

Abstract
Na+-K+-2Cl− cotransporter-1 (NKCC1) mediates the electroneutral transport of Na+, K+, and Cl− and is normally localized to the basolateral membrane of polarized epithelial cells. We recently reported the first known solute carrier family 12 member 2 (SLC12A2) mutation (we call NKCC1-DFX) that causes epithelial dysfunction in an undiagnosed disease program case. The heterozygous mutation leads to truncation of the COOH-terminal tail of the cotransporter, resulting in both mutant and wild-type cotransporters being mistrafficked to the apical membrane of polarized epithelial cells. Here we demonstrate by using consecutive truncations and site-directed mutagenesis of the COOH-terminal domain of NKCC1 that truncation of NKCC1 COOH domain uncouples the cotransporter from the lateral membrane. We identify a dileucine motif that, when mutated, leads to cotransporter accumulation in the cytoplasm and mistrafficking to the apical/subapical region of epithelial cells, thereby recapitulating the phenotype observed with the patient mutation. We show that truncation deletion and LL substitution mutants are trafficked out of the endoplasmic reticulum and trans-Golgi network but accumulate in early and late endosomes where they are degraded.
Keywords: lysosomes, Matrigel, MDCK cells, Na-K-2Cl cotransport, trafficking
INTRODUCTION
We recently reported the case of a patient with a disease affecting most of her major organs with the exception of the central nervous system (6). She suffers from hypo-insufficiency of most endocrine (thyroid, parathyroid, adrenal, pancreatic) and exocrine (salivary, pancreatic) glands, as well as deficits in lung, stomach, intestine, colon, and bladder functions. Whole exome sequencing revealed amino acid substitutions in several genes and a de novo 11 bp deletion in solute carrier family 12 member 2 (SLC12A2), the gene that encodes the Na-K-2Cl cotransporter, NKCC1 (6). The deletion truncates part of the cytosolic carboxyl-terminal tail of the cotransporter and renders the cotransporter functionally inactive. As only one SLC12A2 allele is affected in the patient, NKCC1 function is reduced. This was measured in fibroblasts isolated from the patient. Interestingly, no dominant-negative effect of mutant on wild-type transporter was observed (6).
NKCC1 is expressed in multiple tissues where it serves a variety of physiological functions (10, 16). In the nervous system, NKCC1 is expressed in young immature neurons and in young and mature sensory neurons where it facilitates Cl− accumulation and GABA depolarization (22, 29). In smooth muscle cells, the transporter participates in Cl−-mediated channel depolarization through accumulation of intracellular Cl− (3). Through its role in dorsal root ganglion neurons-mediated sensory feedback in and/or smooth muscle cells, NKCC1 dysfunction might explain the multi-organ involvement.
Aside from being expressed in neurons and smooth muscle cells, NKCC1 is also expressed in many epithelia where it contributes to the transcellular movement of ions and water (10, 16). In the inner ear, the transporter is located on the basolateral membrane of stria vascularis epithelial cells, where it participates in the secretion of the K+-rich endolymph (33). As a result, NKCC1 knockout mice suffer from inner ear defects, deafness, and imbalance, due to the lack of endolymph secretion (5, 8). In salivary glands, lung, and intestine, NKCC1 is also expressed on the basolateral membrane where it facilitates the replenishment of Cl− from blood, when the anion is secreted by CFTR Cl− channels on the apical membrane (9). As a consequence, NKCC1 knockout mice exhibit a deficit in saliva secretion and intestinal obstruction (7, 8). Interestingly, it is in epithelial cells that we observed a significant dominant-negative effect of the truncated transporter on the wild-type transporter (13). Both mutant and some wild-type transporters were expressed at the apical pole and the apical membrane. This was demonstrated in transfected Madin-Darby canine kidney (MDCK) cells cultured on permeabilized support and in three-dimensional (3D) Matrigel, as well as in tissue isolated from mice engineered to carry the patient mutation (13).
Apical and basolateral sorting of membrane proteins in epithelial cells is a tightly regulated process. While the mechanisms directing membrane proteins to the apical membrane are diverse and complex, sorting to the basolateral membrane is often directed by discrete sorting signals embedded in the protein sequence. The most common basolateral sorting motifs include tyrosine-based (NPxY or Yxxф), dileucine (D/ExxxLL), and monoleucine (EExxxL) motifs (28). Recently, the presence of two leucine residues in alternatively spliced exon 21 of NKCC1 was indicated to be a signal for targeting of the cotransporter to the basolateral membrane (4). Absence of this exon in NKCC1 or of its sequence in NKCC2 was described as being involved in apical targeting of these transporters (4). Because exon 21 is located upstream of the COOH-terminal deletion in the DFX mutant, we sought to identify the determinants of NKCC1-DFX expression at the apical membrane and to identify the structures associated with NKCC1-DFX signal accumulation in cytoplasm. Here we show that exon 21 is not involved in the trafficking of NKCC1 to the basolateral membrane of MDCK cells, but that association with the membrane depended on a dileucine motif located close to the end of the protein. We also identified a nucleolar localization signal that, under normal conditions, is inactive but was activated upon eliminating the last 90 residues of the carboxyl-terminus. Finally, we show that the truncated NKCC1-DFX transporter is properly cleared out of the endoplasmic reticulum (ER) and Golgi, as we saw no evidence of accumulation in these compartments. The transporter was found to be associated with lysosome and apical early endosome markers (lyso-track and Rab5, respectively). No colocalization signal was detected with Rab11 and Rab8. We also show that expression of the dominant-negative myosin Vb tail did not cause accumulation of NKCC1-COOH mutants in the common recycling endosome, indicating that the mutants do not reach the apical membrane via transcytosis from the basolateral membrane. These data provide an additional and new understanding of the trafficking of wild-type and carboxyl-terminal truncated forms of NKCC1 in epithelial cells.
MATERIALS AND METHODS
Reagents.
Prolong Gold Antifade with DAPI was obtained from Life Technologies (Grand Island, NY), PureBlu Hoechst 33342 was from Bio-Rad; Lyso-Tracker Red was from ThermoFisher; ER-Tracker Red-BODIPY TR was from ThermoFisher; BODIPY FL C5-Ceramide Complexed to BSA was from ThermoFisher; and Matrigel was obtained from Corning. The following antibodies were used as primary antibodies: Rab5 (1:200; cat. no. ab18211, Abcam); Rab11 (1:200, cat. no. ab3612, Abcam); Rab8 (1:200, cat. no. LS-B9782, LSBio); Rab7 (1:200, cat. no. Rab7, DHSB); ZO-1 (1:200, R26.4 monoclonal, DHSB); PDXL/Gp135 (1:100, cat. no. 3F2/D8, DHSB); green fluorescent protein (GFP; 1:100, 1C9A5 monoclonal, Vanderbilt VAPR Core); Alexa-Fluor-488 tagged anti-GFP (1:200, cat. no. A-21311, ThermoFisher).
cDNA clones.
Epitope-tagged murine NKCC1-wild type (WT) and NKCC1-DFX constructs were previously described (13). To create serial deletions of NKCC1 COOH-terminal domain and the dileucine motif mutants, complementary oligonucleotides containing a stop codon or LL to AA substitutions in their center were used to mutate NKCC1 cDNA fragments using QuikChange (Agilent, Santa Clara, CA). After sequencing and confirmation of sequence integrity and introduction of stop codon, the fragments were reintroduced within the full-length enhanced GFP (EGFP)-NKCC1 clone in pCDNA3. The stop codons were introduced after Thr 789 (NKCC1/236), Val 852 (NKCC1/237), Arg 915 (NKCC1/238), Gln 1114 (NKCC1/239), and Ile 1160 (NKCC1/240) of mouse NKCC1. The dileucine motifs were Leu 1031-1032 (NKCC1/241) and Leu 1192-1193 (NKCC1/242).
Generation of stable MDCK.
NKCC1-WT, NKCC1-DFX, NKCC1Δexon21, and NKCC1-COOH terminal mutant cDNAs purified with Qiagen Mediprep kit and quantitated by absorbance at 260, 280, and 320 nm were transfected into MDCK cells using Lipofectamine 2000 (Life Technologies). Cells were then transferred in selection media (DMEM supplemented with 5% FBS, 1% penicillin and streptomycin and 500 μg/ml of Geneticin) for 14 days. Stable cells lines were subsequently sorted by FACS. GFP-positive cells were maintained in selection media and expanded.
Transient transfection of MDCK cells with myosin Vb dominant-negative plasmid.
The dominant-negative myosin Vb construct was a gift from Dr. James Goldenring (Vanderbilt University Medical Center). MDCK cells were cultured on MatTeK glass bottom dishes until confluent. Cells were then transfected using Lipofectamine 2000 as described above. After 24 h, the cells were incubated in Hanks’ balanced salt solution containing CaCl2 and MgCl2 (HBSS2+) containing 1 μg/ml of PureBlu Hoechst 33342 (Bio-Rad) for 15 min at 37°C. Cells were then washed twice with HBSS2+, incubated in HBSS2+ containing 2.5% FBS, and imaged live on the Zeiss LSM 880 confocal microscope.
Three-dimensional culture of MDCK cells on Matrigel.
3D cell cultures were performed as previously described (13). Briefly, four-well glass slides (Lab-TEK II) were coated with 85 μl of Matrigel solution (Corning) at 4°C. Matrigel was allowed to solidify for 20 min at 37°C. MDCK cells expressing EGFP-tagged NKCC1-WT, NKCC1-DFX, NKCC1–241, NKCC1–242, and NKCC1–244 mutants were resuspended in complete DMEM at 1 × 104 cells/ml. A volume of 20 μl of cell suspension was added to 200 μl of 2% Matrigel. This cell suspension was then layered on top of the solidified Matrigel and incubated in a humidified incubator at 37°C, 5% CO2. Cells were grown from 6 h to 8 days. Conditioned medium was aspirated every 48 h and replaced with fresh 2% Matrigel in complete DMEM medium. Transparent cysts with hollow lumen were visible under brightfield microscopy by day 4.
Immunofluorescence.
MDCK cells expressing epitope-tagged NKCC1-WT and NKCC1-DFX were cultured on glass coverslips until they reached 100% confluency. Cells were fixed with cold (−20°C) methanol:acetone (1:1) solution, washed, and incubated in blocking buffer (5% BSA, 1% goat serum 0.1% Triton X-100 and 0.1% Tween 20 in HBSS2+) for 2 h and incubated in primary antibodies overnight at 4°C. Both Alexa-Fluor-tagged anti-GFP and anti-ZO-1 were added at the same time. Alternatively, anti-GFP antibody was added simultaneously with anti-Rab5, anti-Rab8, anti-Rab11, or anti-Rab7. Slides were washed 3 × 20 min with HBSS2+ and incubated with fluorophore-conjugated (Cy3 or FITC) secondary antibodies for 2 h. Cells on MatTek dishes were then mounted with Prolong Gold Antifade with DAPI (Invitrogen) and covered with 12 mm round microscope cover glass. Images were captured on a Zeiss LSM 880 laser scanning confocal microscope. Samples were scanned using a ×63 oil objective. Z-stacks slices were set at 0.5-μm intervals. The images were exported as TIFF files using the Zeiss ZEN Lite 2012 software. For 3D cultures, MDCK cysts maintained in four-well Lab-Tek chamber slides were fixed with cold (−20°C) methanol:acetone (1:1) solution, washed, and incubated in blocking buffer (5% BSA, 1% goat serum, 0.1% Triton X-100, and 0.1% Tween 20 in HBSS2+) for 2 h and incubated in primary antibodies overnight at 4°C. Both Alexa-Fluor-tagged anti-GFP and anti-Gp135/podocalyxin were added at the same time. Slides were washed 3 × 20 min with HBSS2+ and incubated with fluorophore-conjugated (Cy3 or FITC) secondary antibodies for 2 h. Chambers were removed from the microscope slide and mounted according to the manufacturer’s protocol. Slides were imaged on the Zeiss LSM 880 as described above.
Live cell imaging.
MDCK cells expressing EGFP-tagged NKCC1-WT and NKCC1 COOH-terminal mutants were cultured in MatTek glass bottom microwell dishes until they reach 100% confluency. For lysosome staining, cells were washed twice with HBSS2+ and incubated in HBSS2+ containing 1 μg/ml of PureBlu Hoechst 33342 (Bio-Rad) and 100 nM Lyso-Tracker Red (ThermoFisher) for 30 min at 37°C. Cells were washed twice with HBSS2+, incubated in HBSS2+ containing 2.5% FBS and imaged. For ER staining, cells were washed twice with HBSS2+ and incubated in HBSS2+ containing 1 μg/ml of PureBlu Hoechst 33342 (Bio-Rad) and 100 μM ER-Tracker Red (BODIPY TR, ThermoFisher) for 30 min at 37°C. Cells were then washed twice with HBSS2+, incubated in HBSS2+ containing 2.5% FBS, and imaged. For Golgi staining, cells were washed twice with ice-cold HBSS2+ and incubated in ice-cold HBSS2+ containing 5 μM BODIPY FL C5-Ceramide Complexed to BSA (ThermoFisher) and incubated at 4°C for 30 min. Cells were then washed twice with HBSS2+ and incubated in fresh HBSS2+ containing 1 μg/ml of PureBlu Hoechst 33342 (Bio-Rad) for 30 min at 37°C. Cells were finally washed twice with HBSS2+, incubated in HBSS2+ containing 2.5% FBS, and imaged. All images were captured live on a Zeiss LSM 880 laser scanning confocal microscope. Samples were scanned using a ×63 oil objective. The images were exported as TIFF files using the Zeiss ZEN Lite 2012 software.
RESULTS
Loss of exon 21 does not target NKCC1 to the apical membrane.
The alternatively spliced exon 21 of NKCC1 (Fig. 1A) has been implicated in the trafficking of the cotransporter to the basolateral membrane of epithelial cells (4). To determine if the 11 bp deletion in exon 22 of NKCC1 affects the splicing of exon 21, we isolated RNA from brain of wild-type, DFX heterozygote, and DFX homozygote mutant mice. The brain was chosen because it is a tissue that expresses both the exon 21-containing and the exon 21-deficient NKCC1 isoforms (23). The generation of the mice has been described in a previous study (13) and was performed using a protocol approved by the Vanderbilt University Institutional Animal Care and Use Committee. We then performed RT-PCR to amplify a fragment of the cotransporter between exons 20 and 23 (Fig. 1A). As seen in Fig. 1B, we observed the larger band, or the exon 21-containing isoform, in mice expressing both one and two copies of the DFX allele, indicating that exon 21 is not preferentially spliced out when exon 22 lacks the 11 base pairs. To further understand the mechanisms mediating the trafficking of NKCC1-DFX to the apical and subapical compartment of polarized epithelial cells, we stably expressed the NKCC1 cDNAs with NH2-terminal EGFP tag in MDCK cells. When these cells were grown in 3D in Matrigel for 8–14 days, wild-type NKCC1 localized to the basolateral membrane while the apical membrane marker gp135/podocalixyn localized to the apical membrane (Fig. 2A). On the other hand, deletion of the 16 amino acids encoded by exon 21 from the COOH-terminal domain of NKCC1 did not affect cotransporter expression at the basolateral membrane (Fig. 2B). Therefore, it is unlikely that the absence of exon 21 sequence in NKCC2 accounts for this transporter being expressed at the apical membrane of thick ascending limb epithelial cells.
Fig. 1.

Exon 21 is not spliced out of the patient truncated NKCC1-DFX cotransporter. A: schematic representation of slc12a2 mRNA region from exon 20 to exon 23. The position of the 11 nucleotide deletion (DFX) and PCR primers (P1 and P2) is shown. B: RT-PCR products spanning exon 21 of solute carrier family 12 member 2 (SLC12A2) were amplified from cDNA template of NKCC1WT/WT, NKCC1WT/DFX and NKCC1DFX/DFX mice. Data are representative of 3 individual experiments where 1 mouse per genotype was analyzed. NKCC1, Na+-K+-2Cl− cotransporter-1; WT, wild type.
Fig. 2.

Loss of solute carrier family 12 member 2 (SLC12A2) exon 21 does not target the cotransporter to the apical membrane. MDCK cells stably transfected with EGFP-NKCC1-WT (A and B) and EGFP-NKCC1Δexon21 (C and D) were grown in 3D on Matrigel for 8 days, fixed and immunostained with anti-Gp135/podocalyxin. Images were captured on Zeiss LSM 880 confocal microscope. Bars = 10 μm. Data are representative of 4 individual experiments where 6 cysts per experiment were imaged (Total, n = 24). 3D, three-dimensional; EGFP, enhanced green fluorescent protein; MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1; WT, wild type.
NKCC1 COOH-terminal domain mutants are trafficked out of the endoplasmic reticulum.
To identify the region(s) of NKCC1 COOH-terminal domain involved in NKCC1 targeting to the basolateral membrane, we created serial EGFP-tagged truncations of the COOH-terminal domain of the cotransporter (Fig. 3). In addition, two dileucine motifs 1031DGGLTLL1037 and 1088DLPPVLL1194 that were previously shown to affect trafficking of the cotransporter out of the ER and Golgi were substituted to alanine residues (Fig. 3). Note that the aspartic acid “D”, located 3-4 residues upstream of the two leucines, is highlighted in bold. To determine if NKCC1 COOH-terminal mutants were trapped in the endoplasmic reticulum (ER), MDCK cells stably transfected with the COOH-terminal mutants were plated on MatTek glass bottom microwell dishes, incubated with ER-Tracker probe, and visualized live. In NKCC1-WT transfected cells, the cotransporter localized to the plasma membrane (Fig. 3A). In contrast, the NKCC1 COOH-terminal mutants appeared mostly cytoplasmic (Fig. 3, B–F and H–J). Note that in MDCK cells transfected with NKCC1–236 mutant; where the cotransporter COOH-terminal domain is almost completely removed; NKCC1 was not detectable, indicating that this mutant was unstable and rapidly degraded (Fig. 3G). Taken together, these data indicate that the NKCC1 COOH-terminal domain mutants are trafficked out of the ER.
Fig. 3.

NKCC1-WT and NKCC1 COOH-terminal mutants are trafficked out of the ER. A: cartoons depicting EGFP-NKCC1 and NKCC1 COOH-terminal mutants. WT, wild-type; 240, Δ46aa; 239, Δ92aa; DFX, Δ187aa, 238 Δ291aa; 237, Δ354aa; 236 Δ417aa; 241, LL1031-1032AA; 242, LL1192-1193AA; 244, LL1031-1032AA + LL1192-1193AA. A–J: MDCK-WT and MDCK transfected with COOH-terminal mutants were cultured in MatTek Glass Bottom Microwell dishes. Cells were stained with PureBlu Hoechst 33342 and ER-Tracker Red and imaged live. Bars = 10 μm. ER, endoplasmic reticulum; EGFP, enhanced green fluorescent protein; MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1.
NKCC1 COOH-terminal mutants also exit the Golgi.
To investigate whether NKCC1 COOH-terminal mutants causes accumulation of the cotransporter in the Golgi, MDCK cells expressing WT and the various COOH-terminal mutants were plated on MatTek glass bottom microwell dishes, incubated with Golgi-Tracker probe and visualized live (Fig. 4). Once again in NKCC1-WT transfected cells, the cotransporter remained at the plasma membrane (Fig. 4A), whereas COOH-terminal NKCC1 mutants mainly localized to the cytoplasm (Fig. 4, B–F and H–J).
Fig. 4.

NKCC1-WT and NKCC1 COOH-terminal mutants are trafficked out of the Golgi. A–J: MDCK-WT and MDCK transfected with NKCC1 COOH-terminal mutants were cultured in MatTek Glass Bottom Microwell dishes. Cells were stained with PureBlu Hoechst 33342 and Golgi-Tracker Red and imaged live. WT, wild-type; 240, Δ46aa; 239, Δ92aa; DFX, Δ187aa, 238 Δ291aa; 237, Δ354aa; 236 Δ417aa; 241, LL1031-1032AA; 242, LL1192-1193AA; 244, LL1031-1032AA + LL1192-1193AA. Bars = 10 μm. GFP, green fluorescent protein; MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+- 2Cl− cotransporter-1. Data are representative of 3 individual experiments where 5 live images per experiment were taken (Total, n = 15).
NKCC1 COOH-terminal mutants are inefficiently targeted or recycled to the plasma membrane and are degraded in lysosomes.
To establish the fate of the NKCC1 COOH-terminal mutants in the cells, we next examined the lysosomal compartment. MDCK cells expressing NKCC1-WT and COOH-terminal mutants were again plated on MatTek glass bottom microwell dishes, incubated with the Lyso-Tracker probe, and visualized live (Fig. 5). In the steady-state condition, wild-type NKCC1 was predominantly at the plasma membrane (Fig. 5A). There was no accumulation of the cotransporter in the cytoplasm and a very small fraction of the cotransporter colocalized with the lysosomal marker (Fig. 5A). In contrast, most NKCC1 COOH-terminal mutants were targeted to the lysosomes (Fig. 5, D and F–J). However, a significant amount of mutant cotransporters were still located in an undefined compartment within the cytoplasm. Furthermore, some mutants seemed to evade excessive lysosomal degradation and caused strong “cytoplasmic” retention, as noted for mutants NKCC1–238, NKCC1–239, and NKCC1–240 (Fig. 5, B, C, and E). It is not clear why these mutants behaved differently.
Fig. 5.

Disruption of NKCC1 COOH-terminal domain targets the cotransporter to the lysosomes. A–J: MDCK-WT and MDCK transfected with NKCC1 COOH-terminal mutants were cultured in MatTek Glass Bottom Microwell dishes. Cells were stained with PureBlu Hoechst 33342 and Lyso-Tracker Red and imaged live. WT, wild-type; 240, Δ46aa; 239, Δ92aa; DFX, Δ187aa, 238 Δ291aa; 237, Δ354aa; 236 Δ417aa; 241, LL1031-1032AA; 242, LL1192-1193AA; 244, LL1031-1032AA + LL1192-1193AA. Bars = 10 μm. GFP, green fluorescent protein; Lyso, lysosomes; MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1. Data are representative of 3 individual experiments where 5 live images per experiment were taken (Total, n = 15).
NKCC1 COOH-terminal domain contains a hidden nucleolar localization signal.
NKCC1 is normally targeted to the plasma membrane of most cells or more precisely, to the basolateral membrane of polarized epithelial cells (as in Figs. 3A and 6A). Deletion of 92 amino acids from the COOH-terminal domain caused a redistribution of NKCC1 from the plasma membrane to subnuclear foci (Fig. 6B). Using a nucleolar localization signal (NoLS) predictive algorithm (http://www.compbio.dundee.ac.uk/www-nod), we discovered a putative nucleolar localization signal in the COOH-terminus of NKCC1 (Fig. 6C), located downstream of the patient mutation. These results indicate that the deletion exposed the NoLS site, resulting in translocation of NKCC1 in the nucleolus. Whether this observation has any physiological significance remains to be determined.
Fig. 6.

NKCC1 COOH-terminal domain contains a putative nucleolus localization signal. A and B: MDCK-WT (A) and MDCK transfected with NKCC1–239 COOH-terminal mutant (B) were cultured in MatTek glass bottom microwell dishes. Cells were stained with PureBlu Hoechst 33342 and imaged live. C: nucleolus predicted sequence and score. WT, wild-type; 239, Δ92aa. Bars = 20 μm. GFP, green fluorescent protein; MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1. The data are representative of more than 10 individual experiments and more than 50 images taken.
Deletions and mutation of NKCC1 COOH-terminal domain uncouple the cotransporter from the plasma membrane.
To identify the region of the COOH-terminus of NKCC1 responsible for maintaining the cotransporter in the basolateral membrane, we quickly polarized MDCK cells transfected with EGFP-tagged wild-type and mutant NKCC1 cDNAs and analyzed the cotransporter localization by confocal microscopy. Confocal projections in XZ plane indicated that all NKCC1 mutants excluding EGFP-NKCC1–241 were uncoupled from the basolateral membrane (Fig. 7), while wild-type NKCC1 expression was again restricted to the plasma membrane. Mutations of COOH-terminal domain caused accumulation of mutant transporters in the cytoplasm (Fig. 7). However, mutation of dileucine motif 1026DGGLTLL1032 to 1026DGGLTAA1032 (EGFP-NKCC1–241) did not affect cotransporter expression in the basolateral membrane. This suggests that the 1026DGGLTLL1032 dileucine motif is not required to target or maintain NKCC1 to the basolateral membrane. In contrast, mutation of dileucine motif 1087DLPPVLL1193 to 1087DLPPVAA1193 (EGFP-NKCC1–242) was sufficient to prevent the cotransporter from associating with the basolateral membrane (Fig. 7). Likewise, mutation of both dileucine motifs (EGFP-NKCC1–244) behaved as EGFP-NKCC1–242 mutation, suggesting that 1087DLPPVLL1193 is the dominant motif required to maintained or target NKCC1 to the basolateral membrane.
Fig. 7.

NKCC1 DDxxxxLL1193–1194 dileucine motif is required to maintain the cotransporter in the basolateral membrane. MDCK cells were polarized on MatTek dishes for 3 days then transiently transfected with EGFP-NKCC1-WT (A), EGFP-NKCC1–240 (B), EGFP-NKCC1–239 (C), EGFP-NKCC1-DFX (D), EGFP-NKCC1–238 (E), EGFP-NKCC1–237 (F), EGFP-NKCC1–236 (G), EGFP-NKCC1–241 (H), EGFP-NKCC1–242 (I), and EGFP-NKCC1–244 (J). Cells were immunostained with anti-GFP and anti-ZO-1 antibody. WT, wild-type; 240, Δ46aa; 239, Δ92aa; DFX, Δ187aa, 238 Δ291aa; 237, Δ354aa; 236 Δ417aa; 241, LL1031-1032AA; 242, LL1192-1193AA; 244, LL1031-1032AA + LL1192-1193AA. Bars = 20 μm. GFP, green fluorescent protein; MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1. Data are representative of 3 individual experiments where 5 live images per experiment were taken (Total, n = 15).
Mutation of the 1087DxxxxLL1193 dileucine motif recapitulates the NKCC1-DFX phenotype.
Given the altered localization of EGFP-NKCC1–241 (1026DGGLTAA1033), EGFP-NKCC1–242 (1087DLPPVAA1193) and EGFP-NKCC1–244 (1026DGGLTAA1032 + 1087DLPPVAA1193) in 2D cultures, we cultured EGFP-NKCC1-WT, EGFP-NKCC1-DFX, EGFP-NKCC1–241, EGFP-NKCC1–242, and EGFP-NKCC1–244 in 3D in Matrigel. As shown in Fig. 8A, EGFP-NKCC1-WT nicely localized to the basolateral membrane, and as previously reported NKCC1-DFX was targeted to the apical membrane and accumulated in the subapical areas, though some basolateral localization remained noticeable (Fig. 8B). Although mutation 1026DGGLTLL1032 to 1026DGGLTAA1032 (EGFP-NKCC1–241) showed some cytoplasmic expression in 2D culture (Fig. 7), this mutant transporter was almost entirely targeted to the basolateral membrane in 3D culture (Fig. 8C). This suggests that this dileucine motif is not required to target or maintain NKCC1 to the basolateral membrane. In contrast, mutation of 1087DLPPVLL1194 dileucine motif to 1087DLPPVAA1193 targeted NKCC1 to the apical and subapical region (Fig. 8D). As in 2D culture, mutation of both dileucine motifs also resulted in accumulation of the cotransporter at the apical membrane (Fig. 8E).
Fig. 8.

Mutation of DDxxxxLL1193–1194 dileucine motif to DDxxxxAA1193–1194 recapitulates NKCC1-DFX phenotype. MDCK cells were stably transfected with EGFP-NKCC1-WT (A), EGFP-NKCC1-DFX (B), EGFP-NKCC1–241 (C), EGFP-NKCC1–242 (D), and EGFP-NKCC1–244 (E). Cells were grown on 3D in Matrigel for 8 days, fixed, and immunostained with anti-Gp135/Podocalyxin. WT, wild-type; DFX, Δ187aa, 241, LL1031-1032AA; 242, LL1192-1193AA; 244, LL1031-1032AA + LL1192-1193AA. Bars = 10 μm. GFP, green fluorescent protein; MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1. Data are representative of 4 individual experiments where 6 cysts per experiment were imaged (Total, n = 24).
NKCC1 1087DxxxxLL1193 dileucine is required to target the cotransporter to the basolateral membrane during apical-to-basolateral polarization.
To better understand the reason of NKCC1 COOH-terminal mutants mis-sorting to the apical membrane, we next examined the trafficking of the cotransporter during the process of epithelial cell apical-to-basolateral polarization. At the single-cell stage, 6 h after plating EGFP-MDCK-WT, EGFP-MDCK-DFX, EGFP-MDCK-241, EGFP-MDCK-242 and EGFP-MDCK-244 on Matrigel, NKCC1-WT and COOH-terminal mutant were localized either at the plasma membrane or in the cytoplasm (Fig. 9, A–E). In contrast, 12 h after plating, although most cells were still at the single-cell stage, the localization of the cotransporter was markedly changed. The WT cotransporter was removed from the cell periphery and the cytoplasm and targeted to a pole of the cell, seemingly pushed by a ring of gp135/podocalyxin (Fig. 9F). Similarly, EGFP-NKCC1–241 was propelled to one pole of the cell under gp135/podocalyxin (Fig. 9H). Interestingly, in MDCK-DFX, MDCK-242, and MDCK-244, this polarization of the cotransporter on one side of the cell was undetectable; while most of the signal remained cytoplasmic (Fig. 9, G, I and J). Normally, a gp135/podocalyxin-EZRIN-NHERF1 complex directs the formation and expansion of the apical membrane. Gp135/podocalyxin containing vesicles were accumulated at the apical membrane initiation site (AMIS), which was later turned into a pre-apical patch (PAP). Remarkably, at the 2-cell stage, NKCC1-WT and NKCC1–241 were brought to the AMIS or PAP, which was now located between the two cells (Fig. 9, K and M). In contrast, although some NKCC1-DFX, NKCC1–242 and NKCC1–244 could be found at the AMIS/PAP site at the 2-cell stage, much of the signal was still scattered in the cytoplasm (Fig. 9, L, N and O). In addition, Gp135/podocalyxin persisted at the cell periphery in cells expressing MDCK-DFX or MDCK-242, 24 h after plating (Fig. 9, L and N, arrowhead). Finally, 48 h after plating or at the 4-cell stage, NKCC1-WT and NKCC1–241 were targeted to the nascent basolateral membranes (Fig. 9, P and R). At that stage, NKCC1-DFX, NKCC1–242, and NKCC1–244 were localized at the budding apical membrane and accumulated in the subapical region of MDCK cysts (Fig. 9, Q, S and T). Finally, in MDCK-DFX, Gp135/podocalyxin vesicles failed to coalesce into one area, potentially giving rise to the multiple lumens that we observed in our previously publication (13).
Fig. 9.

Disruption of NKCC1 COOH-terminal domain disrupts the cotransporter interaction with cell trafficking and polarity machinery. Single-cell suspension of MDCK stably transfected with EGFP-NKCC1-WT (A), EGFP-NKCC1-DFX (B), EGFP-NKCC1–241 (C), EGFP-NKCC1–242 (D), and EGFP-NKCC1–244 (E) were grown on 3D in Matrigel for 6–48 h. We then followed these five MDCK clones overtime (F–T). After each time point, cells were fixed and immunostained with anti-Gp135/podocalyxin. Bar = 10 μm WT, wild-type; DFX, Δ187aa, 241, LL1031-1032AA; 242, LL1192-1193AA; 244, LL1031-1032AA + LL1192-1193AA. Bars = 10 μm. GFP, green fluorescent protein; MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1. Data are representative of 3 individual experiments where 5 cysts per experiment were imaged (Total, n = 15).
NKCC1-DFX does not reach the apical membrane via basolateral-to-apical transcytosis.
Given that the mutants NKCC1-DFX, NKCC1–242, and NKCC1–244 display both apical/subapical and basolateral localization, we next asked whether the endocytic recycling complex could be involved in the transport of wild-type NKCC1 and COOH-terminal mutant cotransporters to the plasma membrane. To test this hypothesis, we transiently transfected MDCK-WT, MDCK-DFX, MDCK-241, MDCK-242, and MDCK-244 cells with a dominant-negative mCherry-tagged myosin Vb tail that lacks motor activity (14). As seen in Fig. 10, myosin Vb was not required for the trafficking of NKCC1-WT (Fig. 10A) or NKCC1–241 (Fig. 10C) to the plasma membrane. Further, none of the other mutants were trapped into myosin-Vb-positive endosomes (Fig. 10, B, D and E), indicating that the Na-K-2Cl cotransporter and its mutants are not trafficked to the plasma membrane via the endocytic recycling complex.
Fig. 10.

NKCC1-DFX is not trafficked to the apical/subapical area via the basolateral-to-apical transcytosis. MDCK stably transfected with EGFP-NKCC1-WT (A), EGFP-NKCC1-DFX (B), EGFP-NKCC1–241 (C), EGFP-NKCC1–242 (D), and EGFP-NKCC1–244 (E) were grown on MatTek Glass Bottom Microwell dishes for 3 days then transiently transfected with RFP-tagged DN-myosin-Vb tail (MyoVb) and imaged live. WT, wild-type; DFX, Δ187aa, 241, LL1031-1032AA; 242, LL1192-1193AA; 244, LL1031-1032AA + LL1192-1193AA. Bars = 10 μm. GFP, green fluorescent protein; RFP, red fluorescent protein; DN, dominant-negative; MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1. Data are representative of 3 individual experiments where 5 live images per experiment were taken (Total, n = 15).
NKCC1-DFX and COOH-terminal mutants localize mainly to Rab5-positive endosomes.
To determine the identity of the endosomal compartment accumulating the NKCC1 COOH-terminal mutants, we examined the colocalization of NKCC1-WT, NKCC1-DFX, NKCC1–241, NKCC1–242 and NKCC1–244 with different endosomal markers (Fig. 11). Unexpectedly, neither the WT cotransporter nor the COOH-terminal mutants colocalized with Rab11a-positive recycling endosomes, which are known to recycle proteins from the common recycling endosome back to plasma membrane of epithelial cells (Fig. 11, A–D). Like Rab11a, Rab8a-positive endosomes that also function as slow recycling endosomes did not show any colocalization with NKCC1-WT, NKCC1-DFX, NKCC1–241, NKCC1–242 and NKCC1–244 (data not shown). Because our previous data indicated that NKCC1-DFX is mis-sorted to the apical membrane and accumulates in the subapical region of polarized epithelial cells, we also investigated colocalization of WT and COOH-terminal NKCC1 mutants with the apical early endosomal marker Rab5 (Fig. 12). Notably, NKCC1-WT and NKCC1–241 (Fig. 12, A and C), which were both properly targeted to the basolateral membrane, did not colocalize with Rab5. In contrast, NKCC1-DFX (Fig. 12B), NKCC1–242 (Fig. 12C), and NKCC1–244 (data not shown) did show expression in Rab5-positive endosomes. These data indicate that colocalization of NKCC1-DFX, NKCC1–242 and NKCC1–244 in apical early endosomes may result from instability of these mutants at the plasma membrane.
Fig. 11.

NKCC1-DFX is not retained in slow recycling Rab11-positive vesicles. MDCK stably transfected with EGFP-NKCC1-WT (A), EGFP-NKCC1-DFX (B), EGFP-NKCC1–241 (C), EGFP-NKCC1–242 (D), and EGFP-NKCC1–244 (E) were grown on MatTek Glass Bottom Microwell dishes for 3 days then fixed and immunostained with anti-Rab11. WT, wild-type; DFX, Δ187aa, 241, LL1031-1032AA; 242, LL1192-1193AA; 244, LL1031-1032AA + LL1192-1193AA. Bars = 10 μm. GFP, green fluorescent protein; MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1. Data are representative of 3 individual experiments where 5 images per experiment were taken (Total, n = 15).
Fig. 12.

NKCC1-DFX accumulates in Rab5-positive vesicles. MDCK stably transfected with EGFP-NKCC1-WT (A), EGFP-NKCC1-DFX (B), EGFP-NKCC1–241 (C), EGFP-NKCC1–242 (D), and EGFP-NKCC1–244 (E) were grown on MatTek Glass Bottom Microwell dishes for 3 days then fixed and immunostained with anti-Rab5. WT, wild-type; DFX, Δ187aa, 241, LL1031-1032AA; 242, LL1192-1193AA; 244, LL1031-1032AA + LL1192-1193AA. Bars = 10 μm. GFP, green fluorescent protein; MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1. Data are representative of 3 individual experiments where 5 images per experiment were taken (Total, n = 15).
DISCUSSION
The major goal of this study was to understand the fate of a truncated mutant of NKCC1, which we had previously shown to be functionally inactive (6) and mistargeted to the apical membrane and apical pole of epithelial cells (13). The mutation is carried by a 16-yr-old female patient that suffers from multi-organ disease (6). The truncation shortens the cytosolic 460 residues carboxyl-terminal tail of the cotransporter by 187 amino acids. The 11-bp deletion occurs in exon 22, directly located downstream of a cassette exon that is alternatively spliced in both mouse and human tissues (23, 32). In 2008, Carmosino and colleagues (4) suggested a role for exon 21 in basolateral targeting of NKCC1. To avoid any possibility of transporters not making it out of the ER or Golgi, they used a chimeric approach between NKCC1 (basolateral) and NKCC2 (apical) transporters. They swapped portions of the COOH-terminus and identified a region unique to NKCC1, encoded by exon 21, as containing a signal for basolateral sorting. The presence of two consecutive leucine residues within exon 21 prompted them into substituting them with alanine residues and the mutant transporter failed to traffic to the basolateral membrane. Based on this work, we wondered if the 11-bp deletion that occurs in exon 22 can affect the splicing of exon 21 and explain the mistargeting of the mutant transporter to the apical membrane. We demonstrated that it was not the case as most of the transcripts in the NKCC1-DFX homozygote mouse still contained exon 21. To our surprise, when we eliminated the sequence encoded by exon 21 in EGFP-NKCC1, the transporter was still targeted to the basolateral membrane. Whether the loss of exon 21 is less disruptive to the structure of NKCC1 than the substitution of the dileucine residues in the Carmosino study is unknown.
Since we could not account for the mistargeting of NKCC1-DFX based on exon 21, we performed serial deletions of the carboxyl-terminus. Elimination of 47 residues already resulted in a phenotype similar to NKCC1-DFX, i.e., loss of expression at the basolateral membrane. Examination of the sequence revealed the presence of a possible dileucine motif (DlppvLL), located 13 residues from the end of the protein. Mutation of the two leucine residues into alanine residues also resulted in loss of basolateral staining. This dileucine motif has previously been described as part of a tetrad that is required for normal processing of the cotransporter out of the ER and Golgi (18). We also identified another putative dileucine motif located 150 residues upstream (DDggltLL). Mutations of leucine residues into alanine in this case had no effect on NKCC1 localization.
An interesting observation made with one of our deletion mutants (NKCC1/239) is the clear and strong localization in the nucleoli. This observation is supported by the identification of a putative nucleolar localization signal upstream of the deleted portion of the COOH-terminus. The fact that neither wild-type nor the other truncated forms of the cotransporter were detected in nucleoli, indicates that this signal is usually hidden, and the deletion uncovered this motif. Whether this motif can be activated under some physiological or pathophysiological conditions remains unknown. Why would a membrane-embedded protein be found in the nucleolus? 3D3/lyric is a membrane protein found in the ER and inner nuclear membrane. Because EGFP-tagged 3D3/lyric was also observed in the nucleolus, the authors concluded that the protein was likely cleaved and lacking the transmembrane domain (30). Because the EGFP moiety on NKCC1 is located at the extreme amino terminus of the cotransporter, we do not believe that the NKCC1 protein observed in the nucleolus is missing the transmembrane domains. Other membrane proteins like TRPM7 (19) and PGRMC1 or progesterone receptor membrane component-1 (31) are also found in the nucleolus. PGRMC1 associates with nucleolin, the most abundant protein of the nucleolus, but a protein that is also found at the cell surface where it interacts with plasma membrane proteins (12).
During epithelial apical-to-basolateral polarization, Gp135/podocalyxin is typically translocated from the periphery to the pre-apical patch (PAP) and remained at the apical membrane when the lumen is formed (1). We found that NKCC1-DFX, delayed the early stage of cyst development with persistent Gp135/podocalyxin at the cell periphery and multiple apical membrane initiation sites. This early abnormality explains why we previously observed an increased number of cysts with multiple lumens with cells expressing the mutant transporter (13). Apical-to-basolateral polarization and single lumen formation in MDCK cysts is regulated by the RhoA-ROCK-myosin pathway (2). It is not clear whether NKCC1 plays a role in RhoA-ROCK-myosin pathway activation. However, ROCK kinase has been shown to phosphorylate LIM kinase, which in turn phosphorylates cofilin and inhibits actin depolymerization (15). A possible connection between NKCC1 and this pathway might involve cofilin as this protein was shown to interact with NKCC1 (26).
To determine the identity of the cellular compartment(s) that contain NKCC1-DFX, we costained MDCK cells transfected with wild-type and mutant cotransporters with several organelle markers. First, we were surprised to see absence of signal associated with ER and/or Golgi with all of our deletion or dileucine mutants, indicating that all transporters had been able to clear the first steps of the biosynthetic pathway. These data obtained using MDCK cells differed from those obtained in HEK293 cells (18). The difference, however, is unlikely due to a difference in cell types as we observed plasma membrane expression of the NKCC1-DFX mutant by immunofluorescence in HeLa cells and by cell surface biotinylation in HEK293 as well as MDCK cells (6, 13). The difference could be due to the load of proteins on the ER when cells are transiently transfected (18) versus stably transfected in this study.
We observed intense colocalization of NKCC1-DFX and other mutant NKCC1 transporters with the lyso-track marker, indicating that these cotransporters are targeted for degradation. This pathway is actively utilized to degrade the cotransporter under physiological conditions. In human colonic epithelium, cholinergic (Ca2+) stimulation of fluid secretion involves recruitment of NKCC1 to the basolateral membrane, activation, internalization, degradation, and reexpression (17, 24). Following stimulation, NKCC1 was found to be colocalized with the lysosomal marker LAMP-1. This represents the classical NKCC1 trafficking pathway as depicted in green in Fig. 13.
Fig. 13.
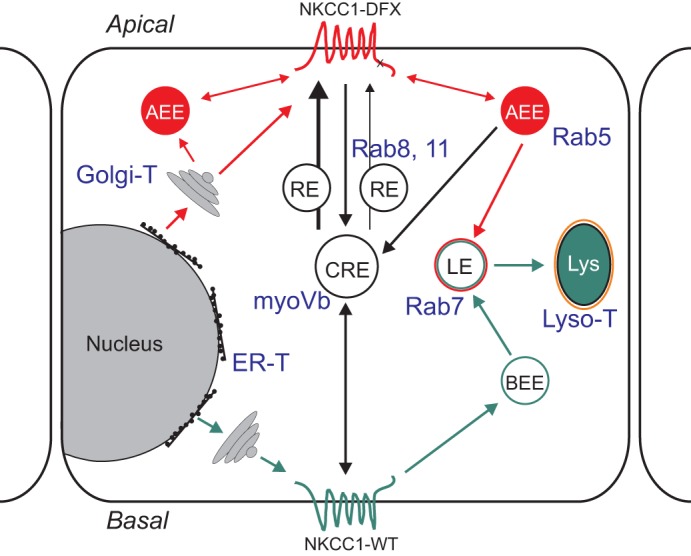
Model depicting the trafficking of NKCC1 wild-type and mutant in polarized epithelial cell. Wild-type NKCC1 (green pathway) is targeted to the basolateral membrane following its exit from the Golgi. The basolateral NKCC1-WT could be endocytosed and trafficked to the basolateral early endosome (BEE) where they can either be recycled to the membrane or targeted to late endosomes and lysosomes for degradation. NKCC1 COOH terminal mutants are trafficked to the apical membrane (red pathway). From the apical membrane these mutants could be endocytosed in apical early endosomes (AEE) and then trafficked to common recycling endosome (CRE), late endosomes and lysosomes. Some proteins could be recycled to the apical membrane from the CRE, or AEE. In blue font: compartment-specific antibodies and dyes used in this study.
Interestingly, we also observed colocalization of NKCC1 mutants with the early endosome marker Rab5, which is found mainly in the early endosomes underneath the apical membrane (27). It is worth noting that Rab5-containing endosomes also recycle caveolin-1 (20), a major component of lipid rafts. We and other have shown that NKCC1 is located in detergent insoluble lipid rafts (11, 13). Rab5 colocalization is consistent with expression of the mutant transporter at the apical membrane and pole observed in our previous study (13) and this study. Some apical membrane proteins in epithelia are first briefly released to the basolateral membrane then trafficked through transcytosis to the apical membrane. This is the case for example for the transferrin receptor (21). Transcytosis from the basolateral membrane to the apical membrane via the common endosome is highly dependent on myosin Vb motor proteins. Coexpression of the dominant-negative myosin Vb tail did not detain NKCC1 mutants in the common recycling endosomes, indicating that these mutants are unlikely targeted to the apical membrane via the common endosome pathway. No costaining was observed with Rab11 and Rab8, which are also known to mediate recycling of proteins from the common endosomes to the plasma membrane (25). Our data therefore indicate that the localization of mutants NKCC1 at the apical pole is most likely due to direct trafficking to the apical membrane and/or early apical endosomes. From there the mutant proteins could be removed from the apical membrane by endocytosis and targeted to apical early recycling endosomes or targeted for degradation in lysosomes (depicted in red in Fig. 13).
GRANTS
This work was supported by NIH Grants GM118944 and DK093501 to E. Delpire. Confocal imaging was performed in part through the use of the Vanderbilt University Medical Center Cell Imaging Shared Resource (supported by NIH Grants CA68485, DK20593, DK58404, DK59637, and EY08126).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.K. and E.D. conceived and designed research; R.K., S.O., and E.D. performed experiments; R.K. and E.D. analyzed data; R.K. and E.D. interpreted results of experiments; R.K. and E.D. prepared figures; R.K., S.O., and E.D. drafted manuscript; R.K., S.O., and E.D. edited and revised manuscript; R.K., S.O., and E.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. James Goldenring, Department of Surgery, Vanderbilt University Medical Center for the myoVb dominant-negative plasmid and for discussions on protein trafficking.
REFERENCES
- 1.Bryant DM, Datta A, Rodríguez-Fraticelli AE, Peränen J, Martín-Belmonte F, Mostov KE. A molecular network for de novo generation of the apical surface and lumen. Nat Cell Biol 12: 1035–1045, 2010. doi: 10.1038/ncb2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bryant DM, Roignot J, Datta A, Overeem AW, Kim M, Yu W, Peng X, Eastburn DJ, Ewald AJ, Werb Z, Mostov KE. A molecular switch for the orientation of epithelial cell polarization. Dev Cell 31: 171–187, 2014. doi: 10.1016/j.devcel.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bulley S, Jaggar JH. Cl− channels in smooth muscle cells. Pflugers Arch 466: 861–872, 2014. [Erratum in Pflugers Arch 466: 873, 2014.] doi: 10.1007/s00424-013-1357-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carmosino M, Giménez I, Caplan M, Forbush B. Exon loss accounts for differential sorting of Na-K-Cl cotransporters in polarized epithelial cells. Mol Biol Cell 19: 4341–4351, 2008. doi: 10.1091/mbc.e08-05-0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delpire E, Lu J, England R, Dull C, Thorne T. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat Genet 22: 192–195, 1999. doi: 10.1038/9713. [DOI] [PubMed] [Google Scholar]
- 6.Delpire E, Wolfe L, Flores B, Koumangoye R, Schornak CC, Omer S, Pusey B, Lau C, Markello T, Adams DR. A patient with multisystem dysfunction carries a truncation mutation in human SLC12A2, the gene encoding the Na-K-2Cl cotransporter, NKCC1. Cold Spring Harb Mol Case Stud 2: a001289, 2016. doi: 10.1101/mcs.a001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans RL, Park K, Turner RJ, Watson GE, Nguyen HV, Dennett MR, Hand AR, Flagella M, Shull GE, Melvin JE. Severe impairment of salivation in Na+/K+/2Cl− cotransporter (NKCC1)-deficient mice. J Biol Chem 275: 26720–26726, 2000. [DOI] [PubMed] [Google Scholar]
- 8.Flagella M, Clarke LL, Miller ML, Erway LC, Giannella RA, Andringa A, Gawenis LR, Kramer J, Duffy JJ, Doetschman T, Lorenz JN, Yamoah EN, Cardell EL, Shull GE. Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J Biol Chem 274: 26946–26955, 1999. doi: 10.1074/jbc.274.38.26946. [DOI] [PubMed] [Google Scholar]
- 9.Frizzell RA, Hanrahan JW. Physiology of epithelial chloride and fluid secretion. Cold Spring Harb Perspect Med 2: a009563, 2012. doi: 10.1101/cshperspect.a009563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gagnon KB, Delpire E. Physiology of SLC12 transporters: lessons from inherited human genetic mutations and genetically engineered mouse knockouts. Am J Physiol Cell Physiol 304: C693–C714, 2013. doi: 10.1152/ajpcell.00350.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartmann AM, Blaesse P, Kranz T, Wenz M, Schindler J, Kaila K, Friauf E, Nothwang HG. Opposite effect of membrane raft perturbation on transport activity of KCC2 and NKCC1. J Neurochem 111: 321–331, 2009. doi: 10.1111/j.1471-4159.2009.06343.x. [DOI] [PubMed] [Google Scholar]
- 12.Jia W, Yao Z, Zhao J, Guan Q, Gao L. New perspectives of physiological and pathological functions of nucleolin (NCL). Life Sci 186: 1–10, 2017. doi: 10.1016/j.lfs.2017.07.025. [DOI] [PubMed] [Google Scholar]
- 13.Koumangoye R, Omer S, Delpire E. Mistargeting of a truncated Na-K-2Cl cotransporter in epithelial cells. Am J Physiol Cell Physiol 315: C258–C276, 2018. doi: 10.1152/ajpcell.00130.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lapierre LA, Kumar R, Hales CM, Navarre J, Bhartur SG, Burnette JO, Provance DWJ Jr, Mercer JA, Bähler M, Goldenring JR. Myosin vb is associated with plasma membrane recycling systems. Mol Biol Cell 12: 1843–1857, 2001. doi: 10.1091/mbc.12.6.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 285: 895–898, 1999. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- 16.Markadieu N, Delpire E. Physiology and pathophysiology of SLC12A1/2 transporters. Pflugers Arch 466: 91–105, 2014. doi: 10.1007/s00424-013-1370-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mykoniatis A, Shen L, Fedor-Chaiken M, Tang J, Tang X, Worrell RT, Delpire E, Turner JR, Matlin KS, Bouyer P, Matthews JB. Phorbol 12-myristate 13-acetate-induced endocytosis of the Na-K-2Cl cotransporter in MDCK cells is associated with a clathrin-dependent pathway. Am J Physiol Cell Physiol 298: C85–C97, 2010. doi: 10.1152/ajpcell.00118.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nezu A, Parvin MN, Turner RJ. A conserved hydrophobic tetrad near the C terminus of the secretory Na+-K+-2Cl− cotransporter (NKCC1) is required for its correct intracellular processing. J Biol Chem 284: 6869–6876, 2009. doi: 10.1074/jbc.M804302200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ogunrinde A, Pereira RD, Beaton N, Lam DH, Whetstone C, Hill CE. Hepatocellular differentiation status is characterized by distinct subnuclear localization and form of the chanzyme TRPM7. Differentiation 96: 15–25, 2017. doi: 10.1016/j.diff.2017.06.001. [DOI] [PubMed] [Google Scholar]
- 20.Pelkmans L, Bürli T, Zerial M, Helenius A. Caveolin-stabilized membrane domains as multifunctional transport and sorting devices in endocytic membrane traffic. Cell 118: 767–780, 2004. doi: 10.1016/j.cell.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 21.Perez Bay AE, Schreiner R, Benedicto I, Rodriguez-Boulan EJ. Galectin-4-mediated transcytosis of transferrin receptor. J Cell Sci 127: 4457–4469, 2014. doi: 10.1242/jcs.153437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plotkin MD, Snyder EY, Hebert SC, Delpire E. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA’s excitatory role in immature brain. J Neurobiol 33: 781–795, 1997. doi:. [DOI] [PubMed] [Google Scholar]
- 23.Randall J, Thorne T, Delpire E. Partial cloning and characterization of Slc12a2: the gene encoding the secretory Na+-K+-2Cl− cotransporter. Am J Physiol Cell Physiol 273: C1267–C1277, 1997. doi: 10.1152/ajpcell.1997.273.4.C1267. [DOI] [PubMed] [Google Scholar]
- 24.Reynolds A, Parris A, Evans LA, Lindqvist S, Sharp P, Lewis M, Tighe R, Williams MR. Dynamic and differential regulation of NKCC1 by calcium and cAMP in the native human colonic epithelium. J Physiol 582: 507–524, 2007. doi: 10.1113/jphysiol.2007.129718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roland JT, Bryant DM, Datta A, Itzen A, Mostov KE, Goldenring JR. Rab GTPase-Myo5B complexes control membrane recycling and epithelial polarization. Proc Natl Acad Sci USA 108: 2789–2794, 2011. doi: 10.1073/pnas.1010754108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schiapparelli P, Guerrero-Cazares H, Magaña-Maldonado R, Hamilla SM, Ganaha S, Goulin Lippi Fernandes E, Huang CH, Aranda-Espinoza H, Devreotes P, Quinones-Hinojosa A. NKCC1 regulates migration ability of glioblastoma cells by modulation of actin dynamics and interacting with cofilin. EBioMedicine 21: 94–103, 2017. doi: 10.1016/j.ebiom.2017.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schnatwinkel C, Christoforidis S, Lindsay MR, Uttenweiler-Joseph S, Wilm M, Parton RG, Zerial M. The Rab5 effector Rabankyrin-5 regulates and coordinates different endocytic mechanisms. PLoS Biol 2: E261, 2004. doi: 10.1371/journal.pbio.0020261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stoops EH, Caplan MJ. Trafficking to the apical and basolateral membranes in polarized epithelial cells. J Am Soc Nephrol 25: 1375–1386, 2014. doi: 10.1681/ASN.2013080883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sung KW, Kirby M, McDonald MP, Lovinger DM, Delpire E. Abnormal GABAA receptor-mediated currents in dorsal root ganglion neurons isolated from Na-K-2Cl cotransporter null mice. J Neurosci 20: 7531–7538, 2000. doi: 10.1523/JNEUROSCI.20-20-07531.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sutherland HG, Lam YW, Briers S, Lamond AI, Bickmore WA. 3D3/lyric: a novel transmembrane protein of the endoplasmic reticulum and nuclear envelope, which is also present in the nucleolus. Exp Cell Res 294: 94–105, 2004. doi: 10.1016/j.yexcr.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 31.Terzaghi L, Luciano AM, Dall’Acqua PC, Modina SC, Peluso JJ, Lodde V. PGRMC1 localization and putative function in the nucleolus of bovine granulosa cells and oocytes. Reproduction 155: 273–282, 2018. doi: 10.1530/REP-17-0534. [DOI] [PubMed] [Google Scholar]
- 32.Vibat CR, Holland MJ, Kang JJ, Putney LK, O’Donnell ME. Quantitation of Na+-K+-2Cl− cotransport splice variants in human tissues using kinetic polymerase chain reaction. Anal Biochem 298: 218–230, 2001. doi: 10.1006/abio.2001.5398. [DOI] [PubMed] [Google Scholar]
- 33.Zdebik AA, Wangemann P, Jentsch TJ. Potassium ion movement in the inner ear: insights from genetic disease and mouse models. Physiology (Bethesda) 24: 307–316, 2009. doi: 10.1152/physiol.00018.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]