

SUMMARY
Transient receptor potential (TRP) channels are regulated by diverse stimuli comprising thermal, chemical, and mechanical modalities. They are also commonly regulated by phosphatidylinositol-4,5-bisphosphate (PIP2), with underlying mechanisms largely unknown. We here revealed an intramolecular interaction of the TRPP3 N and C termini (N-C) that is functionally essential. The interaction was mediated by aromatic Trp81 in pre-S1 domain and cationic Lys568 in TRP-like domain. Structure-function analyses revealed similar N-C interaction in TRPP2 as well as TRPM8/-V1/-C4 via highly conserved tryptophan and lysine/arginine residues. PIP2 bound to cationic residues in TRPP3, including K568, thereby disrupting the N-C interaction and negatively regulating TRPP3. PIP2 had similar negative effects on TRPP2. Interestingly, we found that PIP2 facilitates the N-C interaction in TRPM8/-V1, resulting in channel potentiation. The intramolecular N-C interaction might represent a shared mechanism underlying the gating and PIP2 regulation of TRP channels.
Graphical Abstract
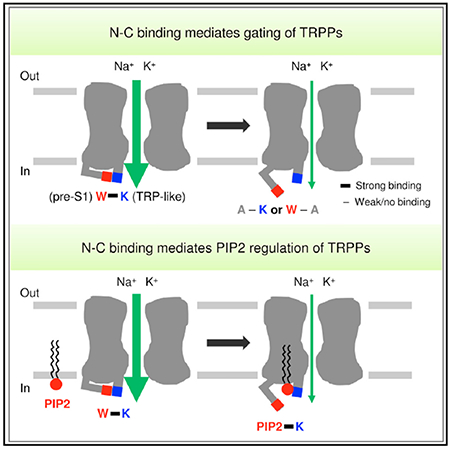
In Brief
Zheng et al. show that an aromatic Trp residue in pre-S1 and a cationic Lys residue in the TRP-like domain of TRP polycystin channels mediate N-C binding, which underlies TRPPs gating and PIP2 regulation. The conservation of these residues suggests that this may be a shared mechanism of TRP channel gating.
INTRODUCTION
Transient receptor potential (TRP) channels are sensors of various physical and chemical stimuli (Montell, 2005). They form a superfamily of cation channels, which is divided into eight subfamilies according to sequence similarities: TRPV (vanilloid); TRPC (canonical); TRPM (melastatin); TRPP (polycystin); TRPA (ankyrin); TRPML (mucolipin); TRPN (no mechanoreceptor potential C); and TRPY (yeast; Venkatachalam and Montell, 2007). TRP channels function as tetramers, in which each subunit contains six transmembrane segments (S1-S6) with S5-loop-S6 forming part of a central pore module and cytosolic N (amino) and C (carboxy) termini (Montell, 2005). A short N-terminal α-helical domain upstream of S1, called the pre-S1 domain, was proposed to be involved in allosteric gating (Saotome et al., 2016; Paulsen et al., 2015; Liao et al., 2013; Huynh et al., 2016). TRPC, TRPV, and TRPM proteins contain in their C terminus almost immediately after S6 a TRP domain characterized by a signature motif WKxxR (also called a TRP box) important for channel activation and PIP2 binding (Ramsey et al., 2006; Valente et al., 2011). The corresponding fragment in TRPA1, which contains a WxxxK motif instead of WKxxR, is termed a TRP-like domain (Paulsen et al., 2015). The TRP and TRP-like domains in structurally resolved TRPs all adopt α-helical configuration, which is parallel to the plasma membrane (Zubcevic et al., 2016; Saotome et al., 2016; Jin et al., 2017; Liao et al., 2013; Paulsen et al., 2015), suggesting that they may have similar functional roles. The corresponding part in resolved TRPP2 represented as a continuation of S6 and was perpendicular to the plasma membrane (Wilkes et al., 2017; Grieben et al., 2017; Shen et al., 2016), but we still called it a TRP-like domain in this manuscript because it also adopted α helix configuration and contained a YxxxK motif. Whereas TRP channels have long been recognized to respond to remarkably diverse stimuli ranging from pH, changes of temperature, light, touch, pheromones, osmolarity, and noxious chemicals, which is well correlated with their various functions in sensory physiology, such as sensations of pain, hot, warmth and cold, taste, pressure, and vision (Clapham et al., 2001; Montell, 2005), it still remains largely unknown as to how these diverse stimuli induce conformational changes resulting in pore opening and channel activation.
Phosphatidylinositol 4,5- bisphosphate (PIP2) is predominately present in the inner leaflet of the cell surface membrane and accounts for more than 99% of the doubly phosphorylated phosphatidylinositol (McLaughlin et al., 2002). Despite low overall sequence similarity, almost all mammalian TRPs are known to be modulated by PIP2 (Rohacs, 2014). Some channels, including TRPV5, TRPV6, TRPM4, TRPM5, and TRPM8, are positively modulated (Rohacs, 2014); others, such as TRPC4 andTRPP2 (Otsuguro et al., 2008; Ma et al., 2005), are negatively regulated, whereas TRPV1 channel function is stimulated or inhibited by PIP2, depending on the experimental conditions (Rohacs et al., 2008; Lukacs et al., 2007). Although PIP2 is known to bind to cationic residues in some TRPs in vitro, including TRPV1, TRPM8, and TRPM4 (Rohacs et al., 2005; Bousova et al., 2015; Poblete et al., 2015), it remains largely unclear as to the structural basis of the PIP2 regulation.
In this study, using two-electrode voltage clamp in Xenopus laevis oocytes, patch clamp in mammalian cells, surface localization, and immunofluorescence, we first identified conserved aromatic residues in the pre-S1 and cationic residues in the TRP/TRP-like domains of TRPP3, TRPP2, TRPV1, TRPM8, and TRPC4 and studied their functional roles. Given the physical proximity between the pre-S1 and TRP/TRP-like domains in resolved TRP structures (Figure S1), by in vitro pull-down, coimmunoprecipitation (co-IP), and blocking peptide strategy, we then examined the physical and functional interaction of the N terminus to C terminus (N-C) that is mediated by an aromatic-cationic residue pair. Further, we examined how PIP2 regulates the N-C binding in TRPP3, TRPP2, TRPV1, and TRPM8 and proposed that the N-C binding might represent as a shared mechanism through which different agonists regulate TRPs channel function.
RESULTS
Interaction between the N and C Termini of TRPP3 and TRPP2 and Functional Importance
Among the N-terminal amino acid (aa) residues M1-T39, S41, and S42 of human TRPP3, only C38 was previously shown to be important for the channel function (Yang et al., 2012; Zheng et al., 2016b). We here wanted to identify aa(s) within I40-L95 of the N terminus that is (are) functionally important. Using Xenopus oocytes expression, the two-electrode voltage-clamp electrophysiology, and Ca2+-induced channel activation as a readout, we found that deleting aa W81-L95, but not deleting aa T43-P50, K51-L80, or replacing I40 by an alanine (A), abolishes the channel activation (Figure 1A). Applying alanine substitution scanning to fragment W81-L95 found that only the mutation at tryptophan (W) 81 abolishes the channel activation (Figure 1B) without affecting the surface membrane targeting (Figures 1H and S2A). Substitution of W81 by an aromatic or anionic residue retained full or partial channel function whereas substitution by leucine (L) or a cationic residue resulted in loss of function (Figures 1C, 1H, and S2A). These data suggest the importance of the aromatic side chain in W81 for TRPP3 channel function. If the aromatic side chain in W81 is near the cationic side chain of an arginine (R) or lysine (K), an energetically favorable π-cation interaction may occur. Interestingly, in TRPV1, a π-cation stacking is observed between the N-terminal W426 (in pre-S1 helix) and C-terminal R701 (in TRP helix; Figure S1), and recently reported TRPs structures revealed physical proximity between the N-terminal pre-S1 and the C-terminal TRP/TRP-like helices (Saotome et al., 2016; Huynh et al., 2016; Liao et al., 2013; Paulsen et al., 2015). Because TRPP3 W81 is located within the highly conserved pre-S1 helix across species (Figure 1D), we carried out alanine substitution in the TRP-like domain (N561-L593) to identify a functionally important cationic residue(s). We found that only mutation K568A, but not K575A, K585A, or K590A, corresponds to loss of function (Figures 1E, 1H, and S2A). Substitution of K568 by an acidic residue (D or E) also led to loss of function whereas substitution by cationic R retained full channel function (Figures 1F, 1H, and S2A). Sequence alignment also showed that K568 is highly conserved across different species (Figure 1G). Taken together, our data suggest that TRPP3 channel function may require the presence of an interaction between the pre-S1 and TRP-like domains that is mediated by the W81-K568 pair through a π-cation interaction.
Figure 1. Roles of the TRPP3 W81 (Pre-S1 Domain) and K568 (TRP-like Domain) Residues in the N-C Interaction and Channel Function.
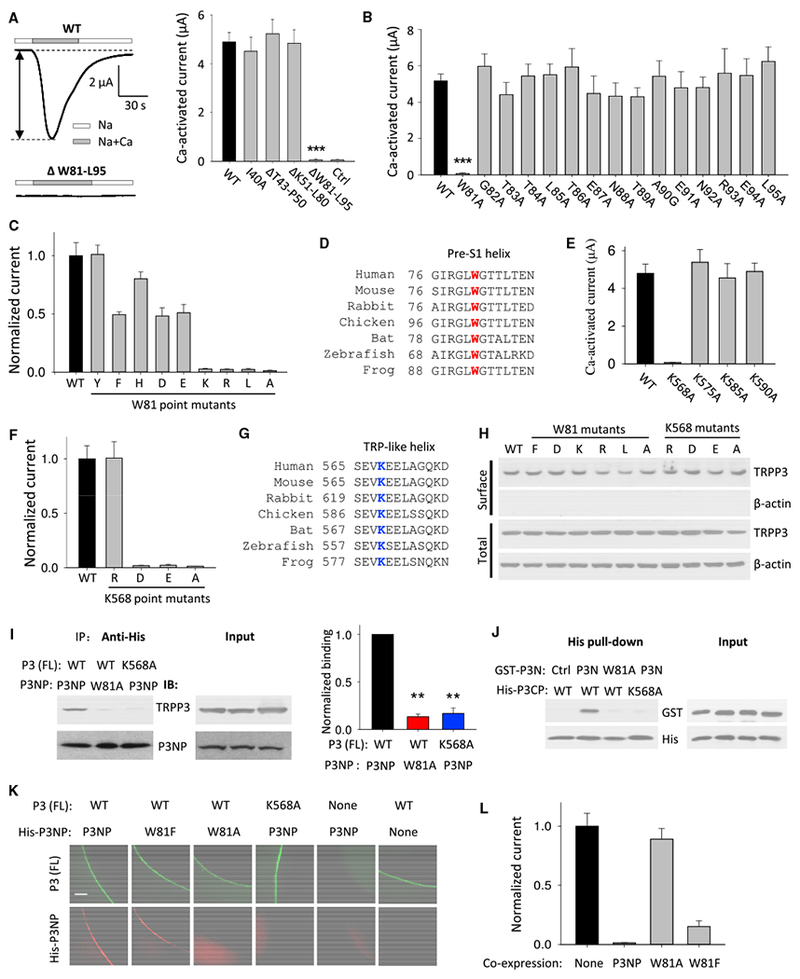
(A) Left panel: representative whole-cell current traces obtained from Xenopus oocytes expressing human TRPP3 WT or mutant DW81-L95 (with the W81-L95 domain deletion), using the two-electrode voltage-clamp technique. Oocytes were voltage clamped at −50 mV, and currents were recorded using the Na+-containing extracellular solution (Na) (100 mM NaCl, 2 mM KCl, 1 mM MgCl2, and 10 mM HEPES [pH 7.5]) or added with 5 mM CaCl2 (Na+Ca). The Ca-activated current (i.e., current at “Na+Ca” – current at “Na”), indicated by the double-arrowed line, was measured to assess TRPP3 channel activity (Chen et al., 1999). Right panel: averaged Ca-activated currents from oocytes expressing TRPP3 WT or an indicated mutant or H2O-injected oocytes (Ctrl) are shown (n = 17–22). Oocytes were from three batches. ***p < 0.001 (compared with WT).
(B) Averaged Ca-activated currents from oocytes expressing TRPP3 WT or an indicated point mutant. ***p < 0.001.
(C) Averaged Ca-activated currents from oocytes expressing TRPP3 WT or an indicated W81 point mutant. Currents were averaged from three independent experiments and normalized to that of WT.
(D) Amino acid sequence alignment of the TRPP3 pre-S1 helix from indicated species, with the conserved residue W highlighted red.
(E) Averaged Ca-activated currents obtained from oocytes expressing TRPP3 WT or an indicated point mutant in TRP-like domain.
(F) Averaged and normalized Ca-activated currents from oocytes expressing TRPP3 WT or an indicated K568 point mutant.
(G) Amino acid sequence alignment of the TRPP3 TRP-like domain from indicated species, with the conserved residue K highlighted blue.
(H) Representative immunoblots of the surface biotinylated (surface) and whole-cell (total) TRPP3 WT or indicated W81 or K568 point mutants.
(I) Left panel: W81-K568 interaction examined with co-IP assays using oocytes co-expressing full-length (FL) TRPP3 and His-tagged TRPP3 N-terminal peptide (His-P3NP; I40-L95). Right panel: data from experiments in left panel were quantified, averaged, and normalized. **p < 0.01; n = 3.
(J) W81-K568 interaction examined with His pull-down assays using the purified GST-tagged human TRPP3 N terminus (GST-P3N; M1-L95) and His-tagged human TRPP3 C-terminal peptide (His-CP; I560-K660) from E. coli. Ctrl, purified GST-human FUBP1 M1-V112 from E. coli.
(K) Colocalization of P3NP with FL TRPP3 examined with co-immunofluorescence assays using oocytes co-expressing FL TRPP3 and His-P3NP. The scale bar represents 50 μm.
(L) Effects of co-expressed P3NP or its point mutants on TRPP3 Ca-activated currents. None, no P3NP expression. Shown are normalized and averaged currents from three independent experiments (n = 15–20).
Data are presented as mean ± SEM. See also Figures S1 and S2.
We next performed co-IP experiments and found that the TRPP3 N-terminal peptide I40-L95 (P3NP) and full-length (FL) TRPP3 are in the same complex and that this interaction is abolished if W81 in P3NP or K568 in FL TRPP3 is substituted by A (Figure 1I), indicating that the W81-K568 pair mediates the interaction. We next performed in vitro pull-down assays using the recombinant and purified glutathione S-transferase (GST)-tagged TRPP3 N terminus M1-L95 (GST-P3N) and the His-tagged C-terminal peptide I560-K660 (His-P3CP). The two peptides directly bound each other, and the binding was abolished in the presence of the W81A or K568A mutation in the respective peptide (Figure 1J), confirming that the W81-K568 pair mediates the N-C binding. The N-C interaction was further supported by whole-oocyte co-immunofluorescence assays: after cRNA injection, peptide P3NP was distributed along the oocyte surface membrane if the FL TRPP3 cRNA was co-injected (Figure 1K). This attachment of P3NP to the surface membrane remained unaffected by the W81F mutation in P3NP but was not observed in the presence of the W81A mutation in P3NP or the K568A mutation in FL TRPP3 or in the absence of FL TRPP3 expression. These co-IP and immunofluorescence data together are in support of the concept that the W81-K568 pair-mediated interaction between P3NP and FL TRPP3 allows P3NP to colocalize with FL TRPP3 along the surface membrane.
We wondered whether the expressed peptide P3NP would compete with the P3NP fragment within the FL TRPP3 protein for binding with the K568 residue in the TRP-like domain of the FL TRPP3, thereby disrupting the N-C binding within FL TRPP3 and abolishing the channel function. Indeed, co-expression of P3NP or P3NP-W81F completely or substantially inhibited the FL TRPP3 channel function, whereas co-expression of mutant peptide P3NP-W81A had no effect (Figure 1L), indicating that P3NP acts as a blocking peptide that inhibits TRPP3 function through disrupting the N-C binding in the FL channel. In summary, our data demonstrate that direct N-C binding does occur in TRPP3 and that residues W81 of the pre-S1 and K568 of the TRP-like domain, through π-cation interaction, are crucial for this interaction, which is required for TRPP3 channel activation.
TRPP3 shares 70% overall sequence similarity with TRPP2 encoded by the PKD2 gene, in which mutations account for 15% of autosomal dominant polycystic kidney disease (ADPKD), and an even higher homology in the pre-S1, TRP-like, and transmembrane domains. The corresponding aromatic and cationic residues in the pre-S1 and TRP-like domains of human TRPP2 are W201 and K688, respectively (Figure 2A). The low basal channel activity of TRPP2 alone impairs reliable recording of currents in oocytes and mammalian cells. However, as the TRPP2 F604P mutation renders TRPP2 in an activated state (Arif Pavel et al., 2016), we used mutant F604P-mediated currents as a function readout and replaced W201 or K688 residue with A in the F604P mutant. No current was detectable for the W201A or K688A mutant, although the overall protein expression and surface membrane targeting were not affected (Figures 2B, 2C, and S2D), indicating that residues W201 and K688 are functionally essential. As in TRPP3, the TRPP2 N-terminal peptide G161-S215 (P2NP) interacted with the FL TRPP2 and the interaction was weakened by the W201A mutation in P2NP or K688A mutation in TRPP2 (Figure 2D). Consistently, fragment P2NP, but not P2NP-W201A, acted as a blocking peptide by drastically reducing the function of TRPP2 F604P (Figure 2E) without affecting its plasma membrane expression (Figure S3E). Overall, our data support the hypothesis that, similar to TRPP3, there exists an interaction between the N and C termini of TRPP2 mediated by W201 and K688 that is essential for TRPP2 channel function.
Figure 2. Roles of the TRPP2 Residues W201 (Pre-S1 Domain) and K688 (TRP-like Domain) in the Channel Function and N-C Binding.
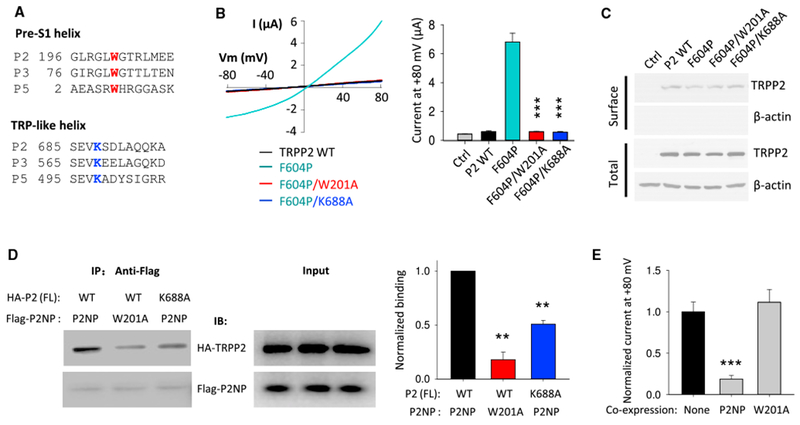
(A) Sequence alignment of pre-S1 and TRP-like helices among human TRPP channels.
(B) Left panel: representative current-voltage (I-V) curves obtained from oocytes expressing human WT or a mutant TRPP2, as indicated, in the presence of a Na+-containing, divalent-free solution (in mM): 100 NaCl; 2 KCl; and 10 HEPES (pH 7.5). Right panel: averaged currents at +80 mV are shown. Ctrl, H2O-injected oocytes. Currents were averaged from 13–17 oocytes of three batches. ***p < 0.001.
(C) Representative immunoblots of surface biotinylated and total proteins of TRPP2 WT and mutants.
(D) Left panel: representative co-IP data showing the effects of TRPP2 W201 and K688 on the N-C interaction using oocytes co-expressing HA-tagged human FL TRPP2 and Flag-tagged TRPP2 N-terminal peptide (Flag-P2NP; G161-S215). Right panel: data from experiments in left panel were quantified, averaged, and normalized. **p < 0.01; n = 3.
(E) Blocking effects of Flag-P2NP and Flag-P2NP-W201A on channel function of FL TRPP2 mutant F604P by use of co-expression. None, no P2NP was co-expressed with FL F604P. Normalized currents at +80 mV were obtained and averaged from three independent experiments. ***p < 0.001.
Data are presented as mean ± SEM. See also Figures S2 and S6.
We recently showed that TRPP2 co-expressed with PKD1, a large transmembrane protein mutated in 85% of ADPKD, in mammalian cells can be activated by WNT molecules, such as WNT9B (Kim et al., 2016). Therefore, we tested the effects of W201A, K688A, ΔY684 (in-frame deletion of residue Y684), and Y684A mutations on WNT9B-activated channel activity of human TRPP2 co-transfected with human PKD1 in Chinese hamster ovary (CHO)-K1 cells. We included Y684 in the analysis because it is located within the TRP-like domain in close proximity to K688. In addition, in-frame deletion of Y684 was first identified as a somatic mutation in a patient with a germline-inactivating mutation inPKD2 (Watnick et al., 2000). However, its functionality has never been tested. Cell surface biotinylation experiments in HEK293 cells, which are more suited for biochemical experiments than CHO-K1 cells because of their high levels of transfectability, did not show significant differences in cell surface expression of the mutants to wild-type (WT) (Figure 3A). Compared to WT TRPP2, mutant W201A or K688A showed substantially reduced WNT9B-activated peak currents (Figures 3B and 3D). Interestingly, the two mutants also exhibited pronounced inactivation and abolished the steady-state currents (Figures 3B, 3C, and 3E), suggesting that each of the W201A and K688A mutations destabilized the channel in an open state induced by WNT9B. In comparison, mutations ΔY684 and Y684A rendered the channel completely insensitive to WNT9B (Figures 3D and 3E). In summary, using WNT9B as an activating ligand, we showed that W201A and K688A mutations suppress channel activity via an apparently similar mechanism, at least based on current kinetics, but distinct from that of ΔY684 or Y684A mutation, which is consistent with the possible engagement of W201 and K688 in a physical interaction during channel activation. The residual peak current in TRPP2 W201A or K688A mutant could be due to the possible contribution of PKD1 to pore formation, which iscurrently unexplored and beyond the scope of this study.
Figure 3. Roles of TRPP2 Residues W201 and K688 in WNT9B-Induced Whole-Cell Currents.
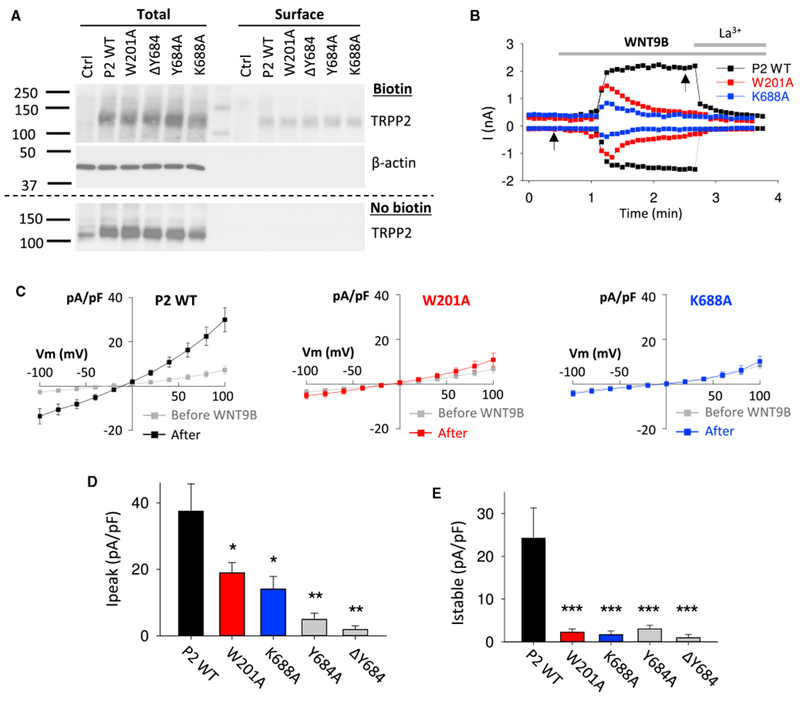
(A) Cell surface expression of WT TRPP2 and mutants W201A, ΛΥ684, Y684A, and K688A in HEK293 cells by transient co-transfection together with PKD1.
(B) Representative whole-cell current traces obtained at +100 and −100 mV before and after extracellular addition of WNT9B (0.5 μg/mL) or La3+ (100 μM) in CHO-K1 cells transiently co-expressing PKD1 with WT TRPP2, the W201A, or K688A mutant. The electrophysiological measurements were performed as described previously (Kim et al., 2016).
(C) Averaged steady-state I-V curves obtained before and ~2 min after application of WNT9B, at time points indicated by arrows in (B). WT TRPP2, n = 7 cells; W201A, n = 11 cells; K688A, n = 7 cells.
(D and E) Averaged peak (D) and steady-state (E) currents induced by WNT9B in CHO-K1 cells under the same experimental conditions as in (B). WT TRPP2, n = 7 cells; W201A, n = 11 cells; ΛY684, n = 8 cells; Y682A, n = 10 cells; K688A, n = 7 cells. *p < 0.05; **p < 0.01; ***p < 0.001.
Data are presented as mean ± SEM.
Characterization of the N-C Binding in TRPM8, TRPV1, and TRPC4
Alignment of the pre-S1 and TRP domains of all members of the TRPM, TRPC, and TRPV subfamilies revealed that the tryptophan and the lysine residues examined in TRPP2 and TRPP3 above are highly conserved (Figures 4A, S4A, and S5A), only with exception of TRPV4, TRPV5, and TRPV6 (Figure S4A). Rat TRPM8 channel mediated large depolarization-activated cation currents in oocytes that were completely abolished by alanine substitution at W682 or R998 (Figure 4B) without affecting the plasma membrane expression (Figure S3A). Consistently, single-channel patch-clamp measurements in HEK293 cells showed that the W682A and R998A mutations substantially decrease the channel open probability and reduce the singlechannel current amplitude to about 60% (Figure S3C). At negative membrane potentials, menthol as a known agonist induced large inward cation currents in oocytes expressing WT TRPM8, but not in those expressing the TRPM8 mutant W682A or R998A (Figure 4C). Similarly, in HEK293 cells expressing TRPM8, Ca2+ entry was increased in the presence of menthol, assessed by ratiometric Fura-2 as Ca2+ indicator, whereas TRPM8 mutant W682A or R998A did not respond (Figure 4D). In summary, our data using both oocytes and HEK293 cells demonstrated that W682 in pre-S1 and R998 in the TRP domain are essential for TRPM8 channel function. As in TRPP3, these two residues are critical for the TRPM8 N-C interaction (Figures 4E and 4F). Either the W682A mutation in the N-terminal peptide N642-K691 (M8NP) or the R998A mutation in the FL TRPM8 completely abolished the interaction of M8NP with the FL TRPM8 protein (Figure 4E). In addition, the W682A mutation in FL TRPM8 or the W998A mutation in the C-terminal peptide G980-F1029 (M8CP) abolished the interaction of M8CP with FL TRPM8 (Figure 4F). Therefore, as in TRPP channels, conserved W682 and R998 in TRPM8 presumably mediated the physical interaction between N and C termini.
Figure 4. Roles of TRPM8 and TRPV1 Aromatic and Cationic Residues in the Pre-S1 and TRP-like Domains, Respectively, in the N-C Binding and Channel Function.
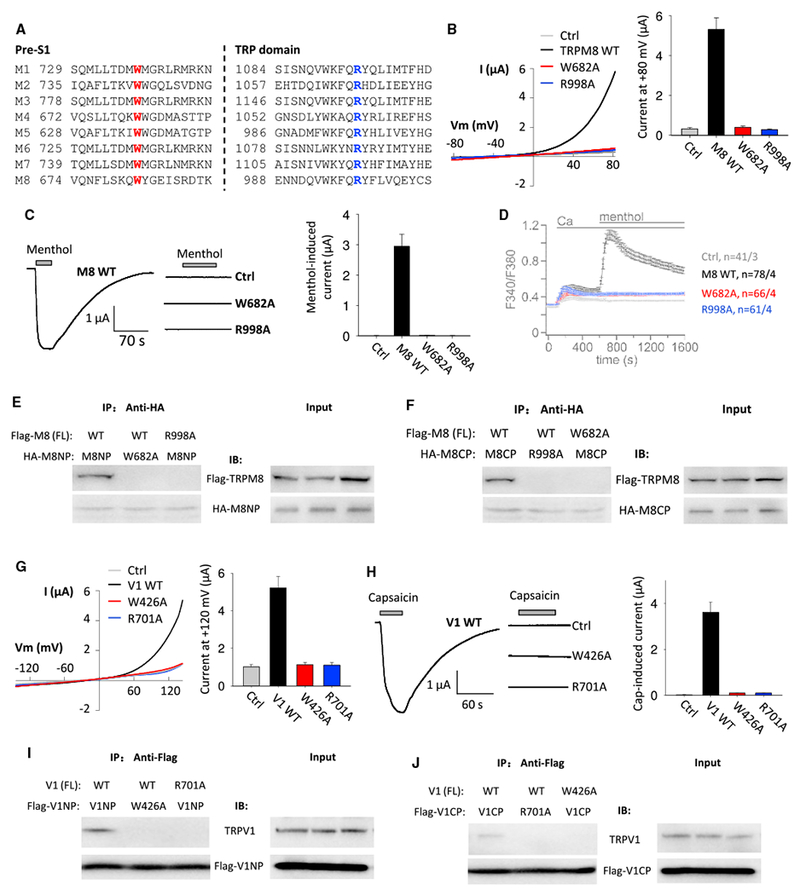
(A) Sequence alignment of human TRPMs pre-S1 and TRP-like domains.
(B) Left panel: representative whole-cell I-V curves obtained from oocytes expressing rat TRPM8 WT or a mutant channel in the presence of Na+-containing extracellular solution at RT. Ctrl, H2O-injected oocytes. Right panel: averaged currents at +80 mV from 12–18 oocytes of three batches are shown.
(C) Left panel: representative current traces obtained at −50 mV from oocytes expressing WT or a mutant TRPM8 before and after addition of 0.5 mM menthol. Right panel: averaged menthol-induced currents from 12–18 oocytes of three batches are shown.
(D) Ca2+-imaging measurements showing averaged fura-2 ratios obtained before and after Ca2+ (2 mM) and menthol (0.5 mM) addition to Na+-containing extracellular solution in HEK293 cells transiently co-expressing GFP with rat WT or a mutant TRPM8 or none (Ctrl) at 37°C.
(E and F) Representative co-IP data using oocytes expression, showing the interaction of Flag-tagged FL TRPM8 with HA-tagged TRPM8 N-terminal peptide (HA-M8NP; N642-K691; E) or C-terminal peptide (HA-M8CP; G980-F1029; F).
(G) Left panel: representative whole-cell I-V curves obtained from oocytes expressing rat WT or a mutant TRPV1 in the presence of the Na+-containing solution at RT. Ctrl, H2O-injected oocytes. Right panel: averaged currents at +120 mV from 10–16 oocytes of three batches are shown.
(H) Left panel: representative current traces obtained at —50 mV in rat WT or a mutant TRPV1 expressing oocytes before and after extracellular addition of capsaicin (15 μM). Right panel: averaged capsaicin-induced currents from 10–16 oocytes of three batches are shown.
(I and J) Representative co-IP data using oocytes expression, showing the interaction of rat FL TRPV1 with Flag-tagged TRPV1 N-terminal peptide (Flag-V1NP; D383-R432; I) or C-terminal peptide (Flag-V1CP; N687-D736; J).
Data are presented as mean ± SEM. See also Figures S3, S4, S5, and S6.
Next, we examined the activity of TRPV1, TRPV1-W426A, and TRPV1-R701A in oocytes in the presence of depolarization or the agonist capsaicin and found that residues W426 and R701 are functionally important (Figures 4G, 4H, S4D, and S4E). The N-terminal peptide D383-R432 (V1NP) and the C-terminal peptide N687-D736 (V1CP) interacted with FL TRPV1 as revealed by co-IP (Figures 4I and 4J). A single alanine substitution either at W426 of V1NP or R701 of FL TRPV1 was sufficient to abolish the interaction (Figure 4I), very much alike as in TRPP3, TRPP2, and TRPM8. Further, mutation W426A in FL TRPV1 or R701A in V1CP abolished the TRPV1-V1NP interaction (Figure 4J). Similar alanine substitution of the corresponding residues in mouse TRPC4, W314 (pre-S1), and R639 (TRP domain), abolished the activity of the constitutively active mutant channel TRPC4-G503S (Beck et al., 2013; Figure S5). Thus, our data on TRPM8, TRPV1 and TRPC4 supported the conclusion made to TRPP channels with respect to the N-C interaction.
Regulation of the TRPs N-C Binding by PIP2
To explore how the N-C interaction can be modulated, we examined the effect of the negatively charged, membrane-anchored phosphoinositide PIP2 on the N-C interaction. We looked into PIP2 because it has been shown to modulate the function of most mammalian TRP channels except of TRPP3, TRPP5, TRPML2, and TRPML3 (Rohacs, 2014). Interestingly, the R998 residue of TRPM8, which pairs with W682 to mediate the N-C binding in TRPM8 (Figures 4B–4F), is part of the TRPM8’s binding pocket for PIP2 (Rohacs et al., 2005). Thus, we next tested whether PIP2 can modulate TRP channel function through affecting the N-C interaction. In oocytes expressing human TRPP3, injection of diC8 PIP2, a H2O-soluble dioctanoyl analog of PIP2 (Rohacs et al., 2005), significantly reduced Ca2+-activated TRPP3 channel activity at −50 mV to 28% ± 4% (Figure 5A). Reversely, by incubating oocytes for 1 hr in the presence of 10 μM wortmannin, a membrane-permeable phosphatidylinositol 4-kinase (PI4K) inhibitor that depletes PIP2 (Zhang et al., 2003; Czirják et al., 2001), Ca2+-activated currents were increased by 1.7 ± 0.2 folds (Figure 5B). At low concentration, wortmannin was shown to selectively inhibit PI3K activity (McNamara and Degterev, 2011), which would increase PIP2. We then treated TRPP3-expressing oocytes with 15 nM wort-mannin and found that the TRPP3 channel activity was little affected (Figure S2C), possibly due to negligible basal PI3K activity in oocytes. Together, these data suggested that TRPP3 is negatively regulated by PIP2.
Figure 5. Inhibition of the TRPP3 Channel Function and N-C Interaction by PIP2.
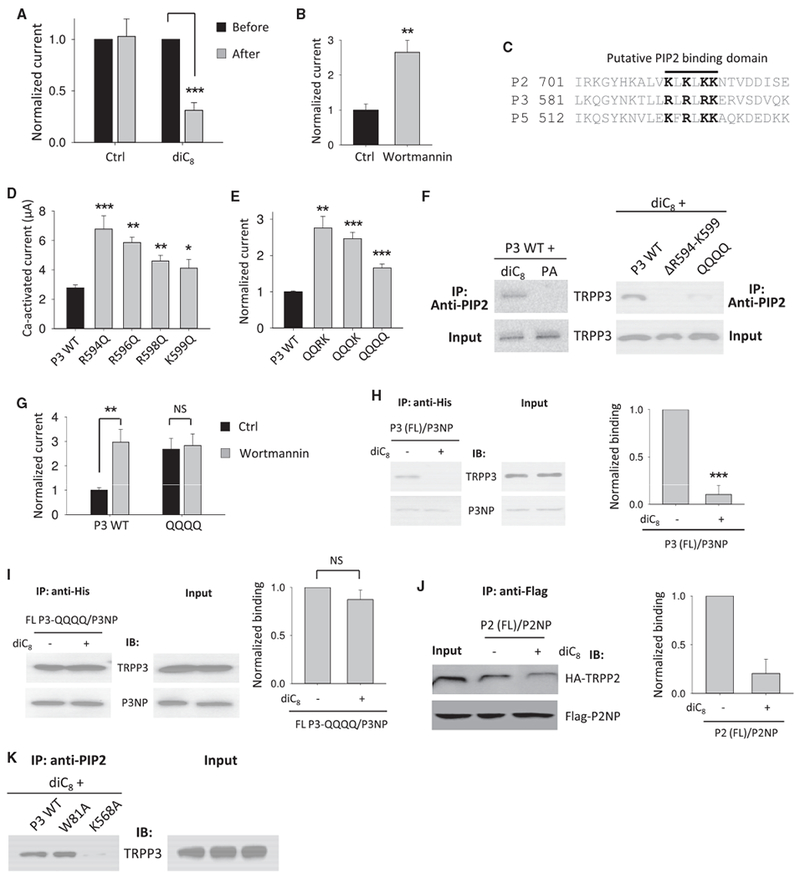
(A) Averaged and normalized Ca-activated currents obtained from TRPP3-expressing oocytes before and after on-site injection of 25 nL water without (Ctrl) or containing diC8 (5 mM). Injection was performed with a third electrode after the initial current measurement. The second current measurement was performed 10 min after the injection. Currents were averaged from three independent experiments (with n = 12–15). ***p < 0.001.
(B) Averaged and normalized Ca-activated currents obtained from TRPP3-expressing oocytes pre-incubated with 10 μM wortmannin or DMSO (Ctrl) for 1 hr before measurements. **p < 0.01.
(C) Alignment of the C-terminal putative PIP2 binding domains in human TRPPs. Conserved cationic residues are highlighted.
(D) Averaged Ca-activated currents obtained from oocytes expressing WT or a mutant TRPP3 (n = 14–18). *p < 0.05; **p < 0.01; and ***p < 0.001.
(E) Averaged and normalized Ca-activated currents. QQRK, R594Q/R596Q double mutant; QQQK, R594Q/R596Q/R598Q triple mutant; QQQQ, R594Q/R596Q/ R598Q/K599Q quadruple mutant. **p < 0.01 and ***p < 0.001.
(F) Left panel: representative co-IP data showing interaction between diC8 PIP2 and TRPP3. Right panel: representative co-IP data show interaction of diC8 PIP2 with WT or a mutant TRPP3 expressed in oocytes. DR594-K599, TRPP3 deleted with fragment R594-K599. diC8 PIP2 or phosphatidic acid (PA, a negative control) was added to cell lysate to a final concentration of 15 μM. An anti-PIP2 antibody (sc-53412) from Santa Cruz Biotechnology was used for immuno-precipitation.
(G) Averaged and normalized Ca-activated currents obtained from oocytes expressing WT TRPP3 or the QQQQ mutant. Oocytes were treated with 10 μM wortmannin or DMSO (Ctrl) for 1 hr before measurements. **p < 0.01; NS, not significant.
(H) Left panel: representative co-IP data showing the effect of diC8 on the interaction of P3NP with FL TRPP3 in oocytes. diC8 was added in the cell lysis buffer to final concentration of 15 μM. Right panel: data from three independent experiments in left panel were quantified, averaged, and normalized. ***p < 0.001.
(I) Left panel: representative co-IP data showing the effect of diC8 on the interaction of P3NP with the TRPP3 QQQQ mutant. Right panel: data from three independent experiments in left panel were quantified, averaged, and normalized (NS, not significant).
(J) Left panel: representative co-IP data showing the effect of diC8 on the interaction of P2NP with FL TRPP2. Right panel: data from three independent experiments in left panel were quantified, averaged, and normalized.
(K) Representative co-IP data showing the interaction of PIP2 with expressed WT or a mutant TRPP3 in oocytes.
Data are presented as mean ± SEM. See also Figure S2.
PIP2 is known to regulate channel function by interacting with cationic residues in the C-terminal TRP domain of several TRPs, including TRPV1, TRPM8, and TRPM5 (Poblete et al., 2015; Rohacs et al., 2005; Nilius et al., 2008). PIP2 was also shown to bind to the TRPM4 C-terminal cationic residue-rich domain, 1136RARDKR1141, located downstream of the TRP domain (Nilius et al., 2006). Sequence alignments revealed the presence of such a cationic residue-rich domain in a number of other TRPs (Nilius et al., 2008), including TRPPs (Figure 5C). We thus wondered whether this domain in the TRPP3 C terminus, 594RLRLRK599 (called a RRRK motif), could be involved in PIP2 binding. Given the vicinity of residues W81 and K568 to the cytoplasmic leaflet of the plasma membrane and proximity of K568 to the RRRK motif, a possible PIP2-TRPP3 interaction could disrupt the TRPP3 N-C binding, thereby inhibiting channel function. We found that replacing one or more of the four cationic residues in TRPP3 594RLRLRK599 with glutamine (Q) significantly increases TRPP3 channel activity (Figures 5D and 5E) without affecting the protein expression (Figure S2B), suggesting that each of these substitutions might have disrupted PIP2-TRPP3 binding and thereby lifted channel inhibition by the endogenous PIP2 present in oocytes. In co-IP experiments, diC8 PIP2 was in the same complex with WT TRPP3 (Figure 5F, left panel), but not with mutant channel proteins in which the motif RRRK was deleted or the four cationic residues were substituted by Q (quadruple glutamine mutant; Figure 5F, right panel). Because no detailed information is available about the sites in PIP2 that bind its antibodies used in our co-IP experiments, how a PIP2-bound antibody can still precipitate TRPP3 remains unknown. Wortmannin treatment had little effect on the activity of the quadruple glutamine mutant channel compared to WT (Figure 5G). In summary, these data show that the RRRK motif confers TRPP3 sensitivity to PIP2, most likely by forming an interaction domain with PIP2, and that the TRPP3-PIP2 interaction has a negative effect on channel activation.
In subsequent co-IP experiments, we found that addition of diC8 PIP2 to the cell lysate disrupts the P3NP-TRPP3 binding in oocytes co-expressing FL TRPP3 and His-P3NP (Figure 5H) but has no effect on the P3NP-TRPP3 binding when motif RRRK was mutated in FL TRPP3 (Figure 5I), suggesting that binding between PIP2 and the RRRK motif is required for PIP2 to disrupt the N-C interaction. Similarly, the inhibitory effect of PIP2 on the N-C binding was also demonstrated in TRPP2 by co-IP of P2NP and FL TRPP2 (Figure 5J), which is consistent with the reported inhibitory effect of PIP2 on TRPP2 channel function (Ma et al., 2005). Because TRPM8 R998 is part of the PIP2 binding pocket (Rohács et al., 2005), we next tested the role of the corresponding residue K568 in TRPP3 for PIP2 binding by co-IP. As shown in Figure 5K, the K568A, but not W81A, mutation in TRPP3 disrupts the PIP2-TRPP3 binding, indicating that K568 is part of the PIP2 binding pocket in TRPP3. In summary, our data together demonstrated that PIP2 binds to TRPP3 through motif RRRK and K568. PIP2 binding to TRPP3 disrupts the W81-K568 pair-mediated N-C binding, which is needed for channel activation. Based on our data on TRPP2 (Figure 5J) and the presence of a similar motif (714KLKLKK719), we propose that TRPP2 and TRPP3 share a similar mechanism of channel modulation by PIP2.
The TRPM8 and TRPV1 proteins possess a similar cationic rich motif and the additional R residue within the TRP domain that mediates binding to the N-terminal W residue. In contrast to TRPP channels, we found by co-IP assays that PIP2 enhances the binding of rat FL TRPM8 with its N-terminal peptide M8NP or C-terminal peptide M8CP (Figures 6A and 6B). This is in agreement with the known stimulatory effect of PIP2 on the TRPM8 channel function (Rohács et al., 2005; Nilius et al., 2008), suggesting that the positive effect of PIP2 on TRPM8 is mediated through stabilizing the interaction between the N and C termini of TRPM8. Using a similar approach, we found that PIP2 promoted N-C binding in TRPV1 (Figures 6C and 6D). This finding was also consistent with the positive regulation of TRPV1 by PIP2 direct binding (Lukacs et al., 2007; Poblete et al., 2015). Overall, our data showed a positive or negative effect of PIP2 on the N-C binding among different TRP channels and suggested that the N-C binding may mediate distinct regulations of PIP2 on the channel activity of TRPs.
Figure 6. Effects of PIP2 on the N-C Interaction in TRPM8 and TRPV1.
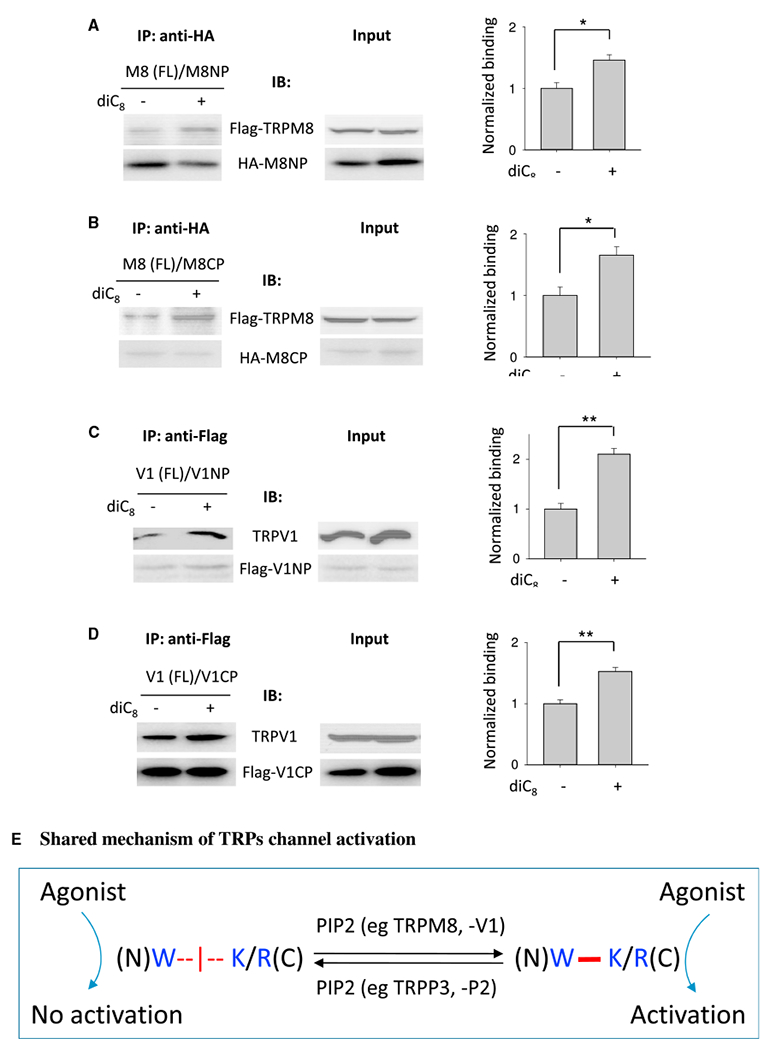
(A and B) Left panels: representative co-IP data showing the effect of diC8 on the interaction between FL TRPM8 and M8NP (A) or M8CP (B) under the same experimental conditions as in Figure 4H. Right panels: data from three independent experiments in left panel were quantified, averaged, and normalized. *p < 0.05.
(C and D) Left panels: representative co-IP data showing the effect of diC8 on the interaction between the FLTRPV1 and V1NP (C) or V1CP (D). Right panels: data from three independent experiments in left panel were quantified, averaged, and normalized. **p < 0.01.
(E) Schematic model showing role of the PIP2-regulated, W-K/R pair-mediated N-C binding in TRP channels activation and function. In this model, the N-C binding is mediated between the conserved aromatic residue Win pre-S1 and the cationic residue K or Rin the TRP/TRP-like domain and is either inhibited (as in TRPP3 and -P2) or enhanced (as in TRPM8 and -V1) by PIP2 through binding to the TRPs protein. The presence of the N-C binding is required for TRPs agonists to activate the channel.
Data are presented as mean ± SEM.
DISCUSSION
In this study, we revealed a gating mechanism involving a physical intramolecular interaction between the N and C termini in TRPP channels. Similar N-C binding was also found in TRPM8, TRPV1, and TRPC4. This N-C binding is mediated by an aromatic tryptophan residue in the pre-S1 domain and a cationic lysine/arginine residue in the TRP/TRP-like domain. We further showed that PIP2 can modulate TRP channel activity by directly interfering with the N-C interaction. The scheme in Figure 6E summarizes these findings, in which (1) the TRP N-C binding is either inhibited or enhanced by PIP2 and (2) an agonist is able to activate the channel when the N-C binding is present, whereas it is not when the N-C binding is absent or weak.
The high resolution of the TRPV1 structure first revealed an intramolecular proximity of the pre-S1 (or S4-S5 linker) and TRP domains (Liao et al., 2013; Cao et al., 2013; Gao et al., 2016). This observation was later confirmed in the structures of TRPA1 (Paulsen et al., 2015), TRPV2 (Huynh et al., 2016; Zubcevic et al., 2016), TRPV6 (Saotome et al., 2016), TRPP2 (Wilkes etal., 2017; Grieben etal., 2017; Shen et al., 2016), and NOMPC (Jin et al., 2017), suggesting that residues in pre-S1, S4-S5 linker, and TRP domains may mediate intramolecular interactions that are important for structural stability, channel gating, or allosteric modulation. In line with this suggestion, the interaction among the pre-S1 linker, pre-S1, and TRP domains was shown to be required for proper folding, assembly, and trafficking of TRPV1 and TRPV4 channels (Garcia-Elias et al., 2015).
The interaction between the TRPV1 W426 (pre-S1) and R701 (TRP domain) identified in the present study was not recognized in previous TRPV1 structures (Liao et al., 2013; Cao et al., 2013; Gao et al., 2016), which instead proposed the Q423-R701 interaction. Comparing TRPV1 structures (PDB: 3J5P; 3J5R; and 3J5Q) in closed (apo) and activated (capsaicin or vanilloid agonist resiniferatoxin [RTX]/spider double-knot toxin [DkTx]-bound) states revealed that Q423 moves away from R701 during the activation (Figure S4F), suggesting that the Q423-R701 interaction might not be required for channel activation. In contrast, residues W426 and R701 remained close to each other during activation (Figure S4F), suggesting the W426-R701 binding may be required for channel function. In support of this, ourfunctional studies revealed that mutant Q423A can still be activated by depolarization and capsaicin (Figures S4B–S4E) whereas W426A and R701A behaved as total loss-of-function mutants (Figures 4G and 4H).
A notable difference between the TRP domain of TRPV1 and TRP-like domain of TRPP2 is that the former is parallel to the plasma membrane whereas the latter represents as a continuation of S6 and is almost perpendicular to the plasma membrane (Liao et al., 2013; Shen et al., 2016). Also, TRPV1 and TRPP2 share little sequence homology in the pre-S1 or TRP/-like domain (Figure S6A). An interesting question would be how the N-C interaction could be conserved in TRPV1 and TRPP2. In TRPV1, the W426 is close (2-amino-acid distance) to S1 whereas R701 is relatively far (14 amino acids) from S6 (Figure S6A). Thus, the parallel orientation of the TRP helix to the plasma membrane would allow R701 to be close enough to interact with W426 (Figure S6B). In contrast, in TRPP2, the W201 is far (14 amino acids) from S1 whereas K688 is close (3 amino acids) to S6 (Figure S6A). In this case, the same orientation of TRP-like helix with that of S6 would allow a W201-K688 interaction (Figure S6B). We speculate that, due to its functional importance for TRP channels, the N-C interaction is evolutionally maintained.
PIP2 was reported to enhance the activation of the TRPM8 channel by its agonist menthol (Liu and Qin, 2005; Rohács et al., 2005), which is consistent with our data (Figure S3B). Neutralization of the conserved cationic residues K995, R998, and R1008 in the TRP domain significantly inhibited the channel function (Rohács et al., 2005). Interestingly, the newly resolved TRPM8 structure (Yin et al., 2018) revealed physical proximity of the pre-S1 and TRP domains in a subunit with melastatin homology region 4 domain from a neighboring subunit. The interfaces between these domains contain several cationic residues, including K995 and R998, which may form the PIP2 binding pocket. The W682 (pre-S1 domain) residue is also located in this pocket and is physically close to R998. Thus, PIP2 binding to TRPM8 could stabilize the N-C interaction (Figures 6A and 6B) that is mediated by the W682 and R998 residues, which may enable menthol to induce channel activation (Figure 6E).
One of the key properties shared by TRPP3 and TRPM8 is that the cationic residue in the TRP domain, K568 of TRPP3 or R998 of TRPM8, mediates both the N-C interaction and binding to PIP2. We showed that PIP2 weakens the N-C binding of TRPP3, presumably by competing for binding to the K residue in the TRP-like domain with the aromatic residue in the pre-S1. This mechanism could be shared with TRPP2. In contrast, in TRPM8, PIP2 surprisingly enhanced the N-C binding. In TRPV1, PIP2 also enhanced the N-C binding, although R701 that pairs with the N-terminal W426 was reported not to be part of the PIP2 binding pocket (Poblete et al., 2015). Significant conformational changes following PIP2 binding in the Kir2.2 K+ channel has previously been reported (Hansen et al., 2011), and similar structural change, induced by PIP2, may lead to a stronger W-R interaction in TRPM8 and TRPV1.
Although PIP2 binding sites in TRP channels were mostly identified in the C terminus, PIP2 was also reported to bind to the N terminus of TRPM1 and TRPM3 (Jirku et al., 2015; Holendova et al., 2012). In fact, a large number of TRP channels were predicted to contain N-terminal PIP2 binding sites (Nilius et al., 2008). The binding of PIP2 to both N and C termini of TRP channels, especially if the binding sites/pocket are close to or overlapped with the N-C interaction sites, may allow PIP2 to efficiently regulate the intramolecular N-C interaction. Interestingly, in TRPV1, functionally identified PIP2 binding sites (Q561, R575, R579, and K688) were indeed close to W426-R701, based on resolved TRPV1 structures (Steinberg et al., 2014).
In summary, this study identified conserved amino acid residues, a tryptophan and a lysine/arginine residue in the N-terminal pre-S1 and the C-terminal TRP/TRP-like domains, respectively, that underlie interaction of the two domains. This N-C binding in TRPs is critical for the channel gating and regulation by PIP2 through direct binding. This intracellular N-C binding may serve as a shared molecular switch that transduces the conformational changes induced by diverse ligands to channel pore opening and closing.
EXPERIMENTAL PROCEDURES
Plasmids, Mutagenesis, Antibodies, and Chemicals
Human Flag-TRPP3 cDNA (GenBank: NM_016112) was cloned into vector pCHGF (Yang et al., 2012) for Xenopus laevis oocyte expression. Hemagglutinin (HA)-tagged human TRPP2 (GenBank: NM_000297) plasmid for Xenopus oocyte expression (Arif Pavel et al., 2016) was a kind gift of Dr. Yong Yu (St. John’s University, NY). Human TRPP2 and PKD1 (GenBank: NM_001009944) for mammalian cell expression was previously described (Kim et al., 2016). Rat TRPM8 (GenBank: NM_134371) plasmid was kindly provided by Dr. David Julius (University of California at San Francisco, CA). Rat TRPV1 (GenBank: NM_031982) in the pGEMEM plasmid was from Dr. Sharona Gordon (University of Washington, WA). All mutations were made with QuikChange Lighting Site-Directed Mutagenesis kit (Agilent Technologies, La Jolla, CA) and confirmed by sequencing. Rabbit antibodies against Flag (D-8) and HA (Y-11) and mouse antibodies against TRPV1 (E-8), GST (B-14), β-actin (C-4), and PIP2 (PIP2 2C11) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse antibody against His (N144/14) for western blotting was from NeuroMab (Davis, CA). Mouse antibody against His (27E8) and rabbit antibody against His (no. 2365) were purchased from Cell Signaling Technology (Danvers, MA) and were used in immunoprecipitation and immunofluorescence assays, respectively. Antibodies against TRPM8 were generated in house. Secondary antibodies were purchased from GE Healthcare (Waukesha, WI). Menthol (TRPM8 agonist) and capsaicin (TRPV1 agonist) were purchased from Sigma (St. Louis, MO). Wortmannin was from InvivoGen (San Diego, CA), and diC8 was purchased from Echelon Biosciences (Salt Lake City, UT).
Xenopus Oocytes Expression
Capped RNAs of TRPP3, TRPP2, TRPM8, and TRPV1 were in vitro transcripted with mMESSAGE mMACHINE kit (Ambion, Austin, TX) and injected (25–50 ng RNA in 50 nL water per oocyte) into Xenopus oocytes prepared as previously described (Zheng et al., 2017). Control oocytes were injected with equal volumes of water. Electrophysiological measurements were performed 1–3 days after injection. The present studywas approved by the Ethical Committee for Animal Experiments of the University of Alberta, and was carried out in accordance with the Guidelines for Research with Experimental Animals of the University of Alberta and the Guide for the Care and Use of Laboratory Animals (NIH Guide) revised in 1996.
Two-Electrode Voltage Clamp
Two-electrode voltage clamp experiments in Xenopus oocytes were performed as described (Yang et al., 2012). Briefly, the two electrodes (capillary pipettes; Warner Instruments, Hamden, CT) impaling an oocyte were filled with 3 M KCl to form a tip resistance of 0.3–2 MΩ. Duration of application of extracellular agonist Ca2+, menthol, or capsaicin was indicated in time course recordings. Currents were recorded using a Geneclamp 500B amplifier and Digidata 1322A AD/DA converter (Molecular Devices, Union City, CA). The pClamp 9 software (Axon Instruments, Union City, CA) was employed for data acquisition and analysis. Currents and voltages were digitally recorded at 200 ms/sample and filtered at 2 kHz through a Bessel filter. SigmaPlot 13 (Systat Software, San Jose, CA) was used for data fitting and plotting.
Oocyte Surface Protein Biotinylation
Xenopus oocytes were washed three times with ice-cold PBS solution followed by incubation with 0.5 mg/mL sulfo-NHS-SS-Biotin (Pierce, Rockford, IL) for 30 min at room temperature (RT). 1 M NH4Cl was used to quench the non-reacted biotin. Oocytes were then washed with ice-cold PBS solution and harvested in ice-cold CelLytic M lysis buffer (Sigma) supplemented with proteinase inhibitor mixture (Thermo Scientific, Waltham, MA). Lysates were incubated at 4°C overnight with gentle shaking upon addition of 100 mL streptavidin (Pierce). The surface protein absorbed by streptavidin was resuspended in a SDS loading buffer and subjected to SDS-PAGE.
Oocytes Immunofluorescence
Whole-mount immunofluorescence assays using Xenopus oocytes were performed as described (Zheng et al., 2017). Briefly, oocytes were washed in PBS, fixed in 4% paraformaldehyde for 15 min, washed three times in PBS plus 50 mM NH4Cl, and then permeabilized with 0.1% Triton X-100 for 4 min. Oocytes werethen blocked in PBS plus 3% skim milk for 30 min and then incubated overnight with indicated primary antibodies, followed by incubation with secondary Alexa-488-conjugated donkey anti-rabbit or Cy3-conjugated goat anti-mouse antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) for 30 min. Oocytes were then mounted in Vectashield (Vector Labs, Burlington, ON) and examined on an AIVI spinning disc confocal microscopy (Cell Imaging Facility, Faculty of Medicine and Dentistry, University of Alberta).
Co-IP
Co-IP experiments were performed using a modified protocol (Zheng et al., 2016a). Briefly, a group of 30 oocytes expressing an indicated TRP channel and/or peptides was washed twice with PBS and solubilized in ice-cold CelLytic-M lysis buffer (Sigma) supplemented with proteinase inhibitor mixture. Supernatants were collected after centrifugation at 16,000 g for 15 min and precleared for 1 hr with protein G-Sepharose (GE Healthcare) and then incubated with indicated antibodies at 4°C overnight. After the addition of 100 μL of 50% protein G-Sepharose, the mixture was incubated for 4 hr or overnight with gentle shaking at 4°C. The immune complexes absorbed to protein G-Sepharose were washed five times with Nonidet P-40 lysis buffer (50 mM Tris [pH 7.5], 150 mM NaCl, and 1% Nonidet P-40) and eluted by SDS loading buffer. Precipitated proteins were analyzed by western blotting using indicated antibodies.
In Vitro His Pull-Down
Purified GST-tagged human TRPP3 N terminus (M1-L95; 2 μg) from E. coli was incubated with the same amount of purified His-tagged human TRPP3 C-terminal fragment (I560-K660) from E. coli in the CelLytic-M lysis buffer (Sigma). The mixture was incubated at RT for 1 hr with gentle shaking, followed by another hr of incubation after addition of 10 μL 50% Ni-NTA agarose bead (QIAGEN, Hilden, Germany). The beads were then washed three times with PBS buffer supplemented with 1% Nonidet P-40, and the remaining proteins were eluted using SDS loading buffer and resolved by SDS-PAGE and transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA). The membrane was then immunoblotted with His and GST antibodies.
Statistical Analyses
Data were analyzed and plotted using Sigmaplot 13 and expressed as mean ± SEM. Student t tests were used to compare two sets of data, whereas one-way ANOVA for multiple comparisons. A probability value (p) of less than 0.05, 0.01, or 0.001 was considered statistically significant and indicated by *, **, and ***, respectively.
Supplementary Material
Highlights.
Conserved Trp in pre-S1 and Lys in TRP-like domains mediate N-C binding in TRPPs
Intramolecular N-C binding is required for activation of TRPPs
PIP2 modulates the N-C binding, conferring its functional regulation of TRPPs
The N-C binding might be a shared mechanism underlying TRPs gating/PIP2 regulation
ACKNOWLEDGMENTS
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) (grant no. RES0012281 to X.Z.C.), the National Natural Science Foundation of China (grant no. 81570648 to X.Z.C.; grant no. 81602448 to J.T.), the Deutsche Forschungsgemeinschaft (DFG) (Sonderforschungsbereich/Transregio 152, P01 to V.F.), and NIH (AR64211 and DK59599 to L.T.). W.Z. was a recipient of the Alberta Innovates-Doctoral Graduate Student Scholarship. R.C. and Q.H. were recipients of the NSERC International Research Training Group Studentship. L.H. was a recipient of the International Research Training Group 1830 Student Scholarship (DFG).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.01.042.
REFERENCES
- Arif Pavel M, Lv C, Ng C, Yang L, Kashyap P, Lam C, Valentino V, Fung HY, Campbell T, Møller SG, et al. (2016). Function and regulation of TRPP2 ion channel revealed by a gain-of-function mutant. Proc. Natl. Acad. Sci. USA 113, E2363–E2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck A, Speicher T, Stoerger C, Sell T, Dettmer V, Jusoh SA, Abdulmughni A, Cavalié A, Philipp SE, Zhu MX, et al. (2013). Conserved gating elements in TRPC4 and TRPC5 channels. J. Biol. Chem. 288, 19471–19483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousova K, Jirku M, Bumba L, Bednarova L, Sulc M, Franek M, Vyklicky L, Vondrasek J, and Teisinger J (2015). PIP2 and PIP3 interact with N-terminus region of TRPM4 channel. Biophys. Chem. 205, 24–32. [DOI] [PubMed] [Google Scholar]
- Cao E, Liao M, Cheng Y, and Julius D (2013). TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504, 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XZ, Vassilev PM, Basora N, Peng JB, Nomura H, Segal Y, Brown EM, Reeders ST, Hediger MA, and Zhou J (1999). Polycystin-L is a calcium-regulated cation channel permeable to calcium ions. Nature 401, 383–386. [DOI] [PubMed] [Google Scholar]
- Clapham DE, Runnels LW, and Strübing C (2001). The TRP ion channel family. Nat. Rev. Neurosci. 2, 387–396. [DOI] [PubMed] [Google Scholar]
- Czirják G, Petheo GL, Spät A, and Enyedi P (2001). Inhibition of TASK-1 potassium channel by phospholipase C. Am. J. Physiol. Cell Physiol. 281, C700–C708. [DOI] [PubMed] [Google Scholar]
- Gao Y, Cao E, Julius D, and Cheng Y (2016). TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 534, 347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Elias A, Berna-Erro A, Rubio-Moscardo F, Pardo-Pastor C, Mrkonjić S, Sepúlveda RV, Vicente R, González-Nilo F, and Valverde MA (2015). Interaction between the linker, pre-S1, and TRP domains determines folding, assembly, and trafficking of TRPV channels. Structure 23, 1404–1413. [DOI] [PubMed] [Google Scholar]
- Grieben M, Pike AC, Shintre CA, Venturi E, El-Ajouz S, Tessitore A, Shrestha L, Mukhopadhyay S, Mahajan P, Chalk R, et al. (2017). Structure of the polycystic kidney disease TRP channel Polycystin-2 (PC2). Nat. Struct. Mol. Biol. 24,114–122. [DOI] [PubMed] [Google Scholar]
- Hansen SB, Tao X, and MacKinnon R (2011). Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 477,495–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holendova B, Grycova L, Jirku M, and Teisinger J (2012). PtdIns(4,5)P2 interacts with CaM binding domains on TRPM3 N-terminus. Channels (Austin) 6, 479–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh KW, Cohen MR, Jiang J, Samanta A, Lodowski DT, Zhou ZH, and Moiseenkova-Bell VY (2016). Structure of the full-length TRPV2 channel by cryo-EM. Nat. Commun. 7, 11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Bulkley D, Guo Y, Zhang W, Guo Z, Huynh W, Wu S, Meltzer S, Cheng T, Jan LY, et al. (2017). Electron cryo-microscopy structure of the mechanotransduction channel NOMPC. Nature 547, 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jirku M, Bumba L, Bednarova L, Kubala M, Sulc M, Franek M, Vyklicky L, Vondrasek J, Teisinger J, and Bousova K (2015). Characterization of the part of N-terminal PIP2 binding site of the TRPM1 channel. Biophys. Chem. 207, 135–142. [DOI] [PubMed] [Google Scholar]
- Kim S, Nie H, Nesin V, Tran U, Outeda P, Bai CX, Keeling J, Maskey D, Watnick T, Wessely O, and Tsiokas L (2016). The polycystin complex mediates Wnt/Ca(2+) signalling. Nat. Cell Biol. 18, 752–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M, Cao E, Julius D, and Cheng Y (2013). Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, and Qin F (2005). Functional control of cold- and menthol-sensitive TRPM8 ion channels by phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 25,1674–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs V, Thyagarajan B, Varnai P, Balla A, Balla T, and Rohacs T (2007). Dual regulation of TRPV1 by phosphoinositides. J. Neurosci. 27, 7070–7080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma R, Li WP, Rundle D, Kong J, Akbarali HI, and Tsiokas L (2005). PKD2 functions as an epidermal growth factor-activated plasma membrane channel. Mol. Cell. Biol. 25, 8285–8298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin S, Wang J, Gambhir A, and Murray D (2002). PIP(2) and proteins: interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 31, 151–175. [DOI] [PubMed] [Google Scholar]
- McNamara CR, and Degterev A (2011). Small-molecule inhibitors of the PI3K signaling network. Future Med. Chem. 3, 549–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell C (2005). The TRP superfamily of cation channels. Sci. STKE 2005, re3.. [DOI] [PubMed] [Google Scholar]
- Nilius B, Mahieu F, Prenen J, Janssens A, Owsianik G, Vennekens R, and Voets T (2006). The Ca2+-activated cation channel TRPM4 is regulated by phosphatidylinositol 4,5-biphosphate. EMBO J. 25, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Owsianik G, and Voets T (2008). Transient receptor potential channels meet phosphoinositides. EMBO J. 27, 2809–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuguro K, Tang J, Tang Y, Xiao R, Freichel M, Tsvilovskyy V, Ito S, Flockerzi V, Zhu MX, and Zholos AV (2008). Isoform-specific inhibition of TRPC4 channel by phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 283, 10026–10036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen CE, Armache JP, Gao Y, Cheng Y, and Julius D (2015). Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 520, 511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poblete H, Oyarzún I, Olivero P, Comer J, Zuñiga M, Sepulveda RV, Báez-Nieto D, González Leon C, González-Nilo F, and Latorre R (2015). Molecular determinants of phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) binding to transient receptor potential V1 (TRPV1) channels. J. Biol. Chem. 290, 2086–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey IS, Delling M, and Clapham DE (2006). An introduction to TRP channels. Annu. Rev. Physiol. 68, 619–647. [DOI] [PubMed] [Google Scholar]
- Rohacs T (2014). Phosphoinositide regulation of TRP channels. Handb. Exp. Pharmacol. 223, 1143–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohács T, Lopes CM, Michailidis I, and Logothetis DE (2005). PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat. Neurosci. 8, 626–634. [DOI] [PubMed] [Google Scholar]
- Rohacs T, Thyagarajan B, and Lukacs V (2008). Phospholipase C mediated modulation of TRPV1 channels. Mol. Neurobiol. 37, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome K, Singh AK, Yelshanskaya MV, and Sobolevsky AI (2016). Crystal structure of the epithelial calcium channel TRPV6. Nature 534, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen PS, Yang X, DeCaen PG, Liu X, Bulkley D, Clapham DE, and Cao E (2016). The structure of the polycystic kidney disease channel PKD2 in lipid nanodiscs. Cell 167, 763–773. e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg X, Lespay-Rebolledo C, and Brauchi S (2014). A structural view of ligand-dependent activation in thermoTRP channels. Front. Physiol. 5,171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente P, Fernández-Carvajal A, Camprubí-Robles M, Gomis A, Quirce S, Viana F, Fernández-Ballester G, González-Ros JM, Belmonte C, Planells-Cases R, and Ferrer-Montiel A (2011). Membrane-tethered peptides patterned after the TRP domain (TRPducins) selectively inhibit TRPV1 channel activity. FASEB J. 25, 1628–1640. [DOI] [PubMed] [Google Scholar]
- Venkatachalam K, and Montell C (2007). TRP channels. Annu. Rev. Biochem. 76, 387–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watnick T, He N, Wang K, Liang Y, Parfrey P, Hefferton D, St George-Hyslop P, Germino G, and Pei Y (2000). Mutations of PKD1 in ADPKD2 cysts suggest a pathogenic effect of trans-heterozygous mutations. Nat. Genet. 25, 143–144. [DOI] [PubMed] [Google Scholar]
- Wilkes M, Madej MG, Kreuter L, Rhinow D, Heinz V, De Sanctis S, Ruppel S, Richter RM, Joos F, Grieben M, et al. (2017). Molecular insights into lipid-assisted Ca2+ regulation of the TRP channel Polycystin-2. Nat. Struct. Mol. Biol. 24, 123–130. [DOI] [PubMed] [Google Scholar]
- Yang J, Wang Q, Zheng W, Tuli J, Li Q, Wu Y, Hussein S, Dai XQ, Shafiei S, Li XG, et al. (2012). Receptor for activated C kinase 1 (RACK1) inhibits function of transient receptor potential (TRP)-type channel Pkd2L1 through physical interaction. J. Biol. Chem. 287, 6551–6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Wu M, Zubcevic L, Borschel WF, Lander GC, and Lee SY (2018). Structure of the cold- and menthol-sensing ion channel TRPM8. Science 359, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Craciun LC, Mirshahi T, Rohács T, Lopes CM, Jin T, and Logothetis DE (2003). PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37, 963–975. [DOI] [PubMed] [Google Scholar]
- Zheng W, Shen F, Hu R, Roy B, Yang J, Wang Q, Zhang F, King JC, Sergi C, Liu SM, et al. (2016a). Far upstream element-binding protein 1 binds the 3’ untranslated region of PKD2 and suppresses its translation. J. Am. Soc. Nephrol. 27, 2645–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Yang J, Beauchamp E, Cai R, Hussein S, Hofmann L, Li Q, Flockerzi V, Berthiaume LG, Tang J, and Chen XZ (2016b). Regulation of TRPP3 channel function by N-terminal domain palmitoylation and phosphorylation. J. Biol. Chem. 291, 25678–25691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Hu R, Cai R, Hofmann L, Hu Q, Fatehi M, Long W, Kong T, Tang J, Light P, et al. (2017). Identification and characterization of hydrophobic gate residues in TRP channels. FASEB J, fj.201700599RR. [DOI] [PubMed] [Google Scholar]
- Zubcevic L, Herzik MA Jr., Chung BC, Liu Z, Lander GC, and Lee SY (2016). Cryo-electron microscopy structure of the TRPV2 ion channel. Nat. Struct. Mol. Biol. 23, 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.