

Abstract
Background
Hepatic encephalopathy is a common complication of cirrhosis which results in poor brain functioning. The spectrum of changes associated with hepatic encephalopathy ranges from the clinically 'indiscernible' or minimal hepatic encephalopathy to the clinically 'obvious' or overt hepatic encephalopathy. Flumazenil is a synthetic benzodiazepine antagonist with high affinity for the central benzodiazepine recognition site. Flumazenil may benefit people with hepatic encephalopathy through an indirect negative allosteric modulatory effect on gamma‐aminobutyric acid receptor function. The previous version of this review, which included 13 randomised clinical trials, found no effect of flumazenil on all‐cause mortality, based on an analysis of 10 randomised clinical trials, but found a beneficial effect on hepatic encephalopathy, based on an analysis of eight randomised clinical trials.
Objectives
To evaluate the beneficial and harmful effects of flumazenil versus placebo or no intervention for people with cirrhosis and hepatic encephalopathy.
Search methods
We searched The Cochrane Hepato‐Biliary Group Controlled Trials Register, CENTRAL, MEDLINE, Embase, Science Citation Index Expanded, and LILACS; meeting and conference proceedings; and bibliographies in May 2017.
Selection criteria
We included randomised clinical trials regardless of publication status, blinding, or language in the analyses of benefits and harms, and observational studies in the assessment of harms.
Data collection and analysis
Two review authors extracted data independently. We undertook meta‐analyses and presented results using risk ratios (RR) with 95% confidence intervals (CI) and I2 values as a marker of heterogeneity. We assessed bias control using the Cochrane Hepato‐Biliary Group domains; determined the quality of the evidence using GRADE; evaluated the risk of small‐study effects in regression analyses; and conducted trial sequential, subgroup, and sensitivity analyses.
Main results
We identified 14 eligible randomised clinical trials with 867 participants, the majority of whom had an acute episode of overt hepatic encephalopathy. In addition, we identified one ongoing randomised clinical trial. We were unable to gather outcome data from 2 randomised clinical trials with 25 participants. Thus, our analyses include 842 participants from 12 randomised clinical trials comparing flumazenil versus placebo. We classified one randomised clinical trial at low risk of bias in the overall assessment and the remaining randomised clinical trials at high risk of bias. The duration of follow‐up ranged from a few minutes to two weeks, but it was less than one day in the majority of the trials.
In total, 32/433 (7.4%) participants allocated to flumazenil versus 38/409 (9.3%) participants allocated to placebo died (RR 0.75, 95% CI 0.48 to 1.16; 11 randomised clinical trials; low quality evidence). The Trial Sequential Analysis and the one randomised clinical trial assessed as low risk of bias (RR 0.76, 95% CI 0.37 to 1.53) found no beneficial or harmful effects of flumazenil on all‐cause mortality. The methods used to evaluate hepatic encephalopathy included several different clinical scales, electrophysiological variables, and psychometric tests. Flumazenil was associated with a beneficial effect on hepatic encephalopathy when including all randomised clinical trials (RR 0.75, 95% CI 0.71 to 0.80; 824 participants; 9 randomised clinical trials; low quality evidence), or just the trial at low risk of bias (RR 0.78, 95% CI 0.72 to 0.84; 527 participants). The Trial Sequential Analysis supported a beneficial effect of flumazenil on hepatic encephalopathy. The randomised clinical trials included little information about causes of death and little information on non‐fatal serious adverse events.
Authors' conclusions
We found low quality evidence suggesting a short‐term beneficial effect of flumazenil on hepatic encephalopathy in people with cirrhosis, but no evidence of an effect on all‐cause mortality. Additional evidence from large, high quality randomised clinical trials is needed to evaluate the potential benefits and harms of flumazenil in people with cirrhosis and hepatic encephalopathy.
Keywords: Humans, Acute Disease, Antidotes, Antidotes/therapeutic use, Chronic Disease, Flumazenil, Flumazenil/therapeutic use, GABA Modulators, GABA Modulators/therapeutic use, GABA‐A Receptor Antagonists, Hepatic Encephalopathy, Hepatic Encephalopathy/drug therapy, Randomized Controlled Trials as Topic
Flumazenil versus placebo or no intervention for people with cirrhosis and hepatic encephalopathy
Background
What is hepatic encephalopathy?
Cirrhosis is a chronic disorder of the liver. People with cirrhosis may develop hepatic encephalopathy, a condition which results in poor brain functioning. In some people, there are obvious clinical features of disturbed brain functioning (overt hepatic encephalopathy); these changes may be short‐lived or persist for long periods of time. In other people, there are no obvious clinical changes but some aspects of brain function, such as attention and the ability to perform complex tasks are impaired when tested (minimal hepatic encephalopathy). The reason people develop hepatic encephalopathy is complex but changes in brain neurotransmitters, which are the chemical messengers which allow nerve cells to communicate with one another, may play a role. The neurotransmitter gamma aminobutyric acid (GABA) is responsible for slowing or inhibiting brain activity and is thought to play a particularly important role.
What is flumazenil?
Flumazenil is a medicine that acts on one of the GABA receptors in the brain to modify its effects on these specialised cells and so may benefit people with hepatic encephalopathy. It has to be given into a vein (intravenous) and its effects do not last for more than a few hours.
Review question
We investigated the use of flumazenil for the treatment of hepatic encephalopathy in people with cirrhosis by reviewing clinical trials in which people were randomly allocated to treatment with flumazenil or an inactive dummy/placebo or no specific intervention.
Search date
We searched medical databases and conducted manual searches in May 2017.
Study funding sources
Five of the included randomised clinical trials received support from pharmaceutical companies.
Study characteristics
We included 14 randomised clinical trials with 867 participants. All randomised clinical trials compared intravenous infusion of flumazenil versus an inactive placebo (dummy infusion, e.g. a salt solution). The duration of treatment ranged from 10 minutes to 72 hours. Ten randomised clinical trials included participants with overt hepatic encephalopathy; three included participants with minimal hepatic encephalopathy; and one randomised clinical trial included participants with overt or minimal hepatic encephalopathy.
Key results
The analyses showed no effect of flumazenil on all‐cause mortality (deaths of any cause) compared with placebo. People who received flumazenil were more likely to recover from their hepatic encephalopathy than people given a placebo. We found little information about serious side effects.
Quality of the evidence
Overall, the evidence for the effect of flumazenil on hepatic encephalopathy was of low quality; only one randomised clinical trial included had a low risk of bias.
Summary of findings
Summary of findings for the main comparison.
Flumazenil versus placebo for people with cirrhosis and hepatic encephalopathy
| Flumazenil versus placebo for people with cirrhosis and hepatic encephalopathy | ||||||
|
Patient or population: people with hepatic encephalopathy Setting: hospital Intervention: flumazenil Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with flumazenil | |||||
| All‐cause mortality follow‐up: range 1 day to 2 weeks | Study population | RR 0.75 (0.48 to 1.16) | 842 (11 RCTs) | ⊕⊕⊝⊝ Low 1,2 | The only RCT with low risk of bias found no effect of flumazenil on all‐cause mortality (RR 0.76, 95% CI 0.37 to 1.53). The Trial Sequential Analysis found insufficient evidence to support or refute an intervention benefit/harm. | |
| 93 per 1000 | 70 per 1000 (45 to 108) | |||||
| Hepatic encephalopathy | Study population | RR 0.75 (0.71 to 0.80) | 824 (9 RCTs) | ⊕⊕⊝⊝ Low 1,2 | The only RCT with a low risk of bias reported a beneficial effect of flumazenil on hepatic encephalopathy (RR 0.78, 95% CI 0.72 to 0.84; Barbaro 1998). The Trial Sequential Analysis found that flumazenil was associated with a beneficial effect on hepatic encephalopathy (Figure 1). The methods used to assess this outcome varied considerably (Table 3) and the duration of follow‐up was very short in the majority of RCTs. | |
| 933 per 1000 | 700 per 1000 (662 to 746) | |||||
| Serious adverse events | See comment | See comment | Not estimable | 842 (11 RCTs) | ⊕⊕⊝⊝ Low 1,2 | All‐cause mortality was the only serious adverse event reported for both the intervention and control group (Table 8). The narrative text in 4 RCTs described that causes of death included liver failure, progressive liver disease, and infections. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RCT: randomised clinical trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Downgraded due to risk of bias: only one RCT had a low risk of bias.
2 Downgraded due to imprecision: wide confidence intervals.
Figure 1.

Trial Sequential Analysis of randomised clinical trials evaluating flumazenil versus placebo for people with hepatic encephalopathy. The outcome is all‐cause mortality. The original meta‐analysis included 11 randomised clinical trials with 842 participants. The Trial Sequential Analysis ignored three randomised clinical trials due to insufficient information size (Cadranel 1995; Gyr 1996; Zhu 1998). The analysis was made with alpha 3%, power 90%, relative risk reduction 20%, assumed control risk 10%, and diversity 10%. The blue line (Z‐curve) corresponds to the cumulative meta‐analysis, the black horizontal line is the conventional boundary (3% level of significance), and the inward sloping green line is the Trial Sequential Monitoring Boundary. Futility boundaries are ignored because the information is insufficient. The analysis found no evidence to support or refute a beneficial or harmful effect of flumazenil on mortality.
Background
Description of the condition
Hepatic encephalopathy is the term used to describe the spectrum of neuropsychiatric changes that can occur in people with cirrhosis. The joint guideline from the European Association for the Study of the Liver (EASL) and the American Association for the Study of Liver Diseases (AASLD) defines hepatic encephalopathy as brain dysfunction associated with liver insufficiency or portal systemic shunting (EASL/AASLD guideline 2014a; EASL/AASLD guideline 2014b).
Clinically apparent orovert hepatic encephalopathy manifests as a neuropsychiatric syndrome encompassing a wide spectrum of mental and motor disorders (Weissenborn 1998; Ferenci 2002). Events such as gastrointestinal bleeding, infection, and alcohol misuse can trigger this so‐called acute or episodic hepatic encephalopathy. Fifty per cent of instances occur with no obvious precipitating cause. Episodes may recur. Between episodes, people may return to their baseline neuropsychiatric status or retain a degree of impairment (Bajaj 2010). Less frequently, people present with persistent neuropsychiatric abnormalities, which are always present to some degree, but which may vary in seriousness. The clinical features of overt hepatic encephalopathy ranges from subtle alterations in personality, intellectual capacity and cognitive function to more profound alterations in motor function and consciousness leading to deep coma (Weissenborn 1998). Other abnormalities include impaired psychometric performance (Schomerus 1998; Randolph 2009), disturbed neurophysiological function (Parsons‐Smith 1957; Chu 1997), altered cerebral neurochemical/neurotransmitter homeostasis (Taylor‐Robinson 1994), reductions in global and regional cerebral blood flow and metabolism (O'Carroll 1991), and changes in cerebral fluid homeostasis (Haussinger 2000). In general, the degree of impairment in these parameters increases as the clinical condition worsens. Minimal hepatic encephalopathy (in the older literature 'subclinical' or 'latent') refers to people with cirrhosis who are 'clinically normal,' but who show abnormalities in neuropsychometric or neurophysiological performance (Ferenci 2002). Hepatic encephalopathy, whether minimal or overt, is associated with impairment in the performance of complex tasks, such as driving (Schomerus 1981; Bajaj 2009; Kircheis 2009), and a detrimental effect on health‐related quality of life (Groeneweg 1998), safety (Roman 2011), neurocognitive function post‐transplantation (Sotil 2009), and survival (Bustamante 1999; D'Amico 2006; Stewart 2007). About 42% of people with cirrhosis are alive one year after their first episode of hepatic encephalopathy but only 23% are alive after three years (Bustamante 1999). Thus, more than 50% die within one year and more than 75% die within three years. Hepatic encephalopathy also poses a substantial burden for the carers of affected people (Bajaj 2011) and on healthcare systems (Poordad 2007; Stepanova 2012).
Prevalence of hepatic encephalopathy
The prevalence of hepatic encephalopathy varies. About 10% to 14% of people with cirrhosis have overt hepatic encephalopathy at the time they are first diagnosed as having liver disease (Saunders 1981). In people with decompensated cirrhosis the prevalence of overt hepatic encephalopathy is around 20% (D'Amico 1986; de Jongh 1992; Zipprich 2012). The cumulated incidence of overt hepatic encephalopathy is as high as 40% (Randolph 2009; Bajaj 2011). The prevalence of minimal hepatic encephalopathy varies but may be as high as 50% (Lauridsen 2011).
Diagnosing hepatic encephalopathy
There is no gold standard for the diagnosis of hepatic encephalopathy. A detailed neuropsychiatric history and examination is important to identify suggestive abnormalities while eliminating other potential causes of similar cerebral changes (Montagnese 2004). The West Haven or Conn criteria are commonly used to assess and grade the mental state (Conn 1977), while the Glasgow Coma Scale is used to grade the level of consciousness (Teasdale 1974). People with hepatic encephalopathy show impairment on a range of psychometric tests. People with minimal hepatic encephalopathy show deficits in attention, visuo‐spatial abilities, fine motor skills, and memory while other cognitive functions are relatively well preserved. People with overt hepatic encephalopathy show additional disturbances in psychomotor speed, executive function, and concentration. The Psychometric Hepatic Encephalopathy Score, which employs five paper and pencil tests to assess attention, visual perception and visuo‐constructive abilities is widely used in the assessment of psychometric change in people with cirrhosis (Schomerus 1998; Weissenborn 2001). People with hepatic encephalopathy may have a number of neurophysiological abnormalities (Guérit 2009). The electroencephalogram, which primarily reflects cortical neuronal activity, may show progressive slowing of background activity and abnormal wave morphology. The brain responses, or evoked potentials, to stimuli such as light and sounds may show abnormal slowing or abnormal wave forms. Other potential diagnostic techniques include the Critical Flicker Fusion Frequency (Kircheis 2002), and the Inhibitory Control Test (Bajaj 2008). Blood ammonia concentrations are not routinely measured to diagnose hepatic encephalopathy but they are sometimes monitored in clinical trials.
Description of the intervention
The pathogenesis of hepatic encephalopathy is complex and incompletely understood. It is associated with a general depression in cerebral function and a shift in balance between inhibitory and excitatory neurotransmission favouring inhibition. Gamma aminobutyric acid (GABA) is the major inhibitory neurotransmitter in the brain; it binds to a specific receptor; the GABA‐A‐complex, which also has neurosteroid and benzodiazepine modulatory sites (Butterworth 2016). People with hepatic encephalopathy have been shown to have increased 'GABA tone'. Use of a benzodiazepine receptor antagonist may counteract the increased tone and benefit people with hepatic encephalopathy (Ahboucha 2008).
Flumazenil competitively inhibits the activity at the benzodiazepine recognition site on the GABA‐A receptor complex, but lacks major intrinsic pharmacological or behavioural activity (Whitwam 1995). Flumazenil is used to treat benzodiazepine overdose and in the reversal of anaesthesia. Following intravenous administration, flumazenil distributes extensively in the extravascular space with an initial distribution half‐life of 4 to 11 minutes and a terminal half‐life of 40 to 80 minutes. Maximum plasma concentrations are reached at around 50 minutes. Flumazenil is completely metabolised primarily by hepatic metabolism and has a relatively high hepatic extraction ratio (Amrein 1990). In people with moderate liver dysfunction, the mean total clearance is decreased to 40% to 60%. In people with severe liver dysfunction, clearance is decreased to 25%. This results in a prolongation of the half‐life from 50 minutes in healthy volunteers to 1.3 hours in people with moderate hepatic impairment and 2.4 hours in people with severe hepatic dysfunction (Amrein 1990). Nevertheless, even in people with severe liver disease, its duration of action remains relatively short.
How the intervention might work
Glutamate dehydrogenase synthesises GABA from glutamate in presynaptic nerves. GABA binds to a specific receptor, which is embedded in the postsynaptic neural membrane. This receptor is part of a larger GABA‐A receptor complex, which also has binding sites for benzodiazepines, barbiturates, and neurosteroids. The binding of any of these ligands opens a chloride channel; the influx of chloride ions results in hyperpolarisation of the postsynaptic membrane and neuro‐inhibition. Neurosteroids are synthesised in the brain, primarily in astrocytes, and mediates an increased GABA‐A tone, which is associated with hepatic encephalopathy. Neurosteroid synthesis is mediated via activation of translocator protein, a mitochondrial neuroglial cholesterol‐transporter protein previously (Butterworth 2016). Translocator protein sites (previously known as 'peripheral‐type' or mitochondrial) benzodiazepine receptors. Neurosteroids such as 3α‐5α‐tetrahydroprogesterone (allopregnanolone) are potent, endogenous, positive allosteric modulators of both the GABA and benzodiazepine sites on the GABA‐A receptor complex. Autopsy and imaging studies show consistent upregulation of translocator protein sites in people with hepatic encephalopathy. This upregulation is most likely mediated by ammonia and manganese (Ahboucha 2008), both of which accumulate as a result of hepatocellular failure and portal‐systemic shunting of blood. In addition, cerebrospinal fluid and autopsied brains of people with hepatic encephalopathy have increased concentrations of agonist ligands such as diazepam‐binding inhibitor and octadecaneuropeptide, which modulate the function of translocator protein sites (Córdoba 2002). People who die from hepatic encephalopathy have increased cerebral concentrations of allopregnanolone, which modulates components of the GABA‐A receptors. The neurosteroids may also act synergistically with other neurotoxins such as ammonia and benzodiazepine‐like compounds to further modulate GABA‐A receptor function. The net effect is an increase in GABA‐A tone and neural inhibition. Involvement of the GABA system in the pathogenesis of hepatic encephalopathy is consistent with the increased sensitivity to benzodiazepines observed in these patients (Batki 1987).
Flumazenil is a selective, synthetic benzodiazepine antagonist with high affinity for the benzodiazepine recognition site but is itself devoid of intrinsic activity. This compound functions as an antagonist of positive and negative modulators acting at benzodiazepine recognition sites located in the GABA‐A receptor (Vicini 1987). Flumazenil may exert an indirect negative allosteric modulatory effect on GABA‐A receptor function by reducing the facilitatory action of the central benzodiazepine receptor on GABA‐related opening of the chloride ion‐channel and in turn the excessive inhibitory effect.
Why it is important to do this review
The resource utilisation associated with the management of people with hepatic encephalopathy continues to escalate (Poordad 2007; Stepanova 2012). The costs of treatment and rehabilitation are increasing year on year (Neff 2010). The Identification of effective interventions which will facilitate the management of people with hepatic encephalopathy is clearly important. A number of randomised clinical trials have assessed the effects of flumazenil in people with cirrhosis and hepatic encephalopathy (Klotz 1989; Hermant 1991; Cadranel 1995; Gooday 1995; Gyr 1996; Amodio 1997; Barbaro 1998; Giger‐Mateeva 1999; Dursun 2003). The numbers of the randomised clinical trials and of the included participants is small and several studies used a cross‐over design, which hampers the analyses of clinical outcomes such as mortality and morbidity. A meta‐analysis undertaken in 2002 found that use of flumazenil may be associated with clinical and electroencephalographic improvement in people with cirrhosis and hepatic encephalopathy (Goulenok 2002). The previous versions of this review included 13 randomised clinical trials (Als‐Nielsen 2001; Als‐Nielsen 2004) and found no beneficial effect of flumazenil on all‐cause mortality, but a potential beneficial effect on manifestations of hepatic encephalopathy. We have updated this review based on current recommendations (Gluud 2017).
Objectives
To evaluate the beneficial and harmful effects of flumazenil versus placebo or no intervention for people with cirrhosis and hepatic encephalopathy.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised clinical trials regardless of their publication status, language, or blinding in our primary analyses. If, during the selection of trials, we identified observational studies (i.e. quasi‐randomised studies, cohort studies, or patient reports) that reported adverse events caused by, or associated with, the interventions in our review, we included these studies for a review of the adverse events. We did not specifically search for observational studies for inclusion in this review, which is a known limitation.
Types of participants
Randomised clinical trials evaluating participants with cirrhosis and hepatic encephalopathy, irrespective of the aetiology and severity of the underlying liver disease. Included participants could have overt or minimal hepatic encephalopathy. If we identified trials including subsets of relevant participants with cirrhosis as well as participants without cirrhosis, we planned to exclude these trials in sensitivity analyses.
Types of interventions
Flumazenil at any dose, duration, or mode of administration versus placebo or no intervention.
Types of outcome measures
We assessed all outcomes at the maximum duration of follow‐up.
Primary outcomes
All cause‐mortality.
Hepatic encephalopathy (number of participants without improved manifestations).
Serious adverse events defined as any untoward medical occurrence that resulted in death, was life‐threatening, required hospitalisation, led to prolongation of the existing hospitalisation, or resulted in persistent or significant disability (ICH‐GCP 1997). We analysed serious adverse events as a composite outcome (Gluud 2017).
Secondary outcomes
Non‐serious adverse events defined as any adverse event that did not fulfil the criteria for a serious adverse event.
Health‐related quality of life.
Exploratory outcomes
Number Connection Test results.
Search methods for identification of studies
The last search update was May 2017.
Electronic searches
We searched The Cochrane Hepato‐Biliary Group Controlled Trials Register (Gluud 2017), Cochrane Central Register of Controlled Trials (CENTRAL; 2017, Issue 2) in the Cochrane Library (searched May 2017), MEDLINE Ovid (1946 to May 2017), Embase Ovid (1974 to May 2017), Science Citation Index Expanded (Web of Science; 1900 to May 2017) (Royle 2003), and LILACS (Bireme; 1982 to May 2017) using the strategy described in Appendix 1. We did not have access to Chinese or Japanese databases. We plan to search both Chinese and Japanese databases in future updates, should they become available to us via the Cochrane Hepato‐Biliary Group.
Searching other resources
We searched the reference lists of papers identified in the electronic searches and wrote to authors of the identified clinical trials and relevant pharmaceutical companies. We searched the conference proceedings of the European Association for the Study of the Liver (EASL), the United European Gastroenterology Week (UEGW), the American Gastroenterological Association (AGA), the American Association for the Study of Liver Diseases (AASLD), the International Society for Hepatic Encephalopathy and Nitrogen metabolism (ISHEN), the World Health Organization (WHO) online trial meta‐register (apps.who.int/trialsearch/), and Google Scholar using the search terms cirrhosis AND flumazenil.
Data collection and analysis
Selection of studies
Three review authors (ETG, MYM, and LLG), working independently, read the updated electronic searches, performed additional handsearches, and listed potentially eligible trials. All authors read the potentially eligible trials and participated in the final selection of those to be included in the analyses. For trials reported in more than one publication, we selected the paper reporting the longest duration of follow‐up as the primary reference. We listed details of all included trials in a 'Characteristics of included studies' table and listed all excluded studies with the reason for their exclusion in a 'Characteristics of excluded studies table'.
Data extraction and management
All review authors participated in data extraction and at least two review authors independently evaluated each randomised clinical trial. We resolved disagreements through discussion and sought key unpublished information that was missing from published trial reports, through correspondence with the primary investigators of included randomised clinical trials.
Where we were not able to gather sufficient data (number of events and participants) from the text and tables of the included reports of randomised clinical trials or from correspondence with investigators we attempted to extrapolate data , where possible from any contained graphical material.
We gathered data on the following:
Trials: design (cross‐over or parallel), setting (number of clinical sites), country of origin, inclusion period;
Participants: mean age, proportion of men, type of hepatic encephalopathy, proportion with cirrhosis, proportion with alcoholic liver disease, proportion with viral hepatitis;
Interventions: type, dose, duration of therapy, mode of administration;
Outcomes: outcomes assessed, criteria used in the assessment of hepatic encephalopathy.
Assessment of risk of bias in included studies
We assessed bias control using the domains described in the Cochrane Hepato‐Biliary Group Module (Gluud 2017) and classified the risk of bias for separate domains as high, unclear, or low. We also included an overall assessment of bias control as described below:
Allocation sequence generation
Low risk of bias: sequence generation achieved using computer random number generation or a random number table. Drawing lots, tossing a coin, shuffling cards, or throwing dice were adequate if performed by an independent person not otherwise involved in the trial.
Unclear risk of bias: not described.
High risk of bias: sequence generation method was not random.
Allocation concealment
Low risk of bias: used a central and independent randomisation unit or similar adequate method (e.g. serially numbered opaque sealed envelopes) to ensure that the allocation sequence was unknown to the investigators (Hrobjartsson 2001; Savovic 2012a; Savovic 2012b).
Unclear risk of bias: allocation not described.
High risk of bias: allocation sequence was likely to be known to the investigators who assigned the participants.
Blinding of participants and personnel
Low risk of bias: blinding of participants and personnel performed adequately using a placebo. We defined lack of blinding as not likely to affect the evaluation of mortality.
Unclear risk of bias: insufficient information to assess blinding.
High risk of bias: no blinding or incomplete blinding.
Blinding of outcome assessors
Low risk of bias: blinding of outcome assessors performed adequately using a placebo. We defined lack of blinding as not likely to affect the evaluation of mortality (Hrobjartsson 2001; Savovic 2012a; Savovic 2012b).
Unclear risk of bias: there was insufficient information to blinding.
High risk of bias: no blinding or incomplete blinding.
Incomplete outcome data
Low risk of bias: missing data were unlikely to make treatment effects depart from plausible values. The investigators used sufficient methods, such as intention‐to‐treat analyses with multiple imputations or carry‐forward analyses to handle missing data.
Unclear risk of bias: there was insufficient information to assess missing data.
High risk of bias: the results were likely to be biased due to missing data.
Selective outcome reporting
Low risk of bias: the trial reported clinically relevant outcomes (all‐cause mortality, hepatic encephalopathy, and serious adverse events). If we had access to the original trial protocol, the outcomes selected should have been those described in the protocol. If we obtained information from a trial registry (such as www.clinicaltrials.gov), we only used that information if the investigators registered the trial before inclusion of the first participant.
Unclear risk of bias: predefined relevant outcomes were not reported fully or the reporting was unclear.
High risk of bias: one or more predefined outcomes were not reported.
For‐profit bias
Low risk of bias: the trial appeared free of industry sponsorship or other type of for‐profit support.
Unclear risk of bias: no information on clinical trial support or sponsorship.
High risk of bias: the trial was sponsored by industry or received other support (such as provision of study drugs).
Other bias
Low risk of bias: the trial appeared free of other biases including: medicinal dosing problems or follow‐up (as defined below).
Unclear risk of bias: the trial may or may not have been free of other domains that could put it at risk of bias.
High risk of bias: there were other factors in the trial that could put it at risk of bias such as the administration of inappropriate treatments being given to the controls (e.g. an inappropriate dose) or follow‐up (e.g. the trial included different follow‐up schedules for participants in the allocation groups).
Overall bias assessment
Low risk of bias: all domains were low risk of bias using the definitions described above.
High risk of bias: one or more of the bias domains were of unclear or high risk of bias.
Measures of treatment effect
We analysed dichotomous data using risk ratios (RR) and continuous data using mean differences (MD), both with 95% confidence intervals (CI).
Unit of analysis issues
Due to the fluctuating nature of hepatic encephalopathy and the nature of our primary outcomes, we included randomised clinical trials using a parallel‐arm design and the first treatment period from cross‐over trials.
Dealing with missing data
We extracted data on all participants randomised to allow intention‐to‐treat analyses. We planned to undertake analyses to evaluate the influence of missing data (Higgins 2008) including worst‐case scenario analysis, and extreme worst‐case and best‐case scenario analyses (Gluud 2017). However, we did not identify randomised clinical trials with missing outcome data.
Assessment of heterogeneity
We assessed heterogeneity through visual inspection of the forest plots and expressed heterogeneity as I2 values using the following thresholds: 0% to 40% (unimportant), 40% to 60% (moderate), 60% to 80% (substantial), and greater than 80% (considerable). We included the information in our 'Summary of findings' tables.
Assessment of reporting biases
For meta‐analyses with at least 10 randomised clinical trials (meta‐analysis evaluating all‐cause mortality), we planned to prepare funnel plots and regression analyses of funnel plot asymmetry (Harbord 2006). However, our analyses included fewer than 10 randomised clinical trials.
Data synthesis
We performed the analyses using Review Manager 5 (RevMan 2014), STATA (Stata 14), and Trial Sequential Analysis (TSA 2011).
Meta‐analysis
We performed random‐effects and fixed‐effect meta‐analyses. The estimates of the random‐effects and fixed‐effect meta‐analyses were similar for all analyses. Therefore, we assumed that any small‐study effects had little effect on the intervention effect estimate. For random‐effects models, precision decreased with increasing heterogeneity and CIs widened correspondingly. Accordingly, the random‐effects model provided the most conservative (and a more correct) estimate of the intervention effect. Accordingly, we report the results of our analyses based on random‐effects meta‐analyses.
Trial Sequential Analysis
We performed Trial Sequential Analysis to evaluate the risk of errors and to evaluate futility (Wetterslev 2008; Thorlund 2011; Wetterslev 2017). We defined the required information size as the number of participants needed to detect or reject an intervention effect. We set alpha to 3% and power to 90% in all analyses. We used the model‐based diversity and repeated all analyses with a diversity increased by 10%. Due to the lack of randomised clinical trials assessed at low risk of bias, we were only able to conduct the analyses with inclusion of all randomised clinical trials (regardless of bias control).
All‐cause mortality: based on one large randomised clinical trial evaluating flumazenil (Barbaro 1998), and cohort studies evaluating the prognosis of people with hepatic encephalopathy (EASL/AASLD guideline 2014a; EASL/AASLD guideline 2014b), as well as the results of our meta‐analysis, we set the relative risk reduction to 10%, the control group risk to 20%, and increased diversity to 10%.
Hepatic encephalopathy: based on the Cochrane Hepato‐Biliary Group recommendations, we conducted the analysis with a relative risk reduction of 20% (corresponding to the upper 95% CI) and reduced the control group risk from the observed 93% to 60% (set lower than the observed control group risk).
Serious adverse events: we were only able to identify serious adverse events that were fatal. Accordingly, our analysis of all‐cause mortality and serious adverse events included the same numbers.
Subgroup analysis and investigation of heterogeneity
We performed subgroup analyses to investigate heterogeneity in randomised clinical trials based on the:
Type of hepatic encephalopathy;
Inclusion of participants with cirrhosis or acute liver failure
The trial design;
Duration of follow‐up.
Only one randomised clinical trial had a low risk of bias in the overall assessment.
Sensitivity analysis
We planned to perform worst‐case scenario analyses, but none of the trials reported missing outcome data.
'Summary of findings' table
We used GRADEpro (GRADEpro 2008) to generate a 'Summary of findings' table with information about outcomes, risk of bias, and results of the meta‐analyses. We used the GRADE system to evaluate the quality of the evidence for outcomes reported in the review considering the within‐study risk of bias (methodological quality), indirectness of evidence, diversity (heterogeneity), imprecision of effect estimate, and risk of publication bias.
Results
Description of studies
We included 14 randomised clinical trials in our analyses of benefits and harms (Klotz 1989; Hermant 1991; Pomier‐Layrargues 1994; Cadranel 1995; Gooday 1995; Van der Rijt 1995; Gyr 1996; Amodio 1997; Barbaro 1998; Zhu 1998; Giger‐Mateeva 1999; Lacetti 2000; Dursun 2003; Li 2009). We identified one randomised clinical trial which is currently ongoing (Yale 2014). We are unable to determine if this ongoing randomised clinical trial will be eligible for inclusion in the review. We excluded one randomised clinical trial and 2 non‐randomised studies from the primary analyses, but included them in the evaluation of harms (Marsepoil 1990; Kapczinski 1995;Jia 1999). For additional information, see Characteristics of included studies; Characteristics of excluded studies; and Characteristics of ongoing studies tables.
Results of the search
We identified 249 references in the electronic and manual searches (Figure 2). After exclusion of duplicates and references to papers that did not describe clinical trials assessing benzodiazepine receptor antagonists for hepatic encephalopathy, we retrieved 33 references for further assessment. Fourteen randomised clinical trials described in 25 references fulfilled our inclusion criteria (Klotz 1989; Hermant 1991; Pomier‐Layrargues 1994; Cadranel 1995; Gooday 1995; Van der Rijt 1995; Gyr 1996; Amodio 1997; Barbaro 1998; Zhu 1998; Giger‐Mateeva 1999; Lacetti 2000; Dursun 2003; Li 2009). In addition, we identified one ongoing randomised clinical trial with an estimated completion date of June 2017 (Yale 2014).
Figure 2.
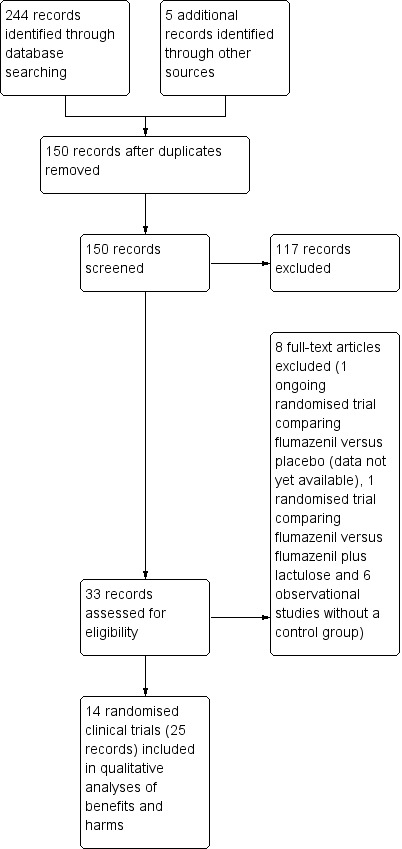
Study flow diagram for identification and selection of randomised clinical trials.
Two trials were letters (Klotz 1989; Hermant 1991), and 12 were full‐text articles (Pomier‐Layrargues 1994; Cadranel 1995; Gooday 1995; Van der Rijt 1995; Gyr 1996; Amodio 1997; Barbaro 1998; Zhu 1998; Giger‐Mateeva 1999; Lacetti 2000; Dursun 2003; Li 2009). The language of the publications was English (Klotz 1989; Pomier‐Layrargues 1994; Cadranel 1995; Gooday 1995; Van der Rijt 1995; Gyr 1996; Amodio 1997; Barbaro 1998; Giger‐Mateeva 1999; Lacetti 2000; Dursun 2003), Chinese (Zhu 1998; Li 2009), or French (Hermant 1991).
Included studies
Six trials used a parallel group (Hermant 1991; Gyr 1996; Zhu 1998; Lacetti 2000; Dursun 2003; Li 2009), and eight used a cross‐over design (Klotz 1989; Pomier‐Layrargues 1994; Cadranel 1995; Gooday 1995; Van der Rijt 1995; Amodio 1997; Barbaro 1998; Giger‐Mateeva 1999). In 3 cross‐over trials, only people classified as non‐responders participated in the second cross‐over treatment period (Pomier‐Layrargues 1994; Cadranel 1995; Barbaro 1998). Six of the cross‐over trials included a washout period, which ranged from 3 minutes to 1 week (Pomier‐Layrargues 1994; Gooday 1995; Van der Rijt 1995; Amodio 1997; Barbaro 1998; Giger‐Mateeva 1999).
The investigators diagnosed hepatic encephalopathy using clinical scores, electrophysiological techniques, and psychometric tests (Table 3; Table 4; Table 5). Nine randomised clinical trials included participants with overt hepatic encephalopathy (Klotz 1989; Hermant 1991; Pomier‐Layrargues 1994; Cadranel 1995; Van der Rijt 1995; Barbaro 1998; Zhu 1998; Lacetti 2000; Li 2009). Six of these randomised clinical trials described precipitating events; the most common was gastrointestinal bleeding (Table 6). Three randomised clinical trials included participants with minimal hepatic encephalopathy (Gooday 1995; Amodio 1997; Giger‐Mateeva 1999), while one included participants with either minimal or overt hepatic encephalopathy (Dursun 2003). The majority of the participants in the 14 included studies had cirrhosis although 30 (33%) of the 90 participants included in two of the trials had fulminant hepatic failure (Van der Rijt 1995; Li 2009).
Table 1.
Definition of improved manifestations of hepatic encephalopathy
| Trial | Type of hepatic encephalopathy | Neuropsychiatric assessment | Definition of overall improvement |
| Amodio 1997 | Minimal | Number Connection Test and Brainstem Auditory Evoked Response. | Investigators did not define or assess the number of participants with an overall improvement. |
| Barbaro 1998 | Overt | Mental status assessed using a clinical scale (Table 4), Modified Glasgow Coma Scale (Table 4), and electroencephalography (Table 5). | Improvement in clinical scores or electroencephalography. |
| Cadranel 1995 | Overt | Mental status assessed using a clinical scale (Table 4), and electroencephalography (Table 5). | Improvement in clinical score or electroencephalography. |
| Dursun 2003 | Minimal or overt | Mental status assessed using a clinical scale (Table 4), Number Connection Test results, and electroencephalography (Table 5). | Improvement based on the clinical score and Number Connection Test results. |
| Giger‐Mateeva 1999 | Minimal | Number Connection Test and brainstem auditory evoked response. | Not defined. The investigators included a post‐hoc subjective assessment of alertness. |
| Gooday 1995 | Minimal | Simple and complex reaction time, verbal memory, psychomotor speed, short‐term and working memory. | Improvement in psychomotor speed evaluated using change in reaction time; Investigators did not define or assess the number of participants with overall improvement. . |
| Gyr 1996 | Overt | Mental status (Table 4), and electroencephalography (Table 5). | Clinically relevant improvement defined as a 2‐point improvement in clinical score at any time during treatment compared with baseline. The investigators also reported improvement defined using the clinical scale score (mean for all individual observations). |
| Hermant 1991 | Overt | Glasgow Coma Scale (Table 4), and electroencephalography. | A 2‐point improvement in the Glasgow Coma Score and electroencephalography. |
| Klotz 1989 | Overt | Clinical assessment (score not described). | Improvement in clinical status |
| Lacetti 2000 | Overt | Mental status (scale not specified) and Glasgow Coma Scale (Table 4). | Investigators originally classified participants as Grade III to IV coma. Method of assessment not stipulated. The trial report defined 'clinically relevant improvement' as primary outcome defined as a 3‐point improvement in Glasgow Coma Score. |
| Li 2009 | Overt | Glasgow Coma Scale (Table 4), and electroencephalography | Improvement in the Glasgow Coma Score of ≥ 3 points. |
| Pomier‐Layrargues 1994 | Overt | Modified Glasgow Coma Score (Table 4), and electroencephalography. | Improvement in ≥ 2 items on modified Glasgow Coma Score within 1 hour after the end of treatment. |
| Van der Rijt 1995 | Overt | Clinical scale (Table 4). | A ≥ 1 ‐point decrease in severity of hepatic encephalopathy. |
| Zhu 1998 | Overt | Clinical scale (Table 4). | Overall improvement in hepatic encephalopathy based on clinical grade. |
RCT: randomised clinical trial.
Table 2.
Neuropsychiatric assessment scales
| Scale (Grippon 1988) used in Cadranel 1995; Barbaro 1998. | |
| I | Euphoria or depression, mild confusion, slowness, disorder in sleep rhythm. |
| II | Drowsiness, inappropriate behaviour, accentuation of stage I. |
| III | Stupor; participant sleeps most of the time, but is rousable; incoherent speech; marked confusion. |
| IVa | Coma, co‐ordinated response to painful stimuli. |
| IVb | Coma, hyperextension, and pronosupination after painful stimuli. |
| IVc | Coma, no response to painful stimuli. |
| V | Clinical decerebration. |
| Scale (Fitz 1998) used in Dursun 2003. | |
| Subclinical | Normal examination with subtle changes in psychometric or Number Connection Tests. |
| I | Impaired attention, irritability, depression, or personality changes. |
| II | Drowsiness, behavioural changes, sleep disorders, and poor memory. |
| III | Confusion, disorientation, somnolence, and amnesia. |
| Scale (Jones 1988) used in Gyr 1996. | |
| ‐ | Clinical assessment criteria consisted of the anamnestic criterion: disorders of sleep pattern (insomnia, hypersomnia, inversion of sleep rhythm) in combination with assessment of the level of consciousness (1 to 4 as described below). Score items weighted so major disturbances of consciousness (portal systemic encephalopathy stage III and IV) were associated with scores of ≥ 11. Portal systemic encephalopathy stage II defined as scores of 5 to 10 and stage I of 3 to 4. |
| 1 | Light disturbance of consciousness if ≥ 1 of following symptoms were present: drowsiness (tendency to fall asleep but wake up spontaneously or in response to normal voice or light), intermittent or permanent disorientation, retardation of ability to perform mental tasks (serial subtractions of sevens), mood disorder, inappropriate behaviour. |
| 2 | Somnolence (arousable to physical stimuli such as mild prodding or shaking only). |
| 3 | Stupor (localised motor response to pain). |
| 4 | Coma (unarousability, no or unlocalised motor reactions to painful stimuli). |
| Scale (no reference provided in paper) used in Van der Rijt 1995. | |
| 1 | Presence of ≥ 2 of following abnormalities: inverted sleep pattern, disturbed memory, impaired calculation (serial sevens), slowness of speech, or flapping tremor. |
| 2 | Presence of ≥ 2 of following: lethargy, time disorientation, or flapping tremor. |
| 3 | Presence of ≥ 2 of following: a state in which person had to be stimulated repetitively to open his/her eyes or execute commands, disorientation in terms of place and disorientation with respect to person. |
| 4 | Coma. |
| Scale (Conn 1977) used in Zhu 1998. | |
| 1 | Trivial lack of awareness, euphoria or anxiety, shortened attention span, impaired performance of addition or subtraction. |
| 2 | Lethargy or apathy, minimal disorientation for time or place, subtle personality change, inappropriate behaviour. |
| 3 | Somnolence to semistupor, but responsive to verbal stimuli; confusion; gross disorientation. |
| 4 | Coma. |
| Glasgow Coma Scale (CGS) (Teasdale 1974) used in Hermant 1991; Lacetti 2000; Dursun 2003; Li 2009. | |
| Scores | Eye opening (E):
Verbal response (V):
Motor response (M):
|
| Grading |
|
| Modified Glasgow Coma Scale (Pappas 1983) used in Pomier‐Layrargues 1994; Barbaro 1998. | |
| Scores |
|
Table 3.
Assessment of electroencephalography changes
| Electroencephalography grading/Fischer classification (Nusinovici 1977 and Spehlman 1991) used in Hermant 1991; Pomier‐Layrargues 1994; Cadranel 1995; Barbaro 1998. | |
| I | Irregular background activity (theta and alpha). |
| II | Continuous theta activity, bursts of delta waves. |
| III | Prevalent delta activity; polyphasic transients sharp and slow wave complexes. |
| IVa | Continuous delta activity; abundant sharp and slow wave complexes; electroencephalography reactivity present. |
| IVb | Slower activity (delta and some polyphasic transients); electroencephalography reactivity = 0. |
| IVc | Discontinuous activity with silent periods. |
| V | Flat. |
| Electroencephalography grading (Parsons‐Smith 1957) used in Dursun 2003. | |
| A | Generalised suppression of alpha rhythm and its frequent replacement by faster potentials in all leads. The tracings in this grade are generally flat and featureless. |
| B | Alpha rhythm very unstable and disturbed by random waves at 5‐7 per second over both hemispheres. Rhythms most often seen over temporal lobes. In many cases with underlying fast activity. |
| C | Alpha rhythm still seen, but disturbed over both hemispheres by medium‐voltage 5‐6 per second waves. These occur in runs, are not paroxysmal, and do not usually block to eye opening although blocking may occur. Rhythms are particularly well seen over temporal and frontal lobes. |
| D | 5‐6 per second rhythms seen in grade C are now constant in all areas and replace all other cortical activity recorded on electroencephalogram. Appearance of this abnormality in a patient presenting with only slight neuropsychiatric symptoms is very striking. |
| E | 5 to 6 per second rhythms replaced by frontally preponderant bi‐lateral synchronous 2 per second rhythms, which spread backwards over hemispheres. At times, 6 per second rhythms might reappear, but special features of records are occurrence of these diencephalic discharges. |
| Electroencephalography grading (Kennedy 1973) used in Gyr 1996. | |
| 0 | 8 to 12 per second basic rhythm, mean dominant frequency > 8 per second, % theta < 20. |
| 1 | Sudden shifts between normal alpha frequency (around 9 or 10 per second) and slow substitutes (6‐8 per second); mean dominant frequency > 7 per second, % theta > 35. |
| 2 | Diffuse slow activity posterior alpha rhythm seen occasionally, mean dominant frequency 5 to 7 per second, % theta > 60. |
| 3 | Dominant slow activity in all areas, mean dominant frequency 3 to 5 per second, % delta 70. |
| 4 | Bilaterally synchronous, 2‐3 per second waves, predominating over frontal lobes and spreading backwards to occipital lobes; occasional short‐lived appearance of faster rhythms (5 or 6 per second) or voltage depression, mean dominant frequency < 3 per second, % delta 70. |
| Electroencephalography grading (Markand 1984) used in Van der Rijt 1995. | |
| 0 | Background activity consisting of alpha rhythm. |
| 1 | Alpha rhythm with some scattered theta waves. |
| 2 | Background activity of theta activity intermixed with some delta and alpha frequencies. |
| 3 | Background of delta polymorphic activity of high amplitude with spontaneous variability. |
| 4 | Delta activity of relatively small amplitude. |
Table 4.
Precipitating factors
| Trial |
Participants (n) |
Precipitating factors (n) |
| Barbaro 1998 | 527 | Gastrointestinal bleeding (352), surgery (95), sepsis (45), dehydration (6), unknown (29). |
| Cadranel 1995 | 14 | Gastrointestinal bleeding (4), sepsis (7), alcoholic hepatitis (3), portal vein thrombosis (1), viral hepatitis (1), unknown (2). |
| Lacetti 2000 | 54 | Gastrointestinal bleeding (31), sepsis (7), drugs (11), surgery (1). |
| Pomier‐Layrargues 1994 | 21 | Gastrointestinal bleeding (7), sepsis (2), dehydration (1), surgery (2), none (9), portacaval shunting (4). |
| Van der Rijt 1995 | 18 | Hepatitis (5), acute exacerbation in cirrhosis (2), partial hepatectomy (1). |
| Zhu 1998 | 25 | Gastrointestinal bleeding (13), protein overload (6), infection (2), wounds (1), unknown (3). |
n: number of participants.
Recent ingestion of benzodiazepines was a stipulated exclusion criterion in 10 randomised clinical trials with exclusion periods ranging from 3 days to 3 months (Hermant 1991; Pomier‐Layrargues 1994; Gooday 1995; Van der Rijt 1995; Amodio 1997; Barbaro 1998; Zhu 1998; Giger‐Mateeva 1999; Lacetti 2000; Dursun 2003; Table 7). Nine randomised clinical trials tested for blood/urine benzodiazepines at baseline (Hermant 1991; Pomier‐Layrargues 1994; Cadranel 1995; Van der Rijt 1995; Gyr 1996; Amodio 1997; Barbaro 1998; Giger‐Mateeva 1999; Lacetti 2000); three randomised clinical trials stipulated negative screening for blood/urine benzodiazepines as an inclusion criterion (Hermant 1991; Van der Rijt 1995; Lacetti 2000), while the proportion who tested positive for benzodiazepines in the other 6 randomised clinical trials ranged from 1.9% to 21%. The remaining five randomised clinical trials either stated that they did not measure blood/urine benzodiazepines or else did not mention testing (Table 7).
Table 5.
Baseline screening for benzodiazepines in trial participants
| Trial | Required period free of benzodiazepines before inclusion | Baseline screening for benzodiazepines | Screening method and detection level | Negative testing at baseline an inclusion criterion | Proportion testing positive for benzodiazepines at baseline |
| Amodio 1997 | 2 weeks | Yes |
|
No | 0% |
| Barbaro 1998 | 4 days | Yes |
|
No | 1.9% |
| Cadranel 1995 | Not reported | Yes |
|
No | 21.4% |
| Dursun 2003 | 3 days | No | Not reported | Not reported | Not reported |
| Giger‐Mateeva 1999 | 3 months | Yes |
|
No | 0% |
| Gooday 1995 | 1 month | No | Not reported | Not reported | Not reported |
| Gyr 1996 | Yes, but length not specified | Yes |
Post‐hoc analysis
|
No | 8.2% on screening tests Flumazenil 11%; placebo 5% 12/49 samples for more sensitive testing lost |
| Hermant 1991 | Not reported | Yes | Not reported | Yes | 0% |
| Klotz 1989 | Not reported | No | Not reported | Not reported | Not reported |
| Lacetti 2000 | 2 weeks | Yes |
Post‐hoc analysis
|
Yes | 0% |
| Li 2009 | Not reported | No | Not reported | N/A | N/A |
| Pomier‐Layrargues 1994 | 3 days | Yes |
Post‐hoc analysis
|
No | 19% |
| Van der Rijt 1995 | Recent | Yes |
|
Yes | 0% |
| Zhu 1998 | 7 days | No | Not reported | Not reported | Not reported |
All randomised clinical trials compared intravenous flumazenil versus placebo. The daily dose of flumazenil ranged from 0.2 mg to 6.5 mg and the total dose from 0.2 mg to 19.5 mg. The duration of treatment ranged from 10 minutes to 72 hours. Five randomised clinical trials evaluated the intervention effects at the end of the intervention (Klotz 1989; Hermant 1991; Amodio 1997; Giger‐Mateeva 1999; Dursun 2003). Four randomised clinical trials evaluated clinical outcomes at maximum of 24 to 72 hours after the intervention (Pomier‐Layrargues 1994; Cadranel 1995; Van der Rijt 1995; Lacetti 2000). The remaining five randomised clinical trials followed participants from 4 days to 4 weeks (Gooday 1995; Gyr 1996; Barbaro 1998; Zhu 1998; Li 2009).
The trials involving participants with overt hepatic encephalopathy defined overall improvement of hepatic encephalopathy based on a clinical assessment of mental status and the electroencephalogram (Table 3). The trials involving participants with minimal hepatic encephalopathy based their assessment of overall improvement on a subjective assessment of 'alertness' (Giger‐Mateeva 1999); Number Connection Test results (Dursun 2003), the Simple Reaction Time test results (Gooday 1995), or electroencephalography (Van der Rijt 1995).
Excluded studies
We excluded one cross‐over randomised clinical trial evaluating cognitive function and anxiety in people with alcohol‐related or non‐alcohol‐related cirrhosis (Kapczinski 1995), one randomised clinical trial evaluating flumazenil alone or with lactulose (Wu 2001), and seven observational studies (Grimm 1988; Bansky 1989; Marsepoil 1990; Devictor 1995; Ozyilkan 1997; Golubovic 1999; Jia 1999). None of the excluded studies reported data that allowed analysis of serious adverse events.
One double‐blind, cross‐over trial involved participants with cirrhosis who were liver transplant candidates (Kapczinski 1995). The objective of the trial was to evaluate the differential effects of flumazenil versus placebo on cognitive function and anxiety in 10 people with alcohol‐related cirrhosis, 10 people with non‐alcohol‐related cirrhosis, and 10 healthy volunteers. None of the included participants had evidence of overt hepatic encephalopathy. The investigators evaluated a range of psychometric tests and reported the results as group mean values. The trial report did not provide information about the number of participants with abnormal test results, but stated that participants with cirrhosis performed worse than people in the control group on several tests including verbal recall, and on reaction time tasks. Treatment with flumazenil had no effect on the test results, but induced anxiety in the participants with non‐alcoholic cirrhosis.
Two prospective non‐randomised observational studies involved 47 participants with cirrhosis and overt hepatic encephalopathy (Marsepoil 1990; Jia 1999). The first study included 25 participants with alcohol‐related cirrhosis and acute hepatic encephalopathy, 13 of whom received flumazenil in a dose of 0.2 mg intravenously every 10 minutes until clinical improvement up to a maximum total dose of 2 mg followed by a continuous maintenance infusion of 0.3 mg. per hour for 48 hours.(Marsepoil 1990). The second study included 22 participants with cirrhosis and overt hepatic encephalopathy (Jia 1999), 12 of whom received flumazenil while the remaining 10 received a traditional Chinese medicine, both infused intravenously. The trial report stated that two participants in the flumazenil group died of liver failure. The report did not mention deaths in the control group.
A randomised clinical trial involving 20 participants with cirrhosis and overt hepatic encephalopathy, 12 of whom were treated with repeated bolus injections of flumazenil combined with lactulose administered as a retention enema while a further eight participants received flumazenil alone (Wu 2001). The study authors defined improvement of hepatic encephalopathy as a three‐point or greater reduction in the Conn Score within six hours after the administration of the interventions. Seven (58%) of the 12 participants in the flumazenil plus lactulose group and 4 (50%) of the eight participants in the flumazenil alone group showed improvement. There were no reported deaths or adverse events.
We excluded five additional observational studies, involving 59 participants, 12 (20%) of whom were children (Grimm 1988; Bansky 1989; Devictor 1995; Ozyilkan 1997; Golubovic 1999). Two studies included participants with fulminant hepatic failure (Grimm 1988; Devictor 1995), One study, involving 14 participants with cirrhosis and overt hepatic encephalopathy reported an improvement in mental status in 71% of participants within minutes of receiving flumazenil which lasted for one to two hours; six participants died (Bansky 1989). Another study, including 10 participants with cirrhosis and severe hepatic encephalopathy reported transient improvements in the manifestations of hepatic encephalopathy in 80% of the included participants; six died within one year (Golubovic 1999). A third study evaluated the effects of incremental intravenous boluses of flumazenil in 11 participants with cirrhosis in whom baseline somatosensory evoked potentials were abnormal; four (36%) showed a improvement in evoked potentials with flumazenil (Ozyilkan 1997). One prospective study, involving 15 adults and two children, reported transient improvement in hepatic encephalopathy, after administration of flumazenil, in four (44%) of nine participants with fulminant hepatic failure and five (63%) of eight with cirrhosis (Grimm 1988). In a study undertaken exclusively in children with fulminant liver failure, awaiting emergency liver transplantation, flumazenil had a transient beneficial effect on arousal in one child (Devictor 1995).
Risk of bias in included studies
We assessed the risk of bias based on published information and on additional information from the trial investigators (Figure 3).
Figure 3.
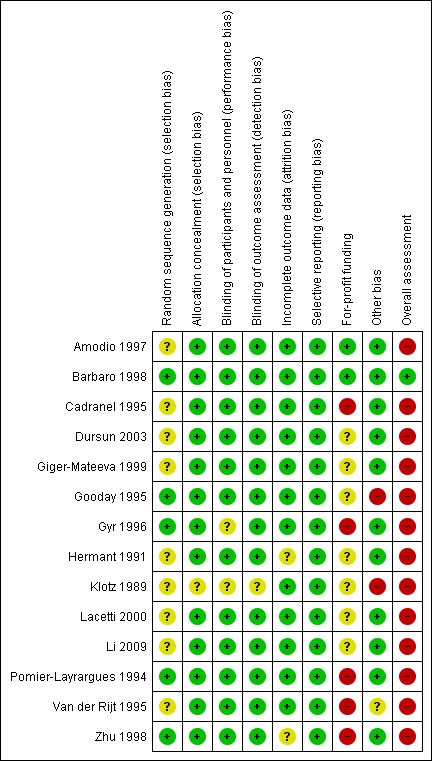
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
We classified 4 randomised clinical trials with adequate allocation sequence generation and allocation concealment at low risk of selection bias (Gooday 1995; Gyr 1996; Barbaro 1998; Zhu 1998). The remaining 10 trials used an adequate method to conceal the allocation, but they did not describe the allocation sequence generation (Klotz 1989; Hermant 1991; Pomier‐Layrargues 1994; Cadranel 1995; Van der Rijt 1995; Amodio 1997; Giger‐Mateeva 1999; Lacetti 2000; Dursun 2003; Li 2009).
Blinding
We classified all randomised clinical trials as having a low risk of performance and detection bias as they were double‐blind and placebo‐controlled with blinding of participants, personnel, and outcome assessors (Klotz 1989; Hermant 1991; Pomier‐Layrargues 1994; Cadranel 1995; Gooday 1995; Van der Rijt 1995; Gyr 1996; Amodio 1997; Barbaro 1998; Zhu 1998; Giger‐Mateeva 1999; Lacetti 2000; Dursun 2003; Li 2009).
Incomplete outcome data
Two randomised clinical trials gave the impression that there were no missing outcome data although this was not specifically stated (Hermant 1991; Zhu 1998). The remaining trials had no missing outcome data and included all participants in the reported analyses (Klotz 1989; Pomier‐Layrargues 1994; Cadranel 1995; Gooday 1995; Van der Rijt 1995; Gyr 1996; Amodio 1997; Barbaro 1998; Giger‐Mateeva 1999; Lacetti 2000; Dursun 2003; Li 2009).
Selective reporting
We did not have access to protocols and were, therefore, unable to evaluate any potential differences between outcomes described in protocols compared with trial publications. All randomised clinical trials included a description of the outcomes all‐cause mortality and hepatic encephalopathy (Klotz 1989; Hermant 1991; Pomier‐Layrargues 1994; Cadranel 1995; Gooday 1995; Van der Rijt 1995; Gyr 1996; Amodio 1997; Barbaro 1998; Zhu 1998; Giger‐Mateeva 1999; Lacetti 2000; Dursun 2003; Li 2009). Therefore, we classified all trials at low risk of reporting bias.
For‐profit funding
Pharmaceutical companies provided financial or other support for 5 of the randomised clinical trials (Pomier‐Layrargues 1994; Van der Rijt 1995; Cadranel 1995; Gyr 1996; Zhu 1998). Seven trials did not provide information about funding (Klotz 1989; Hermant 1991; Gooday 1995; Giger‐Mateeva 1999; Lacetti 2000; Dursun 2003; Li 2009). The remaining 2 trials did not receive support from pharmaceutical companies (Amodio 1997; Barbaro 1998).
Other potential sources of bias
One randomised clinical trial simplified the intervention regimen and assessment of outcomes after the inclusion of 9 of 18 participants (Van der Rijt 1995). We classified this trial at unclear risk of other bias and the remaining trials at low risk of bias (Klotz 1989; Hermant 1991; Pomier‐Layrargues 1994; Cadranel 1995; Gooday 1995; Gyr 1996; Amodio 1997; Barbaro 1998; Zhu 1998; Giger‐Mateeva 1999; Lacetti 2000; Dursun 2003).
Overall risk of bias
We classified one randomised clinical trial at low risk of bias for all domains (Barbaro 1998), and the remaining trials at high risk of bias (Klotz 1989; Hermant 1991; Pomier‐Layrargues 1994; Cadranel 1995; Gooday 1995; Van der Rijt 1995; Gyr 1996; Amodio 1997; Zhu 1998; Giger‐Mateeva 1999; Lacetti 2000; Dursun 2003; Li 2009).
Effects of interventions
See: Table 1
The total number of participants was 867. Three cross‐over randomised clinical trials did not report outcomes for the first intervention period (Gooday 1995; Amodio 1997; Giger‐Mateeva 1999);we received information about the number of participants and all‐cause mortality rates during the first allocation period for one of these trials (Gooday 1995), but not for the remaining two (Amodio 1997; Giger‐Mateeva 1999). We were able to gather data from the first allocation period for the 5 remaining cross‐over trials and required data from all of the parallel‐arm trials. Accordingly, our analyses included 842 participants.
Primary outcomes
All‐cause mortality
In total, 32/433 participants allocated to flumazenil versus 38/409 participants allocated to placebo died (RR 0.75, 95% CI 0.48 to 1.16; 11 trials; I2 = 0%; Analysis 1.1). The trial classified as low risk of bias found no beneficial or detrimental effect of flumazenil on all‐cause mortality (RR 0.76, 95% CI 0.37 to 1.53; Analysis 1.2). There was no evidence of small‐study effects (P = 0.31). The Trial Sequential Analyses ignored three randomised clinical trials due to insufficient information indicating that we have insufficient evidence to support or refute an effect of flumazenil on all‐cause mortality (Cadranel 1995; Gyr 1996; Zhu 1998; Figure 4). The trials including participants with minimal hepatic encephalopathy did not report any deaths (Gooday 1995; Dursun 2003; Analysis 1.1). There were no differences between trials involving participants with cirrhosis compared with trials involving participants with cirrhosis or fulminant hepatic failure (Analysis 1.11). Additional analyses showed no differences between trials using a cross‐over or a parallel‐arm design (Analysis 1.3); or between trials with short‐term or long‐term (> 1 day) follow‐up (Analysis 1.4).
Analysis 1.1.
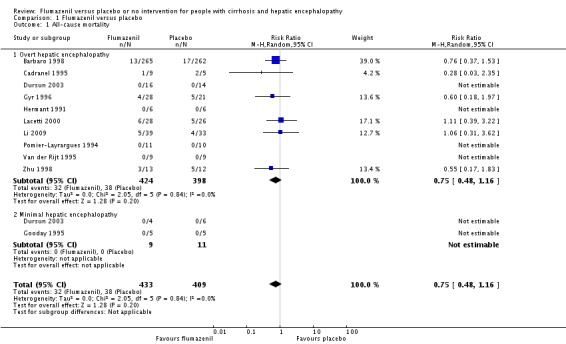
Comparison 1 Flumazenil versus placebo, Outcome 1 All‐cause mortality.
Analysis 1.2.
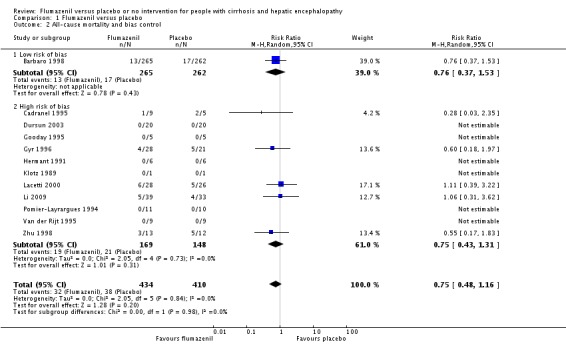
Comparison 1 Flumazenil versus placebo, Outcome 2 All‐cause mortality and bias control.
Figure 4.
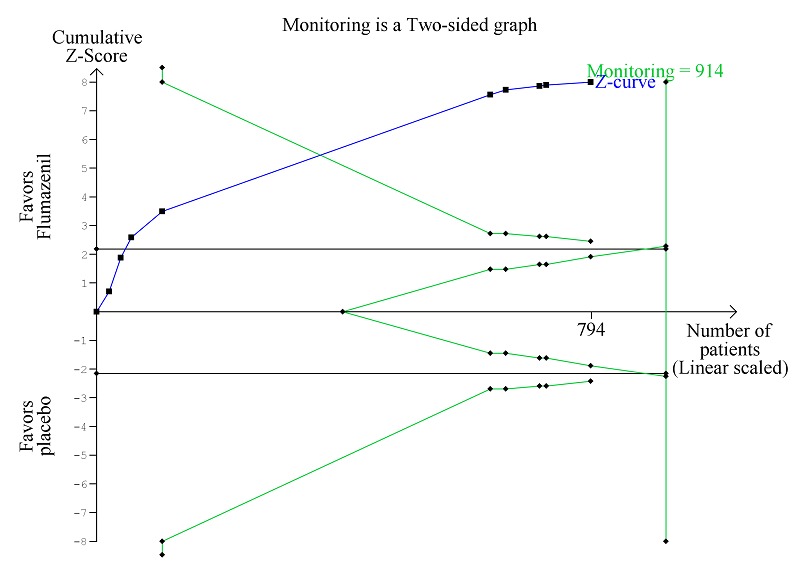
Trial Sequential Analysis of randomised clinical trials evaluating flumazenil versus placebo for people with cirrhosis and hepatic encephalopathy. The outcome is hepatic encephalopathy. The original meta‐analysis included 11 randomised clinical trials with 824 participants. The Trial Sequential Analysis is made with alpha 3%, power 90%, relative risk reduction 20%, assumed control risk 60%, and diversity 10%. The blue line (Z‐curve) corresponds to the cumulative meta‐analysis, the black horizontal line is the conventional boundary (3% level of significance), and the inward sloping green line is the Trial Sequential Monitoring Boundary. The analysis found that the Z‐curve crossed the monitoring boundary before reaching the diversity‐adjusted required information size of 914 participants.
Analysis 1.11.
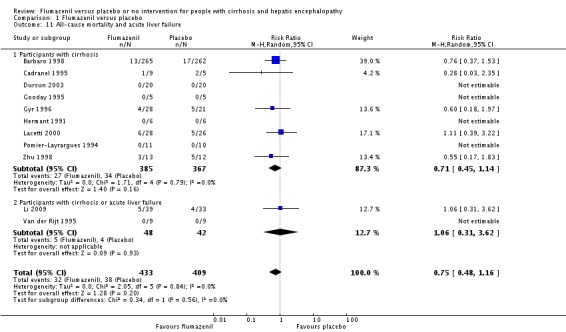
Comparison 1 Flumazenil versus placebo, Outcome 11 All‐cause mortality and acute liver failure.
Analysis 1.3.
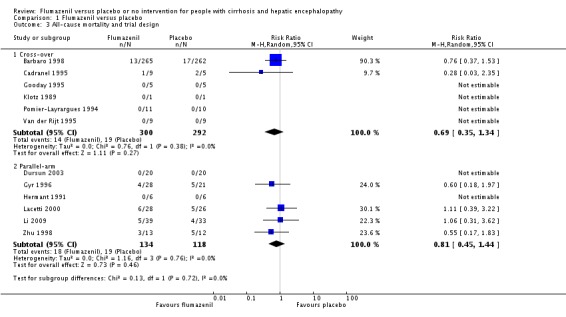
Comparison 1 Flumazenil versus placebo, Outcome 3 All‐cause mortality and trial design.
Analysis 1.4.
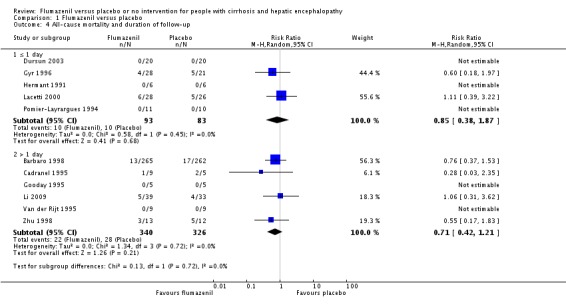
Comparison 1 Flumazenil versus placebo, Outcome 4 All‐cause mortality and duration of follow‐up.
Hepatic encephalopathy
Analysis of the data on 824 participants involved in nine randomised clinical trials showed that flumazenil was associated with a beneficial effect on hepatic encephalopathy (RR 0.75, 95% CI 0.71 to 0.80; I2 = 0%; Analysis 1.5). The analysis only included 10 participants with minimal hepatic encephalopathy. The trial classified as having a low risk of bias found a beneficial effect of flumazenil on hepatic encephalopathy (RR 0.78, 95% CI 0.72 to 0.84; Analysis 1.6). In the Trial Sequential Analysis, the Z‐curve crossed the monitoring boundary (see Figure 4 for additional information). The analysis found that the diversity adjusted information size was 914 participants. The information size was 1028 participants when we increase the diversity to 20% (the model based diversity was 0%). The subgroup analyses showed no differences between subgroups of trials including participants with cirrhosis or fulminant hepatic failure (Analysis 1.9), or trials stratified by their design (Analysis 1.7), or their duration of follow‐up (Analysis 1.8).
Analysis 1.5.
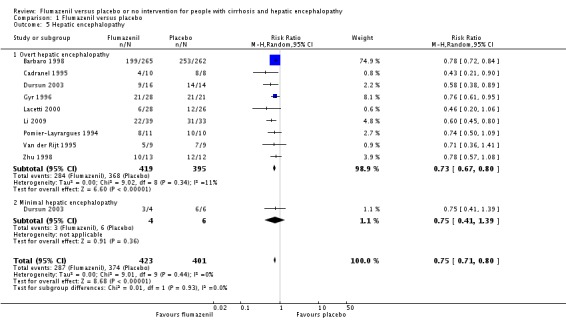
Comparison 1 Flumazenil versus placebo, Outcome 5 Hepatic encephalopathy.
Analysis 1.6.
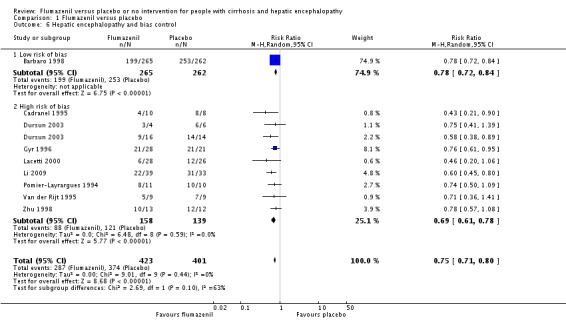
Comparison 1 Flumazenil versus placebo, Outcome 6 Hepatic encephalopathy and bias control.
Analysis 1.9.
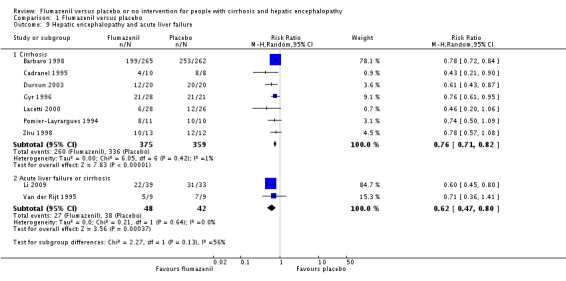
Comparison 1 Flumazenil versus placebo, Outcome 9 Hepatic encephalopathy and acute liver failure.
Analysis 1.7.
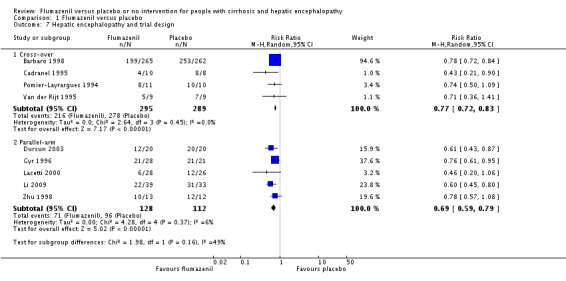
Comparison 1 Flumazenil versus placebo, Outcome 7 Hepatic encephalopathy and trial design.
Analysis 1.8.
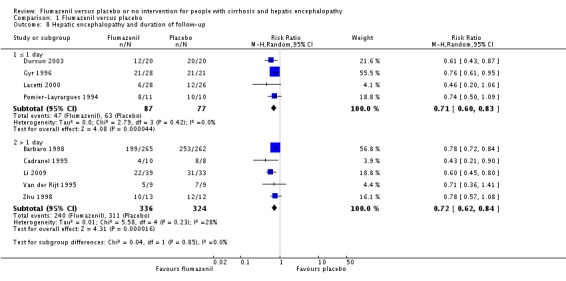
Comparison 1 Flumazenil versus placebo, Outcome 8 Hepatic encephalopathy and duration of follow‐up.
Serious adverse events
We were only able to conduct analyses for fatal serious adverse event (Table 8). In the largest randomised clinical trial (Barbaro 1998), 13 nonresponders in the flumazenil group and 17 nonresponders in the placebo group died within 3 to 4 days (range 2 to 6). The causes of death were septic shock (n = 20); hypovolaemic shock (n = 8) and lactic acidosis (n = 2), but information was not provided on the number of deaths by cause in each group. In a smaller randomised clinical trial, 4 of 28 participants allocated to flumazenil and 5 of 21 participants allocated to placebo died within 4 weeks of the trial (Gyr 1996). One participant in the placebo group died with respiratory failure during the course of the trial, but the causes of death in the remaining eight participants were not provided. Three trials reported all‐cause mortality in the flumazenil and control groups without providing information about the cause of death (Zhu 1998; Lacetti 2000; Li 2009). None of the included participants experienced seizures.
Table 6.
Serious adverse events
| Trial | Number of participants | Included in analyses of serious adverse events | Data included in primary analysis | Serious adverse events |
| Amodio 1997 | 13 | No | Cross‐over RCT. Data from the first treatment period not described. | Publication does not describe any deaths or other serious adverse events. |
| Barbaro 1998 | 527 | Yes | Cross‐over RCT. Data from the first treatment period included. | Thirteen non‐responders in the flumazenil group and 17 non responders in the placebo group died 3 to 4 days (range 2‐6) after randomisation. The causes of dead were septic shock (20 participants); hypovolaemic shock (8 participants) and lactic acidosis (2 participants) but information was not provided on the number of deaths by cause in each group. |
| Cadranel 1995 | 14 | Yes | Cross‐over RCT. Data from the first treatment period included. | One of 12 responders died from septic shock on day 4 and 2 of 6 non‐responders died from septic shock (day 2) and lactic acidosis (day 4) but information is not provided on the groups to which they a were allocated. |
| Dursun 2003 | 40 | Yes | Parallel‐arm RCT. We included all participants in the analyses. | Publication did not describe any deaths or other serious adverse events. |
| Giger‐Mateeva 1999 | 10 | No | Cross‐over RCT. Data from the first treatment period not described. | Publication did not describe any deaths or other serious adverse events. |
| Gooday 1995 | 10 | Yes | Cross‐over RCT. Data from the first treatment period included. | Publication did not describe any deaths or other serious adverse events. |
| Gyr 1996 | 49 | Yes | Parallel‐arm RCT. We included all participants in the analyses. | Four of 28 participants allocated to flumazenil and 5 of 21 allocated to placebo died within 4 weeks of the trial. One participant in the placebo group died with respiratory failure during the course of the study. The authors described participants as having severe liver disease suggesting that the cause of death in the remaining 8 participants may have been cirrhosis‐related although this is not specifically stated. The investigators classified the remaining adverse events viz flushing, nausea, vomiting, and irritability, which were experienced by 4 participants, as non‐serious. |
| Hermant 1991 | 12 | Yes | Parallel‐arm RCT. We included all participants in the analyses. | Publication did not describe any deaths or other serious adverse events. |
| Klotz 1989 | 2 | No | Cross‐over RCT. Data from the first treatment period were not described. | Publication does not describe any deaths or other serious adverse events. |
| Lacetti 2000 | 54 | Yes | Parallel‐arm RCT. We include all participants in the analyses. | Six of 28 participants in the flumazenil group and 5 of 26 in the control group died. The causes of death were not provided. |
| Li 2009 | 72 | Yes | Parallel‐arm RCT. The included participants had hepatic encephalopathy associated with cirrhosis or acute liver failure. Data were not provide separately for the 2 groups. | Five of 39 participants in the flumazenil group and 4 of 33 participants in the control group died. The causes of death were not provided. |
| Pomier‐Layrargues 1994 | 21 | Yes | Cross‐over RCT. Data from the first treatment period were included. | Publication did not describe any deaths or other serious adverse events. |
| Van der Rijt 1995 | 18 | Yes | Cross‐over RCT. The included participants had hepatic encephalopathy associated with cirrhosis or acute liver failure. Data were not provide separately for the 2 groups.Data from the first treatment period were included. | Publication did not describe any deaths or other serious adverse events. Two participants with fulminant hepatic failure underwent orthotopic liver transplantation on day one of the study |
| Zhu 1998 | 25 | Yes | Parallel‐arm RCT. We included all participants in the analyses. | Three of 13 participants in the flumazenil group and 5 of 12 participants in the control group died. The causes of death were not provided |
RCT: randomised clinical trial.
Secondary outcome measures
None of the included trials assessed health‐related quality of life.
We were unable to conduct a meta‐analysis of non‐serious adverse events. Four randomised clinical trials reported that none of the included participants experienced non‐serious adverse events (Hermant 1991; Barbaro 1998; Lacetti 2000; Dursun 2003). In one trial, four participants in the flumazenil group experienced nausea, vomiting, flushing, or irritability (Gyr 1996); the total number of participants with the individual adverse events was not described. Two participants in one trial had transient palpitations, but the intervention group was not specified (Zhu 1998). One cross‐over trials reported that one in 10 participants felt drowsy, possibly after flumazenil infusion (Giger‐Mateeva 1999).
Exploratory outcomes
We were able to include Number Connection Test results from one randomised clinical trial with 40 participants. The trial found a MD of ‐3.79 seconds (95% CI ‐32.14 to 24.56; Analysis 1.10).
Analysis 1.10.
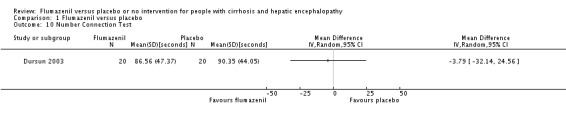
Comparison 1 Flumazenil versus placebo, Outcome 10 Number Connection Test.
'Summary of findings' table
As shown in the Table 1, we downgraded the strength of the evidence to low based on methodological concerns.
Discussion
Summary of main results
This review included 14 randomised clinical trials published between 1989 and 2009. The primary meta‐analyses showed no beneficial or detrimental effect of flumazenil on all‐cause mortality, but it showed a potential short‐term beneficial effect of flumazenil on the manifestations of hepatic encephalopathy. We found little evidence suggesting that flumazenil was associated with serious adverse effects, However, the reporting of serious and non‐serious adverse events was generally incomplete or unclear, and our analyses of adverse events may be subject to outcome reporting bias. Based on methodological concerns, we classified the strength of the evidence as low. Therefore, our review remains inconclusive.
Overall completeness and applicability of evidence
Flumazenil is a short‐acting specific benzodiazepine antagonist which acts by inhibiting activity at the benzodiazepine recognition site on the GABA/benzodiazepine receptor complex. Its oral bioavailability is poor and hence intravenous administration is necessary (Brogden 1991). The main indication for flumazenil is the reversal of benzodiazepine overdose or prolonged anaesthesia. Based on pharmacological studies, the onset of its effect is rapid and its duration of action is short (Brogden 1991). In healthy people, the half‐life is 50 minutes. Thus, repeated low intravenous doses or continuous infusion is needed if the clinical situation requires a longer lasting effect (Hood 2014). Hepatic encephalopathy is characterised by an increase in GABA‐A tone which is the rationale for use of flumazenil in this condition.
The most important outcomes for people with cirrhosis and hepatic encephalopathy include mortality, morbidity, adverse events, and health‐related quality of life (Bajaj 2011). We found no detrimental or beneficial effect on all‐cause mortality or adverse events but a potential short‐term effect on the manifestations of hepatic encephalopathy. There was no reported information on health‐related quality of life. However, the applicability of the evidence will be limited because of the need for intravenous administration. Most trials evaluated single or repeated bolus injections (Klotz 1989; Hermant 1991; Pomier‐Layrargues 1994; Cadranel 1995; Gooday 1995; Amodio 1997; Barbaro 1998; Zhu 1998; Giger‐Mateeva 1999; Lacetti 2000). One trial assessed the effect of a 72‐hour infusion of flumazenil (Van der Rijt 1995), but the investigators changed the intervention to bolus injections because participants found the three‐day infusion too stressful. None of the trials compared different doses or different modes of administration. We found no effect of the dose of flumazenil on the estimated effect on all‐cause mortality or hepatic encephalopathy. However, based on the limited number of events and trials, important clinical differences may have been overlooked. The half‐life of flumazenil is prolonged up to 2.4 hours in people with moderate to severe hepatic decompensation, hence providing some prolongation of action (Amrein 1990). However, we found no further or additional effects of flumazenil when analysing trials with more than 24‐hour follow‐up.
The majority of the included randomised clinical trials enrolled participants with cirrhosis and an acute episode of hepatic encephalopathy although the severity varied between trials. Episodes of hepatic encephalopathy often develop in response to a precipitating event such as gastrointestinal bleeding, which was the most common precipitant identified in the included trials in this review. Identification and treatment of precipitating factors is key to the management of affected people (EASL/AASLD guideline 2014a; EASL/AASLD guideline 2014b). We did not have sufficient data to assess potential difference between precipitated and non‐precipitated hepatic encephalopathy. None of the included participants had surgically created or transjugular intrahepatic portosystemic shunts. Likewise, the review contained very little information about people with recurrent or persistent (chronic) hepatic encephalopathy. Two trials included a small number of participants with fulminant hepatic failure (Van der Rijt 1995; Li 2009). This condition is infrequent in clinical practice. We were unable to gather data that allowed us to evaluate any differential effects of flumazenil on hepatic encephalopathy associated with acute or chronic liver failure. However, subgroup analyses based on aggregated data found no difference between trials including or not including participants with fulminant hepatic failure. Nevertheless, it is likely that the pathophysiology of hepatic encephalopathy in participants with acute liver failure differs from that associated with cirrhosis. Three trials enrolled people with minimal hepatic encephalopathy (Gooday 1995; Amodio 1997; Giger‐Mateeva 1999), while a further trial included participants with both minimal and low‐grade acute hepatic encephalopathy (Dursun 2003). In these four trials, the objective appears to be more mechanistic than therapeutic, but based on our subgroup analysis, we found no difference in outcomes between overt and minimal hepatic encephalopathy. However, the diagnostic end points for hepatic encephalopathy in these trials were very different, namely clinical assessment versus psychometry, and the number of trials was small so that statistical differences may be overlooked.
Prior intake of benzodiazepines may influence the effects of flumazenil. Ten randomised clinical trial stipulated prior ingestion of benzodiazepines as an exclusion criterion (Hermant 1991; Pomier‐Layrargues 1994; Gooday 1995; Van der Rijt 1995; Amodio 1997; Barbaro 1998; Zhu 1998; Giger‐Mateeva 1999; Lacetti 2000; Dursun 2003), while nine trials undertook baseline screening for benzodiazepines (Hermant 1991; Pomier‐Layrargues 1994; Cadranel 1995; Van der Rijt 1995; Gyr 1996; Amodio 1997; Barbaro 1998; Giger‐Mateeva 1999; Lacetti 2000). Three randomised clinical trials stipulated negative testing for benzodiazepines as an inclusion criterion (Hermant 1991; Van der Rijt 1995; Lacetti 2000). In the remaining six trials, in which baseline testing was undertaken, it was unclear whether the results were available at the time of randomisation or whether participants were included irrespective of the results; in two of these trials, none of the participants tested positive (Amodio 1997; Giger‐Mateeva 1999), while in the remaining four trials, between 1.9% and 21.4% of participants tested positive (Pomier‐Layrargues 1994; Cadranel 1995; Gyr 1996; Barbaro 1998). Thus, the exclusion of participants based on reports of non‐ingestion of benzodiazepines is clearly unreliable. In all four of these trials, the assessment of outcomes in relation to the presence/absence of benzodiazepines were related to the response to flumazenil rather than to intervention allocation (Pomier‐Layrargues 1994; Cadranel 1995; Gyr 1996; Barbaro 1998). All four showed that the majority of participants who responded to flumazenil did not have detectable circulating benzodiazepines whereas some non‐responders had measurable quantities of these substances in their blood. The trials concluded that the presence of benzodiazepines was not predictive of participants' responses to flumazenil. We did not find this information in 5 trials, which should be considered when evaluating their results (Klotz 1989; Gooday 1995; Zhu 1998; Dursun 2003; Li 2009).
Administration of flumazenil by sublingual lozenge and topical cream has also been tested, albeit very selectively (Rye 2012); the preparations are not generally available nor applicable for use in most clinical settings and is not tested in clinical trials evaluating participants with hepatic encephalopathy.
On the basis of this review, the use of flumazenil in the management of hepatic encephalopathy would be limited. It might be of value in people with severe hepatic encephalopathy not responding to usual management to facilitate procedures or to allow assessment of cognitive status in potential transplant candidates if other conditions, such as hypoxic injury, are suspected.
Quality of the evidence
The review is limited because of methodological and statistical issues relating to the included randomised clinical trials. Seven of the 14 included trial used a cross‐over design (Pomier‐Layrargues 1994; Cadranel 1995; Gooday 1995; Van der Rijt 1995; Amodio 1997; Barbaro 1998; Giger‐Mateeva 1999). While this design is suitable for evaluating interventions which are predicted to have a temporary effect on chronic stable conditions, it is not suitable for evaluating interventions which have a short‐term effect in unstable conditions (Rosenkranz 2015). The majority of trials in this review included participants with an acute episode of hepatic encephalopathy, and thus, a cross‐over design is not appropriate. The problem was further compounded since some of the trials used a modified design with cross‐over of only the non‐responders to the second treatment period. Therefore, inclusion of both periods in our analyses would introduce potential bias and would have posed statistical problems. We included data from the first period of the seven cross‐over trials, obtaining the data which correspond to those available from the more suitable parallel‐armed randomised clinical trials. The drawback of the strategy is loss of information from the second period. A further problem with the cross‐over design is that it precludes an assessment of risk of relapse.
Only one trial was at low risk of bias in the overall assessment (Barbaro 1998). The trial included the largest number of participants and had a weight of 39% in the analysis of mortality and 56% in the analysis of hepatic encephalopathy. We found no difference between this trial and the remaining trials with a high risk of bias for the two outcomes based on the test for subgroup differences; exclusion of the trial did not change the overall conclusion. We found no evidence of publication bias or other small‐study effects and between‐trial heterogeneity was negligible. Nevertheless, the CIs were wide. Although these findings support the quality of the evidence, we still have potential problems with the use of the cross‐over design. We classified the quality of the evidence as low.
While most trials reported mortality and hepatic encephalopathy, the quality of the reporting of both non‐fatal serious and non‐serious adverse events was low. We looked for additional information about harms in observational studies, but were unable to retrieve and analyse adverse events in the studies identified.
Potential biases in the review process
We attempted to minimise possible selection biases by using a comprehensive search strategy. Searches in electronic databases were combined with extensive handsearches. In addition, we also searched conference proceedings and abstract books. We think it likely that we have not missed published trials, but we cannot exclude the possibility that we have missed unpublished trials. The intervention is not high‐profile and it is possible that negative trials, particularly if small, will not have appeared as abstracts at conferences or been published in full. However, our meta‐regression analyses showed no evidence of publication bias or other dissemination biases.
Agreements and disagreements with other studies or reviews
One meta‐analysis undertaken in 2002 (Goulenok 2002) included 6 randomised clinical trials with 641 participants (Pomier‐Layrargues 1994; Cadranel 1995; Van der Rijt 1995; Groeneweg 1996; Gyr 1996; Barbaro 1998). The mean percentages of people with clinical improvement (5 trials) were 27% in treated groups and 3% in placebo groups. We believe that two of these involve the same population of participants (Groeneweg 1996; Gyr 1996). The first trial included 49 participants with mild to moderate (Grade I to III) hepatic encephalopathy (Gyr 1996), while the second trial, which is described by the authors as an ancillary study, reported electroencephalography data from 32 of the original 49 participants (Groeneweg 1996). We excluded the second trial from this review.
The previous version of this review included 13 randomised clinical trials with 805 participants (Als‐Nielsen 2004). This earlier review found no effect of flumazenil on all‐cause mortality based on an analysis of 10 trials and a beneficial effect on hepatic encephalopathy based on an analysis of 8 trials with a risk difference of 0.28 (95% CI 0.20 to 0.37). We have updated the review by inclusion of two additional randomised clinical trials in the analyses of benefits, and the addition of one previously included randomised clinical trial and two observational studies in our assessment of harms. Our analysis of all‐cause mortality included 13 trials and the analysis of hepatic encephalopathy included 9 trials. In agreement with the previous review, we found no effect on all‐cause mortality and a beneficial effect on hepatic encephalopathy.
Authors' conclusions
This review includes randomised clinical trials evaluating the treatment of hepatic encephalopathy. The analyses found some evidence that flumazenil may be associated with a short‐term effect on hepatic encephalopathy, but no beneficial effect on important clinical outcomes such as all‐cause mortality, serious adverse events, or health‐related quality of life. Likewise, we are unable to determine the risk of non‐fatal serious or non‐serious adverse events based on the available evidence.
We used the EPICOT format to define the implications of our review for research (Brown 2006). Overall, the evidence is insufficient and additional evidence from randomised clinical trials is needed to evaluate whether flumazenil has a clinically relevant effect on hepatic encephalopathy.
Evidence (what is the current state of the evidence?): this review includes 14 randomised clinical trials and found low quality evidence that flumazenil may have a beneficial short‐term effect on hepatic encephalopathy. The evidence concerning all‐cause mortality, non‐fatal serious adverse events, and non‐serious adverse events is insufficient.
Participants (what is the population of interest?): the largest body of evidence evaluated people with cirrhosis and an acute episode of overt hepatic encephalopathy. Only a relatively small proportion had minimal hepatic encephalopathy and chronic overt hepatic encephalopathy; very few had acute liver failure.
Interventions (what are the interventions of interest?): flumazenil.
Comparisons (what are the comparisons of interest?): placebo‐controlled randomised clinical trials.
Outcomes (what are the outcomes of interest?): all‐cause mortality, hepatic encephalopathy, and adverse events; evidence evaluating the effect on health‐related quality of life is also needed.
Time stamp (date of literature search): May 2017.
Acknowledgements
We thank Dr Guiseppe Barbaro and Dr Ronan O'Carroll for providing us with more detailed information on the randomised clinical trials they conducted; Roche Pharma for supplying us with literature from their internal database on flumazenil; Dr Jianping Liu for translating a Chinese publication; and Sarah Klingenberg for performing the electronic literature searches. We also thank Dr Bodil Als‐Nielsen and Dr Christian Gluud who participated in the previous version of this review but were unable to participate in the update due to other commitments.
Cochrane Review Group funding acknowledgement: the Danish State is the largest single funder of The Cochrane Hepato‐Biliary Group through its investment in The Copenhagen Trial Unit, Centre for Clinical Intervention Research, Rigshospitalet, Copenhagen University Hospital, Denmark. Disclaimer: the views and opinions expressed in this review are those of the authors and do not necessarily reflect those of the Danish State or The Copenhagen Trial Unit.
Appendices
Appendix 1. Search strategies
| Database | Time span | Search strategy |
| Cochrane Hepato‐Biliary Group Controlled Trials Register | May 2017. | (((benzodiazepine receptor OR GABA) AND (antagonist* OR blocking agent*)) OR flumazenil OR flumazepil) |
| Cochrane Central Register of Controlled Trials (CENTRAL) in the Cochrane Library | 2017, Issue 2. | #1 MeSH descriptor: [GABA Antagonists] explode all trees #2 MeSH descriptor: [Flumazenil] explode all trees #3 ((benzodiazepine receptor or GABA) and (antagonist* or blocking agent*)) or flumaze*il #4 #1 or #2 or #3 #5 MeSH descriptor: [Liver Cirrhosis] explode all trees #6 MeSH descriptor: [Hepatic Encephalopathy] explode all trees #7 liver cirrhosis or hepatic encephalopathy #8 #5 or #6 or #7 #9 #4 and #8 |
| MEDLINE Ovid | 1946 to May 2017. | 1. exp GABA Antagonists/ 2. exp Flumazenil/ 3. (((benzodiazepine receptor or GABA) and (antagonist* or blocking agent*)) or flumaze*il).mp. [mp=protocol supplementary concept, rare disease supplementary concept, title, original title, abstract, name of substance word, subject heading word, unique identifier] 4. 1 or 2 or 3 5. exp Liver Cirrhosis/ 6. exp Hepatic Encephalopathy/ 7. (liver cirrhosis or hepatic encephalopathy).mp. [mp=protocol supplementary concept, rare disease supplementary concept, title, original title, abstract, name of substance word, subject heading word, unique identifier] 8. 5 or 6 or 7 9. 4 and 8 10. (random* or blind* or placebo* or meta‐analysis).mp. [mp=protocol supplementary concept, rare disease supplementary concept, title, original title, abstract, name of substance word, subject heading word, unique identifier] 11. 9 and 10 |
| Embase Ovid | 1974 to May 2017. | 1. exp benzodiazepine receptor blocking agent/ 2. exp 4 aminobutyric acid receptor blocking agent/ 3. exp FLUMAZENIL/ 4. (((benzodiazepine receptor or GABA) and (antagonist* or blocking agent*)) or flumaze*il).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer] 5. 1 or 2 or 3 or 4 6. exp liver cirrhosis/ 7. exp hepatic encephalopathy/ 8. (liver cirrhosis or hepatic encephalopathy).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer] 9. 6 or 7 or 8 10. 5 and 9 11. (random* or blind* or placebo* or meta‐analysis).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer] 12. 10 and 11 |
| Science Citation Index Expanded (Web of Science) | 1900 to May 2017. | #5 #4 AND #3 #4 TS=(random* or blind* or placebo* or meta‐analysis) #3 #2 AND #1 #2 TS=(liver cirrhosis or hepatic encephalopathy) #1 TS=(((benzodiazepine receptor or GABA) and (antagonist* or blocking agent*)) or flumaze*il) |
| LILACS (Bireme) | 1982 to May 2017. | (benzodiazepine receptor or GABA) and (antagonist$ or blocking agent$)) or flumaze$ [Words] and hepatic encephalopath$ [Words] |
Data and analyses
Comparison 1.
Flumazenil versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 All‐cause mortality | 11 | 842 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.48, 1.16] |
| 1.1 Overt hepatic encephalopathy | 10 | 822 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.48, 1.16] |
| 1.2 Minimal hepatic encephalopathy | 2 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2 All‐cause mortality and bias control | 12 | 844 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.48, 1.16] |
| 2.1 Low risk of bias | 1 | 527 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.37, 1.53] |
| 2.2 High risk of bias | 11 | 317 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.43, 1.31] |
| 3 All‐cause mortality and trial design | 12 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Cross‐over | 6 | 592 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.35, 1.34] |
| 3.2 Parallel‐arm | 6 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.45, 1.44] |
| 4 All‐cause mortality and duration of follow‐up | 11 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 ≤ 1 day | 5 | 176 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.38, 1.87] |
| 4.2 > 1 day | 6 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.42, 1.21] |
| 5 Hepatic encephalopathy | 9 | 824 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.71, 0.80] |
| 5.1 Overt hepatic encephalopathy | 9 | 814 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.67, 0.80] |
| 5.2 Minimal hepatic encephalopathy | 1 | 10 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.41, 1.39] |
| 6 Hepatic encephalopathy and bias control | 9 | 824 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.71, 0.80] |
| 6.1 Low risk of bias | 1 | 527 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.72, 0.84] |
| 6.2 High risk of bias | 8 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.61, 0.78] |
| 7 Hepatic encephalopathy and trial design | 9 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 Cross‐over | 4 | 584 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.72, 0.83] |
| 7.2 Parallel‐arm | 5 | 240 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.59, 0.79] |
| 8 Hepatic encephalopathy and duration of follow‐up | 9 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 ≤ 1 day | 4 | 164 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.60, 0.83] |
| 8.2 > 1 day | 5 | 660 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.62, 0.84] |
| 9 Hepatic encephalopathy and acute liver failure | 9 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 Cirrhosis | 7 | 734 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.71, 0.82] |
| 9.2 Acute liver failure or cirrhosis | 2 | 90 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.47, 0.80] |
| 10 Number Connection Test | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 11 All‐cause mortality and acute liver failure | 11 | 842 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.48, 1.16] |
| 11.1 Participants with cirrhosis | 9 | 752 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.45, 1.14] |
| 11.2 Participants with cirrhosis or acute liver failure | 2 | 90 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.31, 3.62] |
What's new
Last assessed as up‐to‐date: 5 May 2017.
| Date | Event | Description |
|---|---|---|
| 4 May 2017 | New search has been performed | Searches updated. |
| 14 October 2016 | New search has been performed | The review methods and analyses are revised based on the recent recommendations of the Cochrane Hepato‐Biliary Group, the MECIR guidelines, and the Cochrane Handbook for Reviews of Interventions. |
| 14 October 2016 | New search has been performed | Title change. Previously, the review was published with the title "Benzodiazepine receptor antagonists for hepatic encephalopathy". |
| 9 May 2016 | New citation required but conclusions have not changed | We included two additional randomised clinical trials (RCTs) in the analyses of benefits and harms and excluded one RCT from the analyses of benefits. The latter RCT and two additional observational studies are included in analyses of harms. |
Differences between protocol and review
The review has been extensively revised compared to the original protocol and the previously published version of this current review (Als‐Nielsen 2004). The changes mainly reflect the current recommendations (Gluud 2017).
In the previous review (Als‐Nielsen 2004), the primary outcomes included 'recovery' from hepatic encephalopathy defined as complete resolution of symptoms and 'improvement' of hepatic encephalopathy. We removed outcome 'recovery.' The term may be misleading as episodes may recur. Furthermore, people may show some degree of impairment between episodes (Bajaj 2010). Based on current guidelines (Gluud 2017), we assessed 'improvement' as number of participants 'without improvement of hepatic encephalopathy.' We now include all‐cause mortality as a primary outcome rather than 'survival.'
The previous version of the review included data from both periods of cross‐over trials. In this review, we only included data from the first treatment period because hepatic encephalopathy is fluctuating condition and because participants may die early in the randomised clinical trials.
We now report the results of meta‐analyses using risk ratios, instead of risk differences, and include observational studies to improve our assessment of serious adverse events, Trial Sequential Analyses, and regression analysis (Harbord test) to evaluate the risk of small‐study effects. The bias assessment is also updated.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | Double‐blind, single‐centre, placebo‐controlled RCT. Cross‐over design: all participants underwent both intervention periods (received flumazenil and placebo). |
|
| Participants | 13 participants with cirrhosis with no evidence of overt hepatic encephalopathy but with abnormal brainstem evoked potentials (5 participants) or prolonged Number Connection Test times (6 participants), or both at baseline corresponding to a diagnosis of minimal hepatic encephalopathy. Mean ± SD age: flumazenil/placebo: 54 ± 7 years. Proportion of men: 77%. Aetiology of cirrhosis: alcohol 77%; hepatitis B/C 15%. Proportion testing positive for benzodiazepines at baseline (Table 7): 0%. |
|
| Interventions |
Intervention comparison: intravenous bolus flumazenil 1 mg followed by 4 boluses of 0.5 mg every 30 minutes versus placebo (saline). Total dose of flumazenil: 3 mg. Washout period: 72 hours before cross‐over to the alternative arm. Cointerventions: none described. |
|
| Outcomes | Outcomes included in meta‐analyses: none. | |
| Neuropsychiatric assessment |
Baseline and post infusion:
|
|
| Inclusion period (date) | Not described. | |
| Country | Italy. | |
| Notes | Included data: RCT did not describe outcomes for first intervention period. Therefore, we were unable to include the trial in our meta‐analyses. The study report includes 2 tables containing data for the 5 participants with abnormal evoked potentials at baseline and the 6 participants with abnormal Number Connection Test times. There were no change in the group mean variables after flumazenil. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described. |
| Allocation concealment (selection bias) | Low risk | Concealed drug containers. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding of participants and personnel. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of outcome assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data. |
| Selective reporting (reporting bias) | Low risk | Trial described clinically relevant outcomes. We had no access to information about outcomes described in the original protocol or information in trial registries. |
| For‐profit funding | Low risk | Funding from the Italian Liver Foundation. |
| Other bias | Low risk | No other biases. |
| Overall assessment | High risk | High risk of bias. |
| Methods | Double‐blind, multi‐centre, placebo‐controlled RCT. Cross‐over design: investigators crossed‐over participants who did not respond to intervention during first period (remained in Grade III or IVa coma) to alternative intervention. |
|
| Participants | 527 participants with cirrhosis and overt hepatic encephalopathy (Grades III or IVa; Table 4), admitted to an intensive care unit. Diagnostic criteria corresponded to acute hepatic encephalopathy. Precipitating factors are described (Table 6). Mean ± SD age (grade III/IVa): flumazenil: 56 ± 11.5/53 ± 12 years; placebo: 48 ± 20/55 ± 13.5 years. Proportion of men: 69%. Aetiology of cirrhosis: alcohol 40%; hepatitis B/C 59%. Proportion testing positive for benzodiazepines at baseline (Table 7): 10/527 (1.9%) participants. |
|
| Interventions |
Intervention comparison: intravenous infusion flumazenil 1 mg given over 3 to 5 minutes versus placebo (isotonic saline). Total dose of flumazenil: 1 mg. Cointerventions: lactulose 30 mL every 6 hours via nasogastric tube; antibiotics were given to 22 participants with sepsis in the flumazenil group and 8 in the placebo group |
|
| Outcomes | Outcomes included in meta‐analyses: mortality, hepatic encephalopathy (Table 3), and serious adverse events (Table 8) assessed for a maximum of 4 days after randomisation. | |
| Neuropsychiatric assessment |
Baseline and post infusion: |
|
| Inclusion period (date) | January 1993 to December 1997. | |
| Country | Italy. | |
| Notes | Included data: Serum benzodiazepines were detected in 10 participants (4 with Grade III and 6 with Grade IVa coma). The published paper provides no information about the distribution of these participants to flumazenil or placebo during the first intervention period. Therefore, we were unable to exclude these participants from the analyses. The trial reported on several serious adverse events (Table 8). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated sequential list of block‐randomised assignments. |
| Allocation concealment (selection bias) | Low risk | Concealed ampoules of flumazenil and placebo. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding of participants and personnel using placebo. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of outcome assessment using placebo. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data and all participants included in analyses. |
| Selective reporting (reporting bias) | Low risk | Trial described clinically relevant outcomes. We had no access to information about outcomes described in the original protocol or information in trial registries. |
| For‐profit funding | Low risk | No information provided. |
| Other bias | Low risk | No other biases. |
| Overall assessment | Low risk | Low risk of bias. |
| Methods | Double‐blind, single‐centre, placebo‐controlled RCT. Cross‐over design: participants who did not respond after 10 minutes during the first period received the alternative intervention. |
|
| Participants | 14 participants with cirrhosis experiencing 18 separate episodes of acute hepatic encephalopathy classified as Grade II to IV (Table 4). Precipitating factors are described (Table 6). Mean ± SD age: whole group 54.8 ± 7.7 years. Proportion of men: 71%. Aetiology of cirrhosis: alcohol 71%; hepatitis B/C 29%. Proportion testing positive for benzodiazepines at baseline (Table 7): 3/14 (21.4%) participants. |
|
| Interventions |
Intervention comparison: continuous intravenous infusion flumazenil 0.1 mg/mL at 1 mL/minute flumazenil versus placebo (sodium edetate 1 mg). Investigators stopped the infusion after 10 minutes if participants showed improvement in electroencephalography or coma grade. Total dose of flumazenil: 1 mg. Cointerventions: none described. |
|
| Outcomes | Outcomes included in meta‐analyses: mortality, hepatic encephalopathy (Table 3), and serious adverse events (Table 8) assessed after maximum of 3 days. | |
| Neuropsychiatric assessment |
Baseline and post infusion: |
|
| Inclusion period (date) | May 1988 to May 1990. | |
| Country | France. | |
| Notes | Included data: the trial included 14 participants who between them experienced 18 episodes of acute hepatic encephalopathy. 1 participant entered the trial once and 1 entered the trial 3 times. We included data from the first intervention period in our analyses. The published report described the number of participants who died after the second treatment period only (Table 8). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported. |
| Allocation concealment (selection bias) | Low risk | Concealed drug vials. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding of participants and personnel. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of outcome assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Outcome data were complete. |
| Selective reporting (reporting bias) | Low risk | Trial described clinically relevant outcomes. We had no access to information about outcomes described in original protocol or information in trial registries. |
| For‐profit funding | High risk | Hoffmann‐La Roche Ltd. supplied flumazenil and placebo. |
| Other bias | Low risk | No other biases. |
| Overall assessment | High risk | High risk of bias. |
| Methods | Double‐blind, single‐centre, placebo‐controlled, parallel‐arm RCT. | |
| Participants | 40 participants with cirrhosis and hepatic encephalopathy classified as subclinical (corresponding to minimal; 10 participants) or overt Grade I to III (30 participants; Table 4). Type of overt hepatic encephalopathy (acute or chronic) not specified. Mean ± SD age: flumazenil: 44.5 ± 12.9 years; placebo: 43.7 ± 11.9 years. Proportion of men: 73%. Aetiology of cirrhosis: alcohol 0%; hepatitis B/C 100%. Proportion testing positive for benzodiazepines at baseline (Table 7): investigators did not screen for benzodiazepines. |
|
| Interventions |
Intervention comparison: intravenous infusion flumazenil 1 mg/hour for 5 hours versus placebo (saline) administered similarly. Total dose of flumazenil: 5 mg. Cointerventions: lactulose 30 mL 6‐hourly |
|
| Outcomes | Outcomes included in meta‐analyses: mortality, hepatic encephalopathy (Table 3), serious adverse events (Table 8), and Number Connection Test assessed after a maximum of 5 hours. | |
| Neuropsychiatric assessment |
Baseline and post infusion:
|
|
| Inclusion period (date) | December 1999 to January 2002. | |
| Country | Turkey. | |
| Notes | Included data: the trial report included information about participants with minimal and overt hepatic encephalopathy. We have analysed these 2 groups separately. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported. |
| Allocation concealment (selection bias) | Low risk | Concealed drug containers. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding of participants and personnel. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of outcome assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data and all participants are included in analyses. |
| Selective reporting (reporting bias) | Low risk | Trial describes clinically relevant outcomes. We had no access to information about outcomes described in the original protocol or information in trial registries. |
| For‐profit funding | Unclear risk | No information provided. |
| Other bias | Low risk | No other biases. |
| Overall assessment | High risk | High risk of bias. |
| Methods | Double‐blind, single‐centre, placebo‐controlled RCT. Cross‐over design: investigators crossed over all participants to the alternative intervention. |
|
| Participants | 10 participants with cirrhosis and no clinical evidence of overt hepatic encephalopathy; 5 participants had minimal hepatic encephalopathy based on the finding of either abnormal visual evoked potentials or Number Connection Test results. Age (range): 40 to 60 years. Proportion of men: 80%. Aetiology of cirrhosis: alcohol 30%; hepatitis B/C 70%. Proportion testing positive for benzodiazepines at baseline (Table 7): 0%. |
|
| Interventions |
Intervention comparison: intravenous infusion flumazenil 1 mg over 2 minutes versus placebo. Total dose of flumazenil: 1 mg. Washout period: 4 hours. Cointerventions: none reported. |
|
| Outcomes | Outcomes included in meta‐analyses: none. | |
| Neuropsychiatric assessment |
At baseline and post infusion:
|
|
| Inclusion period (date) | Not described. | |
| Country | The Netherlands. | |
| Notes | Included data: trial did not include separate information about the first allocation period. Therefore, we were unable to include the trial in our meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described. |
| Allocation concealment (selection bias) | Low risk | Concealed drug containers. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding of participants and personnel. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of outcome assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data. |
| Selective reporting (reporting bias) | Low risk | The trial describes clinically relevant outcomes. We had no access to information about outcomes described in the original protocol or information in trial registries. |
| For‐profit funding | Unclear risk | No information provided. |
| Other bias | Low risk | No other biases. |
| Overall assessment | High risk | High risk of bias. |
| Methods | Double‐blind, single‐centre, placebo‐controlled RCT. Cross‐over design: all participants were crossed over to alternative intervention. |
|
| Participants | 10 participants with cirrhosis and subclinical (corresponding to minimal) hepatic encephalopathy diagnosed based on a score on the Digit Symbol Substitution test of < 1 SD of the age‐matched normative mean. Mean age ± SD: 53.9 ± 7.4 years. Proportion of men: 80%. Aetiology of cirrhosis: alcohol 60%; hepatitis B/C 20%. Proportion testing positive for benzodiazepines at baseline (Table 7): apparently not performed (not specifically stated). |
|
| Interventions |
Intervention comparison: intravenous infusion flumazenil 0.2 mg over an unspecified time versus placebo (saline). Total dose of flumazenil: 0.2 mg. Washout period: 1 week. Cointerventions: none described. |
|
| Outcomes | Outcomes included in meta‐analyses: mortality and serious adverse events (Table 8) assessed at end of the intervention. | |
| Neuropsychiatric assessment |
At baseline:
At baseline and post infusion:
Duration of follow‐up and timing of tests not described. |
|
| Inclusion period (date) | Not described. | |
| Country | UK. | |
| Notes | Included data: we received additional (unpublished) information about the trial methods and number of participants allocated to flumazenil/placebo during the first allocation period via email in 2003 when conducting the previous version of this review. The trial did not evaluate the number of participants with an overall improvement in hepatic encephalopathy. Therefore, we were unable to include the trial in our analyses of this outcome measure. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number table. |
| Allocation concealment (selection bias) | Low risk | Serially numbered opaque sealed envelopes used in administration of concealed drug containers. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding of participants and personnel. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of outcome assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data. |
| Selective reporting (reporting bias) | Low risk | Trial described clinically relevant outcomes. We had no access to information about outcomes described in the original protocol or information in trial registries. |
| For‐profit funding | Unclear risk | No information provided. |
| Other bias | High risk | Primary investigators described a significant drug by order and group by drug by order interaction. |
| Overall assessment | High risk | High risk of bias. |
| Methods | Double‐blind, multi‐centre, parallel‐arm, placebo controlled RCT. | |
| Participants | 49 participants with cirrhosis and chronic overt hepatic encephalopathy (Grades I to III; Table 4). Mean age ± SD: flumazenil: 55.5 ± 9.4 years; placebo: 53.6 ± 10.3 years. Proportion of men: 69%. Aetiology of cirrhosis: alcohol 51%; hepatitis B/C 35%. Proportion testing positive for benzodiazepines at baseline (Table 7): 11% in flumazenil group; 5% in placebo group. |
|
| Interventions |
Intervention comparison: intravenous boluses of flumazenil 0.4 mg, 0.8 mg, and 1 mg at 1‐minute intervals followed by a 3‐hour infusion of flumazenil 1 mg/hour versus placebo (saline). Total dose of flumazenil: 5.2 mg. Cointerventions: none reported. |
|
| Outcomes | Outcomes included in meta‐analyses: mortality, hepatic encephalopathy (Table 3), and serious adverse events (Table 8) assessed after a maximum of 4 weeks. | |
| Neuropsychiatric assessment |
Baseline and post infusion:
|
|
| Inclusion period (date) | Not reported. | |
| Country | Switzerland (primary), France, Germany, Italy, Canada, the Netherlands, the UK, and Korea. | |
| Notes | Included data: authors reported intention‐to‐treat analyses including all participants randomised and a per‐protocol analysis excluding protocol violators (25 participants). We included data on all participants in our analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated list of random numbers. |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes used in double‐blind administration of flumazenil and placebo. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Blinding of participants and personnel. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of outcome assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data. |
| Selective reporting (reporting bias) | Low risk | Trial described clinically relevant outcomes. We had no access to information about outcomes described in the original protocol or information in trial registries. |
| For‐profit funding | High risk | Support from Hoffmann‐La Roche Ltd. |
| Other bias | Low risk | No other biases. |
| Overall assessment | High risk | High risk of bias. |
| Methods | Double‐blind, parallel‐arm, single‐centre, placebo‐controlled RCT. | |
| Participants | 12 participants with cirrhosis and an acute episode of hepatic encephalopathy defined as Grade IIIa with severely abnormal electroencephalography changes, but a Glasgow Coma Score of < 12 (Table 4). Proportion of men: not reported. Mean age ± SD: whole group 58.2 ± 5.4 years. Aetiology of liver disease: not reported. Proportion testing positive for benzodiazepines at baseline (Table 7): 0%. |
|
| Interventions |
Intervention comparison: intravenous infusion flumazenil 0.2 mg/kg over 10 minutes versus placebo (saline). Total dose of flumazenil: 0.2 mg/kg. Cointerventions: none described. |
|
| Outcomes | Outcomes included in meta‐analyses: mortality (Table 8) and serious adverse events (Table 8). | |
| Neuropsychiatric assessment |
Baseline and post infusion: The timing of assessments post infusion was not described. |
|
| Inclusion period (date) | Not described. | |
| Country | France. | |
| Notes |
Included data: the trial report did not specifically state the number of participants with (or without) improvement in hepatic encephalopathy separately for the allocation groups. Therefore, we were unable to include the trial in the analysis of this outcome measure. Article published in French (full translation available). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported. |
| Allocation concealment (selection bias) | Low risk | Concealed drug containers. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding of participants and personnel. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of outcome assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Trial report gave the impression that there were no missing outcome data although this was not specifically stated. |
| Selective reporting (reporting bias) | Low risk | Trial described clinically relevant outcomes. We had no access to information about outcomes described in the original protocol or information in trial registries. |
| For‐profit funding | Unclear risk | No information provided. |
| Other bias | Low risk | No other biases. |
| Overall assessment | High risk | High risk of bias. |
| Methods | Double‐blind, cross‐over, single‐centre, placebo‐controlled RCT. | |
| Participants | 2 participants with cirrhosis and stable hepatic encephalopathy (Grade III). Description corresponded to chronic overt hepatic encephalopathy although this was not specifically stated. Proportion of men: not reported. Mean age: not reported. Aetiology of liver disease: alcohol 100%. Proportion testing positive for benzodiazepines at baseline (Table 7): apparently not performed (not specifically stated). |
|
| Interventions |
Intervention comparison: intravenous infusion flumazenil 1 mg over 1 minute versus placebo. Total dose of flumazenil: 1 mg. Washout period: not specified. Cointerventions: not reported. |
|
| Outcomes | Outcomes included in meta‐analyses: mortality and serious adverse events (Table 8) assessed for a maximum of 2 hours post interventions. | |
| Neuropsychiatric assessment | Clinical assessment of mental status: assessed after 2 hours (no specific score; timing not specified). | |
| Inclusion period (date) | Not described. | |
| Country | Germany. | |
| Notes | Included data: investigators described the design as cross‐over but did not provide data from the first intervention period. Therefore, we were unable to include the trial in our analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not described. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Trial described as double blind and placebo controlled. However, trial only reported as a letter and the type of placebo (or mode of administration) is not described. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Trial described as double blind and placebo controlled. However, trial only reported as a letter and the type of placebo (or mode of administration) is not clearly described. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data and all participants included in analyses. |
| Selective reporting (reporting bias) | Low risk | Trial gave impression that participants survived although this was not specifically stated. We had no access to information about outcomes described in the original protocol or information in trial registries. |
| For‐profit funding | Unclear risk | No information provided. |
| Other bias | High risk | Trial only included 2 participants. |
| Overall assessment | High risk | High risk of bias. |
| Methods | Double‐blind, single‐centre, parallel‐arm placebo‐controlled RCT. | |
| Participants | 54 participants with cirrhosis and acute hepatic encephalopathy (Grade III or IV). Precipitating factors are described (Table 6). Mean age ± SD: flumazenil: 59.6 ± 6.0 years; placebo: 57.7 ± 5.4 years. Proportion of men: 54%. Aetiology of cirrhosis: hepatitis B/C 100%. Proportion testing positive for benzodiazepines at baseline (Table 7): 0%. |
|
| Interventions |
Intervention comparison: intravenous infusion flumazenil 0.4 mg/mL at 10 mL/minute for 5 minutes versus placebo (saline). Total dose of flumazenil: 2 mg. Cointerventions: lactulose enemas; branch‐chain amino acids. |
|
| Outcomes | Outcomes included in meta‐analyses: mortality, hepatic encephalopathy (Table 3), and serious adverse events (Table 8) assessed for maximum of 24 hours after intervention. | |
| Neuropsychiatric assessment |
Baseline and post infusion:
|
|
| Inclusion period (date) | January 1997 to December 1997. | |
| Country | Italy. | |
| Notes |
Included data: we included data on all participants in our analyses. Notes about the design: investigators repeated the intervention once after 3 hours in non‐responders (no improvement in neurological status) or immediately if they detected an improvement followed by a relapse. The report did not include information about the number of participants who received a second infusion. In the results section of the report the investigators stipulate that the second infusion was the same as the one first received. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported. |
| Allocation concealment (selection bias) | Low risk | Double‐blind administration of flumazenil and placebo. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding of participants and personnel using placebo. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of outcome assessment using placebo. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data and all participants were included in the analyses. |
| Selective reporting (reporting bias) | Low risk | Trial described clinically relevant outcomes. We had no access to information about outcomes described in the original protocol or information in trial registries. |
| For‐profit funding | Unclear risk | No information provided. |
| Other bias | Low risk | No other biases. |
| Overall assessment | High risk | High risk of bias. |
| Methods | Double‐blind, single‐centre, parallel‐arm RCT. | |
| Participants | 72 participants with overt hepatic encephalopathy (Grade III or IV) associated with cirrhosis (65%) or fulminant hepatic failure (35%). Diagnostic criteria for participants with cirrhosis corresponded to acute hepatic encephalopathy. Mean age ± SD: flumazenil: 55.4 ± 6.6 years; placebo: 56.8 ± 7.9 years. Proportion of men: 63%. Aetiology of cirrhosis: not reported. Proportion testing positive for benzodiazepines at baseline (Table 7): apparently not performed (not specifically stated). |
|
| Interventions |
Intervention comparison: slow intravenous injection flumazenil 0.5 mg followed by intravenous infusion of flumazenil 1 mg of over 30 minutes versus placebo (saline). Total dose of flumazenil: 1.5 mg. Cointerventions: lactulose enemas, L‐ornithine L‐aspartate |
|
| Outcomes | Outcomes included in meta‐analyses: mortality, hepatic encephalopathy (Table 3), and serious adverse events (Table 8) assessed after a maximum of 2 weeks. | |
| Neuropsychiatric assessment |
Baseline and post infusion:
|
|
| Inclusion period (date) | May 2006 to July 2008. | |
| Country | China. | |
| Notes | Included data: the trial report did not provide separate information on participants with cirrhosis and participants with acute liver failure. Therefore, we conducted a sensitivity analysis excluding this trial. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported. |
| Allocation concealment (selection bias) | Low risk | Double‐blind administration of flumazenil and placebo. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding of participants and personnel. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of outcome assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data and all participants included in the analyses. |
| Selective reporting (reporting bias) | Low risk | Trial described clinically relevant outcomes. We had no access to information about outcomes described in original protocol or information in trial registries. |
| For‐profit funding | Unclear risk | No information provided. |
| Other bias | Low risk | No other biases. |
| Overall assessment | High risk | High risk of bias. |
| Methods | Double‐blind, single‐centre, cross‐over, placebo‐controlled RCT. Cross‐over design: investigators only crossed over participants who remained in Grade IV coma, 24 hours after the first study period. |
|
| Participants | 21 participants with cirrhosis and acute hepatic encephalopathy (Grade IV). Precipitating factors are described (Table 6). Mean age ± SD: flumazenil: 52.7 ± 5.4 years; placebo: 57.4 ± 9.0 years. Proportion of men: 81%. Aetiology of cirrhosis: alcohol 62%; hepatitis B/C 5%. Proportion testing positive for benzodiazepines at baseline (Table 7): 4/21 (19%) participants. |
|
| Interventions |
Intervention comparison: intravenous infusion flumazenil 2 mg over 5 minutes versus placebo (saline). Total dose of flumazenil: 2 mg. Washout period: 24 hours. Cointerventions: lactulose 30 mL 4 times daily. |
|
| Outcomes | Outcomes included in meta‐analyses: mortality, hepatic encephalopathy (Table 3), and serious adverse events (Table 8) assessed after a maximum follow‐up of 24 hours. | |
| Neuropsychiatric assessment |
Baseline and post infusion: |
|
| Inclusion period (date) | March 1988 to February 1992. | |
| Country | Canada. | |
| Notes | Included data: we only included data from the first treatment period in our analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random numbers. |
| Allocation concealment (selection bias) | Low risk | Blinded administration of flumazenil or placebo. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding of participants and personnel. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of outcome assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data and all participants included in analyses. |
| Selective reporting (reporting bias) | Low risk | Trial described clinically relevant outcomes. We had no access to information about outcomes described in the original protocol or information in trial registries. |
| For‐profit funding | High risk | Technical assistance from Hoffmann‐La Roche Ltd., Canada and Nutley, NJ, USA. |
| Other bias | Low risk | No other biases. |
| Overall assessment | High risk | High risk of bias. |
| Methods | Double‐blind, single‐centre, cross‐over, placebo‐controlled RCT. Cross‐over design: all participants (except 2 who underwent transplantation) received flumazenil and placebo. |
|
| Participants | 18 participants with hepatic encephalopathy secondary to acute liver failure (28%) or cirrhosis (82%), who had an arterial blood ammonia > 30 μmol/L, and an abnormal electroencephalography despite at least 24 hours of treatment with a low protein diet and lactulose alone or with neomycin. Precipitating factors are described (Table 6). Mean age ± SD: whole group 48.56 ± 14.67 years. Proportion of men: 39%. Aetiology of cirrhosis: alcohol 38%; hepatitis B/C 15%. Proportion testing positive for benzodiazepines at baseline (Table 7): 0%. |
|
| Interventions |
Intervention comparison 1: First 9 participants: intravenous infusion flumazenil 0.1 mg/minute over 10 minutes; 4 hours later given a bolus injection flumazenil 0.5 mg followed by a continuous infusion of flumazenil 0.25 mg/hour for 3 days versus infusion vehicle alone. Total dose of flumazenil: 19.5 mg. Washout period: 24 hours. Intervention comparison 2: Second 9 participants: intravenous infusion of flumazenil 0.1 mg/minute over 10 minutes versus infusion vehicle alone. Total dose of flumazenil: 19.5 mg. Washout period 24 hours. Cointerventions: protein restriction, lactulose alone or with neomycin. |
|
| Outcomes | Outcomes included in meta‐analyses: mortality, hepatic encephalopathy (Table 3), and serious adverse events (Table 8) assessed after a maximum of 3 days. | |
| Neuropsychiatric assessment |
First 9 participants: baseline and post infusion:
Second 9 participants: baseline and post infusion: |
|
| Inclusion period (date) | February 1987 to February 1990. | |
| Country | The Netherlands. | |
| Notes | Included data: Two patients were withdrawn on day one of the study to undergo liver transplantation thus only 16 people took part in the full cross‐over study. The study involved people with hepatic encephalopathy associated with cirrhosis and with acute liver failure; the trial data were not provided separately for these two groups so no separate analysis can be performed by aetiology of the hepatic encephalopathy. We only include data from the first treatment period in our analyses | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported. |
| Allocation concealment (selection bias) | Low risk | Used concealed drug containers. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding of participants and personnel. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of outcome assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data and all participants were included in analyses. |
| Selective reporting (reporting bias) | Low risk | Trial described clinically relevant outcomes. We had no access to information about outcomes described in original protocol or information in trial registries. |
| For‐profit funding | High risk | Support provided by Hoffmann‐La Roche B.V., Mijdrecht, The Netherlands. |
| Other bias | Unclear risk | Investigators simplified the intervention regimen after inclusion of the first 9 participants as the second period of the 72‐hour infusion was too demanding for participants. The effect that this has on bias control was unclear. |
| Overall assessment | High risk | High risk of bias. |
| Methods | Double‐blind, single‐centre, parallel‐arm, placebo‐controlled RCT. | |
| Participants | 25 participants with cirrhosis and overt hepatic encephalopathy (Grade II to IV). Precipitating factors are described (Table 6). Mean age ± SD: flumazenil: 62.2 ± 2.7 years; placebo: 52.2 ± 3.3 years. Proportion of men: 69%. Aetiology of cirrhosis: alcohol 80%; hepatitis B/C 12%. Proportion testing positive for benzodiazepines at baseline (Table 7): not conducted (not specifically stated). |
|
| Interventions |
Intervention comparison: intravenous infusion flumazenil 1 mg over 5 minutes versus placebo (saline). Total dose of flumazenil: 1 mg. Cointerventions: intravenous branched‐chain amino acids. |
|
| Outcomes | Outcomes included in meta‐analyses: mortality, hepatic encephalopathy (Table 3), and serious adverse events (Table 8) assessed for a maximum of 2 weeks (until death or discharge). | |
| Neuropsychiatric assessment |
Baseline and post infusion:
|
|
| Inclusion period (date) | April 1995 to March 1996. | |
| Country | China. | |
| Notes | Included data: all participants were included in the analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random numbers with stratified block randomisation. |
| Allocation concealment (selection bias) | Low risk | Administration of concealed drug containers with sealed, opaque, serially numbered envelopes. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding of participants and personnel. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of outcome assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Missing outcome data not described. |
| Selective reporting (reporting bias) | Low risk | Trial described clinically relevant outcomes. We had no access to information about outcomes described in the original protocol or information in trial registries. |
| For‐profit funding | High risk | Roche supplied the flumazenil. |
| Other bias | Low risk | No other biases. |
| Overall assessment | High risk | High risk of bias. |
RCT: randomised clinical trial; SD: standard deviation.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Bansky 1989 | Prospective study including 14 participants with cirrhosis and overt hepatic encephalopathy. The investigators reported an improvement in mental status in 71% of participants within minutes of receiving intravenous flumazenil lasting for 1 to 2 hours. Participants also received lactulose . Six participants died. The study was excluded as it did not include a control group. |
| Devictor 1995 | Prospective study evaluating 7 children with fulminant hepatic failure awaiting emergency liver transplantation. The investigators reported that flumazenil injection led to a transient improvement in mental status in 1 child but had no effect on mental status in the remaining six. The study was excluded as none of the participants had hepatic encephalopathy associated with cirrhosis and it did not include a control group. |
| Golubovic 1999 | Prospective study including 10 participants with alcohol‐related cirrhosis and overt hepatic encephalopathy classified as grade IV based on an assessment of mental status, electroencephalography, and visual evoked responses. The investigators reported an improvement in mental status in 8/10 participants. Six participants died within 1 year. The study was excluded as it did not include a control group. |
| Grimm 1988 | Prospective study including 17 participants (2 children) with hepatic encephalopathy associated with acute liver failure (9 participants) or cirrhosis (8 participants). Cointerventions included lactulose, branched‐chain amino acids, antibiotics, diuretics, histamine‐receptor antagonists, human albumin, and fresh frozen plasma. Transient improvement in the manifestations of hepatic encephalopathy was seen following flumazenil in 4 (44%) participants with fulminant hepatic failure and 5 (63%) with cirrhosis. Mortality was not reported. This study was excluded as it did not include a control group. |
| Jia 1999 | Open, single‐centre, non‐randomised study which is included in the sensitivity analyses of serious adverse events. The study involved 22 participants with cirrhosis and overt hepatic encephalopathy (Grades I‐III using West Haven criteria) recruited between April 1996 and September 1997. Intervention comparison: 12 participants received an intravenous bolus of flumazenil 0.5 mg followed by an intravenous infusion of flumazenil in a dose of 1.0 mg over 4 hours for an unspecified period of time. The remaining 10 participants received Xing‐Nao‐Jing, a traditional Chinese medicine also given as an intravenous infusion. Total dose of flumazenil: 1.5 mg. Outcomes: The study report stated that 2 participants in the flumazenil group died of liver failure but the time of death in relation to the intervention was not specified and study did not specifically state if there were any deaths in control group. The article was published in Chinese but a translation was available. The study was excluded as it did not contain a control group. |
| Kapczinski 1995 | Double‐blind, cross‐over, placebo‐controlled, single‐centre randomised clinical trial including 20 liver transplant candidates with cirrhosis. The trial is included in the sensitivity analyses of serious adverse events. The main objective of trial was to evaluate the differential effects of flumazenil on cognitive function and anxiety in people with alcohol‐related (10 participants) or non‐alcohol‐related (10 participants) cirrhosis. None of the included participants had evidence of overt hepatic encephalopathy. The investigators evaluated a range of psychometric tests and reported the results as group mean values. No information was provided about the number of participants with abnormal test results. Proportion of men: 60%. Mean ± SD age: alcohol‐related cirrhosis: 47.7 ± 10.5 years; non‐alcoholic cirrhosis: 48.4 ± 11.7 years. Proportion testing positive for benzodiazepines at baseline: not tested Intervention: intravenous infusion flumazenil 0.1 mg/minute for 10 minutes then 0.05 mg/minute for 20 minutes versus placebo (saline). Total dose of flumazenil: 2 mg. Washout period: 60 minutes. Outcomes: The investigators reported changes in psychometric tests for participants with alcohol‐related or non‐alcohol‐related cirrhosis without providing an overall estimate of numbers with (or without) improved manifestations. The trial did not report any deaths or serious adverse events. The study was excluded because none of the participants had hepatic encephalopathy. |
| Marsepoil 1990 | Open, single‐centre, prospective, non‐randomised study included in the sensitivity analyses of serious adverse events at 48 hours. The study involved 25 participants with alcohol‐related cirrhosis and acute hepatic encephalopathy, 13 of whom received flumazenil. The proportion of men and the mean age of participants was not reported. Proportion testing positive for benzodiazepines at baseline: not mentioned Intervention: intravenous bolus of flumazenil 0.2 mg every ten minutes until improvement in clinical status up to a maximum total dose of 2 mg followed by a continuous maintenance infusion of 0.3 mg. per hour for 48 hours. Total dose of flumazenil: maximum 16.4 mg. Outcomes: .The Investigators reported that the mortality rates were similar in the flumazenil and control groups, but did not provide information on the number of participants who died. Published in French but a translation was available.The study was excluded as it was not randomised. |
| Ozyilkan 1997 | Prospective study evaluating the effect of 30‐minute, incremental intravenous boluses of flumazenil in 11 participants with cirrhosis (6 stage 0,4 stage one, one stage 2 hepatic encephalopathy) in whom baseline somatosensory evoked potentials were abnormal. Four patients (36%) showed a clear improvement in evoked potentials with flumazenil. Mortality was not described. The study was excluded as it did not include a control group. |
| Wu 2001 | Randomised clinical trial comparing intravenous flumazenil plus lactulose enemas versus flumazenil alone. A total of 20 participants (18 men) with cirrhosis and hepatic encephalopathy were included. The maximum dose of flumazenil was 9 mg. The dose was adjusted based on the clinical effect. Investigators assessed hepatic encephalopathy based on Conn Criteria and defined an improvement from Grade IV to I within 6 hours as clinically significant. None of the included participants died and all 12 in the flumazenil plus lactulose group and all 8 in the flumazenil group showed improved manifestations of hepatic encephalopathy. The study was excluded as there was no placebo arm. |
.
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | Treatment of Hepatic Encephalopathy with Flumazenil and Change in Cortical Gamma Aminobutyric Acid Levels in MRS [magnetic resonance spectroscopy]. |
| Methods | Randomised clinical trial. |
| Participants | Participants with non‐alcoholic cirrhosis and hepatic encephalopathy. |
| Interventions | Flumazenil and placebo. |
| Outcomes | Recovery from hepatic encephalopathy and change in cortical gamma aminobutyric acid levels. |
| Starting date | November 2014. |
| Contact information | Deanna Martin, deanna.martin@yale.edu and Amanda Brennan amanda.brennan@yale.edu. |
| Notes | Estimated completion date: June 2017. |
Contributions of authors
LLG: drafted review and completed the statistical analyses. LLG and MYM: validated the extracted data and refined the drafting of the review. All authors participated in the selection of randomised clinical trials and extraction of data; interpretation of the results and in the critical revision of the review; and approved of the final version before submission.
Peer Reviewers: Manuel Romero‐Gómez, Spain; R Todd Frederick, USA. Contact Editor: Genaro D'Amico, Italy. Sign‐off Editor: Christian Gluud, Denmark.
Sources of support
Internal sources
Copenhagen Trial Unit, Denmark.
External sources
No sources of support supplied
Declarations of interest
LLG: acted as investigator in studies funded by Norgine, Abbvie, Intercept, and Merck; received funding for travel expenses from Novo Nordisk; and received funding for lectures from Eli Lilly and Norgine. MYM: no conflicts of interest. ETG: no conflicts of interest. MLA: no conflicts of interest.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
- Amodio P, Marchetti P, Comacchio F, Beghi A, Piccolo F, Merkel C, et al. Effects of flumazenil on subclinical hepatic encephalopathy (SHE): preliminary data. Italian Journal of Gastroenterology 1993;23:179. [Google Scholar]; Amodio P, Marchetti P, Comacchio F, Beghi A, Piccolo F, Merkel C, et al. Effects of flumazenil on subclinical hepatic encephalopathy. Journal of Hepatology 1993;18(Suppl 1):88. [Google Scholar]; Amodio P, Marchetti P, Piccolo F, Beghi A, Comacchio F, Carraro P, et al. The effect of flumazenil on subclinical psychometric or neurophysiological alterations in cirrhotic patients: a double‐blind placebo‐controlled study. Clinical Physiology 1997;17:533‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Barbaro G, Lorenzo G, Soldini M, Giancaspro G, Bellomo G, Belloni G, et al. Flumazenil for hepatic encephalopathy grade III and IVa in patients with cirrhosis: an Italian multicenter double‐blind, placebo‐controlled, crossover study. Hepatology (Baltimore, Md.) 1998;28:374‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]; Barbaro G, Lorenzo G, Soldini M, Marziali M, Bellomo G, Belloni G, et al. Flumazenil for hepatic coma in patients with liver cirrhosis: an Italian multicentre double‐blind, placebo‐controlled, crossover study. European Journal of Emergency Medicine 1998;5:213‐8. [MEDLINE: ] [PubMed] [Google Scholar]
- Cadranel JF, Younsi M, Pidoux B, Zylberberg P, Benhamou Y, Valla D, et al. Flumazenil therapy for hepatic encephalopathy in cirrhotic patients: a double‐blind pragmatic randomised, placebo study. European Journal of Gastroenterology and Hepatology 1995;7:325‐9. [MEDLINE: ] [PubMed] [Google Scholar]; Cadranel JF, Younsi M, Pidoux B, Zylberberg P, Benhamou Y, Valla D, et al. Immediate improvement of hepatic encephalopathy (HE) in cirrhotic patients by flumazenil. Results of a double‐blind crossover study. Journal of Hepatology 1991;13(Suppl 2):104. 1918873 [Google Scholar]; Younsi M, Cadranel JF, Pidoux B, Zylberberg P, Valla D, Benhamou Y, et al. The immediate effect on the clinical grade and electroencephalogram of cirrhotic patients with hepatic encephalopathy [Effets immediats du flumazenil sur le degre clinique et electroencephalographique de l'encephalopathie hepatique chez le cirrhotique]. Gastroenterologie Clinique et Biologique 1991;15:A216. [Google Scholar]
- Dursun M, Caliskan M, Canoruc F, Aluclu U, Canoruc N, Tuzcu A, et al. The efficacy of flumazenil in subclinical to mild hepatic encephalopathic ambulatory patients. A prospective, randomised, double‐blind, placebo‐controlled study. Swiss Medical Weekly 2003;133:118‐23. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Giger‐Mateeva VI, Reits D, Liberov B, Jones EA, Spekreijse H. The effect of flumazenil on visual event‐related potentials of clinically non‐encephalopathic patients with cirrhosis. Neuroscience Letters 1999;276:173‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]; Jones EA, Giger‐Mateeva VI, Reits D, Riemslag FC, Liberov B, Spekrijse H. Visual event‐related potentials in cirrhotic patients without overt encephalopathy: the effects of flumazenil. Metabolic Brain Disease 2001;16:43‐53. [PUBMED: 11726088] [DOI] [PubMed] [Google Scholar]
- Gooday R, Hayes PC, Bzeizi K, O'Carrol RE. Benzodiazepine receptor antagonism improves reaction time in latent hepatic encephalopathy. Psychopharmacology 1995;119:295‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Groeneweg M, Gyr K, Amrein R, Scollo‐Lavizzari G, Williams R, Yoo JY, et al. Effect of flumazenil on the electroencephalogram of patients with portosystemic encephalopathy. Results of a double‐blind, randomised, placebo‐controlled multicentre trial. Electroencephalography and Clinical Neurophysiology 1996;98:29‐34. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]; Gyr K, Meier R, Häussler J, Boulétreau P, Fleig WE, Gatta A, et al. Evaluation of the efficacy and safety of flumazenil in the treatment of portal systemic encephalopathy: a double blind, randomised, placebo controlled multicentre study. Gut 1996;39:319‐24. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]; Lotterer E, Hoppe M, Balzer C, Fleig WE. Short‐term effects of flumazenil in early stage of portosystemic encephalopathy (PSE): a placebo‐controlled, prospective, randomized study. Gastroenterology 2001;120:376‐77. [Google Scholar]; Meier R, Gyr K, Häussler R, the PSE‐Study Group. Treatment of portosystemic encephalopathy with the benzodiazepine‐receptor antagonist flumazenil (a randomised, double‐blind, placebo‐controlled, multicenter study. Gastroenterology 1994;106:A942. [Google Scholar]
- Hermant JL, Levacher S, Frenkel AL, Blaise M, Volter F, Pourriat JL. The clinical and electroencephalographic effects of flumazenil in acute hepatic encephalopathy [Effets cliniques et electroencephalographiques du flumazenil dans l'encephalopathie hepatique aigue]. Annales Francaises d'Anaesthesie et de Reanimation 1991;10:R172. [MEDLINE: ] [Google Scholar]
- Klotz U, Walker S. Flumazenil and hepatic encephalopathy. Lancet 1989;1:155‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Lacetti M, Manes G, Uomo G, Lioniello M, Rabitti PG, Balzano A. Flumazenil in the treatment of acute hepatic encephalopathy in cirrhotic patients: a double blind randomised placebo controlled study. Digestive and Liver Disease 2000;32:335‐8. [EMBASE: 2000243814] [DOI] [PubMed] [Google Scholar]
- Li F, Lei L, Wei L. Clinical study of flumazenil on severe hepatic encephalopathy. Practical Pharmacy and Clinical Remedies 2009;12:79‐81. [Google Scholar]
- Pomier‐Layrargues G, Giguère JF, Lavoie J, Perney P, Gagnon S, D'Amour M, et al. Flumazenil in cirrhotic patients in hepatic coma: a randomised double‐blind placebo‐controlled crossover trial. Hepatology (Baltimore, Md.) 1994;19:32‐7. [MEDLINE: ] [PubMed] [Google Scholar]
- Rijt CC, Schalm SW, Meulstee J, Stijnen T. Flumazenil therapy for hepatic encephalopathy. A double‐blind cross over study. Gastroenterologie Clinique et Biologique 1995;19:572‐80. [MEDLINE: ] [PubMed] [Google Scholar]; Rijt CCD. Hepatic Encephalopathy: Clinical and Experimental Studies. Alblasserdam, Netherlands: Offsetdrukkerij Haveka BV, 1991. [repub.eur.nl/.../911106_Rijt,Carolina] [Google Scholar]; Rijt CCD, Schalm SW, Meulstee J, Stijnen T. Flumazenil therapy for hepatic encephalopathy: a double‐blind cross‐over study. Hepatology (Baltimore, Md.) 1989;10:4. [PubMed] [Google Scholar]
- Zhu C, Wang J, Liu T. Flumazenil in the treatment of cirrhotic patients with hepatic encephalopathy: a randomised doubled‐blind clinical trial. Chinese Journal of Digestion 1998;18:355‐8. [Google Scholar]
References to studies excluded from this review
- Bansky G, Meier PJ, Riederer E, Walser H, Ziegler WH, Schmid M. Effects of the benzodiazepine receptor antagonist flumazenil in hepatic encephalopathy in humans. Gastroenterology 1989;97:744‐50. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Devictor D, Tahiri C, Lanchier C, Navelet Y, Durand P, Rousset A. Flumazenil in the treatment of hepatic encephalopathy in children with fulminant liver failure. Intensive Care Medicine 1995;21:253‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Golubovic G, Vlahovic A, Tomasevic R, Burg L, Bojovic V. Effects of flumazenil (benzodiazepine antagonist) in hepatic coma. Archives of Gastroenterohepatology 1999;18:32‐5. [EMBASE: 1999234651] [Google Scholar]
- Grimm G, Ferenci P, Katzenschlager R, Madl C, Schneeweiss B, Laggner AN, et al. Improvement of hepatic encephalopathy treated with flumazenil. Lancet 1988;2:1392‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Jia L, Li YY, Wu HS, Se QZ. A prospective, controlled study on flumazenil therapy for portal‐systemic shunt encephalopathy. Chinese Journal of Hepatology 1999;7:56. [Google Scholar]
- Kapczinski F, Sherman D, Williams R, Lader M, Curran V. Differential effects of flumazenil in alcoholic and nonalcoholic cirrhotic patients. Psychopharmacology 1995;120:220‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Marsepoil T, Hermant JL, Ho P, Blin F, Levesque P. Treatment of hepatic encephalopathy with flumazenil [Traitement de l’encephalopathie hepatique par le flumazenil]. Annales Francaises d'Anesthesie et de Reanimation 1990;9:399‐400. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Ozyilkan E, Kahraman H, Onar M, Kesim G, Arik Z. Evoked potentials and the effect of flumazenil in patients with liver cirrhosis. East African Medical Journal 1997;74:210‐2. [MEDLINE: ] [PubMed] [Google Scholar]
- Wu J, Luo H. Flumazenil and lactulose combination therapy for hepatic encephalopathy. Chinese Journal of Modern Medicine 2001;10:103‐4. [Google Scholar]
References to ongoing studies
- Treatment of Hepatic Encephalopathy with Flumazenil and Change in Cortical Gamma Aminobutyric Acid Levels in MRS [magnetic resonance spectroscopy].. Ongoing study November 2014..
Additional references
- Ahboucha S, Butterworth RF. The neurosteroid system: implication in the pathophysiology of hepatic encephalopathy. Neurochemistry International 2008;52:575‐87. [DOI] [PubMed] [Google Scholar]
- Amrein R, Hetzel W. Pharmacology of Dormicum (midazolam) and Anexate (flumazenil). Acta Anaesthesiologica Scandinavica. Supplementum 1990;92:6‐15. [PubMed] [Google Scholar]
- Bajaj JS, Hafeezullah M, Franco J, Varma RR, Hoffmann RG, Knox JF, et al. Inhibitory control test for the diagnosis of minimal hepatic encephalopathy. Gastroenterology 2008;135:1591‐600. [DOI] [PubMed] [Google Scholar]
- Bajaj JS, Wade JB, Sanyal AJ. Spectrum of neurocognitive impairment in cirrhosis: implications for the assessment of hepatic encephalopathy. Hepatology (Baltimore, Md.) 2009;50:2014‐21. [PUBMED: 19787808] [DOI] [PubMed] [Google Scholar]
- Bajaj JS, Schubert CM, Heuman DM, Wade JB, Gibson DP, Topaz A, et al. Persistence of cognitive impairment after resolution of overt hepatic encephalopathy. Gastroenterology 2010;138:2332‐40. [PUBMED: 20178797] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj JS, Cordoba J, Mullen KD, Amodio P, Shawcross DL, Butterworth RF, et al. Review article: the design of clinical trials in hepatic encephalopathy ‐ an International Society for Hepatic Encephalopathy and Nitrogen Metabolism (ISHEN) consensus statement. Alimentary Pharmacology & Therapeutics 2011;33:739‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batki G, Fisch HU, Karlaganis G, Minder C, Bircher J. Mechanism of the selective response of cirrhotics to benzodiazepines. Model experiments with triazolam. Hepatology (Baltimore, Md.) 1987;7:629‐38. [DOI] [PubMed] [Google Scholar]
- Brogden RN, Goa KL. Flumazenil. A reappraisal of its pharmacological properties and therapeutic efficacy as a benzodiazepine antagonist. Drugs 1991;42:1061‐89. [PUBMED: 1724638] [DOI] [PubMed] [Google Scholar]
- Brown P, Brunnhuber K, Chalkidou K, Chalmers I, Clarke M, Fenton M, et al. How to formulate research recommendations. BMJ (Clinical Research Ed.) 2006;333:804‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante J, Rimola A, Ventura PJ, Navasa M, Cirera I, Reggiardo V, et al. Prognostic significance of hepatic encephalopathy in patients with cirrhosis. Journal of Hepatology 1999;30:890‐5. [PUBMED: 10365817] [DOI] [PubMed] [Google Scholar]
- Butterworth R. Neurosteroids in hepatic encephalopathy: Novel insights and new therapeutic opportunities.. Journal of Steroid Biochemistry and Molecular Biology 2016;160:94‐97. [DOI] [PubMed] [Google Scholar]
- Chu NS, Yang SS, Liaw YF. Evoked potentials in liver diseases. Journal of Gastroenterology and Hepatology 1997;12:S288‐93. [PUBMED: 9407349] [DOI] [PubMed] [Google Scholar]
- Conn HO, Leevy CM, Vlahcevic ZR, Rodgers JB, Maddrey WC, Seeff L, et al. Comparison of lactulose and neomycin in the treatment of chronic portal‐systemic encephalopathy. A double blind controlled trial. Gastroenterology 1977;72:573‐83. [PUBMED: 14049] [PubMed] [Google Scholar]
- Córdoba J, Sanpedro F, Alonso J, Rovira A. 1H magnetic resonance in the study of hepatic encephalopathy in humans. Metabolic Brain Disease 2002;17:415‐29. [DOI] [PubMed] [Google Scholar]
- D'Amico G, Morabito A, Pagliaro L, Marubini E. Survival and prognostic indicators in compensated and decompensated cirrhosis. Digestive Diseases and Sciences 1986;31:468‐75. [PUBMED: 3009109] [DOI] [PubMed] [Google Scholar]
- D'Amico G, Garcia‐Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. Journal of Hepatology 2006;44:217‐31. [DOI] [PubMed] [Google Scholar]
- Jongh FE, Janssen HL, Man RA, Hop WC, Schalm SW, Blankenstein M. Survival and prognostic indicators in hepatitis B surface antigen‐positive cirrhosis of the liver. Gastroenterology 1992;103:1630‐5. [PUBMED: 1426884] [DOI] [PubMed] [Google Scholar]
- American Association for the Study of Liver Diseases, European Association for the Study of the Liver. Hepatic encephalopathy in chronic liver disease: 2014 practice guideline by the European Association for the Study of the Liver and the American Association for the Study of Liver Diseases. Journal of Hepatology 2014;61:642‐59. [PUBMED: 25015420] [DOI] [PubMed] [Google Scholar]
- Vilstrup H, Amodio P, Bajaj J, Cordoba J, Ferenci P, Mullen KD, et al. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver. Hepatology (Baltimore, Md.) 2014;60:715‐35. [PUBMED: 25042402] [DOI] [PubMed] [Google Scholar]
- Ferenci P. Hepatic encephalopathy ‐ definition, nomenclature, diagnosis, and quantification: final report of the working party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology (Baltimore, Md.) 2002;35:716‐21. [DOI] [PubMed] [Google Scholar]
- Fitz G. Systemic complications of liver disease. In: Feldman M, Scharschmidt BF, Sleisenger MH editor(s). Sleisenger and Fortran's Gastrointestinal and Liver Disease. Pathology, Diagnosis, Management. 5th Edition. Philadelphia (PA): WB Saunders Company, 1998:1334‐54. [Google Scholar]
- Gluud C, Nikolova D, Klingenberg SL. Cochrane Hepato‐Biliary Group. About Cochrane (Cochrane Review Groups (CRGs)) 2017, Issue 6. Art. No.: LIVER. [PubMed]
- Goulenok C, Bernard B, Cadranel JF, Thabut D, Martino V, Opolon P, et al. Flumazenil vs. placebo in hepatic encephalopathy in patients with cirrhosis: a meta‐analysis. Alimentary Pharmacology & Therapeutics 2002;16:361‐72. [DOI] [PubMed] [Google Scholar]
- Brozek J, Oxman A, Schünemann H. GRADEpro. Version 3.2 for Windows. Grade Working Group, 2008.
- Grippon P, Opolon P. Acute liver failure [Insuffisance hépatique aiguë]. Encyclopedia Medicine Chirurgie 1988;7014:14. [Google Scholar]
- Groeneweg M, Gyr K, Amrein R, Scollo‐Lavizzari G, Williams R, Yoo JY, et al. Effect of flumazenil on the electroencephalogram of patients with portosystemic encephalopathy. Results of a double blind, randomised, placebo‐controlled multicentre trial. Electroencephalography and Clinical Neurophysiology 1996;98(1):29‐34. [DOI] [PubMed] [Google Scholar]
- Groeneweg M, Quero JC, Bruijn I, Hartmann IJ, Essink‐bot ML, Hop WC, et al. Subclinical hepatic encephalopathy impairs daily functioning. Hepatology (Baltimore, Md.) 1998;28:45‐9. [PUBMED: 9657095] [DOI] [PubMed] [Google Scholar]
- Guérit JM, Amantini A, Fischer C, Kaplan PW, Mecarelli O, Schnitzler A, et al. Neurophysiological investigations of hepatic encephalopathy: ISHEN practice guidelines. Liver International 2009;29:789‐96. [PUBMED: 19638107] [DOI] [PubMed] [Google Scholar]
- Harbord RM, Egger M, Sterne JA. A modified test for small‐study effects in meta‐analyses of controlled trials with binary endpoints. Statistics in Medicine 2006;25:3443‐57. [PUBMED: 16345038] [DOI] [PubMed] [Google Scholar]
- Haussinger D, Kircheis G, Fischer R, Schliess F, vom Dahl S. Hepatic encephalopathy in chronic liver disease: a clinical manifestation of astrocyte swelling and low‐grade cerebral edema?. Journal of Hepatology 2000;32:1035‐8. [PUBMED: 10898326] [DOI] [PubMed] [Google Scholar]
- Higgins J, White IR, Wood AM. Imputation methods for missing outcome data in meta‐analysis of clinical trials. Clinical Trials 2008;5:225‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood SD, Norman A, Hince DA, Melichar JK, Hulse GK. Benzodiazepine dependence and its treatment with low dose flumazenil. British Journal of Clinical Pharmacology 2014;77:285‐94. [PUBMED: 23126253] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrobjartsson A, Gotzsche PC. Is the placebo powerless? An analysis of clinical trials comparing placebo with no treatment. New England Journal of Medicine 2001;344:1594‐602. [PUBMED: 11372012] [DOI] [PubMed] [Google Scholar]
- International Conference on Harmonisation Expert Working Group. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline. Guideline for Good Clinical Practice CFR & ICH Guidelines. Vol. 1, Philadelphia (PA): Barnett International/PAREXEL, 1997. [Google Scholar]
- Jones EA, Gammal SA. Hepatic encephalopathy. In: Arias IM, Jakoby WB, Popper H editor(s). The Liver: Biology and Pathobiology. 2nd Edition. New York (NY): Raven Press, 1988. [Google Scholar]
- Kennedy J, Parbhoo SP, MacGillivray B, Sherlock S. Effect of extracorporeal liver perfusion on the electroencephalogram of patients in coma due to acute liver failure. Quarterly Journal of Medicine 1973;42:549‐61. [PubMed] [Google Scholar]
- Kircheis G, Wettstein M, Timmermann L, Schnitzler A, Haussinger D. Critical flicker frequency for quantification of low‐grade hepatic encephalopathy. Hepatology (Baltimore, Md.) 2002;35:357‐66. [DOI] [PubMed] [Google Scholar]
- Kircheis G, Knoche A, Hilger N, Manhart F, Schnitzler A, Schulze H, et al. Hepatic encephalopathy and fitness to drive. Gastroenterology 2009;137:1706‐15. [PUBMED: 19686744] [DOI] [PubMed] [Google Scholar]
- Lauridsen MM, Jepsen P, Vilstrup H. Critical flicker frequency and continuous reaction times for the diagnosis of minimal hepatic encephalopathy: a comparative study of 154 patients with liver disease. Metabolic Brain Disease 2011;26:135‐9. [PUBMED: 21484318] [DOI] [PubMed] [Google Scholar]
- Markand ON. Electroencephalography in diffuse encephalopathies. Journal of Clinical Neurophysiology 1984;1:357‐407. [PUBMED: 6242404] [PubMed] [Google Scholar]
- Montagnese S, Amodio P, Morgan MY. Methods for diagnosing hepatic encephalopathy in patients with cirrhosis: a multidimensional approach. Metabolic Brain Disease 2004;19:281‐312. [DOI] [PubMed] [Google Scholar]
- Neff G. Pharmacoeconomics of hepatic encephalopathy. Pharmacotherapy 2010;30:28S‐32S. [PUBMED: 20412038] [DOI] [PubMed] [Google Scholar]
- Nusinovici V, Crubille C, Opolon P, Touboul JP, Darnis F, Caroli J. Fulminant hepatitis: an experience based on 137 cases. II. Course and prognosis. Gastroenterologie Clinique et Biologique 1977;1:875‐86. [PubMed] [Google Scholar]
- O'Carroll RE, Hayes PC, Ebmeier KP, Dougall N, Murray C, Best JJ, et al. Regional cerebral blood flow and cognitive function in patients with chronic liver disease. Lancet 1991;337:1250‐3. [PUBMED: 1674063] [DOI] [PubMed] [Google Scholar]
- Pappas SC, Jones EA. Methods of assessing hepatic encephalopathy. Seminars in Liver Disease 1983;3:297‐307. [DOI] [PubMed] [Google Scholar]
- Parsons‐Smith BG, Summerskill WH, Dawson AM, Sherlock S. The electroencephalograph in liver disease. Lancet 1957;273:867‐71. [PUBMED: 13482229] [DOI] [PubMed] [Google Scholar]
- Poordad FF. Review article: the burden of hepatic encephalopathy. Alimentary Pharmacology & Therapeutics 2007;25:3‐9. [PUBMED: 17295846] [DOI] [PubMed] [Google Scholar]
- Randolph C, Hilsabeck R, Kato A, Kharbanda P, Li YY, Mapelli D, et al. Neuropsychological assessment of hepatic encephalopathy: ISHEN practice guidelines. Liver International 2009;29:629‐35. [PUBMED: 19302444] [DOI] [PubMed] [Google Scholar]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
- Roman E, Córdoba J, Torrens M, Torras X, Villanueva C, Vargas V, et al. Minimal hepatic encephalopathy is associated with falls. American Journal of Gastroenterology 2011;106:476‐82. [DOI] [PubMed] [Google Scholar]
- Rosenkranz GK. Analysis of cross‐over studies with missing data. Statistical Methods in Medical Research 2015;24:420‐33. [PUBMED: 24501227] [DOI] [PubMed] [Google Scholar]
- Royle P, Milne R. Literature searching for randomized controlled trials used in Cochrane reviews: rapid versus exhaustive searches. International Journal of Technology Assessment in Health Care 2003;19:591‐603. [DOI] [PubMed] [Google Scholar]
- Rye DB, Bliwise DL, Parker K, Trotti LM, Saini P, Fairley J, et al. Modulation of vigilance in the primary hypersomnias by endogenous enhancement of GABAA receptors. Science Translational Medicine 2012;4:161‐51. [DOI] [PubMed] [Google Scholar]
- Saunders JB, Walters JR, Davies AP, Paton A. A 20‐year prospective study of cirrhosis. British Medical Journal (Clinical Research Ed.) 1981;282:263‐6. [PUBMED: 6779978] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savovic J, Jones H, Altman D, Harris R, Juni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomised controlled trials: combined analysis of meta‐epidemiological studies. Health Technology Assessment 2012;16:1‐82. [PUBMED: 22989478] [DOI] [PubMed] [Google Scholar]
- Savovic J, Jones HE, Altman DG, Harris RJ, Juni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomized, controlled trials. Annals of Internal Medicine 2012;157:429‐38. [PUBMED: 22945832] [DOI] [PubMed] [Google Scholar]
- Schomerus H, Hamster W, Blunck H, Reinhard U, Mayer K, Dolle W. Latent portasystemic encephalopathy. I. Nature of cerebral functional defects and their effect on fitness to drive. Digestive Diseases and Sciences 1981;26:622‐30. [PUBMED: 7249898] [DOI] [PubMed] [Google Scholar]
- Schomerus H, Hamster W. Neuropsychological aspects of portal‐systemic encephalopathy. Metabolic Brain Disease 1998;13:361‐77. [PUBMED: 10206827] [DOI] [PubMed] [Google Scholar]
- Sotil EU, Gottstein J, Ayala E, Randolph C, Blei AT. Impact of preoperative overt hepatic encephalopathy on neurocognitive function after liver transplantation. Liver Transplantation 2009;15:184‐92. [DOI] [PubMed] [Google Scholar]
- Spehlman R. EEG Primer. 2nd Edition. New York (NY): Elsevier, 1991. [Google Scholar]
- Stata Corp. Stata 14. Texas, USA: Stata Corp, 2007.
- Stepanova M, Mishra A, Venkatesan C, Younossi ZM. In‐hospital mortality and economic burden associated with hepatic encephalopathy in the United States from 2005 to 2009. Clinical Gastroenterology and Hepatology 2012;10:1034‐41. [PUBMED: 22642955] [DOI] [PubMed] [Google Scholar]
- Stewart CA, Malinchoc M, Kim WR, Kamath PS. Hepatic encephalopathy as a predictor of survival in patients with end‐stage liver disease. Liver Transplantation 2007;13:1366‐71. [DOI] [PubMed] [Google Scholar]
- Taylor‐Robinson SD, Sargentoni J, Marcus CD, Morgan MY, Bryant DJ. Regional variations in cerebral proton spectroscopy in patients with chronic hepatic encephalopathy. Metabolic Brain Disease 1994;9:347‐59. [PUBMED: 7898401] [DOI] [PubMed] [Google Scholar]
- Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet 1974;13:81‐4. [DOI] [PubMed] [Google Scholar]
- Thorlund K, Engstrøm J, Wetterslev J, Brok J, Imberger G, Gluud C. User manual for Trial Sequential Analysis (TSA), 2011. ctu.dk/tsa/files/tsa_manual.pdf (accessed 20 February 2017).
- Copenhagen Trial Unit. TSA ‐ Trial Sequential Analysis. Version 0.9 Beta. Copenhagen: Copenhagen Trial Unit, 2011.
- Vicini S, Alho H, Costa E, Mienville JM, Santi MR, Vaccarino FM. Modulation of y‐aminobutyric acid‐mediated inhibitory synaptic currents in dissociated cortical cell cultures. Proceedings of the National Academy of Sciences of the United States of America 1987;83:9269‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenborn K. Diagnosis of encephalopathy. Digestion 1998;59:22‐4. [DOI] [PubMed] [Google Scholar]
- Weissenborn K, Heidenreich S, Ennen J, Rückert N, Hecker H, et al. Attention deficits in minimal hepatic encephalopathy. Metabolic Brain Disease 2001;16:13‐9. [PUBMED: 11726083] [DOI] [PubMed] [Google Scholar]
- Wetterslev J, Thorlund K, Brok J, Gluud C. Trial sequential analysis may establish when firm evidence is reached in cumulative meta‐analysis. Journal of Clinical Epidemiology 2008;61:64‐75. [DOI] [PubMed] [Google Scholar]
- Wetterslev J, Jakobsen JC, Gluud C. Trial Sequential Analysis in systematic reviews with meta‐analysis. BMC Medical Research Methodology 2017;17(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwam JG, Amrein R. Pharmacology of flumazenil. Acta Anaesthesiologica Scandinavica. Supplementum 1995;108:3‐14. [DOI] [PubMed] [Google Scholar]
- Zipprich A, Garcia‐Tsao G, Rogowski S, Fleig WE, Seufferlein T, Dollinger MM. Prognostic indicators of survival in patients with compensated and decompensated cirrhosis. Liver International 2012;32:1407‐14. [PUBMED: 22679906] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
- Als‐Nielsen B, Kjaergaard LL, Gluud C. Benzodiazepine receptor antagonists for hepatic encephalopathy. Cochrane Database of Systematic Reviews 2001, Issue 4. [DOI: 10.1002/14651858.CD002798.pub2; PUBMED: 15106178] [DOI] [PubMed] [Google Scholar]
- Als‐Nielsen B, Gluud LL, Gluud C. Benzodiazepine receptor antagonists for hepatic encephalopathy. Cochrane Database of Systematic Reviews 2004, Issue 2. [DOI: 10.1002/14651858.CD002798.pub2; PUBMED: 15106178] [DOI] [PubMed] [Google Scholar]