H, 2 × CH2), 4.95–4.68 (m, 4H, galactose-H1, OH, CH2), 4.10 (s, 3H, OCH3), 3.77 (s, 1H), 3.68 (s, 1H), 3.54–3.49 (m, 2H), 3.26–3.17 (m, 4H); 13C NMR (100 MHz, DMSO-d6): δ 151.42, 150.31, 148.11, 145.72, 142.88, 142.04, 137.89, 133.33, 131.23, 127.01, 124.94, 124.50, 122.46, 120.83, 120.70, 108.90, 105.91, 102.52, 88.52, 78.83, 74.17, 69.60, 68.88, 60.85, 57.54, 55.85, 26.83; ESI-TOF HRMS (m/z): calcd for C28H29N4O9+, [M – Cl]+, 565.1929; found, 565.1931.
2,3,4,6-Tetra-O-acetyl-α-d-mannopyranosyl azide (19α)
To the solution of 1,2,3,4,6-penta-O-acetyl-d-mannopyranose 17 (2.0 g, 5.13 mmol) in dry CH2Cl2 (10 mL) was added TMSN3 (1.35 mL, 10.26 mmol) followed by SnCl4 (0.30 mL, 2.56 mmol) at 0 °C under a nitrogen atmosphere, and the reaction mixture was allowed to warm to room temperature and stirred for 2 h. After TLC showed the disappearance of the starting material, the mixture was diluted with CH2Cl2 (40 mL) and washed with saturated NaHCO3 solution (50 mL × 3). The organic phase was dried with anhydrous Na2SO4, the solvent was evaporated, and the resultant crude oil was purified by column chromatography (petroleum ether/ethyl acetate, 2/1, v/v) to give 19α (1.68 g, 88%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 5.39 (d, J = 1.2 Hz, 1H, H-1), 5.32–5.23 (m, 2H, H-3, H-4), 5.16 (t, J = 2.0 Hz, 1H, H-2), 4.30 (dd, J = 5.6, 12.4 Hz, 1H, H-6), 4.19–4.11 (m, 2H, H-5, H-6′), 2.17 (s, 3H, CH3CO), 2.12 (s, 3H, CH3CO), 2.06 (s, 3H, CH3CO), 2.00 (s, 3H, CH3CO); 13C NMR (100 MHz, CDCl3): δ 170.57, 169.84, 169.72, 169.61, 87.48, 70.67, 69.21, 68.26, 65.67, 62.17, 20.80, 20.70, 20.66, 20.59; ESI-TOF HRMS (m/z): calcd for C14H19N3O9Na, [M + Na]+, 396.1014; found, 396.1011; calcd for C14H23N4O9, [M + NH4]+, 391.1460; found, 391.1458.
9-O-(1-(α-d-mannopyranosyl)-4-methylene-1H-1,2,3-triazole)berberine chloride (3)
Compound 3 was achieved from 19α (200 mg, 0.536 mmol) and 8 (176 mg, 0.447 mmol) as a yellow solid (137 mg, 52%), using the same procedure for the synthesis of 1. 1H NMR (400 MHz, DMSO-d6): δ 9.64 (s, 1H, CH N), 8.92 (s, 1H, ArH), 8.45 (s, 1H, triazole-H), 8.21 (d, J = 9.2 Hz, 1H, ArH), 8.03 (d, J = 8.8 Hz, 1H, ArH), 7.78 (s, 1H, ArH), 7.09 (s, 1H, ArH), 6.18 (s, 2H, –OCH2O–), 5.90 (d, J = 3.2 Hz, 1H, mannose-H1), 5.49 (s, 2H, CH2), 5.27–5.23 (m, 1H, OH), 5.07–5.03 (m, 2H, CH2), 4.89 (s, 2H, CH2), 4.60 (t, J = 5.2 Hz, 1H, OH), 4.39 (s, 1H), 4.10 (s, 3H, OCH3), 3.76 (s, 1H), 3.60–3.50 (m, 3H), 3.25–3.15 (m, 3H); 13C NMR (100 MHz, DMSO-d6): δ 151.36, 150.32, 148.15, 145.55, 142.00, 137.98, 133.40, 131.12, 127.03, 125.41, 124.49, 122.40, 120.88, 120.77, 108.89, 105.92, 102.55, 86.16, 78.86, 71.64, 68.50, 68.00, 66.93, 61.09, 57.54, 55.87, 26.83; ESI-TOF HRMS (m/z): calcd for C28H29N4O9+, [M – Cl]+, 565.1929; found, 565.1935.
2,3,4-Tri-O-acetyl-α-l-rhamnopyranosyl azide (22)
Compound 22 was achieved from 21 (5.0 g, 15.06 mmol) as a colorless oil (3.80 g, 80%), using the same procedure for the synthesis of 19α . 1H NMR (400 MHz, CDCl3): δ 5.31 (d, J = 1.2 Hz, 1H, H1), 5.21 (dd, J = 3.2, 10 Hz, 1H), 5.15–5.14 (m, 1H), 5.09 (t, J = 10 Hz, 1H), 4.07–4.00 (m, 1H), 2.16 (s, 3H, CH3CO), 2.06 (s, 3H, CH3CO), 1.99 (s, 3H, CH3CO), 1.29 (d, J = 8.4 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 169.92, 169.85, 169.82, 87.51, 70.50, 69.50, 68.63, 68.30, 20.83, 20.76, 20.64, 17.46; ESI-TOF HRMS (m/z): calcd for C12H17N3O7Na, [M + Na]+, 338.0959; found, 338.0964.
9-O-(1-(α-l-rhamnopyranosyl)-4-methylene-1H-1,2,3-triazole)berberine chloride (4)
Compound 4 was achieved from 22 (169 mg, 0.536 mmol) and 8 (176 mg, 0.447 mmol) as a yellow solid (138 mg, 44%), using the same procedure for the synthesis of 1. 1H NMR (400 MHz, DMSO-d6): δ 9.65 (s, 1H, CH N), 8.92 (s, 1H, ArH), 8.42 (s, 1H, triazole-H), 8.22 (d, J = 9.2 Hz, 1H, ArH), 8.00 (d, J = 9.2 Hz, 1H, ArH), 7.78 (s, 1H, ArH), 7.09 (s, 1H, ArH), 6.17 (s, 2H, –OCH2O–), 5.88 (d, J = 2.0 Hz, 1H, rhamnopyranose-H1), 5.48 (s, 2H, CH2), 5.26 (d, J = 4.8 Hz, 1H), 5.02 (t, J = 5.6, 8.4 Hz, 2H, CH2), 4.89 (t, J = 6.0 Hz, 2H, CH2), 4.39–4.36 (m, 1H), 4.10 (s, 3H, CH3), 3.77–3.73 (m, 1H), 3.19 (t, J = 6.4 Hz, 2H), 1.10 (d, J = 6.4 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 151.43, 150.33, 148.16, 145.59, 141.89, 137.98, 133.37, 131.12, 127.05, 125.64, 124.51, 122.45, 120.87, 120.76, 108.89, 105.91, 102.56, 86.50, 72.83, 72.42, 71.44, 68.81, 66.82, 57.54, 55.85, 26.83, 18.19; ESI-TOF HRMS (m/z): calcd for C28H29N4O8+, [M – Cl]+, 549.1980; found, 549.1986.
2,3,5-Tri-O-acetyl-β-d-ribofuranosyl azide (25)
Compound 25 was achieved from 24 (5.0 g, 15.72 mmol) as a white solid (4.21 g, 89%), using the same procedure for the synthesis of 19α . 1H NMR (400 MHz, CDCl3): δ 5.36 (d, J = 2.0 Hz, 1H), 5.33 (dd, J = 4.8, 6.4 Hz, 1H), 5.14 (dd, J = 2.0, 4.4 Hz, 1H), 4.41 (dd, J = 3.2, 12.0 Hz, 1H), 4.37–4.34 (m, 1H), 4.14 (dd, J = 4.0, 12.4 Hz, 1H, H-5), 2.13 (s, 3H, CH3CO), 2.12 (s, 3H, CH3CO), 2.08 (s, 3H, CH3CO); 13C NMR (100 MHz, CDCl3): δ 170.55169.52, 169.38, 92.69, 79.41, 74.51, 70.50, 63.03, 20.68, 20.51, 20.46; ESI-TOF HRMS (m/z): calcd for C11H15N3O7Na, [M + Na]+, 324.0808; found, 324.0813.
9-O-(1-(β-d-ribofuranosyl)-4-methylene-1H-1,2,3-triazole)berberine chloride (5)
Compound 5 was achieved from 25 (161 mg, 0.536 mmol) and 8 (176 mg, 0.447 mmol) as a yellow solid (150 mg, 49%), using the same procedure for the synthesis of 1. 1H NMR (400 MHz, DMSO-d6): δ 9.65 (s, 1H, CH N), 8.93 (s, 1H, ArH), 8.53 (s, 1H, triazole-H), 8.24 (d, J = 8.4 Hz, 1H, ArH), 8.03 (d, J = 8.8 Hz, 1H, ArH), 7.79 (s, 1H, ArH), 7.09 (s, 1H, ArH), 6.18 (s, 2H, –OCH2O–), 5.94 (d, J = 4.4 Hz, 1H, H1), 5.56 (d, J = 6.0 Hz, 1H), 5.47 (s, 2H), 5.28 (d, J = 4.4 Hz, 1H), 4.98 (t, J = 3.6 Hz, 1H), 4.89 (t, J = 4.2 Hz, 2H), 4.36–4.32 (m, 1H), 4.10 (s, 3H, CH3), 3.96 (s, 1H), 3.61–3.56 (m, 1H), 3.52–3.46 (m, 2H), 3.19 (t, J = 6.4 Hz, 2H); 13C NMR (100 MHz, DMSO-d6): δ 151.32, 150.34, 148.17, 145.58, 142.09, 137.96, 133.39, 131.13, 127.05, 124.48, 124.21, 122.38, 120.86, 120.76, 108.90, 105.91, 102.56, 92.55, 86.41, 75.69, 70.78, 67.07, 61.72, 57.53, 55.86, 26.83; ESI-TOF HRMS (m/z): calcd for C27H27N4O8+, [M – Cl]+, 535.1823; found, 535.1828.
Measurement of cytotoxicity by the CCK-8 assay
HepG2 cells were maintained in DMEM cell medium containing 10% heat-inactivated fetal bovine serum (FBS) and 100 μg mL–1 penicillin/streptomycin in a humidified incubator (Thermo Scientific, Series 8000 WJ) in a 5% CO2 atmosphere at 37 °C. Cells were collected with 0.25% trypsin solution when the cell confluency was approximately 80–90%. Then, the HepG2 cells were plated on 96-well tissue culture plates with a density of 1 × 105 cells per mL. After 12 hours of incubation, the media were removed and replaced with fresh media. All the compounds were dissolved in 0.5 % DMSO–water solution for cell treatment, the cells were treated with different concentrations of the compounds (0.2, 1.0, 5.0, 25, 125.0, and 625 μg mL–1) for 12 hours, and the negative control was incubated without the addition of any compounds. After that, 20 μL of CCK-8 solution (5 mg mL–1) was added to each well and the cells were cultured for 4 hours. The absorbance was measured by MR spectroscopy (DYNEX, Series1SPA-0093) at 570 nm. Furthermore, the absorbance of different concentrations (0.2, 1.0, 5.0, 25, 125.0, and 625 μg mL–1) of the compounds was also measured to eliminate the influence of the colors of the drugs. The IC50 values were defined as the drug concentration that inhibits 50% cell growth by setting the viability of untreated cells as 100%.
Investigation of anti-diabetic activity on HepG2 cell lines
HepG2 cells were suspended in the DMEM cell medium containing 10% heat-inactivated fetal bovine serum (FBS) and 100 μg mL–1 penicillin/streptomycin. Then, cells were seeded in 96-well plates with a density of 2 × 105 cells in each well, and cultured for 12 hours in a humidified incubator at 37 °C in a 5% CO2 atmosphere. Thereafter, all the cells were divided into four groups, including the blank control group, solvent control group (5 % DMSO), berberine chloride group, and carbohydrate modified berberine derivative group. Every berberine derivative was divided into three groups based on dosage (0.2, 1.0, and 5.0 μg mL–1), and the solvent control group and berberine chloride group have only one dosage (5.0 μg mL–1). After two days of incubation, the glucose concentrations of each well were determined by the glucose oxidase method.40 The values of glucose consumption (GC) were calculated by the glucose concentrations of blank wells subtracting the remaining glucose in cell-treated wells.
Conflicts of interest
The authors declare no competing interests.
Supplementary Material
Acknowledgments
This research was supported by grants from the Natural Science Foundation of Shandong Province (No. ZR2018PB007) and the Natural Science Foundation of China (No. 81602982).
Footnotes
†Electronic supplementary information (ESI) available: 1H and 13C NMR spectra. See DOI: 10.1039/c9md00036d
References
- Kumar A., Ekavali, Chopra K., Mukherjee M., Pottabathini R., Dhull D. K. Eur. J. Pharmacol. 2015;761:288–297. doi: 10.1016/j.ejphar.2015.05.068. [DOI] [PubMed] [Google Scholar]
- Zhang M., Chen L. Acta Pharm. Sin. B. 2012;2:379–386. [Google Scholar]
- Wang J., Yang T., Chen H., Xu Y.-N., Yu L.-F., Liu T., Tang J., Yi Z., Yang C.-G., Xue W., Yang F. Eur. J. Med. Chem. 2017;127:424–433. doi: 10.1016/j.ejmech.2017.01.012. [DOI] [PubMed] [Google Scholar]
- Ding Y., Ye X., Zhu J., Zhu X., Li X., Chen B. J. Funct. Foods. 2014;7:229–237. [Google Scholar]
- Bian X., He L., Yang G. Bioorg. Med. Chem. Lett. 2006;16:1380–1383. doi: 10.1016/j.bmcl.2005.11.045. [DOI] [PubMed] [Google Scholar]
- Liao G., Zhou Z., Guo Z. Chem. Commun. 2015;51:9647–9650. doi: 10.1039/c5cc01794g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Wang T., Xu S., Lin A., Yao H., Xie W., Zhu Z., Xu J. Eur. J. Med. Chem. 2017;132:173–183. doi: 10.1016/j.ejmech.2017.03.027. [DOI] [PubMed] [Google Scholar]
- Gautam R., Jachak S. M. Med. Res. Rev. 2009;29:767–820. doi: 10.1002/med.20156. [DOI] [PubMed] [Google Scholar]
- Bahar M., Deng Y., Zhu X., He S., Pandharkar T., Drew M. E., Navarro-Vázquez A., Anklin C., Gil R. R., Doskotch R. W., Werbovetz K. A., Kinghorn A. D. Bioorg. Med. Chem. Lett. 2011;21:2606–2610. doi: 10.1016/j.bmcl.2011.01.101. [DOI] [PubMed] [Google Scholar]
- Maeng H.-J., Yoo H.-J., Kim I.-W., Song I.-S., Chung S.-J., Shim C.-K. J. Pharm. Sci. 2002;91:2614–2621. doi: 10.1002/jps.10268. [DOI] [PubMed] [Google Scholar]
- Lan J., Zhao Y., Dong F., Yan Z., Zheng W., Fan J., Sun G. J. Ethnopharmacol. 2015;161:69–81. doi: 10.1016/j.jep.2014.09.049. [DOI] [PubMed] [Google Scholar]
- Pan G. Y., Huang Z. J., Wang G. J., Fawcett J. P., Liu X. D., Zhao X. C., Sun J. G., Xie Y. Y. Planta Med. 2003;69:632–636. doi: 10.1055/s-2003-41121. [DOI] [PubMed] [Google Scholar]
- Cheng Z., Pang T., Gu M., Gao A.-H., Xie C.-M., Li J.-Y., Nan F.-J., Li J. Biochim. Biophys. Acta. 2006;1760:1682–1689. doi: 10.1016/j.bbagen.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Hawley J. A., Lessard S. J. Acta Physiol. 2007;192:127–135. doi: 10.1111/j.1748-1716.2007.01783.x. [DOI] [PubMed] [Google Scholar]
- Turner N., Li J.-Y., Gosby A., To S. W. C., Cheng Z., Miyoshi H., Taketo M. M., Cooney G. J., Kraegen E. W., James D. E., Hu L.-H., Li J., Ye J.-M. Diabetes. 2008;57:1414–1418. doi: 10.2337/db07-1552. [DOI] [PubMed] [Google Scholar]
- Rosen E. D., MacDougald O. A. Nat. Rev. Mol. Cell Biol. 2006;7:885–896. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- Pham T. P. T., Kwon J., Shin J. Biochem. Biophys. Res. Commun. 2011;413:376–382. doi: 10.1016/j.bbrc.2011.08.110. [DOI] [PubMed] [Google Scholar]
- Xia X., Yan J., Shen Y., Tang K., Yin J., Zhang Y., Yang D., Liang H., Ye J., Weng J. PLoS One. 2011;6:e16556. doi: 10.1371/journal.pone.0016556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Wang X., Yin W., Liu Z., Zhou M., Xiao D., Liu Y., Peng D. Bioorg. Med. Chem. Lett. 2016;26:4799–4803. doi: 10.1016/j.bmcl.2016.08.027. [DOI] [PubMed] [Google Scholar]
- Chen Z., Ye X., Yi J., Chen X., Li X. Med. Chem. Res. 2012;21:1641–1646. [Google Scholar]
- Cheng Z., Chen A.-F., Wu F., Sheng L., Zhang H.-K., Gu M., Li Y.-Y., Zhang L.-N., Hu L.-H., Li J.-Y., Li J. Bioorg. Med. Chem. 2010;18:5915–5924. doi: 10.1016/j.bmc.2010.06.085. [DOI] [PubMed] [Google Scholar]
- Shan Y.-Q., Ren G., Wang Y.-X., Pang J., Zhao Z.-Y., Yao J., You X.-F., Si S.-Y., Song D.-Q., Kong W.-J., Jiang J.-D. Metabolism. 2013;62:446–456. doi: 10.1016/j.metabol.2012.09.009. [DOI] [PubMed] [Google Scholar]
- Qu X., Khutoryanskiy V. V., Stewart A., Rahman S., Papahadjopoulos-Sternberg B., Dufes C., McCarthy D., Wilson C. G., Lyons R., Carter K. C., Schätzlein A., Uchegbu I. F. Biomacromolecules. 2006;7:3452–3459. doi: 10.1021/bm0604000. [DOI] [PubMed] [Google Scholar]
- Gruner S. A. W., Locardi E., Lohof E., Kessler H. Chem. Rev. 2002;102:491–514. doi: 10.1021/cr0004409. [DOI] [PubMed] [Google Scholar]
- Wang K., Chai L., Feng X., Liu Z., Liu H., Ding L., Qiu F. J. Pharm. Biomed. Anal. 2017;139:73–86. doi: 10.1016/j.jpba.2017.02.038. [DOI] [PubMed] [Google Scholar]
- Ma J.-Y., Feng R., Tan X.-S., Ma C., Shou J.-W., Fu J., Huang M., He C.-Y., Chen S.-N., Zhao Z.-X., He W.-Y., Wang Y., Jiang J.-D. J. Pharm. Sci. 2013;102:4181–4192. doi: 10.1002/jps.23718. [DOI] [PubMed] [Google Scholar]
- He X.-P., Zeng Y.-L., Zang Y., Li J., Field R. A., Chen G.-R. Carbohydr. Res. 2016;429:1–22. doi: 10.1016/j.carres.2016.03.022. [DOI] [PubMed] [Google Scholar]
- Shi A., Huang L., Lu C., He F., Li X. Bioorg. Med. Chem. 2011;19:2298–2305. doi: 10.1016/j.bmc.2011.02.025. [DOI] [PubMed] [Google Scholar]
- Ishiwata A., Akao H., Ito Y. Org. Lett. 2006;8:5525–5528. doi: 10.1021/ol062198j. [DOI] [PubMed] [Google Scholar]
- Williams R. J., McGill N. W., White J. M., Williams S. J. J. Carbohydr. Chem. 2010;29:236–263. [Google Scholar]
- Capon B. Chem. Rev. 1969;69:407–498. [Google Scholar]
- Lowary T. L. J. Carbohydr. Chem. 2002;21:691–722. [Google Scholar]
- Wang L., Feng S., Wang S., Li H., Guo Z., Gu G. J. Org. Chem. 2017;82:12085–12096. doi: 10.1021/acs.joc.7b01817. [DOI] [PubMed] [Google Scholar]
- Carrington R., Shaw G., Wilson D. V. J. Chem. Soc. 1965:6864–6870. [Google Scholar]
- Li L., Lin B., Yang Z., Zhang L., Zhang L. Tetrahedron Lett. 2008;49:4491–4493. [Google Scholar]
- Liu B., Wang G., Yang J., Pan X., Yang Z., Zang L. PLoS One. 2011;6:e21416. doi: 10.1371/journal.pone.0021416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge P. W., Li Y. F., Matsa E., Wu H., Ong S.-G., Sharma A., Holmström A., Chang A. C., Coronado M. J., Ebert A. D., Knowles J. W., Telli M. L., Witteles R. M., Blau H. M., Bernstein D., Altman R. B., Wu J. C. Nat. Med. 2016;22:547. doi: 10.1038/nm.4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Wei J., Xue R., Wu J.-D., Zhao W., Wang Z.-Z., Wang S.-K., Zhou Z.-X., Song D.-Q., Wang Y.-M., Pan H.-N., Kong W.-J., Jiang J.-D. Metabolism. 2010;59:285–292. doi: 10.1016/j.metabol.2009.07.029. [DOI] [PubMed] [Google Scholar]
- Huang Q., Chen L., Teng H., Song H., Wu X., Xu M. J. Funct. Foods. 2015;19:487–494. [Google Scholar]
- Kadish A. H., Litle R. L., Sternberg J. C. Clin. Chem. 1968;14:116–131. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.