

Abstract
Chimpanzees (Pan troglodytes) and bonobos (Pan paniscus) are the closest living relatives of humans, but the two species show distinct behavioral and physiological differences, particularly regarding female reproduction. Despite their recent rapid decline, the demographic histories of the two species have been different during the past 1–2 Myr, likely having an impact on their genomic diversity. Here, we analyze the inferred functional consequences of genetic variation across 69 individuals, making use of the most complete data set of genomes in the Pan clade to date. We test to which extent the demographic history influences the efficacy of purifying selection in these species. We find that small historical effective population sizes (Ne) correlate not only with low levels of genetic diversity but also with a larger number of deleterious alleles in homozygosity and an increased proportion of deleterious changes at low frequencies. To investigate the putative genetic basis for phenotypic differences between chimpanzees and bonobos, we exploit the catalog of putatively deleterious protein-coding changes in each lineage. We show that bonobo-specific nonsynonymous changes are enriched in genes related to age at menarche in humans, suggesting that the prominent physiological differences in the female reproductive system between chimpanzees and bonobos might be explained, in part, by putatively adaptive changes on the bonobo lineage.
Keywords: bonobo genome, bonobo reproduction, comparative genomics, deleteriousness
Introduction
Chimpanzees (Pan troglodytes) and bonobos (Pan paniscus) are two closely related species with an estimated divergence time of 1–2 Ma and share a most recent common ancestor with humans 5–13 Ma (Fischer et al. 2011; Langergraber and Prüfer 2012; Prado-Martinez et al. 2013; de Manuel et al. 2016). They are interesting species to study genetic changes in comparison to humans for at least two reasons: First, they have experienced different demographic histories since their divergence. For example, the population history of bonobos is marked by population bottlenecks in the past and a smaller long-term small effective population size (Ne) (Prado-Martinez et al. 2013; de Manuel et al. 2016). They are also known to have been geographically isolated in central Africa for a long time (nowadays Democratic Republic Congo) (Thompson 2003; Takemoto et al. 2017), which is expected to increase their genetic homogeneity. This is in stark contrast to the history of chimpanzees, which inhabited a much wider region across Africa (from Tanzania to Guinea, at the east and west edges of Africa). Most chimpanzee subspecies, except of the western population, are thought to have had a larger population size and to have maintained a greater level of genetic diversity compared with bonobos (de Manuel et al. 2016). The western chimpanzees are thought to have spread from a very small ancestral population (Shimada et al. 2004; Sá et al. 2013), whereas central chimpanzees appear to have been more continuously inhabiting central Africa (de Manuel et al. 2016).
A second reason why comparing the two sister species is interesting is that, despite many similarities, they show recognizable phenotypic differences likely due to genetic differences. One of their most distinct differences is that bonobos, but not chimpanzees, have a prolonged maximal sexual swelling, where females appear to be estrous even when they are not ovulating (e.g., during lactating period; Thompson-Handler et al. 1984; Furuichi 1987). Maximal or exaggerated sexual swellings in the perineal part around their ovulation period is a distinct feature of the Pan clade among the great apes (Nunn 1999), but bonobos are different from chimpanzees in that they present it even when they are not ovulating, and more often than female chimpanzees do. This phenotype is likely to have a physiological mechanism, and indeed at the physiological level, measuring urinary testosterone hormonal levels, it has been shown that female bonobos are on average 3 years younger than their chimpanzee counterparts when they experience the onset of puberty, which is an important clue for their differential sexual development (Behringer et al. 2014). It has been suggested that these differences are a source of different social dynamics between bonobos and chimpanzees. Chimpanzees show a clear hierarchy and frequent violence among males, whereas bonobos show more egalitarian interactions (Nishida 1983; Goodall 1986; Furuichi 1987; Idani 1990), frequent female–female sexual interactions (Kuroda 1980) and high female social status (Furuichi 1987), compared with female chimpanzees. Prolonged maximal sexual swellings in bonobos have been suggested as a mechanism to explain the egalitarian and peaceful social dynamics in bonobos, first by lowering the operational sex ratio (the temporal ratio of adult males to estrous females in the group; Randall Parish 1994; Furuichi 2011) and second by strengthening female–female sexual interactions (females are more attracted to other females that show sexual swellings, which builds female affiliative relationships and a strong social stance in a male-philopatric society; Ryu et al. 2015). Considering that the high social status of bonobo females is also viewed as a driving force of selection on nonaggressive males (Hare et al. 2012), the prolonged sexual swelling can be considered a key aspect of bonobo-specific evolutionary features. Here, we investigate the genetic differences between these species, as they are likely to they underlie their physiological differences. We use derived alleles in each lineage, which are the obvious candidate changes for such lineage-specific traits, particularly nonsynonymous changes and other genetic changes with potential functional impact.
Bonobos have a smaller Ne (11,900–23,800) than central chimpanzees (24,400–48,700). However, chimpanzee subspecies differ substantially, with the Ne of central chimpanzees being about four times larger than that of western chimpanzees (9,800–19,500) (Prado-Martinez et al. 2013) (summarized in supplementary table S2, Supplementary Material online). Whether different demographic histories influence the efficacy of purifying selection in populations has been a debated topic in the field of population genomics. Many studies have shown that in human populations with small Ne and with population bottlenecks, genetic diversity is lower (the number of heterozygous genotypes per individual; Lohmueller et al. 2008; Kidd et al. 2012; Torkamani et al. 2012; Hodgkinson et al. 2013), whereas the rate of random fixation of derived alleles is higher (the proportion of fixed substitutions and number of homozygous derived alleles per individual; Lohmueller et al. 2008; Kidd et al. 2012; Torkamani et al. 2012) than in populations that have maintained a larger Ne. These patterns are generally explained by the effects of random genetic drift, in the framework of the neutral theory (Kimura 1968), which assumes most genetic variation to be neutral or nearly neutral (Ohta 1972, 1973). In order to assess the efficacy of purifying selection, it is crucial to consider the proportion of deleterious changes, particularly at high frequencies within the population. It has been inferred that almost half of all nonsynonymous single nucleotide changes (SNCs) in a single genome are deleterious (Subramanian and Lambert 2012). Such deleterious allele changes are, for example, predicted to alter protein function, which is typically strongly conserved across species, or are associated with an increased risk of disease; hence, they are proposed to be under purifying selection that reduces their probability of fixation in the population (Jukes and Kimura 1984). Slightly deleterious changes are particularly interesting because they are relatively well tolerated, appear as polymorphisms (Henn et al. 2016), and thus could be informative about the effects of purifying selection. We expect a larger proportion of slightly deleterious SNCs at high frequencies in a population with relaxed purifying selection pressure than in one with more efficient purifying selection, in agreement with the nearly neutral theory (Ohta 1972, 1973). This hypothesis has been tested in different species, including archaic and modern humans (Lohmueller et al. 2008; Castellano et al. 2014), dogs (Marsden et al. 2016), and rice populations (Liu et al. 2017). Several studies have also analyzed the efficacy of purifying selection in the Pan clade: Using the exomes of central, eastern and western chimpanzees, Bataillon et al. (2015) showed that the efficacy of natural selection correlates with past Ne, in agreement with Cagan et al. (2016), where lineage-specific selection signatures from all the great ape genomes led to the same conclusion. On the other hand, de Valles-Ibáñez et al. (2016) interpreted data of various great ape species, including the very similar eastern and Nigerian–Cameroon chimpanzees (Prado-Martinez et al. 2013), that the load of loss-of-function (LoF) mutations, which probably have severe consequences, are not influenced by the demographic history. However, given the differences in demographic history across chimpanzee subspecies, it seems important to include all four recognized subspecies, particularly the central and western chimpanzee populations, which had the largest and the smallest Ne, respectively (Prado-Martinez et al. 2013). Bonobos in turn experienced population bottlenecks and are believed to have maintained a small Ne since their split from chimpanzees (Prado-Martinez et al. 2013). Comparing the mutational load in all the chimpanzee lineages and bonobos together based on their well-known demographic history, which has not yet been studied, would provide us with a comprehensive picture on the interplay between purifying selection and the demographic history.
In this study, we examine 59 chimpanzee and 10 bonobo genomes (de Manuel et al. 2016) to investigate the accumulation of putatively deleterious derived alleles, with the goal of testing how their demographic histories have shaped the distribution of such alleles. We analyze changes in the bonobo and chimpanzee lineages, assessing the enrichment of genes associated with different phenotypic traits using human a GWAS database. We interpret an enrichment in genes related to these traits as indication of a genetic basis for lineage-specific differences between the two Pan species.
Materials and Methods
Data Preparation
Chimpanzee and bonobo genomes used in this study were generated in previous studies (Prado-Martinez et al. 2013; de Manuel et al. 2016). The data consist of 59 chimpanzee and 10 bonobo genomes sequenced to high coverage, including four chimpanzee populations: 18 central, 20 eastern, 10 Nigerian–Cameroon, and 11 western chimpanzees (supplementary fig. S1 and table S1, Supplementary Material online). All genomes were mapped to the human genome assembly hg19 (ENSEMBL GRCh37.75), and previously published genotype calls for SNCs—variable sites across all the genomes of the Pan populations—in mappable regions on the autosomes were used (de Manuel et al. 2016). We applied the ENCODE 20mer uniqueness filter and required all the heterozygous loci to be allele balanced, with ratios of the raw reads between 0.25 and 0.75, in order to exclude sites with potential biases or contamination. Across 69 individuals, the initial VCF file contained 36,299,697 loci in total. After applying all filters, we used 33,946,246 loci where at least one individual shows a reliable genotype, whereas only 5,795,261 loci have information for all 69 individuals. For the analysis with eight individuals per population at highest quality, we exclude samples which could have had a small percentage of human contamination. To do so, we counted positions where all Pan individuals carried an allele different from the human reference allele in homozygous state, with the exception of one single sequencing read in one individual. For the same individual, we required at least five sequencing reads to match the derived allele in other Pan individuals to assign the single human-like sequencing read as contamination. We then selected individuals with fewer than 10,000 such observations across the entire genome, choosing those individuals with the highest coverage (supplementary table S1, Supplementary Material online). For these analyses, we only considered sites where high-quality genotypes were available for all individuals. Furthermore, where stated, a subset of ten central chimpanzees for a fair comparison with bonobos was used (supplementary table S1, Supplementary Material online). In the analysis of GWAS-associated genes described below, a more permissive population-wise filtering was applied, using sites at which at least 50% of individuals in each species (chimpanzee and bonobo) pass the above filters, and one of the lineages carries the derived allele at more than 90% frequency, whereas the other population carries less than 10% of the derived allele. Population-wise frequencies were calculated as proportions of the observed counts.
The functional effects for each SNC were inferred using the ENSEMBL Variant Effect Predictor v83 (McLaren et al. 2016) on all segregating sites across the 69 individuals. Nonsynonymous and synonymous SNCs were used as defined by Variant Effect Predictor, and the following functional categories were retrieved from the ENSEMBL annotation: 5′ UTR, 3′ UTR, regulatory elements, splice sites, transcription factor binding sites, upstream variants, and downstream variants. Each variant can carry multiple annotations, and all annotation categories are treated separately in the subsequent analyses. In rare cases where a variant overlaps with multiple genes, both genes would be associated with that variant. Genomic regions and positions were analyzed using R/Bioconductor (Huber et al. 2015) and the packages biomaRt (Durinck et al. 2005) and GenomicRanges (Lawrence et al. 2013). This catalog of variation and functional inference in Pan species is available online at Figshare (http://doi.org/10.6084/m9.figshare.7855850).
Terminology
“Derived change” refers to the allele state different from the reference allele state. In this study, the human genome was used as reference, and because it is an outgroup to the Pan clade it represents closely the ancestral state at Pan-specific mutations. “Ancestral state” refers to the allele state same as the human reference allele state. “Lineage-specific derived changes” refers to the derived changes fixed or almost fixed (>=90%) in one lineage (either chimpanzee or bonobo), where the other lineage appears fixed or almost fixed for the ancestral state (<10% derived). Doing so, we avoid neglecting actual fixed sites which appear not fixed due to sequencing errors and include sites which are influenced by gene flow between populations.
Simulations
We used the forward-simulator SFS_CODE (Hernandez 2008), in order generate three populations diverging from one source population, following the demographic parameters of bonobos, central and western chimpanzees, which we find most relevant in interpreting our observation in relationship of the demographic history and the mutational load. We have generated 8 individuals per population of bonobos, central and western chimpanzees, for one locus of 10,000 base pair length in 200 iterations. We used the simplified Ne in Prado-Martinez et al. (2013), with a divergence of 40,000 generations between chimpanzees and bonobos, and 28,000 generations between central and western chimpanzees. We simulated neutral fragments (sfs_code 3 1 -TS 0 0 1 -Td 0 P 0 10 -Td 0 P 1 22 -TS 28 1 2 -Td 28 P 1 2.7 -Td 28 P 2 0.45 -TE 40 -W 0 -L 1 10000 -n 8 -a N –theta 0.001 –rho 0.001) and fragments under purifying selection with a gamma of 0.17 for 70% of the sites (Bataillon et al. 2015) (sfs_code 3 1 -TS 0 0 1 -Td 0 P 0 10 -Td 0 P 1 22 -TS 28 1 2 -Td 28 P 1 2.7 -Td 28 P 2 0.45 -TE 40 -W 1 0.17 0 0.7 -L 1 10000 -n 8 -a C –theta 0.001 –rho 0.001).
Deleteriousness Assessment
We used the following methods to assess deleteriousness: Grantham score, C-score, GWAVA, SIFT, PolyPhen-2, and Genomic Evolutionary Rate Profiling (GERP) score. We did so in order to avoid a bias coming from a particular method and ensure that our results are robust to the method, some based on conservation across different taxa, others on physicochemical property or known consequences of the variants. More specifically, the Grantham score (Grantham 1974; Li et al. 1984) represents only the physical/chemical properties of amino acid changes. A custom cutoff 150 (>150) was used to determine deleterious/radical changes. The C-score (Kircher et al. 2014) measures deleteriousness on a genome-wide scale for both coding and noncoding variants, integrating a variety of known functional information. A cutoff of 10 was used to select the top 10% most deleterious changes. GWAVA (Ritchie et al. 2014) predicts the functional impact of genetic variants, based on genome-wide properties. A custom cutoff at 0.5 (>0.5) was used to select potentially functional changes. SIFT (Kumar et al. 2009) predicts the deleteriousness of amino acid changes, based on the degree of conservation inferred from sequence alignments. All changes diagnosed as “deleterious” was analyzed. PolyPhen-2 (Polymorphism Phenotyping v2) (Adzhubei et al. 2010) predicts the possible impact of amino acid changes on the structure and function using both physical property and multiple sequence alignments. All changes diagnosed as “probably_damaging” were analyzed. Finally, GERP scores (Davydov et al. 2010) compare, based on multiple alignments, the number of observed substitutions to the number of hypothetical substitutions given that they are neutral changes. A cutoff of 4 (>4) was used to select the changes with “large” functional consequences. They assume a deficit of observed substitutions as “Rejected Substitutions,” a natural measure of constraint on the element. For our analyses, we used precomputed base-wise scores for hg19 (http://mendel.stanford.edu/SidowLab/downloads/gerp/; last accessed March 17, 2019). Neutral loci were defined as described in Gronau et al. (2011).
Analysis of Nonsynonymous Lineage-Specific SNCs
We used the software FUNC (Prüfer et al. 2007) to determine an enrichment of Gene Ontology terms. We ranked the genes by the number of all lineage-specific nonsynonymous changes, divided by the number of lineage-specific deleterious SNCs. In order to assess whether particular gene categories are enriched for lineage-specific deleterious SNCs in each lineage, we used the Wilcoxon rank test.
In order to formally assess an enrichment of lineage-specific nonsynonymous changes in gene clusters associated with known phenotypic traits in humans, we retrieved genome-wide association data from the NHGRI-EBI GWAS Catalog (MacArthur et al. 2017), containing 2,385 associated traits. We analyzed all the associated genes in the data, which have protein-coding SNCs either on the chimpanzee or bonobo lineage, respectively. We used the permissive set of nonsynonymous SNCs (Data Preparation). For each trait from each study (“Disease trait”), we counted the number of nonsynonymous SNCs on each lineage, and performed a contingency table significance test (G-test) against the total number of nonsynonymous SNCs, compared with the total number of genes associated with the trait and the total number of protein-coding genes. This test determines an enrichment of this trait considering the number of protein-coding changes. We also performed a G-test between the total numbers of all nonsynonymous SNCs on each lineage compared with the numbers of nonsynonymous SNCs on each lineage falling in genes associated with the trait, in order to determine whether or not the difference between the two species is significant. In both cases, a P value cutoff of 0.1 was applied. These cutoffs are permissive, considering that the data rely on independent studies on different cohorts, with high significance cutoffs within each study. This also results in a large number of tests with 0 observation counts. Finally, we performed an empirical enrichment test, by creating 500 random sets of genes with similar length as the genes associated with each trait (±10% of the length of each gene), and counting the number of lineage-specific nonsynonymous SNCs in each random set. Here, we require 90% of random sets to contain fewer nonsynonymous SNCs in a given lineage than the real set of associated genes, to determine a trait significant. We note that this test is analogous to a false discovery rate, because it empirically determines how often we would expect GWAS loci to fall in the genes of interest. Performing the same analysis on a randomly drawn set of genes did not show an enrichment of any GWAS trait, suggesting that no significant enrichment is expected after applying these three tests. Finally, we filter for traits with at least 10 associated loci in order to restrict the analysis to multigenic traits, and we report only significant traits where at least three genes carry nonsynonymous SNCs.
We screened the genes which have lineage-specific nonsynonymous SNCs at high frequency, and confirmed their expression in chimpanzees and bonobos, using the data set from Brawand et al. (2011). The RNA sequencing data were mapped to the human genome assembly hg19 using tophat2 (Kim et al. 2013), and gene expression was estimated with samtools (Li et al. 2009) and htseq-count (Anders et al. 2015), which measures the total count of fragments falling in each gene. Genes were defined as expressed when either lineage showed a log 2-normalized expression value larger than 3 (more than two fragments mapped to the whole gene).
Results
Neutrality Index in Populations
Previous studies have shown that the historical Ne has been lowest in western chimpanzees and highest in central chimpanzees (Won and Hey 2005; Prado-Martinez et al. 2013; de Manuel et al. 2016; Kuhlwilm et al. 2016) (supplementary table S2, Supplementary Material online). Accordingly, we observe differences in measures like average heterozygosity per base pair across each population, which is indeed highest in central chimpanzees (0.0014) and lowest in western chimpanzees (0.0006) (supplementary table S2, Supplementary Material online). To study the overall impact of these Ne differences on the efficacy of selection, we used inferred fixed derived (D) and polymorphic (P) nonsynonymous (n) and synonymous (s) alleles in samples of equal size of eight individuals from each population, to calculate a population-wise version of the neutrality index (NI) (Rand and Kann 1996), which is the ratio of Pn/Ps and Dn/Ds. The NI quantifies the direction and degree of natural selection, based on the assumption that synonymous substitutions are neutral. In the classical NI, a value >1 indicates an excess of nonsynonymous polymorphism over fixed derived sites, implying negative selection, whereas a value <1 indicates an excess of fixed nonsynonymous alleles and may imply positive selection. We find that the NI across autosomes in western chimpanzees (1.51) is higher than in the other chimpanzee subspecies (1.19–1.28), caused by an excess of nonsynonymous over synonymous polymorphisms (fig. 1A and supplementary table S3, Supplementary Material online). This could be explained by reduced efficacy of purifying selection in this population as the result of a low Ne (Eyre-Walker 2006) that allows slightly deleterious alleles to accumulate as polymorphisms in the population. The direction of selection (DoS) is another statistic that uses Pn, Ps, Dn, and Ds to quantify the accumulation of slightly deleterious mutations as a measure of reduced efficacy of purifying selection (Stoletzki and Eyre-Walker 2011). Here, when calculating the DoS, we observe the lowest value for western chimpanzees (−0.1) and the highest for central chimpanzees (−0.04) (fig. 1B and supplementary table S2, Supplementary Material online), which suggests that positive selection is not abundant in these populations (no positive values) and that a larger proportion of deleterious sites is segregating in western chimpanzees compared with central chimpanzees (more negative value). In bonobos, the NI is higher (and DoS smaller) than in all nonwestern chimpanzees, suggesting a stronger accumulation of slightly deleterious alleles. When calculating the NI per gene, as in previous studies (Rand and Kann 1996), this pattern is confirmed (supplementary table S2, Supplementary Material online). However, we caution that only 131 genes carry a sufficient number of informative sites in these populations. The excess of potentially deleterious polymorphism in western chimpanzees is not exclusive for protein-coding changes, and it is present also in different categories of noncoding sites in functional elements, such as 5′ UTRs and the upstream and downstream regions of genes (fig. 1B and supplementary table S2, Supplementary Material online). This suggests that, also in noncoding loci, the efficacy of purifying selection was lowest in populations with smaller historical Ne.
Fig. 1.
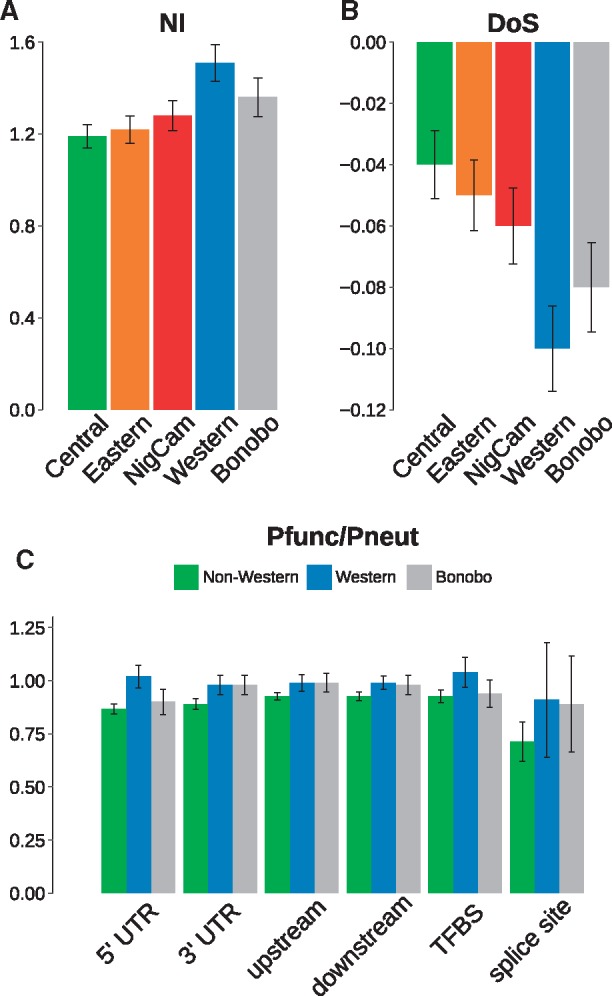
—Patterns of functional and neutral alleles. (A) Neutrality index in the five populations, with 95% confidence intervals across 22 chromosomes. (B) Direction of selection-test in the five populations, with 95% confidence intervals across 22 chromosomes. (C) Genome-wide ratio of functional to neutral polymorphism (Pfunc/Pneut) in different functional categories, for western chimpanzees, bonobos, and the average of three nonwestern chimpanzee populations; with 95% confidence intervals across 22 chromosomes.
Distributions of Deleterious Changes
We analyzed the population-wise ratio of deleterious-to-neutral derived alleles across the site frequency spectrum (SFS) (fig. 2), using eight individuals from each population. Deleterious alleles in figure 2 were defined each by GERP (Davydov et al. 2010) and PolyPhen-2 (Adzhubei et al. 2010), which represents genome-wide and protein-coding predictions, respectively. We have used four other methods for diagnosis of deleteriousness of allele changes, in order to assess robustness of the methods. We note that phylogeny-based approaches, such as C-score and GERP scores, may have a bias in western chimpanzees, because this subspecies was used for the reference chimpanzee genome. Methods based on protein sequence and structure, such as SIFT and PolyPhen-2, could avoid such a bias. The Grantham score, on the other hand, measures only the chemico-physical changes of amino acids and might be the most conservative non phylogeny-based approach. We generally observe a much higher proportion of deleterious derived alleles at high frequencies in bonobos than chimpanzees (fig. 2 and supplementary fig. S2, Supplementary Material online). When stratifying alleles into singletons versus nonsingletons, the deleterious-to-neutral ratio in nonsingletons is highest or second highest in bonobos, except for Grantham scores, compared with chimpanzee populations (supplementary table S5, Supplementary Material online). This suggests that bonobos, which experienced long-term small Ne and bottlenecks since the split from chimpanzees, have accumulated proportionately more deleterious alleles at high frequencies than chimpanzee populations. At low frequencies, we observe all noncentral chimpanzee populations having higher proportions of deleterious derived alleles, with western chimpanzee being particularly high. These patterns are generally similar using other deleteriousness estimates (supplementary fig. S2, Supplementary Material online). When simulating data based on the demographic history of bonobos, central and western chimpanzees, we also find that proportionately more deleterious changes than neutral changes accumulate in singletons and at low frequencies in the western chimpanzee–like population in comparison to the central chimpanzee–like population, which is in agreement with our data (supplementary fig. S16, Supplementary Material online).
Fig. 2.

—Ratio of site frequency spectra (SFS) of deleterious-to-neutral derived alleles. Relative proportion of deleterious derived allele frequencies in each population, defined by GERP (Davydov et al., 2010) and POLYPHEN-2 (Adzhubei et al. 2010), respectively, compared with derived allele frequencies in neutral genomic regions, defined by Gronau et al. (2011). For comparison, eight individuals from each population were selected.
Another way to assess the efficacy of purifying selection is to investigate the effects of population size on the individual mutational load. We estimated the mutational load, defined as the number of sites with putatively deleterious derived alleles per individual. We stratified them into two different classes, by counting either only heterozygous sites or only homozygous sites. When only heterozygous sites are considered, the western chimpanzee population shows the lowest level of mutational load among chimpanzee populations (fig. 3A). This is significantly lower than in the other chimpanzee populations (P < 0.001; Wilcoxon rank test) and is largely due to their low genetic diversity. However, when only homozygous sites are considered, the mutational load of western chimpanzees increases drastically (fig. 3B) and becomes significantly higher than that of central chimpanzees (P < 0.001; Wilcoxon rank test). This is probably because slightly deleterious alleles more often reach high frequencies, and are observed in homozygosity in populations with small Ne, because purifying selection is less efficient. Regarding the other chimpanzee populations, the mutational load shows a gradient with a nonsignificant correlation trend with their long-term Ne (de Manuel et al. 2016) and heterozygosity (supplementary table S2, Supplementary Material online), negative in homozygous sites (R = −0.87, P = 0.05, Spearman correlation test) and positive in heterozygous sites (R = 0.97, P = 0.004), using Grantham score. This pattern is broadly similar throughout other classes of sites, including synonymous changes, in agreement with previous observations (Lohmueller et al. 2008) (supplementary figs. S9–S14, Supplementary Material online). This pattern appears less clear using C-score and GERP (R = 0.05, P = 0.93 and R = 0.2, P = 0.74, respectively, for homozygous mutational load, Spearman correlation test). This might be due to the reference chimpanzee genome used in these methods being a western chimpanzee, as that could lower these scores for the derived changes in western chimpanzees. Still, the distributions of the heterozygous and homozygous mutational load of central chimpanzees are different from the other three chimpanzee populations in C-score and GERP (P < 0.0001 and P = 0.009, respectively; Wilcoxon rank test). Simulated data also appear to show mutational load positively correlating with the Ne in heterozygous loci, and negatively in homozygous loci (supplementary fig. S18, Supplementary Material online), even though the statistical significance is rather weak (R = 0.86 P = 0.33 and R = −0.86 P = 0.33, respectively, both in deleterious and neutral changes, Spearman correlation test).
Fig. 3.
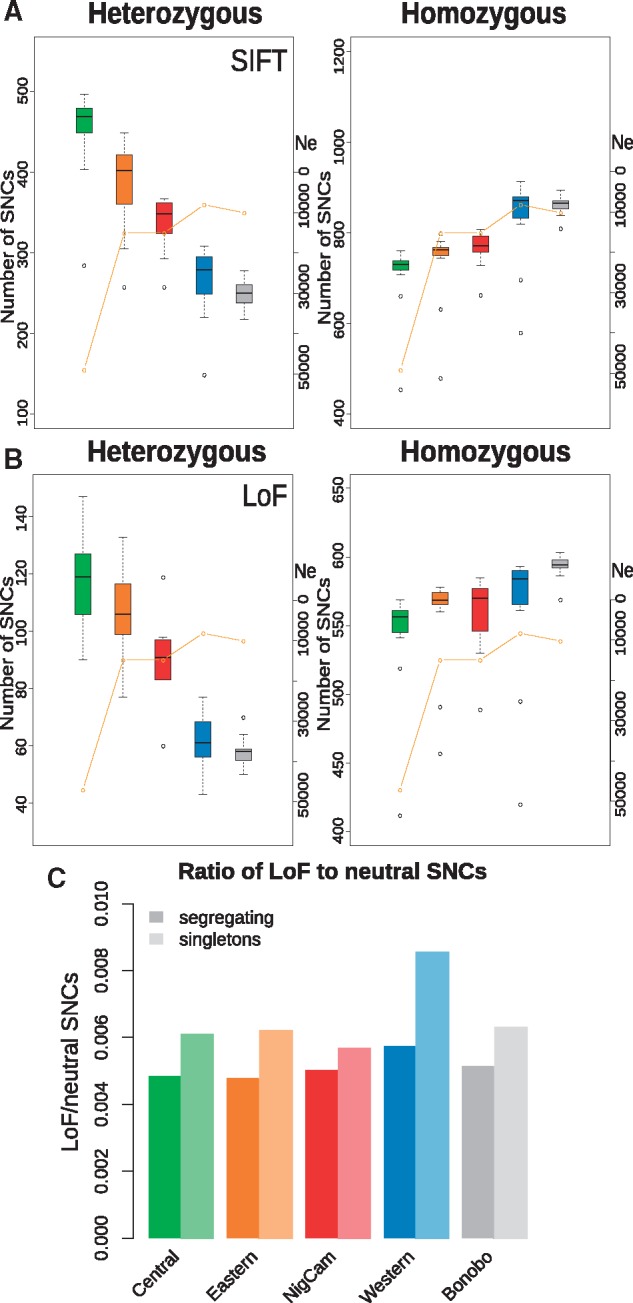
—Mutational load and LoF variants. (A) Individual mutational load in each population, considering only heterozygous sites (left) and only homozygous sites (right). These are the counts of deleterious derived alleles in each individual in each population, as defined by SIFT. Orange lines indicate the trend of Ne. Boxes and whisker diagrams represent lower quartile, median, and upper quartile. (B) Individual numbers of LoF variants, considering only heterozygous sites (left), and only homozygous sites (right). (C) Ratio of LoF variants to neutral variants in each population.
Protein-Truncating Variants
We also assessed the patterns in protein-truncating variants in each population by estimating the mutational load in LoF mutations, defined as the number of LoF derived alleles per individual. These SNCs may be considered as most likely disruptive for protein function and hence a straightforward measure for evaluating the efficacy of purifying selection. In our data, the patterns of mutational load in LoF mutations follow the patterns in other categories of deleterious mutations (fig. 3B). With higher Ne, the load tends to increase in heterozygous sites (r = 0.786 with P = 0.115). On average, chimpanzee populations carry between 61 and 118 heterozygous LoF alleles, and between 547 and 560 as homozygotes (fig. 3C). In comparison, the average mutational load of LoF mutations in modern human populations is 85 for heterozygous sites (Lek et al. 2016), which is in agreement with previous observations of a slightly higher number of heterozygous LoF mutations in chimpanzees than humans, but smaller than in gorillas (Xue et al. 2015; de Valles-Ibáñez et al. 2016). We do not directly compare the number of homozygous sites to those in modern humans, because in Lek et al. (2016) only polymorphisms within the human lineage were used, whereas in our analysis, not only polymorphisms but also fixed differences between the two Pan species were measured (de Manuel et al. 2016).
When considering the same numbers of individuals per population, we observe twice as many fixed LoF mutations in western chimpanzees, and a 2.7-fold increase of fixed LoF mutations in bonobos, compared with central chimpanzees (data not shown). Conversely, the number of LoF singletons is three times higher in central chimpanzees than western chimpanzees, whereas the other chimpanzee populations are similar to the western chimpanzees, as expected by their background levels of genetic diversity. However, in western chimpanzees, the proportion of LoF mutations to neutral mutations is higher in polymorphisms than in fixed variants (P < 0.01; G-test). Also, the proportion of polymorphic LoF to neutral sites is higher in western chimpanzees than central chimpanzees (P = 0.012, G-test). This effect is particularly pronounced in singletons (fig. 3C), suggesting that LoF mutations are more often tolerated in western chimpanzees compared with the other populations, which could be due to less efficient purifying selection.
An Overview of Lineage-Specific Changes
We assessed lineage-specific SNCs for their predicted functional effect, because these are candidates for functional changes explaining lineage-specific traits. In table 1, we provide an overview of these SNCs, stratified by annotation category, when using ten individuals each from the bonobo and central chimpanzee population. This shows that bonobos have, on average, about 2-fold more lineage-specific changes than central chimpanzees. Not surprisingly, this ratio is even higher when using 10 bonobos and the 59 chimpanzees (supplementary table S4, Supplementary Material online).
Table 1.
Summary Statistics of the Total Number of Lineage-Specific Changes in Each Species
| Total Number of Derived Changes | ≥90% |
100% |
||
|---|---|---|---|---|
| Bonobos | Chimpanzees | Bonobos | Chimpanzees | |
| 1,267,164 | 618,150 | 1,193,455 | 520,330 | |
| Annotation category | Bonobos | Chimpanzees | Bonobos | Chimpanzees |
| 3′ UTR variant | 11,983 | 5,923 | 11,352 | 5,012 |
| 5′ UTR variant | 1,861 | 912 | 1,758 | 794 |
| Intergenic variant | 488,378 | 240,355 | 458,804 | 202,127 |
| Mature miRNA variant | 10 | 0 | 7 | 0 |
| Missense variant | 2,714 | 1,329 | 2,557 | 1,175 |
| Regulatory region variant | 170,851 | 82,425 | 161,009 | 69,786 |
| Start lost | 9 | 7 | 9 | 5 |
| Stop gained | 30 | 10 | 27 | 10 |
| Stop lost | 14 | 5 | 14 | 4 |
| Synonymous variant | 3,420 | 1,719 | 3,239 | 1,450 |
| TF binding site variant | 1,213 | 583 | 1,140 | 481 |
| Deleterious derived changes | Grantham | C-score | GWAVA | SIFT | PolyPhen-2 | GERP |
| Bonobos | 162 | 107,064 | 2,316 | 214 | 70 | 15,486 |
| Chimpanzees | 74 | 21,517 | 1,056 | 102 | 49 | 2,365 |
Note.—Number of alleles derived in one species that are fixed ancestral in the other species. ≥90% means higher than 90% allele frequency and 100% means fixed in the population. The first three rows summarize the total numbers of annotated derived alleles in each category in each population. The bottom three rows summarize the numbers of deleterious derived alleles annotated with each method, which are in high frequency in each population.
We assessed the deleteriousness of the lineage-specific SNCs as a proxy for functional and phenotypic consequences, using the six deleteriousness measures described above (Materials and Methods). This produced a catalog of genes with lineage-specific deleterious SNCs among protein-coding changes, and of genes carrying the 50 most deleterious lineage-specific SNCs in genome-wide inferences across these measures (supplementary tables S5 and S6, Supplementary Material online), in total affecting 78 genes in chimpanzees and 244 genes in bonobos. In bonobos, five of these genes are, according to the literature, involved in female reproduction: ABCA13 (Nymoen et al. 2015), ESPL1 (Gurvits et al. 2017), KIF14 (Singel et al. 2014), LVRN (Singel et al. 2014), and MAP4 (Nystad et al. 2014), whereas six are involved in male reproduction: ACSBG2 (Daisuke et al. 2009), GALNTL5 (Fraisl et al. 2006), NME8 (Takasaki et al. 2014), WBP2NL (Sadek et al. 2001), WDR63 (Wu et al. 2007), and ZFHX3 (Hozumi et al. 2008). In chimpanzees, we identified only one gene related to female reproduction (TOP2A, Hering et al. 2014; Konecny et al. 2010; Tubbs et al. 2009) and one gene involved in male reproduction (FANCL, Wong 2003). These genes are expressed in both Pan lineage cell lines (supplementary tables S9 and S10, Supplementary Material online), which implies that those changes might harbor functional differences. We also found that, in both species, multiple genes carry lineage-specific deleterious SNCs related to immunity, optic, heart and nervous system (supplementary tables S6 and S7, Supplementary Material online). Furthermore, we find nine protein-truncating variants in bonobos, and only two in chimpanzees (supplementary table S8, Supplementary Material online).
To explore the putative effect of lineage-specific SNCs on phenotypes, we explored the NHGRI-EBI GWAS Catalog (MacArthur et al. 2017). We find one lineage-specific derived SNC that is almost fixed in bonobos but absent in chimpanzees, which is polymorphic in humans and associated with the trait “economic and social preference.” Chimpanzees carry the ancestral “risk allele” rs12606301-G. On the other hand, alleles that are at high frequency in chimpanzees, but absent in bonobos, are the human protective alleles rs17356907-G for breast cancer and rs3757247-A for both Vitiligo and type I diabetes, and the risk alleles rs1872992-G for “BMI-adjusted waist-hip ratio and physical activity measurement” and rs60945108-A for “physical activity.”
Enrichment of Deleterious and Nonsynonymous SNCs
To test if these lineage-specific deleterious SNCs are enriched in any particular gene family or pathway, we performed a formal Gene Ontology enrichment test. The results suggest that, on the bonobo lineage, among others, there is an enrichment in biological categories like “homophilic cell adhesion via plasma membrane adhesion molecules,” “steroid hormone mediated signaling pathway,” and “cell morphogenesis” (supplementary table S12, Supplementary Material online). On the other hand, in the chimpanzee lineage, we find an enrichment in categories such as “ionotropic glutamate receptor signaling pathway,” “positive regulation of GTPase activity,” and neuron-related categories like “positive regulation of axonogenesis” (supplementary table S12, Supplementary Material online).
In order to explore in further detail genes that are associated with polygenic traits, we performed a systematic enrichment screen for 2,385 traits from genome-wide association studies in humans (MacArthur et al. 2017), using a more permissive set of derived nonsynonymous SNCs at high frequency (Materials and Methods). This analysis is based on the nearest genes to the associated site in humans, because most associated human SNCs are not segregating in the Pan data set. We find 17 unique traits enriched for nonsynonymous SNCs on the chimpanzee lineage, and 5 unique traits on the bonobo lineage (supplementary table S13, Supplementary Material online), among them “Menarche (age at onset).” This suggests that in bonobos there is an enrichment of nonsynonymous changes in genes affecting this female reproduction-related trait. Other categories for which we find an enrichment include “Cognitive performance,” “Parkinson’s disease,” “Urinary albumin-to-creatinine ratio,” and “Obesity-related traits,” which might reflect changes in bonobos related to cognitive abilities and metabolism. Genes with nonsynonymous SNCs on the chimpanzee lineage are enriched, among others, for associations to traits involving body mass index and height, as well as “Schizophrenia” and “Loneliness.”
The finding of an excess of lineage-specific nonsynonymous SNCs in genes associated with age at menarche suggests that we can identify genetic changes that may underlie the physiological differences between the two Pan species in terms of some female reproduction traits. We further investigated this trait using the 307 protein-coding genes associated with age at menarche in the most recent, most comprehensive metaanalysis of this trait to date (MacArthur et al. 2017), which was not included in the GWAS database. Here, we observe a significant proportion of menarche-associated genes with bonobo-specific nonsynonymous SNCs (P = 0.0025, G-test, supplementary table S14, Supplementary Material online). This observation is even more pronounced when considering that 73 SNCs (supplementary table S15, Supplementary Material online) fall into these 55 candidate genes (P = 0.001, G-test). This number of nonsynonymous changes was not observed across 1,000 random sets of genes of similar length as the menarche-associated genes (fig. 4). No enrichment of such changes is found on the chimpanzee lineage (P = 0.48, G-test). We conclude that menarche-related genes seem to have acquired more nonsynonymous SNCs than expected on the bonobo lineage.
Fig. 4.
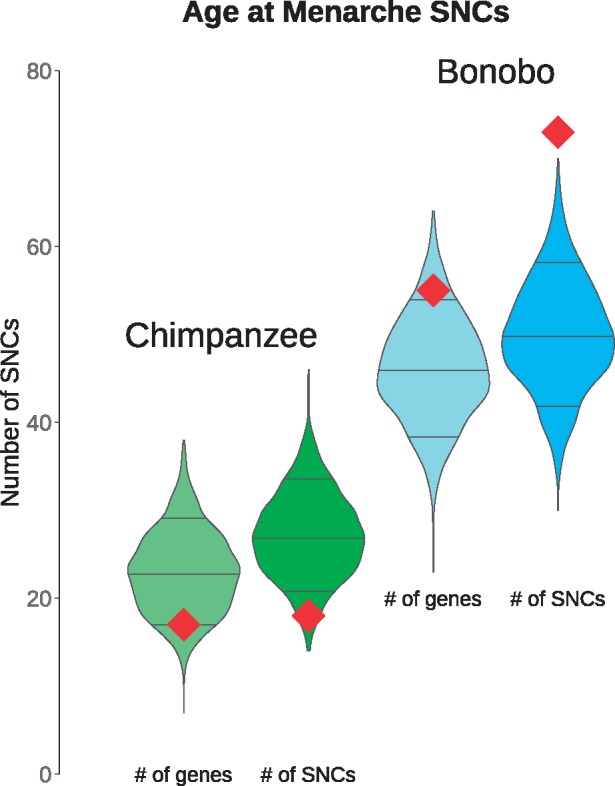
—Enrichment of nonsynonymous SNCs in genes associated with age at menarche in humans. The distributions of the overlap of genes with nonsynonymous SNCs with 1,000 random sets of 307 genes of similar coding length (±10%) as the menarche-associated genes are shown, with the observed values marked as red squares. From the left, the number of genes and SNCs with chimpanzee-specific changes, the number genes and SNCs with bonobo-specific changes. Lines represent median values, upper and lower deciles.
Discussion
We made use of the genetic variation across 10 bonobo and 59 chimpanzee high-coverage genomes (Day et al. 2017), which is the most comprehensive genomic data to date for the two Pan species, including all four known subspecies of chimpanzees. We use the predicted effects of single-nucleotide variants to analyze differences in the distribution of deleterious alleles between populations, stratified by protein-coding, noncoding functional and LoF SNCs. We present a catalog of SNCs that are lineage-specific and determine associations to known functions in either species especially for nonsynonymous variants, because these are likely to underlie phenotypic differences.
Effective Population Size Influences the Distribution of Deleterious Alleles
We investigated the efficacy of purifying selection in relation to the demographic history of populations, by making use of deleterious derived alleles. Our results show that the population-wise NI is highest in the western chimpanzee population compared with the other chimpanzee populations and bonobos (fig. 1A). We also observe that the proportions of deleterious derived alleles, in comparison to neutral derived alleles, are highest in the western chimpanzee population across different noncoding functional element categories (fig. 1C). The ratios of deleterious derived allele frequencies to neutral derived allele frequencies using six different deleteriousness prediction methods (fig. 2 and supplementary fig. S2, Supplementary Material online) show that in populations which experienced a long-term small Ne and population bottlenecks, proportionately more deleterious derived alleles segregate in the population. The proportion of deleterious derived allele frequencies in bonobos is higher at high frequencies compared with chimpanzees, which might be the consequence of a long-term small Ne and multiple population bottlenecks after the split from chimpanzees (de Manuel et al. 2016). At low frequencies, the proportion of deleterious derived allele frequencies is much higher in noncentral chimpanzee populations compared with central chimpanzees, likely the consequence of a more recent small Ne and population bottlenecks in noncentral chimpanzee populations (de Manuel et al. 2016). These observations are predicted by the nearly neutral theory (Won and Hey 2005; Prado-Martinez et al. 2013), that is, slightly deleterious mutations, which would rather be selected against than be selected for by natural selection (Ohta 1972), but minor enough to be compensated by other mechanisms, could behave like neutral mutations and more easily become fixed in populations with small Ne. These results are also similar to the report that proportionately more deleterious derived alleles are observed in Neandertal exomes compared with modern human exomes, which diverged from each other within a similar time range as chimpanzee subpopulations did, and between exomes from Eurasian and African modern humans (Castellano et al. 2014; Prüfer et al. 2014). Proportionately more deleterious derived alleles at low frequency in archaic humans and in Eurasians might be comparable to the patterns among chimpanzees, with a more recent experience of small Ne and population bottlenecks, allowing deleterious mutations to segregate. These results agree with a better efficacy to remove deleterious alleles in larger populations, as observed previously across great apes (Fu et al. 2012), and with previous reports for modern humans, dogs, and rice (Cagan et al. 2016).
On the other hand, we compare the number of homozygous to heterozygous deleterious derived alleles in each individual across populations. This clearly shows a population-wise separation in the distribution of individual deleterious load (fig. 3A). Heterozygous sites are strongly influenced by rare variants, and a proxy for the population diversity. Homozygous derived sites, on the other hand, are likely influenced by long-term Ne and population bottlenecks and dominated by fixed or high frequency derived alleles. Although there is an ongoing discussion on this matter (Charlesworth 2013; Lohmueller 2014a, 2014b ; Simons et al. 2014; Kim and Lohmueller 2015), population size and genetic drift seem to influence the distribution of changes in homozygous positions. This is interpreted as the influence of small Ne and population bottlenecks, which causes disproportionate shifts of deleterious derived changes to high frequencies in a population (Lohmueller 2014a, 2014b; Simons et al. 2014). Our results on heterozygous sites suggest that the central chimpanzee population carries the largest genetic diversity, and on the homozygous sites that western chimpanzees carry the highest level of homozygous mutational load, similar to bonobos. We conclude that the population history generally affects the way derived changes are distributed across homo- and hetero-zygous sites in the genome in our closest living relatives. Interestingly, among chimpanzees, which are generally understood as a species with a large genetic diversity (Prado-Martinez et al. 2013), the western chimpanzee population appears to be rather similar to bonobos in their mutational load, which highlights its experience of small Ne and population drift similar to bonobos, after their split from the other chimpanzee lineages.
Previously, de Valles-Ibáñez et al. (2016) analyzed the effect of population size on the deleterious burden based on LoF mutations in the great apes, suggesting it to be very weak. Here, we improve the inferences with a fine-scale analysis of the Pan clade and by increasing the number of individuals, which is critical for using the same number of individuals in each population when we stratify by frequency. It is important that we could include the western and central chimpanzee populations, which had the smallest and largest historical population sizes among the chimpanzee populations, respectively (Kuhlwilm et al. 2016). The deleterious load in LoF mutations in our data agrees well with the patterns in other categories of deleterious alleles: Western chimpanzees carry more fixed and homozygous LoF alleles and a relative excess of LoF over neutral singletons. These observations generally support the effect of a low Ne in western chimpanzees and bonobos, whereas the higher Ne in central chimpanzees leads to a larger number of disruptive mutations in singletons, which are subsequently removed from the population by purifying selection. Based on these lines of evidence, from the genome-wide measures of neutrality to the proportions of deleterious changes across frequencies to mutational load and LoF mutations, and in agreement with the observations in Cagan et al. (2016), we conclude that the small Ne in the past is a good proxy of a reduced efficacy of purifying selection in the Pan clade. We note that gene flow between chimpanzees and bonobos (de Manuel et al. 2016) might have an influence on our analysis. However, given the small extent of introgressed material (<0.25% per individual), this seems unlikely (Nye et al. 2018). It will be necessary to study much larger cohorts of chimpanzees from the different subspecies to more directly compare our observations to humans, with sample sizes several orders of magnitude larger.
The Genetic Basis of Lineage-Specific Phenotypes
Across all deleterious categories and all loci, bonobos carry a substantially larger number of lineage-specific SNCs. This reflects that in bonobos, which experienced a long-term small Ne in the past, due to genetic drift more alleles reached high frequencies or fixation. We assume that deleterious and protein-altering SNCs are likely to harbor functional consequences, possibly resulting in phenotypic changes. The concept of deleteriousness of an allele often represents conservation across species, because most new mutations are deleterious and thus removed from the population. Yet, novel functional changes would have arisen from such mutations. We present a catalog of genes with such changes, and suggest from literature that several of these in bonobos have functions in male and female reproduction, immunity or the nervous system (supplementary table S6, Supplementary Material online).
The presence of putatively functional lineage-specific SNCs is not an immediate evidence for adaptive evolution. However, we note that several genes with nonsynonymous lineage-specific SNCs (supplementary tables S9 and S10, Supplementary Material online) have been reported to show signatures of positive selection in bonobos (Cagan et al. 2016): PCDH15, IQCA1, CCDC149, and SLC36A1 in the Fay and Wu’s H test, and CFH and ULK4 in the HKA-based test (supplementary table S11, Supplementary Material online). PCDH15 (with two nonsynonymous changes in bonobos, one of them predicted to be deleterious by SIFT and PolyPhen-2) is involved in retinal and cochlear function (Jacobson et al. 2008), and ULK4 has been associated with schizophrenia and neuronal function (Lang et al. 2014). Among the genes with nonsynonymous lineage-specific SNCs in chimpanzees, MUC13 and ADGB were described to show signatures of positive selection (ELS test; Cagan et al. 2016).
One of the most prominent differences between chimpanzees and bonobos is found in female reproduction, with female bonobos having prolonged maximal sexual swellings, which female chimpanzees do not have, and female bonobos experiencing an earlier onset of their reproductive age. We hypothesized that SNCs on the bonobo lineage influencing the female reproductive system might underlie the phenotypic differences of the two species. Genome-wide signatures of strong positive selection (Cagan et al. 2016) found no enrichment in female reproduction-related genes, which could be due to the limited power of these methods to detect selection early after the divergence of the two species. However, we demonstrate that the trait “age at menarche” is among only five traits significantly enriched for bonobo-specific nonsynonymous changes. Furthermore, the most complete data set of genes associated with this trait shows an even stronger enrichment in bonobos, with 73 protein-coding SNCs in 55 genes (supplementary table S15, Supplementary Material online). Most of these genes (98%) are expressed in primary tissues of chimpanzees and bonobos (supplementary table S15, Supplementary Material online), suggesting that they are functional in the Pan clade. Among the 307 menarche-related genes, five (ATE1, DLGAP1, CSMD1, LRP1B, and TRPC6) have been reported to show signatures of positive selection (Cagan et al. 2016), and one (HLA-DQB1) of balancing selection in bonobos. Among these, the Low Density Lipoprotein Receptor LRP1B carries three protein-coding changes, and CSMD1 carries one (supplementary table S15, Supplementary Material online).
To our knowledge, this is the first time that this complex trait in female bonobos has been investigated using genetic data, with the limitation that only ten bonobo genomes are available. Bonobos are an understudied species in population genetics, hence fine-scaled studies of their population structure and sequencing of more individuals would improve power in future studies. Despite age at menarche being the associated trait in humans, these genes encompass broader functions in the female reproductive system, rather than controlling only age at menarche. Hence, we interpret this as an enrichment of functional changes in female reproduction-related genes during the evolutionary history of bonobos on a polygenic scale, with LRP1B and CSMD1 being good candidates to have the strongest influence. Our results are in agreement with suggestions that the prominent physiological differences between chimpanzee and bonobo female sexual swellings could be due to derived features in bonobos (Wrangham 1993), and suggest that it might have been adaptive on the bonobo lineage. These bonobo-specific nonsynonymous changes in menarche-related genes deserve further investigation on the functional level, which would serve as the foundation for a better understanding of the female reproductive system. The sexual swelling in female bonobos has a profound influence on their biology and group dynamics (Hohmann and Fruth 2000; Furuichi 2011), which can be considered typical for bonobo-specific behavior and sociality. Because other relevant traits show an enrichment of changes on the bonobo lineage as well (e.g., in behavior- and cognition-related genes), these deserve further investigation in future studies.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Marc de Manuel for help with data preparation and technical advice. This work was supported by the Max Planck Society (to A.M.A.), BFU2017-86471-P (MINECO/FEDER, UE) (to T.M.-B.), U01 (MH106874 to T.M.-B.), Howard Hughes International Early Career (to T.M.-B.), Obra Social “La Caixa” (to T.M.-B.), Secretaria d’Universitats i Recerca del Departament d’Economia i Coneixement de la Generalitat de Catalunya (to T.M.-B.), and a Deutsche Forschungsgemeinschaft (DFG) fellowship (KU 3467/1-1 to M.K.).
Author Contributions
T.M.-B., A.M.A., and M.K. conceived the project and wrote the article. S.H. and M.K. analyzed data and wrote the article.
Literature Cited
- Adzhubei IA, et al. 2010. A method and server for predicting damaging missense mutations. Nat Methods. 7(4):248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W.. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31(2):166.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataillon T, et al. 2015. Inference of purifying and positive selection in three subspecies of chimpanzees (Pan troglodytes) from exome sequencing. Genome Biol Evol. 7(4):1122–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer V, Deschner T, Deimel C, Stevens JMG, Hohmann G.. 2014. Age-related changes in urinary testosterone levels suggest differences in puberty onset and divergent life history strategies in bonobos and chimpanzees. Horm Behav. 66(3):525–533. [DOI] [PubMed] [Google Scholar]
- Brawand D, et al. 2011. The evolution of gene expression levels in mammalian organs [SupMat]. Nature 478(7369):343–348. [DOI] [PubMed] [Google Scholar]
- Cagan A, et al. 2016. Natural selection in the great apes. Mol Biol Evol. 33(12):3268–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano S, et al. 2014. Patterns of coding variation in the complete exomes of three Neandertals. Proc Natl Acad Sci U S A. 111(18):6666–6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. 2013. Why we are not dead one hundred times over. Evolution (N Y). 67:3354–3361. [DOI] [PubMed] [Google Scholar]
- Daisuke A, et al. 2009. Overexpression of class III beta-tubulin predicts good response to taxane-based chemotherapy in ovarian clear cell adenocarcinoma. Clin Cancer Res. 15:1473–1480. [DOI] [PubMed] [Google Scholar]
- Davydov EV, et al. 2010. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol. 6(12):e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day FR, et al. 2017. Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nat Genet. 49(6):834–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Manuel M, et al. 2016. Chimpanzee genomic diversity reveals ancient admixture with bonobos. Science 354(6311):477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Valles-Ibáñez G, et al. 2016. Genetic load of loss-of-function polymorphic variants in great apes. Genome Biol Evol. 8(3):871–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, et al. 2005. BioMart and bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21(16):3439–3440. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A. 2006. The genomic rate of adaptive evolution. Trends Ecol Evol. 21(10):569–575. [DOI] [PubMed] [Google Scholar]
- Fischer A, et al. 2011. Bonobos fall within the genomic variation of chimpanzees. PLoS One 6(6):e21605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraisl P, et al. 2006. A novel mammalian bubblegum-related acyl-CoA synthetase restricted to testes and possibly involved in spermatogenesis. Arch Biochem Biophys. 451(1):23–33. [DOI] [PubMed] [Google Scholar]
- Fu W, et al. 2012. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493:216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuichi T. 1987. Sexual swelling, receptivity, and grouping of wild pygmy chimpanzee females at Wamba, Zaïre. Primates 28(3):309–318. [Google Scholar]
- Furuichi T. 2011. Female contributions to the peaceful nature of bonobo society. Evol Anthropol. 20(4):131–142. [DOI] [PubMed] [Google Scholar]
- Goodall J. 1986. The chimpanzees of gombe: patterns of behavior. Cambridge: Harvard University Press. [Google Scholar]
- Grantham R. 1974. Amino acid difference formula to help explain protein evolution. Science 185(4154):862–864. [DOI] [PubMed] [Google Scholar]
- Gronau I, Hubisz MJ, Gulko B, Danko CG, Siepel A.. 2011. Bayesian inference of ancient human demography from individual genome sequences. Nat Genet. 43(10):1031–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurvits N, et al. 2017. Separase is a marker for prognosis and mitotic activity in breast cancer. Br J Cancer 117(9):1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare B, Wobber V, Wrangham R.. 2012. The self-domestication hypothesis: evolution of bonobo psychology is due to selection against aggression. Anim Behav. 83(3):573–585. [Google Scholar]
- Henn BM, et al. 2016. Distance from sub-Saharan Africa predicts mutational load in diverse human genomes. Proc Natl Acad Sci U S A. 113(4):E440–E449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering D, Olenski K, Kaminski S.. 2014. Genome-wide association study for sperm concentration in Holstein-Friesian bulls. Reprod Domest Anim. 49(6):1008–1014. [DOI] [PubMed] [Google Scholar]
- Hernandez RD. 2008. A flexible forward simulator for populations subject to selection and demography. Bioinformatics 24(23):2786–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkinson A, et al. 2013. Selective constraint, background selection, and mutation accumulation variability within and between human populations. BMC Genomics. 14(1):495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann G, Fruth B.. 2000. Use and function of genital contacts among female bonobos. Anim Behav. 60(1):107–120. [DOI] [PubMed] [Google Scholar]
- Hozumi A, Padma P, Toda T, Ide H, Inaba K.. 2008. Molecular characterization of axonemal proteins and signaling molecules responsible for chemoattractant-induced sperm activation in Ciona intestinalis. Cell Motil Cytoskeleton 65(3):249–267. [DOI] [PubMed] [Google Scholar]
- Huber W, et al. 2015. Orchestrating high-throughput genomic analysis with bioconductor. Nat Methods. 12(2):115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idani G. 1990. Relations between unit-groups of bonobos at Wamba, Zaire: encounters and temporary fusions. Afr Study Monogr. 11(3): 153–186. [Google Scholar]
- Jacobson SG, et al. 2008. Usher syndromes due to MYO7A, PCDH15, USH2A or GPR98 mutations share retinal disease mechanism. Hum Mol Genet. 17(15):2405–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jukes TH, Kimura M.. 1984. Evolutionary constraints and the neutral theory. J Mol Evol. 21(1):90–92. [DOI] [PubMed] [Google Scholar]
- Kidd JM, et al. 2012. Population genetic inference from personal genome data: impact of ancestry and admixture on human genomic variation. Am J Hum Genet. 91(4):660–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BY, Lohmueller KE.. 2015. Selection and reduced population size cannot explain higher amounts of Neandertal ancestry in East Asian than in European human populations. Am J Hum Genet. 96(3):454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, et al. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14(4):R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. 1968. Evolutionary rate at the molecular level. Nature 217(5129):624–626. [DOI] [PubMed] [Google Scholar]
- Kircher M, et al. 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 46(3):310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konecny GE, et al. 2010. Association between HER2, TOP2A, and response to anthracycline-based preoperative chemotherapy in high-risk primary breast cancer. Breast Cancer Res Treat. 120(2):481–489. [DOI] [PubMed] [Google Scholar]
- Kuhlwilm M, et al. 2016. Evolution and demography of the great apes. Curr Opin Genet Dev. 41:124–129. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC.. 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 4(7):1073–1081. [DOI] [PubMed] [Google Scholar]
- Kuroda S. 1980. Social behavior of the pygmy chimpanzees. Primates 21(2):181–197. [Google Scholar]
- Lang B, et al. 2014. Recurrent deletions of ULK4 in schizophrenia: a gene crucial for neuritogenesis and neuronal motility. J Cell Sci. 127(3):630–640. [DOI] [PubMed] [Google Scholar]
- Langergraber K, Prüfer K.. 2012. Generation times in wild chimpanzees and gorillas suggest earlier divergence times in great ape and human evolution. Proc Natl Acad Sci U S A. 109:15716–15721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, et al. 2013. Software for computing and annotating genomic ranges. PLoS Comput Biol. 9(8):e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536(7616):285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W-H, Wu C-I, Luo C-C.. 1984. Nonrandomness of point mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications. J Mol Evol. 21(1):58–71. [DOI] [PubMed] [Google Scholar]
- Liu Q, Zhou Y, Morrell PL, Gaut BS.. 2017. Deleterious variants in Asian rice and the potential cost of domestication. Mol Biol Evol. 34(4):908–924. [DOI] [PubMed] [Google Scholar]
- Lohmueller KE. 2014a. The distribution of deleterious genetic variation in human populations. Curr Opin Genet Dev. 29:139–146. [DOI] [PubMed] [Google Scholar]
- Lohmueller KE. 2014b. The impact of population demography and selection on the genetic architecture of complex traits. PLoS Genet. 10(5): e1004379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller KE, et al. 2008. Proportionally more deleterious genetic variation in European than in African populations. Nature 451(7181):994–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur J, et al. 2017. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 45(D1):D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden CD, et al. 2016. Bottlenecks and selective sweeps during domestication have increased deleterious genetic variation in dogs. Proc Natl Acad Sci U S A. 113(1):152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, et al. 2016. The Ensembl Variant Effect Predictor. Genome Biol. 17(1):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida T. 1983. Alpha status and agonistic alliance in wild chimpanzees (Pan troglodytes schweinfurthii). Primates 24(3):318–336. [Google Scholar]
- Nunn CL. 1999. The evolution of exaggerated sexual swellings in primates and the graded-signal hypothesis. Anim Behav. 58(2):229–246. [DOI] [PubMed] [Google Scholar]
- Nye J, et al. 2018. Selection in the introgressed regions of the chimpanzee genome. Genome Biol Evol. 10(4):1132–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nymoen DA, Holth A, Hetland Falkenthal TE, Tropé CG, Davidson B.. 2015. CIAPIN1 and ABCA13 are markers of poor survival in metastatic ovarian serous carcinoma. Mol Cancer 14(1):44.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystad M, Sitras V, Larsen M, Acharya G.. 2014. Placental expression of aminopeptidase-Q (laeverin) and its role in the pathophysiology of preeclampsia. Am J Obstet Gynecol. 211(6):686.e1–686.e31. [DOI] [PubMed] [Google Scholar]
- Ohta T. 1972. Population size and rate of evolution. J Mol Evol. 1(4):305–314. [DOI] [PubMed] [Google Scholar]
- Ohta T. 1973. Slightly deleterious mutant substitutions in evolution. Nature 246(5428):96–98. [DOI] [PubMed] [Google Scholar]
- Prado-Martinez J, et al. 2013. Great ape genetic diversity and population history. Nature 499(7459):471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prüfer K, et al. 2007. FUNC: a package for detecting significant associations between gene sets and ontological annotations. BMC Bioinformatics 8:41.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prüfer K, et al. 2014. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505(7481):43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand DM, Kann LM.. 1996. Excess amino acid polymorphism in mitochondrial DNA: contrasts among genes from Drosophila, mice, and humans. Mol Biol Evol. 13(6):735–748. [DOI] [PubMed] [Google Scholar]
- Randall Parish A. 1994. Sex and food control in the ‘uncommon chimpanzee’: How bonobo females overcome a phylogenetic legacy of male dominance. Ethol Sociobiol. 15(3):157–179. [Google Scholar]
- Ritchie GRS, Dunham I, Zeggini E, Flicek P.. 2014. Functional annotation of noncoding sequence variants. Nat Methods. 11(3):294–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu H, Hill DA, Furuichi T.. 2015. Prolonged maximal sexual swelling in wild bonobos facilitates affiliative interactions between females. Behaviour 152(3–4):285–311. [Google Scholar]
- Sá RM, et al. 2013. Gastrointestinal symbionts of chimpanzees in Cantanhez National Park, Guinea-Bissau with respect to habitat fragmentation. Am J Primatol. 75(10):1032–1041. [DOI] [PubMed] [Google Scholar]
- Sadek CM, et al. 2001. Sptrx-2, a fusion protein composed of one thioredoxin and three tandemly repeated NDP-kinase domains is expressed in human testis germ cells. Genes Cells 6(12):1077–1090. [DOI] [PubMed] [Google Scholar]
- Shimada MK, et al. 2004. Mitochondrial DNA genealogy of chimpanzees in the Nimba mountains and Bossou, West Africa. Am J Primatol. 64(3):261–275. [DOI] [PubMed] [Google Scholar]
- Simons YB, Turchin MC, Pritchard JK, Sella G.. 2014. The deleterious mutation load is insensitive to recent population history. Nat Genet. 46(3):220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singel SM, et al. 2014. KIF14 promotes AKT phosphorylation and contributes to chemoresistance in triple-negative breast cancer. Neoplasia 16(3):247–256.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoletzki N, Eyre-Walker A.. 2011. Estimation of the neutrality index. Mol Biol Evol. 28(1):63–70. [DOI] [PubMed] [Google Scholar]
- Subramanian S, Lambert DM.. 2012. Selective constraints determine the time dependency of molecular rates for human nuclear genomes. Genome Biol Evol. 4(11):1127–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasaki N, et al. 2014. A heterozygous mutation of GALNTL5 affects male infertility with impairment of sperm motility. Proc Natl Acad Sci U S A. 111(3):1120–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto H, et al. 2017. The mitochondrial ancestor of bonobos and the origin of their major haplogroups. PLoS One 12(5):e0174851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JAM. 2003. A model of the biogeographical journey from Proto-pan to Pan paniscus. Primates 44:191–197. [DOI] [PubMed] [Google Scholar]
- Thompson-Handler N, Malenky RK, Badrian N.. 1984. Sexual behavior of Pan paniscus under natural conditions in the Lomako Forest, Equateur, Zaire In: The pygmy chimpanzee. Boston (MA: ): Springer; p. 347–368. [Google Scholar]
- Torkamani A, et al. 2012. Clinical implications of human population differences in genome-wide rates of functional genotypes. Front Genet. 3:211.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs R, et al. 2009. Outcome of patients with early-stage breast cancer treated with doxorubicin-based adjuvant chemotherapy as a function of HER2 and TOP2A status. J Clin Oncol. 27(24):3881–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won Y-J, Hey J.. 2005. Divergence population genetics of chimpanzees. Mol Biol Evol. 22(2):297–307. [DOI] [PubMed] [Google Scholar]
- Wong J. 2003. Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet. 12(16):2063–2076. [DOI] [PubMed] [Google Scholar]
- Wrangham RW. 1993. The evolution of sexuality in chimpanzees and bonobos. Hum Nat. 4(1):47–79. [DOI] [PubMed] [Google Scholar]
- Wu ATH, et al. 2007. PAWP, a sperm-specific WW domain-binding protein, promotes meiotic resumption and pronuclear development during fertilization. J Biol Chem. 282(16):12164–12175. [DOI] [PubMed] [Google Scholar]
- Xue Y, et al. 2015. Mountain gorilla genomes reveal the impact of long-term population decline and inbreeding. Science 348(6231):242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.