

Abstract
Background
Trihelix transcription factors (TTFs) are photoresponsive proteins that have a representative three-helix structure (helix-loop-helix-loop-helix). Members of this gene family have been reported to play roles in many plant processes.
Results
In this study, we performed a functional and evolutionary analysis of the TTFs in Moso bamboo (Phyllostachys edulis). A total of 35 genes were identified and grouped into five subfamilies (GT-1, GT-γ, GT-2, SIP1 and SH4) according to their structural properties. Gene structure analysis showed that most genes in the PeTTF family had fewer introns. A unique motif (Motif 16) to the GT-γ subfamily was identified by conserved motif analysis. Promoter analysis revealed various cis-acting elements related to plant growth and development, abiotic and biotic stresses, and phytohormone responses. Data for the 35 Moso bamboo TTF genes were used to generate heat maps, which indicated that these genes were expressed in different tissues or developmental stages. Most of the TTF genes identified here had high expression in leaves and panicles according to the expression profile analysis. The expression levels of the TTF members in young leaves were studied using quantitative real-time PCR to determine their tissue specificity and stress-related expression patterns to help functionally characterize individual members.
Conclusions
The results indicated that members of the TTF gene family may be involved in plant responses to stress conditions. Additionally, PeTTF29 was shown to be located in the nucleus by subcellular localization analysis and to have transcriptional activity in a transcriptional activity assay. Our research provides a comprehensive summary of the PeTTF gene family, including functional and evolutionary perspectives, and provides a basis for functionally characterizing these genes.
Electronic supplementary material
The online version of this article (10.1186/s12870-019-1744-8) contains supplementary material, which is available to authorized users.
Keywords: Trihelix transcription factors, Gene ontology, Phylogenetic analysis, Expression profiling, Moso bamboo, Subcellular localization, Transcriptional activity assay
Background
Transcription factors are a class of DNA-binding proteins that interact specifically with cis-acting elements in the promoter regions of target genes; they generally contain DNA-binding domains, transcriptional activation domains, and nuclear localization signals and can either inhibit or induce the transcription of target genes. [1]. There are more than 60 transcription factor gene families in plants [2]. Recently, many researchers have focused on the trihelix transcription factor (TTF) gene family because its members contain a unique DNA-binding domain including three helical structures (helix-loop-helix-loop-helix). This family is also referred to as the GT transcription factor family, whose core sequence is 5′-G-Pu-(T/A)-A-A-(T/A)-3′ and members of which bind to the photo-responsive element GT in DNA. [3–7].
Since the beginning of this century, whole genome sequencing has been completed for many plants and the important functions of a series of gene families have been determined. Research on the TTF gene family has been gradually carried out, functional investigation of the family has widened, and it has become the subject of much recent attention from biologists. [2, 4, 8–11]. To data, 20, 36, 31, 28, 56, 63 and 59 TTF genes have been identified in chrysanthemum (Chrysanthemum morifolium), tomato (Lycopersicon esculentum), rice (Oryza sativa), Arabidopsis (Arabidopsis thaliana), poplar (Populus), soybean (Glycine max) and maize (Zea mays), respectively [3–7, 12, 13]. The earliest identified TTF gene was the pea GT-1 protein factor, which binds specifically to the GT element in the pea nucleus. Later, Lam et al. cloned the tobacco GT-1 factor and confirmed that it contained three α-helix domains in series [4, 8]. Nevertheless, the TTF gene family in Moso bamboo has not been comprehensively studied.
There is growing evidence that TTF genes play crucial roles in plant growth and development [14], For instance, the development of petals and sepals, and sepal fusion are regulated by the Arabidopsis PETAL LOSS (PTL) genes (GT-2 subfamily) [15]. A previous study on poplar reported that PtaGTL1 was highly homologous to Arabidopsis AtGTL1 and was involved the development of stomata and trichomes [16]. The expression of SHAT1 in the shedding zone is regulated by the triangular leaf transcription factor SH4, which is involved in regulating grain separation traits in rice [17]. Additionally, Arabidopsis At3g10030 was associated with leaf development because the corresponding mutants exhibited leaves that were small, deformed and light green [16]. The key roles TTF genes play in responses to biotic and abiotic stresses have also been well documented [4, 14, 18, 19]. Fang et al. reported that the rice gene GTγ-1 enhanced resistance to salt stress and was slightly induced by ABA treatment and drought stress; in addition, GTγ-2 and GTγ-3 were strongly induced by most abiotic stresses [4]. Furthermore, overexpression in transgenic Arabidopsis plants demonstrated that GmGT-2A and GmGT-2B could enhance tolerance to salt, drought and freezing stresses [20]. On the whole, trihelix transcription factors play important roles in the regulation of developmental processes.
Moso bamboo is a rare species of evergreen tree-like bamboo that grows rapidly [21], and has gradually become widely used and an economically important Gramineae species in China [22]. However, Moso bamboo trees are affected by a variety of environmental stresses, including the rapid spread of pests and drought stress, which lead to significant economic losses. Increasing the resistance of Moso bamboo to these stresses to improve both the quality and quantity of bamboo produced is one of the major aims of researchers working in this area [23, 24].
In this study, we identified 35 members of the PeTTF gene family and conducted various bioinformatics analyses, including phylogenetic, gene structure, expression profile, conserved motif, gene ontology annotation, subcellular localization and promoter cis-acting regulatory element analyses. Moreover, the 35 PeTTF genes were analyzed by qRT-PCR to study their responses to different stresses, including MeJA treatment, and drought and salt stress. We found that all of the genes were stress responsive. The objective of this study was to determine the transcriptional responses of TTF gene family members to various stresses and plant hormone treatments.
Materials and methods
Database searches and identification of TTF family genes in Moso bamboo
The accession numbers of Arabidopsis trihelix family members were acquired from PlantTFDB (http://planttfdb.cbi.pku.edu.cn) [25], which is a comprehensive database that provides a complete list of transcription factors in plant species. Rice trihelix protein sequences were obtained from the rice genome annotation database (http://rice.plantbiology.msu.edu/analyses_search_locus.shtml) [26].
The genome sequence of Moso bamboo was downloaded through the Bamboo Genome Database (http://www.bamboogdb.org/). A hidden Markov model (HMM) profile based on the TTF (PF13837) domain was used to perform a local BLAST search (E-value-5) of the Pfam database (http://pfam.janelia.org/search/sequence) [18, 27, 28]. All of the selected Moso bamboo sequences were filtered, leaving only candidate genes containing known conserved domains, which were checked using the Pfam database (http://pfam.janelia.org/), the NCBI Conserved Domain database (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi), and the SMART database (http://smart.embl-heidelberg.de/). ExPASy (http://www.expasy.ch/tools/pi_tool.html) was used to determine the amino acid sequence, open reading frame (ORF) length, molecular weight (MW) and isoelectric point (pI) [29] and perform bioinformatics analysis of each Moso bamboo TTF gene. Additionally, gene identifiers and genomic/coding/protein sequences were acquired from the Bamboo Genome Database (http://www.bamboogdb.org/).
Phylogenetic analysis
Further multiple sequence alignments of all TTF proteins of Moso bamboo were performed using ClustalX 2.11. At the same time, sequence alignments between Moso bamboo, rice and Arabidopsis were also carried out [30, 31]. Phylogenetic trees were constructed in MEGA6.0 [32] using the neighbor-joining method with 1000 repeats used for bootstrap analysis. We obtained the protein sequence of PeTTF gene in the Bamboo Genome Database (http://www.bamboogdb.org/), and then we used the sequence information to construct phylogenetic trees.
Gene structure and conserved motif analysis
The exon/intron organization of individual TTF genes was determined using the GSDS program (http://gsds.cbi.pku.edu.cn/) [33] to compare each cDNA to its corresponding genomic DNA sequence. Motifs shared between related proteins within the PeTTF family were identified using the MEME motif search tool (http://meme-suite.org/tools/meme) [34], with the maximum number of motifs identified set at 20 and the maximum width at 300 aa. The sequences of the TTF subfamily members were examined using the DNAMAN software and manually modified, with the conserved motifs annotated according to the MEME analysis.
Identification of homologous and calculation of Ka/Ks values
Paralogous and orthologous genes were identified according to Blanc and Wolfe [35]. For each species, full-pairing nucleotide sequence similarity searches were performed with the transcribed sequences using BlastN [36]. Sequences that had over 300 bp aligned and showed ≥40% identity were defined as paralogous. To identify putative orthologs between two species (A and B), each sequence of species A was searched against all the sequences of species B using BlastN. Then, each sequence from species B was searched against all sequences from species A. If two sequences were the best hit to each other and if more than 300 bp of the two sequences was aligned, the two sequences were defined as orthologous.
Multiple sequence alignment of the homologous (orthologous and paralogous) TTF gene pairs was performed using the ClustalX software (version 2.11) software. The sequences were then further aligned using MEGA6.0 and the DnaSP 5 software [37, 38] was used to calculate the Ks and Ka substitution rates. The Ks rate was considered to be an indicator of the time of gene-pair duplication events, and the divergence times (T) were calculated using the Ks value of the λ permutation for each synonymous site per year according to the formula T = Ks/2λ (λ = 6.5 × 10− 9) [21, 39, 40]. Then, the Ka/Ks ratios for all homologous gene pairs were determined using a sliding window analysis with the window size set at 150 bp and the step size at 9 bp.
Expression profile analysis
Expression profile data for each gene was downloaded from the NCBI short read (SRA) database (https://trace.ncbi.nlm.nih.gov/Traces/sra/?study=ERP001341), and then the original RNA-seq reads were trimmed using BioProject ERP001341 to eliminate low quality base calls (Q < 20) and adapter sequences with the pipeline Fastq clean [41]. The differentially expressed genes were then examined by the pipeline tophat2 with default parameters to map the matched clean reads to the P. bridla reference genome [42]. Finally, the Heatmapper Plus tool was used to produce heatmaps from the whole-genome microarray data for three biological replicates of seven different tissue specimens including L (leaf), P1 (early panicle), P2 (advanced panicle), R (root), RH (rhizome), S1 (20 cm shoots) and S2 (50 cm shoots) at various developmental phases [43].
Promoter cis-acting regulatory element analysis, gene ontology (GO) annotation and subcellular localization prediction
The 2000 bp sequence upstream/downstream of each predicted gene coding region was obtained from the Bamboo Genome Database (http:// www.bamboogdb.org/) [44] and analyzed using PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) [45]. The PlantCARE database is a tool for analyzing plant cis-acting regulatory elements and promoter sequences to identify specific motifs for plant growth and development, phytohormone responses and abiotic and biotic stress responses.
Functional annotation of the TTF genes was performed using the Gene Ontology database (GO; www.geneontology.org) [46].
The subcellular localization of the proteins was predicted using WOLF PSORT (http://www.genscript.com/psort.html) [47].
Plant materials, growth conditions, and stress treatments
The seeds of Moso bamboo from Tianmu Mountain National Nature Reserve in Zhejiang Province were planted in black soil and vermiculite. During their development and growth, the plants were grown in a greenhouse at 25/27 °C and 80% humidity with 16/8 h of light/dark. The plants were manure once every 3 days and watered once a day and cultured for 3 months.
For drought and salt stress treatments, the plants were treated with a 20% PEG-6000 solution [48] and 200 mM NaCl, respectively [49]. The effects of injury were simulated at 25 °C using 100 μM MeJA (Sigma, St. Louis, MO, USA) solution [12]. The plants we treated with stress were annual Moso bamboo. The specific process was as follows: i, the required treatment solution was configured; ii, the configured solution was poured into the pot; iii, 6 periods were collected (0, 1, 3, 6, 12 and 24 h) of Moso bamboo leaves, with 0 h being the control; iv, leaf tissue was immediately frozen in liquid nitrogen and stored at − 80 °C for RNA extraction.
RNA isolation and quantitative real-time PCR (qRT-PCR) analysis
To confirm the expression levels of the TTF genes in Moso bamboo, qRT-PCR analysis using SYBR-green fluorescence was performed for each member. Total RNA was extracted from the leaf, young leaf samples using Trizol reagent (Sangon Biotech, Shanghai, China) according to the manufacturer’s instructions; and an optimized modified cetyl trimethyl ammonium bromide procedure [24] was used to extracted total RNA from Stem, Shoot, root, and Rhizome; gel electrophoresis (1% agar) was used to examine the integrity of the RNA. Then, First-strand cDNA was synthesized using a PrimeScriptTM RT Reagent Kit (TaKaRa). Gene-specific primers were designed using Primer Express 3.0 and the NCBI primer Blast tool and the internal control was performed using the tonoplast intrinsic protein 41 (TIP41) [50].
qRT-PCR was carried out in 20 μl volumes consisting of 10 μl 2× SYBR® PremixExTaq™ (TaKaRa), 0.4 μl 50× ROX Reference Dye, 2 μl diluted cDNA template, 0.8 μl of each specific primer, and 6 μl ddH2O. The cycling parameters were 95 °C for 30 s followed by 40 cycles of 95 °C denaturation for 5 s and annealing at 55–60 °C annealing for 34 s. Three biological and three technical replicates were included for each sample. The 2−ΔΔCT [51] method was used to calculate the relative expression level of each gene with expression normalized to the 0 h time point, which was set at 1 [52]. Statistical analyses were conducted using the GraphPad software [53].
Subcellular localization assay
The full-length CDS of PeTTF29 was cloned from bamboo, and then the PeTTF29 coding region was amplified by PCR using the primers 5′-(tgc-ACTAGT-ATGGAGGGGAATTTGC)-3′ and 5′-(cgc-CCCGGG-CTTCTTTGGTTTGACA)-3′ to introduce SpeI and SmaI sites, respectively. The product was then cloned into the pCambia1305 vector (Clontech, Beijing, China) containing the CaMV 35S promoter and the GFP gene to produce a PeTTF29-GFP construct. The constructed PeTTF29-GFP vector was inserted into Agrobacterium tumefaciens EHA105 by freeze-thawing. The suspension was infiltrated into the Nicotiana tabacum leaves using an injection method and the green fluorescent protein (GFP) fluorescence was observed using confocal microscopy [54]. pCambia1305 with only constitutive GFP was used as a control vector.
Transcription activation assay
The full-length CDS of PeTTF29 was cloned from bamboo, and then the PeTTF29 coding region was amplified by PCR using the primers 5′-(tgc-GAATTC-ATGGAGGGGAATTTGC)-3′ and 5′-(cgc-CTGCAG-AATCTTCTTTGGTTTG)-3′ to introduce EcoRI and PstI sites, respectively. The constructed vector plasmids (target gene, negative control and positive control vector) were transformed into prepared yeast competent cells, and plated on SD/Trp − (as control) and SD/Trp−/His−/Ade−/X-α-gal media. Each plate was neatly divided into three regions containing the target gene, negative control and positive control. The autologous activity of the PeTTF29 gene was verified by streaking.
Results
Identification of TTF genes in Moso bamboo
We identified 35 members of the PeTTF gene family, designated PeTTF1 to PeTTF35. Information about the Moso bamboo TTF genes, including their gene identifiers, locations, gene lengths, and CDS lengths, is shown in Table 1. The CDSs ranged from 477 bp (PeTTF35) to 1935 (PeTTF24) bp in length, with an average length of 1097 bp, and the predicted protein lengths ranged from 158 (PeTTF35) to 644 (PeTTF24) aa in length, with an average length of 364 aa. The predicted molecular weights ranged from 6250.04 Da (PeTTF17) to 70,113.15 Da (PeTTF31), with a mean value of 39,553.91 Da. The pI values of all PeTTF gene products were below 11, with most falling between 5.0 and 10.0. The pI values of two predicted proteins (PeTTF33 and PeTTF35) were below 5.0, and only two proteins had a pI above 10.0 (PeTTF2 and PeTTF8).
Table 1.
Detailed information about the predicted PeTTF genes
| Name | Gene ID | Location | ORF length (bp) | Predicted Protein | Exons | ||
|---|---|---|---|---|---|---|---|
| Size (aa) | MW (Da) | pI | |||||
| PeTTF1 | PH01000001G0500 | PH01000001:348404–353,246 | 1392 | 463 | 51,339.99 | 6.51 | 6 |
| PeTTF2 | PH01000010G0450 | PH01000010:333451–334,674 | 837 | 278 | 31,526.86 | 10.51 | 2 |
| PeTTF3 | PH01000016G1050 | PH01000016:832203–835,604 | 978 | 325 | 35,416.69 | 6.71 | 4 |
| PeTTF4 | PH01000017G1770 | PH01000017:1223108–1,227,861 | 1632 | 543 | 57,598.78 | 6.66 | 4 |
| PeTTF5 | PH01000019G0430 | PH01000019:338352–344,501 | 1776 | 591 | 64,500.43 | 8.28 | 7 |
| PeTTF6 | PH01000019G0630 | PH01000019:498070–502,996 | 1251 | 416 | 44,129.17 | 6.53 | 6 |
| PeTTF7 | PH01000020G1490 | PH01000020:1016528–1,021,991 | 1161 | 386 | 42,551.58 | 6.42 | 5 |
| PeTTF8 | PH01000023G2170 | PH01000023:1389503–1,390,380 | 591 | 196 | 21,556.89 | 10.89 | 1 |
| PeTTF9 | PH01000042G1290 | PH01000042:820202–822,366 | 1632 | 543 | 59,326.99 | 5.33 | 1 |
| PeTTF10 | PH01000110G0890 | PH01000110:616131–618,576 | 522 | 173 | 18,904.82 | 10.00 | 1 |
| PeTTF11 | PH01000225G1300 | PH01000225:784587–789,545 | 813 | 270 | 31,899.86 | 8.69 | 2 |
| PeTTF12 | PH01000265G1050 | PH01000265:729361–733,771 | 1266 | 421 | 45,424.90 | 9.18 | 8 |
| PeTTF13 | PH01000551G0750 | PH01000551:454513–457,660 | 1317 | 438 | 49,565.35 | 6.18 | 1 |
| PeTTF14 | PH01000749G0800 | PH01000749:497748–502,389 | 816 | 271 | 29,897.93 | 9.04 | 6 |
| PeTTF15 | PH01000778G0550 | PH01000778:319370–322,783 | 942 | 313 | 34,196.71 | 9.96 | 1 |
| PeTTF16 | PH01000823G0680 | PH01000823:401827–404,100 | 1140 | 379 | 43,468.32 | 9.03 | 1 |
| PeTTF17 | PH01000907G0490 | PH01000907:299223–301,005 | 690 | 229 | 6250.04 | 9.30 | 2 |
| PeTTF18 | PH01001050G0190 | PH01001050:145304–146,705 | 609 | 202 | 22,559.24 | 9.84 | 2 |
| PeTTF19 | PH01001160G0450 | PH01001160:304848–307,615 | 732 | 243 | 27,274.24 | 6.72 | 3 |
| PeTTF20 | PH01001191G0080 | PH01001191:45575–48,488 | 1026 | 341 | 34,296.75 | 6.88 | 1 |
| PeTTF21 | PH01001215G0530 | PH01001215:388790–394,765 | 1695 | 564 | 62,006.92 | 9.62 | 6 |
| PeTTF22 | PH01001227G0510 | PH01001227:318856–320,192 | 981 | 326 | 35,750.42 | 6.81 | 1 |
| PeTTF23 | PH01001447G0230 | PH01001447:145073–149,866 | 1059 | 352 | 37,733.79 | 5.19 | 3 |
| PeTTF24 | PH01001451G0080 | PH01001451:28239–32,258 | 1935 | 644 | 70,109.82 | 5.76 | 3 |
| PeTTF25 | PH01001538G0330 | PH01001538:231626–234,572 | 1047 | 348 | 38,256.31 | 6.22 | 3 |
| PeTTF26 | PH01001567G0130 | PH01001567:89093–92,345 | 756 | 251 | 27,067.65 | 9.81 | 1 |
| PeTTF27 | PH01001778G0330 | PH01001778:262830–265,479 | 1287 | 428 | 48,983.77 | 6.27 | 1 |
| PeTTF28 | PH01002213G0310 | PH01002213:194429–199,841 | 1485 | 494 | 54,202.70 | 8.38 | 7 |
| PeTTF29 | PH01002648G0060 | PH01002648:37644–39,402 | 1200 | 399 | 46,125.49 | 6.13 | 1 |
| PeTTF30 | PH01003510G0200 | PH01003510:123649–126,664 | 1329 | 442 | 50,199.08 | 6.35 | 1 |
| PeTTF31 | PH01008287G0010 | PH01008287:1698–5966 | 1926 | 641 | 70,113.15 | 5.60 | 3 |
| PeTTF32 | PH01011203G0010 | PH01011203:229–1679 | 600 | 199 | 22,030.79 | 6.53 | 3 |
| PeTTF33 | PH01047850G0010 | PH01047850:55–1624 | 897 | 298 | 31,134.39 | 4.85 | 3 |
| PeTTF34 | PH01107020G0010 | PH01107020:53–1034 | 624 | 207 | 21,063.61 | 9.94 | 3 |
| PeTTF35 | PH01153193G0010 | PH01153193:1–820 | 477 | 158 | 17,923.39 | 4.68 | 2 |
Phylogenetic analysis of the TTF genes in Moso bamboo
To explore the phylogenetic relationships among the TTF proteins in rice, Arabidopsis and Moso bamboo, we constructed a neighbor-joining phylogenetic tree with ClustalX using 94 TTF sequences, including 31, 28 and 35 sequences from rice, Arabidopsis and Moso bamboo, respectively. The characteristics of these genes are listed in Additional file 1: Table S1. The phylogenetic tree clearly divided the 94 TTF genes into five distinct subfamilies according to the bootstrap support and evolutionary distances (Fig. 1). The SIP1 subfamily contained the largest number of genes, followed by GT-2, GT-γ and SH4; GT-1 had the fewest TTF genes. The percentage of each subfamily derived from each of the three plant species was calculated (Fig. 2) The GT-γ subfamily was evenly distributed among the three species while two of the larger subfamilies (SIP1 and GT-2) contained mostly Moso bamboo genes. However, the GT-γ and SH4 subfamilies were highly represented by Arabidopsis and rice genes, respectively.
Fig. 1.

Phylogeny of TTFs from Moso bamboo, rice and Arabidopsis. The 35 PeTTF genes, 31 OsTTF genes and 28 AtTTF genes are clustered into five subfamilies. Details of the TTF genes from Arabidopsis and rice are listed in Additional file 1: Table S1. The tree was generated with the Clustal X 2.0 software using the neighbor-joining (N-J) method
Fig. 2.

Comparison of TTF family members from Moso bamboo, rice and Arabidopsis. Different colors represent the different species, and the percentage of each subfamily in each species is shown
We identified paralogous and orthologous genes from the three species, which are listed in Table 2. Fifteen pairs of orthologous genes were identified in rice and Moso bamboo, but there were no orthologs between Moso bamboo and Arabidopsis. Thus, we concluded that the TTF genes in the two monocots (Moso bamboo and rice) had closer relationships than with those of the dicotyledons (Arabidopsis), which was consistent with the evolutionary relationship between dicots and monocots.
Table 2.
Paralogous (Pe-Pe) and orthologous (Pe-Os and Pt-At) gene pairs
| Pe-Pe | Pe-Os |
|---|---|
| PeTTF2/PeTTF10 | PeTTF3/LOC_Os04g32590 |
| PeTTF3/PeTTF23 | PeTTF7/LOC_Os04g40930 |
| PeTTF4/PeTTF21 | PeTTF8/LOC_Os04g45940 |
| PeTTF5/PeTTF14 | PeTTF9/LOC_Os04g45750 |
| PeTTF7/PeTTF16 | PeTTF11/LOC_Os04g51320 |
| PeTTF15/PeTTF26 | PeTTF12/LOC_Os04g33300 |
| PeTTF18/PeTTF32 | PeTTF13/LOC_Os12g06640 |
| PeTTF19/PeTTF25 | PeTTF15/LOC_Os05g48320 |
| PeTTF20/PeTTF22 | PeTTF16/LOC_Os05g03740 |
| PeTTF24/PeTTF31 | PeTTF17/LOC_Os01g21590 |
| PeTTF27/PeTTF30 | PeTTF21/LOC_Os03g02240 |
| PeTTF33/PeTTF35 | PeTTF23/LOC_Os02g31160 |
| PeTTF26/LOC_Os05g48690 | |
| PeTTF29/LOC_Os02g33770 | |
| PeTTF34/LOC_Os10g37240 |
Gene structure and conserved motifs in Moso bamboo
Gene family evolution depends on the diversification of gene structures. The coding sequence of individual TTF genes in Moso bamboo were analyzed, and the similarity/difference among different subfamilies was analyzed according to exon/intron structure and evolutionary tree (Fig. 3). Similar exon/intron structures were observed in closely related genes of the same subfamily, with PeTTF13, − 27, − 29 and − 30 of the GT-γ subfamily containing only one exon. The results showed that 34% of the PeTTF genes (PeTTF8, − 9, − 10, − 13, − 15, − 16, − 20, − 22, − 26, − 27, − 29, − 30 and − 37) had no introns, while the remaining genes contained 1–7 introns. To better understand the exon/intron structures of the paralogous genes, we further analyzed the 12 pairs of paralogous genes. Among these genes, five pairs (PeTTF15/− 26, PeTTF19/− 25, PeTTF20/− 22, PeTTF24/− 31 and PeTTF33/− 35) showed the same number of exons. These results may indicate that these genes have similar functions.
Fig. 3.

Phylogenetic relationships and gene structures of TTF genes in Moso bamboo. Left: Based on the results of sequence alignment, using the NJ method to construct the Phylogenetic tree of TTFs. Five different colors represented different subfamilies. Right: Exons, introns and untranslated regions (UTRs) are indicated by yellow rectangles, gray lines and blue rectangles, respectively
The diversity of the TTF gene family was further examined using the MEME motif search tool (http://meme-suite.org/tools/meme). Twenty motifs were identified, the specific information for which is shown in Additional file 2: Table S2. As shown in Fig. 4, some subfamilies contained less than five motifs (the SH4, GT-1 and SIP1 subfamilies). Almost all TTF genes contained Motif 1 and Motif 2. All members of the GT-γ and SIP1 subfamilies had similar motifs (Fig. 4), indicating that the PeTTF protein sequences were highly conserved within subfamilies.
Fig. 4.

Schematic representation of 20 conserved motifs in the TTF genes. Using the online MEME program, the conserved motifs in the TTF genes were identified. Different colored boxes represented different specific motifs. The length of each box in the figure does not represent the actual motif size
Strong purifying selection in Moso bamboo
To further explore the evolutionary patterns and divergence of the TTF gene family in Moso bamboo, 12 paralogous pairs of Moso bamboo genes were identified. Then, the timing of duplication events was calculated using the formula T = Ks/2λ [55]. The average Ks values and estimated time for each duplication event among the TTF genes of Moso bamboo are listed in Table 3. According to the Ks values, the 12 pairs of duplicate genes were divided into the following groups. The first group included PeTTF7/− 16 and PeTTF19/− 25. These two paralogous pairs had similar Ks values, with an average value of about 1.26, and the corresponding time of the duplication events was about 97 Mya. The second group included PeTTF3/− 23, PeTTF15/− 26 and PeTTF33/− 35. The Ks values of these three paralogous pairs were similar, with an average of about 0.63, and the corresponding time of the duplication events was about 48.12 Mya. The Ks values of the remaining seven pairs were similar to the average value of 0.21, corresponding to duplication events approximately 16.28 Mya. In addition, only three paralogous pairs had duplication events that occurred earlier than the divergence of Moso bamboo (12 Mya), indicating that most of the TTF gene duplication events occurred earlier than the speciation of Moso bamboo.
Table 3.
Ka/Ks analysis and estimated divergence time of PeTTF genes
| Duplicated TTF gene pairs |
Ka | Ks | Ka/Ks | Duplication data (MY) |
|---|---|---|---|---|
| PeTTF2–10 | 0.19628 | 0.28456 | 0.6898 | 21.89 |
| PeTTF3–23 | 0.3001 | 0.57003 | 0.5265 | 43.85 |
| PeTTF4–21 | 0.40385 | 0.30886 | 1.3076 | 23.76 |
| PeTTF5–14 | 0.24701 | 0.26566 | 0.9298 | 20.44 |
| PeTTF7–16 | 2.05006 | 1.33589 | 1.5346 | 102.76 |
| PeTTF15–26 | 0.37904 | 0.7751 | 0.4890 | 59.62 |
| PeTTF18–32 | 0.22935 | 0.26615 | 0.8617 | 20.47 |
| PeTTF19–25 | 2.3437 | 1.19812 | 1.9561 | 92.16 |
| PeTTF20–22 | 0.03489 | 0.15364 | 0.2271 | 11.82 |
| PeTTF24–31 | 0.06509 | 0.10698 | 0.6084 | 8.22 |
| PeTTF27–30 | 0.03282 | 0.0955 | 0.3437 | 7.35 |
| PeTTF33–35 | 1.26582 | 0.53139 | 2.3821 | 40.88 |
We also calculated the pairwise Ka (non-synonymous)/Ks ratios of repeated non-TTF genes (flanking genes) between repeated regions containing TTFs in Moso bamboo. A previous study indicated that a Ka/Ks ratio = 1 suggests neutral selection, while a Ka/Ks ratio > 1 indicates positive selection and a Ka/Ks ratio < 1 indicates negative selection or genetic purification (Additional file 7: Table S5). To investigate the selection pressure during the evolution of the PeTTF family, Ka and Ks replacement rates for each duplicated pair of PeTTF genes were calculated (Table 3). These results showed that the Ka/Ks rates of four of the 12 pairs of paralogous genes were greater than 1, suggesting that a small number of TTF genes had undergone positive selection, which can indicate the production of new genes. The other paralogous genes had undergone strong negative selection, indicating that most TTF genes are evolving more slowly.
Additionally, we performed a sliding-window analysis of the PeTTF gene fragments to better investigate the Ka/Ks ratios of different loci in the coding sequence (Additional file 3: Figure S1). These results indicated that the TTF gene family had undergone strong purifying selection (Ka/Ks < < 1) during the process of evolution.
Expression profile analysis of TTF gene family
Data for the 35 TTF genes was obtained from the NCBI short read archive (SRA) database (https://trace.ncbi.nlm.nih.gov/Traces/sra/?study=ERP001341). In the data, most of the Moso bamboo TTF genes showed tissue-specific expression patterns, with 9, 10 and 11 genes expressed highly in the leaf (L), early panicle (P1) and advanced panicle (P2), respectively, suggesting that they might play important roles in plant growth. Two genes (PeTTF19 and PeTTF35) were highly expressed in the leaf, early panicle, advanced panicle, root, rhizome, 20 cm shoot and 50 cm shoot. Most paralogous genes had similar expression patterns, such as PeTTF19/− 25 and PeTTF33/− 35 (Fig. 5). However, the duplication of genes potentially results in differences in expression patterns, revealing their different evolutionary fates. Here, for example, PeTTF19 was highly expressed in the leaf (L), early panicle (P1), advanced panicle (P2), root (R), rhizome (RH), 20 cm shoot (S1) and 50 cm shoot (S2), while PeTTF25 expression was comparatively low in those tissues.
Fig. 5.
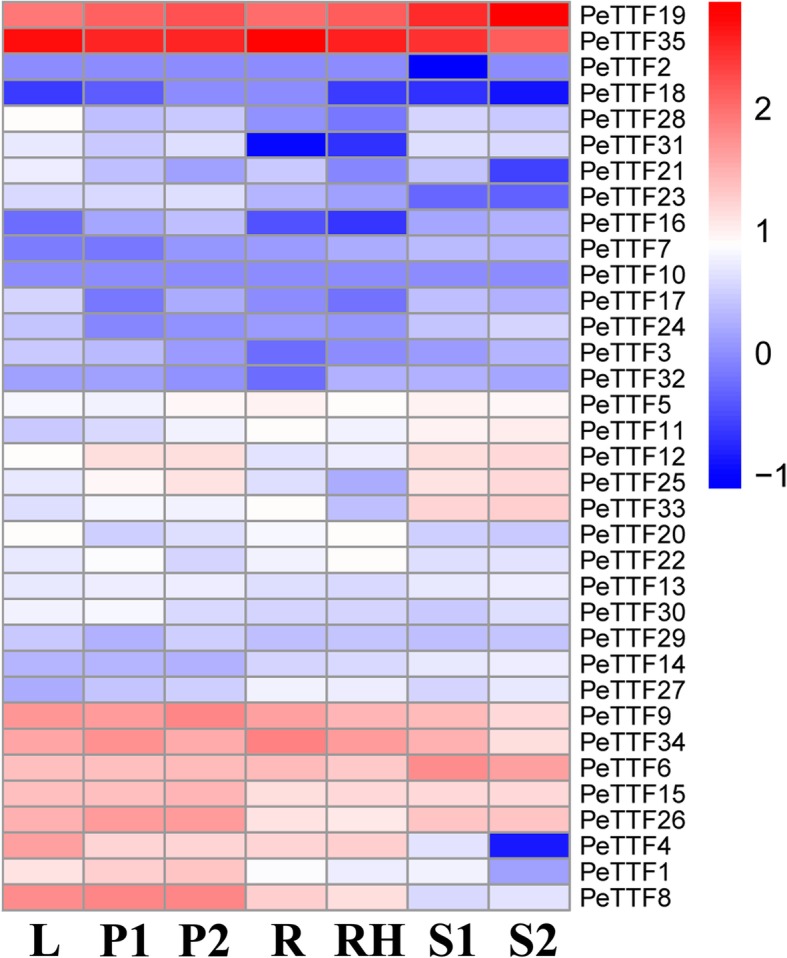
Expression profiles of poplar TTF genes in different tissues and developmental stages. Heatmap shows the hierarchical clustering of the 35 PeTTF genes among the different tissues. The abbreviation represents specific developmental stages: leaf (L); early panicle (P1); advanced panicle (P2); root (R); rhizome (RH); 20 cm shoot (S1); 50 cm shoot (S2)
To obtain an overview of the TTF gene expression profiles, the expression patterns in six tissues, including young leaves, roots, rhizomes, stems, shoots and leaves, were studied by quantitative real-time PCR (qRT-PCR). This showed that the expression of most PeTTF genes was significantly higher in leaves (28.6%), young leaves (25.7%) and shoots (28.6%) (Fig. 6). According to the comparative analysis of paralogous genes, two pairs of paralogous genes showed similar expression patterns in different tissues (PeTTF4/− 21 and PeTTF7/− 16). In contrast, the remaining 10 paralogous pairs (PeTTF2/− 10, PeTTF3/− 23, PeTTF5/− 14, PeTTF15/− 26, PeTTF18/− 32, PeTTF19/− 25, PeTTF20/− 22, PeTTF24/− 31, PeTTF27/− 30 and PeTTF33/− 35) showed different expression in different tissues.
Fig. 6.

Expression profiles of poplar TTF genes across different tissues and developmental stages as revealed by qRT-PCR. Y-axis: relative expression levels; X-axis: the time course of stress treatments; Error bars, 6 ± SE. The abbreviation represents specific developmental stages: leaf (L); young leaf (YL); root (R); rhizome (RH); shoot (S); stem (St)
Promoter analysis of TTF genes in Moso bamboo
To examine the TTF gene expression regulatory mechanisms, the DNA sequences 2000 bp upstream/downstream of the TTF genes were queried against the PlantCARE database. The TTF genes contained four types of cis-elements. The first was phytohormone responsive elements such as the GARE-motif, TATC-box, P-box, TGACG-motif, TCA-element and ABRE. The cis-acting regulatory elements involved in responses to methyl jasmonic acid (MeJA, 47.08%) were the CGTCA-motif (25.29%) and TGACG-motif (21.79%). The cis-acting regulatory elements involved in salicylic acid (SA, 11.67%) responses included the TCA-element (11.28%) and SARE (0.39%). SA and MeJA both play key roles in plant defense signaling [56, 57]. Thus, some TTF genes may be involved in pathogen resistance. The cis-acting regulatory elements involved in abscisic acid (ABA, 19.46%) responses included motif II b (2.72%) and the ABRE (16.74%). Gibberellin (GA, 14.01%) regulatory elements included the GARE-motif (10.89%), TATC-box (1.17%) and P-box (1.95%). Auxin (IAA, 6.23%) responsive cis-acting regulatory elements included the AuxRR-core (1.95%) and TGA-element (4.28%). ABA, GA and IAA all play major roles in plant growth and survival. Therefore, they may regulate the expression of some TTF genes (Fig. 7). The second type of cis-element was abiotic and biotic stress responsive elements, which included heat stress responsive (HSE, 6.82%), defense and stress responsive (TC-rich repeats, 9.54%), anaerobic-induced (ARE, 13.18%), anoxia-specific inducible (GC-motif, 19.54%) and drought-inducible (MBS, 30.47%) elements (Fig. 7). In addition to these, other phytohormone responsive (e.g. ERE) and abiotic and biotic stress responsive (e.g. wound and low-temperature) elements were found in the TTF gene family (Fig. 7). The third class was plant growth and development elements, including endosperm expression cis-acting regulatory elements (Skn-1_motif and GCN4_motif), a cis-acting regulatory element related to meristem-specific activation (CCGTCC-box) and circadian control cis-acting elements (circadian), which were highly represented in the PeTTF genes. The final class consisted of light responsive elements; for example, the G-box, GAG-motif and GT1-motif (Additional file 4: Table S3).
Fig. 7.

Cis-acting element analysis of the promoter regions of PeTTF genes. a Number of each cis-acting element in the promoter regions of PeTTF genes. b and c Statistics for the total number of PeTTF genes involved in different processes. Based on the functional annotation, the cis-acting elements were classified into two major classes: phytohormone responsive and abiotic and biotic stress-related cis-acting elements
Gene ontology (GO) annotation analysis and subcellular localization prediction
The association of 35 TTF genes with different biological processes was analyzed by GO annotation. In the molecular function category (Additional file 5: Table S4), six genes (PeTTF1, − 7, − 9, − 21, − 24 and − 31) were annotated with the term ‘chromatin binding’ (Additional file 5: Table S4), but only three genes (PeTTF3, − 7 and − 23) were annotated with ‘DNA binding’. In the biological process category, three genes (PeTTF5, − 12 and − 14) were associated with cellular amino acid biosynthetic processes.
Subcellular localization analysis of the TTF gene products was also performed (Additional file 7: Table S5) (Fig. 8). Nineteen gene products were located in the nucleus (55.88%, nucleus), 11 were localized to chloroplasts (31.43%, chloroplast), three were situated in the mitochondria (8.56%, mitochondria), and only one was localized to the cytosol (2.86%, cytosol).
Fig. 8.
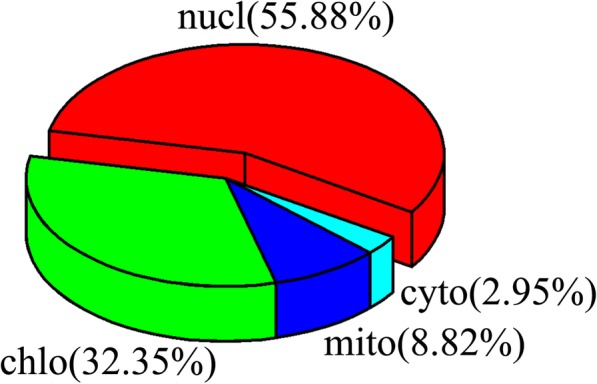
Subcellular localization of the TTF genes in Moso bamboo. Different colors represent different positions. Nucl, nucleus; chlo, chloroplast; cyto, cytoplasm; mito, mitochondrion
Expression analysis of Moso bamboo TTF genes by qRT-PCR
During plant growth, various adverse environmental effects such as drought, disease and insect predation can influence plant development. Many stress-related genes can help manage responses to these adverse conditions. Primers for qRT-PCR were designed for the TTF genes and are listed in Additional file 6: Table S6. As the expression of TTF genes has previously been shown to be sensitive to MeJA treatment [12], qRT-PCR was used to examine the expression patterns of the 35 TTF genes in response to MeJA treatment in Moso bamboo. Most of the PeTTF genes were affected by MeJA but PeTTF29 had the highest expression at 12 h (> 10-fold) (Fig. 9). In total, 17 genes were upregulated, while the PeTTF4 gene was downregulated at all times. In the early stages, at 1 h after treatment, PeTTF5, − 13 and − 24 showed peak expression levels (> 10-fold, 4-fold and 24-fold, respectively). PeTTF1, − 2, − 8, − 16, − 17, − 18, − 19, − 20, − 21, − 22, − 23, − 30 and − 35 showed their highest expression levels after 6 h of MeJA treatment. Among these genes, the expression levels of PeTTF20 and − 23 were more than 20 times that of the control. PeTTF3 and − 28 were expressed relatively highly after 3 h of treatment (> 4-fold and 20-fold, respectively) and peak values for PeTTF6, − 7, − 12, − 14 and − 34 were observed after 24 h of treatment (all were upregulate at least three-fold) (Fig. 9). Furthermore, most paralogous pairs showed similar expression patterns; for example, PeTTF15 and − 26 were both upregulated, with their highest expression levels at 1 h (> 2-fold). As shown in Fig. 9, the expression patterns of PeTTF4 and − 21 were reversed.
Fig. 9.

Expression patterns of all selected genes in Moso bamboo under MeJA treatment, as revealed by qRT-PCR. Y-axis: relative expression levels; X-axis: the time course of stress treatments; Error bars, 6 ± SE
We also treated Moso bamboo seedlings with 20% PEG6000 solution and 200 mM NaCl to simulate drought and salt conditions, and observed TTF gene expression. Five genes (PeTTF24, − 26, − 30, − 31 and − 32) were upregulated in response to PEG (drought) treatment while the other genes were downregulated (Fig. 10). High expression of PeTTF17, − 23, − 24, − 26, − 27, − 29, − 31 and − 35 was observed during the early stages of treatment (1 h), but only PeTTF24 expression was more than 15 times the control value; the expression of the others was more than 2-fold. At 3 h, PeTTF30 and PeTTF32 had the highest expression (> 20-fold and 5-fold, respectively). PeTTF28 and PeTTF33 had the highest expression at 6 h (> 4-fold). PeTTF24, − 28, − 30, − 32, and − 33 were also strongly upregulated (> 4-fold) (Fig. 10).
Fig. 10.

Expression patterns of all selected genes in Moso bamboo under PEG treatment, as revealed by qRT-PCR. Y-axis: relative expression levels; X-axis: the time course of stress treatments; Error bars, 6 ± SE
In the salt treatment, only five genes (PeTTF4, − 5, − 6, − 7 and − 9) were upregulated, but some genes were upregulated in specific periods; for example, PeTTF2 was upregulated from 0 to 6 h and downregulated from 6 to 12 h (Fig. 11). The expression of 11 genes (PeTTF2, − 4, − 5, − 7, − 9, − 10, − 11, − 12, − 18, − 23 and − 25) peaked 1 h after treatment, among which PeTTF2, − 4, − 5, − 7, and − 23 were highly expressed (> 3-fold) (Fig. 11). These results suggested that some PeTTF genes play significant roles in the regulation of insect injury, drought and salt stress responses (Fig. 11).
Fig. 11.

Expression patterns of all selected genes in Moso bamboo under salt treatment, as revealed by qRT-PCR. Y-axis: relative expression levels; X-axis: the time course of stress treatments; Error bars, 6 ± SE
Subcellular localization of PeTTF29
PeTTF29 was predicted to be located in the nucleus by subcellular localization analysis. To confirm this prediction, a PeTTF29-GFP expression vector was transformed into tobacco. In the transgenic plants, the GFP signal was observed only in the nucleus (Fig. 12), whereas the plants transformed with constitutively expressed GFP control vectors show green fluorescence throughout the cells. This results indicated that PeTTF29 was a nuclear protein.
Fig. 12.
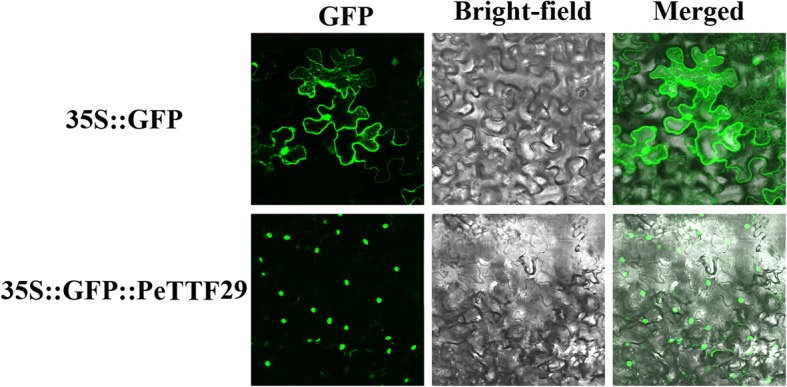
Nuclear localization of PeTTF29. The 35S::GFP::PeTTF29 construct and the control vector 1305(35S::GFP) were transformed into Nicotiana tabacum leaves. The GFP signals in root cells were observed by confocal microscopy
Transcription activation assay of PeTTF29
The yeast yeast fusion expression vector PeTTF29-pGBKT7 was constructed and transformed into yeast cells to detect transcriptional activity. Yeast cells containing the full-length PeTTF29 fragment grew well on the SD/Trp − plate and were also able to grow well on the SD/Trp−/His−/Ade−/X-α-gal plate and produce a blue color reaction; the positive control showed the same results (Fig. 13). However, although the negative control was able to grow well on the SD/Trp − plate, it was unable to grow on the SD/Trp−/His−/Ade−/X-α-gal plate and did not produce a blue reaction. The above results indicated that PeTTF29 was active in the yeast H2GOLD.
Fig. 13.
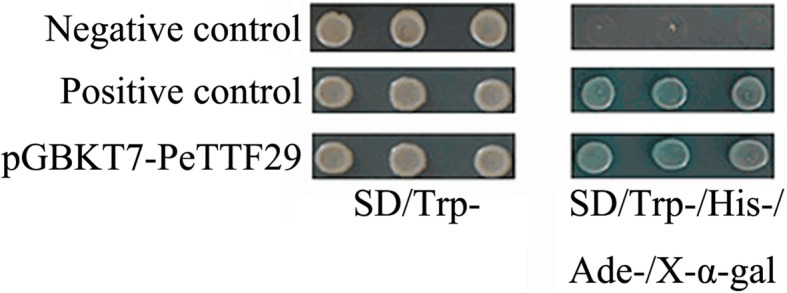
Transcriptional activity analysis of PeTTF29. Positive control, pGBKT7–53 and pGBKT7-T; Negative control, pGBKT7
Discussion
Previous studies of the TTF gene family have been reported in various plant species including Arabidopsis, rice, tomato, and chrysanthemum [3–7, 12, 13]. However, this family has never been examined in depth in Moso bamboo. Therefore, in the present study, we performed a genome-wide analysis of the TTF gene family in Moso bamboo and used qRT-PCR to investigate its antagonistic regulation.
In total, 35 TTF gene members were identified in Moso bamboo (Table 1). According to their phylogenetic relationships with TTF proteins from Arabidopsis and rice (Additional file 1: Table S1), these genes were divided into five subfamilies (SIP1, GT-1, SH4, GT-2, and GT-γ) (Fig. 1). The relatively short branches and intervals indicated that these proteins were highly conserved and that mutations were not common in their evolution, suggesting that they have similar functions. In addition, many TTF genes were clustered together at the ends of the branches, and high sequence similarity was observed between certain gene pairs. This also indicted that these genes may have similar functions.
The 35 genes contained varying numbers of exons and introns, indicating the diversity of the TTF gene family in Moso bamboo (Fig. 3). Nevertheless, in terms of the length of exons or the number of introns, the most closely related genes in the same subfamily shared similar gene structures. Some genes had unique exon/intron structures; for example, some Arabidopsis and rice genes had 16 or more introns [3, 6], while the Moso bamboo genes had substantially less introns (less than seven), indicating that the PeTTF genes may have lost introns or the rice and Arabidopsis genes have obtained introns. Twelve genes in Moso bamboo lacked introns, whereas a maximum of nine genes in Arabidopsis and rice had no introns, indicating that either Moso bamboo genes had lost exons or the rice and Arabidopsis genes had gained exons during the evolutionary process. Furthermore, MEME analysis showed that TTF protein-specific functions could be conferred by a specific sequence motif present in each subfamily. Overall, the similarity of the genetic structures and motif compositions of most TTF genes in each subfamily supported the phylogenetic analysis.
Genome duplication events can allow genes to evolve new functions, so they play a key role in increasing the genomic content and the diversification of gene functions. Moso bamboo (35 PeTTF genes) contained more TTF genes than rice (31 OsTTF genes) and Arabidopsis (28 AtTTF genes) [3, 6]. Moso bamboo has the largest genome among the three plants, as well as the largest number of chromosomes (2n = 48), which likely contributed to the greater number of TTF genes in Moso bamboo than in the other two species (as shown in Additional file 1: Table S1) [58]. In our analysis, we identified 12 duplicated gene pairs (Table 2) in Moso bamboo. The Ka/Ks ratios of eight pairs were less than 1 (Table 3), indicating that these genes had been subjected to purifying selection. We used a sliding Ka/Ks window to analyze each pair of TTF paralogs to further study the effects of purifying selection on the 12 paralogous pairs (Additional file 3: Figure S1). This clearly showed that strong purifying selection was experienced by the paralogous genes. Eight genes were under positive selection in particular regions, indicating that the TTF genes of Moso bamboo were also restrained in forward evolution to ensure their stability.
According to expression profile analysis, the 35 TTF genes are expressed in different tissues of Moso bamboo. Previous studies have suggested that TTF genes can regulate light-responsive genes [59, 60], and that the loss of the AtGTL1 gene can affect the efficiency of water use by reducing the stomatal density [61], thereby increasing plant tolerance to water scarcity. These previous studies have shown relationships between TTF genes and leaf stomata or photoreactions. Accordingly, PeTT19 and − 35 were highly expressed in leaves, suggesting that (i) these two genes are associated with photosynthetic genes, or (ii) these genes are related to stomata or photosynthesis. Additionally, plant roots can quickly perceive changes in the soil and issue a series of signals to the branches and leaves to reduce root damage under drought conditions. In our research, PeTTF35 was highly expressed in roots, indicating that this gene might strengthen the root organization to better absorb moisture and adapt to drought conditions. TTF genes expressed in different tissues with the ability to convey resistance could be used to improve plant tolerance to these stresses [62].
Promoter cis-elements play important roles in the biotic and abiotic stress responses of Moso bamboo. In this study, most promoter regions of the TTF genes were found to contain phytohormone and biotic and abiotic stress responsive cis-elements including the GARE-motif, TATC-box, P-box, and MBS. PeTTF7 and − 11 had 11 cis-elements, indicating they have important functions under different stresses. Notably, PeTTF10 contained the most copies of the CGTCA-motif cis-element (7). However, it showed no significant expression change under MeJA treatment by qRT-PCR. This may be because the gene is upregulated after 24 h. PeTTF27 had two MeJA stress-related cis-elements (the CGTCA-motif and TGACG-motif). The qRT-PCR results showed that PeTTF27 was not only induced by SA, but also by ABA and MeJA, indicating that ABA and MeJA responses are closely related to SA responses.
Plants are often threatened by abiotic and biological stresses, which can cause severe damage or even be fatal. However, many genes have been genetically altered to help plants adapt to these stresses. Therefore, on the basis of the expression in different tissues (Fig. 5), seedling leaves were used to detect gene expression and perform qRT-PCR. Figure 5 shows that nine genes (PeTTF4, − 6, − 8, − 9, − 15, − 19, − 26, − 34, and − 35) were highly expressed whereas seven genes (PeTTF4, − 6, − 8, − 9, − 15, − 19, and − 34) were downregulated after PEG treatment, seven genes (PeTTF6, 9, − 15, − 19, − 26, − 34, and − 35) were downregulated after salt treatment and four genes (PeTTF4, − 6, − 9, − 15, and − 26) were downregulated after MeJA treatment. In addition, six genes (PeTTF23, − 24, − 28, − 30, − 32 and − 33) showed low in expression in Fig. 5, but the expression of these genes rapidly increased after PEG treatment. The expression of eight genes (PeTTF1, − 2, − 5, − 7, − 13, − 21, − 22 and − 23) also began to increase rapidly after salt treatment and the expression of 20 genes (PeTTF1, − 2, − 3, − 5, − 7, − 12, − 13, − 16, − 17, − 18, − 20, − 21, − 22, − 23, − 24, − 28, − 29, − 30, − 31, and − 32) began to increase after MeJA treatment. From these results, it can be seen that Moso bamboo TTF genes play an important role in coping with different stresses.
Using qRT-PCR, we analyzed the transcription levels of TTF genes in response to biotic stress (MeJA) and abiotic stress (drought and salt). The results indicated that each PeTTF gene was differentially expressed after the MeJA, drought and salt treatments, which could provide a useful resource for future gene function analysis. Therefore, we used the GraphPad software to visualize the expression patterns of the TTF genes under different stress conditions (Fig. 12), which suggested the TTF gene family in Moso bamboo may play a key role in regulating abiotic/biological stress responses. In this family, similar sequences and expression patterns were observed between most duplicated genes, indicating that the regulatory sequences responsive to stress conditions had not, generally, diverged significantly through evolution following gene replication.
No previous studies on the functions of TTF gene members in Moso bamboo have been reported. Therefore, we screened a potential gene (PeTTF29) associated with abiotic stress in our experiments that was highly homologous to LOC Os02g33770 in rice according to evolutionary analysis. We used Blast (NCBI) to compare the CDSs of these two genes and the results showed that their homology was 80% or more. LOC Os02g33770 has been reported to be associated with abiotic stress responses [4], which suggests that PeTTF29 may also be involved in some abiotic stress responses. To further explore the function of PeTTF29, we conducted subcellular localization and transcriptional activity experiments. These experiments showed that PeTTF29 was localized in the nucleus (Fig. 12) and had transcriptional activity (Fig. 13). Therefore, this gene had the basic properties that most transcription factors possess, indicating that it is a typical transcription factor gene.
Conclusion
In summary, the structural diversity of Moso bamboo TTF proteins indicates they have diverse functions and may be associated with adaptation to different environmental stresses at different developmental stages. We analyzed 35 members of the TTF gene family in Moso bamboo, which were divided into five different subfamilies. Comparisons between the TTF genes in Moso bamboo and those in other model species confirmed their extensive homology and indicated when they had evolved. The TTF gene family was found to have expanded through large-scale gene duplication. These genes have continued to evolve at the protein level while being subjected to strong positive selection. The expression patterns of the 35 TTF genes were analyzed under two abiotic stresses (drought and salt) and one biotic stress (MeJA). Subcellular localization and transcriptional activity experiments showed that PeTTF29 is localized in the nucleus and has transcriptional activity. The results of this study will help to increase our understanding of the TTF family members, including their possible contributions to abiotic stress responses and other putative functions in Moso bamboo growth and development.
Additional files
Table S1. Detailed information about TTF genes in rice and Arabidopsis. (DOCX 25 kb)
Table S2. MEME motif sequences and lengths of TTF gene family proteins in Moso bamboo. (DOCX 22 kb)
Figure S1. Sliding window plots of the TTF genes in Moso bamboo. (TIF 603 kb)
Table S3. Promoter analysis of TTF proteins in Moso bamboo. (XLS 50 kb)
Table S4. Details of the Gene Ontology annotation. (DOCX 21 kb)
Table S6. Design the qRT-PCR primers. (DOCX 25 kb)
Table S5. Subcellular localization of TTF gene in Moso bamboo. (DOCX 21 kb)
Acknowledgements
We thank for the Laboratory of Modern Biotechnology, National Engineering Laboratory of Crop Stress Resistance Breeding and Key Laboratory of Crop Biology of Anhui Province members for their assistance in this study. And thank Prof. Dingqing Tang, School of Forestry and Bio-technology, Zhejiang A & F University, for giving us the great help in seeds collection for this study. We thank Robbie Lewis, MSc, from Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Funding
This study was supported by the National Science and Technology Support Program (2015BAD04B0302) and National Science Foundation of China (No. 31670672).
These funding bodies had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results. Publication costs are defrayed by these funding.
Availability of data and materials
Data from this article were obtained from the NCBI short read (SRA) database (https:// trace.ncbi.nlm.nih.gov/Traces/sra/?study=ERP001341). The genome sequences of Moso bamaoo, rice and Arabidopsis were downloaded from the Bamboo Genome Database (http://www.bamboogdb.org/), Rice Genome Annotation Project database (http://rice.plantbiology.msu.edu/analyses_search_locus.shtml) and PlantTFDB (http://planttfdb.cbi.pku.edu.cn).
Abbreviations
- aa
Amino acids
- ABA
Abscisic acid
- bp
Base pair
- CDS
Coding sequence
- Da
Dalton
- GFP
Green fluorescent protein
- Ka
Number of non-synonymous substitutions per non-synonymous site
- Ks
Number of synonymous substitutions per synonymous site
- MeJA
Methyl jasmonate
- MW
Molecular weight
- MYA
Million years ago
- NJ
Neighbor-joining
- PEG
Polyethylene glycol
- Pi
Isoelectric point
- qRT-PCR
Quantitative real-time PCR
- SA
Salicylic acid
- TTF
Trihelix transcription factors
Authors’ contributions
XRC projected the study, put into effect the mainly bioinformatics analysis, drew up the manuscript. RX carried out the software, and helped to handle figures and tables. HY took part in the experiments and draw up the manuscript .HLL processed of experimental data and joined to amendment the manuscript. YMG took part in the software and draw up the manuscript. MW had a hand in the project of the study and helped to revamp the manuscript. YX conceived and guided the experiment, involved in its project and coordination and helped to draw up the manuscript. All authors read and accepted the final manuscript.
Ethics approval and consent to participate
Our Moso bamboo’s seeds were provided by Prof. Dingqing Tang, School of Forestry and Bio-technology, Zhejiang A & F University.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Xinran Cheng, Email: 921083951@qq.com.
Rui Xiong, Email: 1641593309@qq.com.
Hanwei Yan, Email: qiji19870814@163.com.
Yameng Gao, Email: 2313212262@qq.com.
Huanlong Liu, Email: 847184534@qq.com.
Min Wu, Email: 1050777822@163.com.
Yan Xiang, Phone: +86-0551-65786021, Email: xiangyan@ahau.edu.cn, Email: xiangyanahau@sina.com.
References
- 1.Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K. AP2/ERF family transcription factors in plant abiotic stress responses. Biochim Biophys Acta. 2012;1819:86–96. doi: 10.1016/j.bbagrm.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 2.Kaplan-Levy RN, Brewer PB, Quon T, et al. The trihelix family of transcription factors--light, stress and development. Trends Plant Sci. 2012;17:163–171. doi: 10.1016/j.tplants.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 3.Ali MA, Yasmeen E, Riaz M, et al. Genome-wide analysis of trihelix transcription factor gene family in Arabidopsis thaliana. Pak J Agric Sci. 2016;53:439–448. [Google Scholar]
- 4.Fang Y, Xie K, Xin H, et al. Systematic analysis of GT factor family of rice reveals a novel subfamily involved in stress responses. Mol Genet Genomics Mgg. 2010;283:157. doi: 10.1007/s00438-009-0507-x. [DOI] [PubMed] [Google Scholar]
- 5.Song A, Dan W, Fan Q, et al. Transcriptome-wide identification and expression profiling analysis of Chrysanthemum Trihelix transcription factors. Plant Physiol Biochem. 2016;102:10–16. doi: 10.1016/j.plaphy.2016.02.009. [DOI] [PubMed] [Google Scholar]
- 6.Du H, Huang M, Liu L. The genome wide analysis of GT transcription factors that respond to drought and waterlogging stresses in maize. Euphytica. 2016;208:113–122. doi: 10.1007/s10681-015-1599-5. [DOI] [Google Scholar]
- 7.Yu C, Cai X, Ye Z, et al. Genome-wide identification and expression profiling analysis of trihelix gene family in tomato. Biochem Biophys Res Commun. 2015;468:653. doi: 10.1016/j.bbrc.2015.11.010. [DOI] [PubMed] [Google Scholar]
- 8.Li C, Zhou A, Sang T, Li C, Zhou A, Sang T. Rice domestication by reducing shattering. Science. 2006;311:1936–1939. doi: 10.1126/science.1123604. [DOI] [PubMed] [Google Scholar]
- 9.Z L, ME G, X L, et al. Origin of seed shattering in rice (Oryza sativa L.) Planta. 2007;226:11–20. doi: 10.1007/s00425-006-0460-4. [DOI] [PubMed] [Google Scholar]
- 10.Gao M-J, Lydiate DJ, Li X, et al. Repression of seed maturation genes by a trihelix transcriptional repressor in Arabidopsis seedlings. Plant Cell. 2009;21:54–71. doi: 10.1105/tpc.108.061309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Willmann MR, Mehalick AJ, Packer RL, et al. MicroRNAs regulate the timing of embryo maturation in Arabidopsis. Plant Physiol. 2011;155:1871–1884. doi: 10.1104/pp.110.171355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z, Liu Q, Wang H, et al. Comprehensive analysis of trihelix genes and their expression under biotic and abiotic stresses in Populus trichocarpa. Sci Rep. 2016;6:36274. doi: 10.1038/srep36274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Osorio MB, Bücker-Neto L, Castilhos G, et al. Identification and in silico characterization of soybean trihelix-GT and bHLH transcription factors involved in stress responses. Genet Mol Biol. 2012;35:233–246. doi: 10.1590/S1415-47572012000200005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xi J, Qiu Y, Du L, et al. Plant-specific trihelix transcription factor AtGT2L interacts with calcium/calmodulin and responds to cold and salt stresses. Plant Sci. 2012;185-186:274. doi: 10.1016/j.plantsci.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 15.Brewer PB, Howles PA, Dorian K, et al. PETAL LOSS, a trihelix transcription factor gene, regulates perianth architecture in the Arabidopsis flower. Development. 2004;131:4035. doi: 10.1242/dev.01279. [DOI] [PubMed] [Google Scholar]
- 16.Weng H, Yoo CY, Gosney MJ, et al. Poplar GTL1 is a Ca2+/calmodulin-binding transcription factor that functions in plant water use efficiency and drought tolerance. PLoS One. 2012;7:e32925. doi: 10.1371/journal.pone.0032925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuromori T, Wada T, Kamiya A, et al. A trial of phenome analysis using 4000 ds-insertional mutants in gene-coding regions of Arabidopsis. Plant J. 2006;47:640. doi: 10.1111/j.1365-313X.2006.02808.x. [DOI] [PubMed] [Google Scholar]
- 18.Marchlerbauer A, Lu S, Anderson JB, et al. CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011;39:D225. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tamura K, Peterson D, Peterson N, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie ZM, Zou HF, Lei G, et al. Soybean Trihelix transcription factors GmGT-2A and GmGT-2B improve plant tolerance to abiotic stresses in transgenic Arabidopsis. PLoS One. 2009;4:e6898. doi: 10.1371/journal.pone.0006898. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Peng Z, Lu Y, Li L, et al. The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla) Nat Genet. 2013;45:456. doi: 10.1038/ng.2569. [DOI] [PubMed] [Google Scholar]
- 22.Wu H, Lv H, Li L, et al. Genome-wide analysis of the AP2/ERF transcription factors family and the expression patterns of DREB genes in Moso bamboo (Phyllostachys edulis) PLoS One. 2015;10:e0126657. doi: 10.1371/journal.pone.0126657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Liu H, Zhu D, et al. Genome-wide analysis of VQ motif-containing proteins in Moso bamboo (Phyllostachys edulis) Planta. 2017;246:165. doi: 10.1007/s00425-017-2693-9. [DOI] [PubMed] [Google Scholar]
- 24.Liu H, Min W, Zhu D, et al. Genome-wide analysis of the AAAP gene family in moso bamboo (Phyllostachys edulis) BMC Plant Biol. 2017;17:29. doi: 10.1186/s12870-017-0980-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin J, Zhang H, Kong L, et al. PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2014;42:D1182. doi: 10.1093/nar/gkt1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finn RD, Mistry J, Schusterböckler B, et al. Pfam: clans, web tools and services. Nucleic Acids Res. 2006;34:247–251. doi: 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Letunic I, Doerks T, Bork P. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 2012;40:302–305. doi: 10.1093/nar/gkr931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finn RD, Penelope C, Eberhardt RY, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44:D279. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gasteiger E, Gattiker A, Hoogland C, et al. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 31.Thompson JD, Gibson TJ, Plewniak F, et al. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tamura K, Stecher G, Peterson D, et al. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo AY, Zhu QH, Chen X, et al. GSDS: a gene structure display server. Hereditas. 2007;29:1023. doi: 10.1360/yc-007-1023. [DOI] [PubMed] [Google Scholar]
- 34.Bailey TL, Boden M, Buske FA, et al. MEME suite: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blanc G, Wolfe KH. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell. 2004;16:1667–1678. doi: 10.1105/tpc.021345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search 1997. 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 38.Rozas J. DNA sequence polymorphism analysis using DnaSP. Methods Mol Biol. 2009;537:337. doi: 10.1007/978-1-59745-251-9_17. [DOI] [PubMed] [Google Scholar]
- 39.Cao J, Huang J, Yang Y, et al. Analyses of the oligopeptide transporter gene family in poplar and grape. BMC Genomics. 2011;12:465. doi: 10.1186/1471-2164-12-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y-J, Ma P-F, Li D-Z. High-throughput sequencing of six bamboo chloroplast genomes: phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae) PLoS One. 2011;6:e20596. doi: 10.1371/journal.pone.0020596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang M, Sun H, Fei Z, et al. IEEE international conference on bioinformatics and biomedicine. 2015. Fastq_clean: An optimized pipeline to clean the Illumina sequencing data with quality control; pp. 44–48. [Google Scholar]
- 42.Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc. 2012;7:562. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toufighi K, Brady SM, Austin R, et al. The botany Array resource: e-Northerns, expression angling, and promoter analyses. Plant J. 2005;43:153–163. doi: 10.1111/j.1365-313X.2005.02437.x. [DOI] [PubMed] [Google Scholar]
- 44.Feng Y, Wu J, Zhang Z, et al. Divergence in function and expression of the NOD26-like intrinsic proteins in plants. BMC Genomics. 2009;10:313. doi: 10.1186/1471-2164-10-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wei W, Hu Y, Cui MY, et al. Identification and transcript analysis of the TCP transcription factors in the diploid woodland StrawberryFragaria vesca. Front Plant Sci. 2016;7:1937. doi: 10.3389/fpls.2016.01937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Consortium GO The gene ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–D261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Horton P, Park KJ, Obayashi T, et al. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35:W585. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Min W, Yuan L, Chen D, et al. Genome-wide identification and expression analysis of the IQD gene family in moso bamboo (Phyllostachys edulis) Sci Rep. 2016;6:24520. doi: 10.1038/srep24520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu M, Wu S, Chen Z, et al. Genome-wide survey and expression analysis of the amino acid transporter gene family in poplar. Tree Genet Genomes. 2015;11:83. doi: 10.1007/s11295-015-0908-4. [DOI] [Google Scholar]
- 50.Fan C, Ma J, Guo Q, et al. Selection of reference genes for quantitative real-time PCR in bamboo (Phyllostachys edulis) PLoS One. 2013;8:e56573. doi: 10.1371/journal.pone.0056573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 52.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 53.Bryfczynski SP, Pargas RP. ACM Sigcse Conference on Innovation and Technology in Computer Science Education. 2009. GraphPad: a graph creation tool for CS2/CS7; p. 389. [Google Scholar]
- 54.Cao Y, Han Y, Li D et al. MYB transcription factors in Chinese Pear (Pyrus bretschneideri Rehd.): genome-wide identification, classification, and expression profiling during fruit development. Front Plant Sci. 2016;7:577. [DOI] [PMC free article] [PubMed]
- 55.Hu R, Qi G, Kong Y, et al. Comprehensive analysis of NAC domain transcription factor gene family in Populus trichocarpa. BMC Plant Biol. 2010;10:145. doi: 10.1186/1471-2229-10-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gaffney T, Friedrich L, Vernooij B, et al. Requirement of salicylic acid for the induction of systemic acquired resistance. Science. 1993;261:754–756. doi: 10.1126/science.261.5122.754. [DOI] [PubMed] [Google Scholar]
- 57.Xu Y, Chang P, Liu D, et al. Plant defense genes are synergistically induced by ethylene and methyl Jasmonate. Plant Cell. 1994;6:1077–1085. doi: 10.1105/tpc.6.8.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen R, Li X, Song W, et al. Chromosome atlas of major economic plants genome in China. Tomus 4. Chromosome atlas of various bamboo species. Beijing: Science press xxx; 2003. p. 646. [Google Scholar]
- 59.Green PJ, Kay SA, Chua NH. Sequence-specific interactions of a pea nuclear factor with light-responsive elements upstream of the rbcS-3A gene. EMBO J. 1987;6:2543. doi: 10.1002/j.1460-2075.1987.tb02542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Green PJ, Yong MH, Cuozzo M, et al. Binding site requirements for pea nuclear protein factor GT-1 correlate with sequences required for light-dependent transcriptional activation of the rbcS-3A gene. EMBO J. 1988;7:4035–4044. doi: 10.1002/j.1460-2075.1988.tb03297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoo CY, Pence HE, Jin JB, et al. The Arabidopsis GTL1 transcription factor regulates water use efficiency and drought tolerance by modulating stomatal density via transrepression of SDD1. Plant Cell. 2010;22:4128–4141. doi: 10.1105/tpc.110.078691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Villordon AQ, Ginzberg I, Firon N. Root architecture and root and tuber crop productivity. Trends Plant Sci. 2014;19:419–425. doi: 10.1016/j.tplants.2014.02.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Detailed information about TTF genes in rice and Arabidopsis. (DOCX 25 kb)
Table S2. MEME motif sequences and lengths of TTF gene family proteins in Moso bamboo. (DOCX 22 kb)
Figure S1. Sliding window plots of the TTF genes in Moso bamboo. (TIF 603 kb)
Table S3. Promoter analysis of TTF proteins in Moso bamboo. (XLS 50 kb)
Table S4. Details of the Gene Ontology annotation. (DOCX 21 kb)
Table S6. Design the qRT-PCR primers. (DOCX 25 kb)
Table S5. Subcellular localization of TTF gene in Moso bamboo. (DOCX 21 kb)
Data Availability Statement
Data from this article were obtained from the NCBI short read (SRA) database (https:// trace.ncbi.nlm.nih.gov/Traces/sra/?study=ERP001341). The genome sequences of Moso bamaoo, rice and Arabidopsis were downloaded from the Bamboo Genome Database (http://www.bamboogdb.org/), Rice Genome Annotation Project database (http://rice.plantbiology.msu.edu/analyses_search_locus.shtml) and PlantTFDB (http://planttfdb.cbi.pku.edu.cn).