

Abstract
Background
Major depressive disorder is a common mental disorder affecting a person's mind, behaviour and body. It is expressed as a variety of symptoms and is associated with substantial impairment. Despite a range of pharmacological and non‐pharmacological treatment options, there is still room for improvement of the pharmacological treatment of depression in terms of efficacy and tolerability. The latest available antidepressant is vortioxetine. It is assumed that vortioxetine's antidepressant action is related to a direct modulation of serotonergic receptor activity and inhibition of the serotonin transporter. The mechanism of action is not fully understood, but it is claimed to be novel. Vortioxetine was placed in the category of "Other" antidepressants and may therefore provide an alternative to existing antidepressant drugs.
Objectives
To assess the efficacy and acceptability of vortioxetine compared with placebo and other antidepressant drugs in the treatment of acute depression in adults.
Search methods
We searched Cochrane's Depression, Anxiety and Neurosis Review Group's Specialised Register to May 2016 without applying any restrictions to date, language or publication status. We checked reference lists of relevant studies and reviews, regulatory agency reports and trial databases.
Selection criteria
We included randomised controlled trials comparing the efficacy, tolerability, or both of vortioxetine versus placebo or any other antidepressant agent in the treatment of acute depression in adults.
Data collection and analysis
Two review authors independently selected the studies and extracted data. We extracted data on study characteristics, participant characteristics, intervention details and outcome measures in terms of efficacy, acceptability and tolerability. We analysed intention‐to‐treat (ITT) data only and used risk ratios (RR) as effect sizes for dichotomous data and mean differences (MD) for continuous data with 95% confidence intervals (CI). Meta‐analyses used random‐effects models.
Main results
We included 15 studies (7746 participants) in this review. Seven studies were placebo controlled; eight studies compared vortioxetine to serotonin‐norepinephrine reuptake inhibitors (SNRIs). We were unable to identify any study that compared vortioxetine to antidepressant drugs from other classes, such as selective serotonin reuptake inhibitors (SSRIs).
Vortioxetine may be more effective than placebo across the three efficacy outcomes: response (Mantel‐Haenszel RR 1.35, 95% CI 1.22 to 1.49; 14 studies, 6220 participants), remission (RR 1.32, 95% CI 1.15 to 1.53; 14 studies, 6220 participants) and depressive symptoms measured using the Montgomery‐Åsberg Depression Scale (MADRS) (score range: 0 to 34; higher score means worse outcome: MD ‐2.94, 95% CI ‐4.07 to ‐1.80; 14 studies, 5566 participants). The quality of the evidence was low for response and remission and very low for depressive symptoms. We found no evidence of a difference in total dropout rates (RR 1.05, 95% CI 0.93 to 1.19; 14 studies, 6220 participants). More participants discontinued vortioxetine than placebo because of adverse effects (RR 1.41, 95% CI 1.09 to 1.81; 14 studies, 6220 participants) but fewer discontinued due to inefficacy (RR 0.56, 95% CI 0.34 to 0.90, P = 0.02; 14 studies, 6220 participants). The quality of the evidence for dropouts was moderate.The subgroup and sensitivity analyses did not reveal factors that significantly influenced the results.
In comparison with other antidepressants, very low‐quality evidence from eight studies showed no clinically significant difference between vortioxetine and SNRIs as a class for response (RR 0.91, 95% CI 0.82 to 1.00; 3159 participants) or remission (RR 0.89, 95% CI 0.77 to 1.03; 3155 participants). There was a small difference favouring SNRIs for depressive symptom scores on the MADRS (MD 1.52, 95% CI 0.50 to 2.53; 8 studies, 2807 participants). Very low quality evidence from eight studies (3159 participants) showed no significant differences between vortioxetine and the SNRIs as a class for total dropout rates (RR 0.89, 95% CI 0.73 to 1.08), dropouts due to adverse events (RR 0.74, 95% CI 0.51 to 1.08) and dropouts due to inefficacy (RR 1.52, 95% CI 0.70 to 3.30).
Against individual antidepressants, analyses suggested that vortioxetine may be less effective than duloxetine in terms of response rates (RR 0.86, 95% CI 0.79 to 0.94; 6 studies, 2392 participants) and depressive symptoms scores on the MADRS scale (MD 1.99, 95% CI 1.15 to 2.83; 6 studies; 2106 participants). Against venlafaxine, meta‐analysis of two studies found no statistically significant differences (response: RR 1.03, 95% CI 0.85 to 1.25; 767 participants; depressive symptom scores: MD 0.02, 95% CI ‐2.49 to 2.54; 701 participants). In terms of number of participants reporting at least one adverse effect (tolerability), vortioxetine was better than the SNRIs as a class (RR 0.90, 95% CI 0.86 to 0.94; 8 studies, 3134 participants) and duloxetine (RR 0.89, 95% CI 0.84 to 0.95; 6 studies; 2376 participants). However, the sensitivity analysis casts some doubts on this result, as only two studies used comparable dosing.
We judged none of the studies to have a high risk of bias for any domain, but we rated all studies to have an unclear risk of bias of selective reporting and other biases.
Authors' conclusions
The place of vortioxetine in the treatment of acute depression is unclear. Our analyses showed vortioxetine may be more effective than placebo in terms of response, remission and depressive symptoms, but the clinical relevance of these effects is uncertain. Furthermore, the quality of evidence to support these findings was generally low. In comparison to SNRIs, we found no advantage for vortioxetine. Vortioxetine was less effective than duloxetine, but fewer people reported adverse effects when treated with vortioxetine compared to duloxetine. However, these findings are uncertain and not well supported by evidence. A major limitation of the current evidence is the lack of comparisons with the SSRIs, which are usually recommended as first‐line treatments for acute depression. Studies with direct comparisons to SSRIs are needed to address this gap and may be supplemented by network meta‐analyses to define the role of vortioxetine in the treatment of depression.
Plain language summary
Vortioxetine for the treatment of depression in adults
Why is this review important?
Many people suffer from major depression. Major depression is a serious illness that can cause significant distress both to patients and their families. Major depression affects people's work and relationships, but can also affect people physically, for example by changing concentration or appetite. Available antidepressant medicines are not always effective in treating major depression and may also have unpleasant side effects. This review compares a new antidepressant, vortioxetine, to placebo (a pretend treatment, e.g. sugar tablet) and other antidepressants. It is assumed that vortioxetine works differently from other available antidepressants and it is important to know if it is an effective treatment and a possible alternative for already available treatments.
Who will be interested in this review?
People affected by major depression and their families, general practitioners (GPs), psychiatrists, and pharmacists and other professionals working in adult mental health services.
What questions does this review aim to answer?
Is vortioxetine more effective than placebo in treating individual with an episode of major depression? Is vortioxetine more or less effective than other available antidepressant treatments? Do more or fewer people stay in treatment when treated with vortioxetine compared to placebo or other antidepressants? Do more or fewer people have side effects when treated with vortioxetine compared to other antidepressants?
Which studies were included in the review?
In May 2016, we searched electronic medical databases to find trials that compared vortioxetine to placebo or other antidepressants. We included only studies that used a randomised controlled design (where people were randomly put into one of two or more treatment groups) and had adults (aged over 18 years) with a diagnosis of major depression. We included 15 trials, involving 7746 participants in the review.
What does the evidence from the review tell us?
The quality of the evidence ranged from very low to moderate, depending on the outcome (what symptom or effect was measured) and the comparison. Vortioxetine was more effective than placebo, but it was not more effective than other commonly used antidepressants. The studies found no difference in people stopping their treatment compared to placebo or other antidepressants. Vortioxetine was only compared to one type of medicine (called SNRIs) and not compared to the most frequently prescribed antidepressants. The outcomes varied markedly across studies.
What should happen next?
No firm conclusion on vortioxetine can be made. Vortioxetine was effective in treating acute major depression, but did not show a clear advantage in comparison with some treatments which are already available. Conclusions are also made difficult because comparisons to the most frequently prescribed antidepressants (called SSRIs) are lacking. Furthermore, it is unclear if vortioxetine has an advantage in specific side effects associated with commonly prescribed antidepressants, for example sexual problems. These questions should be addressed in future studies.
Summary of findings
Summary of findings for the main comparison. Vortioxetine compared to Placebo for adults with Major Depressive Disorder.
| Vortioxetine compared to Placebo for adults with Major Depressive Disorder | ||||||
| Patient or population: adults with Major Depressive Disorder Setting: Inpatients and outpatients Intervention: Vortioxetine Comparison: Placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with Placebo | Risk with Vortioxetine | |||||
| Response assessed with: reduction of at least 50% on the HAMD scale or MADRS scale, or any other score 1 or 2 on CGI‐I follow up: range 6 weeks to 8 weeks | Study population | RR 1.35 (1.22 to 1.49) | 6220 (14 RCTs) | ⊕⊕⊝⊝ LOW 1 2 | All studies were sponsored by the pharmaceutical companies that manufacture vortioxetine. Small difference favouring vortioxetine. | |
| 356 per 1,000 | 480 per 1,000 (434 to 530) | |||||
| Total number of drop‐outs follow up: range 6 weeks to 8 weeks | Study population | RR 1.05 (0.93 to 1.19) | 6220 (14 RCTs) | ⊕⊕⊕⊝ MODERATE 1 | No difference between vortioxetine and placebo. | |
| 160 per 1,000 | 168 per 1,000 (149 to 190) | |||||
| Remission assessed with: 7 points or less on the 17‐item HAM‐D and 8 points or less for longer HAM‐D versions; 10 or less points on the MADRS; score 1 or 2 on CGI‐S follow up: range 6 weeks to 8 weeks | Study population | RR 1.33 (1.15 to 1.53) | 6217 (14 RCTs) | ⊕⊕⊝⊝ LOW 1 2 | Small difference favouring vortioxetine. | |
| 224 per 1,000 | 299 per 1,000 (258 to 343) | |||||
| Depressive Symptoms assessed with: MADRS score (score range: 0‐34; higher score means worse outcome) follow up: range 6 weeks to 8 weeks | The change in depressive symptoms score ranged from 10.8 to 15.9 points | The change was 2.94 points higher (1.8 higher to 4.07 higher) | ‐ | 5566 (14 RCTs) | ⊕⊝⊝⊝ VERY LOW 1 3 | Small difference favouring vortioxetine. |
| Drop‐out due to adverse events follow up: range 6 weeks to 8 weeks | Study population | RR 1.41 (1.09 to 1.81) | 6220 (14 RCTs) | ⊕⊕⊕⊝ MODERATE 1 | Small difference favouring placebo. | |
| 38 per 1,000 | 53 per 1,000 (41 to 68) | |||||
| Drop‐out due to inefficacy follow up: range 6 weeks to 8 weeks | Study population | RR 0.56 (0.34 to 0.90) | 6220 (14 RCTs) | ⊕⊕⊕⊝ MODERATE 1 | Small difference favouring vortioxetine. | |
| 31 per 1,000 | 18 per 1,000 (11 to 28) | |||||
| Tolerability | Study population | RR 1.12 (1.07 to 1.16) | 6182 (14 RCTs) | ⊕⊕⊕⊝ MODERATE 1 | Small difference favouring placebo | |
| 564 per 1,000 | 632 per 1,000 (603 to 654) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 A serious risk of bias is present, as about 30% of the studies showed an overall dropout rate above 20%, evidence was downgraded by one level
2 A moderate degree of heterogeneity (I‐squared 30‐60%) is present, evidence was downgraded by one level
3 A substantial degree of heterogeneity (I‐squared 60‐90%) is present, evidence was downgraded by two levels
Summary of findings 2. Vortioxetine compared to SNRIs for adults with Major Depressive Disorder.
| Vortioxetine compared to SNRIs for adults with Major Depressive Disorder | ||||||
| Patient or population: adults with Major Depressive Disorder Setting: Inpatients and outpatients Intervention: Vortioxetine Comparison: SNRIs | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with SNRIs | Risk with Vortioxetine | |||||
| Response assessed with: assessed with: reduction of at least 50% on the HAMD scale or MADRS scale, or any other score 1 or 2 on CGI‐I follow up: range 6 weeks to 8 weeks | Study population | RR 0.91 (0.82 to 1.00) | 3159 (8 RCTs) | ⊕⊝⊝⊝ VERY LOW 1 2 | All studies were sponsored by the pharmaceutical companies that manufacture vortioxetine. No difference between vortioxetine and SNRIs. | |
| 577 per 1,000 | 525 per 1,000 (473 to 577) | |||||
| Total number of drop‐outs follow up: range 6 weeks to 8 weeks | Study population | RR 0.89 (0.73 to 1.08) | 3159 (8 RCTs) | ⊕⊝⊝⊝ VERY LOW 1 3 | No difference between vortioxetine and SNRIs. | |
| 212 per 1,000 | 189 per 1,000 (155 to 229) | |||||
| Remission assessed with: 7 points or less on the 17‐item HAM‐D and 8 points or less for longer HAM‐D versions; 10 or less points on the MADRS; score 1 or 2 on CGI‐S follow up: range 6 weeks to 8 weeks | Study population | RR 0.89 (0.77 to 1.03) | 3155 (8 RCTs) | ⊕⊝⊝⊝ VERY LOW 1 2 | No difference between vortioxetine and SNRIs. | |
| 370 per 1,000 | 329 per 1,000 (285 to 381) | |||||
| Depressive Symptoms assessed with: MADRS score (score range: 0‐34; higher score means worse outcome) follow up: range 6 weeks to 8 weeks | The change in depressive symptoms score ranged from 14.1 to 23.4 points | The change was 1.52 points lower (0.5 lower to 2.53 lower) | ‐ | 2807 (8 RCTs) | ⊕⊝⊝⊝ VERY LOW 1 2 | Small difference favouring SNRIs. |
| Drop‐out due to adverse events follow up: range 6 weeks to 8 weeks | Study population | RR 0.74 (0.51 to 1.08) | 3159 (8 RCTs) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | No difference between vortioxetine and SNRIs. | |
| 97 per 1,000 | 72 per 1,000 (50 to 105) | |||||
| Drop‐out due to inefficacy follow up: range 6 weeks to 8 weeks | Study population | RR 1.52 (0.70 to 3.30) | 3159 (8 RCTs) | ⊕⊝⊝⊝ VERY LOW 1 4 | No difference between vortioxetine and SNRIs. | |
| 14 per 1,000 | 21 per 1,000 (10 to 45) | |||||
| Tolerability | Study population | RR 0.90 (0.86 to 0.94) | 3134 (8 RCTs) | ⊕⊕⊝⊝ LOW 1 | Small difference favouring vortioxetine | |
| 690 per 1,000 | 621 per 1,000 (593 to 648) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 A very serious risk of bias is present as 60% of the studies had more than 20% dropouts overall, evidence was downgraded by two levels
2 A moderate degree of heterogeneity (I‐squared 30‐60%) is present, evidence was downgraded by one level
3 The 95% CI crossed both 1 (no differences) and 0.75 (appreciable benefit for vortioxetine), evidence was downgraded by one level
4 The 95% CI crossed 1 (no differences), 0.75 (appreciable benefit for vortioxetine) and 1.25 (appreciable benefit for SNRIs). Outcome is very imprecise: evidence was downgraded by two levels
Background
Description of the condition
Major depressive disorder (MDD) affects a person's mind, behaviour and body and is expressed in a variety of symptoms. The core features are depressed mood and loss of interest or pleasure. Other diagnostic criteria include significant changes in bodyweight, decreased or increased appetite, sleep disturbances, psychomotor agitation or retardation, fatigue, feelings of worthlessness or guilt, reduced concentration and suicidal ideation (APA 2013). According to the fourth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM‐IV), a person must have symptoms for at least two weeks (APA 2013). The core features of major depression have not been changed from DSM‐IV to DSM‐5. Although there are some differences in the diagnosis of depression between DSM and International Classification of Diseases (ICD), diagnoses of major depression according to DSM‐IV seems to be congruent with severe or moderate depressive episodes according to ICD‐10 (Saito 2010).
MDD is a common mental disorder, but prevalence rates vary markedly across countries. Lifetime prevalence was estimated at 14.6% on average in high‐income countries and 11.1% in low‐income countries, with a female:male ratio of about 2:1 (Bromet 2011). MDD is associated with substantial impairment. According to the Global Burden of Disease study, MDD is the second leading cause of disability worldwide (Vos 2012).
Description of the intervention
A variety of pharmacological and non‐pharmacological treatment options is available for the treatment of MDD. Pharmacological treatment options comprise monoamine oxidase inhibitors (MAOIs), tricyclic antidepressants (TCAs) or heterocyclic antidepressants, selective serotonin re‐uptake inhibitors (SSRIs), serotonin‐norepinephrine (‐noradrenaline) reuptake inhibitors (SNRIs), and other antidepressant agents (e.g. mirtazapine, bupropion, reboxetine, agomelatine), as well herbal products (e.g. hypericum). Current guidelines for depression recommend the use of an antidepressant or psychological treatment for people with moderate depression (APA 2010; NICE 2009) as first‐line treatment. According to these guidelines, the effectiveness between classes of antidepressants is similar. However, SSRIs are recommended over TCAs and MAOIs due to their favourable adverse effect profile (APA 2010; NICE 2009). SSRIs have now become the most prescribed antidepressant class in most parts of the world (Bauer 2008; Grover 2013; Zhang 2013).
Vortioxetine was licensed for the treatment of depression by the Food and Drugs Administration (FDA) in September 2013 in the USA (FDA 2014) and by the European Medicines Agency (EMA) in December 2013 for the EU (EMA 2014). Despite the similarities to SSRIs, the mechanism of action of vortioxetine is claimed to be novel (see How the intervention might work). According to the ATC classification of the World Health Organization (WHO), vortioxetine is placed in the category of "Other" antidepressants (WHO 2016). Due to the recent marketing authorisation, clinical experience and data on clinical use of vortioxetine is very limited at this time.
How the intervention might work
The mechanism of action of vortioxetine is not fully understood, but it is assumed to be related to a direct modulation of serotonergic receptor activity and inhibition of the serotonin transporter. Vortioxetine is an antagonist to 5‐HT3, 5‐HT1D and 5‐HT7 receptors, a partial agonist to the 5‐HT1B receptor and a 5‐HT1A receptor agonist (EMA 2014a). However, it is unclear if and how these mechanism contribute to an antidepressant effect. It is hypothesised that serotonin transporter inhibition, combined with the several other actions of vortioxetine at 5‐HT receptors, mainly at 5‐HT3 receptors, enhances the release of serotonin and modulates the release of other neurotransmitters within various brain circuits (enhanced release of norepinephrine, dopamine, histamine, acetylcholine and glutamate; reduced gamma‐aminobutyric acid (GABA) signalling). These actions could improve the efficiency of information processing in malfunctioning brain circuits by facilitating long‐term potentiation, neuroplasticity and increased firing of pyramidal neurons (Du Jardin 2016; Pehrson 2016; Stahl 2015).
Why it is important to do this review
Despite the common use of antidepressants and the variety of available treatment options, there is still an ongoing debate about the use of antidepressants in general as some studies show modest effects compared to placebo (Kirsch 2008). A significant proportion of people do not achieve remission with current treatments (Pigott 2011). Furthermore, although the adverse effect profile of SSRIs is in general judged favourable over TCAs or MAOIs, many people are dropping out of antidepressant treatment (Pigott 2011), mainly due to adverse effects (Bull 2002). Thus, there is still room for improvement of the pharmacological treatment of depression.
Another subject of debate is the comparative effects of modern antidepressants. One multiple‐treatment meta‐analysis of 12 new‐generation antidepressant drugs concluded that sertraline and escitalopram may be more favourable in terms of efficacy and acceptability compared to the other included antidepressants (Cipriani 2009). Notably, this finding could not be replicated in another comprehensive review (Gartlehner 2011). The latter review concluded that there are no substantial differences in efficacy between the antidepressants, but that antidepressants differ in onset of action and adverse events (Gartlehner 2011).
Vortioxetine was approved in late 2013 for the USA and EU and is currently the latest available antidepressant. Randomised controlled trials (RCTs) comparing vortioxetine to placebo or other antidepressants have been published. Several systematic reviews and meta‐analyses have been published on the efficacy and tolerability on vortioxetine. The first meta‐analysis included seven studies comparing vortioxetine to placebo (Berhan 2014). One systematic narrative review gave an overview of the vortioxetine studies and reported results of 10 RCTs in adults with major depression without pooling the results (Citrome 2014). Two meta‐analyses did not conduct a systematic review of the data. The first pooled analysis selected 11 short‐term placebo‐controlled trials and five long‐term open‐label studies to evaluate the safety and tolerability of vortioxetine (Baldwin 2016). The second pooled analysis analysed a selected subset of five studies to examine the effect of vortioxetine on quality of life (Florea 2015). Furthermore, 11 studies were included in an indirect comparison of vortioxetine, duloxetine, sertraline, vilazodone, levomilnacipram and escitalopram (Citrome 2016). Two other meta‐analyses focused on a subset of a specific dose of vortioxetine 5 mg (Fu 2015) or 10 mg (Li 2016) compared to placebo. However, another meta‐analysis, based on an analysis of 11 studies, found no effect differences related to dosing (Meeker 2015). This meta‐analysis included comparisons of vortioxetine and placebo as well as vortioxetine and active comparators, but it included subtherapeutic doses below 5 mg. In sum, these reviews agree that vortioxetine has a significant advantage compared to placebo in terms of efficacy, but found no advantage compared to other available antidepressants.
A summary of the FDA review of vortioxetine is also available (Zhang 2015). The FDA review included 10 short‐term placebo‐controlled trials and concluded that vortioxetine demonstrated efficacy in six of the included trials, but reported that only vortioxetine 20 mg/day showed superiority over placebo in US trials and showed smaller effects in general in US populations. The most recent meta‐analysis, which was co‐authored by employees of the manufacturers of vortioxetine, analysed an almost identical dataset and included data from treatment arms in the approved dose range of 11 short‐term studies which had results published on ClinicalTrials.gov (Thase 2016). Most analyses were conducted with aggregated data, but additional individual participant data were used. Apparently, the analyses did not follow an a priori defined protocol. However, the analyses are in line with the findings from previous reviews, showing a statistically significant advantage of vortioxetine compared to placebo, but a clear dose‐response relationship could not be established. It also confirms the findings of smaller effects in US studies. Thus, despite several attempts to synthesise available literature on vortioxetine, a practical interpretation of data from current systematic reviews may be limited by comprehensiveness issues (lack of thorough search for unpublished data, selective inclusion of trials in the analyses), and possible conflicts of interest. Our review provides an independent, comprehensive and up‐to‐date summary of the available evidence of the efficacy and acceptability of vortioxetine compared to placebo and other active pharmacological treatment options.
Objectives
To assess the efficacy and acceptability of vortioxetine compared with placebo and other antidepressant drugs in the treatment of acute depression in adults.
Methods
Criteria for considering studies for this review
Types of studies
We included RCTs but excluded quasi‐RCTs. For trials with a cross‐over design, we considered only the results from the first randomisation period. We included cluster‐RCTs if sufficient information was available to account for the clustering (see Unit of analysis issues).
Types of participants
Characteristics
Participants of both sexes, of any ethnicity, and aged 18 years and older.
Diagnosis
Participants with a primary diagnosis of unipolar major depression according to DSM‐III (APA 1980), DSM‐ III‐R (APA 1987), DSM‐IV (APA 1994), DSM‐IV‐TR (APA 2000), DSM‐5 (APA 2013), ICD‐10 (WHO 1992), Feighner (Feighner 1972) or Research Diagnostic Criteria (Spitzer 1978), and Chinese Classification of Mental Disorders (CCMD‐3, Chinese Society of Psychiatry 2001). We excluded studies of people with treatment‐resistant depression, defined as inadequate treatment response to at least four weeks of adequate antidepressant treatment.
Comorbidities
We included studies with people with comorbid psychiatric disorders. We excluded antidepressant trials in people with depression with a serious concomitant physical illness (e.g. myocardial infarction, diabetes, cancer, etc.).
Setting
Any setting.
Subset data
Based on National Institute for Health and Care Excellence (NICE) guidelines for the treatment of depression in adults (NICE 2009), we included studies in which less than 20% of the included participants had bipolar depression.
We included studies with relevant subsets of data (e.g. some participants aged below 18 years) if the data were available for the relevant subset and the randomisation was stratified by the criterion in question. Inclusion of these studies was examined in sensitivity analyses.
Types of interventions
Experimental intervention
Vortioxetine monotherapy. To increase the clinical applicability of the review, we excluded treatment arms employing dosages below the lowest effective dose of 5 mg/day (EMA 2014b). We included treatment arms with fixed and flexible dosing schemes. Fixed doses are set a priori and are independent from participant criteria, while in flexible dosing schemes the dose is adapted according predefined criteria, for example, insufficient response.
Comparator intervention
Placebo.
-
Another antidepressant as monotherapy, including:
conventional TCA or heterocyclic antidepressants (amitriptyline, amoxapine, clomipramine, desipramine, dosulepin/dothiepin, doxepin, imipramine, lofepramine, maprotiline, nortriptyline, protriptyline, trimipramine);
SSRIs (fluoxetine, fluvoxamine, citalopram, paroxetine, escitalopram);
SNRIs (venlafaxine, duloxetine, milnacipran);
MAOIs (phenelzine, isocarboxazide, tranylcypromine, moclobemide, brofaromine);
other antidepressant agents (mirtazapine, bupropion, reboxetine, agomelatine) or non‐conventional antidepressive agents (herbal products such as hypericum).
We applied no restrictions on dosage of the comparators, but we conducted sensitivity analyses and excluded studies with unequal dosing.
Types of outcome measures
We included studies that meet the above inclusion criteria regardless of whether they report on the following outcomes.
Primary outcomes
Response to treatment: the primary efficacy outcome was the number of participants who responded to acute treatment, as defined by a reduction of at least 50% on the Hamilton Depression Rating Scale (HAM‐D) scale (Hamilton 1960) or Montgomery‐Åsberg Depression Scale (MADRS; Montgomery 1979), or any other depression scale, or "much or very much improved" (score 1 or 2) on Clinical Global Impression ‐ Improvement (CGI‐I) (Guy 1970). We did not consider other definitions of response in this Cochrane Review.
Where more than one scale was provided, we gave preference as listed. We used response rate instead of a continuous symptom score for the primary efficacy analysis to make the interpretation of results easier (Guyatt 1998).
Total number of dropouts: primary outcome measuring acceptability was the total number of participants dropping out during the trial as a proportion of the total number of randomised participants.
Secondary outcomes
Remission: number of participants who achieved remission. We defined remission a priori as:
7 points or less on the 17‐item HAM‐D and as 8 points or less for all the other longer versions of HAM‐D;
10 or less points on the MADRS;
"not ill or borderline mentally ill" (score 1 or 2) on Clinical Global Impression ‐ Severity (CGI‐S) (Guy 1970) at endpoint.
We did not consider other definitions of remissions in this Cochrane Review.
Depressive symptoms: endpoint mean scores, or mean change scores at endpoint on HAM‐D, MADRS, or any other depression rating scale score.
Dropouts due to adverse events: number of participants who dropped out due to adverse events during the trial as a proportion of the total number of randomised participants.
Dropouts due to inefficacy: number of participants who dropped out due to inefficacy during the trial as a proportion of the total number of randomised participants.
Tolerability: evaluated using the total number of participants experiencing at least one adverse event.
We collected any data on specific adverse effects and reported these data in tables. However, due to the poor reporting in RCTs (Zorzela 2014), we did not summarise these data in meta‐analyses.
Although the effect on cognition is a relevant outcome for people treated with psychopharmacological treatments, we decided to exclude cognition as an outcome. According to the EMA, cognition was not systematically assessed (EMA 2014a). The EMA report on vortioxetine indicated that only three studies reported this outcome and that a meta‐analysis by the manufacturer of these studies had outcome reporting bias (EMA 2014a). Furthermore, the EMA report pointed out that it was not possible to distinguish between an effect on cognition and a relief of depressive symptoms, because no active comparator was included.
Timing of outcome assessment
Our primary outcomes were the acute phase treatment response (between four and 12 weeks). When studies reported efficacy data at different time points, we gave preference to the time point closest to eight weeks.
Hierarchy of outcome measures
We included efficacy data measured by HAM‐D (Hamilton 1960), MADRS (Montgomery 1979), or any other depression scale. Response or remission rates may also be based on CGI‐I scores (Guy 1970) or a combination of these outcomes. Where more than one criterion was provided, we planned to use the data according to the following hierarchy: HAM‐D, MADRS, other depression scales and CGI and combination of these in cases of remission and response rates. However, due to the reporting of the MADRS (see Description of studies) we gave preference to MADRS outcomes (see Differences between protocol and review). We did not include CGI scores as a continuous outcome. The HAM‐D scale is available in various versions which differ in the number of included items. The versions with 17, 21 or 24 items are the most common and we gave preference to these in this order. Studies may also use different criteria for response or remission. We gave preference according to the list described in the corresponding outcome section (see Primary outcomes; Secondary outcomes).
Search methods for identification of studies
Cochrane Collaboration Depression, Anxiety and Neurosis Review Group's Specialized Register (CCDANCTR)
The Cochrane Collaboration Depression, Anxiety and Neurosis Group (CCDAN) maintain two clinical trials registers at their editorial base in Bristol, UK: a References Register and a Studies Register. The CCDANCTR‐References Register contains over 37,000 reports of RCTs in depression, anxiety and neurosis. Approximately 60% of these references have been tagged to individual, coded trials. The coded trials are held in the CCDANCTR‐Studies Register and records are linked between the two registers using unique Study ID tags. Coding of trials is based on the EU‐PSI coding manual, using a controlled vocabulary (see Cochrane Collaboration Depression, Anxiety & Neurosis Group for further details). Reports of trials for inclusion in the Group's registers are collated from routine (weekly), generic searches of MEDLINE (from 1950), Embase (from 1974) and PsycINFO (from 1967); quarterly searches of the Cochrane Central Register of Controlled Trials (CENTRAL) and review‐specific searches of additional databases. Reports of trials are also sourced from international trials registers via the WHO trials portal (the International Clinical Trials Registry Platform (ICTRP)), pharmaceutical companies, handsearching of key journals, conference proceedings, and other (non‐Cochrane) systematic reviews and meta‐analyses.
Details of CCDAN's generic search strategies (used to identify RCTs) can be found on the Group's website.
Electronic searches
We performed the following electronic searches with no restrictions on date, language or publication status.
CCDANCTR‐Studies Register using the following controlled search terms:
Condition = depress* AND Intervention = Vortioxetine or "Lu AA21004"
CCDANCTR‐References Register using a more sensitive set of free‐text terms to identify additional untagged/uncoded reports of RCTs:
Free‐text = (depress* or dysthymi* or "mood disorder*" or "affective disorder*" or "affective symptom*") and (Vortioxetine or "Lu AA21004" or LuAA21004 or Brintellix)
International trial registries via the WHO trials portal (ICTRP) and ClinicalTrials.gov to identify unpublished or ongoing studies, together with the trial registries of relevant pharmaceutical companies:
Lundbeck Clinical Trials Registry;
Takeda Clinical Study Protocols and Results.
Regulatory databases including those of the FDA in the US (Drugs@FDA) and the EMA (EMA).
Searching other resources
Reference lists
We checked the reference lists of all included studies and relevant systematic reviews to identify additional studies missed from the original electronic searches (e.g. unpublished or in‐press citations). Also, we conducted a cited reference search on the Web of Science.
Correspondence
We contacted trialists and subject experts for information on unpublished or ongoing studies or to request additional trial data.
Data collection and analysis
Selection of studies
Two review authors (GO, MK) independently screened titles and abstracts for inclusion of all studies identified by the search and coded them as 'potentially eligible.' We retrieved the full‐texts of study reports/publications rated as 'potentially eligible' by one or both review authors. Two review authors (GO, MK) independently screened the full‐text articles and identified studies for inclusion, and identified and recorded reasons for exclusion of the ineligible studies (see Characteristics of excluded studies table). We resolved any disagreements through discussion or, if required, by consulting a third review author (CB). We collated multiple reports of the same study and included them a single study.
Data extraction and management
We used a data collection form which had been piloted on at least one study in the review to extract study characteristics and outcome data. Two review authors (CB, MK) independently extracted study characteristics and outcome data from included studies. We extracted the following study characteristics.
Methods: blinding, total duration of study, details of 'run in' periods, number of study centres and location, study setting, withdrawals and date of study.
Participants: sample size, mean age, age range, gender, severity of condition, diagnostic criteria, inclusion criteria and exclusion criteria.
Interventions: intervention, comparison, concomitant medications, excluded medications, dose and dosing scheme (fixed versus flexible).
Outcomes: primary and secondary outcomes specified and collected, and time points reported.
Notes: funding for trial and notable conflicts of interest of trial authors.
We noted in the Characteristics of included studies table if outcome data were not reported in a usable way. We resolved disagreements by consensus or by involving a third review author (CB). One review author (MK) transferred data into Review Manager 5 (RevMan 2014). Two review authors (GO, JB) double checked the data entered for correctness by comparing the data presented in the systematic review with the study reports. A third review author (CB) spot‐checked study characteristics for accuracy against the trial report.
Main planned comparisons
We combined the comparators (see Types of interventions) into classes in the meta‐analyses. Therefore, the main planned comparisons were:
vortioxetine versus placebo;
vortioxetine versus TCAs/heterocyclics;
vortioxetine versus SSRIs;
vortioxetine versus SNRIs;
vortioxetine versus MAOIs;
vortioxetine versus other antidepressant agents.
Wherever suitable, we presented data with substances as subgroups within each class.
Assessment of risk of bias in included studies
Two review authors (MK, GO) independently assessed risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We resolved any disagreements by discussion or by involving a third review author (CB). We assessed the risk of bias in the included studies according to the following domains.
Random sequence generation.
Allocation concealment.
Blinding of participants and personnel.
Blinding of outcome assessment.
Incomplete outcome data.
Selective outcome reporting.
Other bias.
We judged potential sources of bias as 'high,' 'low' or 'unclear' and provided supporting quotation a from the study report together with a justification for our judgement in the 'Risk of bias' table. The risk of bias judgements were summarised across different studies for each of the domains listed. We considered blinding separately for different key outcomes where necessary (e.g. for unblinded outcome assessment, risk of bias for treatment discontinuation may be different than for a participant‐reported scale). Where information on risk of bias relates to unpublished data or correspondence with a trial author, we noted this in the 'Risk of bias' table.
When considering treatment effects, we took into account the risk of bias for the studies that contributed to that outcome.
Measures of treatment effect
Dichotomous data
We analysed dichotomous data as risk ratios (RRs), because RRs are more intuitive in their interpretation than odds ratios (Grant 2014), with 95% confidence intervals (CI). Where reported intention‐to‐treat (ITT) analysis was based on a 'modified' ITT not including all dropouts (e.g. leaving out dropouts without postbaseline assessment), we applied a conservative approach and considered these dropouts as non‐responders or non‐remitters (i.e. assumed they would have experienced the negative outcome by the end of the trial, e.g. failure to respond to treatment).
Continuous data
We analysed continuous data as mean difference (MD) with 95% CI, if all studies reported the necessary data from HAM‐D‐scales as an outcome. In cases that data from different scales were combined, we used standardised mean difference (SMD) with 95% CI. We entered and presented data with a consistent direction of effect. Data were analysed as endpoint data. We combined endpoint and change scores only if data were analysed as MD. Analyses were conducted with ITT data as reported (e.g. data from last observation carried forward (LOCF) or mixed model for repeated measurements (MMRM) methods).
We conducted meta‐analyses only where this was meaningful (i.e. if the treatments, participants and underlying clinical question were similar enough for pooling to make sense).
Unit of analysis issues
Cluster‐randomised controlled trials
Cluster‐RCTs were eligible for inclusion if sufficient information was available to account for the clustering (see Differences between protocol and review).
Cross‐over trials
For trials with a cross‐over design, we considered only the results from a first randomisation period (see Differences between protocol and review).
Studies with multiple treatment groups
If more than one treatment arm was reported in a single trial, we only included the relevant treatment arms. In case of multiple relevant treatment arms (e.g. different dosages), we combined these treatment arms into a single group. For dichotomous outcomes, we summarised data across groups; while for continuous outcomes, we combined means and standard deviations according to Chapter 7 of the Cochrane Handbook for Systematic Reviews of Interventions (Section 7.7.3.8, Higgins 2011).
In case of relevant treatment arms that could not be combined (e.g. different SSRIs as comparators), we divided the sample size of the shared group so that the two arms were treated as independent comparisons.
Dealing with missing data
We contacted study authors or study sponsors to verify key study characteristics and missing outcome data. Attempts to contact authors and sponsors were documented, as well as additional data we received in these correspondences.
In the absence of supplemental data from the authors, we planned to calculate the SDs of the HAM‐D (or any other depression scale) and response/remission rates according to validated imputation methods (Furukawa 2005; Furukawa 2006). We planned to examine the validity of these imputations in sensitivity analyses.
Assessment of heterogeneity
Variations in participants and interventions lead to clinical heterogeneity. We extracted basic study characteristics (see Data collection and analysis) from the studies and described these in the Characteristics of included studies tables. A decision was then made as to whether studies were similar enough to combine in meta‐analyses.
We quantified statistical heterogeneity of the studies using the I2 statistic and Chi2 test. As the Chi2 test is known to have low power, we used P = 0.10 as a threshold for statistical significance. Our interpretation of the I2 statistic followed the recommendation of the Cochrane Handbook for Systematic Reviews of Interventions and we considered the I2 statistic as:
0% to 40%: might not be important;
30% to 60%: may represent moderate heterogeneity;
50% to 90%: may represent substantial heterogeneity;
75% to 100%: considerable heterogeneity.
We also assessed heterogeneity by visual inspection of forest plots.
Assessment of reporting biases
Publication bias was scrutinised by visual inspection of funnel plots if we include more than 10 studies in the analysis of the outcome in question.
Data synthesis
We used a random‐effects model in our primary analysis. We expected some heterogeneity in the studies included and the random‐effects model incorporates the variance between studies in the model. As a result, CIs are wider. The random‐effects model has the highest generalisability in an empirical examination of summary effect measures for meta‐analyses (Furukawa 2002). We routinely examined the robustness of this summary measure by checking the results under a fixed‐effect model. Material differences between the models were reported.
Subgroup analysis and investigation of heterogeneity
We only conducted subgroup and sensitivity analysis for the primary outcomes.
A priori, we planned to perform the following subgroup analyses.
Vortioxetine dosing: fixed versus flexible dosing schemes.
Treatment setting: primary care versus inpatient care versus outpatient care.
Older people (aged more than 65 years): included versus excluded.
Sensitivity analysis
We planned to perform the following sensitivity analyses to examine the robustness of the effect size.
Exclusion of trials with unequal dosing. We defined comparability of doses by comparing the percentage of the maximum licensed daily dose in both groups (e.g. vortioxetine 10 mg/day (50% of 20 mg/day) equals fluoxetine 40 mg/day (50% of 80 mg/day)).
Exclusion of studies that did not employ a double‐blind approach.
Exclusion of studies with subsets of people with bipolar disorders.
Exclusion of trials with dropout rates of more than 20% in one of the treatment arms included.
Exclusion of studies with imputed data.
Exclusion of studied sponsored by the manufacturer of vortioxetine.
'Summary of findings' table
We employed the GRADE approach to interpret findings (Langendam 2013) and used GRADEpro to import data from Review Manager 5 (RevMan 2014) to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined and the sum of available data on the outcomes.
For each comparison, we reported the primary outcomes (response and total number of dropouts) and secondary outcomes (remission, depressive symptoms, dropouts due to adverse events, dropouts due to inefficacy and participants experience at least one adverse event).
Results
Description of studies
Results of the search
The literature search identified 113 records including eight duplicates. We excluded 45 of the remaining 105 records based on the abstracts. We retrieved 60 full‐text articles for detailed examination, which led to the exclusion of 45 records. The majority (35 trials) were secondary publications of already included or excluded trials. Two studies did not meet our inclusion criterion for acute treatment (Jacobsen 2015a; Jacobsen 2015b); two were relapse prevention studies (Boulenger 2012; NCT02371980); one trial did not meet our criterion for depression because it randomised remitted participants and healthy controls only (Browning 2014); and one study randomised participants to vortioxetine or agomelatine after an inadequate response to at least six weeks of SSRI or SNRI treatment (Montgomery 2014). We identified two ongoing studies (NCT02294305; NCT02389816) which may fulfil the inclusion criteria of this review and two studies are awaiting assessment as there are no published results (NCT02272517; NCT02279966). Request of additional information by the manufactures or the authors was not necessary. The literature search was last updated in May 2016 (see Figure 1; Characteristics of excluded studies table).
1.
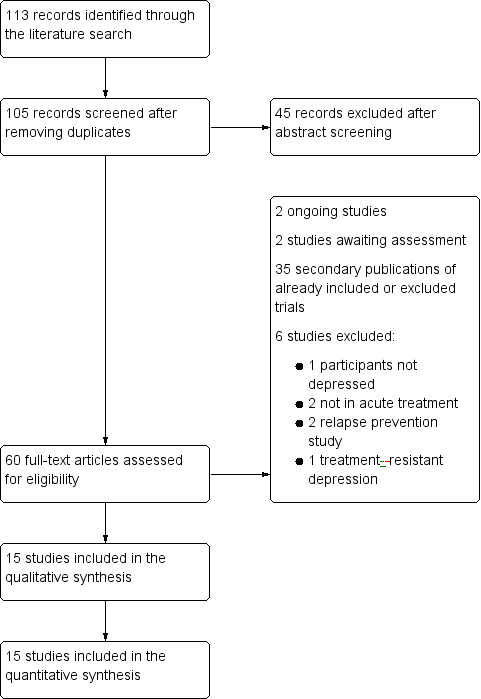
Study flow diagram.
Included studies
Fifteen studies were included in this systematic review (Alvarez 2012; Baldwin 2012; Boulenger 2014; Henigsberg 2012; Jacobsen 2015; Jain 2013; Katona 2012; Mahableshwarkar 2013; Mahableshwarkar 2015a; Mahableshwarkar 2015b; Mahableshwarkar 2015c; McIntyre 2014; NCT01255787; Takeda 2011; Wang 2015). Two of these were unpublished trials carried out by a pharmaceutical company (Takeda) (NCT01255787; Takeda 2011). (See Characteristics of included studies table).
Design
All included studies were randomised trials and applied double‐blind methodology. Seven studies were three‐armed with vortioxetine, an active comparator and placebo (Alvarez 2012; Baldwin 2012; Boulenger 2014; Katona 2012; Mahableshwarkar 2013; Mahableshwarkar 2015a; Mahableshwarkar 2015b). Eight studies were two‐armed, among these seven studies were placebo‐controlled (Henigsberg 2012; Jacobsen 2015; Jain 2013; Mahableshwarkar 2015c; McIntyre 2014; NCT01255787; Takeda 2011), and one study was active controlled only (Wang 2015).
Trial duration
Two studies lasted six weeks (Alvarez 2012; Jain 2013), and 13 studies lasted eight weeks (Baldwin 2012; Boulenger 2014; Henigsberg 2012; Jacobsen 2015; Katona 2012; Mahableshwarkar 2013; Mahableshwarkar 2015a; Mahableshwarkar 2015b; Mahableshwarkar 2015c; McIntyre 2014; NCT01255787; Takeda 2011; Wang 2015).
Sample sizes
Overall, the studies included 7746 participants. Of these, 4134 were randomised to vortioxetine. Of the remaining 3612 participants, 2299 were randomised to placebo, 1313 to SNRIs (344 to venlafaxine and 969 to duloxetine). The mean sample size per arm was 209 participants (range 105 to 448).
Setting
All studies were multicentre trials. Six studies were conducted in a single nation: the USA (Jacobsen 2015; Jain 2013; Mahableshwarkar 2013; Mahableshwarkar 2015a; Mahableshwarkar 2015c) and Japan (Takeda 2011). One multinational study recruited Asian participants only (Wang 2015). The other studies were multinational across continents. An overview of countries where participants were recruited is given in Figure 2.
2.
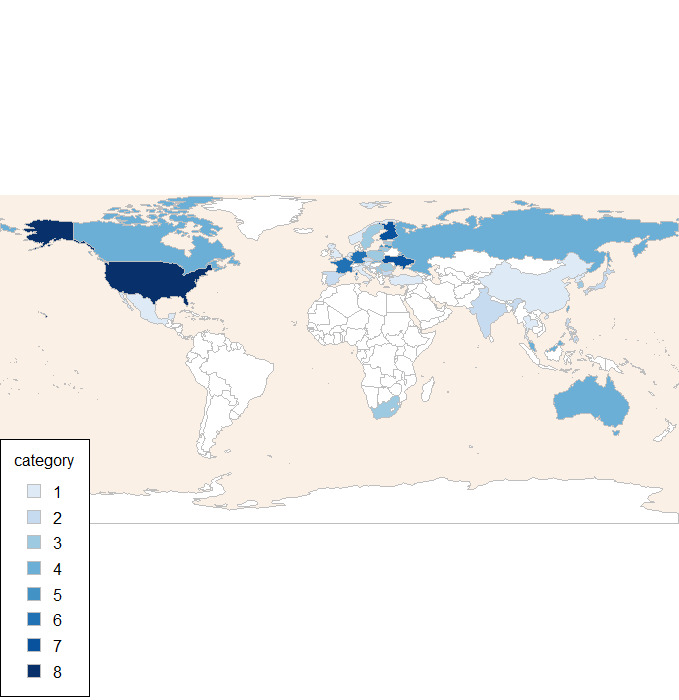
Countries participating in trials. The categories represent the number of studies randomising participants within a country.
Four studies enrolled both inpatients and outpatients (Baldwin 2012; Boulenger 2014; McIntyre 2014; Wang 2015). Three studies recruited exclusively outpatients (Alvarez 2012; Jacobsen 2015; Jain 2013). Eight studies did not explicitly report the setting (Henigsberg 2012; Katona 2012; Mahableshwarkar 2013; Mahableshwarkar 2015a; Mahableshwarkar 2015b; Mahableshwarkar 2015c; NCT01255787; Takeda 2011).
Participants
All studies included participants with a diagnosis of MDD. No trial enrolled people with comorbid psychiatric disorders.
Thirteen studies randomised participants from 18 years of age: eight studies recruited participants aged between 18 and 75 years (Baldwin 2012; Boulenger 2014; Henigsberg 2012; Jacobsen 2015; Jain 2013; Mahableshwarkar 2013; Mahableshwarkar 2015a; Mahableshwarkar 2015c), and four studies recruited between the ages of 18 and 65 years (Alvarez 2012; Mahableshwarkar 2015b; McIntyre 2014; Wang 2015). Two studies recruited participants from 20 years of age: one study between the ages of 20 and 64 years (NCT01255787), and one study between the ages of 20 and 75 years (Takeda 2011). One study included only older participants (Katona 2012; aged 65 to 88 years).
Interventions and comparators
Eight studies compared vortioxetine to SNRIs: two compared vortioxetine to venlafaxine (Alvarez 2012; Wang 2015), and six compared vortioxetine to duloxetine (Baldwin 2012; Boulenger 2014; Katona 2012; Mahableshwarkar 2013; Mahableshwarkar 2015a; Mahableshwarkar 2015b). Seven studies compared vortioxetine to placebo only (Henigsberg 2012; Jacobsen 2015; Jain 2013; McIntyre 2014; Mahableshwarkar 2015c; NCT01255787; Takeda 2011). We found no studies comparing vortioxetine to TCAs/heterocyclics, SSRIs, MAOIs or other antidepressant agents.
One study used a flexible vortioxetine dose scheme (range from 10 mg/day to 20 mg/day) (Mahableshwarkar 2015b). The other 14 trials used a fixed vortioxetine doses scheme (5 mg/day, 10 mg/day, 15 mg/day or 20 mg/day). Two studies applied subtherapeutic dosages of vortioxetine below 5 mg/day (1 mg/day (Henigsberg 2012); 2.5 mg/day (Mahableshwarkar 2013)). These treatment arms were excluded.
Outcomes
All 15 studies provided efficacy data (either as dichotomous or as continuous outcome) and tolerability/acceptability data and could be entered into a meta‐analysis.
Nine studies used the MADRS for their primary outcome measures (Alvarez 2012; Baldwin 2012; Boulenger 2014; Jacobsen 2015; Mahableshwarkar 2015a; Mahableshwarkar 2015c; NCT01255787; Takeda 2011; Wang 2015). Four studies used the HAM‐D‐24 (Henigsberg 2012; Jain 2013; Katona 2012; Mahableshwarkar 2013), two studies used the Digit Symbol Substitution Test (DSST) (Mahableshwarkar 2015b; McIntyre 2014), and one study additionally the Rey Auditory Verbal Learning Test (RAVLT) (McIntyre 2014) for primary outcome measures. For secondary outcomes, the studies used mainly MADRS, CGI‐I, Sheehan Disability Scale (SDS), CGI‐S, HAM‐D‐24, and Hamilton Anxiety Rating Scale (HAM‐A). Two studies also used the HAM‐D‐17 (Henigsberg 2012; Takeda 2011), and two studies used additional cognitive tests (DSST, RAVLT, Trail Making Test ‐ A (TMT‐A), Trail Making Test ‐ B (TMT‐B), Stroop, Perceived Deficits Questionnaire (PDQ), Simple Reaction Time (SRT), Cognitive Reflection Test (CRT), Groton Maze Learning Test (GMLT), Detection Task (DT), Identification Task (IT), and One‐Back Task (Mahableshwarkar 2015b; McIntyre 2014)). All studies reported response rates and remission rates. Thirteen studies defined the response rate as 50% or greater decrease from baseline in MADRS total score and two studies as 50% or greater decrease from baseline in HAM‐D‐24 total score (Jain 2013; Mahableshwarkar 2013). All studies defined the remission rate as MADRS total score of 10 or less.
All studies reported dropouts due to any reason and dropouts due to adverse effects. All but one study (McIntyre 2014) reported dropouts due to inefficacy and the total number of participants who experienced adverse effects.
As expected, the reporting of the individual adverse effects varied markedly. Seven studies reported adverse events if the incidence was at least 5% per arm (Alvarez 2012; Baldwin 2012; Boulenger 2014; Katona 2012; Mahableshwarkar 2015b; McIntyre 2014; Wang 2015). Another seven studies set the threshold at 2% (Henigsberg 2012; Jacobsen 2015; Jain 2013; Mahableshwarkar 2013; Mahableshwarkar 2015a; Mahableshwarkar 2015c; NCT01255787), and one study set the threshold at 0% (Takeda 2011).
Excluded studies
Overall, we excluded six studies (14 references) from the systematic review because they did not meet the inclusion criteria. They were designed as relapse prevention studies (Boulenger 2012; NCT02371980), were not conducted in acute therapy (Jacobsen 2015a; Jacobsen 2015b), recruited randomised remitted participants or healthy controls (Browning 2014), or included participants with a treatment‐resistant depression (Montgomery 2014) (see Figure 1 and Characteristics of excluded studies table).
Studies awaiting classification
Two studies have recently been completed, but have not yet published results (NCT02272517; NCT02279966). Both studies are short‐term randomised, double‐blind trials of eight weeks' duration, which examine the effects of vortioxetine on cognitive functions in people with depression in comparison to an SSRI (see Characteristics of studies awaiting classification table).
Ongoing studies
We identified two ongoing studies (see Characteristics of ongoing studies table). The ongoing studies are short‐term randomised, double‐blind trials of eight weeks' duration (NCT02389816) or 12 weeks' duration (NCT02294305). One study examines the efficacy of vortioxetine for the treatment of depression in people with comorbid social anxiety disorder (NCT02294305). The other study is comparing the efficacy of vortioxetine for the treatment of depression in Japanese people (NCT02389816). One study is ongoing, but not recruiting (NCT02294305), the other is currently recruiting participants (NCT02389816).
Risk of bias in included studies
For graphical representations of the judgements of risk of bias, refer to Figure 3 and Figure 4. Full details of judgements for every included study are presented in the 'Risk of bias' tables within the Characteristics of included studies table.
3.
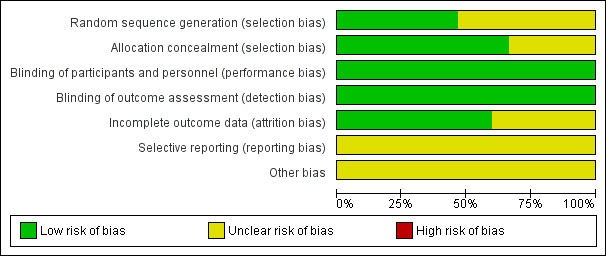
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
4.
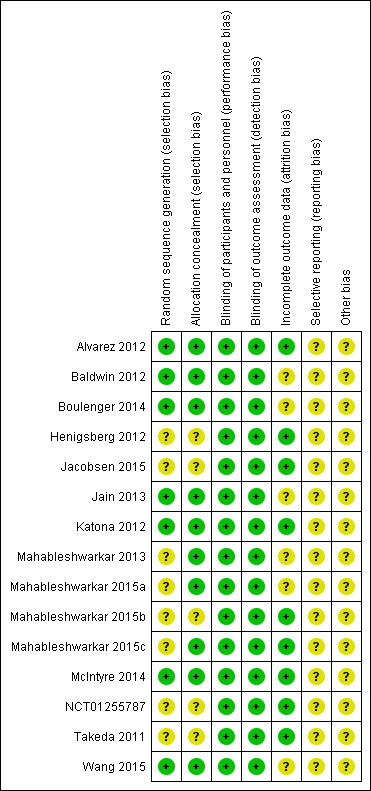
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
We rated none of the studies as having a high risk of bias in any domain, but we rated all studies at unclear risk of bias in at least two domains (see Figure 3 and Figure 4 for summary graphs). All studies were sponsored by the pharmaceutical companies that manufactures vortioxetine (Lundbeck, Takeda), and two of them were unpublished.
Allocation
Eight studies did not report details on sequence generation and were judged at unclear risk of bias (Henigsberg 2012; Jacobsen 2015; Mahableshwarkar 2013; Mahableshwarkar 2015a; Mahableshwarkar 2015b; Mahableshwarkar 2015c; NCT01255787; Takeda 2011). In addition, five studies did not adequately describe allocation concealment (Henigsberg 2012; Jacobsen 2015; Mahableshwarkar 2015b; NCT01255787; Takeda 2011).
Blinding
All RCTs were reported as double‐blind and so were at low risk of bias. All studies used at least identically appearing capsules for blinding.
Incomplete outcome data
Nine studies had a dropout rate below 20% in all treatment arms and so were at low risk of attrition bias (Alvarez 2012; Henigsberg 2012; Jacobsen 2015; Katona 2012; Mahableshwarkar 2015b; Mahableshwarkar 2015c; McIntyre 2014; NCT01255787; Takeda 2011). Of the six remaining studies, two studies had a dropout rate above 20% in the vortioxetine arm (Baldwin 2012; Boulenger 2014), one study in the active control arm (Wang 2015), one study in the placebo arm (Jain 2013), one study in the vortioxetine and in the active control arm (Mahableshwarkar 2015a), and one study in all arms (Mahableshwarkar 2013). The range in these six studies was from 20.1% to 27.4%. These studies were at unclear risk of attrition bias.
Selective reporting
We rated all included studies at unclear risk of selective reporting bias, because published protocols were unavailable. However, publications and entries in clinical trial registers did not reveal discrepancies.
Other potential sources of bias
All studies were sponsored by the pharmaceutical companies that manufactures vortioxetine (Lundbeck, Takeda) and were, therefore, assessed as having an unclear risk of bias.
Effects of interventions
We have reported the results of the present systematic review by grouping the comparators into two classes: placebo and SNRIs. Specific comparators are presented in subgroups where possible. We could not identify relevant studies comparing vortioxetine with TCAs, heterocyclics, SSRIs, MAOIs or other antidepressants.
Comparison 1. Vortioxetine versus placebo
Fourteen studies including 6220 participants contributed data to the comparison of vortioxetine versus placebo (see Table 1). The quality of evidence contributing to all outcomes was rated as moderate to very low, because of high dropout rates and statistical heterogeneity.
Primary outcomes
1.1. Response to treatment
There was evidence that vortioxetine was more effective than placebo (Mantel‐Haenszel RR 1.35, 95% CI 1.22 to 1.49; P < 0.001; 14 studies, 6220 participants). Statistical heterogeneity was substantial between studies (I2 = 60%) (Analysis 1.1; Figure 5).
1.1. Analysis.
Comparison 1 Vortioxetine versus placebo, Outcome 1 Response.
5.
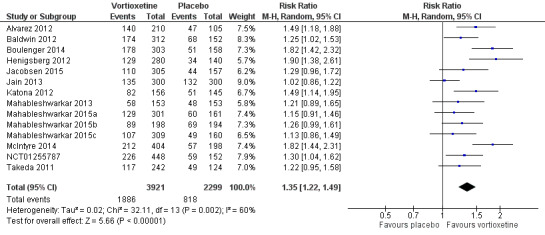
Forest plot of comparison: 1 Vortioxetine versus placebo, outcome: 1.1 Response.
1.2. Total number of dropouts
There was no evidence that vortioxetine was associated with a lower or higher total dropout rate than placebo (RR 1.05, 95% CI 0.93 to 1.19; P = 0.40; 14 studies, 6220 participants). There was no heterogeneity (I2 = 0%) (Analysis 1.2; Figure 6).
1.2. Analysis.
Comparison 1 Vortioxetine versus placebo, Outcome 2 Total number of dropouts.
6.
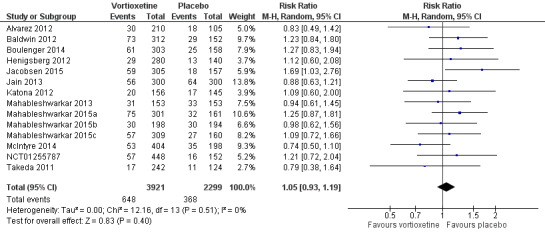
Forest plot of comparison: 1 Vortioxetine versus placebo, outcome: 1.2 Total number of dropouts.
Secondary outcomes
1.3. Achieved remission
There was evidence that more participants achieved remission with vortioxetine than with placebo (RR 1.32, 95% CI 1.15 to 1.53; P < 0.001; 14 studies, 6220 participants). Heterogeneity was substantial between studies (I2 = 58%) (Analysis 1.3).
1.3. Analysis.
Comparison 1 Vortioxetine versus placebo, Outcome 3 Remission.
1.4. Depressive symptoms
There was evidence that vortioxetine was significantly more effective in lowering MADRS score compared to placebo (MD ‐2.94, 95% CI ‐4.07 to ‐1.80, P < 0.001; 14 studies, 5566 participants). Heterogeneity was high between studies (I2 = 79%) (Analysis 1.4).
1.4. Analysis.
Comparison 1 Vortioxetine versus placebo, Outcome 4 Depressive symptoms.
1.5. Dropout due to adverse events
There was evidence that vortioxetine was associated with a higher dropout rate due to adverse events compared to placebo (RR 1.41, 95% CI 1.09 to 1.81; P = 0.008; 14 studies, 6220 participants). There was no heterogeneity (I2 = 0%) (Analysis 1.5).
1.5. Analysis.
Comparison 1 Vortioxetine versus placebo, Outcome 5 Dropout due to adverse events.
1.6. Dropout due to inefficacy
There was evidence that vortioxetine was associated with a lower dropout rate due to inefficacy compared to placebo (RR 0.56, 95% CI 0.34 to 0.90; P = 0.02; 14 studies, 6220 participants). Heterogeneity between studies was moderate (I2 = 41%) (see Analysis 1.6).
1.6. Analysis.
Comparison 1 Vortioxetine versus placebo, Outcome 6 Dropout due to inefficacy.
1.7. Tolerability
There was evidence that more participants experienced adverse effects when treated with vortioxetine than when treated with placebo (RR 1.12, 95% CI 1.07 to 1.16; P < 0.001; 14 studies, 6182 participants). Heterogeneity between studies was low (I2 = 8%) (see Analysis 1.7).
1.7. Analysis.
Comparison 1 Vortioxetine versus placebo, Outcome 7 Tolerability.
Specific adverse effects compared to placebo are reported descriptively in Analysis 1.14. One study reported all adverse effects mentioned (Takeda 2011). Due to the limits of graphs in Review Manager 5 and in line with the majority of studies, we only reported adverse effects with an incidence of 2% or greater in one of the treatment arms for this study. Serious adverse events are reported in Analysis 1.15.
1.14. Analysis.
Comparison 1 Vortioxetine versus placebo, Outcome 14 Adverse events.
1.15. Analysis.
Comparison 1 Vortioxetine versus placebo, Outcome 15 Serious adverse events.
This analysis was not conducted with ITT data according to our conservative approach (see Measures of treatment effect), but with ITT data as reported in the trials.
Comparison 2. Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors
Eight studies including 3159 participants contributed data to the comparison of vortioxetine versus SNRIs (see Table 2). The quality of evidence contributing to all outcomes was very low because of high dropout rates and substantial statistical heterogeneity.
Primary outcomes
2.1. Response to treatment
There was no evidence that vortioxetine was less or more effective than SNRIs as a whole (RR 0.91, 95% CI 0.82 to 1.00; P = 0.06; 8 studies, 3159 participants). Heterogeneity was substantial between studies (I2 = 61%) (Analysis 2.1; Figure 7).
2.1. Analysis.
Comparison 2 Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors (SNRIs), Outcome 1 Response.
7.
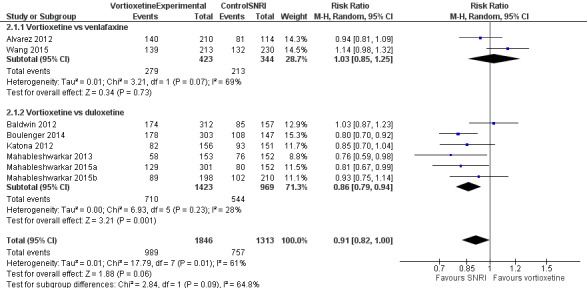
Forest plot of comparison: 3. Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors, outcome: 3.1 Response.
Although there was no statistically significant difference between the specific comparators (Chi2 = 2.84, degrees of freedom (df) = 1, P = 0.09), response rates were significantly lower for vortioxetine compared to duloxetine (RR 0.86, 95% CI 0.79 to 0.94; P = 0.001; 6 studies, 2392 participants; I2 = 28%) while there was no difference in response rates compared to venlafaxine (RR 1.03, 95% CI 0.85 to 1.25; P = 0.73; 2 studies, 767 participants; I2 = 69%).
2.2. Total number of dropouts
There was no evidence that vortioxetine was associated with a lower or higher total dropout rate than SNRIs as a whole (RR 0.89, 95% CI 0.73 to 1.08; P = 0.25; 8 studies, 3159 participants). Heterogeneity between studies was moderate (I2 = 44%) (Analysis 2.2; Figure 8).
2.2. Analysis.
Comparison 2 Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors (SNRIs), Outcome 2 Total number of dropouts.
8.
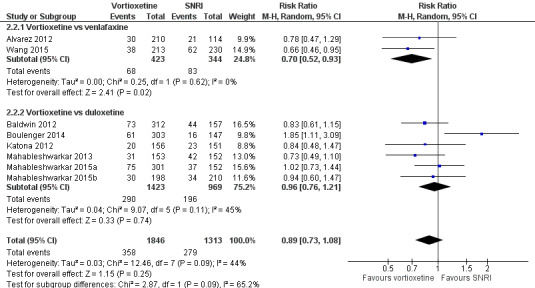
Forest plot of comparison: 3. Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors, outcome: 3.2 Total number of dropouts.
There was no significant difference between trials comparing vortioxetine to duloxetine or venlafaxine (Chi2 = 2.87, df = 1, P = 0.09), but total dropout rates were significantly lower for vortioxetine compared to venlafaxine (RR 0.70, 95% CI 0.52 to 0.93; P = 0.02; 2 studies, 767 participants; I2 = 0%). There was no statistically significant difference between vortioxetine and duloxetine for total dropouts (RR 0.96, 95% CI 0.76 to 1.21; P = 0.74; 6 studies, 2392 participants; I2 = 45%).
Secondary outcomes
2.3. Achieved remission
There was no significant difference in the number of participants who achieved remission between vortioxetine and SNRIs as a whole (RR 0.89, 95% CI 0.77 to 1.03; P = 0.11; 8 studies, 3155 participants). Heterogeneity between studies was substantial (I2 = 57%) (Analysis 2.3).
2.3. Analysis.
Comparison 2 Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors (SNRIs), Outcome 3 Remission.
There was no statistically significant difference between the specific comparators (Chi2 = 1.15, df = 1, P = 0.28) and there were no statistically significant differences in remission rates between vortioxetine and venlafaxine (RR 0.99, 95% CI 0.81 to 1.20, P = 0.88; 2 studies, 767 participants; I2 = 37%) or vortioxetine and duloxetine (RR 0.85, 95% CI 0.70 to 1.02; P = 0.09; 6 studies, 2388 participants; I2 = 58%).
2.4. Depressive symptoms
There was evidence that vortioxetine was less effective in lowering depression scores compared to SNRIs as a whole (MD 1.52, 95% CI 0.50 to 2.53; P = 0.003; 8 studies, 2807 participants). Heterogeneity between studies was moderate (I2 = 50%) (Analysis 2.4).
2.4. Analysis.
Comparison 2 Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors (SNRIs), Outcome 4 Depressive symptoms.
There was no significant difference between trials comparing vortioxetine to duloxetine or venlafaxine (Chi2 = 2.11, df = 1, P = 0.15). Comparing vortioxetine to duloxetine, the depression scores were significantly more reduced by duloxetine (MD 1.99, 95% CI 1.15 to 2.83; P < 0.001; 6 studies, 2106 participants; I2 = 6%). There was no significant difference for this outcome between vortioxetine and venlafaxine (MD 0.02, 95% CI ‐2.49 to 2.54; P = 0.99; 2 studies, 701 participants; I2 = 65%).
2.5. Dropout due to adverse events
There was no evidence that vortioxetine was associated with a lower or higher dropout rate due to adverse events compared to SNRIs as a whole (RR 0.74, 95% CI 0.51 to 1.08; P = 0.12; 8 studies, 3159 participants). Heterogeneity between studies was moderate (I2 = 55%) (Analysis 2.5).
2.5. Analysis.
Comparison 2 Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors (SNRIs), Outcome 5 Dropout due to adverse events.
There was a statistically significant difference between the comparators (Chi2 = 7.07, df = 1, P = 0.008). Dropout rates due to adverse events were significantly lower for vortioxetine compared to venlafaxine (RR 0.42, 95% CI 0.26 to 0.67; P < 0.001; 2 studies; 767 participants; I2 = 0%). There was no statistically significant difference between vortioxetine and duloxetine (RR 0.92, 95% CI 0.65 to 1.31; P = 0.65; 6 studies, 2392 participants; I2 = 30%).
2.6. Dropout due to inefficacy
There was no evidence that vortioxetine was associated with a lower or higher dropout rate due to inefficacy compared to SNRIs as a whole (RR 1.52, 95% CI 0.70 to 3.30; P = 0.29; 8 studies, 3159 participants). Heterogeneity between studies was moderate (I2 = 30%) (Analysis 2.6).
2.6. Analysis.
Comparison 2 Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors (SNRIs), Outcome 6 Dropout due to inefficacy.
There was no significant difference between trials comparing vortioxetine to duloxetine or venlafaxine (Chi2 = 1.29, df = 1, P = 0.26). Furthermore, there were no significant differences in dropout rates due to inefficacy between vortioxetine and venlafaxine (RR 2.68, 95% CI 0.99 to 7.24; P = 0.05; 2 studies, 767 participants; I2 = 0%) or vortioxetine and duloxetine (RR 1.16, 95% CI 0.41 to 3.31; P = 0.78; 6 studies, 2392 participants; I2 = 34%).
2.7. Tolerability
There was evidence that fewer participants experienced adverse effects when treated with vortioxetine than when treated with SNRIs as a whole (RR 0.90, 95% CI 0.86 to 0.94; P < 0.001; 8 studies, 3139 participants). There was no heterogeneity (I2 = 0%) (Analysis 2.7).
2.7. Analysis.
Comparison 2 Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors (SNRIs), Outcome 7 Tolerability.
There was no statistically significant difference between the specific comparators (Chi2 = 0.09, df = 1, P = 0.76). The comparison between vortioxetine and duloxetine showed that fewer participants experienced adverse effects when treated with vortioxetine (RR 0.89, 95% CI 0.84 to 0.95; P < 0.001; 6 studies, 2376 participants; I2 = 18%). There was no significant difference between vortioxetine and venlafaxine (RR 0.91, 95% CI 0.82 to 1.00; P = 0.06; 2 studies, 758 participants; I2 = 0%).
Specific adverse effects compared to SNRIs are reported descriptively in Analysis 2.16. Specific serious adverse events are reported in Analysis 2.17.
2.16. Analysis.
Comparison 2 Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors (SNRIs), Outcome 16 Adverse events.
2.17. Analysis.
Comparison 2 Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors (SNRIs), Outcome 17 Serious adverse events.
This analysis used ITT data as reported in the trials.
Subgroup analyses
Comparison 1. Vortioxetine versus placebo
Fixed versus flexible dosing schemes
Two studies compared placebo to a flexible dose of vortioxetine (Alvarez 2012; Mahableshwarkar 2015b). There were no significant differences between the subgroups in terms of treatment response (test for subgroup differences: Chi2 = 0.05, df = 1, P = 0.82; Analysis 1.8) and total number of dropouts (Chi2 = 0.70, df = 1, P = 0.40; Analysis 1.9).
1.8. Analysis.
Comparison 1 Vortioxetine versus placebo, Outcome 8 Subgroup analysis: fixed vs flexible dosing ‐ response.
1.9. Analysis.
Comparison 1 Vortioxetine versus placebo, Outcome 9 Subgroup analysis: fixed vs flexible dosing ‐ total number of dropouts.
Treatment setting: primary care versus inpatient care versus outpatient care
We found no studies in primary care settings and all studies including inpatients also included outpatients, so it was impossible to conduct this subgroup analysis.
Older people (aged greater than 65 years): included versus excluded
We excluded four studies from this subgroup analysis, because it was unclear if older participants were included (Henigsberg 2012; Jain 2013; Mahableshwarkar 2013; Takeda 2011). Four studies excluded older participants (Alvarez 2012; Mahableshwarkar 2015b; McIntyre 2014; NCT01255787). One study recruited only older participants (Katona 2012). There were no differences in response rates between the subgroups (Chi2 = 0.52, df = 1, P = 0.47; Analysis 1.10), but dropout rates differed significantly (Chi2 = 5.02, df = 1, P = 0.02; Analysis 1.11). In the studies including older participants, the dropout rates were significantly lower in the placebo groups (RR 1.25, 95% CI 1.05 to 1.49; P = 0.01; I2 = 0%). The number of total dropouts was not significantly different compared to placebo in the studies excluding older participants (RR 0.90, 95% CI 0.71 to 1.13; P = 0.36; I2 = 0%).
1.10. Analysis.
Comparison 1 Vortioxetine versus placebo, Outcome 10 Subgroup analysis: inclusion of older (aged > 65 years) participants ‐ response.
1.11. Analysis.
Comparison 1 Vortioxetine versus placebo, Outcome 11 Subgroup analysis: inclusion of older (aged > 65 years) participants ‐ total number of dropouts.
Comparison 2. Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors
Fixed versus flexible dosing schemes
One study compared a flexible dose of vortioxetine 10 mg to 20 mg versus a fixed dose of duloxetine 60 mg/day (Mahableshwarkar 2015b). All other studies used a fixed dose scheme. The study with a flexible dose found no significant differences in terms of response rates (Chi2 = 0.03, df = 1, P = 0.86; Analysis 2.8) and total dropouts (Chi2 = 0.04, df = 1, P = 0.84; Analysis 2.9), as compared to the fixed‐dose studies.
2.8. Analysis.
Comparison 2 Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors (SNRIs), Outcome 8 Subgroup analysis: fixed vs flexible dosing ‐ response.
2.9. Analysis.
Comparison 2 Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors (SNRIs), Outcome 9 Subgroup analysis: fixed vs flexible dosing ‐ total number of dropouts.
Treatment setting: primary care versus inpatient care versus outpatient care
We found no studies in primary care settings and all studies including inpatients also included outpatients, so it was impossible to conduct this subgroup analysis.
Older participants (aged greater than 65 years): included versus excluded
We excluded one study from this analysis, because it was unclear if older participants were included in the study population (Mahableshwarkar 2013). Three studies comparing vortioxetine to an SNRI excluded older participants (Alvarez 2012; Mahableshwarkar 2015b; Wang 2015). There was no significant difference between the subgroups in response rates (test for subgroup differences: Chi2 = 2.52, df = 1, P = 0.11; Analysis 2.10) and total dropout rates (test for subgroup differences: Chi2 = 2.38, df = 1, P = 0.12; Analysis 2.11).
2.10. Analysis.
Comparison 2 Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors (SNRIs), Outcome 10 Subgroup analysis: inclusion of older (aged > 65 years) participants ‐ response.
2.11. Analysis.
Comparison 2 Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors (SNRIs), Outcome 11 Subgroup analysis: inclusion of older (aged > 65 years) participants ‐ total number of dropouts.
Sensitivity analyses
The following sensitivity analyses were defined a priori.
Exclusion of trials with unequal dosing
This sensitivity analysis is only meaningful for the comparisons of vortioxetine with active comparators.
In six studies, vortioxetine and control antidepressants were compared using unequal doses. Two studies compared vortioxetine 5 mg/day (25% of the maximum dose) to duloxetine 60 mg/day (50% of the maximum dose) (Katona 2012; Mahableshwarkar 2013), and one compared vortioxetine 5 mg/day or 10 mg/day (50% of the maximum dose) to venlafaxine 225 mg/day (100% of the maximum dose for moderately depressed outpatients) (Alvarez 2012). Two studies used higher vortioxetine doses and compared vortioxetine 15 mg/day or 20 mg/day (100% of the maximum dose) to duloxetine 60 mg/day (50% of the maximum dose) (Boulenger 2014; Mahableshwarkar 2015a). One study compared flexible dosing of vortioxetine to a fixed dose of duloxetine (Mahableshwarkar 2015b). To increase the usability of the analysis, the analysis was conducted by grouping the comparisons into subgroups according to the direction of the imbalance in dosing.
The analysis of response rates revealed statistically significant differences between the groups (Chi² = 14.99, df = 3, P = 0.002; Analysis 2.12). The two trials with fair (or comparable) dosing found no differences between vortioxetine and SNRIs (RR 1.09, 95% CI 0.97 to 1.22; P = 0.13; 912 participants; I2 = 0%), but the studies using higher and lower vortioxetine doses than comparator showed significantly higher response rates in participants treated with SNRIs (higher: RR 0.80, 95% CI 0.72 to 0.90; P < 0.001; 2 studies, 903 participants; I2 = 0%; lower: RR 0.87, 95% CI 0.78 to 0.98; P = 0.02; 3 studies, 936 participants; I2 = 9%).
2.12. Analysis.
Comparison 2 Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors (SNRIs), Outcome 12 Sensitivity analysis ‐ unequal dosing ‐ response.
The analysis of the total number of dropouts found no statistically significant differences between the subgroups (Chi2 = 3.68, df = 3, P = 0.30; Analysis 2.13). In the trials with fair (or comparable) dosing, vortioxetine showed statistically significant lower total dropout rate than SNRIs as a whole (RR 0.75, 95% CI 0.59 to 0.96; P = 0.02; 2 studies, 912 participants; I2 = 0%). The studies using higher vortioxetine doses, lower vortioxetine doses and flexible versus fixed dosing found no statistically significant dropout rates between vortioxetine and SNRIs (higher dose: RR 1.33, 95% CI 0.74 to 2.39; P = 0.33; 2 studies, 903 participants; I2 = 72%; lower vortioxetine dose: RR 0.77, 95% CI 0.59 to 1.02; P = 0.06; 3 studies, 936 participants; I2 = 0%; flexible versus fixed dosing: RR 0.94, 95% CI 0.60 to 1.47; P = 0.77; 1 study, 408 participants).
2.13. Analysis.
Comparison 2 Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors (SNRIs), Outcome 13 Sensitivity analysis ‐ unequal dosing ‐ total number of dropouts.
Exclusion of studies that did not employ a double‐blind approach
All our included studies were double‐blinded.
Exclusion of studies with subsets of participants with bipolar disorders
None of the included studies randomised people with bipolar disorder.
Exclusion of trials with dropout rates of more than 20% in one of the treatment arm included
This sensitivity analyses were performed for the comparisons of vortioxetine with placebo and vortioxetine with SNRIs.
Six studies reported overall dropout rates of more than 20% and were excluded (Baldwin 2012; Boulenger 2014; Jain 2013; Mahableshwarkar 2013; Mahableshwarkar 2015a; Wang 2015).
The analyses of response rates showed no significant differences between dropout rates below and above 20% for the comparison between vortioxetine and placebo (Chi2 = 1.00, df = 1, P = 0.32; Analysis 1.12) and the comparison between vortioxetine and SNRIs (Chi2 = 0, df = 1, P = 0.95; Analysis 2.14).
1.12. Analysis.
Comparison 1 Vortioxetine versus placebo, Outcome 12 Sensitivity analysis ‐ exclusion > 20% dropouts ‐ response.
2.14. Analysis.
Comparison 2 Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors (SNRIs), Outcome 14 Sensitivity analysis ‐ exclusion > 20% dropouts ‐ response.
The analyses of the total number of dropouts found no statistically significant differences between the subgroups for the comparison between vortioxetine and placebo (Chi2 = 0.21, df = 1, P = 0.64; Analysis 1.13) and the comparison between vortioxetine and SNRIs (Chi2 = 0.11, df = 1, P = 0.74; Analysis 2.15).
1.13. Analysis.
Comparison 1 Vortioxetine versus placebo, Outcome 13 Sensitivity analysis ‐ exclusion > 20% dropouts ‐ total number of dropouts.
2.15. Analysis.
Comparison 2 Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors (SNRIs), Outcome 15 Sensitivity analysis ‐ exclusion > 20% dropouts ‐ total number of dropouts.
Exclusion of studies with imputed data
There is no need for this sensitivity analysis, because the analysis did not include imputed data.
Exclusion of studied sponsored by the manufacturer of vortioxetine
All studies were conducted by the manufacturers of vortioxetine, therefore this sensitivity analysis is not meaningful.
Reporting bias
We examined the funnel plot for the primary outcomes for the comparison of vortioxetine and placebo only, because the comparison with SNRIs did not contain more than 10 studies. Both funnel plots were inconclusive. We were able to identify two unpublished trials (NCT01255787; Takeda 2011).
Discussion
Summary of main results
Our review included 15 studies (7746 participants). Statistically, vortioxetine was more effective than placebo. This advantage was consistent across the three efficacy outcomes of response, remission and depressive symptoms, although the quality of the evidence was low for response and remission and very low for depressive symptoms (see Table 1). Clinically, there is uncertainty on the relevance of these differences. According to some authors, statistically significant differences in response and remission rates should be accepted as clinically relevant (Montgomery 2009), but in the present review we estimated a RR of 1.35 for response and 1.32 for remission, which correspond to a number needed to treat for an additional beneficial outcome (NNTB) of 8 (95% CI 5 to 12) for response and 13 (95% CI 8 to 29) for remission (assuming a baseline response and remission rate of 356/1000 for response and 224/1000 for remission; see Table 1). Thus, doctors would need to treat 8 (95% CI 5 to 12) people with vortioxetine, rather than placebo, for one additional responding patient and 13 people (95% CI 8 to 29) for one additional remitting patient. Furthermore, the difference in change scores between vortioxetine and placebo was estimated to be fewer than 3 points on the MADRS at study endpoint. Some authors suggested that a difference of 2 points on HAM‐D or MADRS is already clinically relevant (Montgomery 2009), but others have shown that an improvement of up to 3 points on the HAM‐D‐17 corresponded to "no change" on CGI (Leucht 2013). Clearly, this casts uncertainty on the real added value of vortioxetine. We found no statistically significant difference in terms of total dropout rates, although more participants discontinued vortioxetine because of adverse effects, while significantly more participants discontinued placebo because of inefficacy. The subgroup or sensitivity analysis revealed no factors that significantly influenced the results.
In comparison with other antidepressants, very low quality of evidence found no clinically significant advantage in efficacy of vortioxetine over the SNRIs as a class (see Table 2). Against individual antidepressants, the analyses of response rates and change in depressive symptoms scores suggested that vortioxetine may be less effective than duloxetine, although in terms of remission rates, there was no difference. Against venlafaxine, meta‐analysis of two studies found no statistically significant differences. In terms of tolerability, our analyses of total dropout rates and dropouts due to adverse events or inefficacy found no significant differences between vortioxetine and the SNRIs as a class. In terms of number of participants reporting at least one adverse effect, vortioxetine was better than the SNRIs as a class and duloxetine. However, the sensitivity analysis casts some doubts on this result, as only two studies used comparable dosing.
Overall completeness and applicability of evidence
All studies included were conducted in highly selected populations, excluding, for example, people with psychiatric comorbidities and suicidal ideation, which are commonly seen in everyday practice. Thus, the external validity of our results may be limited. Furthermore, we were unable to identify studies that compared vortioxetine to pharmacological classes other than SNRI, which is a major limitation in terms of applicability of the evidence in routine care, as the SSRIs are the most commonly prescribed antidepressants as first‐line treatment.
In accordance with the protocol, we did not conduct meta‐analysis on adverse events and did not include scales assessing accompanying symptoms, although these aspects may be of paramount relevance in clinical practice, often guiding the selection of an antidepressant (Gartlehner 2012). Our list of adverse events shows that there is considerable variance in naming, grouping and reporting adverse events; similarly, functional outcomes or accompanying symptoms were erratically assessed and reported. Thus, meta‐analyses of these outcomes cannot be informative and may rather be misleading in informing clinical practice.
Quality of the evidence
We chose seven outcomes for assessing the quality of evidence and to construct the 'Summary of findings' tables (the chosen outcomes being: response, total number of dropouts, remission, depressive symptoms, dropouts due to adverse events, dropouts due to inefficacy, tolerability expressed as patient experiencing as least one adverse event). We constructed two separate 'Summary of findings' tables: one for vortioxetine compared to placebo (Table 1), and one for vortioxetine compared to SNRIs (Table 2).
The quality of the evidence for the comparisons of vortioxetine and placebo was low for remission and response rates and very low for the analysis of depressive symptoms, showing uncertainty in the magnitude of effect. The quality was downgraded because of high dropout rates in some of the trials or because of statistical heterogeneity. We rated the quality of the evidence with respect to treatment discontinuation as moderate for all three outcomes in studies of vortioxetine versus placebo (Table 1).
For studies comparing vortioxetine versus the SNRIs, the quality of the evidence was very low for all outcomes (Table 2). The quality was downgraded because of high dropout rates and substantial statistical heterogeneity.
Study limitations (risk of bias)
All studies employed proper double‐blind procedures and adequate masking of the outcome assessment. However, we judged that all outcomes in studies comparing vortioxetine versus placebo had a serious risk of bias, as about 30% of studies showed an overall dropout rate above 20%. We downgraded the quality of evidence by one level accordingly. In studies comparing vortioxetine versus the SNRIs, the risk of bias was very serious for all outcomes, as about 60% of studies had a dropout rate above 20%.
Consistency of effect
In studies comparing vortioxetine versus placebo, there was a moderate degree of heterogeneity (I2 = 30% to 60%) (Schünemann 2013) for the outcomes 'response' and 'remission.' The quality of evidence was downgraded by one level. There was a substantial degree of heterogeneity (I2 = 60% to 90%) for the outcome 'depressive symptoms.' For this reason, we downgraded the quality of evidence by two levels for these outcomes. In the comparison vortioxetine versus SNRIs, there was a moderate degree of heterogeneity for four outcomes, namely 'response,' 'remission,' 'depressive symptoms' and 'dropouts due to adverse events.' We downgraded the quality of evidence by one level. Reasons underpinning this statistical heterogeneity are uncertain. In general, included studies were homogeneous in terms of clinical features of participants, as the vast majority enrolled participants with the same diagnosis and within the same range of age. Also trial duration was very similar between studies, while the characteristics of the setting differed between studies (see Included studies).
Indirectness
We judged that there was no serious risk of indirectness, as the diagnostic criteria, the characteristics of participants, type of interventions and outcomes largely matched those of interest. Most studies were conducted in high‐ and middle‐income countries, especially in the US. Evidence for people from large parts of the world, for example, Africa, South America and the Middle East was lacking (see Figure 2).
Imprecision
For studies comparing vortioxetine versus placebo, there was no serious risk of imprecision, because of the relatively large number of participants and events. However, for studies comparing vortioxetine versus SNRIs, we downgraded the outcomes 'total dropouts' and 'dropouts due to adverse events' by one level for serious risk of imprecision, as the 95% CI crossed both 1 (no differences) and 0.75 (appreciable benefit for vortioxetine) (according to Guyatt 2011). The outcome 'dropouts due to inefficacy' was downgraded by two levels for very serious risk of imprecision, as the 95% CI crossed 1 (no differences), 0.75 (appreciable benefit for vortioxetine) and 1.25 (appreciable benefit for SNRIs).
Publication bias
Visual inspection of the funnel plot did not suggest a relevant risk of publication bias. Our review included some unpublished studies and this may have reduced the impact of publication bias. As experts in the field were contacted during the search phase, it is unlikely that relevant studies were overlooked. However, although the search was thorough, there is still the possibility of not having identified unpublished studies, considering that standardised procedures to perform this type of search are lacking (Chan 2012), and that all studies were sponsored by the pharmaceutical company.
Potential biases in the review process
Although we judged our funnel plots to be inconclusive and we cannot rule out that we missed studies, we consider that our study sample is rather comprehensive and less prone to publication bias in comparison to, for example, reviews of agomelatine (Guaiana 2013; Koesters 2013). We included two unpublished trials only, but, for example, the failed trial and the three negative trials listed in the FDA medical review (CDER 2013) have been published and are included in our review (Baldwin 2012; Jain 2013; Mahableshwarkar 2013; Mahableshwarkar 2015c). However, there is some evidence for a sponsorship bias in antidepressant research favouring the experimental drug (Barbui 2004; Weinmann 2008). Our analysis contained only trials conducted by the manufacturers, so we cannot rule out a sponsorship bias in favour of vortioxetine in our data. Furthermore, our analysis combined different doses of vortioxetine, which may have increased heterogeneity of the effects. However, considering that previous reviews found no significant dose‐response effects (Meeker 2015; Thase 2016), this approach may be justified. Furthermore, doses may also vary considerably across participants within study arms and it therefore may be necessary to review these effects based on individual participant data.
Agreements and disagreements with other studies or reviews
Our review is in line with previous reviews of vortioxetine (e.g. Meeker 2015; Thase 2016; Zhang 2015) showing that vortioxetine has a statistically significant advantage compared to placebo in terms of efficacy, but did not show an advantage compared to SNRIs. However, considering the quality of the evidence and other potential sources of bias, we suggest caution in the interpretation of efficacy data. This cautious interpretation is in line with findings from previous reviews, for example by not finding a clear dose‐response relationship (Meeker 2015; Thase 2016), and the noteworthy finding that only high doses (20 mg/day) of vortioxetine were significantly superior to placebo in US populations (Thase 2016; Zhang 2015). Furthermore, the findings that duloxetine was more effective than vortioxetine, and that there was no difference in terms of efficacy between vortioxetine and venlafaxine, are inconsistent with the evidence showing the superior efficacy of venlafaxine over duloxetine (Cipriani 2009; Schueler 2011). We are aware of the limitations of such an indirect comparison, but these inconsistencies raise some questions about the validity of the findings.
Authors' conclusions
Implications for practice.
The place of vortioxetine in the treatment of acute depression is unclear. Our analysis showed vortioxetine to be more effective than placebo in terms of response, remission and depressive symptoms, but the clinical relevance of these effects is uncertain, because of the small magnitude of effect, the poor quality of evidence and the highly selected participants enrolled In comparison to serotonin‐norepinephrine reuptake inhibitor (SNRI), there was no advantage for vortioxetine, and no studies compared vortioxetine with any of the selective serotonin reuptake inhibitor (SSRIs). Therefore, the available data leave uncertainty on whether vortioxetine is similarly effective, more effective or even less effective in comparison with these reference antidepressants. Vortioxetine might be less effective than duloxetine, but may have advantages over the SNRIs in terms of tolerability profile, as fewer participants reported adverse effects when treated with vortioxetine compared to SNRIs. According to the US Food and Drug Administration (FDA), the adverse effect profile might be similar to that of the SSRIs (Zhang 2015).
Implications for research.
A major limitation of the current evidence is the lack of comparisons with the SSRIs, which are usually recommended as first‐line treatments for acute depression. Therefore, direct comparisons to these agents may help better determine the place of vortioxetine in the treatment of depression. Two studies that compared vortioxetine to an SSRI have recently been completed and may cushion the limitations, although both trials focus on the effects on cognition (see Studies awaiting classification). Future studies should also address and report adverse effects of competitive treatment options using more standardised approaches, to ascertain if vortioxetine is better tolerated than other antidepressants. Furthermore, advanced meta‐analytical approaches, such as individual participant data meta‐analyses should be conducted to investigate whether a dose‐response effect exists, and whether efficacy is moderated by trial characteristics, including countries and settings, a challenging issue which received little attention so far (Zhang 2015). The paucity of direct head‐to‐head comparisons between vortioxetine and other antidepressants would suggest a requirement for multiple‐treatment meta‐analyses attempting to address how this new antidepressant compares, in terms of efficacy and tolerability, with SSRIs.
Acknowledgements
Other contributions
We thank Sarah Dawson (CCMD Trials Search Co‐ordinator) for developing and writing the search strategy and Sabine Reiser for help with the data management and for additional data checks.
The CCMD Group produces standard text for use in the Methods section of their reviews. We have used this text and adapted it as required.
CRG funding acknowledgement
The National Institute for Health Research (NIHR) is the largest single funder of the CCMD Group.
Disclaimer
The views and opinions expressed herein are those of the review authors and do not necessarily reflect those of the NIHR, National Health Service (NHS) or the Department of Health.
Data and analyses
Comparison 1. Vortioxetine versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Response | 14 | 6220 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [1.22, 1.49] |
| 2 Total number of dropouts | 14 | 6220 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.93, 1.19] |
| 3 Remission | 14 | 6220 | Risk Ratio (M‐H, Random, 95% CI) | 1.32 [1.15, 1.53] |
| 4 Depressive symptoms | 14 | 5566 | Mean Difference (IV, Random, 95% CI) | ‐2.94 [‐4.07, ‐1.80] |
| 5 Dropout due to adverse events | 14 | 6220 | Risk Ratio (M‐H, Random, 95% CI) | 1.41 [1.09, 1.81] |
| 6 Dropout due to inefficacy | 14 | 6220 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.34, 0.90] |
| 7 Tolerability | 14 | 6182 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [1.07, 1.16] |
| 8 Subgroup analysis: fixed vs flexible dosing ‐ response | 14 | 6220 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [1.22, 1.49] |
| 8.1 Fixed dose | 12 | 5513 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [1.19, 1.52] |
| 8.2 Flexible dose | 2 | 707 | Risk Ratio (M‐H, Random, 95% CI) | 1.38 [1.16, 1.63] |
| 9 Subgroup analysis: fixed vs flexible dosing ‐ total number of dropouts | 14 | 6220 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.93, 1.19] |
| 9.1 Fixed dose | 12 | 5513 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.94, 1.22] |
| 9.2 Flexible dose | 2 | 707 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.64, 1.30] |
| 10 Subgroup analysis: inclusion of older (aged > 65 years) participants ‐ response | 10 | 4525 | Risk Ratio (M‐H, Random, 95% CI) | 1.38 [1.24, 1.54] |
| 10.1 Older participants included | 6 | 2616 | Risk Ratio (M‐H, Random, 95% CI) | 1.34 [1.15, 1.55] |
| 10.2 Older participants excluded | 4 | 1909 | Risk Ratio (M‐H, Random, 95% CI) | 1.45 [1.23, 1.71] |
| 11 Subgroup analysis: inclusion of older (aged > 65 years) participants ‐ total number of dropouts | 10 | 4528 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.96, 1.28] |
| 11.1 Older participants included | 6 | 2619 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [1.05, 1.49] |
| 11.2 Older participants excluded | 4 | 1909 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.71, 1.13] |
| 12 Sensitivity analysis ‐ exclusion > 20% dropouts ‐ response | 14 | 6220 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [1.22, 1.49] |
| 12.1 < 20% dropouts | 9 | 3927 | Risk Ratio (M‐H, Random, 95% CI) | 1.41 [1.26, 1.57] |
| 12.2 > 20% dropouts | 5 | 2293 | Risk Ratio (M‐H, Random, 95% CI) | 1.26 [1.03, 1.52] |
| 13 Sensitivity analysis ‐ exclusion > 20% dropouts ‐ total number of dropouts | 14 | 6220 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.93, 1.19] |
| 13.1 < 20% dropout | 9 | 3927 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.86, 1.21] |
| 13.2 > 20% dropout | 5 | 2293 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.91, 1.28] |
| 14 Adverse events | 14 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.1 Constipation | 11 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.2 Nausea | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.3 Diarrhoea | 13 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.4 Dry mouth | 12 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.5 Vomiting | 9 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.6 Abdominal pain upper | 6 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.7 Dyspepsia | 7 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.8 Gastro‐oesophageal reflux disease | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.9 Abdominal discomfort | 4 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.10 Abdominal pain | 3 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.11 Stomach discomfort | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.12 Flatulence | 5 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.13 Fatigue | 12 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.14 Pain | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.15 Thirst | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.16 Irritability | 5 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.17 Decreased appetite | 6 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.18 Increased appetite | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.19 Anorexia | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.20 Headache | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.21 Dizziness | 13 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.22 Somnolence | 10 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.23 Tremor | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.24 Sedation | 4 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.25 Dysgeusia | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.26 Hypoaesthesia | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.27 Poor‐quality sleep | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.28 Ejaculation delayed (men) | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.29 Erectile dysfunction (men) | 3 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.30 Dysmenorrhoea | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.31 Hyperhidrosis | 9 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.32 Pruritus generalised | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.33 Pruritus | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.34 Vision blurred | 5 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.35 Nasopharyngitis | 11 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.36 Influenza | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.37 Respiratory tract infection | 6 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.38 Gastroenteritis viral | 3 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.39 Sinusitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.40 Urinary tract infection | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.41 Bronchitis | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.42 Anorgasmia | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.43 Insomnia | 9 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.44 Restlessness | 3 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.45 Abnormal dreams | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.46 Anxiety | 4 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.47 Depression | 3 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.48 Libido decreased | 3 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.49 Orgasm abnormal | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.50 Nightmare | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.51 Middle insomnia | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.52 Suicidal ideation | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.53 Tachycardia | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.54 Palpitations | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.55 Tinnitus | 3 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.56 Back pain | 7 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.57 Arthralgia | 4 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.58 Myalgia | 3 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.59 Musculoskeletal pain | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.60 Muscle spasms | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.61 Pain in extremity | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.62 Hypertension | 4 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.63 Hot flush | 3 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.64 Fall | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.65 Ligament sprain | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.66 Muscle strain | 3 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.67 Accidental overdose | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.68 Road traffic accident | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.69 Nasal congestion | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.70 Yawning | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.71 Rhinorrhoea | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.72 Oropharyngeal pain | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.73 Weight decreased | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.74 Blood pressure increased | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.75 Hepatic function abnormal | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15 Serious adverse events | 14 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.1 Varicella zoster infection | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.2 Kidney infection | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.3 Herpes zoster infection | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.4 Puncture site infection | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.5 Gastroenteritis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.6 Pyelonephritis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.7 Brain tumour | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.8 Gallbladder cancer | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.9 Colon cancer | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.10 Laryngeal cancer | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.11 Renal cell carcinoma | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.12 Bile duct cancer | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.13 Prostate cancer | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.14 Breast cancer recurrent | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.15 Worsening of major depressive disorder | 6 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.16 Suicidal ideation | 3 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.17 Suicide attempt | 5 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.18 Intentional self‐injury | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.19 Self‐injurious behaviour | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.20 Panic attack | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.21 Suicidal behaviour | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.22 Middle ear effusion | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.23 Jaundice cholestatic | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.24 Cholecystitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.25 Pelvic fracture | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.26 Intentional overdose | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.27 Lumbar vertebral fracture | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.28 Injury | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.29 Hip fracture | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.30 Head injury | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.31 Stress fracture | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.32 Craniocerebral injury | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.33 Subdural haematoma | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.34 Brain contusion | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.35 Road traffic accident | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.36 Serotonin syndrome | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.37 Dizziness | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.38 Cerebrovascular accident | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.39 Convulsion | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.40 Transient ischaemic attack | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.41 Lumbar radiculopathy | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.42 Syncope | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.43 Cerebral haematoma | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.44 Subarachnoid haemorrhage | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.45 Adenomyosis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.46 Vaginal haemorrhage | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.47 Pulmonary embolism | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.48 Blood pressure decreased | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.49 Tachycardia | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.50 Acute myocardial infarction | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.51 Atrial fibrillation | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.52 Coronary artery disease | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.53 Pancreatitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.54 Hiatus hernia | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.55 Drug hypersensitivity | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.56 Abortion spontaneous | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.57 Ectopic pregnancy | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.58 Abortion missed | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.59 Abortion induced | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.60 Type 1 diabetes mellitus | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.61 Hypertension | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.62 Renal colic | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 2. Vortioxetine versus serotonin‐norepinephrine reuptake inhibitors (SNRIs).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Response | 8 | 3159 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.82, 1.00] |
| 1.1 Vortioxetine vs venlafaxine | 2 | 767 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.85, 1.25] |
| 1.2 Vortioxetine vs duloxetine | 6 | 2392 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.79, 0.94] |
| 2 Total number of dropouts | 8 | 3159 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.73, 1.08] |
| 2.1 Vortioxetine vs venlafaxine | 2 | 767 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.52, 0.93] |
| 2.2 Vortioxetine vs duloxetine | 6 | 2392 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.76, 1.21] |
| 3 Remission | 8 | 3155 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.77, 1.03] |
| 3.1 Vortioxetine vs venlafaxine | 2 | 767 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.81, 1.20] |
| 3.2 Vortioxetine vs duloxetine | 6 | 2388 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.70, 1.02] |
| 4 Depressive symptoms | 8 | 2807 | Mean Difference (IV, Random, 95% CI) | 1.52 [0.50, 2.53] |
| 4.1 Vortioxetine vs venlafaxine | 2 | 701 | Mean Difference (IV, Random, 95% CI) | 0.02 [‐2.49, 2.54] |
| 4.2 Vortioxetine vs duloxetine | 6 | 2106 | Mean Difference (IV, Random, 95% CI) | 1.99 [1.15, 2.83] |
| 5 Dropout due to adverse events | 8 | 3159 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.51, 1.08] |
| 5.1 Vortioxetine vs venlafaxine | 2 | 767 | Risk Ratio (M‐H, Random, 95% CI) | 0.42 [0.26, 0.67] |
| 5.2 Vortioxetine vs duloxetine | 6 | 2392 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.65, 1.31] |
| 6 Dropout due to inefficacy | 8 | 3159 | Risk Ratio (M‐H, Random, 95% CI) | 1.52 [0.70, 3.30] |
| 6.1 Vortioxetine vs venlafaxine | 2 | 767 | Risk Ratio (M‐H, Random, 95% CI) | 2.68 [0.99, 7.24] |
| 6.2 Vortioxetine vs duloxetine | 6 | 2392 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.41, 3.31] |
| 7 Tolerability | 8 | 3134 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.86, 0.94] |
| 7.1 Vortioxetine vs venlafaxine | 2 | 758 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.82, 1.00] |
| 7.2 Vortioxetine vs duloxetine | 6 | 2376 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.84, 0.95] |
| 8 Subgroup analysis: fixed vs flexible dosing ‐ response | 8 | 3159 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.82, 1.00] |
| 8.1 Fixed dosing | 7 | 2751 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.81, 1.01] |
| 8.2 Flexible dosing | 1 | 408 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.75, 1.14] |
| 9 Subgroup analysis: fixed vs flexible dosing ‐ total number of dropouts | 8 | 3159 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.73, 1.08] |
| 9.1 Fixed dosing | 7 | 2751 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.71, 1.11] |
| 9.2 Flexible dosing | 1 | 408 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.60, 1.47] |
| 10 Subgroup analysis: inclusion of older (aged > 65 years) participants ‐ response | 7 | 2850 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.83, 1.03] |
| 10.1 Older participants included | 4 | 1675 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.77, 0.98] |
| 10.2 Older participants excluded | 3 | 1175 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.88, 1.15] |
| 11 Subgroup analysis: inclusion of older (aged > 65 years) participants ‐ total number of dropouts | 7 | 2854 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.74, 1.15] |
| 11.1 Older participants included | 4 | 1679 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.76, 1.45] |
| 11.2 Older participants excluded | 3 | 1175 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.60, 0.97] |
| 12 Sensitivity analysis ‐ unequal dosing ‐ response | 8 | 3159 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.82, 1.00] |
| 12.1 Equal dosing | 2 | 912 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.97, 1.22] |
| 12.2 Vortioxetine dose higher | 2 | 903 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.72, 0.90] |
| 12.3 Vortioxetine dose lower | 3 | 936 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.78, 0.98] |
| 12.4 Flexible vs fixed dosing | 1 | 408 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.75, 1.14] |
| 13 Sensitivity analysis ‐ unequal dosing ‐ total number of dropouts | 8 | 3159 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.73, 1.08] |
| 13.1 Equal dosing | 2 | 912 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.59, 0.96] |
| 13.2 Vortioxetine dose higher | 2 | 903 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.74, 2.39] |
| 13.3 Vortioxetine dose lower | 3 | 936 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.59, 1.02] |
| 13.4 Flexible vs fixed dosing | 1 | 408 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.60, 1.47] |
| 14 Sensitivity analysis ‐ exclusion > 20% dropouts ‐ response | 8 | 3159 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.82, 1.00] |
| 14.1 < 20% dropouts | 3 | 1039 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.82, 1.01] |
| 14.2 > 20% dropouts | 5 | 2120 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.77, 1.07] |
| 15 Sensitivity analysis ‐ exclusion > 20% dropouts ‐ total number of dropouts | 8 | 3159 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.73, 1.08] |
| 15.1 < 20% dropouts | 3 | 1039 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.64, 1.14] |
| 15.2 > 20% dropouts | 5 | 2120 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.68, 1.24] |
| 16 Adverse events | 8 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16.1 Constipation | 6 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.2 Diarrhoea | 7 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.3 Dry mouth | 8 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.4 Nausea | 8 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.5 Vomiting | 4 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.6 Abdominal pain upper | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.7 Dyspepsia | 3 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.8 Abdominal discomfort | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.9 Abdominal pain | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.10 Stomach discomfort | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.11 Flatulence | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.12 Fatigue | 6 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.13 Irritability | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.14 Decreased appetite | 6 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.15 Anorexia | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.16 Dizziness | 8 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.17 Headache | 8 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.18 Somnolence | 4 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.19 Tremor | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.20 Sedation | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.21 Dysgeusia | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.22 Poor quality sleep | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.23 Ejaculation delayed (men) | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.24 Erectile dysfunction (men) | 3 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.25 Hyperhidrosis | 6 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.26 Vision blurred | 3 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.27 Nasopharyngitis | 4 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.28 Upper respiratory tract infection | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.29 Gastroenteritis viral | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.30 Urinary tract infection bacterial | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.31 Anorgasmia | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.32 Insomnia | 5 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.33 Restlessness | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.34 Abnormal dreams | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.35 Anxiety | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.36 Depression | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.37 Libido decreased | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.38 Orgasm abnormal | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.39 Nightmare | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.40 Middle insomnia | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.41 Palpitations | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.42 Tinnitus | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.43 Back pain | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.44 Arthralgia | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.45 Myalgia | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.46 Musculoskeletal pain | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.47 Muscle spasms | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.48 Pain in extremity | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.49 Hot flush | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.50 Fall | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.51 Muscle strain | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.52 Accidental overdose | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.53 Yawning | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.54 Rhinorrhoea | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.55 Weight decreased | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.56 Blood pressure increased | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.57 Heart rate increased | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17 Serious adverse events | 8 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 17.1 Varicella zoster infection | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.2 Brain tumour | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.3 Gallbladder cancer | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.4 Bile duct cancer | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.5 Prostate cancer | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.6 Worsening of major depressive disorder | 6 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.7 Suicidal ideation | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.8 Suicide attempt | 3 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.9 Intentional self‐injury | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.10 Self‐injurious behaviour | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.11 Panic attack | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.12 Anxiety | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.13 Middle ear effusion | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.14 Vertigo positional | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.15 Jaundice cholestatic | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.16 Pelvic fracture | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.17 Intentional overdose | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.18 Lumbar vertebral fracture | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.19 Hip fracture | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.20 Head injury | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.21 Stress fracture | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.22 Craniocerebral injury | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.23 Subdural haematoma | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.24 Asthenia | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.25 Serotonin syndrome | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.26 Dizziness | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.27 Convulsion | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.28 Transient ischaemic attack | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.29 Adenomyosis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.30 Vaginal haemorrhage | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.31 Varicocele | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.32 Pulmonary embolism | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.33 Blood pressure decreased | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.34 Acute myocardial infarction | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.35 Atrial fibrillation | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.36 Coronary artery disease | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.37 Abortion induced | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.38 Ligament sprain | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Alvarez 2012.
| Methods | Study design: double‐blind, randomised, placebo‐controlled, active reference study. | |
| Participants |
Diagnosis: MDE as primary diagnosis according to DSM‐IV‐TR criteria; current MDE duration ≥ 3 months and < 12 months; MADRS total score ≥ 30. Method of diagnosis: MINI. Age: 18‐65 years. Sex: 63% women. Location: multinational (Australia, Austria, Canada, Czech Republic, Finland, France, Italy, Malaysia, Slovakia, Spain, Sweden). Comorbidities: none. Adjunctive therapy: people receiving formal behaviour therapy or systematic psychotherapy excluded. Adjunctive medication: occasional use of zolpidem, zopiclone and zaleplon for insomnia allowed. Sample size: vortioxetine 5 mg/d: 109; vortioxetine 10 mg/d: 101; venlafaxine: 114; placebo: 105. |
|
| Interventions | Participants were randomly assigned to 1 of 4 treatments:
6‐week double‐blind treatment period. Randomised participants were given 1‐week wallet cards at each visit and were instructed to take 2 capsules per day, orally, at the same time every day (preferably in the morning). Efficacy and tolerability assessed at screening, baseline, and after 1, 2, 3, 4, 5 and 6 weeks. Participants who completed the 6‐week double‐blind period entered a 2‐week double‐blind taper period. |
|
| Outcomes |
Time points for assessment: 1, 2, 3, 4, 5 and 6 weeks' treatment and 4 weeks' follow‐up. Primary outcome:
Secondary outcomes:
|
|
| Notes |
Date of study: 2006‐2007. Funding source: H. Lundbeck A/S. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients who met the selection criteria at the baseline visit were assigned to double‐blind treatment according to a computer‐generated randomisation." |
| Allocation concealment (selection bias) | Low risk | Quote: "The details of the randomisation series were unknown to any of the investigators and were contained in a set of sealed opaque envelopes." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "All investigators, study personnel and participants were blinded to treatment assignment for the duration of the entire study. The randomisation code was broken for one patient (accidentally) who had completed the study before this was discovered, and was therefore not withdrawn from the study." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Equal dropout rates < 20%; ITT analyses used. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Drug company sponsored trial. |
Baldwin 2012.
| Methods | Study design: randomised, double‐blind, parallel‐group, placebo‐controlled, duloxetine‐referenced, fixed‐dose study. | |
| Participants |
Diagnosis: MDE as primary diagnosis according to DSM‐IV‐TR criteria; moderate‐to‐severe depression; current MDE duration ≥ 3 months; MADRS total score ≥ 26. Method of diagnosis: MINI. Age: range 18‐74 years. Sex: 68% women. Location: multinational (Australia, Bulgaria, Canada, the Czech Republic, Estonia, Finland, France, Hong Kong, India, Republic of Korea, Latvia, Lithuania, Malaysia, Philippines, Romania, Slovakia, Spain, Taiwan, Turkey and Ukraine). Comorbidities: none. Adjunctive therapy: people receiving formal psychological treatment excluded. Adjunctive medication: occasional use of zolpidem, zopiclone and zaleplon for severe insomnia allowed (maximum 2 days/week, not the night before a study visit). Sample size: vortioxetine 2.5 mg/d: 155; vortioxetine 5 mg/d: 159; vortioxetine 10 mg/d: 153 ; duloxetine: 157; placebo: 152. Setting: inpatients and outpatients. |
|
| Interventions | Participants were randomly assigned to 1 of 5 treatments:
8‐week double‐blind treatment period. Efficacy and tolerability assessed at screening, baseline, and after 1, 2, 4, 6 and 8 weeks. Participants who completed the 8‐week double‐blind period could continue in a 52‐week open‐label, flexible‐dose extension study. Study medication given as capsules of identical appearance. Randomised participants instructed to take 1 capsule per day, orally, at the same time every day (preferably in the morning). |
|
| Outcomes |
Time points for assessment: 1, 2, 4, 6 and 8 weeks' treatment and 4 weeks' follow‐up. Primary outcome:
Secondary outcomes:
|
|
| Notes |
Date of study: 2008‐2009. Funding source: H. Lundbeck A/S. Participants not participating in the continuation study entered a 1‐week, double‐blind taper period, during which those on placebo remained on placebo; participants taking vortioxetine 2.5 mg/day, 5 mg/day or 10 mg/day switched abruptly to placebo and participants taking duloxetine 60 mg/day received duloxetine 30 mg/day. The same regimen was offered to participants who withdrew from the study before completion of the acute treatment period. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...a computer‐generated randomisation list." |
| Allocation concealment (selection bias) | Low risk | Central randomisation. Quote: "The details of the randomisation series were unknown to investigators and contained in a set of sealed opaque envelopes." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "All investigators, trial personnel and patients were blinded..." "capsules of identical appearance." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Dropout rate > 20% in the vortioxetine arm. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Drug company sponsored trial. |
Boulenger 2014.
| Methods | Study design: double‐blind, randomised, fixed‐dose, placebo‐controlled, active‐referenced study. | |
| Participants |
Diagnosis: recurrent MDD as the primary diagnosis according to DSM‐IV‐TR criteria (classification code 296.3x); current episode of MDE > 3 months; MADRS total score ≥ 26; CGI‐S score ≥ 4. Method of diagnosis: MINI. Age: range 18‐75 years. Sex: 66% women. Location: multinational (Belgium, Estonia, Finland, France, Germany, Latvia, Lithuania, Norway, Russia, Slovakia, South Africa, Sweden and Ukraine). Comorbidities: none. Adjunctive therapy: people receiving formal psychological treatment excluded. Adjunctive medication: occasional use of zolpidem, zopiclone and zaleplon for severe insomnia allowed (maximum 2 days/week, not the night before a study visit). Sample size: vortioxetine 15 mg/d: 152; vortioxetine 20 mg/d: 151; duloxetine: 147; placebo: 158. Setting: inpatients and outpatients. |
|
| Interventions | Participants were randomly assigned to 1 of 4 treatments:
8‐week double‐blind treatment period. Participants in vortioxetine group received vortioxetine 10 mg/day in week 1 and 15 mg/day or 20 mg/day in weeks 2‐8. Participants in duloxetine group received duloxetine 30 mg/day in week 1 and 60 mg/day in weeks 2‐8. Participants seen weekly during the first 2 weeks of treatment and then every 2 weeks. Study medication given as capsules of identical appearance. Following randomisation, participants instructed to take 1 capsule per day, orally, preferably in the morning. |
|
| Outcomes |
Time points for assessment: pre‐ and post‐treatment and follow‐up 4 weeks after completion or after withdrawal. Primary outcome:
Secondary outcomes:
|
|
| Notes |
Date of study: 2010‐2011. Funding source: H. Lundbeck A/S. Participants who withdrew were seen for a withdrawal visit as soon as possible and were offered a down‐taper regimen. Those who completed the 8‐week, double‐blind treatment period entered a 2‐week, double‐blind discontinuation period. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Eligible patients were assigned to double‐blind treatment according to a randomisation list that was computer generated by H. Lundbeck A/S." |
| Allocation concealment (selection bias) | Low risk | Quote: "The details of the randomisation series were contained in a set of sealed opaque envelopes." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "All investigators, trial personnel and patients were blinded to treatment assignment [...]. The randomisation code was not broken for any patient during the study" "capsules of identical appearance." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Dropout rate > 20% in the vortioxetine arm. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Drug company sponsored trial. |
Henigsberg 2012.
| Methods | Study design: multicentre, randomised, double‐blind, parallel‐group, placebo‐controlled, fixed‐dose study. | |
| Participants |
Diagnosis: primary diagnosis of MDE according to DSM‐IV‐TR criteria; current MDE ≥ 3 months; MADRS total score ≥ 26. Method of diagnosis: MINI. Age: range 18‐75 years. Sex: 61% women. Location: multinational (Australia, Croatia, France, Germany, Latvia, Lithuania, Malaysia, Netherlands, Poland, Republic of Korea, Russia, South Africa, Taiwan and Ukraine). Comorbidities: none. Adjunctive therapy: not reported. Adjunctive medication: not reported. Sample size: vortioxetine 1 mg/d: 140; vortioxetine 5 mg/d: 140; vortioxetine: 10 mg/d: 140; placebo: 140. Setting: unclear. |
|
| Interventions | Participants were randomly assigned to 1 of 4 treatments:
8‐week, double‐blind treatment period. All study medication identical in appearance and dispensed using unique identification numbers. Participants returned to study site for assessments at baseline and weeks 1, 2, 4, 6 and 8. |
|
| Outcomes |
Time points for assessment: 1, 2, 4, 6 and 8 weeks' treatment and 4 weeks' follow‐up. Primary outcome:
Secondary outcomes:
|
|
| Notes |
Date of study: August 2008 to August 2009. Funding source: Takeda; H. Lundbeck A/S. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned;" no details reported. |
| Allocation concealment (selection bias) | Unclear risk | Not discussed. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "All study medication was identical in appearance and dispensed using unique identification numbers." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low and equal dropout rates; ITT analyses used. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Drug company sponsored trial. |
Jacobsen 2015.
| Methods | Study design: phase 3, randomised, double‐blind, parallel‐group, placebo‐controlled, fixed‐dose study. | |
| Participants |
Diagnosis: recurrent MDE as primary diagnosis according to the American Psychiatric Association Diagnostic and DSM‐IV‐TR criteria; MADRS total score ≥ 26 at screening and baseline visits; CGI‐S score ≥ 4 at screening and baseline visits. Method of diagnosis: SCID. Age: range 18‐75 years. Sex: 73% women. Location: US. Comorbidities: none. Adjunctive therapy: not reported. Adjunctive medication: not reported. Sample size: vortioxetine 10 mg/d: 155; vortioxetine 20 mg/d: 150; placebo: 157. Setting: unclear. |
|
| Interventions | Participants were randomly assigned to 1 of 3 treatments:
Treatment remained undisclosed to participant and study doctor throughout study. Participants in vortioxetine group 20 mg/d received vortioxetine 10 mg/day in week 1 and 20 mg/day in weeks 2‐8. Study medication given as capsules in identical blister packs. All participants took 1 capsule orally at the same time each day throughout study. Overall duration 13 weeks. |
|
| Outcomes |
Time points for assessment: 8 weeks' treatment and 4 weeks' follow‐up. Primary outcome:
Secondary outcomes:
|
|
| Notes |
Date of study: 2010 to 2012. Funding source: Takeda. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised," no details reported. |
| Allocation concealment (selection bias) | Unclear risk | No details reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "identical blister packs to maintain blinding." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Equal dropout rates below 20%; ITT analyses used. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Drug company sponsored trial. |
Jain 2013.
| Methods | Study design: multicentre, randomised, double‐blind, parallel‐group, placebo‐controlled, fixed‐dose study. | |
| Participants |
Diagnosis: MDE as primary diagnosis according to DSM‐IV‐TR criteria; reported duration of current MDE ≥ 3 months; MADRS total score ≥ 30. Method of diagnosis: MINI. Age: range 18‐75 years. Sex: 58% women. Location: US. Comorbidities: none. Adjunctive therapy: not reported. Adjunctive medication: none. Sample size: vortioxetine: 300; placebo: 300. Setting: outpatients. |
|
| Interventions | Participants were randomly assigned to 1 of 2 treatments:
6‐week double‐blind treatment period. Centralised computer system used for participant randomisation and study medication assignments. Participants returned to study site weekly to receive wallet cards of either vortioxetine 5 mg or placebo capsules (beginning at baseline) and for efficacy and safety assessments. Blinding of all participants maintained throughout study. All study medication identical in appearance and dispensed using unique identification numbers. |
|
| Outcomes |
Time points for assessment: 6‐week treatment period and a safety follow‐up 4 weeks after the last dose of double‐blind study drug. Primary outcome:
Secondary outcomes:
|
|
| Notes |
Date of study: April‐November 2008. Funding source: Takeda; H. Lundbeck A/S. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A centralised computer system was used for subject randomisation and study medication assignments." |
| Allocation concealment (selection bias) | Low risk | Quote: "A centralized computer system was used for subject randomisation and study medication assignments." "All study medication was identical in appearance and dispensed using unique identification numbers." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Blinding of all participants was maintained throughout the study. All study medication was identical in appearance and dispensed using unique identification numbers." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Dropout rate > 20% in placebo arm. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Drug company sponsored trial. |
Katona 2012.
| Methods | Study design: randomised, double‐blind, parallel‐group, placebo‐controlled, duloxetine‐referenced, fixed‐dose study. | |
| Participants |
Diagnosis: clinical diagnosis of recurrent MDE according the DSM‐IV‐TR criteria; reported duration of current episode ≥ 4 weeks; MADRS total score ≥ 26; ≥ 1 previous MDE before age of 60 years. Method of diagnosis: MINI. Age: range 65‐88 years. Sex: 66% women. Location: multinational (Canada, Finland, France, Germany, Sweden, Ukraine, and the US). Comorbidities: none. Adjunctive therapy: people receiving formal behaviour therapy or systematic psychotherapy excluded. Adjunctive medication: antiarrhythmics, antihypertensives (except metoprolol and class 1C antiarrhythmics), proton pump inhibitors and aspirin as antiplatelet treatment allowed; occasional use of zolpidem, zopiclone and zaleplon for insomnia allowed. Sample size: vortioxetine: 156; duloxetine: 151; placebo: 145. Setting: unclear. |
|
| Interventions | Participants were randomly assigned to 1 of 3 treatments:
8‐week double‐blind treatment period. Participants were seen weekly during first 2 weeks of treatment and then every 2 weeks. Study medication given as capsules of identical appearance. Following randomisation, participants instructed to take 1 capsule per day, orally, preferably in morning. |
|
| Outcomes |
Time points for assessment: 8 weeks' treatment and follow‐up 4 weeks after completion or withdrawal. Primary outcome:
Secondary outcomes:
|
|
| Notes |
Date of study: 2009‐2010. Funding source: H. Lundbeck A/S. Those who completed the 8‐week, double‐blind treatment entered a 1‐week, double‐blind, taper‐down period. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Eligible patients were assigned to double‐blind treatment according to a computer‐generated randomisation list." |
| Allocation concealment (selection bias) | Low risk | Quote: "The details of the randomisation series were contained in a set of sealed opaque envelopes." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quotes: "All investigators, trial personnel and patients were blinded to treatment assignment for the duration of the study." "Study medication was given as capsules of identical appearance." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropouts equally distributed; MMRM and LOCF methods applied. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Drug company sponsored trial. |
Mahableshwarkar 2013.
| Methods | Study design: randomised, double‐blind, parallel‐group, placebo‐controlled, active‐referenced, fixed‐dose study. | |
| Participants |
Diagnosis: MDE as primary diagnosis according to DSM‐IV‐TR criteria; reported duration of current MDE ≥ 3 months. Method of diagnosis: not reported. Age: range 18‐75 years. Sex: 63% women. Location: US. Comorbidities: none. Adjunctive therapy: not reported. Adjunctive medication: not reported. Sample size: vortioxetine 2.5 mg/d: 153; vortioxetine 5 mg/d: 153; duloxetine: 152; placebo: 153. Setting: unclear. |
|
| Interventions | Participants were randomly assigned to 1 of 4 treatments:
8‐week treatment period. Randomisation schedule generated by Takeda Global Research & Development, Inc., and investigators informed of each participant's coded treatment allocation by an interactive voice‐activated system. All study drugs administered as identical‐looking capsules and both participants and investigators blinded to treatment allocation. |
|
| Outcomes |
Time points for assessment: 8‐week double‐blind treatment period and 4‐week safety follow‐up period. Primary outcome:
Secondary outcomes:
|
|
| Notes |
Date of study: 2008. Funding source: Takeda; H. Lundbeck A/S. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details regarding the randomisation available. |
| Allocation concealment (selection bias) | Low risk | Quote: "The randomisation schedule was generated by Takeda Global Research & Development, Inc., and investigators were informed of each patient's coded treatment allocation by an interactive voice‐activated system." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "identical looking capsules and both participants and investigators were blinded." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Dropout rates > 20% in all arms. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Drug company sponsored trial. |
Mahableshwarkar 2015a.
| Methods | Study design: phase 3, multicentre, randomised, double‐blind, parallel‐group, placebo‐controlled, duloxetine‐referenced, fixed‐dose study. | |
| Participants |
Diagnosis: MDE recurrent as primary diagnosis according to DSM‐IV‐TR criteria (classification code 296.3x) confirmed by SCID; reported duration of current MDE ≥ 3 months; MADRS total score ≥ 26 at screening and baseline visits; CGI‐S score ≥ 4 at screening and baseline visits. Method of diagnosis: SCID. Age: range 18‐75 years. Sex: 74% women. Location: US. Comorbidities: none. Adjunctive therapy: people receiving formal cognitive or behavioural therapy or systematic psychotherapy excluded. Adjunctive medication: none. Sample size: vortioxetine 15 mg/d: 147; vortioxetine 20 mg/d: 154; duloxetine: 152; placebo: 161. Setting: unclear. |
|
| Interventions | Participants were randomly assigned to 1 of 4 treatments:
8‐week double‐blind treatment period. Eligible people randomised (1:1:1:1) to receive placebo, vortioxetine 15 mg, vortioxetine 20 mg or duloxetine 60 mg once daily during 8‐week, double‐blind treatment period, using an interactive voice response system. Following randomisation, doses were uptitrated after first week of double‐blind period. Participants assigned to receive vortioxetine 15 mg/day or 20 mg/day received a 10‐mg dose for first week of 8‐week study, and participants assigned to receive duloxetine 60 mg/day received a 30‐mg dose for first week. |
|
| Outcomes |
Time points for assessment: 8 weeks' treatment. Primary outcome:
Secondary outcomes:
|
|
| Notes |
Date of study: 2010‐2012. Funding source: Takeda. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Procedure not described in detail. |
| Allocation concealment (selection bias) | Low risk | Central randomisation; "interactive voice response system." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind; placebo ‐ matching capsules administered. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Dropout rates > 20% in vortioxetine and control group. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Drug company sponsored trial. |
Mahableshwarkar 2015b.
| Methods | Study design: randomised, double‐blind, parallel‐group, placebo‐controlled, active‐referenced, flexible‐dose study. | |
| Participants |
Diagnosis: recurrent MDD as primary diagnosis according to DSM‐IV‐TR criteria (classification code 296.3x). Current MDE confirmed using Mini International Neuropsychiatric Interview V6.0.0; MADRS total score ≥ 26 at screening and baseline; reported duration of current MDE ≥ 3 months. Method of diagnosis: MINI, past medical records. Age: range 18‐65 years. Sex: 65% women. Location: multinational (Bulgaria, Finland, Germany, Poland, Russia Federation, Ukraine and US). Comorbidities: none. Adjunctive therapy: people receiving formal cognitive or behavioural therapy or systematic psychotherapy excluded. Adjunctive medication: not reported. Sample size: vortioxetine: 198; duloxetine: 210; placebo: 194. Setting: unclear. |
|
| Interventions | Participants were randomly assigned to 1 of 3 treatments:
|
|
| Outcomes |
Time points for assessment: 8‐weeks' treatment period, 1‐week taper‐down period. Primary outcome:
Secondary outcomes:
|
|
| Notes |
Date of study: 2012‐2014. Funding source: Takeda. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation not described. |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double blind," "matching capsules." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Equal and low dropout rates; ITT analysis used. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Drug company sponsored trial. |
Mahableshwarkar 2015c.
| Methods | Study design: phase 3, randomised, double‐blind, parallel‐group, placebo‐controlled, fixed‐dose study. | |
| Participants |
Diagnosis: MDE recurrent as primary diagnosis according to DSM‐IV‐TR criteria; reported duration of current MDE ≥ 3 months; MADRS total score ≥ 26 at screening and baseline; CGI‐S score ≥ 4 at screening and baseline. Method of diagnosis: SCID ‐ Clinical Trial version. Age: range 18‐75 years. Sex: 70% women. Location: US. Comorbidities: none. Adjunctive therapy: people receiving formal cognitive or behavioural therapy or systematic psychotherapy excluded. Adjunctive medication: not reported. Sample size: vortioxetine 10 mg/d: 157; vortioxetine 15 mg/d: 152; placebo: 160. Setting: unclear. |
|
| Interventions | Participants were randomly assigned to 1 of 3 treatments:
All participants took 1 capsule at the same time each day throughout the study. Overall time to participate up to 14 weeks. Participants made 7 visits to clinic. |
|
| Outcomes |
Time points for assessment: 8 weeks' treatment period and 4 weeks later safety follow‐up. Primary outcome:
Secondary outcomes:
|
|
| Notes |
Date of study: 2010‐2012. Funding source: Takeda. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised", no details reported. |
| Allocation concealment (selection bias) | Low risk | Central randomisation, "interactive voice system." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Placebo‐matching capsules administered. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Equal and low dropout rates; ITT analysis used. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Drug company sponsored trial. |
McIntyre 2014.
| Methods | Study design: randomised, double‐blind, parallel‐group, placebo‐controlled, fixed‐dose study. | |
| Participants |
Diagnosis: recurrent MDD according to DSM‐IV‐TR criteria (classification code 296.3x). Current MDE confirmed using MINI; MADRS total score ≥ 26; reported duration of current MDE ≥ 3 months. Method of diagnosis: MINI. Age: range 18‐65 years. Sex: 66% women. Location: multinational (Australia, Canada, Finland, France, Germany, Latvia, Mexico, Serbia, Slovakia, South Africa, Ukraine, and US). Comorbidities: none. Adjunctive therapy: people receiving formal psychological treatments excluded. Adjunctive medication: antiarrhythmics, antihypertensives (except metoprolol, carvedilol, timolol and class 1C antiarrhythmics) and proton pump inhibitors (except omeprazole and cimetidine) allowed; episodic use of zolpidem, zopiclone or zaleplon for severe insomnia allowed for maximum 2 days per week, but not the night before a study visit. Sample size: vortioxetine 10 mg/d: 195; vortioxetine 20 mg/d: 207; placebo: 196. Setting: inpatients and outpatients. |
|
| Interventions | Participants were randomly assigned to 1 of 3 treatments:
8‐week double‐blind treatment period. Participants seen at baseline and weeks 1, 4 and 8. Treatments given as capsules of identical appearance. Following randomisation, participants took 1 capsule per day, orally, preferably in morning. |
|
| Outcomes |
Time points for assessment: 8 weeks' treatment period and 4 weeks' follow‐up. Primary outcome:
Secondary outcomes:
|
|
| Notes |
Date of study: 2011‐2013. Funding source: H. Lundbeck A/S. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Eligible patients were assigned to double‐blind treatment according to a randomisation list that was computer generated by H. Lundbeck A/S." |
| Allocation concealment (selection bias) | Low risk | Quote: "The details of the randomisation series were contained in a set of sealed opaque envelopes [...]. The randomisation code was not broken for any patient during the study." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Study medications were given as capsules of identical appearance;" double‐blind trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Equal dropout rates < 20%, ITT analyses used. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Drug company sponsored trial. |
NCT01255787.
| Methods | Study design: randomised, double‐blind, placebo‐controlled, dose‐ranging study (fixed dose). | |
| Participants |
Diagnosis: MDD as primary diagnosis according to DSM‐IV‐TR criteria (classification code 296.2x and 296.3x); reported duration of current MDE ≥ 3 months at screening visit; MADRS total score ≥ 26 at screening and baseline; CGI‐S score ≥ 4 at screening and baseline. Method of diagnosis: MINI. Age: range 20‐64 years. Sex: 63% women. Location: multinational (Croatia, Finland, Germany, Hong Kong, India, Japan, Korea, Republic of, Latvia, Malaysia, Philippines, Poland, Romania, Russian Federation, Serbia, Taiwan, Ukraine). Comorbidities: none. Adjunctive therapy: people receiving formal cognitive or behavioural therapy or systematic psychotherapy excluded. Adjunctive medication: not reported. Sample size: vortioxetine 5 mg/d: 144; vortioxetine 10 mg/d: 150; vortioxetine 20 mg/d: 154; placebo: 152. Setting: unclear. |
|
| Interventions | Participants were randomly assigned to 1 of 4 treatments:
All participants took 1 capsule at the same time each day throughout study. Participants made weekly visits to clinic during the first 2 weeks of 8‐week treatment period and then every 2 weeks up to the end of the 8‐week treatment period. |
|
| Outcomes |
Time points for assessment: 8 weeks' treatment period and 4 weeks' follow‐up. Primary outcome:
Secondary outcomes:
|
|
| Notes |
Date of study: 2010‐2012. Funding source: Takeda. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised," no details reported. |
| Allocation concealment (selection bias) | Unclear risk | No details reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Placebo (dummy inactive pill) ‐ this was a capsule that looked like the study drug but had no active ingredient." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Equal and low dropout rates; ITT analysis used. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Drug company sponsored trial. |
Takeda 2011.
| Methods | Study design: randomised, double‐blind, placebo‐controlled, parallel‐group, phase 3 study. | |
| Participants |
Diagnosis: MDD as primary diagnosis according to DSM‐IV‐TR criteria (classification code 296.2x and 296.3x); reported duration of current MDE ≥ 3 months at screening visit; MADRS total score ≥ 26 at screening and baseline visits; CGI‐S score ≥ 4 at screening and baseline visits. Method of diagnosis: not reported. Age: range 20‐75 years. Sex: 47% women. Location: Japan. Comorbidities: none. Adjunctive therapy: not reported. Adjunctive medication: not reported. Sample size: vortioxetine 5 mg/d: 119; vortioxetine 10 mg/d: 123; placebo: 124. Setting: unclear. |
|
| Interventions | Participants were randomly assigned to 1 of 3 treatments:
1‐week screening period, 8‐week double‐blind treatment period, 4‐week safety follow‐up. Duration of study 13 weeks in total. |
|
| Outcomes |
Time points for assessment: 8 weeks' treatment period and 4 weeks' follow‐up. Primary outcome:
Secondary outcomes:
|
|
| Notes |
Date of study: 2011‐2012. Funding source: Takeda. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised," no details reported. |
| Allocation concealment (selection bias) | Unclear risk | No details reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Placebo‐matching capsules were administered. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Equal and low dropout rates; ITT analysis used. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Drug company sponsored trial. |
Wang 2015.
| Methods | Study design: randomised, double‐blind, parallel‐group, active‐comparator, fixed‐dose study. | |
| Participants |
Diagnosis: recurrent MDD as primary diagnosis according to DSM‐IV‐TR criteria. Current MDE confirmed using MINI; MADRS total score ≥ 26; CGI‐S score ≥ 4; reported duration of current MDE ≥ 3 months. Method of diagnosis: MINI. Age: range 19‐65 years. Sex: 60% women. Location: multinational (China, South Korea, Taiwan and Thailand). Comorbidities: none. Adjunctive therapy: people receiving formal cognitive or behavioural therapy or systematic psychotherapy excluded. Adjunctive medication: occasional use of zolpidem, zopiclone and zaleplon for severe insomnia allowed (maximum 2 days/week, not the night before a study visit). Sample size: vortioxetine: 213; venlafaxine: 230. Setting: inpatients and outpatients. |
|
| Interventions | Participants were randomly assigned to 1 of 2 treatments:
8‐week double‐blind treatment. Participants seen at baseline and weeks 1, 2, 4, 6 and 8. Participants who completed 8‐week treatment period entered a 1‐week double‐blind down‐taper period, during which participants in vortioxetine group received placebo and participants in venlafaxine XR group received 75 mg/day. |
|
| Outcomes |
Time points for assessment: 8 weeks' double‐blind treatment and follow‐up 4 weeks later. Primary outcome:
Secondary outcomes:
|
|
| Notes |
Date of study: 2012‐2013. Funding source: H. Lundbeck A/S. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The IVRS randomly allocated the patient a randomisation number according to a randomisation list that was generated by H. Lundbeck A/S, the manufacturer of vortioxetine." |
| Allocation concealment (selection bias) | Low risk | Quote: "using an interactive voice/web response system." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "...medication was given as venlafaxine XR capsules or encapsulated vortioxetine tablets of identical appearance;" "The randomisation code was not broken for any patient." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Dropout rate > 20% in control (venlafaxine) group. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | Drug company sponsored trial. |
ASEX: Arizona Sexual Experience Scale; C‐SSRS: Columbia Suicide Severity Rating Scale; CGI‐I: Clinical Global Impression ‐ Improvement; CGI‐S: Clinical Global Impression ‐ Severity; CRT: Cognitive Reflection Test; DSM‐IV‐TR: Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision; DSST: Digit Symbol Substitution Test; DT: Detection Task; GDS: Geriatrics Depression Scale; GMLT: Groton Maze Learning Test; HAD: Hospital Anxiety and Depression; HAM‐A: Hamilton Anxiety Rating Scale; HAM‐D: Hamilton Depression Rating Scale; HAM‐D‐24: 24‐item Hamilton Depression Scale; IT: Identification Task; ITT: intention to treat; LOCF: last observation carried forward; MADRS: Montgomery‐Åsberg Depression Scale; MDD: major depressive disorder; MDE: major depressive episode; MINI: Mini International Neuropsychiatric Interview; MMRM: mixed model for repeated measurement; PDQ: Perceived Deficits Questionnaire; RAVLT: Rey Auditory Verbal Learning Task; SCID: Structured Clinical Interview for DSM Disorders; SDS: Sheehan Disability Scale; SF‐36: 36‐Item Short‐Form Health Survey; SRT: Simple Reaction Time; TMT‐A: Trail Making Test A; TMT‐B: Trail Making Test B.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Boulenger 2012 | Relapse prevention study. |
| Browning 2014 | Functional magnetic resonance imaging study that randomised remitted participants and healthy controls. |
| Jacobsen 2015a | Compared switching to vortioxetine vs escitalopram in participants who responded to citalopram, paroxetine or sertraline. Treatment and dosage had to be stable for 8 weeks before randomisation. |
| Jacobsen 2015b | Open‐label extension study including people completing Mahableshwarkar 2015a; Jacobsen 2015; or Mahableshwarkar 2015c. |
| Montgomery 2014 | Compared switching to vortioxetine vs agomelatine in people with inadequate response to ≥ 6 weeks' treatment with selective serotonin re‐uptake inhibitors (citalopram, escitalopram, paroxetine) or serotonin‐norepinephrine reuptake inhibitors (duloxetine, venlafaxine). |
| NCT02371980 | Relapse prevention study (ongoing). |
Characteristics of studies awaiting assessment [ordered by study ID]
NCT02272517.
| Methods | 8‐week, randomised, double‐blind, active controlled trial. |
| Participants | Recurrent MDD as the primary diagnosis according to DSM‐IV‐TR criteria confirmed using MINI; PHQ‐9 score ≥ 14; MADRS total score ≥ 22; CGI‐S score ≥ 4; duration of current episode ≤ 1 year. Age: 18‐65 years. Location: Finland, Germany, Serbia and Slovakia. |
| Interventions | Vortioxetine: 10‐20 mg/day. Escitalopram: 10‐20 mg/day. |
| Outcomes | Primary outcome: change in DSST. Secondary outcomes: various cognitive test, change in PDQ‐D, change in PHQ‐9 (depressive symptoms), CGI‐S, CGI‐I, C‐SSRS. |
| Notes |
NCT02279966.
| Methods | 8‐week, randomised, double‐blind, placebo‐ and active‐controlled trial. |
| Participants | MDD as primary diagnosis according to DSM‐IV‐TR criteria confirmed using MINI; MADRS total score ≥ 26; duration of current episode ≥ 3 months. Age: 18‐65 years. Location: Estonia, Finland, Germany and Lithuania. |
| Interventions | Vortioxetine 10 mg/day. Paroxetine 20 mg/day. Placebo. |
| Outcomes | Primary outcome: change in DSST (number of correct symbols). Secondary outcomes: various cognitive tests, change in PDQ‐D, change in PHQ‐9 (depressive symptoms), CGI‐S, CGI‐I, C‐SSRS, MADRS. |
| Notes |
C‐SSRS: Columbia Suicide Severity Rating Scale; CGI‐I: Clinical Global Impression ‐ Improvement; CGI‐S: Clinical Global Impression ‐ Severity; DSM‐IV‐TR: Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision; DSST: Digit Symbol Substitution Test; MADRS: Montgomery‐Åsberg Depression Scale; MDD: major depressive disorder; MINI: Mini International Neuropsychiatric Interview; PDQ‐D: Perceived Deficits Questionnaire ‐ Depression; PHQ‐9: Patient Health Questionnaire‐9.
Characteristics of ongoing studies [ordered by study ID]
NCT02294305.
| Trial name or title | Vortioxetine versus Placebo in Major Depressive Disorder Comorbid with Social Anxiety Disorder. |
| Methods | 12‐week, randomised, double‐blind, placebo‐controlled trial. |
| Participants | MDD and SAD according to DSM‐V criteria confirmed using MINI; MADRS total score ≥ 26; CGI‐S (composite for MDD and SAD) ≥ 4; duration of current depressive episode ≥ 4 weeks; duration of SAD ≥ 6 months. Age: 18‐70 years. Location: US. |
| Interventions | Vortioxetine 10‐20 mg/day. Placebo. |
| Outcomes | Primary outcome: CGI‐I responder rate. Secondary outcomes: change in MADRS total score, change in Liebowitz Social Anxiety Scale total score. |
| Starting date | |
| Contact information | |
| Notes |
NCT02389816.
| Trial name or title | A Phase 3 Study of Lu AA21004 in Patients with Major Depressive Disorder. |
| Methods | 8‐week, randomised, double‐blind, placebo‐controlled trial. |
| Participants | Recurrent MDD as primary diagnosis according to DSM‐IV‐TR; MADRS total score ≥ 26; HAM‐D‐17 total score ≥ 18; CGI‐S ≥ 4; duration of current depressive episode 3‐12 months. Age: 20‐75 years. Location: Japan. |
| Interventions | Vortioxetine 10 mg/day. Vortioxetine 20 mg/day. Placebo. |
| Outcomes | Primary outcome: change from baseline in MADRS. Secondary outcomes: MADRS response and remission, change in HAM‐D, CGI, SDS, DSST and PDQ‐5. |
| Starting date | April 2015. |
| Contact information | Takeda Study Registration Call Center, tel: +1‐800‐778‐2860 (USA and EU); email: medicalinformation@tpna.com. |
| Notes |
CGI: Clinical Global Impression; CGI‐I: Clinical Global Impression ‐ Improvement; CGI‐S: Clinical Global Impression ‐ Severity; DSM‐IV‐TR: Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision; DSM‐V: Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition; DSST: Digit Symbol Substitution Test; HAM‐D: Hamilton Depression Scale; HAM‐D‐17: 17‐item Hamilton Depression Scale; MADRS: Montgomery‐Åsberg Depression Scale; MDD: major depressive disorder; MINI: Mini International Neuropsychiatric Interview; PDQ‐5: Perceived Deficits Questionnaire; SAD: social anxiety disorder; SDS: Sheehan Disability Scale.
Differences between protocol and review
Our protocol stated that we would prefer Hamilton Depression Rating Scale (HAM‐D) data to Montgomery‐Åsberg Depression Scale (MADRS) data. However, all studies used the MADRS to assess depressive symptoms and eight studies did not use the HAM‐D scale (Boulenger 2014; Jacobsen 2015; Mahableshwarkar 2015a; Mahableshwarkar 2015b; Mahableshwarkar 2015c; McIntyre 2014; NCT01255787; Wang 2015). Therefore, to decrease heterogeneity across trials and to be able use a mean difference as a summary score instead of a standardised mean difference, we decided to give preference to the MADRS data.
The protocol stated that "We will include cluster‐RCTs [randomised controlled trials] if direct effect estimates are provided that account for the clustering or if sufficient information is were available to account for the clustering that allow an approximation in accordance to the method suggested in the Cochrane Handbook for Systematic Reviews of Interventions (Section 16.3.4, Higgins 2011). The method suggested by Higgins 2011 requires an estimate of the intracluster correlation coefficient (ICC), which can be thought of the 'similarity' of individuals within the same cluster and often has to be obtained from an external source. The idea of the approximation is to correct the sample size of the trial to its 'effective sample size' by taking a 'design effect' into account. We excluded cluster randomisation with insufficient information for an approximation." As no relevant cluster controlled trials were identified, we shortened the 'Method' section accordingly.
The protocol stated that trials with cross‐over designs will be included, but no such studies could be identified.
We decided post‐hoc to calculate NNTBs for response and remission to facilitate the interpretation of the effects for these outcomes.
Contributions of authors
MK formulated the idea.
MK, GO and CB wrote the protocol.
GO and MK selected the studies, extracted the data and assessed the risk of bias.
JB double‐checked data entry and analyses.
MK analysed the data and wrote the first draft of the review.
CB resolved disagreements on study inclusion and provided supervision of data extraction and analyses.
GO and GG created the 'Summary of findings' tables and wrote major parts of the 'Quality of the evidence' section.
All authors reviewed the drafts, contributed to the final text and approved the final version of the review.
Sources of support
Internal sources
None, Other.
External sources
None, Other.
Declarations of interest
MK: none.
GO: none.
GG: none.
JB: none.
CB: none.
New
References
References to studies included in this review
Alvarez 2012 {published data only}
- Alvarez E, Perez V, Dragheim M, Loft H, Artigas F. A double‐blind, randomized, placebo‐controlled, active reference study of Lu AA21004 in patients with major depressive disorder. International Journal of Neuropsychopharmacology 2012;15(5):589‐600. [Lundbeck 11492A; NCT00839423] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT00839423. Randomised placebo‐controlled venlafaxine‐referenced study of efficacy and safety of 5 and 10 mg of vortioxetine (Lu AA21004) in acute treatment of major depressive disorder in adults. clinicaltrials.gov/ct2/show/NCT00839423 Date first received: 6 February 2009.
Baldwin 2012 {published data only}
- Baldwin D, Loft H, Dragheim M. A randomised, double‐blind, placebo controlled, duloxetine‐referenced, fixed‐dose study of three dosages of Lu AA21004 in MDD treatment. European Neuropsychopharmacology 2011;21(Suppl 3):S390. [DOI] [PubMed] [Google Scholar]
- Baldwin DS, Loft H, Dragheim M. A randomised, double‐blind, placebo controlled, duloxetine‐referenced, fixed‐dose study of three dosages of Lu AA21004 in acute treatment of major depressive disorder (MDD). European Neuropsychopharmacology 2012;22(7):482‐91. [Lundbeck 11984A; NCT00635219] [DOI] [PubMed] [Google Scholar]
- NCT00635219. Randomised placebo‐controlled duloxetine‐referenced efficacy and safety study of 2.5, 5 and 10 mg of vortioxetine (Lu AA21004) in acute treatment of major depressive disorder. clinicaltrials.gov/ct2/show/NCT00635219 Date first received: 3 March 2008.
Boulenger 2014 {published data only}
- Boulenger J‐P, Loft H, Olsen CK. Efficacy and safety of vortioxetine (Lu AA21004), 15 and 20 mg/day: a randomized, double‐blind, placebo‐controlled, duloxetine‐referenced study in the acute treatment of adult patients with major depressive disorder. International Clinical Psychopharmacology 2014;29(3):138‐49. [NCT01140906] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01140906. A randomised, double‐blind, parallel‐group, placebo‐controlled, duloxetine‐referenced, fixed‐dose study evaluating the efficacy and safety of Lu AA21004 (15 and 20 mg/day) in the acute treatment of adult patients with major depressive disorder. clinicaltrials.gov/show/NCT01140906 Date first received: 9 June 2010.
Henigsberg 2012 {published data only}
- Henigsberg N, Mahableshwarkar A, Jacobsen P, Chen Y, Thase ME. Efficacy and tolerability of multiple doses of LU AA21004 in an 8‐week trial of adults with major depressive disorder. European Neuropsychopharmacology 2011;21(Suppl 3):S393. [DOI] [PubMed] [Google Scholar]
- Henigsberg N, Mahableshwarkar AR, Jacobsen P, Chen Y, Thase ME. A randomized, double‐blind, placebo‐controlled 8‐week trial of the efficacy and tolerability of multiple doses of Lu AA21004 in adults with major depressive disorder. Journal of Clinical Psychology 2012;73(7):953‐9. [DOI] [PubMed] [Google Scholar]
- NCT00707980. A long‐term, open‐label, flexible‐dose, extension study evaluating the safety and tolerability of Lu AA21004 in subjects with major depressive disorder. clinicaltrials.gov/ct2/show/NCT00707980 Date first received: 27 June 2008.
- NCT00735709. A randomized, double‐blind, parallel‐group, placebo‐controlled, fixed‐dose study comparing the efficacy and safety of 3 doses of Lu AA21004 in acute treatment of adults with major depressive disorder. clinicaltrials.gov/show/NCT00735709 Date first received: 14 August 2008.
Jacobsen 2015 {published data only}
- Jacobsen PL, Mahableshwarkar AR, Serenko M, Chan S, Trivedi MH. A randomized, double‐blind, placebo‐controlled study of the efficacy and safety of vortioxetine 10 mg and 20 mg in adults with major depressive disorder. Journal of Clinical Psychiatry 2015;76(5):575‐82. [DOI] [PubMed] [Google Scholar]
- NCT01163266. A phase 3, randomized, double‐blind, parallel‐group, placebo‐controlled, fixed‐dose study comparing the efficacy and safety of 2 doses (10 and 20 mg) of Lu AA21004 in acute treatment of adults with major depressive disorder. clinicaltrials.gov/show/NCT01163266 Date first received: 14 July 2010.
Jain 2013 {published data only}
- Jain R, Mahableshwarkar AR, Jacobsen PL, Chen Y, Thase ME. A randomized, double‐blind, placebo‐controlled 6‐wk trial of the efficacy and tolerability of 5 mg vortioxetine in adults with major depressive disorder. International Journal of Neuropsychopharmacology 2013;16(2):313‐21. [DOI] [PubMed] [Google Scholar]
- NCT00672958. A randomized, double‐blind, parallel‐group, placebo‐controlled, fixed‐dose study comparing the efficacy and safety of Lu AA21004 versus placebo in acute treatment of adults with major depressive disorder. clinicaltrials.gov/show/NCT00672958 Date first received: 2 May 2008.
Katona 2012 {published data only}
- Katona C, Hansen T, Olsen CK. A randomised, double‐blind, placebo controlled, active‐referenced study of the multimodal antidepressant Lu AA21004 in the treatment of elderly depressed patients. European Neuropsychopharmacology 2012;22:S258‐9. [Google Scholar]
- Katona C, Hansen T, Olsen CK. A randomized, double‐blind, placebo‐controlled, duloxetine‐referenced, fixed‐dose study comparing the efficacy and safety of Lu AA21004 in elderly patients with major depressive disorder. International Clinical Psychopharmacology 2012;27(4):215‐23. [Lundbeck 12541A; NCT00811252] [DOI] [PubMed] [Google Scholar]
- NCT00811252. Randomised placebo‐controlled duloxetine‐referenced study of efficacy and safety of 5 mg of vortioxetine (Lu AA21004) in acute treatment of major depressive disorder in elderly patients. clinicaltrials.gov/ct2/show/NCT00811252 Date first received: 17 December 2008.
Mahableshwarkar 2013 {published data only}
- Mahableshwarkar AR, Jacobsen PL, Chen Y. A randomized, double‐blind trial of 2.5mg and 5mg vortioxetine (Lu AA21004) versus placebo for 8 weeks in adults with major depressive disorder. Current Medical Research and Opinion 2013;29(3):217‐26. [NCT00672620] [DOI] [PubMed] [Google Scholar]
- NCT00672620. A randomized, double‐blind, parallel‐group, placebo‐controlled, active‐referenced, fixed‐dose study comparing the efficacy and safety of 2 doses of Lu AA21004 in acute treatment of adults with major depressive disorder. clinicaltrials.gov/show/NCT00672620 Date first received: 2 May 2008.
- NCT00707980. A long‐term, open‐label, flexible‐dose, extension study evaluating the safety and tolerability of Lu AA21004 in subjects with major depressive disorder. clinicaltrials.gov/ct2/show/NCT00707980 Date first received: 27 June 2008.
Mahableshwarkar 2015a {published data only}
- Mahableshwarkar AR, Jacobsen PL, Chen Y, Serenko M, Trivedi MH. A randomized, double‐blind, duloxetine‐referenced study comparing efficacy and tolerability of 2 fixed doses of vortioxetine in the acute treatment of adults with MDD. Psychopharmacology 2015;232(12):2061‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01153009. A phase 3, randomized, double‐blind, parallel‐group, placebo‐controlled, duloxetine‐referenced, fixed‐dose study comparing the efficacy and safety of 2 doses (15 and 20 mg) of Lu AA21004 in acute treatment of adults with major depressive disorder. clinicaltrials.gov/show/NCT01153009 Date first received: 28 June 2010.
Mahableshwarkar 2015b {published data only}
- Jacobson W, Harvey P, Merikle E, Zhong W, Nomikos G, Olsen CK, et al. Impact of vortioxetine on functional capacity in MDD patients with subjective cognitive dysfunction: performance on the University of California San Diego performance‐based skills assessment (UPSA). 54th Annual Meeting of the American College of Neuropsychopharmacology, ACNP; 2015 Dec 6‐10; Hollywood (FL). 2015:S150‐1.
- Jacobson W, Olsen C, Mahableshwarkar A, Yinzhong C, Keefe R. Effect of vortioxetine on functional capacity in patients with major depressive disorder with self‐reported cognitive dysfunction. 28th European College of Neuropsychopharmacology, ECNP Congress; 2015 29 Aug ‐ 1 Sept; Amsterdam Netherlands. 2015:S460.
- Keefe R, Mahableshwarkar A, Zajecka J, Jacobson W, Chen Y. Efficacy of vortioxetine on cognitive function in patients with major depressive disorder: cognitive test performance results: from a randomized, double‐blind, duloxetine‐referenced, placebo‐controlled. Neuropsychopharmacology 2014;39:S389‐90. [Google Scholar]
- Mahableshwarkar AR, Zajecka J, Jacobson W, Chen Y, Keefe RS. A randomized, placebo‐controlled, active‐reference, double‐blind, flexible‐dose study of the efficacy of vortioxetine on cognitive function in major depressive disorder. Neuropsychopharmacology 2015;40(8):2025‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01564862. A randomized, double‐blind, parallel‐group, placebo‐controlled, active‐referenced, flexible dose study on the efficacy of Lu AA21004 on cognitive dysfunction in adult subjects with major depressive disorder (MDD). clinicaltrials.gov/show/NCT01564862 Date first received: 26 March 2012.
Mahableshwarkar 2015c {published data only}
- Mahableshwarkar AR, Jacobsen PL, Serenko M, Chen Y, Trivedi MH. A randomized, double‐blind, placebo‐controlled study of the efficacy and safety of 2 doses of vortioxetine in adults with major depressive disorder. Journal of Clinical Psychiatry 2015;76(5):583‐91. [DOI] [PubMed] [Google Scholar]
- NCT01179516. A phase 3, randomized, double‐blind, parallel‐group, placebo‐controlled, fixed‐dose study comparing the efficacy and safety of 2 doses (10 and 15 mg) of Lu AA21004 in acute treatment of adults with major depressive disorder. clinicaltrials.gov/show/NCT01179516 Date first received: 9 August 2010.
McIntyre 2014 {published data only}
- Harrison J. Assessment of cognitive dysfunction in patients with depression ‐ which test is best?. 28th European College of Neuropsychopharmacology, ECNP Congress; 2015 29 Aug ‐ 1 Sept; Amsterdam Netherlands. 2015:S669.
- McIntyre RS, Lophaven S, Olsen CK. A randomized, double‐blind, placebo‐controlled study of vortioxetine on cognitive function in depressed adults. International Journal of Neuropsychopharmacology 2014;17(10):1557‐67. [NCT01422213] [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre RS, Lophaven S, Olsen CK. Randomized, double‐blind, placebo‐controlled study of the efficacy of vortioxetine on cognitive dysfunction in adult patients with major depressive disorder (MDD). 52nd Annual Meeting of the American College of Neuropsychopharmacology, ACNP; 2013 Dec 8‐12; Hollywood (FL). 2013:S380‐1.
- NCT01422213. Randomised, double‐blind, parallel‐group, placebo‐controlled, fixed dose study on the efficacy of [vortioxetine] Lu AA21004 on cognitive dysfunction in adult patients with major depressive disorder (MDD). clinicaltrials.gov/show/NCT01422213 Date first received: 22 August 2011.
NCT01255787 {published data only}
- Euctr2010‐022257‐41‐Lv. A multinational, randomized, double‐blind, placebo‐controlled, dose ranging study to assess the efficacy and safety of Lu AA21004 in patients with major depressive disorder ‐ efficacy and safety of Lu AA21004 for treatment of major depressive disorder. www.clinicaltrialsregister.eu/ctr‐search/search?query=eudract_number:2010‐022257‐41 2010.
- NCT01255787. A multinational, randomized, double‐blind, placebo‐controlled, dose ranging study to assess the efficacy and safety of Lu AA21004 in patients with major depressive disorder. clinicaltrials.gov/show/NCT01255787 Date first received: 6 December 2010.
Takeda 2011 {published data only}
- Jprn‐Japiccti‐111492. A randomized, double‐blind, placebo‐controlled, phase III study to assess the efficacy and safety of LuAA21004 in patients with major depressive disorder. www.clinicaltrials.jp/user/showCteDetailE.jsp?japicId=JapicCTI‐111492 Date first received: 9 May 2011.
- NCT01355081. A randomized, double‐blind, placebo‐controlled, parallel‐group, phase III study to assess the efficacy and safety of Lu AA21004 in patients with major depressive disorder. clinicaltrials.gov/show/NCT01355081 Date first received: 16 May 2011.
Wang 2015 {published data only}
- Kct0000432. Randomised, double‐blind, parallel‐group, active‐comparator (venlafaxine extended release), fixed‐dose study of LuAA21004 in major depressive disorder in Asian countries. cris.nih.go.kr/cris/en/search/search_result_st01.jsp?seq=2532 Date first approved: 16 August 2011.
- NCT01571453. Randomised, double‐blind, parallel‐group, active‐comparator (venlafaxine extended release), fixed‐dose study of [vortioxetine] Lu AA21004 in major depressive disorder in Asian countries. clinicaltrials.gov/show/NCT01571453 Date first received: 28 March 2012.
- Wang G, Gislum M, Filippov G, Montgomery S. Comparison of vortioxetine versus venlafaxine XR in adults in Asia with major depressive disorder: a randomized, double‐blind study. Current Medical Research and Opinion 2015;31(4):785‐94. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Boulenger 2012 {published data only}
- Boulenger J‐P, Loft H, Florea I. A randomized clinical study of Lu AA21004 in the prevention of relapse in patients with major depressive disorder. Journal of Psychopharmacology (Oxford, England) 2012;26(11):1408‐16. [DOI] [PubMed] [Google Scholar]
- NCT00596817. Efficacy of Lu AA21004 in the prevention of relapse of major depressive episodes. clinicaltrials.gov/ct2/show/NCT00596817 Date first received: January 8, 2008.
Browning 2014 {published data only}
- Browning M, Smith J, Conen S, Smallman R, Buchbjerg J, Larsen Kg, et al. Vortioxetine reduces BOLD signal during performance of the N‐back task in subjects remitted from depression and healthy control participants. 28th European College of Neuropsychopharmacology, ECNP Congress; 2015 Aug 29 to Sep 1; Amsterdam, Netherlands. 2015:S314‐5.
- Browning M, Smith J, Conen S, Smallman R, Buchbjerg J, Larsen Kg, et al. Vortioxetine reduces bold signal during performance of the N‐Back task in subjects remitted from depression and healthy control participants [abstract]. 53rd Annual Meeting of the American College of Neuropsychopharmacology, ACNP; 2014 Dec 7‐11; Phoenix (AZ). 2014:S480.
- Conen S, McKie S, Smallman RP, Dutta A, Dawson Gr, Smith J, et al. Effects of vortioxetine on resting‐state activity in subjects remitted from depression and healthy controls. 28th European College of Neuropsychopharmacology, ECNP Congress; 2015 Aug 29 to Sep 1; Amsterdam, Netherlands. 2015:S442.
- Dawson G, Conen S, McKie S, Smallman R, Smith J, Browning M, et al. Effects of vortioxetine on resting‐state activity in subjects remitted from depression and healthy controls. 53rd Annual Meeting of the American College of Neuropsychopharmacology, ACNP; 2014 Dec 7‐11; Phoenix (AZ). 2014:S214‐5.
Jacobsen 2015a {published data only}
- Jacobsen PL, Mahableshwarkar AR, Chen Y, Chrones L, Clayton AH. Effect of vortioxetine vs. escitalopram on sexual functioning in adults with well‐treated major depressive disorder experiencing SSRI‐induced sexual dysfunction. Journal of Sexual Medicine 2015;12(10):2036‐48. [DOI] [PubMed] [Google Scholar]
Jacobsen 2015b {published data only}
- Jacobsen PL, Harper L, Chrones L, Chan S, Mahableshwarkar AR. Safety and tolerability of vortioxetine (15 and 20 mg) in patients with major depressive disorder: results of an open‐label, flexible‐dose, 52‐week extension study. International Clinical Psychopharmacology 2015;30(5):255‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Montgomery 2014 {published data only}
- Haggstrom L, Nielsen RZ, Danchenko N, Poulsen L. A randomised, double‐blind, study of vortioxetine versus agomelatine in adults with major depressive disorder (MDD) with inadequate response to SSRI/SNRI treatment. 26th European College of Neuropsychopharmacology, ECNP Congress; 2013 Oct 5‐9; Barcelona, Spain. 2013:S412.
- Haggstrom L, Nielsen RZ, Dragheim M. Randomized, double‐blind, study of vortioxetine versus agomelatine in adults with MDD after inadequate response to SSRI or SNRI treatment. European Psychiatry 2013;28(Suppl 1):3009. [Google Scholar]
- Montgomery SA, Nielsen RZ, Poulsen LH, Haggstrom L. A randomised, double‐blind study in adults with major depressive disorder with an inadequate response to a single course of selective serotonin reuptake inhibitor or serotonin‐noradrenaline reuptake inhibitor treatment switched to vortioxetine or agomelatine. Human Psychopharmacology 2014;29(5):470‐82. [EUCTR2011‐002362‐21; NCT01488071] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01488071. A randomised, double‐blind, parallel‐group, active‐controlled, flexible dose study evaluating the effects of Lu AA21004 versus agomelatine in adult patients suffering from major depressive disorder with inadequate response to antidepressant treatment. clinicaltrials.gov/show/NCT01488071 Date first received: 29 November 2011.
- Papakostas G, Dragheim M, Nielsen RZ. Efficacy and tolerability of vortioxetine is independent of previous treatment in MDD patients switched after an inadequate response. European Neuropsychopharmacology 2014;24:S466. [DOI] [PubMed] [Google Scholar]
NCT02371980 {published data only}
- NCT02371980. A randomized, double‐blind, placebo‐controlled, phase 4, relapse prevention study evaluating the efficacy and safety of vortioxetine (5, 10 and 20 mg) in adults with major depressive disorder. clinicaltrials.gov/show/NCT02371980 Date first received: 20 February 2015.
References to studies awaiting assessment
NCT02272517 {published data only}
- NCT02272517. An interventional, randomised, double‐blind, parallel‐group, active‐comparator, flexible‐dose study on the efficacy of vortioxetine versus escitalopram on cognitive dysfunction in patients with inadequate response to current antidepressant treatment of major depressive disorder. clinicaltrials.gov/show/NCT02272517 Date first received: 21 October 2014.
NCT02279966 {published data only}
- NCT02279966. An interventional, randomised, double‐blind, parallel‐group, placebo‐controlled, active‐referenced (paroxetine), fixed‐dose study on the efficacy of vortioxetine on cognitive dysfunction in working patients with major depressive disorder. clinicaltrials.gov/show/NCT02279966 Date first received: 21 October 2014.
References to ongoing studies
NCT02294305 {published data only}
- NCT02294305. Vortioxetine versus placebo in major depressive disorder comorbid with social anxiety disorder. clinicaltrials.gov/show/NCT02294305 Date first received: 11 November 2014.
NCT02389816 {published data only}
- Jprn‐Japiccti‐152831. A randomized, double‐blind, placebo‐controlled, parallel‐group, phase III study to assess the efficacy and safety of Lu AA21004 in patients with major depressive disorder. www.clinicaltrials.jp/user/showCteDetailE.jsp?japicId=JapicCTI‐152831 Date first received: 4 March 2015.
- NCT02389816. A randomized, double‐blind, placebo‐controlled, parallel‐group, phase III study to assess the efficacy and safety of Lu AA21004 in patients with major depressive disorder. clinicaltrials.gov/show/NCT02389816 Date first received: 10 March 2015.
Additional references
APA 1980
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM‐III). 3rd Edition. Washington (DC): American Psychiatric Association, 1980. [Google Scholar]
APA 1987
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM‐III‐R). 3rd Edition. Washington (DC): American Psychiatric Association, 1987. [Google Scholar]
APA 1994
- American Psychiatric Association. .. Diagnostic and Statistical Manual of Mental Disorders (DSM‐IV). 4th Edition. Washington (DC): American Psychiatric Association, 1994. [Google Scholar]
APA 2000
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM‐IV‐TR). 4th Edition. Washington (DC): American Psychiatric Association, 2000. [Google Scholar]
APA 2010
- American Psychiatric Association. American Psychiatric Association Practice Guidelines for the Treatment of Patients with Major Depressive Disorder. 3rd Edition. Arlington (VA): American Psychiatric Publishing, Inc, 2010. [Google Scholar]
APA 2013
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM‐5. 5th Edition. Washington (DC): American Psychiatric Association, 2013. [Google Scholar]
Baldwin 2016
- Baldwin DS, Chrones L, Florea I, Nielsen R, Nomikos Gg, Palo W, et al. The safety and tolerability of vortioxetine: analysis of data from randomized placebo‐controlled trials and open‐label extension studies. Journal of Psychopharmacology (Oxford, England) 2016;30(3):242‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Barbui 2004
- Barbui C, Cipriani A, Brambilla P, Hotopf M. "Wish bias" in antidepressant drug trials?. Journal of Clinical Psychopharmacology 2004;24(2):126‐30. [DOI] [PubMed] [Google Scholar]
Bauer 2008
- Bauer M, Monz BU, Montejo AL, Quail D, Dantchev N, Demyttenaere K, et al. Prescribing patterns of antidepressants in Europe: results from the Factors Influencing Depression Endpoints Research (FINDER) study. European Psychiatry 2008;23(1):66‐73. [DOI] [PubMed] [Google Scholar]
Berhan 2014
- Berhan A, Barker A. Vortioxetine in the treatment of adult patients with major depressive disorder: a meta‐analysis of randomized double‐blind controlled trials. BMC Psychiatry 2014;14:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bromet 2011
- Bromet E, Andrade L, Hwang I, Sampson NA, Alonso J, Girolamo G, et al. Cross‐national epidemiology of DSM‐IV major depressive episode. BMC Medicine 2011;9:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bull 2002
- Bull SA, Hunkeler EM, Lee JY, Rowland CR, Williamson TE, Schwab JR, et al. Discontinuing or switching selective serotonin‐reuptake inhibitors. Annals of Pharmacotherapy 2002;36(4):578‐84. [DOI] [PubMed] [Google Scholar]
CDER 2013
- Center for Drug Evaluation and Research. Application number: 204447Orig1s000. Medical review(s), 2013. www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204447orig1s000medr.pdf (accessed 27 July 2016).
Chan 2012
- Chan AW. Out of sight but not out of mind: how to search for unpublished clinical trial evidence. BMJ 2012;3(344):d8013. [DOI] [PubMed] [Google Scholar]
Chinese Society of Psychiatry 2001
- Chinese Society of Psychiatry. The Chinese Classification and Diagnostic Criteria of Mental Disorders Version 3 (CCMD‐3) 2001. www.21jk.com.cn/english/ (accessed 2 February 2015).
Cipriani 2009
- Cipriani A, Furukawa TA, Salanti G, Geddes JR, Higgins JPT, Churchill R, et al. Comparative efficacy and acceptability of 12 new‐generation antidepressants: a multiple‐treatments meta‐analysis. Lancet 2009;373(9665):746‐58. [DOI] [PubMed] [Google Scholar]
Citrome 2014
- Citrome L. Vortioxetine for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant ‐ what is the number needed to treat, number needed to harm and likelihood to be helped or harmed?. International Journal of Clinical Practice 2014;68(1):60‐82. [DOI] [PubMed] [Google Scholar]
Citrome 2016
- Citrome L. Vortioxetine for major depressive disorder: an indirect comparison with duloxetine, escitalopram, levomilnacipran, sertraline, venlafaxine, and vilazodone, using number needed to treat, number needed to harm, and likelihood to be helped or harmed. Journal of Affective Disorders 2016;196:225‐33. [DOI] [PubMed] [Google Scholar]
Du Jardin 2016
- Du Jardin KG, Liebenberg N, Muller HK, Elfving B, Sanchez C, Wegener G. Differential interaction with the serotonin system by S‐ketamine, vortioxetine, and fluoxetine in a genetic rat model of depression. Psychopharmacology 2016;233(14):2813‐25. [DOI] [PubMed] [Google Scholar]
EMA 2014
- European Medical Agency. Brintellix: EPAR‐Summary for the public. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Summary_for_the_public/human/002717/WC500159448.pdf 2014, (accessed 21 June 2017).
EMA 2014a
- European Medicines Agency. Brintellix EPAR ‐ public assessment report 2014. www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/002717/WC500159447.pdf (accessed 2 February 2015).
EMA 2014b
- European Medicines Agency. Brintellix: EPAR ‐ product information 2014. www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Product_Information/human/002717/WC500159449.pdf (accessed 2 February 2015).
FDA 2014
- Us. Food, Drug Administration. Novel New Drugs 2013 Summary. https://www.fda.gov/downloads/drugs/developmentapprovalprocess/druginnovation/ucm381803.pdf 2014, (accessed 21 June 2017).
Feighner 1972
- Feighner JP, Robins E, Guze SB, Woodruff RA, Winokur G, Munoz R. Diagnostic criteria for use in psychiatric research. Archives of General Psychiatry 1972;26(1):57‐63. [DOI] [PubMed] [Google Scholar]
Florea 2015
- Florea I, Loft H, Danchenko N, Rive B, Brignone M, Merikle E, et al. Vortioxetine effects on overall patient functioning in patients with major depressive disorder. 28th European College of Neuropsychopharmacology, ECNP Congress; 2015 Aug 29 to Sep 1; Amsterdam, Netherlands. 2015:S436.
Fu 2015
- Fu J, Chen Y. The efficacy and safety of 5 mg/d Vortioxetine compared to placebo for major depressive disorder: a meta‐analysis. Psychopharmacology 2015;232(1):7‐16. [DOI] [PubMed] [Google Scholar]
Furukawa 2002
- Furukawa TA, Guyatt GH, Griffith LE. Can we individualize the 'number needed to treat'? An empirical study of summary effect measures in meta‐analyses. International Journal of Epidemiology 2002;31(1):72‐6. [DOI] [PubMed] [Google Scholar]
Furukawa 2005
- Furukawa TA, Cipriani A, Barbui C, Brambilla P, Watanabe N. Imputing response rates from means and standard deviations in meta‐analyses. International Clinical Psychopharmacology 2005;20(1):49‐52. [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(1):7‐10. [DOI] [PubMed] [Google Scholar]
Gartlehner 2011
- Gartlehner G, Hansen RA, Morgan LC, Thaler K, Lux L, Noord M, et al. Comparative benefits and harms of second‐generation antidepressants for treating major depressive disorder: an updated meta‐analysis. Annals of Internal Medicine 2011;155(11):772‐85. [DOI] [PubMed] [Google Scholar]
Gartlehner 2012
- Gartlehner G, Thaler K, Hill S, Hansen RA. How should primary care doctors select which antidepressants to administer?. Current Psychiatry Reports 2012;14(4):360‐9. [DOI] [PubMed] [Google Scholar]
GRADEpro [Computer program]
- GRADE Working Group, McMaster University. GRADEpro. Hamilton (ON): GRADE Working Group, McMaster University, 2014.
Grant 2014
- Grant RL. Converting an odds ratio to a range of plausible relative risks for better communication of research findings. BMJ (Clinical Research Ed.) 2014;348:f7450. [DOI] [PubMed] [Google Scholar]
Grover 2013
- Grover S, Avasth A, Kalita K, Dalal PK, Rao GP, Chadda RK, et al. IPS multicentric study: antidepressant prescription patterns. Indian Journal of Psychiatry 2013;55(1):41‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Guaiana 2013
- Guaiana Giuseppe, Gupta Sumeet, Chiodo Debbie, Davies Simon J C, Haederle Katja, Koesters Markus. Agomelatine versus other antidepressive agents for major depression. Cochrane Database of Systematic Reviews 2013, Issue 12. [DOI: 10.1002/14651858.CD008851.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Guy 1970
- Guy W, Bonato RR. Manual for the ECDEU Assessment Battery 2. Chevy Chase (MD): National Institute of Mental Health, 1970. [Google Scholar]
Guyatt 1998
- Guyatt GH, Juniper EF, Walter SD, Griffith LE, Goldstein RS. Interpreting treatment effects in randomised trials. BMJ 1998;316(7132):690‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Guyatt 2011
- Guyatt GH, Oxman AD, Kunz R, Brozek J, Alonso‐Coello P, Rind D, et al. GRADE guidelines 6. Rating the quality of evidence ‐ imprecision. Journal of Clinical Epidemiology 2011;64(12):1283‐93. [DOI: 10.1016/j.jclinepi.2011.01.012] [DOI] [PubMed] [Google Scholar]
Hamilton 1960
- Hamilton M. A rating scale for depression. Journal of Neurology, Neurosurgery and Psychiatry 1960;23:56‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Kirsch 2008
- Kirsch I, Deacon BJ, Huedo‐Medina TB, Scoboria A, Moore TJ, Johnson BT. Initial severity and antidepressant benefits: a meta‐analysis of data submitted to the Food and Drug Administration. PLoS Medicine 2008;5(2):e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Koesters 2013
- Koesters M, Guaiana G, Cipriani A, Becker T, Barbui C. Agomelatine efficacy and acceptability revisited: systematic review and meta‐analysis of published and unpublished randomised trials. British Journal of Psychiatry 2013;203(3):179‐87. [DOI] [PubMed] [Google Scholar]
Langendam 2013
- Langendam MW, Akl EA, Dahm P, Glasziou P, Guyatt G, Schünemann HJ. Assessing and presenting summaries of evidence in Cochrane Reviews. Systematic Reviews 2013;2:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Leucht 2013
- Leucht S, Fennema H, Engel R, Kaspers‐Janssen M, Lepping P, Szegedi A. What does the HAMD mean?. Journal of Affective Disorders 2013;148(2‐3):243‐8. [DOI] [PubMed] [Google Scholar]
Li 2016
- Li G, Wang X, Ma D. The efficacy and safety of 10 mg vortioxetine in the treatment of major depressive disorder: a meta‐analysis of randomized controlled trials. Neuropsychiatric Disease and Treatment 2016;12:523‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Meeker 2015
- Meeker AS, Herink MC, Haxby DG, Hartung DM. The safety and efficacy of vortioxetine for acute treatment of major depressive disorder: a systematic review and meta‐analysis. Systematic Reviews 2015;4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Montgomery 1979
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. British Journal of Psychiatry 1979;134:382‐9. [DOI] [PubMed] [Google Scholar]
Montgomery 2009
- Montgomery SA, Moller H‐J. Is the significant superiority of escitalopram compared with other antidepressants clinically relevant?. International Clinical Psychopharmacology 2009;24(3):111‐8. [DOI] [PubMed] [Google Scholar]
NICE 2009
- National Institute for Health and Care Excellence. Depression in Adults. CG90. London (UK): National Institute for Health and Care Excellence, 2009. [Google Scholar]
Pehrson 2016
- Pehrson AL, Jeyarajah T, Sanchez C. Regional distribution of serotonergic receptors: a systems neuroscience perspective on the downstream effects of the multimodal‐acting antidepressant vortioxetine on excitatory and inhibitory neurotransmission. CNS Spectrums 2016;21(2):162‐83. [DOI] [PubMed] [Google Scholar]
Pigott 2011
- Pigott HE. STAR*D: a tale and trail of bias. Ethical Human Psychology and Psychiatry 2011;13(1):6‐28. [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Saito 2010
- Saito M, Iwata N, Kawakami N, Matsuyama Y, Ono Y, Nakane Y, et al. Evaluation of the DSM‐IV and ICD‐10 criteria for depressive disorders in a community population in Japan using item response theory. International Journal of Methods in Psychiatric Research 2010;19(4):211‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schueler 2011
- Schueler YB, Koesters M, Wieseler B, Grouven U, Kromp M, Kerekes MF, et al. A systematic review of duloxetine and venlafaxine in major depression, including unpublished data. Acta Psychiatrica Scandinavica 2011;123(4):247‐65. [DOI] [PubMed] [Google Scholar]
Schünemann 2013
- Schünemann H, Brożek J, Guyatt G, Oxman A, editor(s). GRADE handbook. Handbook for grading the quality of evidence and the strength of recommendations using the GRADE approach. [updated October 2013]. The GRADE Working Group, 2013. Available from gdt.guidelinedevelopment.org/app/handbook/handbook.html.
Spitzer 1978
- Spitzer RL, Endicott J, Robins E. Research diagnostic criteria: rationale and reliability. Archives of General Psychiatry 1978;35(6):773‐82. [DOI] [PubMed] [Google Scholar]
Stahl 2015
- Stahl SM. Modes and nodes explain the mechanism of action of vortioxetine, a multimodal agent (MMA): blocking 5HT3 receptors enhances release of serotonin, norepinephrine, and acetylcholine. CNS Spectrums 2015;20(5):455‐9. [DOI] [PubMed] [Google Scholar]
Thase 2016
- Thase ME, Mahableshwarkar AR, Dragheim M, Loft H, Vieta E. A meta‐analysis of randomized, placebo‐controlled trials of vortioxetine for the treatment of major depressive disorder in adults. European Neuropsychopharmacology 2016;26(6):979‐93. [DOI] [PubMed] [Google Scholar]
Vos 2012
- Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990‐2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012; Vol. 380, issue 9859:2163‐96. [DOI] [PMC free article] [PubMed]
Weinmann 2008
- Weinmann S, Becker T, Koesters M. Re‐evaluation of the efficacy and tolerability of venlafaxine vs SSRI: meta‐analysis. Psychopharmacology 2008;196(4):511‐20. [DOI] [PubMed] [Google Scholar]
WHO 1992
- World Health Organization (WHO). The ICD‐10 Classification of Mental and Behavioural Disorders. Geneva (Switzerland): WHO, 1992. [Google Scholar]
WHO 2016
- WHO Collaborating Centre for Drug Statistics Methodology. ATC/DDD Index 2017. https://www.whocc.no/atc_ddd_index/?code=N06AX26 2016 (accessed 21 June 2017).
Zhang 2013
- Zhang Y, Becker T, Kösters M. Preliminary study of patterns of medication use for depression treatment in China. Asia‐Pacific Psychiatry: Official Journal of the Pacific Rim College of Psychiatrists 2013;5(4):231‐6. [DOI] [PubMed] [Google Scholar]
Zhang 2015
- Zhang J, Mathis MV, Sellers JW, Kordzakhia G, Jackson AJ, Dow A, et al. The US Food and Drug Administration's perspective on the new antidepressant vortioxetine. Journal of Clinical Psychiatry 2015;76(1):8‐14. [DOI] [PubMed] [Google Scholar]
Zorzela 2014
- Zorzela L, Golder S, Liu Y, Pilkington K, Hartling L, Joffe A, et al. Quality of reporting in systematic reviews of adverse events: systematic review. BMJ (Clinical Research Ed.) 2014;348:f7668. [DOI] [PMC free article] [PubMed] [Google Scholar]