

Abstract
Diffuse axonal injury (DAI) is an important consequence of traumatic brain injury (TBI). At the moment of trauma, axons rarely disconnect, but undergo cytoskeletal disruption and transport interruption leading to protein accumulation within swellings. The amyloid precursor protein (APP) accumulates rapidly and the standard histological evaluation of axonal pathology relies upon its detection. APP+ swellings first appear as varicosities along intact axons, which can ultimately undergo secondary disconnection to leave a terminal “axon bulb” at the disconnected, proximal end. However, sites of disconnection are difficult to determine with certainty using standard, thin tissue sections, thus limiting the comprehensive evaluation of axon degeneration. The tissue‐clearing technique, CLARITY, permits three‐dimensional visualization of axons that would otherwise be out of plane in standard tissue sections. Here, we examined the morphology and connection status of APP+ swellings using CLARITY at 6 h, 24 h, 1 week and 1 month following the controlled cortical impact (CCI) model of TBI in mice. Remarkably, many APP+ swellings that appeared as terminal bulbs when viewed in standard 8‐µm‐thick regions of tissue were instead revealed to be varicose swellings along intact axons when three dimensions were fully visible. Moreover, the percentage of these potentially viable axon swellings differed with survival from injury and may represent the delayed onset of distinct mechanisms of degeneration. Even at 1‐month post‐CCI, ~10% of apparently terminal bulbs were revealed as connected by CLARITY and are thus potentially salvageable. Intriguingly, the diameter of swellings decreased with survival, including varicosities along intact axons, and may reflect reversal of, or reduced, axonal transport interruption in the chronic setting. These data indicate that APP immunohistochemistry on standard thickness tissue sections overestimates axon disconnection, particularly acutely post‐injury. Evaluating cleared tissue demonstrates a surprisingly delayed process of axon disconnection and thus longer window of therapeutic opportunity than previously appreciated. Intriguingly, a subset of axon swellings may also be capable of recovery.
Keywords: traumatic brain injury, TBI, axonal pathology, CLARITY, amyloid precursor protein, axon degeneration, diffuse axonal injury
Introduction
Diffuse axonal injury (DAI) is a common and important consequence of traumatic brain injury (TBI) and is associated with substantial morbidity 1, 3, 4, 28, 36, 37, 74. The progression of axonal pathology after TBI was originally characterized following severe injury and was thought to persist no longer than a few months 1, 3, 4. However, evidence of axonal injury has now been observed across the spectrum of TBI severity, including mild TBI or concussion, and axon degeneration has been observed many months and even years after injury in both humans and experimental models 6, 14, 32, 35, 37.
While axons rarely disconnect at the instant of trauma, the axonal cytoskeleton can undergo immediate mechanical disruption, including primary breaking of axonal microtubules 84, 85. In addition, TBI can induce progressive loss of ionic homeostasis in injured axons, including a massive influx of calcium ions that can drive calpain‐mediated cytoskeletal degeneration and metabolic crisis 10, 13, 33, 39, 44, 45, 55, 66, 67, 71, 81, 87, 88. Adding further complexity, downstream events following severe TBI, including brain swelling and ischemic change, can be further injurious to axons 22, 23, 29, 31, 35, 64. All of these events can induce interruption of axonal transport leading to accumulation of transported cargos including the amyloid precursor protein (APP) and other proteins, which in turn causes swelling of the injured axons 15, 53, 80. Indeed, identification of APP immunoreactive (APP+) swollen axonal profiles in white matter has become the “gold standard” histological method to identify axonal pathology after TBI 26, 36, 69.
Initially appearing as tortuous or periodic, APP+ axonal swellings appear along axons that have not yet disconnected at the site of injury and are often referred to as “axonal varicosities.” It is thought that as some swellings increase in size and/or there is substantial proteolysis of the cytoskeleton, the axolemma can pinch off causing disconnection 56, 59. The swelling at the site of disconnection is often described as a terminal “axonal bulb” (previously “retraction ball”), beyond which the distal axon undergoes Wallerian degeneration 1, 2, 52, 54, 55, 57, 60, 61, 76, 77, 83. However, as standard paraffin tissue sections are typically only 6–10 microns thick, it is unclear whether purported axonal bulbs are truly disconnected, or if the axon continues out of plane from the relatively two‐dimensional view.
The advent of tissue clearing techniques including CLARITY 17 can circumvent this limitation by allowing three‐dimensional visualization of axon trajectories across what would otherwise be many tissue sections in standard histological approaches. By creating a hydrogel tissue hybrid and then selectively removing lipids from tissue, CLARITY produces optical transparency of thick or even whole‐brain samples that can be subjected to immunostaining 17. Here, we utilized CLARITY to permit a fully three‐dimensional view of the progression of axonal pathology following TBI. Specifically, we examined the temporal changes in morphology of transport‐interrupted axons following controlled cortical impact (CCI) in the mouse up to 1‐month post‐injury. This model relies on dynamic deformation of the cortical surface where primarily compressive forces induce mechanical injury to axons. Notably, these forces also cause a surface contusion with associated regional brain swelling, ischemic injury and inflammatory processes that may further compromise axons and recapitulate multiple aspects of the complex pathology of severe TBI. After first identifying apparently terminal APP+ axonal bulbs on 8‐µm‐thick regions of tissue, the true morphology and connection status of swellings was then re‐evaluated with all three dimensions fully visible through thick slices of cleared tissue. Results indicate that standard histological approaches overestimate the true extent of disconnected axonal bulbs relative to varicosities at all time points examined. Moreover, TBI induced a complex and multiphasic temporal course of axon swelling and disconnection post‐CCI that is not only surprisingly protracted, but may have critical implications for the timing of potential therapeutic interventions and indicates that some swollen axons may be capable of recovery.
Methods
All animal experiments were conducted in accordance with protocols approved by the University of Pennsylvania Institutional Animal Care and Use Committee and in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Mice were housed in a temperature and humidity controlled facility with a 12‐h light/12‐h dark cycle. Animal health was ensured via an established health monitoring program at the institution. Housed with 3–4 littermates, mice had free access to food and water throughout the duration of the study.
Controlled cortical impact (CCI) model of TBI
Twelve‐week‐old CD1 male mice (39.46 ± 2.38 g) (Charles River Laboratories, Wilmington, MA) were subjected to the established CCI model of TBI within a controlled laboratory environment as described in detail previously 79. Briefly, following induction using inhaled 5% isoflurane, mice were placed in a stereotaxic frame and maintenance anesthesia achieved using 2% isoflurane delivered via nose cone. Preemptive analgesia (buprenorphine: 0.1 mg/kg) was administered subcutaneously. Under aseptic conditions, a 5‐mm craniectomy was performed midway between lambda and bregma and immediately lateral to the sagittal suture on the right side. With the head securely fixed, a cortical impact was performed at a 45° angle directly onto the intact dura using a metal impactor with a beveled 3‐mm diameter tip. The impact was driven via the release of pressurized nitrogen to a depth of 1 mm at a velocity of 5 m/s (CCI device model AMS‐201: AmScien Instruments, Richmond, VA, USA). Following clinical assessment, the incision site was sutured followed by withdrawal of anesthesia and appropriate post‐surgical monitoring. All injuries were performed between 9 am–12 pm. Mice were survived for 6 h, 24 h, 1 week and 1 month post‐CCI and compared with sham animals that underwent identical procedures absent the cortical impact. Animals were assigned to groups randomly and in advance. No animals were excluded. Based on existing data, particularly the small variability in the outcomes observed among the animals, the proposed number of animals was anticipated to provide adequate power 49, 90. All subsequent histological experiments, analyses and quantification were performed blind to the injury status of the animal.
Tissue handling procedures
At the study endpoint, ketamine/xylazine/acepromazine (75/15/2 mg/kg, i.p.) was administered and following adequate anesthesia, animals were transcardially perfused with an initial flush of chilled 1X phosphate buffered saline (PBS) followed immediately by appropriate handling procedures to permit either:
Standard Histological Analysis using Formalin Fixed Paraffin Embedded (FFPE) Tissue (CCI: n = 4 per survival group; Shams: n = 3 with 24 h survival; n = 4 with 1‐month survival). Animals were transcardially perfused with chilled 10% neutral buffered formalin (NBF), and the brains extracted and post‐fixed in NBF for 24 h at 4°C. Brains were subsequently blocked in the coronal plane and processed to paraffin using standard techniques. Eight micrometer thick sections were obtained at the midpoint of the impact site using a rotary microtome.
CLARITY (CCI: n = 4 per survival group; Shams: n = 3 with 24 h survival): Passive tissue clearance was performed as originally described by Chung et al 17. Briefly, animals underwent transcardial perfusion with 20 mL of chilled hydrogel monomer solution (4% Acrylamide/0.05% Bis‐Acrylamide (Bio‐Rad Laboratories, Hercules, CA, USA)/0.25% VA‐044 Initiator (Wako Chemicals USA, Richmond, VA, USA)/4% paraformaldehyde/1X PBS). Brains were extracted and post‐fixed for a further 72 h in hydrogel solution at 4°C. The samples, immersed in hydrogel solution, were placed within a chamber where air was removed by vacuum and replaced with nitrogen as described 17. Subsequent incubation in a water bath was performed at 37°C for 3 h until the hydrogel monomer solution had polymerized. Brains were dissected into 2‐mm‐thick blocks in the coronal plane before being immersed in 50 mL of clearing solution (4% sodium dodecyl sulphate, 200 mM boric acid in deionized water, p.H 8.5) and incubated at 37°C in a water bath. The solution was replaced every 3 days until tissue samples appeared transparent (Figure 1A,B). Samples were then rinsed in 0.1% TritonX in 1X PBS (PBST) and subsequently stored at 4°C in PBST with sodium azide.
Figure 1.
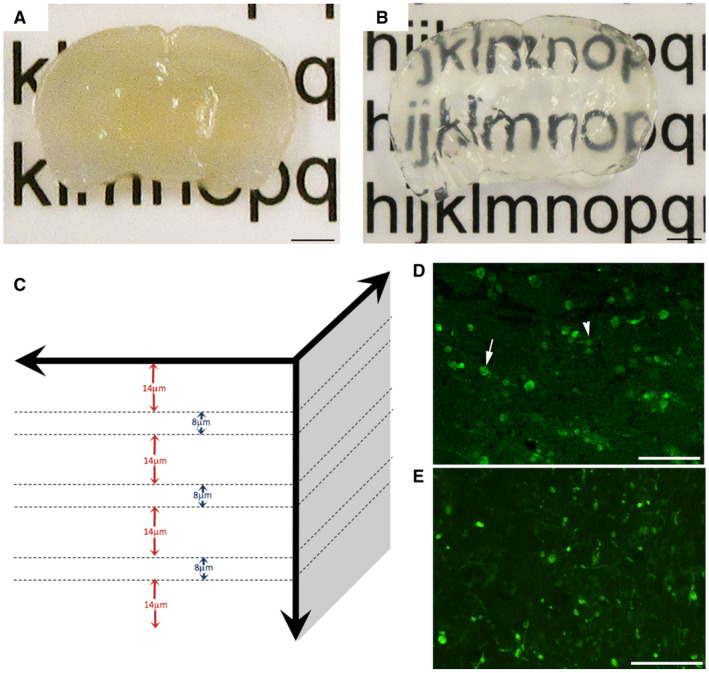
Evaluation of axonal pathology using the CLARITY technique. Representative example of 2‐mm‐thick block of uninjured mouse brain sectioned in the coronal plane (A) before and (B) after tissue clearing. (C) Diagram demonstrating the approach to analysis of cleared tissue. Specifically, 8‐µm regions of tissue were flattened (maximum‐projection function) to simulate traditional formalin‐fixed paraffin‐embedded (FFPE) tissue sections. Bulbs identified within these regions were re‐evaluated with the full 3D of tissue up to 100 µm to determine their true morphology and connectivity status. Fourteen micrometer gaps were included to avoid measurement of the same bulbs twice (based on maximal bulb size). (D) Representative example of standard 8µm‐thick FFPE mouse tissue section immunofluorescently labeled with APP. Axonal swellings can be observed in the peri‐contusional region at 24 h following CCI. Note, while the vast majority of APP swellings appear as disconnected, terminal axonal bulbs (arrow), occasional varicosities can be observed with two clear projections from a single swelling along an intact axon (arrowhead). (E) APP immunoreactive axonal pathology observed peri‐contusionally at 24 h post‐CCI on 8‐µm‐thick maximum projection images using cleared tissue appears indistinguishable from standard FFPE immunofluorescent sections in (D). Scale bars: (A,B) 2 mm, (D,E) 50 µm.
Standard histological examinations of FFPE tissue
To characterize and verify reproducibility of the injury model, standard FFPE tissue was examined following staining with both hematoxylin and eosin (H&E) and Luxol Fast Blue/Cresyl Violet (LFB/CV) as described previously 35. The presence of brain atrophy over time was determined by measuring the area of the ipsilateral (injured) hemisphere as a percentage of the contralateral (uninjured) hemisphere using whole brain coronal sections at the epicenter of the impact site. Measurements were performed using ImageScope software (Leica Biosystems, Wetzlar, Germany) using whole section, 20X high‐resolution digital scans acquired using the Aperio ScanScope (Leica Biosystems, Wetzlar, Germany).
Immunohistochemical labeling of FFPE tissue was also performed to confirm the presence of axonal pathology using standard techniques as previously described 35, 37. Briefly, following deparaffinization and rehydration, endogenous peroxidase activity tissue was quenched using 3% aqueous hydrogen peroxide (15 minutes). Antigen retrieval was performed in a microwave pressure cooker with immersion in Tris EDTA buffer. Subsequent blocking was performed for 30 minutes in 1% normal horse serum (Vector Labs, Burlingame, CA, USA) in Optimax buffer (BioGenex, San Ramon, CA, USA). An additional blocking step was performed using M.O.M. blocking reagent (Vector Labs, Burlingame, CA, USA) for labeling using mouse monoclonal antibodies on mouse tissue. Incubation with the primary antibodies was performed at 4°C for 20 h. Specifically, to identify axonal pathology, sections were labeled with an antibody reactive for the N‐terminal amino acids 66–81 of the amyloid precursor protein (APP) (Clone 22C11 37, 75: Millipore, Billerica, MA) at 1:95k if visualized via 3,3′‐diaminobenzidine (DAB) or 1:1K for immunofluorescence studies, and confirmed with goat anti‐full length APP 70 at 1:1K (DAB). To identify normal axons, anti‐neurofilament‐heavy (NF‐H) (Aves Lab, Tigard, OR, USA) was applied at 1:500 (immunofluorescence). After rinsing, sections were incubated with the appropriate biotinylated secondary antibody for 30 minutes followed by avidin‐biotin complex and visualization achieved using DAB (all reagents: Vector Labs, Burlingame, CA, USA). Alternatively, the corresponding Alexa Fluor (Invitrogen, Carlsbad, CA, USA) fluorescent secondary antibody was applied at 1:500 in a 2% species‐specific serum solution for 2 h at room temperature. Sections were rinsed and either dehydrated and cleared in xylenes before coverslipping in cytoseal or alternatively coverslipped using an aqueous mounting medium (Dako, Carpinteria, CA, USA). Positive control tissue for APP IHC included sections of contused mouse tissue with previously established axonal pathology. Omission of subsets of primary antibodies was performed in positive control material to control for non‐specific binding.
Immunofluorescent labeling of mouse brain tissue following CLARITY
Following CLARITY procedures, tissue was incubated in mouse anti‐APP (22C11) primary antibody (1:100; Millipore, Billerica, MA, USA) and chicken anti‐NFH primary antibody (1:50; Aves Lab, Tigard, OR, USA) in PBST for 72 h at 37°C. Samples were then rinsed in PBST for 3 days at room temperature on a rocker before subsequent incubation in Alexa Fluor 488 donkey anti‐mouse secondary antibody and Alexa Fluor 568 goat anti‐chicken secondary antibodies (1:100; Invitrogen, Carlsbad, CA, USA), for 4 days at 37°C. Samples were again rinsed in PBST for 3 days as above and stored in PBST with sodium azide until imaging.
Confocal imaging
CLARITY tissue was immersed in FocusClear (Cedarlane, Burlington, NC, USA) prior to confocal imaging. Notably, consistent with what has been described previously, the process of passive clearance results in tissue expansion, specifically during lipid removal. In contrast, tissue shrinkage can occur with immersion in FocusClear. Thus, in order to permit appropriate comparisons between samples and groups, all tissue, including from controls, was handled using standardized protocols. Critically, this included a standardized pre‐incubation in FocusClear for 1 h immediately prior to imaging.
To minimize sample movement, tissue was positioned within a 2‐mm‐deep PDMS rectangle glued to a No. 1 coverslip and submerged in FocusClear (Cederlane Labs, Burlington, NC, USA). Tissue was visualized using a confocal Nikon Eclipse Ti microscope (Nikon, Tokyo, Japan). High‐resolution confocal imaging was performed immediately adjacent to the focal contusion in an area measuring 630 × 630 µm (1024 × 1024 pixels) at 1‐µm z‐step intervals through 100 µm of the tissue. FFPE tissue was also imaged throughout the 8‐µm tissue depth using the confocal microscope as above.
Quantification of CLARITY immunofluorescence findings
First, we sought to determine whether disconnected or terminal axonal bulbs identified in standard 8‐µm‐thick tissue sections are indeed truly disconnected axon terminals when viewed with the addition of a third dimension in space.
To achieve this, the peri‐contusional 3D CLARITY confocal images (630 × 630 × 100 µm) at 6 h, 24 h, 1‐week and 1‐month post‐CCI (n = 4 per group) were first split into individual z‐steps of 1µm thickness using Volocity 6.3 software (PerkinElemer, Waltham, MA, USA). Next, sets of eight consecutive z‐steps were compressed to form 8µm‐thick images (via the max projection function) to represent the thickness of tissue typically examined when using standard FFPE sections.
These 8µm‐thick regions of cleared tissue were then examined for the presence of axonal bulbs. Consistent with historical descriptions, axon bulbs were defined as swollen spherical or ovoid axonal profiles that are APP immunoreactive and appear disconnected. An axon was considered swollen if its maximal diameter was greater than 3 μm (at least 50% greater than the maximal diameter of normal axons as determined by the measurement of normal axons identified with NF‐H IHC (n = 336 axons) in cleared tissue from two sham mice).
Note, to avoid assessing individual bulbs twice, a 14µm‐deep gap was omitted between each 8‐µm‐thick image where apparently terminal bulbs were first identified. Similarly, the first 14 µm was omitted from the top and bottom of the sample to ensure complete visualization of all bulbs when viewed in 3D (Figure 1C). This 14‐µm distance was determined based on maximal observed bulb size across all time points. The percentage of swellings that appeared terminal in these 8µm‐thick “FFPE equivalent” images in cleared tissue were compared to actual APP stained 8µm‐thick FFPE sections to ensure they were representative (Figure 1D,E).
Next, all apparently disconnected axonal bulbs identified on 8‐µm‐thick cleared tissue regions were subsequently re‐examined in 3D, with the full thickness of tissue visualized in the Z‐axis through a 100µm depth of the imaged 3D CLARITY samples. Bulbs re‐assessed in 3D were then designated as either: (i) confirmed as a disconnected or terminal axonal bulb with one or no visible projections or (ii) re‐designated as a connected varicose axonal swelling, having two clearly visible projections along an intact portion of axon. The maximal diameter of all swellings was also measured using the line measurement tool in Volocity software (PerkinElemer, Waltham, MA, USA).
Statistical analyses
The area of ipsilateral cortex as a percentage of the area of contralateral cortex was compared between groups as a metric of brain atrophy using ANOVA with Bonferroni correction for multiple comparisons. As was group comparisons of the percentage of apparent bulbs on 8‐µm analyses that were revealed to have retained connectivity on 3D analysis, thus representing periodic varicose swellings. The average bulb diameter was compared between groups using the linear mixed effects model. This model takes into account the fact that each mouse had multiple measures by including a subject‐specific random effect. Finally, logistic regression was used to assess associations of bulb diameter with the presence of revealed connectivity. Odds ratio with 95% confidence interval (CI) is reported.
Image processing for video
All supplemental videos were generated using Nikon NIS Elements AR software, Version 4.4 (Nikon, Tokyo, Japan). Regions of interest were selected on ND2 image files using the 3D volumetric view function and the video generated using the NIS Movie Maker function. Depth encoding was achieved (Video S1) by selecting the depth encoded max intensity projection tool. All supplemental video files were saved as AVI image file formats (.avi) using MPEG compression and subsequently converted to .mov files. Final QuickTime files were generated with a resolution of 1280*720, video bitrate of 2500 kbps, frame rate of 30 fps, and aspect ratio of 16:9.
Results
Clinical and neuropathological characterization of CCI in mice
Immediately following CCI, no apnea beyond 30 seconds or clinical evidence of seizure was observed in any animals. All mice were ambulatory within 5–10 minutes of isoflurane withdrawal and resumed normal eating/drinking/grooming behaviors within 1 hour.
Standard (FFPE) histological characterization of mice (n = 4 per group) revealed neuropathologies consistent with historical descriptions of the model. Specifically, acutely at 6 and 24 hours following CCI, a focal hemorrhagic contusion could be observed of approximately 5 mm in diameter on the right cortical surface. Both grossly and on H&E examination, contusion could be observed throughout the full cortical thickness (Figure 2B,C). By 1 week to 1 month following CCI, on both gross and histological (H&E and LFB/CV) examination, significant and progressive cavitation of the lesion site could be observed (Figure 2D,E,H). Consistent with previous descriptions, measurements of brain atrophy demonstrated a significant and progressive loss of tissue out to 1‐month post‐injury (Figure 2F) 34, 40. APP IHC revealed extensive axonal pathology with tortuous varicose profiles and extensive axonal bulbs at 6 hours to 1‐week post‐CCI in peri‐contusional cortex, underlying corpus callosum, hippocampus, as well as more minimally in the ipsilateral thalamus (Figure 2I). By 1‐month post‐injury, while comparatively minimal, APP positive axonal bulbs and varicosities could still be observed in the lesion site, predominantly surrounding the cavity and minimally in the underlying thalamus (Figure 2J). APP immunoreactive axonal pathology was absent in sham animals.
Figure 2.
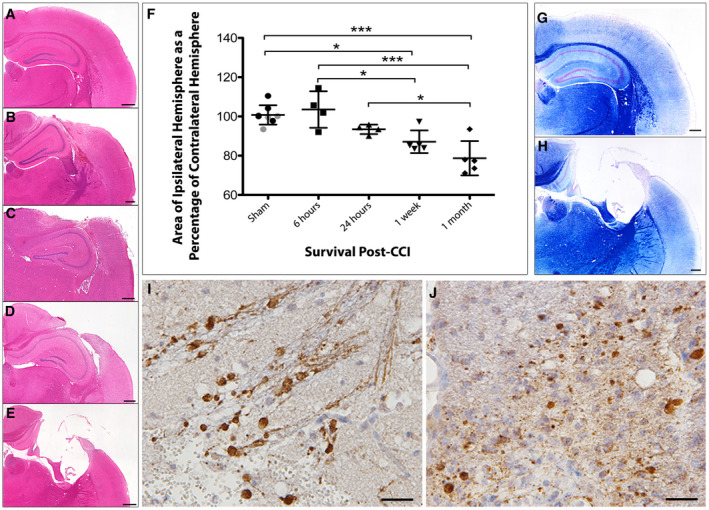
Progressive atrophy and associated axonal pathology post‐CCI in standard FFPE tissue. (A) H&E staining on sham animals reveals an absence of any focal pathology and a morphologically normal cortical surface. (B–E) H&E staining performed (B) 6 h, (C) 24 h, (D) 1 week and (E) 1 month after CCI injury reveals acute cortical contusion and intraparenchymal hemorrhage at the site of impact as well as progressive atrophy of the ipsilateral hemisphere. (F) Tissue atrophy quantified by measuring the area of the injured ipsilateral hemisphere as a percentage of the contralateral hemisphere. All individual data are presented in addition to the mean percentage area ± SD. In the sham group, gray data points indicate mice sacrificed 24 h after sham injury and black data points indicate mice sacrificed 1 month following sham injury, indicating an absence of change caused by craniectomy. (G–H) Luxol fast blue staining indicates loss of white matter and major cortical tissue loss at (H) 1 month following injury compared to (G) sham. (I–J) Swollen axonal profiles and varicosities immunroeactive for APP at (I) 24 h and (J) 1‐month post‐CCI. Scale bars: (A–E, G–H) 400 µm, (I–J) 25 µm. *P < 0.05, ***P < 0.001.
3D CLARITY reveals that axons are more connected post‐CCI than previously appreciated using standard histological approaches
Consistent with FFPE tissue, at all time points post‐injury within cleared tissue, APP accumulation could be observed in injured or degenerating axons. Within 8‐µm‐thick (FFPE‐equivalent) tissue regions, APP accumulated within tortuous varicosities along axons that nonetheless remained intact or connected. However, much more frequently, APP accumulation was also observed in terminal axonal bulbs where only a single proximal process could be observed feeding into a disconnected, swollen axon terminus. In some cases, swellings appeared isolated with no clear projections (Figure 3D, Video [Link], [Link]). Importantly, there was no difference in the percentage of all axonal swellings that appeared terminal between images of standard 8‐µm‐thick FFPE and images of 8‐µm‐thick regions within CLARITY samples (n = 4 per group at 24 h survival P = 0.59).
Figure 3.
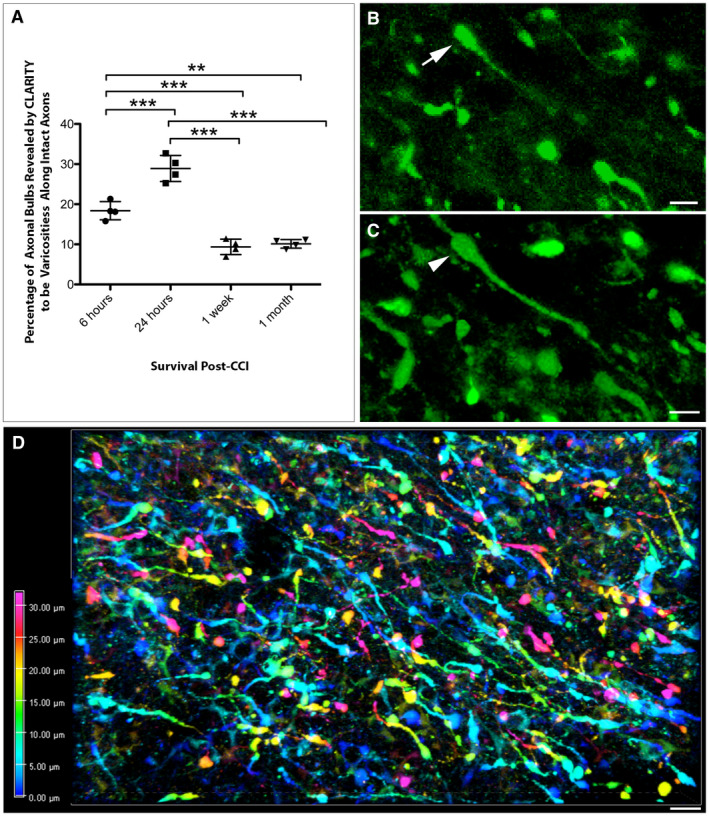
CLARITY reveals less axonal disconnection than previously thought following CCI. (A) Graph showing the percentage of APP+ swellings that appeared terminally disconnected when viewed in standard 8‐µm‐thick regions of tissue that were revealed to be connected varicose axonal swellings when all three dimensions were fully visible using CLARITY (**P ≤ 0.01, ***P ≤ 0.001). (B) Representative example of an APP+ apparent terminal axonal bulb (arrow) observed on an 8‐µm‐thick region of cleared tissue (maximum projection) at 24 h post‐CCI. (C) The same region of tissue as (B) re‐evaluated with all three dimensions now visible reveals that the apparent bulb is in fact a swelling that has two clear projections and thus represents a varicose swelling along an intact region of axon (arrowhead). (Alpha‐blended image) (D) Depth encoded three‐dimensional region of extensive APP+ axonal pathology in the peri‐contusional region 24 h post‐CCI. Extensive and large axonal swellings can be observed with complex morphologies comprising both disconnected axonal bulbs and varicosities along the intact length of axons. Scale bars: (B,C) 10 µm, (D) 50 µm.
However, when the same apparently disconnected axonal bulbs observed in 8µm‐thick regions were re‐evaluated with the inclusion of the full third dimension in space (up to 100µm‐deep using the entire CLARITY image stack), a more complete appreciation of both axonal morphology and true axonal connectivity was ascertained. Specifically, in many instances these apparent axonal bulbs were revealed to have two clear processes and thus actually represented periodic varicose swellings along the length of an intact and thus potentially viable axon (Figure 3B,C, Video S3).
This was notably observed at all time points post‐CCI. Specifically, 18.38 ± 2.27% of apparently terminal axonal bulbs identified on 8‐µm‐thick regions at 6 h post‐injury were revealed as intact varicosities when viewed in 3D up to 100‐µm‐deep. Intriguingly, by 24 h post‐CCI this number significantly increased, with 28.91 ± 3.25% of presumed disconnected bulbs revealed to be varicosities (P = 0.0002) (Figure 3A).
In contrast, by 1 week only 9.36 ± 1.92% of bulbs were revealed to be varicosities, a significant decrease from both 6 (P = 0.0007) and 24 h (P < 0.0001). Remarkably, this number remained stable out to 1‐month post‐CCI at 10.13 ± 1.07% [again significantly decreased from 6 h (P = 0.0015) and 24 h (P < 0.0001), but not significantly different from the 1‐week survival time point (P > 0.9999)] (Figure 3A).
Axonal swellings decrease in size with increased survival post‐CCI
The diameter of axonal swellings was also observed to progressively decrease with survival post‐CCI. This result was true in both swellings that were confirmed to be true axonal bulbs and those that were revealed to be connected varicose swellings (Figure 4). Specifically, of the disconnected axonal bulbs, the mean diameter of swellings was significantly greater at 6 h post‐CCI (7.85 ± 0.43 µm) vs. those at 1 week (5.73 ± 0.68 µm, P < 0.001) and 1 month (5.11 ± 0.52 µm, P < 0.001). In addition, bulbs at 24 h post‐CCI (7.01 ± 1.05 µm) were significantly larger in diameter than those at 1 week (P = 0.042) and 1 month (P = 0.001), although did not differ from the 6 h time point (P = 0.13). No differences in diameter were observed between 1 week and 1 month (P = 0.15) (Figure 4A).
Figure 4.
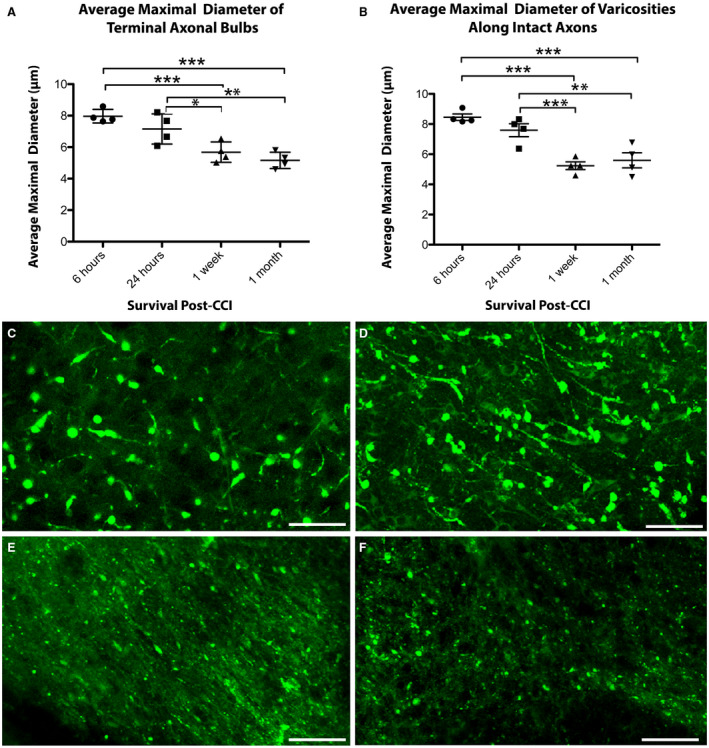
The size of axonal swellings decreases with increased survival post‐CCI. The average maximal diameter of axonal swellings of both (A) terminally disconnected bulbs and (B) axonal varicosities along intact axons decreases over time following CCI. (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). (C–F) Representative examples of the relative size of axonal swellings in the peri‐contusional region in cleared tissue at (C) 6 h, (D) 24 h, (E) 1‐week and (F) 1‐month post‐CCI. Scale bars: (C–F) 20 µm.
Interestingly, apparent bulbs that were revealed to be varicosities in 3D (i.e., still connected both to both the proximal and distal axon) also underwent a progressive decrease in their maximal diameter over time, suggesting possible recovery. Swellings at 6 h post‐CCI (8.46 ± 0.42 µm) were greater in diameter than those at 1 week (5.24 ± 0.52 µm, P < 0.001) and 1 month (5.59 ± 1.00 µm, P < 0.001). In addition, swellings at 24 h post‐CCI (7.59 ± 0.86 µm) were also significantly larger in diameter than those at 1 week (5.24 ± 0.52 µm, P < 0.001) and 1 month (5.59 ± 1.00 µm, P = 0.003), although did not differ from the 6 h time point (P = 0.059). No differences in mean diameter were observed between 1 week and 1 month (P = 0.64) (Figure 4B).
Decreased maximal diameter of axonal swellings is associated with disconnection
While bulb diameter decreased over time regardless of whether swellings were disconnected terminal bulbs or periodic varicose swellings, further analysis using logistic regression revealed that the diameter of individual axonal swellings was predictive of whether swellings were disconnected axonal bulbs or periodic varicose swellings along intact axons.
Specifically, axonal varicosities that initially appeared as terminal axonal bulbs on 8‐µm‐thick regions, were more likely to be of increased diameter vs. those swellings that were confirmed as disconnected axonal bulbs at 6 h (P = 0.018; OR 1.12; 95% CI 1.02–1.23), 24 h (P = 0.011; OR 1.09; 95% CI 1.02–1.16) and 1 month (P = 0.018; OR 1.12; 95% CI 1.02–1.23) post‐injury. However, there was no significant difference at the 1‐week time point (P = 0.24; OR 0.93; 95% CI 0.83–1.05).
While disconnection was associated with decreased overall diameter as described, individual data per mouse indicates that swellings with the smallest diameters occur less frequently in varicosities along intact axons at all time points. In contrast, while swellings that were terminal had a wider range of sizes, this included many of the smallest swellings that may represent the small bead‐like fragmentation that occurs in Wallerian degeneration.
Interestingly, by 1 week and 1 month, the extent of large swellings appears to be decreased in connected axons relative to those that are disconnected, with swellings having a narrower range of diameters, clustered in the lower range (Figure 5A–D). While this may reflect a less severely injured population of axons, it may also reflect recovery of transport.
Figure 5.
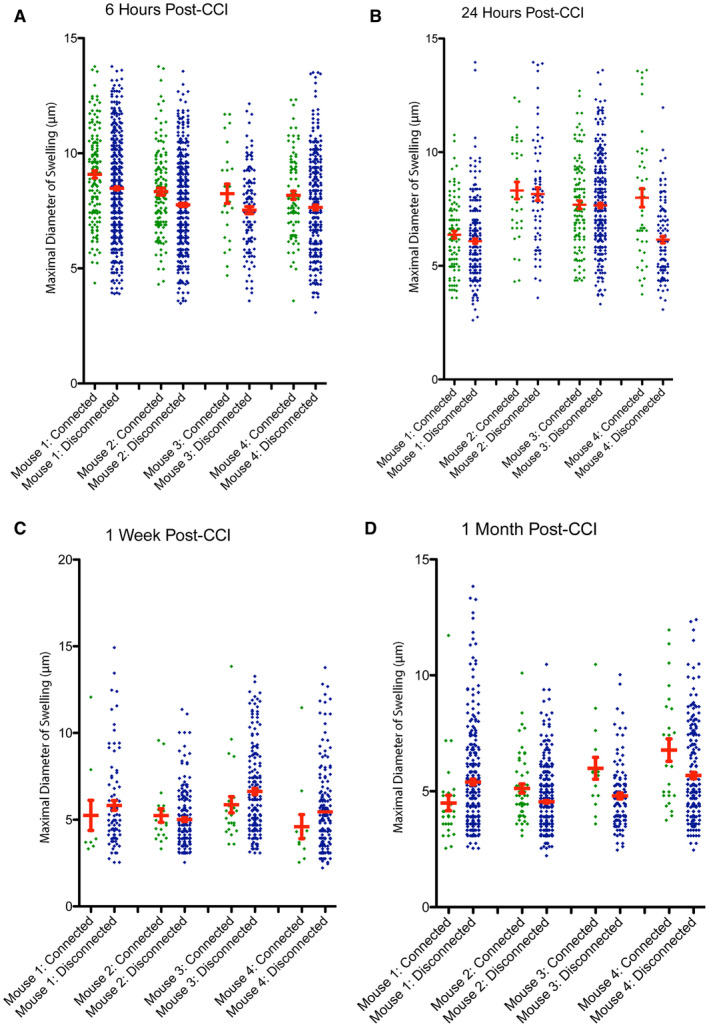
The size of axonal swellings and connection status with survival. The maximal diameter for all individual disconnected, terminal axonal bulbs and connected axonal varicosities shown for each mouse at (A) 6 h, (B) 24 h, (C) 1‐week and (D) 1‐month post‐CCI. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
Using the novel tissue clearing technology, CLARITY, the nature of axonal pathology after TBI in mice was found to be morphologically diverse and followed a surprisingly protracted time course. This complex array of swollen axonal phenotypes and their differential evolution post‐TBI suggests multiple distinct mechanisms of degeneration, and intriguingly, possible reduced axonal transport interruption or recovery over time in some injured axons. Importantly, axonal swellings were observed along potentially salvageable, intact axons even at 1‐month post‐TBI, indicating a surprisingly long window of therapeutic opportunity.
By visualizing axonal pathology across three dimensions in space, we demonstrate that the extent of axonal disconnection is greatly overestimated when examined using conventional histological approaches on thin tissue sections. Specifically, 3D analyses utilizing thick blocks of cleared tissue, reveal that up to ~30% of axonal swellings appear to maintain connectivity post‐CCI that were previously undetected with standard methods. While APP has emerged as a sensitive and early marker of axonal pathology 25, 37, 69, it is unknown whether all APP positive axonal swellings ultimately progress toward terminal disconnection, or whether a subset of these swellings are capable of undergoing repair with restoration of normal transport and function. Thus, an accurate appreciation of axon disconnection is critical when assessing both the degree of injury, as well as quantifying and interpreting histopathological outcome from experimental intervention.
Interestingly, the misidentification of varicose swellings as terminally disconnected axonal bulbs was not static over time, but rather had a variable temporal evolution out to 1‐month post‐injury. Specifically, at 6 h post‐CCI, ~20% of the swollen axons that appeared as terminal bulbs on thin sections were instead revealed to be varicosities along intact axons when viewed in full 3D. Curiously, this number increased significantly to ~30% at 24 h, before declining to ~10% at 1 week and 1 month following injury. These data suggest that CCI causes an early (within 6 h) and severe form of axonal degeneration with rapid transport interruption and disconnection, possibly caused by direct mechanical perturbation of axonal cytoskeletal structure. This rapid disconnection appears to be followed shortly thereafter by a more protracted onset of axonal swellings, which may represent an emerging population of less‐damaged axons undergoing a slower rate of APP accumulation and disconnection. In contrast, the delayed appearance of these swellings may also be caused by the multiple downstream pathophysiological processes including intra‐axonal ionic imbalance, progressive cytoskeletal degradation, metabolic failure, oxidative stress and lipid peroxidation as described extensively following both in vivo and in vitro models of TBI 11, 12, 18, 19, 20, 33, 36, 37, 43, 47, 48, 50, 55, 66, 67, 74, 80, 81, 84, 85, 87, 88.
Understanding the temporal course of axonal disconnection in relation to specific mechanisms of degeneration will be critical for the optimization of timed therapeutic intervention. Remarkably, these data indicate that even 1 month following injury, a proportion of axonal swellings were not terminally disconnected and thus potentially recoverable. Previous studies have shown that tissue loss resulting from experimental contusions produced by focal impact models in rodents, including the CCI model, gradually increases over time, resulting in a dramatic and progressive atrophy persistent for up to 1 year after TBI 7, 8, 40, 72. This may be of particular clinical importance given that ongoing progressive brain atrophy has also been observed radiologically following brain injury in humans 5, 21, 86. Moreover, APP immunoreactive axonal pathology was recently demonstrated in association with marked atrophy in the corpus callosum, even many years after severe TBI in humans 35.
In addition to exploring the temporal course of axonal swelling and disconnection, examinations of cleared tissue revealed that the size of swellings also decreased with survival, regardless of whether swellings were terminally disconnected or remained attached to the distal axon. This may simply reflect an overall reduction in axonal transport in severely damaged tissue, where multiple points of interruption limit the volume of APP available to feed into downstream swellings. More comprehensive sampling or whole‐brain analysis of tracts will be important to further explore the relationship between serial swellings. While it is also possible that downstream Wallerian degeneration can induce smaller swellings along the fragmenting distal axon, recent examination of Thy1‐YFP‐H mice subjected to an impact model of TBI describes an absence of APP accumulation in distal axonal swellings otherwise identified by yellow fluorescent protein 30.
It is particularly noteworthy that swellings along intact axons, even at 1 week and 1‐month post‐injury, are smaller than those observed acutely. Indeed, swellings along intact axons were notably clustered in the smaller size range when compared to disconnected bulbs, which typically encompassed a wider range of diameters. While this observation may indicate a population of less severely transport‐interrupted axons with reduced disconnection, it also supports the intriguing possibility that some swellings decrease in size over time via recovery mechanisms that reinstate transport. While recovery of individual axonal swellings has yet to be directly observed post‐TBI, temporal examinations of an impact model of TBI in rats suggest both physiological and morphological recovery is possible, with variable recovery reported between myelinated and unmyelinated axonal subpopulations 62, 63.
Observations in this study regarding the trajectory of axonal swelling and disconnection are dependent on the pathological events specific to the CCI injury model. Given both the biomechanical and neuropathological features of CCI, findings can most reasonably be extrapolated to the axonal pathology observed in contused human brain tissue. Specifically, CCI relies upon controlled contact loading to the dural surface to cause dynamic deformation of brain tissue over the course of ~10‐30 milliseconds to induce the primary pathology of focal hemorrhagic cortical contusion 34, 79. Consistent with multiple previous reports 34, 41, extensive peri‐contusional APP+ axonal pathology was observed immediately below the impact site, as well as more minimal axon damage in the underlying hippocampus and thalamus. Much of the early axonal pathology is likely caused by the primary compressive forces caused by impact and associated downstream cascades. Specifically, tissue within the peri‐contusional region also displays neuronal death, regional brain swelling and edema, ischemic change, blood–brain barrier disruption, as well as diverse glial and inflammatory responses 34, 40, 79 that together comprise a complex pathological milieu that may be further detrimental to axons. Indeed, it has long been described that APP+ axonal pathology can occur independently as a result of prolonged ischemia or secondary to the vascular complications of raised intracranial pressure 22, 23, 29, 31, 35, 64. As such, vascular compromise within the contused and peri‐contusional tissue may contribute independently to acute axon degeneration following CCI.
An important source of axonal pathology in human TBI is that caused by rotational forces because of rapid acceleration/deceleration of the head at the moment of injury 24, 36, 37, 46, 65, 73, 78. Because of its large mass, the human brain is inherently vulnerable to this type of injury that contributes significantly to acute clinical outcome 24. However, acceleration/deceleration forces are absent during the fixed‐head CCI model, and the degree of scaling required to generate clinically relevant rotational forces in the smaller and lissencephalic mouse brain are not feasible 34. Seminal studies have identified that forces from rotational accelerations are critical for the direct mechanical perturbation of axons to cause diffuse traumatic axonal injury 24, 37, 38, 46, 73. While not possible to examine using rodent cortical impact models as described here, determining how clinically relevant rotational forces influence the trajectory of axon disconnection will be important to explore using appropriate models with a larger brain mass 37.
Furthermore, recent examination of both animal models and post‐mortem human tissue demonstrates that APP does not label all swollen axons. Historically, multiple proteins have been identified accumulating in axonal swellings including various neurofilament subtypes 9, 10, 11, 13, 16, 27, 51, 58, 66, 82, 89, an N‐terminal fragment of the alpha‐spectrin protein (SNTF) 13, 37, 45, 66, 67, 87 and ubiquitin 68. However, APP was selected as a robust and early marker for diagnostic purposes and remains the “gold‐standard” approach 25, 36, 69. Both specific neurofilament subtypes and SNTF have been shown to label distinct populations of APP‐negative axonal swellings 37, 42, 82. Similarly, YFP+ axons in transgenic mice also displayed swellings that were APP‐negative 30, 90. Thus, while APP labeling has been shown to identify the greatest number of swellings relative to other markers 37, it nonetheless fails to detect the entire population of injured axons within the models and time points examined thus far. Further elucidating whether these APP‐negative axonal swellings have a different time course of degeneration and disconnection may permit a more comprehensive and informed approach to the neuropathological assessment of axonal pathology.
Here, we have demonstrated that 3D temporal examinations of post‐traumatic axonal swelling and disconnection reveals novel insights into the timing and morphologies of axon degeneration beyond what can be observed with traditional histological approaches. Indeed, this approach provides unique opportunities for the further exploration of specific mechanisms and whole‐brain effects of axonal injury. Understanding the temporal progression of axonal swelling may inform not only the optimal timing of early interventions, but suggests that future therapies may have utility beyond the acute phase of injury.
Conflicts of Interest
No authors have any conflicts of interest.
Supporting information
Video S1. Video of 3D volume of tissue showing peri‐contusional APP immunoreactive axonal pathology in cleared tissue 24 h following CCI. Color gradation denotes the depth in the z‐plane. Note the extensive pathology displaying a complex array of morphologies including disconnected axonal bulbs and periodic axonal varicosities, often with multiple swellings along the length of injured axons.
Video S2. Video showing peri‐contusional APP immunoreactive axonal profiles (green) through the z‐plane in continuous 1‐µm z‐steps (50 µm total). Axonal bulbs can be followed through the tissue with varying morphologies and connectivity status. Note that some neuronal soma are also APP immunoreactive, though clearly differentiated from axonal swellings by the presence of a nucleus.
Video S3. Video displaying a single post‐CCI APP immunoreactive (green) varicose axon at high magnification tracked through the z‐plane in 1µm increments. While standard FFPE‐equivalent analysis identifies the two large axonal swellings as terminal bulbs, CLARITY analysis reveals these two bulbs are in fact connected and part of the same axon as periodic varicose swellings.
Acknowledgments
Research reported in this publication was supported by the National Institutes of Health under award numbers R01NS092398, TL1TR001880, and R01NS101108. In addition, work was supported in part by Merit Review, Award No. RX002705 (JAW) from the United States (U.S.) Department of Veterans Affairs, Rehabilitation Research and Development Service Program. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the U.S. Department of Veterans Affairs or the United States Government.
References
- 1. Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McLellan DR (1989) Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology 15:49–59. [DOI] [PubMed] [Google Scholar]
- 2. Adams JH, Doyle D, Graham DI, Lawrence AE, Mclellan DR (1984) Diffuse axonal injury in head injuries caused by a fall. Lancet 2:1420–1422. [DOI] [PubMed] [Google Scholar]
- 3. Adams JH, Graham DI, Gennarelli TA, Maxwell WL (1991) Diffuse axonal injury in non‐missile head injury. J Neurol Neurosurg Psychiatry 54:481–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Adams JH, Graham DI, Murray LS, Scott G (1982) Diffuse axonal injury due to nonmissile head injury in humans: an analysis of 45 cases. Ann Neurol 12:557–563. [DOI] [PubMed] [Google Scholar]
- 5. Anderson CV, Bigler ED (1995) Ventricular dilation, cortical atrophy, and neuropsychological outcome following traumatic brain injury. J Neuropsychiatry Clin Neurosci 7:42–48. [DOI] [PubMed] [Google Scholar]
- 6. Blumbergs PC, Scott G, Manavis J, Wainwright H, Simpson DA, McLean AJ (1994) Staining of amyloid precursor protein to study axonal damage in mild head injury. Lancet 344:1055–1056. [DOI] [PubMed] [Google Scholar]
- 7. Bramlett HM, Dietrich WD (2002) Quantitative structural changes in white and gray matter 1 year following traumatic brain injury in rats. Acta Neuropathol 103:607–614. [DOI] [PubMed] [Google Scholar]
- 8. Bramlett HM, Dietrich WD, Green EJ, Busto R (1997) Chronic histopathological consequences of fluid‐percussion brain injury in rats: effects of post‐traumatic hypothermia. Acta Neuropathol 93:190–199. [DOI] [PubMed] [Google Scholar]
- 9. Buki A, Farkas O, Doczi T, Povlishock JT (2003) Preinjury administration of the calpain inhibitor MDL‐28170 attenuates traumatically induced axonal injury. J Neurotrauma 20:261–268. [DOI] [PubMed] [Google Scholar]
- 10. Buki A, Koizumi H, Povlishock JT (1999) Moderate posttraumatic hypothermia decreases early calpain‐mediated proteolysis and concomitant cytoskeletal compromise in traumatic axonal injury. Exp Neurol 159:319–328. [DOI] [PubMed] [Google Scholar]
- 11. Buki A, Okonkwo DO, Povlishock JT (1999) Postinjury cyclosporin A administration limits axonal damage and disconnection in traumatic brain injury. J Neurotrauma 16:511–521. [DOI] [PubMed] [Google Scholar]
- 12. Buki A, Povlishock JT (2006) All roads lead to disconnection?–Traumatic axonal injury revisited. Acta Neurochir 148:181–193; discussion 93–94. [DOI] [PubMed] [Google Scholar]
- 13. Buki A, Siman R, Trojanowski JQ, Povlishock JT (1999) The role of calpain‐mediated spectrin proteolysis in traumatically induced axonal injury. J Neuropathol Exp Neurol 58:365–375. [DOI] [PubMed] [Google Scholar]
- 14. Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH (2009) A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long‐term survivors of traumatic brain injury. Brain Pathol 19:214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Christman CW, Grady MS, Walker SA, Holloway KL, Povlishock JT (1994) Ultrastructural studies of diffuse axonal injury in humans. J Neurotrauma 11:173–186. [DOI] [PubMed] [Google Scholar]
- 16. Christman CW, Salvant JB Jr, Walker SA, Povlishock JT (1997) Characterization of a prolonged regenerative attempt by diffusely injured axons following traumatic brain injury in adult cat: a light and electron microscopic immunocytochemical study. Acta Neuropathol 94:329–337. [DOI] [PubMed] [Google Scholar]
- 17. Chung K, Wallace J, Kim SY, Kalyanasundaram S, Andalman AS, Davidson TJ et al (2013) Structural and molecular interrogation of intact biological systems. Nature 497:332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deng‐Bryant Y, Singh IN, Carrico KM, Hall ED (2008) Neuroprotective effects of tempol, a catalytic scavenger of peroxynitrite‐derived free radicals, in a mouse traumatic brain injury model. J Cereb Blood Flow Metab 28:1114–1126. [DOI] [PubMed] [Google Scholar]
- 19. Deng Y, Thompson BM, Gao X, Hall ED (2007) Temporal relationship of peroxynitrite‐induced oxidative damage, calpain‐mediated cytoskeletal degradation and neurodegeneration after traumatic brain injury. Exp Neurol 205:154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fujita M, Oda Y, Wei EP, Povlishock JT (2011) The combination of either tempol or FK506 with delayed hypothermia: implications for traumatically induced microvascular and axonal protection. J Neurotrauma 28:1209–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gale SD, Johnson SC, Bigler ED, Blatter DD (1995) Nonspecific white matter degeneration following traumatic brain injury. J Int Neuropsychol Soc 1:17–28. [DOI] [PubMed] [Google Scholar]
- 22. Geddes JF, Vowles GH, Beer TW, Ellison DW (1997) The diagnosis of diffuse axonal injury: implications for forensic practice. Neuropathol Appl Neurobiol 23:339–347. [PubMed] [Google Scholar]
- 23. Geddes JF, Whitwell HL, Graham DI (2000) Traumatic axonal injury: practical issues for diagnosis in medicolegal cases. Neuropathol Appl Neurobiol 26:105–116. [DOI] [PubMed] [Google Scholar]
- 24. Gennarelli TA, Thibault LE, Adams JH, Graham DI, Thompson CJ, Marcincin RP (1982) Diffuse axonal injury and traumatic coma in the primate. Ann Neurol 12:564–574. [DOI] [PubMed] [Google Scholar]
- 25. Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW (1993) Beta‐amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett 160:139–144. [DOI] [PubMed] [Google Scholar]
- 26. Gentleman SM, Roberts GW, Gennarelli TA, Maxwell WL, Adams JH, Kerr S et al (1995) Axonal injury: a universal consequence of fatal closed head injury? Acta Neuropathol 89:537–543. [DOI] [PubMed] [Google Scholar]
- 27. Grady MS, McLaughlin MR, Christman CW, Valadka AB, Fligner CL, Povlishock JT (1993) The use of antibodies targeted against the neurofilament subunits for the detection of diffuse axonal injury in humans. J Neuropathol Exp Neurol 52:143–152. [DOI] [PubMed] [Google Scholar]
- 28. Graham DI, Adams JH, Nicoll JA, Maxwell WL, Gennarelli TA (1995) The nature, distribution and causes of traumatic brain injury. Brain Pathol 5:397–406. [DOI] [PubMed] [Google Scholar]
- 29. Graham DI, Smith C, Reichard R, Leclercq PD, Gentleman SM (2004) Trials and tribulations of using beta‐amyloid precursor protein immunohistochemistry to evaluate traumatic brain injury in adults. Forensic Sci Int 146:89–96. [DOI] [PubMed] [Google Scholar]
- 30. Greer JE, McGinn MJ, Povlishock JT (2011) Diffuse traumatic axonal injury in the mouse induces atrophy, c‐Jun activation, and axonal outgrowth in the axotomized neuronal population. J Neurosci 31:5089–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hayashi T, Ago K, Ago M, Ogata M (2009) Two patterns of beta‐amyloid precursor protein (APP) immunoreactivity in cases of blunt head injury. Leg Med (Tokyo) 11(Suppl. 1):S171–173. [DOI] [PubMed] [Google Scholar]
- 32. Iwata A, Chen XH, McIntosh TK, Browne KD, Smith DH (2002) Long‐term accumulation of amyloid‐beta in axons following brain trauma without persistent upregulation of amyloid precursor protein genes. J Neuropathol Exp Neurol 61:1056–1068. [DOI] [PubMed] [Google Scholar]
- 33. Iwata A, Stys PK, Wolf JA, Chen XH, Taylor AG, Meaney DF et al (2004) Traumatic axonal injury induces proteolytic cleavage of the voltage‐gated sodium channels modulated by tetrodotoxin and protease inhibitors. J Neurosci 24:4605–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Johnson VE, Meaney DF, Cullen DK, Smith DH (2015) Animal models of traumatic brain injury. Handb Clin Neurol 127:115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W (2013) Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 136:28–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Johnson VE, Stewart W, Smith DH (2013) Axonal pathology in traumatic brain injury. Exp Neurol 246:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Johnson VE, Stewart W, Weber MT, Cullen DK, Siman R, Smith DH (2016) SNTF immunostaining reveals previously undetected axonal pathology in traumatic brain injury. Acta Neuropathol 131:115–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Johnson VE, Weber MT, Xiao R, Cullen DK, Meaney DF, Stewart W et al (2018) Mechanical disruption of the blood‐brain barrier following experimental concussion. Acta Neuropathol 135:711–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kampfl A, Posmantur R, Nixon R, Grynspan F, Zhao X, Liu SJ et al (1996) mu‐calpain activation and calpain‐mediated cytoskeletal proteolysis following traumatic brain injury. J Neurochem 67:1575–1583. [DOI] [PubMed] [Google Scholar]
- 40. Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI (2014) Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol 73:14–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mac Donald CL, Dikranian K, Song SK, Bayly PV, Holtzman DM, Brody DL (2007) Detection of traumatic axonal injury with diffusion tensor imaging in a mouse model of traumatic brain injury. Exp Neurol 205:116–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marmarou CR, Walker SA, Davis CL, Povlishock JT (2005) Quantitative analysis of the relationship between intra‐ axonal neurofilament compaction and impaired axonal transport following diffuse traumatic brain injury. J Neurotrauma 22:1066–1080. [DOI] [PubMed] [Google Scholar]
- 43. Maxwell WL, Domleo A, McColl G, Jafari SS, Graham DI (2003) Post‐acute alterations in the axonal cytoskeleton after traumatic axonal injury. J Neurotrauma 20:151–168. [DOI] [PubMed] [Google Scholar]
- 44. Maxwell WL, Watt C, Pediani JD, Graham DI, Adams JH, Gennarelli TA (1991) Localisation of calcium ions and calcium‐ATPase activity within myelinated nerve fibres of the adult guinea‐pig optic nerve. J Anat 176:71–79. [PMC free article] [PubMed] [Google Scholar]
- 45. McGinn MJ, Kelley BJ, Akinyi L, Oli MW, Liu MC, Hayes RL et al (2009) Biochemical, structural, and biomarker evidence for calpain‐mediated cytoskeletal change after diffuse brain injury uncomplicated by contusion. J Neuropathol Exp Neurol 68:241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Meaney DF, Smith DH, Shreiber DI, Bain AC, Miller RT, Ross DT et al (1995) Biomechanical analysis of experimental diffuse axonal injury. J Neurotrauma 12:689–694. [DOI] [PubMed] [Google Scholar]
- 47. Mustafa AG, Singh IN, Wang J, Carrico KM, Hall ED (2010) Mitochondrial protection after traumatic brain injury by scavenging lipid peroxyl radicals. J Neurochem 114:271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mustafa AG, Wang JA, Carrico KM, Hall ED (2011) Pharmacological inhibition of lipid peroxidation attenuates calpain‐mediated cytoskeletal degradation after traumatic brain injury. J Neurochem 117:579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nakagawa Y, Nakamura M, McIntosh TK, Rodriguez A, Berlin JA, Smith DH et al (1999) Traumatic brain injury in young, amyloid‐beta peptide overexpressing transgenic mice induces marked ipsilateral hippocampal atrophy and diminished Abeta deposition during aging. J Comp Neurol 411:390–398. [PubMed] [Google Scholar]
- 50. Okonkwo DO, Povlishock JT (1999) An intrathecal bolus of cyclosporin A before injury preserves mitochondrial integrity and attenuates axonal disruption in traumatic brain injury. J Cereb Blood Flow Metab 19:443–451. [DOI] [PubMed] [Google Scholar]
- 51. Pettus EH, Christman CW, Giebel ML, Povlishock JT (1994) Traumatically induced altered membrane permeability: its relationship to traumatically induced reactive axonal change. J Neurotrauma 11:507–522. [DOI] [PubMed] [Google Scholar]
- 52. Povlishock JT (1992) Traumatically induced axonal injury: pathogenesis and pathobiological implications. Brain Pathol 2:1–12. [PubMed] [Google Scholar]
- 53. Povlishock JT, Becker DP (1985) Fate of reactive axonal swellings induced by head injury. Lab Invest 52:540–552. [PubMed] [Google Scholar]
- 54. Povlishock JT, Becker DP, Cheng CLY, Vaughan GW (1983) Axonal change in minor head injury. J Neuropathol Exp Neurol 42:225–242. [DOI] [PubMed] [Google Scholar]
- 55. Povlishock JT, Buki A, Koiziumi H, Stone J, Okonkwo DO (1999) Initiating mechanisms involved in the pathobiology of traumatically induced axonal injury and interventions targeted at blunting their progression. Acta Neurochir Suppl 73:15–20. [DOI] [PubMed] [Google Scholar]
- 56. Povlishock JT, Christman CW (1995) The pathobiology of traumatically induced axonal injury in animals and humans: a review of current thoughts. J Neurotrauma 12:555–564. [DOI] [PubMed] [Google Scholar]
- 57. Povlishock JT, Katz DI (2005) Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil 20:76–94. [DOI] [PubMed] [Google Scholar]
- 58. Povlishock JT, Marmarou A, McIntosh T, Trojanowski JQ, Moroi J (1997) Impact acceleration injury in the rat: evidence for focal axolemmal change and related neurofilament sidearm alteration. J Neuropathol Exp Neurol 56:347–359. [PubMed] [Google Scholar]
- 59. Povlishock JT, Pettus EH (1996) Traumatically induced axonal damage: evidence for enduring changes in axolemmal permeability with associated cytoskeletal change. Acta Neurochir Suppl 66:81–86. [DOI] [PubMed] [Google Scholar]
- 60. Ramon Y, Cajal S (1928) Degeneration and Regeneration in the Nervous System. Oxford University Press: Oxford. [Google Scholar]
- 61. Rand CW, Courville CB (1946) Histologic changes in the brain in cases of fatal injury to the head; alterations in nerve cells. Arch Neurol Psychiatry 55:79–110. [DOI] [PubMed] [Google Scholar]
- 62. Reeves TM, Phillips LL, Povlishock JT (2005) Myelinated and unmyelinated axons of the corpus callosum differ in vulnerability and functional recovery following traumatic brain injury. Exp Neurol 196:126–137. [DOI] [PubMed] [Google Scholar]
- 63. Reeves TM, Smith TL, Williamson JC, Phillips LL (2012) Unmyelinated axons show selective rostrocaudal pathology in the corpus callosum after traumatic brain injury. J Neuropathol Exp Neurol 71:198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Reichard RR, Smith C, Graham DI (2005) The significance of beta‐APP immunoreactivity in forensic practice. Neuropathol Appl Neurobiol 31:304–313. [DOI] [PubMed] [Google Scholar]
- 65. Ross DT, Meaney DF, Sabol MK, Smith DH, Gennarelli TA (1994) Distribution of forebrain diffuse axonal injury following inertial closed head injury in miniature swine. Exp Neurol 126:291–299. [DOI] [PubMed] [Google Scholar]
- 66. Saatman KE, Abai B, Grosvenor A, Vorwerk CK, Smith DH, Meaney DF (2003) Traumatic axonal injury results in biphasic calpain activation and retrograde transport impairment in mice. J Cereb Blood Flow Metab 23:34–42. [DOI] [PubMed] [Google Scholar]
- 67. Saatman KE, Bozyczko‐Coyne D, Marcy V, Siman R, McIntosh TK (1996) Prolonged calpain‐mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol 55:850–860. [DOI] [PubMed] [Google Scholar]
- 68. Schweitzer JB, Park MR, Einhaus SL, Robertson JT (1993) Ubiquitin marks the reactive swellings of diffuse axonal injury. Acta Neuropathol 85:503–507. [DOI] [PubMed] [Google Scholar]
- 69. Sherriff FE, Bridges LR, Sivaloganathan S (1994) Early detection of axonal injury after human head trauma using immunocytochemistry for beta‐amyloid precursor protein. Acta Neuropathol (Berl) 87:55–62. [DOI] [PubMed] [Google Scholar]
- 70. Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM et al (1992) Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science 258:126–129. [DOI] [PubMed] [Google Scholar]
- 71. Singleton RH, Stone JR, Okonkwo DO, Pellicane AJ, Povlishock JT (2001) The immunophilin ligand FK506 attenuates axonal injury in an impact‐acceleration model of traumatic brain injury. J Neurotrauma 18:607–614. [DOI] [PubMed] [Google Scholar]
- 72. Smith DH, Chen XH, Pierce JE, Wolf JA, Trojanowski JQ, Graham DI et al (1997) Progressive atrophy and neuron death for one year following brain trauma in the rat. J Neurotrauma 14:715–727. [DOI] [PubMed] [Google Scholar]
- 73. Smith DH, Chen XH, Xu BN, McIntosh TK, Gennarelli TA, Meaney DF (1997) Characterization of diffuse axonal pathology and selective hippocampal damage following inertial brain trauma in the pig. J Neuropathol Exp Neurol 56:822–834. [PubMed] [Google Scholar]
- 74. Smith DH, Hicks R, Povlishock JT (2013) Therapy development for diffuse axonal injury. J Neurotrauma 30:307–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Smith DH, Johnson VE, Stewart W (2013) Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol 9:211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Smith DH, Meaney DF (2000) Axonal damage in traumatic brain injury. Neuroscientist 6:483–495. [Google Scholar]
- 77. Smith DH, Meaney DF, Shull WH (2003) Diffuse axonal injury in head trauma. J Head Trauma Rehabil 18:307–316. [DOI] [PubMed] [Google Scholar]
- 78. Smith DH, Nonaka M, Miller R, Leoni M, Chen XH, Alsop D et al (2000) Immediate coma following inertial brain injury dependent on axonal damage in the brainstem. J Neurosurg 93:315–322. [DOI] [PubMed] [Google Scholar]
- 79. Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF et al (1995) A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma 12:169–178. [DOI] [PubMed] [Google Scholar]
- 80. Smith DH, Wolf JA, Lusardi TA, Lee VM, Meaney DF (1999) High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. J Neurosci 19:4263–4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Staal JA, Dickson TC, Gasperini R, Liu Y, Foa L, Vickers JC (2010) Initial calcium release from intracellular stores followed by calcium dysregulation is linked to secondary axotomy following transient axonal stretch injury. J Neurochem 112:1147–1155. [DOI] [PubMed] [Google Scholar]
- 82. Stone JR, Singleton RH, Povlishock JT (2001) Intra‐axonal neurofilament compaction does not evoke local axonal swelling in all traumatically injured axons. Exp Neurol 172:320–331. [DOI] [PubMed] [Google Scholar]
- 83. Strich SJ (1956) Diffuse degeneration of the cerebral white matter in severe dementia following head injury. J Neurol Neurosurg Psychiatry 19:163–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tang‐Schomer MD, Johnson VE, Baas PW, Stewart W, Smith DH (2012) Partial interruption of axonal transport due to microtubule breakage accounts for the formation of periodic varicosities after traumatic axonal injury. Exp Neurol 233:364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tang‐Schomer MD, Patel AR, Baas PW, Smith DH (2010) Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule disassembly, and axon degeneration. FASEB J 24:1401–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Tomaiuolo F, Bivona U, Lerch JP, DiPaola M, Carlesimo GA, Ciurli P et al (2012) Memory and anatomical change in severe non missile traumatic brain injury: approximately 1 vs. approximately 8 years follow‐up. Brain Res Bull 87:373–382. [DOI] [PubMed] [Google Scholar]
- 87. von Reyn CR, Spaethling JM, Mesfin MN, Ma M, Neumar RW, Smith DH et al (2009) Calpain mediates proteolysis of the voltage‐gated sodium channel alpha‐subunit. J Neurosci 29:10350–10356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH (2001) Traumatic axonal injury induces calcium influx modulated by tetrodotoxin‐sensitive sodium channels. J Neurosci 21:1923–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yaghmai A, Povlishock J (1992) Traumatically induced reactive change as visualized through the use of monoclonal antibodies targeted to neurofilament subunits. J Neuropathol Exp Neurol 51:158–176. [DOI] [PubMed] [Google Scholar]
- 90. Ziogas NK, Koliatsos VE (2018) Primary traumatic axonopathy in mice subjected to impact acceleration: a reappraisal of pathology and mechanisms with high‐resolution anatomical methods. J Neurosci 38:4031–4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Video of 3D volume of tissue showing peri‐contusional APP immunoreactive axonal pathology in cleared tissue 24 h following CCI. Color gradation denotes the depth in the z‐plane. Note the extensive pathology displaying a complex array of morphologies including disconnected axonal bulbs and periodic axonal varicosities, often with multiple swellings along the length of injured axons.
Video S2. Video showing peri‐contusional APP immunoreactive axonal profiles (green) through the z‐plane in continuous 1‐µm z‐steps (50 µm total). Axonal bulbs can be followed through the tissue with varying morphologies and connectivity status. Note that some neuronal soma are also APP immunoreactive, though clearly differentiated from axonal swellings by the presence of a nucleus.
Video S3. Video displaying a single post‐CCI APP immunoreactive (green) varicose axon at high magnification tracked through the z‐plane in 1µm increments. While standard FFPE‐equivalent analysis identifies the two large axonal swellings as terminal bulbs, CLARITY analysis reveals these two bulbs are in fact connected and part of the same axon as periodic varicose swellings.