

Abstract
Stimulation of metabotropic Gq-coupled purinergic P2Y2 receptors decreases activity of the epithelial Na+ channel (ENaC) in renal principal cells of the distal nephron. The physiological consequences of P2Y2 receptor signaling disruption in the P2Y2 receptor knockout mouse are decreased Na+ excretion and increased arterial blood pressure. However, because of the global nature of this knockout model, the quantitative contribution of ENaC and distal nephron compared with that of upstream renal vascular and tubular elements to changes in urinary excretion and arterial blood pressure is obscure. Moreover, it is uncertain whether stimulation of P2Y2 receptor inhibition of ENaC is sufficient to drive renal (urinary) Na+ excretion (UNaV). Here, using a pharmacogenetic approach and selective agonism of the P2Y2 receptor, we test the sufficiency of targeted stimulation of Gq signaling in principal cells of the distal nephron and P2Y2 receptors to increase UNaV. Selective stimulation of the P2Y2 receptor with the ligand MRS2768 decreased ENaC activity in freshly isolated tubules (as assessed by patch-clamp electrophysiology) and increased UNaV (as assessed in metabolic cages). Similarly, selective agonism of hM3Dq-designer receptors exclusively activated by designer drugs (DREADD) restrictively expressed in principal cells of the distal nephron with clozapine-N-oxide decreased ENaC activity and, consequently, increased UNaV. Clozapine-N-oxide, when applied to control littermates, failed to affect ENaC and UNaV. This study represents the first use of pharmacogenetic (DREADD) technology in the renal tubule and demonstrated that selective activation of the P2Y2 receptor and Gq signaling in principal cells is sufficient to promote renal salt excretion.
Keywords: aldosterone, collecting duct, hypertension, INS45973, MRS2768, renal physiology, sodium excretion, transport
INTRODUCTION
Na+ balance is recognized to influence arterial blood pressure, where natriuresis and antinatriuresis decrease and increase arterial blood pressure, respectively. Na+ balance, in part, reflects Na+ consumption minus Na+ excretion. Na+ is excreted chiefly by the kidneys (1, 4). Consequently, inappropriate Na+ excretion by the kidneys causes disordered arterial blood pressure (13, 43, 51).
Humans, similar to other terrestrial vertebrates, fine tune urinary Na+ excretion (UNaV) by modulating epithelial Na+ channel (ENaC) activity. ENaC is a noninactivating, nonvoltage-gated, Na+-selective ion channel expressed in tight epithelial cells capable of vectorial Na+ transport (7, 8, 19). ENaC shares no similarity with voltage-gated Na+ (Nav) channels, which are encoded by SCN genes, found in the neurons and muscle; rather, it is a member of the degenerin/ENaC family of ion channels encoded by SCNN/ACCN genes.
In the kidneys, ENaC is restrictively expressed in luminal plasma membranes of the principal cells lining the distal nephron, including the latter portion of the distal convoluted tubule, connecting tubule (CNT), and collecting duct. This location makes ENaC the final arbiter of UNaV in the mammalian kidney.
ENaC activity is under the discretionary control of hormones and autocrine/paracrine factors that match Na+ excretion with Na+ intake in the maintenance of arterial blood pressure. Consequently, mutations in ENaC and upstream modulators of this channel that cause gain or loss of function lead to inappropriate UNaV and dependent pathological changes in arterial blood pressure (9, 13, 18, 43–45). Liddle’s syndrome and autosomal recessive pseudohypoaldosteronism type 1 (PHA1) are examples of dominant and recessive forms of familial hypertension and renal salt wasting, respectively, caused by mutation of ENaC. Autosomal dominant PHA1 is a Mendelian form of renal salt wasting caused by a loss of function mutation of the receptor for the natriferic mineralocorticoid steroid hormone aldosterone.
Aldosterone is the primary systemic hormone that modulates ENaC activity. It does so as the final hormone in the renin-ANG II-aldosterone system (RAAS). This hormone cascade controls blood pressure through feedback regulation, where a fall in arterial blood pressure stimulates renin secretion to increase plasma ANG II and aldosterone concentrations with stimulation of ENaC, causing an antinatriuresis in the protection of arterial blood pressure. Several contemporary drugs used to treat elevated blood pressure, including angiotensin-converting enzyme inhibitors, ANG II receptor blockers, and mineralocorticoid receptor antagonists, interdict the RAAS.
In addition to aldosterone, other systemic hormones, including arginine vasopressin, are also capable of influencing blood pressure by modulating Na+ and water balance, in part, through regulation of ENaC (23). Indeed, ENaC is widely recognized as a common end effector of many humoral factors that influence blood pressure. We and others have demonstrated that ENaC activity and, consequently, UNaV and arterial blood pressure are influenced by paracrine factors in the urine. Urinary ATP, via metabotropic Gq-coupled purinergic P2Y2 receptors (P2RY2) in the luminal plasma membrane of principal cells, decreases ENaC activity (22, 30, 31, 33, 38, 47, 50).
Compelling evidence supports the idea that increases in urinary flow stimulate ATP secretion into the urine from tubular epithelial cells to include cells in the distal nephron (6, 11, 12, 26, 33, 46, 47, 49, 50). Increases in blood pressure and volume expansion are factors that can increase urine flow. As demonstrated in the P2Y2 receptor knockout mouse, loss of P2Y2 receptor signaling causes inappropriate Na+ retention and, consequently, pathological increases in blood pressure (30, 33, 38, 39, 47). Purinergic regulation of ENaC acts in parallel with regulation by aldosterone and is as quantitatively important to the control of arterial blood pressure as this steroid hormone.
While the consequences of disrupting purinergic inhibition of ENaC are known to some degree of certainty, whether stimulation of purinergic inhibition of ENaC is sufficient to drive UNaV remains obscure. Moreover, because the preponderance of evidence supporting purinergic effects on UNaV and arterial blood pressure comes from a global P2Y2 receptor knockout model, the quantitative contribution of the distal nephron versus upstream tubular and vascular elements to these effects remains a subject of debate.
The P2Y2 receptor is a canonical seven-transmembrane-spanning G protein-coupled receptor. This receptor is coupled to Gq. In principal cells, as in other cells expressing this receptor, stimulation of the P2Y2 receptor activates Gq signaling. ENaC activity decreases as a function of hydrolysis of luminal plasma membrane phosphatidylinositol (4,5)-bisphosphate by Gq-stimulated phospholipase C (PLC) (17, 30–34, 36).
Recently, designer receptors exclusively activated by designer drugs (DREADDs) have been used to selectively stimulate Gq-PLC signaling in a targeted manner in specific cells. This technology uses pharmacogenetics to combine conditional knockin of a modified G protein-coupled receptor, such as the hM3Dq receptor, with selective stimulation of this modified receptor by an artificial ligand, such as clozapine-N-oxide (CNO), to control Gq-PLC signaling in a cell-specific manner (2, 16, 27, 29, 42, 53). Similar to activation of the P2Y2 receptor by ATP, activation of hM3Dq by CNO stimulates Gq-PLC signaling, causing hydrolysis of phosphatidylinositol (4,5)-bisphosphate and the dependent production of inositol (1,4,5)-trisphosphate and diacylglycerol.
Here, we used a pharmacogenetic approach to test the sufficiency of targeted activation of Gq signaling in principal cells of the distal nephron to increase UNaV. This study represents the first use of DREADD technology in the renal tubule and only the second use of this technology in the kidney (16). We found that selective activation of Gq signaling in principal cells is sufficient to promote UNaV dependent on inhibition of ENaC in a manner akin to selective agonism of the P2Y2 receptor. Such a finding elaborates the central role of purinergic inhibition of ENaC in principal cells in the control of UNaV and arterial blood pressure and identifies this signaling system as a possible therapeutic target to control pathological elevation of arterial blood pressure.
METHODS
Animal care and use.
All animal use and welfare adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Texas Health Science Center at San Antonio. Mice were housed and cared for in the Laboratory Animal Resources facility at the University of Texas Health Science Center at San Antonio, which is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care and licensed by the United States Department of Agriculture. Results involving animals were in compliance with Animal Research: Reporting of In Vivo Experiments guidelines (14).
Young healthy adult (~2 mo old) male and female mice (18.60 ± 0.11 g body wt) were used in approximately equal proportions. Within a specific genotype, assignment of mice to the experimental and control groups was randomized within the limitations imposed by mouse social hierarchies and interactions and with the consideration that each cohort contained at least 3 mice/cage. All mice, in both acclimation and experimental periods, were maintained on a normal 12:12-h light-dark cycle at room temperature; they had free access to water and chow and were housed socially with littermates and peers. All experimental maneuvers, including treatment with CNO in the drinking water and intraperitoneal injection (in 100–200 μl of saline) of MRS2768 (uridine-5′-tetraphosphate δ-phenyl ester), were performed within a constant time window (midmorning) in the laboratory, where mice were housed during the acclimation and experimental periods. The primary end points for experiments involving mice in these studies were 24-h urinary excretion over a 3-day acclimation period and 7-day experimental period followed by humane euthanasia and collection of kidneys for immunohistochemistry (IHC) and immunofluorescence (IF) experiments and microdissection of renal nephrons compatible with electrophysiological assessment of ENaC activity in the split-open CNT and cortical collecting duct (CCD).
Creation of the principal cell Gq-DREADD mouse.
Targeted expression of Gq-DREADD in renal principal cells was achieved by crossing hemizygous male B6N;129-Tg(CAG-CHRM3*,-mCitrine)1Ute/J [formerly known as Gt(ROSA)26Sortm2(CAG-CHRM3*,-mCitrine)Ute/J] mice harboring the CAG-LSL-Gq-DREADD (formerly known as R26-LSL-Gq-DREADD, R26-LSL-hM3Dq-DREADD, or R26-hM3Dq/mCitrine) transgene (stock no. 026220, Jackson Laboratories, Bar Harbor, ME) (54) with hemizygous female B6.Cg-Tg(Aqp2-cre)1Dek/J mice (gifted to us by our collaborator Dr. D. Kohan, University of Utah Health Science Center, Salt Lake City, UT) (5, 15, 28, 37, 41, 48). The line was continued by backcrossing Gq-DREADD:Aqp2-cre-positive female to Gq-DREADD male mice. Gq-DREADD:Aqp2-cre-positive (PC-Gq-DREADD) mice were used for experiments. Aqp2-cre-negative littermates lacking or harboring Gq-DREADD were used as controls. Similarly Aqp2-cre-positive mice lacking Gq-DREADD also served as controls. No noticeable difference in behavior, body weight (17.60 ± 0.06 g at 2 mo of age), pathology, or any other gross attribute was observed between PC-Gq-DREADD and B6 mice or littermate controls.
The Gq-DREADD transgene was identified using the forward 5′-CGCCACCATGTACCCATAC-3′ and reverse 5′-GTGGTACCGTCTGGAGAGGA-3′ PCR primers, producing an expected 204-bp product. The Aqp2-cre transgene was identified with the forward 5′-CTCTGCAGGAACTGGTGCTGG-3′ and reverse 5′-GCGAACATCTTCAGGTTCTGCGG-3′ PCR primers, producing an expected 673-bp product. The forward 5′-AAGGGAGCTGCAGTGGAGTA-3′ and reverse 5′-CCGAAAATCTGTGGGAAGTC-3′ PCR primers, producing an expected 297-bp product, were used as controls for these genotyping reactions.
Split-open tubule preparation and single channel patch-clamp electrophysiology.
The split-open tubule preparation was prepared as previously described (3, 23, 24). Briefly, the mouse kidney was sectioned transversely. Segments of the CNT and CCD were manually microdissected with forceps and adhered to a glass chip coated with polylysine. These chips were transferred to an inverted microscope, where the tubule was split open with sharpened pipettes. Single channel patch-clamp electrophysiology in the cell-attached configuration was then performed on the luminal plasma membranes of principal cells in the region having the exposed lumen. Channel activity (open channel probability multiplied by channel number) was calculated as previously described (23, 24, 30, 33). Tubules were isolated from mice maintained on a Na+-free (<0.01% Na+) diet (custom diet TD.90228, Teklad, Envigo, Indianapolis, IN) and water ad libitum for 5–7 days. This protocol maximized the initial activity of ENaC, creating the ideal condition to identify significant decreases in activity.
Metabolic cage experiments.
Metabolic cage experiments followed previously published protocols (3, 5, 15). Briefly, age- and weight-matched mice were housed socially at 3 mice/cage in metabolic cages (Techniplast, Buguggiate, Italy) and allowed to acclimate with free access to water for 3 days. During this acclimation period, mice were initially maintained with standard chow (0.32% Na+) but, on day 2 of the acclimation period, were switched to a Na+-free (<0.01% Na+) diet (custom diet TD.90228, Teklad, Envigo). During the acclimation period and subsequent 7-day experimental period, urine was collected every 24 h to enable quantification of urinary volume and urinary Na+ concentration (UNa) with drug treatment on days 2 and 3 of the experimental period. Collection surfaces in contact with urine were coated with Sigmacote (Sigma-Aldrich, St. Louis, MO), and urine was collected under light mineral oil to increase the precision of these measurements by reducing resistance to flow to the final collecting reservoir and to minimize loss due to evaporation. UNa was quantified with a flame photometer (Jenway, Staffordshire, UK).
IHC and IF.
Freshly isolated kidneys perfused with ice-cold PBS were fixed in 4% paraformaldehyde on ice. Kidney tissue was embedded in paraffin and sliced into 5-µm sections. Rabbit anti-green fluorescent protein (GFP; 1:1,000 dilution, catalog no. A-11122, Invitrogen, ThermoFisher Scientific, Waltham, MA) and goat anti-aquaporin 2 (AQP2; 1:200 dilution, catalog no. sc-9882, Santa Cruz Biotechnology, Dallas, TX) antibodies were used for these experiments. Sections were deparaffinized, rehydrated, and stained following standard IHC protocols (https://www.agilent.com/cs/library/technicaloverviews/public/08002_ihc_staining_methods.pdf; https://vectorlabs.com/media/docs/brochures/IHC_Guide.L.pdf). Antigen retrieval was not required for 3-amino-9-ethylcarbazole and 3,3′-diaminobenzidine staining with these antibodies. Stained images were acquired on an inverted microscope (model TE2000-U, Nikon Instruments, Melville, NY) equipped with a digital camera.
For IF experiments, slices were probed with the anti-GFP and anti-AQP2 antibodies described above at 4°C overnight following standard protocols (23). Slices were washed and then incubated with the appropriate fluorophore-linked secondary antibodies (Alexa Fluor 568-anti-rabbit IgG and Alexa Fluor 488-anti-goat IgG, Invitrogen, ThermoFisher Scientific), and IF images were collected on a fluorescence microscope (Axiovert 200M, Carl Zeiss Microscopy, Thornwood, NY).
Statistics.
Values are means ± SE. Data were compared by t-test, one-way ANOVA, or a z-test. P = 0.05 was used as the cutoff for statistical significance.
RESULTS
Selective agonism of the P2Y2 receptor decreases ENaC activity and increases UNaV.
We began this study by testing 1) if selective agonism of the P2Y2 receptor could decrease ENaC activity in an ex vivo isolated tubule preparation and 2) if this decrease in ENaC activity resulted in an increase in UNaV. Luminal plasma membranes of principal cells in freshly isolated distal nephrons from C57BL/6 mice were subjected to cell-attached, single channel patch-clamp electrophysiology in the voltage-clamp mode to test the former. Analysis of 24-h urinary flow and UNa from B6 mice treated with the selective P2Y2 receptor agonist MRS2768 or vehicle was used to test the latter.
Figure 1 shows a representative continuous-current trace from a patch-clamped luminal plasma membrane of a principal cell in an isolated split-open tubule from a B6 mouse containing multiple ENaC before and after addition of 1 μM MRS2768 as well as summary data from experiments on luminal plasma membranes in principal cells from tubules isolated from B6 mice. Isolated tubules were treated with vehicle or 1 μM MRS2768 for ~30 min before being patched. Mice were maintained on a Na+-free diet for 5–7 days before patch-clamp analysis. ENaC activity was significantly lower in MRS2768-treated tubules (0.48 ± 0.08) than in vehicle-treated tubules (1.25 ± 0.21) and untreated tubules (1.45 ± 0.24, n = 8 from 3 different mice; not shown).
Fig. 1.

Selective stimulation of the P2Y2 receptor in principal cells decreases epithelial Na+ channel (ENaC) activity. A: gap-free current trace of ENaC activity in a cell-attached patch on the apical plasma membrane of a principal cell in a split-open aldosterone-sensitive distal nephron from a C57BL/6 mouse. The tubule was isolated from a mouse maintained on a Na+-free diet for 1 wk. The patch was voltage clamped to −60 mV with downward inward current, and the closed state current level is denoted by a dashed line. The patch contained ≥7 ENaC. The trace shows time points before and after addition of 1 μM MRS2768 (arrow) to the extracellular bath solution. For the purpose of presentation, the trace was filtered at 40 Hz and slight corrections were made to baseline to adjust for slow current drifts. B: means ± SE (black circles) and individual data points (gray circles) for ENaC activity [open channel probability (NPo; where N is the number of channels in a patch and Po is the probability that these channels are open)] from experiments in tubules treated with vehicle (n = 11 from 3 different mice) or MRS2768 (n = 8 from 3 different mice) for 30 min before being patched. Mice of both sexes were used in approximately equal proportions. *Significantly lower than vehicle (by two-tailed t-test).
UNaV in B6 mice treated with vehicle or 5.0 mg/kg MRS2768 is shown in Fig. 2. Mice were initially acclimated with a regular-Na+ diet and then switched to a Na+-free diet, during which they subsequently received vehicle or MRS2768. The switch to a Na+-free diet creates a condition where conservation of Na+ is maximized during the experimental period due to a significant reduction in UNaV from 9.49 ± 0.64 (not shown) to 0.06 ± 0.02 μmol·h−1·mouse−1 and from 10.56 ±0.70 (not shown) to 0.06 ± 0.003 μmol·h−1·mouse−1 for vehicle- and MRS2768-treated cohorts, respectively. Urinary volumes were 2.01 ± 0.28, 1.81 ± 0.36, 2.31 ± 0.0.30, and 1.50 ± 0.26 ml·h−1·mouse−1, respectively. UNaV was significantly increased in the MRS2768-treated group, to 0.66 ± 0.03 μmol·h−1·mouse−1 (volume: 1.53 ± 0.09 ml·h−1·mouse−1) but not in the vehicle-treated group (0.14 ± 0.05 μmol·h−1·mouse−1, volume: 1.81 ± 0.31 ml·h−1·mouse−1) compared with before drug treatment. However, UNaV was not different between experimental and control cohorts maintained on standard chow and the Na+-free diet, with a significant difference between groups only after administration of MRS2768 versus vehicle.
Fig. 2.

Selective stimulation of the P2Y2 receptor increases urinary Na+ excretion (UNaV). Individual data points and means ± SE are shown for 24-h Na+ excretion for B6 mice maintained on a Na+-free diet before and after treatment with vehicle (n = 7 cages with 3 animals/cage for a total of 21 female mice, each tested once) or MRS2768 (n = 7 cages with 3 animals/cage for a total of 6 male and 15 female mice, each tested once). *Significantly greater than Na+-free diet and vehicle treatment (by one-way ANOVA with a Tukey’s honest significant difference post hoc test).
The results shown in Figs. 1 and 2 are consistent with selective activation of the P2Y2 receptor causing a decrease in ENaC activity and a natriuresis. It is logical to argue that the latter, increased UNaV, is dependent, in part, on the former, decreased ENaC activity. However, these experiments do not discriminate between the quantitative contributions of decreases in ENaC activity in principal cells and the contributions of effects on upstream tubular and vascular elements that also are capable of contributing to a natriuresis and/or an antinatriuresis to UNaV. To identify a primary site of action and the chief final effector underpinning a P2Y2 receptor-dependent increase in UNaV, we selectively stimulated P2Y2 receptor signaling exclusively in principal cells. We used a pharmacogenetics approach in a genetically modified mouse model that conditionally expresses Gq-DREADD in principal cells. CNO selectively and exclusively stimulates Gq signaling in principal cells in this PC-Gq-DREADD mouse model (2, 29, 42, 54). Recall that stimulation of the P2Y2 receptor decreases ENaC activity in principal cells through a signal transduction pathway that includes Gq-activated PLC (30–36).
The PC-Gq-DREADD mouse.
The micrograph shown in Fig. 3 shows the PCR products of a representative genotyping experiment on two typical PC-Gq-DREADD mice (Fig. 3, lanes 1 and 2 and lanes 3 and 4) and a littermate control (Fig. 3, lanes 5 and 6). PC-Gq-DREADD mice were positive for Gq-DREADD and Aqp2-cre transgenes (Fig. 3, lanes 1 and 3), whereas the littermate control mouse was negative for the Aqp2-cre transgene (lane 5). The Gq-DREADD transgene was identified using PCR primers that produced tge expected 204-bp product. The Aqp2-cre transgene was identified using PCR primers that produced the expected 673-bp product. Positive controls for these genotyping reactions produced a 297-bp product (Fig. 3, lanes 2, 4, and 6).
Fig. 3.
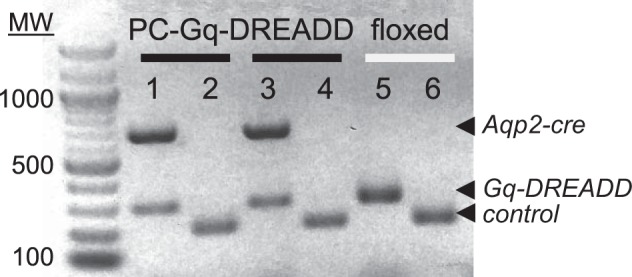
Genotype of the PC-Gq-designer receptors exclusively activated by designer drugs (DREADD mouse). Shown is an inverted image of a representative gel (n > 20) containing typical products from genotyping reactions of two PC-Gq-DREADD mice (lanes 1 and 2 and lanes 3 and 4) and one littermate control mouse (lanes 5 and 6). Expected products for the Aqp2-cre and Gq-DREADD transgenes and internal control are indicated with arrowheads. For presentation purposes, contrast and brightness were adjusted: the image was inverted (black to white) to maximize clarity without changing content. MW, molecular weight.
After Cre recombinase-mediated removal of an upstream floxed-STOP cassette, the Rosa-CAG-LSL-HA-hM3Dq-pta-mCitrine conditional allele expresses CAG promoter-driven Gq-DREADD and mCitrine (54). Figure 4 shows representative fluorescence micrographs from typical IF experiments on kidney sections from the cortex of PC-Gq-DREADD and littermate control mice. These sections were immunoprobed with anti-GFP and anti-AQP2 antibodies and the appropriate fluorophore-conjugated secondary antibodies to identify the enhanced GFP derivative, mCitrine, and principal cells, respectively. As expected, AQP2 was present in the luminal plasma membranes of many of the epithelial cells lining these segments of the nephron. This identifies these tubular epithelial cells as principal cells and these nephron segments as the CNT/CCD. Within these and all other sections from the PC-Gq-DREADD mouse, mCitrine was restrictively expressed only in AQP2-positive principal cells. In littermate control mice, mCitrine was not expressed in any cell. The merged image shown in Fig. 4 shows the marked overlap of mCitrine and AQP2 expression in principal cells of the PC-Gq-DREADD mouse.
Fig. 4.

Gq-designer receptors exclusively activated by designer drugs (DREADD) is restrictively expressed in principal cells of the PC-Gq-DREADD mouse. Shown are representative fluorescence micrographs of cortical renal sections from two PC-Gq-DREADD mice (top and middle) and one littermate control mouse (floxed; bottom) stained with anti-green fluorescent protein antibody (red; left) identifying mCitrine and anti-aquaporin 2 (AQP2) antibody (green; middle). Merged images are shown at right. For presentation purposes, sharpness, contrast, and brightness were adjusted to maximize clarity without changing content.
Confirming observations from IF experiments, mCitrine was also expressed in tubular epithelial cells in IHC experiments on sections of the cortex from PC-Gq-DREADD but not littermate control mice when probed with anti-AQP2 and anti-GFP antibodies (Fig. 5A). Chromogenic identification of conjugated horseradish peroxidase with 3-amino-9-ethylcarbazole for anti-AQP2 and 3,3′-diaminobenzidine for anti-GFP antibodies was used, along with counterstaining of nuclei with hematoxylin, for these IHC experiments. As shown in Fig. 5B, 28 ± 7.5% and 22 ± 2.9% of cortical tubular epithelial cells were positive for AQP2 in control littermate and PC-Gq-DREADD mice, respectively. These proportions were not different. In comparison, 0% and 21 ± 1.3% of cortical tubular epithelial cells were positive for mCitrine in control littermate and PC-Gq-DREADD mice, respectively. The proportion of cortical tubular epithelial cells expressing mCitrine was significantly greater in the PC-Gq-DREADD group.
Fig. 5.

Gq-designer receptors exclusively activated by designer drugs (DREADD) expression in principal cells of the PC-Gq-DREADD mouse is robust. A: representative immunohistochemistry images of aquaporin 2 (AQP2) and mCitrine expression in renal cortical sections stained with anti-AQP2 (top) and anti-green fluorescent protein (bottom) antibodies from control littermates (left) and PC-Gq-DREADD mice (right). For presentation purposes, sharpness, contrast, and brightness were adjusted to maximize clarity without changing content. B: individual data points per section counted and means ± SE for percentages of tubular epithelial cells in cortical sections from control littermates and PC-Gq-DREADD mice that expressed AQP2 (n > 1,900 cells counted in 4 and 8 slices, respectively) and mCitrine (n > 600 cells counted in 14 slices for each genotype). Sections are from both male and female mice. *Significantly greater than control littermates (by a z-test).
These IF and IHC results clearly document that mCitrine and, thus, Gq-DREADD are coexpressed with AQP2 in principal cells of PC-Gq-DREADD but not littermate control mice. Moreover, these results are consistent with widespread and abundant expression of Gq-DREADD in principal cells of the PC-Gq-DREADD mouse.
Targeted activation of Gq-DREADD in principal cells decreases ENaC activity and increases Na+ excretion.
With the PC-Gq-DREADD model in hand, we next tested 1) if selective stimulation of Gq signaling in principal cells was sufficient to decrease ENaC activity and 2) if pharmacogenetic activation of Gq signaling in principal cells was sufficient to cause a natriuresis. As noted above, quantification of ENaC activity by patch-clamp analysis and quantification of UNaV were used, respectively, to test these questions.
Figure 6 shows current traces of the ENaC in patch-clamped luminal plasma membranes of principal cells in isolated split-open tubules from control (Fig. 6A) and PC-Gq-DREADD (Fig. 6B) mice before and after addition of 5 μM CNO. For these experiments, the CNT and CCD were isolated from mice maintained on a Na+-free diet for 5–7 days before being patched. Figure 6C shows a summary graph of results from similar experiments where treatment with CNO for ~40 min significantly decreased ENaC activity in tubules isolated from PC-Gq-DREADD mice (from 0.86 ± 0.15 to 0.18 ± 0.03) but not littermate controls (from 0.68 ± 0.21 to 0.69 ± 0.16). ENaC activity without treatment in tubules from the PC-Gq-DREADD mouse was not different from that in controls, and treatment with CNO had no effect on tubules from littermate control mice. However, after CNO treatment, ENaC activity was significantly lower in tubules from PC-Gq-DREADD than CNO-treated littermate control mice and untreated tubules from PC-Gq-DREADD mice.
Fig. 6.

Pharmacogenetic activation of Gq signaling in principal cells decreases epithelial Na+ channel (ENaC) activity. A and B: current traces of ENaC activity in cell-attached patches on apical plasma membranes of principal cells in split-open aldosterone-sensitive distal nephrons from littermate control (A) and PC-Gq-designer receptors exclusively activated by designer drugs (DREADD; B) mice before and after addition of 5 μM clozapine-N-oxide (CNO; arrow) to extracellular bath solution. Tubules were isolated from mice maintained on a Na+-free diet for 5–7 days, and patches were voltage clamped to −60 mV with downward inward current. The closed state current level is denoted by a dashed line. For the purpose of presentation, current traces were filtered at 40 Hz, slight corrections were made to baselines to adjust for slow current drifts, and the very noisy periods (2.5–10 s) during manual addition of drug to the bath were removed (clearly marked by hatched bars). C: means ± SE (black circles) and individual data points (gray circles) for ENaC activity [probability of open channel (NPo; where N is the number of channels in a patch and Po is the probability that these channels are open)] in tubules from untreated (n = 10 from 3 different mice) and 5 μM CNO-treated (~40 min, n = 10 from 3 different mice) littermate control mice and untreated (n = 9 from 3 different mice) and 5 μM CNO-treated (~40 min, n = 10 from 3 different mice) PC-Gq-DREADD mice. Mice of both sexes were used in approximately equal proportions. *Significantly lower than all other groups (by one-way ANOVA with Tukey’s honest significant difference post hoc test).
UNaV in PC-Gq-DREADD and littermate control mice treated with 0.025 mg/ml CNO in drinking water is shown in Fig. 7. As mentioned above, mice were acclimated for 3 days before the 7-day experimental period. CNO was added to the drinking water on day 2 of the experimental period. Also, mice were switched from regular chow to the Na+-free diet on day 2 of the acclimation period, causing a significant reduction in UNaV from 9.10 ± 0.37 to 0.08 ± 0.01 μmol·h−1·mouse−1 in PC-Gq-DREADD mice and from 10.68 ± 0.89 to 0.10 ± 0.02 μmol·h−1·mouse−1 in littermate control mice. Urinary volumes were 1.7 ± 0.23, 1.69 ± 0.26, 2.26 ± 0.22, and 1.96 ±0.28 ml·h−1·mouse−1, respectively. Na+ excretion was not different between these groups before and after the switch to the Na+-free diet. At 24 h after treatment with CNO, UNaV was significantly increased compared with before treatment in PC-Gq-DREADD mice (2.53 ± 0.30 μmol·h−1·mouse−1, volume: 1.74 ± 0.31 ml·h−1·mouse−1) but not in littermate control mice (0.12 ± 0.03 μmol·h−1·mouse−1, volume: 1.17 ± 0.15 ml·h−1·mouse−1). UNaV 24 h after CNO treatment was also significantly greater in PC-Gq-DREADD than littermate control mice at the same time point.
Fig. 7.

Pharmacogenetic activation of Gq signaling in principal cells causes an increase in urinary Na+ excretion (UNaV). Individual data points and means ± SE of 24-h UNaV are shown for control littermate (n = 7 cages with 3 animals/cage for a total of 15 female and 6 male mice, each tested once) and PC-Gq-designer receptors exclusively activated by designer drugs (DREADD) (n = 7 cages with 3 animals/cage for a total of 12 female and 9 male mice, each tested once) mice maintained with a regular diet, a Na+-free diet (n = 7 cages, for a total of 15 female and 6 male and 12 female and 9 male mice, respectively), and a Na+-free diet + clozapine-N-oxide (CNO; n = 4 and 5 cages with 3 animals/cage for a total of 6 and 9 female and 6 and 6 male mice, respectively, each tested once). *Significantly lower than the control diet; **significantly higher than Na+-free diet and CNO-treated littermate mice (by one-way ANOVA with Tukey’s honest significant difference post hoc test).
The results shown in Figs. 6 and 7 are consistent with selective activation of Gq signaling exclusively in principal cells being sufficient to decrease ENaC activity and cause a dependent natriuresis. Moreover, they identify a reduction in ENaC activity by Gq-dependent signaling in principal cells as a mechanism capable of eliciting a natriuresis similar to that resulting from more widespread activation of P2Y2 receptor signaling.
DISCUSSION
The present study tested if inhibition of ENaC by P2Y2 receptor-Gq signaling in principal cells is sufficient to cause a natriuresis. As a consequence, it definitively identified organ, cell, and protein targets capable of translating purinergic actions on blood pressure.
Changes in UNaV affect blood pressure. UNaV is changed by effects on glomerular filtration rate (GFR) and tubular transport. Vascular smooth muscle and tubular epithelial cells are effectors that control filtration and transport, respectively.
In the global P2Y2 receptor knockout mouse model, blood pressure is elevated in the presence of hypokalemia, decreased fractional Na+ excretion, and suppressed renin and aldosterone levels, although GFR is normal (33, 38, 39, 46). Such observations phenocopy those from mice and humans harboring gain of function mutations in ENaC: Liddle’s syndrome (9, 10, 18, 20, 44). In Liddle’s syndrome, suppression of renin and aldosterone levels is a futile compensatory response to an increase in blood pressure resulting from an end-organ defect. The associated hypokalemia is a manifestation of avid K+ secretion in the maintenance of charge electroneutrality across the distal nephron as Na+ is robustly, but improperly (considering the blood pressure state), reabsorbed here. The fact that K+ excretion is robust, even in the presence of suppressed aldosterone, in both Liddle’s sydrome and the P2Y2 receptor knockout mouse directly points to ENaC hyperactivity. This was subsequently confirmed for both models (9, 20, 30, 33, 44, 47). That GFR is not affected by global deletion of the P2Y2 receptor, although these animals have elevated blood pressure and decreased fractional excretion of Na+, which, recall, is solely a manifestation of tubular transport, is consistent with a tubulopathy, rather than a vascular defect, being the chief cause of the blood pressure phenotype in the P2Y2 receptor knockout mouse. However, while compelling, this evidence has been argued to be indirect with respect to identifying the chief organ and the cell and protein targets responsible for elevated blood pressure in the absence of normal P2Y2 receptor function.
Here, we show that targeted stimulation of the P2Y2 receptor with the selective agonist MRS2768 is sufficient to evoke Na+ excretion when the need to conserve Na+ is high. We also found that MRS2768 decreases ENaC activity in isolated split-open tubules, suggesting one possible cause for this increase in Na+ excretion. These findings are in agreement with similar previous observations that 1) ATP and UTP decrease ENaC activity in native principal cells via P2Y2 receptor-Gq signaling (6, 25, 26, 30, 33, 47); 2) selective P2Y2 receptor agonism with INS45973 decreases blood pressure, increases Na+ excretion, and increases fractional excretion of fluid and Na+ in wild-type, but not P2Y2 receptor knockout, mice; and 3) INS45973 is without effect on GFR in both wild-type and knockout mice (39, 40). The effects of both MRS2768 in the present study and INS45973 in this prior study on renal excretion mirror those of the ENaC inhibitor amiloride: they promote a natriuresis without an accompanying change in GFR. This suggests to us a primary defect in the tubule but does not preclude additional vascular defects contributing to the blood pressure phenotype of the P2Y2 receptor knockout mouse. Indeed, in the earlier study using INS45973 and the original study of the P2Y2 receptor knockout mouse (38, 39), it was suggested that a vascular effect was primarily responsible for changes in excretion and blood pressure.
From our earlier work, as discussed above, we know that ENaC activity in principal cells is decreased by purines via P2Y2 receptor-Gq signaling and that loss of this inhibition contributes to an inappropriate decrease in UNaV and, consequently, an increase in blood pressure (26, 30, 33, 46, 47, 49). This inhibitory purinergic regulation of ENaC via P2Y2 receptor signaling functions, in parallel with the RAAS for salt-dependent increases in blood pressure in the P2Y2 receptor knockout mouse, is further exacerbated when mineralocorticoid levels are clamped at a high level (30, 33, 46). The salt-dependent increase in blood pressure in the P2Y2 receptor knockout mouse, like that in connexin (Cx)30 and large-conductance Ca2+-activated K+ channel containing the β4-subunit (BK-β4) knockout mice, is reversed by the ENaC inhibitor amiloride (6, 26, 46, 52). Cx30 hemichannels in the luminal plasma membranes of intercalated cells have been shown to be a conduit for flow-sensitive ATP secretions into the urine of the distal nephron (21, 26, 46, 47, 49), and recent findings are consistent with BK-β4 functioning as important flow sensors capable of influencing ATP secretions from intercalated cells (6, 11, 52). Deletion of Cx30 and BK-β4 phenocopy gain of function of ENaC and loss of function of the P2Y2 receptor. Findings from this earlier body of work argue that decreases in ENaC activity in response to activation of inhibitory purinergic signaling in principal cells of the distal nephron should be sufficient to promote a natriuresis, even when the need to conserve Na+ is maximized.
We confirm here, as widely appreciated, that long-term restriction of dietary Na+ markedly decreases UNaV. Under such conditions, targeted activation of Gq signaling selectively in principal cells of the distal nephron with a pharmacogenetics approach causes an inappropriate natriuresis, considering the Na+ intake of these animals. This natriuresis is caused by a decrease in ENaC activity, specifically in principal cells. The most logical conclusion that can be drawn from such observations is that activation of P2Y2 receptor-Gq signaling in principal cells is sufficient, in and of itself, to evoke a natriuresis by decreasing ENaC activity. Such a conclusion suggests that perhaps this pathway, when broken, contributes to pathological elevations in blood pressure and is an appropriate target in certain instances to manage increases in blood pressure. As shown by the present study, we know that stimulation of purinergic inhibition of ENaC is powerful enough to evoke a natriuresis under conditions that should severely limit such manifestations. Thus, the P2Y2 receptor-Gq-ENaC axis in the distal nephron may be a reasonable target for therapy to treat elevated blood pressure and, when defective, a possible causative agent in certain instances for pathological elevation of blood pressure.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01 DK-113816 and American Heart Association Grant 17GRNT32920002 (to J. D. Stockand).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
E.M. and J.D.S. conceived and designed research; E.M. and F.S. performed experiments; E.M., F.S., and J.D.S. analyzed data; E.M., F.S., and J.D.S. interpreted results of experiments; E.M. and J.D.S. prepared figures; E.M. and J.D.S. drafted manuscript; E.M. and J.D.S. edited and revised manuscript; E.M., F.S., and J.D.S. approved final version of manuscript.
REFERENCES
- 1.Alpern RJ, Moe OW, Caplan MJ. Seldin and Giebisch's The Kidney. Physiology and Pathophysiology (5th ed). Amsterdam: Elsevier, 2013 [Google Scholar]
- 2.Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci USA 104: 5163–5168, 2007. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berman JM, Mironova E, Stockand JD. Physiological regulation of the epithelial Na+ channel by casein kinase II. Am J Physiol Renal Physiol 314: F367–F372, 2018. doi: 10.1152/ajprenal.00469.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boron WF, Boulpaep EL. Medical Physiology. Philadelphia, PA: Elsevier, 2017, p. xii. [Google Scholar]
- 5.Bugaj V, Mironova E, Kohan DE, Stockand JD. Collecting duct-specific endothelin B receptor knockout increases ENaC activity. Am J Physiol Cell Physiol 302: C188–C194, 2012. doi: 10.1152/ajpcell.00301.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bugaj V, Sansom SC, Wen D, Hatcher LI, Stockand JD, Mironova E. Flow-sensitive K+-coupled ATP secretion modulates activity of the epithelial Na+ channel in the distal nephron. J Biol Chem 287: 38552–38558, 2012. doi: 10.1074/jbc.M112.408476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Canessa CM, Horisberger JD, Rossier BC. Epithelial sodium channel related to proteins involved in neurodegeneration. Nature 361: 467–470, 1993. doi: 10.1038/361467a0. [DOI] [PubMed] [Google Scholar]
- 8.Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367: 463–467, 1994. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 9.Hansson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Lu Y, Canessa C, Iwasaki T, Rossier B, Lifton RP. Hypertension caused by a truncated epithelial sodium channel γ-subunit: genetic heterogeneity of Liddle syndrome. Nat Genet 11: 76–82, 1995. doi: 10.1038/ng0995-76. [DOI] [PubMed] [Google Scholar]
- 10.Hansson JH, Schild L, Lu Y, Wilson TA, Gautschi I, Shimkets R, Nelson-Williams C, Rossier BC, Lifton RP. A de novo missense mutation of the β-subunit of the epithelial sodium channel causes hypertension and Liddle syndrome, identifying a proline-rich segment critical for regulation of channel activity. Proc Natl Acad Sci USA 92: 11495–11499, 1995. doi: 10.1073/pnas.92.25.11495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holtzclaw JD, Cornelius RJ, Hatcher LI, Sansom SC. Coupled ATP and potassium efflux from intercalated cells. Am J Physiol Renal Physiol 300: F1319–F1326, 2011. doi: 10.1152/ajprenal.00112.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holtzclaw JD, Grimm PR, Sansom SC. Role of BK channels in hypertension and potassium secretion. Curr Opin Nephrol Hypertens 20: 512–517, 2011. doi: 10.1097/MNH.0b013e3283488889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hummler E. Implication of ENaC in salt-sensitive hypertension. J Steroid Biochem Mol Biol 69: 385–390, 1999. doi: 10.1016/S0960-0760(99)00073-4. [DOI] [PubMed] [Google Scholar]
- 14.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 8: e1000412, 2010. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kittikulsuth W, Stuart D, Van Hoek AN, Stockand JD, Bugaj V, Mironova E, Blount MA, Kohan DE. Lack of an effect of collecting duct-specific deletion of adenylyl cyclase 3 on renal Na+ and water excretion or arterial pressure. Am J Physiol Renal Physiol 306: F597–F607, 2014. doi: 10.1152/ajprenal.00505.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koehler S, Brähler S, Kuczkowski A, Binz J, Hackl MJ, Hagmann H, Höhne M, Vogt MC, Wunderlich CM, Wunderlich FT, Schweda F, Schermer B, Benzing T, Brinkkoetter PT. Single and transient Ca2+ peaks in podocytes do not induce changes in glomerular filtration and perfusion. Sci Rep 6: 35400, 2016. doi: 10.1038/srep35400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kunzelmann K, Bachhuber T, Regeer R, Markovich D, Sun J, Schreiber R. Purinergic inhibition of the epithelial Na+ transport via hydrolysis of PIP2. FASEB J 19: 142–143, 2005. doi: 10.1096/fj.04-2314fje. [DOI] [PubMed] [Google Scholar]
- 18.Liddle G, Bledsoe T, Coppage WJ. A familial renal disorder simulating primary aldosteronism but with negligible aldosterone secretion. Trans Assoc Am Physicians 76: 199–213, 1963. [Google Scholar]
- 19.Lingueglia E, Voilley N, Waldmann R, Lazdunski M, Barbry P. Expression cloning of an epithelial amiloride-sensitive Na+ channel. A new channel type with homologies to Caenorhabditis elegans degenerins. FEBS Lett 318: 95–99, 1993. doi: 10.1016/0014-5793(93)81336-X. [DOI] [PubMed] [Google Scholar]
- 20.Lu C, Pribanic S, Debonneville A, Jiang C, Rotin D. The PY motif of ENaC, mutated in Liddle syndrome, regulates channel internalization, sorting and mobilization from subapical pool. Traffic 8: 1246–1264, 2007. doi: 10.1111/j.1600-0854.2007.00602.x. [DOI] [PubMed] [Google Scholar]
- 21.McCulloch F, Chambrey R, Eladari D, Peti-Peterdi J. Localization of connexin 30 in the luminal membrane of cells in the distal nephron. Am J Physiol Renal Physiol 289: F1304–F1312, 2005. doi: 10.1152/ajprenal.00203.2005. [DOI] [PubMed] [Google Scholar]
- 22.Mironova E, Boiko N, Bugaj V, Kucher V, Stockand JD. Regulation of Na+ excretion and arterial blood pressure by purinergic signalling intrinsic to the distal nephron: consequences and mechanisms. Acta Physiol (Oxf) 213: 213–221, 2015. doi: 10.1111/apha.12372. [DOI] [PubMed] [Google Scholar]
- 23.Mironova E, Bugaj V, Roos KP, Kohan DE, Stockand JD. Aldosterone-independent regulation of the epithelial Na+ channel (ENaC) by vasopressin in adrenalectomized mice. Proc Natl Acad Sci USA 109: 10095–10100, 2012. doi: 10.1073/pnas.1201978109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mironova E, Bugay V, Pochynyuk O, Staruschenko A, Stockand JD. Recording ion channels in isolated, split-opened tubules. Methods Mol Biol 998: 341–353, 2013. doi: 10.1007/978-1-62703-351-0_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mironova E, Lynch IJ, Berman JM, Gumz ML, Stockand JD, Wingo CS. ENaC activity in the cortical collecting duct of HKα1 H+,K+-ATPase knockout mice is uncoupled from Na+ intake. Am J Physiol Renal Physiol 312: F1073–F1080, 2017. doi: 10.1152/ajprenal.00401.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mironova E, Peti-Peterdi J, Bugaj V, Stockand JD. Diminished paracrine regulation of the epithelial Na+ channel by purinergic signaling in mice lacking connexin 30. J Biol Chem 286: 1054–1060, 2011. doi: 10.1074/jbc.M110.176552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakajima K, Wess J. Design and functional characterization of a novel, arrestin-biased designer G protein-coupled receptor. Mol Pharmacol 82: 575–582, 2012. doi: 10.1124/mol.112.080358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nelson RD, Stricklett P, Gustafson C, Stevens A, Ausiello D, Brown D, Kohan DE. Expression of an AQP2 Cre recombinase transgene in kidney and male reproductive system of transgenic mice. Am J Physiol Cell Physiol 275: C216–C226, 1998. doi: 10.1152/ajpcell.1998.275.1.C216. [DOI] [PubMed] [Google Scholar]
- 29.Pei Y, Dong S, Roth BL. Generation of designer receptors exclusively activated by designer drugs (DREADDs) using directed molecular evolution. Curr Protoc Neurosci Chapter 4: 33, 2010. doi: 10.1002/0471142301.ns0433s50. [DOI] [PubMed] [Google Scholar]
- 30.Pochynyuk O, Bugaj V, Rieg T, Insel PA, Mironova E, Vallon V, Stockand JD. Paracrine regulation of the epithelial Na+ channel in the mammalian collecting duct by purinergic P2Y2 receptor tone. J Biol Chem 283: 36599–36607, 2008. doi: 10.1074/jbc.M807129200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pochynyuk O, Bugaj V, Stockand JD. Physiologic regulation of the epithelial sodium channel by phosphatidylinositides. Curr Opin Nephrol Hypertens 17: 533–540, 2008. doi: 10.1097/MNH.0b013e328308fff3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pochynyuk O, Bugaj V, Vandewalle A, Stockand JD. Purinergic control of apical plasma membrane PI(4,5)P2 levels sets ENaC activity in principal cells. Am J Physiol Renal Physiol 294: F38–F46, 2008. doi: 10.1152/ajprenal.00403.2007. [DOI] [PubMed] [Google Scholar]
- 33.Pochynyuk O, Rieg T, Bugaj V, Schroth J, Fridman A, Boss GR, Insel PA, Stockand JD, Vallon V. Dietary Na+ inhibits the open probability of the epithelial sodium channel in the kidney by enhancing apical P2Y2-receptor tone. FASEB J 24: 2056–2065, 2010. doi: 10.1096/fj.09-151506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pochynyuk O, Tong Q, Medina J, Vandewalle A, Staruschenko A, Bugaj V, Stockand JD. Molecular determinants of PI(4,5)P2 and PI(3,4,5)P3 regulation of the epithelial Na+ channel. J Gen Physiol 130: 399–413, 2007. doi: 10.1085/jgp.200709800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pochynyuk O, Tong Q, Staruschenko A, Ma HP, Stockand JD. Regulation of the epithelial Na+ channel (ENaC) by phosphatidylinositides. Am J Physiol Renal Physiol 290: F949–F957, 2006. doi: 10.1152/ajprenal.00386.2005. [DOI] [PubMed] [Google Scholar]
- 36.Pochynyuk O, Tong Q, Staruschenko A, Stockand JD. Binding and direct activation of the epithelial Na+ channel (ENaC) by phosphatidylinositides. J Physiol 580: 365–372, 2007. doi: 10.1113/jphysiol.2006.127449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramkumar N, Stuart D, Mironova E, Abraham N, Gao Y, Wang S, Lakshmipathi J, Stockand JD, Kohan DE. Collecting duct principal, but not intercalated, cell prorenin receptor regulates renal sodium and water excretion. Am J Physiol Renal Physiol 315: F607–F617, 2018. doi: 10.1152/ajprenal.00122.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rieg T, Bundey RA, Chen Y, Deschenes G, Junger W, Insel PA, Vallon V. Mice lacking P2Y2 receptors have salt-resistant hypertension and facilitated renal Na+ and water reabsorption. FASEB J 21: 3717–3726, 2007. doi: 10.1096/fj.07-8807com. [DOI] [PubMed] [Google Scholar]
- 39.Rieg T, Gerasimova M, Boyer JL, Insel PA, Vallon V. P2Y2 receptor activation decreases blood pressure and increases renal Na+ excretion. Am J Physiol Regul Integr Comp Physiol 301: R510–R518, 2011. doi: 10.1152/ajpregu.00148.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rieg T, Schroth J, Insel PA, Broyer J, Vallon V. Defining roles for P2Y2 and P2Y4 receptors on blood pressure and urinary excretion (Abstract). FASEB J 24: 606.3, 2010. [Google Scholar]
- 41.Roos KP, Bugaj V, Mironova E, Stockand JD, Ramkumar N, Rees S, Kohan DE. Adenylyl cyclase VI mediates vasopressin-stimulated ENaC activity. J Am Soc Nephrol 24: 218–227, 2013. doi: 10.1681/ASN.2012050449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roth BL. DREADDs for neuroscientists. Neuron 89: 683–694, 2016. doi: 10.1016/j.neuron.2016.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rotin D, Schild L. ENaC and its regulatory proteins as drug targets for blood pressure control. Curr Drug Targets 9: 709–716, 2008. doi: 10.2174/138945008785132367. [DOI] [PubMed] [Google Scholar]
- 44.Schild L. The ENaC channel as the primary determinant of two human diseases: Liddle syndrome and pseudohypoaldosteronism. Nephrologie 17: 395–400, 1996. [PubMed] [Google Scholar]
- 45.Schild L, Kellenberger S. Structure function relationships of ENaC and its role in sodium handling. Adv Exp Med Biol 502: 305–314, 2001. doi: 10.1007/978-1-4757-3401-0_20. [DOI] [PubMed] [Google Scholar]
- 46.Sipos A, Vargas SL, Toma I, Hanner F, Willecke K, Peti-Peterdi J. Connexin 30 deficiency impairs renal tubular ATP release and pressure natriuresis. J Am Soc Nephrol 20: 1724–1732, 2009. doi: 10.1681/ASN.2008101099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stockand JD, Mironova E, Bugaj V, Rieg T, Insel PA, Vallon V, Peti-Peterdi J, Pochynyuk O. Purinergic inhibition of ENaC produces aldosterone escape. J Am Soc Nephrol 21: 1903–1911, 2010. doi: 10.1681/ASN.2010040377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stricklett PK, Nelson RD, Kohan DE. Targeting collecting tubules using the aquaporin-2 promoter. Exp Nephrol 7: 67–74, 1999. doi: 10.1159/000020587. [DOI] [PubMed] [Google Scholar]
- 49.Svenningsen P, Burford JL, Peti-Peterdi J. ATP releasing connexin 30 hemichannels mediate flow-induced calcium signaling in the collecting duct. Front Physiol 4: 292, 2013. doi: 10.3389/fphys.2013.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vallon V, Stockand J, Rieg T. P2Y receptors and kidney function. Wiley Interdiscip Rev Membr Transp Signal 1: 731–742, 2012. doi: 10.1002/wmts.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verouti SN, Boscardin E, Hummler E, Frateschi S. Regulation of blood pressure and renal function by NCC and ENaC: lessons from genetically engineered mice. Curr Opin Pharmacol 21: 60–72, 2015. doi: 10.1016/j.coph.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 52.Wen D, Cornelius RJ, Rivero-Hernandez D, Yuan Y, Li H, Weinstein AM, Sansom SC. Relation between BK-α/β4-mediated potassium secretion and ENaC-mediated sodium reabsorption. Kidney Int 86: 139–145, 2014. doi: 10.1038/ki.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wess J, Nakajima K, Jain S. Novel designer receptors to probe GPCR signaling and physiology. Trends Pharmacol Sci 34: 385–392, 2013. doi: 10.1016/j.tips.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu H, Aryal DK, Olsen RH, Urban DJ, Swearingen A, Forbes S, Roth BL, Hochgeschwender U. Cre-dependent DREADD (designer receptors exclusively activated by designer drugs) mice. Genesis 54: 439–446, 2016. doi: 10.1002/dvg.22949. [DOI] [PMC free article] [PubMed] [Google Scholar]