

Abstract
Background
The ataxia telangiectasia and Rad3-related (ATR) checkpoint kinase 1 (CHK1) pathway plays an essential role in suppressing replication stress from DNA damage and oncogene activation.
Main body
Preclinical studies have shown that cancer cells with defective DNA repair mechanisms or cell cycle checkpoints may be particularly sensitive to ATR inhibitors. Preclinical and clinical data from early-phase trials on three ATR inhibitors (M6620, AZD6738, and BAY1895344), either as monotherapy or in combination, were reviewed.
Conclusion
Data from ATR inhibitor-based combinational trials might lead to future expansion of this therapy to homologous recombination repair pathway-proficient cancers and potentially serve as a rescue therapy for patients who have progressed through poly ADP-ribose polymerase inhibitors.
Keywords: ATR, Replication stress, DNA damage, ATR inhibitors, Cancer
Background
DNA damage response (DDR) is a complex interconnected signaling network that is essential to defend human genome integrity against a variety of exogenous and endogenous genotoxic insults, such as ultraviolet radiation, ionizing radiation, or reactive oxygen species. Ataxia telangiectasia-mutated (ATM) checkpoint kinase 2 (CHK2) and ataxia telangiectasia and Rad3-related (ATR) checkpoint kinase 1 (CHK1) signals are two key pathways to initiate DDR. In response to DNA double-strand (dsDNA) breaks, the MRE11/NBS1/RAD5 complex activates the ATM-CHK2 kinase, which stabilizes p53 through phosphorylation and arrests the cell cycle at the G1/S phase checkpoint [1, 2]. When single-strand DNA (ssDNA) is produced at sites of DNA damage or stressed replication forks, replication protein A-coated ssDNA mobilizes ATR and its binding partner, ATR interacting protein (ATRIP) [3, 4]. CHK1 is subsequently phosphorylated by ATR on Ser-317 and Ser-345 [5]. Inhibitory phosphorylation by CHK1 of the phosphatase CDC25A and its subsequent proteasomal degradation leads to a decrease in CDK2 activity during the S phase [6], triggering the intra-S phase and G2/M phase checkpoints [7–9]. Given that an extended ssDNA of a stalled replication fork is a common feature of replication stress, ATR also plays a key role in replication stress response. After ssDNA is coated by RPA, ATR is recruited along with its obligatory partner, ATRIP, to initiate replication stress response. ATR-ATRIP complex activation requires TOPBP1, the trimeric RAD9-RAD1-HUS1 (9-1-1) complex, and Ewing’s tumor-associated antigen 1 (ETAA1). ATR, along with protein adaptors such as Claspin or 9-1-1 complex and interacting nuclear orphan (RHINO), subsequently phosphorylates a multitude of targets, including CHK1 [3, 17]. The downstream targets of the activated ATR-CHK1 axis are essential in suppressing replication stress [18]. ATM knock out mice have a similar ataxia telangiectasia phenotype with a high incidence of lymphoma [19], whereas homozygous elimination of ATR leads to chromosome breaks, proliferative failure in culture, and early embryonic lethality [20].
In vitro and in vivo studies demonstrated cross talks between the ATM and ATR pathways [10–12]. ATM-mediated dsDNA break processing results in regions of RPA-coated ssDNA that are then recognized by ATR. ATR is subsequently activated in response to dsDNA breaks in an ATM-dependent manner [21–23]. UV and hydroxyurea, potent activators of ATR, were also shown to phosphorylate and activate ATM in an ATR-dependent manner [24]. Depletion of ATR with doxycycline-inducible lentiviral system in ATM-deficient cells caused severe G2/M checkpoint attenuation and synthetic lethality following ionizing radiation [16]. Inhibition of ATR with small molecule inhibitor selectively sensitized ATM or p53-deficient cancer cells to cisplatin [13–15].
Several features of cancer cells may sensitize them to inhibitors that target ATR-CHK1. First, ATR-CHK1-mediated signaling is often particularly evident in cells with a defective G1 checkpoint that was caused by a mutation in p53 or a loss of retinoblastoma protein. Mutations in p53 have been reported as potential resistance mechanisms to cytotoxic chemotherapy or targeted therapies such as poly (ADP-ribose) polymerase (PARP) inhibitors [25]. In addition, premature entering into mitosis phase after abrogation of G1 and/or G2 checkpoints will lead to mitotic catastrophe, which is characterized by the formation of nuclear envelopes around individual clusters of missegregated chromosomes [26]. Blocking ATR activity with small molecular inhibitors either alone or combined with DNA-damaging agents can lead to mitotic catastrophe of cancer cells and p53-independent cell death [13, 27]. ATR and/or CHK1 suppress replication stress that arises from causes such as DNA damage and oncogene activation. Combining ATR inhibitors with DNA-damaging radiation or chemotherapy could lead to synthetic lethality, particularly in cancer cells that harbor overexpression of oncogenes like Myc [28]. Currently, there are three different ATR inhibitors under early-phase clinical development (Fig. 1). These are M6620, AZD6738, and BAY1895344. In this mini-review, we discuss the emerging clinical data that have been reported through the development of these ATR inhibitors as either mono- or combinational therapies (Table 1).
Fig. 1.
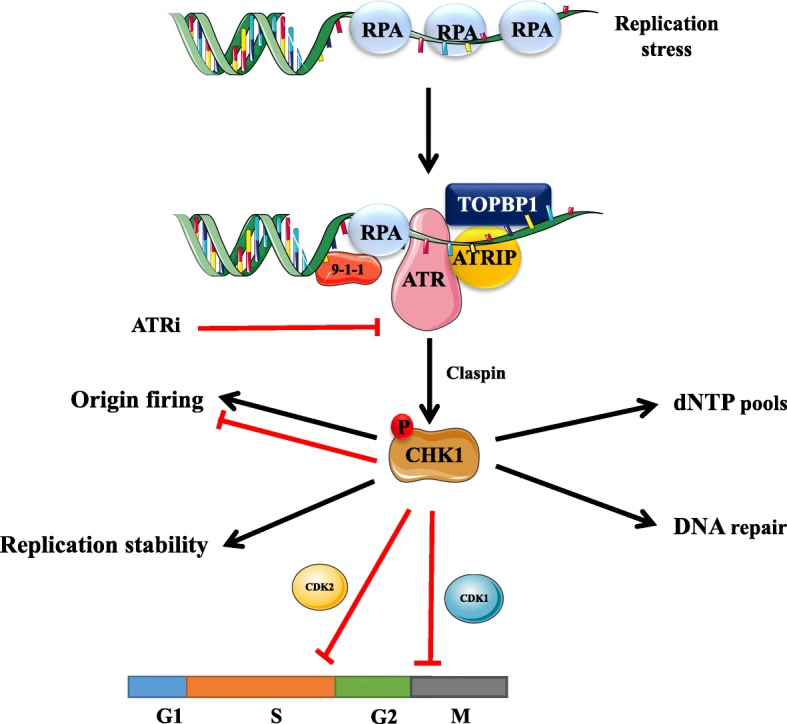
Replication stress induced ATR-CHK1 activation. ATR is activated by replication protein A (RPA)-coated single-stranded DNA (ssDNA) that arises at stalled replication fork or resected DNA double-strand break (DSB), particularly at ssDNA and double-stranded DNA (dsDNA) juncture. The recruitment of ATR-interacting protein (ATRIP) leads to recognition of ATR and RPA-ssDNA complex. Subsequently, it incorporates Rad9-Rad1-hus1 (9-1-1) and DNA topoisomerase 2-binding protein 1 (TOPBP1), leading to ATR activation. Mediated by adaptor protein claspin, ATR phosphorylates checkpoint kinase 1 (CHK1). The activation of CHK1 can prevent genomic instability. The mechanisms are either promoting or inhibiting the initiation of DNA replication (origin firing), ensuring sufficient supply of deoxynucleotides (dNTPs) pool, stabilizing replication fork and DNA repair. Its downstream molecules, cyclin-dependent kinase (CDK) 1 and CDK2, suppresses G2-M transition and slows down S phase
Table 1.
Summary of ATR inhibitor-based clinical trials
| ATRi | Target cancer type | Treatment | Phase | Biomarker selection | Efficacy and toxicity | Identifier |
|---|---|---|---|---|---|---|
| M6620 (formerly VX-970, IV) | Advanced solid tumor | Alone or with carboplatin/paclitaxel | I | Gr 3/4: 5–21% 1 pt had PR | NCT03309150 | |
| Advanced solid tumor | Gemcitabine, cisplatin, etoposide, or carboplatin | I | TP53 mutation of ATM loss | Gem, Gr 3/4, 50%; PFS, 8.0–29.3 weeks Cis, Gr 3/4, 46%; PFS, 4.1 months |
NCT02157792 | |
| Advanced solid tumor | Irinotecan | I | NCT02595931 | |||
| Small-cell cancers | Topotecan | I/II | DDR pathway mutations | Gr 3/4, 10–19%; PR + SD, 42.8% | NCT02487095 | |
| Urothelial carcinoma | Cisplatin or gemcitabine | I/II | p53, p21, and ERCC2 mutations | NCT02567409 | ||
| Ovarian cancer | Carboplatin + gemcitabine | I/II | DNA damage assay, HRR mutations | NCT02627443 | ||
| Ovarian cancer | Gemcitabine | II | NCT02595892 | |||
| mCRPC | Carboplatin ± docetaxel | II | NCT03517969 | |||
| Advanced solid tumor | Cisplatin + veliparib | I | DNA damage and apoptotic assay | Gr 3/4, 4–31%; 3 pts had PR | NCT02723864 | |
| HNSCC | Cisplatin + XRT | I | DNA damage assay | NCT02567422 | ||
| Brain metastases | Whole brain XRT | I | ATR, CHK1, RAD51, cyclin E, DNA-PK assay | NCT02589522 | ||
| M4344 (oral) | Advanced solid tumor | Carboplatin, gemcitabine, or cisplatin | I | NCT02278250 | ||
| AZD6738 (oral) | CLL, PLL or B-cell lymphoma | Alone | I | ATR targeted inhibition biomarker | NCT01955668 | |
| HNSCC | Alone | I | TH1/IFNγ gene and TIL state | NCT03022409 | ||
| Refractory CLL | Acalabrutinib | I | NCT03328273 | |||
| Advanced solid tumor | Paclitaxel | I | NCT02630199 | |||
| Advanced solid tumor | Carboplatin, olaparib, or durvalumab | I/II | ATM deficiency | Carbo, Gr 3/4, 27–33%; 3 pts had PR Ola, Gr 3/4, 4–7%; 2 pts BRCA-mut had PR Dur, no Gr 3/4; 1 pt had PR, 1 had CR |
NCT02264678 | |
| TNBC | Olaparib | II | HRR mutations | NCT03330847 | ||
| Advanced tumor | Olaparib | II | NCT02576444 | |||
| SCLC | Olaparib | II | NCT03428607 | |||
| Ovarian cancer | Olaparib | II | NCT03462342 | |||
| NSCLC | Durvalumab | II | NCT03334617 | |||
| Advanced solid tumor | XRT | I | NCT02223923 | |||
| BAY1895344 | Solid tumor and lymphoma | Alone | I | NCT03188965 |
Abbreviations: CLL chronic lymphocytic leukemia, DDR DNA damage response, Gr grade, HNSCC head and neck squamous cell carcinoma, HRR homologous recombination repair, mCRPC metastatic castration-resistant prostate cancer, NSCLC non–small cell lung cancer, PFS progression-free survival, PLL prolymphocytic leukemia, PR partial response, SCLC small cell lung cancer, SD stable disease, TNBC triple-negative breast cancer, XRT X-ray radiotherapy
ATR inhibitors as monotherapy
M6620 (formerly VX-970 or berzosertib), developed by EMD Serono, is a first-in-class ATR inhibitor that has been tested in human models. It has been shown to enhance tumor DNA replication fork collapse when combined with cisplatin and gemcitabine in vitro [27, 29]. M6620 is well tolerated, and no associated dose-limiting toxicities (DLTs) or grade 3/4 adverse events (AEs) were observed during the subsequent phase 1 study [30]. The recommended phase 2 dose (RP2D) for M6620 monotherapy is 240 mg/m2 given once weekly via intravenous infusion [31]. CHK1 phosphorylation, a marker for ATR inhibition, was observed to be reduced by 73 to 90% with M6620 in the pre- and posttreatment paired biopsies [31]. One colorectal cancer patient with ATM gene loss achieved complete response and remained on single-agent M6620 for more than 20 months [30]. This clinical observation is consistent with the preclinical data on the synthetic lethality between ATM and ATR inhibitors [13–16]. M4344 (formerly VX-803), an oral ATR inhibitor, is currently under phase 1 evaluation as a single agent and in combination with cytotoxic chemotherapy for advanced solid tumors (NCT02278250).
AZD6738 is an orally active ATR inhibitor developed by AstraZeneca that has been shown in vitro to inhibit ATR kinase activity at IC50 of 1 nM and CHK1 phosphorylation at IC50 of 74 nM [13]. AZD6738 was shown to suppress both solid and hematological cell lines with an IC50 of less than 1 μM [14, 32, 33]. Synthetic lethality was observed with AZD6783 in gastric cancer, non-small cell lung cancer (NSCLC), and chronic lymphocytic leukemia (CLL) cell lines that were ATM deficient [13, 32]. Furthermore, AZD6783 sensitized TP53-deficient CLL cells to chemotherapy and ibrutinib [32]. Combining AZD6738 with WEE inhibitor AZD1775 led to mitosis catastrophe and inactivated Rad 51-mediated homologous recombination in triple-negative breast cancer cell lines [33]. On the basis of these preclinical data, AZD6378 was tested as a monotherapy in a phase 1 study for 11q-deleted or ATM-deficient relapsed/refractory CLL (NCT01955668). One arm of this completed phase 1 study investigated the safety and tolerability of AZD6378 among patients with relapsed/refractory CLL, polymorphic leukemia, or B cell lymphoma. The ongoing phase 1 Patriot study aims to identify the maximum tolerated dose (MTD) of AZD6783 alone and in combination with palliative radiation in advanced-stage solid tumors (NCT02223923) [34].
BAY1895344 is an ATR inhibitor developed by Bayer that is used to inhibit the proliferation of human cancer cell lines with a median IC50 of 78 nM. Most sensitive cell lines are characterized by mutations of the ATM-associated pathway [14]. The synergy between BAY1895334 and radium-223, an FDA-approved alpha radiopharmaceutical, was observed in a bone metastasis xenograft model of castration-resistant prostate cancer [14]. The phase 1 study with BAY1895344 in advanced solid tumors and lymphoma is currently at the dose-expansion phase (NCT03188965).
ATR inhibitors with DNA-damaging cytotoxic chemotherapy
Potential synergy in cell killing was observed when an ATR inhibitor was combined with DNA-damaging chemotherapy in preclinical models. When adding AZD6738 to cisplatin, enhanced cytotoxicity was noted in NSCLC cell lines and cell line xenografts with intact ATM signaling [13]. Potent synergy in cell killing was noted after combining cisplatin with AZD6738 in ATM-deficient NSCLC xenografts [13]. Potential synergies in cancer cell killing were also reported when AZD6738 was combined with cisplatin or gemcitabine in preclinical models of breast cancer [35] and pancreatic cancer [36]. In 2016, Yap et al. reported phase 1 dose escalation data on investigations of AZD6738/carboplatin, AZD6738/olaparib, and AZD6738/durvalumab combinations in advanced solid tumors [37]. AZD6738 at 40 mg given twice daily on days 1 and 2, along with carboplatin at an area under the curve of 5 given on day 1, was the recommended RP2D. Twenty-seven patients were enrolled in the study. Grade 3 toxicities included thrombocytopenia (36.4%), neutropenia (27.3%), and anemia (33.3%). Three patients (9.1%) with ATM-deficient ovarian, cervical, and rectal cancers achieved partial remission [37]. The dosing and safety of the other two combinations are addressed below.
The ATR inhibitor and carboplatin (area under the curve of 5 at day 1) combination was also tested with M6620 in a phase 1 study reported by O’Carrigan et al. [31]. M6620 given intravenously on days 2 and 9 of a 21-day cycle was tolerated at a higher dose (90 mg/m2) with fewer associated grade 3/4 AEs than oral AZD6738. Grade 3/4 neutropenia was observed in four patients (21%), and grade 3/4 thrombocytopenia was reported in one (5%). One patient with BRCA1-mutated, platinum-refractory, PARP inhibitor–resistant ovarian cancer achieved partial response (PR) for 6 months [31, 38]. Preliminary data for the dose-expansion cohort of the M6620/cisplatin combination (NCT02157792) were presented at the San Antonio Breast Cancer Symposium in December 2017. In this expansion cohort, patients with metastatic triple-negative breast cancer (TNBC) were given M6620 at 140 mg/m2 on days 2 and 9 and cisplatin at 75 mg/m2 on day 1 of every 21-day cycle. Thirty-five females were enrolled in this study, 18 of whom were confirmed to have BRCA1/2 wild-type TNBC. Median progression-free survival was 4.1 months, and preliminary unconfirmed objective response was 38.9%. Grade 3/4-related AEs occurred among 16 patients (45.7%) [39].
M6620 was also tested in combination with gemcitabine in a phase 1 study on advanced solid tumors. The RP2D and schedule were M6620 given at 210 mg/m2 on days 2, 9, and 16 along with gemcitabine at 1000 mg/m2 given on days 1 and 8 of each 21-day cycle. Grade 3/4 toxicity was observed in 25 out of 50 patients. PR was noted in one out of four breast cancer cases and in one out of six NSCLC cases. Median progression-free survival ranged from 8.3 to 29.3 weeks (NCT02157792) [40].
Enhanced sensitivity to a topoisomerase I inhibitor was observed in ATR-depleted preclinical models. On the basis of this observation, Thomas et al. conducted a phase 1 study (NCT02487095) in which M6620 combined with topotecan was used to treat 21 patients with advanced solid tumors [41]. The maximal planned dose was reached with M6620 at 210 mg/m2 given on days 2 and 5 plus topotecan at 1.25 mg/m2 given on days 1 to 5 of every 21-day cycle. Grade 3/4 toxicities were mainly myelosuppression related, including anemia (19%), leukopenia (19%), neutropenia (19%), lymphopenia (14%), and thrombocytopenia (10%). Two cases of PR and seven cases of stable disease (SD) were observed, which accounted for the 42.8% disease control rate. Moreover, three out of five patients (60%) with platinum-refractory small-cell lung cancer had PR or prolonged SD [41]. Pharmacodynamic studies showed preliminary evidence of enhanced DNA double-strand breaks in response to this combination.
As in the clinical development of the PARP inhibitor/DNA-damaging chemotherapy combination, cytopenias were the main grade 3/4 toxicities in early-phase trials combining an ATR inhibitor with cytotoxic chemotherapy. Nevertheless, M6620 was better tolerated when combined with a topoisomerase inhibitor than with platinum chemotherapy. Small-cell lung cancer is characterized with high frequency of p53 and Rb1 loss, genomic instability, and high mutation burden [42]. The disease is either refractory to platinum-based chemotherapy, or more often, initially responds to the treatment but subsequently becomes resistant. The refractory/resistance mechanism is not fully understood. The mutation status of P53 and Rb1 might be associated with refractory/resistance to chemotherapy [43]. Other factors including SLFN11 gene silencing are also considered to be a potential mechanism of chemoresistance [44]. The combination of M6620 with a topoisomerase inhibitor seems to have great potential in platinum-refractory/resistance small-cell lung cancer.
This use of this combination in platinum-refractory small-cell lung cancer would be a breakthrough in the treatment of this rare and aggressive cancer.
ATR inhibitors with radiotherapy
Ionizing radiation is known to cause a variety of DNA damages, including double-strand and single-strand DNA breaks. To repair radiation-induced DNA damage, cell cycle checkpoint activation is required, as it facilitates the time necessary for damaged-DNA reparations. Blocking ATR activity would not only impair DNA repair but would also block cell cycle checkpoint activation. This double blockade in the DDR against ionizing radiation has been proven to be effective in cancer cell killing in several preclinical models. AZD6738 was shown to radiosensitize multiple cancer cell lines regardless of the p53 and BRCA status [45]. A similar effect was observed with M6620 in pancreatic cancer [46] and lymphoma [47] cell lines. The phase 1 study PATRIOT (NCT02223923) uses a 3 + 3 design to test dose escalation of AZD6738, first as a monotherapy and then in combination with 20 Gy in ten fractions of palliative radiotherapy. The radiation dose will be escalated after the MTD of AZD6738 is reached. The expansion cohorts will add maintenance doses of AZD6738 until disease progression [34]. M6620 is also being studied in combination with whole brain radiation among patients with brain metastases from NSCLC (NCT02589522) and in combination with concurrent chemoradiation with cisplatin for head and neck squamous cell carcinoma (NCT02567422).
ATR inhibitors with immune checkpoint inhibitors
Emerging preclinical evidence indicates that ATR inhibitors can block the programmed death-ligand 1 upregulation on cancer cell surfaces and mitigate the tumor infiltration of regulatory T cells after treatment with radiation or cisplatin [48]. The safety and tolerability of combining ATR inhibitor with anti-programmed death-ligand 1 therapy are being tested in an ongoing phase 1 study [37]. In this study, AZD6738 was given at 80 mg twice daily during the 14-day monotherapy run, which was followed by AZD6738 given on days 22 to 28 concomitantly with durvalumab 1500 mg on days 1 and 28. One patient with squamous cell carcinoma of the larynx achieved PR, and one patient with NSCLC potentially obtained complete response [37]. This dose and schedule were well tolerated with no DLTs observed. This combination is being evaluated in a phase 2 multi-arm umbrella study specifically for ATM-deficient NSCLC [49]. On the basis of having a good tolerability, the ATR inhibitor/immune checkpoint inhibitor combination could be developed as a trimodality therapy by adding treatments such as ionizing radiation.
ATR inhibitors with PARP inhibitors
RNA interference (RNAi)-mediated depletion or inhibition of ATR has been shown to sensitize ovarian cancer cells to cisplatin, topotecan, gemcitabine, and the PARP inhibitor veliparib (ABT-888) [50]. Moreover, an ATR inhibitor further enhanced the killing of BRCA1-depleted ovarian cancer cells by cisplatin, topotecan, and veliparib [50]. Amplification of ATR and CHK1 genes was noted in ovarian cancers with genomic instability. Inactivating Rad51 in the homologous recombination repair (HR) pathway led to differential sensitivity of MCF-7 and Hela cells to ATR and CHK1 inhibitors, implicating ATR and CHK1 as potential drug targets for HR-defective cancers [51]. More recently, ATR was shown to control the abundance of HR factors, largely via CHK1-dependent transcription and promotion of specific HR protein stabilization. Long-term inhibition of ATR signaling severely impaired the ability of cells to use HR-mediated DNA repair [52]. Collectively, these preclinical studies provide the rationale for using an ATR inhibitor/PARP inhibitor combination in HR-proficient and HR-deficient cancer cells.
Twenty-seven patients were enrolled in the AZD6738 and olaparib arm of the phase 1 AZD6738 combinational study reported by Yap et al. at the 2016 EORTC-NCI-AACR Molecular Targets and Cancer Therapeutics Symposium [37]. The RP2D included AZD6738 at 160 mg daily from days 1 to 7 and olaparib at 300 mg twice a day from days 1 to 28. Two patients with BRCA-mutant TNBC achieved PR with this regimen [37]. M6620 was evaluated in combination with veliparib and cisplatin in a phase 1 study, with the intention to induce a BRCA null-like phenotype. When the preliminary data were reported in 2018, the MTD was not yet reached and the study was enrolling at dose level (DL) 7 with cisplatin at 40 mg/m2 on days 1 and 8, M6620 at 210 mg/m2 on days 2 and 9, and veliparib at 200 mg taken orally twice daily from days 1 to 3 and 8 to 10 [53]. The associated grade 3/4 AEs included hypophosphatemia (4%), thrombocytopenia (31%), leukopenia (22%), and lymphopenia (18%). PR was achieved in 3 out of 22 patients (13.6%), including 1 with BRCA wild-type ovarian cancer. SD was observed in 12 out of 22 patients (54.5%) [53].
Further clinical investigation of the PARP inhibitor/ATR inhibitor combination has been extended to multiple phase 2 trials, including the VIOLETTE study on TNBC [54], the SUKSES-N2 study on small-cell lung cancer (NCT03428607), and the CAPRI study on ovarian cancer (NCT03462342). The VIOLETTE study will be stratified on the basis of HR gene alterations and, the CAPRI study will be stratified on the grounds of platinum sensitivity. The OLAPCO study (NCT02576444) is a biomarker-enriched multi-arm olaparib-based combination study. The AZD6738 and olaparib arm requires the preselection of tumors with mutations in HR-DNA repair genes.
Conclusion
Although the ATR-CHK1 pathway in DDR has been studied for decades, it was not until recently that the small molecule inhibitors of ATR were developed for the clinical setting [55]. Inhibition of ATR with an ATR inhibitor either as a monotherapy or in combination with DNA- damaging chemotherapy drugs, ionizing radiation, immune checkpoint blockers, or PARP inhibitors is being tested in early-phase clinical trials in advanced solid tumors and hematological malignancies. Safety and tolerability have been reported for M6620 and AZ6738. Phase 2 combination trials are ongoing. Emerging data from these early-phase studies support the preclinical observations of the synthetic lethality of ATR inhibitors in ATM-deficient cancers. Currently, there are no data on whether lack of functional p53 or the replication stress induced by overexpression of oncogenes such as c-MYC can serve as predicative biomarkers for ATR inhibitor monotherapy. Other than predicative biomarkers, data coming from ATR inhibitor-based combinational trials could shed light on whether we can expand this therapy to HR-proficient cancers and whether this approach can serve as a rescue therapy for patients who have progressed through PARP inhibitors.
Acknowledgements
We thank Paul Fletcher and Daley Drucker (Moffitt Cancer Center) for editorial assistance. They were not compensated beyond their regular salary.
Funding
Not applicable
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ATM
Ataxia telangiectasia mutated
- ATR
Ataxia telangiectasia and Rad3 related
- ATRIP
ATR interacting protein
- CHK1
Checkpoint kinase 1
- CHK2
Checkpoint kinase 2
- CLL
Chronic lymphocytic leukemia
- DDR
DNA damage response
- HR
Homologous recombination repair
- MTD
Maximum tolerated dose
- NSCLC
Non-small cell lung cancer
- PARP
Polymerase
- PR
Partial response
- RNAi
RNA interference
- SD
Stable disease
- ssDNA
Single-strand DNA
- TNBC
Triple-negative breast cancer
Authors’ contributions
JZ wrote the final version of the manuscript. LM contributed to the write up of the manuscript and generated the figure and table. KH contributed to the planning and writing up of the manuscript. JZ contributed to the write up of the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
Dr. Zhang has received consulting and speaker honorarium from AstraZeneca and consulting honorarium from Bayer.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lavin MF. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene. 2007;26(56):7749–7758. doi: 10.1038/sj.onc.1210880. [DOI] [PubMed] [Google Scholar]
- 2.Qiu Z, Oleinick NL, Zhang J. ATR/CHK1 inhibitors and cancer therapy. Radiotherapy Oncol. 2018;126(3):450–464. doi: 10.1016/j.radonc.2017.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16(1):2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science (New York, NY) 2003;300(5625):1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y, Vidanes G, Lin YC, Mori S, Siede W. Characterization of a Saccharomyces cerevisiae homologue of Schizosaccharomyces pombe Chk1 involved in DNA-damage-induced M-phase arrest. Mol Gen Genet. 2000;262(6):1132–1146. doi: 10.1007/PL00008656. [DOI] [PubMed] [Google Scholar]
- 6.Busino L, Chiesa M, Draetta GF, Donzelli M. Cdc25A phosphatase: combinatorial phosphorylation, ubiquitylation and proteolysis. Oncogene. 2004;23(11):2050–2056. doi: 10.1038/sj.onc.1207394. [DOI] [PubMed] [Google Scholar]
- 7.Bahassi EM, Ovesen JL, Riesenberg AL, Bernstein WZ, Hasty PE, Stambrook PJ. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene. 2008;27(28):3977–3985. doi: 10.1038/onc.2008.17. [DOI] [PubMed] [Google Scholar]
- 8.Dai Y, Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res. 2010;16(2):376–383. doi: 10.1158/1078-0432.CCR-09-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma CX, Janetka JW, Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol Med. 2011;17(2):88–96. doi: 10.1016/j.molmed.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 11.Zaugg K, Su YW, Reilly PT, Moolani Y, Cheung CC, Hakem R, et al. Cross-talk between Chk1 and Chk2 in double-mutant thymocytes. Proc Natl Acad Sci U S A. 2007;104(10):3805–10. doi: 10.1073/pnas.0611584104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zannini L, Delia D, Buscemi G. CHK2 kinase in the DNA damage response and beyond. J Mol Cell Biol. 2014;6(6):442–457. doi: 10.1093/jmcb/mju045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vendetti FP, Lau A, Schamus S, Conrads TP, O'Connor MJ, Bakkenist CJ. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget. 2015;6(42):44289–44305. doi: 10.18632/oncotarget.6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wengner AM, Siemeister G, Luecking U, Lefranc J, Lienau P, Deeg G, et al. Abstract 836: ATR inhibitor BAY 1895344 shows potent anti-tumor efficacy in monotherapy and strong combination potential with the targeted alpha therapy Radium-223 dichloride in preclinical tumor models. Cancer Res. 2017;77(13 Supplement):836. doi: 10.1158/1538-7445.AM2017-836. [DOI] [Google Scholar]
- 15.Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7(7):428–430. doi: 10.1038/nchembio.573. [DOI] [PubMed] [Google Scholar]
- 16.Cui Y, Palii SS, Innes CL, Paules RS. Depletion of ATR selectively sensitizes ATM-deficient human mammary epithelial cells to ionizing radiation and DNA-damaging agents. Cell Cycle (Georgetown, Tex) 2014;13(22):3541–3550. doi: 10.4161/15384101.2014.960729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saldivar JC, Cortez D, Cimprich KA. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol. 2017;18(10):622–636. doi: 10.1038/nrm.2017.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wagner SA, Oehler H, Voigt A, Dalic D, Freiwald A, Serve H, et al. ATR inhibition rewires cellular signaling networks induced by replication stress. Proteomics. 2016;16(3):402–416. doi: 10.1002/pmic.201500172. [DOI] [PubMed] [Google Scholar]
- 19.Genik PC, Bielefeldt-Ohmann H, Liu X, Story MD, Ding L, Bush JM, et al. Strain background determines lymphoma incidence in Atm knockout mice. Neoplasia (New York, NY) 2014;16(2):129–136. doi: 10.1593/neo.131980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14(4):397–402. [PMC free article] [PubMed] [Google Scholar]
- 21.Adams KE, Medhurst AL, Dart DA, Lakin ND. Recruitment of ATR to sites of ionising radiation-induced DNA damage requires ATM and components of the MRN protein complex. Oncogene. 2006;25(28):3894–3904. doi: 10.1038/sj.onc.1209426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8(1):37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 23.Cuadrado M, Martinez-Pastor B, Murga M, Toledo LI, Gutierrez-Martinez P, Lopez E, et al. ATM regulates ATR chromatin loading in response to DNA double-strand breaks. J Exp Med. 2006;203(2):297–303. doi: 10.1084/jem.20051923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stiff T, Walker SA, Cerosaletti K, Goodarzi AA, Petermann E, Concannon P, et al. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. EMBO J. 2006;25(24):5775–5782. doi: 10.1038/sj.emboj.7601446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med. 2013;19(11):1381–1388. doi: 10.1038/nm.3369. [DOI] [PubMed] [Google Scholar]
- 26.Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ. 2008;15(7):1153–1162. doi: 10.1038/cdd.2008.47. [DOI] [PubMed] [Google Scholar]
- 27.Hall AB, Newsome D, Wang Y, Boucher DM, Eustace B, Gu Y, et al. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget. 2014;5(14):5674–5685. doi: 10.18632/oncotarget.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshida GJ. Correction to: Emerging roles of Myc in stem cell biology and novel tumor therapies. J Exp Clin Cancer Res. 2018;37(1):285. doi: 10.1186/s13046-018-0964-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fokas E, Prevo R, Hammond EM, Brunner TB, McKenna WG, Muschel RJ. Targeting ATR in DNA damage response and cancer therapeutics. Cancer Treat Rev. 2014;40(1):109–117. doi: 10.1016/j.ctrv.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 30.Yap TA, Luken MJM, O'Carrigan B, Roda D, Papadatos-Pastos D, Lorente D, et al. Abstract PR14: phase I trial of first-in-class ataxia telangiectasia-mutated and Rad3-related (ATR) inhibitor VX-970 as monotherapy (mono) or in combination with carboplatin (CP) in advanced cancer patients (pts) with preliminary evidence of target modulation and antitumor activity. Mol Cancer Ther. 2015;14(12 Supplement 2):PR14–PRPR. doi: 10.1158/1535-7163.TARG-15-PR14. [DOI] [Google Scholar]
- 31.O'Carrigan B, Luken MJM, Papadatos-Pastos D, Brown J, Tunariu N, Lopez RP, et al. Phase I trial of a first-in-class ATR inhibitor VX-970 as monotherapy (mono) or in combination (combo) with carboplatin (CP) incorporating pharmacodynamics (PD) studies. J Clin Oncol. 2016;34(15_suppl):2504. doi: 10.1200/JCO.2016.34.15_suppl.2504. [DOI] [Google Scholar]
- 32.Kwok M, Davies N, Agathanggelou A, Smith E, Oldreive C, Petermann E, et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood. 2016;127(5):582–595. doi: 10.1182/blood-2015-05-644872. [DOI] [PubMed] [Google Scholar]
- 33.Jin J, Fang H, Yang F, Ji W, Guan N, Sun Z, et al. Combined inhibition of ATR and WEE1 as a novel therapeutic strategy in triple-negative breast cancer. Neoplasia (New York, NY). 2018;20(5):478–488. doi: 10.1016/j.neo.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dillon MT, Boylan Z, Smith D, Guevara J, Mohammed K, Peckitt C, et al. PATRIOT: a phase I study to assess the tolerability, safety and biological effects of a specific ataxia telangiectasia and Rad3-related (ATR) inhibitor (AZD6738) as a single agent and in combination with palliative radiation therapy in patients with solid tumours. Clin Transl Radiation Oncol. 2018;12:16–20. doi: 10.1016/j.ctro.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim HJ, Min A, Im SA, Jang H, Lee KH, Lau A, et al. Anti-tumor activity of the ATR inhibitor AZD6738 in HER2 positive breast cancer cells. Int J Cancer. 2017;140(1):109–119. doi: 10.1002/ijc.30373. [DOI] [PubMed] [Google Scholar]
- 36.Wallez Y, Dunlop CR, Johnson TI, Koh SB, Fornari C, Yates JWT, et al. The ATR inhibitor AZD6738 synergizes with gemcitabine in vitro and in vivo to induce pancreatic ductal adenocarcinoma regression. Mol Cancer Ther. 2018;17(8):1670-82. 10.1158/1535-7163.MCT-18-0010. Epub 2018 Jun 11. [DOI] [PMC free article] [PubMed]
- 37.Yap TA, Krebs MG, Postel-Vinay S, Bang YJ, El-Khoueiry A, Abida W, et al. Phase I modular study of AZD6738, a novel oral, potent and selective ataxia telangiectasia Rad3-related (ATR) inhibitor in combination (combo) with carboplatin, olaparib or durvalumab in patients (pts) with advanced cancers. Eur J Cancer. 2016;69:S2. doi: 10.1016/S0959-8049(16)32607-7. [DOI] [Google Scholar]
- 38.Sundar R, Brown J, Ingles Russo A, Yap TA. Targeting ATR in cancer medicine. Curr Probl Cancer. 2017;41(4):302–315. doi: 10.1016/j.currproblcancer.2017.05.002. [DOI] [PubMed] [Google Scholar]
- 39.Telli M, Lord S, Dean E, Abramson V, Arkenau H-T, Murias C, et al. Abstract OT2–07-07: ATR inhibitor M6620 (formerly VX-970) with cisplatin in metastatic triple-negative breast cancer: preliminary results from a phase 1 dose expansion cohort ( NCT02157792) Cancer Research. 2018;78(4 Supplement):OT2–07--OT2. [Google Scholar]
- 40.Plummer ER, Dean EJ, Evans TRJ, Greystoke A, Herbschleb K, Ranson M, et al. Phase I trial of first-in-class ATR inhibitor VX-970 in combination with gemcitabine (Gem) in advanced solid tumors ( NCT02157792) J Clin Oncol. 2016;34(15_suppl):2513. doi: 10.1200/JCO.2016.34.15_suppl.2513. [DOI] [Google Scholar]
- 41.Thomas A, Redon CE, Sciuto L, Padiernos E, Ji J, Lee M-J, et al. Phase I study of ATR inhibitor M6620 in combination with topotecan in patients with advanced solid tumors. J Clin Oncol. 2018;36(16):1594–1602. doi: 10.1200/JCO.2017.76.6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524(7563):47–53. doi: 10.1038/nature14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dowlati A, Lipka MB, McColl K, Dabir S, Behtaj M, Kresak A, et al. Clinical correlation of extensive-stage small-cell lung cancer genomics. Ann Oncol. 2016;27(4):642–647. doi: 10.1093/annonc/mdw005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gardner EE, Lok BH, Schneeberger VE, Desmeules P, Miles LA, Arnold PK, et al. Chemosensitive relapse in small cell lung cancer proceeds through an EZH2-SLFN11 axis. Cancer Cell. 2017;31(2):286–299. doi: 10.1016/j.ccell.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dillon MT, Barker HE, Pedersen M, Hafsi H, Bhide SA, Newbold KL, et al. Radiosensitization by the ATR inhibitor AZD6738 through generation of acentric micronuclei. Mol Cancer Ther. 2017;16(1):25–34. doi: 10.1158/1535-7163.MCT-16-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fokas E, Prevo R, Pollard JR, Reaper PM, Charlton PA, Cornelissen B, et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012;3:e441. doi: 10.1038/cddis.2012.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Biskup E, Naym DG, Gniadecki R. Small-molecule inhibitors of ataxia telangiectasia and Rad3 related kinase (ATR) sensitize lymphoma cells to UVA radiation. J Dermatol Sci. 2016;84(3):239–247. doi: 10.1016/j.jdermsci.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 48.Sun LL, Yang RY, Li CW, Chen MK, Shao B, Hsu JM, et al. Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T cell-mediated killing. Am J Cancer Res. 2018;8(7):1307–1316. [PMC free article] [PubMed] [Google Scholar]
- 49.Heymach J, Thomas M, Besse B, Forde PM, Awad MM, Goss GD, et al. An open-label, multidrug, biomarker-directed, multicentre phase II umbrella study in patients with non-small cell lung cancer, who progressed on an anti-PD-1/PD-L1 containing therapy (HUDSON) J Clin Oncol. 2018;36(15_suppl):TPS3120–TPS3TPS. doi: 10.1200/JCO.2018.36.15_suppl.TPS3120. [DOI] [Google Scholar]
- 50.Huntoon CJ, Flatten KS, Wahner Hendrickson AE, Huehls AM, Sutor SL, Kaufmann SH, et al. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 2013;73(12):3683–3691. doi: 10.1158/0008-5472.CAN-13-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krajewska M, Fehrmann RS, Schoonen PM, Labib S, de Vries EG, Franke L, et al. ATR inhibition preferentially targets homologous recombination-deficient tumor cells. Oncogene. 2015;34(26):3474–3481. doi: 10.1038/onc.2014.276. [DOI] [PubMed] [Google Scholar]
- 52.Kim D, Liu Y, Oberly S, Freire R, Smolka MB. ATR-mediated proteome remodeling is a major determinant of homologous recombination capacity in cancer cells. Nucleic Acids Res. 2018;46(16):8311–8325. doi: 10.1093/nar/gky625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coyne GHOS, Do KT, Kummar S, Takebe N, Quinn MF, Piha-Paul SA, et al. Phase I trial of the triplet M6620 (formerly VX970) + veliparib + cisplatin in patients with advanced solid tumors. J Clin Oncol. 2018;36(15_suppl):2549. doi: 10.1200/JCO.2018.36.15_suppl.2549. [DOI] [Google Scholar]
- 54.Tutt A, Stephens C, Frewer P, Pierce A, Rhee J, So K, et al. VIOLETTE: A randomized phase II study to assess DNA damage response inhibitors in combination with olaparib (Ola) vs Ola monotherapy in patients (pts) with metastatic, triple-negative breast cancer (TNBC) stratified by alterations in homologous recombination repair (HRR)-related genes. J Clin Oncol. 2018;36(15_suppl):TPS1116–TPS1TPS. doi: 10.1200/JCO.2018.36.15_suppl.TPS1116. [DOI] [Google Scholar]
- 55.Lecona E, Fernandez-Capetillo O. Targeting ATR in cancer. Nat Rev Cancer. 2018;18(9):586–595. doi: 10.1038/s41568-018-0034-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.