

Abstract
The second messenger, cAMP, is highly compartmentalized to facilitate signaling specificity. Extracellular vesicles (EVs) are submicron, intact vesicles released from many cell types that can act as biomarkers or be involved in cell-to-cell communication. Although it is well recognized that EVs encapsulate functional proteins and RNAs/miRNAs, currently it is unclear whether cyclic nucleotides are encapsulated within EVs to provide an additional second messenger compartment. Using ultracentrifugation, EVs were isolated from the culture medium of unstimulated systemic and pulmonary endothelial cells. EVs were also isolated from pulmonary microvascular endothelial cells (PMVECs) following stimulation of transmembrane adenylyl cyclase (AC) in the presence or absence of the phosphodiesterase 4 inhibitor rolipram over time. Whereas cAMP was detected in EVs isolated from endothelial cells derived from different vascular beds, it was highest in EVs isolated from PMVECs. Treatment of PMVECs with agents that increase near-membrane cAMP led to an increase in cAMP within corresponding EVs, yet there was no increase in EV number. Elevated cell cAMP, measured by whole cell measurements, peaked 15 min after treatment, yet in EVs the peak increase in cAMP was delayed until 60 min after cell stimulation. Cyclic AMP was also increased in EVs collected from the perfusate of isolated rat lungs stimulated with isoproterenol and rolipram, thus corroborating cell culture findings. When added to unperturbed confluent PMVECs, EVs containing elevated cAMP were not barrier disruptive like cytosolic cAMP but maintained monolayer resistance. In conclusion, PMVECs release EVs containing cAMP, providing an additional compartment to cAMP signaling.
Keywords: cyclic adenosine monophosphate, endothelium, extracellular vesicles, isolated lung, phosphodiesterase
INTRODUCTION
The second messenger cyclic AMP (cAMP) regulates a panoply of cellular and tissue responses, such as metabolism, endothelial barrier function, muscle contraction, and proliferation (43). Membrane-delimited adenylyl cyclases (ACs) generate this diffusible second messenger in response to activation of either Gαs-protein coupled receptors or ACs directly (17). In turn, cAMP activates downstream effectors, such as protein kinase A (PKA), exchange protein activated by cAMP (Epac), cyclic nucleotide gated (CNG) channels and hyperpolarization-activated cyclic nucleotide-gated (HCN) channels. The duration and amplitude of the cAMP signal is regulated not only by production but also by its degradation through phosphodiesterase (PDE) activity, which hydrolyzes cAMP to 5′-AMP. Growing evidence supports the highly compartmentalized nature of cAMP signals within the cell; thus, although PDEs hydrolyze cAMP to terminate the second messenger signal, they also act to spatially confine the signal by forming a functional barricade that acts to hydrolyze the signal before it can diffuse to distant sites. In this way, PDEs control not only amplitude and duration but also spatial distribution of the signal (44). Ultimately, these confined compartments are important in maintaining signaling specificity and tissue responses.
In addition to the intracellular cAMP pool that regulates intracellular signaling, cAMP can also be extruded into the extracellular space, where it contributes directly to intercellular communication (24). Early studies revealed a probenecid-sensitive mechanism for cAMP extrusion from pigeon erythrocytes and was followed by discoveries that glucagon induced an increase in cAMP concentration in the blood and urine due to release of cAMP by the liver (8, 10). Other tissues release cAMP into the blood to regulate coagulation and cardiomyocyte function and as a biomarker of leukemia (35, 37, 41, 48, 51). More recently, secreted vesicles provide a novel dimension to extracellular cell-cell communication, yet currently the role of these vesicles as carriers of cAMP has not been investigated.
Cellular material can be released from cells through the secretion of mobile vesicles encapsulated within a lipid bilayer. Collectively, these heterogeneous mobile vesicles are known as extracellular vesicles (EVs) and include exosomes, microparticles or microvesicles, and apoptotic bodies (13, 22). Exosomes are formed via the invagination of an endosomal membrane, which captures cytosolic components within intraluminal vesicles. Subsequently, endosomes give rise to multivesicular bodies, which fuse with the plasma membrane to release these intraluminal vesicles, exosomes, into the extracellular space. In contrast, microparticles form when the plasma membrane pinches off directly encapsulating cytosolic components but bears receptors and surface markers from the plasma membrane of the parent cell. Finally, apoptotic bodies are released during cell death following activation of the apoptotic pathway. EVs are released constitutively during normal cellular homeostasis.
Release of EVs can also be induced in response to stimuli such as inflammation, angiogenesis, or thrombosis, and in turn, EVs have been implicated in many diseases, including cardiovascular disease, cancer, and neurodegenerative disorders, as well as lung diseases, such as acute lung injury, COPD, and pulmonary hypertension (2, 5, 7, 16, 31, 47). Many different cell types release EVs, and when released from the apical surface of endothelial cells these vesicles are released into the blood (22). EVs can display surface markers from the parent cell, such as selectins and integrins, and have the ability to activate downstream cells through expression of adhesion molecules or receptors. In addition, EVs carry bioactive molecules from the parent cell, such as microRNA, proteins, and lipids (4, 9, 12, 16, 18, 39). Collectively, the surface markers and cargo carried by the EVs can activate target cells or deliver cargo to them.
Although it is well recognized that cAMP is extruded from cells into blood, urine, and cerebrospinal fluid (21, 24), it is unclear whether vascular endothelial cells release cAMP encapsulated within EVs. Indeed, EVs provide a novel mechanism for the release of cAMP signals from a cell. Thus, the goal of this study was to determine whether endothelial cells release EVs that contain cAMP and whether PMVECs or the isolated lung could be stimulated to increase cAMP within these EVs.
MATERIALS AND METHODS
Cell Culture
Pulmonary microvascular endothelial cells (PMVECs), pulmonary artery endothelial cells (PAECs), and aortic endothelial cells of rat origin were obtained from the Cell Culture Core, Center for Lung Biology at the University of South Alabama. Isolation and characterization of these cells has been described previously (28). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Mediatech, Manassas, VA) with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals; Lawrenceville, GA) and 1% penicillin-streptomycin solution (Invitrogen, Carlsbad, CA) at 37°C in 21% O2 and 5% CO2.
Cell Treatments
Cells were seeded in sterile, 150-mm culture dishes, and upon confluence growth media were replaced with serum-free media. The cells were returned to the incubator for 10 min and subsequently treated with a combination of forskolin (NKH477; Tocris Bioscience, Bristol, UK) or isoproterenol (Sigma-Aldrich, St. Louis, MO) and/or rolipram (Sigma-Aldrich, St. Louis, MO) or appropriate vehicle controls (NKH477 and isoproterenol are in water while rolipram is in DMSO), as indicated for the appropriate time interval and as described previously (45). For multidrug resistant protein (MRP) inhibitor studies, following 15-min serum starvation, MK571 (20 µM) was added to the cells for an additional 5 min before isoproterenol and rolipram treatment for an additional 60 min. Following drug or vehicle control treatment over time, the media were collected for EV isolation. Cell lysates were collected for cAMP and protein analysis.
Isolation of EVs and Media
EVs were isolated from the culture media or isolated lung perfusate using a low-speed followed by high-speed centrifugation protocol, as described previously (46). The supernatant from the first 10-min, 1,000-g centrifugation, which removed dead cells and cellular debris, underwent ultracentrifugation at 100,000 g for 1 h at 4°C (Avanti J-30i; Beckman Coulter, Fullerton, CA). The media supernatant was removed and retained while the intact EV pellet was resuspended in buffer for cAMP, protein analysis, EV counts, or transmission electron microscopy (TEM). For transelectrical resistance experiments, the EV pellet underwent an additional rinse and ultracentrifugation step to remove residual drugs.
EV and Whole Cell cAMP
Two separate techniques were used for cAMP analysis.
cAMP enzyme immunoassay.
For cAMP-enzyme immunoassay (EIA), the EV pellet was resuspended in 1 N HCl and normalized with appropriate volume of 1 N NaOH. Similarly, cells were lysed in 1 N HCl and 1 N NaOH, as described previously (36, 45). Cyclic AMP-EIA was performed in triplicate following the manufacturer’s instructions (Cayman Chemical), and levels were normalized to protein content (bicinchoninic acid protein assay kit; Sigma).
Cyclic AMP analysis by HPLC-MS/MS.
For high-performance liquid chromatography-mass spectrometry/mass spectrometry (HPLC-MS/MS) cAMP analysis, the EV pellet was resuspended in ice cold extraction solution consisting of acetonitrile-methanol-water [2:2:1 (vol/vol/vol)], as described previously (3). Once the culture media were removed, the cells were lysed in ice-cold extraction solution. Once resuspended in extraction solution, the EV and cell lysate samples were heated for 20 min at 98°C. After cooling, the samples were centrifuged at 20,000 g for 10 min, and the pellet was resuspended in 100 mM NaOH and protein analysis performed (Pierce bicinchoninic acid protein assay kit; Thermofisher). Supernatants from the 20,000-g centrifugation were transferred into separate centrifuge tubes and samples evaporated completely using speed vacuum. The residue in each tube was dissolved in water, and cAMP quantification was performed by HPLC-MS/MS following separation on an Aglient (Waldbronn, Germany) 1100 series, as described in detail (3).
EV Counts By Flow Cytometry
The EV pellet was resuspended in 1 ml of PBS, and counts were performed using flow cytometry (BD FACSCanto II, BD FACSDiva Software) in the University of South Alabama College of Medicine Flow Cytometry Laboratory, as described previously (46). Only particles <1 µM in diameter were counted in the analysis. Particle counts were obtained for each sample and normalized with the addition of a known amount of CountBright absolute counting beads (7 µm in diameter; Molecular Probes, Eugene, OR) per sample volume.
Isolated Lung Studies
Lungs from male Sprague-Dawley rats (Charles River Laboratories International,) were isolated as described previously (34, 36, 45). All experimental procedures were performed in accordance with current provisions of the US Animal Welfare Act and were approved by the Institutional Animal Care and Use Committee of the University of South Alabama. Briefly, animals were anesthetized with pentobarbital sodium (60 mg/kg ip), a tracheotomy catheter was inserted, and the lungs were ventilated with room air until the heart was cannulated, at which time the lungs were ventilated with 21% O2, 5% CO2, and 74% N2. After a median sternotomy, heparin was administered (200 U) and allowed to circulate. The pulmonary artery was cannulated via the right ventricle and the circulation completed via cannulation of the left ventricle. The lungs were perfused at constant flow at 37°C with phosphate-buffered saline solution (PBS) containing (in mM) 119 NaCl, 4.7 KCl, 1.17 MgSO4·7 H2O, 22.6 NaHCO3, 1.18 KH2PO4, 5.5 glucose, and 3.2 CaCl2·2 H2O and 4% albumin for osmotic stabilization. The lungs and heart were removed en bloc. After flushing, isoproterenol (1 µM) and rolipram (10 µM) or vehicle control were perfused through the pulmonary vasculature for 1 h, at which time the perfusate was collected for EV isolation. One lung was used for each experiment, with a total of four independent lungs for vehicle control and four independent lungs for isoproterenol and rolipram treatment.
Transendothelial Electrical Resistance
Transendothelial electrical resistance (TER) studies were performed as described previously (34, 36). In brief, PMVECs were seeded onto polycarbonate wells containing small evaporated gold electrodes in series with a large gold counterelectrode (10−3 cm, 8W10E+; Applied Biophysics, Troy, NY). Resistance across the monolayer was measured using an electrical cell impedance sensing (ECIS) system (Applied Biophysics) in a humidified chamber at 37°C, 5% CO2, and 21% O2. Upon confluence, the culture media were exchanged for serum-free medium, and a stable baseline TER was recorded over several hours (data not shown). Following a stable baseline, vehicle control or EVs isolated from PMVECs treated with either vehicle control or isoproterenol and rolipram (60 min) were added, and TER was monitored over several hours. Data for each microelectrode was normalized to baseline. Four separate experiments were performed with at least two wells per condition for each experiment.
Transmission Electron Microscopy
Following 60-min isoproterenol (1 µM) and rolipram (10 µM) versus vehicle control, EVs were isolated from PMVEC media (as described above). The EV suspension was fixed in 2% formaldehyde, deposited on Formvar-carbon coated EM grids (Electron Microscopic Sciences, Hatfield, PA), and allowed to adsorb onto the grid for 20 min in a dry environment. PBS was added onto the grid and blotted dry with filter paper, followed by a 5-min incubation with 1% glutaraldehyde. After eight washes with water, the grids were double stained with UranyLess/Lead citrate (Electron Microscopic Sciences, Hatfield, PA) according to the manufacturer’s instructions. Samples were imaged using LEOL 1400 Transmission Electron Microscope (Peabody, MA).
Graphing and Statistical Analysis
Analytical data represent analysis of at least three independent experiments and are reported as means ± SE. Comparison between data groups was performed using a one-way or two-way ANOVA with the Tukey post hoc test or Student’s t-test as appropriate, and values of P < 0.05 were considered significant. Data were graphed using GraphPad Prism (GraphPad Software, La Jolla, CA).
RESULTS
Systemic and Pulmonary Endothelial Cells Release cAMP Containing EVs
Endothelial cells from organ-specific vascular beds and segment-specific regions of vascular beds within the same organ exhibit molecular and functional heterogeneity (20). To determine whether EVs released from these unique vascular beds contain the signaling molecule cAMP, EVs were collected from endothelial cells isolated from either the systemic circulation or segment-specific regions of the pulmonary circulation. Aortic endothelial cells line conduit arteries in the systemic circulation. In the pulmonary circulation, PMVECs line the capillary bed where gas exchange occurs and PAECs line conduit arteries. Each endothelial phenotype has measurable, basal, whole cell cAMP, yet PMVECs have a higher basal cAMP compared with aortic endothelial cells and PAECs (Fig. 1A). Furthermore, systemic and pulmonary endothelial cells constitutively release EVs that contain cAMP; however, EVs released from PMVECs contain significantly higher cAMP compared with either aortic endothelial cells or PAECs (Fig. 1B). Thus, endothelial cells from different vascular beds or segment-specific regions within the same vascular bed exhibit heterogeneity not only in the basal whole cell cAMP but also in the amount of cAMP released into EVs.
Fig. 1.

Extracellular vesicles (EVs) derived from endothelial cells of different vascular beds contain cAMP. cAMP was determined in whole cell lysate (A) and EVs (B) collected from unstimulated endothelial cells isolated from either the systemic circulation (aortic endothelial cells) or different vascular segments of pulmonary circulation, i.e., pulmonary endothelial cells: pulmonary microvascular endothelial cells (MV) or pulmonary artery endothelial cells (artery). EVs were isolated from the culture media, as described in methods. cAMP was normalized to protein content (n = 3; **P < 0.01 and ***P < 0.001, 1-way ANOVA with the Tukey post hoc test). ● and ○, Individual data points.
To test whether free cAMP is pelleted during this high-speed centrifugation, control experiments were performed in which free cAMP was added to untreated media and centrifuged at 100,000 g for 1 h. Following centrifugation of this cAMP-spiked media, no cAMP could be recovered within a pellet, suggesting that cAMP was indeed encapsulated within EVs (data not shown). Thus, although PMVECs constitutively release cAMP-rich EVs, it is unclear whether PMVECs can be stimulated to increase EV-cAMP levels.
Increasing cAMP in PMVECs Leads to an Increase in EV cAMP and not EV Number
To examine whether near-membrane cAMP is packaged into EVs following stimulation of transmembrane AC, PMVECs were treated with forskolin (100 µM) for 15 min in the absence or presence of the PDE4 inhibitor rolipram (10 µM) (14, 15, 50). Forskolin increased whole cell cAMP in PMVECs, which was synergistically increased with the combination of both forskolin and rolipram (Fig. 2A). Although there was no significant increase in cAMP within EVs collected from PMVECs stimulated with either forskolin or rolipram alone, there was a significant increase in cAMP in EVs collected from PMVECs stimulated with forskolin and rolipram combined (Fig. 2B). To determine whether activation transmembrane AC through G protein coupling similarly increased EV-cAMP, PMVECs were stimulated with isoproterenol alone or in combination with rolipram. EIA is routinely used for cAMP measurements, but recently, a mass spectrometry approach has been developed (6). Thus, in these studies we used either EIA (Fig. 3, A and B) or mass spectrometry (Fig. 3, C and D) to measure cAMP. Whereas isoproterenol alone had no effect on whole cell- or EV-cAMP, 15-min stimulation of PMVECs with the combination of isoproterenol and rolipram led to a significant increase in both whole cell- (Fig. 3, A and C) and EV-cAMP (Fig. 3, B and D).
Fig. 2.

Direct activation of transmembrane adenylyl cyclase (AC) in PMVECs increases cellular and extracellular vesicle (EV)-cAMP. Pulmonary microvascular endothelial cells were stimulated with or without forskolin (100 µM) in the presence or absence of rolipram (10 µM) for 15 min and cell lysates and EV isolated. cAMP in the whole cell lysates (A) or EVs (B) was analyzed and normalized to protein content (n = 3; ***P < 0.001, **P < 0.01, and *P < 0.05, 1-way ANOVA with the Tukey post hoc test). ● and ○, Individual data points.
Fig. 3.

Isoproterenol activation of transmembrane adenylyl cyclase (AC) in pulmonary microvascular endothelial cells (PMVECs) increases cellular and extracellular vesicle (EV)-cAMP. PMVECs were stimulated for 15 min with or without isoproterenol in the presence or absence of rolipram and cell lysates (A and C) and EVs (B and D) analyzed for cAMP by either EIA (A and B) or mass spectrometry (C and D). cAMP was normalized to protein content (n = 3; ***P < 0.001, **P < 0.01, 1-way ANOVA with the Tukey post hoc test). ● and ○, Individual data points.
In addition to cAMP, the mass spectrometry approach reports the presence of other canonical cyclic nucleotides, such as cGMP, and noncanonical cyclic nucleotides, including cCMP and cUMP. In these studies, whereas cGMP, cCMP, or cUMP is detected in cell lysates of unstimulated PMVECs, there was no isoproterenol and/or rolipram stimulated increase. Neither cGMP, cCMP, nor cUMP was detected in EVs collected from isoproterenol and/or rolipram-stimulated cells (data not shown). To confirm these negative findings, we simultaneously analyzed cell lysates from PMVECs treated with Type Three Secretion System competent Pseudomonas aeruginosa expressing the soluble AC exotoxin, ExoY (P. aeruginosa ExoY+). Our findings corroborated previous findings that both canonical cyclic nucleotides cAMP and cGMP as well as noncanonical cyclic nucleotides cCMP and cUMP were detected in PMVEC cell lysates inoculated for 4 h with P. aeruginosa ExoY+ (data not shown) (33). Thus, collectively, the data from the P. aeruginosa ExoY+ studies strengthen our data that only cAMP is detected in PMVECs following stimulation of transmembrane AC.
The increase in cAMP detected in EVs could be accounted for by an increase in the number of EVs released following stimulation; thus, EVs counts were performed following 15-min stimulation with isoproterenol, rolipram, or the combination of both. These studies revealed that there was no significant increase in EV number released from control versus stimulated cells (Fig. 4) and support the finding that in PMVECs cAMP generated in the near membrane compartment is encapsulated into EVs.
Fig. 4.
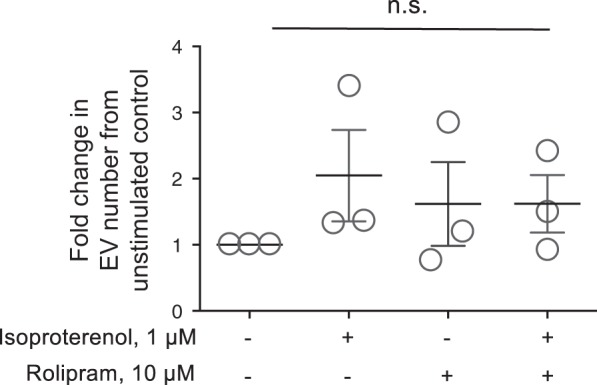
Isoproterenol activation of transmembrane adenylyl cyclase (AC) does not increase the number of extracellular vesicle (EV) released. Pulmonary microvascular endothelial cells were stimulated without or with isoproterenol (1 µM) in the presence or absence of rolipram (10 µM), and EVs were isolated as described in materials and methods. EV counts were determined by flow cytometry of particles <1 µM in diameter [n = 3; not significant (NS), 1-way ANOVA with the Tukey post hoc test]. ○, Individual data points.
EV-cAMP Increases Over Time
To understand the temporal nature of cAMP released into EVs, PMVECs were stimulated with isoproterenol and rolipram over time (0, 5, 15, 30, and 60 min). An increase in whole cell cAMP was detected 5 min after stimulation with isoproterenol and rolipram and peaked 15 min after stimulation; however, following 30-min stimulation with isoproterenol and rolipram, cAMP levels did not further increase but began to decrease (Fig. 5A). Indeed, 15 min after stimulation, whole cell cAMP increased 150-fold above baseline and declined to a 38-fold increase above baseline 60 min after isoproterenol and rolipram stimulation. While EV-cAMP increased only 26-fold above baseline at the 15-min time point, 60 min after stimulation of cells with isoproterenol and rolipram, EV-cAMP increased 170-fold above baseline (Fig. 5B). Thus, these studies reveal a temporal delay in cAMP packaging into EVs; whereas the peak increase in whole cell cAMP occurs 15 min after agonist stimulation, the maximum increase in cAMP packaged into EVs occurs 60 min after stimulation of PMVECs with isoproterenol and rolipram (Fig. 5C).
Fig. 5.

Agonist stimulation increases extracellular vesicle (EV)-cAMP over time. A and B: pulmonary microvascular endothelial cells were stimulated with isoproterenol and rolipram over time (0–60 min), and cell lysates (A) and EVs (B) were analyzed for cAMP and normalized to protein concentration. C: the ratio of cAMP/mg protein from EVs vs. cell lysate for each time point (n = 5; ***P < 0.001, **P < 0.01, and *P < 0.05 vs. baseline, Student’s t-test). ●, ○, and ■, Individual data points.
Isolation of EVs using high-speed centrifugation provides a sample containing a multitude of various-sized vesicles (11). Thus, TEM was used to characterize the size of this vesicle population and consider differences between the two treatment groups. Figure 6, A–D, clearly demonstrates the presence of exosomes (≤150 nm) as well as microparticles (≥100 nm up to 1 μM). Indeed, these particles exhibit the expected morphology and size for both exosomes and microparticles. Treatment with isoproterenol and rolipram (Fig. 6, C and D) did not alter the size distribution of the EVs compared with control EVs (Fig. 6, A and B).
Fig. 6.

Transmission electron microscopy (TEM) analysis of extracellular vesicles (EVs). EVs isolated from either unstimulated or isoproterenol- (1 µM) and rolipram-stimulated (10 µM) (60 min) pulmonary microvascular endothelial cells were fixed and counterstained for TEM. EVs from both unstimulated (A and B) and isoproterenol- and rolipram-stimulated (C and D) pulmonary microvascular endothelial cells exhibit characteristics of intact, heterogenous vesicles. The EVs range in size from exosomes (≤150 nm) to microparticles (≥200 nm up to 1 μM). Representative images from 6 independent preparations for each condition.
cAMP is Extruded from PMVECs Through Multidrug-Resistant Proteins
Cyclic AMP efflux from the intracellular space into the surrounding extracellular fluid occurs in a variety of cells and is mediated by ATP-dependent transporters. These multidrug-resistant protein (MPR) transporters are sensitive to probenecid and MK571. When PMVECs were stimulated with isoproterenol and rolipram for 60 min in the presence of MK571 (20 µM), there was no further increase in whole cell cAMP, yet there was a significant decrease in cAMP effluxed into the media (Fig. 7). Indeed, MK571 induced a >90% decrease in cAMP in the EV-depleted media compared with untreated cells, yet MK571 only attenuated EV-cAMP by 40%. Thus, although inhibiting MRPs blocked cAMP efflux into the extracellular media, cAMP was still packaged into EVs.
Fig. 7.
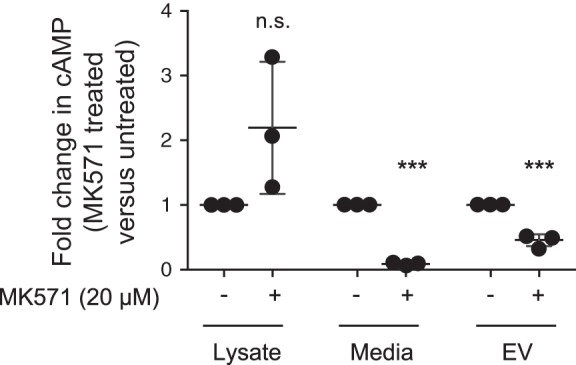
MK571 attenuates extracellular vesicle (EV) cAMP as well as cAMP efflux. Pulmonary microvascular endothelial cells were pretreated with or without MK571 (20 µM) for 5 min before stimulation with isoproterenol (1 µM) and rolipram (10 µM) for 60 min. For pretreated cells, MK571 (20 µM) was maintained throughout the isoproterenol and rolipram treatment. The cell lysate, media, and EVs were collected for cAMP and protein analysis and the ratio of untreated vs. MK571 treated levels of cAMP normalized to protein calculated for each experimental condition [n = 3; ***P < 0.01 vs. without MK571 and not significant (NS); Student’s t-test]. ●, Individual data points.
Cyclic AMP is Detected in EVs from the Perfusate of the Isolated Lung
To determine whether EVs released from the intact organ contain cAMP, isolated rat lungs were perfused in the presence and absence of isoproterenol and rolipram and perfusate EVs analyzed for cAMP. When isolated rat lungs were perfused with isoproterenol and rolipram, there was a significant increase in EV-cAMP compared with vehicle control (Fig. 8). Thus, when the pulmonary circulation is stimulated with agonists to increase cAMP, the lung releases cAMP-containing EVs.
Fig. 8.
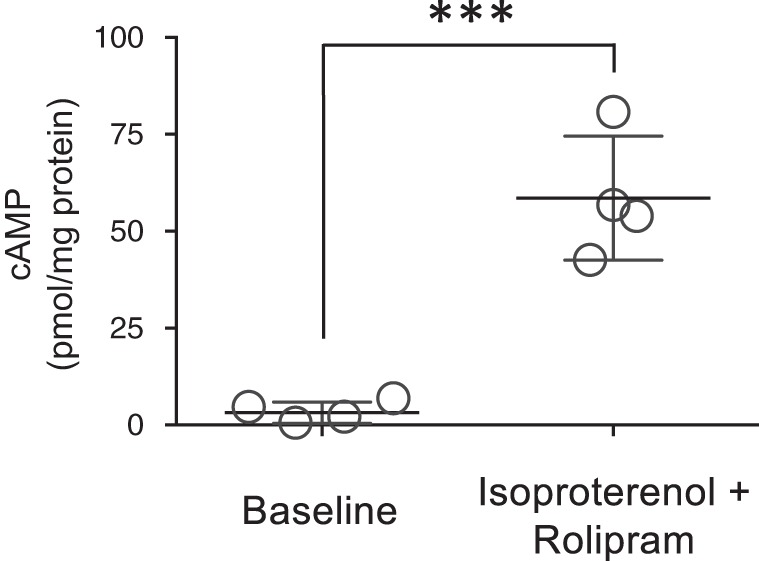
Extracellular vesicles (EVs) from isolated rat lungs perfused with isoproterenol and rolipram contain elevated cAMP. Isolated rat lungs were perfused with or without (i.e vehicle control) isoproterenol (1 µM) and rolipram (10 µM) for 1 h and the perfusate collected for isolation of EVs as described in materials and methods. EVs were analyzed for cAMP and normalized to protein concentration (n = 4 separate lungs/experimental condition; *P < 0.001 vs. vehicle control, Student’s t-test). ○, Individual data points.
Cyclic AMP-Containing EVs Do Not Change PMVEC Monolayer Resistance
Cyclic AMP has long been known to regulate pulmonary endothelial barrier integrity (43). To examine the role of cAMP encapsulated within EVs in regulation of the PMVEC monolayer, EVs were isolated from either unstimulated (vehicle control) or isoproterenol- and rolipram-treated PMVECs. These freshly isolated control or cAMP-containing EVs were added to a confluent naive monolayer, and the change in monolayer resistance was recorded over time. DMEM was added as the EV-vehicle control throughout the ECIS experiments. Although the baseline resistance of the unperturbed PMVEC monolayer increased over the 3-h time course, there was no significant difference in the transelectrical resistance between control versus cAMP-encapsulated EVs (Fig. 9).
Fig. 9.
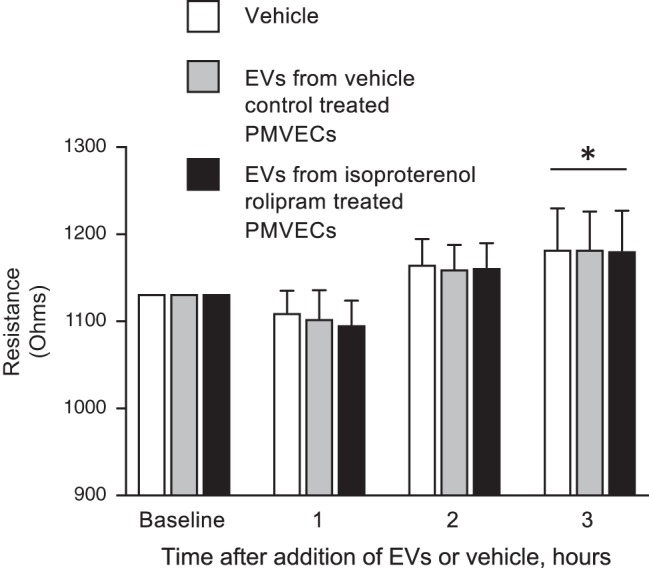
Extracellular vesicles (EVs) from unstimulated (vehicle control) as well as isoproterenol and rolipram stimulated pulmonary microvascular endothelial cells (PMVECs) do not alter transendothelial electrical resistance. EVs collected (as described in materials and methods) from PMVECs stimulated with isoproterenol (1 µM) and rolipram (10 µM) or vehicle control for 60 min were applied to confluent PMVEC monolayers and the change in resistance recorded over time. DMEM was used as the EV vehicle control. EVs collected from vehicle control or isoproterenol- and rolipram-treated PMVECs did not alter monolayer resistance from vehicle control-treated PMVECs (data represent n = 4, with each individual experiment performed as n = 2 or 3; *P < 0.05, 2-way ANOVA with Tukey post hoc test).
DISCUSSION
In aggregate, these data reveal a new paradigm for EVs and compartmentalized cAMP signaling; although cAMP is extruded into the extracellular space, these studies reveal that circulating cAMP can also be packaged into EVs. Previously, extracellular cAMP measurements would have captured both free, circulating cAMP as well as cAMP encapsulated in EVs. Thus, here we have identified a second extracellular compartment of cAMP: cAMP packaged into EVs.
Within the intracellular environment, cAMP signals are highly organized into spatially restricted compartments. These constrained compartments permit the cAMP signal to reach a threshold amplitude necessary for activation of downstream targets while maintaining signaling specificity. Indeed, in pulmonary endothelial cells, cAMP generated by transmembrane ACs is restricted to the subplasma membrane compartment by PDE activity and physical barriers, such as the endoplasmic reticulum (29, 40). Importantly, this spatially confined near-membrane cAMP strengthens the endothelial barrier; in contrast, cAMP within the cytosolic compartment disrupts the endothelial barrier (42–44). Not surprisingly, EVs were long thought of as cellular debris or as a means of eliminating “unwanted” compounds from a cell, as described for caspase-3 and activated protein C (1, 26, 30, 38). Thus, although still unresolved, eliminating cAMP from the near-membrane compartment through packaging excess “unwanted” signal into EVs could be an additional mechanism to preserve this near-membrane barrier-protective cAMP pool and limit the possibility that near-membrane cAMP diffuses into the cytosolic compartment, where it could become barrier disruptive. Thus, in this way, EVs may act to facilitate compartmentalized cAMP signals.
Despite EVs initially being considered as merely cellular debris, there is increasing evidence that these submicron vesicles are important in horizontal transfer of molecules between cells and play an important role in cell-cell communication and regulation of cellular and tissue responses. Efflux of cAMP through MRPs leads to “free” cAMP within the extracellular space. The cAMP/adenosine pathway has been described in several cell and tissue types in which cAMP is sequentially converted to adenosine (25). First, ecto-phosphodiesterase hydrolyzes cAMP to 5′-AMP, followed by conversion to adenosine by ecto-5′-nucleotidase. Thus, “free” cAMP has a limited half-life in the extracellular space; however, unlike “free” cAMP, EV-cAMP is encapsulated within a lipid bilayer and protected from ecto-enzymes, thereby potentially preventing its degradation and allowing the cAMP signal itself to be transduced back to the cell of origin or nearby cell or to be carried in the blood to an adjacent or distant cell. It is interesting to speculate that cAMP-loaded EVs could deliver this second messenger to downstream cells in the pulmonary venous bed, the heart, or, more distally, to the systemic circulation. Whether these vesicles do indeed bind and fuse with target cells to transfer cAMP and whether there is sufficient delivery of cAMP to activate downstream signaling in a target cell is yet to be determined.
The EV-protein, -small RNA, -DNA content can be altered following stimulation of cells, tissues, or the whole animal in response to various physiological and pathophysiological stimuli. Indeed, EV number is often increased during cell stimulation. For example, in COPD, EVs increase in number and are thought to play a role in the pathogenesis of the disease (27). Data presented here are the first reports that cAMP-containing EVs are constitutively released into the circulation. Upon isoproterenol and rolipram treatment, PMVEC-cAMP rapidly increases, as does cAMP within EVs released from these cells. Elevated cellular cAMP occurs rapidly, within 5 min after stimulation. Interestingly, this rapid change in cellular cAMP is rapidly reflected within EVs, which similarly show an increase in cAMP over this same time frame. These findings suggest that EV contents rapidly respond to reflect the changes in the cellular environment. Although many stimuli alter EV contents, there is usually an associated increase in EV number. At the time point tested here, there was no significant change in EV number; indeed, Mirzapoiazova et al. (32) similarly showed that stimulation of pulmonary microvascular endothelial cells did not increase EV number until after 3 h stimulation. Thus, the rapid increase in near-membrane cAMP is rapidly packaged into newly synthesized EVs released into the media; however, although cAMP in the cell peaks at 15 min, there is a steady decline in cellular cAMP over the 60-min time course. Meanwhile, the cAMP in the EVs continues to increase over the 60-min time course. Receptor desensitization facilitates signal termination, but here the temporal attenuation of the cAMP signal could also be due to transfer of cAMP into EVs. Thus, these studies allow us to more clearly define how EV contents can change over a relatively short time frame in response to a rapidly increasing stimulus.
Not surprisingly, neither cGMP nor the noncanonical cyclic nucleotides increased with either AC activation, inhibition of PDE4, or the combination of both; however, whereas cGMP, cCMP, and cUMP are detected in PMVECs under basal conditions, neither cGMP nor the noncanonical cyclic nucleotides are packaged into unstimulated or transmembrane AC-stimulated PMVEC-derived EVs. The reason for this specific cAMP packaging into EVs is unknown but could be due to spatial restraint such that cGMP, cCMP, and cUMP are not in close proximity to the site of EV generation.
Our experimental approach does not facilitate the separation of microparticles from exosomes, and so currently, it is not clear whether cAMP is transferred into just one vesicle population or all different types of endothelial derived vesicles. The proximity of multivesicular bodies to the subplasma membrane space could either limit or facilitate the transfer of cAMP into exosomes, for example. Future studies aimed at refining the isolation of different populations of EVs may reveal a subset of EVs that are even more enriched in this second messenger.
Within the pulmonary endothelium, activation of transmembrane AC generates a near-membrane cAMP pool that attenuates barrier disruption induced by various inflammatory mediators. In contrast, bicarbonate stimulation of soluble AC10, which lacks transmembrane domains and localizes to the cytosol, generates cAMP within the cytosolic compartment that is required for LPS-induced endothelial barrier disruption (36, 43). Whereas some studies suggest activation of transmembrane AC and elevations in near-membrane cAMP strengthen the basal unperturbed endothelial barrier, other studies report that elevations in near-membrane cAMP are not sufficient to alter the integrity of a basal intact monolayer (19, 23, 49). Meanwhile, our previous studies reveal that bicarbonate stimulation of soluble AC10 decreases PMVEC barrier integrity. Thus, cAMP-containing EVs act in a similar manner to near-membrane cAMP by maintaining a stable intact PMVEC monolayer and do not mimic the barrier-disruptive properties that are characteristic of cytosolic cAMP. Future studies will determine whether cAMP-containing EVs can rescue a disrupted endothelial monolayer.
In summary, these studies provide evidence for a fourth cAMP compartment in PMVECs. Two of these compartments exist within the cells and have been described previously, near membrane and cytosolic compartments, and have opposing physiological responses on endothelial barrier. Herein, we identify a second extracellular compartment beyond the previously described “free” cAMP, i.e., cAMP encapsulated within EVs. Future investigations will identify the downstream target of this fourth compartment, cAMP-EVs, and their effect in target cells.
GRANTS
This work was supported in part by research funding from the Parker B. Francis Fellowship; American Heart Association (AHA) Grant GRNT7430039; National Heart, Lung, and Blood Institute (NHLBI) Grants R01-HL-121513 and HL-66299 (to S. L. Sayner); AHA Grant 12SDG9270020 and NHLBI Grant R01 HL133066 (to N. N. Bauer); and the NHLBI Grant T32-HL-076125 (to L. A. Blair and A. K. Scruggs).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.L.S. and N.N.B. conceived and designed research; S.L.S., C.-S.C., M.E.M., K.C.R., C.Z., A.K.S., T.Y., L.A.B., J.A.K., R.S., and V.K. performed experiments; S.L.S., C.-S.C., M.E.M., A.K.S., and J.A.K. analyzed data; S.L.S., C.-S.C., M.E.M., and J.A.K. interpreted results of experiments; S.L.S., C.-S.C., M.E.M., K.C.R., A.K.S., and T.Y. prepared figures; S.L.S. drafted manuscript; S.L.S. and C.-S.C. edited and revised manuscript; S.L.S., C.-S.C., M.E.M., J.A.K., R.S., V.K., and N.N.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Nikola Valkov and Brandon Hartman for assistance with TEM imaging.
REFERENCES
- 1.Abid Hussein MN, Böing AN, Sturk A, Hau CM, Nieuwland R. Inhibition of microparticle release triggers endothelial cell apoptosis and detachment. Thromb Haemost 98: 1096–1107, 2007. doi: 10.1160/TH05-04-0231. [DOI] [PubMed] [Google Scholar]
- 2.Amabile N, Guignabert C, Montani D, Yeghiazarians Y, Boulanger CM, Humbert M. Cellular microparticles in the pathogenesis of pulmonary hypertension. Eur Respir J 42: 272–279, 2013. doi: 10.1183/09031936.00087212. [DOI] [PubMed] [Google Scholar]
- 3.Bähre H, Hartwig C, Munder A, Wolter S, Stelzer T, Schirmer B, Beckert U, Frank DW, Tümmler B, Kaever V, Seifert R. cCMP and cUMP occur in vivo. Biochem Biophys Res Commun 460: 909–914, 2015. doi: 10.1016/j.bbrc.2015.03.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bakouboula B, Morel O, Faure A, Zobairi F, Jesel L, Trinh A, Zupan M, Canuet M, Grunebaum L, Brunette A, Desprez D, Chabot F, Weitzenblum E, Freyssinet JM, Chaouat A, Toti F. Procoagulant membrane microparticles correlate with the severity of pulmonary arterial hypertension. Am J Respir Crit Care Med 177: 536–543, 2008. doi: 10.1164/rccm.200706-840OC. [DOI] [PubMed] [Google Scholar]
- 5.Bastarache JA, Fremont RD, Kropski JA, Bossert FR, Ware LB. Procoagulant alveolar microparticles in the lungs of patients with acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol 297: L1035–L1041, 2009. doi: 10.1152/ajplung.00214.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beste KY, Burhenne H, Kaever V, Stasch JP, Seifert R. Nucleotidyl cyclase activity of soluble guanylyl cyclase α1β1. Biochemistry 51: 194–204, 2012. doi: 10.1021/bi201259y. [DOI] [PubMed] [Google Scholar]
- 7.Blair LA, Haven AK, Bauer NN. Circulating microparticles in severe pulmonary arterial hypertension increase intercellular adhesion molecule-1 expression selectively in pulmonary artery endothelium. Respir Res 17: 133, 2016. doi: 10.1186/s12931-016-0445-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broadus AE, Kaminsky NI, Northcutt RC, Hardman JG, Sutherland EW, Liddle GW. Effects of glucagon on adenosine 3′,5′-monophosphate and guanosine 3′,5′-monophosphate in human plasma and urine. J Clin Invest 49: 2237–2245, 1970. doi: 10.1172/JCI106442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brodsky SV, Zhang F, Nasjletti A, Goligorsky MS. Endothelium-derived microparticles impair endothelial function in vitro. Am J Physiol Heart Circ Physiol 286: H1910–H1915, 2004. doi: 10.1152/ajpheart.01172.2003. [DOI] [PubMed] [Google Scholar]
- 10.Butcher RW, Sutherland EW. Adenosine 3′,5′-phosphate in biological materials. I. Purification and properties of cyclic 3′,5′-nucleotide phosphodiesterase and use of this enzyme to characterize adenosine 3′,5′-phosphate in human urine. J Biol Chem 237: 1244–1250, 1962. [PubMed] [Google Scholar]
- 11.Chuo ST, Chien JC, Lai CP. Imaging extracellular vesicles: current and emerging methods. J Biomed Sci 25: 91, 2018. doi: 10.1186/s12929-018-0494-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collier ME, Mah PM, Xiao Y, Maraveyas A, Ettelaie C. Microparticle-associated tissue factor is recycled by endothelial cells resulting in enhanced surface tissue factor activity. Thromb Haemost 110: 966–976, 2013. doi: 10.1160/TH13-01-0055. [DOI] [PubMed] [Google Scholar]
- 13.Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 30: 255–289, 2014. doi: 10.1146/annurev-cellbio-101512-122326. [DOI] [PubMed] [Google Scholar]
- 14.Creighton J, Zhu B, Alexeyev M, Stevens T. Spectrin-anchored phosphodiesterase 4D4 restricts cAMP from disrupting microtubules and inducing endothelial cell gap formation. J Cell Sci 121: 110–119, 2008. doi: 10.1242/jcs.011692. [DOI] [PubMed] [Google Scholar]
- 15.Creighton JR, Masada N, Cooper DM, Stevens T. Coordinate regulation of membrane cAMP by Ca2+-inhibited adenylyl cyclase and phosphodiesterase activities. Am J Physiol Lung Cell Mol Physiol 284: L100–L107, 2003. doi: 10.1152/ajplung.00083.2002. [DOI] [PubMed] [Google Scholar]
- 16.Densmore JC, Signorino PR, Ou J, Hatoum OA, Rowe JJ, Shi Y, Kaul S, Jones DW, Sabina RE, Pritchard KA Jr, Guice KS, Oldham KT. Endothelium-derived microparticles induce endothelial dysfunction and acute lung injury. Shock 26: 464–471, 2006. doi: 10.1097/01.shk.0000228791.10550.36. [DOI] [PubMed] [Google Scholar]
- 17.Dessauer CW, Watts VJ, Ostrom RS, Conti M, Dove S, Seifert R. International Union of Basic and Clinical Pharmacology. CI. Structures and Small Molecule Modulators of Mammalian Adenylyl Cyclases. Pharmacol Rev 69: 93–139, 2017. doi: 10.1124/pr.116.013078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dignat-George F, Boulanger CM. The many faces of endothelial microparticles. Arterioscler Thromb Vasc Biol 31: 27–33, 2011. doi: 10.1161/ATVBAHA.110.218123. [DOI] [PubMed] [Google Scholar]
- 19.Farrukh IS, Gurtner GH, Michael JR. Pharmacological modification of pulmonary vascular injury: possible role of cAMP. J Appl Physiol (1985) 62: 47–54, 1987. doi: 10.1152/jappl.1987.62.1.47. [DOI] [PubMed] [Google Scholar]
- 20.Gebb S, Stevens T. On lung endothelial cell heterogeneity. Microvasc Res 68: 1–12, 2004. doi: 10.1016/j.mvr.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 21.Godinho RO, Duarte T, Pacini ES. New perspectives in signaling mediated by receptors coupled to stimulatory G protein: the emerging significance of cAMP efflux and extracellular cAMP-adenosine pathway. Front Pharmacol 6: 58, 2015. doi: 10.3389/fphar.2015.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hargett LA, Bauer NN. On the origin of microparticles: From “platelet dust” to mediators of intercellular communication. Pulm Circ 3: 329–340, 2013. doi: 10.4103/2045-8932.114760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He P, Zeng M, Curry FE. Dominant role of cAMP in regulation of microvessel permeability. Am J Physiol Heart Circ Physiol 278: H1124–H1133, 2000. doi: 10.1152/ajpheart.2000.278.4.H1124. [DOI] [PubMed] [Google Scholar]
- 24.Hofer AM, Lefkimmiatis K. Extracellular calcium and cAMP: second messengers as “third messengers”? Physiology (Bethesda) 22: 320–327, 2007. doi: 10.1152/physiol.00019.2007. [DOI] [PubMed] [Google Scholar]
- 25.Jackson EK, Mi Z, Gillespie DG, Dubey RK. Metabolism of cAMP to adenosine in the renal vasculature. J Pharmacol Exp Ther 283: 177–182, 1997. [PubMed] [Google Scholar]
- 26.Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J Biol Chem 262: 9412–9420, 1987. [PubMed] [Google Scholar]
- 27.Kadota T, Fujita Y, Yoshioka Y, Araya J, Kuwano K, Ochiya T. Extracellular Vesicles in Chronic Obstructive Pulmonary Disease. Int J Mol Sci 17: 1801, 2016. doi: 10.3390/ijms17111801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.King J, Hamil T, Creighton J, Wu S, Bhat P, McDonald F, Stevens T. Structural and functional characteristics of lung macro- and microvascular endothelial cell phenotypes. Microvasc Res 67: 139–151, 2004. doi: 10.1016/j.mvr.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 29.Lefkimmiatis K, Zaccolo M. cAMP signaling in subcellular compartments. Pharmacol Ther 143: 295–304, 2014. doi: 10.1016/j.pharmthera.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malloci M, Perdomo L, Veerasamy M, Andriantsitohaina R, Simard G, Martinez MC. Extracellular vesicles: mechanisms in human health and disease. Antioxid Redox Signal 30: 813–856, 2019. doi: 10.1089/ars.2017.7265. [DOI] [PubMed] [Google Scholar]
- 31.McVey M, Tabuchi A, Kuebler WM. Microparticles and acute lung injury. Am J Physiol Lung Cell Mol Physiol 303: L364–L381, 2012. doi: 10.1152/ajplung.00354.2011. [DOI] [PubMed] [Google Scholar]
- 32.Mirzapoiazova T, Lennon FE, Mambetsariev B, Allen M, Riehm J, Poroyko VA, Singleton PA. Extracellular Vesicles from Caveolin-Enriched Microdomains Regulate Hyaluronan-Mediated Sustained Vascular Integrity. Int J Cell Biol 2015: 1–11, 2015. doi: 10.1155/2015/481493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morrow KA, Seifert R, Kaever V, Britain AL, Sayner SL, Ochoa CD, Cioffi EA, Frank DW, Rich TC, Stevens T. Heterogeneity of pulmonary endothelial cyclic nucleotide response to Pseudomonas aeruginosa ExoY infection. Am J Physiol Lung Cell Mol Physiol 309: L1199–L1207, 2015. doi: 10.1152/ajplung.00165.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nickols J, Obiako B, Ramila KC, Putinta K, Schilling S, Sayner SL. Lipopolysaccharide-induced pulmonary endothelial barrier disruption and lung edema: critical role for bicarbonate stimulation of AC10. Am J Physiol Lung Cell Mol Physiol 309: L1430–L1437, 2015. doi: 10.1152/ajplung.00067.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Brien JA, Strange RC. The release of 3′:5′-cyclic monophosphate from the isolated perfused rat heart. Biochem J 152: 429–432, 1975. doi: 10.1042/bj1520429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Obiako B, Calchary W, Xu N, Kunstadt R, Richardson B, Nix J, Sayner SL. Bicarbonate disruption of the pulmonary endothelial barrier via activation of endogenous soluble adenylyl cyclase, isoform 10. Am J Physiol Lung Cell Mol Physiol 305: L185–L192, 2013. doi: 10.1152/ajplung.00392.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peracchi M, Lombardi L, Maiolo AT, Bamonti-Catena F, Toschi V, Chiorboli O, Mozzana R, Polli EE. Plasma and urine cyclic nucleotide levels in patients with acute and chronic leukemia. Blood 61: 429–434, 1983. [PubMed] [Google Scholar]
- 38.Pérez-Casal M, Downey C, Cutillas-Moreno B, Zuzel M, Fukudome K, Toh CH. Microparticle-associated endothelial protein C receptor and the induction of cytoprotective and anti-inflammatory effects. Haematologica 94: 387–394, 2009. doi: 10.3324/haematol.13547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rautou PE, Vion AC, Amabile N, Chironi G, Simon A, Tedgui A, Boulanger CM. Microparticles, vascular function, and atherothrombosis. Circ Res 109: 593–606, 2011. doi: 10.1161/CIRCRESAHA.110.233163. [DOI] [PubMed] [Google Scholar]
- 40.Rich TC, Fagan KA, Tse TE, Schaack J, Cooper DM, Karpen JW. A uniform extracellular stimulus triggers distinct cAMP signals in different compartments of a simple cell. Proc Natl Acad Sci USA 98: 13049–13054, 2001. [Erratum in Proc Natl Acad Sci 98: 14744, 2001.] 10.1073/pnas.221381398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sassi Y, Ahles A, Truong DJ, Baqi Y, Lee SY, Husse B, Hulot JS, Foinquinos A, Thum T, Müller CE, Dendorfer A, Laggerbauer B, Engelhardt S. Cardiac myocyte-secreted cAMP exerts paracrine action via adenosine receptor activation. J Clin Invest 124: 5385–5397, 2014. doi: 10.1172/JCI74349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sayner S, Stevens T. Soluble adenylate cyclase reveals the significance of compartmentalized cAMP on endothelial cell barrier function. Biochem Soc Trans 34: 492–494, 2006. doi: 10.1042/BST0340492. [DOI] [PubMed] [Google Scholar]
- 43.Sayner SL. Emerging themes of cAMP regulation of the pulmonary endothelial barrier. Am J Physiol Lung Cell Mol Physiol 300: L667–L678, 2011. doi: 10.1152/ajplung.00433.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sayner SL, Balczon R, Frank DW, Cooper DM, Stevens T. Filamin A is a phosphorylation target of membrane but not cytosolic adenylyl cyclase activity. Am J Physiol Lung Cell Mol Physiol 301: L117–L124, 2011. doi: 10.1152/ajplung.00417.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sayner SL, Frank DW, King J, Chen H, VandeWaa J, Stevens T. Paradoxical cAMP-induced lung endothelial hyperpermeability revealed by Pseudomonas aeruginosa ExoY. Circ Res 95: 196–203, 2004. doi: 10.1161/01.RES.0000134922.25721.d9. [DOI] [PubMed] [Google Scholar]
- 46.Scruggs AK, Cioffi EA, Cioffi DL, King JA, Bauer NN. Lectin-Based Characterization of Vascular Cell Microparticle Glycocalyx. PLoS One 10: e0135533, 2015. doi: 10.1371/journal.pone.0135533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Serban KA, Rezania S, Petrusca DN, Poirier C, Cao D, Justice MJ, Patel M, Tsvetkova I, Kamocki K, Mikosz A, Schweitzer KS, Jacobson S, Cardoso A, Carlesso N, Hubbard WC, Kechris K, Dragnea B, Berdyshev EV, McClintock J, Petrache I. Structural and functional characterization of endothelial microparticles released by cigarette smoke. Sci Rep 6: 31596, 2016. doi: 10.1038/srep31596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh RP, Chansouria JP, Singh JN, Singh VP, Udupa KN. Circulating and urinary cAMP levels in chronic and acute myeloid leukemia. Indian J Cancer 21: 122–125, 1984. [PubMed] [Google Scholar]
- 49.Stelzner TJ, Weil JV, O’Brien RF. Role of cyclic adenosine monophosphate in the induction of endothelial barrier properties. J Cell Physiol 139: 157–166, 1989. doi: 10.1002/jcp.1041390122. [DOI] [PubMed] [Google Scholar]
- 50.Stevens T, Creighton J, Thompson WJ. Control of cAMP in lung endothelial cell phenotypes. Implications for control of barrier function. Am J Physiol 277: L119–L126, 1999. doi: 10.1152/ajplung.1999.277.1.L119. [DOI] [PubMed] [Google Scholar]
- 51.Vendetti S, Patrizio M, Riccomi A, De Magistris MT. Human CD4+ T lymphocytes with increased intracellular cAMP levels exert regulatory functions by releasing extracellular cAMP. J Leukoc Biol 80: 880–888, 2006. doi: 10.1189/jlb.0106072. [DOI] [PubMed] [Google Scholar]