

Abstract
Background
Haloperidol used alone is recommended to help calm situations of aggression or agitation for people with psychosis. It is widely accessible and may be the only antipsychotic medication available in limited‐resource areas.
Objectives
To examine whether haloperidol alone is an effective treatment for psychosis‐induced aggression or agitation, wherein clinicians are required to intervene to prevent harm to self and others.
Search methods
We searched the Cochrane Schizophrenia Group's Study‐Based Register of Trials (26th May 2016). This register is compiled by systematic searches of major resources (including AMED, BIOSIS CINAHL, Embase, MEDLINE, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates, handsearches, grey literature, and conference proceedings, with no language, date, document type, or publication status limitations for inclusion of records into the register.
Selection criteria
Randomised controlled trials (RCTs) involving people exhibiting aggression and/or agitation thought to be due to psychosis, allocated rapid use of haloperidol alone (by any route), compared with any other treatment. Outcomes of interest included tranquillisation or asleep by 30 minutes, repeated need for rapid tranquillisation within 24 hours, specific behaviours (threat or injury to others/self), adverse effects. We included trials meeting our selection criteria and providing useable data.
Data collection and analysis
We independently inspected all citations from searches, identified relevant abstracts, and independently extracted data from all included studies. For binary data we calculated risk ratio (RR), for continuous data we calculated mean difference (MD), and for cognitive outcomes we derived standardised mean difference (SMD) effect sizes, all with 95% confidence intervals (CI) and using a fixed‐effect model. We assessed risk of bias for the included studies and used the GRADE approach to produce 'Summary of findings' tables which included our pre‐specified main outcomes of interest.
Main results
We found nine new RCTs from the 2016 update search, giving a total of 41 included studies and 24 comparisons. Few studies were undertaken in circumstances that reflect real‐world practice, and, with notable exceptions, most were small and carried considerable risk of bias. Due to the large number of comparisons, we can only present a summary of main results.
Compared with placebo, more people in the haloperidol group were asleep at two hours (2 RCTs, n=220, RR 0.88, 95%CI 0.82 to 0.95, very low‐quality evidence) and experienced dystonia (2 RCTs, n=207, RR 7.49, 95%CI 0.93 to 60.21, very low‐quality evidence).
Compared with aripiprazole, people in the haloperidol group required fewer injections than those in the aripiprazole group (2 RCTs, n=473, RR 0.78, 95%CI 0.62 to 0.99, low‐quality evidence). More people in the haloperidol group experienced dystonia (2 RCTs, n=477, RR 6.63, 95%CI 1.52 to 28.86, very low‐quality evidence).
Four trials (n=207) compared haloperidol with lorazepam with no significant differences with regard to number of participants asleep at one hour (1 RCT, n=60, RR 1.05, 95%CI 0.76 to 1.44, very low‐quality of evidence) or those requiring additional injections (1 RCT, n=66, RR 1.14, 95%CI 0.91 to 1.43, very low‐quality of evidence).
Haloperidol's adverse effects were not offset by addition of lorazepam (e.g. dystonia 1 RCT, n=67, RR 8.25, 95%CI 0.46 to 147.45, very low‐quality of evidence).
Addition of promethazine was investigated in two trials (n=376). More people in the haloperidol group were not tranquil or asleep by 20 minutes (1 RCT, n=316, RR 1.60, 95%CI 1.18 to 2.16, moderate‐quality evidence). Acute dystonia was too common in the haloperidol alone group for the trial to continue beyond the interim analysis (1 RCT, n=316, RR 19.48, 95%CI 1.14 to 331.92, low‐quality evidence).
Authors' conclusions
Additional data from new studies does not alter previous conclusions of this review. If no other alternative exists, sole use of intramuscular haloperidol could be life‐saving. Where additional drugs are available, sole use of haloperidol for extreme emergency could be considered unethical. Addition of the sedating promethazine has support from better‐grade evidence from within randomised trials. Use of an alternative antipsychotic drug is only partially supported by fragmented and poor‐grade evidence. Adding a benzodiazepine to haloperidol does not have strong evidence of benefit and carries risk of additional harm.
After six decades of use for emergency rapid tranquillisation, this is still an area in need of good independent trials relevant to real‐world practice.
Plain language summary
Haloperidol as a means of calming people who are aggressive or agitated due to psychosis
Aim of review
This review looks at whether haloperidol is effective for treating people who are agitated and aggressive as a result of having psychosis.
Background
People with psychosis may experience hearing voices (hallucinations) or abnormal thoughts (delusions), which can make the person frightened, distressed and agitated (restless, excitable or irritable), leading sometimes to aggressive behaviour. This poses a significant challenge for mental health professionals, who have to quickly choose the best available treatment to prevent the risk of harm to both patient and/or others.
Haloperidol is a medication used for treating people with psychosis that can be taken by mouth or injected. As well as being an antipsychotic (preventing psychosis), it also calms people down or helps them to sleep.
Searches
In 2011 and 2016, the information specialist of the Cochrane Schizophrenia Group searched their specialised register for randomised trials that looked at the effects of giving haloperidol compared with either placebo or other treatments to people who are aggressive or agitated because they were experiencing psychosis.
Results
Forty‐one studies are now included in the review but information in these is of low‐quality. Main results show that when compared with placebo or no treatment, more people having haloperidol were asleep after two hours. However, evidence is not strong. Results are made more complex by large variety of available treatments (24 comparisons).
Conclusions
The review authors conclude there is weak evidence that haloperidol calms people down and helps manage difficult situations. However, this is not based on good‐quality trails and therefore health professionals and people with mental health problems are left without clear evidence‐based guidance. In some situations, haloperidol may be the only choice, but this is far from ideal because although haloperidol is effective at calming people down it has side effects (e.g. restlessness, shaking of the head, hands and body, heart problems). These side effects can be just as distressing as psychosis and may act as a barrier that stops people coming back for future treatment. More research is needed to help people consider and understand which medication is better at calming people down; has fewer side effects; works quickly and rapidly; and can be used at lower dosages (or less frequent injections).
This plain language summary is based on a summary written by a consumer Ben Gray from Rethink.
Summary of findings
Summary of findings for the main comparison. HALOPERIDOL compared to PLACEBO/NIL for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared with PLACEBO/NIL for psychosis‐induced aggression or agitation | ||||||
| Patient or population: patients with psychosis‐induced aggression or agitation Setting: inpatients; multi‐centre. Intervention: HALOPERIDOL Comparison: PLACEBO/NIL | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| PLACEBO/NIL | HALOPERIDOL | |||||
| Tranquillisation or asleep not asleep at 2 hours | Low1 | RR 0.88 (0.82 to 0.95) | 220 (2 studies) | ⊕⊝⊝⊝ very low2,3,4 | ||
| 965 per 1000 | 849 per 1000 (791 to 917) | |||||
| Moderate1 | ||||||
| 980 per 1000 | 862 per 1000 (804 to 931) | |||||
| High1 | ||||||
| 995 per 1000 | 876 per 1000 (816 to 945) | |||||
| Repeated need for tranquillisation needing additional injection within 24 hours | Low1 | RR 0.51 (0.42 to 0.62) | 660 (4 studies) | ⊕⊕⊝⊝ low2,4 | ||
| 400 per 1000 | 204 per 1000 (168 to 248) | |||||
| Moderate1 | ||||||
| 600 per 1000 | 306 per 1000 (252 to 372) | |||||
| High1 | ||||||
| 800 per 1000 | 408 per 1000 (336 to 496) | |||||
| Specific behaviour ‐ threat or injury to self or others average change score (ABS, high = worse) at 2 hours | The mean specific behaviour ‐ threat or injury to self or others in the intervention groups was 4.48 lower (5.78 to 3.18 lower) | 474 (3 studies) | ⊕⊝⊝⊝ very low2,4,5 | |||
| Global outcome ‐ overall improvement (not any improvement) | Low1 | RR 0.28 (0.08 to 1.07) | 40 (1 study) | ⊕⊕⊝⊝ low2,6 | ||
| 200 per 1000 | 56 per 1000 (8 to 414) | |||||
| Moderate1 | ||||||
| 350 per 1000 | 98 per 1000 (14 to 724) | |||||
| High1 | ||||||
| 500 per 1000 | 140 per 1000 (20 to 1000) | |||||
| Adverse effects: Specific ‐ dystonia within 24 hours | Moderate1 | RR 7.49 (0.93 to 60.21) | 207 (2 studies) | ⊕⊝⊝⊝ very low2,4,6 | ||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| High1 | ||||||
| 10 per 1000 | 75 per 1000 (9 to 602) | |||||
| Economic outcome ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Control risk ‐ moderate risk is roughly equal to that of the control group. 2 Risk of bias: rated 'serious' ‐ method of randomisation not described, allocation concealment not stated, incomplete outcome data, sponsored by drug company. 3 Indirectness: rated 'serious' ‐ no measure for tranquil or asleep at 30 minutes, therefore had to use not asleep at 2 hours. 4 Publication bias: rated 'strongly suspected' ‐ sponsored by drug company. 5 Indirectness: rated 'serious' ‐ not threat of injury to self or others, scale derived data. 6 Imprecision: rated 'serious' ‐ 95% confidence interval is wide.
Summary of findings 2. HALOPERIDOL compared to OTHER ANTIPSYCHOTIC: a. ARIPIPRAZOLE for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared with OTHER ANTIPSYCHOTICS: a. ARIPIPRAZOLE for psychosis‐induced aggression or agitation | ||||||
| Patient or population: patients with psychosis‐induced aggression or agitation Setting: multi‐centre. Intervention: HALOPERIDOL Comparison: OTHER ANTIPSYCHOTICS: a. ARIPIPRAZOLE | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| OTHER ANTIPSYCHOTICS: a. ARIPIPRAZOLE | HALOPERIDOL | |||||
| Tranquillisation or asleep ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Repeated need for rapid tranquillisation ‐ needing additional injection | Low1 | RR 0.78 (0.62 to 0.99) | 473 (2 studies) | ⊕⊕⊝⊝ low2,3 | ||
| 200 per 1000 | 156 per 1000 (124 to 198) | |||||
| Moderate1 | ||||||
| 400 per 1000 | 312 per 1000 (248 to 396) | |||||
| High1 | ||||||
| 650 per 1000 | 507 per 1000 (403 to 644) | |||||
| Specific behaviours ‐ agitation: CABS average change score at 2 hours | The mean specific behaviours ‐ agitation: average change score at 2 hours in the intervention groups was 0.55 lower (2.10 lower to 1.01 higher) | 470 (2 studies) | ⊕⊝⊝⊝ very low1,2,4 | |||
| Global outcomes: need for benzodiazepine 2 | Low1 | RR 1.26 (0.74 to 2.16) | 477 (2 studies) | ⊕⊝⊝⊝ very low2,3,5 | ||
| 50 per 1000 | 63 per 1000 (37 to 108) | |||||
| Moderate1 | ||||||
| 100 per 1000 | 126 per 1000 (74 to 216) | |||||
| High1 | ||||||
| 200 per 1000 | 252 per 1000 (148 to 432) | |||||
| Adverse effects: any serious, specific ‐ dystonia during 24 hours | Low6 | RR 6.63 (1.52 to 28.86) | 477 (2 studies) | ⊕⊝⊝⊝ very low3,7,8 | ||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate6 | ||||||
| 50 per 1000 | 332 per 1000 (76 to 1000) | |||||
| High6 | ||||||
| 100 per 1000 | 663 per 1000 (152 to 1000) | |||||
| Economic outcome ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Moderate risk roughly equates with that of the trial control group. 2 Risk of bias: rated 'severe' ‐ method of randomisation not described, possible that blinding was broken in Bristol Myers 2005, adverse effects only reported when reported by 5% of participants, sponsored by drug company. 3 Publication bias: rated 'strongly suspected' ‐ sponsored by drug company. 4 Indirectness: rated 'serious' ‐ scale‐derived data, not threat or injury to self or others as stated in protocol. 5 Indirectness: rated 'serious' ‐ no measure for global outcome, overall improvement, therefore global improvement is inferred by need for benzodiazepine. 6 Low risk equates with that of trial control group. 7 Risk of bias: rated 'serious' ‐ method of randomisation not described, incomplete outcome data, adverse effects only reported when reported by at least 5% of participants, sponsored by drug company. 8 Imprecision: rated 'very serious' ‐ 95% confidence intervals are wide.
Summary of findings 3. HALOPERIDOL compared to OTHER ANTIPSYCHOTIC: b. CHLORPROMAZINE for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared with OTHER ANTIPSYCHOTIC: b. CHLORPROMAZINE for psychosis‐induced aggression or agitation | ||||||
| Patient or population: patients with psychosis‐induced aggression or agitation Setting: inpatients. Intervention: HALOPERIDOL Comparison: OTHER ANTIPSYCHOTIC: b. CHLORPROMAZINE | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| OTHER ANTIPSYCHOTIC: b. CHLORPROMAZINE | HALOPERIDOL | |||||
| Tranquillisation or asleep Not tranquil or asleep | Low1 | RR 1.93 (1.04 to 3.60) | 39 (1 study) | ⊕⊝⊝⊝ very low2,3,4 | ||
| 200 per 1000 | 386 per 1000 (208 to 720) | |||||
| Moderate1 | ||||||
| 500 per 1000 | 965 per 1000 (520 to 1000) | |||||
| High1 | ||||||
| 700 per 1000 | 1000 per 1000 (728 to 1000) | |||||
| Repeated need for rapid tranquillisation ‐ more that 1 injection | Low1 | RR 1.07 (0.89 to 1.28) | 30 (1 study) | ⊕⊝⊝⊝ very low2,3,4 | ||
| 800 per 1000 | 856 per 1000 (712 to 1000) | |||||
| Moderate1 | ||||||
| 900 per 1000 | 963 per 1000 (801 to 1000) | |||||
| High1 | ||||||
| 990 per 1000 | 1000 per 1000 (881 to 1000) | |||||
| Specific behaviour ‐ threat or injury of harm to self or others ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Global outcome ‐ not any improvement | Low1 | RR 0.15 (0.05 to 0.49) | 89 (2 studies) | ⊕⊝⊝⊝ very low2,3 | ||
| 1000 per 1000 | 150 per 1000 (50 to 490) | |||||
| Moderate1 | ||||||
| 300 per 1000 | 45 per 1000 (15 to 147) | |||||
| High1 | ||||||
| 500 per 1000 | 75 per 1000 (25 to 245) | |||||
| Adverse effects: specific ‐ cardiovascular ‐ hypotension | Low1 | RR 0.51 (0.01 to 2.60) | 30 (1 study) | ⊕⊝⊝⊝ very low2,3,4 | ||
| 50 per 1000 | 10 per 1000 (0 to 192) | |||||
| Moderate1 | ||||||
| 150 per 1000 | 30 per 1000 (1 to 577) | |||||
| High1 | ||||||
| 250 per 1000 | 50 per 1000 (2 to 962) | |||||
| Adverse effects: specific ‐ central nervous system ‐ seizures | Low1 | RR 0.33 (0.01 to 7.58) | 30 (1 study) | ⊕⊝⊝⊝ very low2,3,4 | ||
| 10 per 1000 | 3 per 1000 (0 to 76) | |||||
| Moderate1 | ||||||
| 50 per 1000 | 17 per 1000 (0 to 379) | |||||
| High1 | ||||||
| 150 per 1000 | 50 per 1000 (1 to 1000) | |||||
| Economic outcome ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Control risk ‐ moderate risk roughly equates to that of the control group 2 Risk of bias: rated 'serious' ‐ not explicitly described as randomised, missing data was not imputed using appropriate methods such as LOCF, small study. 3 Imprecision: rated 'serious' ‐ small study. 4 Publication bias: rated 'strongly suspected' ‐ small study (15 participants per group).
Summary of findings 4. HALOPERIDOL compared to OTHER ANTIPSYCHOTIC: c. DROPERIDOL for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared to OTHER ANTIPSYCHOTIC: c. DROPERIDOL for psychosis‐induced aggression or agitation | ||||||
| Patient or population: psychosis‐induced aggression or agitation Setting: inpatients; emergency room. Intervention: HALOPERIDOL Comparison: OTHER ANTIPSYCHOTIC: c. DROPERIDOL | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with OTHER ANTIPSYCHOTIC: c. DROPERIDOL | Risk with HALOPERIDOL | |||||
| Tranquillisation or asleep ‐ not asleep up to 2 hours | Study population | RR 1.07 (0.44 to 2.60) | 228 (1 RCT) | ⊕⊕⊕⊝ moderate1 | ||
| 82 per 1.000 | 88 per 1.000 (36 to 213) | |||||
| Repeated need for rapid tranquillisation ‐ more than 1 injection | Low | RR 2.38 (1.27 to 4.47) | 255 (2 RCTs) | ⊕⊕⊕⊝ moderate2 | ||
| 50 per 1.000 | 119 per 1.000 (64 to 224) | |||||
| Moderate | ||||||
| 70 per 1.000 | 167 per 1.000 (89 to 313) | |||||
| High | ||||||
| 360 per 1.000 | 857 per 1.000 (457 to 1.000) | |||||
| Specific behaviour ‐ threat or injury to self or others ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| Global outcome ‐ need for benzodiazepine | Study population | RR 0.31 (0.07 to 1.44) | 228 (1 RCT) | ⊕⊕⊕⊝ moderate1 | ||
| 59 per 1.000 | 18 per 1.000 (4 to 85) | |||||
| Adverse effects: specific ‐ dystonia | Study population | RR 0.86 (0.11 to 6.56) | 255 (2 RCTs) | ⊕⊕⊕⊝ moderate5 | ||
| 8 per 1.000 | 7 per 1.000 (1 to 51) | |||||
| Economic outcome ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 CI include no effect
2 Risk of bias: rated 'serious' ‐ method of randomisation not reported, described as double blind but no further information given regarding rater blinding, small short study.
3 Publication bias: rated 'strongly suspected' ‐ small study.
4 Moderate risk roughly equates with that of the trial control group,
5 Adverse effects ‐ imprecision ‐ 95% confidence interval.
6 Adverse effects ‐ publication bias ‐ small study.
Summary of findings 5. HALOPERIDOL compared to OTHER ANTIPSYCHOTIC: d. LOXAPINE for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared with OTHER ANTIPSYCHOTICS: d. LOXAPINE for psychosis‐induced aggression or agitation | ||||||
| Patient or population: patients to psychosis‐induced aggression or agitation Setting: inpatients; emergency room. Intervention: HALOPERIDOL Comparison: OTHER ANTIPSYCHOTICS: d. LOXAPINE | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| OTHER ANTIPSYCHOTICS: d. LOXAPINE | HALOPERIDOL | |||||
| Tranquillisation or asleep ‐ not asleep by 12 hours | Low1 | RR 4.31 (0.54 to 34.48) | 54 (1 study) | ⊕⊝⊝⊝ very low2,3,4 | ||
| 10 per 1000 | 43 per 1000 (5 to 345) | |||||
| Moderate1 | ||||||
| 50 per 1000 | 215 per 1000 (27 to 1000) | |||||
| High1 | ||||||
| 100 per 1000 | 431 per 1000 (54 to 1000) | |||||
| Global outcome: specific Not sedated at 60 minutes * | Low1 | RR 3.50 (0.86 to 14.18) | 30 (1 study) | ⊕⊝⊝⊝ very low2,4,5,6 | * data for prespecified outcome Repeated need for rapid tranquillisation were not reported | |
| 10 per 1000 | 57 per 1000 (9 to 345) | |||||
| Moderate1 | ||||||
| 100 per 1000 | 569 per 1000 (94 to 1000) | |||||
| High1 | ||||||
| 200 per 1000 | 1000 per 1000 (188 to 1000) | |||||
| Specific behaviour ‐ aggression ‐ no overall improvement | Low1 | RR 1.10 (0.69 to 1.76) | 30 (1 study) | ⊕⊝⊝⊝ very low2,4,6,7 | ||
| 500 per 1000 | 550 per 1000 (345 to 880) | |||||
| Moderate1 | ||||||
| 650 per 1000 | 715 per 1000 (448 to 1000) | |||||
| High1 | ||||||
| 800 per 1000 | 880 per 1000 (552 to 1000) | |||||
| Global outcome: no change at 48 hours CGI | Low1 | RR 0.93 (0.14 to 6.15) | 56 (1 study) | ⊕⊕⊝⊝ low2,4 | ||
| 10 per 1000 | 9 per 1000 (1 to 62) | |||||
| Moderate1 | ||||||
| 70 per 1000 | 65 per 1000 (10 to 431) | |||||
| High1 | ||||||
| 140 per 1000 | 130 per 1000 (20 to 861) | |||||
| Adverse effects: Specific ‐ dystonia between 0‐3 days (IM phase) | Low1 | RR 0.94 (0.06 to 13.93) | 35 (1 study) | ⊕⊝⊝⊝ very low2,4 | ||
| 10 per 1000 | 9 per 1000 (1 to 139) | |||||
| Moderate1 | ||||||
| 50 per 1000 | 47 per 1000 (3 to 697) | |||||
| High1 | ||||||
| 100 per 1000 | 94 per 1000 (6 to 1000) | |||||
| Adverse effects: Specific ‐ rigidity between 0‐3 days (IM phase) | Low1 | RR 0.88 (0.65 to 1.19) | 35 (1 study) | ⊕⊝⊝⊝ very low2,4,6 | ||
| 750 per 1000 | 660 per 1000 (487 to 893) | |||||
| Moderate1 | ||||||
| 850 per 1000 | 748 per 1000 (552 to 1000) | |||||
| High1 | ||||||
| 950 per 1000 | 836 per 1000 (617 to 1000) | |||||
| Economic outcome ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Moderate risk is roughly equal to that of the control group. 2 Risk of bias: rated 'serious' ‐ method of randomisation not described, allocation concealment not stated, staff administering medication were not blind to the intervention, the number of participants reported as leaving the study at certain time points is inconsistent. 3 Indirectness: rated 'serious' ‐ reported at 12 hours rather than 30 minutes as stated in protocol. 4 Imprecision: rated 'serious' ‐ 95% confidence interval is wide. 5 Indirectness: rated 'serious' ‐ no data on repeated need for rapid tranquillisation, therefore inferred from the data 'not sedated at 60 minutes'. 6 Publication bias: rated 'strongly suspected' ‐ small study. 7 Indirectness: rated 'serious' ‐ not able to use threat or injury to self or others as stated in the protocol because this was not reported.
Summary of findings 6. HALOPERIDOL compared to OTHER ANTIPSYCHOTIC: e. OLANZAPINE for psychosis induced aggression or agitation.
| HALOPERIDOL compared to OTHER ANTIPSYCHOTIC: e. OLANZAPINE for psychosis induced aggression or agitation | ||||||
| Patient or population: psychosis induced aggression or agitation Setting: Emergency room; multi‐centre. Intervention: HALOPERIDOL Comparison: OTHER ANTIPSYCHOTIC: e. OLANZAPINE | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with OTHER ANTIPSYCHOTIC: e. OLANZAPINE | Risk with HALOPERIDOL | |||||
| Tranquilisation or asleep assessed with: not asleep up to 2 hours | Low | RR 1.16 (1.02 to 1.32) | 257 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 2 3 | ||
| 200 per 1.000 | 232 per 1.000 (204 to 264) | |||||
| Moderate | ||||||
| 700 per 1.000 | 812 per 1.000 (714 to 924) | |||||
| High | ||||||
| 900 per 1.000 | 1000 per 1.000 (918 to 1.000) | |||||
| Repeated need for tranquillisation assessed with: needing an additional injection by 24 hours | Low | RR 1.06 (0.75 to 1.51) | 392 (3 RCTs) | ⊕⊝⊝⊝ VERY LOW 2 | ||
| 50 per 1.000 | 53 per 1.000 (38 to 76) | |||||
| Moderate | ||||||
| 200 per 1.000 | 212 per 1.000 (150 to 302) | |||||
| High | ||||||
| 350 per 1.000 | 371 per 1.000 (262 to 529) | |||||
| Specific behaviour ‐ threat or injury to self or others assessed with: less than 40% reduction in PANSS‐EC rated at 2 hours | Low | RR 0.96 (0.58 to 1.58) | 45 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 2 5 | ||
| 200 per 1.000 | 192 per 1.000 (116 to 316) | |||||
| Moderate | ||||||
| 400 per 1.000 | 384 per 1.000 (232 to 632) | |||||
| High | ||||||
| 600 per 1.000 | 576 per 1.000 (348 to 948) | |||||
| Global outcome ‐ need for benzodiazepine during 24 hours | Low | RR 1.05 (0.63 to 1.74) | 343 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW 6 7 | ||
| 50 per 1.000 | 53 per 1.000 (32 to 87) | |||||
| Moderate | ||||||
| 150 per 1.000 | 158 per 1.000 (95 to 261) | |||||
| High | ||||||
| 250 per 1.000 | 263 per 1.000 (158 to 435) | |||||
| Adverse effects: Specific ‐ dystonia during 24 hours | Low | RR 12.92 (1.67 to 99.78) | 343 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW 6 7 | ||
| 0 per 1.000 | 0 per 1.000 (0 to 0) | |||||
| Moderate | ||||||
| 0 per 1.000 | 0 per 1.000 (0 to 0) | |||||
| High | ||||||
| 5 per 1.000 | 65 per 1.000 (8 to 499) | |||||
| Economic outcome ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No studies reported this outcome. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Moderate risk is roughly equal to that of the control group.
2 Risk of bias: rated 'very serious' ‐ method of randomisation not reported, allocation concealment not stated, described as double blind but no further details are given regarding blinding, selective reporting, sponsored by drug company.
3 Indirectness: rated 'serious' ‐ tranquil at 30 minutes is not measured, therefore used not asleep at 2 hour.
4 Publication bias: rated 'strongly suspected' ‐ sponsored by drug company.
5 Indirectness: rated 'serious' ‐ not threat or injury to self or others as stated in protocol, therefore inferred further aggressive behaviour by less than a 40% reduction in PANSS‐EC score.
6 Risk of bias: rated 'serious' ‐ method of randomisation is not reported, allocation concealment not stated, described as double blind but no further information given, incomplete outcome data, selective reporting, sponsored by drug company.
7 Imprecision: rated 'serious' ‐ 95% confidence interval is wide.
Summary of findings 7. HALOPERIDOL compared to OTHER ANTIPSYCHOTIC: f. PERPHENAZINE for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared with OTHER ANTIPSYCHOTIC: f. PERPHENAZINE for psychosis‐induced aggression or agitation | ||||||
| Patient or population: patients with psychosis‐induced aggression or agitation Setting: inpatients. Intervention: HALOPERIDOL Comparison: OTHER ANTIPSYCHOTIC: f. PERPHENAZINE | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| OTHER ANTIPSYCHOTIC: f. PERPHENAZINE | HALOPERIDOL | |||||
| Tranquillisation or asleep ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Repeated need for rapid tranquillisation ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Specific behaviour ‐ injury or threat of self to others ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Global outcome: general ‐ no improvement | Low1 | RR 0.46 (0.04 to 4.68) | 44 (1 study) | ⊕⊝⊝⊝ very low2,3 | ||
| 10 per 1000 | 5 per 1000 (0 to 47) | |||||
| Moderate1 | ||||||
| 100 per 1000 | 46 per 1000 (4 to 468) | |||||
| High1 | ||||||
| 200 per 1000 | 92 per 1000 (8 to 936) | |||||
| Adverse effect: Specific ‐ hypotensive episode | Low1 | RR 0.31 (0.01 to 7.12) | 44 (1 study) | ⊕⊝⊝⊝ very low2,3,4 | ||
| 10 per 1000 | 3 per 1000 (0 to 71) | |||||
| Moderate1 | ||||||
| 50 per 1000 | 15 per 1000 (0 to 356) | |||||
| High1 | ||||||
| 100 per 1000 | 31 per 1000 (1 to 712) | |||||
| Adverse effect ‐ discontinued due to EPS | Low1 | RR 1.83 (0.18 to 18.7) | 44 (1 study) | ⊕⊝⊝⊝ very low2,3,5 | ||
| 10 per 1000 | 18 per 1000 (2 to 187) | |||||
| Moderate1 | ||||||
| 50 per 1000 | 92 per 1000 (9 to 935) | |||||
| High1 | ||||||
| 100 per 1000 | 183 per 1000 (18 to 1000) | |||||
| Economic outcome ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Moderate risk is roughly equal to that of the trial control group. 2 Risk of bias: rated 'serious' ‐ method or randomisation not described, allocation concealment not stated, described as double‐blind but no further details given regarding rater blinding, author suspects that only the adverse effects severe enough to warrant discontinuation were reported. 3 Publication bias: rated 'strongly suspected' ‐ small study. 4 Imprecision: rated 'serious' ‐ 95% confidence interval is wide, small study. 5 Indirectness: rated 'serious' ‐ not specific adverse effect ‐ only have data for the number of people discontinued due to EPS, not the number of people who had EPS in general.
Summary of findings 8. HALOPERIDOL compared to OTHER ANTIPSYCHOTICS: g. QUETIAPINE for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared to OTHER ANTIPSYCHOTICS: g. QUETIAPINE for psychosis‐induced aggression or agitation | ||||||
| Patient or population: psychosis‐induced aggression or agitation Setting: inpatients. Intervention: HALOPERIDOL Comparison: OTHER ANTIPSYCHOTICS: g. QUETIAPINE | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with OTHER ANTIPSYCHOTICS: g. QUETIAPINE | Risk with HALOPERIDOL | |||||
| Tranquillisation or asleep ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| Repeated need for tranquillisation ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| Specific behaviour ‐ agitation assessed with: change score at 24 hours on the PANSS‐EC | MD 0.10 higher (0.56 lower to 0.76 higher) | ‐ | 80 (1 RCT) | ⊕⊕⊝⊝ low1 | ||
| Global outcome ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| Adverse effects assessed with: one or more drug‐related adverse events | Low | RR 1.81 (1.19 to 2.77) | 80 (1 RCT) | ⊕⊕⊝⊝ low1 | ||
| 200 per 1.000 | 362 per 1.000 (238 to 554) | |||||
| Moderate | ||||||
| 400 per 1.000 | 724 per 1.000 (476 to 1.000) | |||||
| High | ||||||
| 600 per 1.000 | 1000 per 1.000 (714 to 1.000) | |||||
| Economic outcome ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Imprecision: rated 'very serious' ‐ small study, 95% CI are wide.
Summary of findings 9. HALOPERIDOL compared to OTHER ANTIPSYCHOTIC: h. RISPERIDONE for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared with OTHER ANTIPSYCHOTIC: g. RISPERIDONE for psychosis‐induced aggression or agitation | ||||||
| Patient or population: patients with psychosis‐induced aggression or agitation Setting: inpatients; emergency room; multi‐centre. Intervention: HALOPERIDOL Comparison: OTHER ANTIPSYCHOTIC: g. RISPERIDONE | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| OTHER ANTIPSYCHOTIC: g. RISPERIDONE | HALOPERIDOL | |||||
| Tranquillisation or asleep ‐ not asleep | Low1 | RR 0.84 (0.74 to 0.95) | 162 (1 study) | ⊕⊕⊕⊝ moderate2 | ||
| 700 per 1000 | 588 per 1000 (518 to 665) | |||||
| Moderate1 | ||||||
| 900 per 1000 | 756 per 1000 (666 to 855) | |||||
| High1 | ||||||
| 1000 per 1000 | 840 per 1000 (740 to 950) | |||||
| Repeated need for rapid tranquillisation ‐ needing additional benzodiazepine | Low | RR 0.98 (0.65 to 1.47) | 286 (2 studies) | ⊕⊕⊝⊝ low2,3 | ||
| 100 per 1000 | 98 per 1000 (65 to 147) | |||||
| Moderate | ||||||
| 200 per 1000 | 196 per 1000 (130 to 294) | |||||
| High | ||||||
| 300 per 1000 | 294 per 1000 (195 to 441) | |||||
| Specific behaviours ‐ agitation >50% reduction in PANSS‐EC score Follow‐up: 1 days | Low1 | RR 1.15 (0.6 to 2.22) | 124 (1 study) | ⊕⊕⊝⊝ low2,4 | ||
| 50 per 1000 | 58 per 1000 (30 to 111) | |||||
| Moderate1 | ||||||
| 200 per 1000 | 230 per 1000 (120 to 444) | |||||
| High1 | ||||||
| 350 per 1000 | 402 per 1000 (210 to 777) | |||||
|
Global outcome ‐ rated as severe ‐ CGI‐S Follow‐up: 1 days |
Low1 | RR 0.89 (0.51 to 1.58) | 162 (1 study) | ⊕⊝⊝⊝ very low2,3,4 | ||
| 100 per 1000 | 89 per 1000 (51 to 158) | |||||
| Moderate1 | ||||||
| 200 per 1000 | 178 per 1000 (102 to 316) | |||||
| High1 | ||||||
| 300 per 1000 | 267 per 1000 (153 to 474) | |||||
| Adverse effects: Specific ‐ EPS during 24 hours | Low1 | RR 1.6 (0.55 to 4.62) | 124 (1 study) | ⊕⊕⊝⊝ low2,4 | ||
| 10 per 1000 | 16 per 1000 (6 to 46) | |||||
| Moderate1 | ||||||
| 100 per 1000 | 160 per 1000 (55 to 462) | |||||
| High1 | ||||||
| 200 per 1000 | 320 per 1000 (110 to 924) | |||||
| Adverse effects: Specific ‐ acute dystonia during 24 hours | Low1 | RR 5 (0.24 to 102.07) | 286 (2 studies) | ⊕⊝⊝⊝ very low2,3,4 | ||
| 10 per 1000 | 50 per 1000 (2 to 1000) | |||||
| Moderate1 | ||||||
| 20 per 1000 | 100 per 1000 (5 to 1000) | |||||
| High1 | ||||||
| 30 per 1000 | 150 per 1000 (7 to 1000) | |||||
| Economic outcome ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Moderate risk is roughly equal to that of the trial control group. 2 Risk of bias: rated 'serious' ‐ method of randomisation not described, single blind, sponsored by drug company. 3 Imprecision: rated 'serious' ‐ few data and confidence intervals wide. 4 Publication bias: rated 'strongly suspected ‐ sponsored by drug company.
Summary of findings 10. HALOPERIDOL compared to OTHER ANTIPSYCHOTIC: i. THIOTHIXENE for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared to OTHER ANTIPSYCHOTIC: h. THIOTHIXENE for psychosis‐induced aggression or agitation | ||||||
| Patient or population: patients with psychosis‐induced aggression or agitation Setting: inpatients; emergency room. Intervention: HALOPERIDOL Comparison: OTHER ANTIPSYCHOTIC: h. THIOTHIXENE | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| OTHER ANTIPSYCHOTIC: h. THIOTHIXENE | HALOPERIDOL | |||||
| Tranquillisation or asleep ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Repeated need for rapid tranquillisation | Low1 | RR 1.07 (0.89 to 1.28) | 30 (1 study) | ⊕⊝⊝⊝ very low2,3 | ||
| 850 per 1000 | 910 per 1000 (756 to 1000) | |||||
| Moderate1 | ||||||
| 900 per 1000 | 963 per 1000 (801 to 1000) | |||||
| High1 | ||||||
| 950 per 1000 | 1000 per 1000 (845 to 1000) | |||||
| Specific behaviour ‐ threat or injury to self or others ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Global outcome No improvement at 1 hour | Low1 | RR 0.50 (0.14 to 1.84) | 44 (1 study) | ⊕⊝⊝⊝ very low2,3,4 | ||
| 50 per 1000 | 25 per 1000 (7 to 92) | |||||
| Moderate1 | ||||||
| 250 per 1000 | 125 per 1000 (35 to 460) | |||||
| High1 | ||||||
| 450 per 1000 | 225 per 1000 (63 to 828) | |||||
| Adverse effects: specific ‐ hypotensive episode | Low | Not estimable | 30 (1 study) | ⊕⊝⊝⊝ very low2,3,4 | ||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Adverse effects: specific ‐ tachycardia | Low1 | RR 0.28 (0.01 to 6.52) | 44 (1 study) | ⊕⊝⊝⊝ very low2,3,4 | ||
| 10 per 1000 | 3 per 1000 (0 to 65) | |||||
| Moderate1 | ||||||
| 50 per 1000 | 14 per 1000 (0 to 326) | |||||
| High1 | ||||||
| 100 per 1000 | 28 per 1000 (1 to 652) | |||||
| Economic outcome ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Control risk ‐ moderate risk roughly equates to that of the trial control group. 2 Risk of bias: rated 'serious' ‐ method of sequence generation not clear, allocation concealment not reported, small study, sponsored by drug company. 3 Publication bias: rated 'strongly suspected' ‐ small study, sponsored by drug company. 4 Imprecision: rated 'serious' ‐ small study, wide confidence intervals.
Summary of findings 11. HALOPERIDOL compared to OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared to OTHER ANTIPSYCHOTIC: i. ZIPRASIDONE for psychosis‐induced aggression or agitation | ||||||
| Patient or population: psychosis‐induced aggression or agitation Setting: inpatients; emergency room; multi‐centre. Intervention: HALOPERIDOL Comparison: OTHER ANTIPSYCHOTIC: i. ZIPRASIDONE | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with OTHER ANTIPSYCHOTIC: i. ZIPRASIDONE | Risk with HALOPERIDOL | |||||
| Tranquilisation or asleep ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No trial reported this outcome. |
| Repeated need for tranquilisation assessed with: need for additional drugs for tranquillisation up to 24 hours | Study population | RR 1.64 (1.07 to 2.53) | 60 (1 RCT) | ⊕⊝⊝⊝ very low1 2 | ||
| 467 per 1.000 | 765 per 1.000 (499 to 1.000) | |||||
| Specific behaviour ‐ agitation assessed with: average endpoint scores at 2 hours on the PANSS‐EC follow up: 72 hours | MD 0.06 higher (1.13 lower to 1.25 higher) | ‐ | 231 (1 RCT) | ⊕⊕⊝⊝ low3 4 | ||
| Global outcome assessed with: CGI‐S ‐ average change score at 72 hours. | MD 0.34 higher (0.13 higher to 0.55 higher) | ‐ | 132 (1 RCT) | ⊕⊝⊝⊝ very low5 | ||
| Adverse effects: Specific ‐ dystonia during 72 hours | Low | RR 10.26 (1.67 to 63.17) | 508 (2 RCTs) | ⊕⊝⊝⊝ very low5 7 | ||
| 2 per 1.000 | 21 per 1.000 (3 to 126) | |||||
| Moderate | ||||||
| 4 per 1.000 | 41 per 1.000 (7 to 253) | |||||
| High | ||||||
| 10 per 1.000 | 103 per 1.000 (17 to 632) | |||||
| Adverse effects: Specific ‐ clinically significant abnormal ECG during 72 hours | Study population | RR 1.01 (0.60 to 1.71) | 376 (1 RCT) | ⊕⊝⊝⊝ very low3 9 | ||
| 127 per 1.000 | 128 per 1.000 (76 to 217) | |||||
| Economic outcome ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No trial reported this outcome. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated 'serious' ‐ method of allocation concealment not stated, blinding of participants uneffective
2 Imprecision: rated 'very serious' ‐ less than 100 patients, 95% CI extends beyond the no effect point
3 Risk of bias: rated 'serious' ‐ method of randomisation is not described, allocation concealment is not stated, single‐blind.
4 Indirectness: rated 'serious' ‐ not threat or injury to self or others, therefore had to use scale derived data for agitation.
5 Risk of bias: rated 'very serious' ‐ allocation of concealment not stated, open trial, adverse effects only reported where they occurred in ≥10% of people, sponsored by drug company.
6 Publication bias: rated 'strongly suspected' ‐ sponsored by drug company.
7 Imprecision: rated 'serious' ‐ 95% confidence intervals are wide.
8 Moderate risk roughly is roughly equal to that of the control group.
9 Indirectness: rated 'serious' ‐ abnormal ECG ‐ not necessarily a serious adverse effect.
Summary of findings 12. HALOPERIDOL compared to OTHER ANTIPSYCHOTIC: k. ZUCLOPENTHIXOL ACETATE for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared with OTHER ANTIPSYCHOTICS: j. ZUCLOPENTHIXOL ACETATE for psychosis‐induced aggression or agitation | ||||||
| Patient or population: patients with psychosis‐induced aggression or agitation Setting: inpatients. Intervention: HALOPERIDOL Comparison: OTHER ANTIPSYCHOTICS: j. ZUCLOPENTHIXOL ACETATE | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| OTHER ANTIPSYCHOTICS: j. ZUCLOPENTHIXOL ACETATE | HALOPERIDOL | |||||
| Tranquillisation or asleep ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Repeated need for rapid tranquillisation ‐ more than 3 injections | Low1 | RR 2.54 (1.19 to 5.46) | 70 (1 study) | ⊕⊝⊝⊝ very low2,3,4 | ||
| 50 per 1000 | 127 per 1000 (60 to 273) | |||||
| Moderate1 | ||||||
| 200 per 1000 | 508 per 1000 (238 to 1000) | |||||
| High1 | ||||||
| 350 per 1000 | 889 per 1000 (417 to 1000) | |||||
| Specific behaviour ‐ threat or injury to self or others ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Global outcome ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Adverse effects: specific ‐ tremor by 7 days | Low1 | RR 4.16 (0.93 to 18.62) | 70 (1 study) | ⊕⊝⊝⊝ very low2,4,5 | ||
| 10 per 1000 | 42 per 1000 (9 to 186) | |||||
| Moderate1 | ||||||
| 50 per 1000 | 208 per 1000 (47 to 931) | |||||
| High1 | ||||||
| 100 per 1000 | 416 per 1000 (93 to 1000) | |||||
| Economic outcome ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Moderate risk is roughly equal to that of the control group. 2 Risk of bias: rated 'serious' ‐ method of randomisation not reported, open label, author suspects that rater was only blind for some outcomes. 3 Indirectness: rated 'serious' ‐ measures more than 3 injections, not just more than 1 injection. 4 Imprecision: rated 'serious' ‐ small study. 5 Indirectness: rated 'serious' ‐ no serious specific adverse effect, therefore used tremor.
Summary of findings 13. HALOPERIDOL compared to BENZODIAZEPINE: a. FLUNITRAZEPAM for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared with BENZODIAZEPINE: a. FLUNITRAZEPAM for psychosis‐induced aggression or agitation | ||||||
| Patient or population: patients with psychosis‐induced aggression or agitation Setting: inpatients. Intervention: HALOPERIDOL Comparison: BENZODIAZEPINE: a. FLUNITRAZEPAM | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| BENZODIAZEPINE: a. FLUNITRAZEPAM | HALOPERIDOL | |||||
| Tranquillisation or asleep ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Repeated need for tranquillisation within 24 hours ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Specific behaviours ‐ aggression 50% reduction in OAS score at 90 minutes | Low1 | RR 1.15 (0.86 to 1.55) | 28 (1 study) | ⊕⊝⊝⊝ very low2,3,4 | ||
| 650 per 1000 | 747 per 1000 (559 to 1000) | |||||
| Moderate1 | ||||||
| 800 per 1000 | 920 per 1000 (688 to 1000) | |||||
| High1 | ||||||
| 950 per 1000 | 1000 per 1000 (817 to 1000) | |||||
| Global outcome ‐ need for restraint or seclusion | See comment | See comment | Not estimable | 28 (1 study) | ⊕⊝⊝⊝ very low2,4,5,6 | No events in either group. |
| Adverse effects: specific ‐ EPS | See comment | See comment | Not estimable | 28 (1 study) | ⊕⊝⊝⊝ very low2,4,6,7 | No events in either group. |
| Economic outcome ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Moderate risk is roughly equal to that of the control group. 2 Risk of bias: rated 'serious' ‐ allocation concealment not stated, described as double‐blind, however the blinding of participants and personnel are not reported, it is not clear whether all side effects are reported, small short study. 3 Indirectness: rated 'serious' ‐ not threat or injury to self or others, inferred from OAS rating scale. 4 Publication bias: rated 'strongly suspected' ‐ small study. 5 Indirectness: rated 'serious' ‐ there is no measure for overall improvement, therefore inferred no overall improvement by need for restraints. 6 Imprecision: rated 'serious' ‐ small study. 7 Indirectness: rated 'serious' ‐ EPS can refer to a number of different symptoms rather than one specific side effect.
Summary of findings 14. HALOPERIDOL compared to BENZODIAZEPINE: b. LORAZEPAM for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared with BENZODIAZEPINE: b. LORAZEPAM for psychosis‐induced aggression or agitation | ||||||
| Patient or population: patients with psychosis‐induced aggression or agitation Setting: inpatients; emergency room. Intervention: HALOPERIDOL Comparison: BENZODIAZEPINE: b. LORAZEPAM | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| BENZODIAZEPINE: b. LORAZEPAM | HALOPERIDOL | |||||
| Tranquillisation or asleep not asleep at 60 minutes | Low1 | RR 1.05 (0.76 to 1.44) | 60 (1 study) | ⊕⊝⊝⊝ very low2,3,4,5 | ||
| 500 per 1000 | 525 per 1000 (380 to 720) | |||||
| Moderate1 | ||||||
| 700 per 1000 | 735 per 1000 (532 to 1000) | |||||
| High1 | ||||||
| 900 per 1000 | 945 per 1000 (684 to 1000) | |||||
| Repeated need for rapid tranquillisation more than 1 injection | Low1 | RR 1.14 (0.91 to 1.43) | 66 (1 study) | ⊕⊝⊝⊝ very low4,5,6 | ||
| 500 per 1000 | 570 per 1000 (455 to 715) | |||||
| Moderate1 | ||||||
| 750 per 1000 | 855 per 1000 (683 to 1000) | |||||
| High1 | ||||||
| 900 per 1000 | 1000 per 1000 (819 to 1000) | |||||
| Specific behaviour ‐ threat or injury to self or others within 24 hours ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Global outcome: no overall improvement ‐ at 60 minutes | Low1 | RR 1.64 (0.54 to 5.03) | 44 (1 study) | ⊕⊝⊝⊝ very low2,4 | ||
| 50 per 1000 | 82 per 1000 (27 to 252) | |||||
| Moderate1 | ||||||
| 150 per 1000 | 246 per 1000 (81 to 755) | |||||
| High1 | ||||||
| 100 per 1000 | 164 per 1000 (54 to 503) | |||||
| Adverse effects: specific ‐ dystonia (only reported if occurred in ≥9% during 24 hours) | Low1 | RR 3.54 (0.42 to 30.03) | 66 (1 study) | ⊕⊝⊝⊝ very low4,5,6 | ||
| 10 per 1000 | 35 per 1000 (4 to 300) | |||||
| Moderate1 | ||||||
| 30 per 1000 | 106 per 1000 (13 to 901) | |||||
| High1 | ||||||
| 50 per 1000 | 177 per 1000 (21 to 1000) | |||||
| Adverse effects: specific ‐ hypertonia/rigidity (only reported if occurred in ≥9%) during 24 hours | Low7 | RR 6.22 (0.33 to 115.91) | 66 (1 study) | ⊕⊝⊝⊝ very low4,5,6 | ||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate7 | ||||||
| 50 per 1000 | 311 per 1000 (17 to 1000) | |||||
| High7 | ||||||
| 100 per 1000 | 622 per 1000 (33 to 1000) | |||||
| Economic outcome ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Moderate risk is roughly equal to that of the trial control group. 2 Risk of bias: rated 'very serious' ‐ not explicitly described as randomised, allocation concealment not stated, incomplete outcome data, sponsored by drug company. 3 Indirectness: rated 'serious' ‐ no measure for tranquillisation or asleep at 30 minutes, therefore had to use 60 minutes. 4 Imprecision: rated 'serious' ‐ small study. 5 Publication bias: rated 'strongly suspected' ‐ small study, sponsored by drug company. 6 Risk of bias: rated 'serious' ‐ allocation concealment is not stated, incomplete outcome data, selective reporting, sponsored by drug company. 7 Low risk is roughly equal to that of the trial control group.
Summary of findings 15. HALOPERIDOL compared to BENZODIAZEPINE: c. MIDAZOLAM for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared with BENZODIAZEPINE: c. MIDAZOLAM for psychosis‐induced aggression or agitation | ||||||
| Patient or population: patients with psychosis‐induced aggression or agitation Setting: emergency room. Intervention: HALOPERIDOL Comparison: BENZODIAZEPINE: c. MIDAZOLAM | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| BENZODIAZEPINE: c. MIDAZOLAM | HALOPERIDOL | |||||
| Tranquillisation or asleep ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Repeated need for rapid tranquillisation ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Specific behaviour ‐ threat or injury to self or others ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Global outcome Need for rescue drug | Low1 | RR 1.14 (0.46 to 2.87) | 84 (1 study) | ⊕⊕⊝⊝ low2,3 | ||
| 50 per 1000 | 57 per 1000 (23 to 143) | |||||
| Moderate1 | ||||||
| 150 per 1000 | 171 per 1000 (69 to 430) | |||||
| High1 | ||||||
| 250 per 1000 | 285 per 1000 (115 to 717) | |||||
| Adverse effects ‐ general ‐ one or more drug‐related adverse effect | Low4 | RR 5.00 (0.25 to 101.11) | 84 (1 study) | ⊕⊕⊝⊝ low3,5 | ||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate4 | ||||||
| 50 per 1000 | 250 per 1000 (12 to 1000) | |||||
| High4 | ||||||
| 100 per 1000 | 500 per 1000 (25 to 1000) | |||||
| Economic outcome ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Medium risk is roughly equal to that of the trial control group. 2 Indirectness: rated 'serious' ‐ no measure for overall improvement, no overall improvement inferred from need for rescue drug. 3 Imprecision: rated 'serious' ‐ 95% confidence interval is wide. 4 Low risk is roughly equal to that of the trial control group. 5 Indirectness: rated 'serious' ‐ no serious adverse effect, therefore used one or more adverse effects.
Summary of findings 16. HALOPERIDOL compared to COMBINATIONS: a. HALOPERIDOL + LEVOMEPROMAZINE for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared with 1. COMBINATIONS ‐ HALOPERIDOL + LEVOMEPROMAZINE for psychosis‐induced aggression or agitation | ||||||
| Patient or population: patients with psychosis‐induced aggression or agitation Setting: inpatients. Intervention: HALOPERIDOL Comparison: COMBINATIONS: a. HALOPERIDOL + LEVOMEPROMAZINE | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| COMBINATIONS: a. HALOPERIDOL + LEVOMEPROMAZINE | HALOPERIDOL | |||||
| Tranquillisation or asleep ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Repeated need for tranquillisation ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Specific behaviours ‐ threat or injury to self or others ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Global outcome: general ‐ no overall improvement | Low1 | RR 8.18 (0.5 to 133.66) | 19 (1 study) | ⊕⊝⊝⊝ very low2,3,4 | ||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate1 | ||||||
| 200 per 1000 | 1000 per 1000 (100 to 1000) | |||||
| High1 | ||||||
| 400 per 1000 | 1000 per 1000 (200 to 1000) | |||||
| Adverse effect: any serious, specific ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Economic outcome ‐ not measured | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Low risk is roughly equal to that of the trial control group. 2 Risk of bias: rated 'very serious' ‐ method of randomisation not described, allocation concealment not stated, open trial, no reporting of the presence or absence of adverse effects in the results, small study. 3 Imprecision: rated 'very serious' ‐ very small study (n = 19), 95% confidence interval is wide. 4 Publication bias: rated 'strongly suspected' ‐ small study.
Summary of findings 17. HALOPERIDOL compared to COMBINATIONS: b. HALOPERIDOL + LORAZEPAM for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared with COMBINATIONS: b. HALOPERIDOL + LORAZEPAM for psychosis‐induced aggression or agitation | ||||||
| Patient or population: patients with psychosis‐induced aggression or agitation Setting: inpatients; emergency room. Intervention: HALOPERIDOL Comparison: COMBINATIONS: b. HALOPERIDOL + LORAZEPAM | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| COMBINATIONS: b. HALOPERIDOL + LORAZEPAM | HALOPERIDOL | |||||
| Tranquillisation or asleep ‐ not asleep by 3 hours | Low1 | RR 1.83 (1.11 to 3.02) | 67 (1 study) | ⊕⊝⊝⊝ very low2,3,4,5 | ||
| 200 per 1000 | 366 per 1000 (222 to 604) | |||||
| Moderate1 | ||||||
| 400 per 1000 | 732 per 1000 (444 to 1000) | |||||
| High1 | ||||||
| 600 per 1000 | 1000 per 1000 (666 to 1000) | |||||
| Repeated need for rapid tranquillisation ‐ needing additional injection during 24 hours | Low1 | RR 1.05 (0.87 to 1.27) | 67 (1 study) | ⊕⊝⊝⊝ very low2,4,5 | ||
| 750 per 1000 | 788 per 1000 (653 to 952) | |||||
| Moderate1 | ||||||
| 850 per 1000 | 892 per 1000 (740 to 1000) | |||||
| High1 | ||||||
| 950 per 1000 | 997 per 1000 (827 to 1000) | |||||
| Specific behaviour ‐ threat or injury to self or others ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | |
| Global outcome: no overall improvement ‐ at 30 minutes | Low1 | RR 2.67 (1.25 to 5.68) | 45 (1 study) | ⊕⊝⊝⊝ very low4,6 | ||
| 50 per 1000 | 134 per 1000 (62 to 284) | |||||
| Moderate1 | ||||||
| 250 per 1000 | 668 per 1000 (312 to 1000) | |||||
| High1 | ||||||
| 450 per 1000 | 1000 per 1000 (562 to 1000) | |||||
| Adverse effects: Specific ‐ dystonia (only reported if occurred in ≥9%) during 24 hours | Low7 | RR 8.25 (0.46 to 147.45) | 67 (1 study) | ⊕⊝⊝⊝ very low2,4,5,8 | ||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate7 | ||||||
| 50 per 1000 | 412 per 1000 (23 to 1000) | |||||
| High7 | ||||||
| 100 per 1000 | 825 per 1000 (46 to 1000) | |||||
| Adverse effects: Specific ‐ hypertonia/rigidity (only reported if occurred in ≥9%) during 24 hours | Low1 | RR 2.74 (0.30 to 25.05) | 67 (1 study) | ⊕⊝⊝⊝ very low2,4,5,8 | ||
| 10 per 1000 | 27 per 1000 (3 to 250) | |||||
| Moderate1 | ||||||
| 30 per 1000 | 82 per 1000 (9 to 751) | |||||
| High1 | ||||||
| 50 per 1000 | 137 per 1000 (15 to 1000) | |||||
| Economic outcome ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Moderate risk is roughly equal to that of the control group. 2 Risk of bias: rated 'serious' ‐ allocation concealment not stated, 2 people excluded from the efficacy analysis following randomisation, adverse effects only reported if they occurred in ≥9% of participants, sponsored by drug company. 3 Indirectness: rated 'serious' ‐ there are no data for 30 minutes, only 3 hours. 4 Imprecision: rated 'serious' ‐ relatively small number of participants in each treatment arm. 5 Publication bias: rated 'strongly suspected' ‐ sponsored by drug company. 6 Risk of bias: rated 'very serious' ‐ method of randomisation not described, allocation concealment not described, open trial, no reporting regarding the presence or absence of adverse effects, duration of the study is not reported. 7 Low risk is roughly equal to that of the control group. 8 Indirectness: rated 'severe' ‐ not reported as 'severe' in this study.
Summary of findings 18. HALOPERIDOL compared to COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared to COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM for psychosis‐induced aggression or agitation | ||||||
| Patient or population: psychosis‐induced aggression or agitation Setting: emergency room. Intervention: HALOPERIDOL Comparison: COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM | Risk with HALOPERIDOL | |||||
| Tranquillisation or asleep ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| Repeated need for tranquillisation assessed with: need for additional drugs for tranquilisation by 12 hours | Study population | RR 0.88 (0.69 to 1.13) | 60 (1 RCT) | ⊕⊝⊝⊝ very low1 2 | ||
| 867 per 1.000 | 763 per 1.000 (598 to 979) | |||||
| Specific behaviour ‐ agitation assessed with: average endpoint scores at 12 hours on OASS | MD 11.20 lower (12.24 lower to 10.16 lower) | ‐ | 60 (1 RCT) | ⊕⊝⊝⊝ very low1 2 | ||
| Global outcome assessed with: additional restraint, seclusion or special observation during 12 hours | Low | RR 0.29 (0.13 to 0.61) | 60 (1 RCT) | ⊕⊝⊝⊝ very low1 2 | ||
| 350 per 1.000 | 102 per 1.000 (46 to 214) | |||||
| Moderate | ||||||
| 700 per 1.000 | 203 per 1.000 (91 to 427) | |||||
| High | ||||||
| 950 per 1.000 | 275 per 1.000 (124 to 580) | |||||
| Adverse effects assessed with: EPS by 12 hours | Study population | RR 1.67 (0.44 to 6.36) | 60 (1 RCT) | ⊕⊝⊝⊝ very low1 2 | ||
| 100 per 1.000 | 167 per 1.000 (44 to 636) | |||||
| Economic outcome ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated 'very serious' ‐ method of allocation not stated, patients instructed to not reveal their allocation although the trial is described as double blind, selective reporting, small study.
2 Imprecision: rated 'very serious' ‐ small study, 95% CI cross the no effect line.
Summary of findings 19. HALOPERIDOL compared to COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared to COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE for psychosis‐induced aggression or agitation | ||||||
| Patient or population: psychosis‐induced aggression or agitation Setting: emergency room. Intervention: HALOPERIDOL Comparison: COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE | Risk with HALOPERIDOL | |||||
| Tranquillisation or asleep assessed with: Not tranquil or asleep at 20 minutes | Study population | RR 1.60 (1.18 to 2.16) | 316 (1 RCT) | ⊕⊕⊕⊝ moderate1 | ||
| 281 per 1.000 | 450 per 1.000 (332 to 608) | |||||
| Repeated need for tranquillisation assessed with: need for additional drugs for tranquillisation by 2 hours | Low | RR 0.78 (0.43 to 1.41) | 376 (2 RCTs) | ⊕⊝⊝⊝ very low1 3 4 | ||
| 50 per 1.000 | 39 per 1.000 (22 to 71) | |||||
| Moderate | ||||||
| 120 per 1.000 | 94 per 1.000 (52 to 169) | |||||
| High | ||||||
| 450 per 1.000 | 351 per 1.000 (194 to 635) | |||||
| Specific behaviours ‐ aggression assessed with: Further aggressive episode within 24 hours | Study population | RR 1.06 (0.68 to 1.65) | 316 (1 RCT) | ⊕⊕⊕⊝ moderate1 | ||
| 194 per 1.000 | 205 per 1.000 (132 to 320) | |||||
| Global outcomes assessed with: restraints needed by 120 minutes | Study population | RR 1.21 (0.84 to 1.76) | 316 (1 RCT) | ⊕⊕⊕⊝ moderate1 5 | ||
| 238 per 1.000 | 287 per 1.000 (199 to 418) | |||||
| Adverse effects: specific ‐ acute dystonia | Low | RR 19.48 (1.14 to 331.92) | 316 (1 RCT) | ⊕⊕⊝⊝ low1 6 | ||
| 0 per 1.000 | 0 per 1.000 (0 to 0) | |||||
| Moderate | ||||||
| 50 per 1.000 | 974 per 1.000 (57 to 1.000) | |||||
| High | ||||||
| 100 per 1.000 | 1000 per 1.000 (114 to 1.000) | |||||
| Adverse effects: specific ‐ seizure | Study population | RR 2.05 (0.19 to 22.39) | 316 (1 RCT) | ⊕⊕⊝⊝ low1 6 | ||
| 6 per 1.000 | 13 per 1.000 (1 to 140) | |||||
| Economic outcome ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No trial reported this outcome. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated 'serious' ‐ open trial.
2 Moderate risk is roughly equal to that of the trial control group.
3 Inconsistency: rated 'very serious' ‐ I2 86%.
4 Publication bias: rated 'strongly suspected' ‐ visual inspection of the funnel plot.
5 Indirectness: overall improvement not reported, therefore global improvement inferred from need for restraints at by 120 minutes.
6 Imprecision: rated 'serious' ‐ 95% confidence intervals are wide.
7 Low risk is roughly equal to that of the trial control group.
Summary of findings 20. HALOPERIDOL compared to COMBINATIONS: e. HALOPERIDOL + RISPERIDONE for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared to COMBINATIONS: e. HALOPERIDOL + RISPERIDONE for psychosis‐induced aggression or agitation | ||||||
| Patient or population: psychosis‐induced aggression or agitation Setting: inpatients. Intervention: HALOPERIDOL Comparison: COMBINATIONS: e. HALOPERIDOL + RISPERIDONE | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with COMBINATIONS: e. HALOPERIDOL + RISPERIDONE | Risk with HALOPERIDOL | |||||
| Tranquillisation or asleep ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| Repeated need for tranquillisation ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| Specific behaviour ‐ agitation assessed with: average endpoint scores at 24 hours on PANSS‐EC | MD 3.82 higher (1.35 higher to 6.29 higher) | ‐ | 100 (1 RCT) | ⊕⊕⊝⊝ low1 2 | ||
|
Global outcome assessed with: no improvement at 72 hours |
Study population | RR 0.38 (0.11 to 1.33) | 100 (1 RCT) | ⊕⊕⊝⊝ low1 2 | ||
| 160 per 1.000 | 61 per 1.000 (18 to 213) | |||||
|
Adverse effects: specific assessed with akathisia during 14 days |
Study population | RR 0.88 (0.48 to 1.60) | 100 (1 RCT) | ⊕⊕⊝⊝ low1 2 | ||
| 320 per 1.000 | 282 per 1.000 (154 to 512) | |||||
| Economic outcome ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated 'serious' ‐ allocation concealment procedures not stated, blinding procedures not stated.
2 Imprecision: rated 'serious' ‐ 95% CIs are wide.
Summary of findings 21. HALOPERIDOL compared to COMBINATIONS: f. OLANZAPINE + MAGNESIUM VALPROATE for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared to COMBINATIONS: f. OLANZAPINE + MAGNESIUM VALPROATE for psychosis‐induced aggression or agitation | ||||||
| Patient or population: psychosis‐induced aggression or agitation Setting: inpatients. Intervention: HALOPERIDOL Comparison: COMBINATIONS: f. OLANZAPINE + MAGNESIUM VALPROATE | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with COMBINATIONS: f. OLANZAPINE + MAGNESIUM VALPROATE | Risk with HALOPERIDOL | |||||
| Tranquillisation or asleep ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| Repeated need for tranquillisation ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| Specific behaviour ‐ agitation assessed with: average endpoint scores at 72 hours on PANSS‐EC | MD 0.04 lower (1.33 lower to 1.25 higher) | ‐ | 64 (1 RCT) | ⊕⊝⊝⊝ very low1 2 | ||
| Global outcome assessed with: no improvement | Study population | RR 1.06 (0.38 to 2.95) | 64 (1 RCT) | ⊕⊝⊝⊝ very low1 2 | ||
| 182 per 1.000 | 193 per 1.000 (69 to 536) | |||||
| Adverse effects assessed with: one or more drug‐related adverse events | Low | RR 3.06 (1.62 to 5.79) | 64 (1 RCT) | ⊕⊝⊝⊝ very low1 2 | ||
| 100 per 1.000 | 306 per 1.000 (162 to 579) | |||||
| Moderate | ||||||
| 240 per 1.000 | 734 per 1.000 (389 to 1.000) | |||||
| High | ||||||
| 500 per 1.000 | 1000 per 1.000 (810 to 1.000) | |||||
| Economic outcome ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated 'serious' ‐ allocation concealment procedures not stated, blinding procedures not stated, small study.
2 Imprecision: rated 'very serious' ‐ small study, 95% CI cross the no effect area.
Summary of findings 22. HALOPERIDOL compared to COMBINATIONS: g. QUETIAPINE + MAGNESIUM VALPROATE for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared with COMBINATIONS: d. QUETIAPINE + MAGNESIUM VALPROATE for psychosis‐induced aggression or agitation | ||||||
| Patient or population: patients with psychosis‐induced aggression or agitation Setting: inpatients. Intervention: HALOPERIDOL Comparison: COMBINATIONS: d. QUETIAPINE + MAGNESIUM VALPROATE | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| COMBINATIONS: d. QUETIAPINE + MAGNESIUM VALPROATE | HALOPERIDOL | |||||
| Tranquillisation or asleep ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Repeated need for tranquillisation ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Specific behaviours ‐ agitation at 3 days average endpoint score (PANSS‐EC) Follow‐up: 8 weeks | The mean specific behaviours ‐ agitation at 3 days in the intervention groups was 0.02 higher (2.31 lower to 2.35 higher) | 60 (1 study) | ⊕⊝⊝⊝ very low1,2 | |||
| Global effect Specific behaviour ‐ agitation ‐ no effect based on PANSS‐EC (< 25% decrease) | Low3 | RR 1.17 (0.44 to 3.06) | 60 (1 study) | ⊕⊝⊝⊝ very low1,4 | ||
| 50 per 1000 | 58 per 1000 (22 to 153) | |||||
| Moderate3 | ||||||
| 200 per 1000 | 234 per 1000 (88 to 612) | |||||
| High3 | ||||||
| 350 per 1000 | 409 per 1000 (154 to 1000) | |||||
| Adverse effects ‐ any specific, serious ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Economic outcome ‐ not reported | See comment | See comment | Not estimable | 0 (0) | See comment | No study reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Risk of bias: rated 'very serious' ‐ method of randomisation is not reported, allocation concealment not stated, open‐label, selective reporting. 2 Indirectness: rated 'very serious' ‐ no data for aggressive outburst with 24 hours, therefore had to use scale‐derived data for agitation at 72 hours. 3 Moderate risk is roughly equal to that of the trial control group. 4 Indirectness: rated 'serious' ‐ no measure for overall improvement, therefore inferred no overall improvement from "no effect" based on the PANSS‐EC.
Summary of findings 23. HALOPERIDOL compared to COMBINATIONS: h. RISPERIDONE + CLONAZEPAM for psychosis induced aggression or agitation.
| HALOPERIDOL compared to COMBINATIONS: h. RISPERIDONE + CLONAZEPAM for psychosis induced aggression or agitation | ||||||
| Patient or population: psychosis induced aggression or agitation Setting: inpatients. Intervention: HALOPERIDOL Comparison: COMBINATIONS: h. RISPERIDONE + CLONAZEPAM | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with COMBINATIONS: h. RISPERIDONE + CLONAZEPAM | Risk with HALOPERIDOL | |||||
| Tranquillisation or asleep assessed with: not asleep at 4 hours | Study population | RR 0.82 (0.66 to 1.03) | 205 (1 RCT) | ⊕⊝⊝⊝ very low1 2 | ||
| 673 per 1.000 | 552 per 1.000 (444 to 693) | |||||
| Repeated need for tranquillisation ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| Specific behaviour ‐ agitation assessed with: average change scores at 2 hours on PANSS‐EC | MD 0.50 lower (3.8 lower to 2.8 higher) | ‐ | 108 (1 RCT) | ⊕⊝⊝⊝ very low1 2 | ||
| Global outcome assessed with: no improvement at 72 hours | Study population | RR 0.60 (0.15 to 2.38) | 100 (1 RCT) | ⊕⊕⊝⊝ low3 4 | ||
| 100 per 1.000 | 60 per 1.000 (15 to 238) | |||||
| Adverse effects assessed with: akathisia during 5 days (only reported if occurred ≥50%) | Low | RR 1.63 (1.12 to 2.38) | 205 (1 RCT) | ⊕⊝⊝⊝ very low1 5 | ||
| 150 per 1.000 | 244 per 1.000 (168 to 357) | |||||
| Moderate | ||||||
| 300 per 1.000 | 489 per 1.000 (336 to 714) | |||||
| High | ||||||
| 500 per 1.000 | 815 per 1.000 (560 to 1.000) | |||||
| Economic outcome ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated 'very serious' ‐ open study, evidence of reporting bias, sponsored trial.
2 Imprecision: rated 'serious' ‐ 95% CI cross the no effect area.
3 Risk of bias: rated 'serious' ‐ allocation concealment and blinding procedures not stated.
4 Imprecision: rated 'serious' ‐ 95% CI are wide.
5 Imprecision: rated 'serious' ‐ 95% CI are wide.
Summary of findings 24. HALOPERIDOL compared to COMBINATIONS: i. ZIPRASIDONE + CLONAZEPAM for psychosis‐induced aggression or agitation.
| HALOPERIDOL compared to COMBINATIONS: i. ZIPRASIDONE + CLONAZEPAM for psychosis‐induced aggression or agitation | ||||||
| Patient or population: psychosis‐induced aggression or agitation Setting: inpatients. Intervention: HALOPERIDOL Comparison: COMBINATIONS: i. ZIPRASIDONE + CLONAZEPAM | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with COMBINATIONS: i. ZIPRASIDONE + CLONAZEPAM | Risk with HALOPERIDOL | |||||
| Tranquillisation or asleep ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| Repeated need for tranquillisation ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| Specific behaviour ‐ agitation assessed with: average endpoint scores at 12 hours on PANSS‐EC | MD 0.28 lower (1.80 lower to 1.24 higher) | ‐ | 71 (1 RCT) | ⊕⊝⊝⊝ very low1 2 | ||
| Global outcome ‐ no improvement ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| Adverse effects ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| Economic outcome ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | No study reported this outcome. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated 'very serious' ‐ randomisation, allocation and blinding procedures not stated, evidence of attrition bias, small study.
2 Imprecision: rated 'very serious' ‐ small study, 95% CI are wide.
Background
Description of the condition
Psychosis is associated with a number of mental disorders, including schizophrenia and bipolar disorder. Symptoms of psychosis include delusions and hallucinations, which can lead some people to become confused, frightened or agitated. Often they can suffer a combination of these distressing emotions (APA 2006). Agitation is characterised by restlessness, excitability and irritability, and for some people, this can result in verbal and physical aggressive behaviour (Mohr 2005). Agitation and aggression within the psychiatric setting imposes a significant challenge to clinicians who, while attempting to make an accurate diagnosis and formulation (Schleifer 2011), have to intervene quickly in order to manage the risk that the service user may present to themselves, other service users and staff (NICE 2005, NICE 2015).
Description of the intervention
Haloperidol was developed in 1958 by the Belgian company Janssen Pharmaceutica, and with the earlier development of chlorpromazine was considered a "psychopharmacological revolution" for the treatment of schizophrenia (López‐Munoz 2009). Newer anti‐psychotic medication has been developed for the day‐to‐day management of symptoms of schizophrenia, however, haloperidol continues to be in wide use, particularly for the management of psychosis‐induced agitation. In clinical practice, where de‐escalation techniques have not been appropriate nor sufficient in managing a person’s agitation and where that person poses a threat to themselves or others, it may be necessary to use medication for rapid tranquillisation (Pratt 2008). Although there may be occasions where it is appropriate to induce sleep, the aim of rapid tranquillisation should usually be to sedate agitated or aggressive people to a point where they are still able to engage in further assessment and be responsive to communication (Parker 2010). If clinically feasible, oral administration is preferred. However, if the person refuses medication or there is no improvement after 30 to 60 minutes, haloperidol in single doses of 5 mg, administered intramuscularly (IM) is a commonly used treatment option (Pereira 2007).
Cardiac problems and extrapyramidal side effects (EPS) such as dystonia and akathisia are associated with conventional antipsychotics such as haloperidol. Experiencing distressing adverse effects may act as a barrier for future engagement with services or treatment (Pratt 2008). The UK’s NICE guidelines recommend that when using haloperidol IM, an antimuscarinic agent such as procyclidine or benzatropine should be administered to counter the potential for EPS. Subsequent to the publication of these guidelines, the manufacturers of Haldol (Janssen‐Cilag Ltd 2010) recommended that a baseline electrocardiogram (ECG) should be undertaken prior to treatment with haloperidol. In addition, they have discontinued the license for intravenous (IV) use and reduced the recommended maximum dose from 90 mg/day to 30 mg/day orally, and 18 mg/day IM (Parker 2010). When Haldol is prescribed outside of these recommendations, this would be considered ‘off label’ and may increase the prescriber’s professional responsibility and potential liability (Pratt 2008). Despite these adverse effects, haloperidol alone or combined with a benzodiazepine was recommended by the NICE guidelines (NICE 2005) and by the American Psychiatric Association (APA 2006), and haloperidol remains on the World Health Organization's Essential Medicine list (WHO 2011, WHO 2015). With the last updated NICE guidelines, recommendation changed indicating either the use of haloperidol combined with promethazine or lorazepam alone for rapid tranquillisation (NICE 2015).
How the intervention might work
Haloperidol is mainly indicated for schizophrenia or other psychoses, mania, violent or dangerously impulsive behaviour, excitement and for short‐term adjunctive management of psychomotor agitation (BNF 2011). Haloperidol is from the butyrophenone family of antipsychotics (neuroleptics) (López‐Munoz 2009). It is thought that haloperidol prevents the occurrence of delusions and hallucinations by blocking the dopamine D2 receptors in the meso‐cortico‐limbic system. In one study, while the D2 dopamine receptor binding was high in the temporal cortex for both haloperidol and atypical antipsychotics, haloperidol induced a significantly higher binding index in the thalamus and striatum than atypical antipsychotics. It is thought that this antidopaminergic activity in the dorsolateral striatum may contribute to the extra pyramidal effects that are associated with typical antipsychotics such as haloperidol (Xiberas 2001).
Why it is important to do this review
The UK's 2005 NICE guidelines recommended that where haloperidol is administered via injection it should be combined with lorazepam when given IM. However, these guidelines also recommended, that, in exceptional circumstances, haloperidol can be given alone by IV injection (NICE 2005). Despite this recommendation, it is difficult to administer IV injections safely in restraint situations and there is evidence that haloperidol alone continues to be given via IM injection (Choudhury 2011). Surveys undertaken with consultant psychiatrists have shown that following the withdrawal of droperidol, haloperidol was the preferred alternative for rapid tranquillisation (Reid 2003) and that over 33% of respondents would be prepared to use doses above the BNF limits (Pereira 2007). Further, since the NICE guideline on the management of disturbed and violent behaviour was published, Janssen‐Cilag Ltd has withdrawn the license for IV use. With the increased prescribing of haloperidol in the UK by 16% (excluding depot injections) between 2004 and 2009 (NHS 2009), the authors felt it timely and justified to examine the evidence regarding the efficacy and safety of prescribing haloperidol alone for treating psychosis‐induced agitation and aggression. This is one of a series of linked completed and maintained reviews and other reviews that are underway (Table 25) that will create a body of evidence assessing the effectiveness of various drugs and their preferred routes of administration for both short‐ and long‐term psychosis‐induced aggression or agitation. In the UK’s 2015 NICE guidelines, either use of haloperidol combined with promethazine or lorazepam alone is now recommended, with the latter preferred when in presence of cardiovascular disease or in case a recent ECG is unavailable (NICE 2015).
1. Other relevant Cochrane reviews.
| Focus of review | Reference |
| Completed and maintained reviews | |
| 'As required' medication regimens for seriously mentally ill people in hospital | Douglas‐Hall 2015 |
| Benzodiazepines for psychosis‐induced aggression or agitation | Gillies 2013 |
| Chlorpromazine for psychosis induced aggression or agitation | Ahmed 2010 |
| Clotiapine for acute psychotic illnesses | Berk 2004 |
| Containment strategies for people with serious mental illness | Muralidharan 2006 |
| Droperidol for acute psychosis | Khokhar 2016 |
| Haloperidol plus promethazine for psychosis‐induced aggression | Huf 2016 |
| Olanzapine IM or velotab for acutely disturbed/agitated people with suspected serious mental illnesses | Belgamwar 2005 |
| Seclusion and restraint for serious mental illnesses | Sailas 2000 |
| Zuclopenthixol acetate for acute schizophrenia and similar serious mental illnesses | Jayakody 2012 |
| Reviews in the process of being completed | |
| Risperidone for psychosis induced aggression or agitation | Ahmed 2011 |
| Haloperidol for long term aggression in psychosis | Khushu 2016 |
| Loxapine inhaler for psychosis‐induced aggression | Vangala 2012 |
| Quetiapine for psychosis‐induced aggression | Wilkie 2012 |
| De‐escalation techniques for psychosis‐induced aggression | Du 2017 |
Objectives
To examine whether haloperidol alone, administered orally, intramuscularly or intravenously, is an effective treatment for psychosis‐induced aggression or agitation, wherein clinicians are required to intervene to prevent harm to self and others.
Methods
Criteria for considering studies for this review
Types of studies
All relevant randomised controlled trials. If a trial was described as 'double‐blind' but implied randomisation, we included such trials in a sensitivity analysis (see Sensitivity analysis). If their inclusion did not result in a substantive difference, they remained in the analyses. If their inclusion resulted in statistically significant differences, we did not add the data from these lower‐quality studies to the results of the better trials, but presented such data within a subcategory. We excluded quasi‐randomised studies, such as those allocating by alternate days of the week.
Types of participants
People exhibiting agitation or aggression (or both) thought to be due to psychotic illness, regardless of age or sex. We included studies that involved participants with other diagnoses, such as drug or alcohol intoxication, organic problems including dementia, learning disability and non‐psychotic mental illness, providing that the number of participants in these groups did not exceed the number of participants with psychosis.
Types of interventions
1. Rapid use of haloperidol
Alone, any dose via any route of administration; compared with rapid use of the following.
a. Placebo or no intervention
We are aware that, in a situation where haloperidol is available, the option of not giving treatment would be problematic. These are, however, valuable and pioneering studies which give us some insight to the absolute value of haloperidol in the emergency situation.
b. Other antipsychotic
Any dose via any route of administration.
c. Benzodiazepine
Any dose via any route of administration.
d. Anticonvulsant alone
Any dose via any route of administration.
e. Combination of drugs
Any dose via any route of administration.
Rapid use of the interventions is defined as administration deemed necessary to calm behaviour or prevent harm to the participant or others.
Types of outcome measures
All outcomes grouped by time: by 30 minutes, up to two hours, up to four hours, up to 24 hours and over 24 hours.
Primary outcomes
1. Tranquilisation or asleep
1.1 Not tranquil or asleep ‐ by up to 30 minutes 1.2. Repeated need for rapid tranquillisation
Secondary outcomes
1. Tranquillisation or asleep
1.1 Not tranquil ‐ over 30 minutes 1.2 Not asleep ‐ over 30 minutes 1.3 Time to tranquillisation/sleep 1.4 Time to tranquillisation 1.5 Time to sleep
2. Specific behaviours
2.1 Self‐harm, including suicide 2.2 Injury to others 2.3 Agitation 2.3.1 Another episode of agitation by 24 hours 2.3.2 No clinically important change in agitation 2.3.3 No change in agitation 2.3.4 Average endpoint in agitation score 2.3.5 Average change in agitation scores 2.4 Aggression 2.4.1 Another episode of aggression by 24 hours 2.4.2 No clinically important change in aggression 2.4.3 No change in aggression 2.4.4 Average endpoint in aggression score 2.4.5 Average change in aggression scores
3. Global outcomes
3.1 No overall improvement 3.2 Use of restraints/seclusion 3.3 Relapse ‐ as defined by each study 3.4 Recurrence of violent incidents 3.5 Needing extra visits from the doctor 3.6 Refusing oral medication 3.7 Not accepting treatment 3.8 Average endpoint score 3.9 Average change score 3.10 Average dose of drug
4. Service outcomes
4.1 Duration of hospital stay 4.2 Re‐admission 4.3 No clinically important engagement with services 4.4 No engagement with services 4.5 Average endpoint engagement score 4.6 Average change in engagement scores
5. Mental state
5.1 No clinically important change in general mental state 5.2 No change in general mental state 5.3 Average endpoint general mental state score 5.4 Average change in engagement scores
6. Adverse effects
6.1 Death 6.2 Any non‐serious general adverse effects 6.3 Any serious, specific adverse effects 6.4 Average endpoint general adverse effect score 6.5 Average change in general adverse effect scores 6.6 No clinically important change in specific adverse effects 6.7 No change in specific adverse effects 6.8 Average endpoint specific adverse effects 6.9 Average change in specific adverse effects
7. Leaving the study early
7.1 For specific reasons 7.2 For general reasons
8. Satisfaction with treatment
8.1 Recipient of treatment not satisfied with treatment 8.2 Recipient of treatment average satisfaction score 8.3 Recipient of treatment average change in satisfaction scores 8.4 Informal treatment provider not satisfied with treatment 8.5 Informal treatment providers' average satisfaction scores 8.6 Informal treatment providers' average change in satisfaction scores 8.7 Professional providers' not satisfied with treatment 8.8 Professional providers' average satisfaction score 8.9 Professional providers' average change in satisfaction scores
9. Acceptance of treatment
9.1 Not accepting treatment 9.2 Average endpoint acceptance score 9.3 Average change in acceptance scores
10. Quality of life
10.1 No clinically important change in quality of life 10.2 Not any change in quality of life 10.3 Average endpoint quality of life score 10.4 Average change in quality of life score 10.5 No clinically important change in specific aspects of quality of life 10.6 No change in specific aspects of quality of life 10.7 Average endpoint specific aspects of quality of life 10.8 Average change in specific aspects of quality of life
11. Economic outcomes
11.1 Direct costs 11.2 Indirect costs
Outcomes used for 'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2008) and used the GRADE profiler (GRADE PRO) to import data from RevMan 5 (Review Manager) to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes that we rated as important to patient‐care and decision making. We selected the following main outcomes for inclusion in the 'Summary of findings' tables.
Tranquillisation or asleep ‐ not tranquil or asleep by 30 minutes.
Tranquillisation or asleep ‐ repeated need for rapid tranquillisation ‐ within 24 hours.
Specific behaviours ‐ threat or injury to others/self.
Global outcomes ‐ overall improvement.
Adverse effects ‐ any serious, specific adverse effects.
Economic outcomes.
Search methods for identification of studies
Electronic searches
Cochrane Schizophrenia Group’s Study‐Based Register of Trials
On May 26, 2016, the information specialist searched the register using the following search strategy which has been developed based on literature review and consulting with the authors of the review:
(*Agitation* OR *Aggression*) in Healthcare Condition AND (*Haloperidol*) in Intervention of STUDY
In such study‐based register, searching the major concept retrieves all the synonym and relevant studies because all the studies have already been organised based on their interventions and linked to the relevant topics.
This register is compiled by systematic searches of major resources (including AMED, BIOSIS CINAHL, Embase, MEDLINE, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates, handsearches, grey literature, and conference proceedings (see Group’s Module). There is no language, date, document type, or publication status limitations for inclusion of records into the register.
For previous searches, please see Appendix 1.
Searching other resources
1. Reference searching
We inspected references of all included studies for further relevant studies.
2. Personal contact
We attempted to contact the first author of each included study for information regarding unpublished trials.
Data collection and analysis
Selection of studies
Review author MJP inspected citations from the searches and identified relevant abstracts. A random 20% sample was independently re‐inspected by CEA to ensure reliability. Where disputes arose, we acquired the full report for more detailed scrutiny. We obtained full reports of the abstracts that met the review criteria and these were inspected by MJP. Again, a random 20% of reports were re‐inspected by CEA, in order to ensure reliable selection. Where it was not possible to resolve disagreement by discussion, we attempted to contact the authors of the study for clarification.
Review authors EGO and XL screened results from 2016 search to identify relevant abstracts. We obtained and inspected full reports of the abstracts meeting the review criteria.
Data extraction and management
1. Extraction
Review author MJP (original search), EGO (2016 search) extracted data from all included studies. In addition, to ensure reliability, CEA independently extracted data from a random sample of these studies, comprising 10% of the total. Again, any disagreement was discussed, decisions documented and, if necessary, we contacted the authors of studies for clarification. If there were any remaining problems, HJ helped clarify issues and these final decisions were documented. We extracted data presented only in graphs and figures whenever possible, but we only included the data if two review authors independently had the same result. We attempted to contact authors through an open‐ended request in order to obtain missing information or for clarification whenever necessary. If studies were multi‐centre, where possible, we extracted data relevant to each component centre separately.
2. Management
2.1 Forms
We extracted data onto standard, simple forms.
2.2 Scale‐derived data
We included continuous data from rating scales only if: a. the psychometric properties of the measuring instrument have been described in a peer‐reviewed journal (Marshall 2000); and b. the measuring instrument has not been written or modified by one of the trialists for that particular trial. Ideally, the measuring instrument should either be i. a self‐report or ii. completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly, therefore, in Description of studies, we recorded whether or not this was the case.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand, calculation of change needs two assessments (baseline and endpoint), which can be difficult in unstable and difficult to measure conditions such as schizophrenia. We decided primarily to use endpoint data, and only use change data if the former were not available. We combined endpoint and change data in the analysis and we used mean differences (MD) rather than standardised mean differences (SMD) throughout (Higgins 2011).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we aimed to apply the following standards to all data before inclusion: a) standard deviations (SDs) and means are reported in the paper or obtainable from the authors; b) when a scale starts from the finite number zero, the SD, when multiplied by two, is less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution, (Altman 1996); c) if a scale started from a positive value (such as the Positive and Negative Syndrome Scale (PANSS), (Kay 1986)), which can have values from 30 to 210), the calculation described above was modified to take the scale starting point into account. In these cases skew is present if 2 SD > (S‐S min), where S is the mean score and 'S min' is the minimum score. Endpoint scores on scales often have a finite start and end point and these rules can be applied. Skewed data pose less of a problem when looking at means if the sample size is large (> 200) and we entered these into the syntheses. We present skewed data from studies of less than 200 participants in ‘Additional tables’ rather than in analyses. When continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not. These were entered into analyses.
2.5 Common measure
To facilitate comparison between trials, if required, we converted variables that can be reported in different metrics, such as time to tranquillisation (e.g. minutes, hours) to a common metric (e.g. minutes).
2.6 Conversion of continuous to binary
Where possible, efforts were made to convert outcome measures to dichotomous data. This was done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the PANSS scale (Kay 1986), this could be considered as a clinically significant response (Leucht 2005; Leucht 2005a). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for haloperidol for psychosis‐induced agitation. Where keeping to this made it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'Not improved'), we reported data where the left of the line indicates an unfavourable outcome. This was noted in the relevant graphs.
Assessment of risk of bias in included studies
Review authors MJP and EGO assessed risk of bias by using the criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) to assess trial quality. This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting. HJ re‐rated 10% of included studies.
If the raters disagreed, or if either rater was in doubt, we decided the final rating by consensus, with the involvement of another member of the review group. Where inadequate details of randomisation and other characteristics of trials were provided, we contacted authors of the studies in order to obtain further information. We reported non‐concurrence in quality assessment, but if disputes arose as to which category a trial was to be allocated, again, we resolved these by discussion.
We noted the level of risk of bias in both the text of the review and in the 'Summary of findings' tables.
Measures of treatment effect
1. Binary data
For binary outcomes, we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000).
2. Continuous data
For continuous outcomes, we estimated mean difference (MD) between groups. We preferred not to calculate effect size measures (SMD). However, if scales of very considerable similarity were used, we presumed there was a small difference in measurement, and we calculated the effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra‐class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992), whereby P values are spuriously low, CIs unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
Where clustering was not accounted for in primary studies, we presented data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. We attempted to contact the first authors of studies to obtain intra‐class correlation coefficients (ICCs) for their clustered data and to adjust for this by using accepted methods (Gulliford 1999). Where clustering had been incorporated into the analysis of primary studies, we presented these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
We sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the ICC [Design effect=1+(m‐1)*ICC] (Donner 2002). If the ICC was not reported it was assumed to be 0.1 (Ukoumunne 1999).
Where cluster studies had been appropriately analysed taking into account ICCs and relevant data documented in the report, synthesis with other studies was possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, if we had included cross‐over trials, we would only have used data from the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Where a study involved more than two treatment arms, where relevant, the additional treatment arms were presented in comparisons. If data were binary these were simply added and combined within the two‐by‐two table. If data were continuous, we combined data following the formula in section 7.7.3.8 (Combining groups) of the Cochrane Handbook for Systematic Reviews of Interventions. Where the additional treatment arms were not relevant, we did not reproduce these data.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). We chose that, for any particular outcome, should more than 50% of data be unaccounted for, we did not reproduce these data or use them within analyses, (except for the outcome 'leaving the study early'). Where more than 50% of those in one arm of a study were lost, but the total loss was less than 50%, we marked such data with (*) to indicate that such a result may well be prone to bias.
2. Binary
In the case where attrition for a binary outcome was between 0% and 50% and where these data were not clearly described, we presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat (ITT) analysis). Those leaving the study early were all assumed to have the same rates of negative outcome as those who completed, with the exception of the outcome of death and adverse effects. For these outcomes, the rate of those who stayed in the study ‐ in that particular arm of the trial ‐ were used for those who did not. We undertook a sensitivity analysis to test how prone the primary outcomes were to change when 'completer' data only were compared with the ITT analysis using the above assumptions.
3. Continuous
3.1 Attrition
In the case where attrition for a continuous outcome was between 0% and 50% and completer‐only data were reported, we reproduced these.
3.2 Standard deviations (SDs)
If SDs were not reported, we first tried to obtain the missing values from the authors. If not available, where there were missing measures of variance for continuous data, but an exact standard error (SE) and confidence intervals (CIs) were available for group means, and either a P value or 't' value were available for differences in mean, we calculated them according to the rules described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011): When only the SE was reported, SDs were calculated by the formula SD = SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) present detailed formulae for estimating SDs from P values, t or F values, CIs, ranges or other statistics. If these formulae did not apply, we calculated the SDs according to a validated imputation method which is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study’s outcome and thus to lose information. Nevertheless, we examined the validity of the imputations in a sensitivity analysis excluding imputed values.
3.3 Last observation carried forward (LOCF)
We anticipated that in some studies the method of LOCF would be employed within the study report. As with all methods of imputation to deal with missing data, LOCF introduces uncertainty about the reliability of the results (Leucht 2007). Therefore, where LOCF data were used in the trial, if less than 50% of the data had been assumed, we reproduced these data and indicated that they were the product of LOCF assumptions.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We inspected all studies for clearly outlying people or situations which we had not predicted would arise. When such situations or participant groups arose, these were discussed.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We inspected all studies for clearly outlying methods which we had not predicted would arise. When such methodological outliers arose, these were discussed.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
We investigated heterogeneity between studies by considering the I2 statistic alongside the Chi2 P value. The I2 statistic provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I2 depends on i. magnitude and direction of effects and ii. strength of evidence for heterogeneity (e.g. P value from Chi2 test, or a CI for I2). We interpreted an I2 estimate greater than or equal to around 50% accompanied by a statistically significant Chi2 statistic, as evidence of substantial levels of heterogeneity (Section 9.5.2 ‐ Higgins 2011). When substantial levels of heterogeneity were found in the primary outcome, we explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
1. Protocol versus full study
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results. These are described in section 10.1 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We tried to locate protocols of included randomised trials. If the protocol was available, outcomes in the protocol and in the published report were compared. If the protocol was not available, outcomes listed in the methods section of the trial report were compared with actual reported results.
2. Funnel plot
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are again described in Section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar sizes. In other cases, where funnel plots were possible, we sought statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. There is, however, a disadvantage to the random‐effects model. It puts added weight onto small studies which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We chose the fixed‐effect model for all analyses. The reader is, however, able to choose to inspect the data using the random‐effects model.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
We did not anticipate any subgroup analyses.
2. Investigation of heterogeneity
If inconsistency was high, we reported it. First, we investigated whether data were entered correctly. Second, if data were correct, we visually inspected the graph and successively removed outlying studies to see if homogeneity was restored. For this review, we decided that should this occur with data contributing to the summary finding of no more than around 10% of the total weighting, we would present the data. If not, we would not pool data but would discuss the issues. We know of no supporting research for this 10% cut‐off but are investigating the use of prediction intervals as an alternative to this unsatisfactory state.
When unanticipated clinical or methodological heterogeneity were obvious, we stated hypotheses regarding these for future reviews or versions of this review. We did not anticipate undertaking analyses relating to these.
Sensitivity analysis
1. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way as to imply randomisation. For the primary outcomes we included these studies, and if there was no substantive difference when the implied randomised studies were added to those with better description of randomisation, then we used all data from these studies.
2. Assumptions for lost binary data
Where assumptions had to be made regarding people lost to follow‐up (see Dealing with missing data), we compared the findings of the primary outcomes when we used our assumption compared with 'completer' data only. Where there was a substantial difference, we reported results and discussed them, but continued to employ our assumption.
Where assumptions had to be made regarding missing SDs data (see Dealing with missing data), we compared the findings on primary outcomes when we used our assumption compared with 'completer' data only. We undertook a sensitivity analysis to test how prone results were to change when 'completer' data only were compared with the imputed data using the above assumption. If there was a substantial difference, we reported results and discussed them, but continued to employ our assumption.
3. Risk of bias
We analysed the effects of excluding trials that were judged to be at high risk of bias across one or more of the domains of randomisation (implied as randomised with no further details available) allocation concealment, blinding and outcome reporting for the meta‐analysis of the primary outcome. If the exclusion of trials at high risk of bias did not substantially alter the direction of effect or the precision of the effect estimates, then we included data from these trials in the analysis.
4. Imputed values
We also undertook a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect in cluster‐randomised trials.
If substantial differences were noted in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we did not pool data from the excluded trials with the other trials contributing to the outcome, but presented them separately.
5. Fixed and random effects
All data was be synthesised using a fixed‐effect model, however, we also synthesised data for the primary outcome using a random‐effects model to evaluate whether this altered the significance of the result.
Results
Description of studies
For substantive descriptions of studies please see the Characteristics of included studies, Characteristics of excluded studies, Characteristics of studies awaiting classification and Characteristics of ongoing studies.
Results of the search
The electronic search (June 2011) yielded 665 citations of potentially eligible studies. After checking titles and abstracts, 209 full‐text papers were obtained for a second assessment. These publications related to 100 different studies (29 included studies, 55 excluded, 15 awaiting classification and one ongoing). The reference lists of the 29 studies to be included were checked, through which we identified another three potential included studies. Again, full‐text papers were obtained and on closer inspection, Reschke 1974 and Fitzgerald 1969 were included and Goldstein 1966 excluded because, although participants, were experiencing psychosis, they were not specifically aggressive or agitated. The reference lists of related Cochrane systematic reviews were also searched, from which we identified Bailine 1987 for inclusion (2011 search, Figure 1).
1.
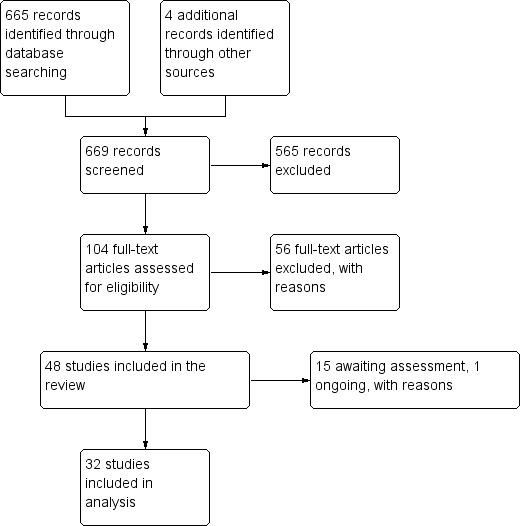
Study flow diagram.
The update search run in 2016 yielded 34 citations, of which nine new studies were included (Figure 2).
2.
Study flow diagram.
Included studies
Details of the 41 studies included in the review are provided in the Characteristics of included studies table.
1. Length of studies
The duration of the studies ranged from two hours (Calver 2015; Dorevitch 1999) to eight weeks (Higashima 2004). The majority were ≦ 72 hours, apart from Garza‐Trevino 1989 and Nobay 2004 which did not report the duration of the study.
2. Participants
2.1 Clinical state
Participants in the studies presented with acute exacerbation of psychotic symptoms. Some studies focused on people whose psychosis had primarily triggered agitation (Battaglia 2002; Breier 2002; Bristol Myers 2004a; Bristol‐Myers 2005b; Brook 2000; Eli Lilly 2004; Fang 2012; Garza‐Trevino 1989; Guo 2007, Higashima 2004; Kinon 2001d; Liang 2013; Lim 2010; Liu 2012; Resnick 1984; Shu 2010; Walther 2014; Yin 2012; Zhou 2014), and other trials included people with both agitation and aggression (Bailine 1987; Baldacara 2011; Battaglia 1997; Calver 2015; Currier 2004; Dorevitch 1999; Fitzgerald 1969; Foster 1997; Fruensgaard 1977; Huf 2007; Kewala 1984; Li 2006; Man 1973; Nobay 2004; Paprocki 1977; Reschke 1974; Ritter 1972; Salzman 1991; Shi 2010; Stotsky 1977; Taymeeyapradit 2002; Tuason 1986). All participants were so disturbed that clinicians felt that they required rapid tranquillisation.
2.2 Diagnosis
Over 80% of participants had a diagnosis of schizophrenia. Other diagnoses included schizoaffective disorder, schizophreniform disorder, bipolar disorder, and 'psychosis not otherwise specified'. A small minority of participants had a diagnosis of organic mental disorder and substance‐induced psychosis, but in no trial was this subgroup the majority or the focus of the study.
2.3. Exclusions
Where exclusion criteria were reported, these often included pregnant or lactating women, people with serious medical illnesses, people who had used certain medications (e.g. anti‐psychotics/anti‐depressants) within a specified period prior to enrolment, people with a known allergy/hypersensitivity to the study drugs and people with alcohol or drug dependency.
2.4 Age
In the 23 studies that reported an age range, participants ranged from 18 to 73 years. Nine studies reported a mean age (mostly around 30), with Garza‐Trevino 1989 being the only study not to report the age of participants.
2.5 Sex
There were 2236 male participants and 1310 participants were women; the sex of 1387 people was not reported.
3. Study size
The study sizes varied with the smallest study having only 19 participants (Higashima 2004) and the largest randomising 448 people (Bristol Myers 2004a).
4. Setting
Where reported, the majority of participants presented at psychiatric emergency departments and were newly admitted inpatients. Interestingly, one study included outpatients (Stotsky 1977).
5. Interventions
It is unsurprising that this old and well‐established drug has been compared with many other interventions. For all but one study (Kinon 2001d ‐ see below), haloperidol and the other drugs were administered by the intramuscular (IM) route. What was surprising, however, was how poorly reported the true doses were. Dose ranges were commonly reported but to stipulate their mean or median was the exception rather than the rule. In several comparisons we have had to make educated guesses and the likely end doses.
5.1 Versus placebo
Trials comparing haloperidol with placebo largely used around 10 mg of the drug given by IM injection.
5.2 Versus other antipsychotics
The aripiprazole comparison involved around 10 mg of haloperidol compared with up to 45 mg of the control drug. The older trials involved in the chlorpromazine comparison used larger doses of haloperidol (10 mg to 20 mg with one trial allowing the option of up to 100 mg/day) with between 100 mg and up to 500 mg of chlorpromazine. In the droperidol trial 10 mg of haloperidol was compared with around 15 mg of droperidol. Although one loxapine versus haloperidol trial did allow for up to 100 mg of haloperidol, more commonly used doses across trials were in the 10 mg to 20 mg range compared with 50 mg to 250 mg of loxapine. The olanzapine dose tended to be around 10 mg to 15 mg, and in these trials haloperidol was given around the 10 mg dose. The Kinon 2001d trial (olanzapine versus haloperidol) is notable for it being the only one that used oral drugs rather then IM route of administration. The perphenazine comparison used doses of both drugs in the mid 20’s mg given by IM injection. The quetiapine trial compared a maximum of 12 mg of haloperidol with up to 750 mg of quetiapine. In the risperidone comparison, haloperidol was used at between about 5 mg and 15 mg and the risperidone between 2 mg and 6 mg oral liquid and oral velotab. The thiothixene trial used higher doses of both drugs (˜30 mg) given by IM injection. The ziprasidone studies used rather high dose haloperidol (20 mg up to 40 mg) and compared this with around 40 mg of ziprasidone oral and IM. Haloperidol dose in the zuclopenthixol acetate trial was also high (˜30 mg) compared with up to ˜100 mg IM of the longer‐acting compound).
5.3 Versus benzodiazepine
The one trial comparing IM flunitrazepam with Im haloperidol used about 1 mg of the benzodiazepine and 5 mg of the antipsychotic. The lorazepam trials, using a combination of oral and IM administration employed doses of around 4 mg to 6 mg of the drug. The comparison dose was around 10 mg of haloperidol. The single midazolam study compared 5 mg with 5 mg (both IM).
5.4 Versus combinations
The haloperidol versus haloperidol plus levomepromazine study were oral treatments, but we remain unclear as to the doses. These seem to be around 5 mg for each compound. For the haloperidol versus haloperidol plus lorazepam comparison, the antipsychotic tended to be given at around the 5 mg level for both groups and the benzodiazepine at 4 mg each. In the haloperidol versus haloperidol plus midazolam study, the antipsychotic at 5 mg was compared with the adjunction of 15 mg of midazolam. In the haloperidol versus haloperidol plus promethazine trial, all treatments were IM. The haloperidol was up to 10 mg and the promethazine up to 50 mg. The haloperidol versus haloperidol plus risperidone study compared 10 mg to 20 mg of haloperidol with 3 mg to 6 mg of risperidone. For the haloperidol versus olanzapine plus magnesium valproate comparison, the first treatment tended to be given at around 15 mg and the second one at respectively 10 mg plus 1.25 mg. In the haloperidol versus quetiapine (IM) plus magnesium valproate (oral) comparison, the dose of haloperidol was up to 30 mg/day and the quetiapine's 800 mg/day. The additional magnesium valproate was dosed at about 0.5 g to 1 g/orally/day. In the haloperidol versus risperidone plus clonazepam comparison, haloperidol was administered at 10 mg to 20, mg whilst risperidone plus clonazepam doses ranged from, respectively, 2 mg to 6 mg plus 0 mg to 8 mg. Finally, the haloperidol versus ziprasidone plus clonazepam study compared up to 30 mg of haloperidol versus up to 160 mg of ziprasidone and up to 6 mg of clonazepam.
6. Outcomes
For the majority of trials, binary data were available with respect to repeated need for tranquillisation, not tranquil or asleep, need for adjunctive medication, leaving the study early and adverse effects. The majority of trials employed continuous scales to measure other clinical outcomes. However, the results of these scale‐derived findings were often presented in graphs, as P values alone, or as means without standard deviations. Much data were unusable for synthesis. Requests for further information from the majority of authors remain unanswered. Dr Dubin (Dubin 1985) kindly answered our request, however was unable to provide us with the standard deviation or standard error. The various rating scales, from which we were able to obtain usable data are listed below.
6.1 Specific behaviour ‐ Agitation
a. Agitated Behavior Scale (ABS/CABS)
The ABS (Corrigan 1989) is a 14‐item scale used for measuring agitation levels. The ABS was originally designed to monitor agitated behaviour in the recovery period after stroke and there are three factor‐based sub‐scores: i. aggression, ii. disinhibition; and iii. lability. High scores indicated higher levels of aggression.
b. Agitation‐Calmness Evaluation Scale (ACES)
The ACES is a single‐item rating scale developed by Eli Lilly and company (Breier 2002). On this scale, 1 = marked agitation, 4 = normal, 9 = unable to be aroused.
c. Positive and Negative Syndrome Scale Excited Component (PANSS‐EC)
The PANSS‐EC is a five‐item scale (excitement, tension, hostility, uncooperativeness, and poor impulse control. The items are rated from one (not present) to seven (extremely severe). Scores range from five to 35, with mean scores ≥ 20 indicating agitation. A high score indicates high levels of agitation (Montoya 2011).
d. Overt Agitation Severity scale (OASS)
The OASS (Yudofsky 1997) is a 12‐item scale used for identification and measurement of agitated behaviours, rated with a intensity score (ranging from one to four); the items are rated in terms of frequency from zero (not present) to four (always present). Total OASS score is the sum of all the Severity Scores (intensity*frequency).
6.2 Specific behaviour ‐ Aggression
a. Overt Aggression Scale (OAS)
The OAS (Yudofsky 1986) is a 16‐item rating scale which aims to measure the intensity of verbal and physical aggression. Clinicians are required to comment on the duration of the aggressive incident as well as the intervention required to control it. High scores are indicative of higher levels of aggression.
6.3 Global state
a. Clinical Global Impression (CGI)
The CGl (Guy 1976) is not a diagnostic tool but rather, enables clinicians to quantify the severity of symptoms of any mental health problem at one point in time. Clinicians are then able to use this to track whether there has been any improvement or worsening of symptoms over time. A seven‐point rating scale is used with high scores indicating increased severity or less recovery.
b. Clinical Global Impression ‐ Severity (CGI‐S)
The CGI‐S (Guy 1976) requires clinicians to consider the severity of a person's symptoms in relation to the clinicians past experience of people with the same diagnosis. Clinicians then have to give a rating from one = normal to seven = extremely ill. High scores indicate increased severity.
c. Clinical Global Impression ‐ Improvement (CGI‐I)
The CGI‐I (Guy 1976) enables clinicians to assess whether a person's symptoms have improved or worsened following an intervention. Based on the clinicians judgement, a rating on a seven‐point scale is given from one (= very much improved) to seven (= very much worse). Therefore, low scores indicate greater improvement.
6.4 Mental state
a. Brief Psychiatric Rating Scale (BPRS)
The Brief Psychiatric Rating Scale was originally developed by Overall and Gorham (Overall 1962) as a 14‐item scale to measure the severity of a range of psychiatric symptoms, including psychosis. This rating scale items evolved over time and now consists of 24 items which can be rated on a seven‐point scale from ‘not present’ to ‘extremely severe’. A high score would suggest poor mental health. It is not clear for the majority of the studies included in this review, which version of the BPRS was used.
b. Positive and Negative Syndrome Scale (PANSS)
The PANSS was developed and published by Kay, Flszbein and Opler (Kay 1987). The PANSS is designed as a brief interview, whereby the severity of 30 symptoms of schizophrenia can be assessed on a scale of one to seven. A high score would indicate more severe symptoms. The PANSS can be divided into separate sub‐scales by focusing on the statements relating to positive symptoms (e.g. hallucinations), negative symptoms (e.g. social withdrawal) or general psychopathology (e.g. anxiety and uncooperativeness).
6.5 Adverse effects
a. Barnes Akathisia Scale (BAS)
The BAS scale was developed by Barnes 1989 and includes items which aim to rate both the observable symptoms which characterise akathisia such as restless movements and also the person's subjective experience, including any distress. The items are rated from zero = normal to three = severe. There is also an item for rating global severity from zero = absent to five = severe. A high score indicates high levels of akathisia.
b. Simpson‐Angus Scale (SAS)
The SAS (Simpson 1970) is a 10‐item scale which measures drug‐induced parkinsonism (extrapyramidal side effects). Each item is scored from zero to four. A high score would indicate increased levels of parkinsonism.
c. Treatment Emergent Symptom Scale (TESS)
The TESS (Guy 1976) is a six‐item scale which is used to assess the occurrence and intensity of treatment‐related adverse effects. A high score indicates worse symptoms. This scale was not reported as a continuous score but studies did glean binary data using this measure (e.g. Li 2006).
7. Missing outcomes
Not one of the studies evaluated satisfaction with care, acceptance of treatment, quality of life or economic outcomes.
8. Funders
Fifteen of the 41 included studies received sponsorship from pharmaceutical companies.
Excluded studies
In total we excluded 66 studies.
Of these, 20 studies had to be excluded based on their method of allocation. NCT00189995 2005 was not conducted because ethical approval was not granted and Daniel 2000, Jones 2001 and Mendelowitz 2004 were literature reviews. Chen 2004, Chen 2008, Fan 2012; Huang 2004, Jiang 2009; Li 2007, Liang 2003; Pan 2005, Pei 2009; Wang 2004 and Wang 2005 were all quasi‐randomised whereby randomisation was based on methods such as allocating to group by hospital number. It is possible that these studies were randomised if allocation of hospital numbers were somehow, designated by a procedure involving chance alone. We were, however, not reassured by the text of the trials, thus, we have excluded this quasi‐randomised group. Currier 2000, Freeman 2009, Kinon 2003, Mantua 2006 and Pedros 2004 used research methods other than a randomised controlled trial, for example a naturalistic observational study or cohort study.
We had to exclude 34 studies on the basis of the characteristics of participants. Singh 1981 and Zapletalek 1986 were not explicitly described as randomised, however, they were reported to be double‐blind and thus remained included on the basis of allocation. They were eventually excluded because although participants were suspected to have psychosis, these people were not overtly agitated or aggressive requiring rapid tranquillisation. We excluded a group of clearly randomised trials for the same reason (Addington 1996; Allen 2007; Beasley 1998; Brook 1998; Chouinard 1991; Colonna 1998; Crandall 2005; Daniel 2003; Daniel 2004; Goldstein 1966; Kane 2000; Kinon 2001a; Mandel 2008; McEvoy 1994; NCT00631722 2008; Pathiraja 1995; Puech 1998; Smith 1974; Teja 1975; Ungvari 1982; Veser 2006). Participants included in Harvey 2004 had mild symptoms of schizophrenia and, again, were not overtly aggressive. We identified two studies where participants were children. We excluded these because either, participants did not display aggression (Faretra 1970), or because less than half of participants were suspected to have psychosis (Asadollahi 2015; Campbell 1982).
In Citrome 2001, Krakowski 2006, Krakowski 2008 and Lewis 2006, participants had a diagnosis of schizophrenia and displayed aggressive behaviour. However, the studies had to be excluded because participants displayed long‐term aggression, rather than aggression within an emergency situation requiring rapid tranquillisation. Participants in Romeo 2009 and Vaisanen 1981 were people with intellectual disability who presented with challenging behaviour rather than psychosis. Thomas 1992 focused on rapid tranquillisation in the emergency setting, but the majority of participants were intoxicated and therefore did not necessarily have psychosis‐induced aggression.
For 10 studies the intervention under investigation did not meet our inclusion criteria. Alexander 2004, Bieniek 1998, Conde 2011, TREC CG, NCT00797277 2008, Simpson 2010 and Srinath 2010 did not include a comparison group which involved giving of haloperidol alone; Vives 2015 did not include a comparison group which involved giving a drug out of haloperidol; Liu 2012b compared haloperidol with a non‐drug methodology of intervention; in Li 2005 there was no specific outcome for agitation.
Finally, two relevant studies were excluded because we found no usable data. Participants were randomised and had a diagnosis of schizophrenia with agitation/aggression. However, in Wan 2005 the scales that were used to assess outcome were not validated and results for haloperidol and chlorpromazine were not presented separately. In Wyant 1990 they used a "modified CGI" for which we could find no evidence of validation and then reported mean data graphically rather than numerically.
Risk of bias in included studies
Although each 'Summary of findings' table describes bias by outcome, an overview is reported here and a graphical impression is seen in Figure 3.
3.
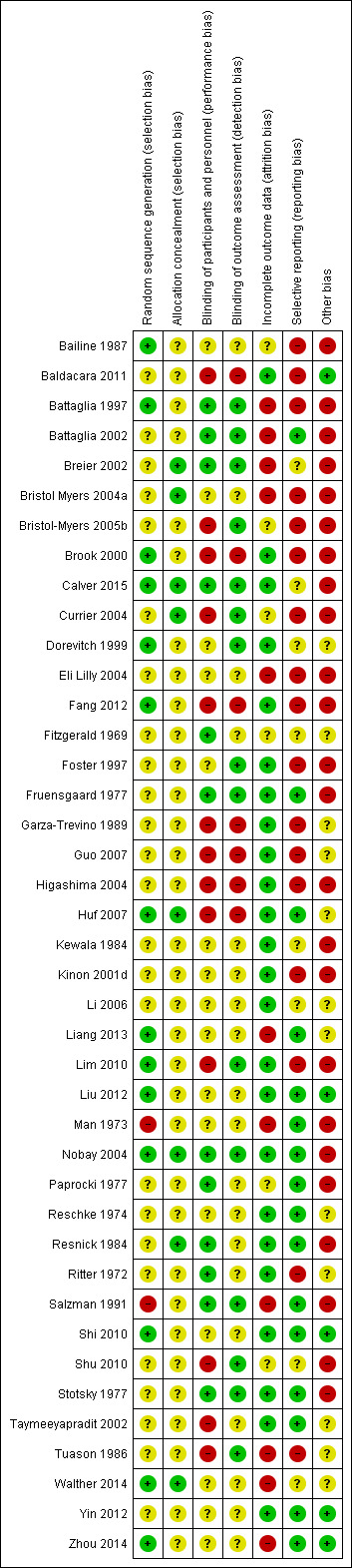
'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Allocation
All but two of the included studies were said to be randomised. Man 1973 and Salzman 1991 were not explicitly described as randomised, however they were described as double‐blind and we felt randomisation was implied. Few of the randomised studies reported information regarding the method of allocation. For the studies that did report further details, methods are described as computer‐generated randomisation (Battaglia 1997; Brook 2000; Huf 2007; Nobay 2004), random number sequence (Bailine 1987; Dorevitch 1999; Liang 2013; Liu 2012; Shi 2010; Zhou 2014), pre‐defined randomisation code (Lim 2010) and randomisation in blocks (Stotsky 1977; Walther 2014).
Blinding
Twenty‐two studies described that they were 'double‐blind' but the variations on how this was carried out were almost as many (Table 26); some studies were reported to be 'single‐blind' (Baldacara 2011; Yin 2012), whilst in five studies it was not stated (Liang 2013; Liu 2012; Shi 2010; Walther 2014; Zhou 2014). Trialists were more or less concerned with issues of performance and detection bias and most 'double‐blind' studies are likely not to have been in reality. Open studies (Brook 2000; Fang 2012; Garza‐Trevino 1989; Guo 2007; Higashima 2004; Huf 2007) recognised that blinded giving of drugs added the possibility that findings may be problematic to apply in routine care. These studies also attempted to use outcomes that were less prone to rater bias.
2. Variations on blinding.
| Description | Study |
| Explicitly referred to how participants, study personnel and raters were blinded + tested blinding. | Stotsky 1977 |
| Explicitly referred to how participants, study personnel and raters were blinded. | Breier 2002, Calver 2015, Nobay 2004 |
| Report that drugs had identical appearance ‐ no further details regarding rater blinding. | Fitzgerald 1969, Fruensgaard 1977, Paprocki 1977, Resnick 1984, Ritter 1972 |
| Explicitly state that rater was blinded ‐ no reference to participant blinding. Not all study personnel could have been blinded because person preparing drug (not blinded), often administered drug. | Bristol‐Myers 2005b |
| Study personnel and raters blinded, not clear if attempts made to blind participants. | Battaglia 1997, Battaglia 2002 |
| Raters blinded ‐ not clear if study personnel and participants were blinded. | Dorevitch 1999, Foster 1997, Salzman 1991 |
| "Double blind" ‐ no further information regarding blinding of participants, study personnel and raters. | Eli Lilly 2004, Kewala 1984, Kinon 2001d, Man 1973 |
| Described as double‐blind, but during oral phase haloperidol described as being in capsule form, and aripiprazole in tablet form, therefore unclear whether drugs had identical appearance. Not explicit if raters were blinded. | Bristol Myers 2004a |
| Described as double‐blind during IM phase ‐ open during oral phase ‐ no further information given regarding blinding. | Reschke 1974 |
| Described as "modified double", whereby staff administering drugs were not blinded, but raters were ‐ not clear if participants were blinded. | Tuason 1986 |
| Single‐blind studies ‐ "open label, rater blind". | Baldacara 2011, Currier 2004, Lim 2010, Shu 2010, Yin 2012 |
| Reported to be single‐blind, but unclear who was blind. | Li 2006 |
| Open‐label and rater blinding for some outcomes (BPRS). | Taymeeyapradit 2002 |
| Rater blinding for assessment of BPRS, but unclear regarding rater blinding for other outcomes and whether participants and study personnel were blind. | Bailine 1987 |
| Open studies | Brook 2000, Fang 2012, Garza‐Trevino 1989, Guo 2007, Higashima 2004, Huf 2007 |
| Not stated | Liang 2013, Liu 2012, Shi 2010, Walther 2014, Zhou 2014 |
BPRS ‐ Brief Psychiatric Rating Scale
Incomplete outcome data
There was no evidence of attrition bias for approximately two thirds of the studies with all participants being accounted for (Baldacara 2011; Brook 2000; Calver 2015; Dorevitch 1999; Fang 2012; Foster 1997; Fruensgaard 1977; Garza‐Trevino 1989; Guo 2007; Higashima 2004; Huf 2007; Kewala 1984; Kinon 2001d; Li 2006; Lim 2010; Liu 2012; Nobay 2004; Reschke 1974; Resnick 1984; Ritter 1972; Shi 2010; Stotsky 1977; Taymeeyapradit 2002; Yin 2012).
In the Bristol‐Myers 2005b, Currier 2004 and Shu 2010 studies, the reasons why participants left early are clearly presented in a table, but the numbers of participants completing different assessments at the same time point vary and reasons are not given. Other studies where attrition is not clearly described are Bailine 1987, Fitzgerald 1969 and Paprocki 1977.
In Battaglia 1997, two people were excluded from the efficacy analysis post randomisation; in Walther 2014 nine people were excluded due to refusal to provide post‐hoc consent and in three studies (Man 1973; Liang 2013; Zhou 2014), although reasons are given for why participants discontinued, results are only presented for those who completed and missing data have not been imputed. Reasons why participants left the study early were not reported in Battaglia 2002, Breier 2002, Bristol Myers 2004a, Eli Lilly 2004, Salzman 1991 and Tuason 1986, Also, attrition approached 50% at 24 hours in Salzman 1991, and was over 50% in Tuason 1986 at 10 days for some outcomes.
Selective reporting
Overall, there was much redundancy in reporting. It is difficult to judge whether this is bias or inadvertent but even‐handed poor reproduction of data. For continuous data, reasons for data being rendered unusable where often that the mean was presented but no variance could be extracted. Also, inexact P values were often presented alone and several studies presented their findings in graphs from which we could not estimate numbers.
Even simple issues were not reported well. For example, use of additional medication can indicate how treatments are not effective and need to be supplemented or that adverse effects are prevalent. The amount of additional benzodiazepine in the Foster 1997 study, the amount of anticholinergic and hypnotic medication in Tuason 1986, and the amount of antiparkinson drugs administered in Lim 2010 and Walther 2014 are not reported. Further, there is inconsistent reporting regarding additional medications in Brook 2000 and Tuason 1986.
In general, there was poor reporting with regard to adverse effects. In many studies it is not clear whether all side effects are reported (Breier 2002; Dorevitch 1999; Fitzgerald 1969; Garza‐Trevino 1989; Higashima 2004; Ritter 1972). Further, in other trials adverse effects are only reported where they occurred in a particular percentage of participants (Battaglia 1997; Bristol Myers 2004a; Bristol‐Myers 2005b; Brook 2000; Currier 2004; Fang 2012; Fitzgerald 1969; Kinon 2001d; Shu 2010). This further undermines methodology that is already weak to pick up adverse effect that may not be common, but could well be important.
Other potential sources of bias
That a study is small or sponsored by industry (or both) is not indicative of bias in itself. There are, however, associations between small trials tending to be positive (Easterbrook 1991), and those sponsored by industry favouring the drug for which they have the pecuniary interest (Lexchin 2003). Many studies in this review were small (Bailine 1987; Dorevitch 1999; Foster 1997; Fruensgaard 1977; Higashima 2004; Man 1973; Paprocki 1977; Resnick 1984; Stotsky 1977; Walther 2014), many were sponsored by industry (Battaglia 1997; Battaglia 2002; Breier 2002; Bristol Myers 2004a; Bristol‐Myers 2005b; Brook 2000; Currier 2004; Eli Lilly 2004; Fang 2012; Kewala 1984; Kinon 2001d; Lim 2010; Salzman 1991; Shu 2010;Stotsky 1977), and one was both (Stotsky 1977).
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9; Table 10; Table 11; Table 12; Table 13; Table 14; Table 15; Table 16; Table 17; Table 18; Table 19; Table 20; Table 21; Table 22; Table 23; Table 24
In the following section, the summary statistic for binary outcome results is reported as risk ratio (RR) and for continuous outcomes, we used mean difference (MD). In all instances 95% confidence intervals (CIs) are reported.
1. COMPARISON 1. HALOPERIDOL versus PLACEBO
Five studies (total n = 700) compared haloperidol with placebo (Battaglia 2002; Breier 2002; Bristol Myers 2004a; Bristol‐Myers 2005b; Reschke 1974).
1.1 Tranquillisation or asleep ‐ asleep
Significantly more people in the haloperidol group were asleep at two hours compared with those in the placebo group (2 RCTs, n = 220, RR 0.88, 95% CI 0.82 to 0.95, I2 = 76%, Analysis 1.1).
1.1. Analysis.
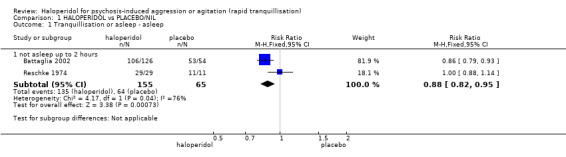
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 1 Tranquillisation or asleep ‐ asleep.
1.2 Repeated need for tranquillisation
More people in the placebo group required repeated rapid tranquillisation compared with those in the haloperidol group (4 RCTs, n = 660, RR 0.51, 95% CI 0.42 to 0.62, Analysis 1.2).
1.2. Analysis.
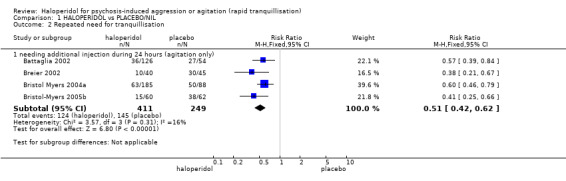
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 2 Repeated need for tranquillisation.
1.3 Specific behaviour ‐ agitation
Two studies (n = 395) found that significantly more people in the haloperidol group were reported as demonstrating at least a 40% reduction in agitation scores at two hours according to the PANSS‐EC response at two hours (RR 1.62, 95% CI 1.28 to 2.07, Analysis 1.3).
1.3. Analysis.
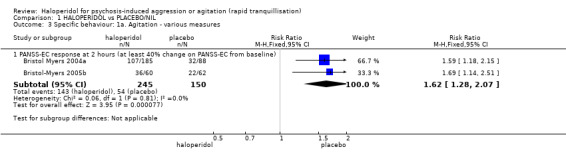
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 3 Specific behaviour: 1a. Agitation ‐ various measures.
Studies used a series of measures at different time periods. By about two hours, participants in the haloperidol group were reported to have experienced a significantly greater reduction in agitation than those in the placebo group according to the Agitated Behavior Scale (ABS) (CABS) rating scale (3 RCTs, n = 474, MD ‐4.48, 95% CI ‐5.78 to ‐3.18). In the same way, again at about two hours, people allocated to placebo had significantly smaller average change scores on the Agitation‐Calmness Evaluation Scale (ACES) compared with those in the haloperidol group (2 RCTs, n = 390, MD 0.83, 95% CI 0.39 to 1.26). One study (n = 199) examined the difference in the Positive and Negative Syndrome Scale Excited Component (PANSS‐EC) scores between the haloperidol and placebo groups in the schizophrenia subgroup, in non‐sedated people based on the ACES and in non‐sedated people. People who received haloperidol had significantly improved scores for agitation when compared with placebo in all three of these subgroups (MD ‐2.57, 95% CI ‐4.05 to ‐1.09, MD ‐2.64, 95% CI ‐3.95 to ‐1.33, and MD ‐2.97, 95% CI ‐4.27 to ‐1.67, respectively, Analysis 1.4). Again, according to average change and endpoint combined scores on the PANSS‐EC, participants in the haloperidol group had significantly reduced scores for agitation (3 RCTs, n = 514, MD ‐3.67, 95% CI ‐4.75 to ‐2.59, Analysis 1.4).
1.4. Analysis.
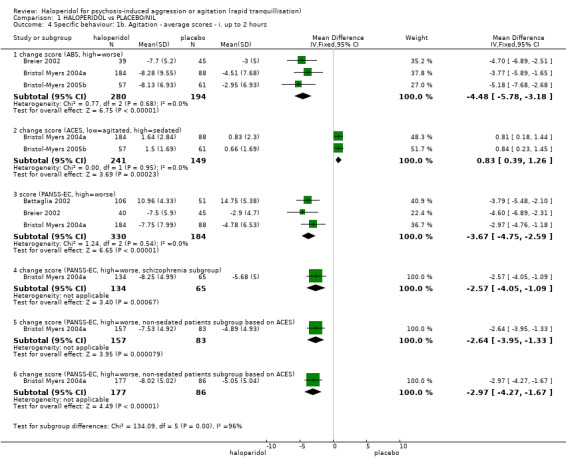
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 4 Specific behaviour: 1b. Agitation ‐ average scores ‐ i. up to 2 hours.
By 24 hours, average change on the ABS (CABS) rating scale was significantly changed in favouring haloperidol (1 RCT, n = 85, MD ‐2.40, 95% CI ‐4.13 to ‐0.67). This was not found for the PANSS‐EC average change scores (1 RCT, n = 85, MD ‐1.40, 95% CI ‐2.97 to 0.17, Analysis 1.5).
1.5. Analysis.
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 5 Specific behaviour: 1c. Agitation ‐ average scores ‐ ii. up to 24 hours.
1.4 Global outcome
Significantly more people in the placebo group exhibited 'not marked improvement' than those who received haloperidol (1 RCT, n = 40, RR 0.61, 95% CI 0.44 to 0.84, Analysis 1.6). Further, people in the placebo group were more likely to be rated as showing 'not any improvement' in global functioning than those receiving haloperidol, however this difference was not statistically significant (1 RCT, n = 40, RR 0.28, 95% CI 0.08 to 1.07, Analysis 1.6).
1.6. Analysis.
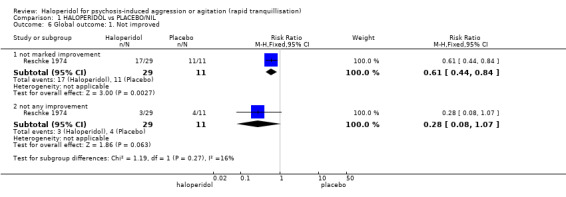
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 6 Global outcome: 1. Not improved.
Four studies found that more people in the placebo group required benzodiazepines (n = 660, RR 0.44, 95% CI 0.31 to 0.62, Analysis 1.7).
1.7. Analysis.
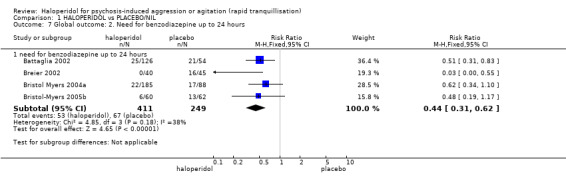
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 7 Global outcome: 2. Need for benzodiazepine up to 24 hours.
Global rating took place around two hours and 24 hours. By two hours people who received haloperidol were reported to have had significantly more symptom improvement according to their average endpoint scores on the Clinical Global Impression ‐ Improvement (CGI‐I), when compared with placebo (2 RCTs, n = 390, MD ‐0.73, 95% CI ‐0.96 to ‐0.51), and in the schizophrenia subgroup (1 RCT, n = 199, MD ‐0.64, 95% CI ‐0.98 to ‐0.30). When the same outcome was measured using average change score on the Clinical Global Impression ‐ Severity (CGI‐S), participants in the haloperidol group were rated as having significantly less severe symptoms than those in the placebo group (2 RCTs, n = 390, MD ‐0.48, 95% CI ‐0.78 to ‐0.18, Analysis 1.8).
1.8. Analysis.
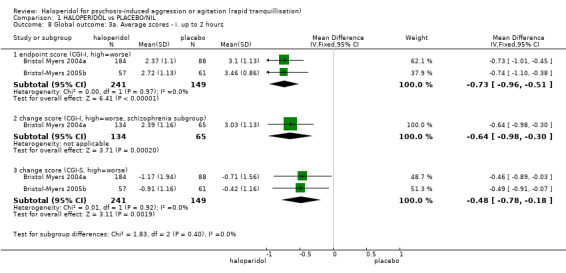
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 8 Global outcome: 3a. Average scores ‐ i. up to 2 hours.
By 24 hours, average change scores on the CGI‐I remained significantly improved (1 RCT, n = 169, MD ‐0.40, 95% CI ‐0.62 to ‐0.18). Using the CGI‐S scale to rate symptom severity at 24 hours, one trial (n = 85) found that although those in the haloperidol group were rated as less severe, the difference was not statistically significant (MD ‐0.20, 95% CI ‐0.46 to 0.06, Analysis 1.9).
1.9. Analysis.
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 9 Global outcome: 3b. Average scores ‐ ii. up to 24 hours.
1.5 Mental state
Participants in the haloperidol group had significantly improved average change scores on the Brief Psychiatric Rating Scale (BPRS) positive sub‐scale when rated at two (3 RCTs, n = 372, MD ‐1.04, 95% CI ‐1.54 to ‐0.54, Analysis 1.10) and 24 hours (2 RCTs, n = 264, MD ‐1.61, 95% CI ‐2.34 to ‐0.89,Analysis 1.11). On the BPRS Total sub‐scale, participants in the haloperidol group had significantly improved scores when rated at two (3 RCTs, n = 371, MD ‐5.69, 95% CI ‐7.38 to ‐4.00, Analysis 1.10) and 24 hours (2 RCTs, n = 264, MD ‐4.81, 95% CI ‐6.82 to ‐2.81, Analysis 1.11).
1.10. Analysis.
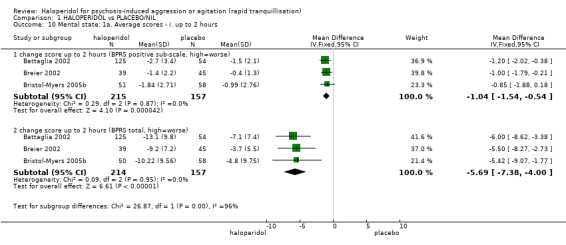
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 10 Mental state: 1a. Average scores ‐ i. up to 2 hours.
1.11. Analysis.
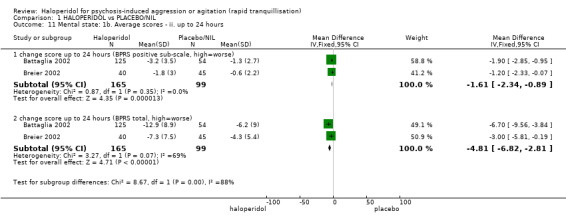
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 11 Mental state: 1b. Average scores ‐ ii. up to 24 hours.
1.6 Adverse effects
1.6.1 General
Bristol Myers 2004a (n = 273) specifically reported that no deaths occurred. Significantly more people in the haloperidol group experienced one or more adverse reactions than those in the placebo group (2 RCTs, n = 395, RR 1.64, 95% CI 1.22 to 2.20). One trial (n = 273) found that people in the haloperidol group were significantly more likely than those in the placebo group to report overall adverse effects at 72 hours (RR 1.78, 95% CI 1.23 to 2.59) and to experience increased severity of adverse effects after the second injection (RR 3.25, 95% CI 1.88 to 5.63, Analysis 1.12).
1.12. Analysis.
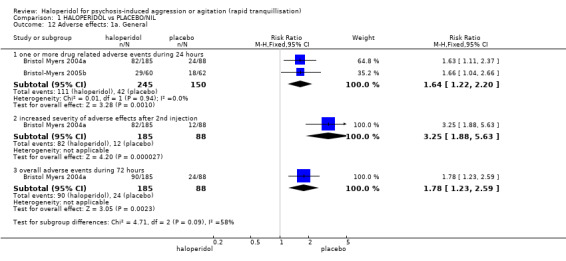
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 12 Adverse effects: 1a. General.
Bristol‐Myers 2005b reported that of all adverse effects reported, only one was rated as 'serious' ‐ this was related to a participant in the placebo group. This difference was not statistically significant (n = 122, RR 0.34, 95% CI 0.01 to 8.29, Analysis 1.13).
1.13. Analysis.
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 13 Adverse effects: 1b. General ‐ serious.
1.6.2 Specific
a. Arousal
Insomnia was reported by people in both the haloperidol and placebo groups and the difference between them was not statistically significant (1 RCT, n = 273, RR 1.31, 95% CI 0.61 to 2.82). Somnolence was reported in both the placebo and haloperidol group and the difference between the groups was not statistically significant (4 RCTs, n = 615, RR 2.28, 95% CI 0.97 to 5.36, Analysis 1.14). Significantly more people in the haloperidol group than the placebo group were reported to be "over sedated" (2 RCTs, n = 313, RR 3.36, 95% CI 1.42 to 7.99. Analysis 1.14).
1.14. Analysis.
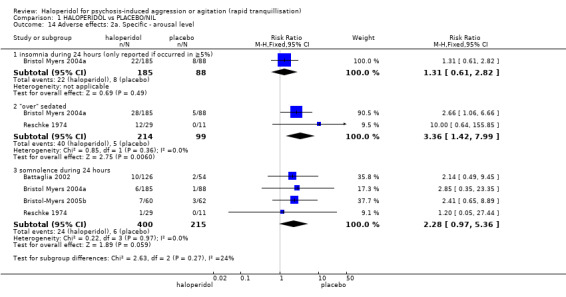
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 14 Adverse effects: 2a. Specific ‐ arousal level.
b. Cardiac
We included dizziness in 'cardiac' adverse effects (2 RCTs, n = 392, RR 1.33, 95% CI 0.48 to 3.65). More specific cardiac effects also did not suggest an effect for the active drug (QTc abnormality, 1 RCT, n = 122, RR 3.10, 95% CI 0.33 to 28.98; sinus tachycardia, 1 RCT, n = 122, RR 3.10, 95% CI 0.33 to 28.98; tachycardia, 1 RCT, n = 122, RR 1.03, 95% CI 0.07 to 16.15, Analysis 1.15). Although in favour of placebo, there were no statistically significant differences between the haloperidol and placebo groups with regards to QTc abnormality according to the average change score (2 RCTs, n = 265, MD 3.63, 95% CI ‐2.67 to 9.93), when rated at 24 hours (Analysis 1.16).
1.15. Analysis.
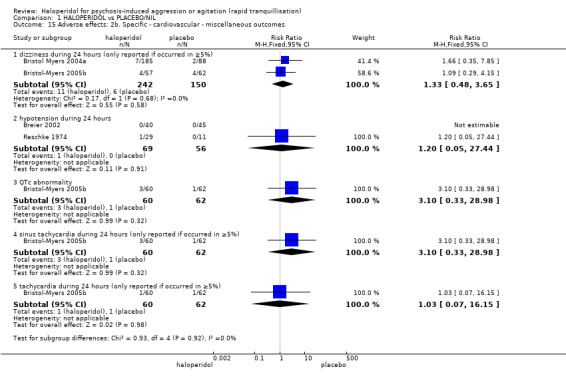
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 15 Adverse effects: 2b. Specific ‐ cardiovascular ‐ miscellaneous outcomes.
1.16. Analysis.
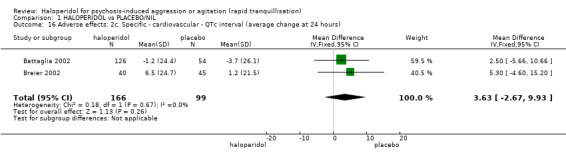
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 16 Adverse effects: 2c. Specific ‐ cardiovascular ‐ QTc interval (average change at 24 hours).
c. Movement disorders
In the haloperidol group, six people reported experiencing akathisia within 24 hours, compared with none in the placebo group. This difference between groups was not statistically significant (1 RCT, n = 122, RR 13.43, 95% CI 0.77 to 233.23). Dystonia was reported in the haloperidol group within 24 hours by six people compared with none in the placebo group; the difference between groups was not statistically significant (2 RCTs, n = 207, RR 7.49, 95% CI 0.93 to 60.21). Significantly more people in the haloperidol group required antiparkinson drugs due to Extrapyramidal Side Effects (EPS) (1 RCT, n = 180, RR 5.57, 95% CI 1.37 to 22.65) with significantly more people experiencing EPS during 24 hours (3 RCTs, n = 398, RR 6.79, 95% CI 2.19 to 21.07) , Analysis 1.17).
1.17. Analysis.
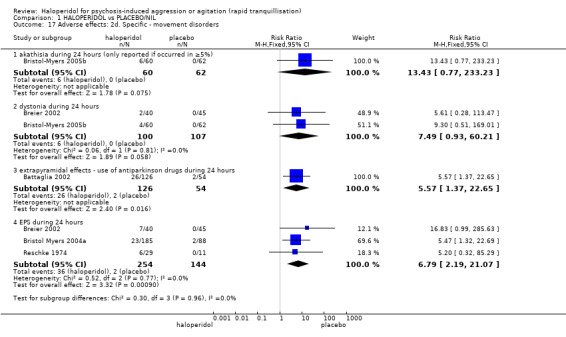
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 17 Adverse effects: 2d. Specific ‐ movement disorders.
There was no statistically significant difference between the average change scores on the Barnes Akathisia Scale (BAS) for participants in the haloperidol group when compared with the placebo group (1 RCT, n = 168, MD 0.09, 95% CI ‐0.17 to 0.35, Analysis 1.18). In one study (n = 167) participants in the haloperidol group experienced significantly higher levels of EPS compared with participants in the placebo group according to their average change scores on the Simpson‐Angus Scale (SAS) (MD 1.89, 95% CI 0.77 to 3.01, Analysis 1.18).
1.18. Analysis.
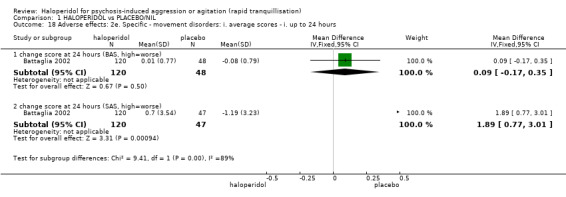
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 18 Adverse effects: 2e. Specific ‐ movement disorders: i. average scores ‐ i. up to 24 hours.
d. Miscellaneous
Two studies (total n = 395) listed a range of effects including agitation (2 RCTs, RR 0.80, 95% CI 0.29 to 2.19), headache (2 RCTs, RR 1.28, 95% CI 0.55 to 3.00), nausea (2 RCTs, RR 0.69, 95% CI 0.13 to 3.67) and vomiting (1 RCT, n = 122, RR 1.03, 95% CI 0.07 to 16.15). Although not statistically significant, two people in the placebo group reported pain at the injection site compared with none in the haloperidol group (1 RCT, n = 122, RR 0.21, 95% CI 0.01 to 4.22). This changed to significantly more people in the haloperidol group complaining of pain at the injection site following the second injection, when compared with the placebo group (44% versus 14%, 1 RCT, n = 273, RR 3.25, 95% CI 1.88 to 5.63, Analysis 1.19).
1.19. Analysis.
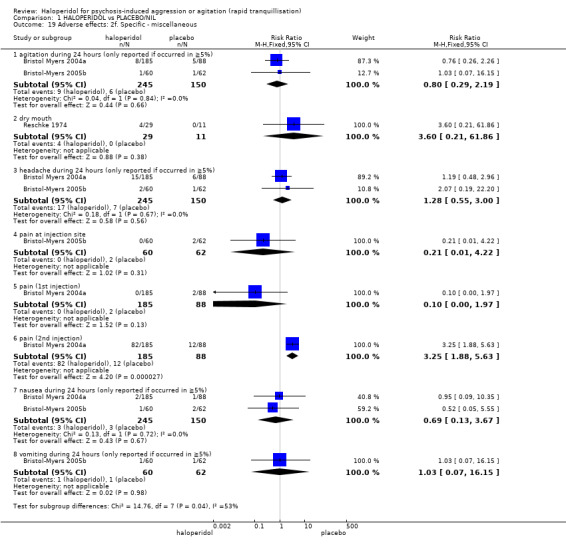
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 19 Adverse effects: 2f. Specific ‐ miscellaneous.
1.7 Leaving the study early
In three studies (total n = 435), although more people in the haloperidol group left the study early (for any reason) when compared with the placebo group, the difference was not statistically significant (RR 1.06, 95% CI 0.53 to 2.11, I2 = 83%). There were significant differences between groups for number of participants who left early due to lack of efficacy (3 RCTs, n = 435, RR 0.12, 95% CI 0.03 to 0.49), but not for reasons of consent (2 RCTs, n = 395, RR 6.20, 95% CI 0.77 to 49.98), adverse effects (3 RCTs, n = 435, RR 1.78, 95% CI 0.21 to 15.39), being asleep (1 RCT, n = 40, RR 1.20, 95% CI 0.05 to 27.44), or 'other' reasons (2 RCTs, n = 395, RR 1.81, 95% CI 0.27 to 12.07). In one study (n = 273), three people in the haloperidol group were withdrawn from the study compared with none in the placebo, although the difference was not statistically significant (RR 3.35, 95% CI 0.17 to 64.15, Analysis 1.20).
1.20. Analysis.
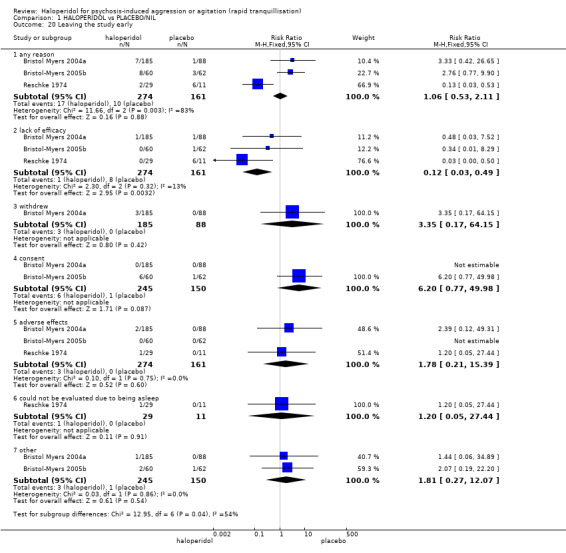
Comparison 1 HALOPERIDOL vs PLACEBO/NIL, Outcome 20 Leaving the study early.
2. COMPARISON 2. HALOPERIDOL versus OTHER ANTIPSYCHOTIC: a. ARIPIPRAZOLE
We found two studies comparing haloperidol with aripiprazole (total n = 599, Bristol Myers 2004a; Bristol‐Myers 2005b).
2.1 Repeated need for rapid tranquillisation
In both studies, people in the haloperidol group required fewer injections than those in the aripiprazole, the difference between the groups being statistically significant (2 RCTs, n = 473, RR 0.78, 95% CI 0.62 to 0.99, Analysis 2.1).
2.1. Analysis.
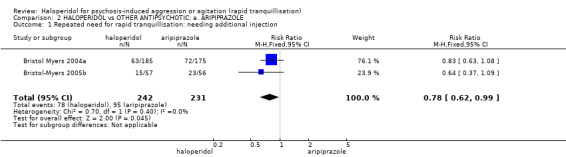
Comparison 2 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: a. ARIPIPRAZOLE, Outcome 1 Repeated need for rapid tranquillisation: needing additional injection.
2.2 Specific behaviours ‐ agitation
In both studies, more people in the aripiprazole group demonstrated at least a 40% reduction in agitation scores (PANSS‐EC) at two hours than in the haloperidol group, though the difference between the groups was not statistically significant (2 RCTs, n = 477, RR 1.07, 95% CI 0.92 to 1.26, Analysis 2.2).
2.2. Analysis.
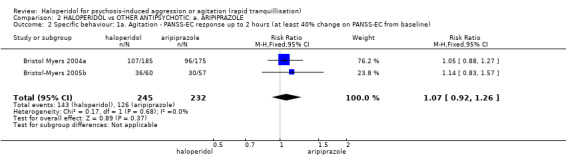
Comparison 2 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: a. ARIPIPRAZOLE, Outcome 2 Specific behaviour: 1a. Agitation ‐ PANSS‐EC response up to 2 hours (at least 40% change on PANSS‐EC from baseline).
At two hours, participants showed similar average change scores according to the Agitation‐Calmness Evaluation Scale (ACES) rating scale (2 RCTs, n = 470, MD 0.12, 95% CI ‐0.30 to 0.55). Also, at two hours, participants showed similar average change scores according to the CABS rating scale (2 RCTs, n = 470, MD ‐0.55, 95% CI ‐2.10 to 1.01). One study found that no differences between the haloperidol and aripiprazole groups in average change scores on the PANSS‐EC (n = 357, MD ‐0.48, 95% CI ‐2.12 to 1.16). This study also examined whether there were differences in various subgroups (schizophrenia subgroup, non‐sedated people based on the ACES and non‐sedated. There were no significant differences between haloperidol and aripiprazole within any of these subgroups (Analysis 2.3).
2.3. Analysis.
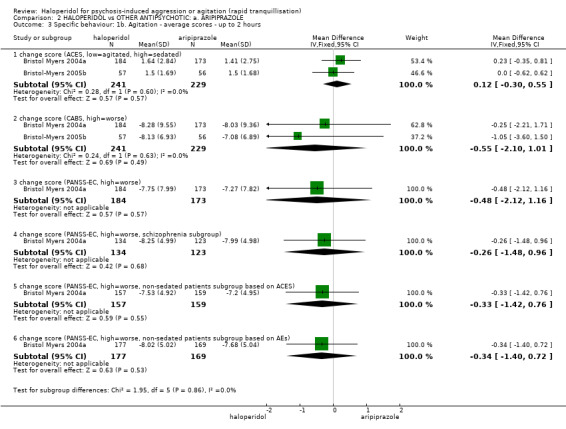
Comparison 2 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: a. ARIPIPRAZOLE, Outcome 3 Specific behaviour: 1b. Agitation ‐ average scores ‐ up to 2 hours.
2.3 Global outcomes
Neither those allocated haloperidol or aripiprazole required additional benzodiazepines (2 RCTs, n = 477, RR 1.26, 95% CI 0.74 to 2.16, Analysis 2.4).
2.4. Analysis.
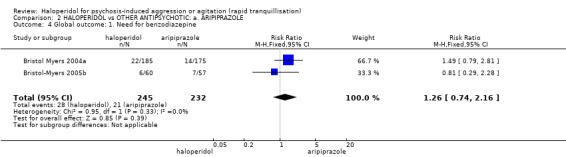
Comparison 2 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: a. ARIPIPRAZOLE, Outcome 4 Global outcome: 1. Need for benzodiazepine.
At two hours, participants in both groups showed similar levels of severity of symptoms according to their average change scores on the CGI‐S rating scale (2 RCTs, n = 470, MD 0.07, 95% CI ‐0.22 to 0.36). On another scale, at the same time, participants showed similar levels of symptom improvement according to their average endpoint scores on the CGI‐I rating scale (2 RCTs, n = 470, MD ‐0.02, 95% CI ‐0.23 to 0.19) . Again, at two hours, people in the haloperidol group (schizophrenia subgroup), were not rated as more improved on the CGI‐I than those in the aripiprazole group (1 RCT, n = 257, MD ‐0.02, 95% CI ‐0.30 to 0.26, Analysis 2.5).
2.5. Analysis.
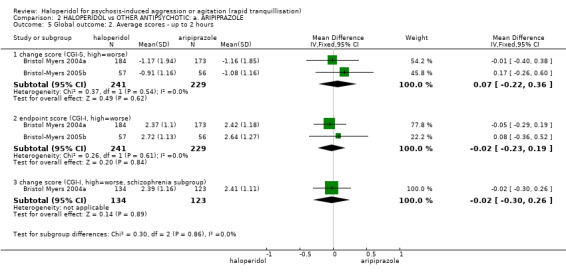
Comparison 2 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: a. ARIPIPRAZOLE, Outcome 5 Global outcome: 2. Average scores ‐ up to 2 hours.
2.4 Mental state
One study (n = 103) found no difference for average change scores for participants on the BPRS (positive sub‐scale MD ‐0.23, 95% CI ‐1.28 to 0.82 and total score n = 102, MD ‐2.03, 95% CI ‐5.76 to 1.70, Analysis 2.6).
2.6. Analysis.
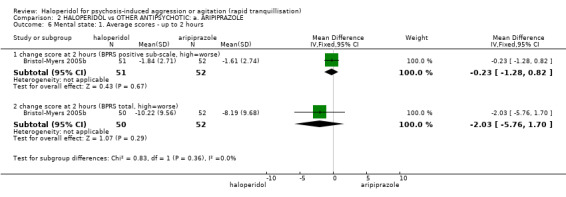
Comparison 2 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: a. ARIPIPRAZOLE, Outcome 6 Mental state: 1. Average scores ‐ up to 2 hours.
2.5 Adverse effects
2.5.1 General
Studies found no difference between groups for 'adverse effects within 24 hours' (2 RCTs, n = 477, RR 1.18, 95% CI 0.95 to 1.46). However, significantly more people in the haloperidol group suffered with increased severity of adverse effects after the second injection (1 RCT, n = 360, RR 1.34, 95% CI 1.03 to 1.74), and overall adverse effects at 72 hours (1 RCT, n = 360, RR 1.33, 95% CI 1.04 to 1.70, Analysis 2.7).
2.7. Analysis.
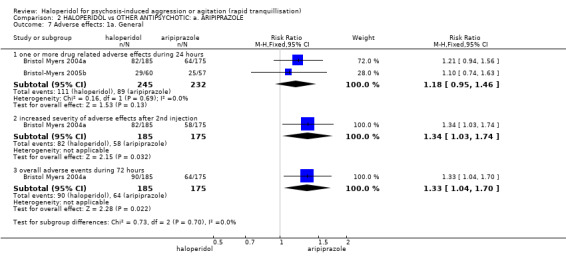
Comparison 2 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: a. ARIPIPRAZOLE, Outcome 7 Adverse effects: 1a. General.
There were no significant differences between the groups with respect to 'serious' adverse effects (Analysis 2.8). In the Bristol Myers 2004a study, it was reported that no participants in either group died during the study. Four people developed severe adverse effects; three in the aripiprazole group compared with none in the haloperidol group (2 RCTs, n = 477, RR 0.57, 95% CI 0.18 to 1.81). In the Bristol Myers 2004a study, one person in the haloperidol group developed severe dystonia and in the Bristol‐Myers 2005b study, one person in the aripiprazole group had a tonic‐clonic seizure.
2.8. Analysis.
Comparison 2 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: a. ARIPIPRAZOLE, Outcome 8 Adverse effects: 1b. General ‐ serious.
2.5.2 Specific
a. Arousal
Significantly more people in the haloperidol group complained of insomnia during the intramuscular (IM) phase (1 RCT, n = 360, RR 2.08, 95% CI 1.01 to 4.27). There were no clear differences between aripiprazole and haloperidol for insomnia during the oral phase (1 RCT, n = 360, RR 0.82, 95% CI 0.47 to 1.44), and for people being recorded as being "over sedated" during the study (1 RCT, n = 360, RR 1.66, 95% CI 0.93 to 2.95). With regards to the various ways of reporting somnolence, again, no difference was apparent (e.g. 1 RCT, n = 360, RR somnolence ‐ between zero to one days 0.71, 95% CI 0.25 to 2.00, Analysis 2.9).
2.9. Analysis.
Comparison 2 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: a. ARIPIPRAZOLE, Outcome 9 Adverse effects: 2a. Specific ‐ arousal level.
b. Cardiac
Similar numbers of people in each group experienced dizziness (2 RCTs, n = 477, RR 0.69, 95% CI 0.33 to 1.48), sinus tachycardia (1 RCT, n = 117, RR 6.66, 95% CI 0.35 to 126.06) and tachycardia (1 RCT, n = 117, RR 0.24, 95% CI 0.03 to 2.06, Analysis 2.10).
2.10. Analysis.
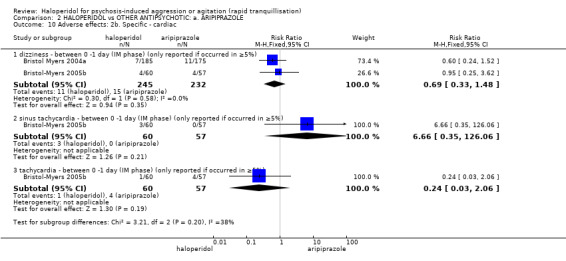
Comparison 2 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: a. ARIPIPRAZOLE, Outcome 10 Adverse effects: 2b. Specific ‐ cardiac.
c. Gastrointestinal problems
Participants in the haloperidol groups complained of significantly more dyspepsia (1 RCT, n = 360, RR 10.41, 95% CI 1.36 to 79.76), and similar problems with diarrhoea (1 RCT, n = 360, RR 0.95, 95% CI 0.28 to 3.21), nausea (1 RCT, n = 360, RR 0.14, 95% CI 0.02 to 1.09), and vomiting (1 RCT, n = 360, RR 0.38, 95% CI 0.07 to 1.92) between three and seven days (Analysis 2.11). .
2.11. Analysis.
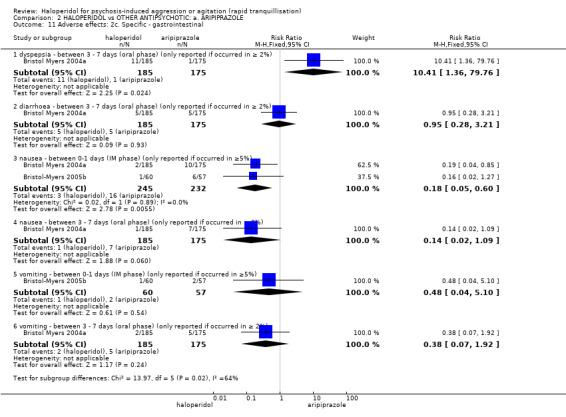
Comparison 2 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: a. ARIPIPRAZOLE, Outcome 11 Adverse effects: 2c. Specific ‐ gastrointestinal.
d. Movement disorder
These all tended to be more common in the group allocated to haloperidol (e.g. 2 RCTs, n = 477, RR akathisia 2.85, 95% CI 0.94 to 8.61). More people in the haloperidol group were reported to have experienced dystonia (2 RCTs, n = 477, RR 6.63, 95% CI 1.52 to 28.86). Significantly more people in the haloperidol group experienced EPS during both the IM (1 RCT, n = 360, RR 9.46, 95% CI 1.22 to 73.13) and oral phase (1 RCT, n = 360, RR 7.09, 95% CI 1.65 to 30.58), and required antiparkinson medication during the oral phase (1 RCT, n = 360, RR 4.94, 95% CI 2.50 to 9.78, Analysis 2.12).
2.12. Analysis.
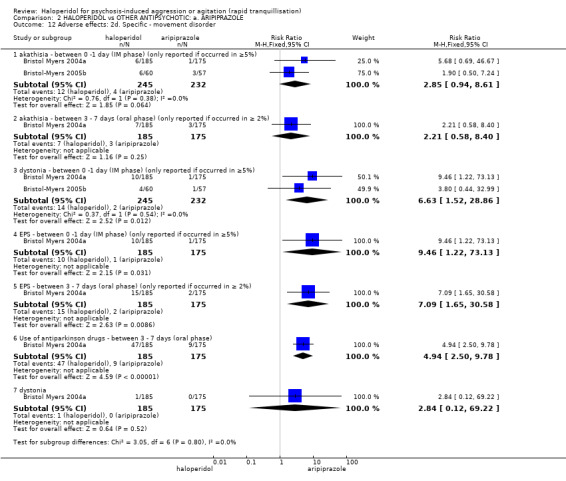
Comparison 2 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: a. ARIPIPRAZOLE, Outcome 12 Adverse effects: 2d. Specific ‐ movement disorder.
e. Miscellaneous
In both studies, participants in both the haloperidol and aripiprazole groups were reported to have agitation (2 RCTs, n = 477, RR 1.22, 95% CI 0.46 to 3.22), and headache (2 RCTs, n = 477, RR 0.85, 95% CI 0.45 to 1.59) between days zero and one. Although not statistically significant, more people in the aripiprazole group reported pain at the injection site following the first injection (1 RCT, n = 360, RR 0.32, 95% CI 0.06 to 1.54, Analysis 2.13).
2.13. Analysis.
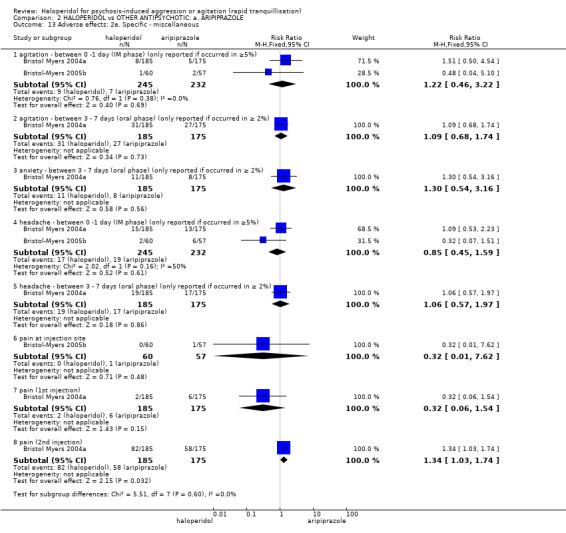
Comparison 2 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: a. ARIPIPRAZOLE, Outcome 13 Adverse effects: 2e. Specific ‐ miscellaneous.
2.6 Leaving the study early
In both studies, more people left the study early in the haloperidol group than in the aripiprazole group, however the difference between groups was not statistically significant (2 RCTs, n = 480, RR 2.07, 95% CI 0.86 to 4.98). When reasons were given these data were also equivocal (e.g. 2 RCTs, n = 480, RR leaving because of adverse effects 0.97, 95% CI 0.17 to 5.59, Analysis 2.14).
2.14. Analysis.
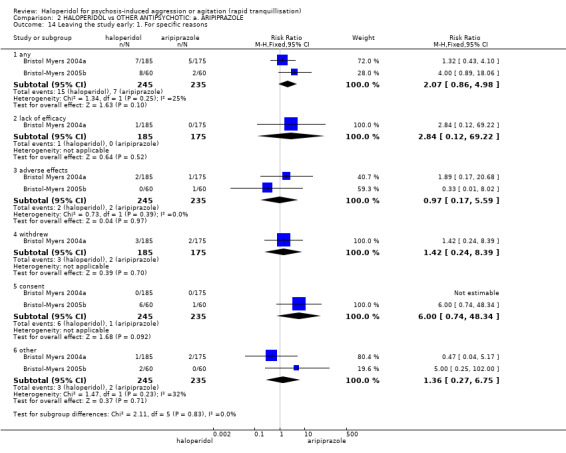
Comparison 2 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: a. ARIPIPRAZOLE, Outcome 14 Leaving the study early: 1. For specific reasons.
3. COMPARISON 3. HALOPERIDOL versus OTHER ANTIPSYCHOTIC: b. CHLORPROMAZINE
3.1 Tranquillisation or asleep ‐ asleep
One small study (n = 39) reported that more people allocated to chlorpromazine did experience sleep by two hours (RR 1.93, 95% CI 1.04 to 3.60, Analysis 3.1).
3.1. Analysis.
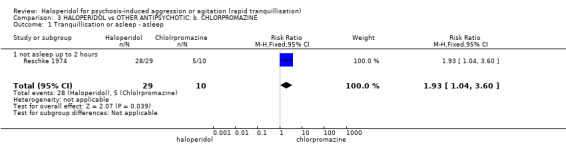
Comparison 3 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: b. CHLORPROMAZINE, Outcome 1 Tranquillisation or asleep ‐ asleep.
3.2 Repeated need for rapid tranquillisation
In the Man 1973 study, all participants in the haloperidol group and all but one participant in the chlorpromazine study required more than one injection, with several in both groups requiring more than three (1 RCT, n = 30, 1.50, 95% CI 0.53 to 4.26, Analysis 3.2).
3.2. Analysis.
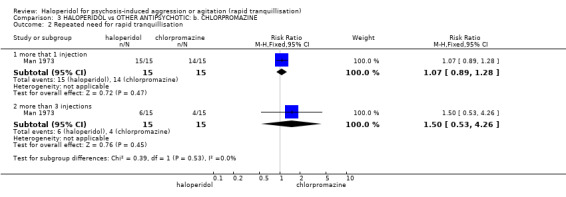
Comparison 3 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: b. CHLORPROMAZINE, Outcome 2 Repeated need for rapid tranquillisation.
3.3 Global outcome ‐ not marked improvement
For the outcome of "not marked improvement" there were no clear differences between groups (2 RCTs, n = 89, RR 0.79, 95% CI 0.61 to 1.02). More people in the chlorpromazine group were rated as showing "not any improvement" (2 RCTs, n = 89, RR 0.15, 95% CI 0.05 to 0.49, Analysis 3.3).
3.3. Analysis.
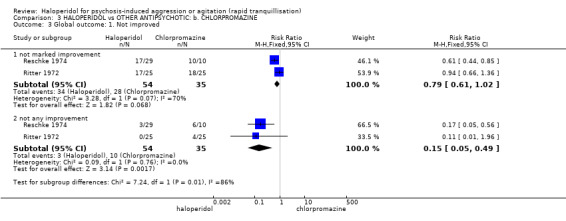
Comparison 3 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: b. CHLORPROMAZINE, Outcome 3 Global outcome: 1. Not improved.
3.4 Adverse effects ‐ specific
Reporting of adverse effects was sporadic. One person developed status epilepticus (1 RCT, n = 30, RR 0.33, 95% CI 0.01 to 7.58), and two people had hypotension (3 RCTs, n = 69, RR 0.51, 95% CI 0.10 to 2.60). In the Man 1973 study (n = 30), there were no reported incidences of EPS or irritation at the injection site in either group.
One person in the chlorpromazine group had an elevated SGPT (serum glutamic pyruvic transaminase) level (1 RCT, n = 30, RR 0.33, 95% CI 0.01 to 7.81) and another in the haloperidol group developed mild leucopenia (1 RCT, n = 50, RR 3.00, 95% CI 0.13 to 70.30, Analysis 3.4).
3.4. Analysis.
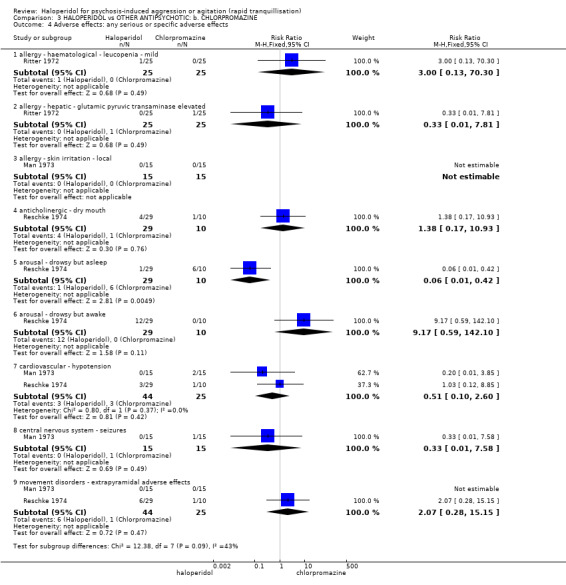
Comparison 3 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: b. CHLORPROMAZINE, Outcome 4 Adverse effects: any serious or specific adverse effects.
3.5 Leaving the study early ‐ for any reason
Few people left these studies early (5%) with little, but significant, difference between groups (4 RCTs, n = 153, RR 0.21, 95% CI 0.07 to 0.71, Analysis 3.5 ).
3.5. Analysis.
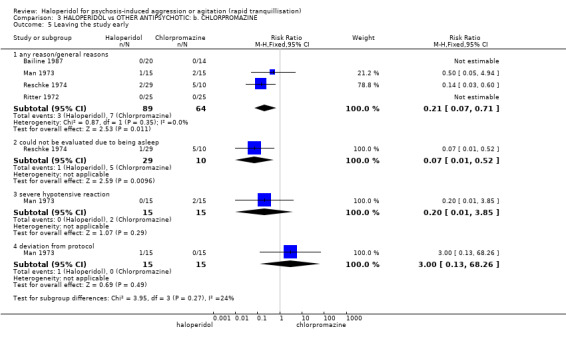
Comparison 3 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: b. CHLORPROMAZINE, Outcome 5 Leaving the study early.
4. COMPARISON 4. HALOPERIDOL versus OTHER ANTIPSYCHOTICS: c. DROPERIDOL
We found two relevant studies (n = 255) (Calver 2015; Resnick 1984).
4.1 Tranquillisation or asleep
There was no statistical significant difference in terms of being not asleep at two hours between the compared groups (1 RCT, n = 228, RR 1.07, 95% CI 0.44 to 2.60, Analysis 4.1). People with haloperidol or droperidol experienced no different time to sleep (1 RCT, n = 114, MD 5.20, 95% CI ‐6.05 to 16.45, Analysis 4.2).
4.1. Analysis.
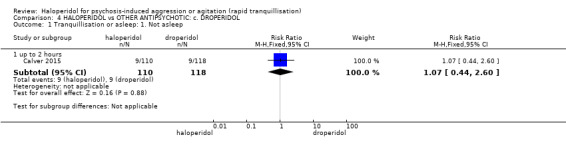
Comparison 4 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: c. DROPERIDOL, Outcome 1 Tranquillisation or asleep: 1. Not asleep.
4.2. Analysis.
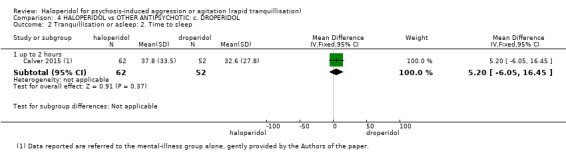
Comparison 4 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: c. DROPERIDOL, Outcome 2 Tranquillisation or asleep: 2. Time to sleep.
4.2 Repeated need for rapid tranquillisation
More people in the haloperidol group required more than one injection compared with those allocated to droperidol (2 RCTs, n = 255, RR 2.38, 95% CI 1.27 to 4.47); however, this was not statistically significant for the outcome of ‘required more than three injections’ (1 RCT, n = 27, RR 2.12, 95% CI 0.09 to 47.68, Analysis 4.3).
4.3. Analysis.
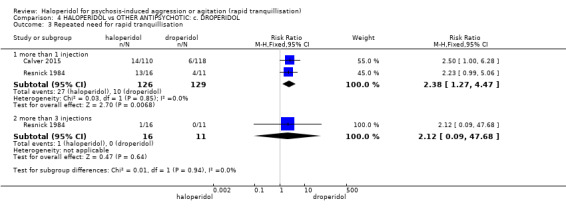
Comparison 4 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: c. DROPERIDOL, Outcome 3 Repeated need for rapid tranquillisation.
4.3 Global outcome ‐ need for benzodiazepine
There was no statistical significant difference in terms of need for adjunctive benzodiazepine between the two comparison groups (1 RCT, n = 228, RR 0.31, 95% CI 0.07 to 1.44, Analysis 4.4).
4.4. Analysis.
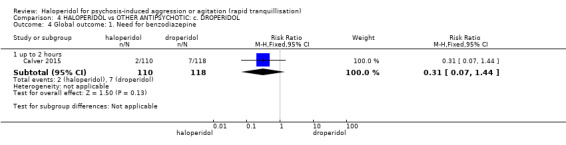
Comparison 4 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: c. DROPERIDOL, Outcome 4 Global outcome: 1. Need for benzodiazepine.
4.4 Adverse effects
4.4.1 General
In the haloperidol group there was only one adverse effects up to two hours, compared with seven in the droperidol group (1 RCT, n = 228, RR 0.15, 95% CI 0.02 to 1.23), involving one person in the haloperidol group and six in the droperidol group (1 RCT, n = 228, Analysis 4.5).
4.5. Analysis.
Comparison 4 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: c. DROPERIDOL, Outcome 5 Adverse effects: 1. General.
4.4.2 Specific
a. Arousal
There were few differences between groups as for being over‐sedated (1 RCT, n = 228, RR 0.36, 95% CI 0.01 to 8.68, Analysis 4.6).
4.6. Analysis.
Comparison 4 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: c. DROPERIDOL, Outcome 6 Adverse effects: 2a. Specific ‐ arousal level.
b. Cardiovascular
One person in the haloperidol group experienced hypotension, compared with four in the droperidol group (1 RCT, n = 228, RR 0.27, 95% CI 0.03 to 2.36, Analysis 4.7).
4.7. Analysis.
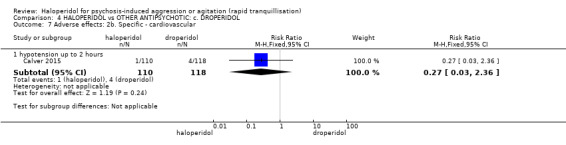
Comparison 4 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: c. DROPERIDOL, Outcome 7 Adverse effects: 2b. Specific ‐ cardiovascular.
c. Movement disorder
One person in the haloperidol group experienced dystonia, compared with one in the droperidol group (2 RCTs, n = 255, RR 0.86, 95% CI 0.11 to 6.56, Analysis 4.8).
4.8. Analysis.
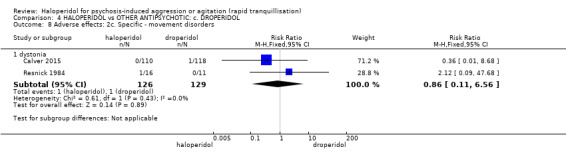
Comparison 4 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: c. DROPERIDOL, Outcome 8 Adverse effects: 2c. Specific ‐ movement disorders.
d. Miscellaneous
There was no difference in terms of desaturation events between the two compared groups (1 RCT, n = 228, RR 0.36, 95% CI 0.01 to 8.68, Analysis 4.9).
4.9. Analysis.
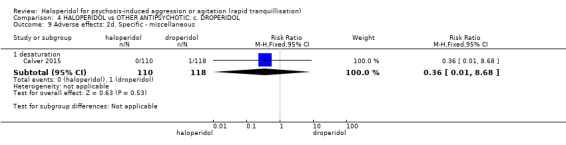
Comparison 4 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: c. DROPERIDOL, Outcome 9 Adverse effects: 2d. Specific ‐ miscellaneous.
5. COMPARISON 5. HALOPERIDOL versus OTHER ANTIPSYCHOTICS: d. LOXAPINE
We found three relevant studies (total n = 119, Fruensgaard 1977; Paprocki 1977; Tuason 1986). Fruensgaard 1977, unlike the other two studies also made sure that both groups of participants were given biperiden, an anticholinergic drug which offsets adverse effects such as movement disorders.
5.1 Tranquillisation or asleep – not asleep at about 12 hours
Loxapine does seem more sedating than haloperidol although the difference was not statistically significant (1 RCT, n = 54, RR 4.31, 95% CI 0.54 to 34.48, Analysis 5.1).
5.1. Analysis.
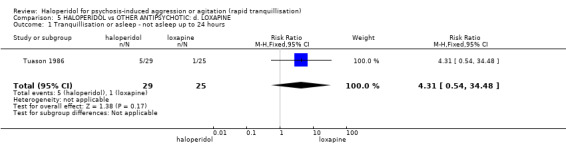
Comparison 5 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: d. LOXAPINE, Outcome 1 Tranquillisation or asleep ‐ not asleep up to 24 hours.
5.2 Specific behaviour
There were very few differences between groups for improvement in levels of agitation (1 RCT, n = 30, RR 1.17, 95% CI 0.51 to 2.66) or levels of aggression (1 RCT, n = 30, RR 1.10, 95% CI 0.69 to 1.76, Analysis 5.2).
5.3 Global outcome
Studies found no differences found between groups at any time point in regards to the number of people who were reported as having ‘no change in global outcome’ (1 RCT, n = 56, RR at 48 hours 0.93, 95% CI 0.14 to 6.15; 1 RCT, n = 54, RR at 72 hours 0.39, 95% CI 0.04 to 3.49 and 1 RCT, n = 35, RR up to 4 weeks 0.40, 95% CI 0.12 to 1.32, Analysis 5.3).
5.3. Analysis.
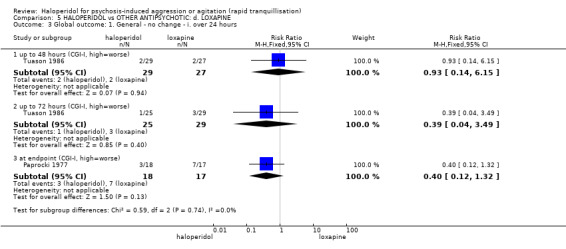
Comparison 5 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: d. LOXAPINE, Outcome 3 Global outcome: 1. General ‐ no change ‐ i. over 24 hours.
Although never statistically significant, more people in the loxapine group were sedated at 30 minutes (1 RCT, n = 30, RR 1.29, 95% CI 0.65 to 2.54), 60 minutes (1 RCT, n = 30, RR 3.50, 95% CI 0.86 to 14.18) and at 120 minutes (1 RCT, n = 30, RR 7.00, 95% CI 0.98 to 50.16) than in the haloperidol group (Analysis 5.4).
5.4. Analysis.
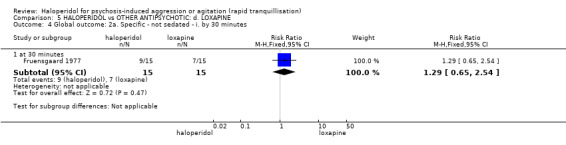
Comparison 5 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: d. LOXAPINE, Outcome 4 Global outcome: 2a. Specific ‐ not sedated ‐ i. by 30 minutes.
5.4 Mental state
One trial found no clear differences between the haloperidol and loxapine in average endpoint scores on the BPRS (n = 52, MD 6.10, 95% CI 4.48 to 7.72, Analysis 5.6).
5.6. Analysis.
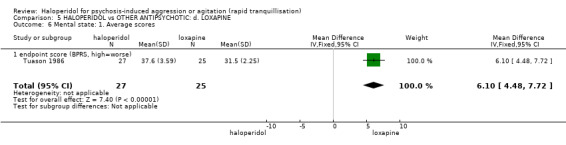
Comparison 5 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: d. LOXAPINE, Outcome 6 Mental state: 1. Average scores.
5.5 Adverse effects
Over 20 different adverse effects are reported including anticholinergic disorders (Analysis 5.8), arousal levels (Analysis 5.9), cardiovascular effects (Analysis 5.10), movement disorders (Analysis 5.11) and several that were difficult to classify (Analysis 5.12), but these are probably best summarised by the general impression from Analysis 5.7 where eight people in the haloperidol group and 10 people in the loxapine group experienced one or more drug‐related adverse effects (1 RCT, n = 30, RR 0.80, 95% CI 0.44 to 1.45).
5.8. Analysis.
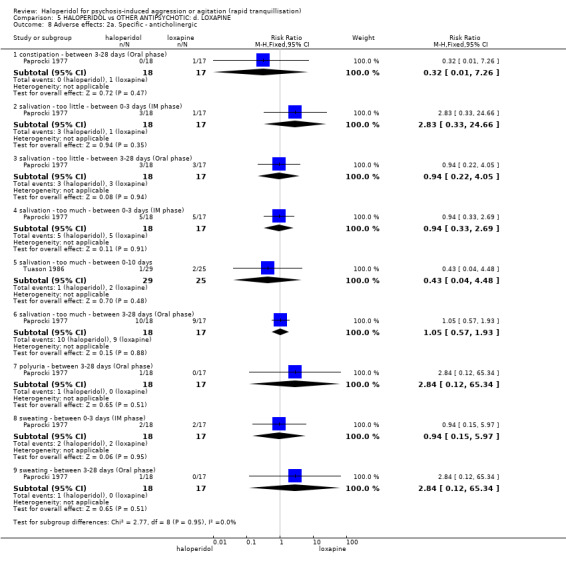
Comparison 5 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: d. LOXAPINE, Outcome 8 Adverse effects: 2a. Specific ‐ anticholinergic.
5.9. Analysis.
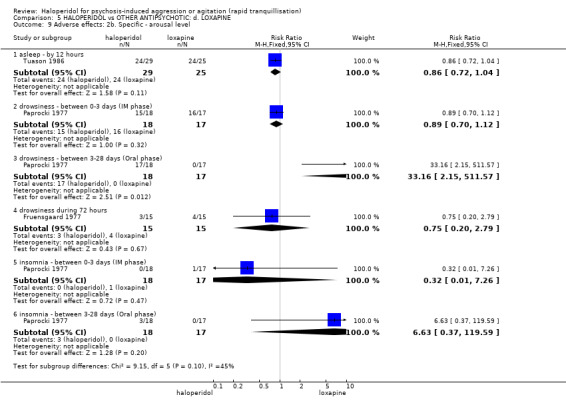
Comparison 5 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: d. LOXAPINE, Outcome 9 Adverse effects: 2b. Specific ‐ arousal level.
5.10. Analysis.
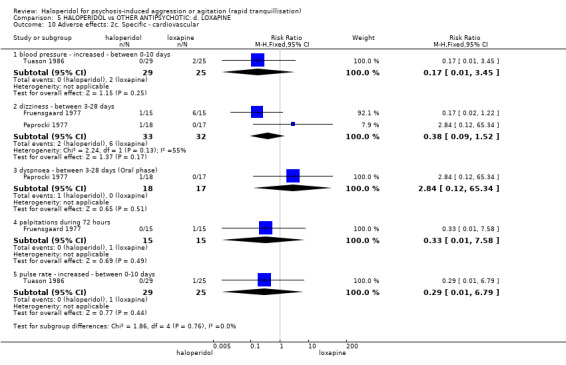
Comparison 5 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: d. LOXAPINE, Outcome 10 Adverse effects: 2c. Specific ‐ cardiovascular.
5.11. Analysis.
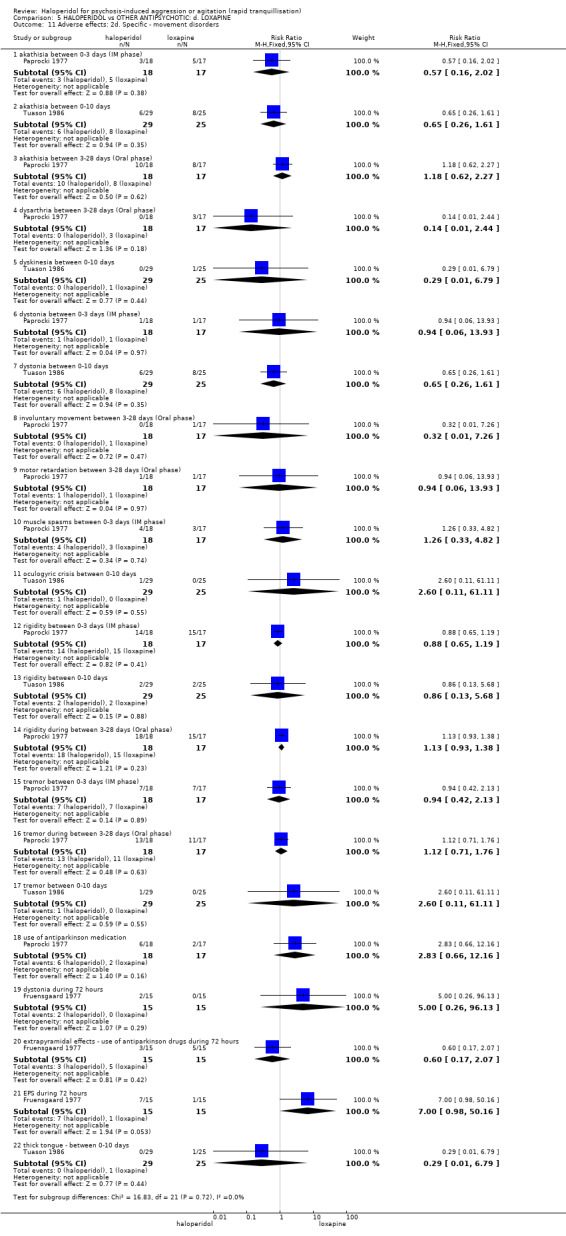
Comparison 5 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: d. LOXAPINE, Outcome 11 Adverse effects: 2d. Specific ‐ movement disorders.
5.12. Analysis.
Comparison 5 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: d. LOXAPINE, Outcome 12 Adverse effects: 2e. Specific ‐ miscellaneous.
5.7. Analysis.
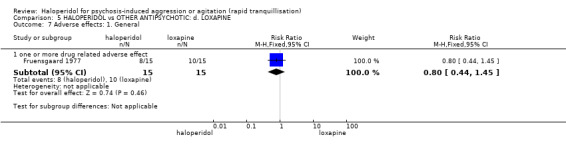
Comparison 5 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: d. LOXAPINE, Outcome 7 Adverse effects: 1. General.
5.6 Leaving the study early
a. General reasons
At no point at anytime did statically more people leave one group than the other (e.g. 1 RCT, n = 35, RR leaving around 72 hours 0.63, 95% CI 0.21 to 1.85). The Tuason 1986 study found no significant differences between the groups for the number of people who discontinued with the study by 10 days (n = 54, RR 1.18, 95% CI 0.67 to 2.07, Analysis 5.13).
5.13. Analysis.
Comparison 5 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: d. LOXAPINE, Outcome 13 Leaving the study early: 1. For general reasons.
b. Specific reasons
In Paprocki 1977 they withdrew one person because of “toxicity” during the IM phase (n = 35, RR 2.84, 95% CI 0.12 to 65.34, Analysis 5.14).
5.14. Analysis.
Comparison 5 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: d. LOXAPINE, Outcome 14 Leaving the study early: 2. Specific reasons.
6. COMPARISON 6. HALOPERIDOL versus OTHER ANTIPSYCHOTICS: e. OLANZAPINE
We found six studies (total n = 720) that compared haloperidol with olanzapine (Baldacara 2011; Battaglia 2002; Breier 2002; Eli Lilly 2004; Kinon 2001d; Walther 2014).
6.1 Tranquillisation or asleep
By two hours significantly more people in the olanzapine group were asleep compared with those allocated haloperidol (1 RCT, n = 257, RR 1.16, 95% CI 1.02 to 1.32, Analysis 6.1).
6.1. Analysis.
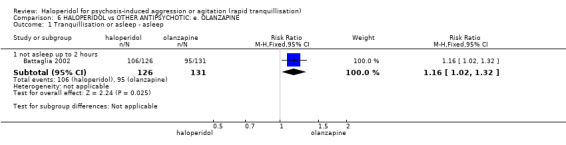
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 1 Tranquillisation or asleep ‐ asleep.
6.2 Repeated need for tranquillisation
The need for additional injection results were statistically significant between the two groups in favour of olanzapine at 12 hours (1 RCT, n = 60, RR 3.29, 95% CI 1.67 to 6.47); however, the two groups results are similar at two hours (1 RCT, n = 60, RR 4.00, 95% CI 0.47 to 33.73) and at 24 hours (3 RCTs, n = 392, RR 1.06, 95% CI 0.75 to 1.51, Analysis 6.2).
6.2. Analysis.
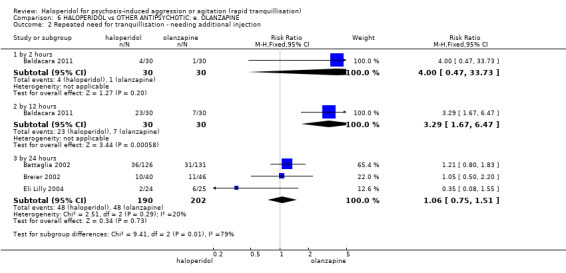
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 2 Repeated need for tranquillisation ‐ needing additional injection.
6.3 Specific behaviour
a. Agitation
At two hours, a similar number of participants in both the haloperidol and olanzapine group were rated as having at least a 40% reduction according to the PANSS‐EC (1 RCT, n = 45, RR 0.96, 95% CI 0.58 to 1.58). Also agitation was rated as an ‘adverse effect’ (1 RCT, n = 49, RR 1.04, 95% CI 0.07 to 15.73), and again reported by 21 days (1 RCT, n = 100, RR 1.08, 95% CI 0.33 to 3.51, Analysis 6.3).
6.3. Analysis.
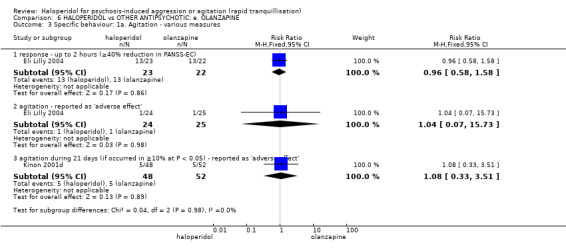
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 3 Specific behaviour: 1a. Agitation ‐ various measures.
By 30 minutes haloperidol and olanzapine showed similar findings at average change scores of ACES (15 minutes: 1 RCT, n = 46, MD 0.50, 95% CI ‐0.35 to 1.35; 30 minutes: 1 RCT, n = 46, MD 0.90, 95% CI ‐0.06 to 1.86) and PANSS‐EC (15 minutes: 1 RCT, n = 46, MD 0.30, 95% CI ‐1.64 to 2.24; 30 minutes: 1 RCT, n = 46, MD 1.30, 95% CI ‐1.16 to 3.76, Analysis 6.4).
6.4. Analysis.
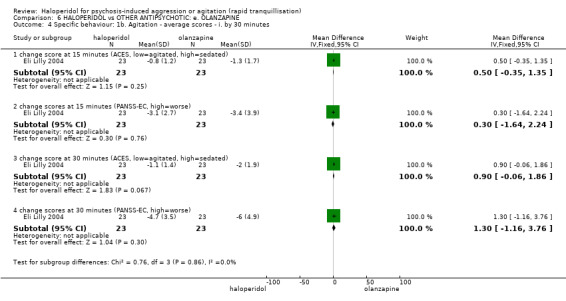
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 4 Specific behaviour: 1b. Agitation ‐ average scores ‐ i. by 30 minutes.
By about two hours, continuous measures of agitation showed similar findings. The ABS change score did favour olanzapine (1 RCT, n = 85, MD 2.70, 95% CI 0.38 to 5.02) and so did the ACES change score at one hour (1 RCT, n = 46, MD 1.20, 95% CI 0.06 to 2.34) and the Overt Agitation Severity Scale (OASS) endpoint score at one hour (1 RCT, n = 60, MD 2.00, 95% CI 1.18 to 2.82) and two hours (1 RCT, n = 60, MD 4.20, 95% CI 3.54 to 4.86). However, the others measurements did not show similar findings (for example, average change and endpoint combined scores PANSS‐EC: 3 RCTs, n = 332, MD ‐0.31, 95% CI ‐1.49 to 0.86, Analysis 6.5).
6.5. Analysis.
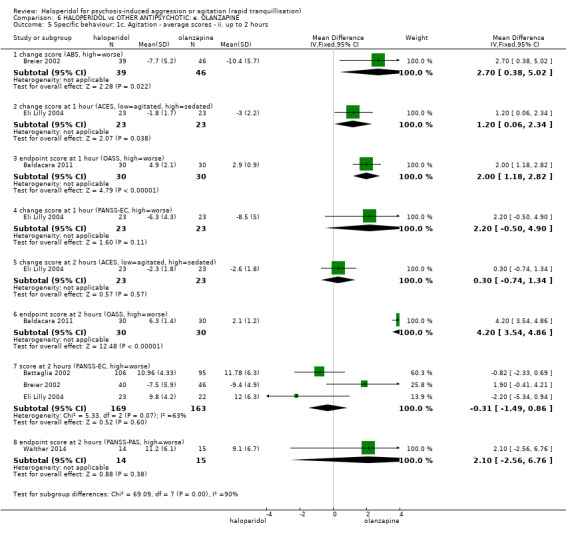
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 5 Specific behaviour: 1c. Agitation ‐ average scores ‐ ii. up to 2 hours.
At four hours, continuous measures of agitation showed similar findings. The average endpoint score on the OASS was statistically significant (1 RCT, n = 60, MD 0.20, 95% CI ‐0.05 to 0.45, Analysis 6.6).
6.6. Analysis.
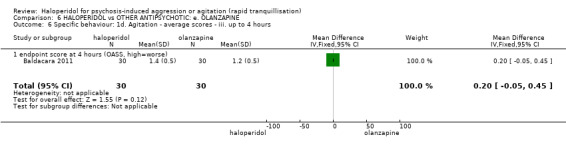
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 6 Specific behaviour: 1d. Agitation ‐ average scores ‐ iii. up to 4 hours.
When it came to 24 hours, continuous measures of agitation showed similar findings. For example, the average change score on the ABS (1 RCT, n = 86, MD 2.40, 95% CI 0.01 to 4.79) and the average endpoint score on the OASS at six hours (1 RCT, n = 60, MD 0.20, 95% CI 0.05 to 0.35) and 12 hours (1 RCT, n = 60, MD 0.90, 95% CI 0.82 to 0.98) were statistically significant, but on the PANSS‐EC it was not (1 RCT, n = 86, MD 1.40, 95% CI ‐0.55 to 3.35, Analysis 6.7).
6.7. Analysis.
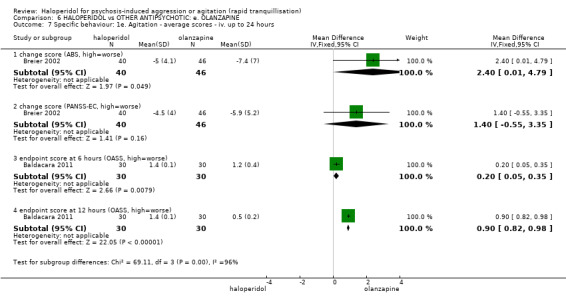
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 7 Specific behaviour: 1e. Agitation ‐ average scores ‐ iv. up to 24 hours.
b. Aggression
The two groups showed no statistical significant difference on the average endpoint OASS score at one hour (1 RCT, n = 60, MD 0.90, 95% CI 0.13 to 1.67) and at two hours (1 RCT, n = 60, MD 0.50, 95% CI ‐0.41 to 1.41, Analysis 6.8).
6.8. Analysis.
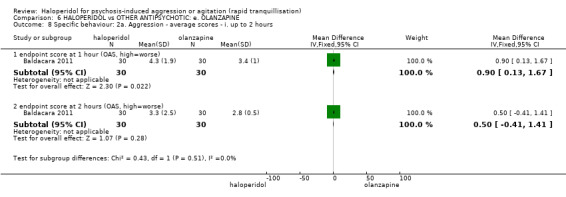
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 8 Specific behaviour: 2a. Aggression ‐ average scores ‐ i. up to 2 hours.
At four hours, the average endpoint score on the OASS showed similar findings (1 RCT, n = 60, MD 0.10, 95% CI ‐0.11 to 0.31, Analysis 6.9).
6.9. Analysis.
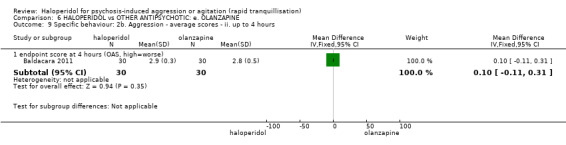
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 9 Specific behaviour: 2b. Aggression ‐ average scores ‐ ii. up to 4 hours.
When it came to 24 hours, the continuous measures of aggression at six hours showed similar findings at the average endpoint OASS score (1 RCT, n = 60, MD ‐0.10, 95% CI ‐0.26 to 0.06); however, at 12 hours the average endpoint OASS score resulted in favour of haloperidol (1 RCT, n = 60, MD ‐0.20, 95% CI ‐0.39 to ‐0.01, Analysis 6.10).
6.10. Analysis.
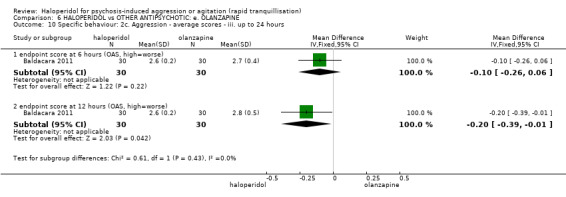
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 10 Specific behaviour: 2c. Aggression ‐ average scores ‐ iii. up to 24 hours.
c. Hostility
This was recorded – again as an adverse effect – but there was not a clear difference between groups (1 RCT, n = 49, RR 0.35, 95% CI 0.01 to 8.12, Analysis 6.11)
6.11. Analysis.
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 11 Specific behaviour: 3. Hostility.
6.4 Global outcome
a. General
There were no significant differences between the groups with regards to the number of people who required additional benzodiazepines during 24 hours (2 RCTs, n = 343, RR 1.05, 95% CI 0.63 to 1.74). This applied up to seven days (1 RCT, n = 100, RR 1.00, 95% CI 0.79 to 1.27), and use of additional restraint, seclusion or special observation (2 RCTs, n = 160, RR 1.49, 95% CI 0.70 to 3.17, Analysis 6.12). In one study (n = 100), there was a significant difference between the haloperidol and olanzapine group for time until the person discontinued the study – it was significantly earlier in the olanzapine group (MD ‐3.48, 95% CI ‐6.28 to ‐0.68). In the same study, the dose of adjunctive lorazepam needed was very similar (MD 0.34, 95% CI ‐0.14 to 0.82, Analysis 6.13).
6.12. Analysis.
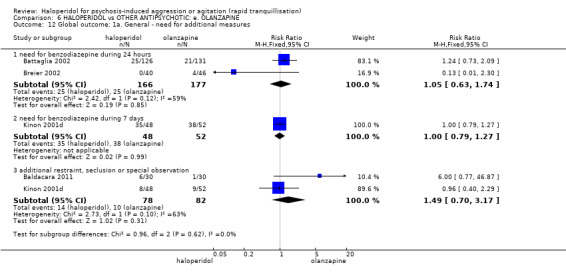
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 12 Global outcome: 1a. General ‐ need for additional measures.
6.13. Analysis.
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 13 Global outcome: 1b. General ‐ time and doses.
People in the haloperidol and olanzapine group were rated as demonstrating similar levels of improvement in global functioning according to average endpoint scores using the CGI‐I when rated at two hours (1 RCT, n = 42, MD ‐0.10, 95% CI ‐0.65 to 0.45, Analysis 6.14), and their average change scores at 24 hours (1 RCT, n = 243, MD 0.00, 95% CI ‐0.20 to 0.20). There was no difference by 24‐72 hours according to average change scores using the CGI‐S (1 RCT, n = 86, MD 0.00, 95% CI ‐0.24 to 0.24, Analysis 6.15). Kinon 2001d used a non‐parametric test and reported that at 21 days, those in the olanzapine group did have a significantly greater change in the CGI‐I score (n = 100, ‐0.38 ± 1.31 and ‐0.2 ± 1 .44, P = 0.25, F test 1.71 df). Further, one study demonstrated similar levels of improvement according to average change scores when using the CGI‐I by 21 days (1 RCT, n= 100, MD 0.36, 95% CI ‐0.18 to 0.90, Analysis 6.15).
6.14. Analysis.
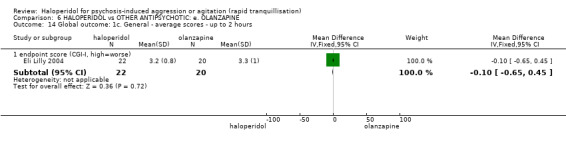
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 14 Global outcome: 1c. General ‐ average scores ‐ up to 2 hours.
6.15. Analysis.
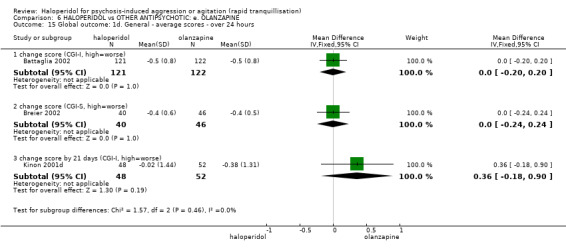
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 15 Global outcome: 1d. General ‐ average scores ‐ over 24 hours.
b. Specific
One study repeatedly recorded levels of arousal (alert to asleep) across several periods of time (one to 24 hours). There were no convincing differences between the two drugs (Analysis 6.16).
6.16. Analysis.
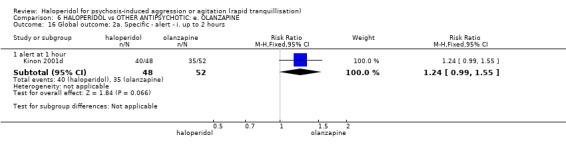
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 16 Global outcome: 2a. Specific ‐ alert ‐ i. up to 2 hours.
6.5 Service use
There was no difference between groups for the outcome of average time to discharge (about 13 days) (1 RCT, n = 100, MD ‐0.60, 95% CI ‐1.85 to 0.65, Analysis 6.28). Time was also measured for the average number of patient‐hours used. These data were skewed and therefore presented in a table (Analysis 6.29).
6.28. Analysis.
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 28 Service use: 1. Average days to discharge.
6.29. Analysis.
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 29 Service use: 2. Average patient‐hours used during 24 hours (skewed data).
| Service use: 2. Average patient‐hours used during 24 hours (skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| days 1‐7 | ||||
| Kinon 2001d | Haloperidol | 2.59 | 6.79 | 48 |
| Kinon 2001d | Olanzapine | 1.57 | 5.52 | 52 |
| days 8‐14 | ||||
| Kinon 2001d | Haloperidol | 0.92 | 4.05 | 48 |
| Kinon 2001d | Olanzapine | 0.33 | 2.23 | 52 |
| days 15‐21 | ||||
| Kinon 2001d | Haloperidol | 0.55 | 2.74 | 48 |
| Kinon 2001d | Olanzapine | 0 | 0 | 52 |
6.6 Mental state
Several adverse effects seem to be better classified in ‘mental state’. Two different studies reported anxiety (n = 49, RR 0.35, 95% CI 0.01 to 8.12), or anxiety by 21 days (n = 100, RR 0.36, 95% CI 0.08 to 1.70). Delusions were also reported as an adverse effect (1 RCT, n = 49, RR 0.35, 95% CI 0.01 to 8.12), as was nervousness (1 RCT, n = 100, RR 2.17, 95% CI 0.70 to 6.74, Analysis 6.30). There were no significant differences between olanzapine and haloperidol with respect to improved mental state according to average change scores on the PANSS Total sub‐scale (1 RCT, n = 100, MD 4.70, 95% CI ‐0.18 to 9.58, Analysis 6.34). There was also few differences according to average change scores on positive sub‐scale of the BPRS at any time period (1 RCT, n = 46) nor on BPRS totals (Analysis 6.31).
6.30. Analysis.
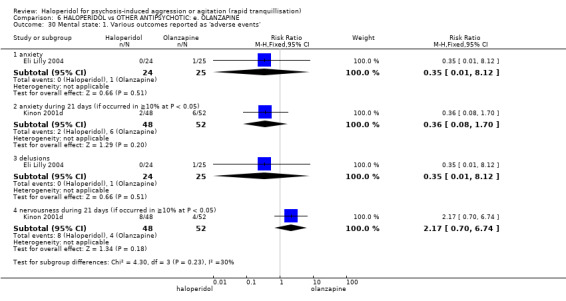
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 30 Mental state: 1. Various outcomes reported as 'adverse events'.
6.34. Analysis.
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 34 Mental state: 2d. Average scores ‐ iv. over 24 hours.
6.31. Analysis.
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 31 Mental state: 2a. Average scores ‐ i. by 30 minutes.
6.7 Adverse effects
6.7.1 General
In three studies significantly more people in the haloperidol group experienced a drug‐related adverse event (3 RCTs, n = 209, RR 1.38, 95% CI 1.09 to 1.76, Analysis 6.35).
6.35. Analysis.
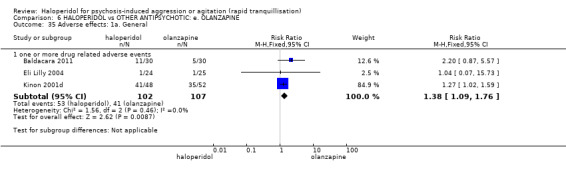
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 35 Adverse effects: 1a. General.
6.7.2 Serious
There was no clear difference in risk of treatment emergent adverse events between groups (1 RCT, n = 49, RR 0.81, 95% CI 0.36 to 1.83). Only a few of these were attributed to the use of the drug. No serious adverse effects nor death were experienced by any participant who received haloperidol or olanzapine (Analysis 6.36).
6.36. Analysis.
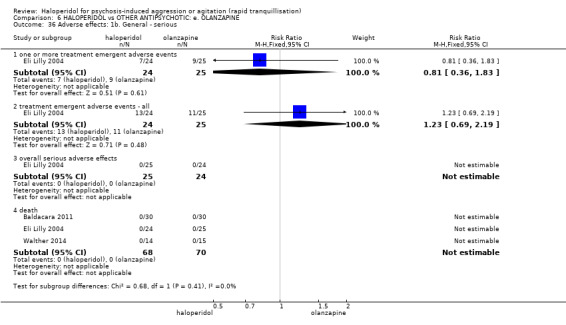
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 36 Adverse effects: 1b. General ‐ serious.
6.7.3 Specific
a. Arousal level
Despite being reported in a way that may have underestimated the overall prevalence, complaints of insomnia and/or somnolence were not different between groups (Analysis 6.38).
6.38. Analysis.
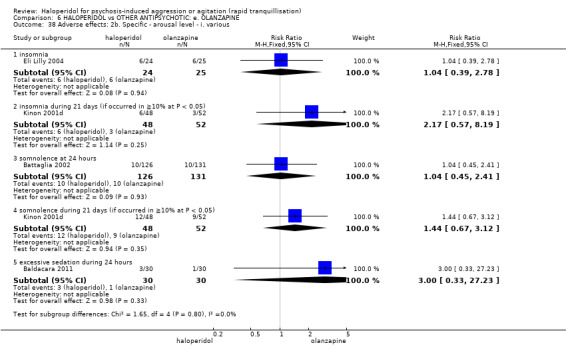
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 38 Adverse effects: 2b. Specific ‐ arousal level ‐ i. various.
b. Cardiovascular
Binary and continuous measures of any cardiovascular change did not show differences between groups (e.g. hypotension 2 RCTs, n = 146, RR 0.27, 95% CI 0.03 to 2.38, Analysis 6.42; change in QT interval 1 RCT, n = 257, MD 1.80, 95% CI ‐3.81 to 7.41, Analysis 6.43).
6.42. Analysis.
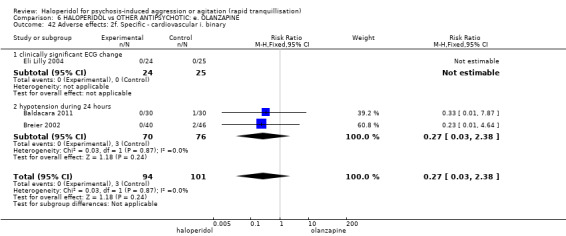
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 42 Adverse effects: 2f. Specific ‐ cardiovascular i. binary.
6.43. Analysis.
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 43 Adverse effects: 2g. Specific ‐ cardiovascular ii. continuous.
c. Movement disorders
Studies reported different movement disorders in several different ways. There was the impression that more people who received haloperidol complained of these effects (e.g. EPS at 24 hours, 3 RCTs, n = 403, RR 8.35, 95% CI 2.27 to 30.63, Analysis 6.45).
6.45. Analysis.
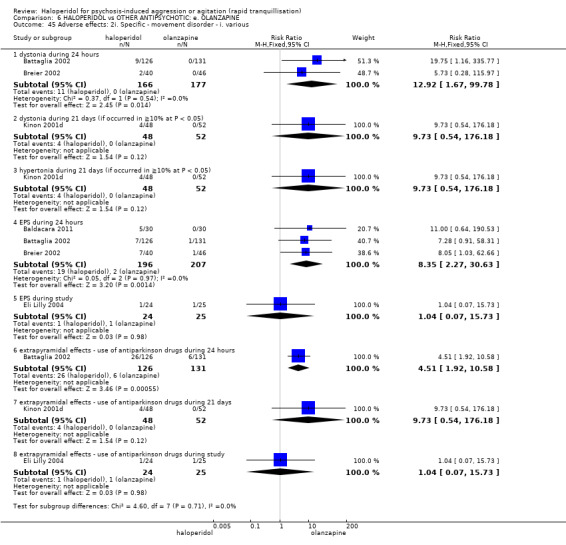
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 45 Adverse effects: 2i. Specific ‐ movement disorder ‐ i. various.
This finding is supported by the continuous measure of SAS – again, this tends to favour olanzapine at 24 hours (1 RCT, n = 242, MD 1.31, 95% CI 0.56 to 2.06, Analysis 6.46). Other skewed SAS endpoint data are not so favourable, but these are presented only in tabular form (Analysis 6.47). This also applies to the akathisia endpoint measure (see Analysis 6.47) and, finally, change scores on the same scale (BAS) at 24 hours favoured olanzapine (1 RCT, n = 241, MD 0.28, 95% CI 0.09 to 0.47, Analysis 6.46).
6.46. Analysis.
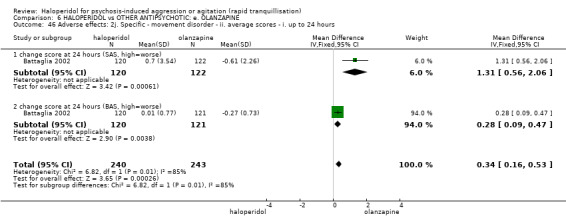
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 46 Adverse effects: 2j. Specific ‐ movement disorder ‐ ii. average scores ‐ i. up to 24 hours.
6.47. Analysis.
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 47 Adverse effects: 2k. Specific ‐ movement disorder ‐ iii. average scores ‐ i. up to 24 hours.
| Adverse effects: 2k. Specific ‐ movement disorder ‐ iii. average scores ‐ i. up to 24 hours | |||||
|---|---|---|---|---|---|
| Study | Intervention | Mean | SD | N | Notes |
| endpoint score at 24 hours (SAS, high=worse) ‐ (skewed data) | |||||
| Eli Lilly 2004 | Haloperidol | 1.4 | 2.8 | 22 | |
| Eli Lilly 2004 | Olanzapine | 2.2 | 3.5 | 20 | |
| endpoint score at 24 hours (BAS, high=worse) ‐ (skewed data) | |||||
| Eli Lilly 2004 | Haloperidol | 1.5 | 1.7 | 22 | |
| Eli Lilly 2004 | Olanzapine | 1.7 | 2.6 | 20 | |
d. Miscellaneous
Reports recorded several other adverse effects, none of which were different in the two groups. These included anticholinergic effects (increased salivation, 1 RCT, n = 100, RR 9.73, 95% CI 0.54 to 176.18, Analysis 6.37), gastric problems (e.g. abdominal pain 1 RCT, n =49, RR 3.12, 95% CI 0.13 to 73.04, Analysis 6.44), and nose bleeds (1 RCT, n = 49, RR 3.12, 95% CI 0.13 to 73.04, Analysis 6.49).
6.37. Analysis.
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 37 Adverse effects: 2a. Specific ‐ anticholinergic.
6.44. Analysis.
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 44 Adverse effects: 2h. Specific ‐ gastrointestinal.
6.49. Analysis.
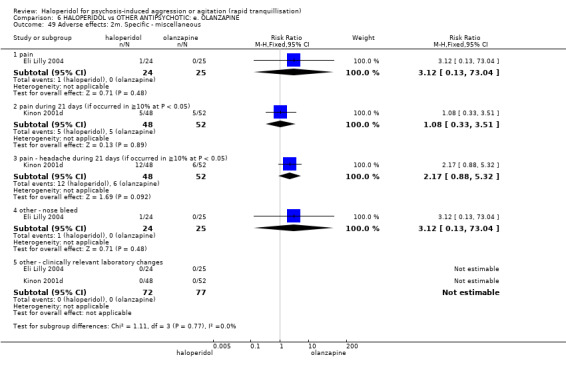
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 49 Adverse effects: 2m. Specific ‐ miscellaneous.
No participants in either group were reported to have had a clinically relevant change in laboratory test results.
6.8 Leaving the study early
About 20% of each group left the study early but slightly fewer in the olanzapine group (2 RCTs, n = 148, RR 1.66, 95% CI 1.04 to 2.65). Power to investigate specific reasons was low but eight people in the haloperidol group, compared with one in the olanzapine group left the study early due to adverse events (1 RCT, n = 100, RR 8.67, 95% CI 1.13 to 66.75, Analysis 6.50).
6.50. Analysis.
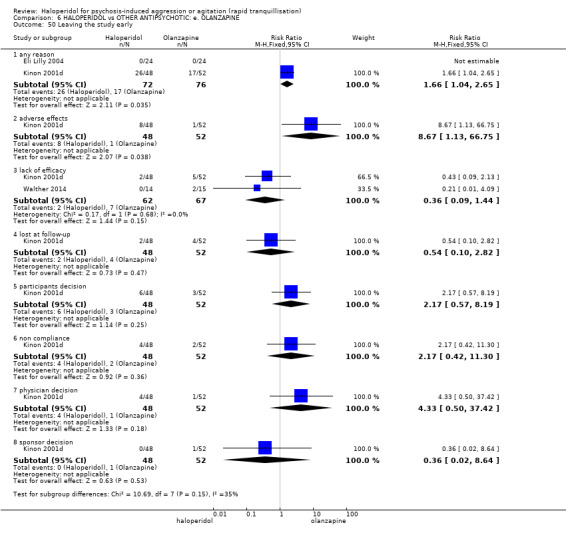
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 50 Leaving the study early.
7. COMPARISON 7. HALOPERDIOL versus OTHER ANTIPSYCHOTICS: f. PERPHENAZINE
We found one relevant study (n = 44, Fitzgerald 1969).
7.1 Global outcome ‐ no improvement
One person in the haloperidol group and two people in the perphenazine were reported to have shown no global improvement (n = 44, RR 0.46, 95% CI 0.04 to 4.68, Analysis 7.1).
7.1. Analysis.
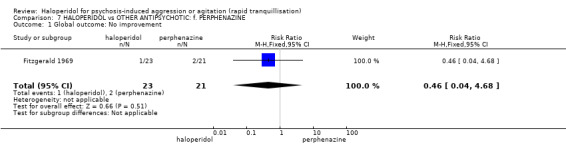
Comparison 7 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: f. PERPHENAZINE, Outcome 1 Global outcome: No improvement.
7.2 Adverse effects
7.2.1 General
Ten people in the haloperidol group and seven in the perphenazine group experienced one or more adverse effect, however, this difference is not statistically significant (n = 44, RR 1.30, 95% CI 0.61 to 2.80, Analysis 7.2). No participants in either group developed clinically significant laboratory changes.
7.2. Analysis.
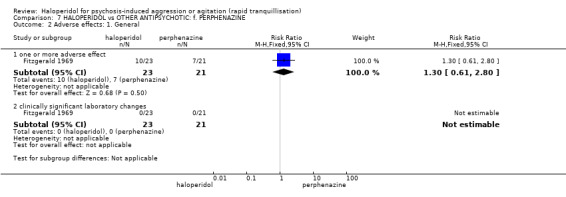
Comparison 7 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: f. PERPHENAZINE, Outcome 2 Adverse effects: 1. General.
7.2.2 Specific
Six people in the haloperidol group and two people in the perphenazine group required antiparkinson medication, but again, this result was not statistically significant (n = 44, RR 2.74, 95% CI 0.62 to 12.12). One person in the perphenazine group developed a hypotensive episode compared with none in the haloperidol group (n = 44, RR 0.31, 95% CI 0.01 to 7.12).
Two people in the haloperidol group and one person in the perphenazine discontinued with the study because they developed EPS (n = 44, RR 1.83, 95% CI 0.18 to 18.70). Also one person was withdrawn from the haloperidol group due to drowsiness/tension and no therapeutic effect (n = 44, RR 2.75, 95% CI 0.12 to 64.04, Analysis 7.3).
7.3. Analysis.
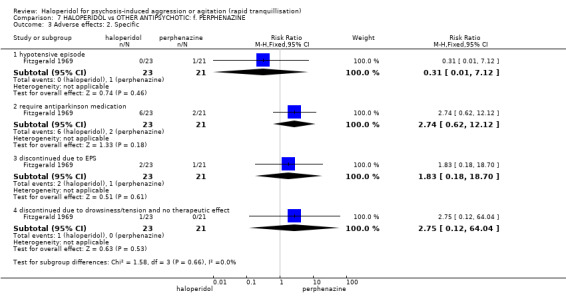
Comparison 7 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: f. PERPHENAZINE, Outcome 3 Adverse effects: 2. Specific.
7.3 Leaving the study early
We felt that we should add these specific reasons into the data on adverse effects (Analysis 7.3), but there were no differences between groups (Analysis 7.4).
7.4. Analysis.
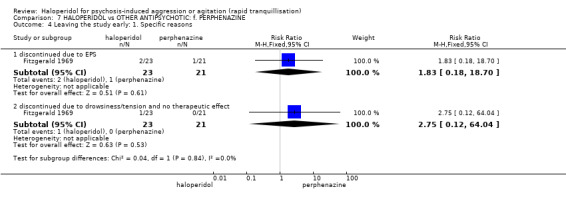
Comparison 7 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: f. PERPHENAZINE, Outcome 4 Leaving the study early: 1. Specific reasons.
8. COMPARISON 8. HALOPERIDOL versus OTHER ANTIPSYCHOTICS: g. QUETIAPINE
We found one relevant study (n = 80, Liang 2013).
8.1 Specific behaviour ‐ agitation
There was no difference between the two groups at the average change score of the PANSS‐EC at 24 hours (MD 0.10, 95% CI ‐0.56 to 0.76, Analysis 8.1).
8.1. Analysis.
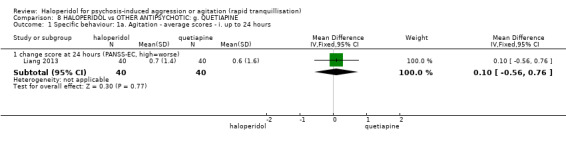
Comparison 8 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: g. QUETIAPINE, Outcome 1 Specific behaviour: 1a. Agitation ‐ average scores ‐ i. up to 24 hours.
Continuous measures over 24 hours showed similar findings, from 72 hours (MD 0.20, 95% CI ‐0.87 to 1.27) to 28 days (MD 0.00, 95% CI ‐1.87 to 1.87, Analysis 8.2).
8.2. Analysis.
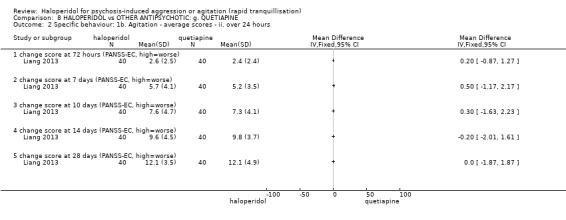
Comparison 8 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: g. QUETIAPINE, Outcome 2 Specific behaviour: 1b. Agitation ‐ average scores ‐ ii. over 24 hours.
8.2 Mental state
No statistically significant difference was found at 28 days in average change score of PANSS (MD ‐0.40, 95% CI ‐7.04 to 6.24), PANSS positive sub‐scale (MD 2.00, 95% CI ‐0.87 to 4.87), and PANSS negative sub‐scale (MD ‐0.60, 95% CI ‐2.27 to 1.07, Analysis 8.3).
8.3. Analysis.
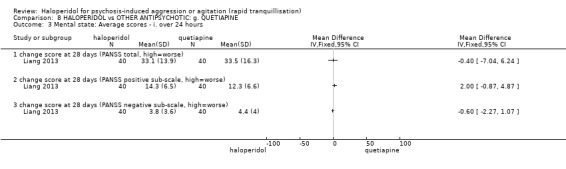
Comparison 8 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: g. QUETIAPINE, Outcome 3 Mental state: Average scores ‐ i. over 24 hours.
8.3 Adverse effects
8.3.1 General
People in the haloperidol group experienced more adverse events in 28 days when comparing with the quetiapine group (RR 1.81, 95% CI 1.19 to 2.77, Analysis 8.4).
8.4. Analysis.
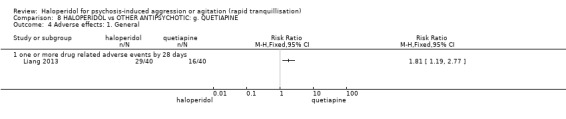
Comparison 8 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: g. QUETIAPINE, Outcome 4 Adverse effects: 1. General.
8.3.2 Specific ‐ movement disorder
Average endpoint SAS scores at 28 days were higher in the haloperidol group (MD 1.30, 95% CI 0.57 to 2.03, Analysis 8.5).
8.5. Analysis.
Comparison 8 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: g. QUETIAPINE, Outcome 5 Adverse effects: 2. Specific ‐ movement disorder ‐ i. average scores.
8.4 Leaving the study early
There were no differences between groups (RR 1.25, 95% CI 0.36 to 4.32, Analysis 8.6).
8.6. Analysis.
Comparison 8 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: g. QUETIAPINE, Outcome 6 Leaving the study early.
9. COMPARISON 9. HALOPERIDOL versus OTHER ANTIPSYCHOTICS: h. RISPERIDONE
We found three relevant studies (n = 314), one of which ‐ Currier 2004 – randomised haloperidol versus risperidone but both groups were given lorazepam (2 mg).
9.1 Tranquillisation or asleep
One trial reported on the outcome of ‘not asleep’ across different time periods (30‐120 minutes). In the company of lorazepam, haloperidol caused more people to be asleep than risperidone (e.g. awake at 30 minutes: 1 RCT, n = 162, RR 0.84, 95% CI 0.74 to 0.95, Analysis 9.1).
9.1. Analysis.
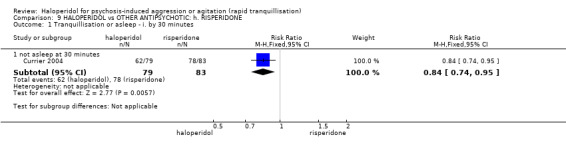
Comparison 9 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE, Outcome 1 Tranquillisation or asleep ‐ i. by 30 minutes.
9.2 Specific behaviour
About 80% of participants in both groups had at least a 50% reduction in agitation scores according to the PANNS‐EC (n = 124, RR 0.96, 95% CI 0.79 to 1.16). By 24 hours, one person in the risperidone group was withdrawn from the study early due to agitation, compared with no one in the haloperidol group (n = 124, RR 0.33, 95% CI 0.01 to 8.03, Analysis 9.3).
9.3. Analysis.
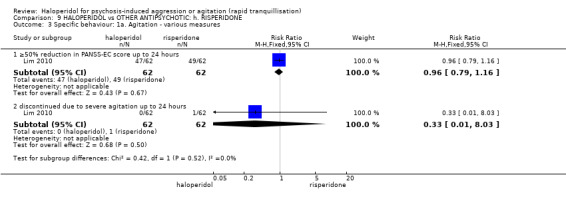
Comparison 9 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE, Outcome 3 Specific behaviour: 1a. Agitation ‐ various measures.
Continuous measures showed similar findings, confirming the absence of a difference between the two groups (1 RCT, n = 28, MD ‐0.40, 95% CI ‐5.22 to 4.42, Analysis 9.4)
9.4. Analysis.
Comparison 9 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE, Outcome 4 Specific behaviour: 1b. Agitation ‐ average scores ‐ i. up to 2 hours.
9.3 Global outcome ‐ need for benzodiazepine
Oddly, in the study that used lorazepam, the great majority of both groups were given additional benzodiazepine. This did not apply to the other trial where benzodiazepine was not part of the routine treatment. There was no difference between groups (Analysis 9.7).
9.7. Analysis.
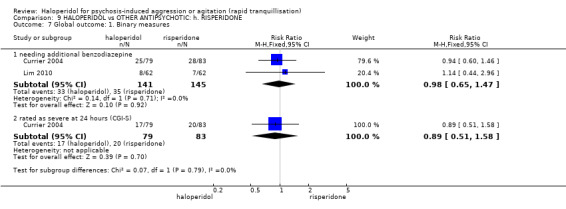
Comparison 9 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE, Outcome 7 Global outcome: 1. Binary measures.
9.4 Adverse effects
9.4.1 General
There were no statistically significant differences between the groups with regard to the number of people who reported adverse effects (e.g. one or more adverse effects during 24 hours, 2 RCTs, n = 286, RR 1.01, 95% CI 0.84 to 1.23, Analysis 9.9).
9.9. Analysis.
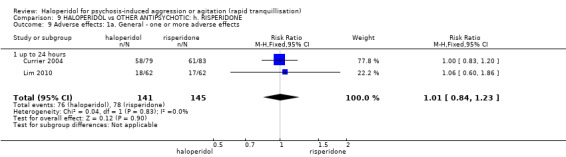
Comparison 9 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE, Outcome 9 Adverse effects: 1a. General ‐ one or more adverse effects.
9.4.2 Specific
Again, levels of arousal suggested that haloperidol was more sedating than risperidone (e.g. sedated at 30 minutes 1 RCT, n = 162, RR 1.90, 95% CI 1.39 to 2.59, Analysis 9.10). Cardiovascular measures were not different except that risperidone seems to marginally increase pulse and haloperidol decrease it (Analysis 9.12).
9.10. Analysis.
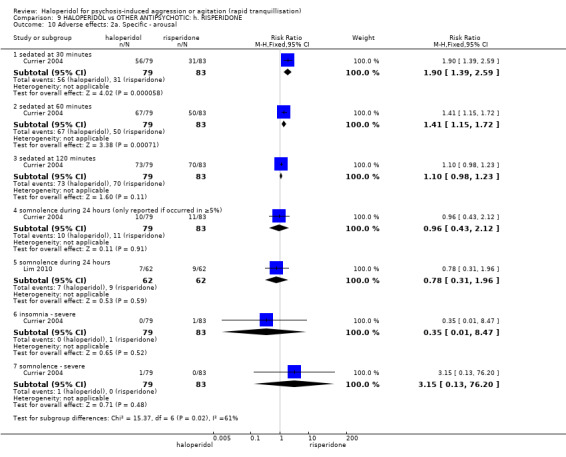
Comparison 9 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE, Outcome 10 Adverse effects: 2a. Specific ‐ arousal.
9.12. Analysis.
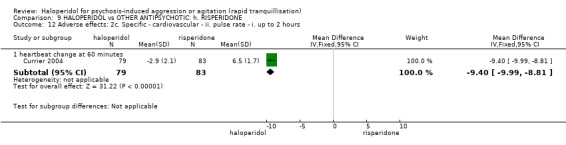
Comparison 9 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE, Outcome 12 Adverse effects: 2c. Specific ‐ cardiovascular ‐ ii. pulse rate ‐ i. up to 2 hours.
Although not statistically significant, eight people the haloperidol group compared with five allocated risperidone suffered EPS (n = 124, RR 1.60, 95% CI 0.55 to 4.62). There was no difference in the levels of acute dystonia between the old and the new drug (about 2%, 2 RCTs, n = 286, RR 1.44, 95% CI 0.28 to 7.28, Analysis 9.14). Continuous measures of akathisia and movement disorders did, however, favour risperidone within 24 hours (Analysis 9.15), finding no difference at 96 hours (Analysis 9.16).
9.14. Analysis.
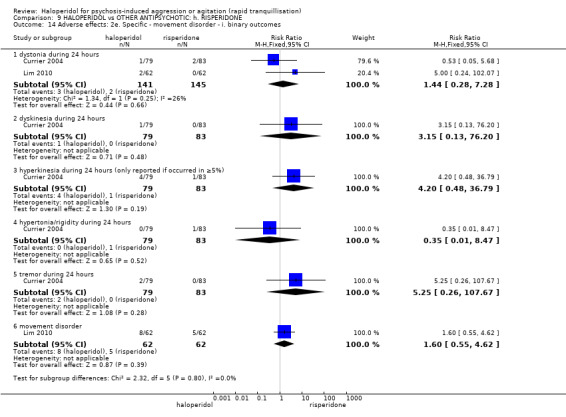
Comparison 9 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE, Outcome 14 Adverse effects: 2e. Specific ‐ movement disorder ‐ i. binary outcomes.
9.15. Analysis.
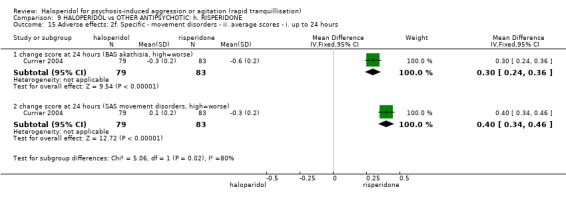
Comparison 9 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE, Outcome 15 Adverse effects: 2f. Specific ‐ movement disorders ‐ ii. average scores ‐ i. up to 24 hours.
9.16. Analysis.
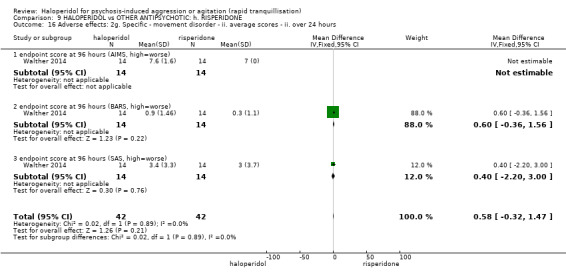
Comparison 9 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE, Outcome 16 Adverse effects: 2g. Specific ‐ movement disorder ‐ ii. average scores ‐ ii. over 24 hours.
Other adverse effects were no different between groups (Analysis 9.17).
9.17. Analysis.
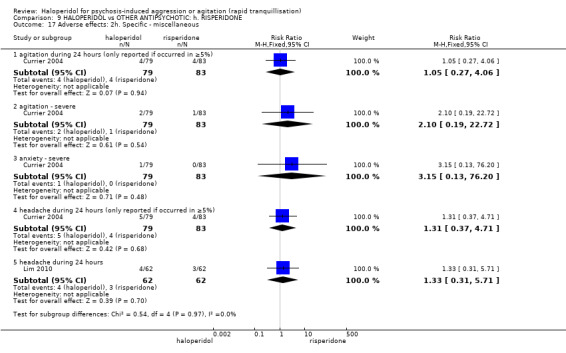
Comparison 9 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE, Outcome 17 Adverse effects: 2h. Specific ‐ miscellaneous.
9.5 Leaving the study early
Again, few people left these studies early with no clear differences between groups (Analysis 9.18).
9.18. Analysis.
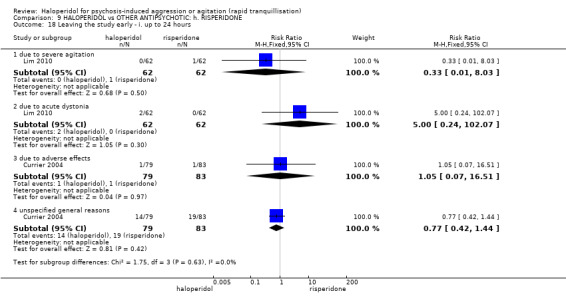
Comparison 9 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE, Outcome 18 Leaving the study early ‐ i. up to 24 hours.
10. COMPARISON 10. HALOPERIDOL versus OTHER ANTIPSYCHOTICS: i. THIOTHIXENE
We found two relevant studies (n = 74, Kewala 1984; Stotsky 1977).
10.1 Repeated need for rapid tranquillisation
There were no significant differences between haloperidol and thiothixene with respect to the number of participants who required more than one injection (1 RCT, n = 30, RR 1.07, 95% CI 0.89 to 1.28) and more than three (1 RCT, n = 30, RR 2.50, 95% CI 0.57 to 10.93, Analysis 10.1).
10.1. Analysis.
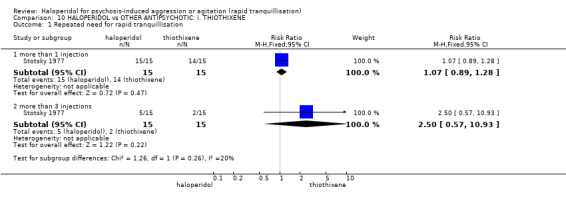
Comparison 10 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: i. THIOTHIXENE, Outcome 1 Repeated need for rapid tranquillisation.
10.2 Specific behaviours
Although agitation was rated as ‘adverse effect’, we have reported it here as it also reflects how we have done so with other comparisons. The study found no difference between those allocated to haloperidol and those given thiothixine (1 RCT, n = 44, RR 0.28, 95% CI 0.01 to 6.52, Analysis 10.2).
10.2. Analysis.
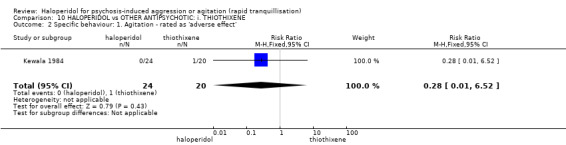
Comparison 10 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: i. THIOTHIXENE, Outcome 2 Specific behaviour: 1. Agitation ‐ rated as 'adverse effect'.
10.3 Global outcome
In one study six people who received haloperidol compared with two people who received thiothixene exhibited no response to the first injection (n = 44, RR 2.50, 95% CI 0.57 to 11.05). This one trial continued to rate people hourly for five hours and never found a clear difference (Analysis 10.3).
10.3. Analysis.
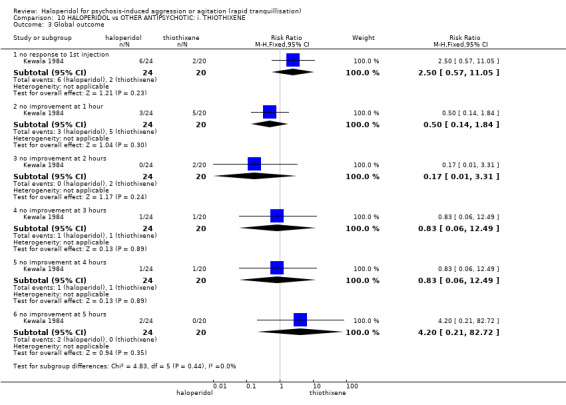
Comparison 10 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: i. THIOTHIXENE, Outcome 3 Global outcome.
10.4 Adverse effects
10.4.1 General
There were no differences between the groups in terms of the number of participants who experienced one or more adverse effects (2 RCTs, n = 74, RR 1.47, 95% CI 0.97 to 2.22, Analysis 10.4).
10.4. Analysis.
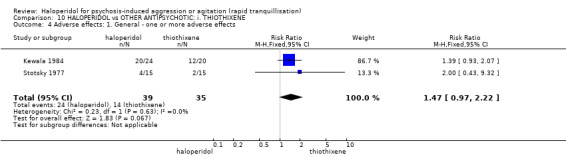
Comparison 10 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: i. THIOTHIXENE, Outcome 4 Adverse effects: 1. General ‐ one or more adverse effects.
10.4.2 Specific
There were no clear differences between the two drugs in terms of movement disorders (e.g. ataxia, 1 RCT, n = 44, RR 0.83, 95% CI 0.06 to 12.49, Analysis 10.5) and a series of other adverse effect outcomes (Analysis 10.6).
10.5. Analysis.
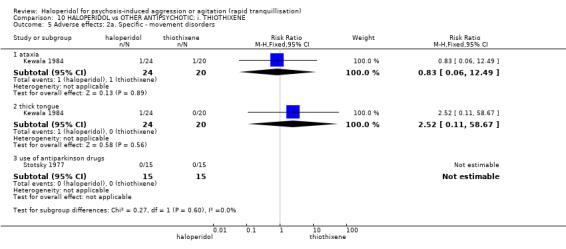
Comparison 10 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: i. THIOTHIXENE, Outcome 5 Adverse effects: 2a. Specific ‐ movement disorders.
10.6. Analysis.
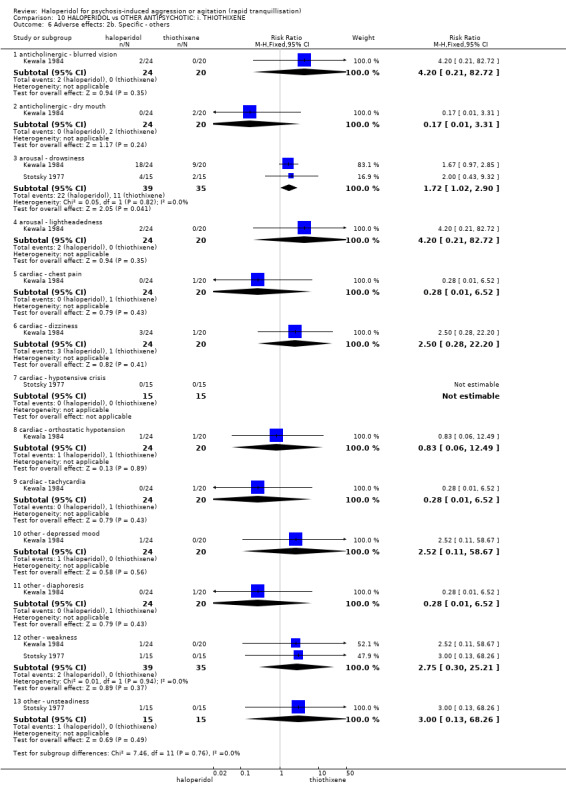
Comparison 10 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: i. THIOTHIXENE, Outcome 6 Adverse effects: 2b. Specific ‐ others.
11. COMPARISON 11. HALOPERIDOL versus OTHER ANTIPSYCHOTICS: j. ZIPRASIDONE
We found five relevant studies (total n = 829, Baldacara 2011; Brook 2000; Li 2006; Shu 2010; Yin 2012).
11.1 Repeated need for tranquillisation
There were no significant differences between haloperidol and ziprasidone in terms of additional drugs for tranquillisation up to two hours (1 RCT, n = 60, RR 0.36, 95% CI 0.13 to 1.01); however, the same outcome up to 24 hours revealed a significant difference in favour of ziprasidone (1 RCT, n = 60, RR 1.64, 95% CI 1.07 to 2.53, Analysis 11.1).
11.1. Analysis.
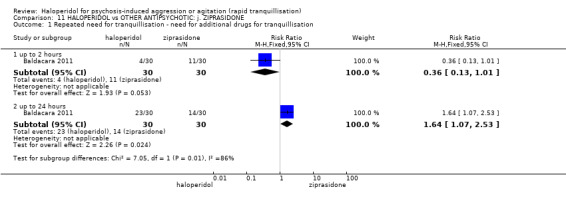
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 1 Repeated need for tranquillisation ‐ need for additional drugs for tranquillisation.
11.2 Specific behaviour
11.2.1 Agitation
In one study (n = 231), there were no significant differences between groups in their average endpoint scores on the PANSS‐EC across any time period from two hours (MD 0.06, 95% CI ‐1.13 to 1.25), right on to 72 hours (MD 0.62, 95% CI ‐0.45 to 1.69, Analysis 11.2).
11.2. Analysis.
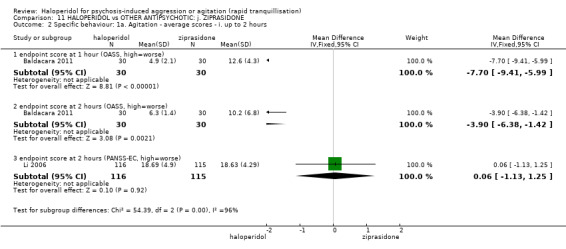
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 2 Specific behaviour: 1a. Agitation ‐ average scores ‐ i. up to 2 hours.
However, in another study (n = 60), there were significant differences between groups in their average endpoint scores on the OASS: in favour of haloperidol at one hour (MD ‐7.70, 95% CI ‐9.41 to ‐5.99), and at two hours (MD ‐3.90, 95% CI ‐6.38 to ‐1.42, Analysis 11.2), in favour of ziprasidone at six hours (MD 0.20, 95% CI 0.05 to 0.35), and at 12 hours (MD 0.90, 95% CI 0.82 to 0.98, Analysis 11.4).
11.4. Analysis.
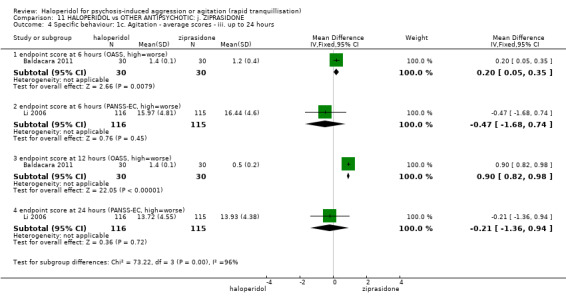
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 4 Specific behaviour: 1c. Agitation ‐ average scores ‐ iii. up to 24 hours.
11.2.2 Aggression
There were no significant differences between groups in their average endpoint scores on the OAS until six hours, when there was a significant difference in favour of ziprasidone (1 RCT, n = 60, MD 0.70, 95% CI 0.60 to 0.80), confirmed at 12 hours (MD 0.20, 95% CI 0.07 to 0.33, Analysis 11.8).
11.8. Analysis.
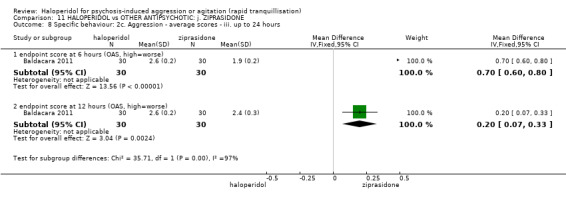
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 8 Specific behaviour: 2c. Aggression ‐ average scores ‐ iii. up to 24 hours.
11.3 Global outcome
In one study (n = 132), there were no significant differences between the groups with regard to the need for anxiolytic drugs (RR 1.11, 95% CI 0.84 to 1.48) or hypnotic medications (RR 0.71, 95% CI 0.20 to 2.50) up to seven days.
Also, there were no significant differences between groups in terms of additional restraint (1 RCT, n = 60, RR 0.60, 95% CI 0.25 to 1.44, Analysis 11.9). Using the CGI‐S, this same study found people in the ziprasidone group had significantly improved average change scores for symptom severity when rated at three days (n = 132, MD 0.34, 95% CI 0.13 to 0.55) and seven days (n = 132, MD 0.51, 95% CI 0.07 to 0.95, Analysis 11.10).
11.9. Analysis.
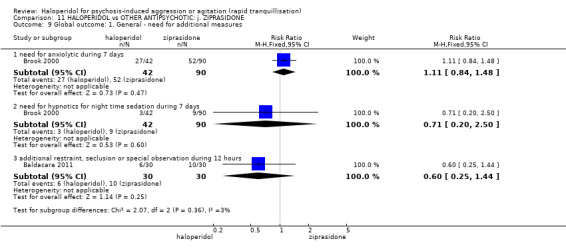
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 9 Global outcome: 1. General ‐ need for additional measures.
11.10. Analysis.
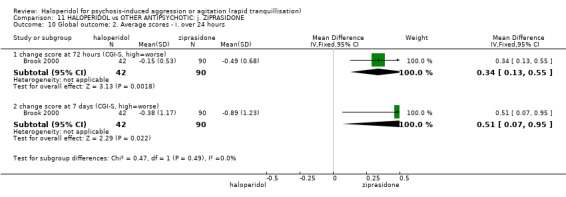
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 10 Global outcome: 2. Average scores ‐ i. over 24 hours.
11.4 Mental state
In one trial (n = 60), people were rated with BPRS at two hours (MD ‐4.25, 95% CI ‐10.16 to 1.66, Analysis 11.11), four hours (MD ‐4.60, 95% CI ‐9.60 to 0.40, Analysis 11.12), 24 hours (MD ‐4.00, 95% CI ‐9.09 to 1.09, Analysis 11.13), and 48 hours (MD ‐2.05, 95% CI ‐6.45 to 2.35, Analysis 11.14); the BPRS was also rated at 72 hours (3 RCTs, n = 511, MD ‐0.43, 95% CI ‐1.93 to 1.07), and at seven days (1 RCT, n = 132, MD 2.93, 95% CI ‐0.81 to 6.67, Analysis 11.14).
11.11. Analysis.
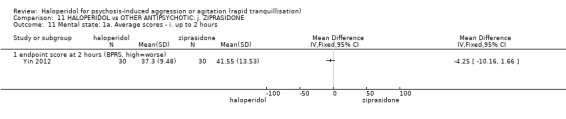
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 11 Mental state: 1a. Average scores ‐ i. up to 2 hours.
11.12. Analysis.
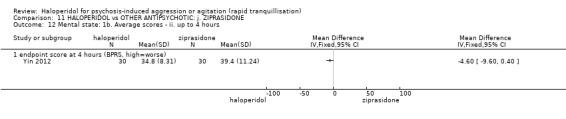
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 12 Mental state: 1b. Average scores ‐ ii. up to 4 hours.
11.13. Analysis.
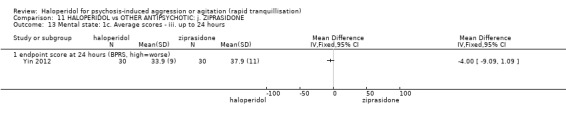
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 13 Mental state: 1c. Average scores ‐ iii. up to 24 hours.
11.14. Analysis.
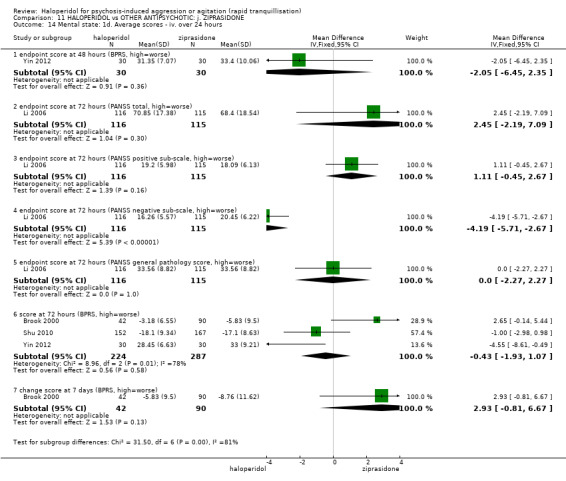
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 14 Mental state: 1d. Average scores ‐ iv. over 24 hours.
Another trial (n = 231) rated people at 72 hours on the PANSS. There were no differences between the groups on the positive sub‐scale (MD 1.11, 95% CI ‐0.45 to 2.67), general pathology score (MD 0.00, 95% CI ‐2.27 to 2.27) or total score (MD 2.45, 95% CI ‐2.19 to 7.09). However, there was a significant difference in favour of haloperidol between groups on the negative sub‐scale (MD ‐4.19, 95% CI ‐5.71 to ‐2.67, Analysis 11.14).
11.5 Adverse effects
For several of the studies reporting of adverse effects only occurred when > 2% or 5% or even 10% of people suffered the effect. This results in under‐reporting.
11.5.1 General
No significant differences were found between the groups in terms of experiencing one or more adverse effects at 12 hours (1 RCT, n = 60, RR 1.38, 95% CI 0.64 to 2.93, Analysis 11.15). At 72 hours, significantly more people in the haloperidol group had experienced one or more adverse effects (4 RCTs, n = 799, RR 1.74, 95% CI 1.47 to 2.06). However, by seven days, the difference between the groups was not statistically significant (1 RCT, n = 132, RR 1.31, 95% CI 0.93 to 1.83, Analysis 11.16).
11.15. Analysis.
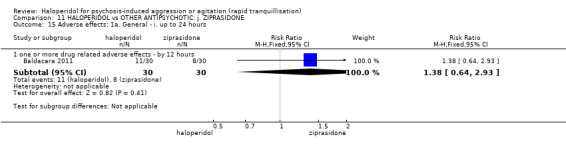
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 15 Adverse effects: 1a. General ‐ i. up to 24 hours.
11.16. Analysis.
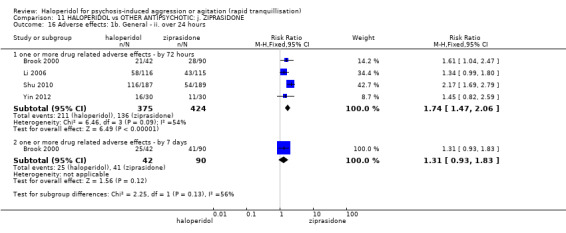
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 16 Adverse effects: 1b. General ‐ ii. over 24 hours.
No one experienced an adverse effect that was thought to be ‘severe’ (Analysis 11.17).
11.17. Analysis.
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 17 Adverse effects: 1c. General ‐ serious.
11.5.2 Specific
a. Anticholinergic
More people with haloperidol experienced episodes of blurred vision (1 RCT, n = 231, RR 3.97, 95% CI 1.15 to 13.68), dry mouth (1 RCT, n = 231, RR 2.97, 95% CI 1.22 to 7.22) and hypersalivation (1 RCT, n = 231, RR 2.55, 95% CI 1.11 to 5.87); no significant differences were found concerning constipation (1 RCT, n = 376, RR 0.40, 95% CI 0.08 to 2.06, Analysis 11.18).
11.18. Analysis.
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 18 Adverse effects: 2a. Specific ‐ anticholinergic.
b. Arousal
There were no differences between haloperidol and ziprasidone using both dichotomous assessments of excessive sedation (1 RCT, n = 60, RR 1.00, 95% CI 0.22 to 4.56), insomnia (1 RCT, n = 376, RR 0.51, 95% CI 0.05 to 5.53), lethargy (1 RCT, n = 231, RR 1.26, 95% CI 0.67 to 2.35) and somnolence (1 RCT, n = 376, RR 1.16, 95% CI 0.43 to 3.12, Analysis 11.19), and continuous rating scales (Analysis 11.20, Analysis 11.21, Analysis 11.22).
11.19. Analysis.
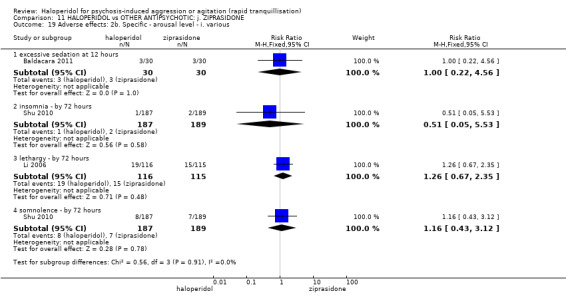
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 19 Adverse effects: 2b. Specific ‐ arousal level ‐ i. various.
11.20. Analysis.
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 20 Adverse effects: 2c. Specific ‐ arousal level ‐ ii. average scores ‐ i. up to 2 hours.
11.21. Analysis.
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 21 Adverse effects: 2d. Specific ‐ arousal level ‐ ii. average scores ‐ ii. up to 4 hours.
11.22. Analysis.
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 22 Adverse effects: 2e. Specific ‐ arousal level ‐ ii. average scores ‐ iii. up to 24 hours.
b. Cardiovascular
Cardiac adverse effects seemed more common in the ziprasidone group (e.g. ECG abnormal by 72 hours 2 RCTs, n = 607, RR 0.38, 95% CI 0.17 to 0.84; tachycardia by 72 hours 3 RCTs, n = 739, RR 0.49, 95% CI 0.23 to 1.04, Analysis 11.23).
11.23. Analysis.
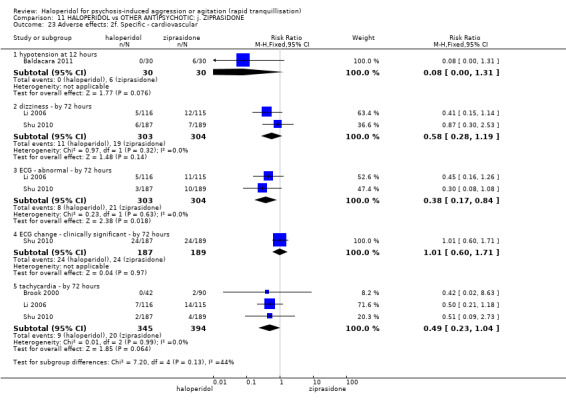
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 23 Adverse effects: 2f. Specific ‐ cardiovascular.
c. Gastrointestinal
This also seems to apply to gastric upset – although not to a level that is statistically significant (e.g. vomiting between zero and three days 1 RCT, n = 132, RR 0.30, 95% CI 0.02 to 5.72; between zero and seven days 1 RCT, n = 132, RR 0.16, 95% CI 0.01 to 2.82, Analysis 11.24).
11.24. Analysis.
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 24 Adverse effects: 2g. Specific ‐ gastrointestinal.
d. Haematological
Abnormal laboratory results were recorded for participants in both groups, but there were no significant differences across a range of measures (e.g. abnormal lab tests – by three days, 2 RCTs, n = 508, RR 0.90, 95% CI 0.69 to 1.17, Analysis 11.25).
11.25. Analysis.
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 25 Adverse effects: 2h. Specific ‐ hematological tests.
e. Movement disorders
Over a wide range of measures, ziprasidone caused fewer movement disorders than haloperidol (e.g. akathisia by 72 hours 3 RCTs, n = 739, RR 2.32, 95% CI 1.34 to 4.01; acute dystonia by 72 hours 2 RCTs, n = 508, RR 10.26, 95% CI 1.67 to 63.17). Overall, more people in the haloperidol group were reported to suffer EPS during 12 hours (1 RCT, n = 60, RR 11.00, 95% CI 0.64 to 190.53) and at a significant level during 72 hours (2 RCTs, n = 508, RR 19.13, 95% CI 7.59 to 48.21) and at seven days (1 RCT, n = 132, RR 34.29, 95% CI 4.70 to 250.02, Analysis 11.26).
11.26. Analysis.
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 26 Adverse effects: 2i. Specific ‐ movement disorders ‐ i. binary measures.
Continuous measures of movement disorders partially supported these findings (Analysis 11.27): a higher incidence of EPS and akathisia in the haloperidol group compared with the ziprasidone group according to the average score on the SAS at three days (2 RCTs, n = 191, MD 3.59, 95% CI 2.15 to 5.03), average change scores at seven days (n = 131, MD 6.10, 95% CI 3.91 to 8.29), and the average change score on the BAS at three days (n = 131, MD 0.47, 95% CI 0.18 to 0.76) and seven days (n = 131, MD 0.90, 95% CI 0.51 to 1.29).
11.27. Analysis.
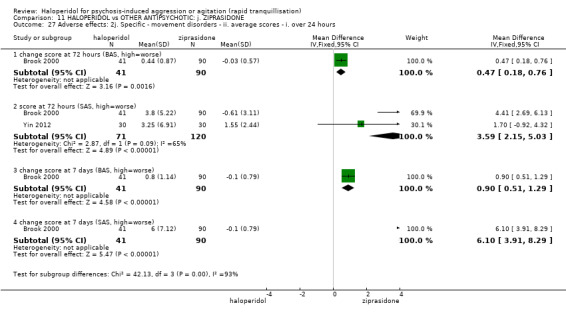
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 27 Adverse effects: 2j. Specific ‐ movement disorders ‐ ii. average scores ‐ i. over 24 hours.
11.6 Leaving the study early
There were no significant differences with regard to the number of participants who left the study early for any reasons during 72 hours (1 RCT, n = 376, RR 1.77, 95% CI 0.53 to 5.94) or within seven days (1 RCT, n = 132, RR 2.14, 95% CI 0.86 to 5.32, Analysis 11.29).
11.29. Analysis.
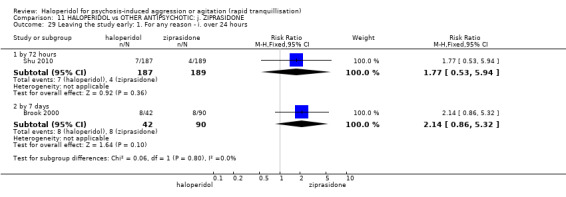
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 29 Leaving the study early: 1. For any reason ‐ i. over 24 hours.
There were also no differences between the groups with regards to the number of participants who left studies early because of adverse effects (by 72 hours, 2 RCTs, n = 508, RR 2.40, 95% CI 0.45 to 12.75; seven days 1 RCT, n = 132, RR 0.54, 95% CI 0.06 to 4.65). There were no differences between number of people who withdrew consent (1 RCT, n = 376, RR 5.05, 95% CI 0.24 to 104.55) or left for any other reason (1 RCT, n = 376, RR 0.20, 95% CI 0.01 to 4.18) by 72 hours (Analysis 11.30).
11.30. Analysis.
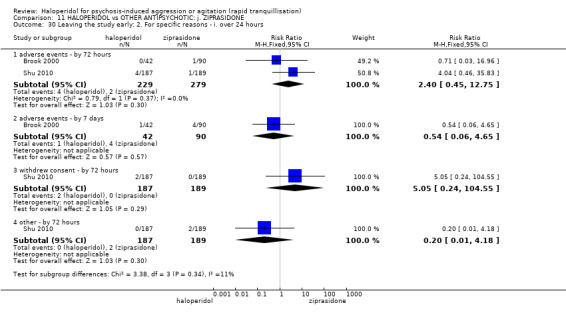
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 30 Leaving the study early: 2. For specific reasons ‐ i. over 24 hours.
12. COMPARISON 12. HALOPERIDOL versus OTHER ANTIPSYCHOTICS: k. ZUCLOPENTHIXOL ACETATE
We found one study (n = 70, Taymeeyapradit 2002).
12.1 Repeated need for tranquillisation
More people in the haloperidol group required more than three injections (n = 70, RR 2.54, 95% CI 1.19 to 5.46, Analysis 12.1).
12.1. Analysis.
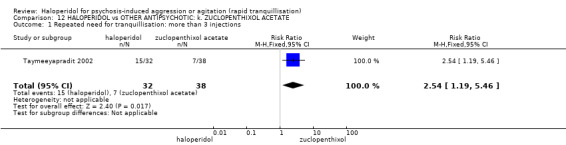
Comparison 12 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: k. ZUCLOPENTHIXOL ACETATE, Outcome 1 Repeated need for tranquillisation: more than 3 injections.
12.2 Adverse effects
One person in the haloperidol group suffered a reaction at the injection site, whereas no one allocated zuclopenthixol experienced this effect (n = 70, RR 3.55, 95% CI 0.15 to 84.14, Analysis 12.3). Seven people allocated to haloperidol and two in the zuclopenthixol acetate group developed a tremor during seven days (n = 70, RR 4.16, 95% CI 0.93 to 18.62, Analysis 12.2).
12.3. Analysis.
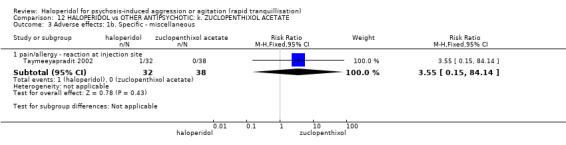
Comparison 12 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: k. ZUCLOPENTHIXOL ACETATE, Outcome 3 Adverse effects: 1b. Specific ‐ miscellaneous.
12.2. Analysis.
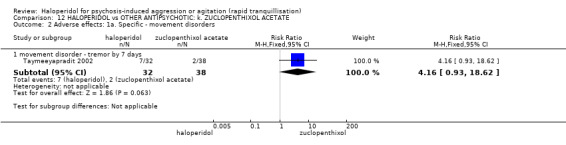
Comparison 12 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: k. ZUCLOPENTHIXOL ACETATE, Outcome 2 Adverse effects: 1a. Specific ‐ movement disorders.
12.3 Leaving the study early
No participants in either group left the study early (Analysis 12.4).
12.4. Analysis.
Comparison 12 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: k. ZUCLOPENTHIXOL ACETATE, Outcome 4 Leaving the study early.
13. HALOPERIDOL versus BENZODIAZEPINE: a. FLUNITRAZEPAM
We found one relevant study (n = 28, Dorevitch 1999).
13.1 Specific behaviours ‐ aggression
The Dorevitch 1999 study found no differences in the number of participants in the haloperidol and flunitrazepam groups who had a ≥ 50% reduction on the OAS by 90 minutes (n = 28, RR 1.15, 95% CI 0.86 to 1.55, Analysis 13.1).
13.1. Analysis.
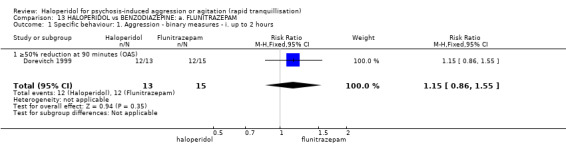
Comparison 13 HALOPERIDOL vs BENZODIAZEPINE: a. FLUNITRAZEPAM, Outcome 1 Specific behaviour: 1. Aggression ‐ binary measures ‐ i. up to 2 hours.
13.2 Global outcome ‐ need for seclusion or restraint
No participants in either the haloperidol or flunitrazepam group required further seclusion or restraint (Analysis 13.2).
13.2. Analysis.
Comparison 13 HALOPERIDOL vs BENZODIAZEPINE: a. FLUNITRAZEPAM, Outcome 2 Global outcome: 1. Need for seclusion or restraint.
13.3 Adverse effects ‐ EPS
No participants in either the haloperidol or flunitrazepam group have experienced extrapyramidal symptoms (Analysis 13.3).
13.3. Analysis.
Comparison 13 HALOPERIDOL vs BENZODIAZEPINE: a. FLUNITRAZEPAM, Outcome 3 Adverse effects: 1. Specific ‐ movement disorders ‐ i. binary measures ‐ i. by 30 minutes.
14. COMPARISON 14. HALOPERIDOL versus BENZODIAZEPINE: b. LORAZEPAM
We found four relevant studies (n = 207, Battaglia 1997; Foster 1997; Garza‐Trevino 1989; Salzman 1991).
14.1 Tranquillisation or asleep ‐ not asleep
There were no significant differences between the haloperidol and lorazepam groups with regard to the number of participants asleep at one hour (1 RCT, n = 60, RR 1.05, 95% CI 0.76 to 1.44). However, by three hours, significantly more people were asleep in the lorazepam group compared with the haloperidol group (1 RCT, n = 66, RR 1.93, 95% CI 1.14 to 3.27, Analysis 14.1).
14.1. Analysis.
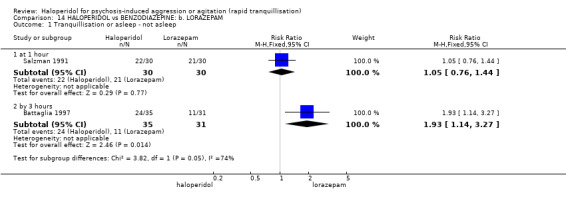
Comparison 14 HALOPERIDOL vs BENZODIAZEPINE: b. LORAZEPAM, Outcome 1 Tranquillisation or asleep ‐ not asleep.
14.2 Repeated need for rapid tranquillisation
There were no differences in numbers requiring more than one and more than three injections (1 RCT, n = 66, RR 1.14, 95% CI 0.91 to 1.43 and RR 1.11, 95% CI 0.50 to 2.45, respectively, Analysis 14.2).
14.2. Analysis.
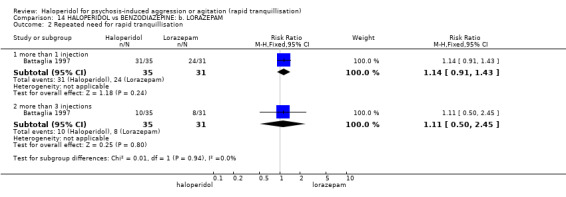
Comparison 14 HALOPERIDOL vs BENZODIAZEPINE: b. LORAZEPAM, Outcome 2 Repeated need for rapid tranquillisation.
14.3 Global outcome ‐ no overall improvement
One trial (n = 44) found no difference between groups for the outcome of ‘showing no improvement at 30 minutes’ (RR 1.10, 95% CI 0.70 to 1.71). By 60 minutes this had not changed (1 RCT, n = 44, RR 1.64, 95% CI 0.54 to 5.03). All participants in both groups had reportedly exhibited global improvement beyond 60 minutes (Analysis 14.3).
14.3. Analysis.
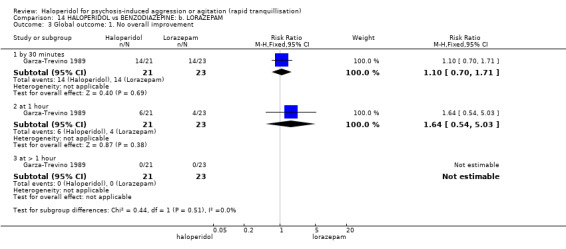
Comparison 14 HALOPERIDOL vs BENZODIAZEPINE: b. LORAZEPAM, Outcome 3 Global outcome: 1. No overall improvement.
14.4 Mental state ‐ average endpoint score
No differences were found between the haloperidol and lorazepam groups for improvement in mental state as measured by BPRS at one hour (1 RCT, n = 37, MD 3.26, 95% CI ‐4.13 to 10.65), and several times up to four hours (Analysis 14.4).
14.4. Analysis.
Comparison 14 HALOPERIDOL vs BENZODIAZEPINE: b. LORAZEPAM, Outcome 4 Mental state: 1a. Average scores ‐ i. up to 2 hours.
14.5 Adverse effects
14.5.1 General
Approximately a third of the participants in both groups experienced one or more drug‐related adverse effect (1 RCT, n = 66, RR 1.13, 95% CI 0.60 to 2.10). No participants in either group were reported to have experienced serious adverse effects (Analysis 14.6).
14.6. Analysis.
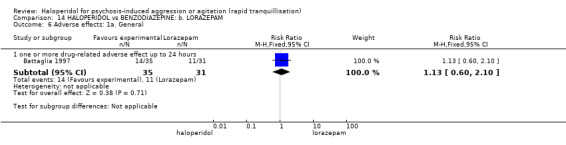
Comparison 14 HALOPERIDOL vs BENZODIAZEPINE: b. LORAZEPAM, Outcome 6 Adverse effects: 1a. General.
14.5.2 Specific
There were no differences between those allocated haloperidol and lorazepam in terms of dry mouth (1 RCT, n = 66, RR 0.53, 95% CI 0.14 to 2.04, Analysis 14.8).
14.8. Analysis.
Comparison 14 HALOPERIDOL vs BENZODIAZEPINE: b. LORAZEPAM, Outcome 8 Adverse effects: 2a. Specific ‐ anticholinergic.
In terms of movement disorders and the emergence of ataxia (1 RCT, n = 66, RR 0.44, 95% CI 0.04 to 4.65), dystonia (1 RCT, n = 66, RR 3.54, 95% CI 0.42 to 30.03), speech disorder (1 RCT,n = 66, RR 1.77, 95% CI 0.35 to 9.01), rigidity (1 RCT, n = 66, RR 6.22, 95% CI 0.33 to 115.91), tremor (1 RCT, n = 66, RR 1.77, 95% CI 0.17 to 18.60) and the need for benztropine (1 RCT, n = 66, RR 1.99, 95% CI 0.68 to 5.83). One study (n = 60) reported that significantly more participants experienced EPS in the haloperidol group compared with the lorazepam group (RR 15.00, 95% CI 2.11 to 106.49, Analysis 14.11).
14.11. Analysis.
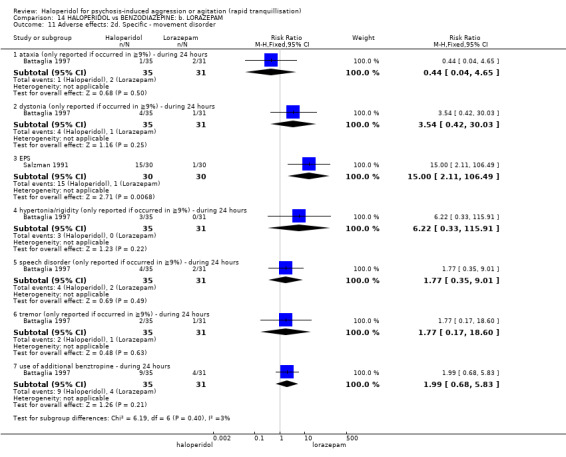
Comparison 14 HALOPERIDOL vs BENZODIAZEPINE: b. LORAZEPAM, Outcome 11 Adverse effects: 2d. Specific ‐ movement disorder.
Some other adverse effects are reported. One study (n = 60) found no differences between the groups in the number of participants asleep at one hour (RR 0.89, 95% CI 0.40 to 1.99). Clonidine was used for hypertension but not significantly more in one group or the other (1 RCT, n = 66, RR 2.67, 95% CI 0.11 to 63.17). Dizziness was not more prevalent in either group (1 RCT, n = 66, RR 0.89, 95% CI 0.19 to 4.07, Analysis 14.9).
14.9. Analysis.
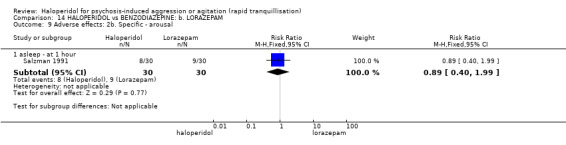
Comparison 14 HALOPERIDOL vs BENZODIAZEPINE: b. LORAZEPAM, Outcome 9 Adverse effects: 2b. Specific ‐ arousal.
15. HALOPERIDOL versus BENZODAIZEPINE: c. MIDAZOLAM
We found one relevant study (n = 84, Nobay 2004).
15.1 Tranquillisation or asleep ‐ average time
Significant differences favouring midazolam over haloperidol were reported for the average time to sedation, however these data were skewed and are presented in tabular form only (Analysis 15.1).
15.1. Analysis.
Comparison 15 HALOPERIDOL vs BENZODIAZEPINE: c. MIDAZOLAM, Outcome 1 Tranquillisation or asleep: average times in minutes ‐ (skewed data).
| Tranquillisation or asleep: average times in minutes ‐ (skewed data) | |||||
|---|---|---|---|---|---|
| Study | Intervention | Average | SD | N | Notes |
| average time to sedation | |||||
| Nobay 2004 | Haloperidol | 28.3 | 25 | 42 | |
| Nobay 2004 | Midazolam | 18.3 | 14 | 42 | |
| average time to arousal | |||||
| Nobay 2004 | Haloperidol | 126.6 | 85 | 42 | |
| Nobay 2004 | Midazolam | 81.9 | 66 | 42 | |
15.2 Global outcome
There were no clear differences between the groups for the number of people requiring a rescue drug (n = 84, RR 1.14, 95% CI 0.46 to 2.87, Analysis 15.2).
15.2. Analysis.
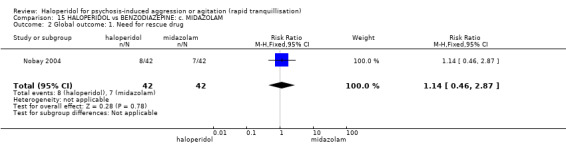
Comparison 15 HALOPERIDOL vs BENZODIAZEPINE: c. MIDAZOLAM, Outcome 2 Global outcome: 1. Need for rescue drug.
15.3 Adverse effects
Two participants were reported to have experienced adverse effects. These were both in the haloperidol group with one person experiencing hypotension (n = 84, RR 3.00, 95% CI 0.13 to 71.61) and the other, apnoea (n = 84, RR 3.00, 95% CI 0.13 to 71.61, Analysis 15.3).
15.3. Analysis.
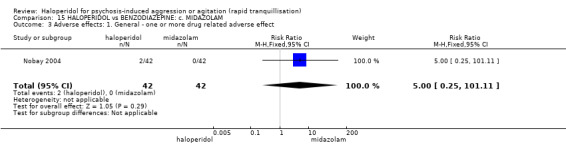
Comparison 15 HALOPERIDOL vs BENZODIAZEPINE: c. MIDAZOLAM, Outcome 3 Adverse effects: 1. General ‐ one or more drug related adverse effect.
16. HALOPERIDOL versus COMBINATIONS: a. HALOPERIDOL + LEVOMEPROMAZINE
We found one study (n = 19, Higashima 2004).
16.1. Global outcome
There was no clear difference for the outcome of ‘not improved’ (1 RCT, n = 19, RR 8.18, 95% CI 0.50 to 133.66, Analysis 16.1).
16.1. Analysis.
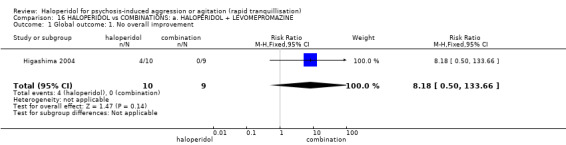
Comparison 16 HALOPERIDOL vs COMBINATIONS: a. HALOPERIDOL + LEVOMEPROMAZINE, Outcome 1 Global outcome: 1. No overall improvement.
17. HALOPERIDOL versus COMBINATIONS: b. HALOPERIDOL + LORAZEPAM
We found two studies that compared haloperidol alone with a combination of haloperidol and lorazepam (n = 113, Battaglia 1997; Garza‐Trevino 1989).
17.1 Tranquillisation or asleep
Significantly more people in the combination group were asleep at three hours than those in the haloperidol alone group (1 RCT, n = 67, RR 1.83, 95% CI 1.11 to 3.02, Analysis 17.1).
17.1. Analysis.
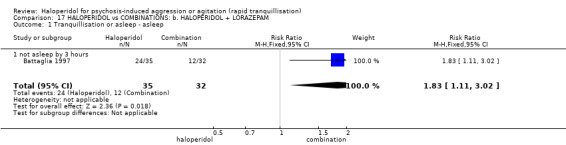
Comparison 17 HALOPERIDOL vs COMBINATIONS: b. HALOPERIDOL + LORAZEPAM, Outcome 1 Tranquillisation or asleep ‐ asleep.
17.2 Repeated need for rapid tranquillisation
Fewer people in the combination group required more than one (1 RCT, n = 67, RR 1.05, 95% CI 0.87 to 1.27) and more than three additional injections within 24 hours than in the haloperidol alone group (1 RCT, n = 67, RR 3.05, 95% CI 0.92 to 10.10, Analysis 17.2).
17.2. Analysis.
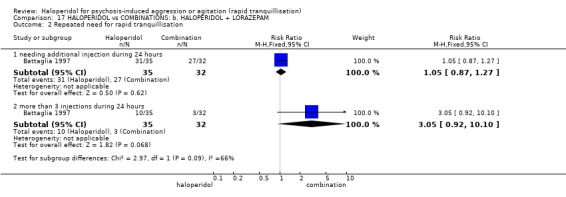
Comparison 17 HALOPERIDOL vs COMBINATIONS: b. HALOPERIDOL + LORAZEPAM, Outcome 2 Repeated need for rapid tranquillisation.
17.3 Global outcome ‐ no overall improvement
Significantly fewer people in the combination group showed no overall improvement by 30 minutes (1 RCT, n = 45, RR 2.67, 95% CI 1.25 to 5.68). By 60 minutes, although six people in the haloperidol alone group continued to show no overall improvement compared with none in the combination group; the difference between the groups was not statistically significant (1 RCT, n = 45, RR 14.77, 95% CI 0.88 to 247.54). No participants in either group showed ‘no overall improvement’ after 60 minutes. All participants in both groups demonstrated overall improvement (Analysis 17.3).
17.3. Analysis.
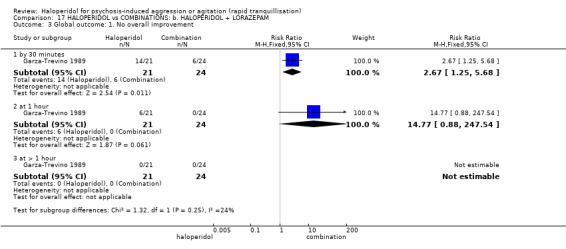
Comparison 17 HALOPERIDOL vs COMBINATIONS: b. HALOPERIDOL + LORAZEPAM, Outcome 3 Global outcome: 1. No overall improvement.
17.4 Adverse effects
17.4.1 General
One study (n = 67) found no difference for numbers experiencing one or more adverse effects (RR 1.16, 95% CI 0.62 to 2.18, Analysis 17.4).
17.4. Analysis.
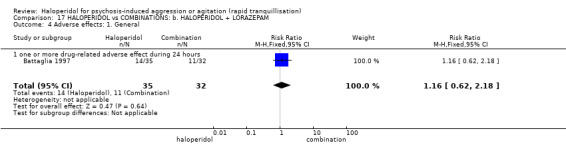
Comparison 17 HALOPERIDOL vs COMBINATIONS: b. HALOPERIDOL + LORAZEPAM, Outcome 4 Adverse effects: 1. General.
17.4.2 Specific
In terms of a series of movement disorders, one study (n = 67) found no statistically significant differences between the groups (e.g. dystonia RR 8.25, 95% CI 0.46 to 147.45; required antiparkinson medication RR 2.74, 95% CI 0.81 to 9.25, Analysis 17.7).
17.7. Analysis.
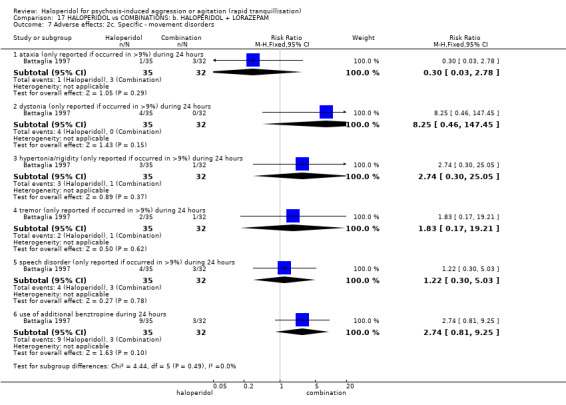
Comparison 17 HALOPERIDOL vs COMBINATIONS: b. HALOPERIDOL + LORAZEPAM, Outcome 7 Adverse effects: 2c. Specific ‐ movement disorders.
The anticholinergic effect of dry mouth was not more common in the haloperidol alone group (1 RCT, n = 67, RR 0.91, 95% CI 0.20 to 4.21, Analysis 17.5). This also applies to cardiovascular effects such as hypertension (1 RCT, n = 67, RR 0.91, 95% CI 0.06 to 14.02) and dizziness (1 RCT, n = 67, RR 1.37, 95% CI 0.24 to 7.69, Analysis 17.6).
17.5. Analysis.
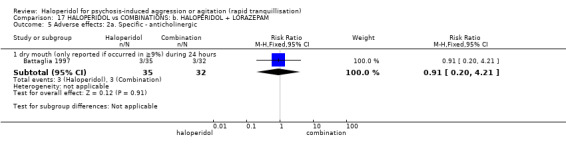
Comparison 17 HALOPERIDOL vs COMBINATIONS: b. HALOPERIDOL + LORAZEPAM, Outcome 5 Adverse effects: 2a. Specific ‐ anticholinergic.
17.6. Analysis.
Comparison 17 HALOPERIDOL vs COMBINATIONS: b. HALOPERIDOL + LORAZEPAM, Outcome 6 Adverse effects: 2b. Specific ‐ cardiovascular.
18. HALOPERIDOL versus COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM
We found one study (n = 60) comparing haloperidol versus haloperidol plus midazolam (Baldacara 2011).
18.1 Repeated need for tranquillisation
There were significant differences between groups in terms of need for additional drugs for tranquillisation at two hours in favour of haloperidol (RR 0.27, 95% CI 0.10 to 0.71); however, this was not confirmed at 12 hours (RR 0.88, 95% CI 0.69 to 1.13, Analysis 18.1).
18.1. Analysis.
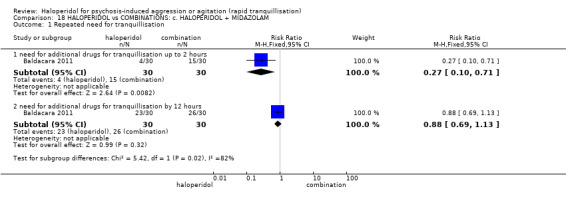
Comparison 18 HALOPERIDOL vs COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM, Outcome 1 Repeated need for tranquillisation.
18.2 Specific behaviour
18.2.1 Agitation
Average endpoint scores on OASS showed a significant difference in favour of haloperidol at one hour (MD ‐8.50, 95% CI ‐9.93 to ‐7.07), at two hours (MD ‐6.70, 95% CI ‐7.46 to ‐5.94, Analysis 18.2), at four hours (MD ‐5.50, 95% CI ‐6.48 to ‐4.52, Analysis 18.3), at six hours (MD ‐3.70, 95% CI ‐4.56 to ‐2.84), and at 12 hours (MD ‐11.20, 95% CI ‐12.24 to ‐10.16, Analysis 18.4).
18.2. Analysis.
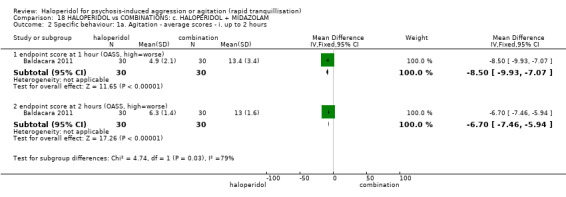
Comparison 18 HALOPERIDOL vs COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM, Outcome 2 Specific behaviour: 1a. Agitation ‐ average scores ‐ i. up to 2 hours.
18.3. Analysis.
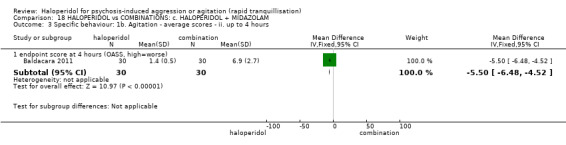
Comparison 18 HALOPERIDOL vs COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM, Outcome 3 Specific behaviour: 1b. Agitation ‐ average scores ‐ ii. up to 4 hours.
18.4. Analysis.
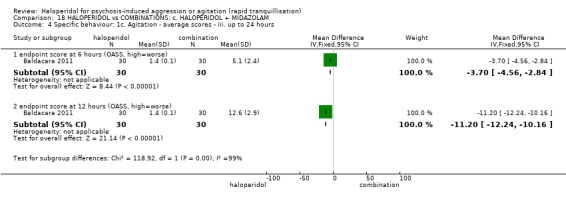
Comparison 18 HALOPERIDOL vs COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM, Outcome 4 Specific behaviour: 1c. Agitation ‐ average scores ‐ iii. up to 24 hours.
18.2.2 Aggression
Continuous measurement on OAS at different rating time points showed a significant advantage of haloperidol over the combination group (Analysis 18.5), with the exception of the rating at four hours (MD ‐0.10, 95% CI ‐0.68 to 0.48, Analysis 18.6) and at six hours (MD 0.40, 95% CI 0.30 to 0.50, Analysis 18.7).
18.5. Analysis.
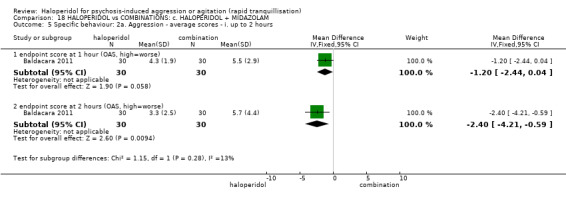
Comparison 18 HALOPERIDOL vs COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM, Outcome 5 Specific behaviour: 2a. Aggression ‐ average scores ‐ i. up to 2 hours.
18.6. Analysis.
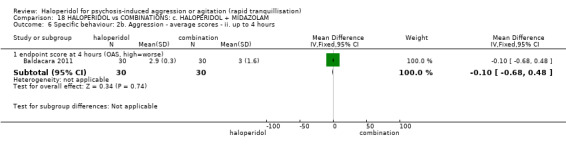
Comparison 18 HALOPERIDOL vs COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM, Outcome 6 Specific behaviour: 2b. Aggression ‐ average scores ‐ ii. up to 4 hours.
18.7. Analysis.
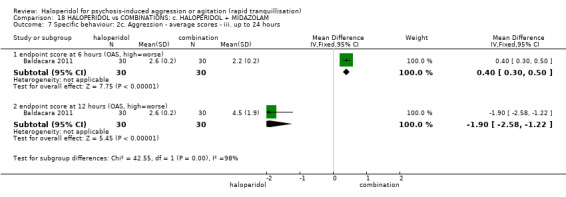
Comparison 18 HALOPERIDOL vs COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM, Outcome 7 Specific behaviour: 2c. Aggression ‐ average scores ‐ iii. up to 24 hours.
18.3 Global outcomes
Six people in the haloperidol group needed additional restraint, seclusion or special observation compared with 21 people in the combination group (1 RCT, n = 60, RR 0.29, 95% CI 0.13 to 0.61, Analysis 18.8).
18.8. Analysis.
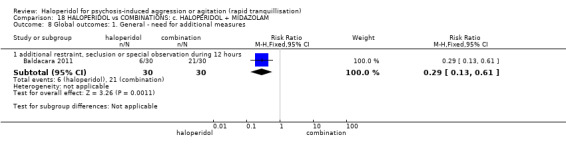
Comparison 18 HALOPERIDOL vs COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM, Outcome 8 Global outcomes: 1. General ‐ need for additional measures.
18.4 Adverse effects
18.4.1 General
There were no significant differences between the compared groups in terms of number of people experiencing one or more adverse effect (RR 0.73, 95% CI 0.41 to 1.32, Analysis 18.9).
18.9. Analysis.
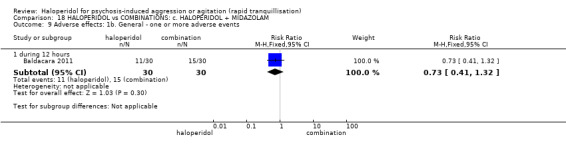
Comparison 18 HALOPERIDOL vs COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM, Outcome 9 Adverse effects: 1b. General ‐ one or more adverse events.
18.4.2 Specific
a. Arousal
More people in the combination group experienced excessive sedation by 12 hours (RR 0.25, 95% CI 0.08 to 0.80, Analysis 18.11). Continuous measurement on the Ramsay Sedation Scale (RSS) at various time points however failed to show differences between the compared groups (Analysis 18.12; Analysis 18.13, Analysis 18.14).
18.11. Analysis.
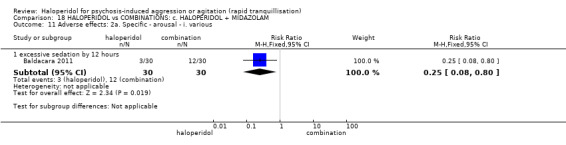
Comparison 18 HALOPERIDOL vs COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM, Outcome 11 Adverse effects: 2a. Specific ‐ arousal ‐ i. various.
18.12. Analysis.
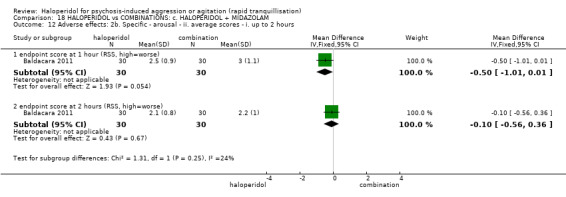
Comparison 18 HALOPERIDOL vs COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM, Outcome 12 Adverse effects: 2b. Specific ‐ arousal ‐ ii. average scores ‐ i. up to 2 hours.
18.13. Analysis.
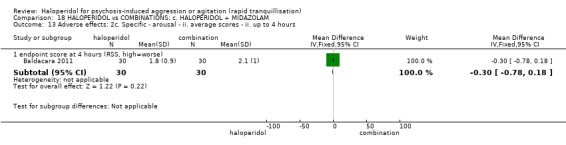
Comparison 18 HALOPERIDOL vs COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM, Outcome 13 Adverse effects: 2c. Specific ‐ arousal ‐ ii. average scores ‐ ii. up to 4 hours.
18.14. Analysis.
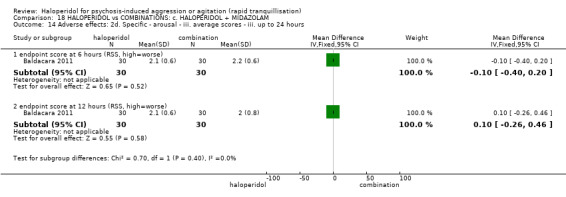
Comparison 18 HALOPERIDOL vs COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM, Outcome 14 Adverse effects: 2d. Specific ‐ arousal ‐ iii. average scores ‐ iii. up to 24 hours.
b. Cardiovascular
Five people in the combination group experienced hypotension episodes compared to no people in the haloperidol group (RR 0.09, 95% CI 0.01 to 1.57, Analysis 18.15).
18.15. Analysis.
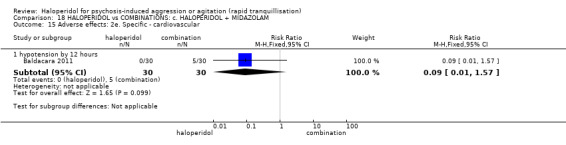
Comparison 18 HALOPERIDOL vs COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM, Outcome 15 Adverse effects: 2e. Specific ‐ cardiovascular.
c. Movement disorders
There were no significant differences in terms of EPS by 12 hours (RR 1.67, 95% CI 0.44 to 6.36, Analysis 18.16).
18.16. Analysis.
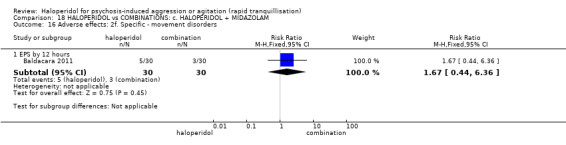
Comparison 18 HALOPERIDOL vs COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM, Outcome 16 Adverse effects: 2f. Specific ‐ movement disorders.
19. HALOPERIDOL versus COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE
We found two relevant studies (n = 376) (Baldacara 2011; Huf 2007).
19.1 Tranquillisation or asleep
More people in the haloperidol group compared with those in the combination group were not tranquil or asleep by 20 minutes (n = 316, RR 1.60, 95% CI 1.18 to 2.16), 40 minutes (n = 316, RR 1.26, 95% CI 0.83 to 1.91), 60 minutes (n = 316, RR 1.42, 95% CI 0.85 to 2.37) and 120 minutes (n = 316, RR 1.32, 95% CI 0.68 to 2.56). The difference between the groups was only statistically significant at 20 minutes (Analysis 19.1; Analysis 19.2).
19.1. Analysis.
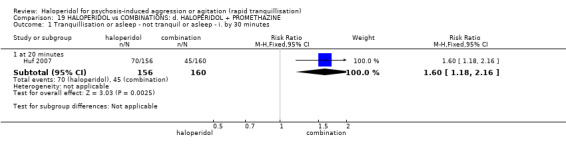
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 1 Tranquillisation or asleep ‐ not tranquil or asleep ‐ i. by 30 minutes.
19.2. Analysis.
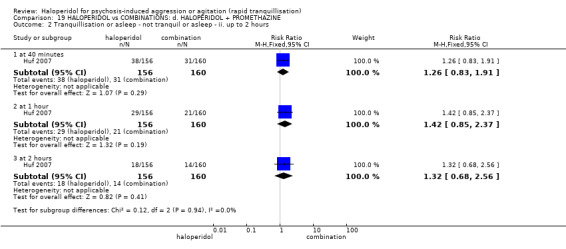
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 2 Tranquillisation or asleep ‐ not tranquil or asleep ‐ ii. up to 2 hours.
19.2 Repeated need for tranquillisation
No more people in the haloperidol alone group required repeat rapid tranquillisation by two hours (2 RCTs, n = 376, RR 0.78, 95% CI 0.43 to 1.41) nor by 12 hours (1 RCT, n = 60, RR 1.10, 95% CI 0.81 to 1.49, Analysis 19.3).
19.3. Analysis.
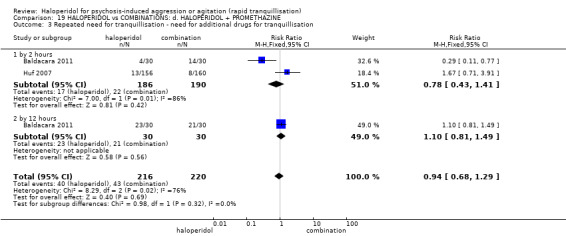
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 3 Repeated need for tranquillisation ‐ need for additional drugs for tranquillisation.
19.3 Specific behaviours
19.3.1 Agitation
When rated on OASS, there was a significant advantage of haloperidol at one hour (1 RCT, n = 60, MD ‐24.50, 95% CI ‐27.32 to ‐21.68) and two hours (1 RCT, n = 60, MD ‐9.40, 95% CI ‐10.39 to ‐8.41, Analysis 19.4), and of combination at four hours (1 RCT, n = 60, MD 3.80, 95% CI 3.27 to 4.33, Analysis 19.5), at six hours (1 RCT, n = 60, MD 2.60, 95% CI 2.13 to 3.07) and at 12 hours (1 RCT, n = 60, MD 0.80, 95% CI 0.55 to 1.05, Analysis 19.6).
19.4. Analysis.
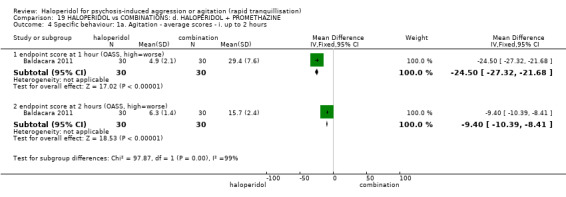
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 4 Specific behaviour: 1a. Agitation ‐ average scores ‐ i. up to 2 hours.
19.5. Analysis.
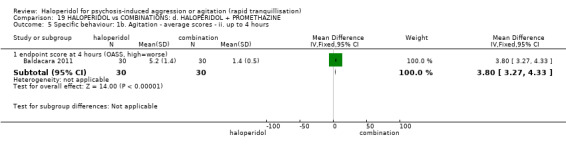
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 5 Specific behaviour: 1b. Agitation ‐ average scores ‐ ii. up to 4 hours.
19.6. Analysis.
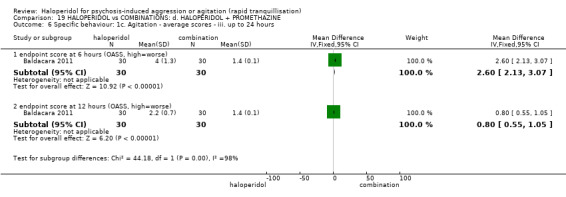
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 6 Specific behaviour: 1c. Agitation ‐ average scores ‐ iii. up to 24 hours.
19.3.2 Aggression
While at continuous rating on OAS there was an significant advantage of haloperidol at one hour (1 RCT, n = 60, MD ‐4.50, 95% CI ‐6.28 to ‐2.72, Analysis 19.7), subsequent rating resulted significantly in favour of combination at four hours (1 RCT, n = 60, MD 0.60, 95% CI 0.49 to 0.71, Analysis 19.8), at six hours (1 RCT, n = 60, MD 1.10, 95% CI 0.91 to 1.29), and at 12 hours (1 RCT, n = 60, MD 1.80, 95% CI 1.67 to 1.93, Analysis 19.9).
19.7. Analysis.
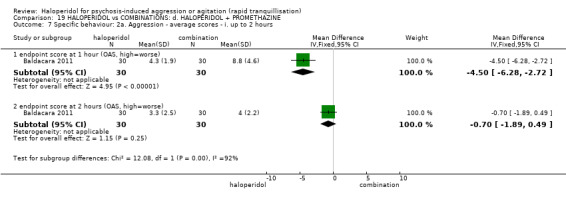
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 7 Specific behaviour: 2a. Aggression ‐ average scores ‐ i. up to 2 hours.
19.8. Analysis.
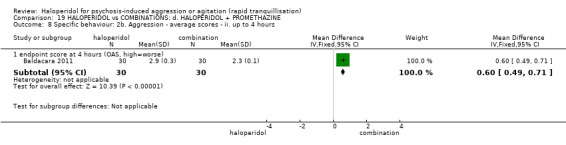
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 8 Specific behaviour: 2b. Aggression ‐ average scores ‐ ii. up to 4 hours.
19.9. Analysis.
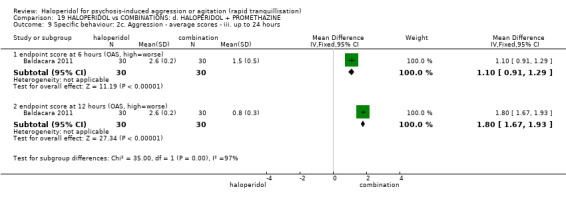
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 9 Specific behaviour: 2c. Aggression ‐ average scores ‐ iii. up to 24 hours.
No more people in the haloperidol alone group exhibited a further aggressive outburst within 24 hours (n = 316, RR 1.06. 95% CI 0.68 to 1.65, Analysis 19.10).
19.10. Analysis.
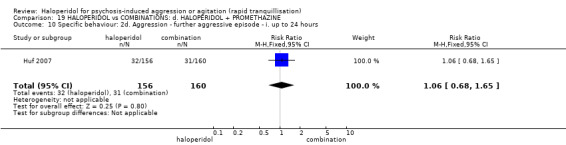
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 10 Specific behaviour: 2d. Aggression ‐ further aggressive episode ‐ i. up to 24 hours.
19.4 Global outcomes
More people in the haloperidol alone group had to be restrained by 120 minutes, however the difference between groups was not statistically significant (n = 316, RR 1.21, 95% CI 0.84 to 1.76, Analysis 19.11). The number of people who refused oral medication was roughly the same in both the haloperidol alone and combination group (n = 316, RR 0.99, 95% CI 0.61 to 1.60). Significantly more people in the haloperidol alone group required extra visits from the doctor (n = 316, RR 1.50, 95% CI 1.05 to 2.14, Analysis 19.12).
19.11. Analysis.
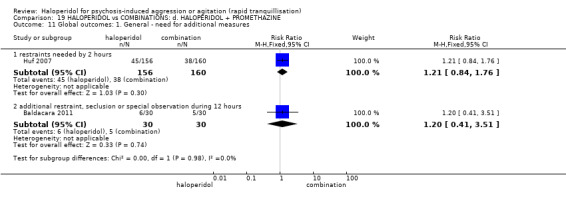
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 11 Global outcomes: 1. General ‐ need for additional measures.
19.12. Analysis.
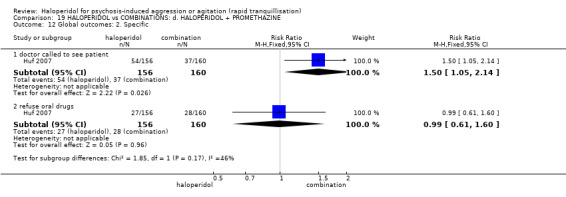
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 12 Global outcomes: 2. Specific.
19.5 Adverse effects
19.5.1 General
Significantly more people in the haloperidol alone group than the combination group experienced one or more adverse effects (2 RCTs, n = 376, RR 2.01, 95% CI 1.07 to 3.80, Analysis 19.13). There were no differences between the compared group in terms of serious adverse events (Analysis 19.14).
19.13. Analysis.
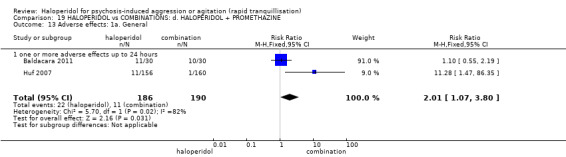
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 13 Adverse effects: 1a. General.
19.14. Analysis.
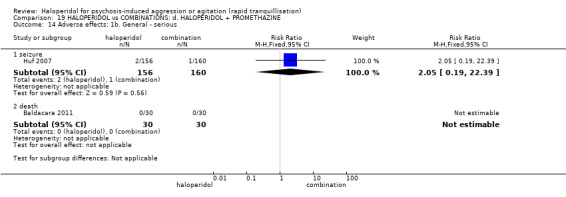
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 14 Adverse effects: 1b. General ‐ serious.
19.5.2 Specific
a. Arousal
No significant differences were found between the compared groups (Analysis 19.15; Analysis 19.16; Analysis 19.17; Analysis 19.18).
19.15. Analysis.
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 15 Adverse effects: 2a. Specific ‐ arousal ‐ i. various.
19.16. Analysis.
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 16 Adverse effects: 2b. Specific ‐ arousal ‐ ii. average scores ‐ i. up to 2 hours.
19.17. Analysis.
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 17 Adverse effects: 2c. Specific ‐ arousal ‐ ii. average scores ‐ ii. up to 4 hours.
19.18. Analysis.
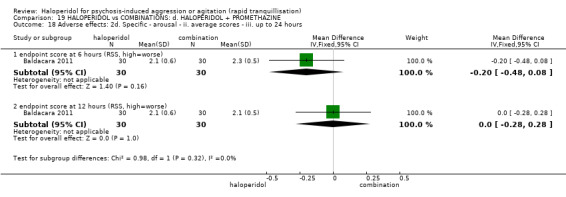
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 18 Adverse effects: 2d. Specific ‐ arousal ‐ ii. average scores ‐ iii. up to 24 hours.
b. Cardiovascular
Three people in the combination group experienced hypotension episodes compared to no people in the haloperidol group (1 RCT, n = 60, RR 0.14, 95% CI 0.01 to 2.65, Analysis 19.19).
19.19. Analysis.
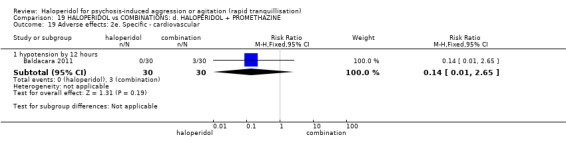
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 19 Adverse effects: 2e. Specific ‐ cardiovascular.
c. Movement disorders
Nine people in the haloperidol alone group experienced acute dystonia compared with no one in the combination group (n = 316, RR 19.48, 95% CI 1.14 to 331.92); there were no differences in terms of number of EPS episodes by 12 hours between the compared groups (1 RCT, n = 60, RR 1.00, 95% CI 0.32 to 3.10, Analysis 19.20).
19.20. Analysis.
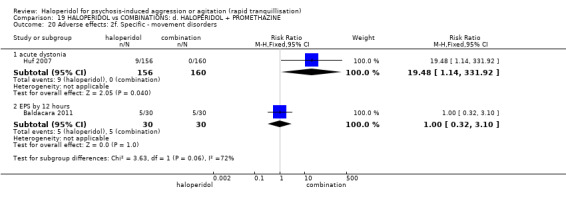
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 20 Adverse effects: 2f. Specific ‐ movement disorders.
19.6 Service outcomes
Less people in the haloperidol alone group were not discharged by 14 days, but this was not a statistically significant difference between groups (n = 316, RR 0.87, 95% CI 0.72 to 1.05, Analysis 19.21).
19.21. Analysis.
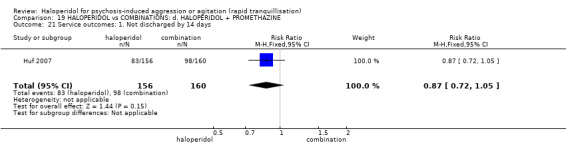
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 21 Service outcomes: 1. Not discharged by 14 days.
19.7 Leaving the study early
Seventeen people from each group left the study early, therefore there were no statistical differences between groups (Analysis 19.22). When specific reasons were cited, again, no differences were clearly shown (e.g. withdrew consent n = 316, RR 1.03, 95% CI 0.06 to 16.25, Analysis 19.22).
19.22. Analysis.
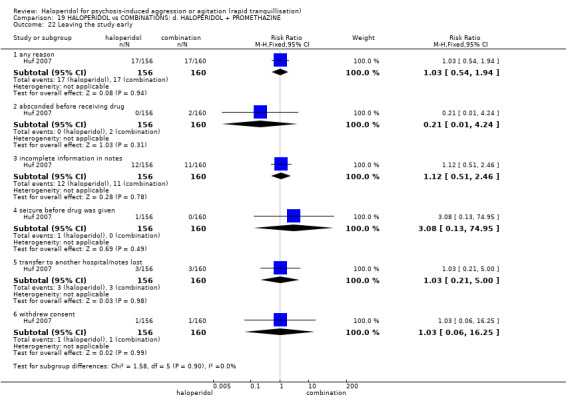
Comparison 19 HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE, Outcome 22 Leaving the study early.
20. HALOPERIDOL versus COMBINATIONS: e. HALOPERIDOL + RISPERIDONE
We found one study (n = 100) (Liu 2012).
20.1 Specific behaviour ‐ agitation
There was a statistically significant difference between the compared groups on the average scores of PANSS‐EC at 24 hours in favour of the comparison group (MD 3.82, 95% CI 1.35 to 6.29, Analysis 20.1); rating at three days, five days, one week and two weeks failed to show a significant difference (Analysis 20.2).
20.1. Analysis.
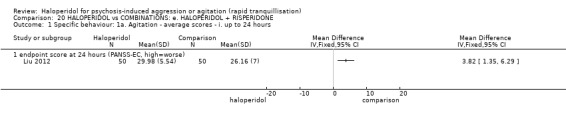
Comparison 20 HALOPERIDOL vs COMBINATIONS: e. HALOPERIDOL + RISPERIDONE, Outcome 1 Specific behaviour: 1a. Agitation ‐ average scores ‐ i. up to 24 hours.
20.2. Analysis.
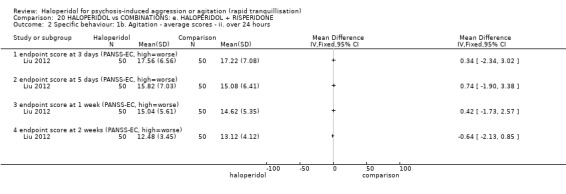
Comparison 20 HALOPERIDOL vs COMBINATIONS: e. HALOPERIDOL + RISPERIDONE, Outcome 2 Specific behaviour: 1b. Agitation ‐ average scores ‐ ii. over 24 hours.
20.2 Global outcomes
More people in the combination group experienced no improvement at 72 hours (RR 0.38, 95% CI 0.11 to 1.33), at five days (RR 0.25, 95% CI 0.03 to 2.16), at one week (RR 0.67, 95% CI 0.12 to 3.82), at two weeks (RR 0.33, 95% CI 0.01 to 7.99) compared to people in the haloperidol group; however, differences were not significant (Analysis 20.3).
20.3. Analysis.
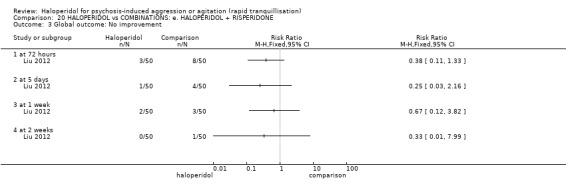
Comparison 20 HALOPERIDOL vs COMBINATIONS: e. HALOPERIDOL + RISPERIDONE, Outcome 3 Global outcome: No improvement.
20.3 Adverse effects
a. Arousal
There were no significant differences in terms of insomnia (RR 1.20, 95% CI 0.39 to 3.68) and somnolence (RR 0.92, 95% CI 0.45 to 1.88, Analysis 20.4).
20.4. Analysis.
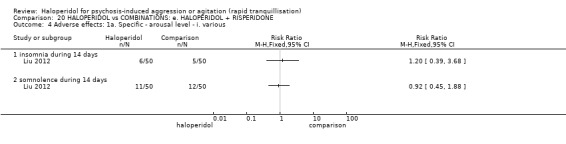
Comparison 20 HALOPERIDOL vs COMBINATIONS: e. HALOPERIDOL + RISPERIDONE, Outcome 4 Adverse effects: 1a. Specific ‐ arousal level ‐ i. various.
b. Movement disorders
There were no significant differences between the compared groups in terms of tremor (RR 0.94, 95% CI 0.54 to 1.65) and akathisia (RR 0.88, 95% CI 0.48 to 1.60) during 14 days (Analysis 20.5).
20.5. Analysis.
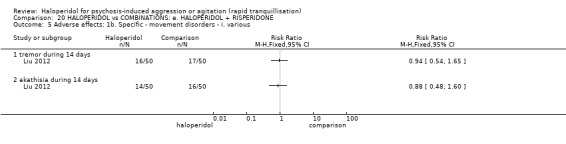
Comparison 20 HALOPERIDOL vs COMBINATIONS: e. HALOPERIDOL + RISPERIDONE, Outcome 5 Adverse effects: 1b. Specific ‐ movement disorders ‐ i. various.
c. Miscellaneous
No significant differences were found when comparing the two groups concerning dry mouth (RR 0.89, 95% CI 0.37 to 2.12), constipation (RR 1.50, 95% CI 0.45 to 4.99), and increased salivation (RR 2.00, 95% CI 0.19 to 21.36) during 14 days (Analysis 20.6).
20.6. Analysis.
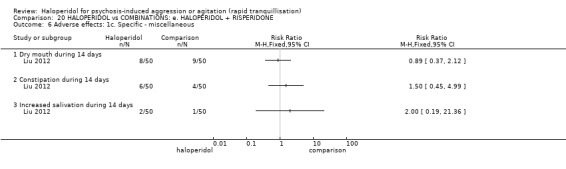
Comparison 20 HALOPERIDOL vs COMBINATIONS: e. HALOPERIDOL + RISPERIDONE, Outcome 6 Adverse effects: 1c. Specific ‐ miscellaneous.
21. HALOPERIDOL versus COMBINATIONS: f. OLANZAPINE + MAGNESIUM VALPROATE
We found one study (n = 64) that compared haloperidol versus olanzapine plus magnesium valproate (Shi 2010).
21.1 Specific behaviour ‐ agitation
There were no significant differences on average endpoint scores on PANSS‐EC at 72 hours (MD ‐0.04, 95% CI ‐1.33 to 1.25); however, rating on PANSS‐EC showed significant differences at seven days (MD 1.61, 95% CI 0.15 to 3.07) and 14 days (MD 1.82, 95% CI 0.61 to 3.03) in favour of combination group (Analysis 21.1).
21.1. Analysis.
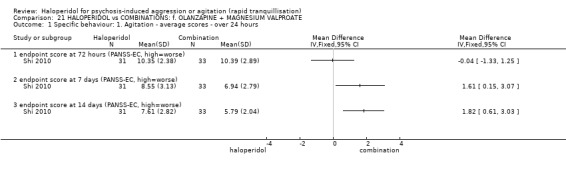
Comparison 21 HALOPERIDOL vs COMBINATIONS: f. OLANZAPINE + MAGNESIUM VALPROATE, Outcome 1 Specific behaviour: 1. Agitation ‐ average scores ‐ over 24 hours.
21.2 Global outcomes ‐ no improvement
The same number of people experienced no improvement (RR 1.06, 95% CI 0.38 to 2.95, Analysis 21.2).
21.2. Analysis.
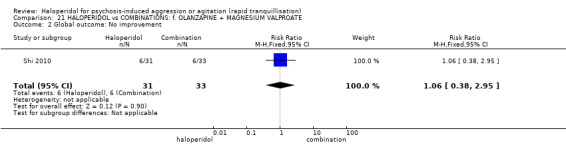
Comparison 21 HALOPERIDOL vs COMBINATIONS: f. OLANZAPINE + MAGNESIUM VALPROATE, Outcome 2 Global outcome: No improvement.
21.3 Mental state
There were no significant differences on average endpoint PANSS scores at 72 hours (MD ‐1.58, 95% CI ‐7.79 to 4.63), at seven days (MD 1.94, 95% CI ‐2.22 to 6.10), and at 14 days (MD 2.42, 95% CI ‐0.86 to 5.70, Analysis 21.3).
21.3. Analysis.
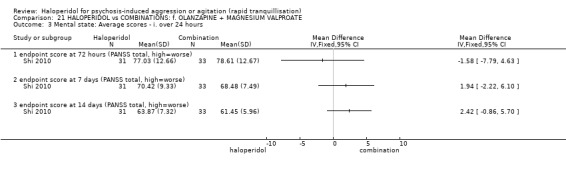
Comparison 21 HALOPERIDOL vs COMBINATIONS: f. OLANZAPINE + MAGNESIUM VALPROATE, Outcome 3 Mental state: Average scores ‐ i. over 24 hours.
21.4 Adverse effects
Few people on the combination group experienced one or more adverse effects (RR 3.06, 95% CI 1.62 to 5.79) compared to haloperidol alone group (Analysis 21.4).
21.4. Analysis.
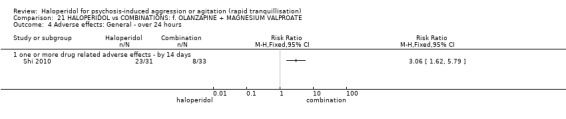
Comparison 21 HALOPERIDOL vs COMBINATIONS: f. OLANZAPINE + MAGNESIUM VALPROATE, Outcome 4 Adverse effects: General ‐ over 24 hours.
22. HALOPERIDOL versus COMBINATIONS: g. QUETIAPINE + MAGNESIUM VALPROATE
We found one study that compared haloperidol with a combination of quetiapine + magnesium valproate (n = 60, Guo 2007).
22.1 Specific behaviour ‐ agitation
Regarding the binary measure of agitation (less than 25% decrease in PANSS‐EC score) there was no difference between groups (n = 60, RR 1.17, 95% CI 0.44 to 3.06, Analysis 22.1). When endpoint scores for agitation were recorded at three, seven and 14 days no differences were apparent (n = 60, MD 14 days 1.24, 95% CI ‐0.49 to 2.97, Analysis 22.2).
22.1. Analysis.
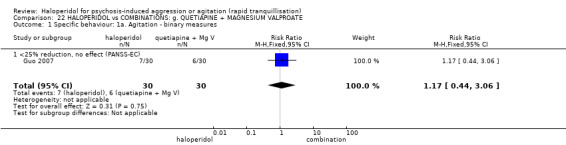
Comparison 22 HALOPERIDOL vs COMBINATIONS: g. QUETIAPINE + MAGNESIUM VALPROATE, Outcome 1 Specific behaviour: 1a. Agitation ‐ binary measures.
22.2. Analysis.
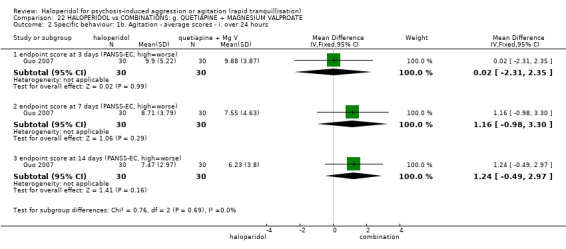
Comparison 22 HALOPERIDOL vs COMBINATIONS: g. QUETIAPINE + MAGNESIUM VALPROATE, Outcome 2 Specific behaviour: 1b. Agitation ‐ average scores ‐ i. over 24 hours.
23. HALOPERIDOL versus COMBINATIONS: h. RISPERIDONE + CLONAZEPAM
We found two studies (n = 305) that compared haloperidol versus risperidone plus clonazepam (Fang 2012; Liu 2012).
23.1 Tranquillisation or asleep
There were no significant differences in the number of people not asleep at two hours (1 RCT, n = 205, RR 1.07, 95% CI 0.82 to 1.39) and at four hours (1 RCT, n = 205, RR 0.82, 95% CI 0.66 to 1.03, Analysis 23.1).
23.1. Analysis.
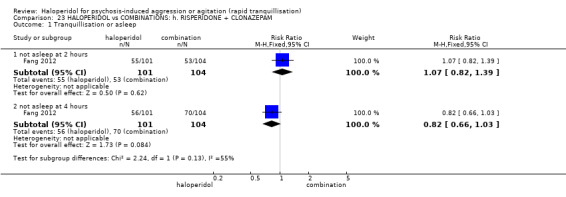
Comparison 23 HALOPERIDOL vs COMBINATIONS: h. RISPERIDONE + CLONAZEPAM, Outcome 1 Tranquillisation or asleep.
23.2 Specific behaviour ‐ agitation
Continuous measurement on average PANSS‐EC scores showed no significant differences between the compared groups at two hours (1 RCT, n = 108, MD ‐0.50, 95% CI ‐3.80 to 2.80, Analysis 23.2), at four hours (1 RCT, n = 126, MD ‐2.40, 95% CI ‐5.20 to 0.40, Analysis 23.3), at 24 hours (2 RCTs, n = 305, MD 0.71, 95% CI ‐0.56 to 1.98, Analysis 23.4), at three days (2 RCTs, n = 305, MD ‐0.41, 95% CI ‐1.66 to 0.83), at five days (2 RCTs, n = 305, MD 0.14, 95% CI ‐1.24 to 1.52), and at two weeks (1 RCT, n = 100, MD ‐0.88, 95% CI ‐2.34 to 0.58); the only exception was at week one in favour of haloperidol (1 RCT, n = 100, MD ‐2.56, 95% CI ‐4.57 to ‐0.55, Analysis 23.5).
23.2. Analysis.
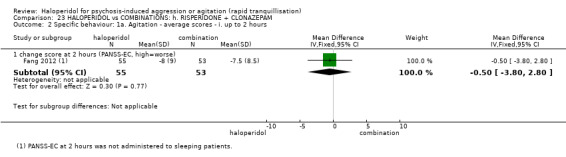
Comparison 23 HALOPERIDOL vs COMBINATIONS: h. RISPERIDONE + CLONAZEPAM, Outcome 2 Specific behaviour: 1a. Agitation ‐ average scores ‐ i. up to 2 hours.
23.3. Analysis.
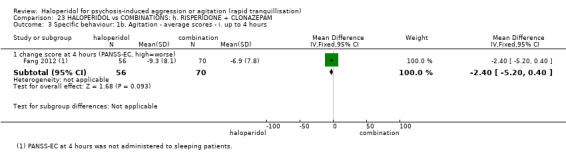
Comparison 23 HALOPERIDOL vs COMBINATIONS: h. RISPERIDONE + CLONAZEPAM, Outcome 3 Specific behaviour: 1b. Agitation ‐ average scores ‐ i. up to 4 hours.
23.4. Analysis.
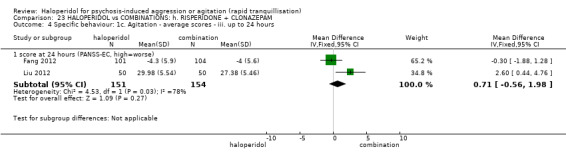
Comparison 23 HALOPERIDOL vs COMBINATIONS: h. RISPERIDONE + CLONAZEPAM, Outcome 4 Specific behaviour: 1c. Agitation ‐ average scores ‐ iii. up to 24 hours.
23.5. Analysis.
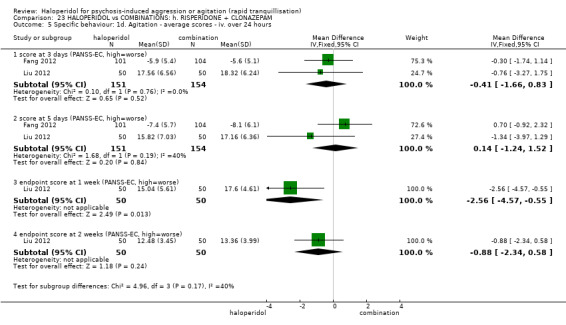
Comparison 23 HALOPERIDOL vs COMBINATIONS: h. RISPERIDONE + CLONAZEPAM, Outcome 5 Specific behaviour: 1d. Agitation ‐ average scores ‐ iv. over 24 hours.
23.3 Global outcomes
More people on the combination group experienced no improvement at 72 hours (RR 0.60, 95% CI 0.15 to 2.38), at five days (RR 0.25, 95% CI 0.03 to 2.16), at one week (RR 0.20, 95% CI 0.01 to 4.06), and at two weeks (RR 0.50, 95% CI 0.10 to 2.61) compared with haloperidol group (Analysis 23.6).
23.6. Analysis.
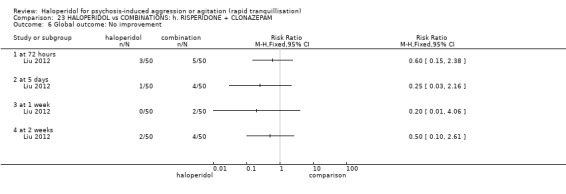
Comparison 23 HALOPERIDOL vs COMBINATIONS: h. RISPERIDONE + CLONAZEPAM, Outcome 6 Global outcome: No improvement.
23.4 Mental state
There were no significant differences on average change score on PANSS at five days (1 RCT, n = 205, MD ‐3.50, 95% CI ‐7.07 to 0.07, Analysis 23.7).
23.7. Analysis.
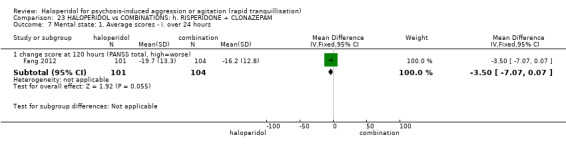
Comparison 23 HALOPERIDOL vs COMBINATIONS: h. RISPERIDONE + CLONAZEPAM, Outcome 7 Mental state: 1. Average scores ‐ i. over 24 hours.
23.5 Adverse effects
23.5.1 General
In the combination group there were fewer adverse events at a significant level (1 RCT, n = 205, RR 1.72, 95% CI 1.29 to 2.29, Analysis 23.8).
23.8. Analysis.
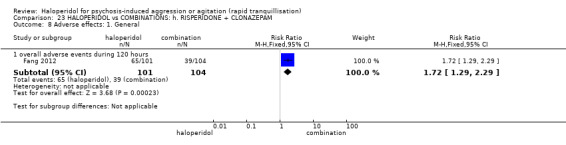
Comparison 23 HALOPERIDOL vs COMBINATIONS: h. RISPERIDONE + CLONAZEPAM, Outcome 8 Adverse effects: 1. General.
23.5.2 Specific
a. Arousal
There were no significant differences concerning arousal adverse events (e.g. insomnia during five days 1 RCT, n = 205, RR 1.54, 95% CI 0.45 to 5.31; insomnia during 14 days 1 RCT, n = 100, RR 0.50, 95% CI 0.20 to 1.23; somnolence during 14 days 1 RCT, n = 100, RR 2.75, 95% CI 0.94 to 8.06) between the compared groups (Analysis 23.9).
23.9. Analysis.
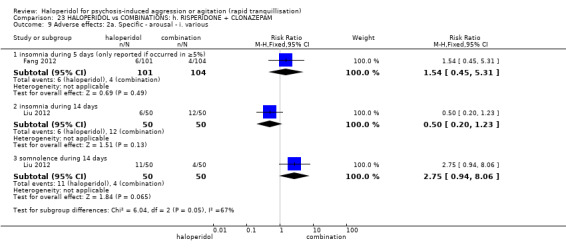
Comparison 23 HALOPERIDOL vs COMBINATIONS: h. RISPERIDONE + CLONAZEPAM, Outcome 9 Adverse effects: 2a. Specific ‐ arousal ‐ i. various.
b. Cardiovascular
There were no significant differences between the groups concerning tachycardia during five days (1 RCT, n = 205, RR 1.29, 95% CI 0.36 to 4.66, Analysis 23.10).
23.10. Analysis.
Comparison 23 HALOPERIDOL vs COMBINATIONS: h. RISPERIDONE + CLONAZEPAM, Outcome 10 Adverse effects: 2b. Specific ‐ cardiovascular ‐ i. various.
c. Movement disorders
People in the haloperidol group experienced more movement disorder‐related adverse events (e.g. akathisia during five days 1 RCT, n = 205, RR 1.63, 95% CI 1.12 to 2.38; EPS during five days 1 RCT, n = 205, RR 2.22, 95% CI 1.52 to 3.23; tremor during 14 days 1 RCT, n = 100, RR 3.20, 95% CI 1.27 to 8.07, Analysis 23.11).
23.11. Analysis.
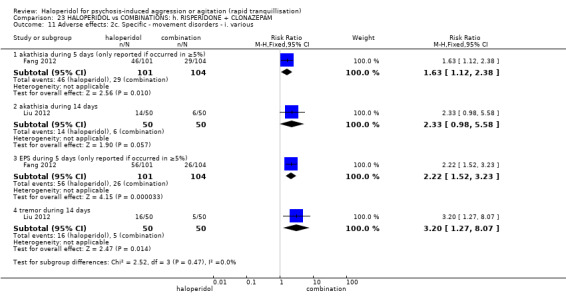
Comparison 23 HALOPERIDOL vs COMBINATIONS: h. RISPERIDONE + CLONAZEPAM, Outcome 11 Adverse effects: 2c. Specific ‐ movement disorders ‐ i. various.
d. Miscellaneous
There were no significant differences in terms of anticholinergic adverse events (Analysis 23.12).
23.12. Analysis.
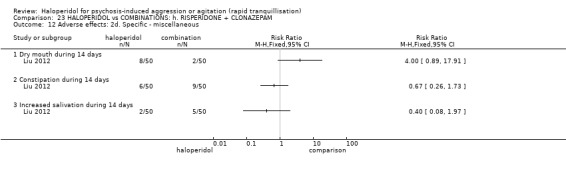
Comparison 23 HALOPERIDOL vs COMBINATIONS: h. RISPERIDONE + CLONAZEPAM, Outcome 12 Adverse effects: 2d. Specific ‐ miscellaneous.
24. HALOPERIDOL versus COMBINATIONS: i. ZIPRASIDONE + CLONAZEPAM
We found 1 study (n = 76) that compared haloperidol versus ziprasidone plus clonazepam (Zhou 2014).
24.1 Specific behaviour ‐ agitation
There were no significant differences on the average endpoint PANSS‐EC scores at 12 hours (MD ‐0.28, 95% CI ‐0.80 to 1.24, Analysis 24.1), at 48 hours (MD ‐1.54, 95% CI ‐3.26 to 0.18), at three days (MD ‐0.42, 95% CI 1.63 to 0.79), at five days (MD ‐0.22, 95% CI ‐1.78 to 1.34), and at seven days (MD ‐0.19, 95% CI ‐1.57 to 1.19, Analysis 24.2).
24.1. Analysis.
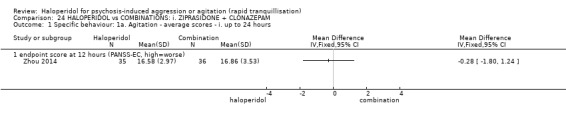
Comparison 24 HALOPERIDOL vs COMBINATIONS: i. ZIPRASIDONE + CLONAZEPAM, Outcome 1 Specific behaviour: 1a. Agitation ‐ average scores ‐ i. up to 24 hours.
24.2. Analysis.
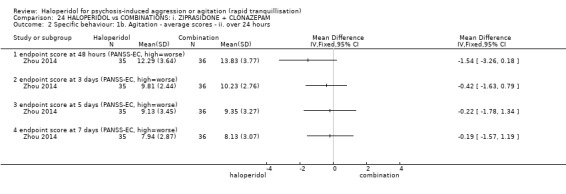
Comparison 24 HALOPERIDOL vs COMBINATIONS: i. ZIPRASIDONE + CLONAZEPAM, Outcome 2 Specific behaviour: 1b. Agitation ‐ average scores ‐ ii. over 24 hours.
24.2 Global outcomes
There were no significant differences on average endpoint CGI‐S scores at seven days (MD 0.00, 95% CI ‐0.26 to 0.26, Analysis 24.3).
24.3. Analysis.

Comparison 24 HALOPERIDOL vs COMBINATIONS: i. ZIPRASIDONE + CLONAZEPAM, Outcome 3 Global outcome: 1. Average scores ‐ i. over 24 hours.
24.3 Mental state
There were no significant differences on average endpoint PANSS total scores at seven days (MD 1.46, 95% CI ‐4.58 to 7.50) and sub‐scales (Analysis 24.4).
24.4. Analysis.
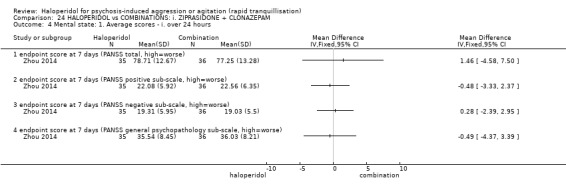
Comparison 24 HALOPERIDOL vs COMBINATIONS: i. ZIPRASIDONE + CLONAZEPAM, Outcome 4 Mental state: 1. Average scores ‐ i. over 24 hours.
24.4 Leaving the study early
Three people in the haloperidol group left the study early compared to two people in the combination group (RR 1.50, 95% CI 0.27 to 8.48, Analysis 24.5).
24.5. Analysis.
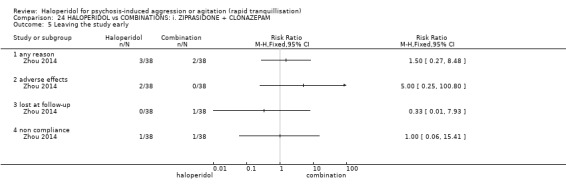
Comparison 24 HALOPERIDOL vs COMBINATIONS: i. ZIPRASIDONE + CLONAZEPAM, Outcome 5 Leaving the study early.
Discussion
Summary of main results
1. COMPARISON 1. HALOPERIDOL versus PLACEBO
It can be seen from the Table 1 that, despite five trials providing data (total n = 700), the overall quality of evidence was not high. No study discussed economic outcomes.
1.1 Tranquillisation or asleep, repeated need for tranquillisation, agitation
When compared with placebo, significantly more people in the haloperidol group were asleep at two hours and demonstrated at least a 40% reduction in Positive and Negative Syndrome Scale Excited Component (PANSS‐EC) agitation score (Bristol Myers 2004a; Bristol‐Myers 2005b). With respect to evidence for rapid tranquillisation, the evidence was downgraded to ‘very low’ for ‘tranquil or asleep’ because there was no measure at 30 minutes, therefore, two hours was the closest time point. We think this is quite distant from the emergency situation and most data for this outcome came from Battaglia 2002 study, in which there is a serious risk of bias because the method of randomisation was not described, allocation concealment was not stated, there were incomplete outcome data and the trial was sponsored by the drug company, which could possibly impact on publication bias (please see Table 1). On the continuous measures of agitation, haloperidol seems to demonstrate a discernable effect. For example, people in the haloperidol group were reported to have experienced a significantly greater reduction in agitation than those in the placebo group at two hours (3 RCTs, n = 474, MD ‐4.48, 95% CI ‐5.78 to ‐3.18, Analysis 1.4). A four‐point reduction may be of clinical meaning, but we could not find this clearly highlighted.
1.2 Global outcome, mental state
Fewer people in the placebo group exhibited ‘marked improvement' than those who received haloperidol (Analysis 1.6), and four studies found that more people in the placebo group required benzodiazepines (Analysis 1.7). All data point to the real effect of use of haloperidol in these difficult and dangerous situations. Mental state data support this impression.
1.3 Adverse effects
More people in the haloperidol group experienced one or more adverse reactions than those in the placebo group (Analysis 1.12). Bristol‐Myers 2005b reported that of all adverse effects reported, only one was rated as 'serious' ‐ this was related to a participant in the placebo group.
More people in the haloperidol group than the placebo group were reported to be "over sedated". Haloperidol did seem to not cause more cardiac problems than placebo but, unsurprisingly, movement disorders were more prevalent. Often, however, and on a range of outcomes, the differences were not statistically significant. However, significantly more people in the haloperidol group did require antiparkinson drugs and experienced Extrapyramidal Side Effects (EPS) (Analysis 1.17).
1.4 Leaving the study early
It is problematic how to interpret data on leaving the study as, in some of these trials, people may not have been fully free to leave. Only 27 of 335 people left early ‐ with no differences between groups.
2. COMPARISON 2. HALOPERIDOL versus OTHER ANTIPSYCHOTIC: a. ARIPIPRAZOLE
This comparison contains the larger trials (two RCTs, total n = 599), but, as can be seem from the Table 2, the selected outcomes are absent or of low or very low quality.
2.1 Repeated need for rapid tranquillisation
Considering how difficult repeated injections are, that use of haloperidol necessitates less is important. The quality of these data is, however, low. We do not have any direct data on tranquillisation, but there is the implication from these data that haloperidol either sedates or tranquillises more effectively than aripiprazole.
2.2 Specific behaviours ‐ agitation
There was no difference between groups at about two hours with about half of each group having a considerable reduction in agitation scores. We graded these data as of very low quality (Table 2). Two hours, is, however, a considerable time after an acute aggressive or agitated event and data at an earlier state would be helpful.
2.3 Global outcomes
All global outcomes – again all around two hours – show no difference between the two drugs (requiring additional benzodiazepines; average change scores on the Clinical Global Impression ‐ Severity (CGI‐S) rating scale; average endpoint scores on the Clinical Global Impression ‐ Improvement (CGI‐I) rating scale; average change scores on the CGI‐I rating scale in the schizophrenia subgroup). This is reassuring that aripiprazole does have some effect but its value in the very acute stage of management is not supported by these data. The few mental state data concur.
2.4 Adverse effects
For some adverse effects haloperidol seems to come off worse than aripiprazole although there was no difference between groups for 'adverse effects within 24 hours'. One person in the haloperidol group developed severe dystonia and one in the aripiprazole group had a tonic‐clonic seizure.
Evidence was not convincing that there were any differences when it came to cardiac or gastrointestinal effects. Movement disorders, however, all tended to be more common in the group allocated to haloperidol and risk of use of antiparkinson medication was increased if a person was allocated to haloperidol. Although this is not often the problem that is most paramount within an emergency, these effects result in an already difficult situation being even more unpleasant. Akathisia (RR nearly 3), for example, can be mistaken for the original agitation and result in administration of yet more haloperidol. These common effects (˜1/3 needed additional antiparkinson medication) are a considerable limitation for the value of haloperidol.
2.5 Leaving the study early
There were no significant differences in the number of people leaving early and, again, the meaning of these data is unclear as we are not sure how at liberty people were to leave the studies by the end of the trial.
Economic outcomes were not measured. Aripiprazole is considerably more expensive than haloperidol.
3. COMPARISON 3. HALOPERIDOL versus OTHER ANTIPSYCHOTICS: b. CHLORPROMAZINE
Although we found four relevant studies, all were small (total n = 153) and we could not rate overall findings more than of very low grade (Table 3), and no important outcome had all trials contributing.
3.1 Tranquillisation or asleep, repeated need for rapid tranquillisation, not marked improvement
One small study (n = 39) of poor quality reported that more people allocated to chlorpromazine did experience sleep by two hours (Analysis 3.1). This does seem to fit with clinical experience.
Another study reported the number of people requiring more than one injection and, despite the suggestion of more sedation in the chlorpromazine group, there was no clear indication that either group avoided more or fewer additional injections – but, again the trial was small and of very limited quality (Analysis 3.2).
Although there was no clear difference for the outcome of "not marked improvement", more people in the chlorpromazine group were rated as showing "not any improvement" (Analysis 3.3). We are unclear if this has meaning or is a chance finding.
3.2 Adverse effects
Reporting of adverse effects was sporadic and therefore, not very helpful. From these data it is not clear that either drug has clear advantage over the other for allergic, cardiovascular, or movement disorders (Analysis 3.4).
3.3 Leaving the study early ‐ for any reason
This was the only outcome where all trials contributed. Few people left these studies early (5%). This could be a function of the coercive aspect of the treatment. Perhaps people were not fully free to leave; significantly fewer people left the haloperidol group (Analysis 3.5).
4. COMPARISON 4. HALOPERIDOL versus OTHER ANTIPSYCHOTICS: c. DROPERIDOL
Droperidol has been taken off the market because of “commercial reasons” (Khokhar 2016). However, there remains interest in its use (Isbister 2010). We found two relevant studies (n = 255) and the outcomes were of moderate grade (higher than others in this review) (Table 4).
4.1 Tranquillisation or asleep, repeated need for rapid tranquillisation
The same amount of people in the haloperidol and droperidol groups did experience sleep by two hours (Analysis 4.1), with no significant differences concerning the time to get asleep (Analysis 4.2).
Droperidol did seem more effective ‐ more people in the haloperidol group required more than one injection, and this held true for the outcome of ‘required more than three injections’ (Analysis 4.3).
4.2 Global outcomes
To note, more people in the droperidol group required more benzodiazepines up to two hours (Analysis 4.4).
4.3 Adverse effects ‐ specific
Droperidol could be equally or more toxic than haloperidol in terms of total adverse events (Analysis 4.5) and specific adverse events (Analysis 4.6; Analysis 4.7; Analysis 4.8; Analysis 4.9).
Further studies are needed; much better evaluation is justified but in times of austerity in health care, it could be a clinically useful option.
5. COMPARISON 5. HALOPERIDOL versus OTHER ANTIPSYCHOTICS: d. LOXAPINE
We found three relevant studies (total n = 119), one of which offset adverse effects using biperiden, but that we could only rate the outcomes as being of low or very low grade (Table 5). Loxapine is widely available in several European countries, Australasia and the USA. It is a shame that we do not have better data on this interesting compound that some consider to be inexpensive, but with 'atypical' properties (Glazer 1999).
5.1 Tranquillisation or asleep, agitation, aggression
Loxapine did seem more sedating than haloperidol although the one study lacked power (n = 54, Analysis 5.1). There were no differences between groups for improvement in levels of agitation or levels of aggression but, again, these outcomes were reported by only one very small study (n = 30, Analysis 5.2).
5.2 Global outcomes, mental state
There are several ways these trials reported global outcomes and measured mental state, but never any real suggestion of an effect favouring one or other drug.
5.3 Adverse effects
Even with the use of biperiden in one study, it might have been expected to see some differences between drugs. None were apparent in these trials.
6. COMPARISON 6. HALOPERIDOL versus OTHER ANTIPSYCHOTICS: e. OLANZAPINE
We found six studies (total n = 720) but no pre‐selected outcome of importance contained data from more than 400 people and all important outcomes we had to rate as being of very low grade (Summary of findings table 6).
6.1 Tranquillisation or asleep, need for more tranquillisation, specific behaviours
Olanzapine does seem more sedating than haloperidol (Analysis 6.1). In terms of requiring additional injections, a slight advantage in favours of olanzapine was demonstrated (Analysis 6.2).
When analysing agitation rating scales, a slight tendency in favour of olanzapine was demonstrated (Analysis 6.3; Analysis 6.4, Analysis 6.5; Analysis 6.6; Analysis 6.7).
In terms of both aggression and hostility, haloperidol and olanzapine showed no difference (Analysis 6.8; Analysis 6.9; Analysis 6.10; Analysis 6.11).
6.2 Global outcome, mental state
Most global outcomes demonstrated no clear difference between the compounds. For example, use of additional restraint, seclusion or special observation was comparable (Analysis 6.12). Olanzapine seems as effective as the older drug. This also seems to apply to the measures of mental state (Analysis 6.30).
6.3 Adverse effects
It was a concern that these industry‐sponsored trials were designed to under‐report adverse effects. It had been stipulated in the design of a trial that adverse effects would be reported only if, for example, more than 10% of people in a group experienced them (Kinon 2001d). Therefore, should 9% of people experienced worrying skin reactions in Kinon 2001d, this could have been omitted from the report.
Nevertheless, it does seem clear that haloperidol does cause more movement disorders than olanzapine. This must be seen as an advantage for the newer drug when being compared with haloperidol used alone.
6.4 Economic
There was no measure for economic outcomes. In older trials this is understandable, but not for comparisons of expensive drugs and the older compounds. It is likely that the expensive olanzapine could save money in the short term ‐ but, disappointingly, this is supposition.
7. COMPARISON 7. HALOPERDIOL versus OTHER ANTIPSYCHOTICS: f. PERPHENAZINE
Considering the recent resurgence of interest in perphenazine (Lieberman 2005), this is an important comparison. The one relevant study (n = 44) at least proves that comparative testing of these two treatments is possible, although outcomes are limited and the grade of evidence largely poor (Table 7). Now that data suggest that perphenazine is a real option for people wanting a more gentle effective antipsychotic drug (Lieberman 2005), there is the possibility that it also would be of value in the emergency situation.
7.1 Global outcome ‐ no improvement
Limited data suggest global impression of improvement is not different between groups (Analysis 7.1).
7.2 Adverse effects
Considerably more people allocated to haloperidol experienced one or more adverse effects (Analysis 7.2) and required antiparkinson medication (Analysis 7.3), but these were not statistically significant in this small trial. Nevertheless, there is a suggestion that perphenazine may have advantages in terms of adverse effects.
This must be an area for future research. However, without the push of industry it is possible that perphenazine will continue to be under evaluated.
8. COMPARISON 8. HALOPERIDOL versus OTHER ANTIPSYCHOTICS: g. QUETIAPINE
We found only one study (n = 80) with the most of the outcomes rated at 28 days and of low grade (Table 8).
8.1 Specific behaviour ‐ agitation
No differences were found between haloperidol and quetiapine across when using rating scales (Analysis 8.1; Analysis 8.2).
8.2 Mental state
Haloperidol and quetiapine showed similar results (Analysis 8.3); however, the Positive and Negative Syndrome Scale (PANSS) and sub‐scales were rated at 28 days, inappropriately with the aim of this review.
8.3 Adverse effects
More people allocated to haloperidol experienced one or more adverse effects (Analysis 8.4); this held true when taking into account movement disorders rating scales (Analysis 8.5).
9. COMPARISON 9. HALOPERIDOL versus OTHER ANTIPSYCHOTICS: h. RISPERIDONE
We found three relevant studies (n = 314), the outcomes of which we had to grade as of moderate, low or very low quality (Table 9).
9.1 Tranquillisation or asleep, agitation, need for more benzodiazepine
We thought the best graded evidence was that, if given haloperidol as well as lorazepam, more people were asleep compared with risperidone – also plus lorazepam (Analysis 9.1). Tranquillisation is often now considered desirable rather than full sedation to sleep, but in certain circumstances the latter may be preferable. The key issue is whether agitation is decreased and, according to PANNS‐EC scores, both medications are effective (Analysis 9.3).
In the trial randomising lorazepam as part of the package of care, more subsequent benzodiazepines were used. There was no difference between groups (Analysis 9.7), just the indication that if benzodiazepines are part of the package of care they will be used again and again. This could be problematic for the cumulative adverse effects.
9.2 Adverse effects
The two drugs did not have many differences as regards adverse effects; one seems as good or as bad as the other (Analysis 9.9). For binary measures of movement disorder no differences were clearly apparent (Analysis 9.14). Continuous measures of akathisia and movement disorders did, however, favour risperidone (Analysis 9.15). These fine‐grain measures may pick up differences not clearly shown by the more blunt binary outcomes. Akathisia is unpleasant and can lead to clinicians misinterpreting that the person is agitated again. Even subtle decreases of akathisia could be valuable in management of agitation and aggression.
9.3 Economic
We found no data on economic outcomes of this comparison. These trials were undertaken at a time when risperidone was not an inexpensive option and omission of this type of data does not seem random.
10. COMPARISON 10. HALOPERIDOL versus OTHER ANTIPSYCHOTICS: i. THIOTHIXINE
The two small studies provided low level data (total n = 74, Table 10).
10.1 Repeated need for rapid tranquillisation, agitation, global outcome
In the two studies that compared haloperidol with thiothixene, there was no outcome measure for tranquillisation or asleep. There were no significant differences between haloperidol and thiothixene with respect to the number of participants who required more than one injection (Analysis 10.1), agitation (Analysis 10.2), and ‘no response to the first injection’ (Analysis 10.3), but the study reporting these outcomes was very small indeed.
10.2 Adverse effects
There were no differences between the groups in terms of the number of participants who experienced one or more adverse effects (2 RCTs, n = 74, RR 1.47, 95% CI 0.97 to 2.22, Analysis 10.4).
The quality of these outcomes was, however, rated as ‘low’ or ‘very low’ because of risk of several biases (Table 10).
11. COMPARISON 11. HALOPERIDOL versus OTHER ANTIPSYCHOTICS: j. ZIPRASIDONE
Ziprasidone is a relatively new drug when compared with others in this set of comparisons. It is clear that the pharmaceutical industry has recognised the paucity of data in this area and that profits are to be made if an expensive drug can be shown to be as effective as the older drug and have fewer adverse effects. We found five relevant studies (total n = 829) but, again we had to grade the quality of outcome evidence as low or very low (Table 11). We have reconsidered as to whether we have been too harsh in judging the quality of evidence but, for the reasons outlined in the Table 11, we feel we have to continue to grade these data as poor. For example, these large studies did not report the outcome of ‘Tranquillisation or asleep’ or even ‘Repeated need for tranquillisation’. In larger trials focused on the coercive treatment of aggressive people we do feel that this is an odd set of omissions.
11.1 Repeated need for tranquillisation, specific behaviours, global outcomes and mental state
All measures are not convincingly better for either drug. The CGI‐S measures are better for the ziprasidone group as are some sub‐scores of mental state measures.
11.2 Adverse effects
The technique of reporting adverse effects only if more than 2%/5% or even 10% of people suffered the effect results in under‐reporting. Haloperidol can be confidently expected to provide frequent adverse effects (Adams 2013), so this device may assist the new drug seem less toxic.
At 72 hours, significantly more people in the haloperidol group had experienced one or more adverse effects but, by seven days, the difference between the groups was not statistically significant (Analysis 11.15).
Ziprasidone does seem to cause fewer movement disorders than haloperidol (Analysis 11.26) and, perhaps less anticholinergic problems. However, even these trials, generated by the company with a pecuniary interest in the outcomes, did nevertheless suggest, that cardiac adverse effects seemed more common in the ziprasidone group (Analysis 11.28). This must be seen as an important concern.
11.28. Analysis.
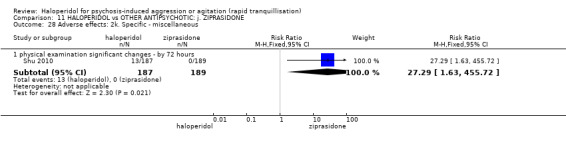
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 28 Adverse effects: 2k. Specific ‐ miscellaneous.
11.3 Economic
In these trials, so obviously comparing an inexpensive with expensive drug we find no economic analysis. This bias by omission casts a further cloud on reporting of all other data.
12. COMPARISON 12. HALOPERIDOL versus OTHER ANTIPSYCHOTICS: k. ZUCLOPENTHIXOL ACETATE
Although we understand that zuclopenthixol acetate may be a choice when rather longer‐term sedation is required that would be expected with haloperidol (Jayakody 2012), we did expect to find more relevant trials. Whether to use haloperidol or zuclopenthixol acetate is a prevalent clinical dilemma in some countries. We found one small study (n = 70), the outcomes of which we had to grade as ‘very low’ (Table 12).
This study reported few outcome with some expected results. For example, in this trial of a little longer duration, more people in the haloperidol group required more than three injections (Analysis 12.1) and there was the suggestion that, at seven days, haloperidol caused more tremor (Analysis 12.2).
This pioneering study is helpful in showing that fair testing could be undertaken for this important comparison. At present, however, best evidence from trials does not support or refute the choice one of these drugs over another in the emergency situation.
13. COMPARISON 13. HALOPERIDOL versus BENZODIAZEPINE: a. FLUNITRAZEPAM
We found one very small relevant study (n = 28) the outcomes of which were graded as being of very low quality (Table 13).
Unsurprisingly with such a small study there were no differences for any outcomes. That even adverse effects were similar highlights how lacking in power this trial was to show real differences. Flunitrazepam may be effective and a real alternative, but this trial does not show this. It does, however, illustrate that trials with this comparison are possible.
14. COMPARISON 14. HALOPERIDOL versus BENZODIAZEPINE: b. LORAZEPAM
Although we found four relevant studies with more than the average number of total participants (n = 207), few outcomes contain data from more than 70 people. The use of haloperidol alone in preference to lorazepam or vice versa is based on such limited evidence of poor quality that this could be considered an embarrassment to a subspecialty that recommends coercive use of these interventions (NICE 2005; NICE 2015).
14.1 Tranquillisation or asleep
Evidence is limited and we had to grade these data as of ‘very poor’ quality (Table 14). There was a suggestion that lorazepam is significantly more sedating after a few hours (Analysis 14.1). This may not be desirable as tranquillisation is frequently the aim without necessarily sedating.
14.2 Other global outcome
There were no differences in numbers requiring more than one and more than three injections and no difference between groups for the outcome of ‘showing no improvement’ at 30 and 60 minutes (Analysis 14.3). Mental state scores on 37 people were the same.
14.3 Adverse effects
These underpowered studies did not highlight many expected differences. However, significantly more participants experienced EPS in the haloperidol group compared with the lorazepam group (Analysis 14.11). Apnoea was not reported.
15. COMPARISON 15. HALOPERIDOL versus BENZODIAZEPINE: c. MIDAZOLAM
We found one small relevant study with data we could use (n = 84), but the limited outcomes we could use for the Table 15 we had to grade as being of low quality.
15.1 Tranquillisation or asleep: average time
Midazolam is a highly effective hypnotic (Olkkola 2008) and the skewed data on tranquillisation did seem to support that it was significantly faster than haloperidol in this clinical situation.
15.2 Adverse effects
Midazolam is known to cause apnoea but in this trial it was a person in the haloperidol group that experienced this (Analysis 15.3). Nevertheless, data from this small study should not be considered conclusive. Other data from better trials (TREC CG; TREC 2003) and other sources suggest that use of midazolam could be accompanied with full resource for control of airways and intravenous reversal of the apneic effects with flumazenil (Olkkola 2008).
16. COMPARISON 16. HALOPERIDOL versus COMBINATIONS: a. HALOPERIDOL + LEVOMEPROMAZINE
We found one very small study (n = 19), the outcomes of which we had to grade as being of very low quality (Table 16).
Clinicians find themselves in an unusual position because of the prevalence of haloperidol and its use for psychosis‐induced aggression but in the recognition of its toxicity. We think it understandable that researchers investigate whether combinations could offset some adverse effects and levomepromazine may do this, but the included trial is too small to illustrate anything except that such a study is possible.
17. COMPARISON 17. HALOPERIDOL versus COMBINATIONS: b. HALOPERIDOL + LORAZEPAM
We were rather surprised to find that there are only two trials of this comparison. We think that this combination is in common use but the two trials have a total of only just over 100 people and we had to grade outcomes all of very low quality (Table 17).
17.1 Tranquillisation or asleep
More people in the combination group were asleep at three hours than those in the haloperidol alone group but these were limited very low‐grade data. Also, three hours is a very long time when a person is acutely disturbed and not an outcome of great clinical utility in regard to the focus of this review.
17.2 Repeated need for rapid tranquillisation
There was some suggestion that the addition of lorazepam did assist the tranquillisation process but no data were conclusive and all were of very poor grade. Significantly fewer people in the combination group exhibited “no overall improvement by 30 minutes”.
17.3 Adverse effects
Overall, there was no difference between the groups. Best evidence suggests that addition of lorazepam does not offset adverse effects. In these small poorly reported and probably poorly conducted trials dystonia was common and could be considered to be at unacceptable levels. People undergoing coercive rapid tranquillisation have enough difficult experiences without the addition of needless distressing adverse effects. We have no data that this will cause further mistrust of healthcare professionals, but it does seem a reasonable assumption.
18. COMPARISON 18. HALOPERIDOL versus COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM
We found one relevant study (n = 60) with outcomes of very low grade only (Table 18).
18.1 Repeated need for tranquillisation, specific behaviours, global outcomes
An overall advantage in favour of haloperidol was shown in terms of agitation and aggression rating scales, and need for seclusion or restraint (Analysis 18.1; Analysis 18.2; Analysis 18.3; Analysis 18.4; Analysis 18.5; Analysis 18.6; Analysis 18.7).
18.2 Adverse effects
Haloperidol and combination data resulted in similar but, again, data from this small study should not be considered conclusive.
19. HALOPERIDOL versus COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE
We found two relevant study (n = 376) and outcomes were graded of moderate quality (higher than others in this review) or low quality (Table 19).
19.1 Tranquillisation or asleep, repeated need for tranquillisation
Haloperidol used on its own clearly is quickly tranquillising, but the combination with promethazine makes is significantly more swift. The difference between being tranquil at 20 and 40 minutes can be of great importance in the clinical front line (Analysis 19.1; Analysis 19.2).
There were no significant differences in terms of requiring additional drugs for tranquillisation (Analysis 19.3).
19.2 Specific behaviours ‐ agitation, aggression
When using agitation and aggression rating scales, an initial advantage for haloperidol was demonstrated (Analysis 19.4; Analysis 19.7); however, this should be replicated by other studies as it appears to come from one trial only of low‐moderate quality (Baldacara 2011).
On other measures of aggression there was not much difference (requiring repeat rapid tranquillisation, exhibiting further aggressive outburst within 24 hours. More people in the haloperidol alone group had to be restrained by 120 minutes, but the difference between groups was not statistically significant. However, significantly more people in the haloperidol alone group required extra visits from the doctor, which is an indication of the clinical concern of the healthcare professionals. Such visits can be for further management of aggression or disturbance, or to address adverse effects.
19.3 Adverse effects
Haloperidol is a potent cause of adverse effects. Significantly more people in the haloperidol alone group than the combination group experienced one or more adverse effects. Acute dystonia – a most distressing effect – was common (n = 316, RR 19.48, 95% CI 1.14 to 331.92, Analysis 19.20), and frequency of occurrence resulted in this trial being terminated prematurely. Use of promethazine offsets the dystonia. The report specifically states that the Steering Group of this trial felt that the guidance from APA in the USA and NICE in the UK was unethical to continue.
20. COMPARISON 20. HALOPERIDOL versus COMBINATIONS: e. HALOPERIDOL + RISPERIDONE
We found one study (n = 100) with outcomes of low‐grade quality (Table 20).
20.1 Specific behaviour, global outcomes
An initial advantage ‐ at 24 hours ‐ for the combination group was demonstrated (Analysis 20.1), even though it was not confirmed by subsequent time points (Analysis 20.2); further studies should be carried out, in particularly focused on the first hours in order to better understand the effect of the combination versus haloperidol.
More people allocated to the haloperidol group showed improvement when compared to the haloperidol + risperidone group, even if this difference was not significant (Analysis 20.3).
20.2 Adverse effects
The two compared groups showed comparable results (Analysis 20.4, Analysis 20.5, Analysis 20.6).
21. COMPARISON 21. HALOPERIDOL versus COMBINATIONS: f. OLANZAPINE + MAGNESIUM VALPROATE
We found one small study (n = 64) with outcomes of very low‐grade quality (Table 21).
21.1 Specific behaviour ‐ agitation
People allocated in the combination group showed a lower level of agitation, rated using the PANSS‐EC, at seven and 14 days; unfortunately, the first time point available in this trial was at 72 hours and showed no differences between the compared groups (Analysis 21.1). These timings are not adequate in order to fulfil the aim of this study, and even more highlight the need for further studies focused on the early effects.
21.2 Global outcomes, mental state
No significant differences were found as an equal number of people showed no improvement in both groups (Analysis 21.2); the PANSS total scores showed a trend over the weeks in advantage for the combination group, even if not significant (Analysis 21.3).
21.3 Adverse effects
The number of people experiencing one or more adverse effects was almost three times in the haloperidol group (Analysis 21.4).
22. COMPARISON 22. HALOPERIDOL versus COMBINATIONS: g. QUETIAPINE + MAGNESIUM VALPROATE
The one study that compared haloperidol with a combination of quetiapine + magnesium valproate was small and outcomes were graded as very low quality (n = 60, Table 22).
Although this one trial was probably underpowered, there did not seem to be a suggestion that the quetiapine‐magnesium valproate combination had any affect over and above that seen with haloperidol alone.
Global effect had to be indirectly inferred from no effect on the PANSS‐EC, and further aggressive behaviour had to be surmised from scale‐derived data for agitation at 72 hours. Risk of bias was rated as serious because the method of randomisation was not reported, allocation concealment was not stated and it was an open‐label trial. There was no measure for economic outcomes. Any use of this quetiapine‐valproate combination must be considered experimental.
23. COMPARISON 23. HALOPERIDOL versus COMBINATIONS: h. RISPERIDONE + CLONAZEPAM
We found two studies (n = 305) and outcomes were graded as of low and very low grade (Table 23).
23.1 Tranquillisation or asleep, specific behaviour
There were no differences when comparing the number of people not asleep in the first four hours (Analysis 23.1) and the PANSS‐EC rating scores (Analysis 23.2; Analysis 23.3; Analysis 23.4; Analysis 23.5).
23.2 Global outcomes, mental state
More people allocated to the haloperidol group experienced an improvement across the explored time points, even though the difference was not significant (Analysis 23.6). At five days when looking at the PANSS total scores, the two groups showed similar results (Analysis 23.7).
23.3 Adverse effects
More adverse events were recorded in the haloperidol group (Analysis 23.8), with a significant predominance of movement disorders‐related adverse effects (Analysis 23.11).
24. COMPARISON 24. HALOPERIDOL versus COMBINATIONS: i. ZIPRASIDONE + CLONAZEPAM
We found one study (n = 76) and outcomes were graded as of very low quality (Table 24).
There did not seem to be a suggestion of superiority of ziprasidone + clonazepam than haloperidol alone; no significant differences were found between the compared groups.
Overall completeness and applicability of evidence
1. Completeness
Despite the great number of comparisons in this review, there are no data on several important questions. For example, although guidelines recommend use of haloperidol intravenously in exceptional circumstances, not one randomised controlled trial was found that investigated the efficacy and safety of haloperidol via this route of administration. For other routes of administration, even where some data do exist, evidence is graded as being of largely poor quality. For example, there is no strong evidence of the clear advantage of olanzapine or risperidone over the older haloperidol. The evidence for perphenazine is not good ether, but it could be seem as an inexpensive option to haloperidol. In addition, not one of the studies evaluated satisfaction with care, acceptance of treatment, quality of life or economic costs. Unfortunately, a large proportion of the efficacy and safety data produced by the trials was left redundant due to poor and/or selective reporting. This is clearly an area of evaluation in which researchers have invested heavily and this leaves those compiling guidance having to use judgements that are based less on good evidence than they should be.
2. Applicability
Many of the included trials were more of the explanatory type of study rather than practical/pragmatic and easily applicable to real‐world practice (Thorpe 2009). Few seem to have been designed with clear clinical needs as a priority. This does not mean that applicability is impossible. It just necessitates a level of conjecture that could have been lessened should more pragmatic trials have been available. Two examples of how trials are near or far from real‐world practice and needs are outlined below.
2.1 Participants seen in real‐world practice
Many of these trials seem to have been undertaken in circumstances very far from real clinical practice. For example, the UK's NICE guidelines recommend that rapid tranquillisation should only be considered "once de‐escalation and other strategies have failed to calm the service user" (NICE 2005, p14) and that “p.r.n. medication can be used as part of a de‐escalation strategy but p.r.n. medication used alone is not de‐escalation.” (NICE 2015, p15). However, in a number of studies, participants (in some cases their legal representatives) were required to provide written informed consent (Battaglia 2002; Breier 2002; Bristol Myers 2004a; Bristol‐Myers 2005b; Brook 2000; Eli Lilly 2004; Kinon 2001d; Currier 2004; Higashima 2004; Lim 2010), Further, participants in many studies all had to undergo a screening period ranging from two to 96 hours (Battaglia 2002; Breier 2002; Bristol Myers 2004a; Bristol‐Myers 2005b; Brook 2000; Currier 2004; Eli Lilly 2004; Fruensgaard 1977; Kinon 2001d; Lim 2010, Shu 2010). Participants in these studies have intervention delayed for screening and are deemed competent to give informed consent ‐ a situation that is not common in the heat of an emergency and makes applicability problematic ‐ a matter of conjecture.
Other studies, however, recognised that conduct of the study had to involve deferred or waived consent ‐ and in this way making it more like clinical practice. In Battaglia 1997, Huf 2007, Nobay 2004 and Walther 2014, informed consent was obtained wherever possible. It was waived in Bailine 1987, Dorevitch 1999 and Foster 1997 because it was acknowledged that if people were that violent or psychologically disorganised to require rapid tranquillisation they would not be likely to be able to give informed consent. In Tuason 1986, informed consent was obtained as soon as participants condition permitted and in Resnick 1984 it was deemed that participants were unable to give informed consent because they were involuntary patients. This last group of trials is likely to be involving people closer to real‐world circumstances.
2.2 Outcomes of value in real‐world practice
Where people are a risk to themselves or others, rapid tranquillisation is often used to prevent the prolonged physical restraint of people, which can result in injury and death. It was therefore surprising that few studies measured tranquillisation, asleep, or global functioning outcomes at time points that one would associate with a rapid intervention. There were only three studies that measured tranquil or asleep on or around 30 minutes (Huf 2007; Currier 2004; Fruensgaard 1977), with a further two measuring at 60 minutes (Kinon 2001d; Salzman 1991), with global improvement being measured at 30 minutes in the Garza‐Trevino 1989 and at 60 minutes in the Kewala 1984 study. Also, not all studies documented simple and important outcomes such as repeated need for rapid tranquillisation. We are therefore left inferring the clinical implications of outcomes that are not recorded in routine care. This is an entirely avoidable situation if those designing trials listened to the needs of clinicians and people who are subject to this coercive care.
Quality of the evidence
1. General
Twenty‐nine of the 41 included studies were published after the first CONSORT guidance (1996). Few studies complied with this internationally agreed standard. Disappointingly, a large proportion of data was rendered unusable, which could have been avoided had the trials been better reported.
2. Specific
Although this review includes 41 studies and approximately 3500 participants, there were 24 different comparisons. Trials were largely very small and meta‐analysis could be undertaken for a minority of comparisons and, even then, for only some outcomes. Overall, the majority of the evidence had to be graded as 'very low', usually because of considerable ‐ and often avoidable ‐ limits in research design, selective reporting and imprecision.
The best available evidence came from the Huf 2007 trial which compared haloperidol with haloperidol plus promethazine, from the Calver 2015 trial which compared haloperidol with droperidol, and from the Nobay 2004 trial that compared haloperidol with midazolam. Nevertheless, these studies also had limitations to their research design which led to the evidence being graded as 'moderate'.
Potential biases in the review process
There are a number of ways in which bias could have been introduced into this review.
i. Double‐data extraction
In some systematic reviews, two people independently inspect all citations obtained from the search and extracted the data to minimise bias. However, this was not the case with this review, with only 20% of the screening of citations and only 10% of the data extraction being performed by two review authors.
ii. Potential conflict of interest
Another source of potential bias is that one of the authors of this review (CEA) was an investigator in the Huf 2007 trial.
Agreements and disagreements with other studies or reviews
The review authors are not aware of any other studies or reviews that have focused solely on use of haloperidol alone for people who are aggressive secondary to serious mental illnesses. Pratt 2008 conducted a systematic review (not meta‐analysis), focusing on the practice of rapid tranquillisation in the UK and concluded that there was a lack of high‐quality evidence.
Authors' conclusions
Implications for practice.
1. For people agitation/aggression due to psychosis
Some clinical guidelines recommend the use of haloperidol, either intramuscularly combined with benzodiazepine or even, in the extreme situation, given intravenously. There is no good‐quality evidence for the latter. These treatments are administered under coercion and people having experienced this very unpleasant situation, when recovered, may wish to enquire just why such a treatment was administered. They could suggest that an advanced directive be stipulated that only treatments given either from a firm evidence base or within the context of a randomised trial ‐ should they fall so ill again. All evidence reported in this review is limited and flawed. Best evidence is probably for the use of haloperidol combined with promethazine; the last update of UK's NICE guidelines supports this evidence (NICE 2015). Evidence of the benefit of haloperidol combined with lorazepam is limited to a few very small trials.
People do not like experiencing dystonia. This is common with haloperidol used with nothing to offset this problem. One larger and independent trial was stopped early because it was felt that the NICE/APA recommended use of haloperidol on its own was unethical (Huf 2007). People with schizophrenia may feel that coercion is enough without use of treatments of dubious ethicality.
2. For clinicians
Use of haloperidol clearly calms difficult situations. Clinicians (as well as people with psychosis and aggression), however, are left vulnerable when evidence is of such limited quality and so thinly spread over many comparisons. In some situations, where a person is clearly both mentally unwell and aggressive, haloperidol may be the only choice ‐ and in this case, a reasonable one. However, where resources are more available, haloperidol used completely on its own does not seem defendable. Haloperidol may be made more sedating by lorazepam but evidence is poor. Adverse effects may be offset by anticholinergics but data are lacking and this is unlikely to happen with lorazepam. Best evidence exists for addition of the sedating antihistamine promethazine. If researchers continue to let down the clinical community by avoiding evaluative studies in this difficult area clinicians should request such investigations formally. Not to have good evidence in this area is difficult to justify.
3. For policy makers
Even in the situation of very limited high‐grade data, guidance has to happen. There is enough evidence to suggest that haloperidol given intramuscularly may be helpful, but probably giving it alone should be avoided. Theoretically anticholinergics are indicated at the same time and this has been best demonstrated by the use of the anticholinergic‐antihistamine promethazine ‐ at least within the context of use of restraints. There is not compelling evidence that newer drugs are better enough to justify their routine use and no economic data whatsoever. More independent trials are indicated.
Implications for research.
1. General
All studies should now comply with the Consolidated Standards of Reporting Trials (CONSORT, Moher 2001). More transparency in the reporting of randomised controlled trials, would enable readers to understand the design, conduct, analysis and interpretation, and to assess the validity of results. Although binary data are easier to interpret, where continuous data are used, some measure of variance should be provided. Data presented in graphs should be accompanied by exact numbers and standard deviations in the text.
The review authors strongly highlight that, despite the recommendations provided by CONSORT reporting guidelines and the first edition of this review, out of nine studies included in this update, only one study (Calver 2015) contributed to a moderate‐grade evidence, whilst the remaining eight studies provided low‐ or very low‐grade evidence only.
2. Specific
This review is an indictment of six decades of research and routine coercive use. Large pragmatic well‐designed randomised trials led by independent researchers, which measure simple and meaningful outcomes such as 'tranquil', 'asleep', 'serious adverse effect', 'needing additional medication', 'further aggressive episodes' are still required. These studies do not need large funds. They just need modest financial support and firm help from ethics committees who understand the need to produce good evidence for practice in this difficult edge of health care. See Table 27 for a suggested design for a study.
3. Suggested design for a trial.
| Methods | Allocation: randomised, clearly described, concealed. Blindness: double, described and tested. Duration: 2 weeks. |
| Participants | Diagnosis: thought to have psychoses. N = 300.* Age: any. Sex: both. History: acutely ill, aggressive. |
| Interventions | 1. Haloperidol IM: dose flexible within recommended limits. N = 150. 2. Benzodiazepine IM: dose flexible within recommended limits. N = 150. |
| Outcomes | All outcomes are grouped by time: by 30 minutes, up to two hours, up to four hours, up to 24 hours and finally over 24 hours. Tranquillisation or asleep (measured at 30 minutes, 2 hours, 4 hours and 24 hours). Mortality. Specific behaviours ‐ self‐harm, including suicide, injury to others, aggression. Global outcomes ‐ overall improvement, use of additional medication, use of restraints/seclusion. Service outcomes ‐ duration of hospital stay, re‐admission. Mental state ‐ no clinically important change in general mental state. Adverse effects ‐ clinically important adverse effects. Leaving the study early ‐ why. Economic outcomes. |
| Notes | * Powered to be able to identify a difference of ˜20% between groups for primary outcome with adequate degree of certainty. |
IM: intramuscular
What's new
| Date | Event | Description |
|---|---|---|
| 7 March 2017 | New citation required but conclusions have not changed | Results of update have not changed the overall conclusions of the review; better evidence on potential droperidol use in the acute setting. 'Summary of findings' tables updated. |
| 17 January 2017 | New search has been performed | Nine new studies added, contributing with data on 1056 new individuals (total number of individuals in the updated review: 3546), six new comparisons, and six updates on previously existing comparisons. |
History
Protocol first published: Issue 10, 2011 Review first published: Issue 11, 2012
| Date | Event | Description |
|---|---|---|
| 26 May 2016 | Amended | Search was updated and 37 new references added to 'Classification Pending References' section of the review. |
| 1 June 2011 | Amended | Cochrane Schizophrenia Group Trial Register searched: 48 studies included in the review, 32 studies included in the analyses. |
Acknowledgements
We would like to thank Gene Amason, Dr Anand, Dr Baldaçara, Dr Dubin, Dr El‐Sayeh, Dr Herrera, Dr Isbister, Dr Kinon, Dr Lerner, Dr Mueller, Dr Resnick, Dr Simon and Dr Spivak for answering our requests for further information regarding studies.
In addition, we would like to thank Sam Roberts at the Cochrane Schizophrenia Group Editorial Base in Nottingham for designing the search strategy, Jun Xia of Systematic Review Solutions (http://www.srs‐org.com/) for data extraction of Chinese papers and correspondence and Claire Irving for her help and patience. Also, Ben Gray for writing the Plain language summary and Hannah Jones for her contributions for the previous version of this review.
The Cochrane Schizophrenia Group Editorial Base in Nottingham produces and maintains standard text for use in the Methods section of their reviews. We have used this text as the basis of what appears here and adapted it as required. Further, this is one of a series of reviews (Table 25). The authors of this review also acknowledge the help and guidance of the structure of the Chlorpromazine for psychosis‐induced aggression or agitation (Ahmed 2010).
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Schizophrenia Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Appendices
Appendix 1. Previous searches
1.1 Search in 2011
1.1.1 Electronic searches
1.1.1.1 Cochrane Schizophrenia Group Trials Register
We searched the register (1st June 2011) using the phrase:
(*haloperi* or *R‐1625* or *haldol* or *alased* or *aloperidi* or *bioperido* or *buterid* or *ceree* or *dozic* or *duraperido* or *fortuna* or *serena* or *serenel* or *seviu* or *sigaperid* or *sylad* or *zafri* in intervention of STUDY) AND (*aggress* or *violen* or *agitat* or *tranq* in title, abstract, index terms of REFERENCE or intervention of STUDY)
This register is compiled by systematic searches of major databases, handsearches and conference proceedings (see group module).
1.1.2 Searching other resources
1.1.2.1 Reference searching
We inspected references of all included studies for further relevant studies.
1.1.2.2 Personal contact
We attempted to contact the first author of each included study for information regarding unpublished trials.
Data and analyses
Comparison 1. HALOPERIDOL vs PLACEBO/NIL.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tranquillisation or asleep ‐ asleep | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 not asleep up to 2 hours | 2 | 220 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.82, 0.95] |
| 2 Repeated need for tranquillisation | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 needing additional injection during 24 hours (agitation only) | 4 | 660 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.42, 0.62] |
| 3 Specific behaviour: 1a. Agitation ‐ various measures | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 PANSS‐EC response at 2 hours (at least 40% change on PANSS‐EC from baseline) | 2 | 395 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.62 [1.28, 2.07] |
| 4 Specific behaviour: 1b. Agitation ‐ average scores ‐ i. up to 2 hours | 4 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 change score (ABS, high=worse) | 3 | 474 | Mean Difference (IV, Fixed, 95% CI) | ‐4.48 [‐5.78, ‐3.18] |
| 4.2 change score (ACES, low=agitated, high=sedated) | 2 | 390 | Mean Difference (IV, Fixed, 95% CI) | 0.83 [0.39, 1.26] |
| 4.3 score (PANSS‐EC, high=worse) | 3 | 514 | Mean Difference (IV, Fixed, 95% CI) | ‐3.67 [‐4.75, ‐2.59] |
| 4.4 change score (PANSS‐EC, high=worse, schizophrenia subgroup) | 1 | 199 | Mean Difference (IV, Fixed, 95% CI) | ‐2.57 [‐4.05, ‐1.09] |
| 4.5 change score (PANSS‐EC, high=worse, non‐sedated patients subgroup based on ACES) | 1 | 240 | Mean Difference (IV, Fixed, 95% CI) | ‐2.64 [‐3.95, ‐1.33] |
| 4.6 change score (PANSS‐EC, high=worse, non‐sedated patients subgroup based on ACES) | 1 | 263 | Mean Difference (IV, Fixed, 95% CI) | ‐2.97 [‐4.27, ‐1.67] |
| 5 Specific behaviour: 1c. Agitation ‐ average scores ‐ ii. up to 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 change score (ABS, high=worse) | 1 | 85 | Mean Difference (IV, Fixed, 95% CI) | ‐2.4 [‐4.13, ‐0.67] |
| 5.2 change score (PANSS‐EC, high=worse) | 1 | 85 | Mean Difference (IV, Fixed, 95% CI) | ‐1.4 [‐2.97, 0.17] |
| 6 Global outcome: 1. Not improved | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 not marked improvement | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.61 [0.44, 0.84] |
| 6.2 not any improvement | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.28 [0.08, 1.07] |
| 7 Global outcome: 2. Need for benzodiazepine up to 24 hours | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 need for benzodiazepine up to 24 hours | 4 | 660 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.44 [0.31, 0.62] |
| 8 Global outcome: 3a. Average scores ‐ i. up to 2 hours | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 8.1 endpoint score (CGI‐I, high=worse) | 2 | 390 | Mean Difference (IV, Fixed, 95% CI) | ‐0.73 [‐0.96, ‐0.51] |
| 8.2 change score (CGI‐I, high=worse, schizophrenia subgroup) | 1 | 199 | Mean Difference (IV, Fixed, 95% CI) | ‐0.64 [‐0.98, ‐0.30] |
| 8.3 change score (CGI‐S, high=worse) | 2 | 390 | Mean Difference (IV, Fixed, 95% CI) | ‐0.48 [‐0.78, ‐0.18] |
| 9 Global outcome: 3b. Average scores ‐ ii. up to 24 hours | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 9.1 change score (CGI‐I, high=worse) | 1 | 169 | Mean Difference (IV, Fixed, 95% CI) | ‐0.4 [‐0.62, ‐0.18] |
| 9.2 change score (CGI‐S, high=worse) | 1 | 85 | Mean Difference (IV, Fixed, 95% CI) | ‐0.2 [‐0.46, 0.06] |
| 10 Mental state: 1a. Average scores ‐ i. up to 2 hours | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.1 change score up to 2 hours (BPRS positive sub‐scale, high=worse) | 3 | 372 | Mean Difference (IV, Fixed, 95% CI) | ‐1.04 [‐1.54, ‐0.54] |
| 10.2 change score up to 2 hours (BPRS total, high=worse) | 3 | 371 | Mean Difference (IV, Fixed, 95% CI) | ‐5.69 [‐7.38, ‐4.00] |
| 11 Mental state: 1b. Average scores ‐ ii. up to 24 hours | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 11.1 change score up to 24 hours (BPRS positive sub‐scale, high=worse) | 2 | 264 | Mean Difference (IV, Fixed, 95% CI) | ‐1.61 [‐2.34, ‐0.89] |
| 11.2 change score up to 24 hours (BPRS total, high=worse) | 2 | 264 | Mean Difference (IV, Fixed, 95% CI) | ‐4.81 [‐6.82, ‐2.81] |
| 12 Adverse effects: 1a. General | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.1 one or more drug related adverse events during 24 hours | 2 | 395 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.64 [1.22, 2.20] |
| 12.2 increased severity of adverse effects after 2nd injection | 1 | 273 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.25 [1.88, 5.63] |
| 12.3 overall adverse events during 72 hours | 1 | 273 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.78 [1.23, 2.59] |
| 13 Adverse effects: 1b. General ‐ serious | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 13.1 death | 1 | 273 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 13.2 rated as serious | 1 | 122 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.01, 8.29] |
| 13.3 tonic clonic seizure | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 14 Adverse effects: 2a. Specific ‐ arousal level | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 14.1 insomnia during 24 hours (only reported if occurred in ≧5%) | 1 | 273 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.31 [0.61, 2.82] |
| 14.2 "over" sedated | 2 | 313 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.36 [1.42, 7.99] |
| 14.3 somnolence during 24 hours | 4 | 615 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.28 [0.97, 5.36] |
| 15 Adverse effects: 2b. Specific ‐ cardiovascular ‐ miscellaneous outcomes | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 15.1 dizziness during 24 hours (only reported if occurred in ≧5%) | 2 | 392 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.48, 3.65] |
| 15.2 hypotension during 24 hours | 2 | 125 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.2 [0.05, 27.44] |
| 15.3 QTc abnormality | 1 | 122 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.1 [0.33, 28.98] |
| 15.4 sinus tachycardia during 24 hours (only reported if occurred in ≥5%) | 1 | 122 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.1 [0.33, 28.98] |
| 15.5 tachycardia during 24 hours (only reported if occurred in ≥5%) | 1 | 122 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.07, 16.15] |
| 16 Adverse effects: 2c. Specific ‐ cardiovascular ‐ QTc interval (average change at 24 hours) | 2 | 265 | Mean Difference (IV, Fixed, 95% CI) | 3.63 [‐2.67, 9.93] |
| 17 Adverse effects: 2d. Specific ‐ movement disorders | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 17.1 akathisia during 24 hours (only reported if occurred in ≥5%) | 1 | 122 | Risk Ratio (M‐H, Fixed, 95% CI) | 13.43 [0.77, 233.23] |
| 17.2 dystonia during 24 hours | 2 | 207 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.49 [0.93, 60.21] |
| 17.3 extrapyramidal effects ‐ use of antiparkinson drugs during 24 hours | 1 | 180 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.57 [1.37, 22.65] |
| 17.4 EPS during 24 hours | 3 | 398 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.79 [2.19, 21.07] |
| 18 Adverse effects: 2e. Specific ‐ movement disorders: i. average scores ‐ i. up to 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.1 change score at 24 hours (BAS, high=worse) | 1 | 168 | Mean Difference (IV, Fixed, 95% CI) | 0.09 [‐0.17, 0.35] |
| 18.2 change score at 24 hours (SAS, high=worse) | 1 | 167 | Mean Difference (IV, Fixed, 95% CI) | 1.89 [0.77, 3.01] |
| 19 Adverse effects: 2f. Specific ‐ miscellaneous | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 19.1 agitation during 24 hours (only reported if occurred in ≧5%) | 2 | 395 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.29, 2.19] |
| 19.2 dry mouth | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.6 [0.21, 61.86] |
| 19.3 headache during 24 hours (only reported if occurred in ≧5%) | 2 | 395 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.28 [0.55, 3.00] |
| 19.4 pain at injection site | 1 | 122 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.21 [0.01, 4.22] |
| 19.5 pain (1st injection) | 1 | 273 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.10 [0.00, 1.97] |
| 19.6 pain (2nd injection) | 1 | 273 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.25 [1.88, 5.63] |
| 19.7 nausea during 24 hours (only reported if occurred in ≧5%) | 2 | 395 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.13, 3.67] |
| 19.8 vomiting during 24 hours (only reported if occurred in ≧5%) | 1 | 122 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.07, 16.15] |
| 20 Leaving the study early | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 20.1 any reason | 3 | 435 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.53, 2.11] |
| 20.2 lack of efficacy | 3 | 435 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.12 [0.03, 0.49] |
| 20.3 withdrew | 1 | 273 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.35 [0.17, 64.15] |
| 20.4 consent | 2 | 395 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.2 [0.77, 49.98] |
| 20.5 adverse effects | 3 | 435 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.78 [0.21, 15.39] |
| 20.6 could not be evaluated due to being asleep | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.2 [0.05, 27.44] |
| 20.7 other | 2 | 395 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.81 [0.27, 12.07] |
Comparison 2. HALOPERIDOL vs OTHER ANTIPSYCHOTIC: a. ARIPIPRAZOLE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Repeated need for rapid tranquillisation: needing additional injection | 2 | 473 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.62, 0.99] |
| 2 Specific behaviour: 1a. Agitation ‐ PANSS‐EC response up to 2 hours (at least 40% change on PANSS‐EC from baseline) | 2 | 477 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.92, 1.26] |
| 3 Specific behaviour: 1b. Agitation ‐ average scores ‐ up to 2 hours | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 change score (ACES, low=agitated, high=sedated) | 2 | 470 | Mean Difference (IV, Fixed, 95% CI) | 0.12 [‐0.30, 0.55] |
| 3.2 change score (CABS, high=worse) | 2 | 470 | Mean Difference (IV, Fixed, 95% CI) | ‐0.55 [‐2.10, 1.01] |
| 3.3 change score (PANSS‐EC, high=worse) | 1 | 357 | Mean Difference (IV, Fixed, 95% CI) | ‐0.48 [‐2.12, 1.16] |
| 3.4 change score (PANSS‐EC, high=worse, schizophrenia subgroup) | 1 | 257 | Mean Difference (IV, Fixed, 95% CI) | ‐0.26 [‐1.48, 0.96] |
| 3.5 change score (PANSS‐EC, high=worse, non‐sedated patients subgroup based on ACES) | 1 | 316 | Mean Difference (IV, Fixed, 95% CI) | ‐0.33 [‐1.42, 0.76] |
| 3.6 change score (PANSS‐EC, high=worse, non‐sedated patients subgroup based on AEs) | 1 | 346 | Mean Difference (IV, Fixed, 95% CI) | ‐0.34 [‐1.40, 0.72] |
| 4 Global outcome: 1. Need for benzodiazepine | 2 | 477 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.74, 2.16] |
| 5 Global outcome: 2. Average scores ‐ up to 2 hours | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 change score (CGI‐S, high=worse) | 2 | 470 | Mean Difference (IV, Fixed, 95% CI) | 0.07 [‐0.22, 0.36] |
| 5.2 endpoint score (CGI‐I, high=worse) | 2 | 470 | Mean Difference (IV, Fixed, 95% CI) | ‐0.02 [‐0.23, 0.19] |
| 5.3 change score (CGI‐I, high=worse, schizophrenia subgroup) | 1 | 257 | Mean Difference (IV, Fixed, 95% CI) | ‐0.02 [‐0.30, 0.26] |
| 6 Mental state: 1. Average scores ‐ up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.1 change score at 2 hours (BPRS positive sub‐scale, high=worse) | 1 | 103 | Mean Difference (IV, Fixed, 95% CI) | ‐0.23 [‐1.28, 0.82] |
| 6.2 change score at 2 hours (BPRS total, high=worse) | 1 | 102 | Mean Difference (IV, Fixed, 95% CI) | ‐2.03 [‐5.76, 1.70] |
| 7 Adverse effects: 1a. General | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 one or more drug related adverse effects during 24 hours | 2 | 477 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.95, 1.46] |
| 7.2 increased severity of adverse effects after 2nd injection | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.34 [1.03, 1.74] |
| 7.3 overall adverse events during 72 hours | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [1.04, 1.70] |
| 8 Adverse effects: 1b. General ‐ serious | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 any | 2 | 477 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.57 [0.18, 1.81] |
| 8.2 tonic clonic seizure | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.62] |
| 8.3 death | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9 Adverse effects: 2a. Specific ‐ arousal level | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 insomnia ‐ between 0‐1 days (IM phase) (only reported if occurred in ≥5%) | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.08 [1.01, 4.27] |
| 9.2 insomnia ‐ between 3 ‐ 7 days (oral phase) (only reported if occurred in ≥ 2%) | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.47, 1.44] |
| 9.3 "over" sedated | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.66 [0.93, 2.95] |
| 9.4 somnolence ‐ between 0‐1 days | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.25, 2.00] |
| 9.5 somnolence ‐ between 0‐1 days (IM phase) (only reported if occurred in ≥5%) | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.22 [0.60, 8.16] |
| 9.6 somnolence ‐ between 3 ‐ 7 days (oral phase) (only reported if occurred in ≥ 2%) | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.84 [0.30, 27.02] |
| 10 Adverse effects: 2b. Specific ‐ cardiac | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 dizziness ‐ between 0 ‐1 day (IM phase) (only reported if occurred in ≥5%) | 2 | 477 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.33, 1.48] |
| 10.2 sinus tachycardia ‐ between 0 ‐1 day (IM phase) (only reported if occurred in ≥5%) | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.66 [0.35, 126.06] |
| 10.3 tachycardia ‐ between 0 ‐1 day (IM phase) (only reported if occurred in ≥5%) | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.03, 2.06] |
| 11 Adverse effects: 2c. Specific ‐ gastrointestinal | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 11.1 dyspepsia ‐ between 3 ‐ 7 days (oral phase) (only reported if occurred in ≥ 2%) | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 10.41 [1.36, 79.76] |
| 11.2 diarrhoea ‐ between 3 ‐ 7 days (oral phase) (only reported if occurred in ≥ 2%) | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.28, 3.21] |
| 11.3 nausea ‐ between 0‐1 days (IM phase) (only reported if occurred in ≥5%) | 2 | 477 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.18 [0.05, 0.60] |
| 11.4 nausea ‐ between 3 ‐ 7 days (oral phase) (only reported if occurred in ≥ 2%) | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.02, 1.09] |
| 11.5 vomiting ‐ between 0‐1 days (IM phase) (only reported if occurred in ≥5%) | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.48 [0.04, 5.10] |
| 11.6 vomiting ‐ between 3 ‐ 7 days (oral phase) (only reported if occurred in ≥ 2%) | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.38 [0.07, 1.92] |
| 12 Adverse effects: 2d. Specific ‐ movement disorder | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.1 akathisia ‐ between 0 ‐1 day (IM phase) (only reported if occurred in ≥5%) | 2 | 477 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.85 [0.94, 8.61] |
| 12.2 akathisia ‐ between 3 ‐ 7 days (oral phase) (only reported if occurred in ≥ 2%) | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.21 [0.58, 8.40] |
| 12.3 dystonia ‐ between 0 ‐1 day (IM phase) (only reported if occurred in ≥5%) | 2 | 477 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.63 [1.52, 28.86] |
| 12.4 EPS ‐ between 0 ‐1 day (IM phase) (only reported if occurred in ≥5%) | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.46 [1.22, 73.13] |
| 12.5 EPS ‐ between 3 ‐ 7 days (oral phase) (only reported if occurred in ≥ 2%) | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.09 [1.65, 30.58] |
| 12.6 Use of antiparkinson drugs ‐ between 3 ‐ 7 days (oral phase) | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.94 [2.50, 9.78] |
| 12.7 dystonia | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.84 [0.12, 69.22] |
| 13 Adverse effects: 2e. Specific ‐ miscellaneous | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 13.1 agitation ‐ between 0 ‐1 day (IM phase) (only reported if occurred in ≥5%) | 2 | 477 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [0.46, 3.22] |
| 13.2 agitation ‐ between 3 ‐ 7 days (oral phase) (only reported if occurred in ≥ 2%) | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.68, 1.74] |
| 13.3 anxiety ‐ between 3 ‐ 7 days (oral phase) (only reported if occurred in ≥ 2%) | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.30 [0.54, 3.16] |
| 13.4 headache ‐ between 0 ‐1 day (IM phase) (only reported if occurred in ≥5%) | 2 | 477 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.45, 1.59] |
| 13.5 headache ‐ between 3 ‐ 7 days (oral phase) (only reported if occurred in ≥ 2%) | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.57, 1.97] |
| 13.6 pain at injection site | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.62] |
| 13.7 pain (1st injection) | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.06, 1.54] |
| 13.8 pain (2nd injection) | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.34 [1.03, 1.74] |
| 14 Leaving the study early: 1. For specific reasons | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 14.1 any | 2 | 480 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.07 [0.86, 4.98] |
| 14.2 lack of efficacy | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.84 [0.12, 69.22] |
| 14.3 adverse effects | 2 | 480 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.17, 5.59] |
| 14.4 withdrew | 1 | 360 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.42 [0.24, 8.39] |
| 14.5 consent | 2 | 480 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.0 [0.74, 48.34] |
| 14.6 other | 2 | 480 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.36 [0.27, 6.75] |
Comparison 3. HALOPERIDOL vs OTHER ANTIPSYCHOTIC: b. CHLORPROMAZINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tranquillisation or asleep ‐ asleep | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.93 [1.04, 3.60] |
| 1.1 not asleep up to 2 hours | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.93 [1.04, 3.60] |
| 2 Repeated need for rapid tranquillisation | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 more that 1 injection | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.89, 1.28] |
| 2.2 more than 3 injections | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.5 [0.53, 4.26] |
| 3 Global outcome: 1. Not improved | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 not marked improvement | 2 | 89 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.61, 1.02] |
| 3.2 not any improvement | 2 | 89 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.15 [0.05, 0.49] |
| 4 Adverse effects: any serious or specific adverse effects | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 allergy ‐ haematological ‐ leucopenia ‐ mild | 1 | 50 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 70.30] |
| 4.2 allergy ‐ hepatic ‐ glutamic pyruvic transaminase elevated | 1 | 50 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 7.81] |
| 4.3 allergy ‐ skin irritation ‐ local | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.4 anticholinergic ‐ dry mouth | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [0.17, 10.93] |
| 4.5 arousal ‐ drowsy but asleep | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.06 [0.01, 0.42] |
| 4.6 arousal ‐ drowsy but awake | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.17 [0.59, 142.10] |
| 4.7 cardiovascular ‐ hypotension | 2 | 69 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.10, 2.60] |
| 4.8 central nervous system ‐ seizures | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 7.58] |
| 4.9 movement disorders ‐ extrapyramidal adverse effects | 2 | 69 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.07 [0.28, 15.15] |
| 5 Leaving the study early | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 any reason/general reasons | 4 | 153 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.21 [0.07, 0.71] |
| 5.2 could not be evaluated due to being asleep | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.07 [0.01, 0.52] |
| 5.3 severe hypotensive reaction | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.2 [0.01, 3.85] |
| 5.4 deviation from protocol | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 68.26] |
Comparison 4. HALOPERIDOL vs OTHER ANTIPSYCHOTIC: c. DROPERIDOL.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tranquillisation or asleep: 1. Not asleep | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 up to 2 hours | 1 | 228 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.44, 2.60] |
| 2 Tranquillisation or asleep: 2. Time to sleep | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 up to 2 hours | 1 | 114 | Mean Difference (IV, Fixed, 95% CI) | 5.20 [‐6.05, 16.45] |
| 3 Repeated need for rapid tranquillisation | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 more than 1 injection | 2 | 255 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.38 [1.27, 4.47] |
| 3.2 more than 3 injections | 1 | 27 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.12 [0.09, 47.68] |
| 4 Global outcome: 1. Need for benzodiazepine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 up to 2 hours | 1 | 228 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.31 [0.07, 1.44] |
| 5 Adverse effects: 1. General | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 overall adverse events up to 2 hours | 1 | 228 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.15 [0.02, 1.23] |
| 5.2 patients with one or more AEs | 1 | 228 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.18 [0.02, 1.46] |
| 6 Adverse effects: 2a. Specific ‐ arousal level | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 "over" sedated | 1 | 228 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.01, 8.68] |
| 7 Adverse effects: 2b. Specific ‐ cardiovascular | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 hypotension up to 2 hours | 1 | 228 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.03, 2.36] |
| 8 Adverse effects: 2c. Specific ‐ movement disorders | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 dystonia | 2 | 255 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.11, 6.56] |
| 9 Adverse effects: 2d. Specific ‐ miscellaneous | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 desaturation | 1 | 228 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.01, 8.68] |
Comparison 5. HALOPERIDOL vs OTHER ANTIPSYCHOTIC: d. LOXAPINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tranquillisation or asleep ‐ not asleep up to 24 hours | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.31 [0.54, 34.48] |
| 2 Specific behaviour: 2. Aggression ‐ various measures | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 no overall improvement | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.1 [0.69, 1.76] |
| 3 Global outcome: 1. General ‐ no change ‐ i. over 24 hours | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 up to 48 hours (CGI‐I, high=worse) | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.14, 6.15] |
| 3.2 up to 72 hours (CGI‐I, high=worse) | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.39 [0.04, 3.49] |
| 3.3 at endpoint (CGI‐I, high=worse) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.40 [0.12, 1.32] |
| 4 Global outcome: 2a. Specific ‐ not sedated ‐ i. by 30 minutes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 at 30 minutes | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.29 [0.65, 2.54] |
| 5 Global outcome: 2b. Specific ‐ not sedated ‐ ii. up to 2 hours | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 at 1 hour | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.5 [0.86, 14.18] |
| 5.2 at 2 hours | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.0 [0.98, 50.16] |
| 6 Mental state: 1. Average scores | 1 | 52 | Mean Difference (IV, Fixed, 95% CI) | 6.10 [4.48, 7.72] |
| 6.1 endpoint score (BPRS, high=worse) | 1 | 52 | Mean Difference (IV, Fixed, 95% CI) | 6.10 [4.48, 7.72] |
| 7 Adverse effects: 1. General | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 one or more drug related adverse effect | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.8 [0.44, 1.45] |
| 8 Adverse effects: 2a. Specific ‐ anticholinergic | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 constipation ‐ between 3‐28 days (Oral phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.26] |
| 8.2 salivation ‐ too little ‐ between 0‐3 days (IM phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.83 [0.33, 24.66] |
| 8.3 salivation ‐ too little ‐ between 3‐28 days (Oral phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.22, 4.05] |
| 8.4 salivation ‐ too much ‐ between 0‐3 days (IM phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.33, 2.69] |
| 8.5 salivation ‐ too much ‐ between 0‐10 days | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.04, 4.48] |
| 8.6 salivation ‐ too much ‐ between 3‐28 days (Oral phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.57, 1.93] |
| 8.7 polyuria ‐ between 3‐28 days (Oral phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.84 [0.12, 65.34] |
| 8.8 sweating ‐ between 0‐3 days (IM phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.15, 5.97] |
| 8.9 sweating ‐ between 3‐28 days (Oral phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.84 [0.12, 65.34] |
| 9 Adverse effects: 2b. Specific ‐ arousal level | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 asleep ‐ by 12 hours | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.72, 1.04] |
| 9.2 drowsiness ‐ between 0‐3 days (IM phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.70, 1.12] |
| 9.3 drowsiness ‐ between 3‐28 days (Oral phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 33.16 [2.15, 511.57] |
| 9.4 drowsiness during 72 hours | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.20, 2.79] |
| 9.5 insomnia ‐ between 0‐3 days (IM phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.26] |
| 9.6 insomnia ‐ between 3‐28 days (Oral phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.63 [0.37, 119.59] |
| 10 Adverse effects: 2c. Specific ‐ cardiovascular | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 blood pressure ‐ increased ‐ between 0‐10 days | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.17 [0.01, 3.45] |
| 10.2 dizziness ‐ between 3‐28 days | 2 | 65 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.38 [0.09, 1.52] |
| 10.3 dyspnoea ‐ between 3‐28 days (Oral phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.84 [0.12, 65.34] |
| 10.4 palpitations during 72 hours | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 7.58] |
| 10.5 pulse rate ‐ increased ‐ between 0‐10 days | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.01, 6.79] |
| 11 Adverse effects: 2d. Specific ‐ movement disorders | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 11.1 akathisia between 0‐3 days (IM phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.57 [0.16, 2.02] |
| 11.2 akathisia between 0‐10 days | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.26, 1.61] |
| 11.3 akathisia between 3‐28 days (Oral phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.62, 2.27] |
| 11.4 dysarthria between 3‐28 days (Oral phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 2.44] |
| 11.5 dyskinesia between 0‐10 days | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.01, 6.79] |
| 11.6 dystonia between 0‐3 days (IM phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.06, 13.93] |
| 11.7 dystonia between 0‐10 days | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.26, 1.61] |
| 11.8 involuntary movement between 3‐28 days (Oral phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.26] |
| 11.9 motor retardation between 3‐28 days (Oral phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.06, 13.93] |
| 11.10 muscle spasms between 0‐3 days (IM phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.33, 4.82] |
| 11.11 oculogyric crisis between 0‐10 days | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.6 [0.11, 61.11] |
| 11.12 rigidity between 0‐3 days (IM phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.65, 1.19] |
| 11.13 rigidity between 0‐10 days | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.13, 5.68] |
| 11.14 rigidity during between 3‐28 days (Oral phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.93, 1.38] |
| 11.15 tremor between 0‐3 days (IM phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.42, 2.13] |
| 11.16 tremor during between 3‐28 days (Oral phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.71, 1.76] |
| 11.17 tremor between 0‐10 days | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.6 [0.11, 61.11] |
| 11.18 use of antiparkinson medication | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.83 [0.66, 12.16] |
| 11.19 dystonia during 72 hours | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.26, 96.13] |
| 11.20 extrapyramidal effects ‐ use of antiparkinson drugs during 72 hours | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.6 [0.17, 2.07] |
| 11.21 EPS during 72 hours | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.0 [0.98, 50.16] |
| 11.22 thick tongue ‐ between 0‐10 days | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.01, 6.79] |
| 12 Adverse effects: 2e. Specific ‐ miscellaneous | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.1 allergy ‐ itch ‐ between 3‐28 days (Oral phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.84 [0.12, 65.34] |
| 12.2 allergy ‐ tissue reaction at injection site ‐ between 0‐10 days | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 12.3 nervousness ‐ between 0‐10 days | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.01, 6.79] |
| 12.4 pain ‐ during 24 hours | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.13, 3.44] |
| 12.5 pain ‐ headache ‐ between 0‐3 days (IM phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.26] |
| 12.6 pain ‐ myalgia ‐ between 3‐28 days (Oral phase) | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.84 [0.12, 65.34] |
| 13 Leaving the study early: 1. For general reasons | 2 | Risk Ratio (M‐H, Fixed, 90% CI) | Subtotals only | |
| 13.1 by 72 hours | 1 | 35 | Risk Ratio (M‐H, Fixed, 90% CI) | 0.63 [0.21, 1.85] |
| 13.2 by 10 days | 1 | 54 | Risk Ratio (M‐H, Fixed, 90% CI) | 1.18 [0.67, 2.07] |
| 13.3 by endpoint | 1 | 35 | Risk Ratio (M‐H, Fixed, 90% CI) | 0.94 [0.42, 2.13] |
| 14 Leaving the study early: 2. Specific reasons | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 14.1 adverse effects by 72 hours | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.84 [0.12, 65.34] |
5.2. Analysis.
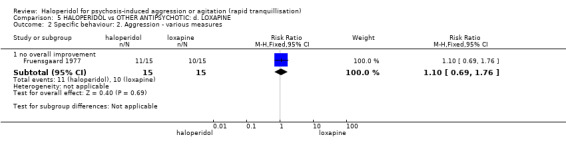
Comparison 5 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: d. LOXAPINE, Outcome 2 Specific behaviour: 2. Aggression ‐ various measures.
5.5. Analysis.
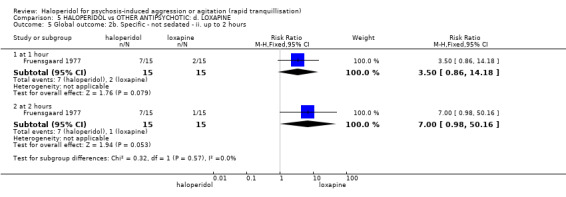
Comparison 5 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: d. LOXAPINE, Outcome 5 Global outcome: 2b. Specific ‐ not sedated ‐ ii. up to 2 hours.
Comparison 6. HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tranquillisation or asleep ‐ asleep | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 not asleep up to 2 hours | 1 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [1.02, 1.32] |
| 2 Repeated need for tranquillisation ‐ needing additional injection | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 by 2 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.0 [0.47, 33.73] |
| 2.2 by 12 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.29 [1.67, 6.47] |
| 2.3 by 24 hours | 3 | 392 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.75, 1.51] |
| 3 Specific behaviour: 1a. Agitation ‐ various measures | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 response ‐ up to 2 hours (≥40% reduction in PANSS‐EC) | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.58, 1.58] |
| 3.2 agitation ‐ reported as 'adverse effect' | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.07, 15.73] |
| 3.3 agitation during 21 days (if occurred in ≧10% at P < 0.05) ‐ reported as 'adverse effect' | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.33, 3.51] |
| 4 Specific behaviour: 1b. Agitation ‐ average scores ‐ i. by 30 minutes | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 change score at 15 minutes (ACES, low=agitated, high=sedated) | 1 | 46 | Mean Difference (IV, Fixed, 95% CI) | 0.5 [‐0.35, 1.35] |
| 4.2 change scores at 15 minutes (PANSS‐EC, high=worse) | 1 | 46 | Mean Difference (IV, Fixed, 95% CI) | 0.30 [‐1.64, 2.24] |
| 4.3 change score at 30 minutes (ACES, low=agitated, high=sedated) | 1 | 46 | Mean Difference (IV, Fixed, 95% CI) | 0.90 [‐0.06, 1.86] |
| 4.4 change scores at 30 minutes (PANSS‐EC, high=worse) | 1 | 46 | Mean Difference (IV, Fixed, 95% CI) | 1.30 [‐1.16, 3.76] |
| 5 Specific behaviour: 1c. Agitation ‐ average scores ‐ ii. up to 2 hours | 5 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 change score (ABS, high=worse) | 1 | 85 | Mean Difference (IV, Fixed, 95% CI) | 2.7 [0.38, 5.02] |
| 5.2 change score at 1 hour (ACES, low=agitated, high=sedated) | 1 | 46 | Mean Difference (IV, Fixed, 95% CI) | 1.2 [0.06, 2.34] |
| 5.3 endpoint score at 1 hour (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 2.00 [1.18, 2.82] |
| 5.4 change score at 1 hour (PANSS‐EC, high=worse) | 1 | 46 | Mean Difference (IV, Fixed, 95% CI) | 2.2 [‐0.50, 4.90] |
| 5.5 change score at 2 hours (ACES, low=agitated, high=sedated) | 1 | 46 | Mean Difference (IV, Fixed, 95% CI) | 0.30 [‐0.74, 1.34] |
| 5.6 endpoint score at 2 hours (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 4.20 [3.54, 4.86] |
| 5.7 score at 2 hours (PANSS‐EC, high=worse) | 3 | 332 | Mean Difference (IV, Fixed, 95% CI) | ‐0.31 [‐1.49, 0.86] |
| 5.8 endpoint score at 2 hours (PANSS‐PAS, high=worse) | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | 2.10 [‐2.56, 6.76] |
| 6 Specific behaviour: 1d. Agitation ‐ average scores ‐ iii. up to 4 hours | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [‐0.05, 0.45] |
| 6.1 endpoint score at 4 hours (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [‐0.05, 0.45] |
| 7 Specific behaviour: 1e. Agitation ‐ average scores ‐ iv. up to 24 hours | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 7.1 change score (ABS, high=worse) | 1 | 86 | Mean Difference (IV, Fixed, 95% CI) | 2.40 [0.01, 4.79] |
| 7.2 change score (PANSS‐EC, high=worse) | 1 | 86 | Mean Difference (IV, Fixed, 95% CI) | 1.40 [‐0.55, 3.35] |
| 7.3 endpoint score at 6 hours (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [0.05, 0.35] |
| 7.4 endpoint score at 12 hours (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.9 [0.82, 0.98] |
| 8 Specific behaviour: 2a. Aggression ‐ average scores ‐ i. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 8.1 endpoint score at 1 hour (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.90 [0.13, 1.67] |
| 8.2 endpoint score at 2 hours (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.5 [‐0.41, 1.41] |
| 9 Specific behaviour: 2b. Aggression ‐ average scores ‐ ii. up to 4 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 9.1 endpoint score at 4 hours (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.10 [‐0.11, 0.31] |
| 10 Specific behaviour: 2c. Aggression ‐ average scores ‐ iii. up to 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.1 endpoint score at 6 hours (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐0.26, 0.06] |
| 10.2 endpoint score at 12 hours (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.20 [‐0.39, ‐0.01] |
| 11 Specific behaviour: 3. Hostility | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.01, 8.12] |
| 12 Global outcome: 1a. General ‐ need for additional measures | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.1 need for benzodiazepine during 24 hours | 2 | 343 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.63, 1.74] |
| 12.2 need for benzodiazepine during 7 days | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.79, 1.27] |
| 12.3 additional restraint, seclusion or special observation | 2 | 160 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.49 [0.70, 3.17] |
| 13 Global outcome: 1b. General ‐ time and doses | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 13.1 time to discontinuation | 1 | 100 | Mean Difference (IV, Fixed, 95% CI) | ‐3.48 [‐6.28, ‐0.68] |
| 13.2 dose of adjunctive lorazepam | 1 | 100 | Mean Difference (IV, Fixed, 95% CI) | 0.34 [‐0.14, 0.82] |
| 14 Global outcome: 1c. General ‐ average scores ‐ up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 14.1 endpoint score (CGI‐I, high=worse) | 1 | 42 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐0.65, 0.45] |
| 15 Global outcome: 1d. General ‐ average scores ‐ over 24 hours | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.1 change score (CGI‐I, high=worse) | 1 | 243 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐0.20, 0.20] |
| 15.2 change score (CGI‐S, high=worse) | 1 | 86 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐0.24, 0.24] |
| 15.3 change score by 21 days (CGI‐I, high=worse) | 1 | 100 | Mean Difference (IV, Fixed, 95% CI) | 0.36 [‐0.18, 0.90] |
| 16 Global outcome: 2a. Specific ‐ alert ‐ i. up to 2 hours | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 16.1 alert at 1 hour | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.99, 1.55] |
| 17 Global outcome: 2b. Specific ‐ alert ‐ ii. up to 4 hours | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 17.1 alert at 4 hours | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.66, 1.42] |
| 18 Global outcome: 2c. Specific ‐ alert ‐ iii. up to 24 hours | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 18.1 alert at 8 hours | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.67, 1.60] |
| 18.2 alert at 16 hours | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.58, 1.78] |
| 18.3 alert at 24 hours | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.60, 1.20] |
| 19 Global outcome: 2d. Specific ‐ tranquil ‐ i. up to 2 hours | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 19.1 tranquil at 1 hour | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.29, 1.34] |
| 20 Global outcome: 2e. Specific ‐ tranquil ‐ ii. up to 4 hours | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 20.1 tranquil at 4 hours | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.84 [0.94, 3.62] |
| 21 Global outcome: 2f. Specific ‐ tranquil ‐ iii. up to 24 hours | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 21.1 tranquil at 8 hours | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.55, 1.87] |
| 21.2 tranquil at 16 hours | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.36, 1.45] |
| 21.3 tranquil at 24 hours | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.50 [0.83, 2.72] |
| 22 Global outcome: 2g. Specific ‐ sedated ‐ i. up to 2 hours | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 22.1 sedated at 1 hour | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.22 [0.01, 4.39] |
| 23 Global outcome: 2h. Specific ‐ sedated ‐ ii. up to 4 hours | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 23.1 sedated at 4 hours | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.18, 1.03] |
| 24 Global outcome: 2i. Specific ‐ sedated ‐ iii. up to 24 hours | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 24.1 sedated at 8 hours | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.49, 2.37] |
| 24.2 sedated at 16 hours | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.47, 1.50] |
| 24.3 sedated at 24 hours | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.19, 1.98] |
| 25 Global outcome: 2j. Specific ‐ asleep ‐ i. up to 2 hours | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 25.1 asleep at 1 hour | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.02, 8.64] |
| 26 Global outcome: 2k. Specific ‐ asleep ‐ ii. up to 4 hours | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 26.1 asleep at 4 hours | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.07, 16.84] |
| 27 Global outcome: 2l. Specific ‐ asleep ‐ iii. up to 24 hours | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 27.1 asleep at 8 hours | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.03, 2.34] |
| 27.2 asleep at 16 hours | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.33 [0.97, 19.40] |
| 27.3 asleep at 24 hours | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.24 [0.14, 77.79] |
| 28 Service use: 1. Average days to discharge | 1 | 100 | Mean Difference (IV, Fixed, 95% CI) | ‐0.60 [‐1.85, 0.65] |
| 29 Service use: 2. Average patient‐hours used during 24 hours (skewed data) | Other data | No numeric data | ||
| 29.1 days 1‐7 | Other data | No numeric data | ||
| 29.2 days 8‐14 | Other data | No numeric data | ||
| 29.3 days 15‐21 | Other data | No numeric data | ||
| 30 Mental state: 1. Various outcomes reported as 'adverse events' | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 30.1 anxiety | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.01, 8.12] |
| 30.2 anxiety during 21 days (if occurred in ≧10% at P < 0.05) | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.08, 1.70] |
| 30.3 delusions | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.01, 8.12] |
| 30.4 nervousness during 21 days (if occurred in ≧10% at P < 0.05) | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.17 [0.70, 6.74] |
| 31 Mental state: 2a. Average scores ‐ i. by 30 minutes | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 31.1 change score at 15 minutes (BPRS positive sub‐scale, high=worse) | 1 | 46 | Mean Difference (IV, Fixed, 95% CI) | ‐0.30 [‐2.11, 1.51] |
| 31.2 change score at 30 minutes (BPRS positive sub‐scale, high=worse) | 1 | 46 | Mean Difference (IV, Fixed, 95% CI) | ‐0.40 [‐2.70, 1.90] |
| 31.3 change score at 15 minutes (BPRS total, high=worse) | 1 | 46 | Mean Difference (IV, Fixed, 95% CI) | ‐0.70 [‐5.42, 4.02] |
| 31.4 change score at 30 minutes (BPRS total, high=worse) | 1 | 46 | Mean Difference (IV, Fixed, 95% CI) | ‐0.30 [‐7.35, 6.75] |
| 32 Mental state: 2b. Average scores ‐ ii. up to 2 hours | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 32.1 change score at 1 hour (BPRS positive sub‐scale, high=worse) | 1 | 46 | Mean Difference (IV, Fixed, 95% CI) | 0.60 [‐2.74, 3.94] |
| 32.2 change score at 2 hours (BPRS positive sub‐scale, high=worse) | 3 | 385 | Mean Difference (IV, Fixed, 95% CI) | 0.28 [‐0.34, 0.89] |
| 32.3 change score at 1 hour (BPRS total, high=worse) | 1 | 46 | Mean Difference (IV, Fixed, 95% CI) | 4.70 [‐5.03, 14.43] |
| 32.4 change score at 2 hours (BPRS total, high=worse) | 3 | 385 | Mean Difference (IV, Fixed, 95% CI) | 1.75 [‐0.12, 3.62] |
| 33 Mental state: 2c. Average scores ‐ iii. up to 24 hours | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 33.1 change score at 24 hours (BPRS positive sub‐scale, high=worse) | 2 | 340 | Mean Difference (IV, Fixed, 95% CI) | ‐0.30 [‐0.97, 0.37] |
| 33.2 change score at 24 hours (BPRS total, high=worse) | 2 | 340 | Mean Difference (IV, Fixed, 95% CI) | 0.47 [‐1.34, 2.29] |
| 34 Mental state: 2d. Average scores ‐ iv. over 24 hours | 1 | 100 | Mean Difference (IV, Fixed, 95% CI) | 4.70 [‐0.18, 9.58] |
| 34.1 change score at 21 days (PANSS total, high=worse) | 1 | 100 | Mean Difference (IV, Fixed, 95% CI) | 4.70 [‐0.18, 9.58] |
| 35 Adverse effects: 1a. General | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 35.1 one or more drug related adverse events | 3 | 209 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [1.09, 1.76] |
| 36 Adverse effects: 1b. General ‐ serious | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 36.1 one or more treatment emergent adverse events | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.36, 1.83] |
| 36.2 treatment emergent adverse events ‐ all | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.23 [0.69, 2.19] |
| 36.3 overall serious adverse effects | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 36.4 death | 3 | 138 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 37 Adverse effects: 2a. Specific ‐ anticholinergic | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 37.1 increased salivation during 21 days (if occurred in ≧10% at P < 0.05) | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.73 [0.54, 176.18] |
| 38 Adverse effects: 2b. Specific ‐ arousal level ‐ i. various | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 38.1 insomnia | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.39, 2.78] |
| 38.2 insomnia during 21 days (if occurred in ≧10% at P < 0.05) | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.17 [0.57, 8.19] |
| 38.3 somnolence at 24 hours | 1 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.45, 2.41] |
| 38.4 somnolence during 21 days (if occurred in ≧10% at P < 0.05) | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.67, 3.12] |
| 38.5 excessive sedation during 24 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.33, 27.23] |
| 39 Adverse effects: 2c. Specific ‐ arousal level ‐ ii. average scores ‐ i. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 39.1 endpoint score at 1 hour (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.30 [‐0.13, 0.73] |
| 39.2 endpoint score at 2 hours (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐0.36, 0.36] |
| 40 Adverse effects: 2d. Specific ‐ arousal level ‐ ii. average scores ‐ ii. up to 4 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 40.1 endpoint score at 4 hours (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.20 [‐0.61, 0.21] |
| 41 Adverse effects: 2e. Specific ‐ arousal level ‐ ii. average scores ‐ iii. up to 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 41.1 endpoint score at 6 hours (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐0.38, 0.18] |
| 41.2 endpoint score at 12 hours (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐0.26, 0.26] |
| 42 Adverse effects: 2f. Specific ‐ cardiovascular i. binary | 3 | 195 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.03, 2.38] |
| 42.1 clinically significant ECG change | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 42.2 hypotension during 24 hours | 2 | 146 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.03, 2.38] |
| 43 Adverse effects: 2g. Specific ‐ cardiovascular ii. continuous | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 43.1 QT interval: average change at 24 hours | 1 | 257 | Mean Difference (IV, Fixed, 95% CI) | 1.8 [‐3.81, 7.41] |
| 43.2 QT interval: average endpoint score at 24 hours | 1 | 86 | Mean Difference (IV, Fixed, 95% CI) | 8.5 [‐3.28, 20.28] |
| 44 Adverse effects: 2h. Specific ‐ gastrointestinal | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 44.1 abdominal pain | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.12 [0.13, 73.04] |
| 44.2 vomiting | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.2 [0.26, 103.03] |
| 45 Adverse effects: 2i. Specific ‐ movement disorder ‐ i. various | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 45.1 dystonia during 24 hours | 2 | 343 | Risk Ratio (M‐H, Fixed, 95% CI) | 12.92 [1.67, 99.78] |
| 45.2 dystonia during 21 days (if occurred in ≧10% at P < 0.05) | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.73 [0.54, 176.18] |
| 45.3 hypertonia during 21 days (if occurred in ≧10% at P < 0.05) | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.73 [0.54, 176.18] |
| 45.4 EPS during 24 hours | 3 | 403 | Risk Ratio (M‐H, Fixed, 95% CI) | 8.35 [2.27, 30.63] |
| 45.5 EPS during study | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.07, 15.73] |
| 45.6 extrapyramidal effects ‐ use of antiparkinson drugs during 24 hours | 1 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.51 [1.92, 10.58] |
| 45.7 extrapyramidal effects ‐ use of antiparkinson drugs during 21 days | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.73 [0.54, 176.18] |
| 45.8 extrapyramidal effects ‐ use of antiparkinson drugs during study | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.07, 15.73] |
| 46 Adverse effects: 2j. Specific ‐ movement disorder ‐ ii. average scores ‐ i. up to 24 hours | 1 | 483 | Mean Difference (IV, Fixed, 95% CI) | 0.34 [0.16, 0.53] |
| 46.1 change score at 24 hours (SAS, high=worse) | 1 | 242 | Mean Difference (IV, Fixed, 95% CI) | 1.31 [0.56, 2.06] |
| 46.2 change score at 24 hours (BAS, high=worse) | 1 | 241 | Mean Difference (IV, Fixed, 95% CI) | 0.28 [0.09, 0.47] |
| 47 Adverse effects: 2k. Specific ‐ movement disorder ‐ iii. average scores ‐ i. up to 24 hours | Other data | No numeric data | ||
| 47.1 endpoint score at 24 hours (SAS, high=worse) ‐ (skewed data) | Other data | No numeric data | ||
| 47.2 endpoint score at 24 hours (BAS, high=worse) ‐ (skewed data) | Other data | No numeric data | ||
| 48 Adverse effects: 2l. Specific ‐ movement disorder ‐ vi. average scores ‐ i. over 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 48.1 endpoint score at 96 hours (AIMS, high=worse) | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [‐0.87, 1.27] |
| 48.2 endpoint score at 96 hours (BARS, high=worse) | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | 0.8 [‐0.01, 1.61] |
| 48.3 endpoint score at 96 hours (SAS, high=worse) | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | 2.2 [‐0.07, 4.47] |
| 49 Adverse effects: 2m. Specific ‐ miscellaneous | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 49.1 pain | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.12 [0.13, 73.04] |
| 49.2 pain during 21 days (if occurred in ≧10% at P < 0.05) | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.33, 3.51] |
| 49.3 pain ‐ headache during 21 days (if occurred in ≧10% at P < 0.05) | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.17 [0.88, 5.32] |
| 49.4 other ‐ nose bleed | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.12 [0.13, 73.04] |
| 49.5 other ‐ clinically relevant laboratory changes | 2 | 149 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 50 Leaving the study early | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 50.1 any reason | 2 | 148 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.66 [1.04, 2.65] |
| 50.2 adverse effects | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 8.67 [1.13, 66.75] |
| 50.3 lack of efficacy | 2 | 129 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.09, 1.44] |
| 50.4 lost at follow‐up | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.54 [0.10, 2.82] |
| 50.5 participants decision | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.17 [0.57, 8.19] |
| 50.6 non compliance | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.17 [0.42, 11.30] |
| 50.7 physician decision | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.33 [0.50, 37.42] |
| 50.8 sponsor decision | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.02, 8.64] |
6.17. Analysis.
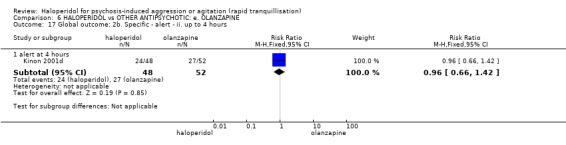
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 17 Global outcome: 2b. Specific ‐ alert ‐ ii. up to 4 hours.
6.18. Analysis.
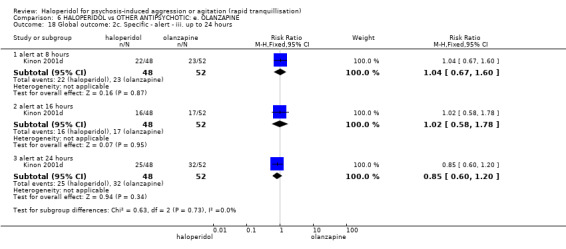
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 18 Global outcome: 2c. Specific ‐ alert ‐ iii. up to 24 hours.
6.19. Analysis.
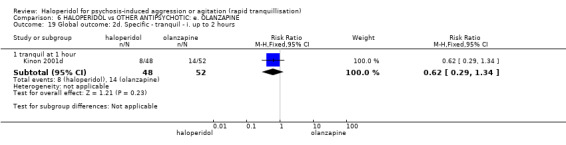
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 19 Global outcome: 2d. Specific ‐ tranquil ‐ i. up to 2 hours.
6.20. Analysis.
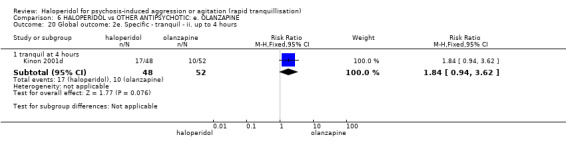
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 20 Global outcome: 2e. Specific ‐ tranquil ‐ ii. up to 4 hours.
6.21. Analysis.
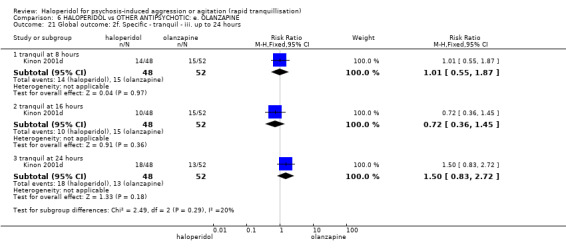
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 21 Global outcome: 2f. Specific ‐ tranquil ‐ iii. up to 24 hours.
6.22. Analysis.
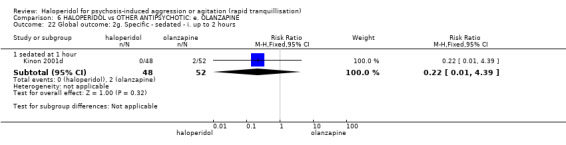
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 22 Global outcome: 2g. Specific ‐ sedated ‐ i. up to 2 hours.
6.23. Analysis.
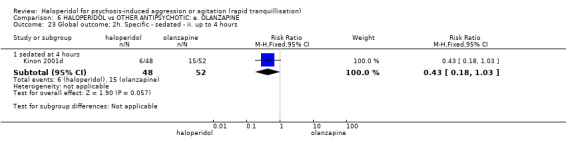
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 23 Global outcome: 2h. Specific ‐ sedated ‐ ii. up to 4 hours.
6.24. Analysis.
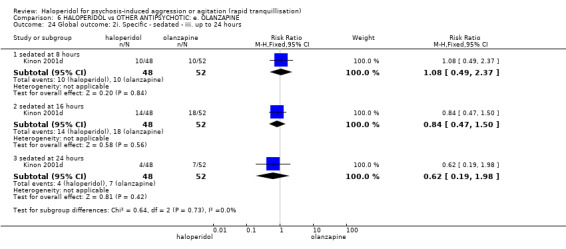
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 24 Global outcome: 2i. Specific ‐ sedated ‐ iii. up to 24 hours.
6.25. Analysis.
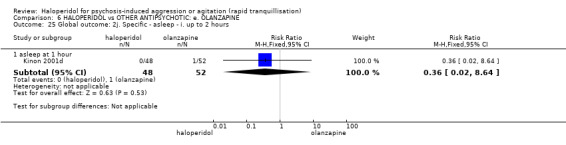
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 25 Global outcome: 2j. Specific ‐ asleep ‐ i. up to 2 hours.
6.26. Analysis.
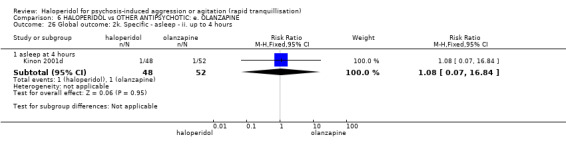
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 26 Global outcome: 2k. Specific ‐ asleep ‐ ii. up to 4 hours.
6.27. Analysis.
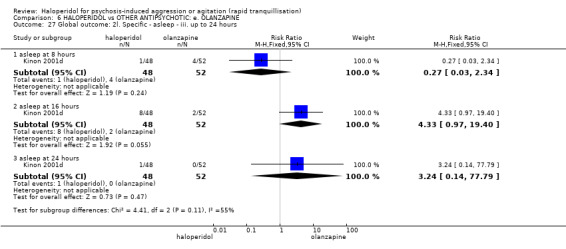
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 27 Global outcome: 2l. Specific ‐ asleep ‐ iii. up to 24 hours.
6.32. Analysis.
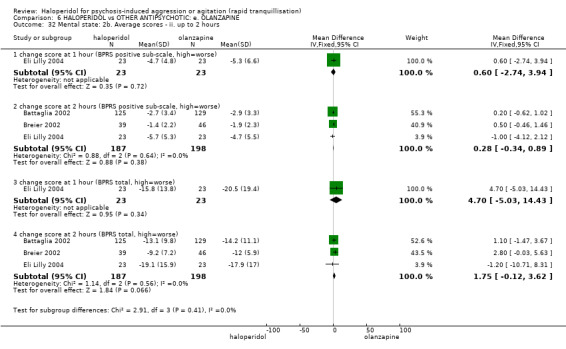
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 32 Mental state: 2b. Average scores ‐ ii. up to 2 hours.
6.33. Analysis.
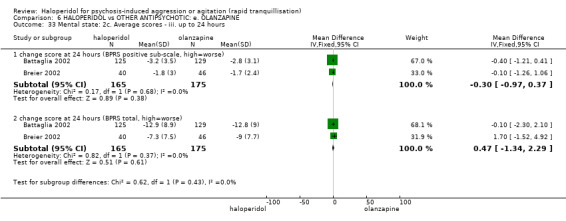
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 33 Mental state: 2c. Average scores ‐ iii. up to 24 hours.
6.39. Analysis.
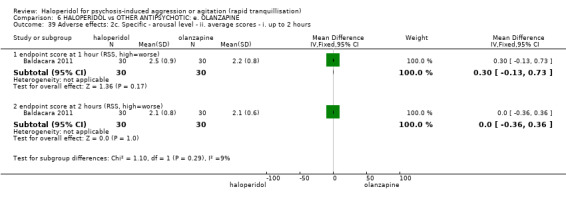
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 39 Adverse effects: 2c. Specific ‐ arousal level ‐ ii. average scores ‐ i. up to 2 hours.
6.40. Analysis.
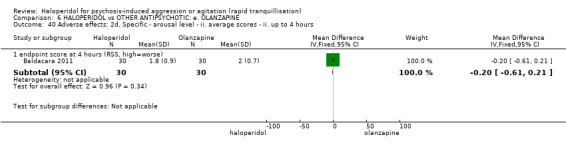
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 40 Adverse effects: 2d. Specific ‐ arousal level ‐ ii. average scores ‐ ii. up to 4 hours.
6.41. Analysis.
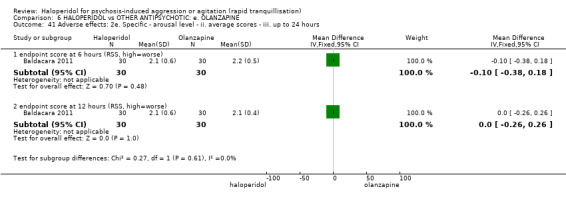
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 41 Adverse effects: 2e. Specific ‐ arousal level ‐ ii. average scores ‐ iii. up to 24 hours.
6.48. Analysis.
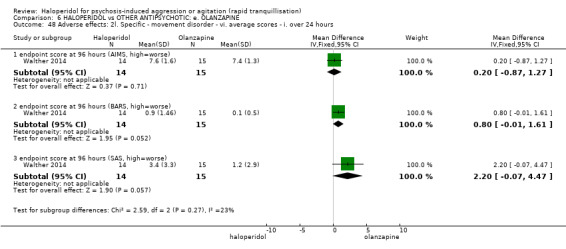
Comparison 6 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: e. OLANZAPINE, Outcome 48 Adverse effects: 2l. Specific ‐ movement disorder ‐ vi. average scores ‐ i. over 24 hours.
Comparison 7. HALOPERIDOL vs OTHER ANTIPSYCHOTIC: f. PERPHENAZINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global outcome: No improvement | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.04, 4.68] |
| 2 Adverse effects: 1. General | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 one or more adverse effect | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.30 [0.61, 2.80] |
| 2.2 clinically significant laboratory changes | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Adverse effects: 2. Specific | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 hypotensive episode | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.31 [0.01, 7.12] |
| 3.2 require antiparkinson medication | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.74 [0.62, 12.12] |
| 3.3 discontinued due to EPS | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.83 [0.18, 18.70] |
| 3.4 discontinued due to drowsiness/tension and no therapeutic effect | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.75 [0.12, 64.04] |
| 4 Leaving the study early: 1. Specific reasons | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 discontinued due to EPS | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.83 [0.18, 18.70] |
| 4.2 discontinued due to drowsiness/tension and no therapeutic effect | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.75 [0.12, 64.04] |
Comparison 8. HALOPERIDOL vs OTHER ANTIPSYCHOTIC: g. QUETIAPINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Specific behaviour: 1a. Agitation ‐ average scores ‐ i. up to 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1 change score at 24 hours (PANSS‐EC, high=worse) | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | 0.10 [‐0.56, 0.76] |
| 2 Specific behaviour: 1b. Agitation ‐ average scores ‐ ii. over 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 change score at 72 hours (PANSS‐EC, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 change score at 7 days (PANSS‐EC, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 change score at 10 days (PANSS‐EC, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.4 change score at 14 days (PANSS‐EC, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.5 change score at 28 days (PANSS‐EC, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Mental state: Average scores ‐ i. over 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 change score at 28 days (PANSS total, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 change score at 28 days (PANSS positive sub‐scale, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 change score at 28 days (PANSS negative sub‐scale, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Adverse effects: 1. General | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 one or more drug related adverse events by 28 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Adverse effects: 2. Specific ‐ movement disorder ‐ i. average scores | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 5.1 endpoint score at 28 days (SAS, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Leaving the study early | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1 any reason | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 adverse event | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 lost at follow‐up | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 9. HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tranquillisation or asleep ‐ i. by 30 minutes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 not asleep at 30 minutes | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.74, 0.95] |
| 2 Tranquillisation or asleep ‐ ii. up to 2 hours | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 not asleep at 1 hour | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.59, 0.92] |
| 2.2 not asleep at 2 hours | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.51, 0.99] |
| 3 Specific behaviour: 1a. Agitation ‐ various measures | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 ≥50% reduction in PANSS‐EC score up to 24 hours | 1 | 124 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.79, 1.16] |
| 3.2 discontinued due to severe agitation up to 24 hours | 1 | 124 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 8.03] |
| 4 Specific behaviour: 1b. Agitation ‐ average scores ‐ i. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 endpoint score at 2 hours (PANSS‐PAS, high=worse) | 1 | 28 | Mean Difference (IV, Fixed, 95% CI) | ‐0.40 [‐5.22, 4.42] |
| 5 Specific behaviour: 2a. Aggression ‐ average scores ‐ i. by 30 minutes | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 change score at 30 minutes (OAS total aggression, high=worse) | 1 | 147 | Mean Difference (IV, Fixed, 95% CI) | ‐0.5 [‐0.58, ‐0.42] |
| 6 Specific behaviour: 2b. Aggression ‐ average scores ‐ ii. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.1 change score at 1 hour (OAS total aggression, high=worse) | 1 | 146 | Mean Difference (IV, Fixed, 95% CI) | ‐0.20 [‐0.27, ‐0.13] |
| 6.2 change score at 2 hours (OAS total aggression, high=worse) | 1 | 145 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐0.13, ‐0.07] |
| 7 Global outcome: 1. Binary measures | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 needing additional benzodiazepine | 2 | 286 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.65, 1.47] |
| 7.2 rated as severe at 24 hours (CGI‐S) | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.51, 1.58] |
| 8 Global outcome: 2. Continuous measures | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 8.1 dose of lorazapam | 1 | 147 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐0.49, 0.29] |
| 8.2 time to additional dose | 1 | 147 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [‐2.17, 2.57] |
| 9 Adverse effects: 1a. General ‐ one or more adverse effects | 2 | 286 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.84, 1.23] |
| 9.1 up to 24 hours | 2 | 286 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.84, 1.23] |
| 10 Adverse effects: 2a. Specific ‐ arousal | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 sedated at 30 minutes | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.90 [1.39, 2.59] |
| 10.2 sedated at 60 minutes | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.41 [1.15, 1.72] |
| 10.3 sedated at 120 minutes | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.98, 1.23] |
| 10.4 somnolence during 24 hours (only reported if occurred in ≥5%) | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.43, 2.12] |
| 10.5 somnolence during 24 hours | 1 | 124 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.31, 1.96] |
| 10.6 insomnia ‐ severe | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.01, 8.47] |
| 10.7 somnolence ‐ severe | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.15 [0.13, 76.20] |
| 11 Adverse effects: 2b. Specific ‐ cardiovascular ‐ i. binary outcomes | 2 | 286 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.46 [0.45, 4.70] |
| 11.1 dizziness during 24 hours | 1 | 124 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.26, 3.82] |
| 11.2 orthostatic hypertension | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.25 [0.26, 107.67] |
| 12 Adverse effects: 2c. Specific ‐ cardiovascular ‐ ii. pulse rate ‐ i. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 12.1 heartbeat change at 60 minutes | 1 | 162 | Mean Difference (IV, Fixed, 95% CI) | ‐9.4 [‐9.99, ‐8.81] |
| 13 Adverse effects: 2d. Specific ‐ cardiovascular ‐ ii. pulse rate ‐ ii. up to 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 13.1 heartbeat change at 8 hours | 1 | 162 | Mean Difference (IV, Fixed, 95% CI) | ‐8.8 [‐9.75, ‐7.85] |
| 14 Adverse effects: 2e. Specific ‐ movement disorder ‐ i. binary outcomes | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 14.1 dystonia during 24 hours | 2 | 286 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.28, 7.28] |
| 14.2 dyskinesia during 24 hours | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.15 [0.13, 76.20] |
| 14.3 hyperkinesia during 24 hours (only reported if occurred in ≥5%) | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.20 [0.48, 36.79] |
| 14.4 hypertonia/rigidity during 24 hours | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.01, 8.47] |
| 14.5 tremor during 24 hours | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.25 [0.26, 107.67] |
| 14.6 movement disorder | 1 | 124 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.6 [0.55, 4.62] |
| 15 Adverse effects: 2f. Specific ‐ movement disorders ‐ ii. average scores ‐ i. up to 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 15.1 change score at 24 hours (BAS akathisia, high=worse) | 1 | 162 | Mean Difference (IV, Fixed, 95% CI) | 0.3 [0.24, 0.36] |
| 15.2 change score at 24 hours (SAS movement disorders, high=worse) | 1 | 162 | Mean Difference (IV, Fixed, 95% CI) | 0.4 [0.34, 0.46] |
| 16 Adverse effects: 2g. Specific ‐ movement disorder ‐ ii. average scores ‐ ii. over 24 hours | 1 | 84 | Mean Difference (IV, Fixed, 95% CI) | 0.58 [‐0.32, 1.47] |
| 16.1 endpoint score at 96 hours (AIMS, high=worse) | 1 | 28 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 16.2 endpoint score at 96 hours (BARS, high=worse) | 1 | 28 | Mean Difference (IV, Fixed, 95% CI) | 0.60 [‐0.36, 1.56] |
| 16.3 endpoint score at 96 hours (SAS, high=worse) | 1 | 28 | Mean Difference (IV, Fixed, 95% CI) | 0.40 [‐2.20, 3.00] |
| 17 Adverse effects: 2h. Specific ‐ miscellaneous | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 17.1 agitation during 24 hours (only reported if occurred in ≥5%) | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.27, 4.06] |
| 17.2 agitation ‐ severe | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.10 [0.19, 22.72] |
| 17.3 anxiety ‐ severe | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.15 [0.13, 76.20] |
| 17.4 headache during 24 hours (only reported if occurred in ≥5%) | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.31 [0.37, 4.71] |
| 17.5 headache during 24 hours | 1 | 124 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.31, 5.71] |
| 18 Leaving the study early ‐ i. up to 24 hours | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 18.1 due to severe agitation | 1 | 124 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 8.03] |
| 18.2 due to acute dystonia | 1 | 124 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.24, 102.07] |
| 18.3 due to adverse effects | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.07, 16.51] |
| 18.4 unspecified general reasons | 1 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.42, 1.44] |
| 19 Leaving the study early ‐ ii. over 24 hours | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.01, 1.89] |
| 19.1 Lack of efficacy at 96 hours | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.01, 1.89] |
9.2. Analysis.
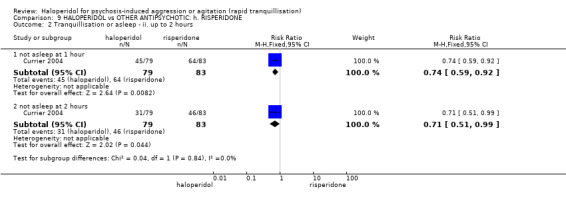
Comparison 9 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE, Outcome 2 Tranquillisation or asleep ‐ ii. up to 2 hours.
9.5. Analysis.
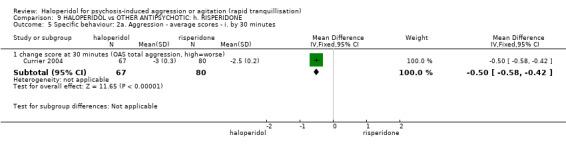
Comparison 9 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE, Outcome 5 Specific behaviour: 2a. Aggression ‐ average scores ‐ i. by 30 minutes.
9.6. Analysis.
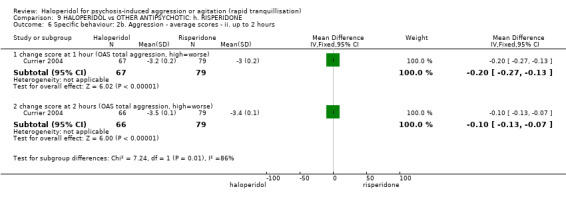
Comparison 9 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE, Outcome 6 Specific behaviour: 2b. Aggression ‐ average scores ‐ ii. up to 2 hours.
9.8. Analysis.
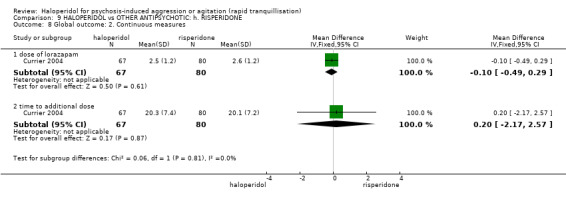
Comparison 9 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE, Outcome 8 Global outcome: 2. Continuous measures.
9.11. Analysis.
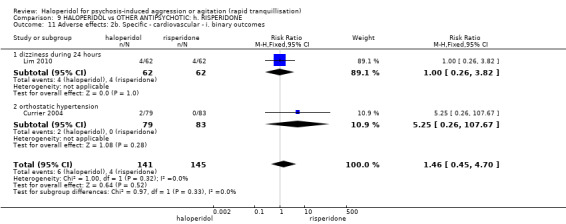
Comparison 9 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE, Outcome 11 Adverse effects: 2b. Specific ‐ cardiovascular ‐ i. binary outcomes.
9.13. Analysis.
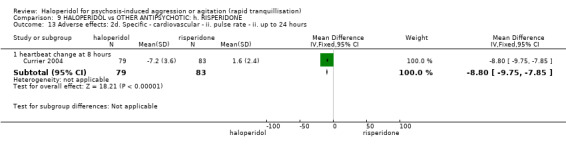
Comparison 9 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE, Outcome 13 Adverse effects: 2d. Specific ‐ cardiovascular ‐ ii. pulse rate ‐ ii. up to 24 hours.
9.19. Analysis.
Comparison 9 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: h. RISPERIDONE, Outcome 19 Leaving the study early ‐ ii. over 24 hours.
Comparison 10. HALOPERIDOL vs OTHER ANTIPSYCHOTIC: i. THIOTHIXENE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Repeated need for rapid tranquillisation | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 more than 1 injection | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.89, 1.28] |
| 1.2 more than 3 injections | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.5 [0.57, 10.93] |
| 2 Specific behaviour: 1. Agitation ‐ rated as 'adverse effect' | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.28 [0.01, 6.52] |
| 3 Global outcome | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 no response to 1st injection | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.5 [0.57, 11.05] |
| 3.2 no improvement at 1 hour | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.5 [0.14, 1.84] |
| 3.3 no improvement at 2 hours | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.17 [0.01, 3.31] |
| 3.4 no improvement at 3 hours | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.06, 12.49] |
| 3.5 no improvement at 4 hours | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.06, 12.49] |
| 3.6 no improvement at 5 hours | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.2 [0.21, 82.72] |
| 4 Adverse effects: 1. General ‐ one or more adverse effects | 2 | 74 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.47 [0.97, 2.22] |
| 5 Adverse effects: 2a. Specific ‐ movement disorders | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 ataxia | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.06, 12.49] |
| 5.2 thick tongue | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.52 [0.11, 58.67] |
| 5.3 use of antiparkinson drugs | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6 Adverse effects: 2b. Specific ‐ others | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 anticholinergic ‐ blurred vision | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.2 [0.21, 82.72] |
| 6.2 anticholinergic ‐ dry mouth | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.17 [0.01, 3.31] |
| 6.3 arousal ‐ drowsiness | 2 | 74 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.72 [1.02, 2.90] |
| 6.4 arousal ‐ lightheadedness | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.2 [0.21, 82.72] |
| 6.5 cardiac ‐ chest pain | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.28 [0.01, 6.52] |
| 6.6 cardiac ‐ dizziness | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.5 [0.28, 22.20] |
| 6.7 cardiac ‐ hypotensive crisis | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6.8 cardiac ‐ orthostatic hypotension | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.06, 12.49] |
| 6.9 cardiac ‐ tachycardia | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.28 [0.01, 6.52] |
| 6.10 other ‐ depressed mood | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.52 [0.11, 58.67] |
| 6.11 other ‐ diaphoresis | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.28 [0.01, 6.52] |
| 6.12 other ‐ weakness | 2 | 74 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.75 [0.30, 25.21] |
| 6.13 other ‐ unsteadiness | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 68.26] |
Comparison 11. HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Repeated need for tranquillisation ‐ need for additional drugs for tranquillisation | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 up to 2 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.13, 1.01] |
| 1.2 up to 24 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.64 [1.07, 2.53] |
| 2 Specific behaviour: 1a. Agitation ‐ average scores ‐ i. up to 2 hours | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 endpoint score at 1 hour (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐7.70 [‐9.41, ‐5.99] |
| 2.2 endpoint score at 2 hours (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐3.90 [‐6.38, ‐1.42] |
| 2.3 endpoint score at 2 hours (PANSS‐EC, high=worse) | 1 | 231 | Mean Difference (IV, Fixed, 95% CI) | 0.06 [‐1.13, 1.25] |
| 3 Specific behaviour: 1b. Agitation ‐ average scores ‐ ii. up to 4 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 endpoint score at 4 hours (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [‐0.05, 0.45] |
| 4 Specific behaviour: 1c. Agitation ‐ average scores ‐ iii. up to 24 hours | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 endpoint score at 6 hours (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [0.05, 0.35] |
| 4.2 endpoint score at 6 hours (PANSS‐EC, high=worse) | 1 | 231 | Mean Difference (IV, Fixed, 95% CI) | ‐0.47 [‐1.68, 0.74] |
| 4.3 endpoint score at 12 hours (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.9 [0.82, 0.98] |
| 4.4 endpoint score at 24 hours (PANSS‐EC, high=worse) | 1 | 231 | Mean Difference (IV, Fixed, 95% CI) | ‐0.21 [‐1.36, 0.94] |
| 5 Specific behaviour: 1d. Agitation ‐ average scores ‐ iv. over 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 endpoint scores at 48 hours (PANSS‐EC, high=worse) | 1 | 231 | Mean Difference (IV, Fixed, 95% CI) | 0.06 [‐1.03, 1.15] |
| 5.2 endpoint scores at 72 hours (PANSS‐EC, high=worse) | 1 | 231 | Mean Difference (IV, Fixed, 95% CI) | 0.62 [‐0.45, 1.69] |
| 6 Specific behaviour: 2a. Aggression ‐ average scores ‐ i. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.1 endpoint score at 1 hour (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐0.77, 0.77] |
| 6.2 endpoint score at 2 hours (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.70 [‐0.25, 1.65] |
| 7 Specific behaviour: 2b. Aggression ‐ average scores ‐ ii. up to 4 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 7.1 endpoint score at 4 hours (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.30 [‐0.04, 0.64] |
| 8 Specific behaviour: 2c. Aggression ‐ average scores ‐ iii. up to 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 8.1 endpoint score at 6 hours (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.70 [0.60, 0.80] |
| 8.2 endpoint score at 12 hours (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [0.07, 0.33] |
| 9 Global outcome: 1. General ‐ need for additional measures | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 need for anxiolytic during 7 days | 1 | 132 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.84, 1.48] |
| 9.2 need for hypnotics for night time sedation during 7 days | 1 | 132 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.20, 2.50] |
| 9.3 additional restraint, seclusion or special observation during 12 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.6 [0.25, 1.44] |
| 10 Global outcome: 2. Average scores ‐ i. over 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.1 change score at 72 hours (CGI‐S, high=worse) | 1 | 132 | Mean Difference (IV, Fixed, 95% CI) | 0.34 [0.13, 0.55] |
| 10.2 change score at 7 days (CGI‐S, high=worse) | 1 | 132 | Mean Difference (IV, Fixed, 95% CI) | 0.51 [0.07, 0.95] |
| 11 Mental state: 1a. Average scores ‐ i. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 11.1 endpoint score at 2 hours (BPRS, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Mental state: 1b. Average scores ‐ ii. up to 4 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 12.1 endpoint score at 4 hours (BPRS, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13 Mental state: 1c. Average scores ‐ iii. up to 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 13.1 endpoint score at 24 hours (BPRS, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14 Mental state: 1d. Average scores ‐ iv. over 24 hours | 4 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 14.1 endpoint score at 48 hours (BPRS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐2.05 [‐6.45, 2.35] |
| 14.2 endpoint score at 72 hours (PANSS total, high=worse) | 1 | 231 | Mean Difference (IV, Fixed, 95% CI) | 2.45 [‐2.19, 7.09] |
| 14.3 endpoint score at 72 hours (PANSS positive sub‐scale, high=worse) | 1 | 231 | Mean Difference (IV, Fixed, 95% CI) | 1.11 [‐0.45, 2.67] |
| 14.4 endpoint score at 72 hours (PANSS negative sub‐scale, high=worse) | 1 | 231 | Mean Difference (IV, Fixed, 95% CI) | ‐4.19 [‐5.71, ‐2.67] |
| 14.5 endpoint score at 72 hours (PANSS general pathology score, high=worse) | 1 | 231 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐2.27, 2.27] |
| 14.6 score at 72 hours (BPRS, high=worse) | 3 | 511 | Mean Difference (IV, Fixed, 95% CI) | ‐0.43 [‐1.93, 1.07] |
| 14.7 change score at 7 days (BPRS, high=worse) | 1 | 132 | Mean Difference (IV, Fixed, 95% CI) | 2.93 [‐0.81, 6.67] |
| 15 Adverse effects: 1a. General ‐ i. up to 24 hours | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 15.1 one or more drug related adverse effects ‐ by 12 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [0.64, 2.93] |
| 16 Adverse effects: 1b. General ‐ ii. over 24 hours | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 16.1 one or more drug related adverse effects ‐ by 72 hours | 4 | 799 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.74 [1.47, 2.06] |
| 16.2 one or more drug related adverse effects ‐ by 7 days | 1 | 132 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.31 [0.93, 1.83] |
| 17 Adverse effects: 1c. General ‐ serious | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 17.1 severe adverse effect ‐ by 72 hours | 1 | 376 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 17.2 death | 2 | 436 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 2.78] |
| 18 Adverse effects: 2a. Specific ‐ anticholinergic | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 18.1 blurred vision by 72 hours | 1 | 231 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.97 [1.15, 13.68] |
| 18.2 constipation by 72 hours | 1 | 376 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.40 [0.08, 2.06] |
| 18.3 dry mouth by 72 hours | 1 | 231 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.97 [1.22, 7.22] |
| 18.4 hypersalivation by 72 hours | 1 | 231 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.55 [1.11, 5.87] |
| 19 Adverse effects: 2b. Specific ‐ arousal level ‐ i. various | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 19.1 excessive sedation at 12 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.22, 4.56] |
| 19.2 insomnia ‐ by 72 hours | 1 | 376 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.05, 5.53] |
| 19.3 lethargy ‐ by 72 hours | 1 | 231 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.67, 2.35] |
| 19.4 somnolence ‐ by 72 hours | 1 | 376 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.43, 3.12] |
| 20 Adverse effects: 2c. Specific ‐ arousal level ‐ ii. average scores ‐ i. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 20.1 endpoint score at 1 hour (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐0.46, 0.46] |
| 20.2 endpoint score at 2 hours (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐0.46, 0.46] |
| 21 Adverse effects: 2d. Specific ‐ arousal level ‐ ii. average scores ‐ ii. up to 4 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 21.1 endpoint score at 4 hours (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.40 [‐0.83, 0.03] |
| 22 Adverse effects: 2e. Specific ‐ arousal level ‐ ii. average scores ‐ iii. up to 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 22.1 endpoint score at 6 hours (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐0.40, 0.20] |
| 22.2 endpoint score at 12 hours (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐0.40, 0.20] |
| 23 Adverse effects: 2f. Specific ‐ cardiovascular | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 23.1 hypotension at 12 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.08 [0.00, 1.31] |
| 23.2 dizziness ‐ by 72 hours | 2 | 607 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.28, 1.19] |
| 23.3 ECG ‐ abnormal ‐ by 72 hours | 2 | 607 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.38 [0.17, 0.84] |
| 23.4 ECG change ‐ clinically significant ‐ by 72 hours | 1 | 376 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.60, 1.71] |
| 23.5 tachycardia ‐ by 72 hours | 3 | 739 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.23, 1.04] |
| 24 Adverse effects: 2g. Specific ‐ gastrointestinal | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 24.1 nausea by 72 hours | 1 | 376 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.40 [0.08, 2.06] |
| 24.2 vomiting by 72 hours | 1 | 132 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.30 [0.02, 5.72] |
| 24.3 vomiting by 7 days | 1 | 132 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.16 [0.01, 2.82] |
| 25 Adverse effects: 2h. Specific ‐ hematological tests | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 25.1 abnormal lab results ‐ by 72 hours | 2 | 508 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.69, 1.17] |
| 25.2 abnormal lab results ‐ by 7 days | 1 | 132 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.47, 2.15] |
| 25.3 aspartate aminotransference ‐ by 72 hours | 1 | 376 | Risk Ratio (M‐H, Fixed, 95% CI) | 11.12 [0.62, 199.64] |
| 25.4 glucose increased ‐ by 24 hours | 1 | 132 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.31, 2.92] |
| 25.5 haemogram abnormal ‐ by 72 hours | 1 | 231 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.17 [0.02, 1.35] |
| 25.6 lactate dehydrogenase increase ‐ by 72 hours | 1 | 231 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.50 [0.05, 5.39] |
| 25.7 liver function abnormal ‐ by 72 hours | 1 | 231 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.50 [0.05, 5.39] |
| 25.8 triglyceride increase ‐ by 72 hours | 1 | 231 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.96 [0.24, 102.14] |
| 26 Adverse effects: 2i. Specific ‐ movement disorders ‐ i. binary measures | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 26.1 akathisia ‐ by 72 hours | 3 | 739 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.32 [1.34, 4.01] |
| 26.2 akathisia ‐ by 7 days | 1 | 132 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.29 [1.13, 16.31] |
| 26.3 dystonia ‐ by 72 hours | 2 | 508 | Risk Ratio (M‐H, Fixed, 95% CI) | 10.26 [1.67, 63.17] |
| 26.4 dystonia ‐ by 7 days | 1 | 132 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.68 [0.76, 9.47] |
| 26.5 EPS ‐ by 12 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 11.0 [0.64, 190.53] |
| 26.6 EPS ‐ by 72 hours | 2 | 508 | Risk Ratio (M‐H, Fixed, 95% CI) | 19.13 [7.59, 48.21] |
| 26.7 EPS ‐ by 7 days | 1 | 132 | Risk Ratio (M‐H, Fixed, 95% CI) | 34.29 [4.70, 250.02] |
| 26.8 extrapyramidal effects ‐ use of antiparkinson drugs ‐ by 7 days | 1 | 132 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.30 [1.82, 5.97] |
| 26.9 hypertonia/rigidity ‐ by 72 hours | 1 | 132 | Risk Ratio (M‐H, Fixed, 95% CI) | 14.81 [0.78, 280.47] |
| 26.10 hypertonia ‐ by 7 days | 1 | 132 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.57 [0.90, 14.25] |
| 26.11 myotonia ‐ by 72 hours | 1 | 231 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.84 [1.85, 8.00] |
| 26.12 pyramidal tract syndrome ‐ by 72 hours | 1 | 376 | Risk Ratio (M‐H, Fixed, 95% CI) | 17.18 [1.00, 295.55] |
| 26.13 reduced movement ‐ by 72 hours | 1 | 231 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.14 [1.17, 3.91] |
| 26.14 torsional spasm ‐ by 72 hours | 1 | 231 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.30 [0.93, 11.70] |
| 26.15 tremor ‐ by 72 hours | 2 | 363 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.64 [1.37, 5.11] |
| 26.16 tremor ‐ by 7 days | 1 | 132 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.29 [0.82, 22.48] |
| 27 Adverse effects: 2j. Specific ‐ movement disorders ‐ ii. average scores ‐ i. over 24 hours | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 27.1 change score at 72 hours (BAS, high=worse) | 1 | 131 | Mean Difference (IV, Fixed, 95% CI) | 0.47 [0.18, 0.76] |
| 27.2 score at 72 hours (SAS, high=worse) | 2 | 191 | Mean Difference (IV, Fixed, 95% CI) | 3.59 [2.15, 5.03] |
| 27.3 change score at 7 days (BAS, high=worse) | 1 | 131 | Mean Difference (IV, Fixed, 95% CI) | 0.9 [0.51, 1.29] |
| 27.4 change score at 7 days (SAS, high=worse) | 1 | 131 | Mean Difference (IV, Fixed, 95% CI) | 6.1 [3.91, 8.29] |
| 28 Adverse effects: 2k. Specific ‐ miscellaneous | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 28.1 physical examination significant changes ‐ by 72 hours | 1 | 376 | Risk Ratio (M‐H, Fixed, 95% CI) | 27.29 [1.63, 455.72] |
| 29 Leaving the study early: 1. For any reason ‐ i. over 24 hours | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 29.1 by 72 hours | 1 | 376 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.77 [0.53, 5.94] |
| 29.2 by 7 days | 1 | 132 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.14 [0.86, 5.32] |
| 30 Leaving the study early: 2. For specific reasons ‐ i. over 24 hours | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 30.1 adverse events ‐ by 72 hours | 2 | 508 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.40 [0.45, 12.75] |
| 30.2 adverse events ‐ by 7 days | 1 | 132 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.54 [0.06, 4.65] |
| 30.3 withdrew consent ‐ by 72 hours | 1 | 376 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.05 [0.24, 104.55] |
| 30.4 other ‐ by 72 hours | 1 | 376 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.20 [0.01, 4.18] |
11.3. Analysis.
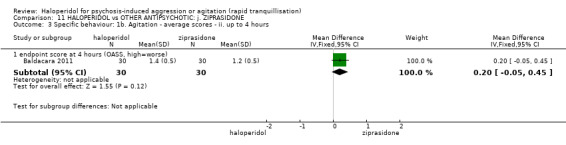
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 3 Specific behaviour: 1b. Agitation ‐ average scores ‐ ii. up to 4 hours.
11.5. Analysis.
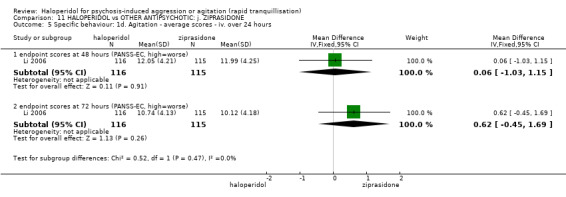
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 5 Specific behaviour: 1d. Agitation ‐ average scores ‐ iv. over 24 hours.
11.6. Analysis.
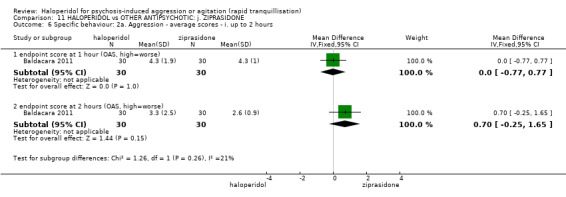
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 6 Specific behaviour: 2a. Aggression ‐ average scores ‐ i. up to 2 hours.
11.7. Analysis.
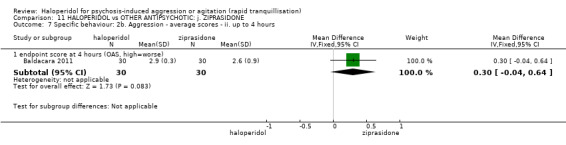
Comparison 11 HALOPERIDOL vs OTHER ANTIPSYCHOTIC: j. ZIPRASIDONE, Outcome 7 Specific behaviour: 2b. Aggression ‐ average scores ‐ ii. up to 4 hours.
Comparison 12. HALOPERIDOL vs OTHER ANTIPSYCHOTIC: k. ZUCLOPENTHIXOL ACETATE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Repeated need for tranquillisation: more than 3 injections | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.54 [1.19, 5.46] |
| 2 Adverse effects: 1a. Specific ‐ movement disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 movement disorder ‐ tremor by 7 days | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.16 [0.93, 18.62] |
| 3 Adverse effects: 1b. Specific ‐ miscellaneous | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 pain/allergy ‐ reaction at injection site | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.55 [0.15, 84.14] |
| 4 Leaving the study early | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 13. HALOPERIDOL vs BENZODIAZEPINE: a. FLUNITRAZEPAM.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Specific behaviour: 1. Aggression ‐ binary measures ‐ i. up to 2 hours | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.86, 1.55] |
| 1.1 ≥50% reduction at 90 minutes (OAS) | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.86, 1.55] |
| 2 Global outcome: 1. Need for seclusion or restraint | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3 Adverse effects: 1. Specific ‐ movement disorders ‐ i. binary measures ‐ i. by 30 minutes | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3.1 EPS by 30 minutes | 1 | 28 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 14. HALOPERIDOL vs BENZODIAZEPINE: b. LORAZEPAM.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tranquillisation or asleep ‐ not asleep | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 at 1 hour | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.76, 1.44] |
| 1.2 by 3 hours | 1 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.93 [1.14, 3.27] |
| 2 Repeated need for rapid tranquillisation | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 more than 1 injection | 1 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.91, 1.43] |
| 2.2 more than 3 injections | 1 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.50, 2.45] |
| 3 Global outcome: 1. No overall improvement | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 by 30 minutes | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.70, 1.71] |
| 3.2 at 1 hour | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.64 [0.54, 5.03] |
| 3.3 at > 1 hour | 1 | 44 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4 Mental state: 1a. Average scores ‐ i. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 endpoint score at 1 hour (BPRS, high=worse) | 1 | 37 | Mean Difference (IV, Fixed, 95% CI) | 3.26 [‐4.13, 10.65] |
| 4.2 endpoint score at 2 hours (BPRS, high=worse) | 1 | 37 | Mean Difference (IV, Fixed, 95% CI) | 4.07 [‐2.62, 10.76] |
| 5 Mental state: 1b. Average scores ‐ ii. up to 4 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 endpoint score at 3 hours (BPRS, high=worse) | 1 | 37 | Mean Difference (IV, Fixed, 95% CI) | 5.03 [‐2.98, 13.04] |
| 5.2 endpoint score at 4 hours (BPRS, high=worse) | 1 | 37 | Mean Difference (IV, Fixed, 95% CI) | 1.73 [‐6.14, 9.60] |
| 6 Adverse effects: 1a. General | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 one or more drug‐related adverse effect up to 24 hours | 1 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.60, 2.10] |
| 7 Adverse effects: 1b. General ‐ serious | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8 Adverse effects: 2a. Specific ‐ anticholinergic | 1 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.53 [0.14, 2.04] |
| 8.1 dry mouth up to 24 hours (only reported if occurred ≥9%) | 1 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.53 [0.14, 2.04] |
| 9 Adverse effects: 2b. Specific ‐ arousal | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 asleep ‐ at 1 hour | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.40, 1.99] |
| 10 Adverse effects: 2c. Specific ‐ cardiovascular | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 dizziness (only reported if occurred in ≧9%) ‐ during 24 hours | 1 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.19, 4.07] |
| 10.2 hypertension ‐ use of additional clonidine ‐ during 24 hours | 1 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.67 [0.11, 63.17] |
| 11 Adverse effects: 2d. Specific ‐ movement disorder | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 11.1 ataxia (only reported if occurred in ≧9%) ‐ during 24 hours | 1 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.44 [0.04, 4.65] |
| 11.2 dystonia (only reported if occurred in ≧9%) ‐ during 24 hours | 1 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.54 [0.42, 30.03] |
| 11.3 EPS | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 15.0 [2.11, 106.49] |
| 11.4 hypertonia/rigidity (only reported if occurred in ≧9%) ‐ during 24 hours | 1 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.22 [0.33, 115.91] |
| 11.5 speech disorder (only reported if occurred in ≧9%) ‐ during 24 hours | 1 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.77 [0.35, 9.01] |
| 11.6 tremor (only reported if occurred in ≧9%) ‐ during 24 hours | 1 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.77 [0.17, 18.60] |
| 11.7 use of additional benztropine ‐ during 24 hours | 1 | 66 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.99 [0.68, 5.83] |
14.5. Analysis.
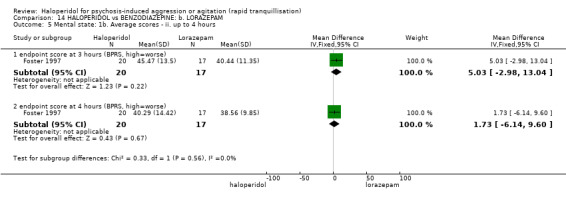
Comparison 14 HALOPERIDOL vs BENZODIAZEPINE: b. LORAZEPAM, Outcome 5 Mental state: 1b. Average scores ‐ ii. up to 4 hours.
14.7. Analysis.
Comparison 14 HALOPERIDOL vs BENZODIAZEPINE: b. LORAZEPAM, Outcome 7 Adverse effects: 1b. General ‐ serious.
14.10. Analysis.
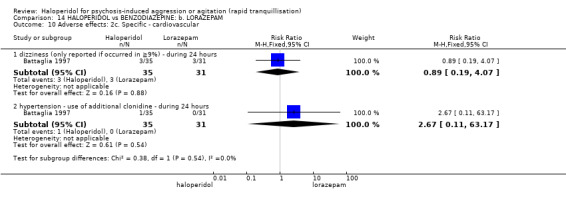
Comparison 14 HALOPERIDOL vs BENZODIAZEPINE: b. LORAZEPAM, Outcome 10 Adverse effects: 2c. Specific ‐ cardiovascular.
Comparison 15. HALOPERIDOL vs BENZODIAZEPINE: c. MIDAZOLAM.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tranquillisation or asleep: average times in minutes ‐ (skewed data) | Other data | No numeric data | ||
| 1.1 average time to sedation | Other data | No numeric data | ||
| 1.2 average time to arousal | Other data | No numeric data | ||
| 2 Global outcome: 1. Need for rescue drug | 1 | 84 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.46, 2.87] |
| 3 Adverse effects: 1. General ‐ one or more drug related adverse effect | 1 | 84 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.25, 101.11] |
| 4 Adverse effects: 2. Specific | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 hypotensive | 1 | 84 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 71.61] |
| 4.2 apnoea | 1 | 84 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 71.61] |
15.4. Analysis.
Comparison 15 HALOPERIDOL vs BENZODIAZEPINE: c. MIDAZOLAM, Outcome 4 Adverse effects: 2. Specific.
Comparison 16. HALOPERIDOL vs COMBINATIONS: a. HALOPERIDOL + LEVOMEPROMAZINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global outcome: 1. No overall improvement | 1 | 19 | Risk Ratio (M‐H, Fixed, 95% CI) | 8.18 [0.50, 133.66] |
Comparison 17. HALOPERIDOL vs COMBINATIONS: b. HALOPERIDOL + LORAZEPAM.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tranquillisation or asleep ‐ asleep | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.83 [1.11, 3.02] |
| 1.1 not asleep by 3 hours | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.83 [1.11, 3.02] |
| 2 Repeated need for rapid tranquillisation | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 needing additional injection during 24 hours | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.87, 1.27] |
| 2.2 more than 3 injections during 24 hours | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.05 [0.92, 10.10] |
| 3 Global outcome: 1. No overall improvement | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 by 30 minutes | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.67 [1.25, 5.68] |
| 3.2 at 1 hour | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 14.77 [0.88, 247.54] |
| 3.3 at > 1 hour | 1 | 45 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4 Adverse effects: 1. General | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.62, 2.18] |
| 4.1 one or more drug‐related adverse effect during 24 hours | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.62, 2.18] |
| 5 Adverse effects: 2a. Specific ‐ anticholinergic | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 dry mouth (only reported if occurred in ≧9%) during 24 hours | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.20, 4.21] |
| 6 Adverse effects: 2b. Specific ‐ cardiovascular | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 dizziness (only reported if occurred in ≧9%) during 24 hours | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.37 [0.24, 7.69] |
| 6.2 hypertension ‐ use of additional clonidine during 24 hours | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.06, 14.02] |
| 7 Adverse effects: 2c. Specific ‐ movement disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 ataxia (only reported if occurred in >9%) during 24 hours | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.30 [0.03, 2.78] |
| 7.2 dystonia (only reported if occurred in >9%) during 24 hours | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 8.25 [0.46, 147.45] |
| 7.3 hypertonia/rigidity (only reported if occurred in >9%) during 24 hours | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.74 [0.30, 25.05] |
| 7.4 tremor (only reported if occurred in >9%) during 24 hours | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.83 [0.17, 19.21] |
| 7.5 speech disorder (only reported if occurred in >9%) during 24 hours | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [0.30, 5.03] |
| 7.6 use of additional benztropine during 24 hours | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.74 [0.81, 9.25] |
Comparison 18. HALOPERIDOL vs COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Repeated need for tranquillisation | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 need for additional drugs for tranquillisation up to 2 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.10, 0.71] |
| 1.2 need for additional drugs for tranquillisation by 12 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.69, 1.13] |
| 2 Specific behaviour: 1a. Agitation ‐ average scores ‐ i. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 endpoint score at 1 hour (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐8.5 [‐9.93, ‐7.07] |
| 2.2 endpoint score at 2 hours (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐6.7 [‐7.46, ‐5.94] |
| 3 Specific behaviour: 1b. Agitation ‐ average scores ‐ ii. up to 4 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 endpoint score at 4 hours (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐5.5 [‐6.48, ‐4.52] |
| 4 Specific behaviour: 1c. Agitation ‐ average scores ‐ iii. up to 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 endpoint score at 6 hours (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐3.70 [‐4.56, ‐2.84] |
| 4.2 endpoint score at 12 hours (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐11.2 [‐12.24, ‐10.16] |
| 5 Specific behaviour: 2a. Aggression ‐ average scores ‐ i. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 endpoint score at 1 hour (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐1.20 [‐2.44, 0.04] |
| 5.2 endpoint score at 2 hours (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐2.40 [‐4.21, ‐0.59] |
| 6 Specific behaviour: 2b. Aggression ‐ average scores ‐ ii. up to 4 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.1 endpoint score at 4 hours (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐0.68, 0.48] |
| 7 Specific behaviour: 2c. Aggression ‐ average scores ‐ iii. up to 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 7.1 endpoint score at 6 hours (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.40 [0.30, 0.50] |
| 7.2 endpoint score at 12 hours (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐1.9 [‐2.58, ‐1.22] |
| 8 Global outcomes: 1. General ‐ need for additional measures | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 additional restraint, seclusion or special observation during 12 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.13, 0.61] |
| 9 Adverse effects: 1b. General ‐ one or more adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 during 12 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.41, 1.32] |
| 10 Adverse effects: 1a. General ‐ serious | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 death | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11 Adverse effects: 2a. Specific ‐ arousal ‐ i. various | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 11.1 excessive sedation by 12 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.25 [0.08, 0.80] |
| 12 Adverse effects: 2b. Specific ‐ arousal ‐ ii. average scores ‐ i. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 12.1 endpoint score at 1 hour (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.5 [‐1.01, 0.01] |
| 12.2 endpoint score at 2 hours (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐0.56, 0.36] |
| 13 Adverse effects: 2c. Specific ‐ arousal ‐ ii. average scores ‐ ii. up to 4 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 13.1 endpoint score at 4 hours (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.30 [‐0.78, 0.18] |
| 14 Adverse effects: 2d. Specific ‐ arousal ‐ iii. average scores ‐ iii. up to 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 14.1 endpoint score at 6 hours (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐0.40, 0.20] |
| 14.2 endpoint score at 12 hours (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.10 [‐0.26, 0.46] |
| 15 Adverse effects: 2e. Specific ‐ cardiovascular | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 15.1 hypotension by 12 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.09 [0.01, 1.57] |
| 16 Adverse effects: 2f. Specific ‐ movement disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 16.1 EPS by 12 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.67 [0.44, 6.36] |
18.10. Analysis.
Comparison 18 HALOPERIDOL vs COMBINATIONS: c. HALOPERIDOL + MIDAZOLAM, Outcome 10 Adverse effects: 1a. General ‐ serious.
Comparison 19. HALOPERIDOL vs COMBINATIONS: d. HALOPERIDOL + PROMETHAZINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tranquillisation or asleep ‐ not tranquil or asleep ‐ i. by 30 minutes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 at 20 minutes | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.60 [1.18, 2.16] |
| 2 Tranquillisation or asleep ‐ not tranquil or asleep ‐ ii. up to 2 hours | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 at 40 minutes | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.83, 1.91] |
| 2.2 at 1 hour | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.42 [0.85, 2.37] |
| 2.3 at 2 hours | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.32 [0.68, 2.56] |
| 3 Repeated need for tranquillisation ‐ need for additional drugs for tranquillisation | 2 | 436 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.68, 1.29] |
| 3.1 by 2 hours | 2 | 376 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.43, 1.41] |
| 3.2 by 12 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.81, 1.49] |
| 4 Specific behaviour: 1a. Agitation ‐ average scores ‐ i. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 endpoint score at 1 hour (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐24.5 [‐27.32, ‐21.68] |
| 4.2 endpoint score at 2 hours (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐9.40 [‐10.39, ‐8.41] |
| 5 Specific behaviour: 1b. Agitation ‐ average scores ‐ ii. up to 4 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 endpoint score at 4 hours (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 3.80 [3.27, 4.33] |
| 6 Specific behaviour: 1c. Agitation ‐ average scores ‐ iii. up to 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.1 endpoint score at 6 hours (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 2.6 [2.13, 3.07] |
| 6.2 endpoint score at 12 hours (OASS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.80 [0.55, 1.05] |
| 7 Specific behaviour: 2a. Aggression ‐ average scores ‐ i. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 7.1 endpoint score at 1 hour (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐4.50 [‐6.28, ‐2.72] |
| 7.2 endpoint score at 2 hours (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.70 [‐1.89, 0.49] |
| 8 Specific behaviour: 2b. Aggression ‐ average scores ‐ ii. up to 4 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 8.1 endpoint score at 4 hours (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.60 [0.49, 0.71] |
| 9 Specific behaviour: 2c. Aggression ‐ average scores ‐ iii. up to 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 9.1 endpoint score at 6 hours (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 1.1 [0.91, 1.29] |
| 9.2 endpoint score at 12 hours (OAS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 1.8 [1.67, 1.93] |
| 10 Specific behaviour: 2d. Aggression ‐ further aggressive episode ‐ i. up to 24 hours | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.68, 1.65] |
| 11 Global outcomes: 1. General ‐ need for additional measures | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 11.1 restraints needed by 2 hours | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.21 [0.84, 1.76] |
| 11.2 additional restraint, seclusion or special observation during 12 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.2 [0.41, 3.51] |
| 12 Global outcomes: 2. Specific | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.1 doctor called to see patient | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.50 [1.05, 2.14] |
| 12.2 refuse oral drugs | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.61, 1.60] |
| 13 Adverse effects: 1a. General | 2 | 376 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.01 [1.07, 3.80] |
| 13.1 one or more adverse effects up to 24 hours | 2 | 376 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.01 [1.07, 3.80] |
| 14 Adverse effects: 1b. General ‐ serious | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 14.1 seizure | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.05 [0.19, 22.39] |
| 14.2 death | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 15 Adverse effects: 2a. Specific ‐ arousal ‐ i. various | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 15.1 excessive sedation by 12 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.33, 27.23] |
| 16 Adverse effects: 2b. Specific ‐ arousal ‐ ii. average scores ‐ i. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 16.1 endpoint score at 1 hour (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.10 [‐0.38, 0.58] |
| 16.2 endpoint score at 2 hours (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐0.50, 0.30] |
| 17 Adverse effects: 2c. Specific ‐ arousal ‐ ii. average scores ‐ ii. up to 4 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 17.1 endpoint score at 4 hours (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.30 [‐0.78, 0.18] |
| 18 Adverse effects: 2d. Specific ‐ arousal ‐ ii. average scores ‐ iii. up to 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.1 endpoint score at 6 hours (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.20 [‐0.48, 0.08] |
| 18.2 endpoint score at 12 hours (RSS, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐0.28, 0.28] |
| 19 Adverse effects: 2e. Specific ‐ cardiovascular | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 19.1 hypotension by 12 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 2.65] |
| 20 Adverse effects: 2f. Specific ‐ movement disorders | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 20.1 acute dystonia | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 19.48 [1.14, 331.92] |
| 20.2 EPS by 12 hours | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.32, 3.10] |
| 21 Service outcomes: 1. Not discharged by 14 days | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.72, 1.05] |
| 22 Leaving the study early | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 22.1 any reason | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.54, 1.94] |
| 22.2 absconded before receiving drug | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.21 [0.01, 4.24] |
| 22.3 incomplete information in notes | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.51, 2.46] |
| 22.4 seizure before drug was given | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.08 [0.13, 74.95] |
| 22.5 transfer to another hospital/notes lost | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.21, 5.00] |
| 22.6 withdrew consent | 1 | 316 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.06, 16.25] |
Comparison 20. HALOPERIDOL vs COMBINATIONS: e. HALOPERIDOL + RISPERIDONE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Specific behaviour: 1a. Agitation ‐ average scores ‐ i. up to 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 endpoint score at 24 hours (PANSS‐EC, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Specific behaviour: 1b. Agitation ‐ average scores ‐ ii. over 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 endpoint score at 3 days (PANSS‐EC, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 endpoint score at 5 days (PANSS‐EC, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 endpoint score at 1 week (PANSS‐EC, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.4 endpoint score at 2 weeks (PANSS‐EC, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Global outcome: No improvement | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 at 72 hours | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 at 5 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 at 1 week | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.4 at 2 weeks | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Adverse effects: 1a. Specific ‐ arousal level ‐ i. various | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 insomnia during 14 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 somnolence during 14 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Adverse effects: 1b. Specific ‐ movement disorders ‐ i. various | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 5.1 tremor during 14 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 akathisia during 14 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Adverse effects: 1c. Specific ‐ miscellaneous | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1 Dry mouth during 14 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Constipation during 14 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 Increased salivation during 14 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 21. HALOPERIDOL vs COMBINATIONS: f. OLANZAPINE + MAGNESIUM VALPROATE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Specific behaviour: 1. Agitation ‐ average scores ‐ over 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 endpoint score at 72 hours (PANSS‐EC, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 endpoint score at 7 days (PANSS‐EC, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 endpoint score at 14 days (PANSS‐EC, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Global outcome: No improvement | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.38, 2.95] |
| 3 Mental state: Average scores ‐ i. over 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 endpoint score at 72 hours (PANSS total, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 endpoint score at 7 days (PANSS total, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 endpoint score at 14 days (PANSS total, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Adverse effects: General ‐ over 24 hours | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 one or more drug related adverse effects ‐ by 14 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 22. HALOPERIDOL vs COMBINATIONS: g. QUETIAPINE + MAGNESIUM VALPROATE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Specific behaviour: 1a. Agitation ‐ binary measures | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.44, 3.06] |
| 1.1 <25% reduction, no effect (PANSS‐EC) | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.44, 3.06] |
| 2 Specific behaviour: 1b. Agitation ‐ average scores ‐ i. over 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 endpoint score at 3 days (PANSS‐EC, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 0.02 [‐2.31, 2.35] |
| 2.2 endpoint score at 7 days (PANSS‐EC, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 1.16 [‐0.98, 3.30] |
| 2.3 endpoint score at 14 days (PANSS‐EC, high=worse) | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | 1.24 [‐0.49, 2.97] |
Comparison 23. HALOPERIDOL vs COMBINATIONS: h. RISPERIDONE + CLONAZEPAM.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tranquillisation or asleep | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 not asleep at 2 hours | 1 | 205 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.82, 1.39] |
| 1.2 not asleep at 4 hours | 1 | 205 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.66, 1.03] |
| 2 Specific behaviour: 1a. Agitation ‐ average scores ‐ i. up to 2 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 change score at 2 hours (PANSS‐EC, high=worse) | 1 | 108 | Mean Difference (IV, Fixed, 95% CI) | ‐0.5 [‐3.80, 2.80] |
| 3 Specific behaviour: 1b. Agitation ‐ average scores ‐ i. up to 4 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 change score at 4 hours (PANSS‐EC, high=worse) | 1 | 126 | Mean Difference (IV, Fixed, 95% CI) | ‐2.40 [‐5.20, 0.40] |
| 4 Specific behaviour: 1c. Agitation ‐ average scores ‐ iii. up to 24 hours | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 score at 24 hours (PANSS‐EC, high=worse) | 2 | 305 | Mean Difference (IV, Fixed, 95% CI) | 0.71 [‐0.56, 1.98] |
| 5 Specific behaviour: 1d. Agitation ‐ average scores ‐ iv. over 24 hours | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 score at 3 days (PANSS‐EC, high=worse) | 2 | 305 | Mean Difference (IV, Fixed, 95% CI) | ‐0.41 [‐1.66, 0.83] |
| 5.2 score at 5 days (PANSS‐EC, high=worse) | 2 | 305 | Mean Difference (IV, Fixed, 95% CI) | 0.14 [‐1.24, 1.52] |
| 5.3 endpoint score at 1 week (PANSS‐EC, high=worse) | 1 | 100 | Mean Difference (IV, Fixed, 95% CI) | ‐2.56 [‐4.57, ‐0.55] |
| 5.4 endpoint score at 2 weeks (PANSS‐EC, high=worse) | 1 | 100 | Mean Difference (IV, Fixed, 95% CI) | ‐0.88 [‐2.34, 0.58] |
| 6 Global outcome: No improvement | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1 at 72 hours | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 at 5 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 at 1 week | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.4 at 2 weeks | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Mental state: 1. Average scores ‐ i. over 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 7.1 change score at 120 hours (PANSS total, high=worse) | 1 | 205 | Mean Difference (IV, Fixed, 95% CI) | ‐3.5 [‐7.07, 0.07] |
| 8 Adverse effects: 1. General | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 overall adverse events during 120 hours | 1 | 205 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.72 [1.29, 2.29] |
| 9 Adverse effects: 2a. Specific ‐ arousal ‐ i. various | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 insomnia during 5 days (only reported if occurred in ≥5%) | 1 | 205 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.54 [0.45, 5.31] |
| 9.2 insomnia during 14 days | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.5 [0.20, 1.23] |
| 9.3 somnolence during 14 days | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.75 [0.94, 8.06] |
| 10 Adverse effects: 2b. Specific ‐ cardiovascular ‐ i. various | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 tachycardia during 120 hours (only reported if occurred in ≥5%) | 1 | 205 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.29 [0.36, 4.66] |
| 11 Adverse effects: 2c. Specific ‐ movement disorders ‐ i. various | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 11.1 akathisia during 5 days (only reported if occurred in ≥5%) | 1 | 205 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.63 [1.12, 2.38] |
| 11.2 akathisia during 14 days | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.33 [0.98, 5.58] |
| 11.3 EPS during 5 days (only reported if occurred in ≥5%) | 1 | 205 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.22 [1.52, 3.23] |
| 11.4 tremor during 14 days | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.2 [1.27, 8.07] |
| 12 Adverse effects: 2d. Specific ‐ miscellaneous | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 12.1 Dry mouth during 14 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.2 Constipation during 14 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.3 Increased salivation during 14 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 24. HALOPERIDOL vs COMBINATIONS: i. ZIPRASIDONE + CLONAZEPAM.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Specific behaviour: 1a. Agitation ‐ average scores ‐ i. up to 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 endpoint score at 12 hours (PANSS‐EC, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Specific behaviour: 1b. Agitation ‐ average scores ‐ ii. over 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 endpoint score at 48 hours (PANSS‐EC, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 endpoint score at 3 days (PANSS‐EC, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 endpoint score at 5 days (PANSS‐EC, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.4 endpoint score at 7 days (PANSS‐EC, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Global outcome: 1. Average scores ‐ i. over 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 endpoint score at 7 days (CGI‐S, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Mental state: 1. Average scores ‐ i. over 24 hours | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4.1 endpoint score at 7 days (PANSS total, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 endpoint score at 7 days (PANSS positive sub‐scale, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 endpoint score at 7 days (PANSS negative sub‐scale, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 endpoint score at 7 days (PANSS general psychopathology sub‐scale, high=worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Leaving the study early | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 5.1 any reason | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 adverse effects | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 lost at follow‐up | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.4 non compliance | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bailine 1987.
| Methods | Allocation: randomised. Blindness: blind for some outcomes. Duration: 72 hours. |
|
| Participants | Diagnosis: Research Diagnostic Criteria (11) diagnosis of schizophrenia, any type, or manic depressive illness, manic type and grossly abnormal behavior. N = 34. Age: mean 25.2 years. Sex: 26 males, 8 females. History: "violent or assaultive or too psychologically disorganized to be maintained on a unit without significant extra supervision." (p.128). Excluded: not reported. Setting: inpatient, USA. |
|
| Interventions | 1. Haloperidol IM: dose 5 mg or 10 mg/IM, maximum cumulative dose of 100 mg during 24 hours (mean number of IMs not reported). N = 20. 2. Chlorpromazine IM: dose 50 mg or 100 mg/IM, maximum cumulative dose of 500 mg during 24 hours (mean number of IMs not reported). N = 14. |
|
| Outcomes | Adverse events: leaving the study early. Unable to use: Mental state: BPRS (mean and overall P value only for significant findings reported, SD/SE/CI not reported). Global state: GAS (not published, mean and overall P value only for significant findings reported, SD/SE/CI not reported). Global state: NOSIE (overall P value only for significantly findings reported, mean/SD/SE/CI not reported). Adverse events: SAS (overall P value only for significantly findings reported, mean/SD/SE/CI not reported). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "random number sequence". |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | BPRS was rater‐blinded. However, it is not clear whether other outcomes were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | It is reported that no one discontinued due to adverse effects, however it does not state if there were discontinuations due to any other reason. |
| Selective reporting (reporting bias) | High risk | Only the significant findings/factors are reported for a number of outcomes. |
| Other bias | High risk | Small short study. |
Baldacara 2011.
| Methods | Allocation: randomised. Blindness: single (rater‐blinded)*. Duration: 12 hours. |
|
| Participants | Diagnosis: DSM‐IV‐TR criteria for bipolar (maniac or mixed episode) or psychotic disorder. N = 150. Age: 18‐50 years. Sex: 91 males, 59 females. History: "admitted to the Psychiatric Emergency Room" (p.31). Excluded: "disorders due to drug abuse, organic disorder, anxiety or personality disorder (DSM‐IV‐TR criteria), failure to agree to participate in the study, incapability of completing all steps and unstable clinical disease" (p.31). Setting: emergency room, São Paulo. |
|
| Interventions | 1. Haloperidol IM: fixed dose 5 mg/IM. N = 30. 2. Olanzapine IM: fixed dose 10 mg/IM. N = 30. 3. Ziprasidone IM: fixed dose 20 mg/IM. N = 30. 4. Haloperidol IM: fixed dose 5 mg/IM + promethazine IM: fixed dose 50 mg/IM. N = 30. 5. Haloperidol IM: fixed dose 5 mg/IM + midazolam IM: fixed dose 15 mg/IM. N = 30. "Only additional doses of haloperidol + promethazine could be used" (p.31). |
|
| Outcomes | Need for additional drugs for tranquillisation. Agitation: OASS**. Aggression: OAS**. Global state: restriction. Adverse events: death, RSS, EPS, excessive sedation, hypotension. |
|
| Notes | *"patients were instructed not to reveal their current treatment to the investigators" (p.31). **endpoint scores at 1 hour are discordant between text (p.32‐33) and Table 2 (p.35); scores from Table 2 were used, due to compatibility with Figure 2 (p.36). Authors were contacted (3rd July 2016, 7th July 2016, 9th July 2016). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomization employed in this clinical trial was allocation by permuted blocks" (p.31); no further informations on the randomisation process. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "study medications were packaged in identical color‐coded boxes" (p.31); stated as double blind, however "patients were instructed not to reveal their current treatment to the investigators" (p.31); no information whether the blindness was effective. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "Patients were assessed by two psychiatrists. The psychiatrists were all masked with regard to the patient’s treatment assignment" (p.31); no information whether the blindness was effective. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete outcome data. |
| Selective reporting (reporting bias) | High risk | Endpoint scores at 1 hour are discordant between text (p.32‐33) and Table 2 (p.35). |
| Other bias | Low risk | None obvious. |
Battaglia 1997.
| Methods | Allocation: randomised. Blindness: double. Duration: 24 hours. |
|
| Participants | Diagnosis: mania (N = 13), psychoactive substance use (N = 16), psychosis not otherwise specified (N = 27), schizophrenia (N = 47), schizophreniform disorder (N = 1).* N = 100.** Age: range 18‐57 years. Sex: 73 males, 25 females. History: "psychosis and behavioral dyscontrol (agitated, aggressive, destructive, assaultive, or restless behavior." Excluded: "clinically obvious alcohol intoxication, central nervous system (CNS) depression, pregnancy, allergic hypersensitivity, delirium, neuroleptic malignant syndrome, airway obstruction, severe hypotension or hypertension, acute narrow angle glaucoma and treatment with a benzodiazepine or neuroleptic within the previous 24 hours (previous two weeks for fluphenazine decanoate and previous 4 weeks for haloperidol decanoate)." Setting: Emergency department, USA hospitals. |
|
| Interventions | 1. Haloperidol IM: dose up to 6 injections of 5 mg (mean number of IMs 2.86). N = 35. 2. Lorazepam IM: dose up to 6 injections of 2 mg (mean number of IMs 2.87). N = 31. 3. Lorazepam IM: dose up to 6 injections of 2 mg + haloperidol 5 mg (mean number of IMs 2.41). N = 32. First 3 injections ‐ at least 1 hour apart, remainder 2 hours apart. Need for subsequent doses made by blinded evaluator. |
|
| Outcomes | Tranquillisation or asleep. Global state: number of additional injections.** Adverse events: EPS, hypertension, others in over ≥ 9% of people. Unable to use ‐ Global state: CGI (reported, out of necessity, only for those awake), Efficacy Index (no data reported). Agitation: ABS (mean, SE/SD not reported, P values of significant findings reported, overall F value). Mental state: MBPRS (modified, unpublished; mean, SE/SD not reported, P values of significant findings reported, overall F value), Alertness Scale (no mean, SE/SD not reported, P values of significant findings reported). Physiological: vital signs (not reported). |
|
| Notes | * 6 people had more than one diagnosis. ** 2 people excluded from efficacy analysis ‐ after enrolment they had received proscribed antipsychotic medication. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Computer‐generated table of random numbers". |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The ED [Emergency Department] psychiatrist who treated and rated the patient remained blinded to the identity of the patient's medication throughout treatment" (p.336). [italic added by reviewers] "Double blind" ‐ participant blinding not specifically mentioned. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The ED [Emergency Department] psychiatrist who treated and rated the patient remained blinded to the identity of the patient's medication throughout treatment" (p.336).[italic added by reviewers]. Not tested. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | "two patients were excluded from the efficacy analysis. It was determined after enrolment that both received proscribed antipsychotic medication before entering the study." |
| Selective reporting (reporting bias) | High risk | More details on level of significance for outcomes with P < 0.05. Adverse effects for central nervous system reported only if present in ≥ 9%. |
| Other bias | High risk | Sponsored by drug company. |
Battaglia 2002.
| Methods | Allocation: randomised. Blindness: double. Duration: 24 hours, preceded by a 2 hour screening period. |
|
| Participants | Diagnosis: DSM‐IV schizophrenia, schizophreniform disorder, or schizoaffective disorder. N = 311 Age: mean 38.2 years. Sex: 204 males, 107 females. History: mean age of illness onset, years (SD); haloperidol 25.1 (8.3), olanzapine 23.5 (8.9), placebo 24.9 (8.0). Excluded: Pregnant or lactating women, people with serious medical illnesses or people currently prescribed antipsychotic medication via depot injection. Setting: Multi‐centre (Australia, Austria, Belgium, Canada, the Czech Republic, France, Greece, Hungary, Israel, the Republic of South Africa, Spain, the UK, and the USA). |
|
| Interventions | 1. Haloperidol IM: dose up to 3 injections of 7.5 mg (mean number of IMs 1.27). N = 126. 2. Olanzapine IM: dose up to 3 injections of 10 mg (mean number of IMs 1.3). N = 131. 3. Placebo IM: up to 3 injections (mean number of IMs ˜ 1.5). N = 54. Need for subsequent doses at the discretion of the investigator. |
|
| Outcomes | Mental state: BPRS. Global state: CGI, number of additional injections, use of benzodiazepine. Adverse effects: SAS, BAS, COSTART, QT changes, use of anticholinergic drugs. Agitation: ACES, PANSS‐EC. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Both drugs and placebo were administered in identical, color‐blinded, translucent standard syringes, and raters and study personnel were blind to treatment assignment" (Wright 2001, p.1149). [italic added by reviewers] |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Both drugs and placebo were administered in identical, color‐blinded, translucent standard syringes, and raters and study personnel were blind to treatment assignment" (Wright 2001, p.1149). [italic added by reviewers] |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The authors only account for 5 of the 26 people who did not complete the 24 hours study. |
| Selective reporting (reporting bias) | Low risk | No indication of selective reporting. |
| Other bias | High risk | Sponsored by the manufacturers of olanzapine. |
Breier 2002.
| Methods | Allocation: randomised. Blindness: double. Duration: 24 hours. |
|
| Participants | Diagnosis: schizophrenia, schizophreniform or schizoaffective disorder. N = 270. Age: range 18‐73 years. Sex: 155 males, 115 females. History: "recently hospitalised with acute agitation." Mean age at onset of illness 25.1 (SD 7.3) years. Excluded: people with "significant medical disorders, including alcohol and/or drug dependency, were excluded from this trial." Setting: Multi‐centre (Croatia, Italy, Romania and South Africa). |
|
| Interventions | 1. Haloperidol IM: dose up to 3 injections of 7.5 mg/IM (mean number of IMs not reported, mean dose 9.9 mg (SD 4.6). N = 40. 2. Olanzapine IM: dose up to 3 injections of 2.5 mg/IM (mean number of IMs not reported, mean dose 4 mg (SD 1.5). N = 48. 3. Olanzapine IM: dose up to 3 injections of 5 mg/IM (mean number of IMs not reported, mean dose 6.9 mg (SD 2.7). N = 45. 4. Olanzapine IM: dose up to 3 injections of 7.5 mg/IM (mean number of IMs not reported, mean dose 9.8 mg (SD 3.8). N = 46. 5. Olanzapine IM: dose up to 3 injections of 10 mg/IM (mean number of IMs not reported, mean dose 12.6 mg (SD 4.9). N = 46*. 6. Placebo IM. N = 50. 2nd injection allowed 2 hours after 1st, 3rd injection allowed 4 hours after 2nd injection. Both to be administered within 20 hours of 1st injection. Need for subsequent doses at the discretion of the investigator. |
|
| Outcomes | Mental state: BPRS Total, BPRS Positive. Global state: CGI‐S, additional injections, need for benzodiazepine. Agitation: ABS, PANSS‐EC. Adverse events: QT interval at 24 hours, hypotension**, acute dystonia**, EPS**. Unable to use*** ‐ Leaving the study early: reasons are not given for why participants did not complete the study. Adverse effects: SAS (mean, SE/SD not reported). Adverse effects: BAS (mean, SE/SD not reported). Adverse effects: need for anticholinergic therapy (olanzapine 10 mg/IM not reported). Adverse effects: QT interval at 2 hours (mean, SE/SD not reported). |
|
| Notes | * For data analysis, where continuous outcomes were used, haloperidol was compared with Olanzapine 10 mg/IM as this is the dose referred to as usual by the BNF (BNF 2011). ** It is possible that this binary data was derived from the BAS scale. *** Additional data are reported by dose but not used in this review (Table 52; Table 53; Table 54; Table 55; Table 56) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Low risk | "Allocation concealed placebo‐controlled trial." Method of randomisation not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "unblinded third‐party personnel, who played no role in evaluating patients, were trained to handle and administer injections in identical, unmarked syringes." "Investigators, patients, clinical carers and raters were blinded." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Investigators, patients, clinical carers and raters were blinded." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Reasons why all participants that left the study early were not reported. |
| Selective reporting (reporting bias) | Unclear risk | It is not clear whether all side effects are reported. |
| Other bias | High risk | Trial sponsored by manufacturers of olanzapine IM. |
4. Adverse effects for 1 mg, 5 mg, 15 mg aripiprazole (Bristol‐Myers 2005).
| AE | Dose | 1 mg (N = 57) | 5 mg (N = 63) | 15 mg (N = 58) |
| At least 1 AE | 28 | 30 | 27 | |
| AE rated as serious | 1 | 2 | 0 | |
| Tachycardia | 3 | 2 | 0 | |
| Sinus tachycardia | 1 | 0 | 0 | |
| Vomiting | 1 | 0 | 3 | |
| Nausea | 0 | 6 | 2 | |
| Dizziness | 4 | 7 | 7 | |
| Headache | 4 | 11 | 8 | |
| Somnolence | 3 | 5 | 6 | |
| Akathisia | 0 | 2 | 0 | |
| Dystonia | 0 | 0 | 1 | |
| Agitation | 0 | 0 | 3 | |
| Pain at injection site | Not reported | 1 | 1 | |
| QTC abnormality | 4 | 4 | 3 | |
AE: adverse event
5. Global outcome ‐ data for olanzapine 2.5 mg, 5 mg and 7.5 mg (Breier 2001).
| Outcome | Dose | Mean | SD | N |
| Mean dose of study drug | 2.5 mg | 4 | 1.5 | 48 |
| 5 mg | 6.9 | 2.7 | 45 | |
| 7.5 mg | 9.8 | 3.8 | 46 | |
| Mean dose of benzodiazepines | 2.5 mg | 3.2 | 1.1 | 48 |
| 5 mg | 2.0 | 0 | 45 | |
| 7.5 mg | 3.0 | 1.4 | 46 |
6. Continuous skewed data rated at 2 hours for olanzapine 2.5 mg, 5 mg and 7.5 mg (Breier 2001).
| Rating scale (Mean change at 2 hr) | Dose | Mean | SD | N |
| Specific behaviour: ACES |
2.5 mg | 1.3 | 1.5 | 48 |
| 5 mg | 2.3 | 1.9 | 45 | |
| 7.5 mg | 2.4 | 1.7 | 46 | |
| Specific behaviour: ABS |
2.5 mg | ‐5.8 | 5.5 | 48 |
| 5 mg | ‐9.0 | 5.5 | 45 | |
| 7.5 mg | ‐10.5 | 5.6 | 46 | |
| Specific behaviour: PANSS‐EC |
2.5 mg | ‐5.5 | 4.6 | 48 |
| 5 mg | ‐8.1 | 5.3 | 45 | |
| 7.5 mg | ‐8.7 | 5 | 46 | |
| Mental state: BPRS Total |
2.5 mg | ‐8.2 | 9.1 | 48 |
| 5 mg | ‐10.4 | 7.5 | 45 | |
| 7.5 mg | ‐12.0 | 7 | 46 | |
| Mental state: BPRS Positive |
2.5 mg | ‐1.5 | 3.1 | 48 |
| 5 mg | ‐1.7 | 2.8 | 45 | |
| 7.5 mg | ‐2.1 | 2.9 | 46 |
ABS: Agitated Behavior Scale ACES: Agitation‐Calmness Evaluation Scale BPRS: Brief Psychiatric Rating Scale PANSS‐EC: Positive and Negative Syndrome Scale Excited Component
7. Continuous skewed data for olanzapine 2.5 mg, 5 mg and 7.5 mg rated at 24 hours (Breier 2001).
| Rating scale (mean change at 24 hr) | Dose | Mean | SD | N |
| Specific behaviour: ACES |
2.5 mg | 0.9 | 0.8 | 48 |
| 5 mg | 1.1 | 1.1 | 45 | |
| 7.5 mg | 1 | 1 | 46 | |
| Specific behaviour: ABS |
2.5 mg | ‐5.7 | 4.2 | 48 |
| 5 mg | ‐6.7 | 5.9 | 45 | |
| 7.5 mg | ‐7.7 | 5.8 | 46 | |
| Specific behaviour: PANSS‐EC |
2.5 mg | ‐4.9 | 4.3 | 48 |
| 5 mg | ‐5.5 | 4.9 | 45 | |
| 7.5 mg | ‐5.5 | 4.1 | 46 | |
| Global outcomes: CGI‐S (24 hr) |
2.5 mg | ‐0.3 | 0.5 | 48 |
| 5 mg | ‐0.5 | 0.8 | 45 | |
| 7.5 mg | ‐0.6 | 0.7 | 46 | |
| Mental state: BPRS Total |
2.5 mg | ‐8.4 | 7.4 | 48 |
| 5 mg | ‐9.2 | 7.8 | 45 | |
| 7.5 mg | ‐9.6 | 7.5 | 46 | |
| Mental state: BPRS Positive |
2.5 mg | ‐1.5 | 2.3 | 48 |
| 5 mg | ‐2.0 | 2.6 | 45 | |
| 7.5 mg | ‐1.9 | 2.7 | 46 |
ABS: Agitated Behavior Scale ACES: Agitation‐Calmness Evaluation Scale BPRS: Brief Psychiatric Rating Scale CGI‐S: Clinical Global Impression – Severity PANSS‐EC: Positive and Negative Syndrome Scale Excited Component
8. Binary data for olanzapine 2.5 mg, 5 mg, 7.5 mg (Breier 2001).
| Dose | 2.5 mg | 5 mg | 7.5 mg | |
| Outcome | N = 48 | N = 45 | N = 46 | |
| Global outcome: > 1 injection | 25 | 17 | 14 | |
| Leaving the study early: lack of efficacy | 0 | 2 | 0 | |
| Adverse effects: EPS | 0 | 0 | 0 | |
| Adverse effects: akathisia | 0 | 2 | 0 | |
| Adverse effects: QT abnormality | 0 | 4 | 2 | |
EPS: Extrapyramidal Side Effects
Bristol Myers 2004a.
| Methods | Allocation: randomised, 2:2:1 ratio. Blindness: double. Duration: 2‐hour screening period, 24 hours initial phase, 4‐day second phase, 35 days follow‐up. |
|
| Participants | Diagnosis: DSM‐IV diagnosis of schizophrenia (N = 324) or schizoaffective disorder (N = 124). N = 448. Age: range 18‐69 years. Sex: males and females. History: voluntarily hospitalised, experiencing acute agitation, mean age of diagnosis ˜ 26.3 years. Excluded: a diagnosis of schizophreniform disorder; a psychiatric disorder other than schizophrenia requiring pharmacotherapy, those at significant risk of suicide, neurological diagnoses (including migraine, Parkinson's disease, epilepsy, Alzheimer's disease, residual of stroke, multiple sclerosis, transient cerebral ischaemic attacks, learning disability, cerebral palsy); a history of seizures, severe head trauma, abnormal ECG, stroke or evidence of other unstable medical conditions or adverse event, substance or alcohol dependence within 2 months of the study, history of neuroleptic malignant syndrome from antipsychotic agents, suspected substance‐induced psychiatric disorder or behavioural disturbance, benzodiazepine or anticholinergics within 4 hours before the first injection of study medication; lack of response to previous antipsychotic medication. Setting: Multi‐centre (USA, Czech Republic, France, Estonia, Latvia, Poland, Croatia, Italy, Puerto Rico, South Africa, and Spain). |
|
| Interventions | 1. Haloperidol: dose 6.5 mg/IM, maximum 3 doses during first 24 hours (mean number of IMs 1.43). N = 185. Followed by haloperidol: max dose 7 mg/oral to 10 mg/oral in 24 hours for 4 days. N = 151. 2. Aripiprazole: dose 10 mg/IM, maximum 3 doses during first 24 hours (mean number of IMs 1.54). N = 175. Followed by aripiprazole: max dose 10 mg/oral to 15 mg/oral in 24 hours for 4 days. N = 153. 3. Placebo IM, maximum 2 doses during first 24 hours. If 3rd dose needed aripiprazole: dose 10 mg/IM administered (mean number of IMs 1.92). N = 88. Followed by aripiprazole: max dose 10 mg/oral to 15 mg/oral in 24 hours for 4 days. N = 76. |
|
| Outcomes | Global state: CGI‐S, CGI‐I, additional medication, need for benzodiazepine. Leaving the study early. Adverse events: concomitant medication, death, ECG, physical examination, vital signs. Leaving the study early. Unable to use: Adverse effects: SAS (SE/SD/CI not reported). Adverse effects: BAS (SE/SD/CI not reported). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Low risk | "Randomization was conducted via a centralized call‐in system and was organized by study center using permuted block randomization". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | During the IM phase, haloperidol, aripiprazole and placebo were delivered in 1.3 mL injections. However, in the oral phase a tablet was used for aripiprazole and a capsule was used for haloperidol and so it is possible that this presented a risk to blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Described as "double blind", however no further details are given. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Reasons why all participants that left the study early were not reported. |
| Selective reporting (reporting bias) | High risk | The author mentions that there is increased glucose levels in both aripiprazole and haloperidol group but does not provide data. The authors only report the side effects that occurred in ≥ 2% of people. |
| Other bias | High risk | Sponsored by the manufacturers of aripiprazole. |
Bristol‐Myers 2005b.
| Methods | Allocation: randomised. Blindness: double. Duration: 24 hours (preceded by 2‐hour screening period). |
|
| Participants | Diagnosis: DSM‐IV diagnosis of schizophrenia (N = 237), schizoaffective disorder (N = 113) or schizophreniform disorder (N = 7). N = 357*. Age: 18‐66 years. Sex: 214 males, 143 females. History: acute agitation. Excluded: psychoactive substance dependence 2 months prior to the start of the study, required involuntary restraint, were suicidal, had a neurological or psychiatric condition other than schizophrenia, schizoaffective disorder or schizophreniform disorder, had a significant medical condition or were known non responders to antipsychotic medication. Setting: Multi‐centre (USA, Canada, Czech Republic, France, Estonia, Latvia, Lithuania, Spain). |
|
| Interventions | 1. Haloperidol IM: dose up to 3 injections of 7.5 mg/IM (mean number of IMs 1.33). N = 60. 2. Aripiprazole IM: dose up to 3 injections of 1 mg/IM (mean number of IMs not reported). N = 57. 3. Aripiprazole IM: dose up to 3 injections of 5 mg/IM (mean number of IMs not reported). N = 63. 4. Aripiprazole IM: dose up to 3 injections of 10 mg/IM (mean number of IMs not reported). N = 57*. 5. Aripiprazole IM: dose up to 3 injections of 15 mg/IM (mean number of IMs not reported). N = 58. 6. Placebo IM (mean number of IMs not reported). N = 50. |
|
| Outcomes | Mental state: BPRS Total and Positive Scores. Global state: CGI‐I, CGI‐S, number of re‐injections*. Agitation: ACES, CABS, PANSS‐EC. Leaving the study early. Adverse events: adverse event reports.Table 57 Unable to use***: Adverse effects: SAS (SE/SD/CI not reported). Adverse effects: BAS (SE/SD/CI not reported). Physiological: QTc is not reported for 9.75 mg. Results of clinical laboratory tests, physical examination, vital sign measurement are not reported. |
|
| Notes | * For data analysis, where continuous outcomes were used, haloperidol was compared with aripiprazole 10 mg/IM as this is the dose referred to as usual by the BNF (BNF 2011). **The number of additional injections needed was not reported individually for each of the aripiprazole doses but given as a range (i.e. 56% to 60% received 1 injection), therefore the author chose the mean value (i.e. 58%). *** Additional data are reported by dose but not used in this review (Table 57;Table 58) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No blinding was used during the preparation of injections and in most cases the person preparing the drug administered the injection. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Study investigators conducting the assessments were blinded to treatment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The reasons for leaving the study early were reported for those participants who did not complete the study. However, the number of participants completing different assessments at the same time point vary and reasons are not given. |
| Selective reporting (reporting bias) | High risk | The authors only report the adverse effects that occurred in ≥ 5% of people. The QTc abnormality for 9.75 mg is not reported. |
| Other bias | High risk | Sponsored by the manufacturers of aripiprazole. |
9. Continous skewed data for aripiprazole 1 mg, 5 mg, 15 mg (Bristol‐Myers 2005).
|
Rating scale (Mean change 2 hr after 1st injection) |
Dose | Mean | SD | N |
| Specific behaviour: ACES |
1 mg | 0.65 | 1.64 | 56 |
| 5 mg | 1.01 | 1.65 | 62 | |
| 15 mg | 0.99 | 1.73 | 58 | |
| Specific behaviour: CABS |
1 mg | ‐5.15 | 6.8 | 56 |
| 5 mg | ‐5.97 | 6.81 | 62 | |
| 15 mg | ‐7.04 | 7.01 | 58 | |
| Global outcomes: CGI‐I |
1 mg | 3.07 | 0.98 | 56 |
| 5 mg | 2.82 | 1.10 | 62 | |
| 15 mg | 2.66 | 1.07 | 58 | |
| Global outcomes: CGI‐S |
1 mg | ‐0.63 | 1.15 | 56 |
| 5 mg | ‐0.82 | 1.12 | 62 | |
| 15 mg | ‐0.99 | 1.17 | 58 | |
| Mental state: BPRS Total |
1 mg | ‐6.53 | 9.61 | 55 |
| 5 mg | ‐8.16 | 9.66 | 61 | |
| 15 mg | ‐8.88 | 9.89 | 56 | |
| Mental state: BPRS Positive |
1 mg | ‐1.20 | 2.72 | 55 |
| 5 mg | ‐1.47 | 2.71 | 61 | |
| 15 mg | ‐1.86 | 2.82 | 57 |
ACES: Agitation‐Calmness Evaluation Scale BPRS: Brief Psychiatric Rating Scale CABS: CABS ‐ Corrigan Agitated Behavior Scale CGI‐I: Clinical Global Impression – Improvement CGI‐S: Clinical Global Impression – Severity
10. Binary data for 1 mg, 5 mg and 15 mg aripiprazole (Bristol‐Myers 2005).
| Dose | 1 mg | 5 mg | 15 mg | |
| Outcome | N | N = 57 | N = 63 | N = 58 |
| PEC response ≥ 40% | 21 | 31 | 32 | |
| Need for rescue medication | 11 | 5 | 12 | |
| Discontinued for any reason | 2 | 3 | 1 | |
| Discontinued due to adverse event | 0 | 0 | 1 | |
| Withdrew consent | 2 | 2 | 0 | |
| Discontinued due to other known cause | 0 | 1 | 0 | |
PEC: Psychiatric Emergency Centre
Brook 2000.
| Methods | Allocation: randomised. Blindness: open. Duration: 7 days. |
|
| Participants | Diagnosis: diagnosis of schizophrenia, schizoaffective disorder, bipolar disorder, brief psychotic disorder, schizophreniform disorder, delusional disorder, or psychotic disorders not otherwise specified as defined by DSM‐III‐R. N = 132. Age: range 19‐66 years. Sex: 123 males, 9 females. History: recently hospitalised with acute psychosis. Excluded: substance‐induced psychosis (confirmed by urinalysis), psychosis related to organic origin, clinically relevant medical illness, abnormal ECG, imminent risk of suicide or homicide, history of substance abuse or dependence in the previous 2 months. Setting: Multi‐centre (7 countries). |
|
| Interventions | 1. Haloperidol: flexible dose 2.5 mg/IM to 10 mg/IM (mean number of IMs not reported), maximum 4 doses in 24 hours for 3 days. Followed by haloperidol: flexible dose 10 mg/oral to 80 mg/oral during 24 hours for 4 days. N = 42. 2. Ziprasidone: dose 10 mg/IM, maximum 4 doses in 24 hours for 3 days (mean number of IMs not reported). Followed by ziprasidone: flexible dose 80 mg/oral to 200 mg/oral in 24 hours for 4 days. N = 90. |
|
| Outcomes | Mental state: BPRS. Global state: CGI‐S. Leaving the study early. Adverse events: COSTART, BAS, modified SAS, investigators assessment of severity, blood pressure, laboratory tests, concomitant medication, ECG, urinalysis, others in ≥ 10 of participants. Unable to use Specific behaviours: BPRS agitation items (not published). Global state: CGI‐I (SE/SD not reported). Sedation score: 5‐point categorical scale (not published, present mean/SD rather than binary data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Computerized randomization assigned". |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open trial. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete outcome data. A table is provided which presents a summary of discontinuations. |
| Selective reporting (reporting bias) | High risk | The number of participants reported as discontinuing due to adverse effects is reported inconsistently (see Brook 1998c table 2 and Brook 2000 table 4). It is not clear whether all adverse events are reported. Brook 2000 table 4 only reports adverse events occurring in ≥10 of people, however there are more adverse events mentioned throughout the text. |
| Other bias | High risk | Sponsored by drug company. |
Calver 2015.
| Methods | Allocation: randomised. Blindness: double. Duration: 2 hours. |
|
| Participants | Diagnosis: mental illness (N = 114), drug‐induced psychosis (N = 70), intoxication (N = 17), threatened self‐harm (N = 6), other/unknown (N = 19) (p.225)*. N = 228. Age: major than 18 years. Sex: 144 males, 84 females. History: admitted involuntarily to the psychiatric intensive care unit from the psychiatric emergency care centre (p.223). Excluded: patients willing to take oral medication for sedation without physical restraint or seclusion. Setting: psychiatry intensive care unit. |
|
| Interventions | 1. Haloperidol: fixed dose 10 mg/IM. N = 110. 2. Droperidol: fixed dose 10 mg/IM. N = 118. |
|
| Outcomes | Tranquillisation or asleep, time to sedation. Need for additional benzodiazepines. Adverse events: respiratory rate less than 12 breaths/min, systolic blood pressure less than 90 mmHg, heart rate less than 60 bpm, oxygen saturation less than 90% or the presence of extrapyramidal side effects. |
|
| Notes | *when available, outcomes are reported for the mental illness sub‐group; gently provided by the authors (contacted 5th July 2016). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Block randomisation was used. Microsoft Excel was used to randomly create blocks of four (ABAB, AABB, etc.) or six (ABABAB, AAABBB, etc.)" (p.224). |
| Allocation concealment (selection bias) | Low risk | "Pre‐packed treatment kits [...] contained either droperidol (10mg in 2ml) or haloperidol (10mg in 2ml) [...] transferred into vials identical" (p.224). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind study. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "labels A or B and not the drug names were known to the investigator. This investigator analysed the data independently and presented this to the other investigators" (p.224). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete outcome data. |
| Selective reporting (reporting bias) | Unclear risk | "In breach of the study protocol midazolam was given nine times simultaneously with the study drug" (p.226); no further evidence of selective reporting. |
| Other bias | High risk | Short study. |
Currier 2004.
| Methods | Allocation: randomised. Blindness: single‐blind (rater‐blinded). Duration: 24 hours. |
|
| Participants | Diagnosis: DSM‐IV diagnosis of paranoid schizophrenia (N = 54), schizoaffective disorder (N = 36), bipolar disorder (N = 13), psychotic disorder not otherwise specified (N = 31), other (N = 28). N = 162. Age: range 18‐65 years. Sex: 105 males, 57 females. History: acute agitation. Excluded: pregnancy, delirium, epilepsy, learning disability, intoxication, symptoms of withdrawal from alcohol or other psychoactive substances, medical illness, treatment with any anti‐psychotic or benzodiazepine within 6 hours of screening, history of neuroleptic malignant syndrome, known hypersensitivity to any of the trial medications, treatment with depot injection, use of disallowed medication. Setting: multi‐centre, USA (24 sites) |
|
| Interventions | 1. Haloperidol: dose 5 mg/IM + lorazepam 2 mg/oral (mean number of IMs not reported). N = 79. 2. Risperidone: dose 2 mg/oral + lorazepam 2 mg/oral (mean number of doses not reported). N = 83. |
|
| Outcomes | Tranquillisation or asleep. Leaving the study early. Global state: CGI‐S, additional lorazepam. Agitation: PANSS‐EC. Aggression: OAS. Adverse effects: BAS, SAS, EPS, heart monitoring, sedation. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Low risk | "A telephone‐based central service was used to randomly assign eligible patients to receive a single dose of oral treatment or IM injection." (p.387) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Single‐blind study. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "independent raters blinded to the treatment arm conducted efficacy assessments". (p.387) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | A flow diagram is provided with the reasons for leaving the study early reported. However, the number of participants completing different assessments at the same time point vary and reasons are not given. |
| Selective reporting (reporting bias) | High risk | The authors only report the side effects that occurred in ≥ 5% of people. |
| Other bias | High risk | Sponsored by drug company (Janssen Phamaceutica). |
Dorevitch 1999.
| Methods | Allocation: randomised. Blindness: double. Duration: 2 hours. |
|
| Participants | Diagnosis: DSM‐IV diagnosis of paranoid schizophrenia (N = 19), schizoaffective disorder (N = 7), bipolar disorder (N = 2). N = 28. Age: range 20‐60 years. Sex: 13 males, 15 females. History: "actively psychotic inpatients". (p.142). Excluded: Not reported. Setting: Psychiatric hospital, Tel Aviv. |
|
| Interventions | 1. Haloperidol: dose 5 mg/IM (mean number of IMs not reported). N = 13. 2. Flunitrazepam: dose 1 mg/IM (mean number of IMs not reported). N = 15. |
|
| Outcomes | Global state: need for seclusion or restraint. Aggression: OAS. Adverse effects. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "assigned by a table of random numbers." (p.142). |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Described as double‐blind, however further details regarding the blinding of participants and personnel are not reported. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "All ratings were completed by the same rater (N.K.), who was blind to the study medications." (p.143). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete outcome data. |
| Selective reporting (reporting bias) | Unclear risk | It is not clear whether all side effects are reported. |
| Other bias | Unclear risk | Small short study. |
Eli Lilly 2004.
| Methods | Allocation: randomised. Blindness: double. Duration: 24‐72 hours. |
|
| Participants | Diagnosis: DSM‐IV diagnosis of schizophrenia. N = 49. Age: range 18‐65* years. Sex: 35 males, 14 females. History: "The mean length of current psychotic episode was 13.8 and 17.9 days in the olanzapine and haloperidol groups respectively." (p.6). Excluded: dementia, documented allergy to study drugs, treatment with injectable depot within 1 injection interval, unstable illnesses where death is anticipated within 1 year, or treatment in intensive care expected within 6 months, previous participation in a Lilly sponsored intra‐muscular olanzapine clinical trial. Setting: multi‐centre, Taiwan. |
|
| Interventions | 1. Haloperidol: dose 7.5 mg/IM (mean number of IMs 1.13). N = 24. 2. Olanzapine: dose 10 mg/IM (mean number of IMs 1.32). N = 25. |
|
| Outcomes | Mental state: BPRS total score, BPRS positive sub‐scale. Global state: CGI‐I, additional medication. Agitation: ACES, PANSS‐EC. Leaving the study early. Adverse effects: SAS, BAS, COSTART, death, laboratory analytes, ECG. Unable to use: Global state: CGI‐S. Adverse effects: change in vital signs (mean, SD CI not reported). |
|
| Notes | *The age difference between the two groups was statistically significant (P = 0.011). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Described as double‐blind, however no further details reported regarding the blinding of participants and personnel are not reported. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Described as double‐blind, however no further details reported regarding rater‐blinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The number of patients that completed the study is unclear: authors assert 45 in a study, 46 participants in the other one out of a possible 49 participants. However, only 42 participants are included in the analysis for the CGI‐I (at the end of the study), SAS, BAS, clinical laboratory evaluations, vital signs and ECG, without explanation for the missing participants. |
| Selective reporting (reporting bias) | High risk | Only the CGI‐S baseline score is reported. |
| Other bias | High risk | Sponsored by the manufacturers of olanzapine. |
Fang 2012.
| Methods | Allocation: randomised. Blindness: open. Duration: 120 hours (as for session I). |
|
| Participants | Diagnosis: DSM‐IV diagnosis of schizophrenia (N = 192), schizophreniform (N = 11) or schizoaffective (N = 0) disorders, no information* (N = 2); PANSS‐EC ≥14; PANSS ≥60. N = 208 screened, 205 randomised. Age: range 18‐45 years. Sex: 99 males, 106 females. History: acute agitation. Excluded: "women pregnant, breastfeeding or planning to become pregnant during the study", "psychotic agitation caused by delirium, epilepsy, mental retardation, or affective disorder", "intoxication or symptoms of withdrawal from alcohol or other psychoactive substances", "serious medical illness", "known hypersensitivity to any of the study medications or no response to risperidone or haloperidol during previous treatment", "treatment with a depot antipsychotic with one cycle of screening", "use of disallowed medication". Setting: multi‐centre (6), China. |
|
| Interventions | 1. Haloperidol: flexible dose IM 10 mg/day to 20 mg/day (mean prescribed daily dose 12.2 ± 3.7 mg), N = 101. 2. Risperidone: flexible dose oral 2 mL/day to 6 mL/day (mean prescribed daily dose 3.4 ± 0.7 mg), N = 104 + clonazepam: flexible dose oral 0 mg/day to 8 mg/day (mean prescribed daily dose 2.9 ± 1.5 mg), N = 99. |
|
| Outcomes | Not asleep. Mental State: PANSS total. Agitation: PANSS‐EC. Adverse events: insomnia, tachycardia, EPS, akathisia. Unable to use: Adverse effects: SAS (mean, SE/SD not reported). Adverse effects: BAS (mean, SE/SD not reported). |
|
| Notes | *Author assume a typing error in the demographic‐clinical "table 1". An attempt to contact the authors was made (2nd July 2016). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "A telephone‐based central service was used to randomly assign eligible patients" (p.108). |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open trial. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete outcome data. |
| Selective reporting (reporting bias) | High risk | SAS and BAS scores at 2, 4, 24, 72, and 120 hours are not reported. Only side effects occurred in ≥ 5% of people are reported. |
| Other bias | High risk | Sponsored by drug company (Xian‐Janssen Pharmaceutical Ltd). |
Fitzgerald 1969.
| Methods | Allocation: randomised. Blindness: double. Duration: 48 hours. |
|
| Participants | Diagnosis: "acute psychotic behavior". "Eleven of the haloperidol recipients and 12 of the perphenazine recipients exhibited paranoid reactions". (p.515). N = 44. Age: mean 41 years (haloperidol group), 36 years (perphenazine group). Sex: 17 males, 27 females. History: "acutely disturbed. Newly admitted patients exhibiting severe flat affect, inappropriateness, agitation, hostility, overactivity or various combinations". (p.515). Excluded: Not reported. Setting: inpatient, USA. |
|
| Interventions | 1. Haloperidol: dose 5 mg/IM (mean number of IMs not reported, mean dose 25.7 mg). N = 23. 2. Perphenazine: dose 5 mg/IM (mean number of IMs not reported, mean dose 27.6 mg). N = 21. |
|
| Outcomes | Global state: clinical observation of global improvement. Leaving the study early. Adverse effects: vital signs, need for anti‐parkinsonian medication. Not usable: Mental state: Target symptoms (unpublished). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "haloperidol and perphenazine were supplied in identically appearing 1‐mL ampoules each containing 5 mg of drug." (p.515). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Described as double‐blind ‐ no further details given regarding rater‐blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | It is reported that 3 participants were discontinued on the medication. According to Table 57, not all participants completed all outcome measures, it is not clear if this is due to attrition or because people were not available for assessment. |
| Selective reporting (reporting bias) | Unclear risk | "Most adverse reactions disappeared with the use of control measures" ‐ author suspects that only the adverse effects severe enough for discontinuation are reported. |
| Other bias | Unclear risk | No clear interested funding. |
Foster 1997.
| Methods | Allocation: randomised. Blindness: double. Duration: 4 hours. |
|
| Participants | Diagnosis: DSM‐III diagnosis of schizophrenia (N = 13), schizoaffective disorder (N = 4), bipolar disorder (N = 13), psychotic disorder not otherwise specified (N = 7). N = 37. Age: range 18‐61 years. Sex: 26 males, 11 females. History: "patients presenting at a psychiatric emergency room service." (p.177). Excluded: Not reported. Setting: psychiatric emergency room, USA. |
|
| Interventions | 1. Haloperidol: dose 5 mg/oral/IM, repeated at 30 minute intervals if necessary for up to 4 hours (mean number of IMs 2.25). N = 17. 2. Lorazepam: dose 2 mg/oral/IM, repeated at 30 minute intervals if necessary for up to 4 hours (mean number of IMs 1.82). N = 20. |
|
| Outcomes | Mental state: BPRS. Unable to use: Global State: need for additional oral/IM medication of study drug (N/SD not reported, mean doses reported). Global state: GCI not published. Behaviour: use of additional benzodiazepine (not reported). Physiological: vital signs (not reported). |
|
| Notes | *Author assumes GCI is the same as CGI. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Described as double‐blind, however it is not clear whether the study drugs appeared identical. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "All raters were unaware of the specific medication received by the patient". (p.176). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete outcome data. |
| Selective reporting (reporting bias) | High risk | "patients continued to receive treatment with a benzodiazepine, if necessary, for the duration of their stay in the emergency department." (p.176). The amount was not reported. |
| Other bias | High risk | Small short study. |
Fruensgaard 1977.
| Methods | Allocation: randomised. Blindness: double. Duration: 72 hours. |
|
| Participants | Diagnosis: acute schizophrenia (N = 12), psychogenic psychosis (N = 18). N = 30. Age: range 19‐65 years. Sex: 7 males, 23 females. History: "newly admitted patients with a diagnosis of acute psychosis and characterized by symptoms such as agitation, excitement, aggressiveness, delusions and hallucinations." (p.257). Onset < 7 days (N = 14), 1 week ‐ 1 month (N = 13), 1‐6 months (N = 3). Excluded: "pregnancy, manic depressive illness, electroconvulsive therapy within the preceding 8 weeks, organic brain syndrome with marked dementia, convulsive disorders, alcoholism or drug dependence, serious impairment of renal, hepatic, cardiovascular or metabolic functions, and present or former increased intra‐ocular pressure."(p.257). Setting: psychiatric hospital, Denmark. |
|
| Interventions | 1. Haloperidol: flexible dose 2.5 mg/IM to 5 mg/IM + biperiden: dose 2.5 mg/IM to 5 mg/IM (mean number of IMs not reported). N = 15. 2. Loxapine: flexible dose 25 mg/IM to 50 mg/IM + biperiden: dose 2.5 mg/IM (mean number of IMs not reported). N = 15. |
|
| Outcomes | Adverse effects: recording of side effects, laboratory tests.. Agitation: observation*. Not able to use: Mental state: BPRS (mean reported, SD/CI not reported). Global state: CGI (mean reported, SD/CI not reported). Adverse effects: blood pressure, pulse rate (reports a decrease in blood pressure/pulse rate in both groups but does not give N/mean/SD/CI). |
|
| Notes | * Scale derived data for agitation/excitation scores was not included as the measure was not validated. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Both drugs were supplied in ampules of identical appearance." (p.257). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Described as double‐blind, however no further details reported regarding rater‐blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete data. |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting. |
| Other bias | High risk | Small study. |
Garza‐Trevino 1989.
| Methods | Allocation: randomised. Blindness: open trial. Duration: not reported. |
|
| Participants | Diagnosis: Schizophrenia (N = 16), mania (N = 22), "atypical psychotic patients" (N = 6), "miscellaneous" (N = 14). N = 68. Age: Not reported. Sex: 41 males, 27 females. History: "judged to require immediate treatment for agitated or assaultive behavior, with either drugs, seclusion or both." (p.1599). Excluded: Not reported. Setting: psychiatric hospital, USA. |
|
| Interventions | 1. Haloperidol: dose 5 mg/IM (mean number of IMs not reported). N = 21. 2. Haloperidol: dose 5 mg/IM + lorazepam 4 mg/IM (mean number of IMs not reported). N = 24. 3. Lorazepam: dose 4 mg/IM (mean number of IMs not reported). N = 23. |
|
| Outcomes | Agitation: VAS. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open trial. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete data. |
| Selective reporting (reporting bias) | High risk | There is no reporting regarding the incidence of adverse effects. |
| Other bias | Unclear risk | The duration of the study is not reported. |
Guo 2007.
| Methods | Allocation: randomised. Blindness: open trial. Duration: 14 days. |
|
| Participants | Diagnosis: CCMD‐3 diagnosis of schizophrenia with agitation/aggression. N = 60. Age: range 18‐49 years. Sex: males and females. History: mean length of illness 56.7 (SD 7.8) months. Excluded: severe physical illness, drug/alcohol dependence. Setting: inpatient, China. |
|
| Interventions | Haloperidol: dose 5 mg/day to 10 mg/day for 3 days, 10 mg/IM/day to 30 mg/IM/day afterwards (mean number of IMs not reported. N = 30. Quetiapine: dose 100 mg/day to 800 mg/day + magnesium valproate dose: 0.5 g/p.o./day to 1 g/p.o./day (mean number of IMs not reported) N = 30. |
|
| Outcomes | Agitation: PANSS‐EC. Not able to use: Global outcome: CGI (not outcome data reported). Adverse effects: TESS (no outcome data reported). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No incomplete outcome data |
| Selective reporting (reporting bias) | High risk | CGI and TESS were measured, but not reported. |
| Other bias | Unclear risk | None obvious. |
Higashima 2004.
| Methods | Allocation: randomised. Blindness: open trial. Duration: 8 weeks. |
|
| Participants | Diagnosis: DSM‐IV diagnosis of schizophrenia. N = 19. Age: mean 29.5 years (monotherapy), mean 26.6 years (combined therapy). Sex: 12 males, 7 females. History: "acute exacerbation of psychotic symptoms." (p.380). Excluded: Not reported. Setting: Psychiatric hospital, Japan. |
|
| Interventions | Haloperidol: average endpoint dose 5.4 (2.7) mg/p.o./day. N = 10. Haloperidol + levomepromazine: average endpoint dose ratio 1:10, 5.4 (2.4) mg/p.o./day. N = 9. |
|
| Outcomes | Global state: no overall improvement. Unable to use: Mental state: BPRS. Adverse effects: not reported. Adverse effects: the quantity of anticholinergic medication prescribed was not reported. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open trial. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete data. |
| Selective reporting (reporting bias) | High risk | The discussion describes how the combination drug "produced moderate sedation without any cardiovascular adverse effect". However, there is no other reporting regarding the incidence adverse effects. |
| Other bias | High risk | Small N study. |
Huf 2007.
| Methods | Allocation: randomised. Blindness: open trial. Duration: 14 days. |
|
| Participants | Diagnosis: psychosis (N = 244), substance misuse (N = 58), others (N = 11), no information (N = 3). N = 316. Age: mean 40.1 years. Sex: males. History: people presenting at a psychiatric emergency room service. 1st attendance (N = 59), previous attendance (N = 224), unknown history (N = 33). Excluded: where the "clinician believed that one of the treatment options represented an additional risk for the patient." (p.3). Setting: psychiatric emergency room, Brazil. |
|
| Interventions | 1. Haloperidol: dose 5 mg/IM to 10 mg/IM. N = 156. 2. Haloperidol: dose 5 mg/IM to 10 mg/IM + promethazine 25 mg/IM to 50 mg/IM. N = 160. |
|
| Outcomes | Tranquillisation or asleep: tranquil/asleep, time to tranquillisation/sleep, need for additional tranquillising drug. Specific behaviours: another episode of aggression by 24 hours. Global state: use of restraints, needing extra visits from the doctor, refusing oral medication. Leaving the study early. Service outcomes: duration of hospital stay. Adverse effects: serious, specific adverse effects. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated randomised sequence was applied to a table of numbers. |
| Allocation concealment (selection bias) | Low risk | "Boxes were consecutively numbered, sealed, opaque, and identical appearance and weight; on the outside was a form with questions to be completed by the attending doctor while "blind" to the contents of the box." (p.2). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open trial. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete outcome data. |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting. |
| Other bias | Unclear risk | The study was stopped early due to adverse effects, therefore it is possible that the available data may be biased. |
Kewala 1984.
| Methods | Allocation: randomised. Blindness: double. Duration: 5 hours. |
|
| Participants | Diagnosis: DSM‐III diagnosis of schizophrenia (N = 13), schizoaffective disorder (N = 4), bipolar disorder (N = 13), psychotic disorder not otherwise specified (N = 7). N = 44. Age: range 19‐56 years. Sex: 21 males, 23 females. History: "newly admitted patients...mean duration of illness 7.75 years." (p.77). Excluded: Not reported. Setting: psychiatric hospital, USA. |
|
| Interventions | 1. Haloperidol: flexible dose 2.5 mg/IM to 10 mg/IM, maximum 4 injections/cumulative dosage of 27.5 mg during 24 hours (mean number of IMs not reported, mean cumulative dose ˜ 12.1 mg). N = 24. 2. Thiothixene: flexible dose 2.5 mg/IM to 10 mg/IM, maximum 4 injections/cumulative dosage of 27.5 mg during 24 hours (mean number of IMs not reported, mean cumulative dose ˜ 16.1 mg). N = 20. |
|
| Outcomes | Global state: global evaluation. Adverse effects. Unable to use: Mental state: BPRS (SE/SD/CI not reported, mean reported). Mental state: Study Symptom Profile (mean, SE/SD not reported, P values of significant findings reported). Physiological: the authors report that there were no abnormal laboratory results, although no data was given. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Described as double‐blind, however no further details reported regarding the blinding of participants and personnel. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Described as double‐blind, however no further details reported regarding rater‐blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete outcome data. |
| Selective reporting (reporting bias) | Unclear risk | No evidence of selective reporting. |
| Other bias | High risk | Recieved grant from Roerig, a division of Phizer Pharmaceuticals (thiothixene manufacturers), small short study. |
Kinon 2001d.
| Methods | Allocation: randomised. Blindness: double. Duration: 21 days. |
|
| Participants | Diagnosis: schizophrenia (N = 56), schizophreniform disorder (N = 42), schizoaffective disorder (N = 2). N = 100. Age: range 18‐50 years. Sex: 71 males, 29 females. History: "newly admitted patients who were acutely agitated." (p.181). Excluded: pregnant or lactating women, people with "serious unstable illnesses, including hepatic, renal, gastroenterologic, respiratory, cardiovascular, endocrinologic, neurologic, immunologic, or hematologic disease, in which pharmacotherapy posed a substantial clinical risk or confounded diagnosis were excluded from this trial." (p.181). Setting: not reported. |
|
| Interventions | 1. Haloperidol: accelerated dose titration 10 mg/p.o. to 20 mg/p.o. during 24 hours. N = 48. 2. Olanzapine: accelerated dose titration 10 mg/p.o. to 20 mg/p.o. during 24 hours. N = 52. |
|
| Outcomes | Mental state: PANSS total, need for increased dose of study drug. Global state: CGI‐I, time in restraints, seclusion and special observation, need for increased dose of study drug, use of benzodiazepine. Agitation: Seclusion and special observation. Leaving the study early. Adverse effects: Tranquillisation Scale, recording rates of discontinuation, recording treatment emergent adverse events in ≥10% of participants, need for parkinsonian drugs. Unable to use: Global state: CGI (between‐group data was described as not significant but figures not reported ‐ only within‐group data reported), CGI‐S (not reported). Agitation: OASS (only present the data from 2 items of the scale), NOSIE (between‐group data were described as not significant but figures not reported ‐ only within‐group data reported), PANSS Agitation sub‐scale (only present data from certain items of the PANSS), SAS (between‐group data were described as not significant but figures not reported ‐ only within‐group data reported). Adverse effects: BAS (between‐group data was described as not significant but figures not reported ‐ only within‐group data reported), vital signs (not reported), laboratory analytes (described as no clinically relevant changes, although no data given), weight gain (mean, P values of significant findings, overall F value reported, SD/SE/CI not reported). Satisfaction: DAI‐10 (significant within‐group data reported, for between‐group P values of non significant findings reported, overall F value). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Described as double‐blind, however no further details reported regarding the blinding of participants and personnel. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Described as double‐blind, however no further details reported regarding rater‐blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete outcome data. |
| Selective reporting (reporting bias) | High risk | Significant findings from the within‐group data are presented on a number of occasions, whilst acknowledging but omitting the non‐significant between‐group findings. The OASS is a 16 item rating scale, yet only the results for two items where the olanzapine group had significantly greater improvement than the haloperidol group were reported. Treatment‐emergent adverse effects were only reported if they occurred with a frequency ≥10% or P< .05 through the end of the study. |
| Other bias | High risk | Sponsored by the manufacturers of olanzapine. |
Li 2006.
| Methods | Allocation: randomised. Blindness: single. Duration: 72 hours. |
|
| Participants | Diagnosis: CCMD‐3 diagnosis of schizophrenia with agitation/aggression. N = 231. Age: mean 34 (SD 12) years. Sex: males and females. History: mean length of illness, haloperidol group 30 (± 68) months, ziprasidone 18 (± 35) months. Excluded: severe physical or neurological impairment/diseases; drug or alcohol abuse, dependence, pregnant women. Setting: not reported. |
|
| Interventions | 1. Haloperidol: flexible dose 5 mg/IM to 10 mg/IM, repeated every 4‐6 hours if necessary, maximum dose 30 mg during 24 hours. N = 116. 2. Ziprasidone: flexible dose 10 mg/IM to 20 mg/IM, repeated every 4‐6 hours if necessary, maximum dose 40 mg during 24 hours. N = 115. |
|
| Outcomes | Mental state: PANSS endpoint. Agitation: PANSS‐EC. Adverse effects: TESS. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Single‐blind – however the author did not state who had been blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Single‐blind – however the author did not state who had been blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No incomplete data. |
| Selective reporting (reporting bias) | Unclear risk | TESS and SAS were used for assessment of adverse events. No continuous data were reported from either scales, but binary outcomes for adverse events were reported. |
| Other bias | Unclear risk | None obvious. |
Liang 2013.
| Methods | Allocation: randomised. Blinding: not stated. Duration: 28 days. |
|
| Participants | Diagnosis: ICD‐10 diagnosis of schizophrenia. N = 80. Age: range 18‐60 years. Sex: 42 males, 38 females. History: acute agitation and excitement; length of Illness 7.1 ± 7.7 (quetiapine group), 7.0 ± 6.3 (haloperidol group). Excluded: "pregnancy, serious physical sickness, addiction to alcohol or drugs, intolerant to quetiapine, haloperidol or orally taking medicines, taking quetiapine or haloperidol or other long‐term schizophrenia medicines one month before recruitment, neutrophil cells ≤ 1.5× 109/L". Setting: inpatients, China. |
|
| Interventions | 1. Haloperidol: dose 2 mg/oral/day to 4 mg/oral/day, by 3rd day 6 mg/day, by 7th day 10 mg/day to 12 mg/day. N = 40. 2. Quetiapine: dose 50 100 mg/oral/day to 100 mg/oral/day, by 3rd day 300 mg/day, by 7th day 600 mg/day to 750 mg/day. N = 40. |
|
| Outcomes | Mental state: PANSS total and sub‐scale. Agitation: PANSS‐EC. Adverse effects: general. Adverse effects: movement ‐ SAS. Dropout from the study. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomised by using random number table". |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | A total of 5 dropped out in haloperidol group (adverse events = 4, lost to contact = 1) and 4 dropped out in quetiapine group (adverse events = 1, lost to contact = 3); missing data have not been imputed using appropriate methods such as LOCF. |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting. |
| Other bias | Unclear risk | None obvious. |
Lim 2010.
| Methods | Allocation: randomised. Blindness: open‐label, rater‐blinded. Duration: 24 hours. |
|
| Participants | Diagnosis: DSM‐IV diagnosis of schizophrenia (N = 78), bipolar disorder (N = 43), others (N = 3). N = 124. Age: range 18‐65 years. Sex: 66 males, 58 females. History: "acute psychotic agitation in the emergency room." (p.82). Excluded: neurological disorder, severe medical disease, alcohol or other psychoactive substance misusers, history of neuromalignant syndrome or hypersensitivity to trial medications, pregnant or lactating women, people treated with antipsychotics or benzodiazepines within 6 hours to the start of the trial or with depot antipsychotic within one treatment cycle of enrolment. Setting: psychiatric emergency room, Korea. |
|
| Interventions | 1. Haloperidol: dose 5 mg/IM, maximum 15 mg during 24 hours (mean number of IMs not reported). N = 62. 2. Risperidone: dose 2 mg/p.o, maximum 6 mg during 24 hours (mean dose not reported). N = 62. |
|
| Outcomes | Global state: need for benzodiazepine. Agitation: PANSS‐EC. Leaving the study early. Adverse effects. Unable to use: Mental state: PANSS Total (mean, SE/SD not reported, P values of significant findings reported, overall F value). Mental state: YMRS Total (mean, SE/SD/CI not reported). Global state: CGI‐S (mean, SE/SD not reported, P values of significant findings reported, overall F value). Adverse effects: use of antiparkinsonian drugs not reported. Adverse effects: AIMS (mean, SE/SD/CI not reported). Adverse effects: SAS (mean, SE/SD/CI not reported). Behaviour: Behaviorally Anchored Rating Scale (mean, SE/SD/CI not reported). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were assigned "according to a predefined randomization code that was balanced to ensure even distribution of patients in each treatment group." (p.82). |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This was an open‐label study where one group received IM and the other group received an orodispersible tablet. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The study was rater‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There is no evidence of incomplete outcome data. The authors give reasons for the participants who discontinued the study. |
| Selective reporting (reporting bias) | High risk | The authors report that antiparkinsonian drugs could be given at the lowest effective dose, however the amount administered is not described in the results. |
| Other bias | High risk | Sponsored by drug company (Janssen Phamaceutica Korea). |
Liu 2012.
| Methods | Allocation: randomised. Blinding: not stated. Duration: 2 weeks. |
|
| Participants | Diagnosis: CCMD‐3 diagnosis of schizophrenia. N = 150. Age: range 18‐59 years. Sex: 69 males, 81 females. History: agitated behaviour; length of Illness 0‐ 30 months. Excluded: "serious physical sickness, additive to alcohol and drugs, intolerant to schizophrenia treatment, pregnancy". Setting: inpatients, China. |
|
| Interventions | 1. Haloperidol: dose 10 mg/IM/day to 20 mg/IM/day (mean dose 16.15 ± 4.67 mg/day). N = 50. 2. Risperidone: dose 3 mg/oral/day to 6 mg/oral/day (mean dose 4.57 ± 0.65 mg/day) + haloperidol dose 10 mg/IM/day to 20 mg/IM/day (mean dose 15.73 ± 4.26 mg/day). N = 50. 3. Risperidone: dose 3 mg/oral/day to 6 mg/oral/day + clonazepam dose 3 mg/IM/day to 6 mg/IM/day (mean dose 4.05 ± 0.81 mg/day). N = 50. All IM injections changed to oral administration by 10 days. |
|
| Outcomes | Global state: clinical observation of global improvement. Agitation: PANSS‐EC. Adverse effects: TESS. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomised by using random number table". |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No incomplete data. |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting. |
| Other bias | Low risk | None obvious. |
Man 1973.
| Methods | Allocation: not reported. Blindness: double. Duration: 72 hours. |
|
| Participants | Diagnosis: acute exacerbation of chronic psychosis ‐ “severe agitation with marked psychomotor hyperactivity, extreme assaultiveness, hostility and mania. The patients presented a clear danger to themselves, to other patients and to staff personnel”. N = 30. Age: mean 33 years. Sex: 15 males and 15 females. History: acute agitation, 90% had previous admission to a psychiatric hospital. Excluded: anyone who used anti‐psychotic or anti‐convulsant within one week prior to the trial or anyone who used long lasting drug within one month prior to the trial. Setting: inpatients, China. |
|
| Interventions | 1. Haloperidol: dose 5 mg/IM, repeated every 30 minutes if necessary, maximum 15 mg during 24 hours (mean number of IMs 3.33). N = 15. 2. Chlorpromazine: dose 50 mg/IM, repeated every 30 minutes if necessary, maximum 600 mg during 24 hours (mean number of IMs 2.93). N = 15. |
|
| Outcomes | Global state: need for additional injections. Adverse effects: reporting of EPS. Leaving the study early. Unable to use: Mental state: BPRS (SD/SE/CI not reported, mean not reported). Global state: TSRS (unpublished, mean reported, SD/SE/CI not reported). Adverse effects: it is reported that there were no significant changes in blood pressure, pulse rate and respirations, however no data given. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Not explicitly described as randomised. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Described as double‐blind, however it is unclear whether the syringes appeared identical given the varied doses. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Described as double‐blind, however no further details reported regarding rater blinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The authors give reasons for the participants who discontinued the study. However, the results are only presented for the 27 participants who completed the study. Missing data have not been imputed using appropriate methods such as LOCF. |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting. |
| Other bias | High risk | Small study. |
Nobay 2004.
| Methods | Allocation: randomised. Blindness: double. Duration: not reported. |
|
| Participants | Diagnosis: violent behaviour and severe agitation. N = 111. Age: mean 40.7 years. Sex: not reported. History: prior psychiatric history (N = 54), no psychiatric history (N = 8), unknown psychiatric history (N = 49). Excluded: known allergy to study drugs, hypotensive, unable to maintain own airway, tachycardia, bradycardia, respiratory distress, <18 or pregnant. Setting: emergency psychiatric department, USA. |
|
| Interventions | 1. Haloperidol: fixed dose 5 mg/IM (mean number of IMs not reported). N = 42. 2. Midazolam: fixed dose 5 mg/IM (mean number of IMs not reported). N = 42.* |
|
| Outcomes | Tranquil or asleep: modified TCS. Global state: need for additional medication. Adverse effects. Unable to use: Adverse effects: it is reported that there were no significant differences between groups for changes to blood pressure, heart rate and oxygen. However, data not given. |
|
| Notes | *There was also a lorazepam group, however we cannot include the data because randomisation was broken. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The administered drugs were prepared in the pharmacy using a computer generated randomisation code." (p.745). |
| Allocation concealment (selection bias) | Low risk | "The administered drugs were prepared in the pharmacy using a computer generated randomisation code." (p.745). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The research assistant [rater], the administering physician, and the patient were all blinded to the drug delivered." (p.745). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The research assistant [rater], the administering physician, and the patient were all blinded to the drug delivered." (p.745). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete outcome data. |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting. |
| Other bias | High risk | The duration of the study was not reported, therefore it is difficult to establish how long participants were monitored for adverse effects. |
Paprocki 1977.
| Methods | Allocation: randomised. Blindness: double. Duration: 4‐day IM phase followed by up to 4 week oral phase. |
|
| Participants | Diagnosis: "acute psychosis with manifest disturbed psychotic symptoms characterised by agitation, excitement, aggressiveness, hostility, delusions and hallucinations." (p.81). N = 35. Age: range 18‐49 years. Sex: females. History: onset of present episode: < 1 week (N = 10), > 1 week < 1 month (N = 26)*. Excluded: psychotropic drugs prior to 24 hours of the start of the study, "known hypersensitivity to dibenzazepine compounds, electroconvulsive therapy or insulin coma or subcoma therapy within the preceding eight weeks, presence of organic brain syndrome with marked dementia and inability to communicate during interviews, history of convulsive disorders, alcoholism or drug dependence as a significant feature of clinical history, presence of serious impairment of renal or hepatic function, increased intra ocular pressure or history of narrow angle glaucoma or urinary retention, cardiovascular or metabolic disease, pregnancy suspected or confirmed." (p.82). Setting: emergency psychiatric department, USA. |
|
| Interventions | 1. Haloperidol: dose 2.5 mg/IM to 5 mg/IM, repeated every 6 to 12 hours if necessary (IM phase), dose 5 mg to 15 mg (oral phase) (mean number of IMs not reported, mean daily dose 11.5 mg). N = 18. 2. Loxapine: dose 25 mg/IM to 50 mg/IM, repeated every 6 to 12 hours if necessary (IM phase), dose 50 mg to 150 mg (oral phase) (mean number of IMs not reported, mean daily dose 115.4 mg). N = 17. |
|
| Outcomes | Global state: CGI. Leaving the study early Adverse effects: observations, laboratory tests. Unable to use: Mental state: BPRS (overall P value and level of significance reported, SD/SE/CI not reported). Global state: NOSIE (overall P value and level of significance reported, SD/SE/CI not reported). Adverse effects: sedation score (mean reported, SD/SE/CI not reported). |
|
| Notes | *This would suggest there were 36 participants. It is not clear if this is a typing error because it is reported in the rest of the paper that there were 35 participants. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind ‐ "supplied in 1ml ampules...of identical appearance, individually packaged and labelled with the code number assigned to each patient". Capsules used in the oral phase were also of identical appearance. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Described as double‐blind ‐ no further details given regarding rater blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The number of participants reported is inconsistent (reported at 36 in one instance and 35 at other times) and it is not clear if this is a typing error. |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting. |
| Other bias | High risk | Small study. |
Reschke 1974.
| Methods | Allocation: randomised. Blinding: double for IM phase, open for oral phase. Duration: approximately 2 hours. | |
| Participants | Diagnosis: schizophrenia (no specified criteria).
N = 50.
Sex: 2 males, 48 females.
Age: range 19‐57, mean 35.9 years. History: severe agitation ‐ "Psychiatric emergencies with symptoms such as agitation, excitement, and assaultiveness of such severity that rapid tranquillisation was necessary". Excluded: not reported. Setting: hospital. |
|
| Interventions | 1. Haloperidol IM: dose up to 4 injections of 1 mg (mean number of IMs not reported). N = 10. 2. Haloperidol IM: dose up to 4 injections of 2 mg (mean number of IMs 3.7). N = 11. 3. Haloperidol IM: dose up to 4 injections of 5 mg (mean number of IMs 2.8). N = 8. 4. Chlorpromazine IM: dose up to 4 injections of 25 mg (mean number of IMs not reported). N = 10. 5. Placebo IM: dose up to 4 injections of 1 mg (mean number of IMs not reported). N = 11. |
|
| Outcomes | Not tranquil or asleep: not asleep at 2 hours. Global state: own 5‐point rating scale. Adverse effects. Leaving the study early. Unable to use: Mental state: BPRS and own target symptoms scale (no mean or SD). |
|
| Notes | Data for the haloperidol interventions were combined within the paper for some outcomes, for the binary outcomes that were not already presented in this way we added events for the different doses together following the protocol (see Unit of analysis issues). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Described as double‐blind, however no further details are given. It is not clear whether the study drugs appeared identical. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Described as double‐blind, however no further details reported regarding rater blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete outcome data. |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting. |
| Other bias | Unclear risk | Small study. |
Resnick 1984.
| Methods | Allocation: randomised. Blindness: double. Duration: 24 hours. |
|
| Participants | Diagnosis: acute agitation. N = 27. Age: range 18‐65 years. Sex: not reported. History: involuntary hospitalised. Excluded: intoxicated, known sensitivity to droperidol or haloperidol, evidence of active renal, hepatic, or cardiac disease. Setting: emergency and psychiatric crisis unit, USA. |
|
| Interventions | 1. Haloperidol: dose up to 4 injections of 5 mg (mean number of IMs not reported). N = 16. 2. Droperidol: dose up to 4 injections of 5 mg (mean number of IMs 1.36). N = 11. |
|
| Outcomes | Global state: need for additional injection. Adverse effects: reporting of EPS. Unable to use: Mental state: BPRS (mean/SD/SE/CI not reported). Adverse effects: it is reported that there were no significant changes in blood pressure, pulse rate and respirations, however no data given. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Low risk | "packages of medication were identified only by a code which was kept in the pharmacy until the conclusion of the study" (p.298). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "packages of medication were identified only by a code which was kept in the pharmacy until the conclusion of the study" (p.298). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Described as double‐blind, however no further details reported regarding rater blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete data. |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting. |
| Other bias | High risk | Small short study. |
Ritter 1972.
| Methods | Allocation: randomised, no further details. Blindness: double, identical appearing ampoules. Duration: 48 hours. | |
| Participants | Diagnosis: paranoid schizophrenia (N = 48), involutional melancholia (N = 1), manic‐depression (N = 1).
N = 50.
Age: range 21‐65 years, mean 36 years.
Sex: males. History: all exhibited symptoms such as aggressiveness, assaultiveness or uncooperativeness that disrupted normal hospital operation and interfered with attempts at therapy. Excluded: not reported. Setting: not reported. |
|
| Interventions | 1. Haloperidol: fixed dose 5 mg/IM, repeated every 6‐8 hours as necessary (mean number of IMs not reported). N = 25. 2. Chlorpromazine: fixed dose 50 mg/IM, repeated every 6‐8 hours as necessary (mean number of IMs not reported). N = 25. | |
| Outcomes | Global state: own 5‐point scale. Adverse events: Leucopenia, SGPT increase. Unable to use: Adverse effects: EPS, hypotension (no data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Described as double‐blind ‐ the test drugs were supplied in identically appearing ampoules. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Described as double‐blind, however no further details reported regarding rater blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete outcome data. |
| Selective reporting (reporting bias) | High risk | Do not report all adverse effects. The authors do not report the mean dosages/number of injections needed. |
| Other bias | Unclear risk | None obvious. |
Salzman 1991.
| Methods | Allocation: not reported. Blindness: double. Duration: 48 hours. |
|
| Participants | Diagnosis: schizophrenia (N = 26), schizoaffective disorder (N = 4), bipolar disorder (N = 11), organic mental disorder (N = 6), other (N = 13). N = 60. Age: mean 30.5 years (haloperidol group), mean 37.9 years (lorazepam group). Sex: not reported. History: involuntary hospitalised ‐ "Aggressive, assaultive or disruptive behaviour, thought to be a danger to the patient or other people". Excluded: intoxicated, known sensitivity to droperidol or haloperidol, evidence of active renal, hepatic, or cardiac disease. Setting: emergency and psychiatric crisis unit, USA. |
|
| Interventions | 1. Haloperidol: fixed dose 5 mg/IM (mean number of IMs 1.1). N = 30. 2. Lorazepam: fixed dose 2 mg/IM (mean number of IMs 1.13). N = 30. |
|
| Outcomes | Adverse effects. Unable to use: Mental state: BPRS (mean change given, SD/SE/CI not reported). Global state: CGI‐S (mean change given, SD/SE/CI not reported). Aggression: OAS (mean change given, SD/SE/CI not reported). Global state: additional injection (mean dose given, SD/SE/CI not reported). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Not explicitly described as randomised. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Parenteral medication was prepared in advance by a member of staff who had no direct clinical responsibility for the patient." (p.177). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Raters were blind to the treatment condition received by the patient." (p.178). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | It is reported that the number of participants declined after the baseline assessment because they were discharged or transferred. However, exact numbers are not given and it is not clear if the participants were transferred because they had a positive or negative outcome. Attrition approaching 50% for some outcomes at 24 hours. |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting. |
| Other bias | High risk | Sponsored by drug company. |
Shi 2010.
| Methods | Allocation: randomised. Blindness: not stated. Duration: 2 weeks. |
|
| Participants | Diagnosis: CCMD‐3 diagnosis of schizophrenia. N = 64. Age: range 18‐50 years. Sex: 37 males, 27 females. History: agitation and aggression. Excluded: serious physical illness; addiction to alcohol or drugs; pregnancy. Setting: inpatients, China. |
|
| Interventions | 1. Haloperidol IM (mean dose 15 ± 2.5 mg/day). N = 31. 2. Olanzapine oral (mean dose 10 ± 2.5 mg/day) + magnesium valproate oral (mean 1.25 ± 0.25 g/day). N = 33. |
|
| Outcomes | Global state: clinical observation of global improvement. Mental state: PANSS total score. Agitation: PANSS‐EC. Adverse effects. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomised by using random number table". |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete outcome data. |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting. |
| Other bias | Low risk | None obvious. |
Shu 2010.
| Methods | Allocation: randomised. Blindness: open‐label, rater‐blinded. Duration: 72 hours. |
|
| Participants | Diagnosis: ICD‐10 diagnosis of schizophrenia. N = 376. Age: range 18‐65 years. Sex: males and females. History: mean duration of acute agitation 3.4 days (haloperidol group), 3.2 days (ziprasidone group); mean duration of schizophrenia 4 years (haloperidol group), 5.6 years (ziprasidone group). Excluded: history of clinically significant physical illness especially myocardial infarction, non compensatory heart failure, people receiving an investigational agent in the previous 3 months to screening, us of antipsychotic agents within 12 hours or parenteral benzodiazepines within 4 hours prior to randomisation and during the study. Setting: multi‐centre, China. |
|
| Interventions | 1. Haloperidol: dose 5 mg/IM, maximum 20 mg during 24 hours (mean number of IMs not reported). N = 187. 2. Ziprasidone: flexible dose 10 mg/IM to 20 mg/IM, maximum 40 mg in 24 hours (mean number of IMs not reported). N = 189. |
|
| Outcomes | Mental state: BPRS. Adverse effects: laboratory tests, ECG's, physical examination. Leaving the study early. Unable to use: Specific behaviour: agitation ‐ BPRS agitation items (modified from BPRS, only report LS means; means/SD not reported). Specific behaviour: agitation ‐ BARS (only give upper quartile value and LS means; means/SD not reported). Global state: CGI‐I (only report CI and LS means, means/SD not reported). Global state: CGI‐S (only give upper quartile value and LS means, means/SD not reported). Adverse effects: BAS and SAS (only report LS means). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomized in a 1:1 ratio (in blocks of 4)" (p.179); no further informations on the randomization procedure is stated. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Rater‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The reasons for leaving the study early were reported for those participants who did not complete the study. However, the number of participants completing different assessments at the same time point vary and reasons are not given. |
| Selective reporting (reporting bias) | Unclear risk | Only report side effects experienced by ≥2% of people. |
| Other bias | High risk | Sponsored by drug company. |
Stotsky 1977.
| Methods | Allocation: randomised. Blindness: double. Duration: 6 hours. |
|
| Participants | Diagnosis: schizophrenia (N = 24), bipolar disorder (N = 2), psychotic depressive reaction (N = 4). N = 30. Age: range 18‐66 years. Sex: 13 males, 17 females. History: disturbed, acutely disruptive symptomology outpatients, 16 of whom had prior inpatient treatment, and 21 of whom had a history of outpatient treatment. Excluded: known hypersensitivity to haloperidol or thiothixene, active hepatic, renal or cardiac disease or organic brain disease. Setting: emergency and psychiatric crisis unit, USA. |
|
| Interventions | 1. Haloperidol: flexible dose 4 mg/IM or 8 mg/IM, maximum dose 32 mg during 6 hours (mean number of IMs 3.07, mean dose 15 mg). N = 15. 2. Thiothixene: flexible dose 4 mg/IM or 8 mg/IM, maximum dose 32 mg during 6 hours (mean number of IMs 2.6, mean dose 10.1 mg). N = 15. |
|
| Outcomes | Global state: need for additional injection. Adverse effects. Unable to use: Mental state: BPRS (mean scores given, SD/SE/CI not reported). Mental state: symptom profile (not a validated scale). Adverse effects: vital signs (reports significant between‐group changes but does not give data). Global state: mean dose given (SD/SE/CI not reported). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised in blocks of 10. Method of sequence generation not clear. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Medication was provided in identically appearing 2cc. ampules each containing 4 mg's. Prior to the administration of the drug labels were removed by a person not involved either in the prescription or administration of the drug." (p.968). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Ratings were performed by the psychiatrist ordering the medication who was not aware which drug was administered to the patient. When asked to guess which drug was given, the psychiatrists administering the drug were unable to guess significantly better than chance." (p.968). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete outcome data. |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting. |
| Other bias | High risk | Small short study, sponsored by drug company. |
Taymeeyapradit 2002.
| Methods | Allocation: randomised. Blindness: present for some outcomes. Duration: 7 days. |
|
| Participants | Diagnoses: ICD‐10 diagnosis of schizophrenia with acute exacerbation of symptoms (N = 39), mania (N = 1), other (N = 6). N = 70. Age: range 18‐65 years. Sex: males and females. History: disturbed and aggressive behaviour, unresponsive to verbal intervention. Mean onset of acute episode, 20.09 days (haloperidol group), 13.63 days (zuclopenthixol group). Excluded: psychosis caused by medication, medical condition, learning disability, people who had anti‐psychotic in the previous 2 weeks. Setting: psychiatric hospital, Thailand. |
|
| Interventions | 1. Haloperidol IM: flexible dose 5 mg to 10 mg repeated every 6 hours as required (mean number of IMs 5). N = 32. 2. Zuclopenthixol acetate IM: flexible dose 50 mg to 100 mg repeated every 12 hours as required (mean number of IMs 2). N = 38. Oral antipsychotics and mood stabilisers allowed. |
|
| Outcomes | Global state: additional injections. Adverse effects: presence of tremor, reaction at injection site. Unable to use ‐ Mental state: BPRS, BRMS (means, inadequate data on SD). Global state: CGI (means, no SD). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The BPRS was completed by a psychiatric nurse who was blind to the intervention. However, it is not reported whether the rater for CGI and adverse effects was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete outcome data. |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting. |
| Other bias | Unclear risk | None obvious. |
Tuason 1986.
| Methods | Allocation: randomised. Blinding: “modified double” ‐ staff administering drugs not blinded, assessments blinded. Duration: 24‐72 hours (IM phase) ‐ oral phase data not included. |
|
| Participants | Diagnosis: schizophrenia, acutely psychotic (DSM‐III used beyond 3 days). N = 54*. Age: range 18‐65, mean 35 (SD 10) years. Sex: 33 males, 19 females, 2 not reported. History: newly admitted. Excluded: ill health, co‐existing mental illness condition. Setting: psychiatric hospital, USA. |
|
| Interventions | 1. Haloperidol: dose 5 mg/IM, then flexible dose 2.5 mg/hour/IM to 5 mg/hour/IM, maximum 100 mg/day (mean daily dose reported only for day one). N = 29. 2. Loxapine: dose 25 mg/IM, then flexible dose 12.5 mg/hour/IM to 25 mg/hour/IM, maximum 250 mg/day (mean daily dose reported only for day one). N = 25. Antiparkinsonian drugs and chloral hydrate as required. |
|
| Outcomes | Mental state: BPRS.* Global state: CGI‐I, needing an extended period of medication > 24 hours. Leaving the study early. Adverse effects: sedation ‐ ESBE. Unable to use: Global state: CGI‐S (no usable data). Adverse effects: amount of antiparkinsonian drugs/sleeping tablets administered not reported. |
|
| Notes | *BPRS scores reported with SD. Data thought not to be SD but SE and review authors have converted to SD. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Staff administering the medication were not blind to the intervention. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Rater‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Participants who were discharged/transferred were not followed up. The number of participants reported as leaving the study is contradictory as follows; "On Day 3, 19% of the haloperidol patients were withdrawn from the study while all loxapine patients remained" "the others (6 loxapine and 11 haloperidol) were dropped on Day 2, when all were still rated severely or very severely ill." (p.127). By day 10, over 50% attrition in both groups. |
| Selective reporting (reporting bias) | High risk | The amount of anticholinergic and sleeping medication administered was not reported. Table 57 and 3 present data from 48 hours after the 1st injection, rather than from 24 hours, despite referring to two of the outcomes in the text; "Twenty‐four hours after the first I.M. dose, 72% (18 of 25) of the loxapine patients and 59% (12 of 27) of the haloperidol patients showed moderate or marked improvement" (p.128). |
| Other bias | Unclear risk | None obvious. |
Walther 2014.
| Methods | Allocation: randomised. Blinding: not stated. Duration: 96 hours. |
|
| Participants | Diagnosis: DSM‐IV criteria for schizophrenia (N = 27), schizoaffective (N = 6), or schizophreniform disorder (N = 10); PANSS‐PES ≥20. N = 43*. Age: range 18‐55. Sex: 31 males, 12 females. History: admitted to acute care inpatient unit. Excluded: relevant medical or neurological disorders, pregnancy, ongoing intake of illicit drugs and alcohol. Setting: psychiatric hospital of Bern, Switzerland. |
|
| Interventions | 1. Haloperidol: fixed dose 10 mg/oral for the first day**, then fixed dose 7.5 mg/twice daily/oral***. N = 14. 2. Risperidone: fixed dose 2 mg/oral for the first day**, fixed dose 2 mg/twice daily/oral***, then fixed dose 3 mg/twice daily/oral***. N = 14. 3. Olanzapine: fixed dose 15 mg/oral for the first day**, then fixed dose 20 mg/oral***. N = 15. |
|
| Outcomes | Specific behaviour: agitation ‐ PANSS‐PAS. Adverse effects: general ‐ death. Adverse effects: movement ‐ AIMS. Adverse effects: movement ‐ BARS. Adverse effects: movement ‐ SAS. Leaving the study early. Unable to use: Mental state: PANSS (mean scores and SD/SE/CI not provided). Adverse effects: amount of patients that needed antiparkinsonian drugs/benzodiazepine not reported. |
|
| Notes | *Eligible patients were 52 participants but 9 refused to provide post‐hoc consent. **Additional use of diazepam up to 30 mg was permitted in day 1. ***Additional use of diazepam up to 60 mg was permitted from day 2 to day 5. Authors were contacted (2nd July 2016, 9th July 2016). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization was performed by creating 3 sets of random numbers between 1 and 60 using a computer‐based research randomizer" (p.125). |
| Allocation concealment (selection bias) | Low risk | "The randomization list was locked in the office of the principal investigator (T.J.M.), who was engaged neither in treatment nor in study assessments" (p.125). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 9 participants were excluded post‐randomisation due to refusal to provide post‐hoc consent; their treatment assignment was not provided. |
| Selective reporting (reporting bias) | Unclear risk | PANSS‐PAS scores at 24, 48, 72, and 96 hours not provided. PANSS scores at 96 hours not provided. Number of patients that needed additional BDZ not provided. Number of patients that needed additional biperiden not provided. |
| Other bias | Unclear risk | Small study. |
Yin 2012.
| Methods | Allocation: randomised. Blinding: researcher‐blinded. Duration: 72 hours. |
|
| Participants | Diagnosis: ICD‐10 diagnosis of schizophrenia. N = 60 Age: range 20‐54 years. Sex: 33 males, 27 females. History: acute agitation; length of Illness: 1‐ 23 years. Excluded: "taking ziprasidone 3 months before recruitment; taking other schizophrenia medications 12 hours before recruitment; taking long‐term schizophrenia medication 2 weeks before recruitment; additive to drugs 3 months before recruitment; ECG irregular (QTc > 500msec)". Setting: inpatients, China. |
|
| Interventions | 1. Haloperidol: starting dose 5 mg/IM/day; maximum dose 20 mg. N = 30. 2. Ziprasidone: starting dose 10 mg/IM/day; maximum dose 40 mg. N = 30. |
|
| Outcomes | Mental state: BPRS total. Adverse effects. Adverse effects: movement ‐ SAS. |
|
| Notes | *It is not clear if this paper presents the results from one centre of the Fang 2012 multi‐centre trial. Attempted to contact author to clarify. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised ‐ method of randomisation is not reported. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Researcher blinded to assignment. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No evidence of incomplete outcome data. |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting. |
| Other bias | Low risk | None obvious. |
Zhou 2014.
| Methods | Allocation: randomised. Blinding: not stated. Duration: 4 weeks*. |
|
| Participants | Diagnosis: CCMD‐3 diagnosis of schizophrenia. N = 76. Age: range 16‐60 years. Sex: 39 males, 37 females. History: "excited/agitated symptoms of acute‐stage schizophrenia"; "length of Illness 1‐32 months". Excluded: "serious physical sickness, additive to alcohol and drugs, pregnant, history of taking long‐term schizophrenia medicine, suicide tendency and allergic to treatment". Setting: inpatients, China. |
|
| Interventions | 1. Haloperidol: flexible dose 5 mg/IM/day to 30 mg/IM/day (mean dose 16.4 ± 6.1 mg/day). N = 38. 2. Ziprasidone: flexible dose 40 mg/oral/day to 160 mg/oral/day (mean dose 90.6 ± 20.8 mg/day) + clonazepam flexible dose 2 mg/oral/day to 6 mg/oral/day. N = 38. |
|
| Outcomes | Mental state: PANSS total score, positive sub‐scale, negative sub‐scale, general psychopathology sub‐scale. Agitation: PANSS‐EC. Global outcome: CGI‐S. Leaving the study early. |
|
| Notes | *Data were extracted only for the first 7 days because at the 8th day research group stopped clonazepam and switched from haloperidol to ziprasidone. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomised by using random number table". |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not stated. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | A total of 3 dropped out in haloperidol group (serious adverse events = 2, not following protocol = 1) and 2 dropped out in ziprasidone + clonazepam (lost to contact = 1, not following protocol = 1); missing data have not been imputed using appropriate methods such as LOCF. |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting. |
| Other bias | Low risk | None obvious. |
ABS: Agitated Behaviour Scale ACES: Agitation‐Calmness Evaluation Scale AIMS: Abnormal Involuntary Movement Scale BARS: Behavioural Activity Rating Scale BAS: Barnes Akathisia Scale BDZ: benzodiazepine BPRS: Brief Psychiatric Rating Scale BRMS: CABS: Corrigan Agitated Behavior Scale CCMD: Chinese Classification of Mental Disorders CGI: Clinical Global Impression CGI‐I: Clinical Global Impression ‐ Improvement CGI‐S: Clinical Global Impression ‐ Severity CI: confidence interval COSTART: Coding Symbols and Thesaurus for Adverse Reaction Terms DAI‐10: Drug Attitude Inventory DSM: Diagnostic and Statistical Manual ECG: electrocardiogram EPS: Extrapyramidal Side Effects ESBE: GAS: Global Adjustment Scale ICD: International Classification of Diseases IM: intramuscular LOCF: last observation carried forward LS: MBPRS: Modified Brief Psychiatric Rating Scale NOSIE: Nurses Observation Scale for Inpatient Evaluation OAS: Overt Aggression Scale OASS: Overt Agitation Severity Scale PANSS: Positive and Negative Syndrome Scale PANSS‐EC: Positive and Negative Syndrome Scale Excited Component PANSS‐PAS: Positive and Negative Syndrome Scale Psychotic Agitation Component PANSS‐PES: p.o.: orally RSS: Ramsay Sedation Scale SAS: Simpson‐Angus Scale SD: standard deviation SE: standard error SGPT: serum glutamic pyruvic transaminase TCS: TESS: Treatment Emergent Symptom Scale TSRS: Target Rating Symptom Scale VAS: Visual analogue scale YMRS: Young Mania Rating Scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Addington 1996 | Allocation: randomised. Participants: people with acute schizophrenia, but not specifically overtly aggressive. |
| Alexander 2004 | Allocation: randomised. Participants: acute agitation and dangerous behaviour. Intervention: haloperidol + promethazine vs olanzapine, no haloperidol alone group. |
| Allen 2007 | Allocation: not randomised, review. |
| Asadollahi 2015 | Allocation: randomised. Participants: psychotic disorders (N = 52), mood disorders (N = 23), cognitive impairment (N = 23), adjustment disorders (N = 17), others (infection, substance intoxication or withdrawal) (N = 35), and unknown aetiology (N = 10). Therefore, eligible patients are < 50% of the sample. An attempt to contact the authors for partial outcomes was made (3rd July 2016). |
| Beasley 1998 | Allocation: randomised. Participants: people with first episode psychosis, but not specifically aggressive. |
| Bieniek 1998 | Allocation: randomised. Participants: newly admitted patients to a psychiatric emergency service who were uncooperative, threatening, and unresponsive to verbal interventions. Intervention: haloperidol + lorazepam vs lorazepam, no haloperidol alone group. |
| Brook 1998 | Allocation: randomised. Participants: people with acute psychosis, but not specifically aggressive. |
| Campbell 1982 | Allocation: randomised. Participants: children with aggressive behaviour, but the number of those with suspected psychosis < 50% of the sample. |
| Chen 2004 | Allocation: quasi‐randomised. |
| Chen 2008 | Allocation: quasi‐randomised. |
| Chouinard 1991 | Allocation: randomised. Participants: people with acute psychosis, but not specifically aggressive. |
| Citrome 2001 | Allocation: randomised. Participants: people with schizophrenia and persistent aggressive behaviour, not acute exacerbation of symptoms. |
| Colonna 1998 | Allocation: randomised. Participants: people with acute schizophrenia, but not specifically aggressive. |
| Conde 2011 | Allocation: randomised. Participants: admitted patients to an emergency setting experiencing agitation associated with active psychosis; inpatients experiencing exacerbation or agitation with active psychosis. Intervention: haloperidol + clonazepam vs risperidone + clonazepam, no haloperidol alone group. |
| Crandall 2005 | Allocation: randomised. Participants: people with chronic schizophrenia, acute exacerbation of symptoms, not specifically aggressive. |
| Currier 2000 | Allocation: not randomised. |
| Daniel 2000 | Allocation: not randomised, review. |
| Daniel 2003 | Allocation: randomised. Participants: people with acute schizophrenia, not acutely agitated or aggressive. |
| Daniel 2004 | Allocation: randomised. Participants: people with psychosis, not acutely agitated or aggressive. |
| Fan 2012 | Quasi‐randomised controlled trial: "the participants were assigned by the order of admission". |
| Faretra 1970 | Allocation: randomised. Participants: children with suspected schizophrenia, not specifically aggressive. |
| Freeman 2009 | Allocation: not randomised, retrospective case note study. |
| Goldstein 1966 | Allocation: randomised. Participants: people with acute psychosis, not aggressive. |
| Harvey 2004 | Allocation: randomised. Participants: people with mild symptoms of schizophrenia, not aggressive. |
| Huang 2004 | Allocation: quasi‐randomised. |
| Jiang 2009 | Quasi‐randomised controlled trial: "the participants were assigned by the order of admission". |
| Jones 2001 | Allocation: not randomised, review. |
| Kane 2000 | Allocation: randomised. Participants: people with acute schizophrenia, not specifically aggressive. |
| Kinon 2001a | Allocation: randomised. Participants: people with schizophrenia, but not specifically overtly aggressive. |
| Kinon 2003 | Allocation: not randomised. |
| Krakowski 2006 | Allocation: randomised. Participants: people with schizophrenia and persistent aggressive behaviour, not an emergency situation. |
| Krakowski 2008 | Allocation: randomised. Participants: people with schizophrenia and persistent aggressive behaviour, not an emergency situation. |
| Lewis 2006 | Allocation: randomised. Participants: people with schizophrenia and persistent aggressive behaviour, not an emergency situation. |
| Li 2005 | Allocation: randomised. Participants: people with acute excitement in schizophrenia. Intervention: haloperidol vs tiapride. Outcome: there was no specific outcome for acute agitation (T1 at 7 days). |
| Li 2007 | Allocation: quasi‐randomised. |
| Liang 2003 | Quasi‐randomised controlled trials: "the participants were randomly assigned by the order of admission". |
| Liu 2012b | Allocation: randomised. Participants: people with schizophrenia and acute agitation. Intervention: haloperidol vs modified electroconvulsive therapy (MECT), no comparison drug. |
| Mandel 2008 | Allocation: randomised. Participants: people with schizophrenia, not specifically aggressive. |
| Mantua 2006 | Allocation: not randomised, naturalistic observational study. |
| McEvoy 1994 | Allocation: randomised. Participants: people with schizophrenia, not specifically aggressive. |
| Mendelowitz 2004 | Allocation: not randomised, review. |
| NCT00189995 2005 | Allocation: was to be randomised, but trial not conducted because ethics approval not granted. |
| NCT00631722 2008 | Allocation: randomised. Participants: people with acute schizophrenia, not specifically aggressive. |
| NCT00797277 2008 | Allocation: randomised. Participants: people with schizophrenia and agitation. Intervention: haloperidol plus lorazepam vs olanzapine ‐ no haloperidol alone group. |
| Pan 2005 | Allocation: quasi‐randomised. |
| Pathiraja 1995 | Allocation: randomised. Participants: people with schizophrenia, not specifically aggressive. |
| Pedros 2004 | Allocation: not randomised ‐ cohort study. |
| Pei 2009 | Quasi‐randomised controlled trial: "the participants were assigned by the order of admission". |
| Puech 1998 | Allocation: randomised. Participants: people with acute schizophrenia, not specifically aggressive. |
| Romeo 2009 | Allocation: randomised. Participants: people with intellectual disability and challenging behaviour, not psychosis‐induced aggression. |
| Simpson 2010 | Allocation: randomised. Participants: people with agitation in the emergency setting. Intervention: haloperidol plus benztropine mesylate vs lorazepam vs quetiapine ‐ no haloperidol alone group. |
| Singh 1981 | Allocation: not reported, described as double‐blind. Participants: people with schizophrenia, not acute exacerbation of symptoms. |
| Smith 1974 | Allocation: randomised. Participants: people with senile psychosis, not specifically aggressive. |
| Srinath 2010 | Allocation: randomised. Participants: "patients who presented with acute psychotic agitation". Intervention: haloperidol + promethazine vs lorazepam, no haloperidol alone group. |
| Teja 1975 | Allocation: randomised. Participants: people with chronic schizophrenia, not specifically aggressive. |
| Thomas 1992 | Allocation: randomised. Participants: violent and agitated people, mostly intoxicated, not necessarily psychosis‐induced aggression. |
| TREC CG | Allocation: randomised. Participants: acute agitation and dangerous behaviour. Intervention: haloperidol + promethazine vs midazolam (no haloperidol alone group). |
| Ungvari 1982 | Allocation: randomised. Participants: people with acute schizophrenia, not specifically aggressive. |
| Vaisanen 1981 | Allocation: randomised. Participants: people with intellectual disability and agitation, not psychosis‐induced aggression. |
| Veser 2006 | Allocation: randomised. Participants: people with psychosis‐induced agitation, not specifically aggressive (exclusion criteria included an inability to give informed consent). |
| Vives 2015 | Allocation: randomised. Participants: agitated schizophrenic patients in the emergency setting. Intervention: haloperidol intranasal vs haloperidol intramuscular, no other drugs except haloperidol. |
| Wan 2005 | Allocation: randomised. Participants: people with schizophrenia and agitation/aggression. Intervention: haloperidol vs chlorpromazine vs risperidone + clonazepam. Outcome: excitement and agitation scale (not validated), also the data for the haloperidol and chlorpromazine groups are combined. |
| Wang 2004 | Allocation: quasi‐randomised. |
| Wang 2005 | Allocation: quasi‐randomised. |
| Wyant 1990 | Allocation: randomised. Participants: people with schizophrenia and acute agitation. Intervention: haloperidol vs sodium amytal vs midazolam. Outcome: modified CGI ‐ no usable data, presented in graphs, no binary data. |
| Zapletalek 1986 | Allocation: not reported, described as double‐blind. Participants: people with schizophrenia, not specifically aggressive. |
CGI: Clinical Global Impression vs: versus
Characteristics of studies awaiting assessment [ordered by study ID]
Daniel 2006.
| Methods | Allocation: randomised. Blindness: not stated. Duration: 24‐hour IM phase followed by 4‐day oral phase. |
| Participants | Diagnosis: schizophrenia with agitation. N = 360. Age: range 18‐60 years. Sex: not stated. History: not stated. Exclusion: not stated. Setting: not stated. |
| Interventions | 1. Haloperidol: dose 6.5 mg/IM, maximum 3 injections during 24 hours, followed by haloperidol 7 mg/p.o./day to 10 mg/p.o./day for 4 days. 2. Aripiprazole: dose 10 mg/IM, maximum 3 injections during 24 hours, followed by haloperidol 10 mg/p.o./day to 15 mg/p.o./day for 4 days. |
| Outcomes | Global state: CGI‐I, CGI‐S. Agitation: PANSS‐EC, ACES, CABS. |
| Notes | Conference proceeding, full characteristics and outcome data not reported. Additionally, unclear whether this is the same study as Bristol‐Myers 2005b ‐ attempted to contact Bristol Myers. |
Davis 2008.
| Methods | Allocation: randomised. Blindness: not stated. Duration: 24 hours. |
| Participants | Diagnosis: schizophrenia. N = 62. Age: not stated. Sex: not stated. History: "agitated schizophrenic patient admitted to hospital." Exclusion: not stated. Setting: not stated. |
| Interventions | 1. Haloperidol: dose 5 mg/IM/day to 15 mg/IM/day. N = 21. 2. Haloperidol: dose 5 mg/IM/day to 15 mg/IM/day + clonazepam: dose 2 mg/IM/day to 6 mg/IM/day. N = 21. 3. Clonazepam: dose 2 mg/IM/day to 6 mg/IM/day. N = 20. |
| Outcomes | Mental state: BPRS Total, BPRS Psychosis sub‐scale. Global state: CGI. Agitation: BPRS agitation sub‐scale. Adverse effects. |
| Notes | Conference proceeding, full characteristics and outcome data not reported. Attempted to contact author. |
Dubin 1985.
| Methods | Allocation: randomised. Blindness: single. Duration: not stated. |
| Participants | Diagnosis: schizophrenia (N = 77), mania (N = 37), psychosis aetiology unknown (N = 25), amphetamine intoxication (N = 11), alcohol intoxication (N = 5), brief reactive psychosis (N = 3), brain organic syndrome (N = 1). N = 159. Age: range 18‐65 years. Sex: 77 males and 82 females. History: newly admitted ‐ previous history not stated. Exclusion: pregnancy, central nervous system depression, history of urinary retention or glaucoma, known sensitivity to phenothiazines, conditions such as epilepsy, severe hypotension or hypertension, head injuries, and haematologic, cardiovascular, renal or hepatic impairments, and ingestion of antipsychotic, sedative‐hypnotic, or antianxiety drugs within 24 hours of admission. Setting: psychiatric emergency hospital, USA. |
| Interventions | 1. Haloperidol: dose 5 mg/oral concentrate (mean number of doses 3.8). N = 15. 2. Haloperidol: dose 5 mg/IM (mean number of IMs not reported 3.2). N = 14. 3. Haloperidol: dose 10 mg/oral concentrate (mean number of doses 3.5). N = 14. 4. Haloperidol: dose 10 mg/IM (mean number of IMs 2.2). N = 17. 5. Thiothixene: dose 10 mg/IM (mean number of IMs 3.1). N = 24. 6. Thiothixene: dose 10 mg/oral concentrate (mean number of doses 4.7). N = 14. 7. Thiothixene: dose 20 mg/oral concentrate (mean number of doses 3.5). N = 14. 8. Thioridazine: dose 25 mg/oral suspension (mean number of doses 3.8). N = 16. 9. Thioridazine: dose 50 mg/oral suspension (mean number of doses 3.8). N = 13. 10. Thioridazine: dose 100 mg/oral suspension (mean number of doses 3.2). N = 18. |
| Outcomes | Global state: additional injections. Mental state: BPRS (mean/SD/SE/CI not reported). Adverse effects: % of whole sample experiencing side effects reported.* |
| Notes | *Attempted to contact author to see if there are any usable data available. |
Esmailian 2015.
| Methods | Allocation: randomised. Blindness: double. Duration: not stated. |
| Participants | Diagnosis: patients who referred to the emergency department because of medical diseases, drug poisoning, or trauma, and need the sedative agent to sedation. N = 48. Age: 18‐65 years. Sex: 36 males and 12 females. History: internal (N = 36)*, drug poisoning (N = 2), trauma (N = 10). Exclusion: sensitivity to haloperidol or midazolam, contraindications to these drugs, sympathomimetic agents, agitation because of reversible factors (such as hypotension, hypoxia, and hypoglycaemia), tachycardia or bradycardia, respiratory distress, pregnancy, symptoms of withdrawal syndrome, and receiving sedative agents within the past 12 hours. Setting: emergency department, Iran. |
| Interventions | 1. Haloperidol: fixed dose 5 mg/IM. 2. Midazolam: flexible doses 2.5 mg/IM to 5 mg/IM according to weight. |
| Outcomes | General: time to sedation, time to full consciousness, need for additional doses, need for re‐sedation within 60 minutes. Adverse effects. |
| Notes | *not clear which diagnosis includes; an attempt to contact the authors was made (3rd July 2016). |
Herrera 2005.
| Methods | Allocation: randomised. Blindness: double. Duration: not stated. |
| Participants | Diagnosis: psychosis with agitation and/or violence. N = 20. Age: not stated. Sex: not stated. History: not stated. Exclusion: not stated. Setting: psychiatric emergency department. |
| Interventions | 1. Haloperidol: dose 10 mg/IM. 2. Risperidone: dose 2 mg/liquid. |
| Outcomes | Agitation: PANSS‐EC. |
| Notes | Conference proceeding, full characteristics and outcome data not reported. Attempted to contact author. |
Hsu 2010.
| Methods | Allocation: randomised. Blindness: single. Duration: 24 hours. |
| Participants | Diagnosis: DSM‐IV diagnosis of schizophrenia (N = 20), bipolar disorder (N = 18), schizoaffective disorder (N = 1), delusional disorder or others (N = 3). N = 42. Age: range 18‐65 years. Sex: 20 males and 22 females. History: within 24 hours of admission ‐ previous psychiatric history not stated. Exclusion: "pregnant or lactating women; patients with serious medical illnesses; patients with closed‐angle glaucoma; patients with an allergic reaction to olanzapine, risperidone, or haloperidol; or patients who had received a long‐acting antipsychotic agent injection within 30 days were excluded". Setting: acute medical centre, Taiwan. |
| Interventions | 1. Haloperidol: dose 7.5 mg/IM. N = 11. 2. Risperidone: dose 3 mg/liquid. N = 10. 3. Olanzapine: dose 10 mg/IM. N = 11. 4. Olanzapine: dose 10 mg/velotab. N = 10. |
| Outcomes | Agitation: PANSS‐EC.* |
| Notes | *Attempted to contact author to see if there is any usable data available. |
Istikoglou 2010.
| Methods | Allocation: randomised. Blindness: rater‐blinded. Duration: not stated. |
| Participants | Diagnosis: psychosis with agitation. N = 120. Age: not stated. Sex: males and females. History: not stated. Exclusion: not stated. Setting: psychiatric emergency room, Greece. |
| Interventions | 1. Haloperidol IM (dose not reported). 2. Aripiprazole IM (dose not reported). |
| Outcomes | Mental state: PANSS, YMRS. Global state: CGI‐I. Agitation: AIMS. Adverse effects: SAS. Quality of life: QoL |
| Notes | Conference proceeding, full characteristics and outcome data not reported. Unable to establish contact details at this time. |
Kong 2009.
| Methods | Allocation: randomised. Blindness: open‐label. Duration: 2 hours. |
| Participants | Diagnosis: acutely agitated patients. N = 37. Age: 18 years or older. Sex: not stated. History: "DSM‐IV diagnosis of schizophrenia, schizophreniform disorder, brief psychotic disorder, or schizoaffective disorder, bipolar dosprder" (p.S525). Exclusion: not stated. Setting: psychiatry ward, Inje University, China. |
| Interventions | 1. Haloperidol: fixed dose 5 mg/IM. N = 10. 2. Olanzapine: fixed dose 10 mg/IM. N = 12. 3. Haloperidol: fixed dose 5 mg/IM + lorazepam fixed dose 4 mg/IM. N = 15. |
| Outcomes | Global State: CGI‐S, CGI‐I. Agitation: PANSS‐EC. Adverse effects. Seclusions. |
| Notes | Abstract, full characteristics and outcome data not reported. Additionally, it is probably the same study as Shim 2009 (discordant doses and reported adverse effects). An attempt to contact the authors was made (3rd July 2016). |
Lasic 2006.
| Methods | Allocation: randomised. Blindness: not stated. Duration: up to 3 months. |
| Participants | Diagnosis: ICD X diagnosis of acute exacerbation of schizophrenia or schizoaffective disorder, mania with psychotic features, acute paranoid reaction, or delusional disorders. N = 60. Age: > 18. Sex: males and females. History: not stated. Exclusion: "delirium, epilepsy, or mental retardation; intoxication or symptoms of withdrawal from alcohol or other psychoactive substances; clinical laboratory values indicating serious medical illness; treatment with any antipsychotic or benzodiazepine within 6 hours of screening; a history of neuroleptic malignant syndrome or known hypersensitivity to any of the trial medications; treatment with a depot antipsychotic within 1 treatment cycle of screening and use of disallowed medications". Setting: acute psychiatric inpatient ward, Croatia. |
| Interventions | 1. Haloperidol IM (dose not reported). 2. Risperidol liquid (dose not reported). |
| Outcomes | Mental state: PANSS. Global state: CGI‐I. Agitation: BARS, PANSS agitation cluster. Adverse effects |
| Notes | Conference proceeding, full characteristics and outcome data not reported. Attempted to contact author. |
Li 2007a.
| Methods | Allocation: not clearly stated; "each unit of patients were randomly assigned to one of three treatment groups by random number and hospital serial number". Blindness: double‐blind. Duration: 24 hours. |
| Participants | Diagnosis: CCMD‐3 diagnosis of schizophrenia. N = 45. Age: range 18‐45 years. Sex: males (20) and females (25). History: acute agitation. Exclusion: serious physical illness; additive to alcohol, drugs; taking schizophrenia medication one month before recruitment. Setting: psychiatric inpatient ward, China. |
| Interventions | 1. Haloperidol: dose 5 mg/IM/day to 15 mg/IM/day. N = 15. 2. Haloperidol: dose 5 mg/IM/day to 15 mg/IM/day + clonazepam dose 2 mg/IM/day to 6 mg/IM/day. N = 15. 3. Clonazepam: dose 2 mg/IM/day to 6 mg/IM/day. N = 15. |
| Outcomes | Mental state: BPRS. Adverse effects. Unable to use: CGI: data not available. |
| Notes | Allocation methodology not clearly stated. |
Lu 2006.
| Methods | Allocation: randomised. Blindness: rater‐blinded. Duration: 72 hours. |
| Participants | Diagnosis: CCMD‐3 diagnosis of schizophrenia. N = 60. Age: mean 33.5 (SD 12.3) years. Sex: male and female. History: acute agitation. Excluded: anyone who used anti‐psychotic or anti‐convulsant within one week prior to the trial or anyone who used long lasting drug within one month prior to the trial. Setting: inpatients, China*. |
| Interventions | 1. Haloperidol: dose 5 mg/IM, to 10 mg/IM, maximum dose 30 mg during 24 hours. N = 30. 2. Ziprasidone: dose 10 mg/IM, to 20 mg/IM, maximum dose 40 mg during 24 hours. N = 30. |
| Outcomes | Global state: CGI‐IS. Adverse effects: TESS. Agitation: PANSS‐EC. Unable to use: Adverse effects: the amount of anticholinergic drugs used was not reported. |
| Notes | *It is not clear if this paper presents the results from one centre of the Li 2006 multi‐centre trial. Attempted to contact author to clarify. |
NCT00859872 2009.
| Methods | Allocation: randomised. Blindness: single. Duration: up to 47 days. |
| Participants | Diagnosis: DSM‐IV diagnosis of acute exacerbation of schizophrenia or schizoaffective disorder with agitation. N = not stated. Age: range > 18 < 45 years. Sex: males and females. History: not stated. Exclusion: pregnant or lactating women, serious medical illness, known sensitivity to study medication, treatment with a depot antipsychotic with 1 cycle of screening, use of disallowed medication, psychosis caused by "delirium, epilepsy, mental retardation and affective disorder; intoxication or symptoms of withdrawal from alcohol of other psychoactive substances." Setting: psychiatric inpatient ward, China. |
| Interventions | 1. Haloperidol: dose 5 mg/IM/day to 20 mg/IM/day. 2. Risperidone: dose 2 mg/oral/day to 6 mg/oral/day + clonazepam: dose 4 mg/oral/day to 8 mg/oral/day. |
| Outcomes | Mental state: PANSS. Agitation: PANSS‐EC. |
| Notes | *Protocol, full characteristics and outcome data not reported. Unable to establish contact details at this time. |
NCT00866645 2009.
| Methods | Allocation: randomised. Blindness: single. Duration: 72 hours. |
| Participants | Diagnosis: DSM‐IV diagnosis of schizophrenia or schizophreniform psychosis. N = not stated. Age: range > 18 < 65 years. Sex: males and females. History: not stated. Exclusion: investigator and his/her relatives, participation in another drug trial within 3 months prior to enrolment into this study, pregnant or lactating women, serious medical illness, a significantly abnormal value in ECG or lab results, family history of sudden death, meet the DSM‐IV criteria for substance abuse within 1 year prior to enrolment, regular use of antipsychotics (clozapine within 90 days), antidepressants, mood stabilisers, anti‐epileptics or prolonged‐action preparations within 2 weeks prior to enrolment, use of ECT within 30 days prior to enrolment, systematic use of sulpiride, levosulpiride, or haloperidol therapy within 30 days prior enrolment, history of neuroleptic malignant syndrome, severe EPS or significant tardive dyskinesia, severe suicide attempt, know hypersensitivity to sulpiride, levosulpiride or haloperidol, history of hypersensitivity to more than 2 drugs, use of psychotropics (except for permitted drugs) within 12 hours prior to enrolment, known lack of efficacy to levosulpiride or haloperidol by formal treatment before, organic mental disorders including learning disability, history of psychosurgery treatment, people who could not comply with the study protocol. Setting: psychiatric inpatient ward, China. |
| Interventions | 1. Haloperidol: dose 5 mg/IM, ≥ 4 hours apart, maximum 10 mg during 24 hours. 2. Levosulpiride: dose 50 mg/IM, ≥ 4 hours apart, maximum 100 mg during 24 hours. |
| Outcomes | Mental state: BPRS, PANSS Total. Global State: CGI, CGI‐I, CGI‐S. Agitation: ACES, PANSS‐EC. Adverse effects: BAS, ECG, laboratory tests, physical examination, RSESE. |
| Notes | Protocol, full characteristics and outcome data not reported. Attempted to contact author to enquire whether there is any available data. |
Shim 2009.
| Methods | Allocation: randomised. Blindness: open‐label. Duration: 2 hours. |
| Participants | Diagnosis: acutely agitated patients. N = 37. Age: not stated. Sex: not stated. History: "hospitalized patients with acute psychotic symptom in association with schizophrenia, bipolar I disorder, schizoaffective disorder, schizophreniform disorder, brief psychotic disorder, delusional disorder" (p.S512). Exclusion: not stated. Setting: psychiatry ward, Inje University, China. |
| Interventions | 1. Haloperidol: fixed dose 4 mg/IM. N = 10. 2. Olanzapine: fixed dose 10mg/IM. N = 12. 3. Haloperidol: fixed dose 4 mg/IM + lorazepam fixed dose 5 mg/IM. N = 15. |
| Outcomes | Global State: CGI‐S. Agitation: PANSS‐EC, BARS. Adverse effects. |
| Notes | Abstract, full characteristics and outcome data not reported. Additionally, it is probably the same study as Kong 2009 (discordant doses and reported adverse effects). An attempt to contact the authors was made (3rd July 2016). |
Si 2009.
| Methods | Allocation: randomised. Blindness: open‐label. Duration: 28 days. |
| Participants | Diagnosis: acute exacerbation of schizophrenia with agitation and hostility. N = 80. Age: not stated. Sex: not stated. History: not stated. Exclusion: not stated. Setting: psychiatric inpatient ward, China. |
| Interventions | 1. Haloperidol: dose 10 mg/oral/day to 12 mg/oral/day. N = 40. 2. Quetiapine: dose 600 mg/oral/day to 750 mg/oral/day. N = 40. |
| Outcomes | Mental state: BPRS, PANSS Total. Global State: CGI, CGI‐I, CGI‐S. Agitation: ACES, PANSS‐EC. Adverse effects: BAS, ECG, laboratory tests, physical examination, RSESE. |
| Notes | Conference proceeding, full characteristics and outcome data not reported. Attempted to contact author. |
Slotnick 1971.
| Methods | Allocation: randomised. Blindness: double‐blind. Duration: 48 hours. |
| Participants | Diagnosis: "acutely agitated psychiatric patients". N = 150 (three trials combined). Age: not stated. Sex: not stated. History: not stated. Exclusion: not stated. Setting: not stated. |
| Interventions | 1. Haloperidol IM (dose not reported). N = 75 (three trials combined). 2. Chorpromazine IM (dose not reported). N = 75 (three trials combined). |
| Outcomes | Mental state: BPRS. |
| Notes | Conference proceeding, full characteristics and outcome data for the three trials not reported. Unable to establish contact details at this time. |
Smythies 1982.
| Methods | Allocation: not reported. Blindness: double. Duration: 10 days. |
| Participants | Diagnosis: DSM‐III diagnosis of schizophrenia with acute agitation. N = 16. Age: not stated. Sex: not stated. History: not stated. Exclusion: at risk of pregnancy, significant medical disorders, history of EPS or hypotensive crisis upon administration of other injectable antipsychotic agents, significantly abnormal laboratory test results, recent alcoholism or drug dependency. Setting: inpatient, USA. |
| Interventions | 1. Haloperidol: dose 2.5 mg/IM to 80 mg/IM, for 2 to 10 days. 2. Molindone: dose 12.5 mg/IM to 150 mg/IM, for 4‐7 days. |
| Outcomes | Mental state: BPRS. Global state: CGI. Agitation: TSRS*. |
| Notes | *Attempted to contact author to see if there is any available usable data available. |
Vasquez‐Gomez 2001.
| Methods | Allocation: randomised. Blindness: simple. Duration: not stated. |
| Participants | Diagnosis: DSM‐IV diagnosis of acute psychosis ‐ "schizophrenic or affective". N = 30. Age: range 18‐83 years. Sex: not stated. History: not stated. Exclusion: not stated. Setting: emergency room, Peru. |
| Interventions | 1. Haloperidol: dose 5 mg/IM. N = 15. 2. Midazolam: dose 15 mg/IM. N = 15. |
| Outcomes | Aggression: OAS. |
| Notes | Conference proceeding, full characteristics and outcome data for the three trials not reported. Unable to establish contact details at this time. |
Wang 2005a.
| Methods | Allocation: randomised. Blinding: single‐blind. Duration: 72 hours. |
| Participants | Diagnosis: CCMD‐3 diagnosis of schizophrenia. N = 60. Age: mean 27.40 ± 9.64 years (ziprasidone group), mean 27.40 ± 9.64 years (haloperidol group). Sex: 39 males and 21 females. History: total score of PANSS ≥ 60; total score of excitement component of PANSS ≥14 and at least one section ≥ 4. Exclusion: physical illness; additive to alcohol, drugs; pregnant or plan to be pregnant. Setting: inpatients*, China. |
| Interventions | 1. Haloperidol: dose 5 mg/IM to 10 mg/IM, maximum 30 mg/day. N = 30. 2. Ziprasidone: dose 10 mg/IM to 20 mg/IM, maximum 40 mg/day. N = 30. |
| Outcomes | Mental state: PANSS‐EC. Adverse effects: TESS. Adverse effects: movement ‐ SAS. |
| Notes | *It is not clear if this paper presents the results from one centre of the Li 2006 multi‐centre trial. Attempted to contact author to clarify. |
Wang 2007.
| Methods | Allocation: randomised. Blindness: single. Duration: 72 hours. |
| Participants | Diagnosis: CCND‐3 diagnosis of schizophrenia with agitation/aggression. N = 32. Age: mean 25.8 (SD 10.4) years (haloperidol group), 28.6 (SD 9.4) years (ziprasidone group). Sex: 16 males and 16 females. History: first episode (N = 27), relapse (N = 5). Excluded: direct blood relatives of staff in current trial, participation in other drug trials 30 days prior to enrolment, pregnant or lactating women, severe disease/unstable medical condition, clinically significant abnormal EEG or laboratory results, QTC interval ≥ 450, family history of sudden death, drug abuse in past year, regular user of antipsychotic, antidepressant, depot medication, ziprasidone or haloperidol in past 30 days, people with drug‐induced malignant syndrome, suicidal ideation, or known allergy to ziprasidone or haloperidol. Setting: inpatient, China*. |
| Interventions | 1. Haloperidol: flexible dose 5 mg/IM, to 10 mg/IM, repeated every 4‐6 hours if necessary, maximum dose 30 mg during 24 hours. N = 16. 2. Ziprasidone: flexible dose 10 mg/IM, to 20 mg/IM, repeated every 4‐6 hours if necessary, maximum dose 40 mg during 24 hours. N = 16. |
| Outcomes | Mental state: PANSS. Global State: CGI. Agitation: PANNS‐EC, ACES. Adverse effects: BAS. |
| Notes | *It is not clear if this paper presents the results from one centre of the Li 2006 multi‐centre trial. Attempted to contact author to clarify. |
Yildiz 2003.
| Methods | Allocation: not stated. Blindness: not stated. Duration: 2 hours. |
| Participants | Diagnosis: "acutely agitated psychiatric patients". N = not stated. Age: not stated. Sex: not stated. History: not stated. Excluded: not stated. Setting: acute psychiatry service. |
| Interventions | 1. Haloperidol: flexible dose 2 mg/IM to 5 mg/IM. N = not stated. 2. Risperidone: flexible dose 1 mg/oral to 2 mg/oral. N = not stated. Concomitant use of lorazepam was allowed. |
| Outcomes | Unable to use: Agitation: "four different agitation rating scales", not reported. |
| Notes | Abstract of poser, full characteristics and outcome data not reported. Unable to establish contact details at this time. |
Zhang 2012.
| Methods | Allocation: randomised. Blinding: not stated. Duration: 7 days. |
| Participants | Diagnosis: DSM‐IV diagnosis of schizophrenia. N = 60. Age: range 18‐65 years. Sex: 36 males, 24 females. History: acute agitation; length of Illness 124.7 ± 115.3 months (haloperidol group), 157.5 ± 113.0 months (risperidone+clonazepam group). Excluded: "allergy to compound utilised in the treatment, serious physical sickness, addiction to alcohol or drugs, history of taking long‐term schizophrenia medicine, suicide tendency, pregnancy". Setting: inpatients, China. |
| Interventions | 1. Haloperidol: dose 10 mg/IM/day to 20 mg/IM/day. N = 30. 2. Risperidone: dose 2 mg/oral/day to 6 mg/oral/day + clonazepam dose 2 mg/oral/day to 8 mg/oral/day. N = 30. |
| Outcomes | Mental state: PANSS total and positive sub‐scale. Agitation: PANSS‐EC. Adverse effects: TESS. |
| Notes |
叶萌, 2009.
| Methods | Allocation: randomised. Blindness: single. Duration: 72 hours. |
| Participants | Diagnosis: DSM‐IV diagnosis of schizophrenia. N = 60. Age: mean 28.37 ±1 0.20 years (haloperidol group), mean 27.40 ± 9.63 years (ziprasidone group). Sex: 39 males, 21 females. History: acute agitation. Excluded: "physical illness; addiction to alcohol or drugs; pregnancy or plan to be pregnant". Setting: inpatients, China*. |
| Interventions | 1. Haloperidol: dose 5 mg/IM to 10 mg/IM, maximum 30 mg/day. N = 30. 2. Ziprasidone: dose 10 mg/IM to 20 mg/IM, maximum 40 mg/day. N = 30. |
| Outcomes | Mental state: PANSS total. Agitation: PANSS‐EC. Adverse events. |
| Notes | *It is not clear if this paper presents the results from one centre of the Li 2006 multi‐centre trial. Authors contact unavailable. |
周德祥, 2012.
| Methods | Allocation: randomised. Blindness: not stated. Duration: 7 days. |
| Participants | Diagnosis: CCMD‐3 diagnosis of schizophrenia. N = 65. Age: range 18‐60 years. Sex: 36 males, 29 females. History: acute psychotic agitation; length of Illness of 1.1 to 22.3 months. Excluded: pregnancy, serious physical illness, addiction to alcohol or drugs. Setting: inpatients, China*. |
| Interventions | 1. Haloperidol: dose 10 mg/IM/day to 20 mg/IM/day. N = 32. 2. Risperidone: dose 4 mg/oral/day to 6 mg/oral/day + clonazepam dose 2 mg/oral/day to 4 mg/oral/day. N = 33. |
| Outcomes | Unable to use: Mental state: PANSS total (data not reported). Agitation: PANSS‐EC (data not reported). Adverse events (data not reported). |
| Notes | *It is not clear if this paper presents the results from one centre of the Fang 2012 multi‐centre trial. Authors contact unavailable. |
ACES: Agitation‐Calmness Evaluation Scale AIMS: Abnormal Involuntary Movement Scale BARS: Behavioural Activity Rating ScaleBAS: Barnes Akathisia Scale BPRS: Brief Psychiatric Rating Scale CABS: Corrigan Agitated Behavior Scale CCMD: Chinese Classification of Mental Disorders CGI: Clinical Global Impression CGI‐I: Clinical Global Impression – Improvement CGI‐S: Clinical Global Impression – Severity CI: confidence interval DSM: Diagnostic and Statistical Manual ECG: electrocardiogram ECT: electroconvulsive therapy EPS: Extrapyramidal Side Effects ICD: International Classification of Diseases IM: intramuscular OAS: Overt Aggression Scale PANSS: Positive and Negative Syndrome Scale PANSS‐EC: Positive and Negative Syndrome Scale Excited Component p.o.: by mouth RSESE: Rating Scale for Extrapyramidal Side Effects. SAS: Simpson‐Angus Scale SD: standard deviation SE: standard error TESS: Treatment Emergent Symptom Scale TSRS: TSRS ‐ Target Rating Symptom Scale YMRS ‐ Young Mania Rating Scale
Characteristics of ongoing studies [ordered by study ID]
NCT00838032 2009.
| Trial name or title | Pilot study to evaluate the efficacy and safety of quetiapine fumarate instant‐release (Seroquel IR) in controlling agitation and aggressive symptoms in the acute treatment of patients with schizophrenia |
| Methods | Allocation: randomised. Blindness: single. Duration: 14 days |
| Participants | Diagnosis: CCMD‐3 diagnosis of schizophrenia with agitation. N = not stated. Age: > 18 < 65. Sex: males and females. History: not stated. Exclusion: pregnancy or lactation, risk of suicide, self‐harm or harm to others, known intolerance or lack of response to quetiapine fumarate or haloperidol, use of ketonazole, itraconazole, fluconazole, erythromycin, clarithromycin, troleandomycin, indinavir, nelfinavir, ritonavir, fluvoxamine, saquinavir, phenytoin, carbamazepine, rifampin, St. John's Wort and glucocorticoids two weeks prior to the trial, depot anti‐psychotic within one dosing interval (for the depot) before randomisation, CCMD diagnosis of substance or alcohol dependence at enrolment (exception of caffeine or nicotine), use of opiates, amphetamine, cocaine, cannabis, barbiturates or hallucinogens within four weeks prior, medical conditions that would affect the absorption, distribution, metabolism, or excretion of study treatment, unstable or inadequately treated medical illnesses, involvement in the planning and conduct of the study, previous enrolment or randomisation in the present study, participation in another drug trial within four weeks prior to enrolment or in accordance with local requirements, a person with diabetes mellitus, an absolute neutrophil count of 1.5 x 109 per litre, 2 x higher than normal upper limit of ALT or AST, use of clozapine within 28 days prior to randomisation. Setting: psychiatric inpatient ward, China. |
| Interventions | 1. Haloperidol. 2. Quetiapine. |
| Outcomes | Not stated. |
| Starting date | August 2008. |
| Contact information | Bo Zhang, PhD 13808203275 zb_73@126.com |
| Notes | Protocol, full characteristics and outcome data not reported. Attempted to contact author to enquire whether there are any available data. |
ALT: alanine aminotransferase AST: aspartate transaminase CCMD: Chinese Classification of Mental Disorders
Differences between protocol and review
We have re‐ordered the main outcomes of interest to reflect order of outcomes in Types of outcome measures
We have clarified the time point of secondary outcome 'not tranquil or not asleep' to differentiate these outcomes from the primary outcome 'not tranquil or asleep'.
We have added the sub‐heading 1. Tranquilisation or asleep to categorise the primary outcomes.
Contributions of authors
Edoardo G Ostinelli ‐ screened and retrieved papers (2016 search) against eligibility criteria, appraised quality of papers, extracted data from papers, entered data into RevMan and analysed data, interpreted data and took the lead in writing the 2016 update of the review.
Melanie J Powney ‐ screened and retrieved papers (original search) against eligibility criteria, appraised quality of papers, extracted data from papers, entered data into RevMan and analysed data, interpreted data and took the lead in writing the protocol and review.
Xue Li ‐ screened and retrieved papers (2016 search) against eligibility criteria, extracted data from papers.
Clive E Adams ‐ screened and retrieved papers against eligibility criteria, extracted data from papers, helped write protocol and review (original search and 2016).
Sources of support
Internal sources
-
Università degli Studi di Milano, Milan, Italy.
Employs review author Edoardo G Ostinelli
-
Systematic Review Solutions Ltd, Nottingham, UK.
At the time of writing, employed review author Xue Li
-
University of Nottingham, UK.
Training and support for review author Melanie J Brooke‐Powney
External sources
-
NIHR Cochrane Programme Grant 2011, UK.
Reference number: 10/4001/15
Declarations of interest
Melanie J Powney ‐ none known.
Clive E Adams ‐ was a principle investigator on one trial included in this review (Huf 2007) .
Edoardo G Ostinelli ‐ none known.
Xue Li ‐ at the time of writing the review, Xue was employed by Systematic Review Solutions Ltd, a company that produces sytematic reviews.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Bailine 1987 {published data only}
- Bailine SH, Lesser MS, Krubit G, Ravasz TJ, Davis RA, Kane JM. Comparison of IM haloperidol and IM chlorpromazine in the treatment of acutely psychotic patients. Psychiatric Hospital 1987;18(3):127‐9. [Google Scholar]
Baldacara 2011 {published data only}
- Baldacara L, Sanches M, Cordeiro DC, Jackoswski AP. Rapid tranquilization for agitated patients in emergency psychiatric rooms: A randomized trial of olanzapine, ziprasidone, haloperidol plus promethazine, haloperidol plus midazolam and haloperidol alone. Revista Brasileira de Psiquiatria 2011; Vol. 33, issue 1:30‐9. [DOI] [PubMed]
Battaglia 1997 {published data only}
- Battaglia J, Moss S, Rush J, Kang J, Mendoza R, Leedom L, et al. Haloperidol, lorazepam, or both for psychotic agitation? A multicenter, prospective, double‐blind, emergency department study. American Journal of Emergency Medicine 1997;15(4):335‐40. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Battaglia 2002 {published data only}
- Battaglia J, David SR, Alaka K, Meehan K, Wright P. Calming versus sedative effects of IM olanzapine in agitated patients. Schizophrenia Research 2002;53(3 Suppl 1):183. [DOI] [PubMed] [Google Scholar]
- Battaglia J, Houston JP, Ahl J, Meyers AL, Kaiser CJ. A post hoc analysis of transitioning to oral treatment with olanzapine or haloperidol after 24‐hour intramuscular treatment in acutely agitated adult patients with schizophrenia. Clinical Therapeutics 2005;27(10):1612‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Battaglia J, Lindborg SR, Alaka K, Meehan K, Wright P. Calming versus sedative effects of intramuscular olanzapine in agitated patients. American Journal of Emergency Medicine 2003;21(3):192‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- David SR, Battaglia J, Alaka K, Meehan K, Wright P. Calming versus sedative effects of IM olanzapine in agitated patients. Proceedings of the 11th Congress of the Association of European Psychiatrists; 2002 May 4‐8; Stockholm, Sweden 2002;17(Suppl 1):104s. [Google Scholar]
- David SR, Beasley CM Jr, Alaka K. Analysis of the QTC interval in acutely agitated patients with schizophrenia, bipolar mania, or dementia treated with intramuscular (IM) olanzapine vs. IM placebo or IM haloperidol. European Neuropsychopharmacology 2001;11(3):276. [Google Scholar]
- David SR, Beasley CM Jr, Alaka K. QTC intervals during treatment with IM olanzapine in acutely agitated patients. Schizophrenia Research 2002;53(3 Suppl 1):164. [Google Scholar]
- David SR, Meehan K, Birkett MA, Wright P, Ferchland I, Alaka K, et al. Intramuscular olanzapine versus intramuscular haloperidol and intramuscular placebo: an international double‐blind study in acutely agitated patients with schizophrenia. Schizophrenia Research 2001;49(1‐2):224. [Google Scholar]
- Houston JP, Kaiser CJ, Ahl J. Oral olanzapine transition dose following intramuscular olanzapine treatment. International Congress on Schizophrenia Research; 2005 Apr 2‐5, Savanagh, Georgia USA. 2005.
- King KG, Wright P, Meehan K, Birkett M, David SR, Taylor CC. Transition from intramuscular to oral olanzapine. Proceedings of the 12th World Congress of Psychiatry; 2002 Aug 24‐29; Yokohama, Japan. 2002.
- Lindborg SR, Beasley CM, Alaka K, Taylor CC. Effects of intramuscular olanzapine vs haloperidol and placebo on QTc intervals in acutely agitated patients. Psychiatry Research 2003;119(1‐2):113‐23. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Meehan K, Alaka KJ, Birkett M, Breier A, David S, Ferchland I, et al. Short‐acting intramuscular olanzapine in acutely agitated schizophrenic patients: A randomized double blind trial vs. placebo and haloperidol. Psychosomatics 2001;42(2):197‐8. [Google Scholar]
- Meehan K, Wright P, David SR, Alaka K, Taylor CC, Breier A, et al. Agitation as a treatable symptom common to schizophrenia, acute mania and dementia. Proceedings of the 57th Annual Meeting of the Society of Biological Psychiatry; 2002 May 16‐18; Philadelphia, Pennsylvania, USA. 2002.
- Meehan K, Wright P, David SR, Alaka K, Taylor CC, Breier A, et al. Agitation as a treatable symptom common to schizophrenia, acute mania, and dementia. Schizophrenia Research 2002;53(3 Suppl 1):63. [Google Scholar]
- Wright P, Birkett M, David SR, Meehan K, Ferchland I, Alaka KJ, et al. Double‐blind, placebo‐controlled comparison of intramuscular olanzapine and intramuscular haloperidol in the treatment of acute agitation in schizophrenia. American Journal of Psychiatry 2001;158(7):1149‐51. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Wright P, Birkett M, Ferchland I, David S, Alaka K, Pullen P, et al. A double‐blind study of intramuscular olanzapine, haloperidol and placebo in acutely agitated schizophrenic patients. European Neuropsychopharmacology 2000;10(Suppl 3):S304. [Google Scholar]
- Wright P, Birkett M, Meehan K, Ferchland I, David S, Alaka K, et al. A double‐blind study of intramuscular olanzapine, haloperidol and placebo in acutely agitated schizophrenic patients. International Journal of Neuropsychopharmacology 2000;3(Suppl 1):S139. [Google Scholar]
- Wright P, Lindborg SR, Birkett M, Meehan K, Jones B, Alaka K, et al. Intramuscular olanzapine and intramuscular haloperidol in acute schizophrenia: antipsychotic efficacy and extrapyramidal safety during the first 24 hours of treatment. Canadian Journal of Psychiatry [Revue Canadienne de Psychiatrie] 2003;48(11):716‐21. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Wright P, Meehan K, Birkett M, Lindborg SR, Taylor CC, Morris PBA. A comparison of the efficacy and safety of olanzapine versus haloperidol during transition from intramuscular to oral therapy. Clinical Therapeutics 2003;25(5):1420‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Breier 2002 {published data only}
- Breier A, Meehan K, Birkett M, David S, Ferchland I, Sutton V, et al. A double‐blind, placebo‐controlled dose‐response comparison of intramuscular olanzapine and haloperidol in the treatment of acute agitation in schizophrenia. Archives of General Psychiatry 2002;59(5):441‐8. [DOI] [PubMed] [Google Scholar]
- Breier A, Wright P, Birkett M, David S, Brook S, Meehan K. A double‐blind dose response study comparing intramuscular (IM) olanzapine, haloperidol, and placebo in acutely agitated schizophrenic patients. Biological Psychiatry 2001;49(8):121S. [Google Scholar]
- Breier A, Wright P, Birkett M, Meehan K, David S, Brook S. A double‐blind dose response study comparing intramuscular olanzapine, haloperidol and placebo in acutely agitated schizophrenic patients. Proceedings of the 39th Annual Meeting of the American College of Neuropsychopharmacology; 2000 Dec 10‐14; San Juan, Puerto Rico. 2000.
- Breier AF, Wright P, Birkett M, Meehan K, David SR, Brook S. Intramuscular olanzapine: dose‐related improvement in acutely agitated patients with schizophrenia. Proceedings of the 154th Annual Meeting of the American Psychiatric Association; 2001 May 5‐10; New Orleans, Louisiana, USA. Marathon Multimedia, 2001.
- Smith RC. 10 mg intramuscular olanzapine reduces acute agitation in schizophrenia more effectively than lower doses [Commentary]. Evidence‐Based Mental Health 2003;6(1):27. [MEDLINE: ] [PubMed] [Google Scholar]
- Wright P, Birkett MA, Meehan K, David SR, Brook S, Breier. A double‐blind dose response study comparing intramuscular olanzapine, haloperidol and placebo in acutely agitated schizophrenic patients. Schizophrenia Research 2001;49(1‐2):250‐1. [Google Scholar]
Bristol Myers 2004a {published data only}
- Andrezina R, Josiassen RC, Marcus RN, Oren DA, Manos G, Stock E, et al. Intramuscular aripiprazole for the treatment of acute agitation in patients with schizophrenia or schizoaffective disorder: a double‐blind, placebo‐controlled comparison with intramuscular haloperidol. Psychopharmacology 2006;188(3):281‐92. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Andrezina R, Marcus RN, Oren DA, Manos G, Stock E, Carson WH, et al. Intramuscular aripiprazole or haloperidol and transition to oral therapy in patients with agitation associated with schizophrenia: sub‐analysis of a double‐blind study. Current Medical Research and Opinion 2006;22(11):2209‐19. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Bristol‐Myers S. A randomized, double‐blind comparison of the efficacy and safety of aripiprazole intramuscular formula, haloperidol, or placebo in the treatment of acutely agitated patients with a diagnosis of schizophrenia or schizoaffective disorder. http://www.clinicalstudyresults.org/ 2004.
- Citrome LL, Vester‐Blokland E, Archibald D, McQuade R, Kostic D, Pikalov A, et al. Benefits of a second dose of intramuscular (IM) aripiprazole to control agitation in patients with schizophrenia or bipolar I disorder. Proceedings of the 159th Annual Meeting of the American Psychiatric Association; 2006 May 20‐25, Toronto, Canada. 2006.
- Currier GW, Crandall D, Archibald D, Nyilas M, Kostic D, Pikalov A, et al. Intramuscular (IM) aripiprazole controls agitation in patients with schizophrenia or bipolar disorder without excessive sedation. Proceedings of the 159th Annual Meeting of the American Psychiatric Association; 2006 May 20‐25; Toronto, Canada. 2006.
- Daniel DG, Currier GW, Zimbroff DL, Allen MH, Oren D, Manos G, et al. Efficacy and safety of oral aripiprazole compared with haloperidol in patients transitioning from acute treatment with intramuscular formulations. Journal of Psychiatric Practice 2007;13(3):170‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Daniel DG, Stock EG, Wilber CH, Marcus RN, Carson WH Jr, Manos G, et al. Intramuscular aripiprazole in acutely agitated psychotic patients. Proceedings of the 157th Annual Meeting of the American Psychiatric Association; 2004 May 1‐6; New York, USA. 2004.
- Dillenschneider A, Oren D, Marcus R, Kostic D, McQuade R, Iwamoto T, et al. Comparison of intramuscular aripiprazole and haloperidol with placebo in acutely agitated schizophrenia patients. European Neuropsychopharmacology 2005;15(Suppl 3):S509. [Google Scholar]
- Dillenschneider A, Oren D, Marcus R, Kostic D, McQuade R, Iwamoto T, et al. Intramuscular aripiprazole in acute schizophrenia: a pivotal phase III study. Proceedings of the 18th European College of Neuropsychopharmacology Congress; 2005 Oct 22‐26; Amsterdam, Netherlands 2005.
- McQuade R, Auby P, Oren D, Marcus R, Kostic D, Iwamoto T, et al. Intramuscular aripiprazole in acute schizophrenia: a double‐blind, placebo‐controlled comparison with IM haloperidol. Proceedings of the 13th Biennial Winter Workshop on Schizophrenia Research; 2006 Feb 4‐10; Davos, Switzerland. 2006.
- Oren D, Marcus R, Kostic D, McQuade R, Iwamoto T, Archibald D. Efficacy and safety of intramuscular aripiprazole, haloperidol or PLACEBO in acutely agitated patients with schizophrenia or schizoaffective disorder. Schizophrenia Bulletin 2005;31:499. [Google Scholar]
- Oren D, Marcus R, Kostic D, McQuade R, Iwamoto T, Archibald D. Efficacy and safety of intramuscular aripiprazole, haloperidol or placebo in acutely agitated patients with schizophrenia or schizoaffective disorder. Proceedings of the 158th Annual Meeting of the American Psychiatric Association; 2005 May 21‐26; Atlanta, Georgia, USA 2005.
- Oren DA, Marcus RN, Manos G, Carson WH, McQuade D. Non‐inferiority of intramuscular aripiprazole versus intramuscular haloperidol for the treatment of agitation associated with schizophrenia/schizoaffective disorder. Schizophrenia Bulletin 2007;33(2):451. [Google Scholar]
- Yocca F, Marcus R, Oren D, Manos G, Carson W, Iwamoto T, et al. Intramuscular aripiprazole in acute schizophrenia: a pivotal phase III study. Proceedings of the 158th Annual Meeting of the American Psychiatric Association; 2005 May 21‐26, Atlanta, Georgia, USA 2005.
Bristol‐Myers 2005b {published data only}
- Bristol‐Myers S. Randomized, double‐blind, dose‐ranging study of intramuscular aripiprazole in the treatment of acute agitation in patients with a diagnosis of schizophrenia, schizoaffective, or schizophreniform disorder. http://www.clinicalstudyresults.org/ 2005.
- Currier GW, Crandall D, Archibald D, Nyilas M, Kostic D, Pikalov A, et al. Intramuscular (IM) aripiprazole controls agitation in patients with schizophrenia or bipolar disorder without excessive sedation. Proceedings of the 159th Annual Meeting of the American Psychiatric Association; 2006 May 20‐25; Toronto, Canada. 2006.
- Gismondi R, Daniel D, Stock E, Wilber R, Marcus R, Carson W, et al. Intramuscular aripiprazole treatment for acute agitation in patients with psychosis. European Neuropsychopharmacology 2004;14(Suppl 3):S261. [Google Scholar]
- Kungel M, Czaniera R, Ebrecht M, Werner C, Oren D, McQuade R, et al. Intramuscular aripiprazole for the treatment of acute agitation accompanied with schizophrenia: Subanalysis of a double‐blind controlled study to find the dosage. Proceedings of the 25th Symposium of the Arbeitsgemeinschaft Neuropsychopharmakologie und Pharmakopsychiatrie; 2007 Oct 3‐6; Munich, Germany. 2007.
- Kungel M, Daniel D, Stock E, Wilber R, Marcus R, Carson W, et al. Efficacy and safety of intramuscular aripiprazole in acutely agitated patients with psychosis. Proceedings of the Thematic Conference of the World Psychiatric Association on "Treatments in Psychiatry: An Update"; 2004 Nov 10‐13; Florence, Italy. 2004.
- Oren DA, Manos G, Markovic O, McQuade RD. Intramuscular aripiprazole for the treatment of acute agitation associated with schizophrenia: Sub‐analysis of a double‐blind, controlled, dose‐ranging study. European Psychiatry 2007;22:S124‐S. [Google Scholar]
- Tran‐Johnson TK, Sack DA, Marcus RN, Auby P, McQuade RD, Oren DA. Efficacy and safety of intramuscular aripiprazole in patients with acute agitation: a randomized, double‐blind, placebo‐controlled trial. Journal of Clinical Psychiatry 2007;68(1):111‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Brook 2000 {published data only}
- Brook S, Krams M, Gunn K. Intramuscular (IM) ziprasidone vs. IM haloperidol in patients with acute, non‐organic psychosis. Proceedings of the 9th Congress of the Association of European Psychiatrists; 1998 Sep 20‐24; Copenhagen, Denmark. 1998.
- Brook S, Krams M, Gunn KP. Intramuscular ziprasidone versus intramuscular haloperidol. Proceedings of the 21st Collegium Internationale Neuro‐Psychopharmacologicum Congress; 1998 Jul 12‐16; Glasgow, UK 1998.
- Brook S, Krams M, Gunn KP, and the Ziprasidone IM Study Group. The efficacy and tolerability of intramuscular (IM) ziprasidone versus IM haloperidol in patients with acute non‐organic psychosis. Proceedings of the 151st Annual Meeting of the American Psychiatric Association; 1998 May 30 ‐ Jun 4; Toronto, Ontario, Canada. 1998.
- Brook S, Lucey JV, Gunn KP. Intramuscular ziprasidone compared with intramuscular haloperidol in the treatment of acute psychosis. Journal of Clinical Psychological Medicine [临床精神医学杂志] 2000;61(12):933‐41. [DOI] [PubMed] [Google Scholar]
- Daniel DG, Brook S, Reeves KR. An overview of the efficacy and safety of rapid acting intramuscular ziprasidone. European Neuropsychopharmacology 2000;10(Suppl 3):S239. [Google Scholar]
- Zimbroff DL, Daniel DG, Brook S, Reeves KR. An overview of the efficacy and safety of rapid‐acting intramuscular ziprasidone. Proceedings of the 153rd Annual Meeting of the American Psychiatric Association; 2000 May 13‐18; Chicago, Illinois, USA. 2000.
Calver 2015 {published data only}
Currier 2004 {published data only}
- Currier GW, Chou JC, Feifel D, Bossie CA, Turkoz I, Mahmoud R, et al. Acute treatment of psychotic agitation: a randomized comparison of oral treatment with risperidone and lorazepam versus intramuscular treatment with haloperidol and lorazepam. Journal of Clinical Psychiatry 2004;65(3):386‐94. [MEDLINE: ] [PubMed] [Google Scholar]
Dorevitch 1999 {published data only}
- Dorevitch A, Katz N, Zemishlany Z, Aizenberg D, Weizman A. Intramuscular flunitrazepam versus intramuscular haloperidol in the emergency treatment of aggressive psychotic behavior. American Journal of Psychiatry 1999;156(1):142‐4. [DOI] [PubMed] [Google Scholar]
Eli Lilly 2004 {published data only}
- Chan HY, Ree SC, Su LW, Chen JJ, Chou SY, Chen CK, et al. A double‐blind, randomized comparison study of efficacy and safety of intramuscular olanzapine and intramuscular haloperidol in patients with schizophrenia and acute agitated behavior. Journal of Clinical Psychopharmacology 2014; Vol. 34, issue 3:355‐8. [DOI] [PubMed]
- Eli Lilly. Comparison of intramuscular olanzapine and intramuscular haloperidol in patients with schizophrenia. Eli Lilly and Company Clinical Trial Registry 2004.
- NCT00485901. A double‐blind randomized comparison of the efficacy and safety of intramuscular olanzapine and intramuscular haloperidol in acutely agitated patients with schizophrenia. http://www.clinicaltrials.gov 2007.
Fang 2012 {published data only}
- Fang M, Chen H, Li LH, Wu R, Li Y, Liu L, et al. Comparison of risperidone oral solution and intramuscular haloperidol with the latter shifting to oral therapy for the treatment of acute agitation in patients with schizophrenia. International Clinical Psychopharmacology 2012; Vol. 27, issue 2:107‐13. [DOI] [PubMed]
Fitzgerald 1969 {published data only}
- Fitzgerald CH. A double‐blind comparison of haloperidol with perphenazine in acute psychiatric episodes. Current Therapeutic Research 1969;11(8):515‐9. [PubMed] [Google Scholar]
Foster 1997 {published data only}
- Foster S, Kessel J, Berman ME, Simpson GM. Efficacy of lorazepam and haloperidol for rapid tranquilization in a psychiatric emergency room setting. International Clinical Psychopharmacology 1997;12(3):175‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fruensgaard 1977 {published data only}
- Fruensgaard K, Korsgaard S, Jorgensen H, Jensen K. Loxapine versus haloperidol parenterally in acute psychosis with agitation. A double‐blind study. Acta Psychiatrica Scandinavica 1977;56(4):256‐64. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Garza‐Trevino 1989 {published data only}
- Garza‐Trevino ES, Hollister LE, Overall JE, Alexander WF. Efficacy of combinations of intramuscular antipsychotics and sedative‐hypnotics for control of psychotic agitation. American Journal of Psychiatry 1989;146(12):1598‐601. [DOI] [PubMed] [Google Scholar]
Guo 2007 {published data only}
- Guo C‐R. Study of quetiapine combined with magnesium valproate release tablets in treatment of schizophrenia with symptoms of elation and agitation [奎硫平合并丙戊酸镁缓释片治疗精神分裂症兴奋激越研究]. Linchuang Jingshen Yixue Zazhi [临床精神医学杂志] 2007;17(3):183‐4. [Google Scholar]
Higashima 2004 {published data only}
- Higashima M, Takeda T, Nagasawa T, Hirao N, Oka T, Nakamura M, et al. Combined therapy with low‐potency neuroleptic levomepromazine as an adjunct to haloperidol for agitated patients with acute exacerbation of schizophrenia. European Psychiatry 2004;19(6):380‐1. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Huf 2007 {published data only}
- Barbui C. Intramuscular haloperidol plus promethazine is more effective and safer than haloperidol alone for rapid tranquillisation of agitated mentally ill patients. Evidence‐Based Mental Health 2008;11(3):86‐7. [DOI] [PubMed] [Google Scholar]
- Huf G. Haloperidol plus promethazine versus haloperidol for psychosis induced aggression. Data on file 2004.
- Huf G, Coutinho ESF, Adams CE. Rapid tranquillisation in psychiatric emergency settings in Brazil: pragmatic randomised controlled trial of intramuscular haloperidol versus intramuscular haloperidol plus promethazine. BMJ 2007;335(7625):869‐72. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huf G, Coutinho ESF, Adams CE. The pharmacological management of agitated patients in emergency psychiatric hospitals in Rio de Janeiro, Brazil: the results of two pragmatic randomized clinical trials. Proceedings of the 5th European Congress on Violence in Clinical Psychiatry; 2007 Oct 25‐27; Amsterdam, The Netherlands. 2007.
- ISRCTN83261243. TREC2 ‐ Rapid tranquillisation for agitated patients in emergency psychiatric rooms in Rio de Janeiro. A randomised trial of intramuscular haloperidol versus intramuscular haloperidol + promethazine. http://www.controlled‐trials.com 2005.
- Migon MN, Coutinho ES, Huf G, Adams CE, Cunha GM, Allen MH. Factors associated with the use of physical restraints for agitated patients in psychiatric emergency rooms. General Hospital Psychiatry 2008;30(3):263‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kewala 1984 {published data only}
- Kewala S, Ban TA, Berney SA, Wilson WH. Rapid tranquilization: A comparative study of thiothixene and haloperidol. Progress in Neuro‐Psychopharmacology and Biological Psychiatry 1984;8:77‐83. [DOI] [PubMed] [Google Scholar]
Kinon 2001d {published data only}
- Kinon B, Rotelli MD, Gilmore JA. Efficacy of olanzapine and adjunctive lorazepam to control agitation in schizophrenia. International Journal of Neuropsychopharmacology 2002;5(Suppl 1):S128. [Google Scholar]
- Kinon BJ, Ahl J, Rotelli MD, McMullen E. Efficacy of accelerated dose titration of olanzapine with adjunctive lorazepam to treat acute agitation in schizophrenia. American Journal of Emergency Medicine 2004;22(3):181‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Kinon BJ, Wang L, Rotelli MD, Gilmore JA. The efficacy of olanzapine plus adjunctive lorazepam to control acute agitation in schizophrenia. European Neuropsychopharmacology 2001;11(3):278. [Google Scholar]
Li 2006 {published data only}
- Li L‐H, Zhao J‐P, Xu X‐F. A comparative study of intramuscular ziprasidone and haloperidol in treating acute agitation in schizophrenia [国产齐拉西酮与氟哌啶醇注射液治疗精神分裂症急性激越症状的对照研究]. Chinese Journal of Psychiatry 2006;39(4):216‐9. [Google Scholar]
Liang 2013 {published data only}
- Liang Y, Su Y, Huang J, Tang M, Li K, Yang F, et al. Effect of quetiapine and haloperidol on QTc interval in treating schizophrenic patients with excitement and agitation [喹硫平和氟哌啶醇治疗精神分裂症急性期兴奋激越对QTc间期的影响]. Chinese New Drugs Journal [Zhongguo Xin Yao Za Zhi; 中国新药杂志] 2013; Vol. 22, issue 16:1886‐9+1894.
- Liang Y, Su Y, Huang J, Tang M, Li K, Yang P, et al. A randomized controlled study of effect and safety of quetiapine and haloperidol in treatment of schizophrenic patients with excitement and agitation [喹硫平和氟哌啶醇治疗精神分裂症急性期兴奋激越的多中心随机对照研究]. Chinese Mental Health Journal [Zhongguo Xin Li Wei Sheng Za Zhi; 中国心理卫生杂志] 2013; Vol. 27, issue 4:262‐7.
Lim 2010 {published data only}
- Kim JJ, Lim HK, Lee CU, Lee C, Pae CU, Paik IH. Risperidone orodispersible tablet and intramuscular haloperidol in treatment of acute psychotic agitation. European Neuropsychopharmacology 2007;17(Suppl 4):S467. [DOI] [PubMed] [Google Scholar]
- Lim HK, Kim JJ, Pae CU, Lee CU, Lee C, Paik IH. Comparison of risperidone orodispersible tablet and intramuscular haloperidol in the treatment of acute psychotic agitation: A randomized open, prospective study. Neuropsychobiology 2010;62(2):81‐6. [ISI: 000280037400001] [DOI] [PubMed] [Google Scholar]
Liu 2012 {published data only}
- 刘锟, 刘云, 宁南义, 孙建中, 孙亚军, 孙同勋, et al. Consolidation of short‐term risperidone intramuscular haloperidol in the treatment of acute agitation in schizophrenia randomized controlled study of the efficacy of behavior [Google Translate] [利培酮合并短期肌注氟哌啶醇治疗精神分裂症急性期激越行为疗效的随机对照研究]. 中国医药导报 2012, issue 35:104‐6.
Man 1973 {published data only}
- Man‐Pang L, Chen CH. Rapid tranquilization of acutely psychotic patients with intramuscular haloperidol and chlorpromazine. Psychosomatics 1973;14(1):59‐63. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Nobay 2004 {published data only}
- Nobay F, Simon BC, Levitt MA, Dresden GM. A prospective, double‐blind, randomized trial of midazolam versus haloperidol versus lorazepam in the chemical restraint of violent and severely agitated patients. Academic Emergency Medicine 2004;11(7):744‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Paprocki 1977 {published data only}
- Paprocki J, Versiani M. A double‐blind comparison between loxapine and haloperidol by parenteral route in acute schizophrenia. Current Therapeutic Research 1977;21(1):80‐100. [PubMed] [Google Scholar]
Reschke 1974 {published data only}
- Reshke RW. Parenteral haloperidol for rapid control of severe, disruptive symptoms of acute schizophrenia. Diseases of the Nervous System 1974;35:112‐5. [PubMed] [Google Scholar]
Resnick 1984 {published data only}
- Resnick M, Burton BT. Droperidol vs. haloperidol in the initial management of acutely agitated patients. Journal of Clinical Psychological Medicine [临床精神医学杂志] 1984;45(7):298‐9. [PubMed] [Google Scholar]
Ritter 1972 {published data only}
- Ritter RM, Davidson DE, Robinson TA. Comparison of injectable haloperidol and chlorpromazine. American Journal of Psychiatry 1972;129(1):78‐81. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Salzman 1991 {published data only}
- Salzman C, Solomon D, Miyawaki E, Glassman R, Rood L. Parenteral lorazepam versus parenteral haloperidol for the control of psychotic disruptive behavior. Journal of Clinical Psychiatry 1991;52(4):177‐80. [MEDLINE: ] [PubMed] [Google Scholar]
Shi 2010 {published data only}
- Shi Y, Dai T, Yi P. olanzapine combined magnesium valproate sustained‐release tablets in the treatment of schizophrenia, acute agitation excited [Google Translate] [奥氮平合并丙戊酸镁缓释片治疗精神分裂症急性期兴奋激越研究]. Modern Practical Medicine 2010; Vol. 22, issue 4:398‐9.
Shu 2010 {published data only}
- NCT00723606. A randomized, open‐label, multi‐center study to evaluate the efficacy and safety of intramuscular ziprasidone in patients with agitation. http://www.clinicaltrials.gov 2008.
- Pfizer. A randomized, open label, rater blind, flexible dose multi‐center study comparing the efficacy and safety of intramuscular ziprasidone with haloperidol for three days in patients with agitation of schizophrenia. http://www.clinicalstudyresults.org/documents/company‐study_10505_0.pdf (accessed 13 June 2011).
- Shu L, Zhang H, Wang G, Zhao J, Xie S, Xu X, et al. Intramuscular ziprasidone has improved tolerability over haloperidol and comparable efficacy for control of agitation in schizophrenia in Chinese patients. Schizophrenia Research 2010;117(2‐3):260. [Google Scholar]
- Zhang H, Wang G, Zhao J, Xie S, Xu X, Shi J, et al. Intramuscular ziprasidone versus haloperidol for managing agitation in Chinese patients with schizophrenia. Journal of Clinical Psychopharmacology 2013; Vol. 33, issue 2:178‐85. [DOI] [PubMed]
Stotsky 1977 {published data only}
- Stotsky BA. Relative efficacy of parenteral haloperidol and thiothixene for the emergency treatment of acutely excited and agitated patients. Diseases of the Nervous System 1977;38(12):967‐73. [MEDLINE: ] [PubMed] [Google Scholar]
Taymeeyapradit 2002 {published data only}
- Taymeeyapradit U, Kuasirikul S. Comparative study of the effectiveness of zuclopenthixol acetate and haloperidol in acutely disturbed psychotic patients. Journal of the Medical Association of Thailand 2002;85(12):1301‐8. [MEDLINE: ] [PubMed] [Google Scholar]
Tuason 1986 {published data only}
- Tuason VB. A comparison of parenteral loxapine and haloperidol in hostile and aggressive acutely schizophrenic patients. Journal of Clinical Psychiatry 1986;47(3):126‐9. [MEDLINE: ] [PubMed] [Google Scholar]
Walther 2014 {published data only}
- Walther S, Moggi F, Horn H, Moskvitin K, Abderhalden C, Maier N, et al. Rapid tranquilization of severely agitated patients with schizophrenia spectrum disorders: a naturalistic, rater‐blinded, randomized, controlled study with oral haloperidol, risperidone, and olanzapine. Journal of Clinical Psychopharmacology 2014; Vol. 34, issue 1:124‐8. [DOI] [PubMed]
- Walther S, Moggi F, Horn H, Moskvitin K, Maier N, Abderhalden C, et al. Rapid tranquilization of severely agitated patients with schizophrenia spectrum disorders: A naturalistic, rater‐blinded, randomized controlled study with oral haloperidol, risperidone, and olanzapine. PharmacoPsychiatry 2013; Vol. 46, issue 6:A77. [DOI] [PubMed]
Yin 2012 {published data only}
- 尹析凡, 王刚. 齐拉西酮注射液治疗精神分裂症激越症状的研究. 中国当代医药 2012; Vol. 30:9‐11.
Zhou 2014 {published data only}
- 周升宝, 孙晓丹, 庞琦, 李志梅, 赵继舒, 张峰. 齐拉西酮联合氯硝西泮治疗精神分裂症急性期兴奋激越症状对照研究. 临床心身疾病杂志 2014; Vol. 20, issue 2:17‐20.
References to studies excluded from this review
Addington 1996 {published data only}
- Addington DE, Arvanitis LA. 'Seroquel' (quetiapine): an atypical antipsychotic ‐ results from a multiple fixed dose, placebo‐controlled study. Proceedings of the 4th International Conference on Schizophrenia ‐ 1996: Breaking down the Barriers; 1996 Oct 6‐9; Vancouver, Canada. 1996.
- Arvanitis LA, Sweitzer DE, Goldstein JM, Yeung PP. Serequel reduces aggression in patients with acute exacerbation of schizophrenia. Proceedings of the 11th European College of Neuropsychopharmacology Congress; 1998 Oct 31 ‐ Nov 4; Paris, France. 1998.
- Borison RL, Arvanitist LA, Miller BG. A comparison of five fixed doses 'seroquel' (ICI 204,636) with haloperidol and placebo in patients with schizophrenia. Schizophrenia Research 1996;18(2‐3):132. [Google Scholar]
- Cantillon M. Quetiapine fumarate reduces aggression. Proceedings of the 11th Annual Meeting of the American Association for Geriatric Psychiatry; 1998 Mar 8‐11; San Diego, California, USA. 1998.
- Cantillon M, Arvanitis LA, Miller BG, Kowalcyk. 'Seroquel' (quetiapine fumarate) reduces hositility and aggression in patients with acute schizophrenia. Proceedings of the 36th Annual Meeting of the American College of Neuropsychopharmacology; 1997 Dec 8‐12; Waikoloa, Hawaii, USA. 1997.
- Cantillon M, Goldstein JM. Efficacy of quetiapine fumarate in affective symptoms of schizophrenia. Proceedings of the 151st Annual Meeting of the American Psychiatric Association; 1998 May 30 ‐ Jun 4; Toronto, Ontario, Canada 1998.
- Cantillon M, Goldstein JM. Quetiapine fumarate reduces aggression and hostility in patients with schizophrenia. Proceedings of the 151st Annual Meeting of the American Psychiatric Association; 1998 May 30 ‐ Jun 4; Toronto, Ontario, Canada 1998.
- Chengappa KNR, Goldstein JM, Greenwood M, John V, Levine J. A post hoc analysis of the impact on hostility and agitation of quetiapine and haloperidol among patients with schizophrenia. Clinical Therapeutics 2003;25(2):530‐41. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Hellewell JSE, Cameron‐Hands D, Cantillon M. Seroquel: evidence for efficacy in the treatment of hostility and aggression. Schizophrenia Research 1998;29(1‐2):154. [Google Scholar]
- Hellewell JSE, Goldstein J. Seroquel (quetiapine): efficacy in improving mood, aggression and hostility. Proceedings of the 9th Congress of the Association of European Psychiatrists; 1998 Sep 20‐24; Copenhagen, Denmark. 1998.
- Hong W, Arvanitis L. Reduction of the positive symptoms of schizophrenia by '(ICI 204,636): results from phase II and III clinical trials. Proceedings of the 9th European College of Neuropsychopharmacology Congress; 1996 Sep 21‐25; Amsterdam, Netherlands. 1996.
Alexander 2004 {published data only}
- Alexander J. Lorazepam versus a combination of haloperidol and promethazine in the acute management of agitation and aggression ‐ a randomized controlled trial [thesis]. Guindy, Chennai: Tamilnadu Dr. MGR Medical University, 2003. [Google Scholar]
- Alexander J, John T, Tharyan P, Adams CE. TREC‐India. A second arm of TREC. Schizophrenia Research 2002;53(3 Suppl 1):236. [Google Scholar]
- Alexander J, Tharyan P, Adams C, John T, Mol C, Philip J. Rapid tranquillisation of violent or agitated patients in a psychiatric emergency setting. Pragmatic randomised trial of intramuscular lorazepam v haloperidol plus promethazine. British Journal of Psychiatry 2004;185:63‐9. [DOI] [PubMed] [Google Scholar]
- NCT00455234. Rapid tranquilization of violent or agitated people in psychiatric emergency settings‐ a pragmatic randomized controlled trial of intramuscular olanzapine vs intramuscular haloperidol + promethazine. http://www.clinicaltrials.gov 2007.
- Raveendran NS. Rapid tranquillization of acutely agitated patients: intramuscular olanzapine vs haloperidol + promethazine ‐ pragmatic randomized control trial. Proceedings of the 5th European Congress on Violence in Clinical Psychiatry; 2007 Oct 25‐27; Amsterdam, The Netherlands. 2007.
- Raveendran NS, Tharyan P, Alexander J, Adams CE. Rapid tranquillisation in psychiatric emergency setting in India: pragmatic randomised controlled trial of intramuscular olanzapine versus intramuscular haloperidol plus promethazine. BMJ 2007;335(7625):865‐9. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharyan P. A randomised controlled trial of intra‐muscular lorazepam versus intra‐muscular haloperidol+promethazine in the management of psychotic agitations and aggression. http://www.controlled‐trials.com 2002.
Allen 2007 {published data only}
- Allen M, Talbott S, Eudicone J, McQuade R, Pikalov A. Similar rates of agitation, anxiety and insomnia in sedating and non‐sedating antipsychotics: evaluating clinical trial results with aripiprazole, haloperidol and olanzapine. Schizophrenia Bulletin 2007;33(2):418. [Google Scholar]
Asadollahi 2015 {published data only}
- Asadollahi S, Heidari K, Hatamabadi H, Vafaee R, Yunesian S, Azadbakht A, et al. Efficacy and safety of valproic acid versus haloperidol in patients with acute agitation: results of a randomized, double‐blind, parallel‐group trial. International Clinical Psychopharmacology 2015; Vol. 30, issue 3:142‐50. [DOI] [PubMed]
Beasley 1998 {published data only}
- Beasley CM, Sayler ME, Keisler GM, Potvin JH, Sanger TM, Tollefson GD. The influence of pharmacotherapy on self‐directed and externally‐directed aggression in schizophrenia. Schizophrenia Research 1998;29(1‐2):28. [Google Scholar]
Bieniek 1998 {published data only}
- Bieniek SA, Ownby RL, Penalver A, Dominguez RA. A double‐blind study of lorazepam versus the combination of haloperidol and lorazepam in managing agitation. Pharmacotherapy 1998;18(1):57‐62. [PubMed] [Google Scholar]
Brook 1998 {published data only}
- Brook S, Berk M, Selemani S, Kolloori J, Nzo I. A randomized controlled double blind study of zuclopenthixol acetate compared to haloperidol in acute psychosis. Human Psychopharmacology 1998;13(1):17‐20. [Google Scholar]
Campbell 1982 {published data only}
- Campbell M, Cohen IL, Small AM. Drugs in aggressive behavior. Journal of the American Academy of Child Psychiatry 1982;21(2):107‐17. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Chen 2004 {published data only}
- Chen M‐F, Liu D‐X, Lin J‐Z. The comparison study on curing agitation by haloperidol and chlorpromazine venoclysis [氟哌啶醇与氯丙嗪静滴治疗急性激越的对照研究]. Medical Journal of Chinese Civil Administration 2004;16(10):599. [Google Scholar]
Chen 2008 {published data only}
- Chen J, Yang X, Song A, Gu X, Ma X. Intramuscular ziprasidone versus haloperidol in the treatment of acute agitation in schizophrenia [齐拉西酮注射液与氟哌啶醇注射液治疗精神分裂症急性激越症状的对比研究]. Shanghai Archives of Psychiatry [上海精神医学] [上海精神醫學] 2008;20(6):363‐6. [Google Scholar]
Chouinard 1991 {published data only}
- Chouinard G, Annable L, Turnier L, Holobow N, Szkrumelak N. A double‐blind randomized clinical trial of rapid tranquilization with IM clonazepam and IM haloperidol in agitated psychotic patients with manic symptoms. Canadian Journal of Psychiatry [Revue Canadienne de Psychiatrie] 1993;38(Suppl 4):S114‐21. [MEDLINE: ] [PubMed] [Google Scholar]
- Chouinard G, Safadi G, Beauclair L. The management of acutely schizophrenic patients newly admitted from the emergency room: A double‐blind clinical trial comparing zuclopenthixol acetate and liquid haloperidol. Proceedings of the 5th World Congress of Biological Psychiatry; 1991 Jun 9‐14; Florence, Italy. Amsterdam, Netherlands: Excerpta Medica, 1991:35‐43.
Citrome 2001 {published data only}
- Citrome L, Volavka J, Czobor P, Sheitman B, Lindenmayer J‐P, McEvoy J, et al. Effects of clozapine, olanzapine, risperidone, and haloperidol on hostility among patients with schizophrenia. Psychiatric Services 2001;52(11):1510‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Citrome LL, Volavka J, Czobor P, Nolan K, Lieberman JA, Lindenmayer J‐P, et al. Overt aggression and psychotic symptoms in patients with schizophrenia treated with clozapine, olanzapine, risperidone, or haloperidol. Proceedings of the 156th Annual Meeting of the American Psychiatric Association; 2003 May 17‐22; San Francisco, California, USA. 2003.
- Citrome LL, Volavka J, Czobor P, Sheitman BB, Lindenmayer J‐P, McEvoy JP, et al. Atypical antipsychotics and hostility in schizophrenia: A double‐blind study. Proceedings of the 154th Annual Meeting of the American Psychiatric Association; 2001 May 5‐10; New Orleans, Louisiana, USA. 2001.
- Citrome LL, Volavka J, Czobor P, Sheitman BB, Lindenmayer J‐P, McEvoy JP, et al. Atypical antipsychotics and hostility in schizophrenia: A double‐blind study. Proceedings of the 155th Annual Meeting of the American Psychiatric Association; 2002 May 18‐23; Philadelphia, Pennsylvania, USA. 2002.
- Hoptman MJ, Volavka J, Czobor P, Gerig G, Chakos M, Blocher J, et al. Aggression and quantitative MRI measures of caudate in patients with chronic schizophrenia or schizoaffective disorder. Journal of Neuropsychiatry and Clinical Neurosciences 2006;18(4):509‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volavka J, Czobor P, Nolan K, Sheitman B, Lindenmayer JP, Citrome LMJP, et al. Overt aggression and psychotic symptoms in patients with schizophrenia treated with clozapine, olanzapine, risperidone, or haloperidol. Journal of Clinical Psychopharmacology 2004;24(2):225‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Colonna 1998 {published data only}
- Colonna L, Martin P, Gerard D. Long‐term study of the effectiveness and tolerance of amisulpride versus haloperidol [Etude à long terme de L'efficacité et de la tolérance à l'amisulpride versus haloperidol]. Proceedings of the 96th Congres de Psychiatrie et de Neurologie de Langue Francaise; 1998 May 10‐15; Saint Paul, France. 1998.
Conde 2011 {published data only}
- Conde BL, Sobreveg EE, Sionzon MP. A randomized trial of oral risperidone versus intramuscular haloperidol in the emergency treatment of acute psychotic agitation. ASEAN Journal of Psychiatry 2011; Vol. 12, issue 1:29‐36.
Crandall 2005 {published data only}
- Crandall D, Pikalov A, Kostic D, Kaplita S, Berman R, McQuade R, et al. Is sedation needed for effective reduction of acute schizophrenia symptoms?. Proceedings of the 158th Annual Meeting of the American Psychiatric Association; 2005 May 21‐26; Atlanta, Georgia, USA. 2005.
Currier 2000 {published data only}
- Currier GW, Simpson GA. Risperidone oral solution versus intramuscular haloperidol for control of psychotic agitation. European Neuropsychopharmacology 2000;10(Suppl 3):S296. [Google Scholar]
Daniel 2000 {published data only}
- Daniel DG, Brook S, Reeves KR. An overview of the efficacy and safety of rapid acting intramuscular ziprasidone. European Neuropsychopharmacology 2000;10(Suppl 3):S289. [Google Scholar]
Daniel 2003 {published data only}
- Daniel DG, Brook S, Benattia I. Efficacy of intramuscular ziprasidone without adjunctive benzodiazepines. European Neuropsychopharmacology 2003;13(4):S299. [Google Scholar]
Daniel 2004 {published data only}
- Daniel DG, Zimbroff DL, Swift RH, Harrigan EP. The tolerability of intramuscular ziprasidone and haloperidol treatment and the transition to oral therapy. International Clinical Psychopharmacology 2004;19(1):9‐15. [DOI] [PubMed] [Google Scholar]
- Lucey JV, Brook S, Daniel DG, Reeves KR, Harrigan EP. Intramuscular (IM) ziprasidone: a novel treatment for the short term management of agitated psychotic patients. Schizophrenia Research 2000;41(1):208. [Google Scholar]
- Modell S, Daniel D, Stock E, Wilber R, Marcus R, Carson W, et al. Intramuscular aripiprazole treatment for acute agitation in patients with psychosis. Proceedings of the 24th Collegium Internationale Neuro‐Psychopharmacologicum Congress; 2004 June 20–24; Paris, France 2004.
- Swift RH, Harrigan EP, Cappelleri JC, Kramer D, Chandler LP. Validation of the behavioural activity rating scale (BARS): a novel measure of activity in agitated patients. Journal of Psychiatric Research 2002;36(2):87‐95. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Swift RH, Harrigan EP, Kammen DP. A comparison of intramuscular (IM) ziprasidone with IM haloperidol. Proceedings of the 9th Congress of the Association of European Psychiatrists; 1998 Sep 20‐24; Copenhagen, Denmark. 1998.
- Swift RH, Harrigan EP, Kammen DP. A comparison of intramuscular ziprasidone with intramuscular haloperidol. Proceedings of the 151st Annual Meeting of the American Psychiatric Association; 1998 May 30 ‐ Jun 4; Toronto, Ontario, Canada 1998.
- Swift RH, Harrigan EP, Kammen D, Casey DE. A comparison of fixed dose, intramuscular (IM) ziprasidone with flexible dose, IM haloperidol. Proceedings of the 11th European College of Neuropsychopharmacology Congress; 1998 Oct 31 ‐ Nov 4; Paris, France. 1998.
- Swift RH, Harrigan EP, Van‐Kammen D. A comparison of fixed‐dose intramuscular (IM) ziprasidone with flexible‐dose IM haloperidol. Proceedings of the 21st Collegium Internationale Neuro‐Psychopharmacologicum Congress; 1998 Jul 12‐16; Glasgow, UK. 1998.
- Yocca F, Daniel D, Stock E, Oren D, Marcus R, Carson W, et al. Efficacy and safety of intramuscular aripiprazole treatment for acute agitation in patients with psychosis. Proceedings of the 43rd Annual Meeting of the American College of Neuro‐Psychopharmacology; 2004 Dec 12‐16; San Juan, Puerto Rico. 2004.
Fan 2012 {published data only}
- 樊凌姿, 曹立新. 氯普噻吨注射液与氟哌啶醇注射液治疗精神分裂症兴奋激越对照研究. 中外健康文摘 2012; Vol. 40:31‐2.
Faretra 1970 {published data only}
- Faretra G, Dooher L, Dowling J. Comparison of haloperidol and fluphenazine in disturbed children. American Journal of Psychiatry 1970;126:1670‐3. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Freeman 2009 {published data only}
- Freeman DJ, DiPaula BA, Love RC. Intramuscular haloperidol versus intramuscular olanzapine for treatment of acute agitation: A cost‐minimization study. Pharmacotherapy 2009;29(8):930‐6. [DOI] [PubMed] [Google Scholar]
Goldstein 1966 {published data only}
- Goldstein BJ, Clyde DJ. Haloperidol in controlling the symptoms of acute psychoses. Part II: A double blind evaluation of haloperidol and trifluoperazine. Current Therapeutic Research 1966;8:236‐40. [PubMed] [Google Scholar]
Harvey 2004 {published data only}
- Harvey AT, Flockhart D, Gorski JC, Greenblatt DJ, Burke M, Werder S, et al. Intramuscular haloperidol or lorazepam and QT intervals in schizophrenia. Journal of Clinical Pharmacology 2004;44(10):1173‐84. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Huang 2004 {published data only}
- Huang W‐S, Di X‐L, Yang F‐D. A comparative study of the effect of modern electroconvulsive therapy on the agitated behavior of psychotic patients. Journal of Clinical Psychological Medicine [临床精神医学杂志] 2004;14(5):276‐7. [Google Scholar]
Jiang 2009 {published data only}
- 蒋合萍, 罗毅平. Olanzapine and haloperidol in the treatment of schizophrenia comparative study of excited agitation [Google Translate] [奥氮平与氟哌啶醇治疗精神分裂症兴奋激越症状的对照研究]. Sichuan Mental Health [四川精神卫生] 2009; Vol. 22, issue 1:43‐4.
Jones 2001 {published data only}
- Jones B, Taylor CC, Meehan K. The efficacy of a rapid‐acting intramuscular formulation of olanzapine for positive symptoms. Journal of Clinical Psychological Medicine [临床精神医学杂志] 2001;62(2):22‐4. [MEDLINE: ] [PubMed] [Google Scholar]
Kane 2000 {published data only}
- Kane J, Ingenito G, Ali M. Efficacy of aripiprazole in psychotic disorders: comparison with haloperidol and placebo. Schizophrenia Research 2000;41(1):39. [Google Scholar]
Kinon 2001a {published data only}
- Eli Lilly. Beasley 1996a ‐ olz vs placebo vs hpl (N America) (as supplied 2004). Data on file 2001.
- Eli Lilly. Beasley 1997 ‐ olz vs hpl (international) (as supplied 2004). Data on file 2001.
- Eli Lilly. Tollefson 1997 ‐ olz vs hpl (as supplied 2004). Data on file 2001.
- Kinon BJ, Basson MS, Tollefson GD. Gender‐specific prolactin response to treatment with olanzapine versus haloperidol in schizophrenia. Proceedings of the 9th Biennial Winter Workshop on Schizophrenia; 1998 Feb 7‐13; Davos, Switzerland 1998.
- Kinon BJ, Milton DR, Hill AL, Williamson DJ. Effective resolution of acute presentation of behavioral agitation and positive psychotic symptoms in schizophrenia with olanzapine. Journal of Psychopharmacology 2000;14(3 Suppl):A60. [Google Scholar]
- Kinon BJ, Roychowdhury SM, Milton DR, Hill AL. Effective resolution with olanzapine of acute presentation of behavioral agitation and positive psychotic symptoms in schizophrenia. Journal of Clinical Psychiatry 2001;62(Suppl 2):17‐21. [MEDLINE: ] [PubMed] [Google Scholar]
- Kinon BJ, Wang L, Stauffer VL. A comparison of prolactin concentrations in the elderly during treatment with olanzapine, haloperidol, and risperidone. Proceedings of the 14th Annual Meeting of the American Association for Geriatric Psychiatry; 2001 Feb 23‐26; San Francisco, California, USA. 2001.
Kinon 2003 {published data only}
- Kinon BJ, Stauffer VL, McGuire HC, Kaiser CJ, Dickson RA, Kennedy JS. The effects of antipsychotic drug treatment on prolactin concentrations in elderly patients. Journal of the American Medical Directors Association 2003;4(4):189‐94. [PUBMED: 12837139] [DOI] [PubMed] [Google Scholar]
- Kinon BJ, Wang L, Stauffer VL. A comparison of prolactin concentrations in the elderly during treatment with olanzapine, haloperidol, and risperidone. Proceedings of the 14th Annual Meeting of the American Association for Geriatric Psychiatry; 2001 Feb 23‐26; San Francisco, California, USA. 2001.
Krakowski 2006 {published data only}
- Krakowski M I, Czobor P. Depression and impulsivity as pathways to violence: implications for antiaggressive treatment. Schizophrenia Bulletin 2014; Vol. 40, issue 4:886‐94. [DOI] [PMC free article] [PubMed]
- Krakowski M, Czobor P. Cholesterol and cognition in schizophrenia: A double‐blind study of patients randomized to clozapine, olanzapine and haloperidol. Schizophrenia Research 2011; Vol. 130, issue 1‐3:27‐33. [DOI] [PubMed]
- Krakowski M, Czobor P, Citrome L. Weight gain, metabolic parameters, and the impact of race in aggressive inpatients randomized to double‐blind clozapine, olanzapine or haloperidol. Schizophrenia Research 2009;110(1‐3):95‐102. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Krakowski MI. Clozapine and olanzapine in violent schizophrenics. CRISP database (https://www‐commons.cit.nih.gov/crisp/index.html) (accessed 19th February 2001.
- Krakowski MI, Czobor P. A prospective longitudinal study of cholesterol and aggression in patients randomized to clozapine, olanzapine, and haloperidol. Journal of Clinical Psychopharmacology 2010;30(2):198‐200. [DOI] [PubMed] [Google Scholar]
- Krakowski MI, Czobor P. Executive function predicts response to antiaggression treatment in schizophrenia: A randomized controlled trial. Journal of Clinical Psychiatry 2012; Vol. 73, issue 1:74‐80. [DOI] [PubMed]
- Krakowski MI, Czobor P. Neurocognitive impairment limits the response to treatment of aggression with antipsychotic agents. Schizophrenia Bulletin 2011; Vol. 37:311‐2.
- Krakowski MI, Czobor P, Citrome L, Bark N, Cooper TB. Atypical antipsychotic agents in the treatment of violent patients with schizophrenia and schizoaffective disorder. Archives of General Psychiatry 2006;63(6):622‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- NCT01123408. Clozapine and Olanzapine Treatment of Aggression. http://ClinicalTrials.gov/ct2/show/NCT01123408 2010.
Krakowski 2008 {published data only}
- Krakowski M, Czobor P. A prospective longitudinal study of cholesterol and aggression in patients randomized to clozaplne, olanzapine and haloperidol. Proceedings of the 15th Biennial Winter Workshop in Psychoses; 2009 Nov 15‐18; Barcelona, Spain. 2009. [DOI] [PubMed]
- Krakowski MI, Czobor P, Nolan KA. Atypical antipsychotics, neurocognitive deficits, and aggression in schizophrenic patients. Journal of Clinical Psychopharmacology 2008;28(5):485‐93. [DOI] [PubMed] [Google Scholar]
- Nolan KA, Volavka J, Czobor P, Sheitman B, Lindenmayer J‐P, Citrome LL, e al. Aggression and psychopathology in treatment‐resistant inpatients with schizophrenia and schizoaffective disorder. Journal of Psychiatric Research 2005;39(1):109‐15. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Lewis 2006 {published data only}
- Lewis CF. Haloperidol versus risperidone in the treatment of aggressive psychotic male inmates. Proceedings of the 159th Annual Meeting of the American Psychiatric Association; 2006 May 20‐25; Toronto, Canada. 2006.
- NCT00203775. Efficacy of risperidone versus haloperidol in the treatment of aggression and hostility in psychotic inmates. http://www.clinicaltrials.gov 2005.
Li 2005 {published data only}
- Li GL. Tiapride intramuscular haloperidol in the treatment of acute schizophrenia excited state control study [硫必利肌注与氟哌啶醇治疗精神分裂症急性期兴奋状态的对照研究]. Nervous Diseases and Mental Health [神经疾病与精神卫生] 2005; Vol. 5, issue 6:456‐7.
Li 2007 {published data only}
- Li C, Zhu S, Wang H, Chen H, Yu Y, Liu D, et al. Safety and efficacy of clonazepam, haloperidol and haloperidol combined with clonazepam in the in the treatment of schizophrenia with excitement and agitation [氯硝西泮、氟哌啶醇及其联合治疗对精神分裂症激越症状疗效的比较]. Shanghai Archives of Psychiatry [上海精神医学] [上海精神醫學] 2007;19(3):150‐2. [Google Scholar]
Liang 2003 {published data only}
- 梁芝国, 罗维肖. Clonazepam and haloperidol on patients with acute agitation control study [氯硝西泮和氟哌啶醇联用治疗急性激越的对照研究]. Medical Journal of Chinese Civil Administration 2003; Vol. 15, issue 5:293‐4.
Liu 2012b {published data only}
- 刘庆梅. 无抽搐电休克治疗精神分裂症伴激越行为疗效观察及护理干预. 临床心身疾病杂志 2012; Vol. 18, issue 5:437‐9.
Mandel 2008 {published data only}
- Mandel F, Allen MH, Lombardo I. Early response to intramuscular ziprasidone as a predictor of end point response to oral ziprasidone. Proceedings of the 161st Annual Meeting of the American Psychiatric Association; 2008 May 3‐8; Washington DC, USA. 2008.
Mantua 2006 {published data only}
- Mantua V, Travis MJ, Atakan Z, Isaac MB, Isaac MT, Smith S, et al. Treatment of agitation caused by severe mental illness: data from the South London and Maudsley intensive care units trial evaluation (SLAMICUTE) study. European Neuropsychopharmacology 2006;16(Suppl 4):S420. [Google Scholar]
McEvoy 1994 {published data only}
- McEvoy JP. Efficacy of risperidone on positive features of schizophrenia. Journal of Clinical Psychiatry 1994;55(5 Suppl):18‐21. [MEDLINE: ] [PubMed] [Google Scholar]
Mendelowitz 2004 {published data only}
- Mendelowitz AJ. The utility of intramuscular ziprasidone in the management of acute psychotic agitation. Annals of Clinical Psychiatry 2004;16(3):145‐54. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
NCT00189995 2005 {published data only}
- NCT00189995. Intramuscular clozapine in the management of aggression in schizophrenic patients. http://www.clinicaltrials.gov 2005.
NCT00631722 2008 {published data only}
- NCT00631722. Multicenter, open‐label, randomised, haloperidol‐controlled study to evaluate seroquel as mono‐therapy in the treatment of agitated symptoms in the patients with acute episode of schizophrenia. http://www.clinicaltrials.gov 2008.
NCT00797277 2008 {published data only}
- NCT00797277. IM olanzapine versus IM haloperidol plus lorazepam for acute agitation in schizophrenia. http://www.clinicaltrials.gov 2008.
Pan 2005 {published data only}
- Pan N, Yang X, Mei Q. Large dose of olanzapine in the ictus treatment of agitated symptoms of schizophrenia. Journal of Clinical Psychosomatic Diseases [临床心身疾病杂志] 2005;11(2):113‐5. [Google Scholar]
Pathiraja 1995 {published data only}
- Pathiraja AP, Diamond BI, Borison RL, Meibech RC, Anand R. Relationship between creatine phosphokinase, psychotic symptoms and novel antipsychotic drugs. Schizophrenia Research 1995;15(1‐2):160‐1. [Google Scholar]
Pedros 2004 {published data only}
- Pedros Rosello A, Tenias JM. Neuroleptics election and clinical outcomes in acute psychosis: A comparative study of risperidone versus other neuroleptics [Eleccion de neuroleptico y respuesta clinica en psicosis aguda: Un estudio comparativo de risperidona frente a otros neurolepticos]. Anales de Psiquiatria 2004;20(4):167‐71. [Google Scholar]
Pei 2009 {published data only}
- 裴树景, 杨红卫. Oral risperidone treatment of schizophrenia clinical observation of excited agitation [Google Translate] [利培酮口服液治疗精神分裂症兴奋激越临床观察]. Linchuang Jingshen Yixue Zazhi [临床精神医学杂志] 2009; Vol. 19, issue 6:405‐6.
Puech 1998 {published data only}
- Muller MJ, Wetzel H, Eich FX, Rein W, Puech A, Benkert O, Amisulpride Study Group. Dose‐related effects of amisulpride on five dimensions of psychopathology in patients with acute exacerbation of schizophrenia. Journal of Clinical Psychopharmacology 2002;22(6):554‐60. [DOI] [PubMed] [Google Scholar]
- Puech A, Fleurot O, Rein W, Amisulpride Study Group. Amisulpride, an atypical antipsychotic, in the treatment of acute episodes of schizophrenia: a dose‐ranging study vs. haloperidol. Acta Psychiatrica Scandinavica 1998;98(1):65‐72. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Romeo 2009 {published data only}
- Romeo R, Knapp M, Tyrer P, Crawford M, Oliver‐Africano P. The treatment of challenging behaviour in intellectual disabilities: cost‐effectiveness analysis. Journal of Intellectual Disability Research 2009;53:633‐43. [DOI] [PubMed] [Google Scholar]
- Tyrer P. Neuroleptics in adults with aggressive challenging behaviour and intellectual disability. http://www.controlled‐trials.com 2003.
Simpson 2010 {published data only}
- Simpson G, Mohammad AR, Fauzia S, Campaneau M, Laska E, Ninah R, et al. A comparison of rapid titration quetiapine and haloperidol in agitated adults in an emergency setting. Proceedings of the 163rd Annual Meeting of the American Psychiatric Association; 2010 May 22‐26; New Orleans, LA 2010.
Singh 1981 {published data only}
- Singh MM, Kay SR, Pitman RK. Aggression control and structuring of social relations among recently admitted schizophrenics. Psychiatry Research 1981;5(2):157‐69. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Smith 1974 {published data only}
- Smith GR, Taylor CW, Linkous P. Haloperidol versus thioridazine for the treatment of psychogeriatric patients: A double‐blind clinical trial. Psychosomatics 1974;15(3):134‐8. [Google Scholar]
Srinath 2010 {published data only}
- Srinath G, Shailaja B, Sai PG, Reddy KA. A comparative study of injection haloperidol and promethazine vs. injection lorazepam in controlling acute psychotic agitation. Indian Journal of Psychiatry 2010; Vol. 52:S21.
Teja 1975 {published data only}
- Teja JS, Grey WH, Clum JM, Warren C. Tranquilizers or anti‐depressants for chronic schizophrenics: A long term study. Australian and New Zealand Journal of Psychiatry 1975;9(4):241‐7. [DOI] [PubMed] [Google Scholar]
Thomas 1992 {published data only}
- Thomas HJ, Schwartz E, Petrilli R. Droperidol versus haloperidol for chemical restraint of agitated and combative patients. Annals of Emergency Medicine 1992;21(4):407‐13. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
TREC CG {published data only}
- Coutinho ES, Huf G, Allen MH, Adams CE. Physical restraints for agitated patients in psychiatric emergency hospitals in Rio de Janeiro, Brazil: a predictive model. Schizophrenia Bulletin 2005;31:220. [Google Scholar]
- Huf G. Rapid safe tranquillisation for acutely disturbed people attending public psychiatric emergency clinics in Rio de Janeiro. http://www.controlled‐trials.com 2002.
- Huf G, Coutinho ESF, Adams CE. TREC I. Background. Schizophrenia Research 2002;53(3 Suppl 1):187. [Google Scholar]
- Huf G, Coutinho ESF, Adams CE. TREC III. The protocol and progress of TREC. Schizophrenia Research 2002;53(3 Suppl 1):187. [Google Scholar]
- Huf G, Coutinho ESF, Adams CE. TREC‐Rio trial: A randomised controlled trial for rapid tranquillisation for agitated patients in emergency psychiatric rooms. BMC Psychiatry 2002;2(11):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huf G, Coutinho ESF, Fagundes HM Jr, Carvalho AL, Ramos FA, Keusen AL, Oliveira ES, et al. TREC II. Current practices in managing acutely disturbed patients at three hospitals in Rio de Janeiro, Brazil. Schizophrenia Research 2002;53(3):236‐7. [Google Scholar]
- TREC CG. Rapid tranquillisation for agitated patients in emergency psychiatric rooms: A randomised trial of midazolam versus haloperidol plus promethazine. BMJ 2003;327(7417):708‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ungvari 1982 {published data only}
- Ungvari G, Petho B. High‐dose haloperidol therapy: Its effectiveness and a comparison with electroconvulsive treatment. Journal of Psychiatric Treatment and Evaluation 1982;4:279‐83. [Google Scholar]
Vaisanen 1981 {published data only}
- Vaisanen K, Viukari M, Rimon R, Raisanen P. Haloperidol, thioridazine and placebo in mentally subnormal patients‐serum levels and clinical effects. Acta Psychiatrica Scandinavica 1981;63(3):262‐71. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Veser 2006 {published data only}
- Veser F, Zealburg J, Veser B, Zhu Y, Gharabawi G. Oral risperidone in the management of agitated behavior in emergency settings. European Neuropsychopharmacology 2002;12(Suppl 3):S313. [Google Scholar]
- Veser FH, Veser BD, McMullan JT, Zealberg J, Currier GW. Risperidone versus haloperidol, in combination with lorazepam, in the treatment of acute agitation and psychosis: a pilot, randomized, double‐blind, placebo‐controlled trial. Journal of Psychiatric Practice 2006;12(2):103‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Vives 2015 {published data only}
- Vives Vilagut R, Duno R, Planet N, Garcia G, Payes M, Marinosa M, et al. Testing the intranasal route for administration of haloperidol in emergency room: Generation of efficacy data to support clinical practice. Clinical Therapeutics 2015; Vol. 1:e152.
Wan 2005 {published data only}
- Wan Z, Zhong Z. Curative effect of risperidone with BDZs in the treatment of excitement and agitate with schizophrenia in acute phase. Heath Psychology Journal 2005;13(1):23‐4. [Google Scholar]
Wang 2004 {published data only}
- Wang G, Cai Z‐J, Wang L‐F. A multicenter study of risperidone treatment for acute agitation in patients with schizophrenia. Chinese Journal of Psychiatry 2004;37(2):88‐91. [Google Scholar]
Wang 2005 {published data only}
- Wang H, Guo X, Shen T. Safety and efficacy of quetiapine and haloperidol in the treatment of schizophrenia with excitement and agitation. Shanghai Archives of Psychiatry [上海精神医学] [上海精神醫學] 2005;17(5):271‐4. [Google Scholar]
Wyant 1990 {published data only}
- Wyant M, Diamond BI, O'Neal E, Sloan A, Borison RL. The use of midazolam in acutely agitated psychiatric patients. Psychopharmacology Bulletin 1990;26(1):126‐9. [PubMed] [Google Scholar]
Zapletalek 1986 {published data only}
- Zapletalek M, Tuma I, Libiger J. Zetidoline compared with haloperidol in a double blind clinical study. Activitas Nervosa Superior 1986;28(1):31‐2. [Google Scholar]
References to studies awaiting assessment
Daniel 2006 {published data only}
- Daniel DG, Crandall D, Manos G, McQuade RD, Gutierrez‐Esteinou R, Pikalov AA, et al. Transitioning from intramuscular (IM) to oral aripiprazole in patients with schizophrenia. Proceedings of the 159th Annual Meeting of the American Psychiatric Association; 2006 May 20‐25; Toronto, Canada. 2006.
Davis 2008 {published data only}
- Davis JM, Wang B, He Y, Jin H, Hu Q, Zhang M. The emergency treatment of acutely agitated psychotic schizophrenia patient. International Journal of Neuropsychopharmacology 2008;11(Suppl 1):163. [Google Scholar]
Dubin 1985 {published data only}
- Dubin WR, Waxman HM, Weiss KJ, Ramchandani D, Tavani‐Petrone C. Rapid tranquilization: the efficacy of oral concentrate. Journal of Clinical Psychiatry 1985;46(11):475‐8. [MEDLINE: ] [PubMed] [Google Scholar]
Esmailian 2015 {published data only}
- Esmailian M, Ahmadi O, Taheri M, Zamani M. Comparison of haloperidol and midazolam in restless management of patients referred to the Emergency DepartmeNT: PDF: A double‐blinded, randomized clinical trial. Journal of Research in Medical Sciences 2015; Vol. 20, issue 9:844‐9. [DOI] [PMC free article] [PubMed]
Herrera 2005 {published data only}
- Herrera M. Double‐blind study with risperidone vs haloperidol in schizophrenic patients with agitation and/or aggression. Proceedings of the 8th World Congress of Psychiatry; 2005 Sep 10‐15; Cairo, Egypt. 2005.
Hsu 2010 {published data only}
- Hsu WY, Huang SS, Lee BS, Chiu NY. Comparison of intramuscular olanzapine, orally disintegrating olanzapine tablets, oral risperidone solution, and intramuscular haloperidol in the management of acute agitation in an acute care psychiatric ward in Taiwan. Journal of Clinical Psychopharmacology 2010;30(3):230‐4. [DOI] [PubMed] [Google Scholar]
Istikoglou 2010 {published data only}
- Istikoglou C, Vlavianou A, Foutsitzis D, Theodorakopoulos G, Polonifis C, Kanellos P, et al. Intramuscular aripiprazole versus injectable haloperidol in treatment of acute psychotic agitation. European Neuropsychopharmacology 2010;20(3):S469‐70. [Google Scholar]
Kong 2009 {published data only}
- Kong BG, Shim JC, Lee SK. Comparison of the effects of intramuscular antipsychotics injection in the treatment of acute agitation in psychosis. European Neuropsychopharmacology 2009; Vol. 19:S525‐S6.
Lasic 2006 {published data only}
- Lasic D, Rubesa G, Dodig G, Katavic Z, Glavina T, Zuljan‐Cvitanovic M. Risperidone oral solution, therapeutic possibility in treatment of acute and agitated psychotic patients. European Neuropsychopharmacology 2006;16(Suppl 4):S445. [Google Scholar]
Li 2007a {published data only}
- Li C, Zhu S, Wang H, Chen H, Yu Y, Liu D, et al. Safety and efficacy of clonazepam, haloperidol and haloperidol combined with clonazepam in the [氯硝西泮、氟哌啶醇及其联合治疗对精神分裂症激越症状疗效的比较]. Shanghai Archives of Psychiatry [上海精神医学] [上海精神醫學] 2007; Vol. 19, issue 3:150‐2.
Lu 2006 {published data only}
- Lu D, Ning J, Zhong X. Clinical evaluation on treatment of acute‐agitation in schizophrenia with zipraside injection [甲磺酸齐拉西酮注射液治疗精神分裂症急性激越症状的临床评价]. China Pharmaceuticals 2006;15(4):48‐9. [Google Scholar]
NCT00859872 2009 {published data only}
- NCT00859872. Efficacy and safety of risperidone oral solution combination clonazepam versus haloperidol intramuscular (IM) injection for treatment of acute psychotic agitation in schizophrenia. http://www.clinicaltrials.gov 2009.
NCT00866645 2009 {published data only}
- NCT00866645. A study to evaluate the efficacy and safety of intramuscular levosulpiride in patients with agitation of schizophrenia. http://www.clinicaltrials.gov 2009.
Shim 2009 {published data only}
- Shim JC, Kong BG, Lee SG, Kim YH. The effect of intramuscular olanzapine in the treatment of acute agitation in patient with psychotic symptoms. European Neuropsychopharmacology 2009; Vol. 19:S512‐S3.
Si 2009 {published data only}
- Si T, Zhong HJ, Yun'Ai S, Qin TM, Qing LK, De YP, et al. A multi‐site,open‐label, randomised, haloperidol‐controlled study to evaluate quetiapine as monotherapy in the treatment of agitated acute schizophrenia. Proceedings of the 162nd Annual Meeting of the American Psychiatric Association; 2009 May 16‐21; San Francisco, CA 2009.
Slotnick 1971 {published data only}
- Slotnick VB. Management of the acutely agitated psychiatric patient with parental neuroleptics: The comparitive symptom effectiveness profiles of haloperidol and chlorpromazine. Proceedings of the 5th World Congress of Psychiatry; 1971 Nov 28 ‐ Dec 4; Mexico City, Mexico. 1971.
Smythies 1982 {published data only}
- Smythies JR, Hain J, Crews E, Grayson G. A clinical trial of injectable molindone (Moban(R)). Current Therapeutic Research, Clinical and Experimental 1982;32(5):752‐6. [Google Scholar]
Vasquez‐Gomez 2001 {published data only}
- Vasquez‐Gomez F. The use of midazolam for control of acutely agitated outpatients in psychiatry emergency. Proceedings of the 7th World Congress of Biological Psychiatry; 2001 Jul 1‐6; Berlin, Germany. 2001.
Wang 2005a {published data only}
- Wang R‐C, Li L‐H, Zhao Y‐C. Ziprasidone made in China and haloperidol in treating acute onset of schizophrenia: a randomized, single blinded, double modelling, parallel control study. Chinese Journal of Clinical Rehabilitation 2005; Vol. 9, issue 44:83‐5.
Wang 2007 {published data only}
- Wang J‐C, Xu X‐F, Ouyang H, Li W‐Y. The second phase clinical trials of intramuscular ziprasidone for the treatment of acute psychotic agitation of the schizophrenia [注射用甲磺酸齐拉西酮治疗精神分裂症兴奋躁动状态的Ⅱ期临床试验]. Nervous Diseases and Mental Health [神经疾病与精神卫生] 2007;7(5):364‐70. [Google Scholar]
Yildiz 2003 {published data only}
- Yildiz A, Turgay A, Alpay M, Sachs GS. Risperidone versus haloperidol in adult psychiatric emergencies. Proceedings of the 156th Annual Meeting of the American Psychiatric Association; 2003 May 17‐22; San Francisco, California, USA 2003:Nr259.
Zhang 2012 {published data only}
- 张庆娥, 王刚, 张玲, 王雪, 罗炯, 路亚洲, et al. Control study of acute agitation in schizophrenia risperidone oral solution merger clonazepam tablets treatment [Google Translate] [利培酮口服液合并氯硝西泮片治疗精神分裂症急性激越的对照研究]. 中华医学会第十次全国精神医学学术会议 2012; Vol. 22, issue 02:89‐91.
叶萌, 2009 {published data only}
- 叶萌, 房茂胜, 李乐华, 赵靖平. Domestic ziprasidone mesylate injection in the treatment of acute agitation of schizophrenia [国产甲磺酸齐拉西酮注射液治疗精神分裂症急性激越症状疗效观察]. Journal of the Xinxiang Medical College [新乡医学院学报] 2009; Vol. 26, issue 1:63‐5.
周德祥, 2012 {published data only}
- 周德祥, 蒋幸衍, 徐清, 方馨怡, 夏鸣华, 陆雅娜. 利培酮口服液合并氯硝西泮治疗精神分裂症兴奋激越症状的随机对照研究. 中国健康心理学杂志 2012; Vol. 8:1123‐4.
References to ongoing studies
NCT00838032 2009 {published data only}
- NCT00838032. Pilot study to evaluate the efficacy and safety of quetiapine fumarate instant‐release (seroquel ir) in controlling agitation and aggressive symptoms in the acute treatment of patients with schizophrenia. http://www.clinicaltrials.gov 2009.
Additional references
Adams 2013
- Adams CE, Bergman H, Irving CB, Lawrie S. Haloperidol versus placebo for schizophrenia. Cochrane Database of Systematic Reviews 2013, Issue 11. [DOI: 10.1002/14651858.CD003082.pub3; CD003082] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ahmed 2010
- Ahmed U, Jones H, Adams CE. Chlorpromazine for psychosis induced aggression or agitation. Cochrane Database of Systematic Reviews 2010, Issue 4. [DOI: 10.1002/14651858.CD007445.pub2; PUBMED: 20393959] [DOI] [PubMed] [Google Scholar]
Ahmed 2011
- Ahmed U, Rehman F, Jones H, Adams CE. Risperidone for psychosis induced aggression or agitation. Cochrane Database of Systematic Reviews 2011, Issue 11. [DOI: 10.1002/14651858.CD009412; CD009412] [DOI] [PMC free article] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
APA 2006
- American Psychiatric Association. Practice Guideline for the Treatment of Patients With Schizophrenia. 2nd Edition. Washington DC: American Psychiatric Association, 2006. [DOI: 10.1176/appi.books.9780890423363.45859] [DOI] [Google Scholar]
Barnes 1989
- Barnes TR. A rating scale for drug‐induced akathisia. British Journal of Psychiatry 1989;154:672‐6. [DOI] [PubMed] [Google Scholar]
Belgamwar 2005
- Belgamwar RB, Fenton M. Olanzapine IM or velotab for acutely disturbed/agitated people with suspected serious mental illnesses. Cochrane Database of Systematic Reviews 2005, Issue 2. [DOI: 10.1002/14651858.CD003729.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Berk 2004
- Berk M, Rathbone J, Mandriota‐Carpenter SL. Clotiapine for acute psychotic illnesses. Cochrane Database of Systematic Reviews 2004, Issue 4. [DOI: 10.1002/14651858.CD002304.pub2; CD002304] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bland 1997
- Bland JM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
BNF 2011
- British National Formulary. British National Formulary. 61. NHS, London: Pharmaceutical Press, 2011. [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'etude des indices d'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
Choudhury 2011
- Choudhury R, Dewsbery M, Williams K, Hovey N. Audit of the rapid tranquilization policy in the 2gether NHS foundation trust. Journal of Psychiatric Intensive Care 2011;7(1):55‐61. [Google Scholar]
Corrigan 1989
- Corrigan JD. Development of a scale for assessment of agitation following traumatic brain injury. Journal of Clinical and Experimental Neuropsychology 1989;11(2):261‐77. [DOI] [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Proceedings of the 8th International Cochrane Colloquium; 2000 Oct 25‐28; Cape Town. Cape Town: The Cochrane Collaboration, 2000.
Divine 1992
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Douglas‐Hall 2015
- Douglas‐Hall P, Whicher EV. 'As required' medication regimens for seriously mentally ill people in hospital. Cochrane Database of Systematic Reviews 2015, Issue 12. [DOI: 10.1002/14651858.CD003441.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Du 2017
- Du M, Wang X, Yin S, Shu W, Hao R, Zhao S, Rao H, Yeung WL, Jayaram MB, Xia J. De‐escalation techniques for psychosis‐induced aggression or agitation. Cochrane Database of Systematic Reviews 2017, Issue 4. [DOI: 10.1002/14651858.CD009922.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Easterbrook 1991
- Easterbrook PJ, Berlin JA, Gopalan R, Matthews DR. Publication bias in clinical research. Lancet 1991;337(8746):867‐72. [PUBMED: 1672966] [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(7):7‐10. [DOI] [PubMed] [Google Scholar]
Gillies 2013
- Gillies D, Sampson S, Beck A, Rathbone J. Benzodiazepines for psychosis‐induced aggression or agitation. Cochrane Database of Systematic Reviews 2013, Issue 4. [DOI: 10.1002/14651858.CD003079.pub3] [DOI] [PubMed] [Google Scholar]
Glazer 1999
- Glazer WM. Does loxapine have "atypical" properties? Clinical evidence. Journal of Clinical Psychiatry 1999;60(Suppl 10):42‐6. [PUBMED: 10340686] [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy W. Clinical Global Impression. ECDEU Assessment Manual for Psychopharmacology. revised. National Institute of Mental Health, 1976:217‐21. [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Huf 2016
- Huf G, Alexander J, Gandhi P, Allen MH. Haloperidol plus promethazine for psychosis‐induced aggression. Cochrane Database of Systematic Reviews 2016, Issue 11. [DOI: 10.1002/14651858.CD005146.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Isbister 2010
- Isbister GK, Calver LA, Page CB, Stokes B, Bryant JL, Downes MA. Randomized controlled trial of intramuscular droperidol versus midazolam for violence and acute behavioral disturbance: the DORM study. Annals of Emergency Medicine 2010;56(4):392‐401. [PUBMED: 20868907] [DOI] [PubMed] [Google Scholar]
Janssen‐Cilag Ltd 2010
- Janssen‐Cilag Ltd. Haldol (haloperidol) 5mg/ml Injection. Summary of product characteristics. http://www.medicines.org.uk/emc/medicine/7267/spc/haldol+injection/ (accessed 3 June 2011).
Jayakody 2012
- Jayakody K, Gibson RC, Kumar A, Gunadasa S. Zuclopenthixol acetate for acute schizophrenia and similar serious mental illnesses. Cochrane Database of Systematic Reviews 2012, Issue 4. [DOI: 10.1002/14651858.CD000525.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Kay 1987
- Kay SR, Flszbein A, Opfer LA. The Positive and Negative Syndrome Scale (PANSS) for Schizophrenia. Psychological Reports 1962;10:799‐812. [DOI] [PubMed] [Google Scholar]
Khokhar 2016
- Khokhar MA, Rathbone J. Droperidol for psychosis‐induced aggression or agitation. Cochrane Database of Systematic Reviews 2016, Issue 12. [DOI: 10.1002/14651858.CD002830.pub3; CD002830] [DOI] [PMC free article] [PubMed] [Google Scholar]
Khushu 2016
- Khushu A, Powney MJ. Haloperidol for long‐term aggression in psychosis. Cochrane Database of Systematic Reviews 2016, Issue 11. [DOI: 10.1002/14651858.CD009830.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Leucht 2005
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of brief psychiatric rating scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Leucht 2007
- Leucht S, Engel RR, Bauml J, Davis JM. Is the superior efficacy of new generation antipsychotics an artifact of LOCF?. Schizophrenia Bulletin 2007;33(1):183‐91. [PUBMED: 16905632] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lexchin 2003
- Lexchin J, Bero LA, Djulbegovic B, Clark O. Pharmaceutical industry sponsorship and research outcome and quality: Systematic review. BMJ 2003;326(7400):1167‐70. [PUBMED: 12775614] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lieberman 2005
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. New England Journal of Medicine 2005;353(12):1209‐23. [PUBMED: 16172203] [DOI] [PubMed] [Google Scholar]
López‐Munoz 2009
- López‐Munoz F, Alamo C. The consolidation of neuroleptic therapy: Janssen, the discovery of haloperidol and its introduction into clinical practice. Brain Research Bulletin 2009;79:130–41. [DOI] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Moher 2001
- Moher D, Schulz KF, Altman DG. The CONSORT statement:revised recommendations for improving the quality of reports of parallel‐group randomised trials. Lancet 2001;357(9263):1191‐4. [PubMed] [Google Scholar]
Mohr 2005
- Mohr P, Pecenak J, Svestka J, Swingler D, Treuer T. Treatment of acute agitation in psychotic disorders. Neuro Endocrinology Letters 2005;26(4):327‐35. [PUBMED: 16136016] [PubMed] [Google Scholar]
Montoya 2011
- Montoya A, Valladares A, Lizán L, San L, Escobar R, Paz S. Validation of the Excited Component of the Positive and Negative Syndrome Scale (PANSS‐EC) in a naturalistic sample of 278 patients with acute psychosis and agitation in a psychiatric emergency room. Health Quality Life Outcomes 2011;9:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Muralidharan 2006
- Muralidharan S, Fenton M. Containment strategies for people with serious mental illness. Cochrane Database of Systematic Reviews 2006, Issue 3. [DOI: 10.1002/14651858.CD002084.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
NHS 2009
- National Health Service. The prescribing preview report on antipsychotic drugs, available to general practitioners in August 2009, is reproduced here for readers with an interest in patterns and trends of prescribing. Impact: Prescribing and Dispensing Newsletter. http://www.nhsbsa.nhs.uk/PrescriptionServices/Documents/PrescriptionServices/Impact_Dec_09_8p.pdf (accessed 2nd June 2011):4‐6.
NICE 2005
- National Institute for Clinical Excellence. The short term‐management of disturbed/violent behaviour in inpatient psychiatric settings and emergency departments. Vol. 25, London: NICE, 2005. [Google Scholar]
NICE 2015
- National Institute for Clinical Excellence. Violence and aggression: short‐term management in mental health, health and community settings. nice.org.uk/guidance/ng10 2015.
Olkkola 2008
- Olkkola KT, Ahonen J. Midazolam and other benzodiazepines. Handbook of Experimental Pharmacology 2008;182:335‐60. [PUBMED: 18175099] [DOI] [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The brief psychiatric rating scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Parker 2010
- Parker C, Khwaja MG. What is new in rapid tranquillisation?. Journal of Psychiatric Intensive Care 2010;7(2):1‐11. [Google Scholar]
Pereira 2007
- Pereira S, Fleischhacker W, Allen M. Management of behavioural emergencies. Journal of Psychiatric Intensive Care 2007;2(2):71–83. [Google Scholar]
Pratt 2008
- Pratt P, Chandler‐Oatts J, Nelstrop L, Branford D, Periera S, Johnston S. Establishing gold standard approaches to rapid tranquillisation: A review and discussion of the evidence of the safety and efficacy of medications used. Journal of Psychiatric Intensive Care 2008;4(1‐2):43‐57. [Google Scholar]
Reid 2003
- Reid G, Hughson M. Droperidol dropped; consultants not consulted: A survey of the practice of rapid tranquillisation by consultant psychiatrists in the west of Scotland. Psychiatric Bulletin 2003;27:301‐4. [Google Scholar]
Sailas 2000
- Sailas E, Fenton M. Seclusion and restraint for people with serious mental illnesses. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: 10.1002/14651858.CD001163] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schleifer 2011
- Schleifer JJ. Management of acute agitation in psychosis: an evidence‐based approach in the USA. Advances in Psychiatric Treatment 2011;17:91‐100. [Google Scholar]
Schünemann 2008
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. The Cochrane Collaboration, 2008:359‐83. [Google Scholar]
Simpson 1970
- Simpson GM, Angus JSW. A rating scale for extrapyramidal side effects. Acta Psychiatri Scandinavica. Supplementum 1970;212:11‐9. [DOI] [PubMed] [Google Scholar]
Thorpe 2009
- Thorpe KE, Zwarenstein M, Oxman AD, Treweek S, Furberg CD, Altman DG, et al. A pragmatic‐explanatory continuum indicator summary (PRECIS): a tool to help trial designers. Journal of Clinical Epidemiology 2009;62(5):464‐75. [PUBMED: 19348971] [DOI] [PubMed] [Google Scholar]
TREC 2003
- TREC Collaborative Group. Rapid tranquillisation for agitated patients in emergency psychiatric rooms: a randomised trial of midazolam versus haloperidol plus promethazine. BMJ 2003;327(7417):708‐13. [PUBMED: 14512476] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organistation‐based intervention in health and health care: A systematic review. Health Technology Assessment 1999;3(5):1‐75. [PubMed] [Google Scholar]
Vangala 2012
- Vangala R, Ahmed U, Ahmed R. Loxapine inhaler for psychosis‐induced aggression. Cochrane Database of Systematic Reviews 2012.
WHO 2011
- World Health Organization. WHO Essential Medicines List. http://whqlibdoc.who.int/hq/2011/a95053_eng.pdf (accessed 2nd June 2011).
WHO 2015
- World Health Organization. WHO Model List of Essential Medicines. http://www.who.int/medicines/publications/essentialmedicines/EML_2015_FINAL_amended_NOV2015.pdf?ua=1 2015.
Wilkie 2012
- Wilkie F, Fenton M. Quetiapine for psychosis‐induced aggression or agitation. Cochrane Database of Systematic Reviews 2012, Issue 4. [DOI: 10.1002/14651858.CD009801] [DOI] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]
Xiberas 2001
- Xiberas X, Martinot JL, Mallet L, Artiges E, Loc'h C, Maziere B, et al. Extrastriatal and striatal D2 dopamine receptor blockade with haloperidol or new antipsychotic drugs in patients with schizophrenia. British Journal of Psychiatry 2001;179:503‐8. [DOI] [PubMed] [Google Scholar]
Yudofsky 1986
- Yudofsky SC, Silver JM, Jackson W, Endicott J, Williams D. The Overt Aggression Scale for the objective rating of verbal and physical aggression. American Journal of Psychiatry 1986;143:35‐9. [DOI] [PubMed] [Google Scholar]
Yudofsky 1997
- Yudofsky SC, Kopecky HJ, Kunik M, Silver JM, Endicott J. The Overt Agitation Severity Scale for the objective rating of agitation. Journal of Neuropsychiatry 1997;9(4):541‐8. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Powney 2011
- Powney MJ, Adams CE, Jones H. Haloperidol (rapid tranquilisation) for psychosis induced aggression or agitation. Cochrane Database of Systematic Reviews 2011, Issue 10. [DOI: 10.1002/14651858.CD009377] [DOI] [PubMed] [Google Scholar]
Powney 2012
- Powney MJ, Adams CE, Jones H. Haloperidol for psychosis‐induced aggression or agitation (rapid tranquillisation). Cochrane Database of Systematic Reviews 2012, Issue 11. [DOI: 10.1002/14651858.CD009377.pub2] [DOI] [PubMed] [Google Scholar]