

Abstract
This tutorial describes the development of a population pharmacokinetic (Pop PK) analysis guidance within Pfizer, which strives for improved consistency and efficiency, and a more systematic approach to model building. General recommendations from the Pfizer internal guidance and a suggested workflow for Pop PK model building are discussed. A description is also provided for mechanisms by which conflicting opinions were captured and resolved across the organization to arrive at the final guidance.
CPT: Pharmacometrics & Systems Pharmacology (2013) 2, e51; doi:10.1038/psp.2013.26; advance online publication 3 July 2013
RESULTS
The Pfizer Population Pharmacokinetic Analysis Guidance is included as Supplementary Appendix S1 online. The full content of the guidance and a general workflow are presented in Figure 1 and Figure 2 , respectively, and general recommendations are summarized below. It should be noted that the recommendations in the guidance were based on current best practice and state of knowledge. The guidance will be updated and revised on a regular basis as new methodologies are developed and the model‐building process is refined. The guidance was written with internal and external references to avoid in‐depth technical and theoretical discussion within the guidance itself: the full list of references applicable to the guidance can be found in the Reference section of the Supplementary Appendix S1 online.
Figure 1.

Table of content for the guidance.
Figure 2.
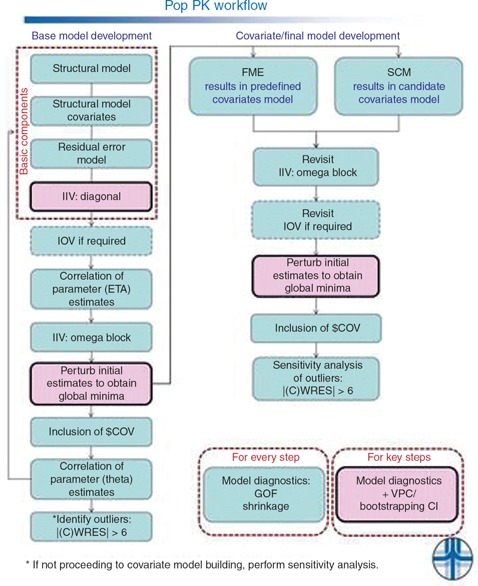
Population pharmacokinetic modeling workflow. CI, confidence interval; $COV, covariance step; CWRES, conditional weighted residuals; FME, full model estimation; GOF, goodness of fit; IIV, interindividual variance; IOV, interoccasional variance; SCM, stepwise covariate modeling; VPC, visual predictive check.
The guidance itself does not address tool‐specific implementation but is primarily focused on outlining the expected population pharmacokinetic (Pop PK) modeling–related processes and procedures that should be undertaken by the analyst. However, guidance recommendations are based on standard tools and relevant terminology, including NONMEM (ICON Development Solutions, Ellicott City, MD), 1 Perl speaks NONMEM (PsN), 2 and Xpose. 3
Points to consider before conducting a Pop PK analysis
Population modeling analysis plan. It is recommended that a population modeling analysis plan (PMAP) be developed to prospectively outline the modeling approach before conducting a Pop PK analysis. In addition, the PMAP should be finalized before database lock if the analysis results are to be included in a regulatory submission. A well‐prepared PMAP should provide an overview of the purpose of the modeling, prior information used, the choice of studies/data to be included for analysis, the proposed modeling approach, and assumptions made. The level of detail required in the PMAP depends on the intended use of the modeling analysis, as the plan in some cases can be considered a “living document,” i.e., updates to the plan can be made as more information becomes available. A PMAP should facilitate writing of the population modeling analysis report (PMAR) in a timely manner upon completion of model development and should be an effective planning tool both for the analyst and for any reviewer to assess whether the original objectives of the analysis were met.
Data check before modeling. An analyst is encouraged to work with the team, including study managers, data managers, and programmers, and to perform periodic data checks during the conduct of each study to ensure that correct information is collected. For example, dates and times for drug administration and PK samples should be recorded in sequential order, and any blank data fields in case report forms should be queried in a timely manner. When draft data becomes available, it is recommended that thorough data checks be conducted in advance of any analyses. Graphical and statistical summaries of dependent variables and demographics, including covariates, should be completed to help with identifying potential errors. In addition, this will help to identify the base structural model and components of the statistical model, as well as potential covariate relationships and outliers.
Below the limit of quantification. It is not uncommon that some concentration data are censored as below the limit of quantification (BLQ) by the bioanalytical laboratory and reported qualitatively in Pop PK data sets. Commonly used approaches for handling BLQ concentrations have been shown to introduce bias in the parameter estimates and to result in model misspecification. 4 , 5 , 6 , 7 , 8 Therefore, the analyst must understand that the method chosen for handling these samples may influence the estimation of PK parameters.
Although defining cutoff values may be arbitrary, it was generally agreed that if <10% of all observations are BLQ, then, with certain provisos about systematic trends in missingness, the missing values should not unduly influence building the base model. However, it is prudent to conduct a sensitivity analysis with inclusion of BLQ values. Likewise, more stringent criteria may be necessary; for example, cases for which trough measurements are important for characterizing the model.
When a substantial proportion of BLQ data is present, it is recommended that BLQ values be retained in the data set. In this case, the recommended approach is based on simultaneous modeling of continuous and categorical data in which the BLQ observations are treated as categorical data, an approach often referred to as the M3 method. 9 Particular caution is needed to avoid bias when producing visual predictive check (VPC) diagnostics in the presence of BLQ samples. 10 VPC methodology has been adapted to address this issue. 10
Pop PK model development
Pop PK base model. The base model is defined as the structural form of the model, such as one‐compartment vs. two‐compartment, and includes the specification of the interindividual and residual random effects, as well as the corresponding covariance structures. It is recommended that the analyst first refine the base structural model and then subsequently refine the residual error model. When prior information is available to identify influential covariates for the PK of a compound, it is recommended that those covariate parameters be included as part of the base structural model. For example, it may be appropriate to include creatinine clearance as a structural model covariate for clearance in the base model for drugs that are primarily cleared by the kidneys. Subsequent covariate model building would then start from the base model, which includes an effect of creatinine clearance on clearance.
In general, it is recommended that the interindividual variance (IIV) term be implemented using an exponential function to maintain positive PK parameter estimates. With reasonable structural and residual error models, a base model can then be tested and refined in an iterative process because of the interplay between different model components including structural, statistical, and covariate models. 11 The IIV should be introduced on the PK parameters for which the estimation of variability can be supported by the data. Consideration should be given to fitting a full‐block omega structure on the base model, followed by inspection of the correlations among the ETAs to guide the development of a parsimonious omega structure. It should be recognized that an extremely small estimate of IIV approaching zero does not necessarily indicate that there is a lack of IIV for the parameter, rather it may simply suggest that the data are not robust enough to estimate the IIV or that there is high shrinkage in the variance due to sparse data. 12
Pop PK final model. The final Pop PK model is defined as the most parsimonious model for which all relevant covariates of interest have been evaluated and retained. It is recommended that systematic procedures be incorporated for covariate model building to improve consistency and harmonization across analyses. Although a covariate selection procedure can result in a model that fits the data well, there are no guarantees that the most parsimonious model will be obtained, especially if collinearity exists among the covariates. Therefore, it is imperative that the scientific (i.e., pharmacologic, biologic, pathophysiologic, clinical, etc.) merits of each covariate be assessed before covariate model‐building procedures.
Two main approaches are recommended for inclusion and evaluation of covariates to arrive at the final model: the full model estimation (FME) 13 and stepwise covariate modeling (SCM) 14 , 15 , 16 , 17 approaches. The FME approach retains all prespecified covariates in the final model (often referred as a full model), and inferences about the evidence in support of a particular covariate effect are based on the confidence intervals around the parameter estimates. The SCM approach includes a forward selection (full covariate model) followed by a backward elimination process (final model). Regardless of the approach used, consideration should be given to further model refinement when the inferences for covariates are not clinically meaningful, even though analysis results indicate that covariate effects may be statistically significant. A list of the commonly used covariate model‐building procedures, along with the pros and cons for each, can be found in the Supplementary Appendix S1 online.
Assessment of model adequacy. Assessment of model adequacy and the assumptions used in model building have been well discussed in regulatory guidances and other literature. 12 , 18 , 19 , 20 , 21 It is recommended that models be evaluated at all stages of development using the following criteria, which include a proposed list of recommended diagnostics:
Successful minimization
Likelihood ratio test
-
Inspection of graphical and numerical diagnostics
Assessment of shrinkage of ETA(η) and EPSILON(ε)
Observations vs. population predicted value (PRED) in both linear and log scales with a line of identity and a regression line
Observations vs. individual PRED in both linear and log scales with a line of identity and a regression line
Weighted residuals (WRES) or conditional WRES vs. PRED
Absolute individual WRES (|IWRES|) vs. individual PRED
WRES or conditional WRES vs. time or time after dose
Observations (or dependent variable), individual PRED, and PRED concentrations vs. time (overlaid and/or side by side)
Histogram and/or QQ plot of IWRES and WRES or conditional WRES
VPCs
Theoretical and technical details of each criteria can be found in the Supplementary Appendix S1 online. To assess model fit across the design space of the available data, it is recommended that all diagnostic plots be stratified by key design features and covariates. The limitations of some of these diagnostics are acknowledged, 21 but it is proposed that these criteria form a minimal cassette of model evaluation techniques. It is recommended that all models be fit with multiple sets (three at a minimum) of perturbed initial estimates to lessen the likelihood of final parameter estimates occurring at a local minimum. Use of the likelihood ratio test as a sole criterion for successful model minimization is not recommended due to an inflated type I error. 6 , 20 , 22 Ultimately, the analyst should assess whether the model adequately describes the data, and whether the model is fit for inferential purposes and for prediction into a suitable population.
Opinion varies as to whether a successful estimate of the variance–covariance matrix (from the covariance step using $COV) is a necessary condition for a robust final model. Certainly, successful completion of the covariance step does not in itself guarantee or convey any robustness or veracity to model inferences; however, for predictive purposes into future populations, a measure of the variance–covariance matrix of fixed and random effects is preferred. When the $COV step fails, it often implies that the model is overparameterized; therefore, the analyst should strive to build models with a successful $COV step. However, it should also be recognized that successful $COV runs may not always be possible due to limitations in the data. Under such circumstances, a successful run with a failed $COV step should not necessarily be discounted, as useful information about parameter estimates may still be provided and the covariance estimate can be better obtained by implementing a bootstrapping method. 23 , 24 , 25
Diagnostics based on the predictions and residuals from the fitted model provide some evidence of model adequacy. However, inappropriately applied, these can sometimes mask model deficiencies and can occasionally be misleading. 21 Simulation‐based diagnostics enable assessment of model fits via generation of new data sets through simulation, followed by an assessment of the current model vs. that simulated data. 21 , 26 , 27 , 28 , 29 A VPC is a model evaluation method that allows the analyst to assess how well the current model describes the observed data by comparing various summary statistics. Although the theoretical and technical details of VPC can be found in the Supplementary Appendix S1 online and associated references, the importance of VPC plot styles is somewhat dependent upon the intended audience and the purpose of the plot. Two VPC plot styles are shown in Figure 3 , including one for nontechnical audiences and one for more technical audiences. The PsN script to generate the VPC and the R script to create these plots are included in the Supplementary Appendix S2 online.
Figure 3.
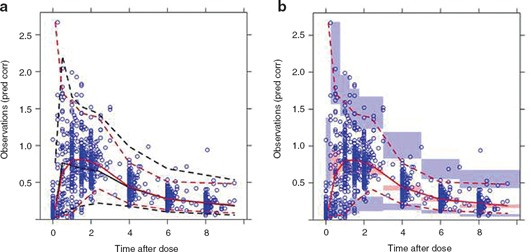
Visual predictive check for (a) nontechnical and (b) technical audiences. Pred Corr, prediction corrected. Blue circles represent observed data. Red lines represent the percentiles of the observed data: solid red lines represent the median; dashed red lines represent the upper and lower percentiles; i.e., 10% and 90%, respectively. The black lines represent the median, upper, and lower percentiles of the simulated data in each bin (aggregating across simulated trials). The shaded regions summarize the percentiles within each bin for each simulated trial. For each simulated trial, the median, lower, and upper percentiles are calculated for each bin, and then a percentile range of these percentiles is shown as the shaded region.
Population modeling analysis report. Once the planned analysis is completed, the results should be summarized in a PMAR. It is important to stress that any assumptions and decisions made during model development be clearly documented in the PMAR. Sensitivity analyses should be performed to test assumptions, and the impact of those assumptions on parameter estimates should be evaluated and documented in the PMAR. For example, missing covariates in a data set could be handled in various ways, such as omission of all records for the subject with the missing covariates or imputation of the missing covariates. The procedure chosen should be specified and documented in the PMAR, and sensitivity analyses should be performed to evaluate the impact of the procedure for handling missing covariates on parameter estimates, i.e., running models with and without those subjects with missing covariates. In addition, any deviations from the PMAP must be clearly delineated in the PMAR, including the specific deviations implemented and rationale.
While the analysis report should summarize the technical aspects of modeling, the clinical meaning of a complicated statistical model should also be addressed in the report and appropriately communicated based on the target audience. As shown in Figure 3 , effective communication on the results of modeling that is well tailored to the target audience would enhance its impact on the decision‐making process in the drug development. 30
DISCUSSION
“A major quest of population pharmacokinetics is to discover which measurable pathophysiologic factors cause changes in the dose–concentration relationship and to estimate the degree to which they do so, so that appropriate dosage adjustments can be made.” Sheiner and Benet 31
The paradigm of model‐based drug development has steered the drug‐development decision‐making process toward a more efficient and quantitative approach over the past three decades. 32 Pharmacometrics is a science that quantifies drug, disease, and trial information to aid efficient drug development, regulatory decisions, and rational drug treatment in patients. Pharmacometric principles are used in model‐based drug development, which has become a standard component of regulatory submissions, as evidenced by the dramatic sixfold increase in the number of new drug applications that include pharmacometric analyses over a recent 9‐year span (from 2000 through to 2008). 33 Pop PK modeling is an integral part of model‐based drug development, representing the major component of pharmacometric analyses submitted. 33 Pop PK analyses aim to characterize the pharmacokinetic properties of a drug and to quantify sources of variability in drug concentrations among individuals by estimating the impact of intrinsic and extrinsic factors that may affect the PK. 18 , 19 Therefore, the results of Pop PK analyses are often included in drug labels, typically supporting dosing recommendations for special populations (e.g., renal/hepatic impairment, pediatrics, elderly, etc.) as well as the clinical relevance of drug–drug interactions.
Despite the rapidly changing environment around this relatively new field, the pharmacometrics community has expended great effort to provide guidelines and standards for these types of analyses. 18 , 19 , 34 , 35 , 36 , 37 , 38 , 39 Guidance from regulatory agencies outlines information that should be included in Pop PK analysis reports. 18 , 19 In contrast to the US Food and Drug Administration (FDA), where there is a Division of Pharmacometrics, European regulatory agencies do not reanalyze a sponsor's data. To that end, the focus of the European Medicines Agency, for example, is to assess whether the model adequately describes the data and whether the inferences made about the model are appropriate based on the information submitted in the sponsor's report. Edholm 40 points out that sponsors still fail to provide sufficiently clear information to enable adequate assessment of models, leading to additional questions and increased review time. Jönsson et al. 41 also note the difficulty in creating more general guidance on modeling and simulation methodology and acceptable uses due to the rapid evolution of techniques and methods. The FDA Population Pharmacokinetics Guidance for Industry was issued in 1999, 18 and although the sections that address expectations for reporting Pop PK analyses continue to be pertinent, it could be argued that methodology, techniques, and software tools have certainly progressed over the interim years. More recent publications provide excellent discussions on the common and fundamental aspects of Pop PK modeling and validation, in addition to quality‐assurance guidelines for population analyses. 34 , 35 , 39 However, to the authors’ best knowledge, a general guidance which addresses the Pop PK modeling process from an initial analysis plan to a final report has not been published in the literature.
Within a large pharmaceutical organization such as Pfizer, the development and implementation of standard procedures for conducting and reporting Pop PK analyses is a step toward increased consistency, high quality output, and industrialization of this key step in model‐based drug development. 42 , 43 Improved consistency should also result in greater efficiency since standardized software tool sets can be exploited, facilitating reuse of code, knowledge sharing, educational training, and easier and more efficient quality control of the work conducted. Therefore, the creation of an internal guidance is expected to enable harmonization and standardization of the Pop PK analysis process and efficient training across the organization. In addition, a written guidance provides the opportunity for rapid updates to reflect best practice in the modeling community. A collaborative forum (wiki pages) was successfully implemented to facilitate discussions, capture differing viewpoints, and assist in collaborative writing of the guidance across the Global Clinical Pharmacology community at Pfizer. The wiki will also serve as an archive and a future source of feedback and input for guidance updates and revisions. Although the scope and focus of this guidance is to provide recommendations for Pop PK analyses, many of the principles outlined here are also applicable to pharmacokinetic and pharmacodynamic analyses.
After release of the guidance, future goals and objectives were developed to continue improvement of the guidance document. The following goals over a 2‐year period were agreed: (i) updating the wiki pages with the current recommendations from the finalized guidance document, (ii) providing standard examples of the Pop PK workflow as described in Figure 2 , (iii) development of a plan to collect data on the utilization of the guidance document within the organization, and (iv) identifying additional topics to be included in a subsequent version of the document. Some additional topics under consideration include model building to address drug absorption, particular issues pertaining to modeling of sparse data, and prediction and bridging of population models to special populations, such as pediatrics. The guidance should also be kept updated with current research and method development in areas such as model diagnostics and covariate model building.
In conclusion, the development of a Pop PK guidance was successfully implemented across a large pharmaceutical organization. This experience may provide helpful advice to those in the industry who are interested in forming their own guidance. Because drug development is a highly regulated area of science, the importance and necessity of implementing systematic, streamlined, and standardized approaches to optimize and harmonize the processes and procedures that contribute to the Pop PK analysis cannot be overstated. As this is an area of continually evolving science and technology, guidances should be considered “living documents” and, as such, should be updated and revised periodically.
METHODS
An internal Pop PK modeling guidance was developed to provide a set of recommendations to assist analysts with the process of Pop PK model building, while maintaining consistency with regulatory guidance, current “best practice”, and methodological advances using available software tools and methods. As the Pfizer Global Clinical Pharmacology organization comprises over 180 colleagues who provide support to research and development in the United States, United Kingdom, Sweden, Japan, and China, the guidance is intended to harmonize, streamline, and optimize practices across the clinical drug‐development spectrum. The breadth of experience in modeling and simulation within the organization is considerable; however, sharing this experience across such a large organization presents logistical problems, not the least of which include time zone issues. Previous internal materials on the topic of model building were used as references in providing the initial position on many key topics within the guidance.
In an effort to capture “standard practice” in Pop PK model building and to serve both as the basis for documentation and training material, a series of discussion group meetings were convened to address key topics such as definition of base, full, and final models; model diagnostics; and model selection. The question‐based approach and output from these discussions were used to initially populate the wiki repository to facilitate discussions across the Pfizer Global Clinical Pharmacology organization. The wiki pages functioned as a platform for collaboration and enabled input from colleagues who were not directly involved with writing the guidance, captured pertinent examples that illustrated some of the discussion points, and maintained a list of links to references, such as training material or manuscripts. This approach was instrumental in allowing colleagues across all sites to gain access to the discussion and the chance to provide input. It also allowed contrasting views and opinions to be captured and provided input for subsequent stages of review.
Following establishment of the wiki pages, an editorial board, consisting of 10 colleagues from different global regions, was organized to compile, consolidate, and incorporate the feedback captured within the wiki pages into a guidance. This editorial board convened weekly to review the collated input from the wiki pages, agreed on recommendations for each topic, and incorporated those recommendations into an integrated document that would form the Pop PK guidance. Each editorial board member was allocated a topic for which he or she was responsible to lead discussions based on comments submitted to the wiki pages. Subsequently, each board member was responsible for producing their appropriate section of the Pop PK guidance document. Once each section was completed, all sections were compiled into a single document, and the editorial board reviewed the guidance in its entirety for consistency across sections. Following approval of the draft guidance by the editorial board, it was distributed to the entire Global Clinical Pharmacology organization, including senior management, for review and feedback. Once all appropriate and relevant feedback were incorporated, the guidance was finalized. At that point, the wiki pages were updated to reflect the recommendations provided in the final guidance, and the original wiki pages, including all comments, were archived for reference purposes.
To illustrate the question‐based approach to generating discussion and how consensus was reached after discussing collated views on wiki pages, an example of the “covariate model building” topic is shared below:
-
Questions on wiki page:
Which covariates should be tested on which parameters? What form of covariate relationships should be evaluated first?
Should we use (automated) covariate selection methods or test all potential covariates simultaneously?
How do we guard against over‐parameterization and collinearity in covariate relationships?
-
Some comments left on wiki page (by the Global Clinical Pharmacology colleagues):
“Never include all covariates!! Bear in mind the questions you want to address. Do any of the covariates warrant altered dosing?”
“I think that we need some discussion on what parameters should be tested on covariates. For example, most of covariates testing is on clearance parameter, some on volume of distribution, and some on ka. Complexity comes in especially when these parameters have high correlations on ETAs, i.e., need an omega block to describe the correlations of the ETAs among these PK parameters.”
“If the final model is a full model in the sense of prespecifying all of the effects you really want to describe, then you're done at this step and your (statistical) inferences are fairly straightforward (with respect, for example, to confidence intervals). If this is a full model that you plan to reduce, then I'm not sure we're interested so much in inference from the full model, are we?”
Subsequent discussions within the Editorial Board
Before addressing covariate model‐building procedures, there was a considerable discussion regarding the definition of “base” and “final” PK models. For example, in defining a “base” model, the discussion focused on whether the base model should include covariates as part of the structural model. Allometric scaling on clearance and V parameters and metabolism‐specific covariates known a priori to affect PK are two examples of covariates that may be included as part of the structural model. There was also discussion regarding the structure of the variance–covariance matrix of random‐effects parameters, as well as which parameters should include random effects in the base model. To resolve these issues, reaching agreement up front on the definition of the base model was helpful. A Pop PK base model not only forms the basis from which to perform covariate model building but can be used to provide initial estimates of individual concentration profiles for assessing initial safety and/or efficacy from early‐phase clinical trials. It was agreed that key covariates should be included in base models, and IIV should be modeled for key model parameters, allowing the model to adequately describe individual profiles for this purpose.
Following agreement on the definitions of “base” and “final” models, the main discussion point around model development from base model to final model was identifying a suitable strategy for covariate identification. Discussion of these topics split the editorial board into two main camps: one group who preferred a method of estimation including prespecified covariates that were suspected a priori of having an influence on PK parameters (FME) and another group who were prepared to use automated covariate selection techniques to screen for statistically significant covariates before model refinement (SCM). An internal survey revealed that these two methods were the most frequently used procedures for incorporating covariates, followed by backward elimination only and Wald's approximation to likelihood ratio test. 44 Both FME and SCM approaches were recommended to cover cases for which there are covariate relationships that are known or strongly suspected to have influence on PK parameters a priori (FME), as well as a method that could be used for more exploratory model building when relationships are not well established (SCM).
Conflict of interest
W.B., M.K.S., P.C., M.A.T., S.R., H.D., J.D., A.R.‐G., K.S., and C.C. are all employees of Pfizer and received salaries and benefits commensurate with employment.
Supporting information
Supplementary Appendix S1
Supplementary Appendix S2
Acknowledgments
The authors acknowledge Pfizer Global Clinical Pharmacology colleagues who provided valuable input, discussion, and feedback to the questions posed on the wiki pages and contributed to internal and external reference materials. The authors also thank Kenneth G Kowalski for his significant contribution to “Points for Consideration: Statistical Aspects of Population Pharmacokinetic and Pharmacodynamic Modeling” given as ref. 1 in Supplementary Appendix S1 online.
References
- 1. Beal, S.L. & Sheiner, L.B. NONMEM User's Guides. NONMEM Project Group (University of California, San Francisco, 1992). [Google Scholar]
- 2. Perl‐speaks‐NONMEM. <http://psn.sourceforge.net/>.
- 3. Jonsson, E.N. & Karlsson, M.O. Xpose–an S‐PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput. Methods Programs Biomed. 58, 51–64 (1999). [DOI] [PubMed] [Google Scholar]
- 4. Ahn, J.E. , Karlsson, M.O. , Dunne, A. & Ludden, T.M. Likelihood based approaches to handling data below the quantification limit using NONMEM VI. J. Pharmacokinet. Pharmacodyn. 35, 401–421 (2008). [DOI] [PubMed] [Google Scholar]
- 5. Bergstrand, M. & Karlsson, M.O. Handling data below the limit of quantification in mixed effect models. AAPS J. 11, 371–380 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Byon, W. , Fletcher, C.V. & Brundage, R.C. Impact of censoring data below an arbitrary quantification limit on structural model misspecification. J. Pharmacokinet. Pharmacodyn. 35, 101–116 (2008). [DOI] [PubMed] [Google Scholar]
- 7. Duval, V. & Karlsson, M.O. Impact of omission or replacement of data below the limit of quantification on parameter estimates in a two‐compartment model. Pharm. Res. 19, 1835–1840 (2002). [DOI] [PubMed] [Google Scholar]
- 8. Hing, J.P. , Woolfrey, S.G. , Greenslade, D. & Wright, P.M. Analysis of toxicokinetic data using NONMEM: impact of quantification limit and replacement strategies for censored data. J. Pharmacokinet. Pharmacodyn. 28, 465–479 (2001). [DOI] [PubMed] [Google Scholar]
- 9. Beal, S.L. Ways to fit a PK model with some data below the quantification limit. J. Pharmacokinet. Pharmacodyn. 28, 481–504 (2001). [DOI] [PubMed] [Google Scholar]
- 10. Bergstrand, M. , Hooker, A.C. & Karlsson, M.O. Visual predictive checks for censored and categorical data. 2009, p 18, Abstr 1604.
- 11. Wade, J.R. , Beal, S.L. & Sambol, N.C. Interaction between structural, statistical, and covariate models in population pharmacokinetic analysis. J. Pharmacokinet. Biopharm. 22, 165–177 (1994). [DOI] [PubMed] [Google Scholar]
- 12. Savic, R.M. & Karlsson, M.O. Importance of shrinkage in empirical bayes estimates for diagnostics: problems and solutions. AAPS J. 11, 558–569 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gastonguay, M. A full model estimation approach for covariate effects: inference based on clinical importance and estimation precision. AAPS J. 6, Abstract W4354 (2004). [Google Scholar]
- 14. Mandema, J.W. , Verotta, D. & Sheiner, L.B. Building population pharmacokinetic--pharmacodynamic models. I. Models for covariate effects. J. Pharmacokinet. Biopharm. 20, 511–528 (1992). [DOI] [PubMed] [Google Scholar]
- 15. Jonsson, E.N. & Karlsson, M.O. Automated covariate model building within NONMEM. Pharm. Res. 15, 1463–1468 (1998). [DOI] [PubMed] [Google Scholar]
- 16. Wahlby, U. , Jonsson, E.N. & Karlsson, M.O. Comparison of stepwise covariate model building strategies in population pharmacokinetic‐pharmacodynamic analysis. AAPS PharmSci. 4, E27 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lindbom, L. , Pihlgren, P. & Jonsson, E.N. PsN‐Toolkit--a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput. Methods Programs Biomed. 79, 241–257 (2005). [DOI] [PubMed] [Google Scholar]
- 18. U.S. Department of Health and Human Services, Food and Drug Administration . Guidance for industry. population pharmacokinetics. <http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/WomensHealthResearch/UCM133184.pdf> (1999).
- 19. European Medicines Agency. Committee for Medicinal Products for Human Use . Guideline on reporting the results of populationa pharmacokinetic analyses. <http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003067.pdf>.
- 20. Wahlby, U. , Jonsson, E.N. & Karlsson, M.O. Assessment of actual significance levels for covariate effects in NONMEM. J. Pharmacokinet. Pharmacodyn. 28, 231–252 (2001). [DOI] [PubMed] [Google Scholar]
- 21. Karlsson, M.O. & Savic, R.M. Diagnosing model diagnostics. Clin. Pharmacol. Ther. 82, 17–20 (2007). [DOI] [PubMed] [Google Scholar]
- 22. Stram, D.O. & Lee, J.W. Variance components testing in the longitudinal mixed effects model. Biometrics 50, 1171–1177 (1994). [PubMed] [Google Scholar]
- 23. Efron, B. Bootstrap methods: another look at the jackknife. The Annal of Statistics 7, 1–26 (1979). [Google Scholar]
- 24. Efron, B. & Tibhirani, R.J. An Introduction to the Bootstrap. (Chapman and Hall/CRC, London, UK, 1994). [Google Scholar]
- 25. Ette, E.I. , Williams, P.J. , Kim, Y.H. , Lane, J.R. , Liu, M.J. & Capparelli, E.V. Model appropriateness and population pharmacokinetic modeling. J. Clin. Pharmacol. 43, 610–623 (2003). [PubMed] [Google Scholar]
- 26. Holford, N. The visual predictive check – superiority to standard diagnostic (Rorschach) plots. 2005, p 14, Abstr 738.
- 27. Karlsson, M.O. & Holford, N. A tutorial on visual predictive checks. 2008, p 17, Abstr 1434.
- 28. Yano, Y. , Beal, S.L. & Sheiner, L.B. Evaluating pharmacokinetic/pharmacodynamic models using the posterior predictive check. J. Pharmacokinet. Pharmacodyn. 28, 171–192 (2001). [DOI] [PubMed] [Google Scholar]
- 29. Bergstrand, M. , Hooker, A.C. , Wallin, J.E. & Karlsson, M.O. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 13, 143–151 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Menon‐Andersen, D. et al Essential pharmacokinetic information for drug dosage decisions: a concise visual presentation in the drug label. Clin. Pharmacol. Ther. 90, 471–474 (2011). [DOI] [PubMed] [Google Scholar]
- 31. Sheiner, L.B. & Benet, L.Z. Premarketing observational studies of population pharmacokinetics of new drugs. Clin. Pharmacol. Ther. 38, 481–487 (1985). [DOI] [PubMed] [Google Scholar]
- 32. Lalonde, R.L. et al Model‐based drug development. Clin. Pharmacol. Ther. 82, 21–32 (2007). [DOI] [PubMed] [Google Scholar]
- 33. Lee, J.Y. , et al Impact of pharmacometric analyses on new drug approval and labelling decisions: a review of 198 submissions between 2000 and 2008. Clin. Pharmacokinet. 50, 627–635 (2011). [DOI] [PubMed] [Google Scholar]
- 34. Kiang, T.K. , Sherwin, C.M. , Spigarelli, M.G. & Ensom, M.H. Fundamentals of population pharmacokinetic modelling: modelling and software. Clin. Pharmacokinet. 51, 515–525 (2012). [DOI] [PubMed] [Google Scholar]
- 35. Bonate, P.L. et al Guidelines for the quality control of population pharmacokinetic‐pharmacodynamic analyses: an industry perspective. AAPS J. 14, 749–758 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grasela, T. The application of systematic analysis for identifying and addressing the needs of the pharmacometric process, 2007, p 16, Abstr 1131. <http://www.page‐meeting.org/?abstract=1131>.
- 37. Mould, D.R. & Upton, R.N. Basic concepts in population modeling, simulation, and model‐based drug development. CPT: Pharmacomet. Syst. Pharmacol. 1, e6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Karlsson, M.O. , Jonsson, E.N. , Wiltse, C.G. & Wade, J.R. Assumption testing in population pharmacokinetic models: illustrated with an analysis of moxonidine data from congestive heart failure patients. J. Pharmacokinet. Biopharm. 26, 207–246 (1998). [DOI] [PubMed] [Google Scholar]
- 39. Sherwin, C.M. , et al Fundamentals of population pharmacokinetic modelling: validation methods. Clin. Pharmacokinet. 51, 573–590 (2012). [DOI] [PubMed] [Google Scholar]
- 40. Edholm, M. Modeling and simulation examples that failed to meet regulator's expectations. European Medicines Agency‐European Federation of Pharmaceutical Industries and Associations Modelling and Simulation Workshop, London, UK, November 2011.
- 41. Jönsson, S. , Henningsson, A. , Edholm, M. , & Salmonson, T. Contribution of modeling and simulation studies in the regulatory review: a European regulatory perspective. In Clinical Trial Simulations: Applications and Trends. AAPS Series. Ch. 2, 15–36 (Springer, New York, 2011). [Google Scholar]
- 42. Romero, K. , et al Pharmacometrics as a discipline is entering the “industrialization” phase: standards, automation, knowledge sharing, and training are critical for future success. J. Clin. Pharmacol. 50, 9S–19S (2010). [DOI] [PubMed] [Google Scholar]
- 43. Manolis, E. , Rohou, S. , Hemmings, R. , Salmonson, T. , Karlsson, M. & Milligan, P.A. The role of modeling and simulation in development and registration of medicinal products: output from the EFPIA/EMA Modeling and Simulation Workshop. CPT: Pharmacomet. Syst. Pharmacol. 2, e31 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kowalski, K.G. & Hutmacher, M.M. Efficient screening of covariates in population models using Wald's approximation to the likelihood ratio test. J. Pharmacokinet. Pharmacodyn. 28, 253–275 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Appendix S1
Supplementary Appendix S2