

SUMMARY
Neuronal chloride levels are developmentally regulated. Early in life, high intracellular concentrations support chloride efflux and depolarization at GABAergic synapses. In mouse, intracellular chloride decreases over the first postnatal week in the somatodendritic compartment, eventually supporting mature, hyperpolarizing GABAergic inhibition. In contrast to this dendritic switch, it is less clear how GABAergic signaling at the axon initial segment (AIS) functions in mature pyramidal cells, as reports of both depolarization and hyperpolarization have been reported in the AIS past the first postnatal week. Here, we show that GABAergic signaling at the AIS of prefrontal pyramidal cells, indeed, switches polarity from depolarizing to hyperpolarizing but does so over a protracted periadolescent period. This is the most delayed maturation in chloride reversal in any structure studied to date and suggests that chandelier cells, which mediate axo-axonic inhibition, play a unique role in the periadolescent maturation of prefrontal circuits.
Graphical Abstract
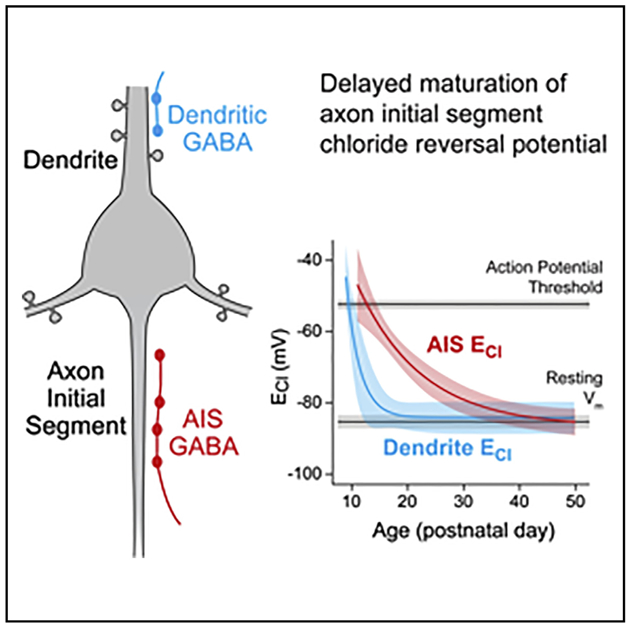
In Brief
Rinetti-Vargas et al. examine the development of chloride reversal potential (ECl), which determines GABAergic synapse polarity, in the axon initial segment of mouse prefrontal layer 2/3 pyramidal cells. They find that axon initial segment ECl hyperpolarizes over a periadolescent development period, eventually matching dendritic values.
INTRODUCTION
Adolescence is a period of development for high-order cognitive functions, including impulse control, long-term planning, and risk evaluation. Prefrontal cortex (PFC) circuits, which are involved in high-order decision making and risk assessment, undergo a high degree of refinement and maturation during adolescence (Johnson et al., 2016; Lewis, 1997). GABAergic inhibitory circuits, including parvalbumin-positive chandelier cells, are a major site of adolescent maturation in the PFC (Taniguchi et al., 2013; Uhlhaas and Singer, 2011; Vandenberg et al., 2015). Chandelier cells are thought to regulate large PFC networks, as their dendrites sample associative inputs from other cortical regions, and their axons diverge to synapse onto hundreds of glutamatergic pyramidal cells (Inan et al., 2013). In these pyramidal cells, chandelier cells synapse exclusively onto the axon initial segment (AIS) (Somogyi et al., 1982), the site of action potential (AP; spike) initiation (Bender and Trussell, 2012). Because of this subcellular targeting, single chandelier cells are thought to inhibit AP generation in large networks of pyramidal cells (Lewis, 2011). However, whether chandelier cells, indeed, inhibit pyramidal cells in mature PFC circuits remains controversial (Dugladze et al., 2012; Glickfeld et al., 2009; Khirug et al., 2008; Massi et al., 2012; Szabadics et al., 2006; Woodruff et al., 2009, 2011).
The concentration of chloride in the postsynaptic cell determines whether chloride-fluxing GABAA receptors depolarize or hyperpolarize postsynaptic cells. This concentration is dependent on chloride transporters that typically increase (sodium potassium chloride cotransporter-1; NKCC1) or decrease (potassium chloride cotransporter-2; KCC2) intracellular chloride (Kahle et al., 2015). Transporter function is developmentally regulated. Early in life (rodent embryonic stages to postnatal week 1; human third trimester), NKCC1 function is high, resulting in GABA-mediated depolarization of postsynaptic cells. Later, NKCC1 function diminishes and KCC2 function increases, resulting in mature, hyperpolarizing GABA synapses (Ben-Ari et al., 2007; Tyzio et al., 2007). Interestingly, the AIS, which is postsynaptic to chandelier synapses, may not follow this developmental progression, as there are conflicting reports that the AIS is depolarized (Khirug et al., 2008; Szabadics et al., 2006; Woodruff et al., 2009) or hyperpolarized by GABA past the first postnatal week (Glickfeld et al., 2009; Wang et al., 2014; Woodruff et al., 2011). Understanding how axo-axonic inhibition regulates pyramidal cell networks during adolescence is critical, as this is a critical period for prefrontal development and a period in which PFC-associated neurological disease, including schizophrenia, is often first diagnosed. Indeed, chandelier cells exhibit marked genetic and anatomical changes during adolescence, and corresponding anatomical changes have been noted in post-mortem tissue from those diagnosed with schizophrenia (Lewis, 2011).
Here, we assessed GABAergic function at the AIS and dendrite of prefrontal pyramidal cells from infancy to adulthood. We identify a previously unrecognized periadolescent critical period for GABAergic maturation at the AIS membrane that is mediated by changes in local chloride transporter function. Moreover, this developmental switch can largely account for previous observations of both depolarizing and hyperpolarizing inhibition at the AIS. This is, to our knowledge, the most developmentally delayed switch in GABAergic polarity within any neuronal structure studied to date and highlights the unique role of chandelier cell inhibition in PFC adolescent development.
RESULTS
Gramicidin-based perforated patch recordings, which preserve endogenous chloride gradients, were made from prefrontal pyramidal cells in acute slices prepared from postnatal day (P)11 to P72 mice, spanning mouse adolescent development (Brust et al., 2015). Pyramidal cells in layer 2/3 were targeted, as these neurons are postsynaptic to both basket and chandelier cells (Le Magueresse and Monyer, 2013). Because postsynaptic ECl (chloride reversal potential) is independent of the source of GABA, we bypassed the presynaptic cell and applied GABA via an iontophoretic pipette, allowing for consistent, spatiotemporally restricted activation of GABAA receptors. In the AIS, GABAA receptors are restricted to ankyrin-G-deficient puncta that are apposed to VGAT-positive boutons (King et al., 2014; Wefelmeyer et al., 2015). Therefore, iontophoresis onto the AIS likely activates pools of synaptic GABAA receptors.
To visualize neurites for GABA iontophoresis, the patch pipette was also loaded with 40 μM carboxyfluorescein-AM (acetoxy-methyl ester), which passes through the membrane seal and is fluorescent only after cleavage by cellular esterases. Dye loading and patch perforation occurred over similar timescales and allowed for visualization of all proximal neurites (Figure 1A). Once neuronal arbors were visualized, iontophoretic pipettes were positioned near the AIS or a dendritic branch. Axonal and dendritic iontophoresis sites equidistant from the soma were chosen to minimize differences in space clamp across these two structures. When possible, pipettes were repositioned to assess both axonal and dendritic ECl in the same cell. Iontophoretic application of GABA activated the receptors with a space constant of 9 μm (amplitude at AIS: 5.2 mV ± 1.1 mV; amplitude 10 μm from AIS: 0.75 mV ± 0.3 mV; n = 4; Figure 1B), and postsynaptic potentials were blocked by the GABAA antagonist SR95531 (10 μM; baseline amplitude: 10.7 mV ± 1 mV; SR amplitude: 0.4 mV ± 0.1 mV; n = 5; Figure 1C). Resting membrane potential (Vrest), input resistance (Rin), and AP threshold were monitored throughout recordings. Experiments were terminated if perforated patches ruptured, which resulted in a sudden increase in IPSP (inhibitory postsynaptic potential) amplitude (pipette solution had an ECl of 0 mV relative to external recording solution) and a redistribution of fluorescence into the recording pipette beyond the site of membrane seal (Figure 1A).
Figure 1. Methods for Assessing ECl in Specific Neuronal Compartments.

(A) Left: maximum intensity z series projection of fluorescein-filled layer 2/3 pyramidal cell in medial PFC. Middle: single optical in plane with the somata detailing fluorescein in cell. Simultaneous scanning differential contrast image is overlaid in grayscale, detailing cell location within PFC and recording pipette. Right: single optical at the AIS (12 μm above soma), highlighting GABA iontophoretic (Ionto) pipette apposed to AIS. Segments of dendrites are also observed.
(B) Left: example of response from iontophoretic delivery at varying distances from the AIS. Pipette was withdrawn perpendicular to the main axis of the axon. Right: average amplitude (Amp) ± SEM of IPSP versus pipette distance (Dist.) from neurite. Gray line indicates exponential fit.
(C) Left: example of response before and after application of SR95531. Right: peak amplitude of IPSPs before and after SR95531. Circles indicate individual cells; bars connect within-cell measurements.
Neurons were held at various membrane potentials with constant current injection, and ECl was determined from linear fits of GABAergic postsynaptic potentials (PSPs) versus membrane potential (2A–2C). Consistent with studies in hippocampus, dendritic ECl was depolarized before P10 (P9 dendritic ECl: −45 mV ± 7.8 mV; n = 3) (Dzhala and Staley, 2003). After P10, dendritic ECl hyperpolarized to Vrest and remained constant through all subsequent ages (Vrest: −85 mV ± 1.6 mV; dendritic ECl: −83 mV ± 1.5 mV; P14–P73, n = 17; mean difference permutation test, p = 0.88; Figure 2D). In contrast to this rapid ECl development in the dendritic compartment, AIS ECl shifted considerably between P11 and P70. Within a single neuron from preadolescent tissue, large differences in ECl were observed between the AIS and dendrite (2B and 2C). At infancy (P11–P20), AIS ECl was close to spike threshold (Vrest: −81.5 mV ± 1.8 mV; AIS ECl: −59.7 mV ± 3.14 mV; AP threshold: −52.1 mV ± 3.14 mV; n = 17; Vrest versus ECl mean difference permutation test, p < 0.001). In preadolescence (P21–P39; n = 15), AIS ECl hyperpolarized but remained depolarizing relative to Vrest (AIS Vrest: −86.9 mV ± 1.5 mV; AIS ECl: −73.1 mV ± 2.4 mV; mean difference permutation test, p < 0.001). Following adolescence (P55–P70; n = 15), AIS ECl continued to hyperpolarize, eventually resulting in inhibition with an ECl comparable to Vrest (i.e., shunting inhibition) (Vrest: −88 mV ± 1.9 mV; AIS ECl: − 84 mV ± 2 mV; AP threshold: −53.8 mV ± 1.6 mV; Vrest versus ECl mean difference permutation test, p = 0.05) (Figure 2D). Given these results, the intracellular chloride concentration at the AIS was calculated to be 5.5 mM ± 0.4 mM, comparable to other reports of mature, perisomatic intracellular Cl− levels (Doyon et al., 2011; Price and Trussell, 2006).
Figure 2. Development of ECl in the Dendrites and AIS.

(A) Maximum intensity z series projection of fluorescein-filled layer 2/3 pyramidal cell in medial PFC. Arrows indicate position of iontophoretic (ionto.) pipettes on the AIS and dendrite (Dend.) at different focal points. Dendrites are distinguished from the axon by the presence of spines.
(B) Left: same neuron as in (A). Slice is visualized using scanning Dodt contrast image (grayscale). Note the focus of the iontophoresis pipette near the AIS. Neuron was held with constant current at multiple membrane potentials to determine ECl at AIS. Gray bar denotes iontophoresis epoch. Note the predominantly depolarizing PSPs. Right: same neuron with iontophoretic pipette relocated to basal dendrite. Note the predominantly hyperpolarizing PSPs that lead to differences in ECl across compartments.
(C) Linear fits to peak of GABA currents versus holding Vm was used to determine local ECl. Data are from the cell in (A). Amp, amplitude.
(D) Summary of AIS (red) and dendritic (blue) ECl values across development. Circles indicate single data points, and curves indicate an exponential fit ± 95% confidence interval. Black lines and gray bars indicate average mean ± 95% confidence interval for spike threshold and Vrest across all recordings.
Multiple mechanisms contribute to chloride homeostasis, including the chloride transporters KCC2 and NKCC1 (Kaila et al., 2014), the chloride channel ClC-2 (Földy et al., 2010; Ratté and Prescott, 2011), and local impermeant anions (Glykys et al., 2014). While it is not possible to manipulate the chloride-buffering capacity of impermeant anions directly (Glykys et al., 2014), the role of ClC-2 and chloride transporters can be assessed experimentally. Since ClC-2 influences intracellular Cl levels, we hypothesized that it may play a role in Cl homeostasis in the mature AIS. Therefore, we measured ECl in the AIS of ClC-2 knockout mice at P52–P57. The ECl in these recordings was not different from that in age-matched controls (ClC-2−/−: −80.8 mV ± 2.3 mV; age-matched controls: −82 mV ± 2.9 mV; n = 4 for each group; mean difference by permutation test with 1,000 replicates, p =0.342), suggesting that ClC-2 does not influence ECl at the AIS.
To assess the roles of NKCC1 and KCC2, we used two complementary techniques. First, we measured overall transporter levels in membrane fractions from PFC at P9, P13–P15, P23, and P83, as these ages capture various periods of dendritic and AIS ECl development (Figure 3). Antibodies were validated by confirming that single bands at the correct molecular weight were observed in membrane fractions. Unfortunately, all antibodies tested had nonspecific binding in total homogenates and were, therefore, deemed unsuitable for immunofluorescence localization of transporters.
Figure 3. Age-Dependent Differences in Membrane NKCC1 and KCC2 Protein Level and Phosphorylation.

(A) Western blot analysis depicts the membrane enrichment of NKCC1 and KCC2 at p82. Anti-Na/K ATPase antibodies and anti-GAPDH antibodies were used as membrane (Mem) and cytosolic (Cyt) markers, respectively. Tot, total.
(B) The total protein and phosphorylation levels of NKCC1 and KCC2 were determined by western blot analysis. Actin was used as a loading control. Membranes were first probed with anti-phospho-NKCC1 antibodies (left) and anti-phospho-KCC2 antibodies (right), as well as anti-actin antibodies. Membranes were then stripped and re-probed with anti-NKCC1 antibodies (left) and anti-KCC2 antibodies (right). P9 samples (data not shown) were run in a separate gel including additional P13–P15 samples.
(C) ImageJ was used for optical density quantification. Data are presented as the average ratio of NKCC1/actin or phospho-NKCC1/actin. All data are normalized to their respective immunoreactivity at P13–P15. Error bars indicate mean ± SEM. ns = 6, 13, 7, and 7 animals for P9, P13–P15, P23, and P82, respectively. Significance was determined using an ANOVA with a Bonferroni post hoc test corrected for multiple comparisons (*p < 0.0083).
(D) Same as for (C), but for KCC2 and pKCC2.
Total NKCC1 levels were stable between P9 and P23 and then decreased to 75% ± 3.5% of baseline levels at P13–P15 (Figure 3; p = 0.0004, ANOVA and Bonferroni post hoc test were used for all western blot analyses). In contrast, KCC2 levels increased dramatically between P9 and P23 (P9: 43% ± 2%, n = 6; P13–P15: 100% ± 3.6%, n = 13; P23: 143% ± 18%, n = 7; p < 0.01, comparing each age group) but were then similar between P23 and P82 (P82: 148% ± 13%, n = 7 each; p = 0.78). Transporter function is regulated by phosphorylation (Nakamura et al., 2015), with phosphorylation at threonine 212 and 217 on NKCC1 and serine 940 on KCC2 known to increase transporter flux (Flemmer et al., 2002; Lee et al., 2007). Within the PFC, phospho-NKCC1 levels were high at P9 but then stabilized between P13 and P82 (P9: 249% ± 34%; P13–P15: 100% ± 6%; P23: 96% ± 16%, P82: 78% ± 18%; p < 0.0001, P9 versus other ages). Phosphorylation levels at serine 940 on KCC2 dipped at P13–P15 relative to P9 or P82 (P9: 217% ± 40%; P13–P15: 100% ± 11%; P23: 143% ± 18%; P82: 148% ± 13%; p <0.005, P13–P15 versus P9 or P82). Combined with the overall low levels of KCC2 expressed at P9, it appears that the KCC2 function generally increases between P9 and P82. Overall, this suggests that transporter levels and function in PFC-derived membranes changes during development, from conditions that favor depolarized ECl (NKCC1 active) to conditions that favor hyperpolarized/shunting ECl (KCC2 active).
While these results are consistent with the gradual hyperpolarization of ECl in the AIS, the AIS constitutes only a small part of the total membrane fraction. Therefore, we next made recordings from <P25 and >P45 neurons and assessed AIS ECl following the block of one or both transporters to gain more direct insight into transporter function within this compartment. Furosemide (1 mM), which blocks both NKCC1 and KCC2 within 10 min of application (Blaesse et al., 2009; Thompson and Gähwiler, 1989), was used first to determine the overall function of AIS chloride transporters. In both age ranges studied, furosemide depolarized ECl relative to baseline controls, though depolarizations were more pronounced in older mice (absolute ECl depolarization from baseline, <P25: 10 mV ± 3.6 mV, n = 6; >P45: 19 mV ± 6.2 mV, n = 5; median difference permutation test, p = 0.007; Figure 4). This suggests that, in the absence of NKCC1 or KCC2 function, chloride equilibrium is, overall, more depolarized, perhaps due to contributions from bicarbonate load, pH, or impermeant anions (Glykys et al., 2014; Kim and Trussell, 2009).
Figure 4. NKCC1 and KCC2 Differentially Regulate Pre- and Post-adolescent AIS ECl.

(A) Example traces of IPSPs from AIS iontophoresis at different ages depict shift of ECl from baseline to post-drug. Furosemide: broad-spectrum NKCC1/KCC2 inhibitor. Bumetanide: NKCC1 inhibitor. VY 0240551: KCC2 inhibitor. Scale bar, 100 ms in all traces. Note the direction of shift for each transporter inhibitor. Vehicle controls (far right) were performed over similar timescales as inhibitor application.
(B) Summary of NKCC1 and/or KCC2 block effect on ECl across development. Values of ECl for each recorded cell were obtained as depicted in (A) and plotted relative to the average AIS ECl developmental profile (Figure 2D). Baseline ECl: small filled circle; post-drug ECl: larger open circle; each pre-post pair is connected with a vertical line. Individual data points from bumetanide preincubation are shown as large, open circles only.
To examine the specific effects of NKCC1 or KCC2 at these ages, we applied more selective inhibitors of each transporter. To selectively inhibit NKCC1, we applied the NKCC1-preferring blocker bumetanide at 10 μM, a concentration that completely blocks NKCC1 but only blocks KCC2 by 20%–30% (Payne, 1997). In young mice, within-cell measurements of ECl were obtained before and 40 min after bumetanide application. In these cells, bumetanide hyperpolarized ECl by −7 mV ± 1.8 mV (ECl pre-bumetanide: −75.7 mV ± 2 mV; E post-bumetanide: −82.7 mV ± 1.8 mV; n = 7; paired mean difference by permutation test, p < 0.001; Figure 4). These results are consistent with NKCC1 acting as a chloride intruder in the preadolescent PFC.
In slices from older mice, we were unable to maintain perforated recordings long enough to obtain baseline and post-bumetanide measurements. Therefore, we preincubated slices in bumetanide for over 1 hr and compared ECl in these cells to untreated, age-matched controls (P54–P58). Here, we found that bumetanide depolarized, rather than hyperpolarized, ECl (ECl bumetanide: −71.6 mV ± 3.1 mV, n = 5; ECl age-matched controls: −80.8 mV ± 2.5 mV, n = 5; permutation test, p < 0.001; Figure 4). This change in bumetanide action may result from partial block of KCC2 in an AIS that lacks NKCC1, though detectable levels of NKCC1 are still present in PFC membranes (Figure 3). Alternatively, NKCC1 may be functioning at these ages as a chloride extruder, as its polarity switches when intracellular chloride is low (Brumback and Staley, 2008). This, in concert with increased KCC2 activity in older animals (Figure 3), could account for ECl hyperpolarization as the AIS develops.
To determine whether KCC2 function increases throughout development at the AIS, we next applied the KCC2-specific inhibitor VU0240551 (VU; 10 μM) for 30 min after assessing baseline ECl within each recording. KCC2 inhibition significantly depolarized cells in slices from old animals (ECl pre-VU0240551: −79.4 mV ± 3 mV; ECl post-VU0240551: −49.2 mV ± 7 mV; n = 5; mean difference permutation test, p < 0.001; Figure 4). Although stronger, the depolarization induced by VU0240551 is similar to that induced by the broad-spectrum transporter blocker furosemide. This effect is consistent with a predominantly active form of KCC2 at older ages, as reported in western blots (Figure 3).
To confirm that the changes in ECl reflect blockage of transporters, and that measurements from perforated patch remain stable over prolonged recording times under different pharmacological paradigms, we performed controls in which ECl was monitored over the same time course of antagonist application. Because ECl would be 0 mV if the perforated patch completely ruptures, any small tear in the seal would likely depolarize ECl. Therefore, controls were performed in >P45 tissue where ECl is hyperpolarized close to Vrest. No marked changes in ECl were observed in these experiments (ECl baseline: −77.5 mV ±1.2 mV; ECl vehicle: −78.5 mV ± 2.8 mV; n = 4; paired mean difference permutation test, p = 0.45), suggesting that the perforated patch can be maintained over the course of transporter blocker application. Thus, these results indicate that both NKCC1 and KCC2 are important regulators of ECl at the AIS and that periadolescent changes in chloride polarity at the AIS are due, in part, to a shift in the dominant transporter regulating local chloride.
DISCUSSION
Early in development, high intracellular chloride allows GABA to depolarize postsynaptic cells, which is important for activitydependent circuit refinement (Kandler, 2004). Here, we find that, unlike dendritically localized GABAA signaling, which matures around birth, GABAergic signaling at the AIS continues to depolarize the cell through preadolescent periods. This is, to our knowledge, the most developmentally delayed switch in GABAergic polarity identified to date.
The polarity of chandelier cell synapses has long been the subject of debate. The first report of chandelier function stemmed from experiments in juvenile rat somatosentory cortex (P20–P35), where perforated-patch techniques were used to show that chandelier cells depolarized pyramidal cells (ECl:−51 mV). Similarly, depolarizing GABAergic events have also been observed in mouse and rat dentate granule cells and cortical layer 2/3 pyramidal cells at P16–P20 (ECl range: −65 mV to −70 mV) (Khirug et al., 2008) and in somatosensory cortical layer 2/3 pyramidal cells at P15–P23 (ECl: −58 mV) (Woodruff et al., 2009) and at P18–P25 (ECl: −51 mV) (Woodruff et al., 2011). Indirect evidence for a depolarized ECl was also observed in P16–P25 amygdala. Here, APs evoked in chandelier cells consistently generated disynaptic excitation (Woodruff et al., 2009). These data align well with findings reported here, where pre-adolescent AIS ECl was consistently depolarizing relative to Vrest in prefrontal layer 2/3 pyramidal cells (Figure 2).
In contrast to these studies, several groups have found that chandelier cells hyperpolarize pyramidal cells. In hippocampus, non-invasive unitary field IPSPs were consistently hyperpolarizating in CA1 from >P42 rats (Glickfeld et al., 2009). In vivo juxtacellular recordings from identified CA3 chandelier cells in anesthetized and awake behaving adult rats showed that activity in chandelier cells was correlated well with pauses in pyramidal cell activity (Viney et al., 2013). Similarly, perforated-patch recordings paired with optogenetic activation of chandelier cell synapses hyperpolarized piriform cortex pyramidal cells in P42–P56 mice (Wang et al., 2014).
Differences in species and brain region examined make it difficult to make direct comparisons across these studies. Here, we made the first measurements of AIS ECl throughout the development of PFC. Remarkably, our results are consistent with those from hippocampus, piriform cortex, amygdala, and other cortical regions, provided that all data are placed in context of the delayed development of ECl at the AIS (Figure 2). Thus, these data largely resolve discrepancies in chandelier cell function.
Two findings contradict those reported here. In rat hippocampus, disynaptic inhibitory postsynaptic currents (IPSCs) were infrequently noted in paired recordings between chandelier cells and pyramidal cells, even when the bulk of chandelier activity recorded via fields was hyperpolarizing (Glickfeld et al., 2009). These same disynaptic events were more commonly observed in adult human cortex excised from patients undergoing menigioma treatment. Thus, these data suggest that, while the majority of initial segments have a hyperpolarized ECl, there are cells within slice preparations in which AIS ECl is presumably depolarized, allowing for the recruitment of an intermediate pyramidal cell, evoking disynaptic activity. Whether this reflects a subpopulation of pyramidal cells in which AIS ECl remains depolarizing in adulthood or is related to the process of slice preparation (e.g., disrupted chloride handling resulting from damage to neurites) remains to be examined.
Overall, transporter levels at the membrane transitioned over periadolescent development from favoring chloride efflux to influx (Figure 3). At the AIS, similar changes in transporter function were identified by blocking NKCC1 and KCC2 at pre- and post-adolescent ages (Figure 4). The development of hyperpolarizing ECl at the AIS appeared to be due largely to increased levels of functional KCC2 at the AIS membrane later in life, as KCC2 antagonism produced a marked change in ECl in >P45 animals. In addition to increased membrane density, PKC-dependent phosphorylation of KCC2 increased at S940. Phosphorylation at this site enhances transporter efficacy and further stabilizes the transporter in the membrane (Lee et al., 2007).
GABAergic function is dynamic, and GABAergic synapse polarity can be augmented by physiological stressors. In the spinal cord, nerve damage is associated with an upregulation of NKCC1 (Pieraut et al., 2007) and a downregulation of KCC2 (Boulenguez et al., 2010). In the brain, some forms of epilepsy, ischemic insults, and trauma are all associated with reductions in KCC2 membrane levels (Kahle et al., 2016; Papp et al., 2008; Shulga et al., 2008). More recently, stress induced by restraint was shown to lead to long-lasting depolarizations in ECl, detectable in ex vivo brain slices (Ostroumov et al., 2016). This change is due in part to a reduction in KCC2 phosphorylation at S940. Whether similar stress- or seizure-induced modifications occur at the AIS-localized KCC2 would be of interest to examine, especially in cases where depolarized AIS ECl is still observed in adulthood (Szabadics et al., 2006).
Compared to other cortical regions, PFC circuit maturation is developmentally delayed. Functionally, a major component of this is the maturation of network synchrony, including oscillations in the gamma frequency range (30–100 Hz). Gamma power increases markedly between late adolescence and early adulthood and is disrupted in neuropsychiatric diseases such as schizophrenia (Uhlhaas et al., 2011). In the PFC, the activation of parvalbumin-positive interneurons, which includes chandelier and basket cells, is sufficient to enhance entrainment of PFC networks to gamma oscillations (Cardin et al., 2009; Cho et al., 2015; Sohal et al., 2009). The delayed development of hyperpolarizing inhibition at the AIS may play a role in the maturation of neuronal synchrony. Chandelier inputs to the AIS are anatomically refined during this time, peaking in density immediately before adolescence, and then decreasing in number throughout adolescence (Cruz et al., 2009). Depolarization at the AIS may be important for this restructuring, similar to the role of depolarizing GABA seen across all synapses in the first postnatal week (Ben-Ari, 2014; Kandler, 2004), but once chloride handling at the AIS matures, chandelier-mediated inhibition would be expected to suppress activity over a large population of pyramidal cells, potentially contributing to the generation of cortical oscillations. Interestingly, deficits in neuronal synchrony and chandelier cell anatomy have been correlated with psychiatric disease (Cruz et al., 2009). In future work, it will be of interest to determine whether parallel changes in chandelier function further contribute to the etiology of these diseases (Ben-Ari et al., 2012).
EXPERIMENTAL PROCEDURES
Animals
All procedures were in accordance with University of California, San Francisco (UCSF), Institutional Animal Care and Use Committee (IACUC) guidelines. C57BL/6J wild-type mice of either sex were used for all developmental and pharmacological recordings, as well as for western blots. ClC-2−/− mice were generated as described previously (Nehrke et al., 2002). Heterozygous mating pairs were bred in house, and full knockout offspring were used for recordings.
Electrophysiology and Imaging
Acute coronal slices (300 mm) containing PFC were prepared from P11–P75 mice, in accordance with UCSF IACUC policies. ClC-2 knockout mice were genotyped by PCR. Slices were prepared in high-HEPES and low-Na solutions to preserve neuronal health in layer 2/3 throughout all ages studied (Ting et al., 2014). HEPES-cutting solution contained (in millimolar): 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 5 Na ascorbate, 2 thiourea, 3 Na pyruvate, 25 glucose, 10 MgSO4 • 7 H2O, and 0.5 CaCl2 • 2 H2O, 92 NaCl (pH 7.3), 300 mOsm, ice cold. NMDG (N-methyl-D-glucamine)-recovery solution contained (in millimolar): 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 5 Na+ ascorbate, 2 thiourea, 3 Na+ pyruvate, 25 glucose, 10 MgSO4 • 7H2O, 0.5 CaCl2 • 2H2O, and 93 NMDG (pH 7.3), with 10 N HCl, 300 mOsm, at 32°C–34°C. Slices were recovered for 15 min in NMDG-recovery solution, followed by a 2-hr incubation in HEPES-cutting solution at room temperature until recording. Recording solution contained (in millimolar): 125 NaCl, 2.5 KCl, 1 MgCl2, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, and 2 CaCl2 (pH 7.3), 300–310 mOsm, at 32°C–34°C. All solutions were bubbled with 5% CO2/95% O2.
Gramicidin-based perforated-patch recordings were made from prefrontal layer 2/3 pyramidal cells. Cells were visualized with Dodt contrast optics and identified based on their laminar position, somatic shape, and presence of dendritic spines. Recording pipettes (Schott 8250 glass) were front-filled with a solution containing (in millimolar): 10 NaCl, 10 K-gluconate, 125 KCl, 15 HEPES, 1.5 CaCl2, 1 MgCl2, and were back-filled with the same solution augmented with gramicidin (3–20 μg/mL) and carboxyfluorescein-AM (40 μM; Invitrogen). Carboxyfluorescein was excited using a two-photon microscope powered by a Mira 900 laser (800 nm) and visualized with R9110 photomultiplier tubes positioned to capture epifluroescence (<660-nm light, no bandpass) and transfluorescence (535/150 bandpass, Semrock) light through a 40× 0.8-NA objective and a 1.4-NA oil-coupled condenser, respectively. Scanning interference contrast images of slice morphology were acquired with an additional photomultiplier tube downstream of a 770-nm longpass filter. After formation of a gigaohm seal, hyperpolarizing current steps (−5 to −30 pA) were applied to monitor Rin for 10–30 min until appropriate recording conditions were obtained. The perforated patch was considered formed and stable when: Vm reached steady state, Rin was <400 MΩ, and Rs was 40–80 MΩ. Data were acquired at 20 kHz and filtered at 10 kHz using a Multiclamp 700B amplifier (Molecular Devices) and custom routines written in IgorPro (Wavemetrics). APs were generated with somatic current injection, and threshold was defined as the peak of the third derivative between the onset of the current pulse and the peak of the first AP generated.
GABA iontophoresis was performed using borosilicate pipettes placed in apposition with visualized neurites (0.5-μm tips, 200 mM GABA in H2O [pH 4]; Dagan ION-100, 80–140 nA application current, −30 nA retention current, 20-ms pulses). Experiments were performed in the presence of CGP55845 (10 mM) to block GABAB receptors. ECl was determined by linear interpolation of IPSP amplitude observed at different membrane potentials (via constant-current injection). Experiments were terminated if perforated patches ruptured, as evident by a sudden increase in IPSP amplitude (perforated patch solution has an ECl of 0 mV relative to external recording solution) and a redistribution of fluorescence into the patch pipette.
Preparation of Membrane Fractions and Western Blot Analysis
The mPFC was dissected bilaterally from ages as noted earlier. Immediately after dissection, tissue was homogenized in a glass homogenizer containing 500 μL ice-cold Krebs-sucrose buffer (containing [in millimolar]: 125 NaCl, 1.2 KCl, 1.2 MgSO4, 1.2 CaCl2, 22 Na2CO3, 1.2 NaH2PO4, 10 glucose, and 320 sucrose [pH 7.4]) in the presence of protease and phosphatase inhibitors. The homogenate was centrifuged at 16,000 × g at 4°C for 20 min to pellet the membrane fraction. The pellet was resuspended in 100 μL RIPA buffer containing protease and phosphatase inhibitors. Total protein concentration was determined using a bicinchoninic acid (BCA) protein assay kit.
Equal amounts of membrane preparation from individual mice (30 μg) were resolved on NuPAGE 4%–12% Bis-Tris gels and transferred onto nitrocellulose membranes. Blots were incubated for 1 hr at room temperature in blocking solution (5% [w/v] nonfat milk in PBS containing 0.1% [v/v] Tween 20) and then incubated overnight at 4°C in a blocking solution including the primary antibodies. Membranes were then washed and probed with horseradish peroxidase (HRP)-conjugated secondary antibodies for 2 hr at room temperature. Bands were visualized using enhanced chemiluminescence (ECL) and quantified in ImageJ (NIH).
Chemicals
SR95531, CGP55845, furosemide, bumetanide, and VU0240551 were from Tocris Bioscience. All other chemicals used for electrophysiology were from Sigma. DMSO (0.001%) was used as vehicle for all drugs and controls. All tubing was replaced after furosemide and bumetanide use.
Antibodies
For western blot, primary antibodies included: rabbit anti-phospho-NKCC1 (1:1,000) and rabbit anti-KCC2 (1:1,000) antibodies (EMD Millipore), rabbit anti-NKCC1 (1:250) (Abcam), rabbit anti-phospho-KCC2 (PhosphoSolutions), and mouse anti-actin (1:10,000) (Sigma). Phosphatase inhibitor cocktails 1 and 2 were from Sigma-Aldrich. Rabbit anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:1,000) antibodies were from Santa Cruz Biotechnology. Mouse anti-Na/K ATPase (1:1,000) antibody was from Affinity Bioreagents. Donkey HRP-conjugated secondary antibodies (1:1,000) were from Jackson ImmunoResearch Laboratories. ECL was from GE Healthcare. NuPAGE 4%–12% Bis-Tris pre-casted gels were from Life Technology. The BCA protein assay kit and the protease inhibitor cocktails were from Thermo Scientific.
Statistics
Unless otherwise stated, all mean differences between two groups were calculated by permutation statistics coded in R Studio. The mean difference between the original two groups was first calculated and then compared to 1,000 simulations for p-value calculations. Paired comparisons were obtained by calculating the mean difference and randomly shifting the sign of the pairs with 1,000 replicates. Regression analyses applied to developmental ECl values were calculated in R Studio and Igor.
Highlights.
Before adolescence, axon initial segment GABAA receptors are depolarizing
Axon initial segment chloride-driving force hyperpolarizes over adolescence
After adolescence, both dendritic and axonal GABAA receptors are inhibitory
Development in KCC2 and NKCC1 function underlies this switch
ACKNOWLEDGMENTS
We are grateful to J.E. Melvin for providing ClC-2 knockout mice and to Drs. M. Scanziani and R. Nicoll and members of the Bender and Ron labs for comments and discussions. This work was supported by NIH grants MH112117 (to K.J.B.) and AA017072 (to K.J.B. and D.R.) and the UCSF Library open access fund.
REFERENCES
- Ben-Ari Y (2014). The GABA excitatory/inhibitory developmental sequence: a personal journey. Neuroscience 279, 187–219. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Gaiarsa JL, Tyzio R, and Khazipov R (2007). GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev 87, 1215–1284. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Khalilov I, Kahle KT, and Cherubini E (2012). The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist 18, 467–486. [DOI] [PubMed] [Google Scholar]
- Bender KJ, and Trussell LO (2012). The physiology of the axon initial segment. Annu. Rev. Neurosci 35, 249–265. [DOI] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, and Kaila K (2009). Cation-chloride cotransporters and neuronal function. Neuron 61, 820–838. [DOI] [PubMed] [Google Scholar]
- Boulenguez P, Liabeuf S, Bos R, Bras H, Jean-Xavier C, Brocard C, Stil A, Darbon P, Cattaert D, Delpire E, et al. (2010). Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat. Med 16, 302–307. [DOI] [PubMed] [Google Scholar]
- Brumback AC, and Staley KJ (2008). Thermodynamic regulation of NKCC1-mediated Cl− cotransport underlies plasticity of GABA(A) signaling in neonatal neurons. J. Neurosci 28, 1301–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brust V, Schindler PM, and Lewejohann L (2015). Lifetime development of behavioural phenotype in the house mouse (Mus musculus). Front. Zool 12 (Suppl 1), S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, and Moore CI (2009). Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 459, 663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KK, Hoch R, Lee AT, Patel T, Rubenstein JL, and Sohal VS (2015). Gamma rhythms link prefrontal interneuron dysfunction with cognitive inflexibility in Dlx5/6(+/−) mice. Neuron 85, 1332–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz DA, Lovallo EM, Stockton S, Rasband M, and Lewis DA (2009). Postnatal development of synaptic structure proteins in pyramidal neuron axon initial segments in monkey prefrontal cortex. J. Comp. Neurol 514, 353–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon N, Prescott SA, Castonguay A, Godin AG, Kröger H, and De Koninck Y (2011). Efficacy of synaptic inhibition depends on multiple, dynamically interacting mechanisms implicated in chloride homeostasis. PLoS Comput. Biol 7, e1002149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugladze T, Schmitz D, Whittington MA, Vida I, and Gloveli T (2012). Segregation of axonal and somatic activity during fast network oscillations. Science 336, 1458–1461. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, and Staley KJ (2003). Excitatory actions of endogenously released GABA contribute to initiation of ictal epileptiform activity in the developing hippocampus. J. Neurosci 23, 1840–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemmer AW, Gimenez I, Dowd BF, Darman RB, and Forbush B (2002). Activation of the Na-K-Cl cotransporter NKCC1 detected with a phospho-specific antibody. J. Biol. Chem 277, 37551–37558. [DOI] [PubMed] [Google Scholar]
- Földy C, Lee SH, Morgan RJ, and Soltesz I (2010). Regulation of fastspiking basket cell synapses by the chloride channel ClC-2. Nat. Neurosci 13, 1047–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickfeld LL, Roberts JD, Somogyi P, and Scanziani M (2009). Interneurons hyperpolarize pyramidal cells along their entire somatodendritic axis. Nat. Neurosci 12, 21–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J, Dzhala V, Egawa K, Balena T, Saponjian Y, Kuchibhotla KV, Bacskai BJ, Kahle KT, Zeuthen T, and Staley KJ (2014). Local impermeant anions establish the neuronal chloride concentration. Science 343, 670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inan M, Blázquez-Llorca L, Merchán-Pérez A, Anderson SA, DeFelipe J, and Yuste R (2013). Dense and overlapping innervation of pyramidal neurons by chandelier cells. J. Neurosci 33, 1907–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CM, Loucks FA, Peckler H, Thomas AW, Janak PH, and Wilbrecht L (2016). Long-range orbitofrontal and amygdala axons show divergent patterns of maturation in the frontal cortex across adolescence. Dev. Cogn. Neurosci 18, 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Khanna AR, Alper SL, Adragna NC, Lauf PK, Sun D, and Delpire E (2015). K-Cl cotransporters, cell volume homeostasis, and neurological disease. Trends Mol. Med 21, 513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Khanna AR, Duan J, Staley KJ, Delpire E, and Poduri A (2016). The KCC2 cotransporter and human epilepsy: getting excited about inhibition. Neuroscientist 22, 555–562. [DOI] [PubMed] [Google Scholar]
- Kaila K, Price TJ, Payne JA, Puskarjov M, and Voipio J (2014). Cationchloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci 15, 637–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandler K (2004). Activity-dependent organization of inhibitory circuits: lessons from the auditory system. Curr. Opin. Neurobiol 14, 96–104. [DOI] [PubMed] [Google Scholar]
- Khirug S, Yamada J, Afzalov R, Voipio J, Khiroug L, and Kaila K (2008). GABAergic depolarization of the axon initial segment in cortical principal neurons is caused by the Na-K-2Cl cotransporter NKCC1. J. Neurosci 28, 4635–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, and Trussell LO (2009). Negative shift in the glycine reversal potential mediated by a Ca2+- and pH-dependent mechanism in interneurons. J. Neurosci 29, 11495–11510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AN, Manning CF, and Trimmer JS (2014). A unique ion channel clustering domain on the axon initial segment of mammalian neurons. J. Comp. Neurol 522, 2594–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Magueresse C, and Monyer H (2013). GABAergic interneurons shape the functional maturation of the cortex. Neuron 77, 388–405. [DOI] [PubMed] [Google Scholar]
- Lee HH, Walker JA, Williams JR, Goodier RJ, Payne JA, and Moss SJ (2007). Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J. Biol. Chem 282, 29777–29784. [DOI] [PubMed] [Google Scholar]
- Lewis DA (1997). Development of the prefrontal cortex during adolescence: insights into vulnerable neural circuits in schizophrenia. Neuropsychopharmacology 16, 385–398. [DOI] [PubMed] [Google Scholar]
- Lewis DA (2011). The chandelier neuron in schizophrenia. Dev. Neurobiol 71, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massi L, Lagler M, Hartwich K, Borhegyi Z, Somogyi P, and Klausberger T (2012). Temporal dynamics of parvalbumin-expressing axo-axonic and basket cells in the rat medial prefrontal cortex in vivo. J. Neurosci 32, 16496–16502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Darnieder LM, Deeb TZ, and Moss SJ (2015). Regulation of GABAARs by phosphorylation. Adv. Pharmacol 72, 97–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehrke K, Arreola J, Nguyen HV, Pilato J, Richardson L, Okunade G, Baggs R, Shull GE, and Melvin JE (2002). Loss of hyperpolarization-activated CI(−) current in salivary acinar cells from Clcn2 knockout mice. J. Biol. Chem 277, 23604–23611. [DOI] [PubMed] [Google Scholar]
- Ostroumov A, Thomas AM, Kimmey BA, Karsch JS, Doyon WM, and Dani JA (2016). Stress increases ethanol self-administration via a shift toward excitatory GABA signaling in the ventral tegmental area. Neuron 92, 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp E, Rivera C, Kaila K, and Freund TF (2008). Relationship between neuronal vulnerability and potassium-chloride cotransporter 2 immunoreactivity in hippocampus following transient forebrain ischemia. Neuroscience 154, 677–689. [DOI] [PubMed] [Google Scholar]
- Payne JA (1997). Functional characterization of the neuronal-specific K-Cl cotransporter: implications for [K+]o regulation. Am. J. Physiol 273, 1516–1525. [DOI] [PubMed] [Google Scholar]
- Pieraut S, Laurent-Matha V, Sar C, Hubert T, Méchaly I, Hilaire C, Mersel M, Delpire E, Valmier J, and Scamps F (2007). NKCC1 phosphorylation stimulates neurite growth of injured adult sensory neurons. J. Neurosci 27, 6751–6759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price GD, and Trussell LO (2006). Estimate of the chloride concentration in a central glutamatergic terminal: a gramicidin perforated-patch study on the calyx of Held. J. Neurosci 26, 11432–11436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratté S, and Prescott SA (2011). ClC-2 channels regulate neuronal excitability, not intracellular chloride levels. J. Neurosci 31, 15838–15843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulga A, Thomas-Crusells J, Sigl T, Blaesse A, Mestres P, Meyer M, Yan Q, Kaila K, Saarma M, Rivera C, and Giehl KM (2008). Posttraumatic GABA(A)-mediated [Ca2+]i increase is essential for the induction of brain-derived neurotrophic factor-dependent survival of mature central neurons. J. Neurosci 28, 6996–7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, and Deisseroth K (2009). Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459, 698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P, Freund TF, and Cowey A (1982). The axo-axonic interneuron in the cerebral cortex of the rat, cat and monkey. Neuroscience 7, 2577–2607. [DOI] [PubMed] [Google Scholar]
- Szabadics J, Varga C, Molnár G, Oláh S, Barzó P, and Tamás G (2006). Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science 311, 233–235. [DOI] [PubMed] [Google Scholar]
- Taniguchi H, Lu J, and Huang ZJ (2013). The spatial and temporal origin of chandelier cells in mouse neocortex. Science 339, 70–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SM, and Gähwiler BH (1989). Activity-dependent disinhibition. II. Effects of extracellular potassium, furosemide, and membrane potential on EClin hippocampal CA3 neurons. J. Neurophysiol 61, 512–523. [DOI] [PubMed] [Google Scholar]
- Ting JT, Daigle TL, Chen Q, and Feng G (2014). Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol. Biol 1183, 221–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyzio R, Holmes GL, Ben-Ari Y, and Khazipov R (2007). Timing of the developmental switch in GABA(A) mediated signaling from excitation to inhibition in CA3 rat hippocampus using gramicidin perforated patch and extracellular recordings. Epilepsia 48 (Suppl 5), 96–105. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ, and Singer W (2011). The development of neural synchrony and large-scale cortical networks during adolescence: relevance for the pathophysiology of schizophrenia and neurodevelopmental hypothesis. Schizophr. Bull 37, 514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlhaas PJ, Pipa G, Neuenschwander S, Wibral M, and Singer W (2011). A new look at gamma? High- (>60 Hz) gamma-band activity in cortical networks: function, mechanisms and impairment. Prog. Biophys. Mol. Bio 105, 14–28. [DOI] [PubMed] [Google Scholar]
- Vandenberg A, Piekarski DJ, Caporale N, Munoz-Cuevas FJ, and Wilbrecht L (2015). Adolescent maturation of inhibitory inputs onto cingulate cortex neurons is cell-type specific and TrkB dependent. Front. Neural Circuits 9, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viney TJ, Lasztoczi B, Katona L, Crump MG, Tukker JJ, Klausberger T, and Somogyi P (2013). Network state-dependent inhibition of identified hippocampal CA3 axo-axonic cells in vivo. Nat. Neurosci 16, 1802–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hooks BM, and Sun QQ (2014). Thorough GABAergic innervation of the entire axon initial segment revealed by an optogenetic ‘laserspritzer’. J. Physiol 592, 4257–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wefelmeyer W, Cattaert D, and Burrone J (2015). Activity-dependent mismatch between axo-axonic synapses and the axon initial segment controls neuronal output. Proc. Natl. Acad. Sci. USA 112, 9757–9762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff A, Xu Q, Anderson SA, and Yuste R (2009). Depolarizing effect of neocortical chandelier neurons. Front. Neural Circuits 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff AR, McGarry LM, Vogels TP, Inan M, Anderson SA, and Yuste R (2011). State-dependent function of neocortical chandelier cells. J. Neurosci 31, 17872–17886. [DOI] [PMC free article] [PubMed] [Google Scholar]