

Abstract
In phylogenetic, the diversity measures as UniFrac, weighted UniFrac, and normalized weighted UniFrac are used to estimate the closeness between two samples of genetic material sequences. These measures are widely used in microbiology to compare microbial communities. Furthermore, when the sample size is large enough, very good results have been obtained experimentally. However, some authors do not suggest using them when the sample size is very small. Recently, it has been mentioned that the weighted UniFrac measure can be seen as the Kantorovich-Rubinstein metric between the corresponding empirical distributions of samples of genetic material. Also, it is well known that the Kantorovich-Rubinstein metric complies the metric definition. However, one of the main reasons to establish it is that the sample size is large enough. The goal of this article is to prove that the diversity measures UniFrac are not metrics when the sample size is very small, which justifies why it must not be used in that case, but yes the Kantorovich-Rubinstein metric.
Keywords: phylogenetic, phylogenetic tree, pseudometric, semimetric
Introduction
Phylogenetic is a field of biology that studies how organisms are related during evolution. The basic principle is that the members of an organism set that descend from the same ancestor share an evolutionary history. A problem in phylogenetic analysis is to determine similarities and differences between genetic material sequences. For example, the study of the degree of difference between two samples A and B of genetic material sequences. For this, the diversity measure UniFrac,1 weighted UniFrac and normalized weighted UniFrac2 have been used.
The above diversity measures are used by several authors in microbiology field to compare genetic material samples. For example, Frank et al3 said the diversity measure UniFrac is used to check whether patients with inflammatory bowel disease present samples from different microbial communities to patients without the disease. According to Costello et al,4 the weighted UniFrac and normalized weighted UniFrac are used to better understand the structure of the microbial community in skin sites and other body habitats between different individuals and at different times, and it is suggested that these trends may reveal how changes in the microbial cause or prevent diseases. Another application of these measures is given in Charlson et al,5 which are used to compare the population of bacteria in the lungs and their relationship with the population of bacteria of the upper respiratory tract, the former in healthy individuals. On the other hand, Ley et al6 said the diversity measure was used to measure the difference between bacterial communities in mice intestines, in order to test the effects of kinship and genotype diversity.
Moreover, from the theoretical point of view, diversity measures UniFrac give rise to other measures, for example, Chang et al7 proposed a new weighting scheme assuming that the sequences are randomly distributed; this scheme is called weighted UniFrac adjusted variance (VAW-UniFrac) and it is proposed as an improvement of weighted UniFrac. Furthermore, the VAW-UniFrac measure is compared to the UniFrac and weighted UniFrac measures to determine which is more efficient. Chen et al8 gave a generalization of the UniFrac diversity measures, this generalization is more usefulness to detect a set of biologically relevant changes than the UniFrac measure.
However, despite its practical application in the microbiology field, in Schloss,9 it is mentioned that ‘A recent simulation study concluded that UniFrac is unsuitable as a distance metric and should not be used for multivariate analysis’ that means, it is not appropriate to use diversity measures UniFrac as metrics and they should not be used in multivariate analysis.
Recently, in Evans and Matsen10 was mentioned that the weighted UniFrac measure is the classical Kantorovich-Rubinstein metric11–14 or Earth Mover Distance15 between the corresponding empirical distribution of samples of genetic material on a phylogenetic tree. The above, under assumption that the sample size is large enough. In this way, McClelland and Koslicki16 propose the earth mover distance UniFrac (EMDUniFrac) and an algorithm to compute it.
In this article, we proof that the original version of diversity measures UniFrac are not metrics but they are pseudosemimetrics. They satisfy the following definition.
Definition 1
Let be a set, a function is a pseudosemimetric in if for all the function satisfies
If , then .
.
The above justifies why UniFrac measures can behave unexpectedly for small samples in multivariate data analysis, but it is not the case when the sample size is large enough. Thus, when the sample size is very small, it is recommended to use EMDUniFrac metric.
The rest of the work is developed as follows. In section “Rooted phylogenetic trees,” the necessary concepts will be given to define diversity measures. In section “Diversity measures UniFrac,” the three versions are defined: UniFrac, weighted UniFrac, and normalized weighted UniFrac, and we will show that they are pseudo-parametric; in this way, we prove that they are not metrics and how they are susceptible to small samples. In section “EMDUniFrac,” the UniFrac measures are estimated for some examples and they are compared with EMDUniFrac metric. Finally, some conclusions will be presented in section “Conclusions.”
Rooted Phylogenetic Trees
The diversity measures are calculated on a given phylogenetic tree. In this section, the concepts related to trees will be defined. They will be useful to address diversity measures UniFrac.
Basic definitions
Warnow17 defines a tree as a connected graph without cycles. A rooted tree is a tree in which a vertex is designated as root. The root in phylogenetic represents the common ancestor in the species represented in the tree . The vertices represented the characteristics that allow to establish the similarities between different species. These characteristics are given by genetic material sequences.
The vertex is parent of and is a child of , if and are vertices in the rooted tree such that . Moreover, a vertex is a leaf if does not have any children and is a binary tree if it has vertices with at most two children.
On the other hand, in a tree , a path from vertex to vertex is the sequence of vertices in the graph such that there exist an edge between the vertex and the next one and so on until , denoted by . A branch is the vertices set and edges that belong to the path that goes from the leaf to the root . We call leaf set of to the set built with different labels that are assigned to tree leaves and denoted by . Additionally, a clade of is a subset in that it contains the leaf set of a subtree , with root in some vertex and it is denoted by and is the clades set such that . The set contains all the singular sets of leaves, a set that contains all the leaves and a clade for each remaining vertex of .
Otherwise, Warnow17 associated the parameter to the edge where denotes the probability of changing state where . A model tree Cavender-Farris-Neyman (CFN) is a pair where is a binary rooted tree with leaf set and gives the values for all edges . Under the CFN model, the number of changes in an edge is modeled by a Poisson random variable with expected value . Then, instead of using the probability substitution in each edge, we will use , with the condition that for all .
Thus, the branch length , denoted by , is a positive number that represents the rate of change between the root and the leaf , it is
Let be the expected number of changes on the way on the tree , it follows that
We can see by the definition that is the matrix distance on the road in a tree, where the path distance between two leaves is the sum of branch length and all branch lengths are positive. The matrix is an additive matrix, which is defined as follows.
Definition 2
A matrix is additive if there is a three with leaf set and the lengths of the edges are non-negative, that is branch length of in is equal to .
Phylogenetic tree construction
To construct a binary rooted phylogenetic tree using two samples and , it is necessary to consider the partial order definition.
Definition 3
A partial order is a binary relation in a set such that for any satisfies
Transitivity: and imply that .
Reflexivity: .
Antisymmetry: and imply that .
Two elements and are compatible if or .
Hasse diagram is a graphic scheme of a partially ordered set. To construct the Hasse diagram of a set, a vertex is created for each element of and a directed edge if and x ≠ y. They are sorted from bottom to top, so the directed edges go up. The directed edges are removed if there is a third vertex such that and .
Let be a rooted phylogenetic tree and the clades set . The sequences and of genetic material are in relation, if if and only if . We can see that the relation is partial order.
Now, we will construct the Hasse diagram by set. A graph is made assigning a vertex for each element in the set and a directed edge from vertex to different vertex if . The smallest subset must be found, and if , we put a directed edge from to . As containment is transitive, if and , so . Therefore, if there are directed edges from to and from to so there are edges from to , and we can remove the directed edge from to without losing information.
The next theorem say that a binary rooted tree is isomorphic to the Hasse diagram built by . It is proven by Warnow.17
Theorem 1
Let be a rooted tree in which each internal node has two children. Then the Hasse diagram built by is isomorphic to . In this way, we can get the binary rooted tree from Hasse diagram built using the set .
In the next section, the diversity measures are addressed in their three versions, UniFrac, weighted UniFrac, and normalized weighted UniFrac. Also, we will show that they satisfy the pseudosemimetric definition and we will give examples where diversity measures do not satisfy the metric definition.
Diversity Measures UniFrac
To define the diversity measures UniFrac, it is considered a binary rooted phylogenetic tree for two samples and of genetic material sequences, where sample has sequences and sample has sequences, not necessarily different, that means that may occur and in each sample could be two or more equal sequences; furthermore, each sample can contain the root or not (common ancestor between species and ). Let be the tree with branches and let be the length for each branch, with , they coincide with the distance from the root () to the sequence () that is in the leaf on branch.
Let be the number of vertices in A that are in branch , analogously, the number of vertices in that are in branch We define
note that they are the proportions of descendant sequences in samples and in the branch, respectively.
Example 1
Consider the rooted tree in Figure 1. It is constructed using samples and , where and . The first branch has the sequences and , where and , so the proportion of descendant sequences in samples and on branch 1 are
Figure 1.
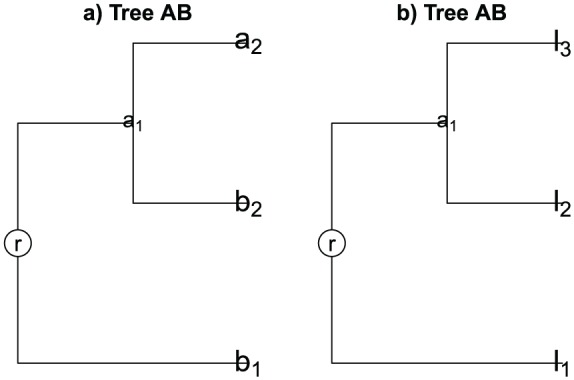
(a) Tree for samples and . (b) Tree for samples and with the label leaves for with ().
| (1) |
respectively. The second branch has the sequences where and , in this way
analogously, the proportions of descendant sequences in third branch are
In later examples, the sequences in leaf on the branch will be denoted by (see Figure 1) in order to follow the given notation. This is because to definite the diversity measures, we need the branch length () whose notation is given for sequences in the leaf.
UniFrac
The diversity measure UniFrac was proposed by Lozupone and Knight1 and it is defined as
| (2) |
where is the indicator function. We can see that the absolute value is or . It is when the branch has sequences in samples or and it is when has two samples.
Example 2
Consider the raised tree in Example 1, with . The proportion of descendant sequences in and are greater than , see the expression (1), then
thus,
The diversity measure UniFrac version ignores the abundant information about sequences, only consider its presence or absence in the branch.
Proposition 1
The diversity measure UniFrac is a pseudosemimetric.
Proof
We will prove that the diversity measure UniFrac satisfies Definition 1. Moreover, we will give an example where it does not satisfy the metric definition.
If , it is for all and , so
for all , thus,
therefore,
2. To prove symmetry, we consider
Then the diversity measure UniFrac satisfies Definition 1. Additionally, we will give an example that does not satisfy the metric definition.
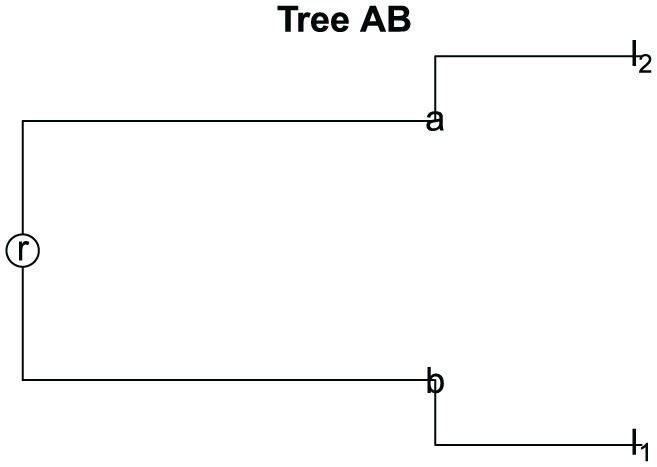
Let be the tree built from two different samples:
where is the root and the sequence l1 ≠ l2 (see Figure 2). The branch is the path from to and the branch the path from to , we have to
Figure 2.

Tree for samples and .
however, we supposed that l1 ≠ l2. So that if , it does not imply that .
2. We consider the samples
and the trees , , and built for samples and , and , and and , respectively (see Figure 3), where
Figure 3.
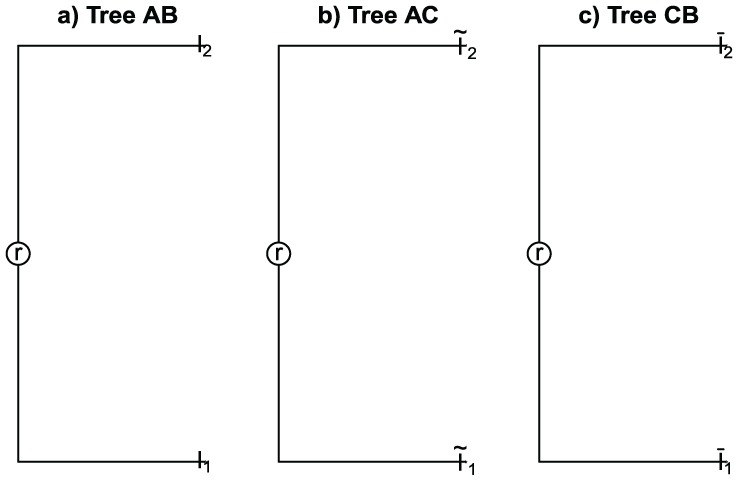
(a) Tree for samples and . (b) Tree for samples and . (c) Tree for samples and .
Moreover, suppose that
| (3) |
If we estimate , , and we have the following:
| (4) |
| (5) |
| (6) |
If the triangle inequality is satisfied and we considered expressions (4) to (6), we have that
where not necessary
It contradicts assumption (3). Then, triangle inequality is not satisfied.
Weighted UniFrac
The weighted UniFrac was proposed by Lozupone et al2 and is denoted by
| (7) |
It uses information about the abundance of the genetic material sequences. If the branch has large length, it means a fast evolution, and it could influence more than other in .
Proposition 2
The weighted UniFrac is a pseudosemimetric.
Proof
We will prove that the weighted UniFrac satisfies Definition 1.
If we suppose that , we have for all and , so
thus,
Therefore,
To prove symmetry, we consider
Then, the weighted UniFrac satisfies Definition 1, but it does not satisfy the metric definition. We show some examples
Consider the different samples
and the tree built for samples and (see Figure 4) that satisfied the next conditions:
Figure 4.
Tree for samples and .
Therefore,
with A ≠ B.
Consider the samples
and the trees , , and built for samples and , and , and and , respectively (see Figure 5), where
Figure 5.
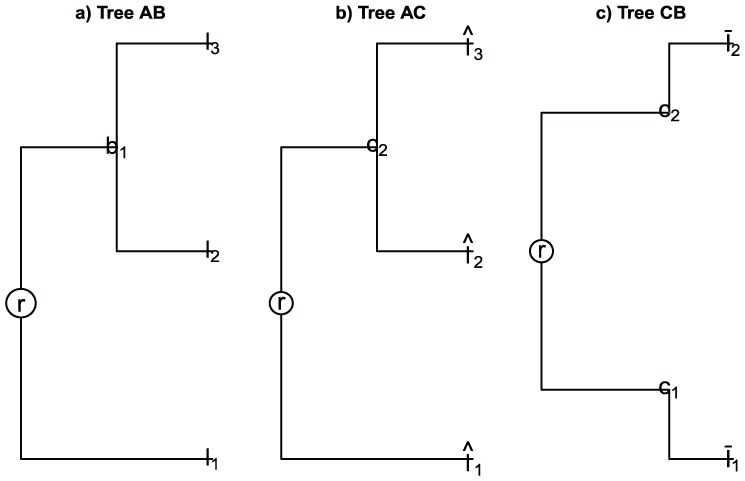
(a) Tree for samples and . (b) Tree for samples and . (c) Tree for samples and .
| (8) |
and also, we assume
| (9) |
| (10) |
From equations (8) and (9), we have that
If we estimate , , and , we have the following:
| (11) |
| (12) |
| (13) |
If the triangle inequality is satisfied, using equalities (11) to (13), we have
where we can get
this contradicts the supposition (10), so the weighted UniFrac does not comply with the triangle inequality.
We proved that weighted UniFrac satisfies Definition 1; however, it is not a metric.
Normalized weighted UniFrac
The normalized weighted UniFrac was proposed by Lozupone et al2 and it is given by
| (14) |
where the normalizing factor is
| (15) |
with the number of different sequences in and the distance from the root to the sequence ; furthermore,
| (16) |
where and are the number of times that the sequence is observed in samples and , respectively.
Example 3
In Example 1, , where the sequences proportions in sample are
and the sequences proportions in sample are
The normalized weighted UniFrac is less sensitive to branches with a long length and is determined by branches with different proportions.
Proposition 3
The normalized weighted UniFrac is a pseudosemimetric.
Proof
We will prove that the normalized weighted diversity measure UniFrac satisfies Definition 1.
1. Analogous to 2. of Proposition 2, we have
2. To prove symmetry, we consider
Thus, the normalized weighted diversity measure UniFrac satisfies with Definition 1. Now, examples where it does not satisfy:
1. We consider the example in item (1) from Proposition 2. Therefore,
2. Consider the samples
and the trees , , and TCB built for samples and , and , and and , respectively (see Figure 6), where
Figure 6.
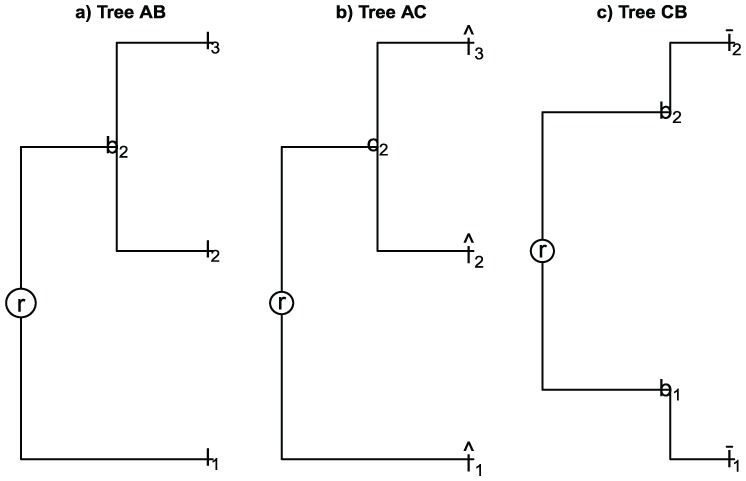
(a) Tree by samples and . (b) Tree by samples and . (c) Tree by samples and .
| (17) |
and additionally assume
| (18) |
| (19) |
Note that equations (17) and (18) imply that
Thus,
| (20) |
| (21) |
| (22) |
with the respective normalizing factor. As the triangle inequality is satisfied using the equalities (20)-(22), we have
from we can get
it contradict the supposition (10). Therefore, the normalized weighted diversity measure UniFrac does not satisfy the triangle inequality.
We proved that normalized weighted diversity measure UniFrac is a pseudosemimetric. Next, we will give examples where we calculated the diversity measures UniFrac on a tree illustrate by McClelland and Koslicki16 and we will compare with EMDUniFrac.
EMDUniFrac
Based on Evans and Matsen,10 the EMDUniFrac is proposed in McClelland and Koslicki,16 Given two samples and of genetic material and their associated abundances, we can estimate two probability distributions and on their phylogenetic tree that represent the fraction of a given sample that appears at each node in . Let be the matrix of all pairwise distances between nodes in and describe the space of all ways in which one community can be transformed into the other. The entry of indicates the total abundance of has been moved from node in sample to node in sample . In this way, the EMDUniFrac is given by
it represents the minimum amount of ‘work’ required to transform the distribution into the distribution along the phylogenetic tree. It has been previously show that EMDUniFrac(P, Q) is equivalent to weighted UniFrac distance when the sample size is large enough.10 However, we will give examples where the EMDUniFrac distance and the diversity measures UniFrac are different between them.
Considerate the tree as in Figure 1(b) in McClelland and Koslicki16 where EMDUniFrac(P, Q) is . We calculate the diversity measure UniFrac on :
Thus,
both under .
It is important to mention the samples size is very small.
The weighted diversity measure UniFrac (see expression 7) on the tree is
thus, can see that
Now, we obtain the normalized weighted UniFrac value as
Thus,
Therefore, the diversity measures UniFrac and EMDUniFrac are different between them. Then, we can say the diversity measures UniFrac are not equal to EMDUniFrac(P, Q) if the samples size is not large enough.
On the other hand, considerate the tree as the Figure 1(b) in McClelland and Koslicki,16 it is built for the different samples and , we calculate the weighted UniFrac measure as :
however, samples and are different. So that if , it does not imply that .
Conclusions
In this article, we prove that diversity measures UniFrac, weighted UniFrac, normalized weighted UniFrac satisfy the positive property, symmetry property, and the implication that if the samples are equal then the diversity measures are zero. On the other hand, examples were presented where the diversity measures mentioned do not comply the metric definition. We prove that diversity measures comply the pseudosemimetric definition.
Although measures UniFrac are used in microbiology as a tool to measure the proximity between samples of genetic material large enough and showing a good performance, as mentioned in the literature,3–7 when the sample size is small, no it is appropriate to use it in that sense. The previous thing due to the lack of the properties previously amended, as Schloss said. In section “EMDUniFrac,” we could see examples where the diversity measures UniFrac and EMDUniFrac are different between them; in this way, we can say the diversity measures UniFrac are not equivalent to EMDUniFrac if the samples size is not large enough. Furthermore, if we calculate the weighted UniFrac for two different small samples, it does not imply that weighted UniFrac is zero. Then an alternative for diversity measures UniFrac is the Kantorovich-Rubinstein metric10 or EMDUniFrac metric.18–20
Acknowledgments
This article was developed under the Project PRODEP UV-PTC-779 of Mexico Government.
Footnotes
Funding:The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: JARA performed the analytic calculations and MLAG supervised the project. Both JARA and MLAG authors contributed to the final version of the manuscript.
ORCID iD: Martha Lorena Avendaño Garrido  https://orcid.org/0000-0001-7956-8958
https://orcid.org/0000-0001-7956-8958
References
- 1. Lozupone CA, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol. 2007;73:1576–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Frank DN, Amand ALS, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104:13780–13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Charlson ES, Bittinger K, Haas AR, et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184:957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102:11070–11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chang Q, Luan Y, Sun F. Variance adjusted weighted UniFrac: a powerful beta diversity measure for comparing communities based on phylogeny. BMC Bioinformatics. 2011;12:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen J, Bittinger K, Charlson ES, et al. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics. 2012;28:2106–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schloss PD. Evaluating different approaches that test whether microbial communities have the same structure. ISME J. 2008;2:265. [DOI] [PubMed] [Google Scholar]
- 10. Evans SN, Matsen FA. The phylogenetic Kantorovich-Rubinstein metric for environmental sequence samples. J R Stat Soc Series B Stat Methodol. 2012;74:569–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rachev ST. Probability Metrics and the Stability of Stochastic Models, Volume 269. Hoboken, NJ: John Wiley & Son Ltd; 1991. [Google Scholar]
- 12. Rachev ST, Rüschendorf L. Mass Transportation Problems, Volume I: Probability and Its Applications. New York, NY: Springer; 1998. [Google Scholar]
- 13. Villani C. Topics in Optimal Transportation. Providence, RI: American Mathematical Society; 2003. [Google Scholar]
- 14. Villani C. Optimal Transport: Old and New. Grundlehren der mathematischen Wissenschaften. Berlin, Germany: Springer; 2008. [Google Scholar]
- 15. Levina E, Bickel P. The earth mover’s distance is the mallows distance: some insights from statistics. In: Proceedings Eighth IEEE International Conference on Computer Vision. ICCV 2001, vol. 2 New York, NY: IEEE; 2001:251–256. [Google Scholar]
- 16. McClelland J, Koslicki D. EMDUnifrac: exact linear time computation of the Unifrac metric and identification of differentially abundant organisms. J Math Biol. 2018;77:935–949. [DOI] [PubMed] [Google Scholar]
- 17. Warnow T. Computational Phylogenetics. An Introduction to Designing Methods for Phylogeny Estimation. Cambridge, UK: Cambridge University Press; 2017. [Google Scholar]
- 18. Srinivasan S, Hoffman NG, Morgan MT, et al. Bacterial communities in women with bacterial vaginosis: high resolution phylogenetic analyses reveal relationships of microbiota to clinical criteria. PLoS ONE. 2012;7:e37818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smith BC, McAndrew T, Chen Z, et al. The cervical microbiome over 7 years and a comparison of methodologies for its characterization. PLoS ONE. 2012;7:e40425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Livermore JA, Mattes TE. Phylogenetic detention of novel cryptomycota in an Iowa (United States) aquifer and from previously collected marine and freshwater targeted high-throughput sequencing sets. Environ Microbiol. 2013;15:2333–2341. [DOI] [PubMed] [Google Scholar]