

Abstract
Choroid plexus epithelial cells (CPECs) secrete cerebrospinal fluid (CSF). They express Na+-K+-ATPase and Na+-K+-2Cl− cotransporter 1 (NKCC1) on their apical membrane, deviating from typical basolateral membrane location in secretory epithelia. Given this peculiarity, the direction of basal net ion fluxes mediated by NKCC1 in CPECs is controversial, and cotransporter function is unclear. Determining the direction of basal NKCC1-mediated fluxes is critical to understanding the function of apical NKCC1. If NKCC1 works in the net efflux mode, it may be directly involved in CSF secretion. Conversely, if NKCC1 works in the net influx mode, it would have an absorptive function, contributing to intracellular Cl− concentration ([Cl−]i) and cell water volume (CWV) maintenance needed for CSF secretion. We resolve this long-standing debate by electron microscopy (EM), live-cell-imaging microscopy (LCIM), and intracellular Na+ and Cl− measurements in single CPECs of NKCC1+/+ and NKCC1−/− mouse. NKCC1-mediated ion and associated water fluxes are tightly linked, thus their direction is inferred by measuring CWV changes. Genetic or pharmacological NKCC1 inactivation produces CPEC shrinkage. EM of NKCC1−/− CPECs in situ shows they are shrunken, forming large dilations of their basolateral extracellular spaces, yet remaining attached by tight junctions. Normarski LCIM shows in vitro CPECs from NKCC1−/− are ~17% smaller than NKCC1+/+. CWV measurements in calcein-loaded CPECs show that bumetanide (10 μM) produces ~16% decrease in CWV in NKCC1+/+ but not in NKCC1−/− CPECs. Our findings suggest that under basal conditions apical NKCC1 is continuously active and works in the net inward flux mode maintaining [Cl−]i and CWV needed for CSF secretion.
Keywords: apical NKCC1, cell volume, choroid plexus epithelial cells, NKCC1−/−, ultrastructure
INTRODUCTION
The choroid plexuses (CPs) are highly vascularized epithelial structures that reside in the brain ventricles, where they float in the surrounding cerebrospinal fluid (CSF) they secrete. Forming the blood-CSF barrier, CPs are composed of a monolayer of cuboidal epithelial cells held together by junctional complexes at their apical side and surrounding a core of fenestrated capillaries facing the basolateral membrane (68). Choroid plexus epithelial cells (CPECs) secrete most of the CSF and regulate its ionic composition through poorly understood mechanisms (9, 71). CPECs are unique among Cl− secretory epithelial cells because the Na+-K+-2Cl− cotransporter NKCC1 (SLC12A1) and the Na+-K+ transporting adenosine triphosphatase (Na+-K+-ATPase) are located on their apical membrane (CSF-facing), deviating from typical basolateral membrane (blood-facing) location (3, 15). Given this noncanonical location, NKCC1 function in CPECs is not understood, and the direction of net ion fluxes mediated by this cotransporter and associated water fluxes under basal physiological conditions is controversial (10, 28, 71).
Understanding NKCC1 function in CPECs is clinically relevant because this protein and the kinases involved in its regulation are potential pharmacological targets to control CSF secretion (52, 103, 105). Pharmacological tools for modulation of CSF secretion are critically needed for the treatment of neurological disorders in which there is an uncontrolled increase in intracranial pressure, such as idiopathic intracranial hypertension (38), hydrocephalus (43, 44), and traumatic and ischemic brain injury (12, 51). If under basal physiological conditions NKCC1 works in the net efflux mode, it may be directly involved in CSF secretion as suggested by some investigators (36, 46, 72, 81, 91, 92). Two sets of observations have been taken as evidence backing up this hypothesis: first, intraventricular bumetanide, an inhibitor of Na+-K+-2Cl− cotransporters (NKCCs), significantly reduces CSF secretion; and, second, estimated intracellular Na+ is higher in CPECs than in other epithelial cells, making NKCC1-mediated net ion efflux thermodynamically feasible.
The evidence for the first assumption originated from experiments showing that intraventricular bumetanide in canines significantly reduced (50%) CSF secretion (35) and Cl− fluxes associated with CSF secretion (37). In these early studies, NKCC was assumed to be localized to the basolateral membrane of CPECs (34), contributing to CSF secretion as in the classic models of Cl− secretory epithelia (23, 24, 87, 88). A new study shows that intraventricular bumetanide (100 µM) in mice decreases CSF production by ~50%, confirming earlier observations in canines (92). The second argument proposed for NKCC1 working in the net efflux mode stems from estimates of intracellular Na+ concentration ([Na+]i) in CPECs, derived from whole CP tissue measurements (36, 46, 62, 90, 92). The relatively high estimates of [Na+]i (30–50 mM) and the observations that bumetanide (10–30 µM) causes 75% inhibition of K+ efflux in rat CP in vitro and that cerebroventricular bumetanide inhibits CSF and Cl− secretion at concentrations of 100 and 10 µM, respectively (35, 37), have been taken together as functional evidence for an apical NKCC that contributes to CSF secretion by working in the net efflux, secretory, mode (46, 92).
Following the cloning of NKCCs, it was definitely established that the cotransporter expressed in CP was NKCC1 (70). Immunolabeling microscopy revealed that NKCC1 was located to the apical membrane of rat (70), mouse (69), and human CPECs (72). This finding required a total reappraisal of the mechanism of CSF secretion and the functional role of apical NKCC1 in this Cl− secretory epithelium. The debate about the directionality of NKCC1-mediated net solute fluxes reached a high point with the proposal by Wu and coworkers (98) that apical NKCC1 is “constitutively active” and mediates net influx of ions in CPECs. They proposed that NKCC1 functions as a K+-reabsorption mechanism from CSF to blood. Accordingly, inhibition of NKCC1 should produce shrinkage of CPECs due to unbalanced solute and water efflux pathways. They tested these hypotheses using isolated single CPECs from rat and estimating cell volume changes by differential interference contrast (DIC) measurement of cross-sectional areas (CSAs). They found that bumetanide at high concentrations (100 µM) caused a 9% decrease in cell volume, whereas increasing external K+ concentration to 25 mM produced a 33% increase in cell volume that was blocked by bumetanide (100 µM) or external Na+ removal.
The experiments of Wu and coworkers (98) have been criticized because of the high concentration of bumetanide used, which, admittedly, can have effects in other transporters expressed in the CP (10). The possibility of nonspecific actions of bumetanide was tested by Brown and coworkers (10, 30). They concluded that the decrease in cell volume reported by Wu and coworkers (98) could be replicated using bumetanide at concentrations of 100 µM but not at 10 µM (10, 30), a concentration known to produce selective and complete block of NKCC1 (3, 83). Thus, based on their results, Brown and coworkers (10, 30) questioned the conclusions of Wu et al. (98) regarding a role of NKCC1 in cell volume maintenance.
In the present study, we address the apical NKCC1 debate reviewed above, by using a combined methodological approach in dissociated CPECs and in in situ CP of NKCC1 knockout (KO) models. Changes in cell water volume (CWV) were measured in single CPECs loaded with the fluorescent dye calcein, a live-cell-imaging fluorescence microscopy method that, unlike DIC, is independent of changes in cell shape and allows “real-time” measurements of CWV in single CPECs with time resolution of <1 Hz and sensitivity of ~1% (4, 5, 14, 79). Intracellular Na+ and Cl− concentrations were measured in single CPECs with fluorescent indicators. DIC live-cell images were also used to measure steady-state CSAs and immunoelectron microscopy was used to determine the subcellular location of NKCC1. Our results show that genetic and pharmacological inactivation of apical NKCC1 produces significant shrinkage of choroid plexus epithelial cells, both in vitro and in situ. Measured [Na+]i and [Cl−]i energetically favor NKCC1 net solute uptake under basal conditions. Inactivation of NKCC1 results in a significant decrease in [Cl−]i. Taken together, these results demonstrate that under basal physiological conditions the cotransporter is continuously active and works in the net inward flux mode maintaining cell water volume and [Cl−]i needed for CSF secretion.
MATERIALS AND METHODS
Animal Husbandry, Treatment, Euthanasia, and Tissue Collection
The use and handling of all animals in this study were approved by the Wright State University Institutional Animal Care and Use Committee, in accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals. Two NKCC1−/− mouse lines were used. One line was generated by deletion of exon 9 of the Slc12a2 gene on C57 black background animals (17). These animals were kindly donated by Prof. Eric Delpire (Vanderbilt University School of Medicine) and were used for electron microscopy (EM) and initial immunolabeling studies. The second line, on a 129 Black Swiss mixed background, came from a colony raised at Wright State University Laboratory Animal Resources facility from breeding pairs generously donated by Prof. Gary Shull (University of Cincinnati College of Medicine). Mice from this colony lacked exon 6 of the Slc12a2 gene (22). These animals were also used in some immunofluorescence experiments and in all of the functional experiments described in this study.
Animals were housed in an American Association for the Accreditation of Laboratory Animal Care (now Association for Assessment and Accreditation of Laboratory Animal Care International)-certified facility, and procedures were completed in accordance with federal guidelines and regulations. Food (Teklad; Envigo, Madison, WI) and tap water were provided ad libitum with water removed at the time of the experiment. Aspen chip bedding (Teklad; Envigo) and nesting material (Nestlets; Ancare, Bellmore, NY) were provided. Lighting was maintained on a 12:12-h light-dark cycle, and ambient temperature was maintained at 23.3 ± 2.2°C. It has been reported that NKCC1−/− mice exhibit hyposalivation in response to muscarinic agonists (21). This condition might become a problem after weaning. To avoid this potential discomfort, most animals were used on or before postnatal day 21 (P21). Animals older than P21 were supplied with moist food after weaning.
To optimize tissue preservation for immunofluorescence and EM microscopy studies, fixation was done in animals that were deeply anesthetized with pentobarbital sodium (50 mg/kg ip). In subsequent histological experiments, animals were anesthetized with Euthasol (pentobarbital sodium and phenytoin sodium), injected intraperitoneally (270 mg/kg). In all cases, deep anesthesia was continuously monitored before thoracotomy, exsanguination, and perfusion. Depth of anesthesia was assessed regularly by testing withdrawal reflexes by pinching of the toes, ears, and tail. Mice for experiments on dissociated CPECs, or those in excess/ill/moribund, were euthanized by CO2 anesthesia followed by decapitation, in accordance with AVMA Guidelines for the Euthanasia of Animals: 2013 Edition (American Veterinary Medical Association, Schaumburg, IL).
Antibodies and Immunofluorescence Microscopy
For immunofluorescence microscopy, deeply anesthetized animals were perfused transcardially with 200 ml of 2–4% paraformaldehyde in 0.1 M PBS, pH 7.3. The brain and other tissues were extracted and postfixed for 1–2.5 h in the same fixative solution and stored at 4°C in 0.1 M PBS with 15% sucrose until used. Frozen sections (cryostat, 20 µm thick or freezing sliding microtome, 50 µm thick) were collected on gelatinized slides (cryostat) or processed free floating (freezing sliding). After wash (3 × 5 min) in 0.01 M PBS with 0.1% Triton X-100 (PBS-T; pH 7.4), sections were blocked with 10% normal donkey or horse serum for 60 min and incubated overnight at 4°C with the corresponding primary antibodies diluted in PBS-T. For detection of NKCC1, we used an affinity-purified polyclonal antibody raised in rabbits against a fusion protein fragment encompassing amino acids 938–1,011 of the carboxy terminus (CT) of mouse NKCC1 (41). We refer to this antibody provided by Prof. Delpire as Kaplan-CT. It recognizes both NKCC1 variants, long (NKCC1a) and short (NKCC1b), when tested with maltose-binding fusion proteins with or without exon 21 (56).
The specificity of the Kaplan-CT antibody toward NKCC1 has been thoroughly validated in NKCC1−/− tissues, including choroid plexus epithelium (56, 69, 93). In the present study, we verified once more that NKCC1 immunoreactivity detected with this antibody in the apical membrane of NKCC1+/+ CPECs disappeared in NKCC1−/−. As positive controls, the same antibody was tested in other cell types where NKCC1 expression is well established. These positive controls included mouse and rat dorsal root ganglion (DRG) neurons (56, 93), the basolateral membrane of α-intercalated cells in the outer medullary collecting duct of kidney (56), and the basolateral membrane of mouse salivary glands (69). The Kaplan-CT NKCC1 antibody was used at dilutions of 1:50 to 1:400 in PBS-T. Sections were incubated with primary antibody overnight at 4°C in a humid chamber and washed 3 × 5 min with PBS-T followed by incubation (2.5 h) with FITC anti-rabbit secondary antibodies (Jackson ImmunoResearch, West Grove, PA) diluted 1:50 in PBS-T. The sections were coverslipped with VECTASHIELD.
NKCC1 was also detected using affinity-purified monoclonal antibody (MAb) T4. The unpurified T4 MAb supernatant came from the Developmental Studies Hybridoma Bank, University of Iowa and was affinity-purified as previously described (6). This MAb was raised against a fusion protein fragment of the CT of the human colonic NKCC1 (55). NKCC1 immunoreactivity detected with this MAb in the apical membrane of NKCC1+/+ CPECs disappeared in NKCC1−/−. Further tests of NKCC1-specific immunodetection using T4 MAb have been recently reported by another group (47). Further, we observed the same pattern of NKCC1 immunoreactivity in CPECs with an antibody (data not shown) raised against the NH2 terminus of NKCC1: α-wNT (56). The α-wNT NKCC1 antibody was also validated against NKCC1−/− CPECs. The Kaplan-CT antibody targeted sequences of amino acids in the COOH terminus of mouse NKCC1, whereas α-wNT targeted an amino acid sequence located in the NH2 terminus of rat NKCC1. All confocal immunofluorescence and ultrastructural studies (see below) were done in CPs of lateral ventricles. Unpublished studies in our laboratory using the α-wNT NKCC1 antibody reveal the same pattern of immunoreactivity in the CP of the third and fourth ventricles.
Electron Microscopy
Two NKCC1−/− mice lacking exon 9 of the Slc12a2 gene (17) and their corresponding controls were perfusion-fixed with 2% glutaraldehyde and 2% paraformaldehyde in 0.1 M phosphate buffer (PB; pH 7.3) and postfixed for 1 h in the same fixative solution. After thorough washes in PBS, pH 7.2, we obtained 50-µm-thick brain sections using a vibratome. The sections were treated with freshly made 2% OsO4 in 0.1 M PB with 5% sucrose for 1 h. After washing the OsO4 with PB, the sections were dehydrated 2 × 5 min in ascending ethanol (EtOH) diluted in double-distilled water (50, 70, and 95%) and 3 × 10 min in 100% EtOH, ending in propylene oxide (2 × 5 min). The dehydrated sections were then infiltrated overnight in resin made up of a mixture of 1:1 propylene oxide and Epon-Araldite. The next day, the sections were flat-embedded in Epon-Araldite between two Teflon-coated coverslips. One percent of “depleted” uranyl acetate was added to the first 70% alcohol step and left staining “en bloc” for 20 min before proceeding with subsequent alcohol dehydration steps. The “wafer” embedded sections were cured at 64°C for 4–6 days and then examined in a microscope. Areas containing choroid plexus were recut and “Super Glued” to the top of an EM beam capsule, and ultrathin sections were obtained (silver-gold approximately 80–90 nm thick) in a Sorvall MT-5000 Ultra Microtome and collected in 200 mesh nickel grids. The sections were then counterstained with standard Reynolds lead citrate and observed in a Phillips EM208S transmission electron microscope at 70 kV. Imaging was done with standard photographic 4 × 5 EM plates. For presentation, all photographic images were scanned in a conventional Hewlett-Packard scanner.
Pre-Embedding Immunoelectron Microscopy
To study the subcellular localization of NKCC1 in CPECs, we used pre-embedding immunoperoxidase avidin-biotin complex (ABC)-diaminobenzidine (DAB) electron microscopy, a method in which immunolabeling is done before embedding, having the advantage that antigenic sites are not modified by osmication and embedding in hydrophobic resins used for maximal ultrastructural definition (73). For pre-embedding immunoelectron microscopy, the animals were perfusion-fixed with a lower concentration of glutaraldehyde to maximize antigen preservation. The fixative mixture thus consisted of 4% paraformaldehyde and 0.25% glutaraldehyde in 0.1 M PB. After extraction, the brains were postfixed for an additional 1–2.5 h in the same fixative and then washed overnight in 15% sucrose in 0.01 M PBS. The following day, 50-µm-thick sections were obtained with a vibratome. To reduce glutaraldehyde fixation and increase antibody penetration, the sections were treated for 15 min with 1% NaBH4 and then thoroughly washed in PBS. After the sections were blocked with normal goat serum (1:10 in 0.1 M PBS), they were incubated overnight at room temperature and under constant agitation with one of two different primary antibodies (Abs) against NKCC1: Kaplan-CT diluted 1:50 in PBS without Triton X-100 or T4 MAb diluted 1:100 in PBS without Triton X-100. The following day, immunoreactive sites were revealed with rabbit or mouse ABC kits (all diluted in 0.01 M PBS; Vector, Burlingame, CA), and peroxidase histochemistry was performed using 0.02% DAB and 0.01% H2O2 diluted in 0.5 M Tris buffer (pH 7.4). Then, the sections were osmicated, dehydrated, and flat-embedded as described above for conventional EM. Ultramicrotomy was performed as for conventional EM, but the ultrathin sections were not counterstained with lead citrate; they were only stained with uranyl acetate in the 70% alcohol step to maximize contrast visualization in the EM of the DAB precipitate labeling of immunoreactivity sites. Thus contrast between EM images in Fig. 1, C and D, and those in Figs. 3 and 4 is different. The sections were observed in the same transmission electron microscope but this time at 60 kV. Photography was done as for conventional EM but using higher-contrast photographic paper to visualize more clearly ultrastructural details below the immunoreaction. The Abs used for pre-embedding immunoelectron microscopy confirmed that NKCC1 immunoreactivity was restricted to the apical membrane of CPECs from NKCC1+/+ and was absent in CPECs of NKCC1−/−.
Fig. 1.

Na+-K+-2Cl− cotransporter 1 (NKCC1) immunolocalization in mouse choroid plexus epithelium (CPE). A: low-magnification confocal image of CPE. NKCC1 immunoreactivity is localized in the apical surface (arrows) of CPE. Apical side of CPECs faces the ventricle (V) and is in direct contact with cerebrospinal fluid. Top left shows adjacent brain tissue facing the ventricle. NKCC1 immunoreactivity is barely detectable in ependymal cells lining the ventricular surface (arrowheads). B: high-magnification confocal image of a segment of CPE showing NKCC1 immunoreactivity in the apical surface (arrows). Note the profiles of cuboidal CPECs. NKCC1 immunoreactivity was undetectable in the basolateral side of the CPE (arrowheads). Calibration bars: A, 100 μm; B, 20 μm. Primary anti-NKCC1 was a polyclonal antibody (Ab) raised in rabbits against a fusion protein fragment of the COOH terminus (CT) of mouse NKCC1 (Kaplan-CT; 1:50), and secondary Ab was FITC anti-rabbit (1:50) in PBS-Triton X-100. C and D: ultrastructural immunolocalization of NKCC1 in mouse CPECs using pre-embedding immunoperoxidase avidin-biotin complex-diaminobenzidine methods and 2 different primary antibodies targeting NKCC1. C: Kaplan-CT Ab. D: T4 monoclonal Ab. Both antibodies reveal NKCC1 is localized in the microvilli (mv) on the apical side of the cell (i.e., the luminal side facing ventricle/cerebrospinal fluid). Examples of the NKCC1 immunoreactivity in mv are indicated by arrows in C and D. NKCC1 immunolabeling was absent in the basolateral membrane indicated by arrowheads in the lower right of C, where the basal lamina (bl) of the basement membrane is indicated with an arrow.
Fig. 3.

Ultrastructural changes of choroid plexus epithelium (CPE) in Na+-K+-2Cl− cotransporter 1 (NKCC1) knockout mouse. A: low-magnification electron micrograph of CPE in wild-type mouse (NKCC1+/+). The CPE is formed by a monolayer of cells, the apical surface of which is packed with microvilli (mv). Cells are held together by intercellular junctions (IJ) at the apical ends of the lateral plasma membrane of contiguous cells. In basal regions, the plasma membrane forms extensive infoldings (inf). Cells are anchored to a basement membrane (bm) composed of connective tissue, capillaries (cap), and a basal lamina (bl). B: choroid plexus epithelial cells (CPECs) of NKCC1−/− mouse exhibit large dilations of the extracellular space between the lateral plasma membrane of contiguous cells (asterisks) and between the basal plasma membranes (arrowheads) and the bl of the bm. CPECs remain attached to each other through apical IJs, and their retracted basal plasma membrane remains anchored to the bl of the bm, forming fingerlike extensions (arrowheads). Microvilli (mv) of these cells appear smaller than in wild-type mice.
Fig. 4.

High-power electron micrograph of choroid plexus of wild-type mouse compared with that of Na+-K+-2Cl− cotransporter 1 (NKCC1) knockout mouse. A: choroid plexus epithelial cells (CPECs) of NKCC1 wild-type mouse (NKCC1+/+) show normal ultrastructure. Their apical (cerebrospinal fluid-facing) surface exhibits abundant microvilli (mv), and the cells are joined together at their apical ends by an intercellular junctional complex: a zonula occludens or tight junction (tj) and, beneath this, a zonula adherens (za) or adherens junction. Beyond this junctional complex, the intercellular space exhibits modest dilations that gradually expand toward the basal pole, at which point the basolateral membrane forms a series of large infoldings. Basal end of the cells (e.g., left bottom corner) is relatively smooth and is anchored to the basal lamina (bl) of the basement membrane. Cytoplasm contains abundant mitochondria (mit), a nucleus (Nuc), and some vesicular bodies (vb). B: CPECs of NKCC1 knockout mouse (NKCC1−/−) are shrunken, and their mv appear less abundant and smaller than wild-type mouse. Shrunken CPECs remain joined through the tj and the za. Beneath the za, there is a large dilation of the intercellular space (asterisk) resulting from cell shrinkage. Basolateral membrane is retracted, and the extracellular spaces in between the basal infoldings are greatly enlarged (stars). Basolateral membrane forms fingerlike extensions that remain attached to the bl of the basement membrane (bm).
Genotyping and Identification of Transcripts of NKCC1 by RT-PCR
Genotyping procedures.
Mice were genotyped by PCR using genomic DNA extracted from 1 to 3 mm of tissue obtained from tail clips during P3–P6. The genotyping procedure for the NKCC1−/− mice strain lacking exon 6 of the Slc12a2 gene was as described by Shull’s group (22), using three primers in the reaction mixture: sense (5′-GGA ACA TTC CAT ACT TAT GAT AGA TG-3′); antisense (5′-CTC ACC TTT GCT TCC CAC TCC ATT CC-3′); and neo (5′-GAC AAT AGC AGG CAT GCT GG-3′). Genealogy and data for each mouse were recorded using GenoPro software (https://www.genopro.com). As for NKCC1−/− lacking exon 9 of the Slc12a2 (17), they arrived at Wright State University Laboratory Animal Resources facility already genotyped from Prof. Delpire’s laboratory.
PCR amplification of NKCC1 transcripts.
The strategy to amplify variants of the Slc12a2 gene, as well as the methods followed, were essentially the same as those described in previous work (56). These variants are the full length (18), here referred to as NKCC1a, and a shorter variant lacking exon 21, here referred to as NKCC1b. In previous work, we (56) called the long-full-length variant NKCC1-L and the short NKCC1-S. The primer sets to coamplify both variants, NKCC1a (502 bp) and NKCC1b (453 bp), were NKCC1-502a/453b: sense (5′-TGT GGT CAT TCG CCT AAA GGA AGG ACT GGA TAT ATC-3′); and antisense (5′-GGA GAA GTC TAT TCG GAA TTT ACT GAG TAA-3′). RT-PCR was done using 1–1.5 µg of total RNA extracted from juvenile/adult (P12–P29) mouse’s brain, choroid plexus, spinal cord, kidney, and DRG tissues. Negative controls were performed replacing cDNA from the respective tissues with water (H2O) and using NKCC1-502a/453b as the PCR primer set. Densitometric analysis was performed using the open source ImageJ software package v1.42 for Linux.
Isolation of Single Choroid Plexus Epithelial Cells
The method for isolation of single CPECs was adapted from that described for rat CPECs (98) and Necturus gallbladder epithelial cells (94). Mice, P10–P21, were anesthetized using CO2 and then euthanized by rapid decapitation. The brain was removed and immediately submersed into a dish containing Dulbecco’s PBS (no Ca2+, no Mg2+, pH 7.1; Life Technologies) at room temperature. The bottom of the dish had a silicone lining to affix the brain with pins and visualize it under a ×20 dissecting microscope. The CP of the fourth ventricle was dissected out with watchmaker forceps (no. 4) and incubated for 1 h in 1 ml of freshly made dissociation medium composed of culture medium (see below) supplemented with protease XIV (0.5 mg/ml; Sigma-Aldrich) and collagenase IV (0.5 mg/ml; Sigma-Aldrich). The culture medium was prepared the day of the experiment and contained DMEM/F-12 (HyClone; Fisher), 10% heat-inactivated FBS (Gibco; Fisher), penicillin-streptavidin (10 U/ml-10 µg/ml; HyClone; Fisher), and human EGF (40 ng/ml). The medium was filtered at room temperature using a 10-ml Luer-Lock syringe attached to a sterile filter (33-mm Millex-GP Xpress PES 0.22 µm; Merck Millipore). Following the incubation period, the dissociation medium was aspirated off, and the CP was rinsed three times with culture medium and suspended in 500-µl culture medium.
To dissociate the CPECs, the CP suspended in culture medium was vigorously shaken with an electronic vortex for ~3 s, after which the cells were gently triturated with fire-polished glass Pasteur pipettes of two different bore diameters (equivalent to the inner diameter of 23- and 32-gauge needles and used in that order), 20 times per bore size. Cells were plated on 25-mm sterile coverslips (no. 72196-25; Electron Microscopy Sciences) previously degreased and cleaned with acetone, 70% EtOH, and double-distilled water and coated with laminin (5 µg/ml; no. L2020; Sigma-Aldrich) and poly-d-lysine (100 µg/ml; no. 354210; BD Bioscience). The coverslips with the plated cells were covered with culture medium (~250 µl) contained in petri dishes and placed in an incubator at 5% CO2-95% air atmosphere, 85–95% humidity, 37°C, for 2–6 h before use for live-cell-imaging experiments.
Solutions
The isosmotic physiological control (ISO) solution contained, in mM, 123.5 NaCl, 3 KCl, 1.0 CaCl2, 1.25 MgCl2, 5 HEPES, and 10 glucose. The pH was adjusted to 7.3 at 25°C with NaOH, and the osmolality was adjusted with sucrose to 290 ± 1% mosmol/kgH2O.
The anisosmotic calibration solutions for CWV measurements were prepared by adjusting the amount of sucrose in the ISO solution to 261 ± 1% mosmol/kgH2O for the 10% hypoosmotic solution and to 319 ± 1% mosmol/kgH2O for the 10% hyperosmotic solution, as previously described (5). The Cl−-free ISO solution (ISO 0Cl−) was made by replacing Cl− with gluconate in the ISO control solution on a mole-for-mole basis, and the pH was adjusted to 7.3. The osmolality of all solutions was measured with a vapor pressure osmometer (model 5520; Wescor Biomedical Systems, Logan, UT). All the solutions were equilibrated with air. Stock solutions of the following chemicals were prepared using dimethyl sulfoxide (DMSO) or water as the initial solvent and then diluted in ISO solution: bumetanide, ouabain, verapamil, calcein-acetoxymethyl ester (AM), Asante NaTRIUM Green-2 (ANG-2), and N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide (MQAE). When DMSO was used as the solvent, its concentration in the final solutions was <1%.
Live-Cell-Imaging Microscopy
Coverslips with attached CPECs were mounted into a fluid perfusion imaging chamber (RC-21BRW; Warner Instruments, Hamden, CT) positioned on the stage of an epifluorescence inverted microscope (Olympus IX81; Olympus, Center Valley, PA) equipped with a Fluor oil-immersion lens (×40, 1.35 numerical aperture; Olympus) and DIC optics. Experimental solutions were perfused at a flow rate of 6 ml/min at 25°C. The fluid volume of the chamber (~300 µl) was exchanged with a half-time of <5 s. The fluorescent dyes calcein (Invitrogen), MQAE (Invitrogen), and ANG-2 (TEFLabs, Austin, TX) were used to measure CWV changes, [Cl−]i, and [Na+]i, respectively. Each dye was excited at its characteristic wavelength and bandwidth (see below) using a monochromator (OptoScan; Cairn Research, Faversham, United Kingdom) equipped with a 75-W xenon arc lamp (Ushio UXL S50A, Ultra High Stability). The excitation light passed through a liquid light guide before entering the microscope optical path and reaching the cells. The emitted fluorescence from the dye-loaded CPECs passed through emission filter cubes specific for the emission wavelength of each dye. A cooled, digital charge-coupled device camera (ORCA 2-ER C4742-95; Hamamatsu, Hamamatsu City, Japan) captured the emitted fluorescence as well as the cell images. Image acquisition, digital pinhole size and position, and fluorescence recording were done using MetaFluor imaging software (Molecular Devices, Sunnyvale, CA).
Measurement of Cell Water Volume Changes in Single Choroid Plexus Epithelial Cells Using Calcein Fluorescence
The method for measuring changes in CWV in single cells with the fluorescent dye calcein has been thoroughly described since its development and subsequent refinements in our laboratory (4, 5, 14, 56, 79). CPECs were loaded by exposure to control ISO solution containing 2 μM calcein-AM along with 20 μM verapamil (Invitrogen). Verapamil was added to inhibit the multidrug-resistant P-glycoprotein 1 (MDR1-Pgp), which is known to pump out calcein and other fluorescent dyes from cells (95). Multidrug-resistant P-glycoprotein is highly expressed in CPECs (76). Verapamil significantly reduced drift due to dye efflux from CPECs. Calcein was excited at 495 ± 3 nm with 80-ms light pulses at a frequency of 0.25 Hz, and emission collected from each cell through a circular digital pinhole region (15 × 15 pixels) was recorded at 535 ± 13 nm. This pinhole area was ~4.7% of the mean cross-sectional area of wild-type (WT) CPECs (~155 µm2). Osmotic water volume calibration was performed for each individual cell with ±10% anisosmotic solutions as described previously (5, 56).
Measurement of Cross-Sectional Area of In Vitro Choroid Plexus Epithelial Cells
Freshly dissociated CPECs from NKCC1+/+ and NKCC1−/− loaded with calcein were imaged using fluorescence and DIC optics to measure their CSA. Each cell within the field was identified with a color-coded pinhole and a number. With the use of the DIC images, the perimeter of each cell, including the brush-border edge, was outlined offline using a computer mouse. Only cells having clearly discernible borders in both DIC and calcein-fluorescent images were included (see below). Likewise, only cells that were loaded with calcein and retained the dye were measured using DIC imaging; it is well-known that cells that do not retain calcein, or do not cleave calcein-AM, are damaged or dead. To overcome the issue of the overlap of the border with the neighboring cells, deconvolution of the fluorescent images was carried out using ImageJ 1.46r (85). Fluorescent images, however, cannot be used to determine CSAs because the fluorescence intensity at the edges of the cells is very dim due to the lower concentration of dye in the thin volumes of the edges, which results in underestimates of CSAs. However, deconvolved fluorescent images were used as a guide to determine the borders between cells in the measurement of the perimeters of DIC images. The CSA of each individual CPEC was calculated using the roipoly function in MATLAB. This function yields the area of each cell in square pixels, which is then converted to square micrometers using the conversion factor 0.40484 µm2/pixel2. The latter is obtained from the imaging system calibration (10 µm/49.7 pixels = 0.201207 µm/pixel).
CSAs were measured in 484 CPECs obtained from 43 NKCC1+/+ mice (19 male and 24 female) and 255 CPECs from 22 NKCC1−/− mice (14 male and 8 female). Initially, all of the CPECs cells were divided into 3 groups according to mouse postnatal age and genotype. For NKCC1+/+, the groups were P10–P14 (108 cells/11 mice), P15–P18 (322 cells/27 mice), and P19–P21 (54 cells/5 mice). Kruskal-Wallis ANOVA showed that there were no significant differences between WT groups of different postnatal ages. For NKCC1−/−, the groups were P10–P14 (34 cells/4 mice), P15–P18 (n = 143 cells/12 mice), and P19–P21 (78 cells/6 mice). Kruskal-Wallis ANOVA showed that there were no significant differences between NKCC1 KO groups of different postnatal ages. Thus all of the cells from WT were considered as coming from the same population for statistical analysis. Likewise, all of the cells from NKCC1−/− were considered as samples from the same population.
Measurement of Intracellular Chloride in Single Choroid Plexus Epithelial Cells
The method to measure [Cl−]i in single CPECs loaded with the fluorescent indicator MQAE (96) was essentially the same that we described in detail for other cell types (56, 79). In brief, CPECs were loaded with MQAE by applying the dye to the imaging chamber at a concentration of 5 mM and with 20 μM verapamil. MQAE was excited at 350 ± 5 nm by exposure to 20-ms light pulses at a frequency of 0.1 Hz. The emission signals (460 ± 25 nm) were sampled at the same frequency. Once loaded, cells were equilibrated in isosmotic (ISO) solution before Cl− depletion by exposure to isosmotic Cl−-free solution (ISO 0Cl−). Once a new steady state was reached in the ISO 0Cl− solution, the CPECs were considered to be depleted of Cl−, i.e., apparent [Cl−]i = 0. The validity of this assumption was discussed in previous work from our laboratory (56). Following Cl− depletion, cell Cl− was recovered by exposure to the ISO control solution. At the end of the experiment, MQAE was calibrated against [Cl−]i using a double-ionophore technique in which the cells were permeabilized with the K+/H+ ionophore nigericin (5 μM; Sigma-Aldrich) and the Cl−/OH− ionophore tributyltin acetate (10 μM; Sigma-Aldrich). Besides these ionophores, the calibration solutions contained, in mM, 10 glucose, 5 HEPES, 120 K+, and [Cl−] + [] = 120. The calibration steps were 0, 20, 40, and 60 mM Cl−. MQAE was then quenched with 150 mM KSCN to determine the background fluorescence. MQAE fluorescence signal drift correction was performed offline by fitting the entire data transient to a straight line. The correlation coefficient of the fit was ≥0.99 for the cell to be included in the analysis (see below). MQAE fluorescence data measured for each individual cell was then converted to [Cl−]i using the Eq. 1 derived from the Stern-Volmer relation:
| (1) |
where F0 is the steady-state fluorescence from each cell measured in ISO 0Cl− solution, Ft is the fluorescence recorded with respect to time, and KSV is the Stern-Volmer quenching constant of MQAE for Cl−. The KSV was calculated for each cell following the four-point calibration procedure outlined above by plotting (F0/Ft) − 1 versus [Cl−]. The slope of this relation is KSV. The average KSV for the intracellular calibrations of MQAE in WT CPECs was 16.9 M−1 [standard deviation (SD) 2.7 M−1; n = 41 cells from 5 mice, 2 female and 3 male], and the average KSV of KO CPECs was 10.3 M−1 (SD 3.7 M−1; n = 45 cells from 7 mice, 3 female and 4 male). These KSV values fall within the range of 5–25 M−1 reported for other cell types (40, 56) and are large enough to obtain reliable calibration plots for [Cl−]i determinations. Furthermore, only cells fulfilling the following criteria for cell viability and MQAE performance were chosen for analysis: 1) calibration plot yielding KSV > 5 M−1; 2) correlation coefficient of calibration plot >0.99; 3) MQAE fluorescence in each calibration solution reached steady state; and 4) MQAE fluorescence in both the ISO control and ISO 0Cl− solutions reached steady state.
The Cl− equilibrium potential (ECl) for each cell was calculated using the Nernst equation:
| (2) |
where [Cl−]i and [Cl−]o are the measured concentrations of intracellular and extracellular Cl−, respectively, z (= −1) is ion valence, R is the gas constant, T is absolute temperature (in kelvin), and F is the Faraday constant.
Measurement of Intracellular Sodium in Single Choroid Plexus Epithelial Cells
Measurements of [Na+]i in single CPECs were done with the fluorescent sodium indicator dye ANG-2 (TEFLabs). Measurements were first attempted using the Na+-sensitive indicator sodium-binding benzofuran isophthalate (SBFI), but CPECs could not be adequately loaded with this indicator. ANG-2 was developed as an alternative Na+ indicator (20); we found that the in situ dissociation constant (Kd) for Na+ of this dye is in the range between 20 and 30 mM, as explained below. Kd values in this range yield reliable measurement of [Na+]i within expected physiological levels (60). ANG-2 has been successfully used in invertebrate epithelial cells (80) and in mammalian cell lines (32). Unlike SBFI that is excited in the UV spectral range (340/380 nm), ANG-2 is excited between 488 and 520 nm (peak excitation is 517 nm), therefore decreasing photodynamic cell damage. ANG-2 peak emission wavelength is between 540 and 550 nm (48). To load the cells, 5 μM ANG-2 (AM) was applied to the coverslip with the cells attached, along with an equal volume of Pluronic F-127 (20% wt/vol) and verapamil (20 μM). During data acquisition, the excitation wavelength was set at 500 ± 5 nm (80-ms excitation light pulses, at 0.1 Hz). Emitted fluorescence was detected at 535 nm and was monitored online as described above for calcein. Emitted fluorescence from each cell was measured through a digital pinhole (20 × 20 pixels) positioned at the center of each imaged cell. The fluorescence intensity of each cell was sampled until reaching a stable baseline. At the end of the observation period, cells were calibrated in situ to determine [Na+]i. At the start of calibration, cells were treated with the nonselective monovalent cation ionophore gramicidin D (7 μM; Sigma-Aldrich) to collapse the Na+ and K+ gradients across the cell membrane (78). The calibration solutions contained, in mM, 10 HEPES, 1.2 Ca2+, 57.4 Cl−, and 65 gluconic acid, with [Na+] + [K+] = 120 mM, pH 7.3, and 290 mosmol/kgH2O osmolality, adjusted with sucrose. The calibration points were 120, 90, 60, 30, 12, and 6 mM Na+. Independent measurements in calcein-loaded cells showed that changes in CWV on exposure to these calibration solutions were negligible. This is an important control inasmuch as ANG-2 is a single-wavelength excitation-emission dye. For analysis, the fluorescence data transient obtained from each individual cell was normalized to the value recorded in the 0Na+ calibration solution (after fluorescence background subtraction) using Eq. 3:
| (3) |
where Ft is the fluorescence with respect to time of the entire transient and F0 is the mean steady-state fluorescence in the 0Na+ calibration solution. The mean ANG-2 normalized fluorescence intensity (F; Eq. 3) measured for each Na+ calibration solution was used to plot the graph [1 − (Ft/F0)] versus [Na+]i, and a regression curve was fitted to the data using the Michaelis-Menten equation:
| (4) |
where the maximal normalized fluorescence (Fmax) and Kd values were obtained for each individual cell, as explained below. The normalized fluorescence data (F) was then converted into [Na+]i using the following equation:
| (5) |
where F = [1 − (Ft/F0)], Fmin is the minimal normalized fluorescence (F = 0), and Fmax and Kd were obtained from the curve fitted to the data points (see Fig. 7B). The mean Kd for Na+ in NKCC1+/+ CPECs was 26.8 mM [SD 9.4 mM, standard error of the mean (SEM) 2.0 mM, n = 21 cells, from 3 mice, 1 male and 2 female] and that in NKCC1−/− cells was 17.1 mM (SD 4.3 mM, SEM 2.0 mM, n = 21 cells, from 3 mice, 1 male and 2 female).
Fig. 7.
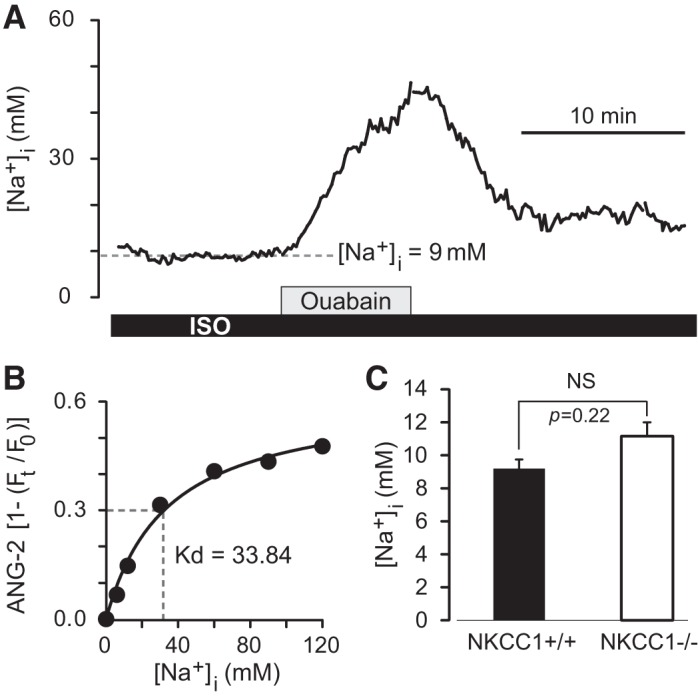
Basal intracellular Na+ concentration ([Na+]i) in single choroid plexus epithelial cells (CPECs) measured with the fluorescent indicator Asante NaTRIUM Green-2 (ANG-2). A: example of a recording of basal [Na+]i in an ANG-2-loaded CPEC from a wild-type (NKCC1+/+) mouse, (postnatal day 21, male). Na+-K+ pump was inhibited with ouabain (1 mM) to test that the dye responded to changes in [Na+]i. Basal [Na+]i in this CPEC was 9 mM and increased to ~45 mM during ouabain exposure at an initial rate (d[Na+]i/dt) of 6.5 mM·min−1. Ouabain-induced increase in [Na+]i was reversible on removal of the pump inhibitor. ISO, isosmotic control solution. B: at the end of the observation period each ANG-2-loaded CPEC was calibrated in situ to determine the apparent Kd for Na+ and the maximal normalized fluorescence intensity value Fmax by fitting the Michaelis-Menten equation to the data points. Minimal normalized fluorescence Fmin, the fluorescence recorded in the 0Na+ calibration solution, was adjusted to 0 (after background subtraction). Graph shows the relative fluorescence intensity [1(Ft/F0)] of ANG-2 as a function of [Na+]i from the CPEC in A. F0, steady-state fluorescence; Ft, fluorescence recorded with respect to time. From the Michaelis-Menten fitted to the data points the apparent Kd for Na+ (33.84 mM) and Fmax (0.61) were obtained. Parameters obtained from each cell were used to convert ANG-2 fluorescence intensities into [Na+]i using Eq. 5. C: basal [Na+]i in NKCC1+/+ and NKCC1−/− CPECs. Average basal [Na+]i in NKCC1+/+ cells (black bar) was 9.2 mM (SD 2.5 mM, SEM 0.6 mM, n = 21 cells, 3 mice, 1 male and 2 female). Average basal [Na+]i in NKCC1−/− cells was 11.1 mM (SD 3.9 mM, SEM 0.8 mM, n = 21 cells, 3 mice, 1 male and 2 female). Difference between means was not significant (NS; P = 0.22).
Statistical Analysis
Statistical analysis was done using nonparametric Student’s t-test. Differences were considered significant when P < 0.05. Data are presented as sample means and their corresponding standard deviation (SD). The standard error of the mean (SEM) is also reported. A two-sample two-tailed unequal-variances t-test (Mann-Whitney) was used for analyzing the statistical parameters of the NKCC1−/− versus NKCC1+/+ CPEC populations. To test the alternative hypothesis that the population means are not equal, α-significance level was 0.001. The null hypothesis that the means of KO and WT population are equal was successfully rejected with a P value (P < 0.0001) indicating that the means of the two populations were indeed different. The t-test was performed in Excel, MATLAB, and GraphPad Prism 5 for verification. Kruskal-Wallis ANOVA was used to see whether there were differences between CSAs of cells obtained from WT or KO mice of three different age groups, P10–P14, P15–P18, and P19–P21. The results showed no statistical differences (P > 0.1) between different age groups (as described above in the section dealing with measurement of CSAs of in vitro CPECs).
RESULTS
Apical Location of NKCC1 in the Microvilli of Choroid Plexus Epithelial Cells
Confocal immunofluorescence of WT mouse choroid plexus using the anti-NKCC1 polyclonal antibody Kaplan-CT (41) showed clear and specific NKCC1 immunoreactivity localized to the apical surface of the choroid plexus epithelium (CPE), i.e., the ventricular CSF-facing surface of the epithelium (Fig. 1, A and B). The specificity of the Kaplan-CT antibody was validated against NKCC1−/− CPE (negative control) and various NKCC1-immunoreactive tissues (positive control; Refs. 56, 69, 93). NKCC1 immunoreactivity of CPE was absent in NKCC1−/− mouse.
Although the localization of NKCC1 in the apical surface of CPECs is well established (56, 69, 70, 72), its subcellular localization is unknown. To address this issue, we used pre-embedding immunoperoxidase-ABC-DAB electron microscopy. NKCC1 immunolabeling was done using Kaplan-CT Ab and T4 monoclonal Ab validated for NKCC1 specificity in CP. As expected from the confocal immunofluorescence observations, both antibodies revealed that NKCC1 is localized in the apical microvilli (mv) and in the apical membrane segments between mv (Fig. 1, C and D), whereas NKCC1 immunolabeling was not detected in the basolateral membrane (Fig. 1C).
Choroid Plexus Epithelial Cells Express Two NKCC1 Splice Variants
Immunolabeling experiments establish the apical location of NKCC1 in CPECs but do not reveal the identity of the NKCC1 variant expressed because there are no variant-specific antibodies. Rat and mouse express two NKCC1 variants, one full length (NKCC1a) and a shorter one (NKCC1b) resulting from exon 21 exclusion (18, 56, 61, 75). Exon 21 encodes for 16 amino acids containing a dileucine motif that has been proposed to determine the basolateral sorting of NKCC1 in secretory epithelia (11). Disruption of this motif targets the cotransporter to the apical membrane of transfected Madin-Darby canine kidney cells. If this mechanism determines the apical polarity of native NKCC1 in CPECs, these cells should only express NKCC1b. To address this question, we used an RT-PCR strategy that allows coamplification of NKCC1a and NKCC1b transcripts (2, 56) and found that in CP, as in kidney, NKCC1b is barely detectable (Fig. 2B). In contrast, other tissues such as DRG neurons, brain, and spinal cord showed robust expression of NKCC1b (Fig. 2B). We used densitometry ratios obtained from coamplified NKCC1a and NKCC1b bands as surrogates of their relative abundance. The results showed that in CP and in kidney the NKCC1a-to-NKCC1b mRNA ratio was ~15 and ~24, respectively, and the difference between these ratios was not statistically significant (P = 0.41), whereas in brain, DRG, and spinal cord, the ratio was ≤5 (Fig. 2C). Comparison between CP and brain, DRG, and spinal cord showed that the difference in NKCC1a-to-NKCC1b mRNA ratios between CP and nervous tissues was highly significant (P < 0.0001). Interestingly, in mouse kidney, NKCC1 is most highly expressed in the basolateral membrane of the terminal inner medullary collecting duct, the afferent arteriole, and in the glomerular and extraglomerular mesangium (41), whereas NKCC1 is strictly localized to the apical membrane of CPECs. Thus, the abundant expression of NKCC1a, which comprises exon 21, cannot explain the apical polarity of the cotransporter in CPECs.
Fig. 2.

Relative mRNA expression of Na+-K+-2Cl− cotransporter 1 (NKCC1) variants, long (NKCC1a) and short (NKCC1b), in brain, choroid plexus, spinal cord, dorsal root ganglion (DRG), and kidney tissues. A: representation of the primer set NKCC1-502a/453b used to coamplify NKCC1a and NKCC1b mRNAs from the tissues indicated. B: representative RT-PCR experiments showing amplicons of expected sizes corresponding to NKCC1a (502 bp) and NKCC1b (453 bp) mRNAs expressed in each tissue. Negative controls were performed using water instead of cDNA (data not shown). Original digital images captured using an UV transilluminator were inverted for clarity to obtain the positive images shown. Gels were cropped to exclude edges and other lanes used to probe for other gene fragments irrelevant to the present study or lanes showing redundant results. Thus, except for the choroid plexus gel shown, the molecular weight standards on each of the other gels were cropped and aligned to the lane showing the NKCC1a/NKCC1b amplicons. To indicate this, a space was added between the lane showing the amplicons and the molecular weight markers. Spinal cord and kidney lanes came from the same gel, and hence the molecular weight markers are the same. C: densitometry ratios obtained from coamplified NKCC1a and NKCC1b bands as a surrogate of their relative abundance. Bars show the mean and standard error of the mean of the results obtained in choroid plexus (n = 14), brain (n = 7), DRG (n = 17), spinal cord (n = 6), and kidney (n = 7), where n = number of animals.
Choroid Plexus Epithelial Cells from NKCC1−/− Mice are Shrunken Both In Situ and In Vitro
In most animal cells, whether polarized or not, NKCC1 mediates net electroneutral ion influx because the net driving force for transport is inward (3, 79, 83). NKCC1-mediated ion transport is tightly linked to transmembrane water fluxes (79). NKCC1-solute-driven osmotic water transport can occur via both aquaporins and the phospholipid bilayer (64). In addition, water translocation can be mediated by NKCC1 cotransport (25, 26, 102). We hypothesize that under basal conditions apical NKCC1 is constantly active, inwardly transporting ions and associated water, thus maintaining the normal and constant CWV needed for secretion of CSF. Hence, deletion of NKCC1 by gene disruption ought to result in CPEC shrinkage due to unbalanced net solute and water fluxes across the CP epithelium. To begin to test this hypothesis, we studied the CPE of NKCC1+/+ and NKCC1−/− mice by transmission electron microscopy. We found that the CPE of NKCC1−/− mice undergoes substantial morphological changes, as shown in the electron micrographs of Figs. 3 and 4, obtained at low and high magnification, respectively.
At low magnification (Fig. 3), the EM images of in situ CPE from NKCC1−/− mice reveal large dilations of the extracellular space between the lateral plasma membrane of adjacent cells (Fig. 3B). The cells remain attached to each other by intercellular junctions (IJ) at the apical side (Fig. 3B). In deeper regions of the epithelium, toward the basal side, the extracellular space dilations are observed between the basal plasma membrane of CPECs and the basal lamina of the basement membrane. Examples of these extracellular space dilations are shown in Figs. 3B and 4B. The retracted basal plasma membrane of CPECs of NKCC1−/− forms fingerlike extensions that remain attached to the basal lamina of the basement membrane. Another ostensible morphological change of CPECs of NKCC1−/− is that their microvilli appear to be smaller than those of NKCC1+/+ animals. The ultrastructural morphological changes found in CPECs of NKCC1−/− can be more clearly seen in the high-power electron micrographs of NKCC1+/+ and NKCC1−/− shown in Fig. 4, A and B, respectively.
The ultrastructural changes of CPECs of NKCC1−/− revealed by EM could be due to CPEC solute and water loss or to an increase in interstitial fluid volume that would squeeze the epithelial cells. To investigate the issue, we used live-cell-imaging microscopy of freshly dissociated CPECs obtained from NKCC1+/+ and NKCC1−/− mice to measure their CSA and estimate their basal steady-state volume. Under these conditions, changes in CPEC volume can only be attributed to changes in cellular solute and water contents. The CPECs plated on coverslips were continuously perfused with control isosmotic solution (ISO) and imaged using DIC microscopy (Fig. 5A). Each cell was uniquely identified with a color-coded number and a digital circular region (Fig. 5, A–C). To verify their viability, the cells were loaded with the fluorescent dye calcein (Fig. 5B). The perimeter of each CPEC was manually traced from the DIC images (cell edges including microvilli) of the cells that were filled with calcein (Fig. 5C) to determine their CSA. The distribution of CSAs of both NKCC1+/+ and NKCC1−/− were Gaussian (Fig. 5D). The mean CSA was 153.6 µm2 for the NKCC1+/+ CPECs (SD 47.3 µm2, SEM 2.2 µm2, n = 484 cells/43 mice), and 135.2 µm2 for the NKCC1−/− (SD 37.6 µm2, SEM 2.4 µm2, n = 255 cells/22 mice). The difference between the mean CSAs of NKCC1+/+ versus NKCC1−/− is 18.4 µm2 (P < 0.0001). Thus, the CSA of NKCC1−/− CPECs is ~12% smaller than that of NKCC1+/+. The percentage of NKCC1−/− cells that are less than the mean of NKCC1+/+ cells is 72.16%. Assuming that CPECs attached to coverslips adopt the shape of hemispheres, from their measured radii it is possible to calculate their volume from the expression (2/3) π r3. The estimated mean volume of CPECs from NKCC1+/+ is ~715 µm3, whereas the mean volume from NKCC1−/− is ~591 µm3. Therefore, NKCC1−/− CPEC volume is ~17% smaller than WT cells.
Fig. 5.

In vitro live-cell-imaging analysis of the cross-sectional areas (CSAs) of freshly dissociated choroid plexus epithelial cells from Na+-K+-2Cl− cotransporter 1 (NKCC1) wild-type (NKCC1+/+) and knockout (NKCC1−/−) mice. A: cells imaged using differential interference contrast were uniquely identified with a number and a small, colored, digital circle. B: same cells shown in A, loaded with the fluorescent dye calcein. The dye served as an indicator of cell viability; cells that did not load or retained the dye were considered dead and omitted from analysis. C: border of each cell was traced with a colored digital profile to determine the CSA. D: histograms of total distribution of CSAs of cells from NKCC1+/+ (left) and NKCC1−/− (right). Red curves are Gaussian approximations fitted to the histograms. Box plots illustrate the spread of CSA measurements, with the median denoted as a red line. Blue box represents the 1st and 3rd quartiles, whiskers above and below the box show the location of the minimum and maximum, and the red circles represent outliers. Mean (x̄) CSA of choroid plexus epithelial cells from NKCC1−/− cells is significantly smaller than that from NKCC1+/+. Difference between means (153.6 − 135.2 µm2) is 18.4 µm2. Thus, the CSA of NKCC1−/− is 18.4 µm2 smaller than that of NKCC1+/+. Difference between the mean CSAs is highly significant (P < 0.0001). Calibration bars in A–C are 10 µm. Examples shown in A–C came from choroid plexus epithelium of the 4th ventricle of a postnatal day 10 NKCC1+/+ mouse.
Pharmacological Inactivation of NKCC1 with Bumetanide Produces Choroid Plexus Epithelial Cell Shrinkage
The results presented so far show that genetic deletion of NKCC1 leads to significant decrease in cell volume of CPECs, both in situ and in vitro. These observations are consistent with the hypothesis that apical NKCC1 is continuously active under basal conditions, inwardly transporting ions and associated water, thereby keeping basal CWV constant. Accordingly, specific pharmacological inhibition of apical NKCC1 with bumetanide should lead to a decrease in basal CWV of CPECs. The half-maximal inhibitory concentration (IC50) of bumetanide for human NKCC1 ranges between 0.16 and 0.68 µM (33, 52, 66). It is generally agreed that the inhibitory effect of bumetanide on NKCC1 is specific at concentrations ≤10 µM (3, 83). At higher concentrations (e.g., >50 µM), bumetanide cannot provide positive proof that a given function is specifically mediated by NKCC1 because it also blocks K+-Cl− cotransporters (KCCs), of which three members [K+-Cl− cotransporter 1 (KCC1; SLC12A4); K+-Cl− cotransporter 3 (KCC3; SLC12A6), splice variant a (KCC3a); and K+-Cl− cotransporter 4 (KCC4; SLC12A7)] are claimed to be expressed in CPECs (39, 42, 50, 57, 67, 92). Thus, we tested the effect of 10 µM bumetanide on basal CWV of calcein-loaded CPECs dissociated from NKCC1+/+ and NKCC1−/− mice, respectively (Fig. 6). On exposure to 10 µM bumetanide, CPECs from NKCC1+/+ mice shrank by 16% of their initial volume (SD 6.4%, SEM 1.7%, n = 14 cells/3 animals) at an initial rate of 3.7%/min (SD 0.4%/min, SEM 0.1%/min). The bumetanide-induced shrinkage reversed during washout of the inhibitor. In contrast, CPECs from NKCC1−/− did not respond to 10 µM bumetanide [the mean CWV change on bumetanide exposure in these cells was −0.4%, (SD 1.3%, SEM 0.4%, n = 10 cells/3 animals), which falls within the noise level of the method].
Fig. 6.

Effect of bumetanide (BUM) on basal cell water volume (CWV) in choroid plexus epithelial cells (CPECs) from Na+-K+-2Cl− cotransporter 1 (NKCC1) wild-type (NKCC1+/+) and knockout (NKCC1−/−) mice. A and B: relative CWV changes with respect to time [(Vt/V0) where V0 is the cell water volume in basal control condition (V0 = 1), and Vt is the percentage change in cell water reached at time t, divided by 100] were measured in single CPECs loaded with calcein, as described in materials and methods. Cells were equilibrated in isosmotic control solution (ISO). Osmotic calibration pulses (±10% anisosmotic) preceded the exposure to BUM (10 µm). Bars at the bottom of each trace indicate the duration of exposure to each solution. All of the solutions had the same osmolality (290 ± 1 mosmol/kgH2O) except for those used for the calibration pulses, which were nominally −10% (hypoosmotic) and +10% (hyperosmotic) with respect to the ISO control solution. A: BUM (10 µm) produced shrinkage in CPECs from NKCC1+/+. In the example shown [NKCC1+/+, postnatal day 18 (P18), male], BUM produced a maximum decrease in CWV of −13.1% at an initial rate of −3.8%/min. Cell recovered its initial volume on returning to the isosmotic (ISO) control solution. BUM exposure was preceded by 2 pulses of osmotic calibration solutions nominally −10 and +10% hypoosmotic and hyperosmotic, respectively. Double-headed gray arrows indicate the points in which the maximum of each response is measured for statistical analysis. B: BUM (10 µm) had no effect on basal CWV of CPECs lacking NKCC1. Example shown is from an NKCC1−/−, P11, female. Thus BUM-induced shrinkage is due to a specific pharmacological inhibition of NKCC1. C: maximum percentage change in CWV (% volume response) produced by 10 µM BUM in NKCC1+/+ and NKCC1−/− CPECs. In NKCC1+/+ cells, BUM produced an average CWV decrease of −16.0% (SD 6.4%; SEM 1.7%, n = 14 cells, 3 mice). BUM had no effect in CWV of NKCC1−/− (mean 0.4%, SD 1.4%, SEM 0.4, n = 10 cells, 3 mice). Difference between means is significant, P < 0.0001. D: percentage change in CWV produced by exposure to ±10% anisosmotic calibration pulses preceding BUM exposure. Amplitudes of the responses were measured as indicated by the double-headed gray arrows in A. Differences between hypoosmotic and hyperosmotic pulses recorded in NKCC1+/+ and NKCC1−/− cells were not statistically significant (NS). Bars in C and D represent the mean % change in CWV response. Error bars represent standard error of the mean (SEM). Black bars in C and D represent data from NKCC1+/+, and white bars correspond to data from NKCC1−/−.
Figure 6 shows representative examples of the effect of 10 µM bumetanide on basal CWV of single CPECs dissociated from NKCC1+/+ (Fig. 6A) and NKCC1−/− (Fig. 6B) mice, respectively. Figure 6, C and D, shows the statistical summary of these results. In these experiments, cells were initially equilibrated with isosmotic control solution (ISO), followed by exposure to osmotic calibration solutions (±10% anisosmotic), re-equilibrated with ISO, and then exposed to bumetanide as indicated by the gray box at the bottom of each trace (Fig. 6, A and B). In the example shown in Fig. 6A, on exposure to 10 µM bumetanide, the cell shrank by 13.1% of its initial volume, at an initial rate of −3.8%/min. On returning to isosmotic (ISO) control solution, the cell recovered its initial volume. Figure 6, B and C, shows that bumetanide did not elicit any changes in CWV in the NKCC1−/− CPECs. This demonstrates that the bumetanide-induced shrinkage of CPECs from NKCC1+/+ is specifically mediated by NKCC1. Furthermore, the nominally ±10% anisosmotic calibration pulses applied before bumetanide followed, as expected, ideal osmometric behavior, and their amplitude was not different when comparing cells from NKCC1+/+ and NKCC1−/− mice (Fig. 6D). Thus the lack of effect of bumetanide in the NKCC1−/− cells cannot be attributed to changes in the osmotic properties of these cells.
Measured Basal Intracellular [Na+] in CPECs Energetically Favors NKCC1 Net Solute Uptake
A key point in the controversy regarding the direction of net ion fluxes mediated by NKCC1 is the basal [Na+]i of CPECs. This is because [Na+]i is a major determinant of the sign and magnitude of the net free energy driving NKCC1 transport, as discussed below. We reasoned that if the driving force for basal cotransport is inward, [Na+]i cannot be as high as previously reported in mammalian CPECs (36, 46, 62). Thus we measured [Na+]i in freshly dissociated CPECs of NKCC1 WT and KO mouse using the Na+ fluorescent indicator ANG-2 (20, 80, 92). Figure 7 shows an example of these measurements and a summary of the results. The mean [Na+]i in CPECs of NKCC1+/+ was 9.2 mM (SD 2.5 mM, SEM 0.6 mM, n = 21 cells, 3 mice), a value that is considerably lower than the 30 to >48 mM previously reported for mammalian CPECs that would energetically favor NKCC1 working in the outward (secretory) mode (36, 46, 92). Interestingly, our value is closer to that obtained from measurements of intracellular Na+ activity using ion-selective microelectrodes in single cells of amphibian CP (84). We also found that the mean [Na+]i in CPECs of NKCC1−/− was 11.1 mM (SD 3.9 mM, SEM 0.8 mM, n = 21 cells, 3 mice), a value that is not significantly different (P = 0.22) to that measured in NKCC1+/+ cells (Fig. 7C). This result shows that the mechanisms that regulate [Na+]i in CPECs remain unchanged in NKCC1−/− cells.
Basal Intracellular [Cl−] in CPECs of NKCC1−/− Mouse is Significantly Lower than in NKCC1+/+
A central hypothesis of the present study is that apical NKCC1 maintains a constant cell water volume of CPECs and a higher-than-equilibrium [Cl−]i needed for transepithelial solute and water transport required for CSF secretion. Accordingly, basal [Cl−]i in NKCC1+/+ should have a value commensurate with these requirements. Furthermore, genetic inactivation of NKCC1 should produce a significant drop in [Cl−]i. We tested these hypotheses by measuring basal [Cl−]i in NKCC1+/+ and NKCC1−/− CPECs using MQAE. Figure 8, A and B, shows representative examples of these measurements. The mean basal [Cl−]i of NKCC1 WT was 60.7 (SD 12.3 mM, SEM 1.9 mM, n = 41 cells/5 mice), and the corresponding ECl calculated from Eq. 2 (see materials and methods) was −20.3 mV (SD 5.2 mV, SEM 0.8 mV). The basal [Cl−]i of NKCC1 KO was 31.4 mM (SD 12.8 mM, SEM 1.9 mM, n = 45 cells/7 mice), and the corresponding ECl was −38.9 mV (SD 10.9 mV, SEM 1.6 mV). The differences between mean [Cl−]i and ECl of NKCC1+/+ versus NKCC1−/− were significantly different (P < 0.0001), as shown in Fig. 8, C and D. The average ECl of NKCC1−/− CPECs (−38.9 ± 1.6 mV) is close to the membrane potential (Em) of NKCC1+/+ mouse CPECs (−39.5 ± 1.5 mV) kept under similar experimental conditions (81) but slightly more depolarized (8.5 mV) than the Em of −47.4 ± 2.1 mV reported by another group (13). These observations indicate that NKCC1 is the main Cl− uptake system in CPECs, maintaining [Cl−]i above its electrochemical equilibrium potential; genetic deletion of NKCC1 results in a significant reduction in [Cl−]i to values close to electrochemical equilibrium (i.e., in NKCC1−/− cells, ECl is close to Em).
Fig. 8.

Intracellular Cl− concentration ([Cl−]i) in single choroid plexus epithelial cells (CPECs) of Na+-K+-2Cl− cotransporter 1 (NKCC1) wild-type (NKCC1+/+) and knockout (NKCC1−/−) mice. Shown are examples of intracellular Cl− transients recorded in single N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide (MQAE)-loaded CPECs dissociated from NKCC1+/+ (A) and NKCC1−/− (B) mice in response to isosmotic removal and return of external Cl−. These type of transients were used to measure basal [Cl−]i and calculate the Cl− equilibrium potential (ECl) for each cell. CPECs were initially equilibrated in isosmotic control solution (ISO). Isosmotic removal of external Cl− (0Cl−) depleted the cells of Cl−. On restoration of external Cl− (ISO), the [Cl−]i recovered to initial values. Basal [Cl−]i for each cell was measured as the difference between the ISO baseline and the 0Cl− steady state, as indicated by the double-headed gray arrows. Basal [Cl−]i in the NKCC1+/+ CPEC shown in A [postnatal day 14 (P14), female] was 62.9 mM, and the ECl calculated from Eq. 2 was −18.9 mV. Basal [Cl−]i in the NKCC1−/− CPEC shown in B (P18, female) was 22.9 mM, and ECl was −44.8 mV. C: basal [Cl−]i in NKCC1+/+ and NKCC1−/− CPECs. Average basal [Cl−]i in NKCC1+/+ cells was 60.7 mM (SD 12.3 mM, SEM 1.9 mM) n = 41 cells/5 mice), and in NKCC1−/− cells it was 31.4 mM (SD 12.8 mM, SEM 1.9 mM, n = 45 cells/7 mice). Difference between means was highly significant (P < 0.0001). D: ECl for the same population of cells shown in C were −20.3 mV (SD 5.2 mV, SEM 0.8 mV) for the NKCC1+/+ and −38.9 mV (SD 10.9 mV, SEM 1.6 mV) for the NKCC1−/−. Difference between means was highly significant (P < 0.0001).
Modeling the Impact of Intracellular [Na+] on the Net Free Energy Driving NKCC1 Transport
NKCC1 transport process is electroneutral; the stoichiometry in vertebrate cells is 1 Na+:1 K+:2 Cl−. The direction of cotransport is determined by the sum of the chemical potential gradients of the transported ions. Therefore the net direction of cotransport depends on the overall net free energy (ΔG) of the system. If ΔG has a negative sign, the direction of cotransport will be inward, i.e., favoring net solute and associated water uptake. When ΔG is 0, the cotransporter is at equilibrium. When ΔG is positive, the direction of cotransport will be outward, favoring net solute and associated water efflux. For a 1 Na+:1 K+:2 Cl− stoichiometry, ΔG is given by the following equation (3):
| (6) |
Thus the driving force for the electroneutral Na+-K+-2Cl− cotransport (ΔµNa,K,Cl) is the sum of the chemical potential differences of Na+ (ΔµNa) and K+ (ΔµK) and twice the Cl− potential difference (ΔµCl), since two Cl− ions are translocated per cycle. From the definition of chemical potential for each of the ions involved, it follows that:
| (7) |
In most vertebrate cells under physiological conditions, ΔµNa,K,Cl is negative, thus favoring net ion and associated water uptake. Computation of ΔµNa,K,Cl for a variety of vertebrate cells ranges from −0.71 kJ/mol in bovine aortic endothelial cells to −8.86 kJ/mol in rabbit ventricular myocytes (79, 83). The rare exception may be some red blood cells that have unusually high [Cl−]i (75–100 mM), where ΔμNa,K,Cl is nearly 0, leaving little if any driving force to run the cotransporter under basal conditions (53). When ΔµNa,K,Cl = 0, the cotransporter is in equilibrium, a condition corresponding to the net flux reversal point. From Eq. 7, it is evident that variations in the chemical gradients of any of the transported ions can alter the direction of transport, but this is particularly critical for [K+]o and [Na+]i. Indeed, relatively small changes in [K+]o or [Na+]i have a major impact on ΔµNa,K,Cl. Given that the CSF [K+] is tightly regulated and kept at ~3.0 mM (7, 8, 31), we modeled the impact of [Na+]i on ΔµNa,K,Cl and thus in the direction of NKCC1-mediated net fluxes and the flux reversal point. We used the intracellular and extracellular ion concentrations reported for rat CPECs (36, 46, 62), for amphibian CPECs (84), and for mouse CPECs measured in the present study and in a recent report (92). Figure 9 shows plots of ΔµNa,K,Cl as a function of [Na+]i. The diagrams on the right of the graphs represent the predicted direction of net ion and associated water fluxes mediated by NKCC1. The filled circles superimposed on each graph correspond to the value of ΔµNa,K,Cl (in kilojoules per mole) for the average [Na+]i (in millimolar concentration) reported in each study (i.e., 48, 30, 31,10.5, and 9.2 mM), as specified at the bottom of the figure and explained in the corresponding legend. The results show that for the 48 mM [Na+]i reported by Johanson and Murphy (36) and the 30 mM [Na+]i reported by Keep et al. (46), the corresponding values of ΔµNa,K,Cl are 3.5 and 0.4 kJ/mol, thus energetically favoring outward NKCC1 cotransport. A similar conclusion was reached in a recent study in mouse CP, in which [Na+]i and ΔµNa,K,Cl were estimated to be 31 mM and 0.448 kJ/mol, respectively (92). In contrast, for the intracellular Na+ activity of 8 mM (corresponding to 10.5 mM [Na+]i) reported by Saito and Wright (84) for amphibian CPECs and the mean [Na+]i of 9.2 mM measured in the present study, the values of ΔµNa,K,Cl are −1.5 and −1.3 kJ/mol, respectively, thus favoring NKCC1 transport in the net uptake mode.
Fig. 9.

Modeling the effect of intracellular Na+ concentration ([Na+]i) on the magnitude and direction of Na+-K+-2Cl− cotransporter 1 (NKCC1)-mediated net fluxes in choroid plexus epithelial cells (CPECs). Graphs represent the net free energy driving NKCC1 transport (ΔμNKCC1) plotted as a function of [Na+]i (5–50 mM) by solving Eq. 7, using the reported values of extra- and intracellular ion concentrations for both mammalian (36, 46, 92) and amphibian (84) CPECs. ΔμNKCC1 computed for each of the reported values of basal [Na+]i (indicated at the bottom of the graphs) is represented by color-filled circles superimposed on each plot. Values reported for mouse CP by Steffensen et al. (92) almost overlap with those of Johanson and Murphy (36) estimated in rat and thus are not plotted for all of the range of [Na+]i. ΔμNKCC1 corresponding to the basal [Na+]i and [Cl−]i measured in the present study in single cells of mouse CP is indicated by the red filled circle. Note that the basal [Na+]i of single CPECs measured directly using the fluorescent indicator dye Asante NaTRIUM Green-2 is significantly lower than that reported by Johanson and Murphy (36), Keep et al. (46), and Steffensen et al. (92), based on flame photometry estimates. Basal [Na+]i measured in the present study, 9.2 mM, is closer to that reported for amphibian CPECs, 10.5 mM, using ion-selective microelectrodes (84). Because of the relatively low [Na+]i of amphibian and mouse CPECs (present study), ΔµNKCC1 has a negative value (green and red filled circles, respectively), indicating that under basal conditions NKCC1 is working in the inward direction and, therefore, it cannot directly contribute to cerebrospinal fluid secretion. Extracellular and intracellular ion concentrations used to compute ΔµNKCC1 were [Na+]o = 154 mM, [K+]o = 3 mM, [Cl−]o = 128 mM, [Na+]i (basal = 48 mM, 5–50 mM modeled range), [K+]i = 145 mM, [Cl−]i = 65 mM, absolute temperature (T) = 310 K, for Johanson and Murphy (36); [Na+]o = 148 mM, [K+]o = 2.9 mM, [Cl−]o = 133 mM, [Na+]i (basal = 30 mM, 5–50 mM modeled range), [K+]i = 120 mM, [Cl−]i = 50 mM, T = 310 K, for Keep et al. (46); [Na+]o = 150 mM, [K+]o = 3 mM, [Cl−]o = 100 mM, [Na+]i (basal = 31 mM), [K+]i = 141 mM, [Cl−]i = 35 mM, T = 310 K, for Steffensen et al. (92); [Na+]o = 110 mM, [K+]o = 2 mM, [Cl−]o = 89 mM, [Na+]i (basal = 10.5 mM, 5 to 50 mM modeled range), [K+]i = 89.5 [Cl−]i = 31.6 mM, T = 295 K, for Saito and Wright (84), and [Na+]o = 126 mM, [K+]o = 3 mM, [Cl−]o = 131 mM, [Na+]i (basal = 9.2 mM, 5–50 mM modeled range), [K+]i = 110 mM, [Cl−]i = 61.9 mM, T = 295 K, for the present study.
DISCUSSION
CPECs secrete CSF through mechanisms that are unclear. CPECs express Na+-K+-ATPase and NKCC1 on their apical membrane domain, diverging from the typical basolateral membrane location in secretory epithelia (3, 15). Although it is well-established that the apical Na+/K+ pump is the major source of Na+ secretion toward the luminal side of the CPE (reviewed in Ref. 71), the function of apical NKCC1 in this secretory epithelium is not understood and has been the subject of much debate (28, 71, 92). The apical location of NKCC1, the inhibitory effect of intraventricular bumetanide on CSF secretion, and the uncertainty regarding the ion gradients that determine the direction of NKCC1 cotransport are the essential points fueling the controversy about the function of NKCC1 in CPECs (71). The key issue in this debate is whether under normal physiological conditions, apical NKCC1 has a secretory versus an absorptive function or a dual absorptive and secretory function. These transport modes (secretory, absorptive, and dual) are not mutually exclusive since NKCC1 cotransport direction is reversible, i.e., net electroneutral cotransport can be inward or outward, depending on the Na+, K+, and Cl− chemical gradients across the cell membrane, as defined in Eq. 7 (3, 24, 83). Determining the direction of net ion fluxes mediated by NKCC1 and associated water fluxes under basal physiological conditions is essential for understanding the function of this cotransporter in CPE. If NKCC1 normally works in the net efflux mode, it may be directly involved in CSF secretion. Conversely, if NKCC1 works in the net influx mode, as it does in the basolateral membrane of other Cl− secreting epithelia, then it would have an absorptive function and could be involved in CSF K+ regulation and maintenance of CWV and [Cl−]i needed for secretion.
Keep and coworkers (46, 91) and, more recently, MacAulay’s group (92) proposed that although apical NKCC-mediated net fluxes could be bidirectional, NKCC1 normally works in the net outward (secretory) mode, as shown in Fig. 9. According to this view, NKCC1 directly contributes to CSF secretion. However, work by Wu et al. (98) suggested that under basal conditions apical NKCC1 functions in the inward mode, absorbing K+ from CSF to blood. In the present work, we resolve this long-standing controversy using a combined methodological approach that includes EM, live-cell-imaging microscopy, single-cell measurements of intracellular free ion concentrations and CWV, as well as pharmacological and molecular inactivation of NKCC1 in NKCC1 WT and KO mice models. The evidence obtained from both in situ and in vitro models shows that under basal conditions NKCC1 normally works in the net inward flux mode maintaining both the [Cl−]i and the water volume of CPECs. We propose that by virtue of being continuously active, transporting ions and associated water in the inward direction, apical NKCC1 maintains [Cl−]i and CWV at a certain level (set point), playing an indirect but critical role in CSF secretion. This explains the counterintuitive observation that pharmacological inhibition of NKCC1 by intracerebroventricular application of bumetanide significantly reduces basal CSF secretion (35, 92) and CSF hypersecretion following posthemorrhagic hydrocephalus (44).
NKCC1 is Densely Packed in the Microvilli of Choroid Plexus Epithelial Cells
Our confocal immunofluorescence results confirm the expression of NKCC1 to the apical domain of mouse CPE (69, 92). The observed pattern of NKCC1 expression in mouse CPE is consistent with that reported in rat and human (70, 72). With the use of immuno-EM with two different anti-NKCC1 antibodies validated against NKCC1−/− tissues, we show for the first time that NKCC1 is densely packed in the microvilli of the apical domain of CPECs (Fig. 1, C and D). NKCC1 localization in microvilli is physiologically significant; microvilli considerably increase the surface area of the apical side of the CPE interfacing with CSF. Morphometric studies show that in adult rat (P30), the total apical surface area of the CPE is 75 cm2, which is about half of the surface area of the cerebral capillaries (155 cm2) forming the blood-brain barrier (45). Besides NKCC1 and the Na+-K+-ATPase, CPECs abundantly express aquaporin 1 (AQP1) in the apical membrane (63, 72). The location of this molecular machinery indicates a vast surface area available for solute and water exchange in the apical membrane domain of CPECs.
NKCC1 Variants and Apical Sorting in Choroid Plexus Epithelium
The mechanisms of apical sorting of NKCC1 are unknown. The COOH terminus of NKCC1a, the full-length variant of NKCC1, contains a dileucine motif encoded by exon 21, which has been proposed to be essential for basolateral sorting of the cotransporter in epithelial cells (11). The shorter splice variant of NKCC1 (NKCC1b) does not have this dileucine motif in its COOH terminus. Experiments in cultured Madin-Darby canine kidney cells transfected with NKCC1 and NKCC2 chimeric constructs show that transfection with chimeras lacking the dileucine motif are targeted to the apical membrane (11). Furthermore, NKCC2 (SLC12A1), which lacks this sorting signal sequence in the COOH terminus, is targeted to the apical membrane of the thick ascending limb cells in the mammalian kidney (11, 16). We addressed the question of whether the alternative splice NKCC1 isoform (NKCC1b), which is abundantly expressed in the brain (75), spinal cord, and DRG neurons (56), is the variant localized to the apical membrane in CPECs. Our results show that mouse CPE expresses both NKCC1a and NKCC1b transcripts. Contrary to what was expected based on the dileucine motif hypothesis (11), mRNA expression analysis showed that the relative abundance of NKCC1a/NKCC1b in CPE is significantly higher than in brain, DRG, and spinal cord tissues but not significantly different to that in kidney (Fig. 2C). Thus the relative expression of NKCC1a/NKCC1b in CPE resembles that in mouse kidney, where NKCC1 is localized to the basolateral membrane of the terminal inner medullary collecting duct (41). Therefore, the predominant expression of NKCC1a over NKCC1b in CPECs cannot explain the apical sorting of the cotransporter in terms of the dileucine motif hypothesis. Further research is needed to elucidate the mechanisms underlying the apical sorting of NKCC1 in CPECs.
Inactivation of NKCC1 Produces Isosmotic Shrinkage of Choroid Plexus Epithelial Cells Both In Situ and In Vitro
In many cell types, it is a well-established fact that NKCC1 offsets the cell shrinkage induced by exposure to hypertonic solutions of unphysiologically high osmolality, usually in the range between 30 and 50% hyperosmotic. Under these conditions, NKCC1 is activated, thereby mediating net ion influx and associated water flux until cell volume is restored, a response known as regulatory volume increase (29). Whether NKCC1 functions to maintain cell volume constant under physiological isosmotic conditions is less clear. In epithelial cells, net solute influx and efflux must be balanced to maintain constant cell solute and water and, therefore, constant cell volume (Fig. 10). Thus, if NKCC1 is a key mechanism for maintenance of cell volume at a constant value under normal conditions, its inactivation is expected to produce cell shrinkage, inasmuch as these cells express solute and water efflux pathways. Thus, at steady state, CWV homeostasis of CPECs is achieved by a delicate balance between solute and water entry across the apical membrane and solute and water exit across the basolateral membrane (Fig. 10, middle). An uncompensated decrease in solute and water entry across the apical membrane will cause a decrease in intracellular solutes (e.g., K+ and Cl−) and water, with consequent cell shrinkage (Fig. 10, left). We propose that unbalanced (uncompensated net efflux) pathways continue working following NKCC1 inactivation until the cells adopt a new shrunken steady state. It is conceivable that these uncompensated efflux pathways are KCCs, given the well-known fact that they become inactivated as cell volume decreases (see below). Therefore, apical NKCC1 inactivation ought to result in CPEC shrinkage. The present results show that NKCC1 gene inactivation indeed results in shrinkage of CPEC, both in situ (Figs. 3 and 4) and in vitro (Fig. 5). EM images of in situ CPE of NKCC1−/− mouse showed that CPECs are shrunken, remaining attached to contiguous cells by intercellular junctions. The basolateral membrane of shrunken CPECs forms fingerlike extensions through which the cells remain anchored to the basal lamina of the basement membrane. The estimated mean cell volume of in vitro CPECs from NKCC1−/− mouse is ~17% smaller than WT cells. Consistent with these observations, pharmacological inhibition of NKCC1 with bumetanide (10 µM) decreases basal CWV by 16% in CPECs isolated from NKCC1+/+. Bumetanide has no effect on the CWV of cells from NKCC1−/−. This lack of effect of bumetanide in the NKCC1−/− CPECs cannot be attributed to changes in the osmotic properties of these cells as demonstrated in the experiments in Fig. 6D. Thus it is safe to conclude that the effect of bumetanide on NKCC1+/+ CWV is due to selective inhibition of NKCC1.
Fig. 10.

Balance of net solute and water fluxes determines choroidal epithelial cell volume. At steady state (cell in the middle), epithelial cell solute and water contents are maintained constant; the rate of apical solute and water entry (blue arrow) equals the rate of basolateral solute and water exit (red arrow), i.e., the net solute and water fluxes are balanced. Steady state can be altered by changes in net solute and water fluxes at 1 of the 2 membrane domains (apical and basolateral). Thus an unbalanced increase in apical net solute and water influx leads to cell swelling (cell on the right), whereas an unbalanced decrease in apical net solute and water influx leads to cell shrinkage (cell on the left). We propose that Na+-K+-2Cl− cotransporter 1 (NKCC1) is a key solute and associated water entry mechanism in the apical membrane of choroid plexus epithelial cells (CPECs). Under basal conditions, NKCC1 is continuously active, transporting ions and associated water in the inward direction, through the apical membrane. These inward fluxes are balanced by basolateral solute and water efflux, thereby maintaining constant cell water volume (and intracellular Cl− concentration). This hypothesis predicts that NKCC1 inactivation results in CPEC shrinkage due to unbalanced solute and water fluxes. Thus CPECs in which NKCC1 is inactivated achieve a new steady-state shrunken volume, and they do not recover their initial volume as long as NKCC1 remains inactivated.
The CPEC shrinkage produced by apical NKCC1 inactivation reveals the existence of uncompensated solute and water exit pathways. The identity of these efflux pathways remains to be determined. It is conceivable that K+, Cl−, and water exit via apical AQP1 and K+ and Cl− efflux pathways expressed in both the apical and the basolateral membrane such as KCC4 (42), KCC3a (67), and KCC1 (92). The transport mechanisms underlying cell volume maintenance of CPECs during transepithelial transport are also unclear. However, given the functional properties of KCC3a, such as its volume and intracellular Cl− sensitivity (59, 104), this cotransporter could work as a basolateral K+ and Cl− efflux pathway, maintaining cell solute and water constant during transepithelial transport, as illustrated in Fig. 11A. Depending on the degree of phosphorylation, KCC3a can be active under isotonic conditions (74, 77). Given the well-known reciprocal regulation of the Na+-K+ 2Cl− and K+-Cl− cotransporters (54), it is conceivable that NKCC1 working in coordination with KCCs maintains the CWV of CPECs at a set point necessary to keep both transport mechanisms active under basal conditions, maintaining cell solute and water constant during transepithelial transport, thus making possible CSF secretion and absorption, as explained in Fig. 11B.
Fig. 11.
Proposed mechanism of apical Na+-K+-2Cl− cotransporter 1 (NKCC1) function in choroid plexus epithelial cells (CPECs) under basal conditions. We propose that by virtue of being continuously active (CA) and working in the inward direction under basal conditions, apical NKCC1 maintains intracellular Cl− concentration ([Cl−]i) and cell water volume (CWV) at a certain level (set point), playing an indirect but critical role in cerebrospinal fluid (CSF) secretion. Intracellular [Cl−] and CWV result from a delicate balance between active Cl− uptake via apical NKCC1 along with associated water [osmotically driven by NKCC1 via aquaporin 1 (AQP1) and/or cotransported by NKCC1] and various Cl− and water efflux pathways, working in a concerted and coordinated manner. Putative Cl− efflux pathways in CPECs are the basolateral K+-Cl− cotransporters (KCC3a and/or KCC1), the apical KCC4, and 2 types of apical Cl− conductances, i.e., the volume-activated (VA) and the inward-rectifying (IR) Cl− channels. This homeostatic molecular machinery contributes to both CSF secretion and K+ absorption from CSF to blood. Pharmacological or genetic inactivation of NKCC1 disrupts the homeostatic equilibrium of CWV and [Cl−]i, thereby resulting in unbalanced Cl− and water efflux leading to cell shrinkage and a subsequent decrease in CSF secretion. A: diagram depicting some of the cotransporters and channels proposed to be involved in the maintenance of CPEC volume and [Cl−]i needed for both CSF secretion and K+ absorption under basal conditions. They include the apical KCC4, NKCC1, the Na+-K+-ATPase, AQP1 water channels, and VA and IR Cl− channels. Mechanisms by which water permeates the basolateral membrane are unknown. B: diagram depicting the factors determining the set point for the basal activity of both NKCC1 and KCCs, which maintain the CWV and [Cl−]i necessary for both simultaneous CSF secretion and K+ absorption. NKCC1 and KCCs cotransport activity (in arbitrary units 0–100) is plotted as a function of relative (normalized) cell water volume [Vo is the basal steady-state cell water volume (to normalize Vo = 1), and Vt is the steady-state cell water volume reached after a change in intracellular solute and water content (Vt/Vo)]. In the ordinate, 100 represents maximal cotransport activity. In the abscissa, basal Vt/Vo = 1.0. NKCC1 and KCCs cotransport activity is reciprocally regulated by numerous interdependent factors that include Vt/Vo, [Cl−]i, and the relative fraction of phosphorylated cotransporter molecules (P*/P), where P* represents the number of phosphorylated cotransporters and P the number of dephosphorylated cotransporters. Phosphorylation of NKCC1 and KCCs is likely to be effected by the WNK-SPS1-related proline/alanine-rich kinase (SPAK)/oxidative stress-responsive kinase 1 (OSR1) kinase complex as in other epithelial cells (1, 86). Sigmoid curves are only for schematic purposes.
In the basic model shown in Fig. 11A, K+ is taken up into CPECs via NKCC1 and the Na+-K+-ATPase and exits via KCC3a and/or other KCCs in the basolateral membrane. Na+ is recycled by apical Na+-K+-ATPase, and Cl− is partly recycled via Cl− channels and KCC4 and/or other KCCs in the apical membrane. Thus NKCC1 together with the Na+-K+-ATPase might play a central role in the reabsorption of K+ from CSF to blood and in the regulation of both CSF and brain interstitial [K+]. In this model, water moves across the apical membrane through AQP1 following osmotic gradients generated by NKCC1 solute fluxes (64) and/or via cotransport mediated by NKCC1 (25, 26, 102). The missing elements in this basic model are the water pathways in the basolateral membrane of CPECs, which remain unknown (65). AQP1, if at all present in the basolateral domain, is barely detectable by immuno-EM (63). Proteins other than AQPs, specifically KCCs, have been proposed to cotransport water (99, 101). Thus basolateral KCCs could cotransport solute (K+ and Cl−) and water. Further studies using validated antibodies against KCCs are needed to establish definitively the molecular identity of the KCC isoforms expressed in the basolateral membrane.
The possible apical location of KCC4 in CPECs (42) deserves further comment. When expressed in Xenopus oocytes, KCC4 exhibits some transport activity under isotonic conditions that increases significantly by cell swelling under hypotonic conditions (58). Compared with other KCC isoforms, KCC4 exhibits the highest activation by hypotonic swelling (58), suggesting that relatively small CWV increases could result in KCC4 activation. Thus apical KCC4 might be involved in K+ and Cl− secretion toward the lumen (CSF side).
Intracellular [Na+] and [Cl−] in Choroid Plexus Epithelial Cells Energetically Favor NKCC1 Working in the Inward Mode under Basal Conditions
A critical issue in the debate about the direction of transport mediated by apical NKCC1 is the [Na+]i of CPECs. From Eq. 7, it is clear that the level of intracellular Na+ is a major determinant of the direction of NKCC1 transport (Fig. 9). The idea that NKCC1 works in the net efflux mode, contributing to CSF secretion, originated from remarkably high estimates of [Na+]i (34–48 mM) derived from flame photometry measurements using pieces of rat CP (36, 46, 62, 89). More recent reports of [Na+]i estimates in mice CPECs, also using pieces of whole CP and flame photometry, yielded similarly high values of [Na+]i (31 mM). As pointed out by Minta and Tsien (60), whole tissue methods to measure [Na+]i such as flame photometry, atomic absorption, and counting of 22Na at isotopic equilibrium, besides destroying the tissue, measure total [Na+]i, which is considerably higher than free [Na+]i: “it is the latter that affects binding equilibria, transmembrane electrochemical gradients, and cell function.” The CP is a complex structure composed of not only CPECs, but also endothelial cells, fibroblasts, and blood cells. Thus whole tissue measurements represent average estimates of different cell populations. Another pitfall of this methodological approach is the uncertainty in estimating the extracellular and intracellular water content. To circumvent these problems, we measured [Na+]i directly, in individually calibrated single CPECs, using the fluorescent indicator dye ANG-2. The results show that that [Na+]i of NKCC1+/+ is 9.2 ± 0.6 mM, a value similar to that reported for amphibian CPECs using intracellular ion-sensitive microelectrodes (84). Furthermore, our measured [Na+]i is within the range found in other epithelial and nonepithelial cells (9–15 mM) using fluorescent indicator dyes or ion-sensitive microelectrodes (e.g., Refs. 19, 27, 82, 99, 106).
As expected, [Na+]i in CPECs from NKCC1−/− was not significantly different to that of NKCC1+/+, suggesting that the mechanisms of intracellular Na+ regulation of CPECs are not affected by deletion of NKCC1. This result is inconsistent with the conclusions of a recent report claiming that pharmacological inhibition of NKCC1 with bumetanide leads to an increase in [Na+]i, an observation that was taken as indicative of outwardly directed NKCC1-mediated Na+ transport (92). However, the latter conclusion was based on semiquantitative determination of changes in [Na+]i inferred from fluorescent signals recorded from pieces of whole CP loaded with the fluorescent Na+ indicator dye SBFI (92). Besides the inherent difficulties in interpreting optical signals from heterogeneous cell populations, bumetanide can interfere with SBFI fluorescence (78).
The basal [Cl−]i we measured in mouse CPECs of NKCC1+/+ using MQAE was 60.7 ± 1.9 mM. This value is close to that estimated for rat CP pieces (36, 62) but higher than the ~31 mM measured with Cl−-sensitive microelectrodes in amphibian CPECs (84, 100) and that measured in mouse CP pieces using a colorimetric method (92). Nevertheless, the [Cl−]i measured in the present study is close to that reported in other mammalian epithelial cells using MQAE, e.g., rabbit salivary glands (49), or Cl−-sensitive microelectrodes, e.g., canine tracheal epithelium (97). Assuming, for the sake of argument, that [Cl−]i was 31 mM, the inward driving force NKCC1 (ΔµNa,K,Cl) would be even larger than that calculated using the measured [Cl−]i of ~61 mM, i.e., −4.6 instead of −1.3 kJ/mol.
In contrast to what we observed for the [Na+]i, the [Cl−]i of CPECs from NKCC1−/− was reduced to half of the value measured in NKCC1+/+ (Fig. 8). The levels of intracellular Cl− in NKCC1−/− cells were close to those expected for passive electrochemical equilibrium. This indicates that NKCC1 maintains [Cl−]i at a higher than equilibrium level, and it is the main Cl− uptake mechanism of these cells. The measured [Na+]i and [Cl−]i in resting CPECs support an inward directed driving force for NKCC1 in mouse CPECs and thus are consistent with the main conclusions of the present study.
We propose that under basal conditions, continuously active apical NKCC1 working in the inward direction maintains [Cl−]i and CPEC water volume at a target level (set point) in coordination with basolateral KCCs (probably KCC3a) and other volume-sensitive/mechanosensitive transporters and channels, e.g., apical KCC4, and/or volume-activated Cl− and K+ channels, which are also kept constantly active. Inhibition of NKCC1 disrupts this solute and water flux balance between apical and basolateral transport mechanisms, resulting in decreased solute and water contents and consequent cell shrinkage, leading to decreased CSF secretion. This cross talk mechanism between apical and basolateral membrane solute and water transport is essential for CSF secretion and K+ absorption by the CPECs.
GRANTS
This study was supported by the following grants to F. J. Alvarez-Leefmans: Dayton Children’s Hospital Foundation (15-425-1/15-0395) and Boonshoft School of Medicine Seed Grant Proteomics Program (229117 RIF). The initial studies were supported by National Institute of Neurological Disorders and Stroke Grant NS-29227. J. M. C. Gregoriades held a National Science Foundation Integrative Graduate Education and Research Traineeship Fellowship in the Biomedical Sciences PhD Program (Grant DGE-0504438, Wright State University).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
F.J.A.-L. conceived and designed research; J.M.C.G., F.J.A., and F.J.A.-L. performed experiments; J.M.C.G., A.M., F.J.A., and F.J.A.-L. analyzed data; J.M.C.G., F.J.A., and F.J.A.-L. interpreted results of experiments; J.M.C.G., A.M., F.J.A., and F.J.A.-L. prepared figures; F.J.A.-L. drafted manuscript; J.M.C.G., F.J.A., and F.J.A.-L. edited and revised manuscript; J.M.C.G., A.M., F.J.A., and F.J.A.-L. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address of F. J. Alvarez: Dept. of Physiology, Emory Univ. School of Medicine, Atlanta, GA.
F. J. Alvarez-Leefmans is grateful to Drs. Robert Lober and Gogi Kumar (Dayton Children’s Hospital) for valuable discussions on the clinical implications of the findings reported in this study, Prof. Joseph P. Y. Kao (Center for Biomedical Engineering and Technology and Department of Physiology, University of Maryland School of Medicine) for useful comments on in vitro calibration and properties of ANG-2, Vishal Faldu for helping with data analysis, Dr. Mauricio Di Fulvio for help with the design of the primers shown in Fig. 2, and Dr. José Mendoza-Sotelo for help with initial immunolabeling studies.
REFERENCES
- 1.Alessi DR, Zhang J, Khanna A, Hochdörfer T, Shang Y, Kahle KT. The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci Signal 7: re3, 2014. doi: 10.1126/scisignal.2005365. [DOI] [PubMed] [Google Scholar]
- 2.Alshahrani S, Alvarez-Leefmans FJ, Di Fulvio M. Expression of the Slc12a1 gene in pancreatic β-cells: molecular characterization and in silico analysis. Cell Physiol Biochem 30: 95–112, 2012. doi: 10.1159/000339050. [DOI] [PubMed] [Google Scholar]
- 3.Alvarez-Leefmans FJ. Intracellular chloride regulation. In: Cell Physiology Sourcebook, edited by Sperelakis N. London: Academic Press-Elsevier, 2012, p. 221–259. [Google Scholar]
- 4.Alvarez-Leefmans FJ, Altamirano J, Crowe WE. Use of ion-selective microelectrodes and fluorescent probes to measure cell volume. In: Measurement and Manipulation of Intracellular Ions, edited by Kraicer J and Dixon SJ. San Diego, CA: Academic Press, 1995, p. 361–391. [Google Scholar]
- 5.Alvarez-Leefmans FJ, Herrera-Pérez JJ, Márquez MS, Blanco VM. Simultaneous measurement of water volume and pH in single cells using BCECF and fluorescence imaging microscopy. Biophys J 90: 608–618, 2006. doi: 10.1529/biophysj.105.069450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alvarez-Leefmans FJ, León-Olea M, Mendoza-Sotelo J, Alvarez FJ, Antón B, Garduño R. Immunolocalization of the Na+-K+-2Cl− cotransporter in peripheral nervous tissue of vertebrates. Neuroscience 104: 569–582, 2001. doi: 10.1016/S0306-4522(01)00091-4. [DOI] [PubMed] [Google Scholar]
- 7.Ames A 3rd, Higashi K, Nesbett FB. Relation of potassium concentration in choroidplexus fluid to that in plasma. J Physiol 181: 506–515, 1965. doi: 10.1113/jphysiol.1965.sp007779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bradbury MW, Kleeman CR. Stability of the potassium content of cerebrospinal fluid and brain. Am J Physiol 213: 519–528, 1967. doi: 10.1152/ajplegacy.1967.213.2.519. [DOI] [PubMed] [Google Scholar]
- 9.Brinker T, Stopa E, Morrison J, Klinge P. A new look at cerebrospinal fluid circulation. Fluids Barriers CNS 11: 10, 2014. doi: 10.1186/2045-8118-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown PD, Davies SL, Millar ID. Ion transport in choroid plexus. In: Physiology and Pathology of Chloride Transporters and Channels in the Nervous System, edited by Alvarez-Leefmans FJ and Delpire E. London: Academic Press-Elsevier, 2009, p. 569–583. [Google Scholar]
- 11.Carmosino M, Giménez I, Caplan M, Forbush B. Exon loss accounts for differential sorting of Na-K-Cl cotransporters in polarized epithelial cells. Mol Biol Cell 19: 4341–4351, 2008. doi: 10.1091/mbc.e08-05-0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen H, Luo J, Kintner DB, Shull GE, Sun D. Na+-dependent chloride transporter (NKCC1−/−) mice exhibit less gray and white matter damage after focal cerebral ischemia. J Cereb Blood Flow Metab 25: 54–66, 2005. doi: 10.1038/sj.jcbfm.9600006. [DOI] [PubMed] [Google Scholar]
- 13.Christensen HL, Păunescu TG, Matchkov V, Barbuskaite D, Brown D, Damkier HH, Praetorius J. The V-ATPase is expressed in the choroid plexus and mediates cAMP-induced intracellular pH alterations. Physiol Rep 5: e13072, 2017. doi: 10.14814/phy2.13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crowe WE, Altamirano J, Huerto L, Alvarez-Leefmans FJ. Volume changes in single N1E-115 neuroblastoma cells measured with a fluorescent probe. Neuroscience 69: 283–296, 1995. doi: 10.1016/0306-4522(95)00219-9. [DOI] [PubMed] [Google Scholar]
- 15.Damkier HH, Brown PD, Praetorius J. Cerebrospinal fluid secretion by the choroid plexus. Physiol Rev 93: 1847–1892, 2013. doi: 10.1152/physrev.00004.2013. [DOI] [PubMed] [Google Scholar]
- 16.Delpire E, Gagnon KB. Na+-K+-2Cl− cotransporter (NKCC) physiological function in nonpolarized cells and transporting epithelia. Compr Physiol 8: 871–901, 2018. doi: 10.1002/cphy.c170018. [DOI] [PubMed] [Google Scholar]
- 17.Delpire E, Lu J, England R, Dull C, Thorne T. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat Genet 22: 192–195, 1999. doi: 10.1038/9713. [DOI] [PubMed] [Google Scholar]
- 18.Delpire E, Rauchman MI, Beier DR, Hebert SC, Gullans SR. Molecular cloning and chromosome localization of a putative basolateral Na+-K+-2Cl− cotransporter from mouse inner medullary collecting duct (mIMCD-3) cells. J Biol Chem 269: 25677–25683, 1994. [PubMed] [Google Scholar]
- 19.Donoso P, Mill JG, O’Neill SC, Eisner DA. Fluorescence measurements of cytoplasmic and mitochondrial sodium concentration in rat ventricular myocytes. J Physiol 448: 493–509, 1992. doi: 10.1113/jphysiol.1992.sp019053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Escamilla PR, Minta A. Cytosolic Fluorescent Ion Indicators. US Patent WO2012040204A2, 2012.
- 21.Evans RL, Park K, Turner RJ, Watson GE, Nguyen HV, Dennett MR, Hand AR, Flagella M, Shull GE, Melvin JE. Severe impairment of salivation in Na+/K+/2Cl− cotransporter (NKCC1)-deficient mice. J Biol Chem 275: 26720–26726, 2000. doi: 10.1074/jbc.M003753200. [DOI] [PubMed] [Google Scholar]
- 22.Flagella M, Clarke LL, Miller ML, Erway LC, Giannella RA, Andringa A, Gawenis LR, Kramer J, Duffy JJ, Doetschman T, Lorenz JN, Yamoah EN, Cardell EL, Shull GE. Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J Biol Chem 274: 26946–26955, 1999. doi: 10.1074/jbc.274.38.26946. [DOI] [PubMed] [Google Scholar]
- 23.Frizzell RA, Hanrahan JW. Physiology of epithelial chloride and fluid secretion. Cold Spring Harb Perspect Med 2: a009563, 2012. doi: 10.1101/cshperspect.a009563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haas M, Forbush B 3rd. The Na-K-Cl cotransporter of secretory epithelia. Annu Rev Physiol 62: 515–534, 2000. doi: 10.1146/annurev.physiol.62.1.515. [DOI] [PubMed] [Google Scholar]
- 25.Hamann S, Herrera-Perez JJ, Bundgaard M, Alvarez-Leefmans FJ, Zeuthen T. Water permeability of Na+-K+-2Cl− cotransporters in mammalian epithelial cells. J Physiol 568: 123–135, 2005. doi: 10.1113/jphysiol.2005.093526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamann S, Herrera-Perez JJ, Zeuthen T, Alvarez-Leefmans FJ. Cotransport of water by the Na+-K+-2Cl− cotransporter NKCC1 in mammalian epithelial cells. J Physiol 588: 4089–4101, 2010. doi: 10.1113/jphysiol.2010.194738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harootunian AT, Kao JP, Eckert BK, Tsien RY. Fluorescence ratio imaging of cytosolic free Na+ in individual fibroblasts and lymphocytes. J Biol Chem 264: 19458–19467, 1989. [PubMed] [Google Scholar]
- 28.Hladky SB, Barrand MA. Fluid and ion transfer across the blood-brain and blood-cerebrospinal fluid barriers; a comparative account of mechanisms and roles. Fluids Barriers CNS 13: 19, 2016. doi: 10.1186/s12987-016-0040-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoffmann EK, Lambert IH, Pedersen SF. Physiology of cell volume regulation in vertebrates. Physiol Rev 89: 193–277, 2009. doi: 10.1152/physrev.00037.2007. [DOI] [PubMed] [Google Scholar]
- 30.Hughes AL, Pakhomova A, Brown PD. Regulatory volume increase in epithelial cells isolated from the mouse fourth ventricle choroid plexus involves Na+-H+ exchange but not Na+-K+-2Cl− cotransport. Brain Res 1323: 1–10, 2010. doi: 10.1016/j.brainres.2009.12.094. [DOI] [PubMed] [Google Scholar]
- 31.Husted RF, Reed DJ. Regulation of cerebrospinal fluid potassium by the cat choroid plexus. J Physiol 259: 213–221, 1976. doi: 10.1113/jphysiol.1976.sp011462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iamshanova O, Mariot P, Lehen’kyi V, Prevarskaya N. Comparison of fluorescence probes for intracellular sodium imaging in prostate cancer cell lines. Eur Biophys J 45: 765–777, 2016. doi: 10.1007/s00249-016-1173-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Isenring P, Jacoby SC, Payne JA, Forbush B 3rd. Comparison of Na-K-Cl cotransporters. NKCC1, NKCC2, and the HEK cell Na-L-Cl cotransporter. J Biol Chem 273: 11295–11301, 1998. doi: 10.1074/jbc.273.18.11295. [DOI] [PubMed] [Google Scholar]
- 34.Javaheri S. Role of NaCl cotransport in cerebrospinal fluid production: effects of loop diuretics. J Appl Physiol (1985) 71: 795–800, 1991. doi: 10.1152/jappl.1991.71.3.795. [DOI] [PubMed] [Google Scholar]
- 35.Javaheri S, Wagner KR. Bumetanide decreases canine cerebrospinal fluid production. In vivo evidence for NaCl cotransport in the central nervous system. J Clin Invest 92: 2257–2261, 1993. doi: 10.1172/JCI116829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johanson CE, Murphy VA. Acetazolamide and insulin alter choroid plexus epithelial cell [Na+], pH, and volume. Am J Physiol Renal Physiol 258: F1538–F1546, 1990. doi: 10.1152/ajprenal.1990.258.6.F1538. [DOI] [PubMed] [Google Scholar]
- 37.Johnson DC, Singer S, Hoop B, Kazemi H. Chloride flux from blood to CSF: inhibition by furosemide and bumetanide. J Appl Physiol (1985) 63: 1591–1600, 1987. doi: 10.1152/jappl.1987.63.4.1591. [DOI] [PubMed] [Google Scholar]
- 38.Kahle KT, Walcott BP, Staley KJ. Resolution of headache and papilledema in idiopathic intracranial hypertension associated with inhibition of Na+-K+-2Cl− cotransport. J Child Neurol 26: 205–208, 2011. doi: 10.1177/0883073810391264. [DOI] [PubMed] [Google Scholar]
- 39.Kanaka C, Ohno K, Okabe A, Kuriyama K, Itoh T, Fukuda A, Sato K. The differential expression patterns of messenger RNAs encoding K-Cl cotransporters (KCC1,2) and Na-K-2Cl cotransporter (NKCC1) in the rat nervous system. Neuroscience 104: 933–946, 2001. doi: 10.1016/S0306-4522(01)00149-X. [DOI] [PubMed] [Google Scholar]
- 40.Kaneko H, Putzier I, Frings S, Kaupp UB, Gensch T. Chloride accumulation in mammalian olfactory sensory neurons. J Neurosci 24: 7931–7938, 2004. doi: 10.1523/JNEUROSCI.2115-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaplan MR, Plotkin MD, Brown D, Hebert SC, Delpire E. Expression of the mouse Na-K-2Cl cotransporter, mBSC2, in the terminal inner medullary collecting duct, the glomerular and extraglomerular mesangium, and the glomerular afferent arteriole. J Clin Invest 98: 723–730, 1996. doi: 10.1172/JCI118844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karadsheh MF, Byun N, Mount DB, Delpire E. Localization of the KCC4 potassium-chloride cotransporter in the nervous system. Neuroscience 123: 381–391, 2004. doi: 10.1016/j.neuroscience.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 43.Karimy JK, Duran D, Hu JK, Gavankar C, Gaillard JR, Bayri Y, Rice H, DiLuna ML, Gerzanich V, Marc Simard J, Kahle KT. Cerebrospinal fluid hypersecretion in pediatric hydrocephalus. Neurosurg Focus 41: E10, 2016. doi: 10.3171/2016.8.FOCUS16278. [DOI] [PubMed] [Google Scholar]
- 44.Karimy JK, Zhang J, Kurland DB, Theriault BC, Duran D, Stokum JA, Furey CG, Zhou X, Mansuri MS, Montejo J, Vera A, DiLuna ML, Delpire E, Alper SL, Gunel M, Gerzanich V, Medzhitov R, Simard JM, Kahle KT. Inflammation-dependent cerebrospinal fluid hypersecretion by the choroid plexus epithelium in posthemorrhagic hydrocephalus. Nat Med 23: 997–1003, 2017. doi: 10.1038/nm.4361. [DOI] [PubMed] [Google Scholar]
- 45.Keep RF, Jones HC. A morphometric study on the development of the lateral ventricle choroid plexus, choroid plexus capillaries and ventricular ependyma in the rat. Brain Res Dev Brain Res 56: 47–53, 1990. doi: 10.1016/0165-3806(90)90163-S. [DOI] [PubMed] [Google Scholar]
- 46.Keep RF, Xiang J, Betz AL. Potassium cotransport at the rat choroid plexus. Am J Physiol Cell Physiol 267: C1616–C1622, 1994. doi: 10.1152/ajpcell.1994.267.6.C1616. [DOI] [PubMed] [Google Scholar]
- 47.Koumangoye R, Omer S, Delpire E. Mistargeting of a truncated Na-K-2Cl cotransporter in epithelial cells. Am J Physiol Cell Physiol 315: C258–C276, 2018. doi: 10.1152/ajpcell.00130.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lamy CM, Chatton JY. Optical probing of sodium dynamics in neurons and astrocytes. Neuroimage 58: 572–578, 2011. doi: 10.1016/j.neuroimage.2011.06.074. [DOI] [PubMed] [Google Scholar]
- 49.Lau KR, Evans RL, Case RM. Intracellular Cl− concentration in striated intralobular ducts from rabbit mandibular salivary glands. Pflügers Arch 427: 24–32, 1994. doi: 10.1007/BF00585938. [DOI] [PubMed] [Google Scholar]
- 50.Le Rouzic P, Ivanov TR, Stanley PJ, Baudoin FM, Chan F, Pinteaux E, Brown PD, Luckman SM. KCC3 and KCC4 expression in rat adult forebrain. Brain Res 1110: 39–45, 2006. doi: 10.1016/j.brainres.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 51.Lu KT, Wu CY, Cheng NC, Wo YY, Yang JT, Yen HH, Yang YL. Inhibition of the Na+-K+-2Cl−-cotransporter in choroid plexus attenuates traumatic brain injury-induced brain edema and neuronal damage. Eur J Pharmacol 548: 99–105, 2006. doi: 10.1016/j.ejphar.2006.07.048. [DOI] [PubMed] [Google Scholar]
- 52.Lykke K, Töllner K, Feit PW, Erker T, MacAulay N, Löscher W. The search for NKCC1-selective drugs for the treatment of epilepsy: structure-function relationship of bumetanide and various bumetanide derivatives in inhibiting the human cation-chloride cotransporter NKCC1A. Epilepsy Behav 59: 42–49, 2016. doi: 10.1016/j.yebeh.2016.03.021. [DOI] [PubMed] [Google Scholar]
- 53.Lytle C. Na+-K+-2Cl− cotransport. In: Red Cell Membrane Transport in Health and Disease, edited by Bernhardt I and Ellory JC. Berlin; New York: Springer, 2003, p. 173–195. [Google Scholar]
- 54.Lytle C, McManus T. Coordinate modulation of Na-K-2Cl cotransport and K-Cl cotransport by cell volume and chloride. Am J Physiol Cell Physiol 283: C1422–C1431, 2002. doi: 10.1152/ajpcell.00130.2002. [DOI] [PubMed] [Google Scholar]
- 55.Lytle C, Xu JC, Biemesderfer D, Forbush B 3rd. Distribution and diversity of Na-K-Cl cotransport proteins: a study with monoclonal antibodies. Am J Physiol Cell Physiol 269: C1496–C1505, 1995. doi: 10.1152/ajpcell.1995.269.6.C1496. [DOI] [PubMed] [Google Scholar]
- 56.Mao S, Garzon-Muvdi T, Di Fulvio M, Chen Y, Delpire E, Alvarez FJ, Alvarez-Leefmans FJ. Molecular and functional expression of cation-chloride cotransporters in dorsal root ganglion neurons during postnatal maturation. J Neurophysiol 108: 834–852, 2012. doi: 10.1152/jn.00970.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marcoux AA, Garneau AP, Frenette-Cotton R, Slimani S, Mac-Way F, Isenring P. Molecular features and physiological roles of K+-Cl− cotransporter 4 (KCC4). Biochim Biophys Acta, Gen Subj 1861: 3154–3166, 2017. doi: 10.1016/j.bbagen.2017.09.007. [DOI] [PubMed] [Google Scholar]
- 58.Mercado A, Song L, Vazquez N, Mount DB, Gamba G. Functional comparison of the K+-Cl− cotransporters KCC1 and KCC4. J Biol Chem 275: 30326–30334, 2000. doi: 10.1074/jbc.M003112200. [DOI] [PubMed] [Google Scholar]
- 59.Mercado A, Vázquez N, Song L, Cortés R, Enck AH, Welch R, Delpire E, Gamba G, Mount DB. NH2-terminal heterogeneity in the KCC3 K+-Cl− cotransporter. Am J Physiol Renal Physiol 289: F1246–F1261, 2005. doi: 10.1152/ajprenal.00464.2004. [DOI] [PubMed] [Google Scholar]
- 60.Minta A, Tsien RY. Fluorescent indicators for cytosolic sodium. J Biol Chem 264: 19449–19457, 1989. [PubMed] [Google Scholar]
- 61.Moore-Hoon ML, Turner RJ. Molecular and topological characterization of the rat parotid Na+-K+-2Cl− cotransporter. Biochim Biophys Acta 1373: 261–269, 1998. doi: 10.1016/S0005-2736(98)00112-6. [DOI] [PubMed] [Google Scholar]
- 62.Murphy VA, Johanson CE. Na+-H+ exchange in choroid plexus and CSF in acute metabolic acidosis or alkalosis. Am J Physiol Renal Physiol 258: F1528–F1537, 1990. doi: 10.1152/ajprenal.1990.258.6.F1528. [DOI] [PubMed] [Google Scholar]
- 63.Nielsen S, Smith BL, Christensen EI, Agre P. Distribution of the aquaporin CHIP in secretory and resorptive epithelia and capillary endothelia. Proc Natl Acad Sci USA 90: 7275–7279, 1993. doi: 10.1073/pnas.90.15.7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oshio K, Watanabe H, Song Y, Verkman AS, Manley GT. Reduced cerebrospinal fluid production and intracranial pressure in mice lacking choroid plexus water channel aquaporin-1. FASEB J 19: 76–78, 2005. doi: 10.1096/fj.04-1711fje. [DOI] [PubMed] [Google Scholar]
- 65.Patyal P, Alvarez-Leefmans FJ. Expression of NKCC1 and aquaporins 4, 7 and 9 in mouse choroid plexus and ependymal cells (Abstract) FASEB J 30: lb621, 2016. [Google Scholar]
- 66.Payne JA, Xu JC, Haas M, Lytle CY, Ward D, Forbush B 3rd. Primary structure, functional expression, and chromosomal localization of the bumetanide-sensitive Na-K-Cl cotransporter in human colon. J Biol Chem 270: 17977–17985, 1995. doi: 10.1074/jbc.270.30.17977. [DOI] [PubMed] [Google Scholar]
- 67.Pearson MM, Lu J, Mount DB, Delpire E. Localization of the K+-Cl− cotransporter, KCC3, in the central and peripheral nervous systems: expression in the choroid plexus, large neurons and white matter tracts. Neuroscience 103: 481–491, 2001. doi: 10.1016/S0306-4522(00)00567-4. [DOI] [PubMed] [Google Scholar]
- 68.Peters A, Palay SL, Webster H. The Fine Structure of the Nervous System: Neurons and Their Supporting Cells. New York: Oxford Univ. Press, 1991, p. xviii. [Google Scholar]
- 69.Piechotta K, Lu J, Delpire E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J Biol Chem 277: 50812–50819, 2002. doi: 10.1074/jbc.M208108200. [DOI] [PubMed] [Google Scholar]
- 70.Plotkin MD, Kaplan MR, Peterson LN, Gullans SR, Hebert SC, Delpire E. Expression of the Na+-K+-2Cl− cotransporter BSC2 in the nervous system. Am J Physiol Cell Physiol 272: C173–C183, 1997. doi: 10.1152/ajpcell.1997.272.1.C173. [DOI] [PubMed] [Google Scholar]
- 71.Praetorius J, Damkier HH. Transport across the choroid plexus epithelium. Am J Physiol Cell Physiol 312: C673–C686, 2017. doi: 10.1152/ajpcell.00041.2017. [DOI] [PubMed] [Google Scholar]
- 72.Praetorius J, Nielsen S. Distribution of sodium transporters and aquaporin-1 in the human choroid plexus. Am J Physiol Cell Physiol 291: C59–C67, 2006. doi: 10.1152/ajpcell.00433.2005. [DOI] [PubMed] [Google Scholar]
- 73.Priestley JV, Alvarez FJ, Averill S. Pre-embedding electron microscopic immunocytochemistry. In: Electron Microscopic Immunocytochemistry, edited by Polak JM and Priestley JV. Oxford, UK: Oxford Univ. Press, 1992, p. 89–121. [Google Scholar]
- 74.Race JE, Makhlouf FN, Logue PJ, Wilson FH, Dunham PB, Holtzman EJ. Molecular cloning and functional characterization of KCC3, a new K-Cl cotransporter. Am J Physiol Cell Physiol 277: C1210–C1219, 1999. doi: 10.1152/ajpcell.1999.277.6.C1210. [DOI] [PubMed] [Google Scholar]
- 75.Randall J, Thorne T, Delpire E. Partial cloning and characterization of Slc12a2: the gene encoding the secretory Na+-K+-2Cl− cotransporter. Am J Physiol Cell Physiol 273: C1267–C1277, 1997. doi: 10.1152/ajpcell.1997.273.4.C1267. [DOI] [PubMed] [Google Scholar]
- 76.Rao VV, Dahlheimer JL, Bardgett ME, Snyder AZ, Finch RA, Sartorelli AC, Piwnica-Worms D. Choroid plexus epithelial expression of MDR1 P glycoprotein and multidrug resistance-associated protein contribute to the blood-cerebrospinal-fluid drug-permeability barrier. Proc Natl Acad Sci USA 96: 3900–3905, 1999. doi: 10.1073/pnas.96.7.3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rinehart J, Maksimova YD, Tanis JE, Stone KL, Hodson CA, Zhang J, Risinger M, Pan W, Wu D, Colangelo CM, Forbush B, Joiner CH, Gulcicek EE, Gallagher PG, Lifton RP. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 138: 525–536, 2009. doi: 10.1016/j.cell.2009.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Robertson MA, Foskett KJ. Fluorescence measurements of cytosolic sodium concentration. In: Measurement and Manipulation of Intracellular Ions, edited by Kraicer J and Dixon SJ. San Diego, CA: Academic Press, 1995, p. 274–288. [Google Scholar]
- 79.Rocha-González HI, Mao S, Alvarez-Leefmans FJ. Na+,K+,2Cl− cotransport and intracellular chloride regulation in rat primary sensory neurons: thermodynamic and kinetic aspects. J Neurophysiol 100: 169–184, 2008. doi: 10.1152/jn.01007.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roder P, Hille C. ANG-2 for quantitative Na+ determination in living cells by time-resolved fluorescence microscopy. Photochem Photobiol Sci 13: 1699–1710, 2014. doi: 10.1039/C4PP00061G. [DOI] [PubMed] [Google Scholar]
- 81.Roepke TK, Kanda VA, Purtell K, King EC, Lerner DJ, Abbott GW. KCNE2 forms potassium channels with KCNA3 and KCNQ1 in the choroid plexus epithelium. FASEB J 25: 4264–4273, 2011. doi: 10.1096/fj.11-187609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rose CR, Ransom BR. Regulation of intracellular sodium in cultured rat hippocampal neurones. J Physiol 499: 573–587, 1997. doi: 10.1113/jphysiol.1997.sp021951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Russell JM. Sodium-potassium-chloride cotransport. Physiol Rev 80: 211–276, 2000. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- 84.Saito Y, Wright EM. Regulation of intracellular chloride in bullfrog choroid plexus. Brain Res 417: 267–272, 1987. doi: 10.1016/0006-8993(87)90451-3. [DOI] [PubMed] [Google Scholar]
- 85.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675, 2012. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shekarabi M, Zhang J, Khanna AR, Ellison DH, Delpire E, Kahle KT. WNK kinase signaling in ion homeostasis and human disease. Cell Metab 25: 285–299, 2017. doi: 10.1016/j.cmet.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 87.Silva P, Solomon RJ, Epstein FH. The rectal gland of Squalus acanthias: a model for the transport of chloride. Kidney Int 49: 1552–1556, 1996. doi: 10.1038/ki.1996.223. [DOI] [PubMed] [Google Scholar]
- 88.Silva P, Stoff J, Field M, Fine L, Forrest JN, Epstein FH. Mechanism of active chloride secretion by shark rectal gland: role of Na-K-ATPase in chloride transport. Am J Physiol Renal Physiol 233: F298–F306, 1977. doi: 10.1152/ajprenal.1977.233.4.F298. [DOI] [PubMed] [Google Scholar]
- 89.Smith QR, Johanson CE. Active transport of chloride by lateral ventricle choroid plexus of the rat. Am J Physiol Renal Physiol 249: F470–F477, 1985. doi: 10.1152/ajprenal.1985.249.4.F470. [DOI] [PubMed] [Google Scholar]
- 90.Smith QR, Woodbury DM, Johanson CE. Uptake of 36Cl and 22Na by the choroid plexus-cerebrospinal fluid system: evidence for active chloride transport by the choroidal epithelium. J Neurochem 37: 107–116, 1981. doi: 10.1111/j.1471-4159.1981.tb05297.x. [DOI] [PubMed] [Google Scholar]
- 91.Spector R, Keep RF, Robert Snodgrass S, Smith QR, Johanson CE. A balanced view of choroid plexus structure and function: focus on adult humans. Exp Neurol 267: 78–86, 2015. doi: 10.1016/j.expneurol.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 92.Steffensen AB, Oernbo EK, Stoica A, Gerkau NJ, Barbuskaite D, Tritsaris K, Rose CR, MacAulay N. Cotransporter-mediated water transport underlying cerebrospinal fluid formation. Nat Commun 9: 2167, 2018. doi: 10.1038/s41467-018-04677-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sung KW, Kirby M, McDonald MP, Lovinger DM, Delpire E. Abnormal GABAA receptor-mediated currents in dorsal root ganglion neurons isolated from Na-K-2Cl cotransporter null mice. J Neurosci 20: 7531–7538, 2000. doi: 10.1523/JNEUROSCI.20-20-07531.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Torres RJ, Altenberg GA, Copello JA, Zampighi G, Reuss L. Preservation of structural and functional polarity in isolated epithelial cells. Am J Physiol Cell Physiol 270: C1864–C1874, 1996. doi: 10.1152/ajpcell.1996.270.6.C1864. [DOI] [PubMed] [Google Scholar]
- 95.Valverde MA, Bond TD, Hardy SP, Taylor JC, Higgins CF, Altamirano J, Alvarez-Leefmans FJ. The multidrug resistance P-glycoprotein modulates cell regulatory volume decrease. EMBO J 15: 4460–4468, 1996. doi: 10.1002/j.1460-2075.1996.tb00823.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Verkman AS. Development and biological applications of chloride-sensitive fluorescent indicators. Am J Physiol Cell Physiol 259: C375–C388, 1990. doi: 10.1152/ajpcell.1990.259.3.C375. [DOI] [PubMed] [Google Scholar]
- 97.Welsh MJ. Intracellular chloride activities in canine tracheal epithelium. Direct evidence for sodium-coupled intracellular chloride accumulation in a chloride-secreting epithelium. J Clin Invest 71: 1392–1401, 1983. doi: 10.1172/JCI110892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wu Q, Delpire E, Hebert SC, Strange K. Functional demonstration of Na+-K+-2Cl− cotransporter activity in isolated, polarized choroid plexus cells. Am J Physiol Cell Physiol 275: C1565–C1572, 1998. doi: 10.1152/ajpcell.1998.275.6.C1565. [DOI] [PubMed] [Google Scholar]
- 99.Zeuthen T. Cotransport of K+, Cl− and H2O by membrane proteins from choroid plexus epithelium of Necturus maculosus. J Physiol 478: 203–219, 1994. doi: 10.1113/jphysiol.1994.sp020243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zeuthen T. The effects of chloride ions on electrodiffusion in the membrane of a leaky epithelium. Studies of intact tissue by microelectrodes. Pflügers Arch 408: 267–274, 1987. doi: 10.1007/BF02181469. [DOI] [PubMed] [Google Scholar]
- 101.Zeuthen T, Gorraitz E, Her K, Wright EM, Loo DD. Structural and functional significance of water permeation through cotransporters. Proc Natl Acad Sci USA 113: E6887–E6894, 2016. doi: 10.1073/pnas.1613744113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zeuthen T, Macaulay N. Cotransport of water by Na+-K+-2Cl− cotransporters expressed in Xenopus oocytes: NKCC1 versus NKCC2. J Physiol 590: 1139–1154, 2012. doi: 10.1113/jphysiol.2011.226316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang J, Deng X, Kahle KT. Leveraging unique structural characteristics of WNK kinases to achieve therapeutic inhibition. Sci Signal 9: pe3, 2016. doi: 10.1126/scisignal.aaj2227. [DOI] [PubMed] [Google Scholar]
- 104.Zhang J, Gao G, Begum G, Wang J, Khanna AR, Shmukler BE, Daubner GM, de Los Heros P, Davies P, Varghese J, Bhuiyan MI, Duan J, Zhang J, Duran D, Alper SL, Sun D, Elledge SJ, Alessi DR, Kahle KT. Functional kinomics establishes a critical node of volume-sensitive cation-Cl− cotransporter regulation in the mammalian brain. Sci Rep 6: 35986, 2016. doi: 10.1038/srep35986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang J, Karimy JK, Delpire E, Kahle KT. Pharmacological targeting of SPAK kinase in disorders of impaired epithelial transport. Expert Opin Ther Targets 21: 795–804, 2017. doi: 10.1080/14728222.2017.1351949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhao H, Muallem S. Na+, K+, and Cl− transport in resting pancreatic acinar cells. J Gen Physiol 106: 1225–1242, 1995. doi: 10.1085/jgp.106.6.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]