

Abstract
Intestinal Niemann-Pick C1 Like 1 (NPC1L1) protein plays a key role in cholesterol absorption. A decrease in NPC1L1 expression has been implicated in lowering plasma cholesterol and mitigating the risk for coronary heart disease. Little is known about the mechanisms responsible for NPC1L1 protein degradation that upon activation may lead to a reduction in NPC1L1 protein levels in intestinal epithelial cells (IECs). In current studies, the human intestinal Caco-2 and HuTu-80 cell lines expressing NPC1L1-hemagglutinin fusion protein were used to investigate the mechanisms of NPC1L1 protein degradation. Incubation with the proteasome inhibitors MG-132 and lactacystin (10 μM, 24 h) significantly increased NPC1L1 protein levels in IECs. Also, the inhibition of the lysosomal pathway with bafilomycin A1 (80 nM, 24 h) resulted in a significant increase in NPC1L1 protein levels. Immunoprecipitation studies showed that NPC1L1 protein is both a poly- and monoubiquinated polypeptide and that the inhibition of the proteasomal pathway remarkably increased the level of the polyubiquinated NPC1L1. The surface expression of NPC1L1 was increased by the inhibition of both proteasomal and lysosomal pathways. Furthermore, the pharmacological inhibition of mitogen-activated protein kinase pathway (PD-98059, 15 μM, 24 h) and siRNA silencing of ERK1/2 resulted in a significant decrease in NPC1L1 protein levels in IECs. In conclusion, our results showed that basal level of intestinal cholesterol transporter NPC1L1 protein is modulated by both ubiquitin proteasome- and lysosome-dependent degradation as well as by ERK1/2-dependent pathway. The modulation of these pathways may provide novel clues for therapeutic intervention to inhibit cholesterol absorption and lower plasma cholesterol.
Keywords: cholesterol absorption, lysosomal degradation, ubiquitin proteasome degradation
INTRODUCTION
The Niemann-Pick type C1 Like 1 (NPC1L1) sterol transporter mediates cholesterol uptake in intestinal epithelial cells and hepatocytes (4). The lack of NPC1L1 in knockout mice resulted in a remarkable reduction in cholesterol absorption (2). Also, NPC1L1 was shown to be the molecular target of the cholesterol absorption inhibitor ezetimibe that is used to lower plasma levels of low-density lipoprotein (LDL) cholesterol (10). These findings highlight the essential roles of NPC1L1 in the maintenance of cholesterol homeostasis. Consistent with this notion are recent findings showing that NPC1L1 knockout mice are protected against diet-induced hypercholesterolemia (8). Additionally, genetic screening of a large cohort of humans showed that individuals with inactivating mutations in NPC1L1 had a significantly lower risk for the development of coronary heart disease (19). Beside the inactivating mutations, multiple genetic variants of NPC1L1 with nonsynonymous sequences were associated with individuals who were having low efficiency of cholesterol absorption and lower baseline of LDL cholesterol (7). Molecular characterization of these variants of NPC1L1 associated with low cholesterol absorption showed that these genetic alterations produced NPC1L1 polypeptides that are less stable and rapidly degraded (24). This observation strongly suggests that the rate of NPC1L1 protein degradation could be one of the limiting factors controlling the efficiency of cholesterol absorption.
Two major protein degradation pathways have been identified in mammalian cells that are important to control the levels of cellular proteins: the ubiquitin proteasome and lysosomal degradation pathways. The ubiquitin proteasome degradation pathway is an ATP-dependent process that uses ubiquitin (76-amino acid polypeptide) as a marker to label cellular proteins for proteolysis. In this process, multiple ubiquitin molecules are attached to a protein to form polyubiquitinated chains that are then recognized and degraded by the proteasome machinery (5). The ubiquitin proteasome degradation pathway has recently been shown to be involved in modulating the expression of cholesterol transporters such as the ATP-binding cassette transporters, ABCA1 and ABCG1, suggesting a role in cholesterol homeostasis (21). There is also evidence that the lysosomal degradation pathway modulates cholesterol balance in the body, such as in the case of the E387K mutant of the low-density lipoprotein receptor (LDLR) that is found in patients with familial hypercholesterolemia. Studies have shown that treatment with bafilomycin A1, the classical inhibitor of the lysosomal degradation pathway, prevented the degradation of E387K LDLR mutant (23) to reduce the associated hypercholesterolemia.
Not only are protein degradation pathways responsible for determining the steady-state levels of cellular proteins, they were also shown to be modulated by signaling pathways, suggesting their role as regulatory mechanisms of protein levels in response to external stimuli. In this regard, previous studies demonstrated the involvement of the MEK/ERK signaling pathway in altering the protein levels and activity of the cholesterol transporter ABCA1 and the scavenger receptor class B type 1 (SR-B1), the protein responsible for the uptake of the high-density lipoprotein particles (17, 25). Given the importance of protein degradation pathways in modulating processes responsible for controlling cholesterol homeostasis, it was of interest to investigate their potential roles in influencing the levels of NPC1L1 protein in intestinal epithelial cells.
In the present study, we showed that the basal cellular level of NPC1L1 protein in intestinal epithelial cells is modulated by both ubiquitin proteasome and lysosomal degradation pathways. We have also shown that inhibition of the mitogen-activated protein (MAP) kinase pathway resulted in a decrease in NPC1L1 protein in intestinal epithelial cells. These degradation pathways of NPC1L1 protein may represent a novel therapeutic target for inhibiting cholesterol absorption and lowering plasma cholesterol.
MATERIALS AND METHODS
Reagents.
The proteasome inhibitors lactacystin and MG-132, lysosome inhibitor bafilomycin A1, and MEK inhibitor PD-98059 were purchased from Enzo Life Sciences (Farmingdale, NY). siRNAs for ERK1 and ERK2 were obtained from Qiagen. Cycloheximide was purchased from Calbiochem. All of the tissue culture supplies and Lipofectamine 2000 were obtained from Life Technologies (Carlsbad, CA). Antibodies against ubiquitin (sc-8017) and hemagglutinin (HA) tag (sc-805) (for immunoprecipitation) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). GAPDH (G9545) and actin (A1978) antibodies were purchased from Sigma-Aldrich (St. Louis, MO). Antibodies against ERK1/2 (4695S) and HA tag (2367S) were purchased from Cell Signaling Technology (Danvers, MA). Protein A/G plus agarose beads were obtained from Santa Cruz Biotechnology. Sulfo-NHS-SS-biotin was obtained from Thermo Scientific (Rockford, IL). Eagle’s minimal essential media (EMEM) was purchased from American Type Culture Collection (ATCC, Manassas, VA). Routine research reagents were purchased from Sigma-Aldrich. The plasmid vector containing human NPC1L1 cDNA fused to a HA tag vector was synthesized as described previously (20). HuTu-80 and Caco-2 cell lines were obtained from ATCC.
Cell culture, transient transfection, and development of stable cell line.
Both wild-type HuTu-80 and Caco-2 cells were grown in EMEM media supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in 5% CO2. HuTu-80 cells were transfected with plasmid vector coding for human NPC1L1-HA fusion protein using Lipofectamine 2000 according to the manufacturer’s instructions. At 48 h after transfection, cells were selected by incubation with 800 µg/ml geneticin (G418; Life Technologies). Transfected clones were pooled and maintained by culturing in media containing G418 at the same concentration. Total proteins were isolated from stably transfected HuTu-80 cells and analyzed for NPC1L1 expression by Western blotting using antibody against HA. Caco-2 cells were transiently transfected with hNPC1L1-HA expression vector using Amaxa nucleofection as previously described (13).
Western blotting.
Total cellular proteins were isolated using cell lysis buffer (Cell Signaling Technology, Danvers, MA) supplemented with a cocktail of protease inhibitors. Protein concentration was determined using the method of Bradford. Equal amounts of protein from the control and treated groups were solubilized in protein sample buffer and boiled for 5 min. Proteins were separated on a 7.5% Tris-glycine SDS polyacrylamide gel and then transferred to nitrocellulose membranes for Western blotting. Membranes were incubated in a blocking buffer containing 5% nonfat dry milk in 1× PBS for 1 h at room temperature. After blocking, membranes were then incubated with the primary antibodies overnight at 4°C and washed subsequently with 1× PBS containing 0.1% Tween 20. The blots were then incubated with horseradish peroxidase-conjugated secondary antibodies, either anti-rabbit IgG (W4018; Promega) or anti-mouse IgG (W4028; Promega), for 1 h at room temperature. The bands were visualized by an enhanced chemiluminescence system according to the manufacturer's instructions (Bio-Rad, Hercules, CA).
Immunoprecipitation.
Protein lysates were prepared and precleared by incubation with protein A/G plus agarose beads for 30 min at 4°C. Precleared protein lysates were incubated overnight with HA antibody at 4°C. Antigen-antibody complexes were then incubated with protein A/G plus agarose beads for 3 h at 4°C, and beads were then separated from the solution and washed with 1× cold PBS. Precipitated proteins were eluted by boiling with 2× Laemmli buffer for 5 min, separated on SDS-PAGE, and then analyzed by Western blotting using anti-HA and anti-ubiquitin antibody.
Cell surface biotinylation.
Biotinylation studies were performed to assess the surface expression of NPC1L1 in stably transfected HuTu-80 cells overexpressing NPC1L1-HA fusion protein as previously described (18). Cell-surface labeling was performed using sulfo-NH-SS-biotin (1.5 mg/ml; Pierce Biotechnology, Rockford, IL) in borate buffer (in mM: 154 NaCl, 7.2 KCl, 1.8 CaCl2, and 10 H3BO3, pH 9.0). Biotinylated proteins were pulled down with NeutrAvidin agarose beads and then released by boiling with 100 μM DTT. Biotinylated proteins were eluted in Laemmli buffer and subjected to SDS-PAGE. The amount of surface, intra-, and total NPC1L1 protein was determined by immunoblotting.
siRNA knockdown.
Expression of ERK1/2 in HuTu-80 cells stably transfected with NPC1L1-HA was attenuated using siRNAs specific for ERK1/2 as previously described (16). Scrambled siRNA was used as negative control. Cells were plated in six-well culture plates and were transiently transfected after 24 h using 100 pmol of siRNA duplexes and Lipofectamine 2000. Transfected cells were harvested at 48 h after transfection, and total proteins were isolated. Silencing was validated by Western blotting using ERK1 and ERK2 specific antibodies.
Statistical analysis.
Results are expressed as means ± SE. Student's t-test and one-way ANOVA post hoc test were used in statistical analysis. Prism 6 (GraphPad) was used for statistical analysis. P ≤ 0.05 was considered statistically significant.
RESULTS
Inhibition of ubiquitin proteasome pathway increases NPC1L1 protein levels.
The transcriptional regulation of NPC1L1 has been well investigated (1, 13, 15). However, little is known about the posttranscriptional pathway(s) responsible for the degradation of the NPC1L1 protein. We first measured the turnover rate of the NPC1L1 protein. Intestinal HuTu-80 cells stably transfected with NPC1L1-HA fusion protein were incubated with cycloheximide (100 μg/ml) to block protein synthesis. As shown in Fig. 1A, the NPC1L1-HA fusion protein was detected as two bands with molecular mass of ~280 and 145 kDa. The blots presented in Fig. 1 show that NPC1L1-HA protein levels were decreased following cycloheximide treatment in a time-dependent manner reaching to ~50% of the control levels (Fig. 1B) after 8 h of incubation. We next examined the role of the ubiquitin proteasome pathway in the degradation of NPC1L1 protein. HuTu-80 cells were treated with either lactacystin (10 μM) or MG-132 (10 μM), potent inhibitors of the ubiquitin proteasome pathway, for 24 h, and then NPC1L1 protein levels were determined by Western blotting. As shown in Fig. 2A, incubation with lactacystin or MG-132 significantly increased NPC1L1 protein levels by approximately threefold (Fig. 2B), suggesting that the ubiquitin proteasome pathway is responsible for NPC1L1 protein degradation and contributes to its steady-state levels in intestinal epithelial cells.
Fig. 1.
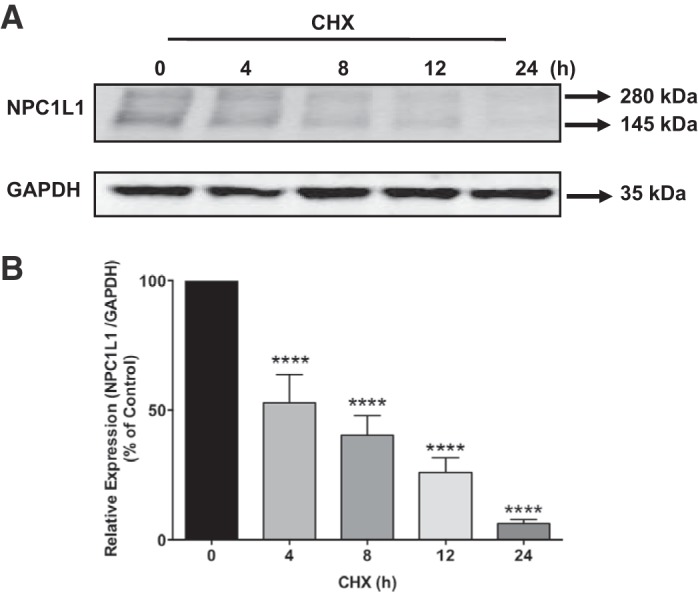
Blocking protein synthesis rapidly decreased Niemann-Pick C1 Like 1 (NPC1L1) protein levels. A: stably transfected HuTu-80 cells were treated with cycloheximide (CHX, 100 μg/ml) for different periods of time, and NPC1L1-hemagglutinin (HA) level was then assessed by Western blotting. B: densitometric analysis of NPC1L1-HA expression in response to treatment with CHX compared with untreated cells (time 0). Data are presented as means ± SE from 5 different experiments. ****P < 0.0001 compared with control.
Fig. 2.
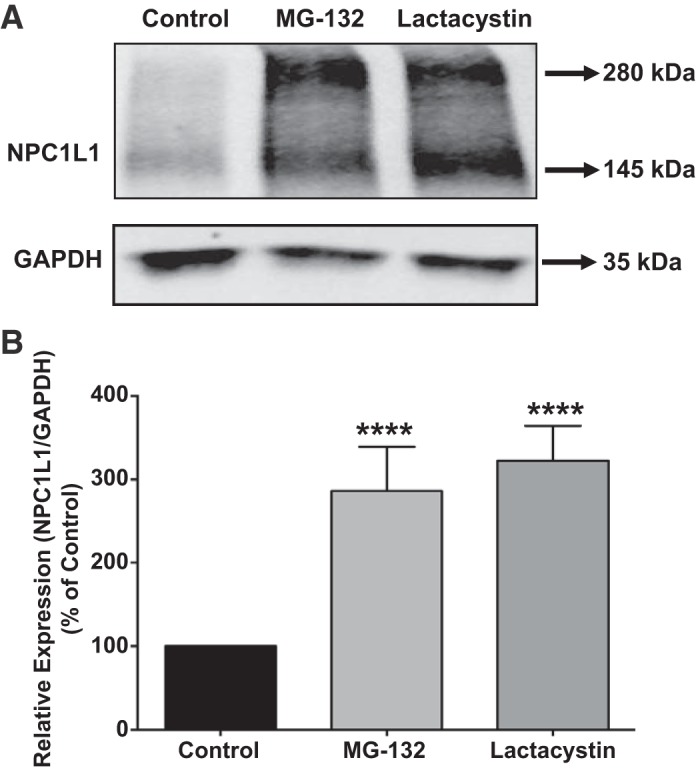
Niemann-Pick C1 Like 1 (NPC1L1) protein is degraded by proteasomal pathway. A: stably transfected human intestinal HuTu-80 cells were treated with proteasome inhibitors, lactacystin (10 μM) or MG-132 (10 μM), for 24 h. Cells were then harvested, and total protein was isolated. NPC1L1 protein expression was detected by Western blotting using anti-hemagglutinin (HA) antibody. GAPDH was used as a loading control. B: densitometric analysis showed a significant increase in NPC1L1 protein expression in both the MG-132- and lactacystin-treated groups compared with control. Data are expressed as percent of control and presented as means ± SE from 3 different experiments. ****P < 0.0001 compared with control.
Blocking lysosomal degradation pathway increases NPC1L1 protein levels.
Cell surface proteins that are internalized by endocytosis may be routed to lysosomes for digestion and degradation in the lysosomes (6). To investigate the involvement of lysosomal pathway in determining the steady-state levels of NPC1L1 protein, stably transfected HuTu-80 cells were treated with 80 nM bafilomycin A1 for 24 h, and NPC1L1 protein levels were analyzed by Western blotting. As shown in Fig. 3A, treatment of HuTu-80 cells with the lysosomal inhibitor resulted in a significant increase in NPC1L1 protein levels compared with control as assessed by Western blotting. Densitometric analysis (Fig. 3B) showed an ~3.5-fold increase in the relative levels of NPC1L1 protein in response to blocking the lysosomal degradation pathway by bafilomycin A1. These data suggested that the lysosomal degradation pathway is also involved in controlling the basal cellular levels of NPC1L1 protein in intestinal epithelial cells.
Fig. 3.
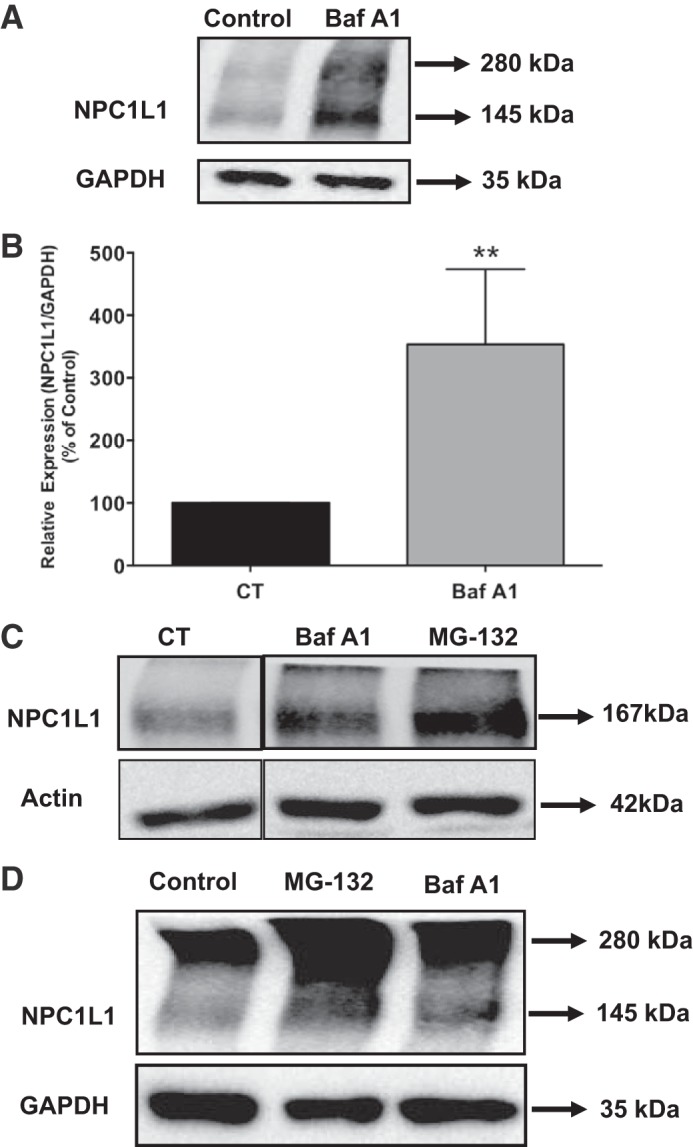
Inhibition of lysosomal pathway increased Niemann-Pick C1 Like 1 (NPC1L1) protein expression. A: stably transfected HuTu-80 cells were treated with bafilomycin A1 (80 nM) for 24 h. Equal amounts of total protein from control and bafilomycin A1-treated groups were resolved on SDS-PAGE and analyzed by Western blotting. B: densitometric analysis showed a significant increase in NPC1L1 protein levels. Data are presented as means ± SE from 3 different experiments. **P < 0.001 compared with control. C: HuTu-80 cells were transfected with green fluorescent protein (GFP)-tagged human (h) NPC1L1 cDNA to generate a stable cell line overexpressing NPC1L1-GFP fusion protein. Cells were treated with the proteasome inhibitor MG-132 (10 μM) and lysosome inhibitor bafilomycin A1 (80 nM) for 24 h. After 24 h, cells were harvested, and total proteins were separated by SDS-PAGE and analyzed by Western blot analysis. D: human intestinal Caco-2 cells were transiently transfected with hemagglutinin (HA)-tagged hNPC1L1. After transfection (24 h), cells were treated with proteasome or lysosome inhibitor MG-132 (10 μM) and bafilomycin A1 (80 nM), respectively, for 24 h followed by Western blot analysis using anti-HA antibody.
To rule out the possible interference of the HA tag in determining the degradation pathway for NPC1L1, we investigated the effects of MG-132 and bafilomycin A1 on NPC1L1 protein fused to green fluorescent protein (GFP) tag and stably transfected in HuTU-80 cells. As shown in Fig. 3C, treatment with inhibitors resulted in a significant increase in NPC1L1-GFP (~167 kDa) protein similar to that observed in NPC1L1-HA protein. These data indicate that the degradation of NPC1L1 protein by ubiquitin proteasome and lysosomal degradation pathways is independent of the tag fused to the protein. We next investigated the roles of the ubiquitin proteasome and lysosomal degradation pathways in controlling the levels of NPC1L1 protein in a different intestinal epithelial cell line. For these experiments, we transiently transfected intestinal Caco-2 cells with NPC1L1-HA fusion protein, treated the cells with MG-132 and bafilomycin A1, and then evaluated their effects on NPC1L1-HA protein expression by Western blotting. As shown in Fig. 3D, inhibiting the ubiquitin proteasome and lysosomal degradation pathways caused a significant increase in the level of NPC1L1 protein similar to that observed in HuTu-80 cells. These data suggested that the effects of the inhibitors of these protein degradation pathways on NPC1L1 protein are not cell line specific.
NPC1L1 protein is ubiquitinated in intestinal epithelial cells.
Cellular proteins that are destined for degradation by ubiquitin proteasome pathway are first labeled by covalent binding to ubiquitin (5). However, studies have shown that ubiquitinated proteins may also be degraded via the lysosomal pathway (3). We, therefore, investigated the ubiquitination of NPC1L1-HA protein in both Caco-2 and HuTu-80 cells and determined whether the pattern of its ubiquitination is affected by the proteasomal or the lysosomal inhibitors. Cells were transiently transfected with NPC1L1-HA expression vector and then treated with MG-132 and bafilomycin A1. NPC1L1-HA was immunoprecipitated using anti-HA antibody, and the level of ubiquitination was assessed by Western blotting using ubiquitin monoclonal antibody. Figure 4 depicts the results after treatment with MG-132 and bafilomycin A1 (Fig. 4A represents the results in HuTu-80, and Fig. 4B depicts the results in Caco-2 cells). As shown in the Fig. 5, A and B, blocking the ubiquitin proteasome pathway with MG-132 caused a remarkable increase in the ubiquitination of the higher-molecular-weight band when the blots were probed with antiubiquitin antibodies. These results suggest that the higher-molecular-weight band of NPC1L1-HA fusion protein represents a polyubiquitinated NPC1L1 polypeptide that is accumulated in response to blocking the ubiquitin proteasome pathway. On the other hand, the same analysis showed that treatment with bafilomycin A1 caused mainly an increase in the ubiquitination of the lower-molecular-mass band (~145 kDa), suggesting an accumulation of a monoubiquitinated NPC1L1 protein. Collectively, these results indicated that NPC1L1 protein is present as mono- and polyubiquitinated polypeptides that are targeted for degradation by the ubiquitin proteasome and lysosomal degradation pathways, respectively.
Fig. 4.

Ubiquitinated Niemann-Pick C1 Like 1 (NPC1L1) polypeptides increased by blocking both the proteasomal and lysosomal degradation pathway. HuTu-80 (A) and Caco-2 cells (B) were transiently transfected with hemagglutinin (HA)-tagged NPC1L1 mammalian expression vector and then treated with MG-132 or bafilomycin A1 for 24 h. NPC1L1-HA was then immunoprecipitated using anti-HA antibody, and the immunoprecipitates were probed with anti-HA antibody and visualized by Western blot analysis. The blots were then stripped and reprobed with the antiubiquitin antibody (n = 3).
Fig. 5.
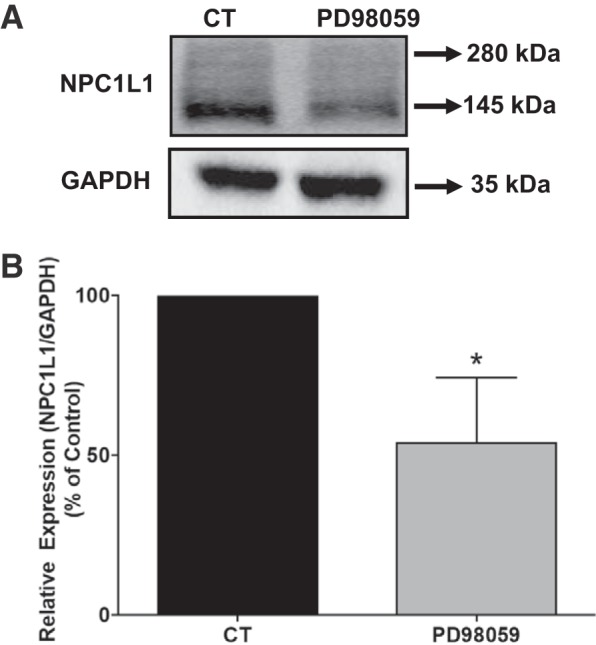
Inhibition of mitogen-activated protein kinase (MAPK)-ERK signaling decreased Niemann-Pick C1 Like 1 (NPC1L1) protein expression. A: stably transfected HuTu-80 cells were treated with MEK inhibitor PD-98059 (15 μM) or vehicle for 24 h, and cell lysates were analyzed for NPC1L1 protein expression by Western blot analysis. B: densitometric analysis showed a decrease in NPC1L1 protein expression in response to PD-98059. Data are presented as means ± SE from 3 different experiments. *P < 0.01 compared with control.
Mitogen-activated protein kinase signaling modulates NPC1L1 protein stability.
Previous studies have shown that inhibition of the mitogen-activated protein kinase (MAPK)-ERK pathway promotes the degradation of SR-B1 protein involved in the uptake of high-density lipoprotein (25). We then examined the potential effects of inhibiting MAPK-ERK1/2 signaling pathway on the level of the NPC1L1 protein. Stably transfected HuTu-80 cells were treated with PD-98059 (inhibitor of MAP kinases MEK1/2) for 24 h, and the level of NPC1L1-HA fusion protein was assessed by Western blotting. As shown in Fig. 5A, treatment of cells with PD-98059 significantly decreased the NPC1L1 protein expression compared with control, indicating the involvement of MAPK pathway in controlling the basal levels of cellular NPC1L1 protein. Densitometric analysis presented in Fig. 5B revealed ~50% reduction in NPC1L1 protein expression in response to treatment with the inhibitor of the MAPK-ERK1/2 kinase. To further support the involvement of MAPK-ERK1/2 signaling pathway in the regulation of NPC1L1 protein expression, we examined the effects of siRNA-mediated attenuation of ERK1 and -2 expression on the levels of cellular NPC1L1 protein. Figure 6, A and B, shows that ERK1/2 protein expression was significantly suppressed by their specific siRNA. The data presented in Fig. 6, A and B, also showed that the decrease in either ERK1 or ERK2 caused a significant reduction in NPC1L1 protein levels. Altogether, these findings strongly suggested that the inhibition of ERK-mediated signaling pathway enhances the degradation of the NPC1L1 protein in intestinal epithelial cells.
Fig. 6.

siRNA attenuation of ERK1/2 expression decreased Niemann-Pick C1 Like 1 (NPC1L1) protein expression. Stably transfected HuTu-80 cells were transfected with ERK2 (A), ERK1 (B), or scrambled siRNA for 48 h, and the protein expressions of NPC1L1, ERK2, and ERK1 were analyzed by Western blotting. NPC1L1 protein levels were quantified relative to GAPDH used as a loading control. Data are presented as means ± SE from 3 different experiments. **P < 0.001 and *P < 0.01 compared with controls.
Inhibiting ubiquitin proteasome or lysosomal pathway modulates surface expression of NPC1L1.
NPC1L1 protein is mainly found on the plasma membrane of intestinal epithelial cells (2). We then investigated the effects of blocking the cellular degradation pathways and inhibiting the MAPK-ERK1/2 signaling on NPC1L1 levels on plasma membrane. Stably transfected HuTu-80 cells were treated with MG-132, bafilomycin A1, or PD-98059 for 24 h, and the levels of NPC1L1 protein on plasma membrane were assessed by cell surface biotinylation. Similar to their effects on total cellular levels of NPC1L1, Fig. 7 shows that treatment with MG-132 and bafilomycin A1 increased surface levels of NPC1L1. Also, treatment with PD-98059 decreased the surface expression of NPC1L1 protein similar to its effect on total cellular NPC1L1. These data suggested that blocking the ubiquitin proteasome or lysosomal pathways and inhibiting the MAPK-ERK1/2 signaling pathway resulted in altering the NPC1L1 protein levels at the plasma membrane concomitant with the respective changes in total cellular levels.
Fig. 7.

Effect of blocking proteasomal or lysosomal degradation pathways or mitogen-activated protein kinase (MAPK) inhibition on the cell surface levels of Niemann-Pick C1 Like 1 (NPC1L1). Stably transfected HuTu-80 cells were treated with MG-132, bafilomycin A1, and PD-98059 for 24 h, and surface biotinylation was performed. Biotinylated proteins were separated on SDS-polyacrylamide gel, and the blots were probed with anti-hemagglutinin (HA) and GAPDH antibodies. Representative blot of 3 separate experiments is shown.
DISCUSSION
The steady-state level of NPC1L1 protein in intestinal epithelial cells is one of the major determinants of cholesterol absorption. The basal level of membrane transporter proteins in enterocytes is determined by a balance between the rate of transcription, translation, and degradation of NPC1L1 mRNA and protein in epithelial cells. Studies have previously focused on the transcriptional regulation of NPC1L1 (1, 12, 13, 15), but little is known about the pathways involved in NPC1L1 degradation under basal conditions. In this study we have demonstrated that basal levels of NPC1L1 protein are increased by the inhibition of the ubiquitin proteasome and the lysosome degradation pathways in intestinal epithelial cells. We have also shown that inhibition of ERK1/2-dependent pathway results in a significant decrease in the level of NPC1L1 protein.
To investigate the protein degradation pathways, we used an in vitro model in which NPC1L1 was exogenously expressed to eliminate any potential confounding effects of the inhibitors of these pathways on gene transcription. Inhibiting protein degradation pathways using proteasome inhibitors MG-132 and lactacystin and lysosomal inhibitor bafilomycin A1 significantly increased the basal levels of NPC1L1 protein. The increase in NPC1L1 protein levels was similar in both NPC1L1-HA-transfected intestinal HuTu-80 cells and intestinal Caco-2 cells, indicating that the observed effects were not cell line specific. Similar increase in NPC1L1 protein expression was also observed in NPC1L1- and GFP-transfected HuTu-80 cells treated with MG-132 and bafilomycin A1, suggesting that the degradation of NPC1L1 was not dependent on the attached tag in the in vitro models used in this study.
Ubiquitination is an enzymatic process that involves the attachment of ubiquitin and labeling cellular proteins for degradation by proteasome and the lysosomal machineries (5). Western blot analysis in the current studies showed two bands for NPC1L1 protein with different molecular masses: a band with a molecular mass of 145 kDa and a band with a higher molecular mass of ~280 kDa. Immunoprecipitation of NPC1L1 followed by Western blot with ubiquitin antibody suggested that both molecular mass bands of the NPC1L1 are ubiquitinated at the basal level. Because the expected molecular mass of the NPC1L1 protein is ~145 kDa, these data suggest that the NPC1L1 is a monoubiquitinated protein of ~145 kDa and a polyubiquitinated protein of ~280 kDa molecular mass in intestinal epithelial cells. It is interesting to note that incubation with the ubiquitin proteasomal (MG-132, lactacystin) and lysosomal inhibitor (bafilomycin A1) significantly increased the two molecular mass bands; however, the association of ubiquitin occurred to much greater extent with the upper band in response to the proteasomal inhibitor. Based on the data, it can be concluded that, although ubiquitinated NPC1L1 polypeptides are degraded by both proteasome and lysosome pathways, the majority of polyubiquitinated NPC1L1 is degraded through the proteasome degradation pathway. In this regard, recent studies suggest that different ubiquitin linkages mark the proteins for different degradation pathways, e.g., lysine 48 (K48) ubiquitin chains code for proteasome degradation, whereas lysine 63 (K63) ubiquitin chains code for endocytosis and lysosomal degradation of a protein (9, 11, 14). It will be interesting in future studies to examine the type of ubiquitin chain attached to NPC1L1 polypeptides.
The involvement of signal transduction pathways in modulating the expression and activity of proteins involved in cholesterol homeostasis has been recently established. Wood et al. (25) demonstrated that inhibition of MAPK reduces SR-B1 protein levels in a peroxisome proliferator-activated receptor-α-dependent manner. Also, Mulay et al. (17) demonstrated that blocking the MAPK-ERK1/2 signaling pathway increased the expression of ABCA1 and ABCG1 cholesterol transporters. These studies signified the role for MAPK signaling in controlling the degradation of receptors and transporters involved in maintaining cholesterol homeostasis. Our current findings also showed that the inhibition of MAPK-ERK1/2 pathway resulted in a decrease in the total cellular level of NPC1L1 protein concomitant with a decrease in its level on the plasma membrane. This finding was further supported by the silencing of ERK1 and ERK2 expression with gene-specific siRNA. These data further confirmed the pharmacological inhibition studies and demonstrated that the decrease in either ERK1 or ERK2 expression was sufficient to reduce the expression of NPC1L1 protein in intestinal epithelial cells. Our results expand on our current knowledge regarding MAPK pathway and show that the ERK1/2-dependent effects on NPC1L1 in intestine may be involved in fine tuning the effects of cholesterol in the liver by decreasing the expression of SR-B1-mediated uptake of cholesterol and also reducing the intestinal load on the liver mediated by NPC1L1. Collectively, the regulation of NPC1L1 by protein degradation pathways adds additional layer of complex regulation of cholesterol homeostasis in the body. Indeed, several studies provided cumulative evidence for the involvement of the ubiquitin proteasome degradation in modulating different aspects of cholesterol metabolism, including synthesis, uptake, and efflux (22). For example, the stability of the 3-hydroxy-3-methylglutaryl-coenzyme A reductase is known to increase as a compensatory mechanism in response to low levels of cholesterol. Moreover, it is possible that NPC1L1 protein stability would also increase in coordination with 3-hydroxy-3-methylglutaryl-coenzyme A reductase to enhance both the uptake and synthesis of cholesterol in cases where cholesterol levels are low. We speculate that processes of NPC1L1 protein degradation in the liver and intestine are coordinated to reduce overall cholesterol intake in the body via decreasing cholesterol uptake in hepatocytes and enterocytes, as one of the major mechanisms to control serum level of cholesterol and prevent hypercholesterolemia.
In conclusion, the findings presented in this study demonstrate that the steady-state levels of the intestinal NPC1L1 cholesterol transporter could be altered by the inhibition of the proteasomal and lysosomal pathways under basal conditions. Furthermore, inhibiting MAPK-ERK1/2 signaling decreased NPC1L1 protein expression. The results regarding the involvement of ERK1/2 kinases in NPC1L1 protein degradation, along with previous findings related to other cholesterol transporters, suggest a molecular link between cellular protein degradation pathways and the maintenance of cholesterol homeostasis in the body that could be exploited for the treatment of hypercholesterolemia.
GRANTS
These studies were supported by a Veterans Affairs (VA) Research Career Scientist Award to W. A. Alrefai and a Merit Award No. BX000152 (W. A. Alrefai). The studies were also partially supported by NIH/National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-109709 (W. A. Alrefai), DK-81858 (P. K. Dudeja), DK-54016 (P. K. Dudeja), DK-92441 (P. K. Dudeja), and DK-98170 (R. K. Gill). P. K. Dudeja is supported by a VA Senior Research Career Scientist Award and by a Merit Award (BX002011). S. Saksena is supported by VA Merit Award (BX002867).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.M. and W.A.A. conceived and designed research; P.M., V.S., Y.Y., T.T., and H.S. performed experiments; P.M., R.K.G., S.S., P.K.D., and W.A.A. analyzed data; P.M., R.K.G., and W.A.A. interpreted results of experiments; P.M. prepared figures; P.M. drafted manuscript; P.M., R.K.G., S.S., P.K.D., and W.A.A. edited and revised manuscript; P.M., Y.Y., T.T., H.S., R.K.G., S.S., P.K.D., and W.A.A. approved final version of manuscript.
REFERENCES
- 1.Alrefai WA, Annaba F, Sarwar Z, Dwivedi A, Saksena S, Singla A, Dudeja PK, Gill RK. Modulation of human Niemann-Pick C1-like 1 gene expression by sterol: role of sterol regulatory element binding protein 2. Am J Physiol Gastrointest Liver Physiol 292: G369–G376, 2007. doi: 10.1152/ajpgi.00306.2006. [DOI] [PubMed] [Google Scholar]
- 2.Altmann SW, Davis HR Jr, Zhu LJ, Yao X, Hoos LM, Tetzloff G, Iyer SP, Maguire M, Golovko A, Zeng M, Wang L, Murgolo N, Graziano MP. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science 303: 1201–1204, 2004. doi: 10.1126/science.1093131. [DOI] [PubMed] [Google Scholar]
- 3.Arancibia-Cárcamo IL, Yuen EY, Muir J, Lumb MJ, Michels G, Saliba RS, Smart TG, Yan Z, Kittler JT, Moss SJ. Ubiquitin-dependent lysosomal targeting of GABA(A) receptors regulates neuronal inhibition. Proc Natl Acad Sci USA 106: 17552–17557, 2009. doi: 10.1073/pnas.0905502106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Betters JL, Yu L. NPC1L1 and cholesterol transport. FEBS Lett 584: 2740–2747, 2010. doi: 10.1016/j.febslet.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonifacino JS, Weissman AM. Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu Rev Cell Dev Biol 14: 19–57, 1998. doi: 10.1146/annurev.cellbio.14.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chevallier J, Chamoun Z, Jiang G, Prestwich G, Sakai N, Matile S, Parton RG, Gruenberg J. Lysobisphosphatidic acid controls endosomal cholesterol levels. J Biol Chem 283: 27871–27880, 2008. doi: 10.1074/jbc.M801463200. [DOI] [PubMed] [Google Scholar]
- 7.Cohen JC, Pertsemlidis A, Fahmi S, Esmail S, Vega GL, Grundy SM, Hobbs HH. Multiple rare variants in NPC1L1 associated with reduced sterol absorption and plasma low-density lipoprotein levels. Proc Natl Acad Sci USA 103: 1810–1815, 2006. doi: 10.1073/pnas.0508483103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davies JP, Scott C, Oishi K, Liapis A, Ioannou YA. Inactivation of NPC1L1 causes multiple lipid transport defects and protects against diet-induced hypercholesterolemia. J Biol Chem 280: 12710–12720, 2005. doi: 10.1074/jbc.M409110200. [DOI] [PubMed] [Google Scholar]
- 9.Ferreira JV, Soares AR, Ramalho JS, Pereira P, Girao H. K63 linked ubiquitin chain formation is a signal for HIF1A degradation by Chaperone-Mediated Autophagy. Sci Rep 5: 10210, 2015. doi: 10.1038/srep10210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Calvo M, Lisnock J, Bull HG, Hawes BE, Burnett DA, Braun MP, Crona JH, Davis HR Jr, Dean DC, Detmers PA, Graziano MP, Hughes M, Macintyre DE, Ogawa A, O’neill KA, Iyer SP, Shevell DE, Smith MM, Tang YS, Makarewicz AM, Ujjainwalla F, Altmann SW, Chapman KT, Thornberry NA. The target of ezetimibe is Niemann-Pick C1-Like 1 (NPC1L1). Proc Natl Acad Sci USA 102: 8132–8137, 2005. doi: 10.1073/pnas.0500269102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang F, Zeng X, Kim W, Balasubramani M, Fortian A, Gygi SP, Yates NA, Sorkin A. Lysine 63-linked polyubiquitination is required for EGF receptor degradation. Proc Natl Acad Sci USA 110: 15722–15727, 2013. doi: 10.1073/pnas.1308014110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iwayanagi Y, Takada T, Suzuki H. HNF4alpha is a crucial modulator of the cholesterol-dependent regulation of NPC1L1. Pharm Res 25: 1134–1141, 2008. doi: 10.1007/s11095-007-9496-9. [DOI] [PubMed] [Google Scholar]
- 13.Kumar P, Malhotra P, Ma K, Singla A, Hedroug O, Saksena S, Dudeja PK, Gill RK, Alrefai WA. SREBP2 mediates the modulation of intestinal NPC1L1 expression by curcumin. Am J Physiol Gastrointest Liver Physiol 301: G148–G155, 2011. doi: 10.1152/ajpgi.00119.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee S, Henderson MJ, Schiffhauer E, Despanie J, Henry K, Kang PW, Walker D, McClure ML, Wilson L, Sorscher EJ, Zeitlin PL. Interference with ubiquitination in CFTR modifies stability of core glycosylated and cell surface pools. Mol Cell Biol 34: 2554–2565, 2014. doi: 10.1128/MCB.01042-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malhotra P, Boddy CS, Soni V, Saksena S, Dudeja PK, Gill RK, Alrefai WA. D-Glucose modulates intestinal Niemann-Pick C1-like 1 (NPC1L1) gene expression via transcriptional regulation. Am J Physiol Gastrointest Liver Physiol 304: G203–G210, 2013. doi: 10.1152/ajpgi.00288.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malhotra P, Soni V, Kumar A, Anbazhagan AN, Dudeja A, Saksena S, Gill RK, Dudeja PK, Alrefai WA. Epigenetic modulation of intestinal cholesterol transporter Niemann-Pick C1-like 1 (NPC1L1) gene expression by DNA methylation. J Biol Chem 289: 23132–23140, 2014. doi: 10.1074/jbc.M113.546283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mulay V, Wood P, Manetsch M, Darabi M, Cairns R, Hoque M, Chan KC, Reverter M, Alvarez-Guaita A, Rye KA, Rentero C, Heeren J, Enrich C, Grewal T. Inhibition of mitogen-activated protein kinase Erk1/2 promotes protein degradation of ATP binding cassette transporters A1 and G1 in CHO and HuH7 cells. PLoS One 8: e62667, 2013. doi: 10.1371/journal.pone.0062667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muthusamy S, Malhotra P, Hosameddin M, Dudeja AK, Borthakur S, Saksena S, Gill RK, Dudeja PK, Alrefai WA. N-glycosylation is essential for ileal ASBT function and protection against proteases. Am J Physiol Cell Physiol 308: C964–C971, 2015. doi: 10.1152/ajpcell.00023.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Myocardial Infarction Genetics Consortium I; Stitziel NO, Won HH, Morrison AC, Peloso GM, Do R, Lange LA, Fontanillas P, Gupta N, Duga S, Goel A, Farrall M, Saleheen D, Ferrario P, Konig I, Asselta R, Merlini PA, Marziliano N, Notarangelo MF, Schick U, Auer P, Assimes TL, Reilly M, Wilensky R, Rader DJ, Hovingh GK, Meitinger T, Kessler T, Kastrati A, Laugwitz KL, Siscovick D, Rotter JI, Hazen SL, Tracy R, Cresci S, Spertus J, Jackson R, Schwartz SM, Natarajan P, Crosby J, Muzny D, Ballantyne C, Rich SS, O’Donnell CJ, Abecasis G, Sunaev S, Nickerson DA, Buring JE, Ridker PM, Chasman DI, Austin E, Kullo IJ, Weeke PE, Shaffer CM, Bastarache LA, Denny JC, Roden DM, Palmer C, Deloukas P, Lin DY, Tang ZZ, Erdmann J, Schunkert H, Danesh J, Marrugat J, Elosua R, Ardissino D, McPherson R, Watkins H, Reiner AP, Wilson JG, Altshuler D, Gibbs RA, Lander ES, Boerwinkle E, Gabriel S, Kathiresan S. Inactivating mutations in NPC1L1 and protection from coronary heart disease. N Engl J Med 371: 2072–2082, 2014. doi: 10.1056/NEJMoa1405386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Narushima K, Takada T, Yamanashi Y, Suzuki H. Niemann-pick C1-like 1 mediates alpha-tocopherol transport. Mol Pharmacol 74: 42–49, 2008. doi: 10.1124/mol.107.043034. [DOI] [PubMed] [Google Scholar]
- 21.Ogura M, Ayaori M, Terao Y, Hisada T, Iizuka M, Takiguchi S, Uto-Kondo H, Yakushiji E, Nakaya K, Sasaki M, Komatsu T, Ozasa H, Ohsuzu F, Ikewaki K. Proteasomal inhibition promotes ATP-binding cassette transporter A1 (ABCA1) and ABCG1 expression and cholesterol efflux from macrophages in vitro and in vivo. Arterioscler Thromb Vasc Biol 31: 1980–1987, 2011. doi: 10.1161/ATVBAHA.111.228478. [DOI] [PubMed] [Google Scholar]
- 22.Sharpe LJ, Cook EC, Zelcer N, Brown AJ. The UPS and downs of cholesterol homeostasis. Trends Biochem Sci 39: 527–535, 2014. doi: 10.1016/j.tibs.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 23.Tveten K, Ranheim T, Berge KE, Leren TP, Kulseth MA. The effect of bafilomycin A1 and protease inhibitors on the degradation and recycling of a Class 5-mutant LDLR. Acta Biochim Biophys Sin (Shanghai) 41: 246–255, 2009. doi: 10.1093/abbs/gmp008. [DOI] [PubMed] [Google Scholar]
- 24.Wang LJ, Wang J, Li N, Ge L, Li BL, Song BL. Molecular characterization of the NPC1L1 variants identified from cholesterol low absorbers. J Biol Chem 286: 7397–7408, 2011. doi: 10.1074/jbc.M110.178368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wood P, Mulay V, Darabi M, Chan KC, Heeren J, Pol A, Lambert G, Rye KA, Enrich C, Grewal T. Ras/mitogen-activated protein kinase (MAPK) signaling modulates protein stability and cell surface expression of scavenger receptor SR-BI. J Biol Chem 286: 23077–23092, 2011. doi: 10.1074/jbc.M111.236398. [DOI] [PMC free article] [PubMed] [Google Scholar]