

A multifaceted approach combining RNA-seq, genomics, and molecular biology allows systematic comparative analysis of pseudogenes in seven plant species.
Abstract
Pseudogenes (Ψs), nonfunctional relatives of functional genes, form by duplication or retrotransposition, and loss of gene function by disabling mutations. Evolutionary analysis provides clues to Ψ origins and effects on gene regulation. However, few systematic studies of plant Ψs have been conducted, hampering comparative analyses. Here, we examined the origin, evolution, and expression patterns of Ψs and their relationships with noncoding sequences in seven angiosperm plants. We identified ∼250,000 Ψs, most of which are more lineage specific than protein-coding genes. The distribution of Ψs on the chromosome indicates that genome recombination may contribute to Ψ elimination. Most Ψs evolve rapidly in terms of sequence and expression levels, showing tissue- or stage-specific expression patterns. We found that a surprisingly large fraction of nontransposable element regulatory noncoding RNAs (microRNAs and long noncoding RNAs) originate from transcription of Ψ proximal upstream regions. We also found that transcription factor binding sites preferentially occur in putative Ψ proximal upstream regions compared with random intergenic regions, suggesting that Ψs have conditioned genome evolution by providing transcription factor binding sites that serve as promoters and enhancers. We therefore propose that rapid rewiring of Ψ transcriptional regulatory regions is a major mechanism driving the origin of novel regulatory modules.
INTRODUCTION
Pseudogenes (Ψs) are disabled copies of protein-coding genes and are often referred to as genomic fossils (Balasubramanian et al., 2009; Sisu et al., 2014). Protein-coding genes become Ψs if degenerated features are present, such as frameshifts, in-frame stop codons, and truncations of full-length genes (Zhang et al., 2003). Depending on the mechanism of the duplication event, Ψs can be classified into two categories: nonprocessed and processed. Nonprocessed Ψs originated from genomic DNA duplication or unequal crossing-over; processed Ψs originated from reverse transcription and integration events (Zhang et al., 2003; Zou et al., 2009). Ψs have been defined as nonfunctional sequences and thus are expected to evolve neutrally (Torrents et al., 2003); consistent with this, the majority of Ψs evolve neutrally in the human (Homo sapiens), worm (Caenorhabditis elegans), and fruitfly (Drosophila melanogaster) genomes (Sisu et al., 2014).
Although Ψs are disabled copies of protein-coding genes, a small fraction of Ψs have been shown to function as versatile regulators in fundamental processes, acting by producing regulatory RNAs (Guo et al., 2009; Wen et al., 2011). For example, several studies suggest that Ψs could serve as sources of endogenous small interfering RNAs (Tam et al., 2008; Watanabe et al., 2008; Wen et al., 2011). Ψs have also been shown to regulate gene expression by sequestering microRNAs (miRNAs; Poliseno et al., 2010). These observations suggest that Ψs play regulatory roles in gene expression and have motivated scientists to investigate the functions of Ψs in different organisms.
Evolutionary analyses of Ψs, including their expression patterns and associations with noncoding RNAs (Guo et al., 2009), have provided important clues into lineage-specific genomic evolutionary histories and the genetic basis of Ψ functions. However, despite growing interest in Ψs, such analyses remain scarce. Further evolutionary studies of Ψs can be informative for identifying the origin and regulation of RNA genes.

The evolutionary forces that affect the chromosomal distribution of Ψs are poorly understood. Genome duplication (paleopolyploidy) is common in flowering plants (Wendel, 2000). The long-term evolution of paleopolyploids often involves extensive genome reorganization and elimination of a large fraction of duplicate genes (Wolfe, 2001). This may produce thousands of Ψs in plant genomes. Recombination has been recognized as one of the key factors that shapes genomic features, such as the elimination/retention of duplicated genes after whole-genome duplication (WGD), distribution of transposable elements (TEs) and genes, and nucleotide variation in eukaryotes (Gaut et al., 2007; Tian et al., 2009; Du et al., 2012). Ψs in the human and fruitfly genomes are enriched in regions of low recombination, but Ψs in worms show the opposite trend (Sisu et al., 2014). These observations may conflict because of differing distributions of recombination events in specific genomes, based on differing distributions of heterochromatin.
Few studies have performed genome-wide, multispecies analyses of Ψs regarding their rates of evolution and surrounding chromatin environment. As a result, little is known about the evolutionary forces that shape the patterns of Ψs in paleopolyploid organisms. The only cross-species comparison of Ψs in plants concerned the identification and evolution of Ψs in Arabidopsis (Arabidopsis thaliana) and rice (Oryza sativa) genomes (Zou et al., 2009). Therefore, the evolution of plant Ψs requires further examination. The availability of complete annotations of several plant genomes, including rice, Arabidopsis, and Populus trichocarpa, allowed us to embark on a comprehensive, cross-species comparison to discover common features of the evolution of Ψs across different organisms. In addition, the recombination data from soybean (Glycine max; Du et al., 2012) provided us with the opportunity to study whether recombination shaped the distribution of Ψs in detail. A surprisingly large fraction of non–TE long noncoding RNA (lncRNA) transcripts originate from transcription at putative Ψ proximal upstream regions, indicating a common mechanism for the origin of novel regulatory modules.
RESULTS
Identification of Ψs in Seven Angiosperm Species
To systematically identify candidate pseudogenic regions in seven species, Arabidopsis, Brachypodium distachyon, soybean, Medicago truncatula, rice, Populus trichocarpa, and Sorghum bicolor, we used a combination of homology searches and stringent filters to minimize noise and increase positive signals (Figure 1). First, repeat sequences in the intergenic regions were masked using RepeatMasker (RM) to avoid alignment errors. We identified 90,000 to 800,000 intergenic homologous contigs with significant similarity (identity ≥ 20%; match length ≥ 5% of the query sequence) to known proteins from the non-redundant database in the seven taxa. We then examined Ψs near the cutoff (length 30 to 107 amino acids; match length coverage ratio, 0.050 to 0.052) and found that eight were WGD-derived Ψs and three were syntenic Ψs located on the syntenic blocks between P. trichocarpa and Arabidopsis (Supplemental Figure 1). By this method, it is possible that we missed some intergenic regions that resemble protein-coding sequences in repeat regions. After stringent filtering, most of the initial homologous contigs did not remain in the final Ψ data set; these may represent artifacts or may be too diverged to display characteristics of Ψs (such as match length ≥ 30 amino acids). The application of these stringent filters retained 5128 to 73,811 putative Ψs per species (Figure 2A; Supplemental Data Sets 1 to 7), and 146 to 2524 of the Ψs are derived from WGD events (Figure 2A). We also found that 11.6 to 25% of the total Ψ pool have introns; these could be Ψs that retained their original intron structure.
Figure 1.

The Ψ Identification Pipeline.
The overall procedure for identifying Ψs from seven plant species: P. trichocarpa, Arabidopsis, B. distachyon, soybean, M. truncatula, rice (O. sativa subsp japonica), and S. bicolor. E-value, BLAST expect value, match length, alignment length.
Figure 2.

Identification of Ψs in Seven Plant Species.
(A) Number of Ψs identified in each genome.
(B) Distribution of genes and the different types of Ψs across chromosomes (Chr) in P. trichocarpa, Arabidopsis, and B. distachyon.
(C) Comparison of evolution between pairs of FG–FG (functional WGDs) and Ψ–FP (closest FPs).
(D) Distribution of sequence similarity to their closest FPs (as a function of Ψ age) for P. trichocarpa. CDS, coding sequence.
(E) Comparative distribution of Ψs and paralogs per gene in P. trichocarpa.
We observed a moderate, but not significant, trend of a higher number of Ψs in larger genomes (Pearson’s correlation = 0.71, P = 0.07), with the most Ψs present in soybean and the fewest in Arabidopsis. S. bicolor is an exception: it has the second largest genome size but a low Ψ number. As expected, we found a strong correlation between the Ψ and protein-coding gene densities in all the seven taxa (Figure 2B). A closer inspection revealed that the distribution of Ψs among the chromosomes is also proportional to the chromosome length (Pearson’s correlation > 0.72, P < 0.02) and gene density (Pearson’s correlation > 0.90, P < 0.02; Figure 2B; Supplemental Figure 2).
Among the species examined, the soybean Ψs appear to be more fragmented than those in other species (Figure 2A). The soybean lineage has undergone two rounds of WGD within the last 60 million years (Myr), with a recent event (∼13 Myr ago) resulting in a highly duplicated genome with nearly 75% of the genes having multiple copies (Schmutz et al., 2010). Thus, the shorter extent of soybean Ψs may result from the rapid gene loss that occurred in the early stages of genome reshaping shortly after the recent WGD (Inoue et al., 2015). Consistent with this, we found that gene pairs of WGD blocks containing Ψs have peaks with a synonymous substitution rate (Ks) of ∼0.13, corresponding to a soybean lineage-specific paleotetraploidization (∼13 million years ago; Supplemental Figure 3). The highest alignment coverage of Ψs to their closest functional paralogs (FPs) was found in P. trichocarpa, which is known to have a slower evolution rate (Tuskan et al., 2006). We determined the evolution rate of the Ψs by estimating the Ks, nonsynonymous substitution rate (Ka), and Ka:Ks ratio between Ψs and their FPs (Figure 2C). In general, the majority of the Ψ–FP pairs had Ka:Ks ratios that were much greater than that of functional WGD (FG–FG) pairs. The median Ka:Ks ratio for FG–FG pairs was <0.40, representing selection on FGs. However, large differences in Ka and Ks for both Ψ–FP and FG–FG were detected across the seven species. We detected lower Ks values for soybean FG–FG pairs compared with other species (Wilcox one-tailed test, P < 0.05), which also suggested a recent WGD event. The sole exception to this was P. trichocarpa, which is known to have a slow molecular clock due to long generation times (Tuskan et al., 2006). Also, the large variation of FG–FG Ks values in P. trichocarpa may indicate divergent selection after WGD (Tuskan et al., 2006). The lowest median Ka value was detected in M. truncatula and the highest were detected in P. trichocarpa.
Asymmetric Elimination Rate of Ancient Full-Length Ψs
Next, we inferred the age of the Ψs by examining their sequence similarity to their FPs. We observed that most species show a stepwise increase in the number of Ψs at similar time points and a stepwise decrease after that time point (Figure 2D; Supplemental Figure 4). By contrast, in B. distachyon, we found a stepwise decrease at most time points. Since Ψs are expected to evolve neutrally (Zou et al., 2009), we examined three type of disablements: insertions, deletions, and stop codons in the Ψs. The average number of deletions was lower than the number of insertions in all species except M. truncatula (Supplemental Figure 5). Of the three kinds of disablement, we observed a higher density of stop codons in P. trichocarpa, M. truncatula, and soybean and a higher density of insertions in Arabidopsis, B. distachyon rice, and S. bicolor.
Considering that Ψs evolve neutrally, the strength of selection at all sites of the ancient full-length Ψ is expected to be identical. When we examined where the Ψ fragments overlap with their FPs, we did, however, observe an asymmetric elimination rate of the ancient full-length Ψ, with peaks of higher retention at both ends (Supplemental Figure 6). When analyzing data simulating random loss, we observed a much more uniform distribution of sequence elimination (Supplemental Figure 7). Our results thus suggest that the 5′ and 3′ ends were under stronger selection than the middle of the ancient Ψs, resulting in small Ψ fragment peaks located at each end.
Dynamic Repertoire of Ψs in Different Species
We compared the distribution pattern of Ψs with their FPs (Figure 2E; Supplemental Figure 8) and found that on average only 23.3% of genes had Ψ counterparts, resulting in a highly uneven distribution of Ψs per gene. By investigating the distribution of paralogs per FP across all seven genomes, we found little overlap between the genes with many paralogs and those with many Ψ counterparts (Figure 2E; Supplemental Figure 8). Interestingly, among all seven species, we found numerous types of FPs that were enriched in Ψs and depleted in paralogs and vice versa. We further assessed the overrepresented or underrepresented function pfam domains of Ψs by examining the annotations of their FPs. In general, defense domain families (leucine-rich repeat, NB-ARC [for nucleotide binding adaptor shared by APAF-1, R proteins, and CED-4]) had significantly overrepresented numbers of Ψs, whereas transcription factor–associated domains were underrepresented (Figure 3). Several domains related to secondary metabolism were overrepresented in Ψs of P. trichocarpa, B. distachyon, M. truncatula, and G. max. The top Ψ domain family in P. trichocarpa was wound-induced protein WI12 (Supplemental Data Set 8), possibly reflecting the family’s rapid evolution (Ma et al., 2013). Interestingly, M. truncatula, soybean, S. bicolor, Arabidopsis, and rice shared reverse transcriptase as their dominant domain, an indication of the activity of retrotransposons.
Figure 3.

Representative Overrepresented and Underrepresented Pfam Domains of Ψs in Seven Plant Species.
Graph shows overrepresented (top) and underrepresented (bottom) Pfam domains, respectively. Colors represent different gene families: orange, defense-related genes; purple, enzymes involved in secondary metabolism; green, kinase; and yellow, transcription factor–associated domains. The degree of shading represents the significance in the Fisher’s exact test (P-value; red, overrepresentation; blue, underrepresentation).
To directly compare Ψs from different species and identify core and specific families shared by all species, we grouped the Ψs into 38,278 families according to the similarity of their FPs (Supplemental Data Set 9). This method detected only 43 core Ψ families (>0.1%) across the seven species, compared with >22% of protein-coding genes and >6% of small RNA primary transcripts (Supplemental Figure 9). Since the two closest species examined were rice and B. distachyon, and any other two species were separated by at least 48 Myr of parallel evolution, we identified only 543 common Ψ families (Supplemental Figure 10). Some of the core Ψ families such as defense genes (leucine-rich repeat) were tandem duplicates, suggesting that these domain families are experiencing repeated gene gain and loss and therefore have higher chances of being present across species.
Recombination Contributes to Ψ Elimination
The majority of Ψs are under no selective constraint and are free to accumulate non-gene-like features such as frameshifts and stop codons. Therefore, we wondered whether recombination rates would affect the pattern of Ψ distribution. Deleterious mutations accumulate more easily in regions with suppressed recombination, such as pericentromeric regions, due to Hill–Robertson effects. We therefore expect to observe an enrichment of Ψs in regions of low recombination and near the pericentromeric regions. As expected, we found that Ψs were relatively enriched in the pericentromeric regions in genomes for which centromere positions were available. In soybean, rice, and Arabidopsis, pericentromeric regions were often associated with low recombination rates (Supplemental Table 1; Fisher’s exact test, P < 0.05). One striking feature of the soybean genome is that 57% of the genomic sequence occurs in pericentromeric regions (Du et al., 2012). Examination of recombination rate and Ψ density in the soybean genome revealed a significant negative correlation (P < 0.03; Pearson’s correlation less than –0.32; Figure 4A), with a negative Spearman’s r in the 0.32 to 0.73 range.
Figure 4.

Recombination Rate Shapes the Pattern of the Elimination of Ψs.
(A) Pearson correlation between recombination rate and the frequency of Ψs in the G. max genome. Each chromosome is divided into 1-Mb bins. The recombination rates in each bin are indicated above the x axis, and the frequency of Ψs is indicated below the x axis. GR, genome recombination.
(B) Comparison of evolution rates of Ψs in chromosomal arms and pericentromeric regions. The statistical analysis was conducted between each set of Ψs by Wilcoxon one-tailed test. An “a” above each column indicates P < 0.001 and “b” indicates P < 0.05.
Several studies have reported asymmetric evolution of protein-coding genes between high and low recombination regions (Hamblin and Aquadro, 1996; Du et al., 2012). Our study extended these analyses to compare the evolution between Ψs and their FPs within various genomic features. We began by aligning the annotated Ψs in the soybean genome to their respective FPs using an empirical codon model and removing low confidence Ψ–FP pairs. Next, the Ka and Ks of each Ψ–FP pair was calculated (Supplemental Data Set 10). We observed a significantly higher Ka for the Ψs in chromosomal arms and FPs in pericentromeric regions (P < 0.001; Figure 4B). By contrast, no significant difference in Ka was observed between Ψ–FP pairs when both were located within chromosomal arms or pericentromeric regions (Figure 4B), suggesting that Ψs in both regions have experienced similar levels of selective constraints. This suggests a higher mutation rate for Ψs in pericentromeric regions, although the median Ka:Ks ratio for Ψs in pericentromeric regions was significantly higher than that of Ψs in chromosome arms (P < 0.05; Figure 4B). Alternatively, if there are differences in age in different genomic compartments, the pace of evolution could be the same across the genome, but in some regions, such as pericentromeric regions, Ψs are simply retained longer, thus accumulating more mutations. Similarly, for FPs in pericentromeric regions, the corresponding Ψs in pericentromeric regions displayed significantly higher Ks values. Overall, based on the data, our findings suggest that genome recombination rates may be an essential contributor to the Ψs elimination.
Cis-Regulatory Elements Are Enriched in the Proximal Upstream Regions of Ψs
Although many Ψs appear to have a high turnover rate and do not encode proteins, some may still produce RNA. To examine the expression of Ψs, we reanalyzed the RNA sequencing (RNA-seq) data from six species and acquired strand-specific RNA-seq data for Populus under four abiotic stress treatments. In each species, we used RNA-seq reads from at least three samples (Supplemental Table 2), and all libraries were prepared with poly(A)-selected RNA. Expression was detected for 75.5% (on average) of the protein-coding genes but for only 32.5% (on average) of the Ψs (significantly fewer by Fisher’s exact test, P < 2e−16). In Arabidopsis, 0.29 to 0.44 of Ψs were expressed in each sample, and in P. trichocarpa 0.02 to 0.11 of Ψs were expressed. Across gene expression profiles, the median expression level of Ψs was significantly lower than that of protein-coding genes in seven species (Wilcox test, P < 2e−16; Figure 5A). We also found that some Ψs showed highly tissue-specific expression (Figure 5C), with the highest median tissue specificity found in M. truncatula. The lowest specificity was detected in B. distachyon, which may be due to its small sample size.
Figure 5.

Enrichment of Cis-Regulatory Elements in the Proximal Upstream Regions of Ψs.
(A) Comparison the maximum expression for protein-coding genes and Ψs.
(B) Pseudo-protein identity, Ka, and Ks and the ratio between Ka and Ks of Ψ–FP pairs.
(C) Tissue specificity of protein-coding genes and Ψs.
(D) Expression Pearson correlation among different gene sets. Genome duplicates were obtained from the Plant Genome Duplication Database (http://chibba.agtec.uga.edu/duplication/). Error bars indicate 95% confidence intervals generated by 1000 bootstrap replicates.
(E) Frequency of in silico–predicted binding sites for different proximal regions (2 kb upstream) of gene set, including genes, old Ψs (pseudo-protein identity < 0.8), Ψs (total Ψ set), young Ψs (pseudo-protein identity ≥ 0.8), and random intergenic regions. Error bars indicate 95% confidence intervals generated by 1000 bootstrap replicates.
(F) Percentage of shared transcription of Ψ families between species pairs with varying divergence times: 47 Myr, B. distachyon versus O. sativa; 48 Myr, S. bicolor versus O. sativa; 52 Myr, G. max versus M. truncatula; 108 Myr, P. trichocarpa versus A. thaliana; and 149 Myr, B. distachyon versus P. trichocarpa.
A detailed analysis revealed that the Ψs with detectable expression (expressed Ψs) tended to have a significantly higher sequence identity to their FPs and a significantly lower Ka, Ks, and Ka:Ks ratio compared with Ψs without detectable expression (nonexpressed Ψs) in the seven species (Wilcox test, P ≤ 0.015; Figure 5B; Supplemental Figure 11). This suggests that the expressed Ψs may be derived from relatively recent duplication events. One possible explanation is that the ancient parental cis-regulatory elements of the expressed Ψs have not completely degenerated. In this case, the expression of the Ψs should be highly associated with the expression of their FPs. As expected, in all seven species, Spearman’s correlation coefficient for Ψ–FP pairs was 0.19, on average, a value that is higher than that of randomly selected gene pairs or Ψ–Ψ pairs but lower than that observed for pairs of WGDs (0.29; Figure 5D).
The low expression levels and high tissue specificity of Ψs raise the question of whether cis-regulatory elements are enriched in the proximal upstream regions (i.e., their promoters) of Ψs. To test this hypothesis, we analyzed the frequency of transcription factor binding sites (TFBSs). Using a genome-wide set of TFBSs predicted in silico, we found that proximal upstream regions of Ψs were more frequently associated with TFBSs compared with random intergenic regions (Figure 5E).
Many transcriptional units are associated with chromatin-modifying complexes, and their expression is affected by histone modifications. We collected published chromatin immunoprecipitation sequencing (ChIP-seq) data profiling three kinds of histone marks in Arabidopsis (Jin et al., 2017) and rice (He et al., 2010), including two positive marks, acetylated histone 3 lysine 9 and trimethylated histone 3 lysine, and one negative mark, trimethylated histone 3 lysine 27. We reanalyzed the data and compared the histone modification marks within Ψs and published lncRNA loci by genomic position. In total, ∼64.9% of Arabidopsis and 33.1% of rice Ψ proximal upstream regions were associated with either positive or negative histone modification peaks in selected samples, which is higher than the association with histone peaks of lncRNAs (Supplemental Table 3). Furthermore, the Populus ChIP-seq data set for transcription factors (including members of class I KNOX, class III HD ZIP, BEL1-like families; Liu et al., 2015) showed that 6.1% of the peaks were associated with Ψ proximal upstream regions. Consistent with this, the frequency of DNase I hypersensitive (DH) peaks, also an essential indicator of cis-regulatory elements (Zhang et al., 2012), was significantly higher than randomly intergenic regions (Supplemental Figure 12). Taken together, these results suggest that cis-regulatory elements are enriched in the proximal upstream regions of Ψs.
We next assessed the evolutionary conservation of Ψ expression patterns. To this end, we first estimated the presence of shared transcriptional activities across species and found that Ψ transcription evolves rapidly. Only ∼52.1% of expressed rice Ψ families were also expressed in B. distachyon, and only ∼52.4% of expressed soybean Ψ families were expressed in M. truncatula (Figure 5F). However, more than 74% of the protein-coding genes from all seven plant species showed conserved expression (Figure 5F). Comparisons of the transcript ratios highlighted this discrepancy in conservation of expression among Ψs and protein-coding genes (Supplemental Figure 13).
Most homologous Ψs that are conserved in syntenic blocks between Arabidopsis and P. trichocarpawere found to be divergently transcribed (Supplemental Data Set 11). For example, Chr1|20148959-20149332 from Arabidopsis and Chr01|13707000-13707886 from P. trichocarpaare syntenic sequences that are conserved between the two species, yet their transcription is not conserved. Chr1|20148959-20149332 is expressed in four tissues of Arabidopsis but is not expressed in P. trichocarpa (Supplemental Figure 14). Overall, these results indicate that rapid transcriptional evolution is a genuine feature of Ψs.
Noncoding RNA Genes Are Associated with mRNA Genes and Ψs
To explore the contribution of Ψs to the makeup and regulation of noncoding RNAs, we compiled a catalog of lncRNA species by combining published and unpublished lncRNA data for species where extensive lncRNA data sets are available (Supplemental Table 4). For the analysis, each lncRNA in the initial pool was required to have a 5′ end that originated from a genomic site and to be at least 200 nucleotides long. Furthermore, to exclude possible association with TEs, we excluded lncRNAs or their proximal regions (2 kb upstream and downstream) that overlap with TEs by 10 bp or more (Supplemental Data Sets 12 to 16). We refer to the remaining lncRNAs as non–TE lncRNAs.
Previous studies showed that the observed lncRNA species vary due to differences in genome sequence and RNA-seq data quantity and quality, as well as differences in the diversity of samples used for sequencing in different species (Necsulea et al., 2014). Inspection of the position of origin for non–TE lncRNAs revealed that the majority were located closer to genes than to Ψs (70.0% on average) and the minority were located closer to Ψs than to genes (Figure 6A).
Figure 6.

Many lncRNAs Are Transcribed from the Proximal Upstream Regions of Protein-Coding Genes and Ψs.
(A) Summary of various types and numbers of lncRNA loci in five species, including P. trichocarpa, Arabidopsis, M. truncatula, O. sativa (O. sativa subsp japonica), and S. bicolor.
(B) Example of lncRNA locus whose 5′ end occurs within 2 kb of the 5′ end of a Ψ (proximal upstream region–associated lncRNA). The x axis represents the linear sequence of genomic DNA, and the y axis represents the total number of RNA-seq mapped reads in root of Arabidopsis. RNA-seq reads that map to lncRNA (blue) and Ψ (red) of genomic DNA are shown separately. The scale is indicated in the top right.
(C) to (H) Subcellular localization of four constructs in tobacco leaf epidermal cells by Agrobacterium-mediated transient expression. The fluorescence signal of the YFP under the putative promoter regions of Chr5|5286482-5287564 (C), Chr2|852936-853952 (D), Chr1|26153967-26154985 (E), and Chr3|16675465-16676590 (F). Fluorescence signal of the negative control, YFP under no promoter (G), and the positive control, fABD2 with YFP fusion under a Cauliflower mosaic virus 35S promoter (H).
(I) Schematic diagram of the constructs. Bar = 10 μm.
Most of the non–TE lncRNAs that were closer to mRNA genes were found to originate within a 2-kb region surrounding the transcriptional start sites of protein-coding genes (ranging from 52.1% in P. trichocarpa to 70.0% in Arabidopsis), while a smaller fraction (37.9% on average) were more distant (>2 kb) from protein-coding genes (Figure 6A). Further examination of the non–TE lncRNAs that were closer to transcriptional start sites of genes (<2 kb) revealed that the majority (62.0% on average; 31.1% in P. trichocarpa and 95.3% in Arabidopsis) were associated with the promoters of protein-coding genes, suggesting that a surprising fraction of lncRNAs originate from the promoters of mRNA genes. A large number of non–TE lncRNA species were found closer to the 5′ end of Ψs than to protein-coding genes (21.1.4% of the total non–TE lncRNA pool on average). A visual inspection of individual genes suggested that many of the lncRNA species are transcribed from the proximal upstream regions of Ψs. For example, a 256-bp lncRNA transcript of Arabidopsis originated ∼1082 bp upstream of the Ψ locus Chr5:5286482-5286293 and was divergently transcribed in relation to the Ψ (Figure 6B). An analysis of the entire non–TE lncRNA population revealed that 12.5 (Arabidopsis) to 22.1% (M. truncatula) of the total non–TE lncRNA pool was associated with proximal upstream regions of Ψs (Figure 6A). For the non–TE lncRNAs that were closer to the Ψs (<2 kb), the majority were associated with the proximal regions of Ψs in all five species, ranging from the smallest proportion in Arabidopsis (71.8%) to the largest in rice (83.5%).
We randomly selected 30 proximal upstream regions that were associated with non–TE lncRNA loci (found in antisense of Ψs) for a transient expression experiment (Supplemental Figure 15; Supplemental Data Sets 17 and 18). The selected sequences were synthesized and cloned to the binary vector 3302Y3 by replacing the cauliflower mosaic virus 35S promoter. Twenty-one yellow fluorescent protein (YFP) signals out of 30 localized to the cell membrane, trichome, and nucleus (Figures 6C to 6F; Supplemental Figure 16). Analysis of the miRNA species suggested that several non–TE miRNAs could also be transcribed from the proximal upstream regions of Ψs (Figure 7). Altogether, these results suggest that a substantial fraction of lncRNAs/miRNAs are transcribed from proximal upstream regions of Ψs.
Figure 7.

Many Non–TE miRNAs Are Transcribed from the Proximal Upstream Regions of Protein-Coding Genes and Ψs. Summary of various types and numbers of non–TE miRNA loci in Seven Plant Species.
DISCUSSION
Ψs Have a High Turnover Rate in Plants
In this study, we performed a systematic investigation of Ψs using seven representative plant species with highly accurate expression profiles, thereby providing an important resource for future studies. Our results suggest that Ψs are highly lineage specific and have a high turnover rate, indicating that a large number of plant Ψs appear to be evolving under relaxed selective constraints and therefore tend to be rapidly eliminated during pseudogenization. Indeed, we detected only 43 core Ψ families (>0.1%) across the seven species, which is substantially lower than the numbers for protein-coding genes and miRNAs. The distributions of Ψs across the genome have been documented in human, worm, and fly genomes (Sisu et al., 2014). However, these analyses do not provide consistent results, and the accumulation, elimination, and distribution of Ψs in local genomic regions have not been comprehensively investigated. Here, we have investigated the distribution of Ψs in three plant species and determined that they are uniformly enriched in the centromeric regions.
We further analyzed the genomic recombination data in soybean, and the results show that the distribution of Ψs is significantly negatively correlated with the local genomic recombination rate, indicating that Ψs are organized along recombinational gradients on chromosomes. Recombination is typically initiated by double-stranded breaks that trigger strand exchange (Schuermann et al., 2005). An increasing body of evidence indicates that recombination plays an essential role in genome evolution by generating mutations (Rattray and Strathern, 2003), increasing microsatellite instability, and contributing to gross chromosomal rearrangements (Pearson et al., 2005). In regions of high recombination, the neutral mutation and rearrangement rates increase, which may help explain the greater rate of loss of Ψs in these regions. Ψs are thus expected to accumulate in regions of low recombination rates. This study provides an in-depth analysis of the distribution and rates of elimination of Ψs in relation to genomic recombination rates in plant genomes. All these findings suggest that genomic recombination is an essential contributor to Ψ elimination.
Using the 90th percentile of the distribution of intron probe intensities as a threshold, multiple Ψs are likely expressed (32.5% on average) but at a lower level compared with functional genes. Ψ expression also tends to be spatially and temporally more restricted than that of functional genes. Additionally, expressed Ψs appear to have lower Ka:Ks ratios and are more complete, indicating that they are derived from relatively recent duplication events. Indeed, the protein sequence identity between expressed Ψs and their FPs is significantly higher than that of other Ψ–FP pairs. The Spearman’s correlation coefficient for pairs of Ψs and their respective FPs is lower than that between WGDs, and at the promoter level, this dynamic includes a loss and gain of TFBSs. Together, the fast-evolving expression patterns, the highly dynamic Ψ families in distinct lineages, and the multiple mechanisms affecting the turnover of Ψs suggest that both sequences and regulatory regions of Ψs can evolve extremely rapidly between closely related plant species.
Intergenic Noncoding RNAs Are Derived from Divergent Ψs
Expressed Ψs are unlikely to only represent transcriptional noise, as many Ψs exhibit specific expression patterns and are associated with abiotic stress (Zou et al., 2009). In our study, a fraction of Ψs are actively transcribed (32.5% on average), and proximal upstream regions of these Ψs are enriched in TFBSs, suggesting the proximal upstream regions of many Ψs are still active and some Ψs may still be active as RNA genes. We further observed extensive divergence of expression patterns between Ψ–FP pairs, suggesting that a vast majority of Ψ–FP pairs have diverged in expression through random degeneration in their cis-regulatory regions. Nonetheless, our collections of expression profiles are by no means complete, and more precise expression data will provide more evidence regarding Ψ expression.
Using the available oligo(dT)-based RNA purification for RNA-seq data led us to focus on polyadenylated Ψs, which are more stable and abundant than nonpolyadenylated transcripts. However, this data set is missing some types of Ψ transcripts. Therefore, the numbers of expressed Ψs are underestimated to some extent. Recent high-throughput efforts to characterize the transcriptomes of eukaryotes have uncovered thousands of lncRNAs (Liu et al., 2012; Qi et al., 2013; Hezroni et al., 2015). lncRNA catalogs are far from exhaustive and also contain false positives (Kapusta et al., 2013), indicating that the complexity of lncRNAs may exceed our current estimates.
The transcriptional control and origin of lncRNAs have been the subject of intense study; yet, most of these investigations have focused on protein-coding genes or transposons (Kapusta et al., 2013; Sigova et al., 2013). A large fraction of lncRNAs are predicted to originate from divergent transcription from promoters of active protein-coding genes based on high-throughput RNA-seq analysis (Sigova et al., 2013). Using strand-specific RNA-seq, we found that many intergenic non–TE lncRNAs and non–TE miRNAs in plant species are divergently transcribed at the proximal upstream regions of Ψs in all seven species. Only a few studies have described several non–TE lncRNAs that originated from Ψs and have essential roles in development and disease (Milligan and Lipovich, 2015). Our study found that on average, 20.2% of non–TE lncRNAs are located within the 2-kb proximal upstream regions of Ψs, and only a minority of these overlap with the Ψ body.
The complexity of plant transcriptomes is further demonstrated by the frequent overlap between different transcript categories or between lncRNAs and other genomic elements. For example, lncRNAs can act as miRNA precursors, or function as miRNA sponges (Tian et al., 2016). The evidence described here reveals that many intergenic non–TE lncRNAs are derived from transcription at the proximal upstream regions of Ψs and provides insight into the evolution of novel regulatory modules.
One implication of this finding is that the transcription of lncRNAs undergoes evolutionary dynamics. Large-scale investigations of these data sets have only recently begun and should provide a rich source of information for additional studies into the functions of these noncoding RNA species and the control of their expression. Thus, the strong association of noncoding RNAs with Ψ proximal upstream regions is probably a common characteristic of plant lncRNA repertoires that distinguish them from those that are derived from genes and transposons. This provides another important mechanism for the origin of noncoding RNAs. Future investigations of lncRNA–Ψ pairs and the lncRNAs described here could provide insights into the contributions of Ψs to transcriptomic complexity.
Do Novel Regulatory Sequences Originate De Novo or from Preexisting Regulatory Sequences?
Understanding the genomic origins of transcriptional novelties can provide insight into the construction of the regulatory system and thus into evolutionary biology. Many noncoding RNA species, such as lncRNAs and miRNAs, are transcribed from intergenic regions within the genome (Xie et al., 2017). The poorly conserved profiles and spatio-temporal expression patterns of lncRNAs raise the following question: What are the mechanisms of sequence evolution leading to the rapid formation and loss of regulatory sites?
New patterns of gene expression could be generated by two main mechanisms: de novo evolution and rewriting of the preexisting regulatory information. The second mechanism may fall into three general categories: transposition, promoter switching, and co-option (Rebeiz et al., 2011). Gene regulation is controlled by coordinated binding of transcription factors at the TFBSs in the promoters of genes. In many species, TFBSs tend to occur as homotypic or heterotypic clusters, possessing complicated regulatory motifs (Gupta and Liu, 2005). The stretches of these intergenic regions in the genome often harbor sequences that contain various TFBSs, and such regions could acquire a series of random point mutations, small indels, or TE transfers that subsequently generate functional regulatory sequences. The high frequency of TFBSs at promoters and the expression patterns of lncRNAs suggest that their transcription is actively regulated overall (Necsulea et al., 2014). However, the extent to which regulatory elements occur de novo is unknown, and we are unaware of any empirical examples of their occurrence.
Compared with the de novo mechanism, generating new expression patterns that are founded on preexisting regulatory sequences seems to be more plausible, based on our findings. This mode of regulatory system evolution is supported by several lines of evidence. TEs are currently thought to provide a common route by which regulatory DNA sequences evolve (Hezroni et al., 2015). In the case of pesticide resistance in fruitfly, gene expression is driven by a preexisting TFBS in TE sequences (Daborn et al., 2002). In addition, transcriptional data from embryonic stem cells show that mRNA genes could share regulatory activity with their adjacent lncRNAs (Sigova et al., 2013). Statistical analysis of 346 cis-regulatory modules in fruitfly show that local sequence duplication is an essential mechanism that transports and produces cis-regulatory information (Nourmohammad and Lässig, 2011). In this study, we found that from 12.5 to 22.1% of the total non–TE lncRNA pool is derived from proximal upstream regions of Ψs. Further analyses show that the proximal upstream regions of Ψs are more enriched in TFBSs and DH peaks than are random intergenic regions. Consistent with this, a number of plant Ψs are likely expressed and show a low expression correlation with their FPs. Studies also indicate that some proximal upstream regions of Ψs are highly active and have the potential to contribute to novel transcriptional systems (Scarola et al., 2015; Ma et al., 2016). Thus, it appears that for lncRNAs and miRNAs, evolution rarely produces novelties from scratch but works on the promiscuous activities that existed previously, and this may reflect a general mechanism whereby new transcripts evolve.
METHODS
Data Set for Populus trichocarpa
Populus trichocarpa plants were grown in a greenhouse under a 16-h-light/8-h-dark photoperiod, with light provided by cool white fluorescent lights (at 250 μmol m−2 s−1 photosynthetic photon flux density [PPFD]). For stress treatments, 12 plants obtained from a single genotype were used for chilling stress (three plants; at 4°C for 6 h, 250 μmol m−2 s−1 PPFD), heat stress (three plants; at 42°C for 6 h, 250 μmol m−2 s−1 PPFD), exposure to 150 mM NaCl, 30% polyethylene glycol 6000 (three plants; for 6 h), and drought stress (three plants; at 25°C, 250 μmol m−2 s−1 PPFD, soil moisture content 15% to 20%). Leaves were collected from P. trichocarpa for RNA extraction with different treatments (three biological replicates per treatment). For expression analyses of genes and Ψs in different species, filtered transcriptome reads were mapped to the corresponding reference genome using hisat2 (Kim et al., 2015), with parameters -q -x -S -p. Gene and Ψ quantification was determined using StringTie (Pertea et al., 2016), with parameter -e -G. To measure the expression specificity of Ψs, the specificity score (Liao and Zhang, 2006) was computed.
TE Annotation
TE annotations used in this study were obtained from the outputs of RM 4.0.6 software (Chen, 2009) with the combined database (Dfam_Consensus-20170127, RepBase-20170127; species parameter: Arabidopsis thaliana [Arabidopsis]; P. trichocarpa: Populus; G. max: Glycine; M. truncatula: Medicago; O. sativa: Oryza; B. distachyon: Brachypodium; S. bicolor: Panicoideae). These RM outputs were filtered to remove non–TE elements (satellites, simple repeats, low complexity, rRNA).
Identification of Ψs in the Seven Taxa
The selected taxa including rosids (Arabidopsis, P. trichocarpa, soybean [Glycine max], and M. truncatula) and monocots (rice [Oryza sativa], B. distachyon, and S. bicolor) were used for Ψ identification. The genome information is provided in Supplemental Table 5. The overall pipeline for identification is outlined in Figure 1 and is generally based on the previous PseudoPipe workflow (Zhang et al., 2006; Zou et al., 2009), with modifications. Generally, the pipeline consisted of five major steps: (1) identify intergenic regions (masked genic and transposon regions) with sequence similarity to known proteins using exonerate; (2) quality control, identity ≥ 20%, match length ≥ 30 amino acids, match length ≥ 5% of the query sequence, and only the best match is retained; (3) link homologous segments into contigs (set I Ψs); (4) realign using tfasty to identify features that disrupt contiguous protein sequences; and (5) distinguish WGD-derived Ψs and set II Ψs.
In the first step, RM-masked genomes were used to mask the genic regions (annotated transcription unit in the genome annotation) and generate a file of intergenic regions. Thus, our following steps of Ψ identification focused on intergenic non–TE regions.
The second step in the annotation pipeline was to identify all regions in the genome that share sequence similarity with any known protein, using exonerate (Slater and Birney, 2005) with parameters --model protein2genome --showquerygff no --showtargetgff yes --maxintron 5000 --showvulgar yes --ryo \"%ti\\t%qi\\t%tS\\t%qS\\t%tl\\t%ql\\t%tab\\t%tae\\t%tal\\t%qab\\t%qae\\t%qal\\t%pi\\n\". In addition to the filters already included in PseudoPipe (overlap > 30 bp between a hit and a functional gene), we did not accept alignments with E-value >1e−5, identity < 20%, match length < 30 amino acids, and match length (proportion aligned) < 5%. Then, the best match of alignment hits was selected in places where a given chromosomal segment had multiple hits.
The third step was to link Ψ contigs based on the distance between the hits on the chromosome (Gc) and the distance on the query protein (Gq). In our workflow, these gaps Gc could arise from low complexity or very decayed regions of the Ψ that were discarded by exonerate. We set this distance to 50 bp.
In the fourth step, the set I Ψs were realigned using a more accurate alignment program, tfasty34, with parameters “-A -m 3 ‘q”. Accurate sequence similarity and annotate positions of disablements (frame shifts and stop codons) as well as insertions and deletions were generated in this step.
In the final step, WGD-derived Ψs were detected using MCScanX (Wang et al., 2012) based on the DAGchainer algorithm (Haas et al., 2004) with parameters -k 50 -g -1 -s 5 -m 25, and blocks with minimum of five gene pairs were selected. We used protein pairs from each organism with a BLASTP E-value of <1e−5 and Ψ–FP pairs as the input data when running MCScanX. Pairs of Ψ–FPs in the syntenic block were considered WGD derived.
Ψ Family Identification
Parent protein-coding gene information was downloaded from the Ensembl database (http://plants.ensembl.org/index.html) and Michigan State University Rice Genome Annotation Project and Phytozome (http://phytozome.jgi.doe.gov/pz/portal.html; Supplemental Table 5). First, pairwise sequence similarities between all input protein sequences from selected species were calculated using BLASTP with an E-value cutoff of 1e−5. Markov clustering of the resulting similarity matrix was used to define the gene family, using an inflation value of 1.5. The Orthomcl clustering results list the gene family members from all plant species. Second, the corresponding Ψs can be grouped into Orthomcl families according to their closest FPs (Supplemental Data Set 9).
We first constructed a phylogenetic tree with the seven species studied. The maximum likelihood phylogenetic tree was generated by RAxML (Stamatakis, 2014) using the PROTGAMMALGF model with 100 bootstrap replicates based on 124 single-copy proteins (Supplemental Data Set 19) that were identified by OrthoMCL (Li et al., 2003). Branch lengths reflect evolutionary divergence times in millions of years as inferred from TimeTree (http://www.timetree.org/). TimeTree assembles the public data from thousands of published studies into a searchable tree of life scaled to time. The median molecule time estimates were selected from this study.
Expression Conservation Analyses
For the qualitative assessment of transcription conservation of Ψs, we analyzed the expression ratio of the total shared Ψs families between the two species across different divergence times: 47 Myr, O. sativa versus B. distachyon; 48 Myr, S. bicolor versus rice; 52 Myr, soybean versus M. truncatula; 108 Myr, P. trichocarpa versus Arabidopsis; and 149 Myr, B. distachyon versus P. trichocarpa . In the analysis, one Ψ family was defined to be expressed if at least one member of the family was expressed.
To study the coexpression patterns of Ψ–FP, WGDs, random gene pairs, and random Ψ pairs, Spearman correlations of expression levels (fragments per kilobase of exon per million reads mapped values) across different samples were calculated.
Measurement of Expression Specificity
To measure the expression specificity of Ψs, the specificity score (Liao and Zhang, 2006) was computed. We let aij be the average expression of gene i in tissue/treatment j. Then, the expression specificity of gene i was given by
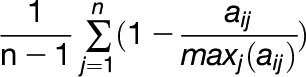 |
where n is the number of tissues or treatments. Thus, if a gene was expressed in only one tissue the score was 1, and if the average expression of a gene was the same in all tissues the score was 0.
Frequency of In Silico–Predicted TFBSs
We used a genome-wide set of transcription factor binding sites of the seven species that were manually curated, nonredundant, and high-quality transcription factor binding motifs derived from experiments (Plant Transcription Factor Database; http://planttfdb.cbi.pku.edu.cn/download.php). Predictions were performed using MEME package (fimo --oc. --verbosity 1 --thresh 1.0E−5). Average frequency of in silico–predicted binding sites was calculated for different categories of proximal regions (2 kb upstream) of genes, including genes, old Ψs (pseudo-protein identity < 0.8), Ψs (total Ψ set), young Ψs (pseudo-protein identity ≥ 0.8), and in random intergenic regions. Frequency of TFBSs refers to the average number of binding sites per fraction of promoters or regions. Error bars indicate 95% confidence intervals generated by 1000 bootstrap replicates.
We examined proximal upstream regions within 2 kb of the annotated start sites or 5ʹ end for all Ψs, genes, and lncRNAs. For analyzing the upstream sequence activity, we used the acetylated histone 3 lysine 9 and trimethylated histone 3 lysine, and one negative mark, trimethylated histone 3 lysine 27, in Arabidopsis and rice. We also analyzed Populus ChIP-seq data for transcription factors (Liu et al., 2015). Regions were labeled as active if ChIP-seq peaks overlapped with them. The frequency of the DH peaks in proximal upstream regions was reanalyzed. The DNase I hypersensitive data of Arabidopsis, B. distachyon, and rice were downloaded from PlantDHS (http://plantdhs.org/). As a control, we also analyzed the frequency DNase I hypersensitive peaks for 2000 randomly generated intergenic regions.
The Evolution Analyses of Ψs
WGD pairs for each organism were detected using MCScanX (Wang et al., 2012), based on the DAGchainer algorithm (Haas et al., 2004). We used protein pairs from each organism with a BLASTP E-value of <1e−5 and blocks with a minimum of five gene pairs were selected. Ψ–FP pairs in the syntenic block were considered WGD derived. To evaluate the level of selective constraint on Ψs, we calculated the Ks and Ka between each Ψ and its parent gene in all selected plant species. First, the protein alignments of Ψ–FP were extracted from the pipeline output and regions representing gaps in any of the aligned sequences were removed. Then, the corresponding codon alignment was obtained on the basis of protein alignment using Python scripts. Second, the evolutionary rates were determined using the yn00 program in the PAML program package (Yang, 1997). Pairs with errors or pairs that were too divergent (Ks > 3) were excluded.
Differences in the dynamics of genome evolution make it difficult to directly estimate the age of Ψs. The Ψ ages were estimated using the sequence similarity to FPs as an indicator. Thus, older Ψs have a lower sequence similarity to their FPs. Three different types of Ψ disabling mutations (insertion, deletions, and stop codons) were extracted from the pipeline and the average defect density per kilobase was calculated for each plant species.
Pfam Domain Analyses of Ψs
We annotated all the Ψs according to their FPs in the Pfam database (Finn et al., 2014). Fisher’s exact test was used to test whether the annotated pfam domains were significantly overrepresented or underrepresented.
Genome Recombination Rate and Ψ Density
The soybean genome recombination rate was obtained from a previous study (Du et al., 2012). Each chromosome was subdivided into 1-Mb bins, and Ψ density and the recombination rate in each chromosome were used for analysis of potential correlation. The genome recombination-suppressed pericentromeric regions were defined based on the comparison of soybean physical and genetics maps as previously described.
Positions of Ψ Overlap with Their FPs
The study was based on the PseudoPipe output; this output provided the alignment position of the Ψ fragments and their FPs. The relative positions of the full-length parental genes were calculated by start/L and end/L, where L is the full-length of FPs, start is the alignment start position, and end is the alignment end position. Thus, the density of the Ψ fragments relative to the position of FPs was calculated. For the randomization test, 1000 randomly generated gene fractions from 100 genes of seven species were aligned to their full-length genes, and their positions were plotted. A different volume of simulated data was also generated in this test.
Association of Non–TE Noncoding RNA and Proximal Upstream Regions of Ψs
The positions of non–TE noncoding transcripts (lncRNAs and miRNAs) relative to the proximal upstream regions of Ψs (2-kb sequences preceding the 5ʹ end of each annotated Ψ sequence) were determined. The location of noncoding transcripts was divided into four categories: (1) proximal upstream region–associated lncRNA loci, (2) gene body–associated lncRNA loci, (3) tail-to-tail lncRNA loci, and (4) distant lncRNA loci. Their relative positions were determined using an in-house Python script. The data for the four categories are provided in Supplemental Data Sets 20 to 24.
For expression analyses of genes and Ψs in different species, filtered transcriptome reads were mapped to the corresponding reference genome using hisat2 (Kim et al., 2015), with parameters -q -x -S -p. Gene and Ψ quantification was conducted using StringTie (Pertea et al., 2016), with parameters -e -G.
Identification of lncRNA Catalogs
For prediction of lncRNAs in Populus, the clean reads were first aligned to the reference genome using hisat2 v2.0.5 (Pertea et al., 2016) with parameters -q -x -U -p --rna-strandness -S . The mapped reads were used to merge and assemble transcripts using samtools v1.3.1 sort function (Li et al., 2009) and cuffcompare package in Cufflinks v2.1.1 (Trapnell et al., 2012) with default parameters. The different sets of lncRNAs, including intergenic, TE-containing, sense, and natural antisense lncRNA transcripts, were identified using Evolinc-I (Nelson et al., 2017) by searching the corresponding repeat database.
Transient Expression in Nicotiana benthamiana
Arabidopsis plants used in this study were the wild-type Columbia ecotype, and genomic DNA of Arabidopsis was extracted for PCR. N. benthamiana plants were grown in a greenhouse at 25°C under long-day conditions (16-h-light/8-h-dark cycle). Four-week-old N. benthamiana plants were used for transient expression experiments. To test the activity of proximal upstream regions of Ψ associated with non–TE lncRNAs, 30 randomly selected proximal upstream regions of expressed Arabidopsis Ψs were synthesized to the binary vector 3302Y3 by replacing the cauliflower mosaic virus 35S promoter (GeneRay; Supplemental Data Set 18). Vector 3302Y3 without any promoter was used as a negative control, and a vector with a YFP-fABD2 fusion with the 35S promoter was used as positive control. Transient expression in N. benthamiana was performed as previously described (Sun et al., 2018). Microscopy analysis was performed for 2 d after infiltration. Fluorescence was observed with an SP5 confocal laser scanning microscope (Leica) and captured with a charge-coupled device camera.
Quantitative RT-PCR Validation of Expression Profiles
Quantitative RT-PCR was performed on a DNA Engine Opticon 2 machine (MJ Research) using the LightCycler FastStart DNA master SYBR Green I kit (Roche). The cDNA template for reactions was reverse transcribed using total RNA extracted from leaves with or without stress treatment. Poplar Actin was used as the internal control for gene expression measurements. The PCR program was as described previously (Zhang et al., 2011). The primers used were Chr1|20148959-20149332F (5′-GTTGTTGGTAACACGACCGC-3′) and Chr1|20148959-20149332R (5′-GTCCGCTCCCATGTTCAAGA-3′) for Arabidopsis and Chr01|13707000-13707886F (5′-TGAGTTTGCCACCACTGGG-3′) and Chr01|13707000-13707886R (5′-ACCTTTCCGGCAGATGGATT-3′) for Populus.
Accession Numbers
Raw data are available for download at the Beijing Institute of Genomics Data center Genome Sequence Archive under accession number CRA000471. Bioinformatics analysis pipelines, singularity images, recipe files, and clear instructions are available online at GitHub (https://github.com/bjfupoplar/PlantPseudo.git). Ψs identified using the PlantPseudo pipeline are reported in Supplemental Data Sets 1 to 7. Non–TE lncRNA data sets are reported in Supplemental Data Sets 12–16. Associations of non–TE lncRNA and Ψ/gene proximal regions are reported in Supplemental Data Set 20. Poplar Actin sequence is available under the accession number EF145577.
Supplemental Data
Supplemental Figure 1. Some Ψs near the cutoff are in syntenic positions between closely related species (P. trichocarpa and Arabidopsis) or in WGD blocks.
Supplemental Figure 2. Distribution of genes and the different types of Ψs across chromosomes.
Supplemental Figure 3. Distribution of Ks values between gene pairs on WGD blocks that contain Ψs.
Supplemental Figure 4. Distribution of Ψs in P. trichocarpa, Arabidopsis, B. distachyon, G. max, M. truncatula, O. sativa (O. sativa subsp japonica), and S. bicolor as a function of age (sequence similarity to parents).
Supplemental Figure 5. Distribution of disablements in Ψs as functions of type and Ψ age.
Supplemental Figure 6. Position of Ψ fragment overlaps with their FPs.
Supplemental Figure 7. Randomly generated gene fractions uniformly overlap with their FPs.
Supplemental Figure 8. Orthologs, paralogs, and families.
Supplemental Figure 9. Dynamic repertoire of Ψs in different species.
Supplemental Figure 10. Common Ψ families of the seven plant species. Seven plant species were used in the study.
Supplemental Figure 11. Recently created Ψs tend to have higher expression values.
Supplemental Figure 12. Frequency of DNase I hypersensitive peaks.
Supplemental Figure 13. Comparison of the transcript ratio between Ψs and protein-coding genes in seven species.
Supplemental Figure 14. Rapid transcriptional evolution of Ψs.
Supplemental Figure 15. Position of randomly selected proximal upstream regions associated with non–TE lncRNA loci.
Supplemental Figure 16. Positive transient expression assays of the other 16 proximal upstream sequences.
Supplemental Table 1. Comparison of the Ψs located on chromosome arms and centromeres.
Supplemental Table 2. RNA-seq data used in this study.
Supplemental Table 3. Association of histone marks with lncRNAs and Ψs.
Supplemental Table 4. lncRNA data used in this study.
Supplemental Table 5. Genomes used in this study.
Supplemental Data Set 1. Ψs identified in P. trichocarpa.
Supplemental Data Set 2. Ψs identified in Arabidopsis.
Supplemental Data Set 3. Ψs identified in B. distachyon.
Supplemental Data Set 4. Ψs identified in G. max.
Supplemental Data Set 5. Ψs identified in M. truncatula.
Supplemental Data Set 6. Ψs identified in O. sativa.
Supplemental Data Set 7. Ψs identified in S. bicolor.
Supplemental Data Set 8. Top 30 pfam domains of Ψs in seven species.
Supplemental Data Set 9. Ψ families shared by all species.
Supplemental Data Set 10. Values for Ka and Ks between Ψs and their functional WGDs.
Supplemental Data Set 11. Syntenic blocks containing Ψ between Arabidopsis and Populus.
Supplemental Data Set 12. Non–TE lncRNA data sets of P. trichocarpa.
Supplemental Data Set 13. Non–TE lncRNA data sets of A. thaliana.
Supplemental Data Set 14. Non–TE lncRNA data sets of B. distachyon.
Supplemental Data Set 15. Non–TE lncRNA data sets of M. truncatula.
Supplemental Data Set 16. Non–TE lncRNA data sets of O. sativa.
Supplemental Data Set 17. Thirty randomly selected proximal upstream regions associated with non–TE lncRNA loci.
Supplemental Data Set 18. Proximal upstream sequences used in the transient expression experiment.
Supplemental Data Set 19. Single-copy genes used in the phylogenetic analysis.
Supplemental Data Set 20. Association of non–TE lncRNA and Ψ/gene proximal regions in P. trichocarpa.
Supplemental Data Set 21. Association of non–TE lncRNA and Ψ/gene proximal regions in Arabidopsis.
Supplemental Data Set 22. Association of non–TE lncRNA and Ψ/gene proximal regions in B. distachyon.
Supplemental Data Set 23. Association of non–TE lncRNA and Ψ/gene proximal regions in M. truncatula.
Supplemental Data Set 24. Association of non–TE lncRNA and Ψ/gene proximal regions in O. sativa.
Acknowledgments
We thank Ronald R. Sederoff (North Carolina State University) for specific suggestions and detailed comments to improve the manuscript. This work was supported by the State “13.5” Key Research Program of China (2016YFD0600102), the Project of the National Natural Science Foundation of China (31600537 and 31670333), Young Elite Scientists Sponsorship Program by CAST (2018QNRC001), and the Program of Introducing Talents of Discipline to Universities (111 project, B13007).
Author Contributions
D.Z. designed the research. J.X. performed the research, analyzed the data, contributed new computational pipeline, and wrote the paper. X.L. performed the transient expression experiment. Y.L., X.L., Y.Z., and D.Z. revised the manuscript. B.L. and P.K.I. provided valuable suggestions to the manuscript. D.Z. obtained funding and is responsible for this article. All authors read and approved the manuscript.
Footnotes
Articles can be viewed without a subscription.
References
- Balasubramanian S., Zheng D., Liu Y.J., Fang G., Frankish A., Carriero N., Robilotto R., Cayting P., Gerstein M. (2009). Comparative analysis of processed ribosomal protein pseudogenes in four mammalian genomes. Genome Biol. 10: R2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N. (2009). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 25: 4.10.1-4.10.14 [DOI] [PubMed] [Google Scholar]
- Daborn P.J., et al. (2002). A single p450 allele associated with insecticide resistance in Drosophila. Science 297: 2253–2256. [DOI] [PubMed] [Google Scholar]
- Du J., Tian Z., Sui Y., Zhao M., Song Q., Cannon S.B., Cregan P., Ma J. (2012). Pericentromeric effects shape the patterns of divergence, retention, and expression of duplicated genes in the paleopolyploid soybean. Plant Cell 24: 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R.D., et al. (2014). Pfam: The protein families database. Nucleic Acids Res. 42: D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaut B.S., Wright S.I., Rizzon C., Dvorak J., Anderson L.K. (2007). Recombination: An underappreciated factor in the evolution of plant genomes. Nat. Rev. Genet. 8: 77–84. [DOI] [PubMed] [Google Scholar]
- Guo X., Zhang Z., Gerstein M.B., Zheng D. (2009). Small RNAs originated from pseudogenes: Cis- or trans-acting? PLOS Comput. Biol. 5: e1000449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M., Liu J.S. (2005). De novo cis-regulatory module elicitation for eukaryotic genomes. Proc. Natl. Acad. Sci. USA 102: 7079–7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas B.J., Delcher A.L., Wortman J.R., Salzberg S.L. (2004). DAGchainer: A tool for mining segmental genome duplications and synteny. Bioinformatics 20: 3643–3646. [DOI] [PubMed] [Google Scholar]
- Hamblin M.T., Aquadro C.F. (1996). High nucleotide sequence variation in a region of low recombination in Drosophila simulans is consistent with the background selection model. Mol. Biol. Evol. 13: 1133–1140. [DOI] [PubMed] [Google Scholar]
- He G., et al. (2010). Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell 22: 17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hezroni H., Koppstein D., Schwartz M.G., Avrutin A., Bartel D.P., Ulitsky I. (2015). Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Reports 11: 1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue J., Sato Y., Sinclair R., Tsukamoto K., Nishida M. (2015). Rapid genome reshaping by multiple-gene loss after whole-genome duplication in teleost fish suggested by mathematical modeling. Proc. Natl. Acad. Sci. USA 112: 14918–14923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J., Tian F., Yang D.C., Meng Y.Q., Kong L., Luo J., Gao G. (2017). PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 45: D1040–D1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapusta A., Kronenberg Z., Lynch V.J., Zhuo X., Ramsay L., Bourque G., Yandell M., Feschotte C. (2013). Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet. 9: e1003470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Langmead B., Salzberg S.L. (2015). HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12: 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. (2009). The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Stoeckert C.J. Jr., Roos D.S. (2003). OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 13: 2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao B.Y., Zhang J. (2006). Evolutionary conservation of expression profiles between human and mouse orthologous genes. Mol. Biol. Evol. 23: 530–540. [DOI] [PubMed] [Google Scholar]
- Liu J., Jung C., Xu J., Wang H., Deng S., Bernad L., Arenas-Huertero C., Chua N.H. (2012). Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 24: 4333–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Ramsay T., Zinkgraf M., Sundell D., Street N.R., Filkov V., Groover A. (2015). A resource for characterizing genome-wide binding and putative target genes of transcription factors expressed during secondary growth and wood formation in Populus. Plant J. 82: 887–898. [DOI] [PubMed] [Google Scholar]
- Ma T., et al. (2013). Genomic insights into salt adaptation in a desert poplar. Nat. Commun. 4: 2797. [DOI] [PubMed] [Google Scholar]
- Ma H.W., Xie M., Sun M., Chen T.Y., Jin R.R., Ma T.S., Chen Q.N., Zhang E.B., He X.Z., De W., Zhang Z.H. (2016). The pseudogene derived long noncoding RNA DUXAP8 promotes gastric cancer cell proliferation and migration via epigenetically silencing PLEKHO1 expression. Oncotarget 8: 52211–52224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan M.J., Lipovich L. (2015). Pseudogene-derived lncRNAs: Emerging regulators of gene expression. Front. Genet. 5: 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Necsulea A., Soumillon M., Warnefors M., Liechti A., Daish T., Zeller U., Baker J.C., Grützner F., Kaessmann H. (2014). The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 505: 635–640. [DOI] [PubMed] [Google Scholar]
- Nelson A.D.L., Devisetty U.K., Palos K., Haug-Baltzell A.K., Lyons E., Beilstein M.A. (2017). Evolinc: a tool for the identification and evolutionary comparison of long intergenic non-coding RNAs. Front. Genet. 8: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nourmohammad A., Lässig M. (2011). Formation of regulatory modules by local sequence duplication. PLOS Comput. Biol. 7: e1002167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson C.E., Nichol Edamura K., Cleary J.D. (2005). Repeat instability: mechanisms of dynamic mutations. Nat. Rev. Genet. 6: 729–742. [DOI] [PubMed] [Google Scholar]
- Pertea M., Kim D., Pertea G.M., Leek J.T., Salzberg S.L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11: 1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliseno L., Salmena L., Zhang J., Carver B., Haveman W.J., Pandolfi P.P. (2010). A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 465: 1033–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X., Xie S., Liu Y., Yi F., Yu J. (2013). Genome-wide annotation of genes and noncoding RNAs of foxtail millet in response to simulated drought stress by deep sequencing. Plant Mol. Biol. 83: 459–473. [DOI] [PubMed] [Google Scholar]
- Rattray A.J., Strathern J.N. (2003). Error-prone DNA polymerases: When making a mistake is the only way to get ahead. Annu. Rev. Genet. 37: 31–66. [DOI] [PubMed] [Google Scholar]
- Rebeiz M., Jikomes N., Kassner V.A., Carroll S.B. (2011). Evolutionary origin of a novel gene expression pattern through co-option of the latent activities of existing regulatory sequences. Proc. Natl. Acad. Sci. USA 108: 10036–10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarola M., Comisso E., Pascolo R., Chiaradia R., Marion R.M., Schneider C., Blasco M.A., Schoeftner S., Benetti R. (2015). Epigenetic silencing of Oct4 by a complex containing SUV39H1 and Oct4 pseudogene lncRNA. Nat. Commun. 6: 7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutz J., et al. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463: 178–183. [DOI] [PubMed] [Google Scholar]
- Schuermann D., Molinier J., Fritsch O., Hohn B. (2005). The dual nature of homologous recombination in plants. Trends Genet. 21: 172–181. [DOI] [PubMed] [Google Scholar]
- Sigova A.A., Mullen A.C., Molinie B., Gupta S., Orlando D.A., Guenther M.G., Almada A.E., Lin C., Sharp P.A., Giallourakis C.C., Young R.A. (2013). Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. Proc. Natl. Acad. Sci. USA 110: 2876–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisu C., et al. (2014). Comparative analysis of pseudogenes across three phyla. Proc. Natl. Acad. Sci. USA 111: 13361–13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater G.S., Birney E. (2005). Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q., Li J., Cheng W., Guo H., Liu X., Gao H. (2018). AtPAP2, a Unique Member of the PAP Family, Functions in the Plasma Membrane. Genes (Basel) 9: E257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam O.H., Aravin A.A., Stein P., Girard A., Murchison E.P., Cheloufi S., Hodges E., Anger M., Sachidanandam R., Schultz R.M., Hannon G.J. (2008). Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature 453: 534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J., Song Y., Du Q., Yang X., Ci D., Chen J., Xie J., Li B., Zhang D. (2016). Population genomic analysis of gibberellin-responsive long non-coding RNAs in Populus. J. Exp. Bot. 67: 2467–2482. [DOI] [PubMed] [Google Scholar]
- Tian Z., Rizzon C., Du J., Zhu L., Bennetzen J.L., Jackson S.A., Gaut B.S., Ma J. (2009). Do genetic recombination and gene density shape the pattern of DNA elimination in rice long terminal repeat retrotransposons? Genome Res. 19: 2221–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrents D., Suyama M., Zdobnov E., Bork P. (2003). A genome-wide survey of human pseudogenes. Genome Res. 13: 2559–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuskan G.A., et al. (2006). The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313: 1596–1604. [DOI] [PubMed] [Google Scholar]
- Wang Y., Tang H., Debarry J.D., Tan X., Li J., Wang X., Lee T.H., Jin H., Marler B., Guo H., Kissinger J.C., Paterson A.H. (2012). MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40: e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T., et al. (2008). Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature 453: 539–543. [DOI] [PubMed] [Google Scholar]
- Wen Y.Z., Zheng L.L., Liao J.Y., Wang M.H., Wei Y., Guo X.M., Qu L.H., Ayala F.J., Lun Z.R. (2011). Pseudogene-derived small interference RNAs regulate gene expression in African Trypanosoma brucei. Proc. Natl. Acad. Sci. USA 108: 8345–8350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel J.F. (2000). Genome evolution in polyploids. Plant Mol. Biol. 42: 225–249. [PubMed] [Google Scholar]
- Wolfe K.H. (2001). Yesterday’s polyploids and the mystery of diploidization. Nat. Rev. Genet. 2: 333–341. [DOI] [PubMed] [Google Scholar]
- Xie J., Yang X., Song Y., Du Q., Li Y., Chen J., Zhang D. (2017). Adaptive evolution and functional innovation of Populus-specific recently evolved microRNAs. New Phytol. 213: 206–219. [DOI] [PubMed] [Google Scholar]
- Yang Z. (1997). PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13: 555–556. [DOI] [PubMed] [Google Scholar]
- Zhang W., Zhang T., Wu Y., Jiang J. (2012). Genome-wide identification of regulatory DNA elements and protein-binding footprints using signatures of open chromatin in Arabidopsis. Plant Cell 24: 2719–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Harrison P.M., Liu Y., Gerstein M. (2003). Millions of years of evolution preserved: A comprehensive catalog of the processed pseudogenes in the human genome. Genome Res. 13: 2541–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Carriero N., Zheng D., Karro J., Harrison P.M., Gerstein M. (2006). PseudoPipe: An automated pseudogene identification pipeline. Bioinformatics 22: 1437–1439. [DOI] [PubMed] [Google Scholar]
- Zhang Z.L., Ogawa M., Fleet C.M., Zentella R., Hu J., Heo J.O., Lim J., Kamiya Y., Yamaguchi S., Sun T.P. (2011). Scarecrow-like 3 promotes gibberellin signaling by antagonizing master growth repressor DELLA in Arabidopsis. Proc. Natl. Acad. Sci. USA 108: 2160–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou C., Lehti-Shiu M.D., Thibaud-Nissen F., Prakash T., Buell C.R., Shiu S.H. (2009). Evolutionary and expression signatures of pseudogenes in Arabidopsis and rice. Plant Physiol. 151: 3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]