

Abstract
Combination antiretroviral therapy (cART) given orally has transformed HIV from a terminal illness to a manageable chronic disease. Yet despite the recent development of newer and more potent drugs for cART and suppression of virus in blood to undetectable levels, residual virus remains in tissues. Upon stopping cART, virus rebounds and progresses to AIDS. Current oral cART regimens have several drawbacks including (1) challenges in patient adherence due to pill fatigue or side-effects, (2) the requirement of life-long daily drug intake, and (3) limited penetration and retention in cells within lymph nodes. Appropriately designed injectable nano-drug combinations that are long-acting and retained in HIV susceptible cells within lymph nodes may address these challenges. While a number of nanomaterials have been investigated for delivery of HIV drugs and drug combinations, key challenges involve developing and scaling delivery systems that provide a drug combination targeted to HIV host cells and tissues where residual virus persists. With validation of the drug-insufficiency hypothesis in lymph nodes, progress has been made in the development of drug combination nanoparticles that are long-acting and targeted to lymph nodes and cells. Unique drug combination nanoparticles (DcNPs) composed of three HIV drugs—lopinavir, ritonavir, and tenofovir—have been shown to provide enhanced drug levels in lymph nodes; and elevated drug-combination levels in HIV-host cells in the blood and plasma for two weeks. This review summarizes the progress in the development of nanoparticle-based drug delivery systems for HIV therapy. It discusses how injectable nanocarriers may be designed to enable delivery of drug combinations that are long-lasting and target-selective in physiological contexts (in vivo) to provide safe and effective use. Consistent drug combination exposure in the sites of residual HIV in tissues and cells may overcome drug insufficiency observed in patients on oral cART.
Keywords: nanomedicine, long-acting, targeted, drug combination, HIV/AIDS
1. Introduction
Since the first reported cases of human immunodeficiency virus infection and acquired immune deficiency syndrome (HIV/AIDS) in 1981 [1, 2], the HIV pandemic continues to persist. The Joint United Nations Programme on HIV/AIDS (UNAIDS) estimated 36.7 million people are currently living with HIV worldwide [3]. With significant US investments in HIV basic and translational research, a number of bio-markers related to disease progression have been validated and well-characterized [4]. Based on the HIV replication life cycle and the known molecular targets of HIV, a significant number of highly potent orally administered antiretroviral (ARV) drugs have been approved and are widely used (Table 1). This pharmacological success represents a major breakthrough in the fight against HIV infection. Treatment with HIV medicines is called antiretroviral therapy (ART). When used daily as prescribed, highly active antiretroviral therapy (HAART) oral drug regimens composed of three or more drugs, or combination antiretroviral therapy (cART), suppress HIV replication and thus limit disease progression. HIV patients are now expected to live to an old age as long as an oral HIV drug regimen can be continued. Thus, oral drug combinations or cART have transformed HIV from a terminal illness to a chronic disease.
Table 1.
| Drug Class | Name (acronym) | FDA Approval |
Half-life (hr) |
Dosage Formb |
References |
|---|---|---|---|---|---|
|
Nucleoside Reverse Transcriptase Inhibitors (NRTIs) |
Zidovudine (ZDV) | 1987 | 1.2 | PO | [153] |
| Didanosine (DDI) | 1991 | 1.5 | PO | [154] | |
| Delayed-release didanosine | 2000 | PO | |||
| Stavudine (d4T) | 1994 | 1.6 | PO | [155] | |
| Lamivudine (3TC) | 1995 | 5.4 | PO | [156] | |
| Abacavir (ABC) | 1998 | 1.3 | PO | [157] | |
| Tenofovir disoproxil fumarate (TDF) | 2001 | 18.3 | PO | [158] | |
| Tenofovir alafenamide (TAF) | 2015 | 51.3 | PO | [159] | |
| Emtricitabine (FTC) | 2003 | 4.8 | PO | [160] | |
|
Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs) |
Efavirenz (EFV) | 1996 | 37.7 | PO | [161] |
| Nevirapine (NVP) | 1996 | 21.5 | PO | [162–166] | |
| Extended-release NVP | 2011 | 45 | PO | ||
| Etravirine (ETR) | 2008 | 30–40 | PO | [167] | |
| Rilpivirine (RPV) | 2011 | 48 | PO | [168] | |
|
Protease Inhibitors (PIs) |
Saquinavir (SQV) | 1995 | 3.6 | PO | [169] |
| Ritonavir (RTV) | 1996 | 3.5 | PO | [170] | |
| Indinavir (IDV) | 1996 | 1.8 | PO | [171] | |
| Nelfinavir (NFV) | 1997 | 4.3 | PO | [169, 172] | |
| Lopinavir (LPV) | 2000 | 5–6 | PO | [142] | |
| Lopinavir (LPV) Oral Pellets | 2015 | 5–6 | PO | [173] | |
| Fosamprenavir (FPV) | 2003 | 4.8 | PO | [174] | |
| Atazanavir (ATV) | 2003 | 7.5 | PO | [175] | |
| Tipranavir (TPV) | 2005 | 2.6 | PO | [176] | |
| Darunavir (DRV) | 2006 | 14.6 | PO | [177] | |
| Fusion Inhibitors | Enfuvirtide (T-20) | 2003 | 2 | SC | [178] |
| Entry Inhibitors | Maraviroc (MVC) | 2007 | 23 | PO | [179] |
|
Pharmacokinetic Enhancers |
Cobicistat (COBI) | 2014 | 4.3 | PO | [175] |
| Integrase Inhibitors | Dolutegravir (DTG) | 2013 | 13.5 | PO | [180] |
| Elvitegravir (EVG) | 2014 | 9.9 | PO | [181] | |
| Raltegravir (RAL) | 2017 | 9.3 | PO | [182] | |
| Bictegravir (BIC) | 2018 | 17.8 | PO | [183] | |
|
Combination HIV Medicines |
3TC + ZDV | 1997 | |||
| LPV + RTV | 2000 | ||||
| ABC + 3TC + ZDV | 2000 | ||||
| ABC + 3TC | 2004 | ||||
| FTC + TDF | 2004 | ||||
| EFV + FTC + TDF | 2006 | ||||
| FTC + RPV + TDF | 2011 | ||||
| QUAD, EVG + COBI + FTC + TDF | 2012 | ||||
| ABC + DTG + 3TC | 2014 | ||||
| DRV + COBI | 2015 | ||||
| EVG + COBI + FTC + TAF | 2015 | ||||
| ATV + COBI | 2015 | ||||
| FTC + RPV + TAF | 2016 | ||||
| FTC + TAF | 2016 | ||||
| DTG + RPV | 2017 | ||||
| BIC + FTC + TAF | 2018 | ||||
Antiretroviral drug names (with their acronym abbreviation) are grouped according to their drug class and fixed-dose combinations (FDCs) are listed at the bottom of the table. Also listed are the year that the drug or FDC was approved, and the terminal plasma half-life for each drug.
Dosage form: PO, oral; SC, subcutaneous.
Although a number of new cART composed of drugs with high potency and optimal treatment regimens have been developed [5], the cure for HIV/AIDS remains elusive [6]. As a member of the lentivirus genus, HIV bears a high mutation rate to evade the host’s immune surveillance. Mono-drug therapies resulted in a high frequency of harboring drug-resistant viruses that are linked to therapeutic failure. Current combination of HIV drugs (in cART) was shown to durably suppress viral replication and reduce drug resistance [7]. Although patients receiving cART have undetectable plasma viral loads, they still carry virus in lymph nodes and other tissues. Viral levels rebound quickly if cART is stopped. Multiple research studies have confirmed that HIV persists in lymph nodes and other tissues of infected animals and patients taking cART even though they have no detectable virus in their blood [8–10]. Importantly, most of the anatomic sites of HIV-infected cells (e.g., lymph nodes, brain, and certain part of the digestive tract) cannot be easily accessed by drugs [10, 11]. Since our initial proposal and report of insufficient HIV drug exposure in cells within the lymph nodes [12], where residual virus persists, the drug-insufficiency hypothesis has been validated by others in prospective studies [9, 10, 13]. Thus, the residual HIV-infected cells in lymphoid tissues could be due to insufficient drug exposure in these tissues after oral cART [9, 10, 13].
After analyzing isolated peripheral blood mononuclear cells (which include CD4+ T lymphocytes) in blood from HIV primates on the oral HIV protease inhibitor indinavir (IDV) who had reached steady-state drug levels in blood after multiple doses of IDV, the intracellular IDV levels in this same cell type from lymph nodes were about 3-fold lower than those found inside the cells from the blood [12–14]. In a recent work, simian immunodeficiency virus (SIV) and HIV tissue burdens in tissues of infected nonhuman primates and lymphoid tissue biopsies from infected humans were quantified in HIV and SIV infections. Although the numbers of virus RNA+ cells substantially decreased after ART, they remained detectable in these tissues, and their persistence was associated with lower drug concentrations in lymphoid tissue than in peripheral blood [9]. These data and other recent reports [10, 13] validated that drug insufficiency could be one of the factors resulting in the residual virus in lymphoid tissues.
As shown in Table 1, the available cART regimens mostly consist of oral dosage forms. A significant fraction of drug given orally may be subjected to (1) first-pass elimination, (2) limited drug absorption via the gut, and (3) premature elimination due to local metabolism or export of absorbed drugs by efflux transporters such as P-glycoprotein (P-gp). Experimental data also suggest that orally administered cART that is found in the blood may not fully penetrate and provide sufficient drug exposure consistently in tissues such as lymph nodes that harbor residual HIV. Therefore, higher doses of cART treatment must continue throughout a patient’s life to control the viral replication and preserve the host’s immune system. However, life-long daily oral administration of cART is associated with several significant limitations such as treatment or pill fatigue, non-adherence to therapy, fluctuating plasma drug levels (i.e., daily peak-and-trough oscillations from near toxic to ineffective levels), low drug distribution into the cells and tissues that harbor residual HIV, drug resistance, and side-effects. Having a new long-acting formulation that incorporates multiple anti-viral therapies in a single administration where the time between each dose is spread out to several weeks or months could significantly enhance the convenience and acceptability for patients. In addition to developing a long-acting cART that provides plasma drug concentrations above therapeutic levels for weeks, it could also reduce fluctuation of drug levels and keep drug resistance in check. Having a drug delivery system capable of targeting ARV drugs to the lymph nodes and tissues with residual HIV could address the tissue drug insufficiency and allow evaluation of the impact of disease outcomes with sufficient and sustained drug exposure in tissues.
The application of nanotechnology in medicine provides numerous possibilities in HIV therapy [15]. Drug delivery systems fabricated on the nanoscale can improve the pharmacological properties of drugs and some have demonstrated the ability to beneficially modify the biodistribution behavior of the loaded drugs in vivo to overcome tissue-drug-insufficiency, enhance the therapeutic efficacy, and reduce drug toxicity [16]. Compared to current oral regimens, nanomedicines that are long-acting and carry drug combinations may allow targeting to HIV host cells and tissues and reduce treatment fatigue in HIV patients [17]. Overall, nanomedicines formulated with safe and effective excipients could be a promising strategy for working towards eradicating HIV/AIDS.
This review focuses on the current state of nano-drug delivery systems in HIV therapy with respect to nano-drug combinations for long-acting and targeted therapy. Where appropriate, we will discuss the ability to target to cells infected by HIV. In the following, we highlight the progress in the development of nanoparticle-based drug delivery systems for HIV therapy. We specifically discuss three strategies employed with nano-carriers for HIV chemotherapy—drug combinations, long-acting, and targeting—which together will help facilitate further development of nanomedicines aimed at maximally suppressing HIV to produce a durable treatment response.
2. Current HIV treatment
With the understanding of HIV-1 pathogenesis and fine details of HIV protein structures and functions defined, ARV drugs targeted to specific viral proteins have been developed. The classes of anti-HIV drugs include (1) viral fusion inhibitors (FIs), which block fusion of virus with the cell membrane and subsequent entry into the host cells; (2) cell entry inhibitors such as CCR5 co-receptor antagonists (CRAs), which bind to HIV-1 co-receptors to inhibit viral entry into host cells; (3) nucleoside/-tide reverse transcriptase inhibitors (NRTIs), which incorporate into the viral DNA for chain-termination; (4) non-nucleoside reverse transcriptase inhibitors (NNRTIs), which interfere with the activity of the reverse transcriptase enzyme; (5) integrase strand transfer inhibitors (INSTIs), which inhibit the integration of viral DNA into the host cell DNA; and (6) protease inhibitors (PIs), which interfere with viral assembly to block viral budding and maturation. Currently, there are over 40 US Food and Drug Administration (FDA)-approved ARV drug products used clinically in the US for the treatment of HIV/AIDS (Table 1) [18].
ART is ideally initiated early in the course of HIV and continues throughout the life of an HIV patient. Current cART regimens include at least two drugs to suppress viral replication and reduce drug resistance [7]. Since the introduction of HAART in the mid-1990s, the annual number of new HIV infections has declined over the past two decades (1997–2014), and HIV-related deaths have fallen by 45% since the peak in 2005 [3]. Individuals who adhere to HAART can achieve durable viral suppression to undetectable levels in their blood, resulting in partial but slow recovery of CD4+ T cell numbers and function, and maintenance of some immune functions. Durable viral suppression can keep individuals remaining healthy and productive as long as ARV drugs are taken consistently and patients are adherent [19].
As a retrovirus, HIV can establish viral “reservoirs” or residual virus to maintain persistent infections. HIV reservoirs harbor integrated, replication-competent provirus within host cellular DNA. The viral genome is transcriptionally silent (“latent”) and virus is refractory to both cART and the immune system; viruses originating from these reservoirs lead to viral rebound in the plasma if treatment is stopped [20–22]. It is increasingly being recognized that latent viral reservoirs can persist by taking advantage of complex immune mechanisms that function to sustain life-long persistence of cellular immune memory [23]. Recently, it was discovered that CD32a is a surface-expressed marker upregulated on about half of the CD4+ T lymphocytes in the blood that harbor latent replication-competent HIV-1 [24], which will help facilitate critical studies on analysing, monitoring, and possibly therapeutically targeting the latent viral reservoir. However, it remains to be determined whether CD32a is a good marker for latently infected cells in lymph nodes, bone marrow, gut, and other tissues.
Researchers are currently employing multiple strategies to find ways to target and destroy latent HIV reservoirs [25]. These include using gene therapy (both the CRISPR-Cas9 nuclease and zinc finger nuclease [27] approaches) to cut out certain HIV genes that have integrated into host cells to inactivate the virus in these HIV-infected immune cells. Gene therapy is also being used to delete the CCR5 surface receptor on HIV host cells that HIV uses to enter into cells [28], and to engineer T cells to express anti-HIV chimeric antigen receptors (CARs) that recognize and eliminate virus-infected cells [29]. A second approach is developing drugs and other methods that reactivate latent HIV reservoirs so the immune system or new therapies can effectively eliminate them (i.e., the “shock and kill” strategy) [30–32]. A third approach is to develop methods to enhance the immune system’s ability to recognize and clear reactivated latent HIV reservoirs [33]. While all these efforts continue, and their particular contribution toward providing effective HIV clearance is being investigated, it is clear that orally administered cART which has limited ability to penetrate and provide sufficient drug exposure in viral reservoirs cannot clear residual HIV [6, 8].
Since current oral ARV drugs fall short of obtaining complete viral clearance, other biologically active substances such as antisense oligonucleotide, ribozyme, siRNA and nuclease strategies for gene therapy [34], and protein- or peptide-based antigens for immunotherapy [35] are under intense research and some have advanced to clinical testing, including experimental vaccines for prevention and treatment, with the hope of clearing HIV or fining a functional cure. However, despite these efforts, there is still no effective anti-HIV vaccine [36] or scalable gene therapy that not only prevents infection but also provides some degree of a functional cure to effectively address the world’s HIV pandemic. To reduce HIV prevalence rates, pre-exposure prophylaxis (PrEP) and post-exposure prophylaxis (PEP) have been proposed as effective strategies to prevent HIV-1 acquisition. Anti-HIV microbicides intended as a PrEP approach are given intravaginally or rectally prior to sexual intercourse [37]. Other implantable devices containing long-acting formulations of current ARV drugs have also been developed for HIV-1 prevention [38–40]. Recently, some oral therapies used in the clinic have been proven to be an effective means to prevent HIV transmission in high-risk populations [41]. Although PrEP has demonstrated satisfactory results in the fight against HIV/AIDS and its prevention, on the treatment spectrum, providing a cure for HIV infection remains elusive. These prevention measures also require frequent dosing. Thus, these strategies face similar compliance issues to those encountered in HIV treatments such as pill fatigue and low patient adherence. Without patient adherence, effective therapies can become ineffective in addressing HIV transmission among injection drug users and some special-needs populations (e.g., infants, adolescents, and elderly). A long-acting therapy is needed to prevent HIV transmission in such populations that are driving the rebound in new HIV infections in some areas of North America [42].
3. Major challenges in treating HIV
Although the development of ARV drug therapy for HIV infection has undergone substantial progress and contributed significantly toward improving disease management, numerous limitations and inconveniences persist for HIV/AIDS patients. Unlike traditional pathogens that infect humans, HIV is a unique retroviral pathogen in that its main target is CD4+ T cells, which are a key coordinator in the human immune system. HIV can hide in the genomes of latently-infected CD4+ memory T cells for years to escape host immune surveillance. HIV may establish persistent infection (where a low frequency of virus-infected cells may reactivate at opportune times during periodic low drug concentrations between each administration of oral dosing regimens) in tissues with limited drug exposure such as the brain in the central nervous system (CNS) and lymph nodes in the lymphatic system. In these tissues, orally administered drugs found in plasma must penetrate through blood-brain or blood-lymph barriers to reach virus within these hard-to-reach tissues. The low drug levels and limited exposure may allow virus to replicate. Under lower drug concentrations, the virus in these tissues could be actively replicating at low levels or some fraction could be integrated into the DNA of slow-replicating memory T cells, as well as in actively-replicating CD4+ T lymphocytes and monocytes/macrophages in these tissues [43]. To maintain inhibition of viral replication, life-long daily administration of cART with three or more different HIV drugs is needed for HIV/AIDS patients, but as mentioned, to date, no cure for HIV has been discovered. As soon as oral cART is discontinued, virus rebounds within 2 to 8 weeks [44–46], even though plasma viral levels were suppressed below the detection limit when HIV patients were in care and adhered to their daily oral drug regimens. This observation is key as it indicates that the inability to clear virus from residual tissues is not due to drug resistance (since durable suppression of plasma viremia is possible with cART), but instead may due to limited drug penetration into these anatomic sites of HIV-infected cells in which drugs cannot easily access [9, 10, 13]. Although long-acting oral dosage forms are reported to provide dosage retention in the stomach for ~1 week in pigs, solid dosage form retention in the stomach may pose stomach discomfort and patient compliance [47]. Drug release in the stomach would then be absorbed into the blood, and would not provide additional targeting value to tissues and cells with residual HIV.
More efficient targeting (or delivery) of ARVs to tissues with residual HIV and CD4+ T cells could enable improved treatment outcomes. Current cART lacks a specific targeting to tissues with residual HIV to inhibit HIV replication (only a minor fraction of the dose administered reaches these tissues). Although plasma viremia can be suppressed successfully with cART, viral levels will rebound if cART is stopped. The existence of drug insufficiency in lymphoid tissues and the CNS after oral dosing has been verified. For example, the intracellular IDV levels in the same cells from lymph nodes were about 3-fold lower than those found inside the cells from blood [12]. Most ARV drugs possess limited permeability across the blood brain barriers (BBB) in neuroAIDS [48]. Strategies to deliver and trap anti-HIV drugs in tissues and cells with residual HIV are urgently needed to overcome the drug insufficiency in patients on oral cART regimens.
The current life-long cART treatment method requires that HIV patients strictly adhere to cART (i.e., daily, twice-, or three-times daily taking of pills) to sustain HIV suppression and improve overall health and survival. The complex daily administration of three or more different drugs could result in missed doses of ARV drugs when patients’ daily lives interfere with this demanding dosing schedule. Non-adherence significantly increases in patients taking large and frequent dose administrations, which may relate to dose-limiting untoward-effects. The challenge in adhering to a demanding daily dosing schedule for life could lead to pill fatigue. Any missed dosing could give residual virus in tissues an opportunity for rebound. Virus rebound, if left unattended, could lead to progression to AIDS. Thus, non-adherence for patients on ART may eventually lead to emergence of drug resistance, therapeutic failure, and disease progression. Upon emergence of drug resistant virus, the ARV drugs lose their ability to control HIV replication and disease progression. Clinicians may use alternative drug combinations to overcome drug resistance. Clearly drug combinations targeted to different HIV targets are needed to provide sustained viral suppression and mono-drug therapy no longer is an acceptable option for modern HIV treatment. Approximately 20% of patients, especially those in youth and adolescent populations, encountered treatment failure even under prescribed cART therapy, almost exclusively due to incomplete adherence [49].
The available cART regimens mostly consist of oral dosage forms, which are also accompanied with some formulation-related problems. For instance, oral pills, tablets, and even suspensions are difficult for children and the elderly to take. It is reported that over 3.2 million children worldwide are infected with HIV, and more acceptable pediatric formulations are critically needed [50]. In these special populations, injectable drug formulations may provide a more consistent drug exposure in acceptable dosage forms and settings. Moreover, many ARV drugs show low bioavailability possibly due to gastrointestinal degradation and first-pass metabolism. The half-lives of most ARVs are short (Table 1), and some of these drugs exhibit poor water solubility. As a result, large and frequent doses are required to maintain minimum effective drug concentrations, which can lead to serious adverse side-effects [51].
Taken together, there is a great need to discover new approaches/drugs for developing non-toxic and highly effective treatments that enable good patient compliance through providing sustained dosing coverage and efficient eradication of residual HIV in cells and tissues. Formulations that employ drug combinations, long-acting pharmacokinetics (PK), and targeting strategies may address the limitations of current oral cART (Fig. 1). Long-acting therapies, which are referred to as sustained-release or extended-release oral dosage forms, have been developed to convert multiple daily doses to only once-a-day dosing to improve patient compliance. Traditional oral sustained drug release platforms could generally act via a drug depot within the gastrointestinal tract, with slow release of native drug from the dosage form. However, oral long-acting therapy has not been feasible in HIV drug therapy as the drugs released from the oral dosage forms only last for a maximum of 2–3 days. Recently, a novel oral device with drug-loaded polymeric arms that unfold and becomes lodged in the stomach has been reported to provide plasma drug levels in rats and pigs for weeks. The ability of this device to retain in the stomach and prevent it from being expelling from the gastrointestinal (GI) tract, combined with drug release from biodegradable polymers, poly(lactic-co-glycolic acid) (PLGA), have provide long-acting plasma drug levels [47]. Several long-acting implantables and injectables are also being developed for HIV PrEP and contraceptives [52]. However, many of these implantables may not be on patients’ preference list. Long-acting injectables, some of which could be done in private and invisible to others, might overcome the adherence challenges and could potentially replace daily pill taking with routine injection visits (or self-administration) [53, 54]. Persons with HIV unable or unwilling to swallow oral tablets or capsules—including persons hospitalized with critical illness, after surgery, or with pill fatigue; children (infants to teenagers who experience peer pressure and ridicule due to their daily pill-taking practice); and the elderly—may benefit from such a strategy. Moreover, injection drug users and others who are non-compliant with daily oral drug regimens could be a key population that would benefit from a long-acting anti-HIV injectable. About 10% of the new HIV infections in the US in 2015 were caused by injection drug use [55], which spreads HIV through the sharing of contaminated needles, sexual partners, and children born to HIV-infected injection drug users. With the US currently experiencing a opioid epidemic, which is projected to worsen over the next decade [56, 57], a long-acting cART injectable may be critical to address a potential increase in the rate of new HIV infections among the injection drug user community. For these reasons, a long-acting therapy is urgently needed and is a key priority on the health agenda of the US National Institutes of Health (NIH) [58].
Fig. 1.

Long-acting, combination and targeted HIV chemotherapy to address the limitations of current oral cART.
Two injectables—long-acting rilpivirine (RPV LA or TMC 278 LA) and long-acting cabotegravir (CAB LA or GSK1265744 LA) [59, 60] particles; each formulation containing a single agent—are currently in clinical development for HIV prevention as individual agents, and for HIV treatment when combined together. These drug particle technologies may allow sustained release of drug from the drug-carrier complexes at the site of (intramuscular) injection for a month or longer, and the slow and sustained release of drug could provide effective plasma drug levels. However, these injectables are single drug formulations. Their drug exposure in tissues and the ability to target to the residual virus, which is pivotal to suppress viral rebound, has not been fully investigated. Only long-acting HIV drug combinations with the ability to target to anatomical locations with residual virus could address the challenges of current cART. Therefore, our research program has focused on developing the targeted long-acting combination antiretroviral therapy (TLC-ART) formulation composed of three or more drugs in combination stabilized by lipid excipients to form nanocomplexes. These TLC-ART formulations carry multiple ARV drugs in a single nanoformulation and have been shown in primates to enhance accumulation of drug combinations in lymph node tissues. Using a combination of the subcutaneous (SC) route of administration and a formulation intended to enhance drug concentrations and exposure in lymphoid tissue, drug-combination nanoparticles composed of three HIV drugs—lopinavir (LPV), ritonavir (RTV), and tenofovir (TFV)—have been shown to provide sustained levels in the lymph nodes for over two weeks, as well as elevated drug-combination levels in HIV host cells found in the blood and nodes [61].
4. Emerging nanotherapeutics for anti-HIV/AIDS chemotherapy
Nanotechnology—which assembles atoms or compound organized structures into the nanometer scale—is gaining interest across scientific disciplines including chemistry, material sciences, and engineering. In pharmaceutical sciences, the idea of making nano-drug formulations is not new. It has been used to formulate drugs into nano-sized crystals, emulsions, colloids, and liposomal formulations in suspension or in solid dosage forms. Recently, nanotechnology-based drug delivery platforms have gained renewed interest—particularly in drug-combination nanoformulation research—to find a way to target a diverse combination of drugs to cells and tissues of interest and extend these drug levels for weeks with only a single dose. Such a challenging goal is necessary to address clinical needs in patient adherence and to overcome the tissue drug insufficiency that occurs in HIV patients on life-long oral daily drug combination therapy. Once such a novel targeted long-acting drug combination treatment platform is established, it could be used to address the most challenging life-threatening and chronic diseases like HIV/AIDS, cancer, cardiovascular disorders, etc.
On the most basic level, nano-drug formulations are attractive for pharmaceutical applications primarily due to their unique surface and scale effects, such as surface charge, particle shape, controllable size, and large surface-to-volume ratio. These carrier systems exhibit properties such as increased drug stability, protection of drug molecules from degradation, efficient penetration of biological barriers, improved cellular uptake and transport, enhanced intestinal absorption and bioavailability, and optimized drug biodistribution to enable efficient delivery of therapeutic agents to target sites and improve toxicity profiles [62]. Leveraging these properties through applying nanosized drug delivery platforms to ARV drugs holds promise to address the limitations of current cART discussed above. Using targeting approaches, drugs loaded in nanoparticles could achieve effective drug concentrations in tissues and cells with residual HIV. Drugs in long-acting nanocarriers could reduce cellular efflux and metabolism to prolong effective drug concentrations in target cells. The side-effects associated with cART may also be ameliorated due to the reduced dosage and enhanced bioavailability and targeting with appropriately designed nanomedicines. Nano-drug delivery systems also have the capacity to incorporate drugs with different physicochemical characteristics to combat drug resistance. Together, these benefits could improve patient compliance and overall therapeutic outcomes. Many published reviews have focused on specific types of nanoparticulate carriers for the treatment and prevention of HIV/AIDS; thus, please refer to them for additional details [63–67]. Below, we first briefly review select nanocarrier platforms for HIV chemotherapy, and then we examine three strategies for nanocarriers that are ideal for HIV chemotherapy—drug combinations, long-acting PK, and targeting to sites of residual HIV. Examples of nanomedicines as defined by their long-acting and targeted profile for combination HIV chemotherapy are listed in Table 2.
Table 2.
Examples of emerging nanomedicines as defined by their long-acting and targeted profile for combination HIV chemotherapy.a
| Category | Delivery system |
Drugb | Particle Size (nm) |
Routec | PK parameters | Targetd | Species Model |
Ref |
|---|---|---|---|---|---|---|---|---|
| Lipid-based | Lipid nanoparticles |
ATV or DRV |
33.6–68.2 | SC | Plasma drug concentrations persisted > 7 days (168 h) |
Plasma | Macaques | [72] |
| Lipid nanoparticles |
LPV, RTV and TFV |
52 | SC | 50-fold higher PBMC concentrations, sustained for 7 days |
Lymph nodes & lymphocyte s |
Macaques | [73] | |
| Lipid-stabilized nanosuspension (TLC-ART101) |
LPV, RTV and TFV |
~50 | SC | Plasma and PBMC intracellular drug levels persisted > 2 weeks t1/2 of TFV and LPV were 65.3 and 476.9 h in plasma, and 169.1 and 151.2 h in PBMCs TFV and LPV drug levels in LNMCs were up to 79-fold higher than those in PBMCs at 24 and 192 h |
Lymph nodes & lymphocyte s |
Macaques | [61] | |
|
Polymer- based |
Lactoferrin nanoparticles |
AZT, EFV and 3TC |
67 | PO | >4-fold increase in AUC and AUMC, 30% increase in the Cmax, >2 fold in the t1/2 of each drug |
Plasma | Wistar rats |
[83] |
| PLGA nanoparticles |
RTV, LPV, and EFV |
262 ± 83.9 | Intracellular peak drug levels continued until day 28 Free drugs eliminated by 2 days |
Plasma | Monocyte s from HIV-1, −2, & HBV(−) humans |
[106] | ||
| Poloxamer 338/drug nanosuspension s (LA rilpivirine) |
RPV | 200 | IM | Plasma t1/2 of about 40 days | Plasma | Adults with HIV- 1 infection |
[60, 107] |
|
| Polysorbate 20 /drug nanosuspension s (LA cabotegravir) |
CAB | 200 | IM/SC | Mean apparent terminal-phase t1/2 was 25 to 54 days |
Plasma | Adults with HIV- 1 infection |
[59, 107] |
|
| Poloxamer- 188/drug nanocrystals (nanoART) |
ATV or RTV |
233–540 | SC | Plasma drug level sustained > 14 days |
Plasma | Mice/ monkeys |
[102, 184] |
|
| Folate acid- poloxamer 147 nanocrystals |
ATV or RTV |
257–433 | IM | Drug bioavailability increased 5- fold, PD activity improved 100- fold |
Folic acid receptor |
HIV-1- infected humanize d mice |
[123, 124] |
|
| Crosslinked PEG- polymethyl methacrylate nanoparticles |
ZDV, 3TC, NVP, and RAL |
137.9 ± 1.3 | / | / | Gut- associated lymphoid tissue |
CD4+ T cells and PBMCs |
[105] | |
|
Inorganic- based |
Gold nanoparticles |
RAL | 1.8 ± 0.32 | IV | Distribution in brain with 333.6 ng Ag/g tissue |
Brain | Female adult Balb/c mice |
[91] |
| Iron oxide nanoparticles coated with amphiphilic polymer |
T-20 | 35.2 ± 2.2 | IV | Entry and trafficking into brain | Brain | Balb/c mice |
[136] | |
| Cell-based | Macrophage loaded poloxamer- 188/drug nanocrystals |
IDV | IV | Continuous drug release for 14 days, could distribute to brain |
Brain | HIVE SCID mice |
[95] | |
| Pro-drug | Self-assembled into spherical vesicles |
AZT and DDI |
174 | IV, IP, PO |
Bioavailability 90.5% and 30.8% for the IP and PO vs IV t1/2 = 15~30 min |
Macrophag e-rich tissues |
Mice | [104] |
Emerging nanomedicines are categorized according to the predominant material a particular drug delivery platform employs. For each drug delivery system, the drug(s) delivered, particle size, route of administration, notable pharmacokinetic (PK) parameters, the target tissue, and the species/model used for investigation are listed.
For definitions of drug abbreviations, please refer to Table 1.
Administration route: IM, intramuscular; IP, intraperitoneal IV, intravenous; PO, oral; SC, subcutaneous.
As defined by the available data published.
4.1. Nanocarriers used in HIV chemotherapy
4.1.1. Lipid-based nanoparticle formulations
Through the exploration of lipid–drug and lipid–protein interactions, lipid-based drug delivery systems (liposomes, lipid-nanoparticles, solid lipid-nanoparticles, and ethosomes) have been developed to enhance therapeutic benefits. Liposomes or lipid vesicles, which are fabricated with concentric lipid bilayers (a biomimetic of cell membranes) to protect drug molecules in the lipid membrane or aqueous core, are a pivotal biocompatible and biodegradable formulation platform for anti-HIV/AIDS drugs. Liposomes offer advantages such as incorporation of both the hydrophilic drugs in the aqueous core and the hydrophobic drugs in the phospholipid bilayer. However, the fraction of water soluble drug encapsulated in the aqueous core, even under saturating lipid concentrations is small, and typically for small vesicles or liposomes in 50–100 nm would be about 1% or less. They also allow surface functionalization to provide drug targeting, and they have low immunogenicity. Four types of liposome-based delivery systems—cationic, anionic, sterically stabilized, and immunoliposomes—have mainly been studied for anti-HIV/AIDS drug delivery [68]. With intricate designs, liposomes can be modified with targeting ligands to enhance intracellular drug delivery and improve drug efficacy Liposomes can also entrap other nanoparticle-entrapped ARV drugs for sustained drug release [70].
Lipid-nanoparticles are lipid–drug complexes that lack a contiguous bilayer. They demonstrate physical stability and high drug-loading of hydrophobic drugs in general, but more challenging in encapsulating hydrophilic molecules. Lipid-nanoparticles exhibit distinct distribution and elimination patterns in the body compared with liposomes [71]. Our research team has initially reported a lipid-nanoparticle containing IDV that enhanced drug levels in all analyzed lymph nodes in primates [12]. Later, we found that the interactions of atazanavir (ATV) and darunavir (DRV) with lipids could facilitate combination delivery [72]. From this, we developed a lipid-stabilized drug-combination nanoparticle (DcNP) containing two PIs, LPV and RTV, and a reverse transcriptase inhibitor TFV, for simultaneous triple-drug delivery to HIV host cells in the blood and lymphatics. These DcNP nanoparticles produced over 50-fold higher intracellular LPV, RTV, and TFV concentrations in lymph nodes of primates compared to free drug. Plasma and intracellular drug levels in blood were enhanced and sustained for 7 days [73]. The long-acting intracellular and plasma exposure over 2 weeks using a similar but optimized injectable combination formulation called TLC-ART101 were further validated and characterized [61].
Solid lipid-nanoparticles (SLNs) contain a solid lipid core that is solid at both room and human body temperatures [74]. They have been used as alternatives to existing lipid particulate systems to modulate drug release profiles [75]. SLNs could be further modified with polymers to overcome the drawbacks associated with solid lipid formulations including limited drug loading capacity, unpredictable drug release profile, and limited solubility of drug in the lipid matrix [76].
Ethosomes—containing phospholipids, alcohol at relatively high concentrations, and water—are other carrier vesicles with promising results for transdermal delivery of anti-HIV/AIDS drugs. IDV which has limited bioavailability via the oral route, was loaded into ethosomes for transdermal delivery. Permeation studies of IDV across human cadaver skin showed significantly enhanced transdermal flux from ethanolic liposomes than from an ethanolic drug solution, conventional liposomes, or a plain drug solution [77]. However, the local effects of ethanol on skin could be a challenge for chronic use in some patients [78].
4.1.2. Polymer-based nanoparticle formulations
Polymer-based nanoparticle platforms have been widely studied for anti-HIV/AIDS treatment with their enhanced drug loading capacity, prolonged circulation time in the bloodstream, easy chemical modification, and convenient surface functionalization. Drugs can be covalently conjugated into polymer, loaded into polymeric micelles by hydrophobic interactions, or encapsulated into polymeric nanoparticles.
Polymer-drug conjugates include water-soluble polymeric carriers, biodegradable linkages, and antiviral cargos. Drugs can also be cleverly designed to undergo “triggered” release from polymer with specific conjugation bonds. The capability of intracellular triggered release of these polymer prodrugs could effectively prolong the circulation half-life and alleviate off-target cytotoxic side-effects. In addition, polymers with polyanionic side-chains, such as acrylic acid or methacrylic acid, have been shown to lead to increased anti-HIV efficacy. Danial et al. synthesized copolymers consisting of sulfonated side chains, hydroxylated side chains, and the reverse transcriptase prodrug lamivudine (3TC) attached via disulfide self-immolative pendent chains by reversible addition–fragmentation chain transfer (RAFT) polymerization [79]. These polymers that carry multiple antiviral cargos could make effective combination agents. They could provide multiple synergistic modes of action against HIV by inhibiting viral entry and also having two different approaches for reverse transcription inhibition. Chitosan, a well-known abundant natural polysaccharide, has also been chosen as a polymeric carrier to deliver stavudine (d4T). Through the Atherton–Todd reaction, a chitosan-O-isopropyl-5’-O-d4T monophosphate conjugated prodrug with a phosphoramide linkage between glucosamine and nucleoside monophosphate was synthesized. These polymer-drug conjugate nanoparticles could be used for sustained drug release and for targeting tissues with residual HIV [80].
Drugs could also be loaded or encapsulated into polymeric nanoparticles without chemical conjugation. A variety of polymers, such as PLGA, polymethylmethacrylate (PMMA), poly(propyleneimine) (PPI) dendrimers, have been used for successful delivery of ARV drugs [66]. PLGA nanoparticles loading maraviroc (MVC), etravirine (ETR), or raltegravir (RAL) were prepared using emulsion-solvent evaporation techniques for synergistic inhibition of both cell-free and cell-associated HIV-1 infection in vitro. These drug combination nanoparticles exhibited a large dose-reduction due to synergistic effects. Higher intracellular drug concentrations were obtained for cells dosed with the triple-ARV-NP combination compared to the equivalent unformulated drugs [81]. For oral administration, Giardiello et al. screened large libraries of solid drug nanoparticles (160 individual components) and arrived at 10 polymers and 16 surfactants as the materials for a potential aqueous pediatric HIV nanotherapy [82]. Such a screening process may offer rapid production, evaluation, and optimization of cost-effective orally-dosed drug nanoparticles to address unmet clinical needs for various diseases. Through suitable mechanical preparation processes, ARV drugs could be made into nanosuspensions for injection use with some polymeric surfactants. Injectable RPV LA and CAB LA were manufactured by conventional wet-bead milling with poloxamer 338 [60] or polysorbate 20 and polyethylene glycol 3350 [59] as wetting agents. The size of these nanosuspensions are about 200 nm.
Proteins have also been used to deliver HIV drugs. Lactoferrin—a multifunctional protein of the transferrin family with a molecular mass of about 80 kDa—is one of the components of the immune system of the body and has a broad spectrum of functional activities such as anti-HIV, antimicrobial, and anti-bacterial activities. Loading HIV drugs onto lactoferrin could make drugs with improved safety, bioavailability, and PK [83, 84].
4.1.3. Inorganic nanoparticle formulations
Inorganic nanoparticles, such as iron oxide nanoparticles (IONPs), gold nanoparticles (AuNPs), sliver nanoparticles (AgNPs), quantum dots, silica nanoparticles, and carbon nanotubes, have been investigated extensively for their potential role in drug delivery. Some of the unique physical and biological properties of these particles may allow development of multi-function particles that could be used for diagnostic and therapeutic (“theranostic”) purposees [85].
Magnetic IONPs have been used to deliver ARV drugs due to their unique features: controllable size distribution, biocompatibility, ability to bind drug and sustain drug release, magnetically-guided site-specific targeting, and the ability to convert magnetic fields to heat energy. Saiyed et al. made 3’-azido-3’-deoxythymidine-5’-triphosphate (AZTTP) bind onto magnetic Fe3O4 nanoparticles by ionic interaction; the magnetic nanoparticle-bound AZTTP maintained its biological activity in peripheral blood mononuclear cells, and active NRTIs were targeted to the brain by applying an external magnetic force [86]. Magnetic IONPs could also be used for magnetic resonance imaging (MRI) to monitor drug distribution to target tissues in vivo Superparamagnetic iron oxide nanoparticles (SPIONs) have shown to have the ability to track HIV-specific cytotoxic T lymphocytes (CTLs) [88], and to increase the cytotoxic potential of HIV-specific CTLs and improve the killing of HIV-infected cells in latent HIV reservoirs by employing magnetic field hyperthermia effects [89].
AuNPs bear attractive characteristics as drug delivery systems for anti-HIV drugs due to their small size that facilitates entry into tissues and cells, controllable shape and geometry to modulate their PK behavior in vivo, and their easy modification with targeting ligands or drugs. AuNPs have been shown to enter different cell types including macrophages [90] and cross the BBB to exert antiviral activity when conjugated to an ARV drug [91].
Lastly, AgNPs have presented remarkable antiviral activity against HIV [92]. They have been applied in a vaginal product to prevent sexually transmitted HIV [93].
4.1.4. Cell-based delivery systems
A variety of cells—erythrocytes, bacteria, dendritic cells, leukocytes, lymphocytes, and macrophages—have recently emerged as biological drug carriers. Cell-mediated drug delivery systems may offer the advantage of integrating drug cargos into functional cells, which may provide a more selective, active, and safe therapeutic strategy for HIV treatment [94]. However, consistency and the ability to produce and scale drug-loaded cells for therapeutic purposes could be challenging.
Monocyte-derived macrophages (MDMs) were used as Trojan horses to deliver drug-loaded nanoparticles to tissues with residual HIV. Loading IDV nanoparticles into murine bone marrow macrophages carried drug across the BBB into HIV-1-infected brain regions [95]. This approach could limit ARV drugs metabolism and excretion while enhancing potent antiretroviral efficacy compared to unformulated drugs. In addition, the nanoparticles could interact with a broad range of activatable proteins that could enhance phagocytosis, secretory functions, and cell migration, facilitating targeted activity and controllable drug release [96].
4.2. Nanomedicine as a platform for drug combinations
Clinicians and scientists have lately used the term cART, which is synonymous with the popular term HAART. The changes reflect the attempt by public health officials to reframe the communication to the public of the efficacy of HIV therapy by emphasizing “combination.” The ideal HIV ART should be user-friendly, affordable, effective at prolonged suppression of HIV replication, well-tolerated/safe, and have no drug-drug interactions. Individual ARV drugs are unable to suppress HIV infection for long durations. The use of monotherapy and dual therapy often leads to mutations and development of drug resistance and results in inadequately supressing viral replication, thus necessitating combination therapy, which became the clinical standard of care in 1996 [97]. Drug combinations act by durably suppressing the propagation of mutations that arise from one of the drug classes. As a result, cART drug regimens consist of a minimum of two active drugs from two classes, and usually contain three or more different drugs. Previous studies using plasma viral load as a primary endpoint did not show a benefit from four-or five-drug therapies targeting more than two viral sites [98, 99]. Although use of cART reduces the risk of developing drug resistance and makes treatment more effective, such drug regimens are complex and they do not eradicate HIV from the body. Patients must take a variety of pills everyday with stringent intake schedules and food and fluid restrictions.
In recent years, drug companies have worked to combine these complex regimens into simpler oral dosage forms, termed oral fixed-dose combinations (FDCs) [100]. For example, TAF has been co-formulated with emtricitabine (FTC), elvitegravir (EVG), and the EVG metabolic enhancer, cobicistat (COBI), into a single-tablet regimen, which is associated with high efficacy and better kidney and bone safety [101]. This greatly reduced the pill burden of patients and improved medication compliance. However, simply blending different drugs together still may be accompanied by the limitations of the distinct PK profiles of each drug that leads to inconsistent in vivo biodistribution and action. Drug-drug interactions and negative synergistic side-effects may also be important issues that remain.
Nanoparticles can provide a unique platform for delivering different drug combinations and synchronizing their targeting and PK. They possess several advantages over conventional formulations that physically blend multiple drugs together, including: (1) improved solubility and bioavailability, (2) prolonged drug circulation half-life, (3) controlled PK of each drug, and (4) increased drug accumulation in tissues and cells with residual HIV through targeting, resulting in enhanced drug efficacy and dose reduction. Many nanoparticulate platforms have been developed for co-delivery of two or three ARV drugs. Some studies prepared each drug-loaded nanoparticle formulation separately and then blended the different formulations together to obtain combination anti-HIV therapy. For example, MVC, ETR, or RAL were incorporated into PLGA nanoparticles using emulsion-solvent evaporation techniques. This nanoparticle combination showed a large dose-reduction compared to the free-drug combination, and demonstrated in vitro inhibition of RT-SHIV virus propagation in macaque cervicovaginal tissue explants [81]. Whether these particles would provide long-acting kinetics in vaginal tissues after dosing is not known. One long-acting injectable nano-formulation for HIV treatment called NanoART, was formed with crystalline drug surrounded by a thin layer of a poloxamer surfactant using high-pressure homogenization. ATV and RTV nanoART were manufactured separately, and the two formulations were mixed together before administering to mice and rhesus macaques [102]. Although these blended nanoparticles could impart synergistic effects, they cannot solve the problem of distinct PK profiles for each drug. In addition, the tedious preparation processes hindered their further development for clinical application.
By considering the physical, pharmacological, and pharmaceutical aspects of the drug and excipient constituents, combination ART could be realized with just one injectable fixed-dose nanoformulation, in which two or more drugs are incorporated inside each nanoparticle. Such an injectable cART nanoparticle could deliver different active pharmaceutical ingredients (APIs) into the same active site (or HIV target cells) and exert synergistic effects. The drugs could be either encapsulated or surface-conjugated to make a single nanoparticle entity. Li et al. developed a cocktail-like drug delivery vehicle using biodegradable PEG-PLA nanoparticles encapsulating the non-nucleoside reverse transcriptase inhibitor (NNRTI) DAAN-14f (14f) and surface-conjugated the HIV-1 fusion inhibitor T1144. These polymeric nanoparticles carrying two ARV drugs achieved enhanced cellular uptake, improved antiviral activity, and prolonged blood circulation times [103]. Jin et al. synthesized a bolaamphiphilic prodrug, zidovudinephosphoryl-deoxycholyl didanosine (ZPDD), by combining zidovudine (AZT) and didanosine (DDI) in one molecule. The ZPDD could self-assemble into spherical vesicles in water. They showed high anti-HIV activity in vitro and targeted macrophage-rich tissues in vivo and rapidly released AZT in target tissues following intravenous administration to mice [104]. Four different ARV drugs—AZT (LogP −0.1), 3TC (LogP −1.3), nevirapine (NVP, LogP 1.75), and RAL (LogP −0.39)—were loaded into stealth hydrolyzable crosslinked PEG-PMMA (PMM) nanoparticles and stealth crosslinked poly-ε-Caprolactone-HEMA (ECA) nanoparticles by dispersion polymerization. The PMM and ECA nanoparticles loaded with these four drugs efficiently inhibited HIV-1 infection in CD4+ T cells and peripheral blood mononuclear cells (PBMCs) in vitro [105]. Although a cART nanoparticle could be realized by chemical modification or polymerization processes, the uncontrollable synthesis steps might cause great difficulty for drug development. Physical combination of excipients with drugs might be easier for manufacturers to develop formulations. Destache et al. showed PLGA nanoparticles could be fabricated containing three hydrophobic ARV drugs—RTV, LPV, and efavirenz (EFV). Drugs were slowly released from PLGA nanoparticles and drug levels in PBMCs isolated from whole blood collection of HIV-1, −2 and hepatitis B seronegative donors were maintained at a high concentration until day 28 without cytotoxicity [106]. Although three drugs could be entrapped inside PLGA nanoparticles simultaneously, it is very difficult to entrap both hydrophobic and hydrophilic drugs together by physical complexation. Recently, we developed a simple, scalable, and characteristic-controlled drug combination in a lipid-stabilized nanosuspension to co-deliver three drugs: TFV, a relatively water soluble compound (LogP −1.6), and LPV and RTV, both relatively hydrophobic (LPV LogP 5.94, RTV LogP 4.24) at physiological pH. This anti-HIV drug combination nanoformulation, which is now referred to as TLC-ART101, has been verified to have long-acting PK and the ability to target lymph nodes in primates [61].
4.3. Long-acting strategies in nanomedicine
Maintenance of cART drug levels in relevant tissue and cell compartments is essential to enable sustained antiviral responses and avoid the development of viral resistance and treatment failure. To solve one of the toughest problems in HIV management—in which people do not adhere to daily pill regimens that their doctors recommend—long-acting drug formulations are under development for both prevention and treatment of HIV infection. Several long-acting injectables and implantables that could replace daily pill taking with routine injections or implantations are under development and are intended for HIV PrEP and contraceptives [52]. Long-acting ARV drugs are also under development for maintenance therapy in HIV patients [107]. This could provide a convenient approach to manage HIV for patients who report pill fatigue.
Nanomedicine could provide sustained-release injectable drug delivery systems that sustain blood drug concentrations for long durations and consequently reduce the dosage levels needed. RPV LA and CAB LA—for HIV prevention as single agents [53, 54] and for HIV treatment when given together [107]—are in a nanocrystal state for intramuscular administration and are absorbed slowly from the injection site into the systemic circulation (Table 3). The PK of RPV in healthy individuals after intramuscular injection demonstrated the time to reach a maximum plasma concentrations (tmax) of 5–8 days in plasma and plasma half-life (t1/2) of about 40 days [108]. The t1/2 of the CAB LA in healthy individuals was 25–40 days [109]. Based on their PK properties, the intended dosing interval for prevention applications for RPV LA is once monthly, and for CAB LA is once every three months. For HIV treatment, the intended dosing interval is monthly or once every two months and requires an oral lead-in induction period prior to initiating the long-acting injectable regimen [107]. Although these two nanoparticles represent important first steps in developing long-acting anti-HIV drugs, they are formulated with only one drug and require separate injections, which patients report are painful [107]. Any missed dosing could also negatively impact drug resistance.
Table 3.
A comparison of long-acting nanoparticle products LA CAB/LA RPV, nanoART, NMDTG, and TLC-ART101 with respect to physical and pharmacokinetic characteristics, and manufacturing process.
| LA CAB a | LA RPV G001 b | NanoART c | NMDTG d | TLC-ART101e | |
|---|---|---|---|---|---|
| Physical properties | |||||
| Characterist ics |
Nanosuspension | Nanosuspension | Nanocrystals | Nanosuspension | Drug combination particles |
| (excipient stabilized drug particles) |
|||||
| Hydrodyna mic size |
200 nm | 200 nm | 233–540 nm | 275–340 nm | ~52 nm |
| Excipients | Mannitol, Polysorbate 20, PEG3350 |
Poloxamer 338, Citric acid |
Poloxamer188 | Unpublished | DSPC, DSPE-mPEG2000 |
| Drugs and solubility |
Cabotegravir (LogP =2.20 ) |
Rilpivirine (LogP =4.32 ) |
Atazanavir (LogP=4.08) or Ritonavir (LogP=4.24) |
Myristoylated dolutegravir prodrug |
Lopinavir (LogP=5.94), Ritonavir (LogP=4.24), and Tenofovir (LogP= −1.6) |
| Drug ratios (molar)c |
- | - | - | - | 4:1:5 |
| Number of Drug |
Single | Single | Single | Single | Triple |
| Manufacturing process | |||||
| Manufactur ing stepsd |
Wet-bead milling | Wet-bead milling | Seperately homogenization then mix together |
One-step synthesis and homogenization |
Mixing three drugs together and homogenization |
| Pharmacokinetic and tissue distribution characteristics | |||||
| Species Model |
Adults with HIV-1 infection |
Adults with HIV-1 infection |
Mice | Rhesus Macaques | Macaques |
| Route | IM/SC | IM | SC | IM | SC |
| t1/2 | 25–54 days | ~ 40 days | 1,152.5 h (ATV) 230.1 h (RTV) |
458.2 h | 476.9 h (LPV), 44.1 h (RTV), 65.3 h (TFV) |
| Tissue distribution |
Low penetration into mucosal tissue |
Substantial levels of RPV in cervico-vaginal fluid and vaginal tissue for 84 days |
At 24, ATV and RTV levels were up to 506- fold higher in tissues |
Prodrug observed in blood leukocytes through day 42. |
TFV and LPV drug levels in LNMCs were up to 79-fold higher than those in PBMCs at 24 and 192 h |
| Stage | Phase III | Phase III | Pre-clinical | Pre-clinical | Pre-clinical |
| Ref | [59] | [60] | [102] | [110] | [61] |
Long-acting cabotegravir
Long-acting rilpivirine
Long-acting nanoformulated antiretroviral therapy
Long-acting nanoformulated dolutegravir prodrug
Targeted long-acting combination antiretroviral therapy 101.
Abbreviations: ATV, atazanavir; LPV, lopinavir; RTV, ritonavir; TFV, tenofovir; IM, intramuscular injection; SC, subcutaneous injection; LNMCs, lymph node mononuclear cells; PBMCs, peripheral blood mononuclear cells
Another long-acting nano-formulation for HIV treatment is NanoART, which is at the preclinical stage [102]. ATVand RTV levels in male BALB/c mice at fourteen days after intramuscular injection of NanART were up to 13-, 41-, and 4,500-fold higher in plasma, tissues, and at the injection site, respectively, than those resulting from unformulated drug. Because ATV and RTV NanoART were manufactured separately and then mixed together before use, the tedious preparation led to the abandonment of this formulation for further research. Recently, the same team led by H Gendelman developed a long-acting nanoformulated, acylated dolutegravir prodrug (NMDTG) formulation. After intramuscular injection of NMDTG to three rhesus macaques, the t1/2 of DTG (after deacylation of NMDTG) was 467 h and 460 h in plasma and PBMCs, respectively [110]. However, the DTG concentrations in PBMCs were consistently lower than those in the plasma following a single administration; this is in contrast to the SC TLC-ART101 drug combination nanosuspension, which achieved persistently higher drug concentrations in lymph node mononuclear cells (LNMCs) and PBMCs compared to those in the plasma over the two-week course of the study (Fig. 2, Table 4). Thus, assuming an analogous TLC-ART formulation with a DTG-containing drug combination would similarly boost intracellular LNMC and PBMC drug concentrations compared to those in the plasma, the TLC-ART101 formulation appears to be better able to target drug combinations that contain both hydrophobic and hydrophilic antiretrovirals to HIV host cells than the NMDTG formulation, which only contains a single potent drug. The cell targeting ability of DcNPs formulated with hydrophobic LPV and RTV and hydrophilic TFV in TLC-ART101 is apparent when the SC injectable TLC-ART101 drug combination effects on cell (PBMC)-to-plasma ratios were compared to the same set of drugs given by current oral dosage forms (Table 5).
Fig. 2.
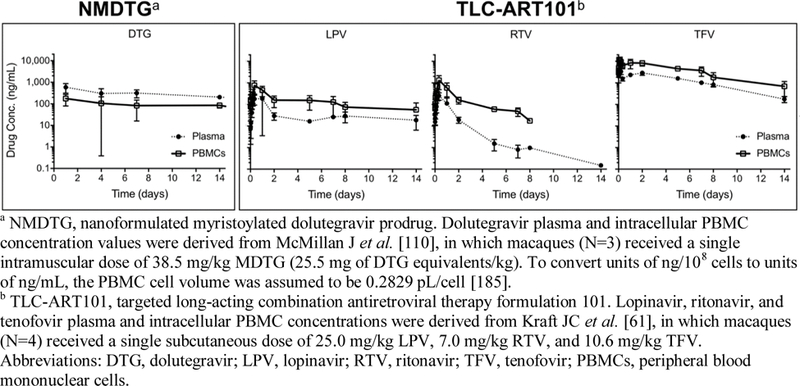
Comparison of different anti-HIV nanoformulations (NMDTG vs. TLC-ART101) and their ability to boost drug levels in HIV host cells in the blood (PBMCs) relative to the plasma following a single injection in primates.
Table 4.
Comparison of different anti-HIV nanoformulations (NMDTG vs. TLC-ART101) and their ability to boost drug levels in HIV host cells in lymph nodes (LNMCs) and the blood (PBMCs) relative to the plasma approximately one week following a single injection in primates.
| NMDTGa | TLC-ART101b | |||
|---|---|---|---|---|
| Active drug analyte | DTG | LPV | RTV | TFV |
| Dose (mg/kg) | 25.5 | 25.0 | 7.0 | 10.6 |
| Route | IM (single drug suspension) |
SC (all 3 drugs in one suspension) | ||
|
LNMC/PBMC ratio (at 8 days) |
NAc | 79.20 | 531.37 | 4.41 |
|
PBMC/plasma ratio (at 7 days) |
0.26 | 4.95 | 59.63 | 3.67 |
NMDTG, nanoformulated myristoylated dolutegravir prodrug. Dolutegravir plasma and intracellular PBMC concentration values were derived from McMillan J et al. [110], in which macaques (N=3) received a single intramuscular dose of 38.5 mg/kg MDTG (25.5 mg of DTG equivalents/kg). To convert units of ng/108 cells to units of ng/mL and calculate the PBMC/plasma ratio at the 168 hr (7 day) time point, the PBMC cell volume was assumed to be 0.2829 pL/cell [185].
TLC-ART101, targeted long-acting combination antiretroviral therapy formulation 101. Lopinavir, ritonavir, and tenofovir plasma and intracellular PBMC concentrations were derived from Kraft JC et al. [61], in which macaques (N=4) received a single subcutaneous dose of 25.0 mg/kg LPV, 7.0 mg/kg RTV, and 10.6 mg/kg TFV.
NA, not available (lymph node drug levels were not investigated).
Abbreviations: DTG, dolutegravir; LPV, lopinavir; RTV, ritonavir; TFV, tenofovir; IM, intramuscular injection; SC, subcutaneous injection; LNMCs, lymph node mononuclear cells; PBMCs, peripheral blood mononuclear cells.
Table 5.
The PBMC-to-plasma drug ratios for all three antiretrovirals, compared to oral dosing reference, after a single subcutaneous dose of a long-acting drug combination formulation (TLC-ART101). The enhanced PBMC-to-plasma ratio following one subcutaneous dose of TLC-ART101 compared to the same drugs given orally suggest cell targeting effects are mediated by the TLC-ART nano-drug combination particles.
| Species | Humans | Monkeys | ||||
|---|---|---|---|---|---|---|
| Reference | [147] | [147] | [186] | [61] | ||
| Route | PO | PO | PO | SC | ||
| Formulation | Kaletra® | Kaletra® | Viread® | TLC-ART101 | ||
| Drug | LPV | RTV | TFV | LPV | RTV | TFV |
| Dose (mg/kg) | 22.9a | 5.7a | 2.4b | 25.0 | 7.0 | 10.6 |
| Time | 16 hr | 16 hr | 24 hr | AUC(0–336hr)c | AUC(0–336hr)c | AUC(0–336hr)c |
|
PBMC Drug Level |
2.7 µg/mL | 0.23 µg/mL | 26 ng/mLd | 44 hr•µg/mL | 35 hr•µg/mL | 1.3 hr•mg/mL |
|
Plasma Drug Level |
10 µg/mL | 0.45 µg/mL | 40 ng/mL | 11 hr•µg/mL | 4.8 hr•µg/mL | 0.42 hr•mg/mL |
|
Ratio of PBMC/Plasma Drug Levels |
0.27 | 0.52 | 0.65 | 4.02 | 7.40 | 3.01 |
The median age of patients in the van Kampen et al. 2010 study was 10 years old. Since dose and median weight were not reported in this study, we used the recommended dose of 800 mg LPV/200 mg RTV administered once daily as Kaletra® for children weighing 35 kg (which is the median weight of children 10 years old) to calculate the mg/kg dose for LPV and RTV given in this study.
The oral dose for TFV in humans was calculated for 300 mg tenofovir disoproxil fumarate (TDF; 519.44 g/mol) and assuming 70 kg human weight as follows: (300 mg TDF/70 kg) x (287.21 g/mol TFV/519.44 g/mol TDF) = 2.4 mg/kg.
Because complete drug concentration-time course profiles over two weeks (0–336 hr) were characterized for LPV, RTV, and TFV in monkeys after TLC-ART101 dosing, AUC(0–336h) values were used to calculate the PBMC/Plasma Ratio for each drug.
Since only tenofovir diphosphate (TFV-DP) drug levels in PBMCs were reported in [186], the steady-state fraction of 61% of total intracellular TFV being TFV-DP was used to estimate the total intracellular TFV level reported in the table (26 ng/mL) [187–190]. For these calculations, the PBMC cell volume was assumed to be 0.2829 pL/cell [185].
AUC(0–336hr), area under the concentration-time curve between time 0 to 336 hours; PBMC, peripheral blood mononuclear cell; PO, oral; SC, subcutaneous; LPV, lopinavir; RTV, ritonavir; TFV, tenofovir.
Since current cART regimens involve two or more drugs in combination, there is an urgent need to develop long-acting triple-drug combination formulations. As mentioned, TLC-ART101 developed by our group is a combination of three anti-HIV drugs—LPV, RTV, and TFV—in a lipid-stabilized nanosuspension, and achieves long-acting effects. In this formulation the HIV drug RTV is intended to boost plasma LPV. Plasma and PBMC intracellular drug levels of LPV and TFV after a single SC dose of TLC-ART101 in macaques persisted for over 2 weeks. PBMC drug exposures were three- to four-fold higher than those in plasma. Apparent terminal t1/2 of LPV and TFV were 476.9 and 65.3 h in plasma, and 151.2 and 169.1 h in PBMCs, respectively [61]. A comparison of long-acting nanoparticle products CAB LA/RPV LA, nanoART, NMDTG, and TLC-ART101 with respect to physical and PK characteristics, and manufacturing process are listed in Table 3.
4.4. Targeting strategies in nanomedicine
4.4.1. Extended plasma drug levels
To date, most approved HIV medicines belong to the class of NRTIs and PIs, which have terminal half-lives on the order of only several hours (Table 1). Due to their low bioavailability from first-pass metabolism and short half-lives, they require frequent administration of large doses. As discussed, nanomedicine has the means to provide controlled-release delivery systems to protect the entrapped drug from degradation, keep the drug molecules in circulation longer, change their biodistribution profiles, prolong their half-lives, enhance intracellular delivery, and consequently enhance efficacy. As a result, nanoformulations have improved the bioavailability of poorly-bioavailable drugs and have provided extended drug levels in the blood, which is a tissue that harbors HIV and requires delivery of effective drug levels to inhibit HIV replication. A lot of work has focused on development of oral nanoformulations to improve drug bioavailability.
In one study, saquinavir (SQV) was loaded into PEGylated liposomes to provide sustained drug release and more efficient delivery and reduced cellular toxicity in Jurkat T-cells compared to free SQV [111]. Conjugating ARV drugs into polymer also provides an option for prolonging the circulation half-life and alleviating cytotoxic side-effects. Skanji et al. designed a glycerolipidic prodrug of DDI (ProddINP) by covalently coupling 1,3-dipalmitoyl lipid through a succinate linker bound to the 5-OH group of the sugar moiety of DDI, and incorporated this prodrug into dipalmitoylphosphatidylcholine (DPPC) bilayers. In rats, this formulation compared to free DDI after oral administration demonstrated an increased DDI blood half-life (3-fold) and enhanced accumulation of the prodrug form at 24 h in numerous organs, especially the intestines [112]. Polymeric nanoparticles have also been used to entrap ARV drugs to improve their PK profiles. Compared with free EFV, EFV-loaded lactoferrin nanoparticles (lacto-EFV-nano) exhibited improved oral bioavailability and improved PK, with a > 3–4-fold increase in the area under the plasma concentration-time curve (AUC), a > 30% increase in the peak plasma concentration of the drug after oral administration (Cmax), and a 2-fold increase in the time to reach Cmax (tmax) and t1/2 [113]. In another study, using methacrylate co-polymer (Eudragit-E100) to engineer nanoparticles of EFV, EFV bioavailability and biodistribution were improved, which resulted in higher drug concentrations in serum and major organs, especially in the brain, compared to the free drug formulation [114].
Although these oral nanoformulations could prolong drug half-lives and increase drug efficacy to some extent, they could not solve the non-adherence problem. People also must take pills daily, or once every two days (which could be even more difficult to adhere to). Long-acting formulations allow sustained release of drug from the drug-carrier complexes at the site of injection for a month or longer. The recently developed long-acting HIV nanoformulations listed in Table 3 all could provide effective plasma drug levels. Long-acting drug delivery systems should not only allow drug to slowly release from the matrices deposited at the site of injection to provide plasma drug concentrations within a therapeutically useful range for an extended time, but they should also enhance intracellular drug exposure in tissues and cells with residual HIV and enhance intracellular drug delivery in HIV host cells. Due to the difficulty in investigation of the targeting ability of nanoformulation to HIV host cells in animal models, most works have been investigated in in vitro cell models.
4.4.2. Extended intracellular drug levels
4.4.2.1. CD4+ T cells
Cellular reservoirs of HIV include latently infected CD4+ T cells, macrophages, and follicular dendritic cells (FDCs). The most prominent and well-characterized of these cellular reservoirs are latently infected CD4+ T-cells. Ramana et al. developed stealth anti-CD4 immunoliposomes containing two ARV drugs (NVP and SQV) that selectively home to HIV infected cells through the CD4 receptor [115]. The drugs delivered via these immunoliposomes inhibited viral proliferation at a significantly lower concentration compared to free drugs. Endsley et al. designed and characterized two peptides selective for HIV host cells expressing CD4 [116]. Using these peptides to modify lipid nanoparticles could provide selective binding and efficient delivery of IDV to CD4-HIV host cells and enhance anti-HIV efficacy of IDV [117]. Kovochich et al. used lipid nanoparticles incorporated with the protein kinase C activator bryostatin-2 (LNP-Bry) to target and activate primary human CD4+ T-cells. The histone deacetylase inhibitor sodium butyrate synergistically enhanced this activation. LNP-Bry could also be loaded with the PI nelfinavir (NFV) to both activate latent virus and inhibit viral spread [118].
4.4.2.2. Macrophages
Macrophages are another type of immune cell that serve as a portal of entry and viral deposit for HIV. AuNPs distribute to diverse organs upon intravenous injection in mice, specifically the liver and spleen, and large portions are internalized by local macrophages [119]. Thus, AuNPs-based nanocarriers were used for targeted delivery of ARV drugs to human primary macrophages in the bloodstream [90]. Gallium, in its nitrate salt form [Ga(NO3)3], is an FDA-approved drug. It is a metal with many similarities to iron. Because many Fe binding proteins are unable to distinguish Ga3+ from Fe3+, the Fe-dependent pathways in a virus could be potentially disrupted using Ga, leading to virus inhibition. Narayanasamy et al. used p40725 polymer to prepare Ga tetraphenyl porphyrin nanoparticles by homogenization and found that the nanoformulation could target macrophages and inhibit growth of both HIV and Mycobacterium tuberculosis (TB) [120]. As mentioned, the Gendelman group has developed NanoART— Poloxamer 188-coated nanocrystals containing the PI ATV. They have demonstrated a nearly exclusive drug depot in macrophages within the liver and spleen following both intraperitoneal and intramuscular injection [121]. Using Poloxamer 407 (P407) to coat ATV nanocrystals they achieved about 1,000-fold higher drug concentrations in cells than those with unformulated drug [122].
As the folate receptor is expressed on macrophages, coating nanomaterials with folic acid (FA) has been an effective means to facilitate particle uptake. Puligujja et al. used FA-modified P407 to prepare FA-modified nanoART and increase cellular drug uptake. Folate coating of nanoART containing ritonavir-boosted atazanavir (ATV/r) demonstrated enhanced cell uptake, retention, and antiretroviral activities without altering cell viability [123, 124]. Folate-coated nanoART enhanced retention in recycling endosomes, which could provide a stable subcellular long-acting drug depot [125]. Similarly, using folate-modified P407 to encapsulate myristoylated abacavir (MABC) prodrug, colocalization of abacavir within all subcellular endosomal compartments was significantly greater compared to non-folate-modified nanoparticles [126].
4.4.3. Extended tissue drug levels
4.4.3.1. Central nervous system
HIV-1 can cross the BBB and enter the CNS at an early stage of infection. This process is known to be mediated via mononuclear phagocytes (i.e., monocytes and blood-borne macrophages) [127]. It is possible that HIV-1-infected brain cells can acquire latency and escape the immune response. Upon appropriate endogenous or exogenous stimulus, these latent cells could produce fresh infectious virions, and thus the CNS could act as a reservoir site for HIV persistence. The chronic presence of HIV in the CNS can contribute to the development of neurological disorders, classified as HIV-1-associated neurocognitive disorder (HAND) [128]. Nearly half of all people infected with HIV show some form of neurological dysfunction. Although current cART drugs can suppress HIV in the peripheral blood, they fail to inhibit viral load in the brain because the BBB can restrict CNS delivery of these drugs. The presence of tight endothelial cell junctions and the expression of efflux transporters such as P-gp in the BBB inhibit the transport of ARV drugs to the brain.
Thus, there is clearly a need to develop new strategies to improve drug delivery across the brain, activate latent HIV in the CNS, and eliminate this HIV reservoir without hampering the integrity of the BBB or CNS. Some drugs are intrinsically more permeable to the BBB than others. For example, at steady-state conditions, plasma-to-cerebrospinal fluid (representing brain levels) of AZT is 1, while this value is much lower for most PIs such as ATV, NFV and IDV, which are substrates of P-gp efflux transporter [129]. The use of nanoparticles for drug delivery across the BBB to the brain may enhance CNS delivery by increasing the local drug gradient at the BBB through passive targeting (by way of long circulation times that enable persistent drug levels in the blood), thus allowing drug trafficking by nonspecific or receptor-mediated endocytosis, and bypassing drug efflux transporters at the BBB [130].
SLNs have been shown to effectively target the CNS through endocytosis due to their lipophilic nature and small size. The single-drug SLN formulations of ATV enhanced drug transport across the endothelial hCMEC/D3 cell monolayer compared to drug solution [131, 132]. In addition, other kinds of nanoparticles may bypass P-gp. For instance, pluronic block copolymers protect the entrapped drug from P-gp efflux and increase the brain’s drug exposure—in a study, Pluronic P85 inhibited the interaction of P-gp with NFV and SQV [133].
Nanoparticles could also be engineered to target the CNS by surface-coupling specific molecules or encapsulating particles in special cellular vehicles. Such engineered nanoparticles could have the ability to overcome the BBB and eventually reach HIV-reservoir sites. One of the most widely studied proteins for targeted CNS drug delivery is transferrin (Tf), which binds to the Tf receptor—which is expressed in the luminal membrane of the capillary endothelium of the Tf-modified nanoparticles could selectively transport drug cargo across the BBB following Tf receptor binding. Tf-coupled submicron lipid emulsions (SLEs) containing IDV demonstrated effective brain delivery by Tf receptor-mediated transcytosis [134]. As the vehicle for virus carriage into the nervous system, macrophages could also be harnessed as an ARV drug carrier. Loading IDV nanoparticles into murine bone marrow macrophages brought about a measurable reduction in the brain viral load in HIV-infected mice [95].
Lastly, magnetic nanoparticles have been directed to deliver drug to the brain through applying an external magnetic force. Jayant et al. developed a layer-by-layer assembly of an anti-HIV drug (TFV) and a latency-breaking agent (vorinostat) on ultrasmall magnetic nanoparticles (MNPs) to achieve effective penetration across the BBB with an external magnetic force [135]. Fiandra et al. also demonstrated the ability of iron oxide nanoparticles coated with poly(methacrylic acid) amphiphilic polymer to enhance the permeation of the high molecular weight molecule enfuvirtide (T-20) across the BBB both in vitro and in vivo [136].
4.4.3.2. Lymph nodes
Viral replication occurs predominantly in peripheral lymphoid organs, and lymph nodes in the gut and throughout the body are key tissues that are infected by HIV-1 during acute and chronic infection [9]. They are sites of viral production, storage of viral particles in immune complexes, and viral persistence. Patients on cART with undetectable levels of virus in their bloodstream still showed virus evolution and trafficking between blood and lymph node tissue compartments [10]. In HIV patients taking cART, virus can be isolated from their lymph nodes. In 2003, by isolation of the peripheral blood [12]. contributions of each source of residual virus remain to be elucidated, it is clear that addressing drug-insufficiency could help reduce the load of residual virus, and enable novel clinical strategies in finding a cure for AIDS. To improve lymphatic HIV drug exposure and eliminate HIV drug insufficiency and disease progression, we pioneered lymph node-targeted HIV drug delivery systems. While the exact physiological mechanisms leading to targeted delivery of drug combinations in cells for an extended time remains to be elucidated in detail, we found that DcNPs are first taken up into lymph vessels from the subucutaneous space, and subsequently undergo lymphatic first-pass distribution and retention throughout the channels of lymph vessel and node network in the body; this retention of drug in lymph vessels and nodes throughout the body is key to the lymphocyte-targeting and long-acting properties of DcNPs. More detailed discussions of a working hypothesis can be found in these recent papers [61, 137, 138]. Reviews by our group describe some important results obtained with lipid nanoparticles for drug delivery to lymph nodes and have further discussion on relevant targeting mechanisms [139–141].
We initially developed 50–90 nm IDV-lipid particles based on the ability of lipid to stabilize water-insoluble IDV at neutral pH. When IDV particles were given subcutaneously to primates, they resulted in greatly enhanced drug concentrations in lymph nodes throughout the body compared to free IDV given via the traditional oral route [12, 14]. Guided by our work with lipid-associated IDV, we further developed drug combination particles containing three drugs—LPV, RTV, and TFV—based on the consideration of increasing the clinical efficacy and reducing the risk of harboring HIV drug-resistant virus. Primates treated with this lipid-associated drug combination had significantly higher drug concentrations both in plasma and intracellularly in lymph node and blood mononuclear cells at 24 h after administration, while only low or undetectable drug levels were found in primates treated with a SC free drug formulation of all three compounds [73]. These lipid nanoparticles extended intracellular drug exposure in lymph nodes to over 7 days in primates after a single SC dose [72]. More recently, we developed TLC-ART101, and its lymph node targeting effects were verified in primates. LPV and TFV drug levels in LNMCs were up to 79-fold higher than those in PBMCs, which were in turn higher than the drug levels in plasma [61]. A single dose of this lipid suspension sustained plasma and PBMC intracellular drug levels for over 2 weeks. By comparison, after oral administration of LPV and RTV [142], or injection of TFV [143], these drugs clear from the body within 24–48 h. Conventional oral dosage forms of these drugs would require 14 consecutive doses over 2 weeks to achieve similar plasma levels, and intracellular drug levels would be less than those in plasma [144–147]. Thus, this nanoparticle platform holds promise as a targeted, long-acting, drug combination HIV treatment that may improve therapeutic outcomes.
5. Perspectives and future directions
Those involved in charting the future evolution of HIV therapeutics should focus on making HIV care more streamlined, client-centered, and effective at clearing HIV from the body. Although the development of HIV nanotherapeutics has the potential to address the limitations of current oral cART, a significant number of challenges remain. Broadly, these challenges can be categorized with respect to pharmaceutics (formulation, drug combinations, scalability, usability), PK (absorption, disposition, safety), and pharmacodynamics (efficacy against disease). At present, most long-acting anti-HIV strategies are focused on optimizing the pharmaceutic and PK aspects in preclinical models; only CAB LA and RPV LA have recently advanced to clinical investigations to assess their pharmacodynamics in humans [107]. However, their actual utility in the clinic and acceptance by patients are being determined in broader populations affected by HIV. Few of the long-acting nano-formulation strategies for HIV chemotherapies proposed thus far include drug combinations or have the ability to accommodate multiple different drug combinations with varying degrees of physicochemical properties. Few also efficiently target drug combinations to HIV host cells throughout the body. As the greater efficacy of cART compared to monotherapy is clear, successfully formulating drug combinations is essential. Without the additional targeting of drug combinations to sites of HIV replication and HIV host cells to address pharmacological deficiencies such as lymphatic drug insufficiency, long-acting strategies may merely help with the adherence challenges of oral cART without addressing the poor penetration of cART drugs into tissues and cells that harbor residual HIV to boost the efficacy of clearing HIV.
As mentioned, cabotegravir (CAB) LA and rilpivirine (RPV) LA, each administered as separate 2 to 3 mL intramuscular injections, both take several days to reach maximum plasma concentrations (median tmax: 9–69 days [18] or 6–11 days [148]). Because of this, an oral lead-in of these drugs prior to starting the injectable regimen is needed to rapidly provide effective plasma levels in order to prevent the development of viral resistance from subtherapeutic concentrations and to ensure patients can tolerate these drugs [107]. This slow rise to peak plasma drug levels suggests the absorption of these drugs into the plasma occurs primarily from sustained release of free drug as a result of the slow dissolution of hydrophobic CAB and RPV nanocrystals in the intramuscular depot, with limited lymphatic uptake, distribution, and retention of the drug-nanocrystals. Free CAB or RPV released into the extracellular fluid from the intramuscular depot is taken up immediately into the rapidly-flowing blood capillaries, bypassing an opportunity to instead be taken up predominantly by lymphatic capillaries from the intramuscular space, distribute through the lymphatics and accumulate in lymph nodes to produce long-acting PK in both the plasma and lymphocytes. This dissolution and absorption behavior of CAB LA and RPV LA is in contrast to drug combination nanoparticle-associated antiretrovirals in TLC-ART101 that cleared readily form the SC injection site, were predominantly taken into lymph capillaries instead of the blood, distributed through lymph vessels to lymph nodes throughout the body, reached peak plasma levels in non-human primates at ~6 to ~8 hours after injection, and had higher drug levels in lymph node and peripheral blood mononuclear cells than in the plasma [61, 137, 138]. A recent MRI study of CAB LA in rats confirmed that both intramuscular and SC depots did not readily clear from the injection site and did not distribute to and accumulate in multiple lymph nodes throughout the rat; moreover, consistent with human data, peak plasma drug levels of these drugs after both routes of injection were only achieved after multiple days and had similar shapes in their plasma absorption curves, indicating similar pathways of absorption (from the injection site directly to the blood) but slightly different rates and extents of absorption (likely due to physiological and dissolution rate differences between the two injection sites) [149]. Furthermore, the intramuscular injection that is being used in humans for CAB LA and RPV LA is associated with significant injection site pain and discomfort [107]. This could be intolerable if these injections are needed throughout a patient’s life. Thus, patients will inevitably have to make a trade-off between the convenience of not having to adhere to daily oral therapy (which can be dispensed anywhere from a 2 to 6 months’ supply of cART at a time) and the inconvenience of this injection discomfort and having to seek health care to be injected on a monthly basis for life.
Due to these limitations, there is still much room for innovations to be made in long-acting anti-HIV nanomedicines. Single, all-in-one, simple, scalable, and user-friendly drug delivery platforms should be established, such as TLC-ART101 [61], that enable targeted long-acting delivery of multiple different drug combinations to plasma and HIV host cells throughout the body. Once established, the human PK (not only plasma, but also tissue distribution and intracellular drug levels over time) of these formulations must be understood in detail. The formulation- and physiological-related mechanisms that drive the long-acting PK should be elucidated (we recently reported on such mechanisms for TLC-ART101 [137]). Finally, the non-inferiority of long-acting cART approaches versus conventional daily oral cART in non-human primate models and humans should be investigated. Currently, there is a debate underway among HIV researchers between the presence [9, 10, 13] and absence [150, 151] of ongoing HIV replication in anatomical sites of residual HIV such as lymphoid tissues in patients who have adhered to long-term cART and have no plasma viremia. In addition, exploration of residual virus in specific locations of key tissues such as (gut, rectal, or other visceral or peripheral) lymph nodes have just begun [9]. Settling this debate through rigorous studies that accurately test both hypotheses head-to-head would benefit the HIV field. It would allow efforts on treatment strategies to focus on the detailed HIV biology that takes place in patients adherent with cART. This could be a major step that is needed for the field to make significant progress toward realizing a cure for HIV/AIDS.
Overall, nanotechnology for ART is still in its early stage of clinical translation. We have been fortunate to experience AIDS changing from a rapidly progressing and fatal illness in the 1980s to a chronic and manageable disease, thanks in large part to the advent of cART and social, political, and economic efforts on the individual and organizational levels (e.g., academic, industrial, public, and non-governmental sectors). Fine details on residual virus sites and their role in rapid virus rebound is just emerging. The next step is to focus our sights on effectively clearing HIV from the residual site in the body (starting with validated sites) using a combination of targeted drug localization and immunotherapeutics. Targeted long-acting drug combination nanomedicines are aimed directly at this goal. Although numerous benefits have been achieved pre-clinically for ART nanomedicines, the scale-up and the cost of large-scale synthesis of these nanoparticle platforms remain as significant challenges. In addition to an expanded understanding of viral-host distribution and pathology, a more detailed understanding of the nanoformulations—including their safety, PK, and efficacy profiles; long-term toxicity; and unwarranted immune responses—needs to be assessed before the true impact of this research on the prevention and treatment of HIV infection can be measured.
6. Conclusion
Despite cART oral drug regimens transforming HIV from a terminal illness to a chronic disease and improving the quality of life for HIV patients, residual replication-competent viruses remain in tissues and leads to viral rebound in the plasma if treatment is stopped. Current oral cART regimens have limited penetration and retention in lymph nodes, require life-long daily administration, and have patient adherence challenges due to pill fatigue and side-effects. The disconnect between drug and virus distribution in the body could be leveraged to enhance viral clearance in tissues that harbor residual HIV. Some progress has been made in the development of nanoparticle-based drug delivery systems for HIV therapy to enable targeted drug delivery, or to enable long-acting PK in the plasma; however, few have been able to integrate these two concepts into a simple and scalable injectable formulation that contains drug combinations. Using the subcutaneous route of dosing and a unique formulation intended to enhance drug concentrations and exposure in tissues that harbor residual HIV, we have developed drug-combination nanoparticles composed of three HIV drugs— LPV, RTV, and TFV—that have been shown to provide sustained drug levels in lymph nodes for over two weeks, with elevated drug-combination levels in HIV-host cells found in the blood and nodes. This new strategy could help facilitate development of long-acting, drug-combination and targeted nanomedicines for overcoming lymphoid tissue drug insufficiency and clearing the residual HIV in lymphoid tissues throughout the body.
Acknowledgements:
We thank members of the University of Washington Targeted Long-Acting Combination Anti-Retroviral Therapy (TLC-ART) Program who have contributed helpful insights during discussions.
Funding:
This work was supported by the National Institutes of Health (grant numbers UM1 AI120176, T32-GM007750, TL1–1TR002318–01), National Natural Science Foundation of China (81571802), Natural Science Foundation of Fujian Province (2016J06020), the Fujian Provincial Youth Top-notch Talent Support Program, the China Scholarship Council (201706655015), and the Fund of the University Overseas Training Program of Jiangsu Province.
Footnotes
Declarations of Interest: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
7 References
- 1.Centers for Disease C, Kaposi’s sarcoma and Pneumocystis pneumonia among homosexual men--New York City and California. MMWR Morb Mortal Wkly Rep, 1981. 30(25): p. 305–8. [PubMed] [Google Scholar]
- 2.Hymes KB, et al. , Kaposi’s sarcoma in homosexual men-a report of eight cases. Lancet, 1981. 2(8247): p. 598–600. [DOI] [PubMed] [Google Scholar]
- 3.UN AIDS Data 2017. http://www.unaids.org/sites/default/files/media_asset/20170720_Data_book_2017_en.pdf, July 2017; Accessed December 12, 2017.
- 4.Pau AK and George JM, Antiretroviral therapy: current drugs. Infect Dis Clin North Am, 2014. 28(3): p. 371–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gunthard HF, et al. , Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2016 Recommendations of the International Antiviral Society-USA Panel. JAMA, 2016. 316(2): p. 191–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richman DD, et al. , The challenge of finding a cure for HIV infection. Science, 2009. 323(5919): p. 1304–7. [DOI] [PubMed] [Google Scholar]
- 7.Cihlar T and Fordyce M, Current status and prospects of HIV treatment. Curr Opin Virol, 2016. 18: p. 50–6. [DOI] [PubMed] [Google Scholar]
- 8.Honeycutt JB, et al. , HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nature Medicine, 2017. 23(5): p. 638–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Estes JD, et al. , Defining total-body AIDS-virus burden with implications for curative strategies. Nat Med, 2017. 23(11): p. 1271–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lorenzo-Redondo R, et al. , Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature, 2016. 530(7588): p. 51–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee S, et al. Integrase and protease inhibitor concentrations in lymph node and gut mucosal tissue (Abstract #407). in Conference on Retroviruses and Opportunistic Infections (CROI) 2017. Seattle, WA. [Google Scholar]
- 12.Kinman L, et al. , Lipid-drug association enhanced HIV-1 protease inhibitor indinavir localization in lymphoid tissues and viral load reduction: a proof of concept study in HIV-2287-infected macaques. J Acquir Immune Defic Syndr, 2003. 34(4): p. 387–97. [DOI] [PubMed] [Google Scholar]
- 13.Fletcher CV, et al. , Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc Natl Acad Sci U S A, 2014. 111(6): p. 2307–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kinman L, et al. , Optimization of lipid-indinavir complexes for localization in lymphoid tissues of HIV-infected macaques. J Acquir Immune Defic Syndr, 2006. 42(2): p. 155–61. [DOI] [PubMed] [Google Scholar]
- 15.Bell IR, et al. , Advances in Integrative Nanomedicine for Improving Infectious Disease Treatment in Public Health. Eur J Integr Med, 2013. 5(2): p. 126–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi J, et al. , Nanotechnology in drug delivery and tissue engineering: from discovery to applications. Nano Lett, 2010. 10(9): p. 3223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siccardi M, et al. , Nanomedicines for HIV therapy. Ther Deliv, 2013. 4(2): p. 153–6. [DOI] [PubMed] [Google Scholar]
- 18.Spreen W, et al. , GSK1265744 pharmacokinetics in plasma and tissue after single-dose long-acting injectable administration in healthy subjects. J Acquir Immune Defic Syndr, 2014. 67(5): p. 481–6. [DOI] [PubMed] [Google Scholar]
- 19.Hoogbruin A, Complementary and alternative therapy (CAT) use and highly active antiretroviral therapy (HAART): current evidence in the literature, 2000–2009. J Clin Nurs, 2011. 20(7–8): p. 925–39. [DOI] [PubMed] [Google Scholar]
- 20.Chun TW, et al. , Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A, 1997. 94(24): p. 13193–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finzi D, et al. , Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science, 1997. 278(5341): p. 1295–300. [DOI] [PubMed] [Google Scholar]
- 22.Wong JK, et al. , Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science, 1997. 278(5341): p. 1291–5. [DOI] [PubMed] [Google Scholar]
- 23.Kuo HH and Lichterfeld M, Recent progress in understanding HIV reservoirs. Curr Opin HIV AIDS, 2018. 13(2): p. 137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Descours B, et al. , CD32a is a marker of a CD4 T-cell HIV reservoir harbouring replication-competent proviruses. Nature, 2017. 543(7646): p. 564–567. [DOI] [PubMed] [Google Scholar]
- 25.Rasmussen TA, et al. , Eliminating the latent HIV reservoir by reactivation strategies: advancing to clinical trials. Hum Vaccin Immunother, 2013. 9(4): p. 790–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaminski R, et al. , Elimination of HIV-1 Genomes from Human T-lymphoid Cells by CRISPR/Cas9 Gene Editing. Sci Rep, 2016. 6: p. 22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qu X, et al. , Zinc-finger-nucleases mediate specific and efficient excision of HIV-1 proviral DNA from infected and latently infected human T cells. Nucleic Acids Res, 2013. 41(16): p. 7771–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tebas P, et al. , Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med, 2014. 370(10): p. 901–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hale M, et al. , Engineering HIV-Resistant, Anti-HIV Chimeric Antigen Receptor T Cells. Mol Ther, 2017. 25(3): p. 570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friedman J, et al. , Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J Virol, 2011. 85(17): p. 9078–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pearson R, et al. , Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J Virol, 2008. 82(24): p. 12291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quivy V, et al. , Synergistic activation of human immunodeficiency virus type 1 promoter activity by NF-kappaB and inhibitors of deacetylases: potential perspectives for the development of therapeutic strategies. J Virol, 2002. 76(21): p. 11091–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shan L, et al. , Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity, 2012. 36(3): p. 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huyghe J, Magdalena S, and Vandekerckhove L, Fight fire with fire: Gene therapy strategies to cure HIV. Expert Rev Anti Infect Ther, 2017. 15(8): p. 747–758. [DOI] [PubMed] [Google Scholar]
- 35.Chupradit K, et al. , Current Peptide and Protein Candidates Challenging HIV Therapy beyond the Vaccine Era. Viruses, 2017. 9(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahmed Y, Tian M, and Gao Y, Development of an anti-HIV vaccine eliciting broadly neutralizing antibodies. AIDS Res Ther, 2017. 14(1): p. 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.das Neves J, et al. , Nanomedicine in the development of anti-HIV microbicides. Adv Drug Deliv Rev, 2016. 103: p. 57–75. [DOI] [PubMed] [Google Scholar]
- 38.Andrews CD and Heneine W, Cabotegravir long-acting for HIV-1 prevention. Curr Opin HIV AIDS, 2015. 10(4): p. 258–63. [DOI] [PubMed] [Google Scholar]
- 39.Landovitz RJ, Kofron R, and McCauley M, The promise and pitfalls of long-acting injectable agents for HIV prevention. Curr Opin HIV AIDS, 2016. 11(1): p. 122–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andrews CD, et al. , A long-acting integrase inhibitor protects female macaques from repeated high-dose intravaginal SHIV challenge. Sci Transl Med, 2015. 7(270): p. 270ra4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grant RM, et al. , Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med, 2010. 363(27): p. 2587–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peters PJ, et al. , HIV Infection Linked to Injection Use of Oxymorphone in Indiana, 2014–2015. N Engl J Med, 2016. 375(3): p. 229–39. [DOI] [PubMed] [Google Scholar]
- 43.Sarmati L, et al. , HIV Replication at Low Copy Number and its Correlation with the HIV Reservoir: A Clinical Perspective. Current Hiv Research, 2015. 13(3): p. 250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Calin R, et al. , Treatment interruption in chronically HIV-infected patients with an ultralow HIV reservoir. AIDS, 2016. 30(5): p. 761–9. [DOI] [PubMed] [Google Scholar]
- 45.Chun TW, et al. , Re-emergence of HIV after stopping therapy. Nature, 1999. 401(6756): p. 874–5. [DOI] [PubMed] [Google Scholar]
- 46.de Jong MD, et al. , Overshoot of HIV-1 viraemia after early discontinuation of antiretroviral treatment. AIDS, 1997. 11(11): p. F79–84. [DOI] [PubMed] [Google Scholar]
- 47.Kirtane AR, et al. , Development of an oral once-weekly drug delivery system for HIV antiretroviral therapy. Nat Commun, 2018. 9(1): p. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nair M, et al. , Getting into the brain: Potential of nanotechnology in the management of NeuroAIDS. Advanced Drug Delivery Reviews, 2016. 103: p. 202–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryscavage PA, et al. , Clinical Outcomes of Adolescents and Young Adults in Adult HIV Care. Jaids-Journal of Acquired Immune Deficiency Syndromes, 2011. 58(2): p. 193–197. [DOI] [PubMed] [Google Scholar]
- 50.Schlatter AF, Deathe AR, and Vreeman RC, The Need for Pediatric Formulations to Treat Children with HIV. AIDS Res Treat, 2016. 2016: p. 1654938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abers MS, Shandera WX, and Kass JS, Neurological and Psychiatric Adverse Effects of Antiretroviral Drugs. Cns Drugs, 2014. 28(2): p. 131–145. [DOI] [PubMed] [Google Scholar]
- 52.Lykins WR, et al. , Long acting systemic HIV pre-exposure prophylaxis: an examination of the field. Drug Deliv Transl Res, 2017. 7(6): p. 805–816. [DOI] [PubMed] [Google Scholar]
- 53.Dolgin E, Long-acting HIV drugs advanced to overcome adherence challenge. Nature Medicine, 2014. 20(4): p. 323–324. [DOI] [PubMed] [Google Scholar]
- 54.Margolis DA and Boffito M, Long-acting antiviral agents for HIV treatment. Current Opinion in Hiv and Aids, 2015. 10(4): p. 246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.HIV and Injection Drug Use (CDC HIV IDU Fact Sheet). https://www.cdc.gov/hiv/pdf/risk/cdc-hiv-idu-fact-sheet.pdf, November 2016; Accessed Dec 19, 2017.
- 56.New data show continuing opioid epidemic in the United States. Press release of the Centers for Disease Control and Prevention Newsroom, https://www.cdc.gov/media/releases/2016/p1216-continuing-opioid-epidemic.html, December 16, 2016; Accessed Dec 19, 2017. [Google Scholar]
- 57.Haffajee RL and Mello MM, Drug Companies’ Liability for the Opioid Epidemic. N Engl J Med, 2017. 377(24): p. 2301–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.NIH Future Directions for HIV Treatment Research https://www.niaid.nih.gov/diseases-conditions/future-hiv-treatment, Accessed Dec 19, 2017.
- 59.Trezza C, et al. , Formulation and pharmacology of long-acting cabotegravir. Curr Opin HIV AIDS, 2015. 10(4): p. 239–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Williams PE, Crauwels HM, and Basstanie ED, Formulation and pharmacology of long-acting rilpivirine. Curr Opin HIV AIDS, 2015. 10(4): p. 233–8. [DOI] [PubMed] [Google Scholar]
- 61.Kraft JC, et al. , Long-acting combination anti-HIV drug suspension enhances and sustains higher drug levels in lymph node cells than in blood cells and plasma. AIDS, 2017. 31(6): p. 765–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Min Y, et al. , Clinical Translation of Nanomedicine. Chem Rev, 2015. 115(19): p. 11147–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gupta U and Jain NK, Non-polymeric nano-carriers in HIV/AIDS drug delivery and targeting. Adv Drug Deliv Rev, 2010. 62(4–5): p. 478–90. [DOI] [PubMed] [Google Scholar]
- 64.Mahajan SD, et al. , Anti-HIV-1 nanotherapeutics: promises and challenges for the future. Int J Nanomedicine, 2012. 7: p. 5301–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Owen A and Rannard S, Strengths, weaknesses, opportunities and challenges for long acting injectable therapies: Insights for applications in HIV therapy. Adv Drug Deliv Rev, 2016. 103: p. 144–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khalil NM, et al. , Potential of polymeric nanoparticles in AIDS treatment and prevention. Expert Opin Drug Deliv, 2011. 8(1): p. 95–112. [DOI] [PubMed] [Google Scholar]
- 67.Nelson AG, et al. , Drug delivery strategies and systems for HIV/AIDS pre-exposure prophylaxis and treatment. J Control Release, 2015. 219: p. 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chopra S, Venkatesan N, and Betageri GV, Liposomes as nanocarriers for anti-HIV therapy. Drug Delivery and Translational Research, 2013. 3(5): p. 471–478. [DOI] [PubMed] [Google Scholar]
- 69.Pollock S, et al. , N-Butyldeoxynojirimycin is a broadly effective anti-HIV therapy significantly enhanced by targeted liposome delivery. Aids, 2008. 22(15): p. 1961–1969. [DOI] [PubMed] [Google Scholar]
- 70.Nayak D, et al. , Stavudine loaded gelatin liposomes for HIV therapy: Preparation, characterization and in vitro cytotoxic evaluation. Mater Sci Eng C Mater Biol Appl, 2017. 73: p. 406–416. [DOI] [PubMed] [Google Scholar]
- 71.Kraft JC, et al. , Emerging Research and Clinical Development Trends of Liposome and Lipid Nanoparticle Drug Delivery Systems. Journal of Pharmaceutical Sciences, 2014. 103(1): p. 29–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Duan J, et al. , Evaluation of atazanavir and darunavir interactions with lipids for developing pH-responsive anti-HIV drug combination nanoparticles. J Pharm Sci, 2014. 103(8): p. 2520–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Freeling JP, et al. , Long-acting three-drug combination anti-HIV nanoparticles enhance drug exposure in primate plasma and cells within lymph nodes and blood. Aids, 2014. 28(17): p. 2625–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Naseri N, Valizadeh H, and Zakeri-Milani P, Solid Lipid Nanoparticles and Nanostructured Lipid Carriers: Structure, Preparation and Application. Advanced Pharmaceutical Bulletin, 2015. 5(3): p. 305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Javan F, et al. , Encapsulation of ritonavir in solid lipid nanoparticles: in-vitro anti-HIV-1 activity using lentiviral particles. Journal of Pharmacy and Pharmacology, 2017. 69(8): p. 1002–1009. [DOI] [PubMed] [Google Scholar]
- 76.K SJ, et al. , Gelatin modified lipid nanoparticles for anti-viral drug delivery. Chem Phys Lipids, 2017. 207(Pt A): p. 24–37. [DOI] [PubMed] [Google Scholar]
- 77.Dubey V, et al. , Enhanced transdermal delivery of an anti-HIV agent via ethanolic liposomes. Nanomedicine, 2010. 6(4): p. 590–6. [DOI] [PubMed] [Google Scholar]
- 78.Chu GJ and Murad A, A case of ethanol-induced systemic allergic dermatitis. Contact Dermatitis, 2017. 76(3): p. 182–184. [DOI] [PubMed] [Google Scholar]
- 79.Danial M, et al. , Triple Activity of Lamivudine Releasing Sulfonated Polymers against HIV-1. Molecular Pharmaceutics, 2016. 13(7): p. 2397–2410. [DOI] [PubMed] [Google Scholar]
- 80.Yang L, et al. , Synthesis, nanosizing and in vitro drug release of a novel anti-HIV polymeric prodrug: chitosan-O-isopropyl-5’-O-d4T monophosphate conjugate. Bioorg Med Chem, 2010. 18(1): p. 117–23. [DOI] [PubMed] [Google Scholar]
- 81.Jiang Y, et al. , Nanoparticle-Based ARV Drug Combinations for Synergistic Inhibition of Cell-Free and Cell-Cell HIV Transmission. Molecular Pharmaceutics, 2015. 12(12): p. 4363–4374. [DOI] [PubMed] [Google Scholar]
- 82.Giardiello M, et al. , Accelerated oral nanomedicine discovery from miniaturized screening to clinical production exemplified by paediatric HIV nanotherapies. Nat Commun, 2016. 7: p. 13184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kumar P, Lakshmi YS, and Kondapi AK, Triple Drug Combination of Zidovudine, Efavirenz and Lamivudine Loaded Lactoferrin Nanoparticles: an Effective Nano First-Line Regimen for HIV Therapy. Pharmaceutical Research, 2017. 34(2): p. 257–268. [DOI] [PubMed] [Google Scholar]
- 84.Kumar P, et al. , Improved Safety, Bioavailability and Pharmacokinetics of Zidovudine through Lactoferrin Nanoparticles during Oral Administration in Rats. Plos One, 2015. 10(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Anselmo AC and Mitragotri S, A Review of Clinical Translation of Inorganic Nanoparticles. AAPS J, 2015. 17(5): p. 1041–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Saiyed ZM, Gandhi NH, and Nair MPN, AZT 5 ‘-triphosphate nanoformulation suppresses human immunodeficiency virus type 1 replication in peripheral blood mononuclear cells. Journal of Neurovirology, 2009. 15(4): p. 343–347. [DOI] [PubMed] [Google Scholar]
- 87.Guo DW, et al. , Small magnetite antiretroviral therapeutic nanoparticle probes for MRI of drug biodistribution. Nanomedicine, 2014. 9(9): p. 1341–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Beer AJ, et al. , Visualization of antigen-specific human cytotoxic T lymphocytes labeled with superparamagnetic iron-oxide particles. European Radiology, 2008. 18(6): p. 1087–1095. [DOI] [PubMed] [Google Scholar]
- 89.Williams JP, et al. , Application of magnetic field hyperthermia and superparamagnetic iron oxide nanoparticles to HIV-1-specific T-cell cytotoxicity. Int J Nanomedicine, 2013. 8: p. 2543–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zazo H, et al. , Gold Nanocarriers for Macrophage-Targeted Therapy of Human Immunodeficiency Virus. Macromol Biosci, 2017. 17(3). [DOI] [PubMed] [Google Scholar]
- 91.Garrido C, et al. , Gold nanoparticles to improve HIV drug delivery. Future Medicinal Chemistry, 2015. 7(9): p. 1097–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lara HH, et al. , Use of silver nanoparticles increased inhibition of cell-associated HIV-1 infection by neutralizing antibodies developed against HIV-1 envelope proteins. Journal of Nanobiotechnology, 2011. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ardestani MS, et al. , Nanosilver based anionic linear globular dendrimer with a special significant antiretroviral activity. Journal of Materials Science-Materials in Medicine, 2015. 26(5). [DOI] [PubMed] [Google Scholar]
- 94.Gutierrez Millan C., et al. , Cell-based drug-delivery platforms. Ther Deliv, 2012. 3(1): p. 25–41. [DOI] [PubMed] [Google Scholar]
- 95.Dou HY, et al. , Macrophage Delivery of Nanoformulated Antiretroviral Drug to the Brain in a Murine Model of NeuroAIDS. Journal of Immunology, 2009. 183(1): p. 661–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Martinez-Skinner AL, et al. , Functional Proteome of Macrophage Carried Nanoformulated Antiretroviral Therapy Demonstrates Enhanced Particle Carrying Capacity. Journal of Proteome Research, 2013. 12(5): p. 2282–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Carpenter CC, et al. , Antiretroviral therapy for HIV infection in 1996. Recommendations of an international panel. International AIDS Society-USA. JAMA, 1996. 276(2): p. 146–54. [PubMed] [Google Scholar]
- 98.Markowitz M, et al. , A randomized open-label study of 3- versus 5-drug combination antiretroviral therapy in newly HIV-1-infected individuals. J Acquir Immune Defic Syndr, 2014. 66(2): p. 140–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shafer RW, et al. , Comparison of four-drug regimens and pairs of sequential three-drug regimens as initial therapy for HIV-1 infection. N Engl J Med, 2003. 349(24): p. 2304–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shey MS, et al. , Co-formulated abacavir-lamivudine-zidovudine for initial treatment of HIV infection and AIDS. Cochrane Database of Systematic Reviews, 2013(3). [DOI] [PMC free article] [PubMed]
- 101.Imaz A and Podzamczer D, Tenofovir alafenamide, emtricitabine, elvitegravir, and cobicistat combination therapy for the treatment of HIV. Expert Review of Anti-Infective Therapy, 2017. 15(3): p. 195–209. [DOI] [PubMed] [Google Scholar]
- 102.Gautam N, et al. , Preclinical Pharmacokinetics and Tissue Distribution of Long-Acting Nanoformulated Antiretroviral Therapy. Antimicrobial Agents and Chemotherapy, 2013. 57(7): p. 3110–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li W, et al. , Co-delivery of HIV-1 entry inhibitor and nonnucleoside reverse transcriptase inhibitor shuttled by nanoparticles: cocktail therapeutic strategy for antiviral therapy. Aids, 2016. 30(6): p. 827–837. [DOI] [PubMed] [Google Scholar]
- 104.Jin YG, et al. , Combination Anti-HIV Therapy with the Self-Assemblies of an Asymmetric Bolaamphiphilic Zidovudine/Didanosine Prodrug. Molecular Pharmaceutics, 2011. 8(3): p. 867–876. [DOI] [PubMed] [Google Scholar]
- 105.Ogunwuyi O, et al. , Antiretroviral Drugs-Loaded Nanoparticles Fabricated by Dispersion Polymerization with Potential for HIV/AIDS Treatment. Infect Dis (Auckl), 2016. 9: p. 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Destache CJ, et al. , Combination antiretroviral drugs in PLGA nanoparticle for HIV-1. Bmc Infectious Diseases, 2009. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Margolis DA, et al. , Long-acting intramuscular cabotegravir and rilpivirine in adults with HIV-1 infection (LATTE-2): 96-week results of a randomised, open-label, phase 2b, non-inferiority trial. Lancet, 2017. 390(10101): p. 1499–1510. [DOI] [PubMed] [Google Scholar]
- 108.Jackson AG, et al. , A compartmental pharmacokinetic evaluation of long-acting rilpivirine in HIV-negative volunteers for pre-exposure prophylaxis. Clin Pharmacol Ther, 2014. 96(3): p. 314–23. [DOI] [PubMed] [Google Scholar]
- 109.Spreen W, et al. , GSK1265744 Pharmacokinetics in Plasma and Tissue After Single-Dose Long-Acting Injectable Administration in Healthy Subjects. Jaids-Journal of Acquired Immune Deficiency Syndromes, 2014. 67(5): p. 481–486. [DOI] [PubMed] [Google Scholar]
- 110.McMillan J, et al. , Pharmacokinetics of a Long-Acting Nanoformulated Dolutegravir Prodrug in Rhesus Macaques. Antimicrob Agents Chemother, 2018. 62(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ramana LN, et al. , Investigation on the stability of saquinavir loaded liposomes: implication on stealth, release characteristics and cytotoxicity. Int J Pharm, 2012. 431(1–2): p. 120–9. [DOI] [PubMed] [Google Scholar]
- 112.Skanji R, et al. , A new nanomedicine based on didanosine glycerolipidic prodrug enhances the long term accumulation of drug in a HIV sanctuary. International Journal of Pharmaceutics, 2011. 414(1–2): p. 285–297. [DOI] [PubMed] [Google Scholar]
- 113.Kumar P, Lakshmi YS, and Kondapi AK, An oral formulation of efavirenz-loaded lactoferrin nanoparticles with improved biodistribution and pharmacokinetic profile. HIV Med, 2017. 18(7): p. 452–462. [DOI] [PubMed] [Google Scholar]
- 114.Hari BNV, et al. , Engineered nanoparticles of Efavirenz using methacrylate co-polymer (Eudragit-E100) and its biological effects in-vivo. Materials Science & Engineering C-Materials for Biological Applications, 2016. 67: p. 522–532. [DOI] [PubMed] [Google Scholar]
- 115.Ramana LN, et al. , Stealth anti-CD4 conjugated immunoliposomes with dual antiretroviral drugs - Modern Trojan horses to combat HIV. European Journal of Pharmaceutics and Biopharmaceutics, 2015. 89: p. 300–311. [DOI] [PubMed] [Google Scholar]
- 116.Endsley AN and Ho RJ, Design and characterization of novel peptide-coated lipid nanoparticles for targeting anti-HIV drug to CD4 expressing cells. AAPS J, 2012. 14(2): p. 225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Endsley AN and Ho RJ, Enhanced anti-HIV efficacy of indinavir after inclusion in CD4-targeted lipid nanoparticles. J Acquir Immune Defic Syndr, 2012. 61(4): p. 417–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kovochich M, Marsden MD, and Zack JA, Activation of Latent HIV Using Drug-Loaded Nanoparticles. Plos One, 2011. 6(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bartneck M, et al. , Peptide-functionalized gold nanorods increase liver injury in hepatitis. ACS Nano, 2012. 6(10): p. 8767–77. [DOI] [PubMed] [Google Scholar]
- 120.Narayanasamy P, Switzer BL, and Britigan BE, Prolonged-acting, Multi-targeting Gallium Nanoparticles Potently Inhibit Growth of Both HIV and Mycobacteria in Co-Infected Human Macrophages. Scientific Reports, 2015. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Martinez-Skinner AL, et al. , Cellular Responses and Tissue Depots for Nanoformulated Antiretroviral Therapy. Plos One, 2015. 10(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Guo D, et al. , Endosomal trafficking of nanoformulated antiretroviral therapy facilitates drug particle carriage and HIV clearance. J Virol, 2014. 88(17): p. 9504–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Puligujja P, et al. , Pharmacodynamics of long-acting folic acid-receptor targeted ritonavir-boosted atazanavir nanoformulations. Biomaterials, 2015. 41: p. 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Puligujja P, et al. , Pharmacodynamics of folic acid receptor targeted antiretroviral nanotherapy in HIV-1-infected humanized mice. Antiviral Research, 2015. 120: p. 85–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Puligujja P, et al. , Macrophage folate receptor-targeted antiretroviral therapy facilitates drug entry, retention, antiretroviral activities and biodistribution for reduction of human immunodeficiency virus infections. Nanomedicine, 2013. 9(8): p. 1263–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Singh D, et al. , Development and characterization of a long-acting nanoformulated abacavir prodrug. Nanomedicine, 2016. 11(15): p. 1913–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gonzalez-Scarano F and Martin-Garcia J, The neuropathogenesis of AIDS. Nat Rev Immunol, 2005. 5(1): p. 69–81. [DOI] [PubMed] [Google Scholar]
- 128.Gray F, et al. , Neuropathology and neurodegeneration in human immunodeficiency virus infection - Pathogenesis of HIV-induced lesions of the brain, correlations with HIV-associated disorders and modifications according to treatments. Clinical Neuropathology, 2001. 20(4): p. 146–155. [PubMed] [Google Scholar]
- 129.Robillard KR, et al. , Role of P-glycoprotein in the distribution of the HIV protease inhibitor atazanavir in the brain and male genital tract. Antimicrob Agents Chemother, 2014. 58(3): p. 1713–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yang H, Nanoparticle-mediated brain-specific drug delivery, imaging, and diagnosis. Pharm Res, 2010. 27(9): p. 1759–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wong HL, Wu XY, and Bendayan R, Nanotechnological advances for the delivery of CNS therapeutics. Advanced Drug Delivery Reviews, 2012. 64(7): p. 686–700. [DOI] [PubMed] [Google Scholar]
- 132.Chattopadhyay N, et al. , Solid lipid nanoparticles enhance the delivery of the HIV protease inhibitor, atazanavir, by a human brain endothelial cell line. Pharm Res, 2008. 25(10): p. 2262–71. [DOI] [PubMed] [Google Scholar]
- 133.Shaik N, Pan G, and Elmquist WF, Interactions of Pluronic Block Copolymers on P-gp Efflux Activity: Experience With HIV-1 Protease Inhibitors. Journal of Pharmaceutical Sciences, 2008. 97(12): p. 5421–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Prabhakar K, et al. , Brain delivery of transferrin coupled indinavir submicron lipid emulsions-Pharmacokinetics and tissue distribution. Colloids and Surfaces B-Biointerfaces, 2011. 86(2): p. 305–313. [DOI] [PubMed] [Google Scholar]
- 135.Jayant RD, et al. , Sustained-release nanoART formulation for the treatment of neuroAIDS. Int J Nanomedicine, 2015. 10: p. 1077–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fiandra L, et al. , Nanoformulation of antiretroviral drugs enhances their penetration across the blood brain barrier in mice. Nanomedicine-Nanotechnology Biology and Medicine, 2015. 11(6): p. 1387–1397. [DOI] [PubMed] [Google Scholar]
- 137.Kraft JC, et al. , Mechanism-based pharmacokinetic (MBPK) models describe the complex plasma kinetics of three antiretrovirals delivered by a long-acting anti-HIV drug combination nanoparticle formulation. J Control Release, 2018. [DOI] [PMC free article] [PubMed]
- 138.Kraft JC, Treuting PM, and Ho RJY, Indocyanine green nanoparticles undergo selective lymphatic uptake, distribution and retention and enable detailed mapping of lymph vessels, nodes and abnormalities. J Drug Target, 2018: p. 1–11. [DOI] [PMC free article] [PubMed]
- 139.Ho RJ, et al. , Systems Approach to targeted and long-acting HIV/AIDS therapy. Drug Deliv Transl Res, 2015. 5(6): p. 531–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shao J, et al. , Nanodrug formulations to enhance HIV drug exposure in lymphoid tissues and cells: clinical significance and potential impact on treatment and eradication of HIV/AIDS. Nanomedicine (Lond), 2016. 11(5): p. 545–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mu Q, et al. , Translation of combination nanodrugs into nanomedicines: lessons learned and future outlook. J Drug Target, 2018: p. 1–13. [DOI] [PMC free article] [PubMed]
- 142.Sham HL, et al. , ABT-378, a highly potent inhibitor of the human immunodeficiency virus protease. Antimicrob Agents Chemother, 1998. 42(12): p. 3218–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Deeks SG, et al. , Safety, pharmacokinetics, and antiretroviral activity of intravenous 9-[2-(R)-(Phosphonomethoxy)propyl]adenine, a novel anti-human immunodeficiency virus (HIV) therapy, in HIV-infected adults. Antimicrob Agents Chemother, 1998. 42(9): p. 2380–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Colombo S, et al. , Intracellular measurements of anti-HIV drugs indinavir, amprenavir, saquinavir, ritonavir, nelfinavir, lopinavir, atazanavir, efavirenz and nevirapine in peripheral blood mononuclear cells by liquid chromatography coupled to tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci, 2005. 819(2): p. 259–76. [DOI] [PubMed] [Google Scholar]
- 145.Colombo S, et al. , Are plasma levels valid surrogates for cellular concentrations of antiretroviral drugs in HIV-infected patients? Ther Drug Monit, 2006. 28(3): p. 332–8. [DOI] [PubMed] [Google Scholar]
- 146.D’Avolio A, et al. , Intracellular accumulation of ritonavir combined with different protease inhibitors and correlations between concentrations in plasma and peripheral blood mononuclear cells. J Antimicrob Chemother, 2013. 68(4): p. 907–10. [DOI] [PubMed] [Google Scholar]
- 147.van Kampen JJ, et al. , Ultra-fast analysis of plasma and intracellular levels of HIV protease inhibitors in children: a clinical application of MALDI mass spectrometry. PLoS One, 2010. 5(7): p. e11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.McGowan I, et al. , Long-acting rilpivirine as potential pre-exposure prophylaxis for HIV-1 prevention (the MWRI-01 study): an open-label, phase 1, compartmental, pharmacokinetic and pharmacodynamic assessment. Lancet HIV, 2016. 3(12): p. e569–e578. [DOI] [PubMed] [Google Scholar]
- 149.Jucker BM, et al. , Multimodal imaging approach to examine biodistribution kinetics of Cabotegravir (GSK1265744) long acting parenteral formulation in rat. J Control Release, 2017. 268: p. 102–112. [DOI] [PubMed] [Google Scholar]
- 150.Asmuth DM, et al. , Tissue Pharmacologic and Virologic Determinants of Duodenal and Rectal Gastrointestinal-Associated Lymphoid Tissue Immune Reconstitution in HIV-Infected Patients Initiating Antiretroviral Therapy. J Infect Dis, 2017. 216(7): p. 813–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Van Zyl GU, et al. , No evidence of HIV replication in children on antiretroviral therapy. J Clin Invest, 2017. 127(10): p. 3827–3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.RxList the internet drug index, R., Inc. Available from: http://www.rxlist.com/drugs/alpha_a.htm.
- 153.Stagg MP, et al. , Clinical pharmacokinetics of 3’-azido-3’-deoxythymidine (zidovudine) and catabolites with formation of a toxic catabolite, 3’-amino-3’-deoxythymidine. Clin Pharmacol Ther, 1992. 51(6): p. 668–76. [DOI] [PubMed] [Google Scholar]
- 154.Singlas E, et al. , Didanosine pharmacokinetics in patients with normal and impaired renal function: influence of hemodialysis. Antimicrob Agents Chemother, 1992. 36(7): p. 1519–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Grasela DM, et al. , Pharmacokinetics of single-dose oral stavudine in subjects with renal impairment and in subjects requiring hemodialysis. Antimicrob Agents Chemother, 2000. 44(8): p. 2149–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Johnson MA, Horak J, and Breuel P, The pharmacokinetics of lamivudine in patients with impaired hepatic function. Eur J Clin Pharmacol, 1998. 54(4): p. 363–6. [DOI] [PubMed] [Google Scholar]
- 157.McDowell JA, et al. , Multiple-dose pharmacokinetics and pharmacodynamics of abacavir alone and in combination with zidovudine in human immunodeficiency virus-infected adults. Antimicrob Agents Chemother, 2000. 44(8): p. 2061–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kearney BP, et al. , Pharmacokinetics and dosing recommendations of tenofovir disoproxil fumarate in hepatic or renal impairment. Clin Pharmacokinet, 2006. 45(11): p. 1115–24. [DOI] [PubMed] [Google Scholar]
- 159.Custodio JM, et al. , Pharmacokinetics and Safety of Tenofovir Alafenamide in HIV-Uninfected Subjects with Severe Renal Impairment. Antimicrob Agents Chemother, 2016. 60(9): p. 5135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lamorde M, et al. , Effect of Food on the Steady-State Pharmacokinetics of Tenofovir and Emtricitabine plus Efavirenz in Ugandan Adults. AIDS Res Treat, 2012 2012: p. 105980. [DOI] [PMC free article] [PubMed]
- 161.To KW, et al. , Pharmacokinetics of plasma efavirenz and CYP2B6 polymorphism in southern Chinese. Ther Drug Monit, 2009. 31(4): p. 527–30. [DOI] [PubMed] [Google Scholar]
- 162.van Heeswijk RP, et al. , The steady-state pharmacokinetics of nevirapine during once daily and twice daily dosing in HIV-1-infected individuals. AIDS, 2000. 14(8): p. F77–82. [DOI] [PubMed] [Google Scholar]
- 163.de Maat MM, et al. , Population pharmacokinetics of nevirapine in an unselected cohort of HIV-1-infected individuals. Br J Clin Pharmacol, 2002. 54(4): p. 378–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Clarke SM, et al. , Pharmacokinetic interactions of nevirapine and methadone and guidelines for use of nevirapine to treat injection drug users. Clin Infect Dis, 2001. 33(9): p. 1595–7. [DOI] [PubMed] [Google Scholar]
- 165.Battegay M, et al. , Bioavailability of extended-release nevirapine 400 and 300 mg in HIV-1: a multicenter, open-label study. Clin Ther, 2011. 33(9): p. 1308–20. [DOI] [PubMed] [Google Scholar]
- 166.Yong CL, et al. , Pharmacokinetic analysis of nevirapine extended release 400 mg once daily vs nevirapine immediate release 200 mg twice daily formulation in treatment-naive patients with HIV-1 infection. HIV Clin Trials, 2017. 18(5–6): p. 189–195. [DOI] [PubMed] [Google Scholar]
- 167.Scholler-Gyure M, et al. , Effects of hepatic impairment on the steady-state pharmacokinetics of etravirine 200 mg BID: an open-label, multiple-dose, controlled Phase I study in adults. Clin Ther, 2010. 32(2): p. 328–37. [DOI] [PubMed] [Google Scholar]
- 168.Crauwels HM, et al. , Impact of food and different meal types on the pharmacokinetics of rilpivirine. J Clin Pharmacol, 2013. 53(8): p. 834–40. [DOI] [PubMed] [Google Scholar]
- 169.Washington CB, et al. , Effect of simultaneous versus staggered dosing on pharmacokinetic interactions of protease inhibitors. Clin Pharmacol Ther, 2003. 73(5): p. 406–16. [DOI] [PubMed] [Google Scholar]
- 170.Hsu A, et al. , Pharmacokinetic-pharmacodynamic analysis of lopinavir-ritonavir in combination with efavirenz and two nucleoside reverse transcriptase inhibitors in extensively pretreated human immunodeficiency virus-infected patients. Antimicrob Agents Chemother, 2003. 47(1): p. 350–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yeh KC, et al. , Single-dose pharmacokinetics of indinavir and the effect of food. Antimicrob Agents Chemother, 1998. 42(2): p. 332–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Damle B, et al. , Pharmacokinetics of nelfinavir in subjects with hepatic impairment. J Clin Pharmacol, 2006. 46(11): p. 1241–9. [DOI] [PubMed] [Google Scholar]
- 173.Musiime V, et al. , The pharmacokinetics and acceptability of lopinavir/ritonavir minitab sprinkles, tablets, and syrups in african HIV-infected children. J Acquir Immune Defic Syndr, 2014. 66(2): p. 148–54. [DOI] [PubMed] [Google Scholar]
- 174.Veronese L, et al. , Single-dose pharmacokinetics of amprenavir, a human immunodeficiency virus type 1 protease inhibitor, in subjects with normal or impaired hepatic function. Antimicrob Agents Chemother, 2000. 44(4): p. 821–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sevinsky H, et al. , A randomized trial in healthy subjects to assess the bioequivalence of an atazanavir/cobicistat fixed-dose combination tablet versus administration as separate agents. Antivir Ther, 2015. 20(5): p. 493–500. [DOI] [PubMed] [Google Scholar]
- 176.MacGregor TR, et al. , Pharmacokinetic characterization of different dose combinations of coadministered tipranavir and ritonavir in healthy volunteers. HIV Clin Trials, 2004. 5(6): p. 371–82. [DOI] [PubMed] [Google Scholar]
- 177.Sekar V, et al. , The effect of different meal types on the pharmacokinetics of darunavir (TMC114)/ritonavir in HIV-negative healthy volunteers. J Clin Pharmacol, 2007. 47(4): p. 479–84. [DOI] [PubMed] [Google Scholar]
- 178.Kilby JM, et al. , The safety, plasma pharmacokinetics, and antiviral activity of subcutaneous enfuvirtide (T-20), a peptide inhibitor of gp41-mediated virus fusion, in HIV-infected adults. AIDS Res Hum Retroviruses, 2002. 18(10): p. 685–93. [DOI] [PubMed] [Google Scholar]
- 179.Abel S, et al. , Assessment of the absorption, metabolism and absolute bioavailability of maraviroc in healthy male subjects. Br J Clin Pharmacol, 2008. 65 Suppl 1: p. 60–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Song I, et al. , Effect of food on the pharmacokinetics of the integrase inhibitor dolutegravir. Antimicrob Agents Chemother, 2012. 56(3): p. 1627–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mathias AA, et al. , Dose-response of ritonavir on hepatic CYP3A activity and elvitegravir oral exposure. Clin Pharmacol Ther, 2009. 85(1): p. 64–70. [DOI] [PubMed] [Google Scholar]
- 182.Iwamoto M, et al. , Lack of a clinically important effect of moderate hepatic insufficiency and severe renal insufficiency on raltegravir pharmacokinetics. Antimicrob Agents Chemother, 2009. 53(5): p. 1747–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Gallant JE, et al. , Antiviral Activity, Safety, and Pharmacokinetics of Bictegravir as 10-Day Monotherapy in HIV-1-Infected Adults. J Acquir Immune Defic Syndr, 2017. 75(1): p. 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Nowacek AS, et al. , Analyses of nanoformulated antiretroviral drug charge, size, shape and content for uptake, drug release and antiviral activities in human monocyte-derived macrophages. Journal of Controlled Release, 2011. 150(2): p. 204–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Simiele M, et al. , Evaluation of the mean corpuscular volume of peripheral blood mononuclear cells of HIV patients by a coulter counter to determine intracellular drug concentrations. Antimicrob Agents Chemother, 2011. 55(6): p. 2976–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Louissaint NA, et al. , Single dose pharmacokinetics of oral tenofovir in plasma, peripheral blood mononuclear cells, colonic tissue, and vaginal tissue. AIDS Res Hum Retroviruses, 2013. 29(11): p. 1443–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Durand-Gasselin L, et al. , Nucleotide analogue prodrug tenofovir disoproxil enhances lymphoid cell loading following oral administration in monkeys. Mol Pharm, 2009. 6(4): p. 1145–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Massud I, et al. , Chemoprophylaxis With Oral Emtricitabine and Tenofovir Alafenamide Combination Protects Macaques From Rectal Simian/Human Immunodeficiency Virus Infection. J Infect Dis, 2016. 214(7): p. 1058–62. [DOI] [PubMed] [Google Scholar]
- 189.Ruane PJ, et al. , Antiviral activity, safety, and pharmacokinetics/pharmacodynamics of tenofovir alafenamide as 10-day monotherapy in HIV-1-positive adults. J Acquir Immune Defic Syndr, 2013. 63(4): p. 449–55. [DOI] [PubMed] [Google Scholar]
- 190.PMDA (Pharmaceutical and Medical Device Agency of Japan) http://www.pmda.go.jp/drugs/2014/P201400148/800155000_22600AMX01325_H100_1.pdf, Complera (FTC/RPV/TFV) Gilead Nonclinical Summary, 2010. Accessed Jan 31, 2018.