

Abstract
Liver injury due to acetaminophen (APAP) overdose is the major cause of acute liver failure in the US. While treatment with N-acetylcysteine (NAC) is the current standard of care for APAP overdose, anecdotal evidence suggests that administration of 4-methylpyrazole (4MP) may be beneficial in the clinic. The objective of the current study was to examine the protective effect of 4MP and its mechanism of action. Male C57BL/6J mice were co-treated with 300mg/kg APAP and 50mg/kg 4MP. The severe liver injury induced by APAP at 6h as indicated by elevated plasma ALT activities, centrilobular necrosis and nuclear DNA fragmentation, was almost completely eliminated by 4MP. In addition, 4MP largely prevented APAP-induced activation of c-jun N-terminal kinase (JNK), mitochondrial translocation of phospho-JNK and Bax, and the release of mitochondrial inter-membrane proteins. Importantly, 4MP inhibited the generation of APAP-protein adducts and formation of APAP-GSH conjugates and attenuated the depletion of the hepatic GSH content. These findings are relevant to humans because 4MP also prevented APAP-induced cell death in primary human hepatocytes. In conclusion, early treatment with 4MP can completely prevent liver injury after APAP overdose by inhibiting cytochrome P450 and preventing generation of the reactive metabolite.
Keywords: Acetaminophen, drug hepatotoxicity, reactive intermediates, mitochondria, N-acetylcysteine
INTRODUCTION
Acetaminophen (APAP) is a widely used analgesic and anti-pyretic drug, which is considered safe at therapeutic doses. However, intentional and unintentional overdoses of APAP cause tens of thousands of emergency department visits and hundreds of deaths due to acute liver failure in the US every year.1 At therapeutic doses, the majority of APAP is conjugated with glucuronic acid (45–55%) and sulfate (20–30%) and eliminated via bile and urine.2 However, a limited amount of the dose (<10%) is metabolized to a reactive metabolite, N-acetyl-p-benzoquinone imine (NAPQI), by cytochrome P450 enzymes, especially Cyp2E1, Cyp1A2 and Cyp3A4.2 Although NAPQI can be detoxified by conjugation with glutathione (GSH), a limited amount binds to cysteine groups of proteins forming protein adducts even at therapeutic doses.3,4 However, an overdose of APAP can lead to liver injury via excess formation of NAPQI, which depletes hepatic GSH levels and forms extensive protein adducts.5 The formation of mitochondrial protein adducts is most critical for initiation of cell death mechanisms.6,7 This leads to mitochondrial dysfunction with formation of reactive oxidant species (ROS) and peroxynitrite inside of mitochondria.8–10 This initial oxidant stress is amplified by a mitogen-activated protein kinase cascade resulting in the activation (phosphorylation) of c-jun N-terminal kinase (JNK) in the cytosol.11,12 Phospho-JNK translocates to the mitochondria and amplifies the mitochondrial oxidant stress and peroxynitrite formation,13,14 which ultimately triggers the opening of the mitochondrial membrane permeability transition pore (MPTP) resulting in the collapse of the membrane potential and cessation of ATP production.15 MPTP-induced matrix swelling leads to the rupture of the outer mitochondrial membrane and the release of intermembrane proteins including apoptosis-inducing factor (AIF) and endonuclease G, which translocate to the nucleus and cause DNA fragmentation.16,17 Extensive mitochondrial dysfunction, declining ATP levels and degradation of the nucleus eventually causes cell necrosis.18 Importantly, most of the mechanisms of APAP-induced cell death identified in mice are applicable to human hepatocytes and patients.19–21
Despite the substantial increase in knowledge of APAP hepatotoxicity, N-acetylcysteine (NAC) remains the only clinically approved antidote against APAP overdose.22 NAC is most effective during the first few hours after an overdose,23 mainly due to the accelerated synthesis of GSH and the effective scavenging of NAPQI leading to reduced protein adduct formation.24,25 However, many patients present to the emergency department late during the metabolism phase when the injury is in progress.26 At that time, the NAC-induced GSH synthesis promotes the recovery of mitochondrial GSH pools, which assist in scavenging ROS and peroxynitrite inside of mitochondria.27
A recent case report described a patient presenting with extremely high plasma APAP levels of 1,100 mg/L, who was initially treated with a single dose of Fomepizole (4-methylpyrazole, 4MP) followed by a standard NAC treatment regimen.28 This led others to suggest that the co-treatment with 4MP was a likely reason for the survival of the patient.29 Although 4MP is a competitive inhibitor of alcohol dehydrogenase,30–32 and as such is clinically used as an antidote to methanol and ethylene glycol poisoning,33 studies with human microsomes and HepG2 cells have demonstrated that 4MP can also inhibit CYP2E134,35 In addition, 4MP attenuated APAP-induced liver injury in rats36,37 but appeared to be not more effective than NAC.37 However, there are some concerns with these animal studies. First, use of a rat model is problematic as excessive doses of APAP are being used resulting in limited necrosis, which questions the relevance of this model for human patients.38 Second, these in vivo studies were purely descriptive with little mechanistic insight into the protective mechanisms of 4MP. However, in order to possibly consider 4MP as adjunct therapy to NAC in the clinic, it is important to understand the mechanisms of protection and the potential advantages and limitations of this treatment. Therefore, the objective of the current study was to assess the protective mechanisms of 4MP in a clinically relevant mouse model in vivo39 and also directly test the efficacy of 4MP in primary human hepatocytes, an in vitro model closest to the human pathophysiology.19 Our results clearly demonstrate that 4MP is a potent inhibitor of the P450 enzymes that metabolically activate APAP and thus is highly effective in protecting against APAP toxicity in mice in vivo and in human hepatocytes. Since the mechanism of action is different from NAC, 4MP could be used as adjunct therapy to the standard of care antidote NAC; however, at this point, the therapeutic window for 4MP is narrow and restricted to the metabolism phase.
MATERIALS AND METHODS
Animals and experimental design
In this study, 8 to 12 weeks old male C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME). Before the experiments, animals were accustomed to a temperature-controlled room with a 12 hours light/dark cycle, food and water ad libitum for 3–5 days. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Kansas Medical Center and performed following the National Research Council for the care and use of laboratory animals’ guidelines.
Mice were fasted for 15 h before being injected i.p. with the standard overdose of 300 mg/kg APAP (Sigma-Aldrich, St. Louis, MO) or saline vehicle.4,10 4MP (Sigma-Aldrich, St. Louis, MO) was dissolved in saline and was co-administered with APAP at a dose of 50 mg/kg as used in previous studies.36,37 Five mice were used per group for each intervention and time point. The animals were euthanized under isoflurane anesthesia 30 min or 6 h post-APAP. Blood and liver tissues were collected for ALT and metabolite measurements, respectively. In addition, liver tissue was used to isolate mitochondria as described.40
Primary human hepatocyte isolation and experiments
Primary human hepatocytes were isolated and plated on collagen-coated dishes by the departmental Cell Isolation Core as described in detail.19 Three hours later, primary hepatocytes were washed with sterile phosphate-buffered saline (PBS) and treated for 24 h with either 10 mM APAP, 2 mM 4MP or both dissolved in William’s medium E supplemented with dexamethasone, insulin and penicillin.19
Biochemical assays
The ALT reagent kit was used to measure alanine aminotransferase (ALT) activity levels in plasma or cells and cell culture supernatant (Pointe Scientific, MI).19 A modified Tietze assay was used to measure total liver glutathione (GSH+GSSG) as described.41 Cytochrome P450 enzyme activity was assessed in liver homogenate using a fluorogenic substrate: 7-ethoxy-4-trifluoromethyl coumarin (7-EFC) (InVitrogen, Carlsbad, CA) as described.42,43
Protein adducts measurements
Liver homogenates were filtered through Bio-Spin 6 columns in order to remove low molecular weight metabolites such as the APAP-GSH and APAP-Cys conjugates. The filtrates were digested overnight at 50°C with 8 U/ml Streptomyces griseus protease solution to free APAP-Cys from the cellular proteins. After digestion, cell lysates were precipitated with 40% trichloroacetic acid for 10 minutes on ice and the supernatant was filtered to collect the protein-derived APAP-Cys containing residues. APAP-Cys was measured with high pressure liquid chromatography and electrochemical detection.4,38
LC-MS/MS determination of APAP metabolites in mouse plasma
APAP was obtained from Sigma-Aldrich. (St. Louis, MO). The standards, 4-acetamidophenyl β-D-glucuronide (APAP-Gluc), 4-acetaminophen sulfate (APAP-Sulf), 3-(N-acetylcysteine) acetaminophen (APAP-NAC), 3-cysteinylacetaminophen trifluoroacetic acid (APAP-Cys), acetaminophen glutathione (APAP-GSH), and the internal standard, 4-acetaminophen sulfate-d3, were obtained from Toronto Research Chemicals (Toronto, Canada). Other chemicals and solvents used were obtained from standard suppliers and were of reagent or analytical grade. Stock solutions of APAP and its conjugated metabolites were prepared at 1 mM in 50:50 water:methanol and stored at -20°C until use.
All mouse plasma samples were thawed on ice. Several plasma samples from untreated mice were pooled and used as blank matrix for preparation of the calibration curve. A 20 μl aliquot of mouse plasma sample was added to 90 μl of methanol (containing 1000 ng/ml of acetaminophen sulfate-d3) to precipitate the proteins. Then, 70 μl of water and 20 μl of 50:50 water:methanol were added. Samples were vortexed for 10 sec and centrifuged for 10 min at 13,400 × g and 4°C. The standards were prepared following the same procedure but replacing the 20 μl of 50:50 water:methanol by 20 μl of working standards in 50:50 water:methanol. The concentration of calibration standards ranged from 0.5 μM to 75 μM for acetaminophen glutathione and 0.25 μM to 75 μM for all the other analytes.
All samples were analyzed by LC-MS/MS with a Waters Acquity Ultra-Performance Liquid Chromatography (UPLC®) system interfaced by electrospray ionization with a Waters Quattro Premier XE triple quadrupole mass spectrometer (Waters Corp., Milford, MA) operated in positive mode with multiple reaction monitoring (MRM) scan type. The following source conditions were applied: 3.0 kV for the capillary voltage, 120°C for the source temperature, 350°C for the desolvation temperature, 50 l/h for the source gas flow and 600 l/h for the desolvation gas flow. The following mass transitions and collision energies (CE) were used to detect the respective analytes: 152>110, CE = 15 V for APAP; 313>208, CE = 20 V for 3-(N-acetylcysteine) acetaminophen; 328>152, CE = 20 V for 4-acetaminophen glucuronide; 457>140, CE = 22 V for acetaminophen glutathione; 232>152, CE = 15 V for 4-acetaminophen sulfate; 271>140, CE = 24 V for 3-cysteinylacetaminophen and 235>155, CE = 15 V for acetaminophen sulfate-d3. The different compounds were separated on a Waters UPLC® HSS T3 column (1.8 μm, 2.1 × 150 mm) maintained at 50°C and using a gradient at 0.4 ml/min flow rate of mobile phase A (6 mM ammonium acetate in water with 0.01% formic acid) and mobile phase B (methanol) as follows: 2% B held for 0.5 min, increased to 75% over 3.5 min followed by an increase to 98% over 0.5 min and held at 98% for 2.0 min. Acetaminophen and its metabolites were quantified by back calculation of a weighted (1/x), linear least squares regression. The regression fit used the analyte/internal standard peak area ratios calculated from the calibration. Acetaminophen sulfate-d3 was used as the internal standard. The MS peaks were integrated using QuanLynx software (version 4.1, Waters Corp., Milford, MA). The limit of detection for the LC-MS/MS method was 0.250 μM for acetaminophen glutathione, 0.125 μM for 4-acetaminophen sulfate and 4-acetaminophen glucuronide, 0.063 μM for 3-cysteinylacetaminophen and 0.025 μM for 3-(N-acetylcysteine) acetaminophen. Samples above the calibration curve were further diluted 100-fold.
Histopathological Procedures
Liver tissue samples were fixed in 10% formalin in phosphate-buffered saline (pH 7.4), embedded in paraffin and cut in 5μm sections for hematoxylin and eosin (H&E) staining to assess hepatic necrosis18 and staining with the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay (In Situ Cell Death Detection assay; Roche, Indianapolis, NJ) to analyze nuclear DNA fragmentation.
Western blotting
Western blotting was performed as described44 with primary antibodies (1:1000 dilutions): rabbit anti-JNK antibody (Cat. # 9252), rabbit antiphospho-JNK antibody (Cat. # 4668), rabbit anti-Bax polyclonal antibody (Cat. # 2772), rabbit anti-AIF antibody (Cell Signaling Technology, Danvers, MA; Cat # 5318) and mouse anti-Smac/DIABLO (BD Biosciences, San Diego, CA; Cat # 612245). For secondary antibody (1:5000 dilutions), an anti-rabbit or anti-mouse horseradish peroxidase-coupled IgG (Santa Cruz) was used. Protein bands of interest were detected by chemiluminescence (GE Bioscience) on the Licor Odyssey imaging system.
Statistics
Results are expressed as mean ± SE. Student’s t-test was used to assess statistical significance between 2 groups. One-way ANOVA and Student-Newman-Keul’s test assisted in comparing variance between multiple groups. ANOVA was performed on rank followed by Dunn’s multiple comparison for non-normally distributed data using SPSS Statistics for Windows,V21.0, IBM Corp., Armonk, NY). P< 0.05 was considered significant for all tests.
RESULTS
4MP protects against APAP-induced hepatic injury
To evaluate the protective potential of 4MP, C57BL/6J mice were treated with 300 mg/kg APAP and co-treated with 50 mg/kg 4MP or the vehicle (saline). At 6 h after APAP administration, the animals developed severe liver injury as indicated by the high plasma ALT levels (Fig. 1A) and extensive necrotic cell death (N) in the areas around the central veins (CV) (Fig. 1B). In addition, extensive nuclear DNA fragmentation occurs in the necrotic areas as shown by the TUNEL assay_(Fig. 1C). Co-treatment with 4MP almost completely eliminated APAP toxicity at 6 h as indicated by the low plasma ALT levels and the absence of detectable necrosis or nuclear DNA damage (Fig. 1A-C).
Figure 1: 4-Methylpyrazole protects against APAP hepatotoxicity.
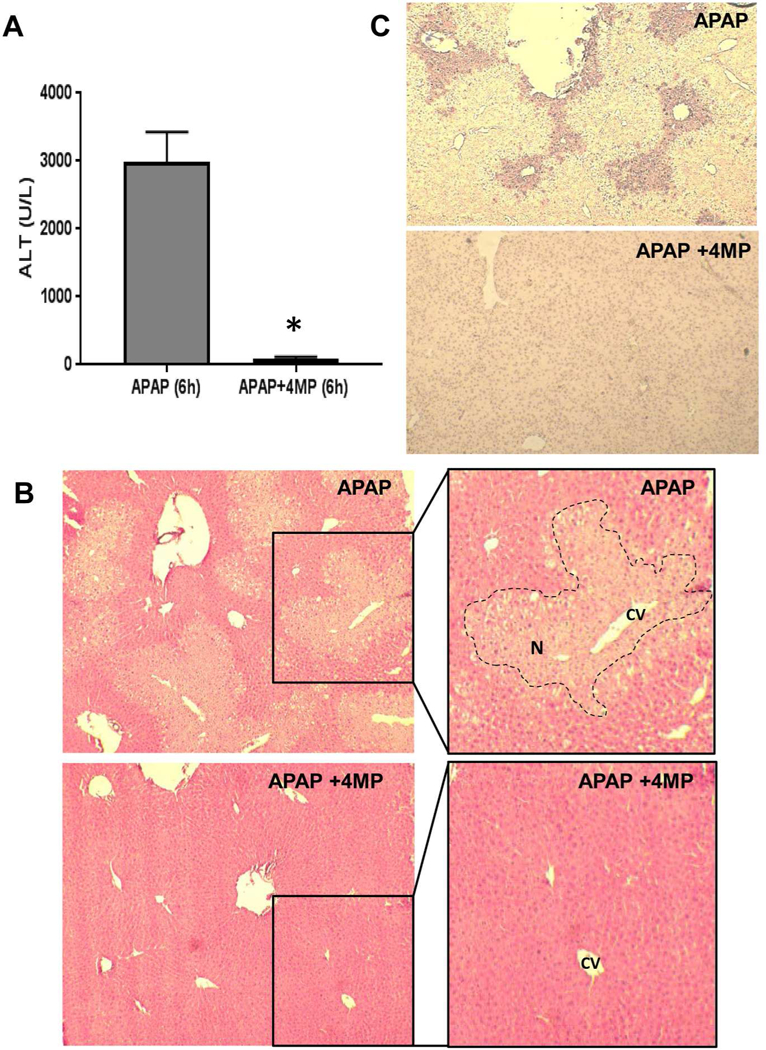
Mice were co-treated with 300 mg/kg APAP and 50 mg/kg 4MP or 10 ml/kg saline. Blood and liver tissues were collected 6 h post APAP. A) Plasma alanine aminotransferase activities. B) H&E staining; magnification ×100 and ×200. The area of necrosis (N) is outlined around the central vein (CV) in the higher magnification image. C) TUNEL staining; magnification ×100. Bars represent mean ± SE for 5 mice per group. *P< 0.05 versus APAP.
APAP hepatotoxicity is critically dependent on the activation of a mitogen-activated protein kinase cascade leading to the activation of JNK in the cytosol and the translocation of P-JNK to the mitochondria.11,12 In support of this hypothesis, there was no activated JNK in the cytosol or mitochondria in control livers; however, in APAP-treated animals, substantial JNK activation and mitochondrial P-JNK translocation was evident (Fig. 2A,B). Co-treatment with 4MP prevented JNK activation and the translocation of P-JNK to the mitochondria (Fig. 2A,B).
Figure 2: 4-Methylpyrazole inhibits JNK phosphorylation and mitochondrial translocation.
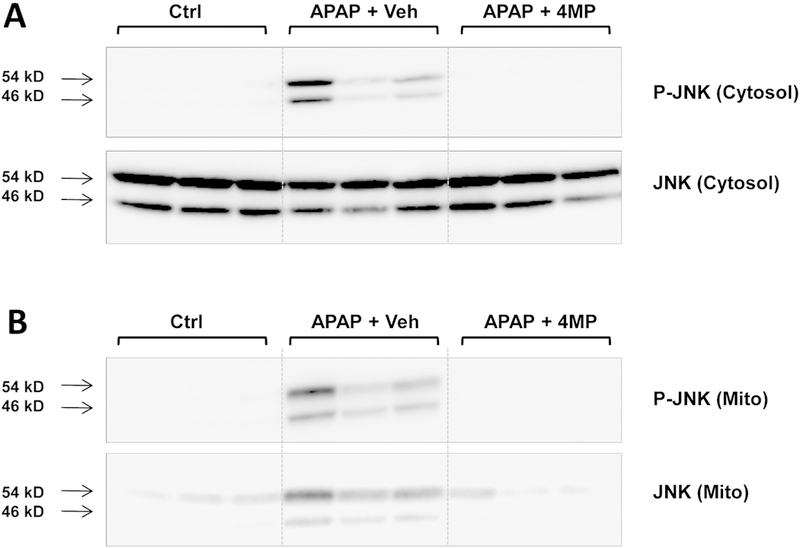
Mice were co-treated with 300 mg/kg APAP and 50 mg/kg 4MP or 10 ml/kg saline. Liver samples collected 6 h post APAP were analyzed for cytosolic (A) or mitochondrial (B) P-JNK and JNK.
Bax translocation to the mitochondria and release of mitochondrial intermembrane proteins such as AIF and Smac are events well described in APAP hepatotoxicity and the release of these mediators are an indicator of mitochondrial dysfunction.16,17,45 Consistent with previous reports, APAP overdose caused translocation of Bax to the mitochondria and induced the release of AIF and Smac into the cytosol (Fig. 3A,B). Co-treatment with 4MP attenuated AIF and Smac release by 80% and 65%, respectively. However, mitochondrial Bax translocation was only inhibited by 30% (Fig. 3A,B).
Figure 3: 4-Methylpyrazole inhibits APAP-induced mitochondrial dysfunction.
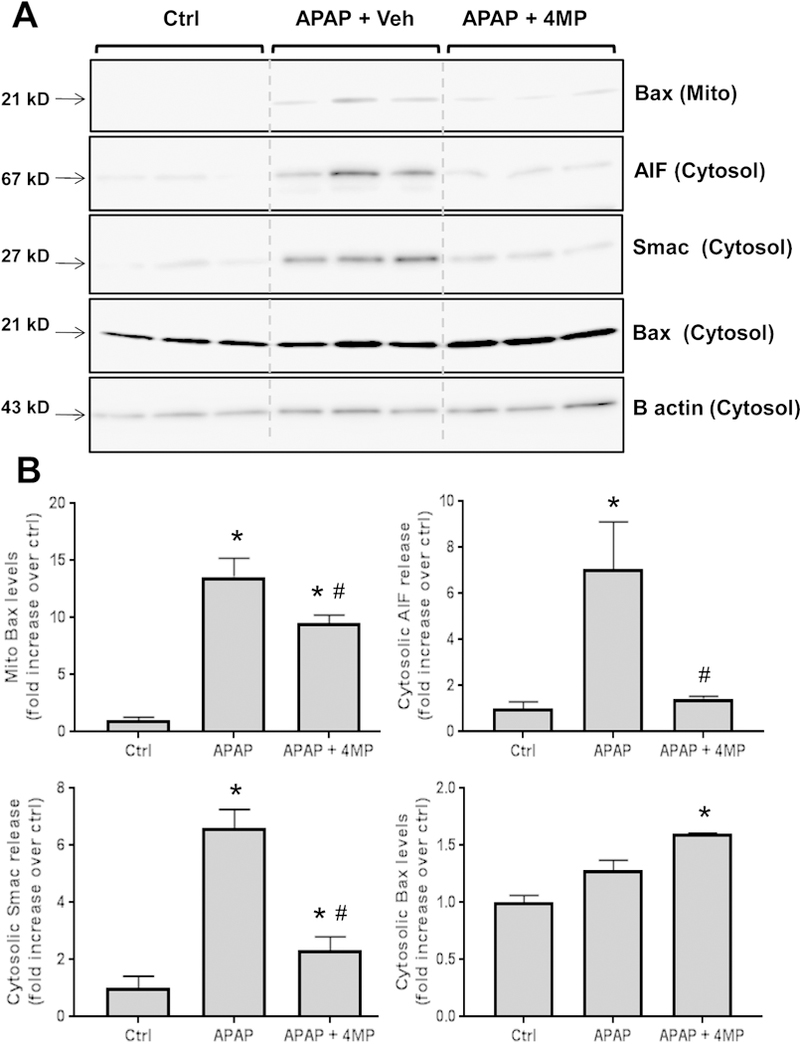
Mice were co-treated with 300 mg/kg APAP and 50 mg/kg 4MP or 10 ml/kg saline. A) Liver samples were collected 6 h post APAP and were analyzed for mitochondrial Bax and cytosolic apoptosis-inducing factor (AIF), second mitochondria-derived activator of caspases (Smac), Bax and beta-actin via western blotting. B) Densitometric analysis of the western blots. Bars represent mean ± SE for 3 mice per group. *P< 0.05 versus Ctrl. #P<0.05 versus APAP
Effect of 4MP on APAP metabolism
Our experiments clearly showed that 4MP treatment effectively protected against APAP-induced liver injury and mitochondrial dysfunction. In order to evaluate if 4MP affected the metabolic activation of APAP, APAP-protein adducts were measured in the 6 h liver samples (Fig. 4A) and the depletion of hepatic GSH levels was evaluated during the first 30 min after APAP injection (Fig. 4B). APAP caused extensive protein adducts formation and depleted hepatic GSH levels by >90% (Fig. 4). However, 4MP treatment almost completely prevented the formation of protein adducts and allowed only a 33% depletion of GSH (Fig. 4). These data suggest that 4MP prevented the metabolic activation of APAP.
Figure 4: 4-Methylpyrazole inhibits APAP protein adducts formation and hepatic GSH depletion.
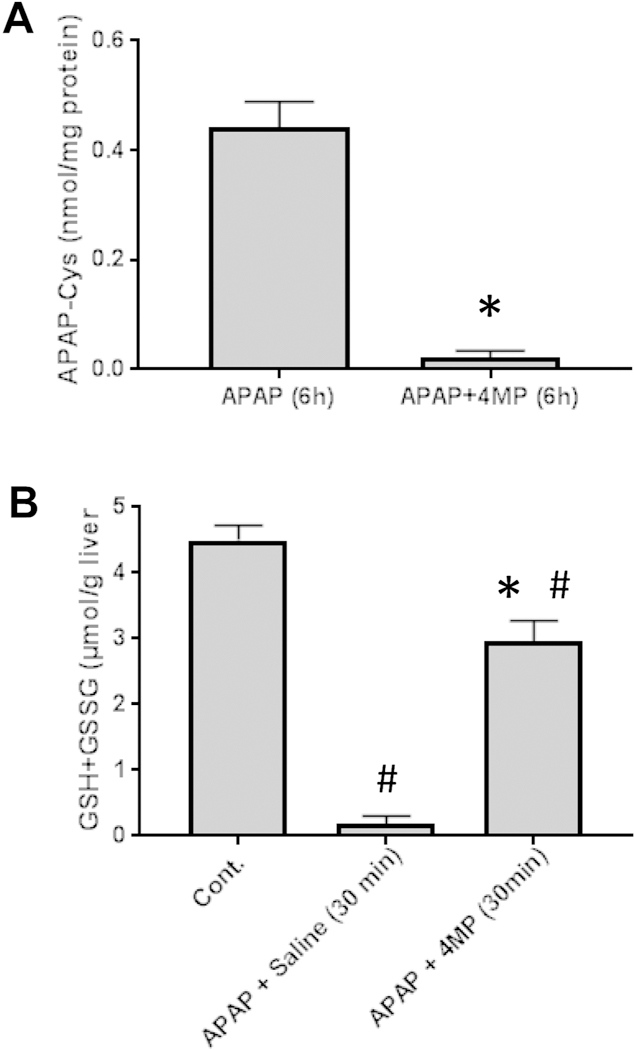
Mice were co-treated with 300 mg/kg APAP and 50 mg/kg 4MP or 10 ml/kg saline. A) Liver samples collected 6 h post APAP and were analyzed for APAP-protein adducts. B) In a separate experiment, liver samples were collected 30 min after APAP treatment and total GSH (GSH+GSSG) was measured. Data represent mean ± SE of n = 5 mice per group. *P< 0.05 versus APAP; #P<0.05 versus Control.
To further evaluate drug metabolism of APAP, the metabolites APAP-glucuronide, APAP-sulfate, APAP-GSH, APAP-Cys and APAP-NAC were measured at 30 min after APAP administration. High levels of un-metabolized APAP and of the glucuronide were detected; however, their levels were not affected by 4MP treatment (Fig. 5A). APAP-sulfate and the cysteine metabolites were substantially lower compared to the glucuronide and all were significantly reduced by 4MP (Fig. 5A,B). Although APAP-sulfate levels were 50% lower in 4MP-treated animals, all metabolites associated with the detoxification of NAPQI by GSH were reduced by 85–100% (Fig. 5B). These data further support the conclusions that 4MP effectively inhibited the formation of reactive metabolite of APAP by P450 enzymes.
Figure 5: Effect of 4-Methylpyrazole on APAP metabolism.
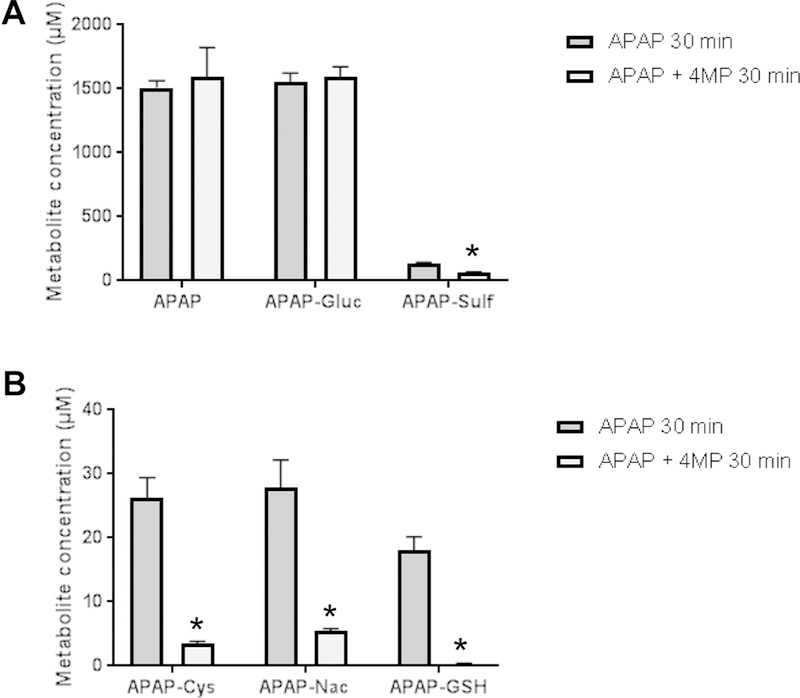
Mice were co-treated with 300 mg/kg APAP and 50 mg/kg 4MP or 10 ml/kg saline. Blood samples were collected at 30 min after APAP and the parent compound and its metabolites were quantified in plasma. A) APAP, APAP-glucuronide and APAP-sulfate. B) APAP-GSH, APAP-Cys and APAP-NAC. Data represent mean ± SE of n = 5 mice per group. *P< 0.05 versus APAP
In order to directly assess if 4MP can inhibit the most relevant P450 enzymes responsible for APAP metabolism, i.e. CYP2E1 and CYP1A2,46 cytochrome P450 enzyme activities were measured in liver homogenate using the fluorogenic substrate 7-ethoxy-4-trifluoromethyl coumarin (7-EFC).42,43 4MP dose-dependently inhibited the P450 enzyme activities measured with 7-EFC (Fig. 6A). In contrast, the clinical antidote NAC did not affect these enzyme activities under similar conditions (Fig.6B).
Figure 6: Effect of 4-Methylpyrazole and N-Acetylcysteine on cytochrome P450 enzyme activity.
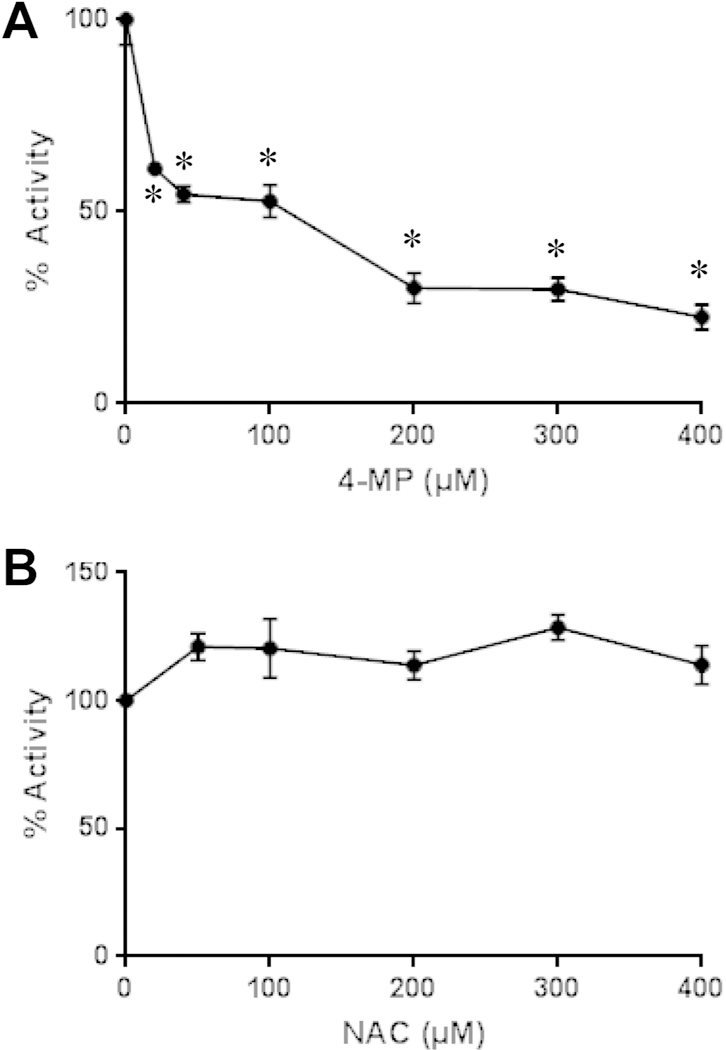
CYP enzyme activity was measured in liver homogenate using the substrate 7-ethoxy-4-trifluoromethyl coumarin (7-EFC). The dose-dependent inhibition of the enzyme activity was assessed with 4MP (A) and NAC (B). Data are expressed in percent of enzyme activity compared to homogenate in the absence of any inhibitor (100%). Data represent mean ± SE of n = 3 separate experiments. *P< 0.05 versus conc. = 0 μM.
4MP protects against APAP-induced hepatotoxicity in primary human hepatocytes
Although the mechanisms of APAP-induced cell death in mouse hepatocytes is relevant for the human pathophysiology,39 the effect of 4MP was evaluated in freshly isolated primary human hepatocytes.19 Exposure of primary human hepatocytes to 10 mM APAP for 24 h resulted in cell death and release of 40% of the cellular ALT into the medium compared to <10% of ALT in controls (Fig. 7A,B). However, in the presence of 2 mM 4MP, APAP-induced cell death was completely prevented as indicated by similar ALT release as untreated cells or cells exposed to 4MP alone (Fig. 7A,B).
Figure 7: 4-Methylpyrazole protects against APAP toxicity in primary human hepatocytes.
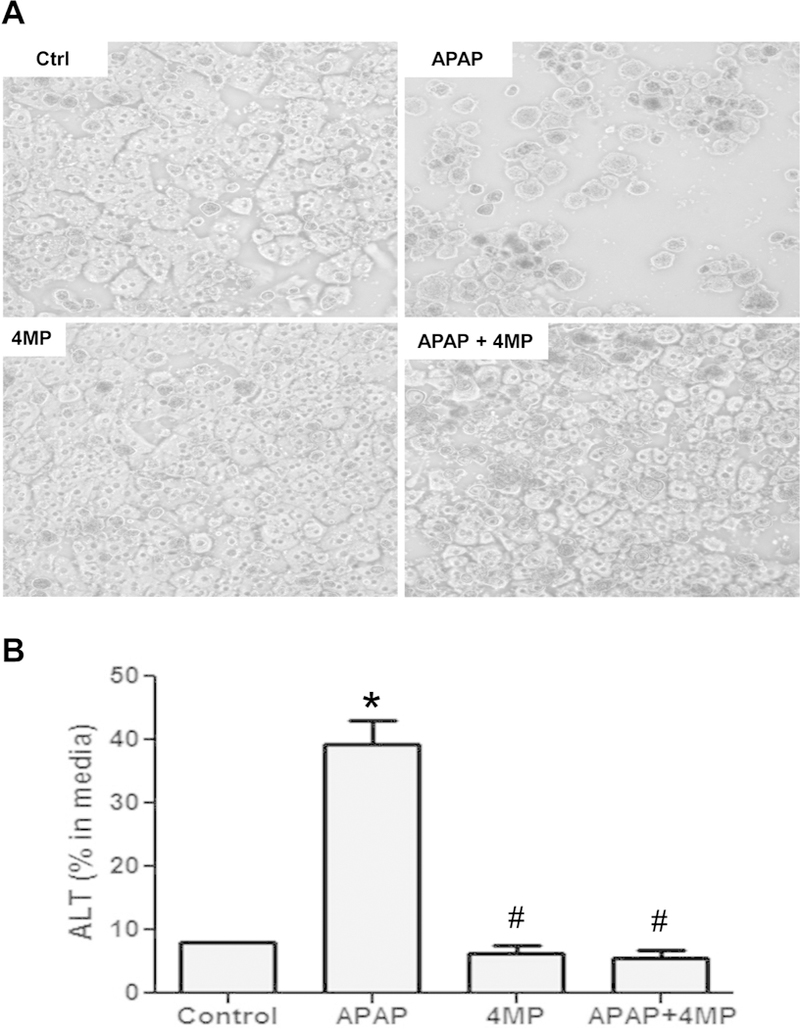
Cells were treated with 10 mM APAP and 2 mM 4MP alone or in combination for 24 h. A) Phase contrast images of control and treated cells. B) Release of alanine aminotransferase (ALT) activity into the culture medium. Bars represent mean ± SE for 3 separate cell isolations. *P<0.05 versus Control #P< 0.05 versus APAP.
DISCUSSION
The purpose of this study was to investigate the efficacy of the alcohol dehydrogenase inhibitor 4MP against APAP hepatotoxicity in a murine model in vivo and in primary human hepatocytes. 4MP is a FDA approved drug against methanol and ethylene glycol poisoning.33 Recent circumstantial evidence advocated for the clinical use of 4MP with APAP poisoning.28,29 However, the benefit of such treatment is not well established.
Our data showed that co-treatment of 4MP and APAP almost completely eliminated the extensive liver injury caused by APAP. This was demonstrated by the dramatically reduced increase in plasma ALT activities and the lack of characteristic centrilobular necrosis. In addition, DNA fragmentation, as detected by the TUNEL assay, was prevented. Because nuclear DNA fragmentation is caused by endonuclease G and AIF released from the mitochondrial intermembrane space,16,17 these findings indicate that 4MP reduced mitochondrial dysfunction. Intermembrane protein release occurs through 2 mechanisms, a Bax pore formation and rupture of the outer mitochondrial membrane during matrix swelling due to the MPTP.45 In fact, 4MP treatment significantly attenuated mitochondrial Bax translocation and substantially reduced both AIF and Smac release from the mitochondria. These results further support the protection of 4MP against APAP-induced mitochondrial dysfunction. Upstream of these events is JNK activation, which is triggered by an initial mitochondrial oxidant stress.13 The phosphorylated form (P-JNK) then translocates to the mitochondria and amplifies the oxidant stress thereby causing the MPTP opening.13–15 The fact that 4MP effectively prevented JNK activation and the mitochondrial translocation of P-JNK suggests that 4MP had to act upstream of these events. Thus, early treatment of 4MP effectively prevented APAP-induced cell necrosis and events that are critically involved in the pathophysiology including nuclear DNA fragmentation, mitochondrial dysfunction and JNK activation in the mouse model. This protection could also be reproduced in freshly isolated, primary human hepatocytes, which show a very similar mechanism of APAP-induced cell death as observed in mouse hepatocytes.19
It is well established that APAP toxicity starts with the formation of the reactive metabolite NAPQI by cytochrome P450 enzymes.47 NAPQI can be scavenged by hepatocellular GSH leading to the formation of APAP-GSH adducts and depletion of hepatic GSH levels.5 At the same time, NAPQI binds to cysteine residues of proteins, especially mitochondrial proteins, which triggers the initial oxidant stress in mitochondria that leads to JNK activation.2,48 Consistent with these previously described events, we observed extensive (>90%) hepatic GSH depletion within 30 min after APAP overdose and the appearance of the APAP-GSH conjugate together with its metabolites of APAP-Cys and APAP-NAC in plasma. In addition, APAP protein adducts were detected. However, 4MP treatment largely prevented GSH depletion, the formation of APAP-GSH conjugates and protein adduct formation. These findings strongly support the conclusion that 4MP inhibits the cytochrome P450-dependent formation of the reactive metabolite NAPQI and thus, prevents the initiation of the toxicity. This conclusion was further supported by an in vitro assay in liver homogenate using the substrate 7-EFC. This substrate mainly tests for CYP2E1 and CYP1A2 activities,42,43 which are the key enzymes for the metabolic activation of APAP in mice.46 4MP dose-dependently inhibited this assay suggesting that 4MP can directly inhibit P450 enzyme activities. Thus, both the results of the in vitro P450 enzyme activity assay and the in vivo results suggest that 4MP protects against APAP hepatotoxicity by preventing the metabolic activation of APAP. At the same time, the formation of the major phase II metabolite APAP-glucuronide was not affected by 4MP. However, there was a clear reduction of the APAP-sulfate concentration. Further studies are needed to assess if 4MP can also directly inhibit sulfotransferase activities. However, under these overdose conditions the APAP-sulfate metabolite represents only about 6% of all metabolites, which is consistent with recent studies in humans.25 Therefore, a 50% reduction of the sulfate conjugation reaction is of limited pathophysiological consequence, in particular, as it is known that the sulfation reaction is saturated even after a therapeutic dose of APAP.49
The recommended loading dose for 4MP to treat patients with methanol or ethylene glycol poisoning is 15 mg/kg.50 The currently used dose of 4MP, which is highly effective against APAP toxicity in mice, is 50 mg/kg, which translates into a human equivalent dose of approximately 4 mg/kg.51 This suggests that the approved clinical dose is almost four times higher than the effective dose in mice. Although a therapeutic dose of 4MP for human APAP overdose patients would need to be established, the fact that the effective dose in mice is well below the safe levels that have been established for other indications, it appears feasible that 4MP can be successfully used in the clinic.
The current standard of care treatment for APAP overdose is NAC,22 which is most useful when given early (<8 h) after the overdose but is still partially effective when given at later time points.23 NAC promotes hepatic GSH synthesis, which enhances the scavenging capacity for NAPQI and attenuates protein adducts formation.24 However, after the metabolism phase, the enhanced GSH synthesis improves the capacity to scavenge peroxynitrite and reactive oxygen species52,53 and an excess of NAC can be converted to Krebs cycle intermediates that support mitochondrial bioenergetics.27 The capacity to affect the toxicity at multiple levels is the reason why NAC is effective over a relatively large time span. However, the down-side of NAC treatment is that it requires the synthesis of GSH, which then can act as a scavenger for NAPQI and for reactive oxygen. Nevertheless, early NAC treatment is equally as effective as shown for 4MP in the current study.24,27,52,53 On the other hand, 4MP acts as direct inhibitor of P450 enzymes, which generate the reactive metabolite. Thus, 4MP affects a different therapeutic target than NAC and therefore, can potentially complement the protective effect of NAC. However, this potential additive effect of 4MP and NAC based on their mechanism of action needs to be investigated in patients under clinical conditions. The only caveat is that the efficacy of 4MP is mostly restricted to the metabolism phase, which means that it is only effective when treating the patients early after an overdose. Many patients who present at later time points with significant liver injury are less likely to benefit from 4MP but NAC may still have some positive impact.23
In summary, our data demonstrated that the early treatment with 4MP provided almost complete protection against APAP-induced liver injury in the mouse model in vivo and in primary human hepatocytes. The therapeutic target of 4MP was the inhibition of cytochrome P450 enzymes with the prevention of reactive metabolite formation and protein binding, which are the most upstream events in the pathophysiology. Although 4MP proved to be highly effective, due to its mechanism of action, the therapeutic window is very narrow and restricted to the very early metabolism phase. However, because it affects a different therapeutic target than the standard of care antidote NAC, it could provide additional benefits for early presenting patients.
ACKNOWLEDGEMENTS
This work was supported in part by a grant from McNeil Consumer Health Care, Inc., the National Institutes of Health grant R01 DK102142, and by grants from the National Institute of General Medical Sciences (P20 GM103549 and P30 GM118247) of the National Institutes of Health.
H. Jaeschke was supported in part by a grant from McNeil Consumer Health, Inc.
Footnotes
CONFLICT OF INTEREST DISCLOSURE
The remaining authors declare that there is no conflict of interest.
REFERENCES
- 1.Yoon E, Babar A, Choudhary M, et al. Acetaminophen-induced hepatotoxicity: a comprehensive update. J Clin Transl Hepatol. 2016; 4: 131–142.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGill MR and Jaeschke H. Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharm Res 2013; 30: 2174–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heard KJ, Green JL, James LP, et al. Acetaminophen-cysteine adducts during therapeutic dosing and following overdose. BMC Gastroenterol 2011; 11: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGill MR, Lebofsky M, Norris HR, et al. Plasma and liver acetaminophen-protein adduct levels in mice after acetaminophen treatment: dose-response, mechanisms, and clinical implications. Toxicol Appl Pharmacol 2013; 269: 240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitchell JR, Jollow DJ, Potter WZ, et al. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187: 211–217. [PubMed] [Google Scholar]
- 6.Tirmenstein MA and Nelson SD. Subcellular binding and effects on calcium homeostasis produced by acetaminophen and a nonhepatotoxic regioisomer, 3’-hydroxyacetanilide, in mouse liver. J Biol Chem 1989; 264: 9814–9819. [PubMed] [Google Scholar]
- 7.Xie Y, McGill MR, Du K, et al. Mitochondrial protein adducts formation and mitochondrial dysfunction during N-acetyl-m-aminophenol (AMAP)-induced hepatotoxicity in primary human hepatocytes. Toxicol Appl Pharmacol 2015; 289: 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyers LL, Beierschmitt WP, Khairallah EA, et al. Acetaminophen-induced inhibition of hepatic mitochondrial respiration in mice. Toxicol Appl Pharmacol 1988; 93: 378–387. [DOI] [PubMed] [Google Scholar]
- 9.Jaeschke H Glutathione disulfide formation and oxidant stress during acetaminophen-induced hepatotoxicity in mice in vivo: the protective effect of allopurinol. J Pharmacol Exp Ther 1990; 255: 935–941. [PubMed] [Google Scholar]
- 10.Cover C, Mansouri A, Knight TR, et al. Peroxynitrite-induced mitochondrial and endonuclease-mediated nuclear DNA damage in acetaminophen hepatotoxicity. J Pharmacol Exp Ther 2005; 315: 879–887. [DOI] [PubMed] [Google Scholar]
- 11.Han D, Shinohara M, Ybanez MD, et al. Signal transduction pathways involved in drug-induced liver injury. Handb Exp Pharmacol 2010; (196): 267–310. [DOI] [PubMed] [Google Scholar]
- 12.Du K, Xie Y, McGill MR, et al. Pathophysiological significance of c-jun N-terminal kinase in acetaminophen hepatotoxicity. Expert Opin Drug Metab Toxicol 2015; 11 : 1769–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanawa N, Shinohara M, Saberi B, et al. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem 2008; 283: 13565–13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saito C, Lemasters JJ and Jaeschke H. c-Jun N-terminal kinase modulates oxidant stress and peroxynitrite formation independent of inducible nitric oxide synthase in acetaminophen hepatotoxicity. Toxicol Appl Pharmacol 2010; 246: 8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kon K, Kim JS, Jaeschke H, et al. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 2004; 40: 1170–1179. [DOI] [PubMed] [Google Scholar]
- 16.Bajt ML, Cover C, Lemasters JJ, et al. Nuclear translocation of endonuclease G and apoptosis-inducing factor during acetaminophen-induced liver cell injury. Toxicol Sci 2006; 94: 217–225. [DOI] [PubMed] [Google Scholar]
- 17.Bajt ML, Ramachandran A, Yan HM, et al. Apoptosis-inducing factor modulates mitochondrial oxidant stress in acetaminophen hepatotoxicity. Toxicol Sci 2011; 122: 598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gujral JS, Knight TR, Farhood A, et al. Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol Sci 2002; 67: 322–328. [DOI] [PubMed] [Google Scholar]
- 19.Xie Y, McGill MR, Dorko K, et al. Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol Appl Pharmacol 2014; 279: 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McGill MR, Sharpe MR, Williams CD, et al. The mechanism underlying acetaminophen- induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest 2012; 122: 1574–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGill MR, Staggs VS, Sharpe MR, et al. Acute Liver Failure Study Group. Serum mitochondrial biomarkers and damage-associated molecular patterns are higher in acetaminophen overdose patients with poor outcome. Hepatology 2014; 60: 1336–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Green JL, Heard KJ, Reynolds KM, et al. Oral and Intravenous acetylcysteine for treatment of acetaminophen toxicity: A systematic review and meta-analysis. West J Emerg Med 2013; 14: 218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smilkstein MJ, Knapp GL, Kulig KW, et al. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319: 1557–1562. [DOI] [PubMed] [Google Scholar]
- 24.Corcoran GB, Racz WJ, Smith CV, et al. Effects of N-acetylcysteine on acetaminophen covalent binding and hepatic necrosis in mice. J Pharmacol Exp Ther 1985; 232: 864–872. [PubMed] [Google Scholar]
- 25.Xie Y, McGill MR, Cook SF, et al. Time course of acetaminophen-protein adducts and acetaminophen metabolites in circulation of overdose patients and in HepaRG cells. Xenobiotica 2015; 45: 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rumack BH, Bateman DN. Acetaminophen and acetylcysteine dose and duration: past, present and future. Clin Toxicol (Phila) 2012; 50: 91–98. [DOI] [PubMed] [Google Scholar]
- 27.Saito C, Zwingmann C and Jaeschke H. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology 2010; 51: 246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zell-Kanter M, Coleman P, Whiteley PM, et al. A gargantuan acetaminophen level in an acidemic patient treated solely with intravenous N-acetylcysteine. Am J Ther 2013; 20: 104–106. [DOI] [PubMed] [Google Scholar]
- 29.Yip L and Heard K. Potential adjunct treatment for high-risk acetaminophen overdose. Clin Toxicol (Phila) 2016; 54: 459. [DOI] [PubMed] [Google Scholar]
- 30.Bekka R, Borron SW, Astier A, et al. Treatment of methanol and isopropanol poisoning with intravenous fomepizole. J Toxicol Clin Toxicol 2001; 39: 59–67. [DOI] [PubMed] [Google Scholar]
- 31.Brent J, McMartin K, Phillips S, et al. Methylpyrazole for Toxic Alcohols Study G. Fomepizole for the treatment of methanol poisoning. N Engl J Med 2001; 344: 424–429. [DOI] [PubMed] [Google Scholar]
- 32.Bestic M, Blackford M and Reed M. Fomepizole: a critical assessment of current dosing recommendations. J Clin Pharmacol 2009; 49: 130–137. [DOI] [PubMed] [Google Scholar]
- 33.Barceloux DG, Krenzelok EP, Olson K, et al. American Academy of Clinical Toxicology Practice Guidelines on the Treatment of Ethylene Glycol Poisoning. Ad Hoc Committee. J Toxicol Clin Toxicol 1999; 37: 537–560. [DOI] [PubMed] [Google Scholar]
- 34.Dai Y and Cederbaum AI. Cytotoxicity of acetaminophen in human cytochrome P4502E1-transfected HepG2 cells. J Pharmacol Exp Ther 1995; 273: 1497–1505. [PubMed] [Google Scholar]
- 35.Hazai E, Vereczkey L and Monostory K. Reduction of toxic metabolite formation of acetaminophen. Biochem Biophys Res Commun 2002; 291: 1089–1094. [DOI] [PubMed] [Google Scholar]
- 36.Brennan RJ, Mankes RF, Lefevre R, et al. 4-Methylpyrazole blocks acetaminophen hepatotoxicity in the rat. Ann Emerg Med 1994; 23: 487–494. [DOI] [PubMed] [Google Scholar]
- 37.Kucukardali Y, Cinan U, Acar HV, et al. Comparison of the therapeutic efficacy of 4-methylpyrazole and N-acetylcysteine on acetaminophen (paracetamol) hepatotoxicity in rats. Curr Med Res Opin 2002; 18: 78–81. [DOI] [PubMed] [Google Scholar]
- 38.McGill MR, Williams CD, Xie Y, et al. Acetaminophen-induced liver injury in rats and mice: comparison of protein adducts, mitochondrial dysfunction, and oxidative stress in the mechanism of toxicity. Toxicol Appl Pharmacol 2012; 264: 387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaeschke H, Xie Y and McGill MR. Acetaminophen-induced liver injury: from animal models to humans. J Clin Transl Hepatol 2014; 2: 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Du K, McGill MR, Xie Y, et al. Resveratrol prevents protein nitration and release of endonucleases from mitochondria during acetaminophen hepatotoxicity. Food Chem Toxicol 2015; 81: 62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jaeschke H and Mitchell JR. Use of isolated perfused organs in hypoxia and ischemia/reperfusion oxidant stress. Methods Enzymol 1990; 186: 752–759. [DOI] [PubMed] [Google Scholar]
- 42.Buters JT, Schiller CD, and Chou RC. A highly sensitive tool for the assay of cytochrome P450 enzyme activity in rat, dog and man. Direct fluorescence monitoring of the deethylation of 7-ethoxy-4-trifluoromethylcoumarin. Biochem Pharmacol 1993; 46:1577–1584. [DOI] [PubMed] [Google Scholar]
- 43.Chang TK, Crespi CL, and Waxman DJ. Determination of CYP2B6 component of 7-ethoxy-4-trifluoromethylcoumarin O-deethylation activity in human liver microsomes. Methods Mol Biol 2006; 320: 97–102. [DOI] [PubMed] [Google Scholar]
- 44.Bajt ML, Lawson JA, Vonderfecht SL, et al. Protection against Fas receptor-mediated apoptosis in hepatocytes and nonparenchymal cells by a caspase-8 inhibitor in vivo: evidence for a postmitochondrial processing of caspase-8. Toxicol Sci 2000; 58: 109–117. [DOI] [PubMed] [Google Scholar]
- 45.Bajt ML, Farhood A, Lemasters JJ, et al. Mitochondrial bax translocation accelerates DNA fragmentation and cell necrosis in a murine model of acetaminophen hepatotoxicity. J Pharmacol Exp Ther 2008; 324: 8–14. [DOI] [PubMed] [Google Scholar]
- 46.Zaher H, Buters JT, Ward JM, et al. Protection against acetaminophen toxicity in CYP1A2 and CYP2E1 double-null mice. Toxicol Appl Pharmacol 1998; 152: 193–199. [DOI] [PubMed] [Google Scholar]
- 47.Nelson SD. Molecular mechanisms of the hepatotoxicity caused by acetaminophen. Semin Liver Dis 1990; 10: 267–278. [DOI] [PubMed] [Google Scholar]
- 48.Du K, Ramachandran A, Jaeschke H. Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol 2016; 10: 148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clements JA, Critchley JA, Prescott LF. The role of sulphate conjugation in the metabolism and disposition of oral and intravenous paracetamol in man. Br J Clin Pharmacol 1984; 18: 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zakharov S, Pelclova D, Navratil T, et al. Fomepizole versus ethanol in the treatment of acute methanol poisoning: Comparison of clinical effectiveness in a mass poisoning outbreak. Clin Toxicol (Phila) 2015; 53: 797–806. [DOI] [PubMed] [Google Scholar]
- 51.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J 2008; 22: 659–661. [DOI] [PubMed] [Google Scholar]
- 52.Knight TR, Ho YS, Farhood A, et al. Peroxynitrite is a critical mediator of acetaminophen hepatotoxicity in murine livers: protection by glutathione. J Pharmacol Exp Ther 2002; 303: 468–475. [DOI] [PubMed] [Google Scholar]
- 53.James LP, McCullough SS, Lamps LW, et al. Effect of N-acetylcysteine on acetaminophen toxicity in mice: relationship to reactive nitrogen and cytokine formation. Toxicol Sci 2003; 75: 458–467. [DOI] [PubMed] [Google Scholar]