

Abstract
Proteins containing intrinsic disorder often form secondary structure upon interaction with a binding partner. Modulating such structures presents an approach for manipulating the resultant functional outcomes. Translational repressor protein 4E-BP1 is an example of an intrinsically disordered protein that forms an a-helix upon binding to its protein ligand, eIF4E. Current biophysical methods for analyzing binding-induced structural changes are low-throughput, require large amounts of sample, or are extremely sensitive to signal interference by the ligand itself. Herein, we describe the discovery and development of a conditionally fluorescent 4E-BP1 peptide that reports structural changes of its helix in high-throughput format. This reporter peptide is based on conditional quenching of fluorescein by thioamides. In this case, fluorescence signal increases as the peptide becomes more ordered. Conversely, destabilization of the α-helix results in decreased fluorescence signal. The low concentration and low volume of peptide required make this approach amenable for high-throughput screening to discover ligands that alter peptide secondary structure.
Keywords: bioorganic chemistry, fluorescent probes, helical structures, high-throughput screening, protein folding
Dynamic conformational change is a critically important occurrence for mediating protein function.[1] From exposing enzyme active sites and initiating catalytic turnover, to driving selectivity in interactions between protein binding partners, modulation of protein structure can yield information about functional fate(s).[1,2] Intrinsically disordered proteins (IDPs) and proteins containing intrinsically disordered regions (IDRs) are exceptionally dynamic, existing as conformational ensembles as opposed to more rigid secondary and tertiary structures.[3] Their structural fluidity contributes to their function as signaling hubs in protein interaction networks that regulate cellular growth and development.[1,2] Within these signaling cascades, binding-induced structuring has been found to be an important driver of protein–protein interaction (PPI) affinity and specificity.[1]
For example, a family of three intrinsically disordered translational repressor proteins known as the eIF4E-binding proteins (4E-BPs) regulate a subset of mRNA transcripts, including Mcl-1, cyclin D1, and c-Myc, that are integral for maintenance of cellular homeostasis.[4] These proteins function by sequestering eIF4E from eIF4G, which shares a similar eIF4E-binding motif, to inhibit the initiation of cap-dependent translation (Figure 1).[5] The 4E-BPs each form a short a-helix upon binding to eIF4E, with helix formation driving binding.[1,2,5] Mechanistic target of rapamycin complex 1 (mTORC1)-dependent phosphorylation of the 4E-BPs regulates their activity,[6] in part by destabilizing the helix,[7] thus indicating that chemical modulation of the helix-forming peptide region could be a promising strategy through which to impart functional control over the protein. This rationale has been used in the design of stapled peptides based on the eIF4E-binding sequences of 4E-BPs,[8] as well as their biological competitor, eIF4G.[9] Stabilization of 4E-BP binding to eIF4E also contributes to the function of the small-molecule modulator of cap-dependent translation, 4EGI-1;[10] however, identifying selective 4E-BP ligands that specifically modulate helicity presents an additional challenge.
Figure 1.
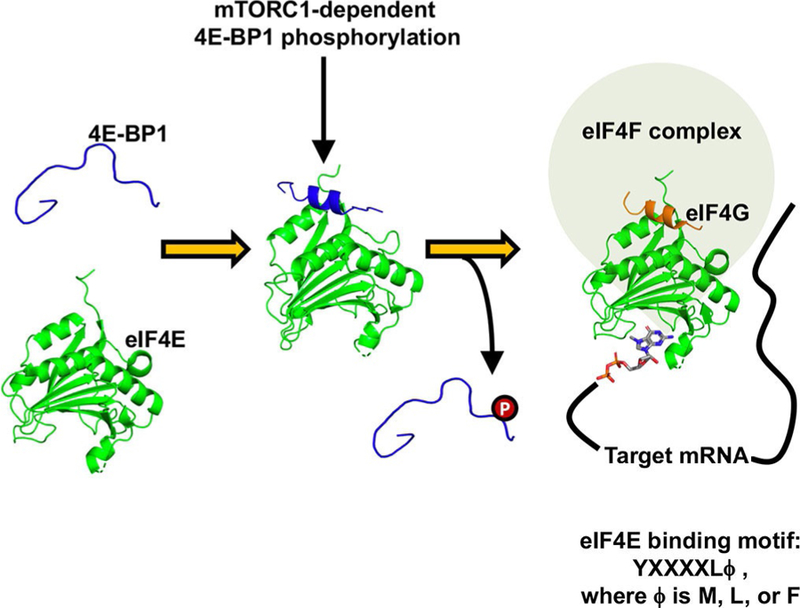
The interaction between 4E-BP1 and eIF4E is driven by the formation of an α-helix upon binding. When this helix is destabilized by mTORC1-dependent phosphorylation, it is outcompeted by the scaffolding protein eIF4G. eIF4E·4E-BP1 peptide complex PDB: 4UED;[11] eIF4E·eIF4G peptide complex PDB: 1EJH.[5]
Commonly used methods for analyzing protein and peptide structure include circular dichroism (CD) and NMR spectroscopies, as well as X-ray crystallography. Although these techniques are powerful and yield detailed information about protein structure, they are also low-throughput, they require large amounts of sample, and data analysis is often challenging, particularly when studying IDPs.[1,2, 12] Thus, there is a need for new approaches to identify modulators of conformational change in a broadly applicable format.
Previous studies have demonstrated the utility of thioamides and their derivatives as minimally perturbing fluorescence quenchers in proteins through a photoinduced electron transfer (PET) mechanism.[13] In contrast to other processes that result in quenched fluorescence through energy transfer, such as FRET, PET quenching does not require spectral overlap of the electron donor and acceptor and occurs over very short distances (≈ 3–5 Å).[14] Inspired by the use of conditional thioamide quenching for sensing protease activity[14a] and conformational dynamics of macromolecules,[14a,15] we have adapted this approach to observe the disorder-to-order transition of the 4E-BP1 “hot-spot” peptide. Herein, we describe the serendipitous discovery and development of a fluorescent peptide reporter that can detect modulation of the 4E-BP1 helix at nanomolar peptide concentrations.
We initially synthesized a 15-amino acid peptide containing residues Thr50–Asn64 of the 4E-BP1 isoform for development of a fluorescence polarization assay. This “hot-spot” peptide contained the eIF4E-binding motif conjugated to fluorescein isothiocyanate (FITC) at the N terminus through a two-β-alanine linker. Interestingly, rather than reporting polarization upon binding to eIF4E, the peptide demonstrated increased fluorescence (data not shown). Because the thiourea motif formed upon FITC coupling (Scheme 1 A) has been shown to quench in a similar way to thioamides,[13a] our hypothesis was that, in a dynamic solution state, this moiety might partially quench the disordered 4E-BP1 peptide’s fluorescence through PET. Any increase in peptide helicity (i.e., upon eIF4E binding) would subsequently reorient the thiourea out of the PET distance, thereby decreasing the effective concentration of quenched peptide and resulting in the observed increase in fluorescence signal (Scheme 1 B). Conversely, a decrease in fluorescence signal would be expected when the helix is destabilized, due to an increase in the effective concentration of the quenched peptide. Such a peptide reporter could greatly impact the way changes in IDP conformation and dynamics are observed, and could be utilized for high-throughput screening of chemical modulators of IDP structure. Thus, we set out to test the hypothesis that we had inadvertently discovered such a reporter.
Scheme 1.

Rationale behind the fluorescent peptide reporter. A) FITC coupling to the N terminus of β-alanine yields a thiourea linkage. B) Working hypothesis for the peptide reporter.
To explore this hypothesis, fluorescence measurements were recorded in the presence and in the absence of 50 % 2,2,2-trifluoroethanol (TFE), a solvent known to induce helical ordering in 4E-BP1 and other peptides.[16] Additionally, we directly compared the effect of TFE on the FITC peptide with its effect on a peptide N-terminally labeled with 5(6)-carboxyfluorescein (FAM), which lacks the thiourea moiety at the peptide–fluorophore linkage. As shown in Figure 2 A, a 2.5-fold increase in fluorescence was observed upon the addition of TFE to a 10 nm solution of the FITC-4E-BP1 peptide. Importantly, on using CD spectroscopy, we confirmed that TFE enhanced the peptide’s helical propensity from 7 % to 33 % (Figure 2 B). Although the FAM-4E-BP1 peptide showed a similar response to TFE by CD, the fluorescence response did not increase to the same extent as in the case of the FITC peptide. A C-terminal FITC-4E-BP1 peptide was also prepared; however, it exhibited little helical propensity even in 50 % TFE (Figure S2 in the Supporting Information). This finding was not surprising because, on the basis of the eIF4E-4E-BP1 peptide structures,[5, 11, 12, 17] C-terminal modification of the 4E-BP1 helix likely disrupts the formation or stability of secondary structure by destabilization of the helical dipole. Together, these results offered preliminary substantiation for our hypothesis that the observed increase in fluorescence signal was linked to helix induction.
Figure 2.
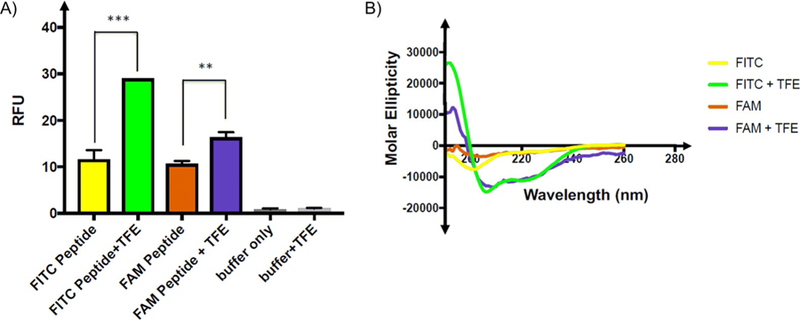
Evaluation of peptide fluorescence quenching by thiourea. A) Relative fluorescence units (RFUs, λex =494, λem =520 nm) of 10 μL of 10 nM FITC- or FAM-conjugated 4E-BP1 peptides in assay buffer (50 mM sodium phosphate pH 7.4, 200 mM NaCl, 1 mM DTT, and 1 mM EDTA) in the absence or in the presence of 50 % TFE. FAM-conjugated peptide fluorescence does not increase in TFE to the same degree as that of the FITC-conjugated peptide. Fluorescence measurements were made in triplicate; p values were generated in GraphPad prism by using two-way ANOVA. *** p =0.001; ** p =0.004. B) FITC-and FAM-conjugated 4E-BP1 peptides exhibit similar increases in helicity in the presence of TFE, as determined by CD spectroscopy (100 μM peptide in 50 mM sodium phosphate buffer, pH 7.4) with or without 50 % TFE at 25 °C.
To provide additional evidence of structure-mediated fluorescence enhancement, we probed a cysteine–aromatic interaction in the 4E-BP1 peptide (Cys62–Phe58, Figure 3 A), which we hypothesized to stabilize helical conformation.[18] Consistent with this model, alkylation of the cysteine residue with iodoacetamide caused a 4.8-fold decrease in peptide fluorescence, which was not altered with 50 % TFE (Figure 3 B). Importantly, these results were corroborated by CD spectroscopy (Figure 3 C). These data not only show the ability of our peptide reporter to detect helix disruption, but also indicate that the TFE-induced increase in fluorescence of the unmodified peptide is not primarily a consequence of nonspecific solvent effects on fluorescein itself, thus confirming that we had, in fact, created a reporter of 4E-BP structure modulation. Armed with such a reporter, we wished to optimize its utility for high-throughput screening (HTS) experiments to identify peptide ligands that specifically induce 4E-BP1 helicity as a mode of action for modulating eIF4E activity and cap-dependent translation.
Figure 3.
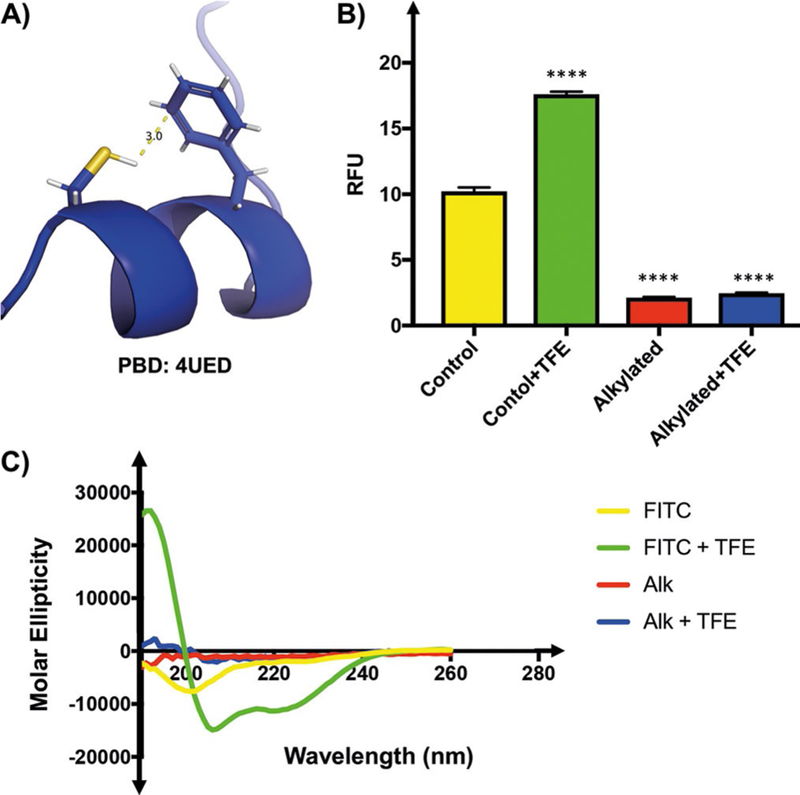
A thiol–aromatic interaction is required for 4E-BP1 helix formation. A) Phe58 and Cys62 enter into a H-bond-like interaction in the 4E-BP1 helix. B) RFUs (λex =494, λem =520 nm) of 10 μL of 10 nm FITC-4E-BP1 peptides in assay buffer in the absence or in the presence of 50 % TFE. Alkylation of Cys62 by iodoacetamide decreased peptide fluorescence relative to the unmodified FITC-4E-BP1 peptide. This decrease in RFUs is unaffected by the presence of 50 % TFE. C) Alkylation of Cys62 by iodoacetamide decreased peptide helicity relative to the unmodified FITC-4E-BP1 peptide. This decrease in helicity was unaffected by the presence of TFE. Percentage helicities of FITC-4E-BP1 peptides were determined by CD spectroscopy (100 μm peptide in 50 mM sodium phosphate buffer pH 7.4) with or without 50 % TFE at 25 °C.
To enable these efforts, we sought to improve the signal-to-background ratio (S/B) of our assay; thus, we explored the effect of incorporating additional thioamide moieties within the peptide sequence.[14a,15a,19] We chose to incorporate thio-leucine in place of Leu59, which is contained within the helix, and thioisoleucine in place of Ile53, which is N-terminal to the helix (Figure 4 A). Fmoc-protected thioamide amino acid precursors were synthesized as described[14a,20] and used in solidphase peptide synthesis. Whereas additional thioamide moieties did decrease the overall fluorescence of the peptides, the S/B was not significantly improved (Figure 4 B). Thus, the native FITC-4E-BP1 peptide reporter was determined to be optimal because it does not require the synthesis of thioamide-containing amino acid precursors.
Figure 4.
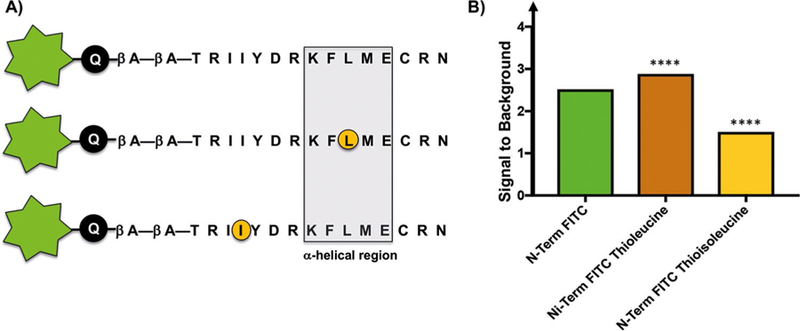
Incorporation of additional thioamide residues into the 4E-BP1 sequence does not significantly improve the signal-to-background ratio. A) Peptide sequences of thioamide-containing peptides (circled in orange). B) Fluorescence signals (λex =494, λem =520 nm) of 10 μL of 10 nM FITC-4E-BP1 peptides in assay buffer in the absence or in the presence of 50 % TFE. Signal-to-background ratios were determined by dividing the fluorescence signal of each peptide in 50 % TFE by its fluorescence signal in assay buffer alone.
To recapitulate our original observation, the FITC-labeled 4E-BP1 peptide was titrated with varying concentrations of MBP-eIF4E. As shown in Figure 5A, a dose-dependent increase in peptide fluorescence was observed. The calculated EC50 from this titration experiment was (93.5 ± 3.0) nM; however, the S/B from this experiment was low (Figure 5 B). We hypothesized that because fluorescein fluorescence is pH-dependent between pH 6.8 and 9.0,[21] raising the pH to 8.5 should improve S/B by increasing the maximum observable signal. Indeed, when the titration experiment was repeated at pH 8.5, a significant increase in S/B was observed (Figure 5 C). Similar results were also found with TFE (Figure S3). Moreover, there was little change in helicity trends observed at the elevated pH as assessed by CD spectroscopy (Figure S4 and Table S1). Important-ly, the measured EC50 value was (65.5 ± 1.4) nM, which is comparable with the reported Kd value of 4E-BP1 peptide binding to eIF4E (50 nM by isothermal calorimetry).[5] We also demonstrated specificity of the signal generation, and no fluorescence enhancement was observed when the peptide was titrated with α-synuclein in assay buffer at pH 8.5. This protein, a known aggregator, is not known to bind to 4E-BP1 (Figure S5). Intriguingly, the maximum fluorescence response for eIF4E titration was found to be much larger than the response with 50 % TFE. We believe this is due to additional hydrophobic contacts that have been experimentally shown to occur between the amino acid side chains of eIF4E and the 4E-BP1 peptide N terminus.[5, 10a,11, 17] These interactions provide additional stabilization of the N terminus, resulting in lower probability of fluorescence quenching, but are not directly involved in the 4E-BP1 helix; however we have not ourselves experimentally precluded the possibility of direct interaction between the fluorophore and MBP-eIF4E.
Figure 5.
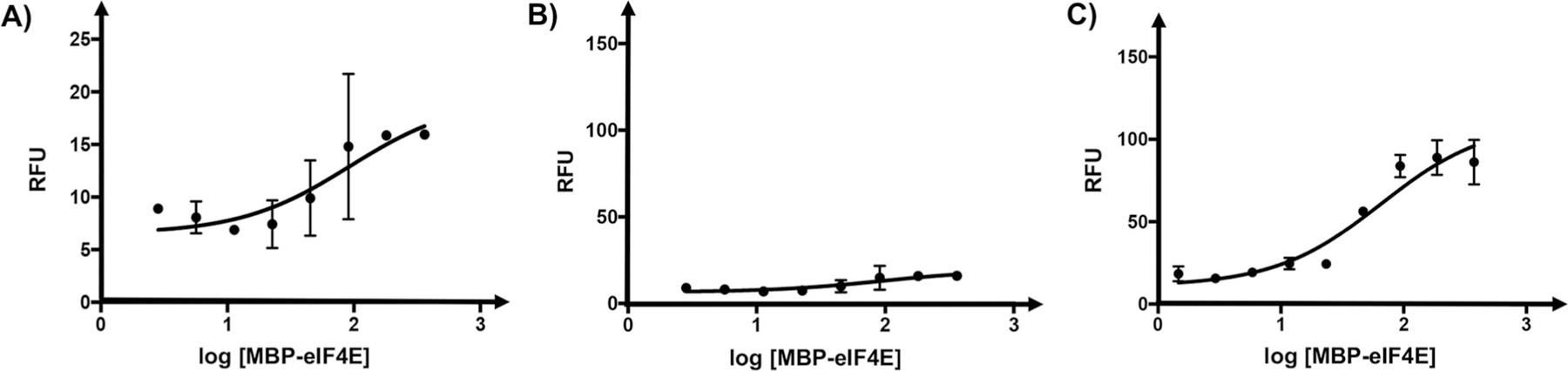
Fluorescent peptide responses to titration with MBP-eIF4E. RFUs (λex =494, λem =520 nm) of FITC-4E-BP1 peptide in assay buffer titrated with varying concentrations of MBP-eIF4E (0–500 nM). Final well volumes for fluorescence readings were 10 mL with a final peptide concentration of 10 nM. A) MBP-eIF4E titration at pH 7.4. B) Data from (A) graphed to scale with (C) show low relative fluorescence of peptide in buffer at pH 7.4. C) MBP-eIF4E titration at pH 8.5 shows an overall improvement both in fluorescence signal and in signal-to-background ratio. Titration experiments were performed in triplicate.
We were then prompted to confirm that a PET mechanism was indeed responsible for the changes in fluorescence observed in our experiments under the optimized buffer conditions. As expected, there was an increase in the fluorescence lifetime of the peptide (Figure S6 A) in 50 % TFE (4.976 ns) relative to that in buffer alone (4.029 ns). In addition, the absorbance spectrum of the peptide was relatively unchanged by the presence of TFE (Figure S6 B). Together, these results are consistent with observations in other PET quenched systems.[15b] Although there was a minute blue shift in peptide absorbance in 50 % TFE (Figure S6 B), this is characteristic of conformational change in fluorophore conjugates.[15b]
Lastly, we tested the robustness of the FITC-4E-BP1 reporter peptide to determine its amenability to HTS for discovery of new inducers of the 4E-BP1 a-helix. To do so, we calculated the Z’ factor, which statistically assesses the dynamic range and standard deviation of the assay.[22] This analysis yielded a Z’ value of 0.7 in a 384-well plate experiment with use of automated liquid handling. Importantly, assays with Z’ values of >0.5 have been described as excellent for HTS efforts. This shows the utility of such a probe for the discovery of secondary structure modulators (Figure 6).
Figure 6.
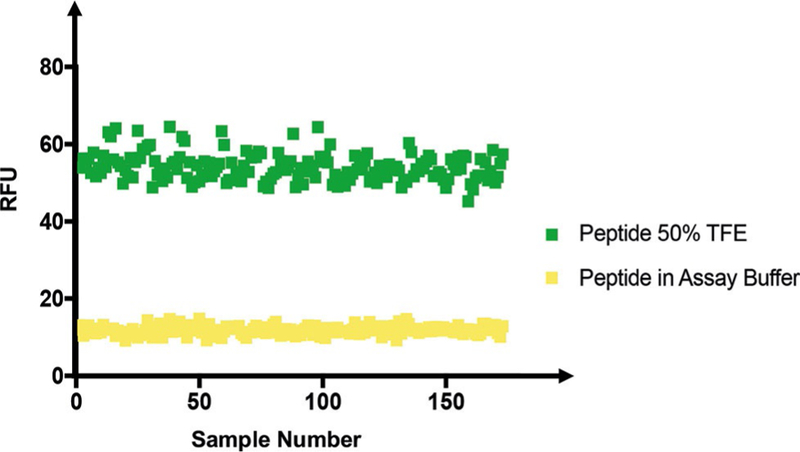
Z’ factor in a 384-well plate. RFUs (λex =494, λem =520 nm) of 10 μL of 10 nM FITC–E-BP1 peptide in assay buffer at pH 8.5 in the absence (nega-tive controls) or in the presence (positive controls) of 50 % TFE.
In conclusion, we have developed a fluorescent 4E-BP1 peptide that can report both induction and stabilization of its a-helix. Additionally, we have used our 4E-BP1 peptide to probe the thiol–aromatic interaction between Phe58 and Cys62 and its impact on 4E-BP1 helix stability. The prevalence of similar binding-induced folding transitions in biological signaling pathways indicates potential utility of a conditionally fluorescent peptide in systems including p27 linker ordering upon binding to CDK4, p53 folding upon MDM2 binding, and structuring of pKID coupled to binding of KIX.[1] Such peptides could be used for the discovery of new modulators of structure and function.
Notably, not all IDPs form static secondary structures. Such “fuzzy complexes” retain varying levels of conformational restriction at the bound IDP’s interaction site.[1, 23] It is likely that a reporter, such as the one we have described, would only be useful in a subset of these systems, dependent upon the relative conformational restriction occurring in the bound state of a given IDP, as well as optimization of thioamide placement. We look forward to application of our rationale in such systems to determine its applicability for studying a more diverse range of IDP PPIs. There is also the possibility to utilize such an approach for more complex systems. For example, native chemical ligation can be used for the incorporation of a fluorophore and quenching thioamide through protein semisynthesis.[24]
Finally, what we have described is an equilibrium assay for a PPI in the nanomolar range; however, many IDPs interact with their ligands through multiple low-affinity sites.[1, 2] In application of our approach to such interactions—those in the millimolar range, for example— there will be two time-dependent processes affecting the equilibrium: the binding kinetics of the PPI and the timescale of PET quenching. PET quenching occurs on a nanosecond timescale, which presumably favors PPIs with slower dissociation rates. Further exploration of PPI kinetics and the correlation to PET quenching studies are needed to determine the range of affinity interactions that can be reliably observed with our platform. With these considerations in mind, our described approach demonstrates great promise as a chemical biology tool for observation of protein structure perturbation in a high-throughput format.
Experimental Section
Assay fluorescence measurements:
Peptides, which were dissolved as stocks (1 mM) in buffer A [sodium phosphate (pH 7.4 or 8.5, 50 mm), NaCl (200 mM), dithiothreitol (DTT; 1 mM), EDTA (1 mM), DMF (30 %, v/v)], were diluted to appropriate working concentrations for subsequent experiments. For fluorescence measurements, peptides were diluted to working solutions (20 nm in assay buffer). For TFE experiments, the working solutions were further di-luted to 10 nm with assay buffer or TFE. Peptide working solution (10 nM, 10 μL) was added to each well, and the fluorescence was read immediately with use of excitation/emission wavelengths of 490/520 nm with a 515 nm cutoff and three scans/well. For MBP-eIF4E and α-synuclein titrations, peptide working solutions (10 nM, 10 μL) were titrated serially with protein solutions (10 μL) for a final peptide concentration of 10 nm and final protein concentrations of 0–500 nM and 0–900 nM for MBP-eIF4E and α-synuclein, respectively. All measurements were performed in triplicate.
Supplementary Material
Acknowledgements
We would like to thank Emily J. Sherman for preliminary synthet-ic efforts relating to the thioamide-containing amino acids. We also gratefully thank Wenhao (Steven) Shao and Prof. Jinsang Kim at the University of Michigan for assistance with fluorescence lifetime measurements. This work was supported through a generous start-up package from the University of Michigan College of Pharmacy, the NIH (R01 CA202018 to A.L.G. and T32 GM008597 to O.T.J.), and the NSF (O.T.J.).
Biography

Amanda Garner received her Ph.D. in chemistry from the University of Pittsburgh, working under the supervision of Prof. Kazunori Koide, and completed NIH-funded postdoctoral studies in the laboratory of Prof. Kim Janda at The Scripps Research Institute. She began her independent career in 2013 in the Department of Medicinal Chemistry at the University of Michigan. Her laboratory uses chemical biology, medicinal chemistry, and molecular and cellular biology approaches to investigate the high-risk/high-reward areas of targeting microRNAs and RNA-protein and protein–protein interactions for probe and drug discovery.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Wright PE, Dyson HJ, Nat. Rev. Mol. Cell Biol 2015, 16, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Dyson HJ, Wright PE, Nat. Rev. Mol. Cell Biol 2005, 6, 197–208. [DOI] [PubMed] [Google Scholar]
- [3].He B, Wang K, Liu Y, Xue B, Uversky VN, Dunker AK, Cell Res 2009, 19, 929–949. [DOI] [PubMed] [Google Scholar]
- [4].a) Martineau Y, Azar R, Bousquet C, Pyronnet S, Oncogene 2013, 32, 671–677; [DOI] [PubMed] [Google Scholar]; b) Averous J, Proud CG, Oncogene 2006, 25, 6423–6435; [DOI] [PubMed] [Google Scholar]; c) Qin X, Jiang B, Zhang Y, Cell Cycle 2016, 15, 781–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Marcotrigiano J, Gingras AC, Sonenberg N, Burley SK, Mol. Cell 1999, 3, 707–716. [DOI] [PubMed] [Google Scholar]
- [6].Gingras AC, Raught B, Gygi SP, Niedzwiecka A, Miron M, Burley SK, Polakiewicz RD, Wyslouch-Cieszynska A, Aebersold R, Sonenberg N, Genes Dev 2001, 15, 2852–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Tait S, Dutta K, Cowburn D, Warwicker J, Doig AJ, McCarthy JE, Proc. Natl. Acad. Sci. USA 2010, 107, 17627–17632; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tomoo K, Abiko F, Miyagawa H, Kitamura K, Ishida T, J. Biochem 2006, 140, 237–246. [DOI] [PubMed] [Google Scholar]
- [8].Gallagher EE, Song JM, Menon A, Chmiel AF, Mishra LD, Mitchell DC, Garner AL, unpublished results
- [9].Lama D, Quah ST, Verma CS, Lakshminarayanan R, Beuerman RW, Lane DP, Brown CJ, Sci. Rep 2013, 3, 3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Sekiyama N, Arthanari H, Papadopoulos E, Rodriguez-Mias RA, Wagner G, Léger-Abraham M, Proc. Natl. Acad. Sci. USA 2015, 112, E4036–E4045; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, Gross JD, Degterev A, Yuan J, Chorev M, Halperin JA, Wagner G, Cell 2007, 128, 257–267. [DOI] [PubMed] [Google Scholar]
- [11].Peter D, Igreja C, Weber R, Wohlbold L, Weiler C, Ebertsch L, Wei-chenrieder O, Izaurralde E, Mol. Cell 2015, 57, 1074–1087. [DOI] [PubMed] [Google Scholar]
- [12].a) Lukhele S, Bah A, Lin H, Sonenberg N, Forman-Kay JD, Structure 2013, 21, 2186–2196; [DOI] [PubMed] [Google Scholar]; b) Bah A, Vernon RM, Siddiqui Z, Krzemin-ski M, Muhandiram R, Zhao C, Sonenberg N, Kay LE, Forman-Kay JD, Nature 2015, 519, 106–109. [DOI] [PubMed] [Google Scholar]
- [13].Goldberg JM, Wissner RF, Klein AM, Petersson EJ, Chem. Commun 2012, 48, 1550–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Goldberg JM, Batjargal S, Chen BS, Petersson EJ, J. Am. Chem. Soc 2013, 135, 18651–18658; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen Y, Tsao K, Keillor JW, Can. J. Chem 2015, 93, 389–398. [Google Scholar]
- [15].a) Petersson EJ, Goldberg JM, Wissner RF, Phys. Chem. Chem. Phys 2014, 16, 6827–6837; [DOI] [PubMed] [Google Scholar]; b) Doose S, Neuweiler H, Sauer M, ChemPhys Chem 2009, 10, 1389–1398. [DOI] [PubMed] [Google Scholar]
- [16].Hackl EV, Biopolymers 2014, 101, 591–602. [DOI] [PubMed] [Google Scholar]
- [17].Volpon L, Osborne MJ, Topisirovic I, Siddiqui N, Borden KL, EMBO J 2006, 25, 5138–5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Forbes CR, Sinha SK, Ganguly HK, Bai S, Yap GP, Patel S, Zondlo NJ, J. Am. Chem. Soc 2017, 139, 1842–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Walters CR, Szantai-Kis DM, Zhang Y, Reinert ZE, Horne WS, Chenoweth DM, Petersson EJ, Chem. Sci 2017, 8, 2868–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Petersson E, Szantai-Kis D, Walters C, Barrett T, Hoang E, Synlett 2017, 28, 1789–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Martin MML, Lindqvist L, Lumin J. 1975, 10, 381–390. [Google Scholar]
- [22].Zhang JH, Chung TD, Oldenburg KR, Biomol J. Screening 1999, 4, 67–73. [DOI] [PubMed] [Google Scholar]
- [23].a) Mollica L, Bessa LM, Hanoulle X, Jensen MR, Blackledge M, Schneider R, Front. Mol. Biosci 2016, 3, 52 ; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Borgia A, Borgia MB, Bugge K, Kissling VM, Heidarsson PO, Fernandes CB, Sottini A, Soranno A, Buholzer KJ, Nettels D, Kragelund BB, Best RB, Schuler B, Nature 2018, 555, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].a) Batjargal S, Huang Y, Wang YJ, Petersson EJ, J. Pept. Sci 2014, 20, 87–91; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang YJ, Szantai-Kis DM, Petersson EJ, Org. Biomol. Chem 2015, 13, 5074–5081. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.