

Abstract
A major ambition of artificial intelligence lies in translating patient data to successful therapies. Machine learning models face particular challenges in biomedicine, however, including handling of extreme data heterogeneity and lack of mechanistic insight into predictions. Here, we argue for “visible” approaches that guide model structure with experimental biology.
Like many fields, biomedicine is in the midst of a data revolution. Comprehensive molecular and clinical datasets–including complete human genomes, gene expression profiles, high-resolution imaging, metabolomics, electronic medical records, and so on–are no longer isolated to a few study participants; in a few years, we will have such comprehensive information for millions of patients (Torkamani et al., 2017). Multiple analysis approaches have been advanced to transform patient data into successful therapies, each with their particular benefits and limitations (Figure 1). Most prominently, the field of machine learning has seen dramatic advances in the past few years (LeCun et al., 2015) with much excitement around the use of many-layered, “deep,” artificial neural networks, inspired by actual neural networks and how the brain processes patterns. After training over many examples, artificial neural networks learn to predict the correct answer –or output –that should be returned for the many possible input patterns. Deep learning approaches have been used to recognize objects in images like dogs, people, and faces and to distinguish good from bad moves in games like chess and Go (Silver et al., 2016).
Figure 1. Current Approaches Relevant to Analysis of Big Biomedical Data.

First subheading (blue): what each of four approaches (A)–(D) accomplish. Second subheading (orange): limitations and challenges for these approaches. Box: this commentary argues for the synthesis of two approaches in particular–machine learning (B) and experimental cell and tissue biology (C)–resulting in an infrastructure for visible intelligence.
Given the parallel advances in biomedical data and computer science, a key question is the extent to which current machine-learning models will be effective at interpreting the massive streams of biomedical information. In particular, will large patient datasets, provided as inputs to deep neural networks or related methods, be sufficient to create the next generation of reliable and precise intelligence infrastructure for understanding and treating disease?
Here, we argue that the answer is no–that the very high complexity of biological systems will intrinsically limit applications of current “black box” machine learning in patient data. As one path forward, we highlight a new generation of “visible” approaches that aim to guide the structure of machine-learning models with an increasingly extensive knowledge of biological mechanism. That is, machine learning will not replace the need for experimental cell and tissue biology; it will be substantially enabled by such knowledge, given the right visible intelligence infrastructure.
Dual Challenges of Data Heterogeneity and Lack of Mechanistic Interpretation
Machine-learning systems face two recognized challenges that become particularly acute in biomedical applications. The first is input heterogeneity. Nearly all types of statistical analysis rely on identifying recurrent patterns in data, which provide rules by which future predictions are made. Problems arise, however, when the same outcomes may result from vastly different inputs. Although such input heterogeneity is a property of many and perhaps all complex systems, biological systems are almost certainly more complex than those addressed by machine learning in other areas. For example, cancer can arise as the result of many different combinations of genetic alterations involving many potential genes, any one of which may be mutated only rarely; as a consequence, each new patient presents a distinct constellation of molecular changes never before seen in nature (Alvarez et al., 2016; Kourou et al., 2014). Similar heterogeneity arises in patient data from nearly all common diseases, including cardiovascular, metabolic, and neurodevelopmental disorders, in which recurrent patterns are elusive, making it difficult to make reliable predictions (Boyle et al., 2017). Even rare, presumably Mendelian, disorders can be modified by myriad genetic modifiers elsewhere. Such heterogeneity has long posed a significant challenge to genetic association studies, which tend to be powered to identify single-locus effects (Figure 1A); it is also a significant challenge for machine learning.
One might consider a brute-force solution to the problem of heterogeneity by profiling ever more subjects to increase the total volume of data. Certainly, technologies like DNA sequencing are now powerful and inexpensive enough that we may soon have complete genomes for most new patients. The total number of patients is finite, however. Even for common conditions like cancer and heart disease, the number of available datasets will saturate at a few million patient examples. While a million may seem large, this number is modest compared to the amounts of data often needed to train a statistical model, compounded by the as-yet-unknown, but undoubtedly high, complexity of biological systems. As a consequence, even after obtaining the genome sequence of every patient, the genetic patterns driving disease may still remain undiscovered by current statistical methods. In contrast, in many other applications of machine learning such as game playing, if the machine runs out of test games for learning, more examples can readily be generated without bound.
A second well-known challenge is that modern machine learning models, including deep neural networks, are black boxes, devices which focus on predicting outputs from inputs without regard for the mechanism or rationale by which a particular outcome is brought about. The game-playing system known as AlphaGo can beat human Go players (Silver et al., 2016), but an examination of its internal structure gives little insight into its moves. Its neural network is subject to extensive mathematical optimization during training, leading to a dense web of neural connections neither tied to an actual system nor based on human reasoning. Similarly, in biomedicine, many machine-learning methods are being developed to predict patient outcomes (Kourou et al., 2014), but these approaches typically do not link predictions to underlying mechanisms (Figure 1B). This is a missed opportunity, as causal mechanistic insights are key to identifying drug targets and advancing basic biological knowledge.
Toward Visible Engines for Machine Learning
A popular toy in the 1950’s was a working model of an automobile engine called the Visible V8, versions of which are still available today (Figure 2A). As with many toy models of cars, the engine turned a crankshaft, useful for driving a car forward. However, the main draw of the Visible V8 was not this final engine output, but its faithful simulation of interacting engine components necessary to bring about this result. The engine was correctly subdivided into parts such as the engine block, cylinder heads, distributor, cooling fan, alternator, and both intake and exhaust manifolds. The block and heads, in turn, contained working models of pistons, spark plugs, cams, and camshafts. Importantly, all these aspects were clearly visible because the entire engine case and its hierarchy of constituent parts were transparent.
Figure 2. Visible Models.
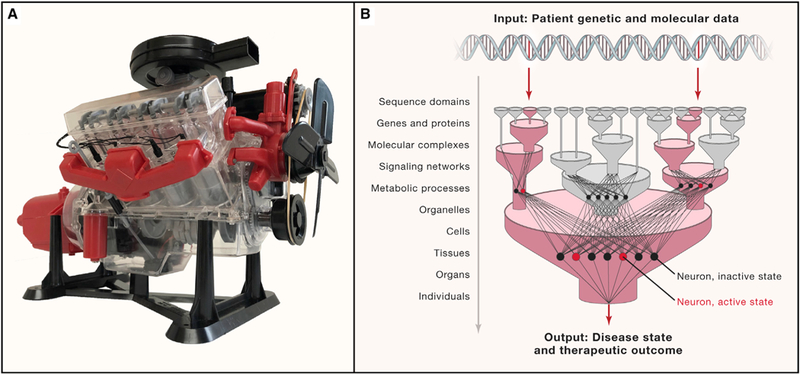
(A) The Visible V8 was a popular quarter-scale model of an internal combustion engine sold by Renwal Model Company starting in 1958. It conveys the concept and value of visible machine learning. Photo: Trey Ideker. (B) Guiding machine learning systems with visible multi-scale biological structure.
Like man-made engines, biological systems are also complex machines whose outputs emerge from a hierarchy of internal components (Simon, 1962). DNA nucleotides assemble to form sequence domains and genes; linear gene sequences encode 3-dimensional protein structures; proteins assemble to create molecular complexes and pathways; pathways occur within organelles and cells; and cells and cell types assemble to form tissues, organs, and individuals. Mapping such structures has classically been the domain of cell and tissue biology, which has developed a spectrum of experimental measurement techniques to characterize biological machines at each scale (Figure 1C). To name a few of the relevant approaches, protein structures are determined using technologies like cryo-electron microscopy; multimeric protein complexes are cataloged systematically by affinity purification tandem mass spectrometry; larger cell structures are tracked dynamically by advanced light microscopy; and the multicellular architecture of tissues is determined increasingly rapidly by single-cell RNA sequencing. Prior information about cell and tissue biology can also be mined from indirect sources such as literature, although consistent literature curation is a difficult problem and misses the large amount of human biology that we do not yet know (Figure 1D).
Unfortunately, basic experimental data types are not usually well connected to analysis of patient data. It is nonetheless easy to see how prior knowledge of biological structure might provide distinct advantages to models capable of incorporating this information–what we here call visible learning–and recent research has begun to prove the concept.
Groundwork toward Visible Machine Learning in Biology
Visible learning relates to a topic called model interpretation, an active research area in the field of artificial intelligence. Generally, model interpretation tries to explain a model’s internal logic after a model has been trained (Ribeiro et al., 2016) or to force the model to have fewer parameters, which makes it easier to interpret (Lei et al., 2016). In biology, the particular need to understand internal mechanisms, along with the ability to probe these mechanisms, has inspired a class of machine-learning models that is guided by prior mechanistic knowledge. This knowledge is often represented by large molecular network structures, which document known mechanistic aspects of cell biology such as interactions among subunits of a protein complex, between receptors and kinases, or among transcriptional regulatory proteins, enhancers, and genes. One way in which these networks have been used to guide learning is in selection of a minimal set of features for prediction, such as prioritizing candidate disease genes based on network proximity to other genes for which sequence variations are strongly associated with disease (Leiserson et al., 2013). Network connections can also nominate a set of genes whose input data should be aggregated to create a composite feature, such as pooling of rarely altered genes in cancer into a single pathway, which, viewed as a single aggregate, is recurrently altered across a patient population (Alvarez et al., 2016; Chuang et al., 2007).
Models with hierarchical (multi-scale) resolution of cell biology are also emerging (Karr et al., 2012), an idea that fits naturally with deep (multi-layered) neural networks. For instance, in formulating a neural network, one might specify the architecture of the intermediate layers–the number of layers, the number of neurons per layer, and which connections between layers are allowed. These neurons, usually hidden inside of the black box, can be made visible by attaching them to actual biological components (Figure 2B). This idea was recently explored by Lin et al. (2017), who predicted cell type and state using a deep neural network for which the structure was determined based on the hierarchical organization of transcriptional regulatory factors in the nucleus. We extended such a strategy to assemble DCell, a model of a basic eukaryotic cell (Ma et al., 2018). DCell uses a deep neural network to translate a list of mutated genes (genotype input) to the resulting cell proliferation rate (phenotype output). Neurons are organized into banks, each of which maps to a distinct biological module within a large hierarchy of known cellular components and processes. A predicted change in cell phenotype can then be interpreted by examining the functional states of underlying cellular components, internal to DCell, whose neuron states are also highly affected.
In building these visible systems, care must be taken to learn from the successes and pitfalls of the vast body of work in biological modeling over the past several decades (Fisher and Henzinger, 2007). For instance, an important lesson is that increased model resolution (e.g., introducing many detailed biochemical parameters) may come at the expense of model scope (e.g., the ability to address all biological elements and generalize to new patient cohorts).
Understanding Biomedical Data with Visible Machine Learning
Visible machine learning offers two advantages in building intelligent models of biomedical systems. First, prior knowledge of biological structure can address the problem of data heterogeneity, since different input patterns, even when entirely distinct from one another (i.e., without common patterns), converge on common higher-order biological processing units corresponding to discrete modular components of cells and tissues. All machine-learning systems perform this type of data compression, or dimensionality reduction. In black-box models, the configuration of hidden layers that is required for sufficient data compression is inferred during the training procedure, typically requiring very large quantities of training data. In contrast, direct incorporation of the biological structure of cells and tissues, such as explored by Lin et al. (2017) and Ma et al. (2018), leads to a ready-made working model of how biological inputs, such as genotype, are compressed to determine outcomes.
Second, models guided by biological structure can be interpreted mechanistically, informing our understanding of the system and suggesting potential therapeutic strategies. Given input patient data, execution of the model not only produces a final output state; it also reveals the states of internal biological systems. The most striking of these internal states provide hypotheses as to the underlying mechanisms governing patient phenotype, which is important because many internal biological states are difficult to measure through direct experimental observations. For example, Alvarez et al. (2016) used a transcriptional regulatory network to translate patient mRNA expression profiles to activities of regulatory proteins, most of which are difficult to interrogate experimentally. Internal states of the model may also indicate biological components that can be targeted by therapeutic interventions or form the basis for in silico testing of treatment combinations.
Goals and Milestones for the Near Future
We conclude with a short summary of milestones that research in visible machine learning might seek to achieve in the relatively near term. First is the advent of diverse algorithms to inform machine-learning systems with prior knowledge of biological structure, along with rigorous testing and validation of such algorithms. These developments may involve the application of existing mathematical approaches, require new frameworks, or both. Second, advances in our understanding of complexity are needed to assess and quantify complexity in biological systems and how it differs across the spectrum of tasks to be addressed by biomedical machine learning models. Third, investments in large-scale experimental biology will greatly expand the type and coverage of data that are available to map biological structures within cells and tissues. Generation of such data may involve new technology development to increase experimental throughput, such as advances in 3D cellular imaging or protein interaction mapping, or scale-up of existing technologies. Fourth, significant advances in computation infrastructure are needed to create high performance computing environments, along with web resources for community model development and distribution. Finally, early routes should be sought for embedding visible machine learning models in the clinic to begin evaluating best practices and validating efficacy for predicting patient outcomes and therapies within and across institutions. Notably, funding agencies have begun to promote research into some of these milestones (e.g., the NIH Data Commons or the DARPA Explainable Artificial Intelligence program), although not always with a focus on biomedicine. Parallel development of these directions will enable a new generation of biomedical machine learning, replacing black-box models that focus on isolated problem domains with visible models that survey general biological systems.
ACKNOWLEDGMENTS
We are indebted to many individuals for conversations that inspired and informed this commentary, including Terry Sejnowski, Andrea Califano, Michael Kramer, and Janusz Dutkowski. We are grateful for the help of Aidan Ideker, Cherie Ng, and Charlotte Curtis in building the Visible V8 engine model shown in Figure 2A. This work was supported by grants from the NIH (R01 HG009979, OT3 TR002026, and P41 GM103504 to T.I. and R01 HG007069 and U24 CA211000 to B.J.R.).
Footnotes
DECLARATION OF INTERESTS
T.I. is co-founder of Data4Cure and has an equity interest. T.I. has an equity interest in Ideaya BioSciences. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. B.J.R. is a founder of Medley Genomics and a member of its board of directors.
REFERENCES
- Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, and Califano A (2016). Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 48, 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle EA, Li YI, and Pritchard JK (2017). An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 169, 1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang H-Y, Lee E, Liu Y-T, Lee D, and Ideker T (2007). Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 3, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher J, and Henzinger TA (2007). Executable cell biology. Nat. Biotechnol. 25, 1239–1249. [DOI] [PubMed] [Google Scholar]
- Karr JR, Sanghvi JC, Macklin DN, Gutschow MV, Jacobs JM, Bolival B Jr., Assad-Garcia N, Glass JI, and Covert MW (2012). A whole-cell computational model predicts phenotype from genotype. Cell 150, 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourou K, Exarchos TP, Exarchos KP, Kara-mouzis MV, and Fotiadis DI (2014). Machine learning applications in cancer prognosis and prediction. Comput. Struct. Biotechnol. J. 13, 8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeCun Y, Bengio Y, and Hinton G (2015). Deep learning. Nature 521, 436–444. [DOI] [PubMed] [Google Scholar]
- Lei T, Barzilay R, and Jaakkola T (2016). Rationalizing neural predictions, arXiv, arXiv:1606.04155. http://arXiv.org/abs/1606.04155. [Google Scholar]
- Leiserson MDM, Eldridge JV, Ramachandran S, and Raphael BJ (2013). Network analysis of GWAS data. Curr. Opin. Genet. Dev. 23, 602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Jain S, Kim H, and Bar-Joseph Z (2017). Using neural networks for reducing the dimensions of single-cell RNA-Seq data. Nucleic Acids Res. 45, e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Yu MK, Fong S, Ono K, Sage E, Dem-chak B, Sharan R, and Ideker T (2018). Using deep learning to model the hierarchical structure and function of a cell. Nat. Methods 15, 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro MT, Singh S, and Guestrin C (2016). Why should I trust you?: Explaining the predictions of any classifier. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, (ACM), pp. 1135–1144. [Google Scholar]
- Silver D, Huang A, Maddison CJ, Guez A, Sifre L, van den Driessche G, Schrittwieser J, Antonoglou I, Panneershelvam V, Lanctot M, et al. (2016). Mastering the game of Go with deep neural networks and tree search. Nature 529, 484–489. [DOI] [PubMed] [Google Scholar]
- Simon HA (1962). The Architecture of Complexity. Proc. Am. Philos. Soc. 106, 467–482. [Google Scholar]
- Torkamani A, Andersen KG, Steinhubl SR, and Topol EJ (2017). High-Definition Medicine. Cell 170, 828–843. [DOI] [PMC free article] [PubMed] [Google Scholar]