

Abstract
Background
Induction of labour is carried out for a variety of indications and using a range of methods. For women at low risk of pregnancy complications, some methods of induction of labour or cervical ripening may be suitable for use in outpatient settings.
Objectives
To examine pharmacological and mechanical interventions to induce labour or ripen the cervix in outpatient settings in terms of effectiveness, maternal satisfaction, healthcare costs and, where information is available, safety.
Search methods
We searched the Cochrane Pregnancy and Childbirth Group’s Trials Register (30 November 2016) and reference lists of retrieved studies.
Selection criteria
We included randomised controlled trials examining outpatient cervical ripening or induction of labour with pharmacological agents or mechanical methods. Cluster trials were eligible for inclusion.
Data collection and analysis
Two review authors independently assessed trials for inclusion and risk of bias, extracted data and checked them for accuracy. We assessed evidence using the GRADE approach.
Main results
This updated review included 34 studies of 11 different methods for labour induction with 5003 randomised women, where women received treatment at home or were sent home after initial treatment and monitoring in hospital.
Studies examined vaginal and intracervical prostaglandin E₂ (PGE₂), vaginal and oral misoprostol, isosorbide mononitrate, mifepristone, oestrogens, amniotomy and acupuncture, compared with placebo, no treatment, or routine care. Trials generally recruited healthy women with a term pregnancy. The risk of bias was mostly low or unclear, however, in 16 trials blinding was unclear or not attempted. In general, limited data were available on the review's main and additional outcomes. Evidence was graded low to moderate quality.
1. Vaginal PGE₂ versus expectant management or placebo (5 studies)
Fewer women in the vaginal PGE₂ group needed additional induction agents to induce labour, however, confidence intervals were wide (risk ratio (RR) 0.52, 95% confidence interval (CI) 0.27 to 0.99; 150 women; 2 trials). There were no clear differences between groups in uterine hyperstimulation (with or without fetal heart rate (FHR) changes) (RR 3.76, 95% CI 0.64 to 22.24; 244 women; 4 studies; low‐quality evidence), caesarean section (RR 0.80, 95% CI 0.49 to 1.31; 288 women; 4 studies; low‐quality evidence), or admission to a neonatal intensive care unit (NICU) (RR 0.32, 95% CI 0.10 to 1.03; 230 infants; 3 studies; low‐quality evidence).
There was no information on vaginal birth within 24, 48 or 72 hours, length of hospital stay, use of emergency services or maternal or caregiver satisfaction. Serious maternal and neonatal morbidity or deaths were not reported.
2. Intracervical PGE₂ versus expectant management or placebo (7 studies)
There was no clear difference between women receiving intracervical PGE₂ and no treatment or placebo in terms of need for additional induction agents (RR 0.98, 95% CI 0.74 to 1.32; 445 women; 3 studies), vaginal birth not achieved within 48 to 72 hours (RR 0.83, 95% CI 0.68 to 1.02; 43 women; 1 study; low‐quality evidence), uterine hyperstimulation (with FHR changes) (RR 2.66, 95% CI 0.63 to 11.25; 488 women; 4 studies; low‐quality evidence), caesarean section (RR 0.90, 95% CI 0.72 to 1.12; 674 women; 7 studies; moderate‐quality evidence), or babies admitted to NICU (RR 1.61, 95% CI 0.43 to 6.05; 215 infants; 3 studies; low‐quality evidence). There were no uterine ruptures in either the PGE₂ group or placebo group.
There was no information on vaginal birth not achieved within 24 hours, length of hospital stay, use of emergency services, mother or caregiver satisfaction, or serious morbidity or neonatal morbidity or perinatal death.
3. Vaginal misoprostol versus placebo (4 studies)
One small study reported on the rate of perinatal death with no clear differences between groups; there were no deaths in the treatment group compared with one stillbirth (reason not reported) in the control group (RR 0.34, 95% CI 0.01 to 8.14; 77 infants; 1 study; low‐quality evidence).
There was no clear difference between groups in rates of uterine hyperstimulation with FHR changes (RR 1.97, 95% CI 0.43 to 9.00; 265 women; 3 studies; low‐quality evidence), caesarean section (RR 0.94, 95% CI 0.61 to 1.46; 325 women; 4 studies; low‐quality evidence), and babies admitted to NICU (RR 0.89, 95% CI 0.54 to 1.47; 325 infants; 4 studies; low‐quality evidence).
There was no information on vaginal birth not achieved within 24, 48 or 72 hours, additional induction agents required, length of hospital stay, use of emergency services, mother or caregiver satisfaction, serious maternal, and other neonatal, morbidity or death.
No substantive differences were found for other comparisons. One small study found that women who received oral misoprostol were more likely to give birth within 24 hours (RR 0.65, 95% CI 0.48 to 0.86; 87 women; 1 study) and were less likely to require additional induction agents (RR 0.60, 95% CI 0.37 to 0.97; 127 women; 2 studies). Women who received mifepristone were also less likely to require additional induction agents (average RR 0.59, 95% CI 0.37 to 0.95; 311 women; 4 studies; I² = 74%); however, this result should be interpreted with caution due to high heterogeneity. One trial each of acupuncture and outpatient amniotomy were included, but few review outcomes were reported.
Authors' conclusions
Induction of labour in outpatient settings appears feasible and important adverse events seem rare, however, in general there is insufficient evidence to detect differences. There was no strong evidence that agents used to induce labour in outpatient settings had an impact (positive or negative) on maternal or neonatal health. There was some evidence that compared to placebo or no treatment, induction agents administered on an outpatient basis reduced the need for further interventions to induce labour, and shortened the interval from intervention to birth.
We do not have sufficient evidence to know which induction methods are preferred by women, the interventions that are most effective and safe to use in outpatient settings, or their cost effectiveness. Further studies where various women‐friendly outpatient protocols are compared head‐to‐head are required. As part of such work, women should be consulted on what sort of management they would prefer.
Keywords: Female; Humans; Pregnancy; Ambulatory Care; Acupuncture Therapy; Acupuncture Therapy/methods; Cesarean Section; Cesarean Section/statistics & numerical data; Dinoprostone; Dinoprostone/administration & dosage; Feasibility Studies; Intensive Care, Neonatal; Intensive Care, Neonatal/statistics & numerical data; Labor, Induced; Labor, Induced/methods; Misoprostol; Misoprostol/administration & dosage; Oxytocics; Randomized Controlled Trials as Topic
Plain language summary
Medications and mechanical interventions for induction of labour in outpatient settings
What is the issue?
Induction of labour (starting labour artificially) is often needed for medical reasons, such as when women have passed their due dates. Different induction methods can be used, such as medications (like prostaglandin E₂, misoprostol or isosorbide mononitrate) or breaking membranes. Inductions are usually carried out in hospital; some methods may be suitable for use with women treated as outpatients, and allowed to go home to wait for labour to progress. We examined the feasibility, effectiveness and safety of outpatient induction, as well as women’s satisfaction and healthcare costs.
Why is this important?
Pregnant women who have reached their due date can be assessed in hospital as outpatients, given the induction treatment followed by monitoring for a short time, and then sent home. Alternatively, they are given the drug or treatment to take at home. Women may be more comfortable waiting for labour to start at home, and outpatient care may be less costly for health services.
What evidence did we find?
This is an updated review that includes six new studies. We included 34 randomised controlled trials involving 5003 pregnant women (search date: November 2016). The women were healthy and at low risk of complications. They were given induction, a fake treatment (placebo) or no treatment. Limited information was available on the outcomes that were of interest, and risk of bias was generally low or unclear. The quality of evidence was judged to be low‐quality, with a few moderate‐quality findings.
Women at term who were induced as outpatients may be less likely to need further induction, compared to women given placebo or no treatment. Medications like vaginal PGE₂, mifepristone and oral misoprostol appear to be effective. No clear differences were reported for excessive activity of the uterus (hyperstimulation), caesarean section or need for neonatal intensive care.
There were too few women in these trials to determine differences in rare events, such as infant deaths or serious illnesses of mothers or babies. The trials did not report on use of emergency services to return to hospital. Some medications caused side effects (such as headaches). Overall, there was little information on costs of different methods.
What does this mean?
For healthy, low‐risk pregnant women at term, outpatient induction and enabling women to return home to wait for labour to start appears to be feasible. Outpatient induction treatments may reduce both need for further drugs and time from treatment to birth. It does not appear to increase the likelihood of needing other interventions in labour. However, there is insufficient evidence to say definitively whether outpatient induction is safe. Future research should focus on which methods women prefer, and are most effective and safe.
Summary of findings
Summary of findings for the main comparison. Vaginal PGE₂ compared to placebo or expectant management for the induction of labour in outpatient settings.
| Vaginal PGE₂ compared to placebo or expectant management for the induction of labour in outpatient settings | ||||||
| Patient or population: women requiring term labour induction Setting: outpatient clinics and hospitals in the USA Intervention: vaginal PGE₂ Comparison: placebo or expectant management | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo or expectant management | Risk with intravaginal PGE₂ gel | |||||
| Vaginal birth not achieved within 24 h | Study population | ‐ | (0 study) | ‐ | No included trial reported this outcome. | |
| see comment | see comment | |||||
| Vaginal birth not achieved in 48 to 72 h | Study population | ‐ | (0 study) | ‐ | No included trial reported this outcome. | |
| see comment | see comment | |||||
| Uterine hyperstimulation (fetal heart rate changes unclear) | Study population | RR 3.76 (0.64 to 22.24) | 244 (4 RCTs) | ⊕⊕⊝⊝ LOW 1 | There were no events in the control group and so it was not possible to calculate the anticipated absolute effects. | |
| see comment | see comment | |||||
| Caesarean section | Study population | RR 0.80 (0.49 to 1.31) | 288 (4 RCTs) | ⊕⊕⊝⊝ LOW 2 | ||
| 196 per 1000 | 157 per 1000 (96 to 257) | |||||
| Serious neonatal morbidity or death | Study population | ‐ | (0 study) | ‐ | No included trial reported this outcome. | |
| see comment | see comment | |||||
| Serious maternal morbidity or death | Study population | ‐ | (0 studies) | ‐ | No included trial reported this outcome. | |
| see comment | see comment | |||||
| Neonatal intensive care unit admission | Study population | RR 0.32 (0.10 to 1.03) | 230 (3 RCTs) | ⊕⊕⊝⊝ LOW 1 | ||
| 93 per 1000 | 30 per 1000 (9 to 96) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Wide confidence interval crossing the line of no effect, few events and small sample size (‐2).
2 Wide confidence interval crossing the line of no effect and small sample size (‐2).
Summary of findings 2. Intracervical PGE₂ compared to placebo for the induction of labour in outpatient settings.
| Intracervical PGE₂ compared to placebo for the induction of labour in outpatient settings | ||||||
| Patient or population: women requiring induction of labour Setting: outpatient clinics and hospitals in the USA Intervention: intracervical PGE₂ Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with intracervical PGE₂ | |||||
| Vaginal birth not achieved within 24 h | Study population | ‐ | (0 study) | ‐ | No included trial reported this outcome. | |
| see comment | see comment | |||||
| Vaginal birth not achieved in 48 to 72 h | Study population | RR 0.83 (0.68 to 1.02) | 43 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | ||
| 1000 per 1000 | 830 per 1000 (680 to 1000) | |||||
| Uterine hyperstimulation (with fetal heart rate changes) | Study population | RR 2.66 (0.63 to 11.25) | 488 (4 RCTs) | ⊕⊕⊝⊝ LOW 1 | ||
| 4 per 1000 | 11 per 1000 (3 to 45) | |||||
| Caesarean section | Study population | RR 0.90 (0.72 to 1.12) | 674 (7 RCTs) | ⊕⊕⊕⊝ MODERATE 2 | ||
| 310 per 1000 | 279 per 1000 (223 to 347) | |||||
| Serious neonatal morbidity or death | Study population | ‐ | (0 study) | ‐ | ||
| see comment | see comment | |||||
| Serious maternal morbidity or death | Study population | ‐ | (0 study) | ‐ | ||
| see comment | see comment | |||||
| Neonatal intensive care unit admission | Study population | RR 1.61 (0.43 to 6.05) | 215 (3 RCTs) | ⊕⊕⊝⊝ LOW 1 | ||
| 28 per 1000 | 44 per 1000 (12 to 167) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; OR: odds ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Wide confidence interval crossing the line of no effect, few events and small sample size (‐2).
2 Wide confidence interval crossing the line of no effect (‐1).
Summary of findings 3. Vaginal misoprostol compared to placebo for the induction of labour in outpatient settings.
| Vaginal misoprostol compared to placebo for the induction of labour in outpatient settings | ||||||
| Patient or population: women requiring induction of labour Setting: outpatient clinics and hospitals in the USA and Nigeria Intervention: Vaginal misoprostol Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with vaginal misoprostol | |||||
| Vaginal birth not achieved within 24 h | Study population | ‐ | (0 study) | ‐ | No included trial reported this outcome. | |
| see comment | see comment | |||||
| Vaginal birth not achieved within 48 and 72 h | Study population | ‐ | (0 study) | ‐ | No included trial reported this outcome. | |
| see comment | see comment | |||||
| Uterine hyperstimulation (with fetal heart rate changes) | Study population | RR 1.97 (0.43 to 9.00) | 265 (3 RCTs) | ⊕⊕⊝⊝ LOW 1 | ||
| 15 per 1000 | 29 per 1000 (6 to 131) | |||||
| Caesarean section | Study population | RR 0.94 (0.61 to 1.46) | 325 (4 RCTs) | ⊕⊕⊝⊝ LOW 2 | ||
| 206 per 1000 | 194 per 1000 (126 to 301) | |||||
| Serious neonatal morbidity or death | Study population | RR 0.34 (0.01 to 8.14) | 77 (1 RCT) | ⊕⊕⊝⊝ LOW 1 | Study reported perinatal deaths. | |
| 26 per 1000 | 9 per 1000 (0 to 209) | |||||
| Serious maternal morbidity or death | Study population | ‐ | (0 study) | ‐ | No included trial reported this outcome. | |
| see comment | see comment | |||||
| Neonatal intensive care unit admission | Study population | RR 0.89 (0.54 to 1.47) | 325 (4 RCTs) | ⊕⊕⊝⊝ LOW 2 | ||
| 147 per 1000 | 131 per 1000 (79 to 216) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; OR: odds ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Wide confidence interval crossing the line of no effect, few events and small sample size (‐2).
2 Wide confidence interval crossing the line of no effect and small sample size (‐2).
Background
Introduction
This Cochrane Review was first published in 2010 and updated in 2017. The review complements existing reviews on labour induction examining effectiveness and safety.
Description of the condition
The number of women whose labours are induced has risen over the past two decades. Rates in the USA and the UK now exceed 20% of all births (Glantz 2003; Kirby 2004; NHS 2014‐15). There is considerable variation in reported induction rates, and the reasons for this variability are often not clear. In some units in the USA, up to half of all births follow induction of labour (Rayburn 2002). Fewer data are available on induction rates in facilities in low‐ and middle‐income countries; however, the World Health Organization (WHO) Global Survey of Maternal and Perinatal Health of facility obstetric practices in 22 countries reported induction rates of 11.4% in eight Latin American countries, 4.4% in seven African countries, and 12.1% in nine Asian countries (Guerra 2009; Vogel 2013).
It has not been definitively shown that increased use of induction has been associated with improvements in maternal, fetal or neonatal outcomes; women who are induced also tend to be less satisfied with their experience of childbirth (Shetty 2005). In this context, and with increasing pressure on healthcare resources, it is particularly important to address questions about how to provide induction of labour safely and effectively, in settings and ways that are acceptable to women, and in the most cost‐effective way possible.
Description of the intervention
A number of pharmacological and mechanical methods of cervical ripening and induction of labour are available, and these have been the focus of a series of Cochrane Reviews that share generic protocols (Hofmeyr 2009). On the basis of these Cochrane Reviews, WHO currently recommends oral misoprostol (25 μg, 2 hourly) and vaginal low‐dose misoprostol (25 μg, 6 hourly) for induction of labour (misoprostol is not recommended for women with a previous caesarean section) (WHO 2011). If prostaglandins are not available, WHO recommends intravenous oxytocin alone for induction. Balloon catheter is recommended for induction, but amniotomy alone is not. The combination of balloon catheter plus oxytocin is recommended as an alternative method of induction of labour when prostaglandins (including misoprostol) are not available or are contra‐indicated. Importantly, the WHO recommendations stipulate the need to assess maternal and fetal well‐being during induction.
In these reviews, the safety and effectiveness of different methods and agents have been examined, but less attention was paid to the setting in which cervical ripening and induction of labour take place. In this review, we brought together some of the studies included in previous reviews, focusing specifically on those studies where labour induction or cervical ripening was carried out in outpatient settings. For most methods of induction, the number of trials carried out in outpatient settings is likely to be small, making it difficult to definitively establish benefits and harms. The purpose of this review was to examine issues such as benefits and harms, health service utilisation, feasibility and women's views about their care. For some interventions, there may be sufficient data to address questions of effectiveness and safety. In this way, this review complements others in the suite rather than simply duplicating findings.
A related review included trials in which the same methods of ripening or induction were compared in outpatient and hospital settings (Kelly 2013).
Induction of labour is carried out for a variety of indications and using a range of pharmacological, mechanical and other methods. The main indication for induction of labour is prolonged pregnancy, and there is evidence from a related Cochrane Review (Gülmezoglu 2012) that for pregnancies which have continued beyond 41 weeks, induction of labour may reduce perinatal mortality. Other inductions are carried out on an individual basis. Most inductions of labour are carried out in inpatient settings. Outpatient procedures may not be safe for women with some risk factors, and some methods may only be feasible and safe in hospital, or in settings with specialised staff and facilities available. For example, outpatient induction is unlikely to be suitable for women with serious medical conditions or complications in the current pregnancy (Sawai 1995). Some women may be unsuitable for home care simply because they live at an unacceptable travelling distance from emergency care facilities.
How the intervention might work
Ideally, the agents or methods used for cervical ripening at home would achieve changes in the cervix similar to the normal physiological changes which promote the 'spontaneous' onset of labour, but without causing uterine contractions (Sawai 1995). Most methods for cervical ripening or induction of labour do have some undesirable side effects, including, on occasions, excessive uterine activity. The consequences of excessive uterine activity as a result of iatrogenic uterine hyperstimulation can be life‐threatening for the mother and fetus.
Sometimes drugs to induce labour can only be administered by intravenous infusion or by repeated injections, or using specialist procedures that cannot easily be carried out in an outpatient setting. Drugs that can be taken orally, or procedures that are simple to perform, and require only limited monitoring, may lend themselves more readily for use in an outpatient setting. At least theoretically, outpatient induction may offer a number of advantages to women, clinical staff and providers of health services. Outpatient induction may be more convenient for and preferred by women; it may reduce hospital bed occupancy, and therefore, be associated with lower healthcare costs.
A number of papers have set out indications for outpatient cervical ripening or induction such as post‐dates pregnancy in women who are otherwise well, and where there have been no signs of fetal distress. Several outpatient induction protocols have been described in observational studies suggesting that such inductions are feasible, safe and acceptable to women (Elliott 1992; McGill 2007; Neale 2002).
Why it is important to do this review
For some methods, and for selected groups of women, induction of labour is already being carried out in outpatient settings. This Cochrane Review brings together evidence from available trials in outpatient settings to assess benefits and harms of outpatient induction, as well as preferences of women and providers, and the feasibility of their use in outpatient settings. To achieve this, we pooled data from trials examining the same methods to address questions of safety.
In the context of this review, the issue of safety is of great importance. At the same time, it is unlikely that safety could be adequately addressed in studies of randomised cohorts. Severe maternal and neonatal mortality and morbidity are likely to be very rare events in the low‐risk population included in studies of outpatient induction. Information on adverse events and the relative safety of outpatient methods is most likely to emerge where there have been several large studies and where the same methods have been directly compared in different settings. Information on rare adverse events takes time to accumulate, but by systematically recording information on adverse events in all the studies included in the review, we may shed some light on this question.
We did not include studies where the same method of cervical ripening or induction of labour was compared in outpatient versus inpatient settings: this has been addressed in a related Cochrane Review (Kelly 2013).
Objectives
To examine pharmacological and mechanical interventions to induce labour or ripen the cervix in outpatient settings in terms of effectiveness, maternal satisfaction, healthcare costs and, where information is available, safety.
This Cochrane Review complements existing Cochrane Reviews on labour induction examining effectiveness and safety.
Methods
Criteria for considering studies for this review
Types of studies
All published and unpublished randomised trials which compared different methods of cervical ripening or induction of labour carried out in outpatient settings were eligible for inclusion. All trials included random allocation to intervention and control groups. We did not include quasi‐randomised trials. We included studies reported in abstracts and brief reports provided that sufficient information was available to allow us to assess eligibility and risk of bias; where such information was not provided we attempted to contact trial authors. We planned to include cluster‐randomised trials if they were otherwise eligible. We did not include cross‐over studies because we did not consider they were appropriate in this topic area.
Types of participants
Pregnant women (with a viable fetus) at or near team (greater than 35 weeks) in an outpatient setting. Specifically, women in whom induction of labour is being considered, but where expectant management is acceptable.
Types of interventions
We included studies examining outpatient cervical ripening or induction of labour with pharmacological agents or mechanical methods. We included studies where different methods of induction of labour in outpatient settings were compared; where a method was compared with a placebo; where a method was compared with expectant management or routine care; or where different doses of the same drug were compared. 'Outpatient' was defined by the trialists and included any cervical ripening or induction of labour intervention (with the exception of membrane sweeping) that can be carried out at home or within community healthcare settings. It also includes a package of care initially provided in hospital (fetal monitoring, drug administration) after which the woman is allowed home until later review or admission in labour. We did not include interventions where women remain in hospital throughout (even if they were in 'day‐care' settings, or in other parts of the hospital, but not formally admitted as inpatients) because a purpose of this review was to examine outcomes where women do not have immediate access to emergency care facilities. Trials comparing inpatient versus outpatient induction of labour were considered in Kelly 2013.
Types of outcome measures
Clinically relevant outcomes for trials of methods of cervical ripening and labour induction have been pre‐specified by two authors of labour induction reviews (Justus Hofmeyr and Zarko Alfirevic) (Hofmeyr 2009). We have used most of these outcomes (relevant to both inpatient and outpatient settings) in this review.
In addition, we attempted to use relevant outcome measures to quantify any cost effectiveness benefits of outpatient ripening.
Main outcomes
Vaginal birth not achieved within 24 hours.
Additional induction agents required.
Length of hospital stay.
Use of emergency services.
Maternal satisfaction
Caregiver satisfaction
Serious neonatal morbidity or perinatal death (composite outcome will include, for example, seizures, birth asphyxia defined by trialists, neonatal encephalopathy, disability in childhood).
Serious maternal morbidity or death (composite outcome will include, for example, uterine rupture, admission to intensive care unit, septicaemia).
Additional outcomes of interest
Additional outcomes of interest related to measures of effectiveness, complications and satisfaction
Measures of effectiveness
Vaginal birth not achieved within 48 and 72 hours.
Randomisation to birth interval.
Oxytocin augmentation.
Pain relief requirements (epidural, opioids).
Complications
Uterine hyperstimulation (with fetal heart rate (FHR) changes).
Uterine hyperstimulation (without FHR changes).
Instrumental vaginal birth.
Caesarean section.
Apgar score < 7 at 5 minutes.
Neonatal intensive care unit admission.
Perinatal death.
Postpartum haemorrhage (as defined by the trial authors).
Serious maternal complications (considered as separate outcomes, e.g. intensive care unit admission, septicaemia, uterine rupture).
Serious neonatal complications (considered as separate outcomes).
In the absence of formal economic evaluation, we had planned to estimate potential cost savings and the impact of interventions used within an outpatient setting. These estimates could involve using some measures of effectiveness and complications in combination with estimates of healthcare provision.
We also included some additional outcomes that may serve as proxy measures of progress towards labour or birth.
Indicators of progress in labour such as: preterm rupture of membranes, diagnosis of active/spontaneous labour, self‐referral back to hospital, Bishop scores at fixed time points post‐randomisation.
Failed induction (as defined by trialists, but excluding the use of oxytocin for augmentation in women already in established labour).
Time to birth including the interval from randomisation to birth; interval to admission along with length of labour.
Side effects or adverse outcomes (not specified above) related or possibly related to the intervention.
Detailed definitions for outcomes
Perinatal and maternal morbidity and mortality are composite outcomes. This is not an ideal solution because some components are clearly less severe than others. It is possible for one intervention to cause more deaths but less severe morbidity. However, in the context of labour induction at term, this is unlikely. All these events are rare, and a modest change in their incidence will be easier to detect if composite outcomes are presented. The incidence of individual components are explored as additional outcomes (see above).
'Uterine rupture' includes all clinically significant ruptures of unscarred or scarred uteri. Trivial scar dehiscence noted incidentally at the time of surgery is excluded.
The terminology of uterine hyperstimulation is problematic (Curtis 1987). In the reviews, the term 'uterine hyperstimulation' is defined as uterine tachysystole (more than 5 contractions per 10 minutes for at least 20 minutes) and uterine hypersystole/hypertonus (a contraction lasting at least 2 minutes).
'Uterine hyperstimulation with FHR changes' is usually defined as uterine hyperstimulation syndrome (tachysystole or hypersystole with FHR changes such as persistent decelerations, tachycardia or decreased short‐term variability). However, due to varied reporting, there is the possibility of subjective bias in the interpretation of these outcomes. Also, it is not always clear from trials if these outcomes are reported in a mutually exclusive manner. More importantly, continuous monitoring is unlikely in an outpatient setting. Therefore, there is a high risk of biased reporting of uterine hyperstimulation (with or without FHR changes). It is possible that bias will favour the outpatient setting (i.e. by failure to recognise mild forms of hyperstimulation without continuous monitoring). On the other hand, clinicians who favour inpatient induction may, in the absence of continuous monitoring, label any maternal description of painful, frequent uterine contractions as hyperstimulation. Therefore, in the absence of blinding, hyperstimulation and other 'soft' outcomes should be interpreted with extreme caution.
While we sought data on all of the outcomes listed above, we documented only those with data in the analysis tables. We included outcomes in the analysis if reasonable measures were taken to minimise observer bias, and data were available according to original treatment allocation.
Search methods for identification of studies
The following methods section of this review is based on a standard template used by Cochrane Pregnancy and Childbirth.
Electronic searches
We searched Cochrane Pregnancy and Childbirth’s Trials Register by contacting their Information Specialist (30 November 2016).
The Register is a database containing over 23,000 reports of controlled trials in the field of pregnancy and childbirth. For full search methods used to populate Pregnancy and Childbirth’s Trials Register including the detailed search strategies for CENTRAL, MEDLINE, Embase and CINAHL; the list of handsearched journals and conference proceedings, and the list of journals reviewed via the current awareness service, please follow this link to the editorial information about the Cochrane Pregnancy and Childbirth in the Cochrane Library and select the ‘Specialized Register ’ section from the options on the left side of the screen.
Briefly, Cochrane Pregnancy and Childbirth’s Trials Register is maintained by their Information Specialist and contains trials identified from:
monthly searches of the Cochrane Central Register of Controlled Trials (CENTRAL);
weekly searches of MEDLINE (Ovid);
weekly searches of Embase (Ovid);
monthly searches of CINAHL (EBSCO);
handsearches of 30 journals and the proceedings of major conferences;
weekly current awareness alerts for a further 44 journals plus monthly BioMed Central email alerts.
Search results are screened by two people and the full text of all relevant trial reports identified through the searching activities described above is reviewed. Based on the intervention described, each trial report is assigned a number that corresponds to a specific Pregnancy and Childbirth review topic (or topics), and is then added to the Register. The Information Specialist searches the Register for each review using this topic number rather than keywords. This results in a more specific search set which has been fully accounted for in the relevant review sections (Included studies; Excluded studies; Studies awaiting classification).
Searching other resources
We searched the reference lists of retrieved studies.
We did not apply any language or date restrictions.
Data collection and analysis
For methods used in the previous version of this review, see Dowswell 2010.
For this update, the following methods were used to assess the 10 reports that were identified as a result of the updated search.
The following methods section of this review is based on a standard template used by Cochrane Pregnancy and Childbirth.
Selection of studies
Two review authors independently assessed all the potential studies identified as a result of the search strategy for inclusion. We resolved any disagreement through discussion or, if required, we consulted a third review author.
Data extraction and management
We designed a form to extract data. For eligible studies, two review authors extracted the data using the agreed form. We resolved discrepancies through discussion or, if required, we consulted the third review author. Data were entered into Review Manager software (RevMan 2014) and checked for accuracy.
When information regarding any of the above was unclear, we planned to contact authors of the original reports to provide further details.
Assessment of risk of bias in included studies
Two review authors independently assessed risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). Any disagreement was resolved by discussion or by involving a third assessor.
(1) Random sequence generation (checking for possible selection bias)
We described for each included study the method used to generate the allocation sequence in sufficient detail to allow an assessment of whether it should produce comparable groups.
We assessed the method as:
low risk of bias (any truly random process, e.g. random number table; computer random number generator);
high risk of bias (any non‐random process, e.g. odd or even date of birth; hospital or clinic record number);
unclear risk of bias.
(2) Allocation concealment (checking for possible selection bias)
We described for each included study the method used to conceal allocation to interventions prior to assignment and assessed whether intervention allocation could have been foreseen in advance of, or during recruitment, or changed after assignment.
We assessed the methods as:
low risk of bias (e.g. telephone or central randomisation; consecutively numbered sealed opaque envelopes);
high risk of bias (open random allocation; unsealed or non‐opaque envelopes, alternation; date of birth);
unclear risk of bias.
(3.1) Blinding of participants and personnel (checking for possible performance bias)
We described for each included study the methods used, if any, to blind study participants and personnel from knowledge of which intervention a participant received. We considered that studies were at low risk of bias if they were blinded, or if we judged that the lack of blinding unlikely to affect results. We assessed blinding separately for different outcomes or classes of outcomes.
We assessed the methods as:
low, high or unclear risk of bias for participants;
low, high or unclear risk of bias for personnel.
(3.2) Blinding of outcome assessment (checking for possible detection bias)
We described for each included study the methods used, if any, to blind outcome assessors from knowledge of which intervention a participant received. We assessed blinding separately for different outcomes or classes of outcomes.
We assessed methods used to blind outcome assessment as:
low, high or unclear risk of bias.
(4) Incomplete outcome data (checking for possible attrition bias due to the amount, nature and handling of incomplete outcome data)
We described for each included study, and for each outcome or class of outcomes, the completeness of data including attrition and exclusions from the analysis. We stated whether attrition and exclusions were reported and the numbers included in the analysis at each stage (compared with the total randomised participants), reasons for attrition or exclusion where reported, and whether missing data were balanced across groups or were related to outcomes. Where sufficient information was reported, or could be supplied by the trial authors, we planned to re‐include missing data in the analyses which we undertook.
We assessed methods as:
low risk of bias (e.g. no missing outcome data; missing outcome data balanced across groups);
high risk of bias (e.g. numbers or reasons for missing data imbalanced across groups; ‘as treated’ analysis done with substantial departure of intervention received from that assigned at randomisation);
unclear risk of bias.
(5) Selective reporting (checking for reporting bias)
We described for each included study how we investigated the possibility of selective outcome reporting bias and what we found.
We assessed the methods as:
low risk of bias (where it is clear that all of the study’s pre‐specified outcomes and all expected outcomes of interest to the review have been reported);
high risk of bias (where not all the study’s pre‐specified outcomes have been reported; one or more reported primary outcomes were not pre‐specified; outcomes of interest are reported incompletely and so cannot be used; study fails to include results of a key outcome that would have been expected to have been reported);
unclear risk of bias.
(6) Other bias (checking for bias due to problems not covered by (1) to (5) above)
We described for each included study any important concerns we had about other possible sources of bias.
(7) Overall risk of bias
We made explicit judgements about whether studies were at high risk of bias, according to the criteria given in the Handbook (Higgins 2011). With reference to (1) to (6) above, we planned to assess the likely magnitude and direction of the bias and whether we considered it is likely to impact on the findings. In future updates, we will explore the impact of the level of bias through undertaking sensitivity analyses (see Sensitivity analysis).
Assessment of the quality of the evidence using the GRADE approach
For this update the quality of the evidence was assessed using the GRADE approach as outlined in the GRADE handbook to assess the quality of the body of evidence relating to the following outcomes for the main comparisons (vaginal PGE₂ versus placebo or expectant management; intracervical PGE₂ versus placebo or expectant management; vaginal misoprostol versus placebo or expectant management):
Vaginal birth not achieved within 24 hours.
Vaginal birth not achieved within 48 and 72 hours.
Uterine hyperstimulation with FHR changes.
Caesarean section.
Serious neonatal morbidity or perinatal death (composite outcome will include, for example, seizures, birth asphyxia defined by trialists, neonatal encephalopathy, disability in childhood).
Serious maternal morbidity or death (composite outcome will include, for example, uterine rupture, admission to intensive care unit, septicaemia).
Neonatal intensive care unit admission.
GRADEpro Guideline Development Tool was used to import data from Review Manager 5.3 (RevMan 2014) to create 'Summary of findings' tables. A summary of the intervention effect and a measure of quality for each of the above outcomes was produced using the GRADE approach. The GRADE approach uses five considerations (study limitations, consistency of effect, imprecision, indirectness and publication bias) to assess the quality of the body of evidence for each outcome. The evidence can be downgraded from 'high quality' by one level for serious (or by 2 levels for very serious) limitations, depending on assessments for risk of bias, indirectness of evidence, serious inconsistency, imprecision of effect estimates or potential publication bias.
Measures of treatment effect
Dichotomous data
We presented results as summary risk ratio with 95% confidence intervals for dichotomous data.
Continuous data
We used the mean difference if outcomes were measured in the same way between trials for continuous data. We used the standardised mean difference to combine trials that measured the same outcome, but used different methods.
Unit of analysis issues
Cluster‐randomised trials
If future searches identify eligible cluster‐randomised trials, we will include these in the analyses along with individually randomised trials. We will adjust their sample sizes or standard errors using the methods described in the Handbook (Section 16.3.4 or 16.3.6 as appropriate) using an estimate of the intracluster correlation co‐efficient (ICC) derived from the trial (if possible), from a similar trial or from a study of a similar population. If we use ICCs from other sources, we will report this and conduct sensitivity analyses to investigate the effect of variation in the ICC. If we identify both cluster‐randomised trials and individually‐randomised trials, we plan to synthesise the relevant information. We will consider it reasonable to combine the results from both if there is little heterogeneity between the study designs and the interaction between the effect of intervention and the choice of randomisation unit is considered to be unlikely.
We will also acknowledge heterogeneity in the randomisation unit and perform a sensitivity analysis to investigate the effects of the randomisation unit.
Cross‐over trials
We decided to exclude cross‐over trials as we did not think this design was appropriate in this topic area.
Other unit of analysis issues
Trials with multiple arms
Two trials had multiple intervention arms ‐ Larmon 2002 and Magann 1998. Larmon 2002 was a three‐arm trial comparing intracervical PGE₂, oestrogen and placebo and is included in more than one comparison. In Magann 1998, one intervention was not eligible for inclusion so data for this were not included. If we identify further multiple‐armed trials in future searches, we will divide the control group in the analysis to avoid double counting, and follow the methods set out in the Handbook (Higgins 2011).
Dealing with missing data
Levels of attrition were noted for included studies. In future updates, if more eligible studies are included, the impact of including studies with high levels of missing data in the overall assessment of treatment effect will be explored by using sensitivity analysis.
For all outcomes, analyses were carried out, as far as possible, on an intention‐to‐treat basis, i.e. we attempted to include all participants randomised to each group in the analyses. The denominator for each outcome in each trial was the number randomised minus any participants whose outcomes were known to be missing.
Assessment of heterogeneity
We assessed statistical heterogeneity in each meta‐analysis using the Tau², I² and Chi² statistics. We regarded heterogeneity as substantial if I² was greater than 30% and either Tau² was greater than zero, or there was a low P value (< 0.10) in the Chi² test for heterogeneity. If we identified substantial heterogeneity (> 30%), we planned to explore the source in pre‐specified subgroup analysis.
Assessment of reporting biases
In future updates, if there are 10 or more studies in the meta‐analysis we will investigate reporting biases (such as publication bias) using funnel plots. We will assess funnel plot asymmetry visually. If asymmetry is suggested by a visual assessment, we will perform exploratory analyses to investigate the source.
Data synthesis
We carried out statistical analysis using the Review Manager software (RevMan 2014). We used fixed‐effect meta‐analysis for combining data where it was reasonable to assume that studies were estimating the same underlying treatment effect: i.e. where trials were examining the same intervention, and the trials’ populations and methods were judged sufficiently similar.
If there was clinical heterogeneity sufficient to expect that the underlying treatment effects differed between trials, or if substantial statistical heterogeneity was detected, we used random‐effects meta‐analysis to produce an overall summary if an average treatment effect across trials was considered clinically meaningful. The random‐effects summary will be treated as the average range of possible treatment effects and we will discuss the clinical implications of treatment effects differing between trials. If the average treatment effect is not clinically meaningful, we will not combine trials. If we used random‐effects analyses, the results were presented as the average treatment effect with 95% confidence intervals, and the estimates of Tau² and I².
Subgroup analysis and investigation of heterogeneity
If we identified substantial heterogeneity, we investigated the source using subgroup analyses. We considered whether an overall summary was meaningful, and if it was, we used random‐effects analysis to produce the effect.
If sufficient data were available, we planned to carry out the following subgroup analyses.
Nulliparous versus multiparous.
Induction indication.
We planned to use only the primary outcomes in subgroup analysis.
We also planned to assess subgroup differences by interaction tests available in RevMan (RevMan 2014), and to report the results of subgroup analyses quoting the Chi² statistic and P value, and the interaction test I² value. However, insufficient data were available to permit any subgroup analyses.
Sensitivity analysis
We planned to carry out sensitivity analyses to explore the effect of trial quality assessed by concealment of allocation, high attrition rates, or both, with poor quality studies being excluded from the analyses in order to assess whether this makes any difference to the overall result. Gaffaney 2009 and Rijnders 2011 were assessed as being at high risk of attrition bias. However, insufficient data were available to permit any sensitivity analyses.
Results
Description of studies
Results of the search
The previous version of this review identified 72 reports, representing 55 separate studies (some trials were reported in more than one published paper) (Dowswell 2010). A total of 28 studies were included in the review, 25 were excluded and two awaiting classification (Ascher‐Walsh 2000; Thakur 2005) (Figure 1).
1.
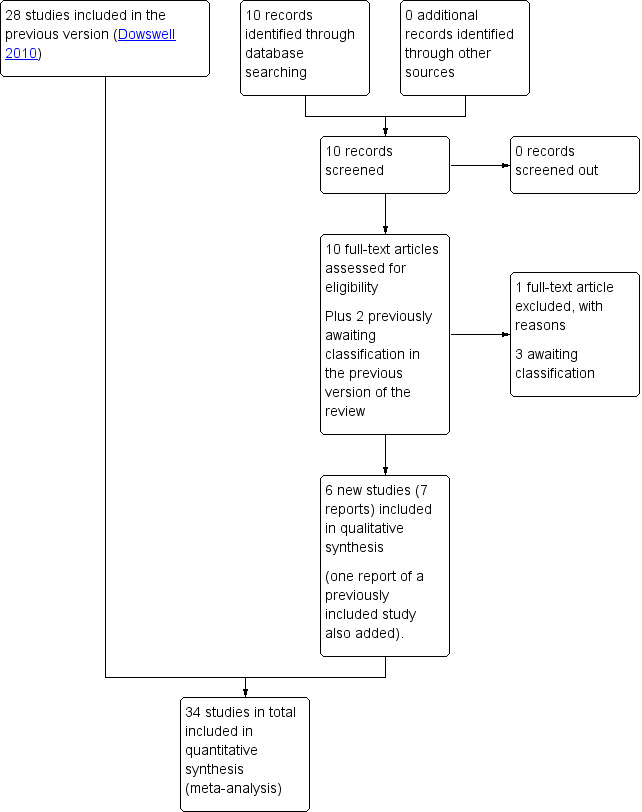
Study flow diagram.
For this update, the search identified an additional 10 reports. We also reassessed two studies that were awaiting further classification in Dowswell 2010. A total of six new studies were included (Agarwal 2012; Attanayake 2014; Gaffaney 2009; Ghanaie 2013; Rijnders 2011; Schmitz 2014), one excluded (Rezk 2014;), and one new study is awaiting classification (Mostaghel 2009). One report was included as an additional report of a study already included in the review (Bollapragada 2006a). Both studies previously awaiting classification (Ascher‐Walsh 2000; Thakur 2005) remain; and ISRCTN47736435 (previously excluded) was included as an additional report of Rijnders 2011. See Characteristics of studies awaiting classification tables.
Included studies
We included 34 studies that involved a total of 5003 women (Characteristics of included studies).
The studies included a variety of different comparisons.
Vaginal prostaglandin (PGE₂) versus expectant management or placebo (5 studies) (Hage 1993; Newman 1997; O'Brien 1995; Sawai 1991; Sawai 1994).
Intracervical prostaglandin (PgE₂) versus expectant management or placebo (7 studies) (Buttino 1990; Gittens 1996; Larmon 2002; Lien 1998; Magann 1998; McKenna 1999; Rayburn 1999).
Vaginal misoprostol versus placebo (4 studies) (Incerpi 2001; McKenna 2004; Oboro 2005; Stitely 2000).
Vaginal misoprostol 25 µg versus 50 µg (1 study) (Kipikasa 2005).
Intracervical prostaglandin (PGE₂) versus vaginal misoprostol (1 study) (Meyer 2005).
Oral misoprostol versus placebo (2 studies) (Gaffaney 2009; Lyons 2001).
Mifepristone versus placebo (5 studies) (Elliott 1998; Frydman 1992; Giacalone 1998; Lelaidier 1994; Stenlund 1999).
Oestrogen versus placebo (1 study) (Larmon 2002).
Vaginal isosorbide mononitrate (IMN) versus placebo (7 studies) (Agarwal 2012; Attanayake 2014; Bollapragada 2006a; Bullarbo 2007; Ghanaie 2013; Habib 2008; Schmitz 2014).
Acupuncture versus routine care (1 study) (Harper 2006).
Outpatient amniotomy for induction versus routine care (1 study) (Rijnders 2011).
In all trials it was intended that women would spend part of the study period at home. In most studies women received the initial treatment in a hospital setting (and frequently underwent a period of surveillance) before discharge home. Women were advised to seek help or return to hospital if any problems arose, if labour commenced, or after a predefined period. In some studies, women self‐administered the study intervention at home, and again were advised to return either if they had concerns, if labour started, or for review after a specified period (e.g. in Bollapragada 2006a women scheduled for labour induction were given vaginal IMN with instructions on self‐administration 48, 32 and 16 hours before the scheduled induction time).
The studies almost invariably recruited healthy women at term. A small number of studies focused on women with particular histories. In the trials by Gittens 1996, Lelaidier 1994 and Rayburn 1999 women who had a previous caesarean birth were recruited; Incerpi 2001 focused on women with insulin‐dependent diabetes and Newman 1997 included women with diabetes along with those requiring induction of labour for post maturity. Two studies (Lelaidier 1994; Rayburn 1999) recruited women who had a previous caesarean section and who were aiming to achieve a vaginal birth. In the remaining studies the main indication for induction of labour was prolonged pregnancy, although recruitment was not always restricted to this group. Six studies included only primiparous women (Bollapragada 2006a; Elliott 1998; Ghanaie 2013; Hage 1993; Harper 2006; Schmitz 2014) and two multiparous women only (Lelaidier 1994; Rayburn 1999).
The main recruitment criterion in all of these studies was that labour had not already started (i.e. women were not having regular painful contractions). Most studies also specified a Bishop score indicating an unfavourable cervix as an inclusion criterion although the definition of an unfavourable cervix (low Bishop score) varied. No studies specifically recruited women where the cervix was favourable. Where it was mentioned, studies invariably recruited women with intact membranes; no studies specifically focused on women with ruptured membranes. Most of these studies specifically mentioned that multiple pregnancies were excluded, and at recruitment it was usually specified as an inclusion criterion that the fetus was in good condition with no signs of distress (e.g. normal fetal heart rate monitoring and normal amniotic fluid volume).
Further information on interventions, participants and inclusion and exclusion criteria are set out in the Characteristics of included studies tables.
Excluded studies
We excluded 25 trials (Characteristics of excluded studies). The main reason for excluding studies was study design.
Four studies used a cross‐over design; we had decided to exclude cross‐over trials as we did not think this design was appropriate in this topic area; in all of these studies the focus was on breast stimulation. Women in the control groups initially received no intervention, while those in intervention groups were asked to stimulate their nipples for a specified time period; after this time period, women then crossed over into the control or intervention arm (Adewole 1993; Di Lieto 1989; Elliott 1984; Salmon 1986).
In three studies (Damania 1988; Griffin 2003; Manidakis 1999) there was too little information on study methods to allow us to ascertain whether group allocation was truly random, or to allow us to carry out an assessment of risk of bias (the studies by Griffin 2003 and Manidakis 1999 were reported in brief abstracts; we attempted to contact the authors for more information without success). Two studies used quasi‐randomisation and were at high risk of bias (Garry 2000; Kadar 1990). Evans 1983 described findings from two separate studies, one of which seemed to be carried out in a hospital setting and included a control group receiving no treatment; a second "outpatient" study did not include a control group; different doses of porcine ovarian relaxin were compared. In the study by Ohel 1996, whilst there seemed to be random allocation to treatment groups, results were not reported by randomisation group, and we were not able to include data in the review. In one study reported in a brief abstract, no original data were reported in the results section (Krammer 1995).
A number of studies focused on interventions that we had either specifically excluded (e.g. Doany 1997; Kaul 2004; Magann 1999; Salamalekis 2000 looked at membrane sweeping), or interventions that are not currently used in clinical practice (extra amniotic saline infusion was examined by Moghtadaei 2007; it was not clear that women in both arms of this trial were discharged home; Spallicci 2007 examined the use of hyaluronidase injection).
In five studies it was not clear that the study was carried out in an outpatient setting or that the women were expected to spend some of the study period at home (Damania 1992; Herabutya 1992; Rayburn 1988; Voss 1996; Ziaei 2003). One study (Rezk 2014) was not conducted in an outpatient setting.
Dorfman 1987 looked at homeopathic preparations (caulophyllum‐arnica‐actea and racemosa‐pulsatilla‐gelsemium) used with the intention of generally preparing women for childbirth rather than for labour induction.
Risk of bias in included studies
Summary of risk bias assessments are presented graphically (Figure 2; Figure 3).
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
3.
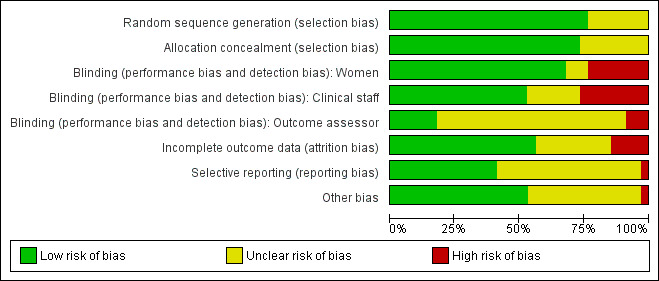
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
We assessed most of the included studies as using adequate methods to generate the randomisation sequence and to conceal group allocation.
Sequence generation was either computer generated or derived from random number tables in 26 of the 34 included studies. In eight trials the methods used to generate the randomisation order were not clear (Elliott 1998; Frydman 1992; Gittens 1996; Hage 1993; Lelaidier 1994; Lyons 2001; Newman 1997; Sawai 1991).
Eighteen studies used either external or pharmacy randomisation services, or identical coded drug packs from pharmacy to conceal group allocation (Bollapragada 2006a; Buttino 1990; Frydman 1992; Gaffaney 2009; Giacalone 1998; Habib 2008; Incerpi 2001; Kipikasa 2005; Lelaidier 1994; Lien 1998; McKenna 1999; McKenna 2004; O'Brien 1995; Stenlund 1999; Rijnders 2011; Sawai 1994; Schmitz 2014; Stitely 2000). Four trials used sealed, opaque, sequentially numbered envelopes to conceal allocation (Bullarbo 2007; Harper 2006; Larmon 2002; Magann 1998). Opaque envelopes were used in Attanayake 2014 and Meyer 2005 , although it was not stated that they were sealed. Oboro 2005 used sealed envelopes, but did not state they were opaque. In nine trials, methods to conceal group allocation were not clear (Agarwal 2012; Elliott 1998; Ghanaie 2013; Gittens 1996; Hage 1993; Lyons 2001; Newman 1997; Rayburn 1999; Sawai 1991).
Blinding
Most (26) of the included studies were placebo controlled, and women and clinical staff were described as blind to group allocation. However, it was not always clear when the randomisation code was broken, so it was difficult to assess whether outcome assessment was carried out by blinded investigators. Eighteen studies were judged to be at low risk of bias by adequately blinding the women and staff (Bollapragada 2006a; Bullarbo 2007; Buttino 1990; Elliott 1998; Frydman 1992; Gaffaney 2009; Giacalone 1998; Habib 2008; Incerpi 2001; Lelaidier 1994; Lien 1998; Lyons 2001; O'Brien 1995; Sawai 1991; Sawai 1994; Schmitz 2014; Stenlund 1999; Stitely 2000). In two of the placebo controlled trials, blinding may not have been convincing; in the Kipikasa 2005 trial women in both groups were given tablet fragments (either an eighth or a quarter of whole tablets) so the tablets may have not appeared identical (at least to staff). In the Larmon 2002 study women may have been blind to intravaginal preparations, but staff are unlikely to have been.
In eight trials women in both arms of the studies were given different interventions; therefore blinding of women and staff was not feasible, or not attempted (Gittens 1996; Harper 2006; Magann 1998; Meyer 2005; Newman 1997; Oboro 2005; Rayburn 1999; Rijnders 2011). The lack of blinding in these studies may have affected some of the outcomes examined in the review.
Outcome assessors were not blinded in three studies (Meyer 2005; Oboro 2005; Rijnders 2011). Six studies reportedly blinded outcome assessors until analysis was completed (Giacalone 1998; Kipikasa 2005; Lien 1998; Schmitz 2014; Stenlund 1999; Stitely 2000). The blinding of outcome assessors was unclear in the remaining studies.
Incomplete outcome data
Loss of women to follow up and missing data were not serious problems in most of the included studies. In 10 studies the levels of attrition were not clear (Elliott 1998; Frydman 1992; Ghanaie 2013; Gittens 1996; Hage 1993; Harper 2006; Incerpi 2001; Kipikasa 2005; Lyons 2001; Newman 1997).
Five trials were assessed as high risk of attrition bias. In the study by Sawai 1994, attrition was approximately 12% and some of the exclusions were for non‐compliance. Attrition was also high in the study by Bollapragada 2006a; in this trial randomisation occurred up to nine days before the initiation of treatment, hence 80 of the 350 women did not start treatment as they had already gone into labour. To reduce risk of bias, the authors reported an intention‐to‐treat analysis (including all women randomised) for the trial's primary outcomes but not for secondary outcomes. In Gaffaney 2009, nine women were excluded post‐randomisation and not included for analysis. In Schmitz 2014, 10 women (5 in each group) were excluded post‐randomisation, and the maternal satisfaction outcome had 23% attrition. Rijnders 2011 and used a satisfaction survey, however, responses were not balanced (221 at home and 183 in hospital). In Kipikasa 2005, there were inconsistencies in figures between the text and the tables, hence rated unclear risk of attrition bias.
Selective reporting
Reporting bias was difficult to assess, as many studies did not pre‐specify all the outcomes that were reported (Frydman 1992; Ghanaie 2013; Gittens 1996; Hage 1993; Incerpi 2001; Kipikasa 2005; Larmon 2002; Lelaidier 1994; Lien 1998; Lyons 2001; Magann 1998; McKenna 1999; McKenna 2004; Newman 1997; O'Brien 1995; Oboro 2005; Rayburn 1999; Stenlund 1999; Stitely 2000). Schmitz 2014 pre‐specified maternal and newborn intensive care unit admission outcomes, but these were not reported.
Other potential sources of bias
In many included studies women were likely to receive other interventions at some stage in their treatment as well as the study allocated intervention (e.g. amniotomy, membrane sweeping, additional medication) and these in turn may have affected other outcomes (e.g. length of labour and rate of caesarean section). Without adequate blinding, it is possible that women in intervention and control groups may have had different co‐interventions, or co‐interventions at different stages. For example, in the study by Harper 2006, women in the intervention group attended for treatment on three occasions, and at these visits (not available to women in the control group) may have been exposed to a range of co‐interventions, or additional tests or observations, that may have had an impact on outcomes.
Other sources of bias included baseline imbalance in parity between groups (Oboro 2005) and imbalance in numbers of randomised women between the treatment and control groups (Elliott 1998).
Effects of interventions
See: Table 1; Table 2; Table 3
1. Vaginal prostaglandin (PGE₂) versus placebo or expectant management: five studies, 335 women
Main outcomes
We included five studies in this comparison (Hage 1993; Newman 1997; O'Brien 1995; Sawai 1991; Sawai 1994). None of the studies collected information on most of the review's main outcomes. We do not have information on the numbers of women achieving vaginal birth within 24 hours, on length of hospital stay, on the use of emergency services or on maternal or caregiver satisfaction. Maternal and perinatal deaths were not reported.
Additional induction agents required
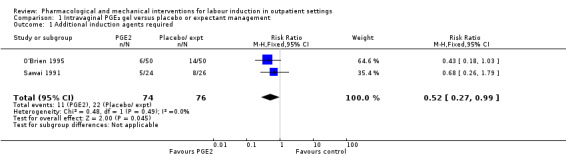
O'Brien 1995 and Sawai 1991 reported the numbers of women requiring further (non‐study) induction agents with fewer women in the PGE₂ group needing further medication to induce labour. While 14.8% of the PGE₂ group needed further induction agents this applied to 28.9% of the control group. However, as only two relatively small studies contributed data for this outcome, confidence intervals were wide and very close to the line of no effect (risk ratio (RR) 0.52, 95% confidence interval (CI) 0.27 to 0.99; 150 women; 2 trials; Analysis 1.1).
1.1. Analysis.
Comparison 1 Intravaginal PGE₂ gel versus placebo or expectant management, Outcome 1 Additional induction agents required.
Additional outcomes of interest
Measures of effectiveness
None of the trials reported vaginal birth not achieved within 48 and 72 hours, randomisation to birth interval, or oxytocin augmentation.
Pain relief requirements (epidural, opioids)
O'Brien 1995 examined the use of epidural; again, there was no strong evidence of any difference between groups (RR 0.83, 95% CI 0.62 to 1.12; 100 women; 1 study; Analysis 1.2).
1.2. Analysis.
Comparison 1 Intravaginal PGE₂ gel versus placebo or expectant management, Outcome 2 Epidural.
Complications
There was no clear evidence of differences between women who received vaginal PGE₂ and placebo or expectant management for the following outcomes:
Uterine hyperstimulation (with or without FHR changes ‐ unclear) (RR 3.76, 95% CI 0.64 to 22.24; 244 women; 4 studies; Analysis 1.3; low‐quality evidence).
Caesarean section (RR 0.80, 95% CI 0.49 to 1.31; 288 women; 4 studies; Analysis 1.4; low‐quality evidence).
Apgar score less than seven at five minutes (RR 0.45, 95% CI 0.07 to 2.93; 180 infants; 2 studies; Analysis 1.5).
Neonatal intensive care unit (NICU) admission (RR 0.32, 95% CI 0.10 to 1.03; 230 infants; 3 studies; Analysis 1.6; low‐quality evidence).
1.3. Analysis.
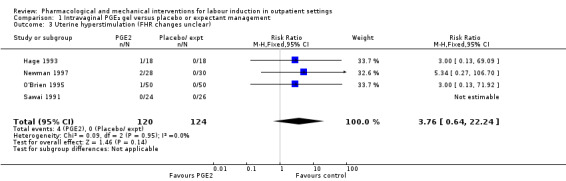
Comparison 1 Intravaginal PGE₂ gel versus placebo or expectant management, Outcome 3 Uterine hyperstimulation (FHR changes unclear).
1.4. Analysis.
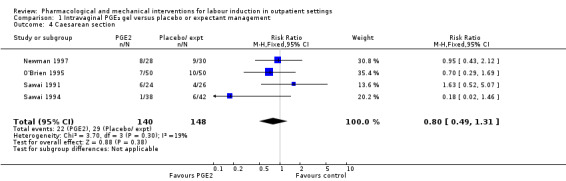
Comparison 1 Intravaginal PGE₂ gel versus placebo or expectant management, Outcome 4 Caesarean section.
1.5. Analysis.
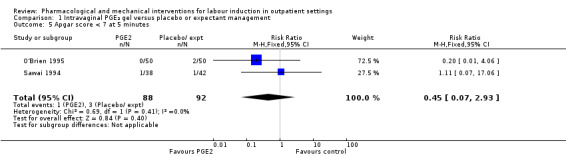
Comparison 1 Intravaginal PGE₂ gel versus placebo or expectant management, Outcome 5 Apgar score < 7 at 5 minutes.
1.6. Analysis.
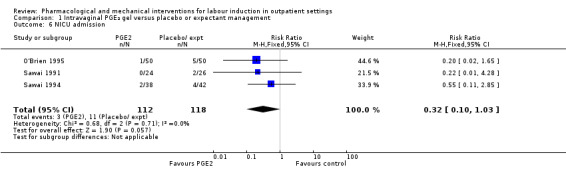
Comparison 1 Intravaginal PGE₂ gel versus placebo or expectant management, Outcome 6 NICU admission.
Serious maternal complications (considered as separate outcomes, e.g. intensive care unit admission, septicaemia, uterine rupture)
There was only limited information on the impact of interventions on the health of mothers and babies. O'Brien 1995 and Sawai 1994 reported rates of chorioamnionitis and results favoured women in the PGE₂ group (RR 0.37, 95% CI 0.15 to 0.90; 180 women; 2 studies; Analysis 1.7). There was no information on the use of antibiotics or on rates of endometritis.
1.7. Analysis.
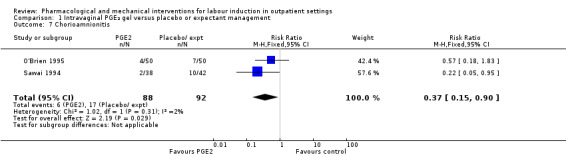
Comparison 1 Intravaginal PGE₂ gel versus placebo or expectant management, Outcome 7 Chorioamnionitis.
Instrumental vaginal birth, perinatal death, postpartum haemorrhage (as defined by the trial authors), and serious neonatal complications (considered as separate outcomes) were not reported by any study.
Non‐prespecified outcomes
While none of these five studies reported the numbers of women achieving vaginal birth within a certain specified period, other 'proxy' measures of progress towards labour or birth were included. Each study reported different outcomes.
Hage 1993 reported on the rate of change in Bishop scores and, compared with women receiving PGE₂, those in the control group were more likely to have score changes of less than three at follow‐up (RR 0.13, 95% CI 0.03 to 0.47; 36 women; 1 study; Analysis 1.8) although it was not clear when follow‐up occurred.
1.8. Analysis.
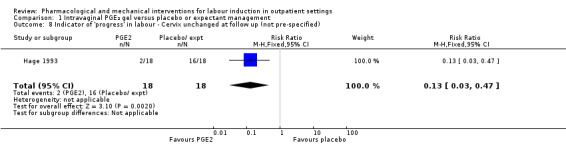
Comparison 1 Intravaginal PGE₂ gel versus placebo or expectant management, Outcome 8 Indicator of 'progress' in labour ‐ Cervix unchanged at follow up (not pre‐specified).
Newman 1997 reported figures for the number of women going into "spontaneous labour" within 48 hours of treatment commencing; it was more likely for labour to start in the PGE₂ group compared with women receiving routine care (RR 6.43, 95% CI 2.12 to 19.48; 58 women; 1 study; Analysis 1.9).
1.9. Analysis.
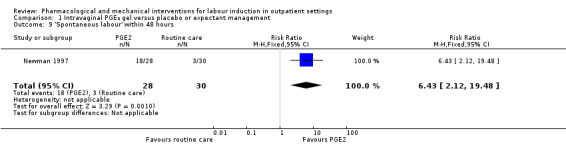
Comparison 1 Intravaginal PGE₂ gel versus placebo or expectant management, Outcome 9 'Spontaneous labour' within 48 hours.
O'Brien 1995 reported that the median interval from study enrolment to birth was four days in the PGE₂ group (range 0 to 28 days) versus 10 days (range 0 to 26 days) in the control group (P = 0.002). The shorter interval between randomisation and birth was reflected in a lower gestational age (weeks) at birth in the intervention group (mean difference (MD) ‐0.60 weeks, 95% CI ‐0.99 to ‐0.21; 100 women; 1 study; Analysis 1.11). It was also reported that, during the five‐day treatment period, compared with controls, more women in the intervention group were admitted to hospital "for labour" (RR 2.70, 95% CI 1.47 to 4.97; 100 women; 1 study; Analysis 1.10), although it was not clear whether this included women in active labour only, or women admitted after premature rupture of membranes (PROM) or for other reasons. The numbers of women diagnosed with post‐term pregnancy was small in both groups (2 women in the intervention group and 3 in the control group).
1.11. Analysis.
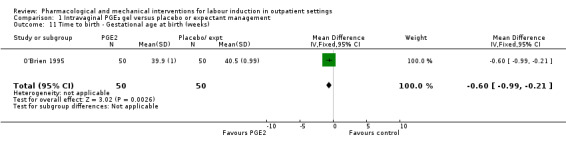
Comparison 1 Intravaginal PGE₂ gel versus placebo or expectant management, Outcome 11 Time to birth ‐ Gestational age at birth (weeks).
1.10. Analysis.
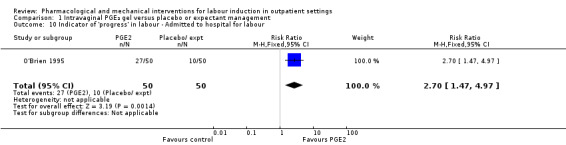
Comparison 1 Intravaginal PGE₂ gel versus placebo or expectant management, Outcome 10 Indicator of 'progress' in labour ‐ Admitted to hospital for labour.
Sawai 1991 described Bishop scores in control and intervention groups at hospital admission, but there were differences between groups at baseline and the authors report no significant differences between groups at follow‐up (data not shown). Sawai 1994 reported the mean gestational age (in days) at hospital admission (although the indications for admission included pregnancy complications as well as signs of the onset of labour). There was not a clear difference between groups (MD ‐2.00 days, 95% CI ‐4.17 to 0.17; 80 women; 1 study; Analysis 1.12).
1.12. Analysis.
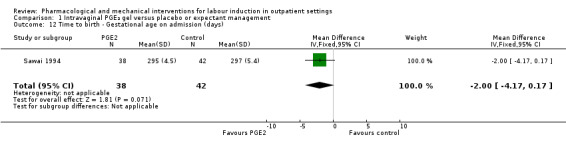
Comparison 1 Intravaginal PGE₂ gel versus placebo or expectant management, Outcome 12 Time to birth ‐ Gestational age on admission (days).
2. Intracervical prostaglandin (PGE₂) versus expectant management or placebo: seven studies, 678 women
Main outcomes
We included seven studies in this comparison (Buttino 1990; Gittens 1996; Larmon 2002; Lien 1998; Magann 1998; McKenna 1999; Rayburn 1999).
Additional induction agents required
Three studies (Lien 1998; McKenna 1999; Rayburn 1999) looked at whether, compared with no treatment or placebo, women receiving intracervical PGE₂ were less likely to need further (non‐study) interventions to induce labour. There was no strong evidence of a difference between groups (RR 0.98, 95% CI 0.74 to 1.32; 445 women; 3 studies; Analysis 2.1). Lien 1998 also examined whether women given intracervical PGE₂ were less likely to receive further doses of prostaglandin to induce labour. Again, there was no evidence to suggest a difference between groups (RR 0.61, 95% CI 0.22 to 1.67; 90 women; 1 study; Analysis 2.2).
2.1. Analysis.
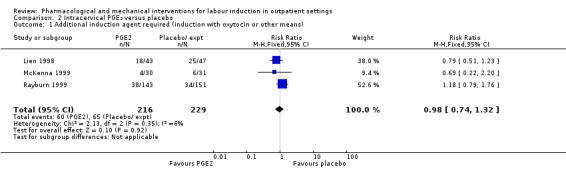
Comparison 2 Intracervical PGE₂ versus placebo, Outcome 1 Additional induction agent required (induction with oxytocin or other means).
2.2. Analysis.
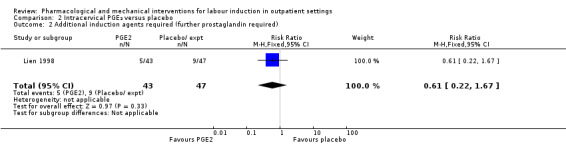
Comparison 2 Intracervical PGE₂ versus placebo, Outcome 2 Additional induction agents required (further prostaglandin required).
Serious maternal morbidity or death (composite outcome will include, for example, uterine rupture, admission to intensive care unit, septicaemia)
Rayburn 1999 reported rates of uterine rupture, and there were no events in either the PGE₂ group or control group participants (Analysis 2.3).
2.3. Analysis.
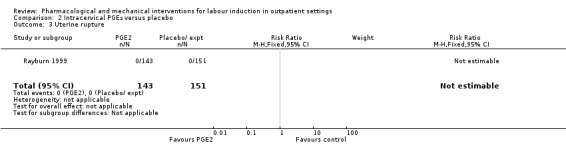
Comparison 2 Intracervical PGE₂ versus placebo, Outcome 3 Uterine rupture.
There was no information on vaginal birth not achieved within 24 hours, length of hospital stay, use of emergency services, maternal or caregiver satisfaction, or serious neonatal morbidity or perinatal death.
Additional outcomes of interest
Measures of effectiveness
Vaginal birth not achieved within 48 and 72 hours
Buttino 1990 reported on the number of women not giving birth within 48 to 72 hours, and although results favoured the PGE₂ group, did not show a clear difference as the confidence intervals just crossed the line of no effect (RR 0.83, 95% CI 0.68 to 1.02; 43 women; 1 study; Analysis 2.4; low‐quality evidence).
2.4. Analysis.
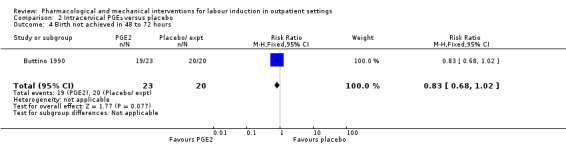
Comparison 2 Intracervical PGE₂ versus placebo, Outcome 4 Birth not achieved in 48 to 72 hours.
Oxytocin augmentation
There was no strong evidence of differences between groups for the number of women who received oxytocin augmentation (RR 0.67, 95% CI 0.40 to 1.12; 84 women; 1 study; Analysis 2.5).
2.5. Analysis.
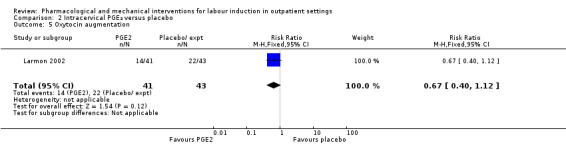
Comparison 2 Intracervical PGE₂ versus placebo, Outcome 5 Oxytocin augmentation.
Randomisation to birth interval, and pain relief requirements were not reported under this comparison.
Complications
The impact of interventions on maternal health were explored in five studies (Buttino 1990; Larmon 2002; Lien 1998; McKenna 1999; Rayburn 1999).
There was no clear evidence of differences between women who received intracervical PGE₂ and placebo or expectant management for the following outcomes:
Uterine hyperstimulation (with FHR changes) (RR 2.66, 95% CI 0.63 to 11.25; 488 women; 4 studies; Analysis 2.6; low‐quality evidence).
Assisted (instrumental) vaginal birth (RR 1.29, 95% CI 0.85 to 1.96; 538 women; 4 studies; Analysis 2.7).
Caesarean section (RR 0.90, 95% CI 0.72 to 1.12; 674 women; 7 studies; Analysis 2.8; moderate‐quality evidence).
Apgar score less than seven at five minutes (RR 0.82, 95% CI 0.42 to 1.60; 515 infants; 4 studies; Analysis 2.9).
NICU admission (RR 1.61, 95% CI 0.43 to 6.05; 215 infants; 3 studies; Analysis 2.10; low‐quality evidence).
Postpartum haemorrhage (RR 3.10, 95% CI 0.13 to 73.16; 61 women; 1 study; Analysis 2.11).
-
Serious maternal complications (considered as separate outcomes, e.g. intensive care unit admission, septicaemia, uterine rupture)
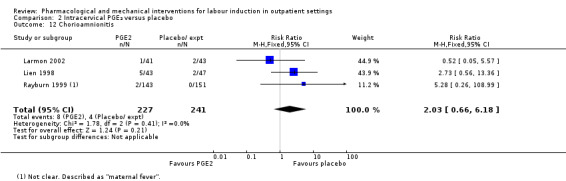
Chorioamnionitis (RR 2.03, 95% CI 0.66 to 6.18; 468 women; 3 studies; Analysis 2.12).
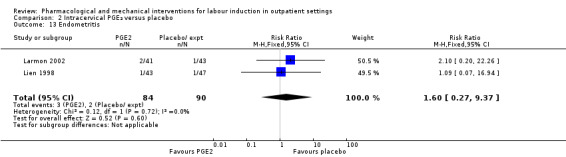
Endometritis (RR 1.60, 95% CI 0.27 to 9.37; 174 women; 2 studies; Analysis 2.13).
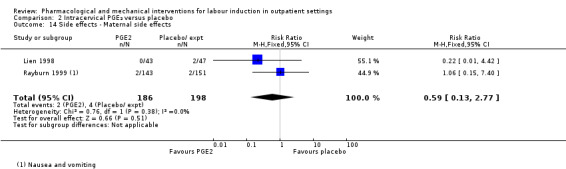
Maternal side effects (RR 0.59, 95% CI 0.13 to 2.77; 384 women; 2 studies; Analysis 2.14).
2.6. Analysis.
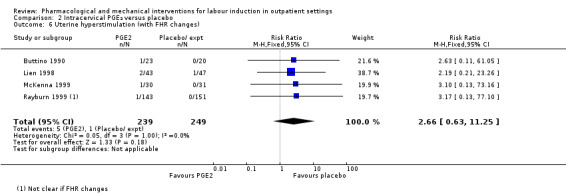
Comparison 2 Intracervical PGE₂ versus placebo, Outcome 6 Uterine hyperstimulation (with FHR changes).
2.7. Analysis.
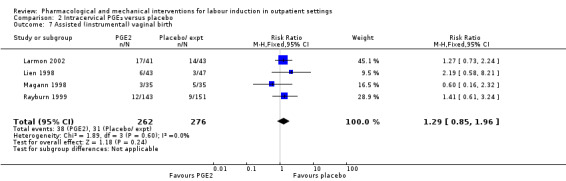
Comparison 2 Intracervical PGE₂ versus placebo, Outcome 7 Assisted (instrumental) vaginal birth.
2.8. Analysis.
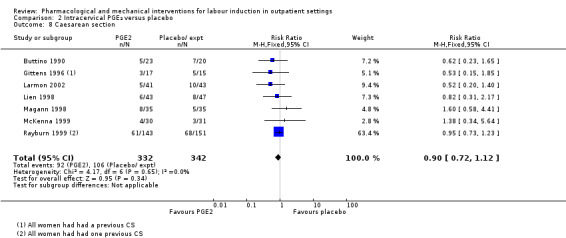
Comparison 2 Intracervical PGE₂ versus placebo, Outcome 8 Caesarean section.
2.9. Analysis.
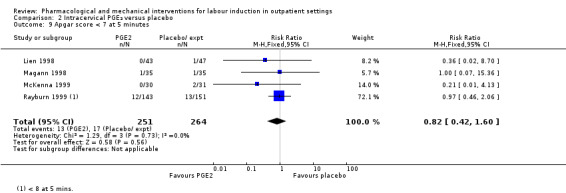
Comparison 2 Intracervical PGE₂ versus placebo, Outcome 9 Apgar score < 7 at 5 minutes.
2.10. Analysis.
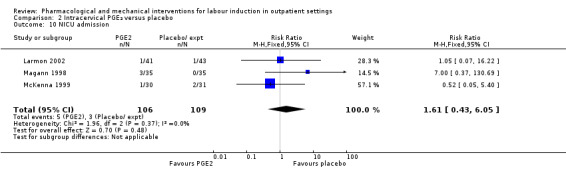
Comparison 2 Intracervical PGE₂ versus placebo, Outcome 10 NICU admission.
2.11. Analysis.
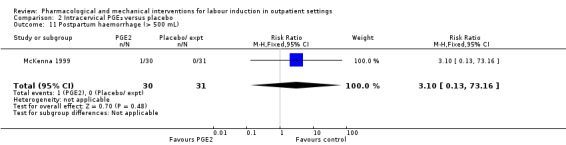
Comparison 2 Intracervical PGE₂ versus placebo, Outcome 11 Postpartum haemorrhage (> 500 mL).
2.12. Analysis.
Comparison 2 Intracervical PGE₂ versus placebo, Outcome 12 Chorioamnionitis.
2.13. Analysis.
Comparison 2 Intracervical PGE₂ versus placebo, Outcome 13 Endometritis.
2.14. Analysis.
Comparison 2 Intracervical PGE₂ versus placebo, Outcome 14 Side effects ‐ Maternal side effects.
The included studies did not provide information on other review outcomes including uterine hyperstimulation (without FHR changes), perinatal death, other serious maternal complications such as admission to intensive care, and other serious neonatal complications such as the use of antibiotics, and neonatal infection.
Non‐prespecified outcomes
All seven studies collected information on progress towards labour and birth; again reported outcomes were different in each study (Buttino 1990; Gittens 1996; Larmon 2002; Lien 1998; Magann 1998; McKenna 1999; Rayburn 1999).
Buttino 1990 and Lien 1998 reported no differences between women in the PGE₂ and control groups in the time interval (days) between the first dose of drug or placebo and birth (SMD ‐0.20 days, 95% CI ‐0.55 to 0.14; 133 women; 2 studies; Analysis 2.15).
2.15. Analysis.
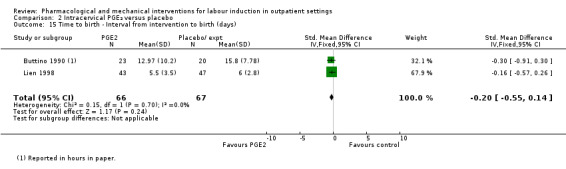
Comparison 2 Intracervical PGE₂ versus placebo, Outcome 15 Time to birth ‐ Interval from intervention to birth (days).
Larmon 2002 found no differences between groups for the median number of days from recruitment to hospital admission (16.8 days for the PGE₂ group versus 15.4 days for the control group (data not shown)). For other outcomes reported in this study (Bishop score on admission, and estimated gestational age on admission) there were no clear differences between groups. However, some women were admitted for induction rather than in labour and it was not clear if these mean figures included all women.
Lien 1998 and Magann 1998 reported the estimated gestational age at birth (in weeks) and found no difference between groups for this outcome (MD ‐0.06 weeks, 95% CI ‐0.35 to 0.23; 156 women; 2 studies; I² = 85%; Tau² = 0.04; Chi² = 6.79; Analysis 2.16) (there was high heterogeneity for this outcome and results should be interpreted with caution). Lien 1998 and Magann 1998 provided information on the number of women requiring induction for 'postdates' pregnancy (women reaching 42 weeks' gestation). In view of high heterogeneity and different clinical management in the two studies, we did not pool results for this outcome but have set out the data in Analysis 2.17. While in the Magann 1998 study more women in the control group required induction (22 of 35 women) compared to the PGE₂ group (7 of 35 women) the results were difficult to interpret as some women had been admitted to hospital for induction at an earlier stage because of changes in Bishop score or for other reasons.
2.16. Analysis.
Comparison 2 Intracervical PGE₂ versus placebo, Outcome 16 Time to birth ‐ Gestational age at birth (weeks).
2.17. Analysis.
Comparison 2 Intracervical PGE₂ versus placebo, Outcome 17 Indicator of 'progress' in labour ‐ Induction for gestational age > 42 weeks.
McKenna 1999 reported the median time from recruitment to admission to hospital; the interval was shorter in the PGE₂ group compared with control group participants (2.5 days versus 7 days, P = 0.02). However, reasons for admission included change in Bishop score, as well as for onset of labour. McKenna 1999 also reported the number of women delivering within two days of commencing treatment; more women gave birth within two days if they had the active treatment (RR 3.10, 95% CI 1.29 to 7.47; 61 women; 1 study; Analysis 2.18).
2.18. Analysis.
Comparison 2 Intracervical PGE₂ versus placebo, Outcome 18 Time to birth ‐ Birth within 48 hours of treatment (all births).
Rayburn 1999 reported the numbers of women delivering at various gestational ages (all deliveries). There were no clear differences between groups at any of the time points measured (data not shown).
3. Vaginal misoprostol versus placebo: four studies, 274 women
Four studies compared vaginal misoprostol with placebo (Incerpi 2001; McKenna 2004; Oboro 2005; Stitely 2000). In all four studies the initial dose of misoprostol was 25 µg; in the study by Incerpi 2001 women received a second dose after three to four days if labour had not commenced, and in the study by Stitely 2000 a second dose was administered after one day.
Main outcomes
Serious neonatal morbidity or perinatal death (composite outcome will include, for example, seizures, birth asphyxia defined by trialists, neonatal encephalopathy, disability in childhood)
For this comparison, only Oboro 2005 reported on the rate of perinatal death with no clear differences between groups; there were no deaths in the active treatment group (N = 38) compared with one stillbirth (reason not reported) in the control group (N = 39) (RR 0.34, 95% CI 0.01 to 8.14; 77 infants; 1 study; Analysis 3.1; low‐quality evidence).
3.1. Analysis.
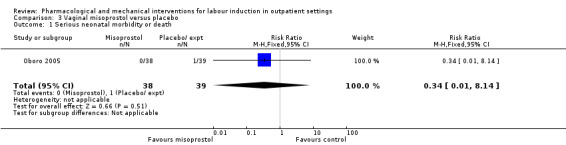
Comparison 3 Vaginal misoprostol versus placebo, Outcome 1 Serious neonatal morbidity or death.
There was no information on other review outcomes such as failure to achieve vaginal birth within 24 hours, additional induction agents required, length of hospital stay, use of emergency services, maternal or caregiver satisfaction, and serious maternal morbidity or death.
Additional outcomes of interest
Measures of effectiveness
Oxytocin augmentation
No study reported rates of oxytocin augmentation between groups; however, mean dose of oxytocin used was reported in Incerpi 2001, and is described in Non‐prespecified outcomes.
Pain relief requirements (epidural, opioids)
McKenna 2004 reported similar numbers of women in each group had epidural anaesthesia (RR 0.98, 95% CI 0.77 to 1.26; 50 women; 1 study; Analysis 3.2). Opioid use was not reported.
3.2. Analysis.
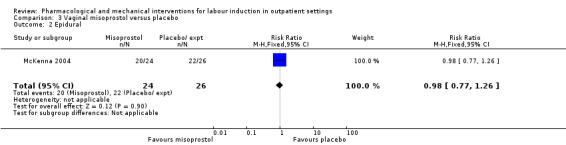
Comparison 3 Vaginal misoprostol versus placebo, Outcome 2 Epidural.
Vaginal birth not achieved within 48 and 72 hours, and randomisation to birth interval were not reported in any study.
Complications
There was little information from these studies on the impact of interventions on mothers' and babies' health.
There was no clear evidence of differences between women who received vaginal misoprostol and placebo for the following outcomes:
Uterine hyperstimulation (with fetal heart rate (FHR) changes) (RR 1.97, 95% CI 0.43 to 9.00; 265 women; 3 studies; Analysis 3.3; low‐quality evidence).
Uterine hyperstimulation (without FHR changes) (RR 3.64, 95% CI 0.15 to 85.97; 137 women; 2 studies; Analysis 3.4).
Instrumental vaginal birth (RR 0.91, 95% CI 0.50 to 1.67; 145 women; 2 studies; Analysis 3.5).
Caesarean section (RR 0.94, 95% CI 0.61 to 1.46; 325 women; 4 studies; Analysis 3.6; low‐quality evidence).
Apgar score less than seven at five minutes (RR 0.21, 95% CI 0.01 to 4.25; 248 infants; 3 studies; Analysis 3.7).
NICU admission (RR 0.89, 95% CI 0.54 to 1.47; 325 infants; 4 studies; Analysis 3.8; low‐quality evidence).
-
Serious neonatal complications (considered as separate outcomes).
Neonatal infection (RR 0.30, 95% CI 0.07 to 1.36; 68 infants; 1 study; Analysis 3.10).
3.3. Analysis.
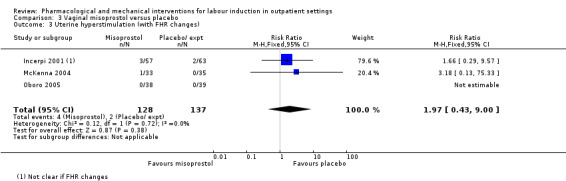
Comparison 3 Vaginal misoprostol versus placebo, Outcome 3 Uterine hyperstimulation (with FHR changes).
3.4. Analysis.
Comparison 3 Vaginal misoprostol versus placebo, Outcome 4 Uterine hyperstimulation (without FHR changes).
3.5. Analysis.
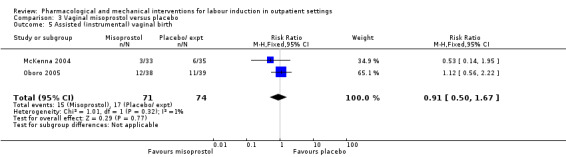
Comparison 3 Vaginal misoprostol versus placebo, Outcome 5 Assisted (instrumental) vaginal birth.
3.6. Analysis.
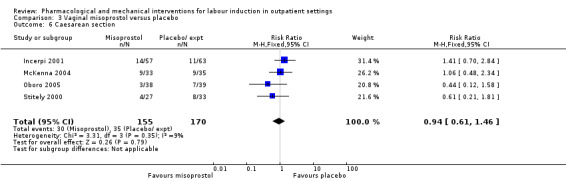
Comparison 3 Vaginal misoprostol versus placebo, Outcome 6 Caesarean section.
3.7. Analysis.
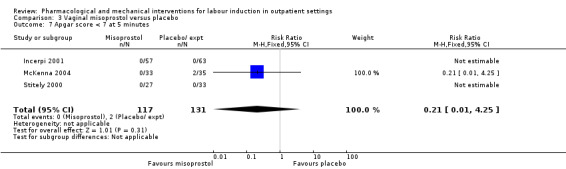
Comparison 3 Vaginal misoprostol versus placebo, Outcome 7 Apgar score < 7 at 5 minutes.
3.8. Analysis.
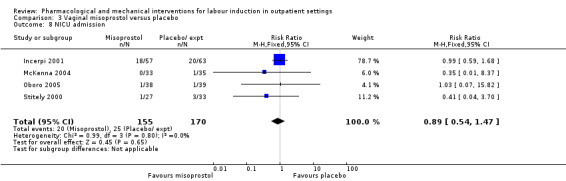
Comparison 3 Vaginal misoprostol versus placebo, Outcome 8 NICU admission.
3.10. Analysis.
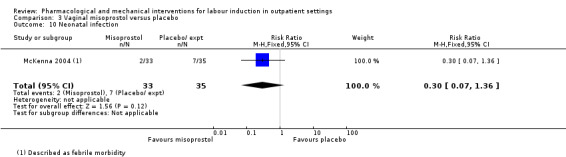
Comparison 3 Vaginal misoprostol versus placebo, Outcome 10 Neonatal infection.
No information was provided in these studies on other review outcomes including postpartum haemorrhage, use of neonatal antibiotics or other maternal or neonatal complications.
Non‐prespecified outcomes
Incerpi 2001 reported the mean dose of oxytocin used for each group; there was no evidence of any difference between the groups (MD 1508.70 mU, 95% CI ‐2357.55 to 5374.95; 72 women; 1 study; Analysis 3.11).
3.11. Analysis.
Comparison 3 Vaginal misoprostol versus placebo, Outcome 11 Indicator of 'progress' in labour ‐ Oxytocin dose used (mU).
Stitely 2000 gave information about the number doses of medication given to the women (MD ‐0.44, 95% CI ‐0.49 to ‐0.39; 60 women; 1 study; Analysis 3.12) and the number of women requiring subsequent doses on study days two and three; fewer women received further doses in the intravaginal misoprostol group (P < 0.01 for both time points: day two RR 0.61, 95% CI 0.43 to 0.87; 60 women; 1 study; Analysis 3.13, day three RR 0.13, 95% CI 0.04 to 0.38; 60 women; 1 study; Analysis 3.14).
3.12. Analysis.
Comparison 3 Vaginal misoprostol versus placebo, Outcome 12 Indicator of 'progress' in labour ‐ Number of medication dose.
3.13. Analysis.
Comparison 3 Vaginal misoprostol versus placebo, Outcome 13 Indicator of 'progress' in labour ‐ Number of women requiring dosing on day 2.
3.14. Analysis.
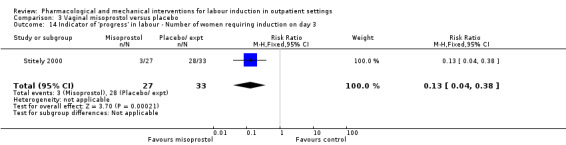
Comparison 3 Vaginal misoprostol versus placebo, Outcome 14 Indicator of 'progress' in labour ‐ Number of women requiring induction on day 3.
Oboro 2005 reported that the interval from the commencement of treatment to hospital admission (in days) was shorter for the misoprostol group both for nulliparous and parous women (MD ‐2.90 days, 95% CI ‐4.99 to ‐0.81; 77 women; 1 study; Analysis 3.15). Data are shown separately for nulliparous and parous women in Analysis 3.16. There was also evidence from this trial that the gestational age at labour (in weeks) was reduced in the misoprostol group compared with women in the control group, with labour approximately a week earlier in the misoprostol group (MD ‐0.80 weeks, 95% CI ‐1.05 to ‐0.55; 77 women; 1 study; Analysis 3.17). There was also evidence that the time to preterm rupture of membranes (in days) was shorter in the misoprostol group (MD ‐2.50 days, 95% CI ‐4.14 to ‐0.86; 77 women; 1 study; Analysis 3.19), although it was not clear whether this was the interval from commencement of treatment or from hospital admission.
3.15. Analysis.
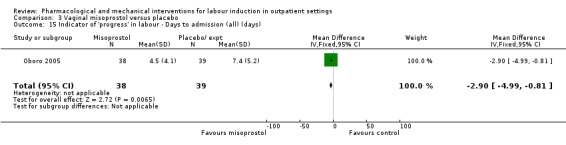
Comparison 3 Vaginal misoprostol versus placebo, Outcome 15 Indicator of 'progress' in labour ‐ Days to admission (all) (days).
3.16. Analysis.
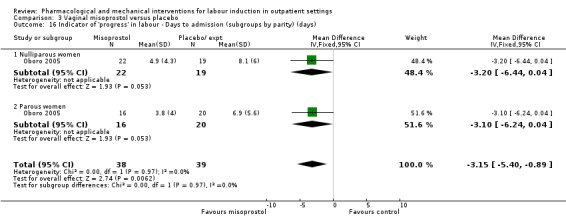
Comparison 3 Vaginal misoprostol versus placebo, Outcome 16 Indicator of 'progress' in labour ‐ Days to admission (subgroups by parity) (days).
3.17. Analysis.
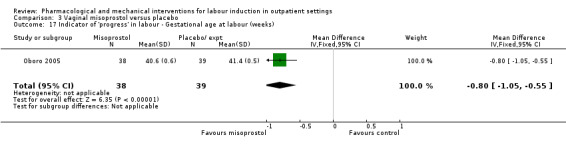
Comparison 3 Vaginal misoprostol versus placebo, Outcome 17 Indicator of 'progress' in labour ‐ Gestational age at labour (weeks).
3.19. Analysis.
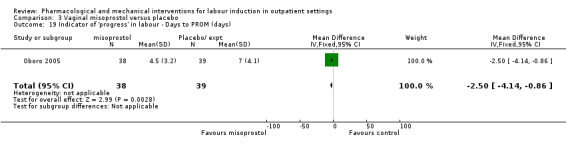
Comparison 3 Vaginal misoprostol versus placebo, Outcome 19 Indicator of 'progress' in labour ‐ Days to PROM (days).
McKenna 2004 provided data on the interval from treatment to vaginal birth (in days); the difference between groups was not clear (MD ‐1.40 days, 95% CI ‐3.51 to 0.71; 50 women; 1 study; Analysis 3.20). McKenna 2004 also reported the mean interval from recruitment to birth (in days), which was less for the misoprostol group compared with women receiving placebo (Analysis 3.21); information was provided separately for nulliparous and multiparous women (Analysis 3.22). It was not clear whether the figures included those women who had caesarean sections or other interventions in labour.
3.20. Analysis.
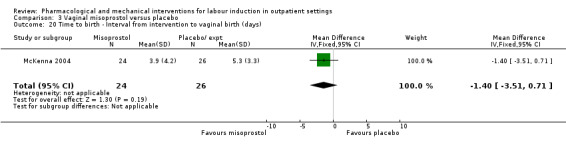
Comparison 3 Vaginal misoprostol versus placebo, Outcome 20 Time to birth ‐ Interval from intervention to vaginal birth (days).
3.21. Analysis.
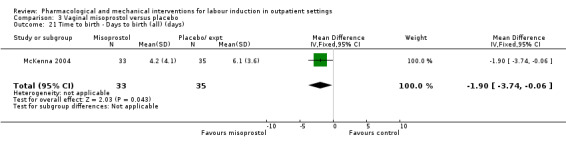
Comparison 3 Vaginal misoprostol versus placebo, Outcome 21 Time to birth ‐ Days to birth (all) (days).
3.22. Analysis.
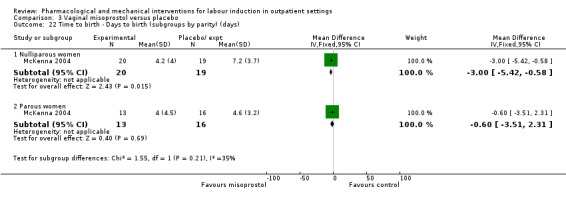
Comparison 3 Vaginal misoprostol versus placebo, Outcome 22 Time to birth ‐ Days to birth (subgroups by parity) (days).
4. Vaginal misoprostol 25 µg versus 50 µg: one study with 52 women
Kipikasa 2005 looked at two different doses of vaginal misoprostol.
Main outcomes
Additional induction agents required
There were no differences between groups in the number of women requiring further induction agents (oxytocin) (RR 2.26, 95% CI 0.22 to 23.33; 49 women; 1 study; Analysis 4.1).
4.1. Analysis.
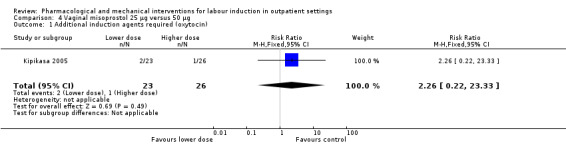
Comparison 4 Vaginal misoprostol 25 µg versus 50 µg, Outcome 1 Additional induction agents required (oxytocin).
There was no information on any other of the review's main outcomes: vaginal birth not achieved within 24 hours, length of hospital stay, use of emergency services, maternal or caregiver satisfaction, serious neonatal morbidity or perinatal death (composite outcome will include, for example, seizures, birth asphyxia defined by trialists, neonatal encephalopathy, disability in childhood), and serious maternal morbidity or death (composite outcome will include, for example, uterine rupture, admission to intensive care unit, septicaemia).
Additional outcomes
Measures of effectiveness
No outcomes of measures of effectiveness were reported: vaginal birth not achieved within 48 and 72 hours, randomisation to birth interval, oxytocin augmentation, pain relief requirements (epidural, opioids).
Complications
There was little difference between groups for the following outcomes:
uterine hyperstimulation ‐ there were no cases of hyperstimulation in either group (Analysis 4.2);
rates of caesarean section (RR 0.94, 95% CI 0.33 to 2.68; 49 women; 1 study; Analysis 4.3); and
NICU admission (RR 0.57, 95% CI 0.05 to 5.83; 49 infants; 1 study; Analysis 4.4).
4.2. Analysis.
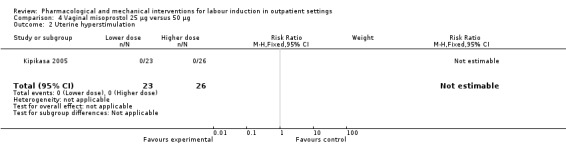
Comparison 4 Vaginal misoprostol 25 µg versus 50 µg, Outcome 2 Uterine hyperstimulation.
4.3. Analysis.
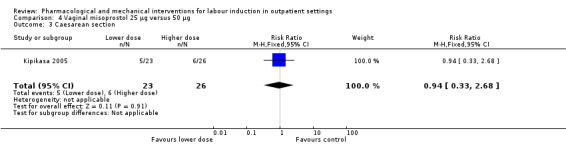
Comparison 4 Vaginal misoprostol 25 µg versus 50 µg, Outcome 3 Caesarean section.
4.4. Analysis.
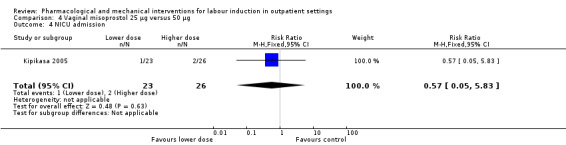
Comparison 4 Vaginal misoprostol 25 µg versus 50 µg, Outcome 4 NICU admission.
Apgar score
One baby in the higher dose group had a low Apgar score (< 6) at five minutes (data not shown).
There was no information on instrumental vaginal birth, perinatal death, postpartum haemorrhage (as defined by the trial authors), serious maternal complications (considered as separate outcomes, e.g. intensive care unit admission, septicaemia, uterine rupture), or serious neonatal complications (considered as separate outcomes).
Non‐prespecified outcomes
The interval to birth (in days) was reported to be shorter in the group receiving the higher dose of misoprostol; with women receiving 50 µg delivering, on average, one and a half days earlier than those receiving 25 µg (MD 1.50 days, 95% CI 1.19 to 1.81; 49 women; 1 study; Analysis 4.5).
4.5. Analysis.
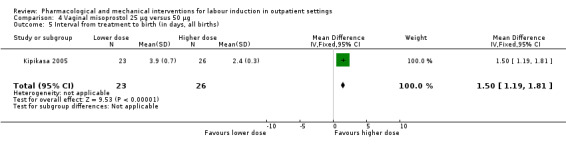
Comparison 4 Vaginal misoprostol 25 µg versus 50 µg, Outcome 5 Interval from treatment to birth (in days, all births).
5. Intracervical PGE₂ versus vaginal misoprostol: one study, 84 women
Main outcomes
One study is included in this comparison between intracervical PGE₂ and vaginal misoprostol (Meyer 2005). None of the review's primary outcomes were considered in this study.
Additional outcomes
Measures of effectiveness
No outcomes of measures of effectiveness were reported.
Complications
There was no strong evidence of differences between intervention and control groups for the following outcomes:
uterine hyperstimulation (with or without FHR changes) (RR 0.26, 95% CI 0.03 to 2.73; 64 women; 1 study; Analysis 5.1);
caesarean section (RR 0.89, 95% CI 0.38 to 2.08; 84 women; 1 study; Analysis 5.2)
Apgar scores less than seven at five minutes (RR 0.33, 95% CI 0.01 to 7.96; 84 infants; 1 study; Analysis 5.3); and
NICU admission (RR 1.25, 95% CI 0.36 to 4.33; 84 women; 1 study; Analysis 5.4).
5.1. Analysis.
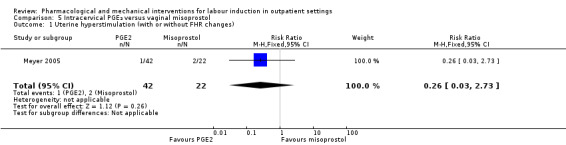
Comparison 5 Intracervical PGE₂ versus vaginal misoprostol, Outcome 1 Uterine hyperstimulation (with or without FHR changes).
5.2. Analysis.
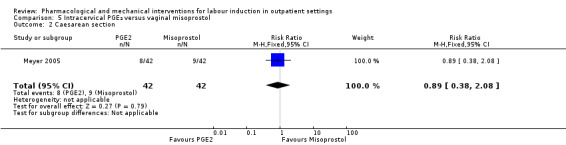
Comparison 5 Intracervical PGE₂ versus vaginal misoprostol, Outcome 2 Caesarean section.
5.3. Analysis.
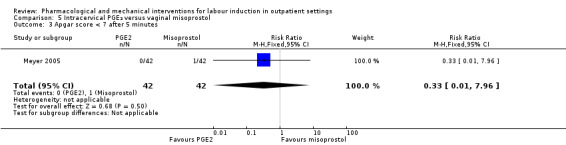
Comparison 5 Intracervical PGE₂ versus vaginal misoprostol, Outcome 3 Apgar score < 7 after 5 minutes.
5.4. Analysis.
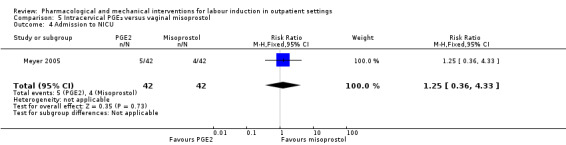
Comparison 5 Intracervical PGE₂ versus vaginal misoprostol, Outcome 4 Admission to NICU.
There was no information on instrumental vaginal birth, perinatal death, postpartum haemorrhage (as defined by the trial authors), serious maternal complications (considered as separate outcomes, e.g. intensive care unit admission, septicaemia, uterine rupture), or serious neonatal complications (considered as separate outcomes).
Non‐prespecified outcomes
It was reported that the proportion of women not requiring oxytocin was 22% in the misoprostol group versus 2% in for those in the PGE₂ group (P = 0.006). The dose of oxytocin used was also reported to be decreased in those women receiving misoprostol (P = 0.008 for cumulative dose of oxytocin) (data not shown) (Meyer 2005).
The interval from the administration of the cervical ripening agent to admission (hours) was shorter for women who received misoprostol (MD 2.50 hours, 95% CI 2.22 to 2.78; 75 women; 1 study; Analysis 5.5), and misoprostol was also reported to increase by 32% the number of women starting labour or with SROM during the ripening period (RR 0.31, 95% CI 0.14 to 0.69; 83 women; 1 study; Analysis 5.6).
5.5. Analysis.
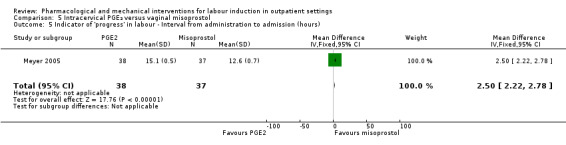
Comparison 5 Intracervical PGE₂ versus vaginal misoprostol, Outcome 5 Indicator of 'progress' in labour ‐ Interval from administration to admission (hours).
5.6. Analysis.
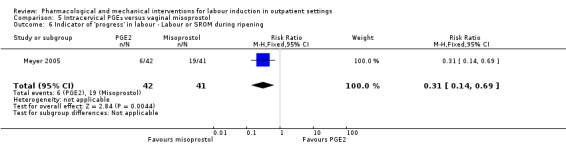
Comparison 5 Intracervical PGE₂ versus vaginal misoprostol, Outcome 6 Indicator of 'progress' in labour ‐ Labour or SROM during ripening.
Misoprostol was reported to increase the number of deliveries within 24 and 48 hours (RR 0.90, 95% CI 0.75 to 1.07; Analysis 5.7; and RR 0.93, 95% CI 0.81 to 1.06; Analysis 5.8 (respectively); 83 women; 1 study), but the differences between groups were not clear.
5.7. Analysis.
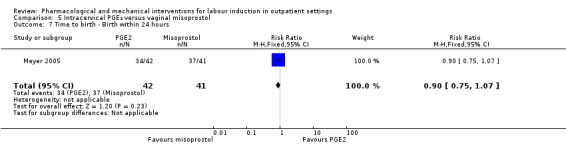
Comparison 5 Intracervical PGE₂ versus vaginal misoprostol, Outcome 7 Time to birth ‐ Birth within 24 hours.
5.8. Analysis.
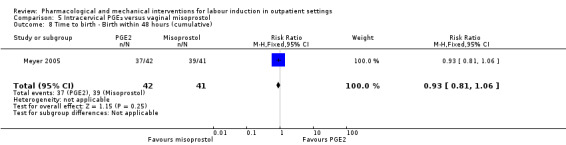
Comparison 5 Intracervical PGE₂ versus vaginal misoprostol, Outcome 8 Time to birth ‐ Birth within 48 hours (cumulative).
6. Oral misoprostol versus placebo: two studies, 127 women
Main outcomes
Two studies were included in this comparison (Gaffaney 2009; Lyons 2001). Gaffaney 2009 was assessed as being at high risk of attrition bias so should have been removed according to the pre‐specified sensitivity analysis. However, there were insufficient studies in this comparison for meaningful sensitivity analysis.
Vaginal birth not achieved within 24 hours
Women in the oral misoprostol group had a higher rate of vaginal birth achieved within 24 hours (RR 0.65, 95% CI 0.48 to 0.86; 87 women; 1 study; Analysis 6.1).
6.1. Analysis.
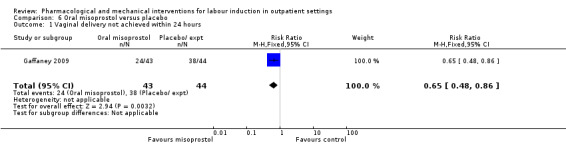
Comparison 6 Oral misoprostol versus placebo, Outcome 1 Vaginal delivery not achieved within 24 hours.
Additional induction agents required
Women in the oral misoprostol group had lower rates of additional induction agents compared to the placebo group (RR 0.60, 95% CI 0.37 to 0.97; 127 women; 2 studies; Analysis 6.2).
6.2. Analysis.
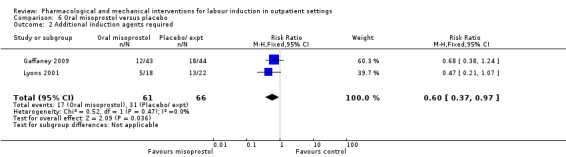
Comparison 6 Oral misoprostol versus placebo, Outcome 2 Additional induction agents required.
There was no information for length of hospital stay, use of emergency services, maternal or caregiver satisfaction, serious neonatal morbidity or perinatal death, or serious maternal morbidity or death.
Additional outcomes
Measures of effectiveness
Oxytocin augmentation
There was no evidence of a difference between groups for women who received oxytocin augmentation (RR 0.81, 95% CI 0.61 to 1.08; 87 women; 1 study; Analysis 6.3).
6.3. Analysis.
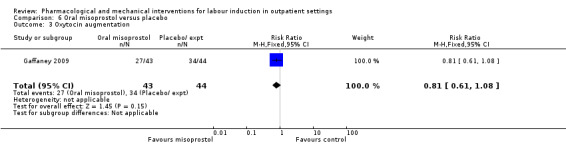
Comparison 6 Oral misoprostol versus placebo, Outcome 3 Oxytocin augmentation.
Vaginal birth not achieved within 48 and 72 hours, randomisation to birth interval, and pain relief requirements (epidural, opioids) were not reported.
Complications
There was no evidence of differences between the misoprostol and placebo groups for the following outcomes:
uterine hyperstimulation with FHR changes (RR 1.53, 95% CI 0.47 to 5.06; 87 women; 1 studies; Analysis 6.4), or where it was unclear if there were FHR changes (RR 0.61, 95% CI 0.06 to 6.21; 40 women; 1 study; Analysis 6.5);
rate of instrumental vaginal birth (RR 0.51, 95% CI 0.17 to 1.57; 87 women; 1 study; Analysis 6.6);
rate of caesarean section (RR 0.62, 95% CI 0.28 to 1.33; 86 women; 1 study; Analysis 6.7);
Apgar scores of less than seven at five minutes (there were none in either group) (Analysis 6.8);
NICU admission (RR 1.02, 95% CI 0.07 to 15.84; 87 infants; 1 study; Analysis 6.9);
postpartum haemorrhage (RR 5.11, 95% CI 0.25 to 103.51; 87 women; 1 study; Analysis 6.10);
-
serious maternal complications:
chorioamnionitis (RR 1.06, 95% CI 0.52 to 2.17; 124 women; 2 studies; Analysis 6.11); and
endometritis (RR 0.51, 95% CI 0.05 to 5.44; 87 women; 1 study; Analysis 6.12).
6.4. Analysis.
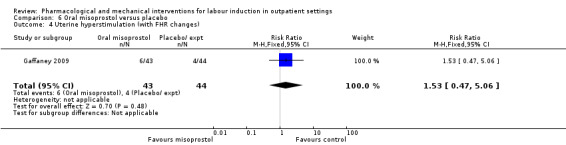
Comparison 6 Oral misoprostol versus placebo, Outcome 4 Uterine hyperstimulation (with FHR changes).
6.5. Analysis.
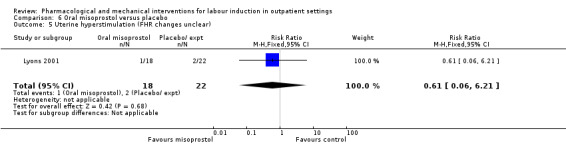
Comparison 6 Oral misoprostol versus placebo, Outcome 5 Uterine hyperstimulation (FHR changes unclear).
6.6. Analysis.
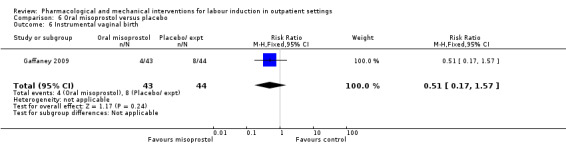
Comparison 6 Oral misoprostol versus placebo, Outcome 6 Instrumental vaginal birth.
6.7. Analysis.
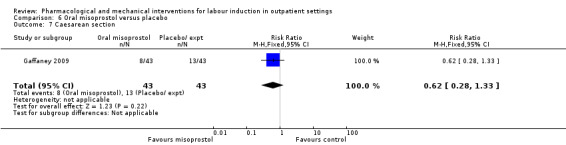
Comparison 6 Oral misoprostol versus placebo, Outcome 7 Caesarean section.
6.8. Analysis.
Comparison 6 Oral misoprostol versus placebo, Outcome 8 Apgar score < 7 at 5 minutes.
6.9. Analysis.
Comparison 6 Oral misoprostol versus placebo, Outcome 9 Neonatal intensive care unit admission.
6.10. Analysis.
Comparison 6 Oral misoprostol versus placebo, Outcome 10 Postpartum haemorrhage.
6.11. Analysis.
Comparison 6 Oral misoprostol versus placebo, Outcome 11 Chorioamnionitis.
6.12. Analysis.
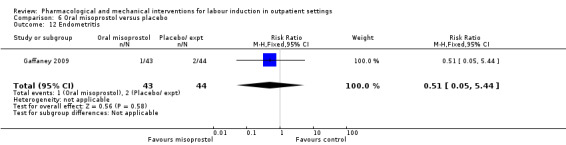
Comparison 6 Oral misoprostol versus placebo, Outcome 12 Endometritis.
Perinatal death, other serious maternal complication, and serious neonatal complications are not reported.
Non‐pre‐specified outcomes
Oral misoprostol may be associated with a reduction in the times (hours) from first dose to active labour and first dose to birth (MD ‐37.08, 95% CI ‐52.44 to ‐21.72; 127 women; 2 studies; Analysis 6.13, and MD ‐37.94, 95% CI ‐57.97 to ‐17.91; 87 women; 1 study; Analysis 6.14), which is reflected in the larger total doses of medication in the placebo group (MD ‐0.51, 95% CI ‐0.92 to ‐0.10; 40 women; 1 study; Analysis 6.15).
6.13. Analysis.
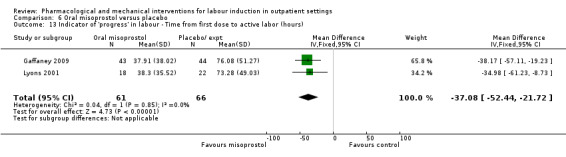
Comparison 6 Oral misoprostol versus placebo, Outcome 13 Indicator of 'progress' in labour ‐ Time from first dose to active labor (hours).
6.14. Analysis.
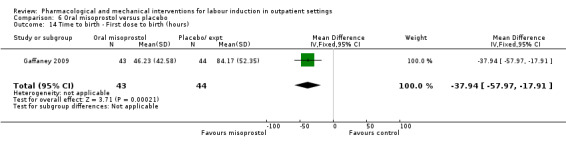
Comparison 6 Oral misoprostol versus placebo, Outcome 14 Time to birth ‐ First dose to birth (hours).
6.15. Analysis.
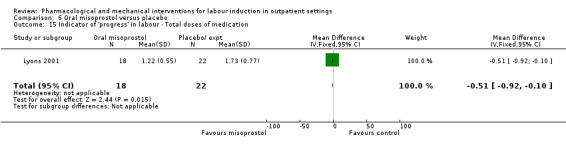
Comparison 6 Oral misoprostol versus placebo, Outcome 15 Indicator of 'progress' in labour ‐ Total doses of medication.
7. Mifepristone versus placebo: five studies, 393 women
We included five studies in this comparison (Elliott 1998; Frydman 1992; Giacalone 1998; Lelaidier 1994; Stenlund 1999).
Main outcomes
Additional induction agents required
Women in the mifepristone group were less likely to require further medication to induce labour compared with those in the control group (average RR 0.59, 95% CI 0.37 to 0.95; 311 women; 4 studies; I² = 74%; Analysis 7.1). However, there was considerable heterogeneity for this outcome (I² = 74%, Tau² = 0.16, Chi² test for heterogeneity P = 0.009). The wide 95% prediction interval (0.08 to 4.39) indicated that this result should be interpreted cautiously as some further study might yield a negative result.
7.1. Analysis.
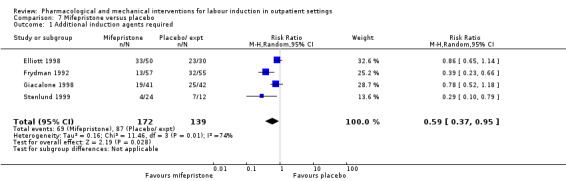
Comparison 7 Mifepristone versus placebo, Outcome 1 Additional induction agents required.
Serious neonatal morbidity or perinatal death (composite outcome will include, for example, seizures, birth asphyxia defined by trialists, neonatal encephalopathy, disability in childhood)
Stenlund 1999 examined serious neonatal morbidity (the number of babies requiring antIconvulsive therapy); there was little difference between groups (RR 1.56, 95% CI 0.07 to 35.67; 36 infants; 1 study; Analysis 7.2). Lelaidier 1994 reported on perinatal mortality and there were no deaths in either group (Analysis 7.9).
7.2. Analysis.
Comparison 7 Mifepristone versus placebo, Outcome 2 Serious neonatal morbidity or death.
7.9. Analysis.
Comparison 7 Mifepristone versus placebo, Outcome 9 Perinatal death.
There was no information on vaginal birth not achieved within 24 hours, length of hospital stay, use of emergency services, maternal or caregiver satisfaction, or serious maternal morbidity or death.
Additional outcomes of interest
Measures of effectiveness
Oxytocin augmentation
There was no evidence that mifepristone had an impact on the number of women who required oxytocin augmentation (RR 0.89, 95% CI 0.63 to 1.26; 116 women; 2 studies; Analysis 7.3).
7.3. Analysis.
Comparison 7 Mifepristone versus placebo, Outcome 3 Oxytocin augmentation.
Pain relief requirements (epidural, opioids)
A similar number of women in each group used epidural anaesthesia (RR 0.87, 95% CI 0.73 to 1.03; 112 women; 1 study; Analysis 7.4).
7.4. Analysis.
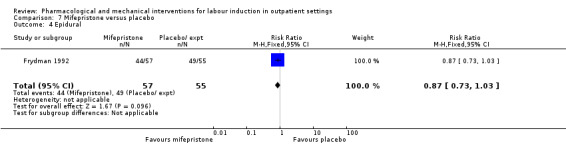
Comparison 7 Mifepristone versus placebo, Outcome 4 Epidural.
Vaginal birth not achieved within 48 and 72 hours, and randomisation to birth interval were not reported.
Complications
There was only limited evidence on the impact of mifepristone on maternal and neonatal health.
There were no clear differences between the groups for the following outcomes:
instrumental vaginal birth (RR 1.35, 95% CI 0.93 to 1.97; 343 women; 5 studies; Analysis 7.5);
caesarean section (RR 0.88, 95% CI 0.62 to 1.25; 343 women; 5 studies; Analysis 7.6);
Apgar score < 7 at five minutes (RR 1.56, 95% CI 0.07 to 35.67; 119 infants; 2 studies; Analysis 7.7);
NICU admission (RR 0.93, 95% CI 0.31 to 2.79; 163 infants; 2 studies; Analysis 7.8)
-
Serious maternal complications:
uterine scar separation (one women in each group) (RR 1.00, 95% CI 0.07 to 14.64; 32 women; 1 study; Analysis 7.10); and
chorioamnionitis (RR 2.00, 95% CI 0.20 to 19.91; 32 women; 1 study; Analysis 7.11).
7.5. Analysis.
Comparison 7 Mifepristone versus placebo, Outcome 5 Assisted (instrumental) vaginal birth.
7.6. Analysis.
Comparison 7 Mifepristone versus placebo, Outcome 6 Caesarean section.
7.7. Analysis.
Comparison 7 Mifepristone versus placebo, Outcome 7 Apgar score < 7 at 5 minutes.
7.8. Analysis.
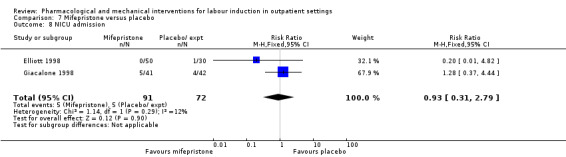
Comparison 7 Mifepristone versus placebo, Outcome 8 NICU admission.
7.10. Analysis.
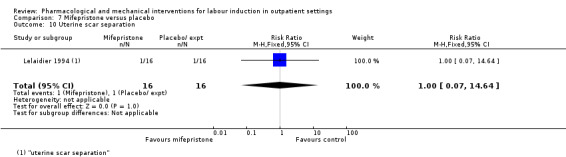
Comparison 7 Mifepristone versus placebo, Outcome 10 Uterine scar separation.
7.11. Analysis.
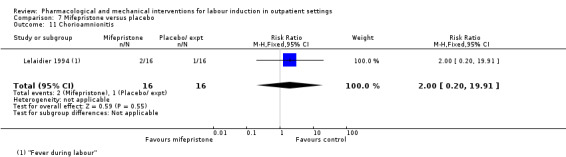
Comparison 7 Mifepristone versus placebo, Outcome 11 Chorioamnionitis.
Uterine hyperstimulation (with and without fetal heart rate (FHR) changes), postpartum haemorrhage (as defined by the trial authors), other serious maternal complications, and serious neonatal complications were not reported.
Non‐prespecified outcomes
Stenlund 1999 reported that during the first 48 hours after treatment started, 83.3% of women with mifepristone and 41.7% with placebo went into labour or had a ripe cervix (RR 2.00, 95% CI 1.00 to 4.00; 36 women; 1 study; Analysis 7.12). The median time to onset of labour from commencing treatment was 24 hours 10 minutes for women who had mifepristone and 52 hours for women with placebo. Giacalone 1998 and Stenlund 1999 looked at failure to achieve changes in the cervix after 24 to 48 hours and here results favoured the mifepristone group (RR 0.36, 95% CI 0.20 to 0.63; 119 women; 2 studies; Analysis 7.13).
7.12. Analysis.
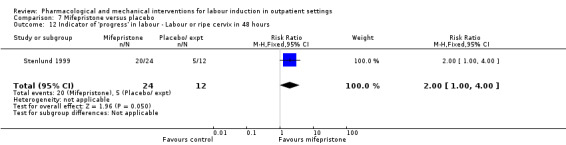
Comparison 7 Mifepristone versus placebo, Outcome 12 Indicator of 'progress' in labour ‐ Labour or ripe cervix in 48 hours.
7.13. Analysis.
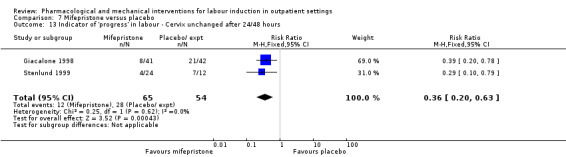
Comparison 7 Mifepristone versus placebo, Outcome 13 Indicator of 'progress' in labour ‐ Cervix unchanged after 24/48 hours.
None of the studies reported on the number of women achieving vaginal birth within 24 hours, but Elliott 1998 described the number of women in spontaneous labour within 72 hours. There was no evidence of a difference between groups receiving mifepristone versus placebo (RR 1.46, 95% CI 0.68 to 3.10; 80 women; 1 study; Analysis 7.14). The time to onset of labour was similar in all three study groups, with a median of 81 hours 15 minutes for placebo, 80 hours 20 minutes for 50 mg mifepristone, and 75 hours 50 minutes for 200 mg mifepristone.
7.14. Analysis.
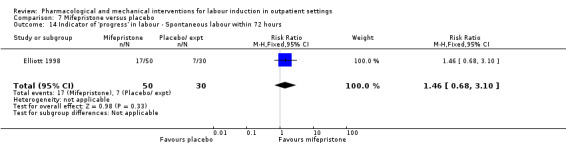
Comparison 7 Mifepristone versus placebo, Outcome 14 Indicator of 'progress' in labour ‐ Spontaneous labour within 72 hours.
Giacalone 1998 reported on "spontaneous labour" within 48 hours and results favoured the mifepristone group (RR 2.05, 95% CI 1.27 to 3.30; 83 women; 1 study; Analysis 7.15). There was a shorter interval between the beginning of treatment and onset of labour, and between treatment and vaginal birth for the mifepristone group (the median interval to labour onset was 31.7 hours for mifepristone group versus 53.9 hours for placebo, and 31.3 hours versus 58.5 hours between treatment and birth; with a reported P = 0.02 for both outcomes). Lelaidier 1994 reported a reduction in oxytocin dose (international units, IU) (MD ‐2.56 IU, 95% CI ‐4.01 to ‐1.11; 32 women; 1 study; Analysis 7.16) and also reported that the interval between the start of treatment and the onset of labour (hours) was shorter in the mifepristone group (MD ‐22.15, 95% CI ‐35.96 to ‐8.34; 32 women; 1 study; Analysis 7.17). In the Frydman 1992 study, mifepristone reduced the total dose (described in French as "international measurement" units, IM) of oxytocin for women having both vaginal (MD ‐2.07 IM, 95% CI ‐3.21 to ‐0.93; 76 women) and caesarean deliveries (MD ‐1.97 IM, 95% CI ‐3.37 to ‐0.57; 36 women); however, this should be interpreted with caution (data not shown).
7.15. Analysis.
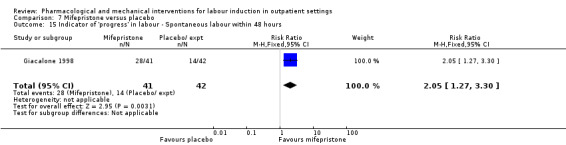
Comparison 7 Mifepristone versus placebo, Outcome 15 Indicator of 'progress' in labour ‐ Spontaneous labour within 48 hours.
7.16. Analysis.
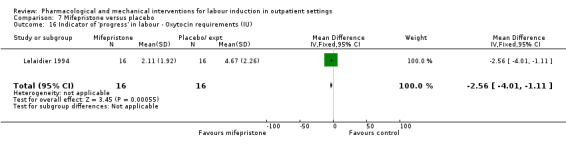
Comparison 7 Mifepristone versus placebo, Outcome 16 Indicator of 'progress' in labour ‐ Oxytocin requirements (IU).
7.17. Analysis.
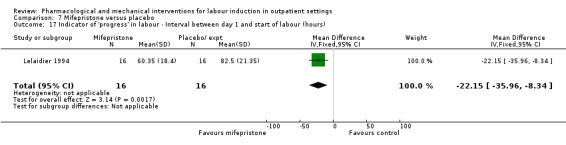
Comparison 7 Mifepristone versus placebo, Outcome 17 Indicator of 'progress' in labour ‐ Interval between day 1 and start of labour (hours).
8. Oestrogen versus placebo: one study, analysis for 87 women
Main outcomes
We included one study (Larmon 2002) in this comparison and there was no information reported on any of the review's main outcomes.
Additional outcomes of interest
Measures of effectiveness
Oxytocin augmentation
Similar numbers of women in each group received oxytocin augmentation (RR 0.93, 95% CI 0.61 to 1.43; 87 women; 1 study; Analysis 8.1)
8.1. Analysis.
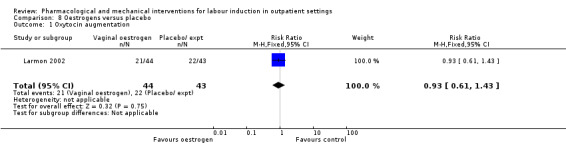
Comparison 8 Oestrogens versus placebo, Outcome 1 Oxytocin augmentation.
There was no information on vaginal birth not achieved within 48 and 72 hours, randomisation to birth interval, and pain relief requirements (epidural, opioids).
Complications
There was no clear differences between the oestrogen and placebo groups for:
instrumental vaginal birth (RR 0.84, 95% CI 0.44 to 1.60; 87 women; 1 study; Analysis 8.2);
caesarean section (RR 1.27, 95% CI 0.63 to 2.58; 87 women; 1 study; Analysis 8.3);
NICU admission (RR 0.98, 95% CI 0.06 to 15.13; 87 infants; 1 study; Analysis 8.4)
-
serious maternal complications:
chorioamnionitis (RR 1.95, 95% CI 0.38 to 10.12; 87 women; 1 study; Analysis 8.5); and
endometritis (RR 2.93, 95% CI 0.32 to 27.10; 87 women; 1 study; Analysis 8.6).
8.2. Analysis.
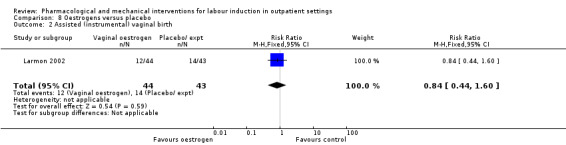
Comparison 8 Oestrogens versus placebo, Outcome 2 Assisted (instrumental) vaginal birth.
8.3. Analysis.
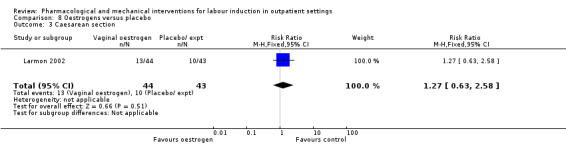
Comparison 8 Oestrogens versus placebo, Outcome 3 Caesarean section.
8.4. Analysis.
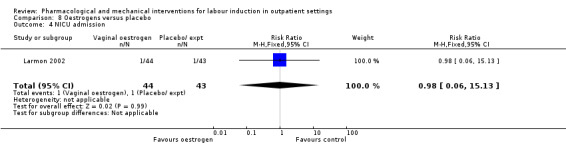
Comparison 8 Oestrogens versus placebo, Outcome 4 NICU admission.
8.5. Analysis.
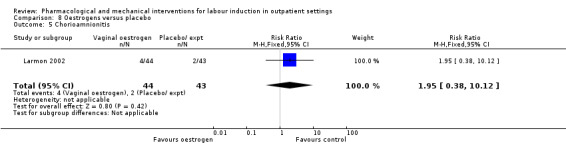
Comparison 8 Oestrogens versus placebo, Outcome 5 Chorioamnionitis.
8.6. Analysis.
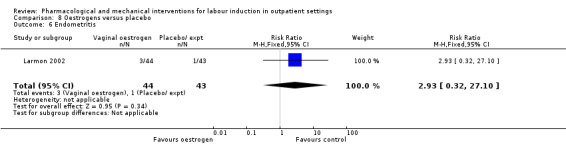
Comparison 8 Oestrogens versus placebo, Outcome 6 Endometritis.
There was no information for uterine hyperstimulation (with or without FHR changes), Apgar score less than seven at five minutes, perinatal death, postpartum haemorrhage, other serious maternal complications, or serious neonatal complications.
9. Vaginal isosorbide mononitrate (IMN) versus placebo: seven studies, 2287 women
We included four trials in this comparison group (Bollapragada 2006a; Bullarbo 2007; Habib 2008; Schmitz 2014). Schmitz 2014 was a large multicenter trial of 1362 women in France which provided additional data for a range of outcomes for this update.
Main outcomes
There was no clear differences between the vaginal isosorbide mononitrate and placebo groups for:
vaginal birth not achieved within 24 hours (RR 0.97, 95% CI 0.83 to 1.15; 238 women; 1 study; Analysis 9.1);
additional induction agents required (average RR 0.87, 95% CI 0.75 to 1.00; 4 studies; I² = 66%; Tau² = 0.01; Chi² = 8.92; Analysis 9.2);
-
serious neonatal morbidity or perinatal death:
perinatal death (average RR 1.61, 95% CI 0.08 to 33.26; 1712 infants; 2 studies; I² = 48%; Tau² = 2.31; Chi² = 1.94; Analysis 9.5);
neonatal trauma (long bone fracture, collarbone fracture, basal skull fracture, brachial plexus palsy, facial nerve palsy, phrenic nerve palsy, or subdural haemorrhage) (RR 0.67, 95% CI 0.19 to 2.37; 1362 infants; 1 study; Analysis 9.6);
neonatal convulsions in first 24 hours (there were no incidences of this outcome in either group; Analysis 9.7);
tracheal ventilation longer than 24 hours (RR 1.01, 95% CI 0.14 to 7.14; 1362 infants; 1 study; Analysis 9.8);
NICU admission for five or more days (RR 0.67, 95% CI 0.19 to 2.37; 1362 infants; 1 study; Analysis 9.9);
neonatal transfer (RR 1.07, 95% CI 0.67 to 1.70; 1362 infants; 1 study; Analysis 9.10);
-
serious maternal morbidity or death:
maternal death (there were no incidences of maternal death in either group; Analysis 9.11);
severe postpartum haemorrhage (RR 1.55, 95% CI 0.78 to 3.09; 1362 women; 1 study; Analysis 9.12);
deep vein thrombosis (RR 3.03, 95% CI 0.12 to 74.16; 1362 women; 1 study; Analysis 9.13);
perinatal death (average RR 1.61, 95% CI 0.08 to 33.26; 1712 infants; 2 studies; I² = 48%; Tau² = 2.31; Chi² = 1.94; Analysis 9.5);
neonatal trauma (long bone fracture, collarbone fracture, basal skull fracture, brachial plexus palsy, facial nerve palsy, phrenic nerve palsy, or subdural haemorrhage) (RR 0.67, 95% CI 0.19 to 2.37; 1362 infants; 1 study; Analysis 9.6);
neonatal convulsions in first 24 hours (there were no incidences of this outcome in either group; Analysis 9.7);
tracheal ventilation longer than 24 hours (RR 1.01, 95% CI 0.14 to 7.14; 1362 infants; 1 study; Analysis 9.8);
NICU admission for five or more days (RR 0.67, 95% CI 0.19 to 2.37; 1362 infants; 1 study; Analysis 9.9);
neonatal transfer (RR 1.07, 95% CI 0.67 to 1.70; 1362 infants; 1 study; Analysis 9.10);
9.1. Analysis.
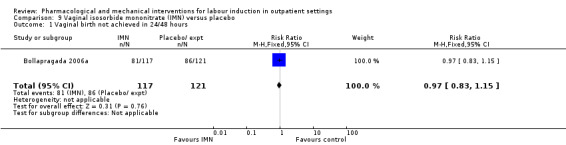
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 1 Vaginal birth not achieved in 24/48 hours.
9.2. Analysis.
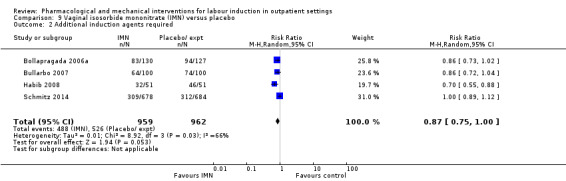
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 2 Additional induction agents required.
9.5. Analysis.
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 5 Perinatal death.
9.6. Analysis.
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 6 Neonatal trauma (long bone fracture, collarbone fracture, basal skull fracture, brachial plexus palsy, facial nerve palsy, phrenic nerve palsy, or subdural haemorrhage).
9.7. Analysis.
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 7 Neonatal convulsions in the first 24 hours.
9.8. Analysis.
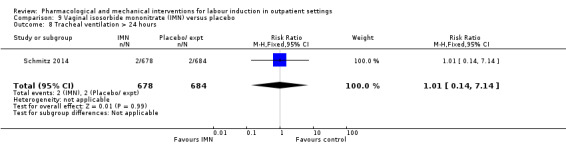
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 8 Tracheal ventilation > 24 hours.
9.9. Analysis.
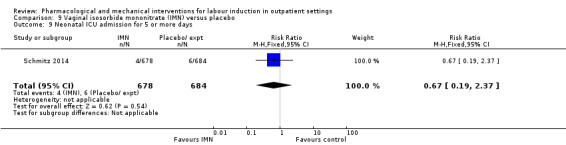
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 9 Neonatal ICU admission for 5 or more days.
9.10. Analysis.
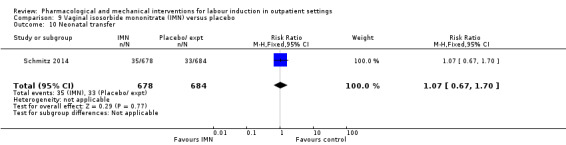
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 10 Neonatal transfer.
9.11. Analysis.
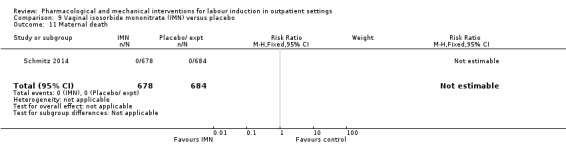
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 11 Maternal death.
9.12. Analysis.
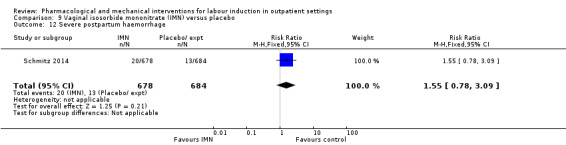
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 12 Severe postpartum haemorrhage.
9.13. Analysis.
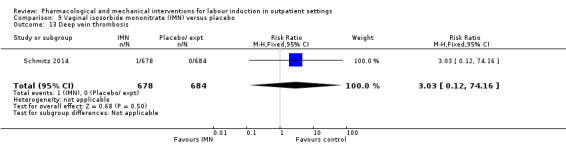
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 13 Deep vein thrombosis.
Maternal satisfaction
In four trials, women were asked to rate their satisfaction, however results could not be meta‐analysed due to differences in how questions were structured. Bullarbo 2007 found no difference in levels of satisfaction between women in the two arms of the trial (Analysis 9.4). Schmitz 2014 asked women to rate their satisfaction, and whether they would recommend the same treatment, finding that more women in the IMN group felt very or extremely satisfied, and would recommend the same treatment (Analysis 9.4). In the study by Bollapragada 2006a, women were asked to rate their satisfaction with the induction process at home. On five of the six measures of satisfaction, women in the placebo group were slightly more satisfied with their care compared with those in the IMN group, although the differences between groups were not large, and the mean scores in both groups suggested general satisfaction (Analysis 9.3). Satisfaction data from Attanayake 2014 could not be meta‐analysed, as only narrative results were provided ‐ they reported that greater than 75% of women in both groups considered the therapy as a good intervention (rather than inpatient therapy), and greater than 85% were happy to use outpatient therapy in a subsequent pregnancy, and would recommend to a friend.
9.4. Analysis.
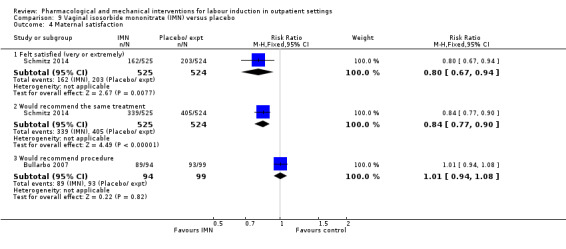
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 4 Maternal satisfaction.
9.3. Analysis.
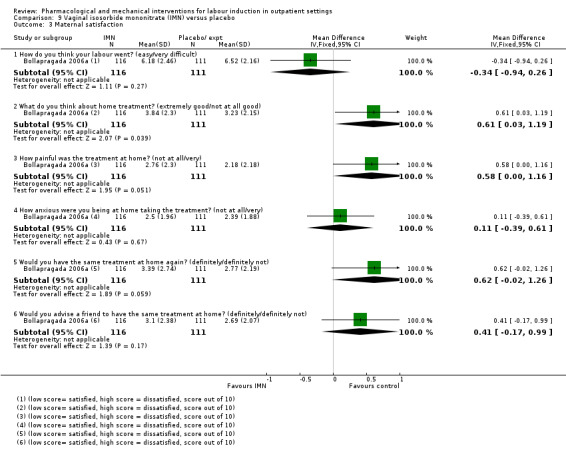
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 3 Maternal satisfaction.
Length of hospital stay, use of emergency services, and caregiver satisfaction were not reported.
Additional outcomes of interest
Measures of effectiveness
There were no clear differences between the groups for the outcomes:
oxytocin augmentation (average RR 0.95, 95% CI 0.78 to 1.14; 1816 women; 3 studies; I² = 72%; Tau² = 0.02; Chi² = 7.11; Analysis 9.14); and
pain relief requirements (epidural) (RR 0.94, 95% CI 0.82 to 1.09; 350 women; 1 study; Analysis 9.15).
9.14. Analysis.
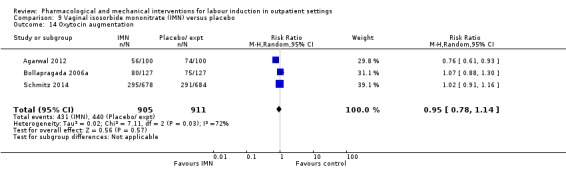
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 14 Oxytocin augmentation.
9.15. Analysis.
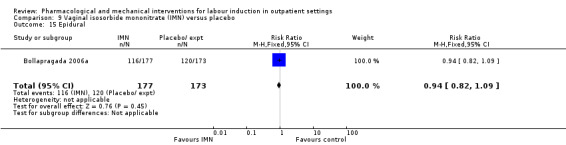
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 15 Epidural.
No information was available for vaginal birth not achieved within 48 and 72 hours, or randomisation to birth interval.
Complications
There was no evidence of clear difference between groups for:
uterine hyperstimulation (with FHR changes) (RR 0.20, 95% CI 0.01 to 4.07; 102 women; 1 study; Analysis 9.16);
uterine hyperstimulation (unclear if with or without FHR changes) (RR 0.09, 95% CI 0.01 to 1.62; 200 women; 1 study; Analysis 9.17);
Instrumental vaginal birth (RR 0.81, 95% CI 0.61 to 1.07; 1712 women; 2 studies; Analysis 9.18);
caesarean section (RR 0.99, 95% CI 0.87 to 1.14; 2286 women; 6 studies; Analysis 9.19);
Apgar score less than seven at five minutes (RR 0.88, 95% CI 0.44 to 1.76; 2214 infants; 5 studies; Analysis 9.20);
NICU admission (RR 0.89, 95% CI 0.59 to 1.36; 1068 infants; 6 studies; Analysis 9.21);
perinatal death (reported in Serious neonatal morbidity or perinatal death);
postpartum haemorrhage (> 500 mL) (RR 1.13, 95% CI 0.95 to 1.36; 2214 women; 5 studies; Analysis 9.22); and
-
serious neonatal complications:
neonatal infection (RR 1.00, 95% CI 0.26 to 3.89; 200 infants; 1 study; Analysis 9.23).
9.16. Analysis.
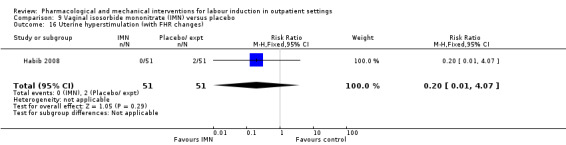
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 16 Uterine hyperstimulation (with FHR changes).
9.17. Analysis.
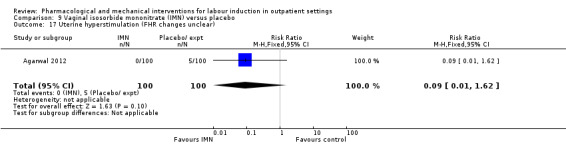
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 17 Uterine hyperstimulation (FHR changes unclear).
9.18. Analysis.
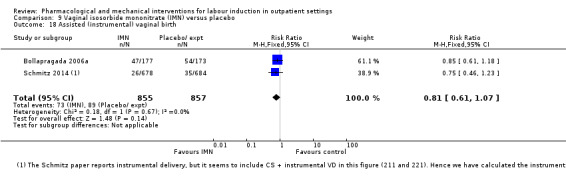
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 18 Assisted (instrumental) vaginal birth.
9.19. Analysis.
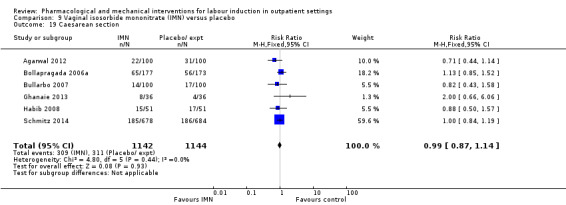
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 19 Caesarean section.
9.20. Analysis.
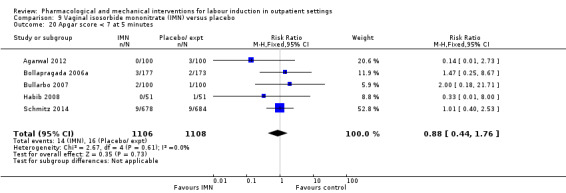
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 20 Apgar score < 7 at 5 minutes.
9.21. Analysis.
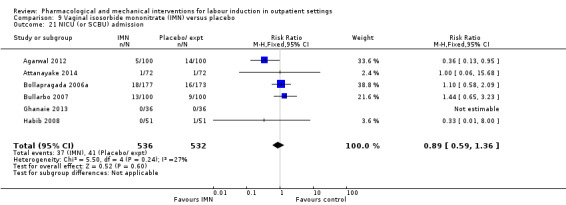
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 21 NICU (or SCBU) admission.
9.22. Analysis.
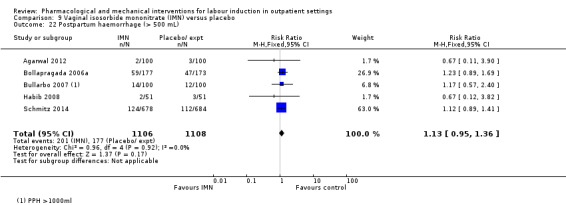
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 22 Postpartum haemorrhage (> 500 mL).
9.23. Analysis.
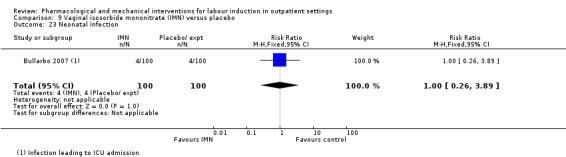
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 23 Neonatal infection.
No other serious maternal complications were reported.
Non‐prespecified outcomes
IMN use was associated with increased side effects, including nausea (RR 2.39, 95% CI 1.54 to 3.70; 1926 women; 4 studies; I² = 37%; Tau² = 0.07; Chi² = 4.78; Analysis 9.24), and particularly headaches (RR 5.45, 95% CI 3.38 to 8.81; 2300 women; 7 studies; I² = 76%; Tau² = 0.21; Chi² = 21.09; Analysis 9.25). In one study 22/112 women in the IMN group reported severe headaches compared with only 1/108 in the placebo group (RR 21.21, 95% CI 2.91 to 154.65; 220 women; 1 study; Analysis 9.26).
9.24. Analysis.
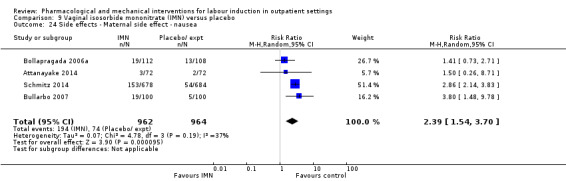
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 24 Side effects ‐ Maternal side effect ‐ nausea.
9.25. Analysis.
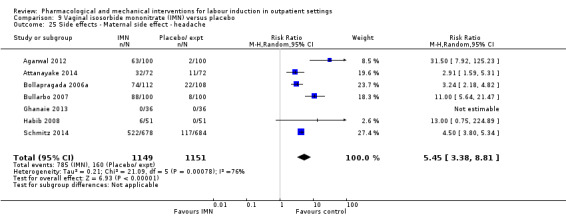
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 25 Side effects ‐ Maternal side effect ‐ headache.
9.26. Analysis.
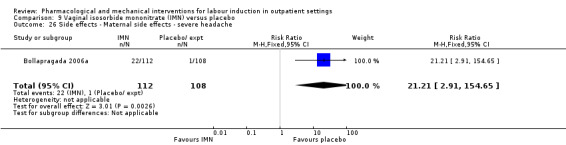
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 26 Side effects ‐ Maternal side effects ‐ severe headache.
Several measures of progress in labour were reported, with one to three trials available per outcome. In general, results indicate that IMN increased the likelihood of being admitted in established labour within 24 hours (RR 2.75, 95% CI 1.29 to 5.88; 200 women; 1 study; Analysis 9.27) and caused changes in the Bishop score (Bishop score < 6 or active labour at 36 hours: RR 3.80, 95% CI 1.54 to 9.40; 102 women; 1 study; Analysis 9.28, Bishop score on admission after treatment: MD 2.73, 95% CI 2.17 to 3.29; 200 women; 1 study; Analysis 9.30. Change in Bishop score: MD 2.76, 95% CI 2.48 to 3.03; 272 women; 2 studies; Analysis 9.31). Time in hours from admission to birth (days) was also reduced (MD ‐4.7 hours, 95% CI ‐6.08 to ‐3.31; I² =42%; Tau² =0.63; 374 women; 3 studies; Analysis 9.29).
9.27. Analysis.
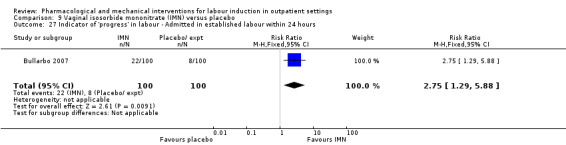
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 27 Indicator of 'progress' in labour ‐ Admitted in established labour within 24 hours.
9.28. Analysis.
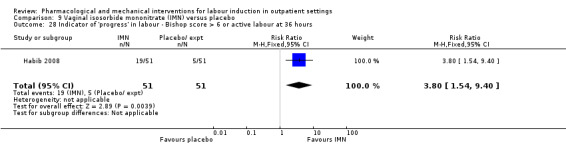
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 28 Indicator of 'progress' in labour ‐ Bishop score > 6 or active labour at 36 hours.
9.30. Analysis.
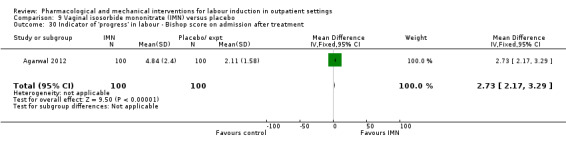
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 30 Indicator of 'progress' in labour ‐ Bishop score on admission after treatment.
9.31. Analysis.
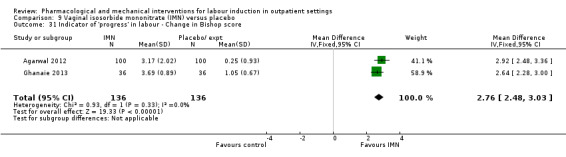
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 31 Indicator of 'progress' in labour ‐ Change in Bishop score.
9.29. Analysis.
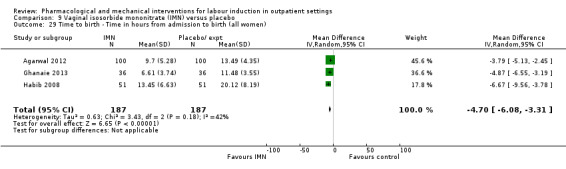
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 29 Time to birth ‐ Time in hours from admission to birth (all women).
However, Agarwal 2012 reported that the interval from onset of labour to birth (hours) was not different between IMN and placebo (MD ‐1.24 hours, 95% CI ‐1.82 to ‐0.66; 200 women; 1 study; Analysis 9.32), and Bollapragada 2006a reported that the rate of cervix unchanged after 48 hours was higher in the IMN group compared to placebo (RR 0.83, 95% CI 0.70 to 0.97; 257 women; 1 study; Analysis 9.33).
9.32. Analysis.
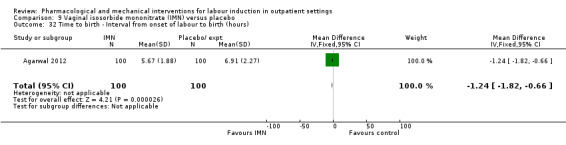
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 32 Time to birth ‐ Interval from onset of labour to birth (hours).
9.33. Analysis.
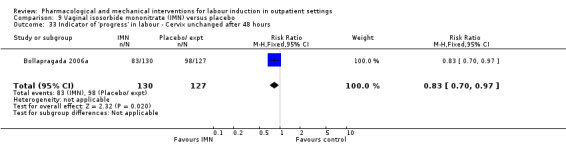
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 33 Indicator of 'progress' in labour ‐ Cervix unchanged after 48 hours.
Bollapragada 2006a also reported the mean interval from hospital admission to birth (hours) for all women (MD ‐0.70 hours, 95% CI ‐6.11 to 4.71; 128 women; 1 study; Analysis 9.34), and for those women having vaginal deliveries, along with the mean change in Bishop scores at 48 hours after baseline; there were no differences between groups for any of these outcomes (data not shown).
9.34. Analysis.
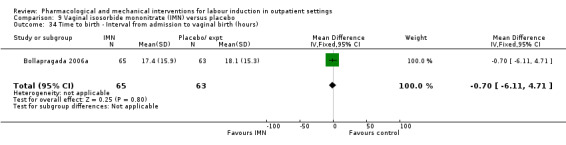
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 34 Time to birth ‐ Interval from admission to vaginal birth (hours).
Bollapragada 2006a collected information on the cost of providing care; the mean overall cost of the care (GBP) package was very similar for women in both groups (MD 11.98 GBP, 95% CI ‐105.34 to 129.30; Analysis 9.35).
9.35. Analysis.
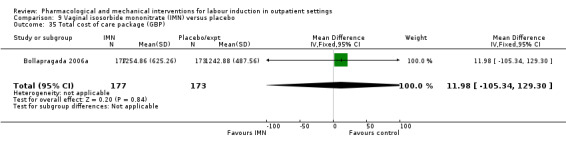
Comparison 9 Vaginal isosorbide mononitrate (IMN) versus placebo, Outcome 35 Total cost of care package (GBP).
10. Acupuncture versus routine care: one study 56 women
Harper 2006 presented limited information relevant to this review. The intervention did not appear to have any impact on the number of women requiring additional agents to induce labour (RR 0.60, 95% CI 0.31 to 1.17; 56 women; 1 study; Analysis 10.1) or having caesarean section (RR 0.43, 95% CI 0.17 to 1.11; 56 women; 1 study; Analysis 10.2). There were no clear differences between groups for women starting labour spontaneously, cervical dilatation at hospital admission, or the mean time from study enrolment to birth (data not shown). No further main or additional outcomes were reported in this study.
10.1. Analysis.
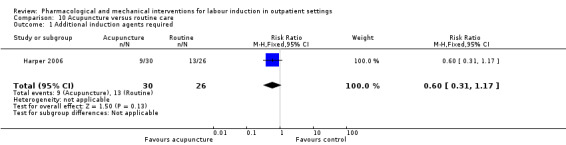
Comparison 10 Acupuncture versus routine care, Outcome 1 Additional induction agents required.
10.2. Analysis.
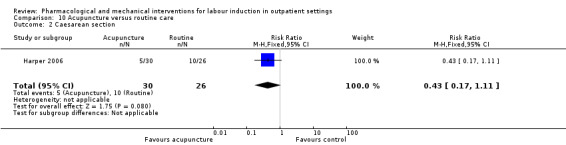
Comparison 10 Acupuncture versus routine care, Outcome 2 Caesarean section.
11. Outpatient amniotomy for induction of labour versus routine care: one study, 521 women
Rijnders 2011 was an unblinded trial that randomised 521 women to amniotomy in an outpatient setting (at home) for induction between 292 and 294 days gestation, or routine care (as per local guidelines, this was referral to an obstetrician for foetal assessment on the morning of day 294). Rijnders 2011 was assessed as being at high risk of attrition bias so should have been removed according to the pre‐specified sensitivity analysis. However as this was the only study in this comparison, sensitivity analysis was not performed.
Main outcomes
Maternal satisfaction
While measures of maternal satisfaction were higher in the amniotomy at home group (look back positively on treatment: RR 1.04, 95% CI 0.97 to 1.10; 404 women; 1 study; Analysis 11.1. Would have preferred other treatment: RR 0.51, 95% CI 0.36 to 0.72; 472 women; 1 study; Analysis 11.2), the proportion of women completing the survey was higher in the amniotomy group (82% versus 73%), and it seems likely that a response bias may have occurred for surveys conducted in home settings.
11.1. Analysis.
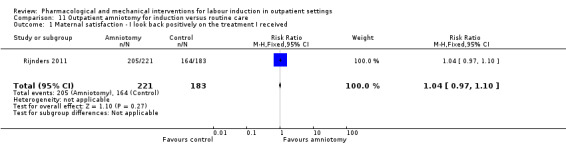
Comparison 11 Outpatient amniotomy for induction versus routine care, Outcome 1 Maternal satisfaction ‐ I look back positively on the treatment I received.
11.2. Analysis.
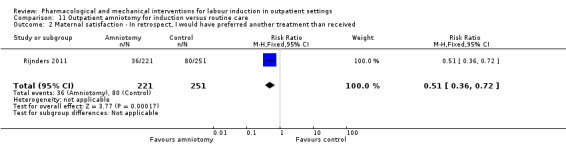
Comparison 11 Outpatient amniotomy for induction versus routine care, Outcome 2 Maternal satisfaction ‐ In retrospect, I would have preferred another treatment than received.
There was no information on vaginal birth not achieved within 24 hours, additional induction agents required, length of hospital stay, use of emergency services, caregiver satisfaction, serious neonatal morbidity or perinatal death (composite outcome will include, for example, seizures, birth asphyxia defined by trialists, neonatal encephalopathy, disability in childhood), or serious maternal morbidity or death (composite outcome will include, for example, uterine rupture, admission to intensive care unit, septicaemia).
Additional outcomes of interest
Measures of effectiveness
Oxytocin augmentation
Fewer women had augmentation or induction or both in the outpatient amniotomy group than in the routine care group (RR 0.83, 95% CI 0.71 to 0.97; 521 women; 1 study; Analysis 11.3).
11.3. Analysis.
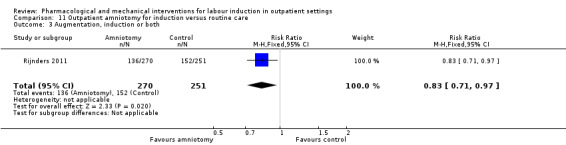
Comparison 11 Outpatient amniotomy for induction versus routine care, Outcome 3 Augmentation, induction or both.
Pain relief requirements (epidural, opioids)
There was little difference between groups for women receiving epidural or opioids or both (RR 0.99, 95% CI 0.76 to 1.30; 521 women; 1 study; Analysis 11.4).
11.4. Analysis.
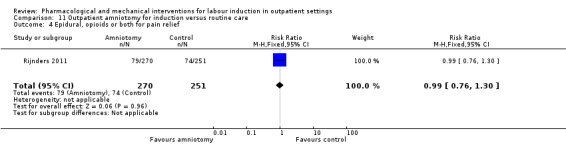
Comparison 11 Outpatient amniotomy for induction versus routine care, Outcome 4 Epidural, opioids or both for pain relief.
Vaginal birth not achieved within 48 and 72 hours and randomisation to birth interval were not reported.
Complications
There were no clear differences between the groups for the following outcomes:
instrumental vaginal birth (RR 0.70, 95% CI 0.46 to 1.08; 521 women; 1 study; Analysis 11.5);
caesarean section (RR 1.20, 95% CI 0.78 to 1.86; 521 women; 1 study; Analysis 11.6);
Apgar score less than seven at five minutes (RR 1.86, 95% CI 0.34 to 10.06; 521 women; 1 study; Analysis 11.7); and
NICU admission (RR 1.09, 95% CI 0.64 to 1.85; 521 women; 1 study; Analysis 11.8).
11.5. Analysis.
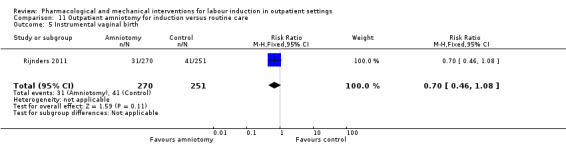
Comparison 11 Outpatient amniotomy for induction versus routine care, Outcome 5 Instrumental vaginal birth.
11.6. Analysis.
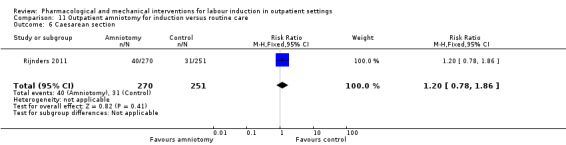
Comparison 11 Outpatient amniotomy for induction versus routine care, Outcome 6 Caesarean section.
11.7. Analysis.
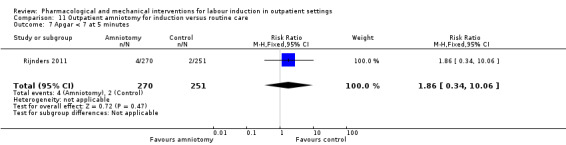
Comparison 11 Outpatient amniotomy for induction versus routine care, Outcome 7 Apgar < 7 at 5 minutes.
11.8. Analysis.
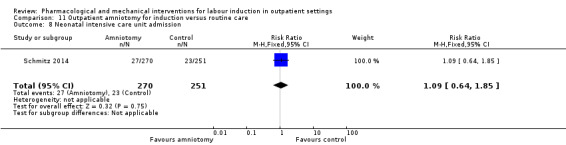
Comparison 11 Outpatient amniotomy for induction versus routine care, Outcome 8 Neonatal intensive care unit admission.
There was no information for uterine hyperstimulation (with FHR changes), uterine hyperstimulation (without FHR changes), perinatal death, postpartum haemorrhage, serious maternal complications (considered as separate outcomes, e.g. intensive care unit admission, septicaemia, uterine rupture), or serious neonatal complications (considered as separate outcomes).
Non‐prespecified outcomes
Mean duration of birth (hours) was very similar in each group (MD 0.40 hours, 95% CI ‐0.72 to 1.52; 521 women; 1 study; Analysis 11.9).
11.9. Analysis.
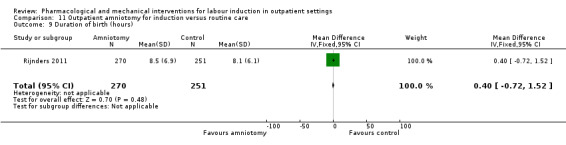
Comparison 11 Outpatient amniotomy for induction versus routine care, Outcome 9 Duration of birth (hours).
Discussion
Summary of main results
The included studies examined 11 different types of interventions in outpatient settings. Overall, the results demonstrate that outpatient induction of labour is feasible and that important adverse events are rare (Table 4; Table 5; Table 6). However, the safety data should be treated with considerable caution. First, very few of the studies provided information on maternal and neonatal death or serious morbidity. It may not be safe to assume that because adverse outcomes were not reported, they did not occur. Further, even where outcomes such as perinatal mortality, maternal complications or serious neonatal morbidity were reported, the finding that there was no apparent difference between groups was not surprising as none of these studies had the statistical power to detect differences for such rare outcomes in relatively low‐risk populations.
1. Uterine hyperstimulation with outpatient inductions.
| Uterine hyperstimulation | |
| PGE₂(vaginal) | |
| Hage 1993 | 1/18 PGE group (FHR status unknown), 0/18 in placebo group |
| Newman 1997 | 2/28 PGE group (FHR status unknown), 0/30 in control group (no treatment) |
| O'Brien 1995 | 1/50 PGE group (normal FHR), 0/50 in placebo group |
| Total | 4/96 PGE, 0/98 in control group |
| PGE₂(intracervical) | |
| Buttino 1990 | 1/23 PGE group (with FHR decelerations), 0/20 in placebo group |
| Lien 1998 | 2/43 PGE group, 1/47 placebo group with FHR deceleration in both |
| McKenna 1999 | 1/30 PGE group (fetal bradycardia), 0/31 placebo group |
| Rayburn 1999 | 1/143 PGE group, 0/151 control (no treatment) with hyperstimulation 11/143 FHR decelerations in PGE group, 12/151 in control |
| Total | 5/239 PGE, 1/249 control with hyperstimulation |
| Intravaginal misoprostol | |
| Stitely 2000 | 2/27 misoprostol group with FHR deceleration, 2/33 placebo group 1/27 misoprostol with tachysystole without FHR changes, 0/33 placebo group |
| Incerpi 2001 | 3/57 misoprostol with hyperstimulation (FHR unknown), 2/63 placebo group 2/57 misoprostol with hypertonus, 5/57 misoprostol with tachysystole, none control |
| McKenna 2004 | 1/33 misoprostol (FHR deceleration), 0/35 placebo group |
| Oral misoprostol | |
| Lyons 2001 | 1/18 misoprostol, 2/22 placebo group (FHR unknown) with hyperstimulation |
| Gaffaney 2009 | 8/43 misoprostol, 4/44 placebo group hyperstimulation syndrome (tachysystole or hypertonus, with FHR changes) |
| Total | 9/61 misoprostol, 6/66 placebo group |
| Mifepristone | |
| Giacalone 1998 | 4/41 mifepristone group, 0/42 placebo group with hypertonia (FHR unknown) |
| Lelaidier 1994 | 0/16 in both groups |
| Total | 4/57 mifepristone, 0/58 placebo with hypertonia |
| IMN | |
| Habib 2008 | 0/51 IMN group, 2/51 placebo group with hyperstimulation (abnormal FHR) 1/51 IMN, 8/51 placebo group with tachysystolia (FHR normal) |
| Agarwal 2012 | 0/100 IMN group, 5/100 placebo group with hyperstimulation (FHR changes unclear) |
| Total | 1/151 IMN group, 15/151 placebo group (hyperstimulation or tachysystolia, ±FHR changes) |
IMN: isosorbide mononitrate; FHR: fetal heart rate
2. Neonatal complications following induction in outpatient setting.
| Neonatal complications | |
| PGE₂vaginal | |
| Sawai 1991 | 0/24 in PGE₂ group; 2/26 in placebo group to NICU |
| Sawai 1994 | 2/38 in PGE₂; 4/42 in placebo group to NICU |
| O'Brien 1995 | 1/50 in PGE₂; 5/50 in placebo group to NICU |
| Total | 3/112 PGE, 11/118 control to NICU |
| PGE₂intracervical | |
| Larmon 2002 | 6/41 PGE group, 8/43 placebo group with complication such as tachypnoea, meconium aspiration, meconium or admission to NICU |
| Magann 1998 | 3/35 PGE₂ versus 0/35 control NICU admission |
| McKenna 1999 | 1/30 PGE, 2/31 placebo group with complication |
| Total | 10/106 PGE, 10/109 controls with neonatal complications/admitted to NICU |
| Vaginal misoprostol | |
| Stitely 2000 | 1/27 misoprostol, 3/33 placebo group to NICU |
| Incerpi 2001 | 18/57 misoprostol, 20/63 placebo group to NICU |
| McKenna 2004 | 0/33 misoprostol, 1/35 placebo group to NICU |
| Oboro 2005 | 1/38 misoprostol, 1/39 control (no treatment) to NICU |
| Gaffaney 2009 | 1/43 misoprostol, 1/44 placebo group to NICU |
| Total | 21/198 misoprostol, 26/214 control to NICU |
| Misoprostol 25 µg versus 50 µg | |
| Kipikasa 2005 | 1/23 25 µg, 2/26 50 µg misoprostol to NICU |
| Intracervical PGE₂versus intravaginal misoprostol | |
| Meyer 2005 | 5/42 PGE, 4/42 misoprostol to NICU |
| Mifepristone | |
| Elliott 1998 | 0/50 mifepristone, 1/30 placebo group to NICU |
| Giacalone 1998 | 5/41 mifepristone, 4/42 control to NICU |
| Total | 5/91 mifepristone, 5/72 control to NICU |
| IMN | |
| Bollapragada 2006a | 18/177 IMN, 16/173 placebo group to NICU |
| Bullarbo 2007 | 13/100 IMN, 9/100 placebo group to NICU |
| Habib 2008 | 0/51 IMN, 1/51 placebo group to NICU |
| Agarwal 2012 | 5/100 IMN, 14/100 placebo group to nursery admission |
| Ghanaie 2013 | 0/36 IMN, 0/36 placebo group to NICU |
| Attanayake 2014 | 1/72 IMN, 1/72 placebo group to NICU |
| Total | 37/536 IMN, 41/532 placebo group to NICU |
| Outpatient amniotomy for induction versus routine care | |
| Rijnders 2011 | 27/270 IMN, 23/251 placebo group to NICU |
NICU: neonatal intensive‐care unit
3. Maternal complications following induction of labour in outpatient setting.
| Maternal complications | |
| Intracervical PGE₂ | |
| Larmon 2002 | 4/41 PGE, 10/43 placebo group with complication such as endometritis, chorioamnionitis and pre‐eclampsia |
| Lien 1998 | 6/43 PGE, 3/47 placebo group with complication such as endometritis and chorioamnionitis |
| McKenna 1999 | 1/30 PGE with PPH, 0/31 placebo group 2/30 PGE, 2/31 placebo group with infection |
| Rayburn 1999 | 8/143 PGE, 7/151 control (no treatment) with endometritis |
| Total | 21/257 PGE₂, 22/272 control with maternal complications |
| Oral misoprostol | |
| Gaffaney 2009 | 8/43 misoprostol group, 9/44 placebo group with chorioamnionitis 1/43 misoprostol group, 2/44 placebo group with endometritis 2/43 misoprostol group, 0/44 placebo group with PPH |
| Total | 11/43 misoprostol group, 11/44 placebo group with maternal complications |
| IMN | |
| Bollapragada 2006a | Blood loss > 500 mL: 59/177 IMN, 47/173 placebo group |
| Bullarbo 2007 | Blood loss > 1000 mL: 14/100 IMN, 12/100 placebo group |
| Habib 2008 | PPH: 2/51 IMN, 3/51 placebo group |
| Agarwal 2012 | 2/100 IMN group, 3/100 placebo group with PPH |
| Ghanaie 2013 | 0/36 IMN group, 0/36 placebo group with need for blood transfusion |
| Schmitz 2014 | 0/678 IMN group, 0/684 placebo group for maternal death 124/678 IMN group, 112/684 placebo group for PPH 20/678 IMN group, 13/684 placebo group for severe PPH 1/678 IMN group, 0/684 placebo group for deep vein thrombosis |
| Total | 202/1142 IMN group, 204/1148 placebo group with maternal complications |
| Outpatient amniotomy | |
| Rijnders 2011 | 26/270 amniotomy group, 29/251 routine care group ‐ mother treated with antibiotics |
PPH: postpartum haemorrhage
There was some evidence that, compared with placebo or no treatment, induction agents reduced the need for further intervention to induce labour, and potentially shorten the interval from intervention to birth. However, we were unable to pool results on outcomes relating to progress in labour, as studies tended to measure a very broad range of outcomes.
There was no evidence that induction agents increased interventions in labour such as operative deliveries. Only five studies (Attanayake 2014; Bollapragada 2006a; Bullarbo 2007; Rijnders 2011; Schmitz 2014) collected information on women's views about the induction process, and overall there was very little information on the costs to health services of different methods of induction of labour in outpatient settings.
Few studies reported on maternal satisfaction. The Bollapragada 2006a trial suggested that women receiving isosorbide mononitrate were less satisfied than controls. This finding may have been associated with the relatively high number of women in the intervention group experiencing unpleasant side effects (particularly headaches) during the treatment period.
Overall completeness and applicability of evidence
It is debatable what would constitute definitive evidence on the effectiveness and safety of various induction protocols in the outpatient (home) environment. The issues that are likely to be important to women and healthcare providers were not adequately addressed in the included trials in this review or a related Cochrane Review comparing home and hospital inductions (Kelly 2013).
Safety
Adverse events in the pregnant population of women who are likely to be eligible for outpatient induction are rare (Table 4; Table 5). There is no consensus on what would be an unacceptable risk of an outpatient induction; views may vary among different healthcare systems and among women, doctors and healthcare commissioners in the same system. Assuming that one additional serious adverse event (e.g. perinatal death/serious morbidity) for every 500 outpatient inductions is considered unacceptable (irrespective of the cost savings made), a very large randomised trial or meta‐analysis including thousands of women would be needed to be able to exclude a possibility of such an excess risk. A trial (or meta‐analysis) of this size designed to exclude such an excess risk (equivalence trial) is unlikely to be funded, irrespective of the method used.
In the absence of adequate safety data from randomised trials, the only pragmatic solution is to rely on observational data from large cohorts with relatively robust surrogate outcomes such as emergency caesarean section for presumed fetal distress or emergency transfer to hospital. A paper from Canada (Salvador 2009) reported on 567 outpatient inductions with no serious complications, but it is not entirely clear what was included in this composite outcome. Other surrogate outcomes, such as uterine hyperstimulation or fetal heart rate abnormalities (which have been reported in some studies (e.g. Ramsey 2005)), may be difficult to interpret unless there are clear definitions of what these outcomes mean. The use of common outcomes with agreed definitions applicable to all healthcare settings would be welcome; see Implications for research.
Experience of women and staff
Outpatient induction may be more convenient for women, who may feel more comfortable at home, and prefer being there rather than in hospital. On the other hand, women may feel worried about the induction process (especially if they live at some distance from emergency facilities) and the induction agent may cause side effects that are distressing, so some women may prefer the reassurance offered by hospital care. We have very limited information on what women would prefer, and no evidence on whether any women were forced to make arrangements for rapid transfer to hospital.
Outcomes such as average time to 'admission in labour' may be difficult to understand if there is no clear definition of what this means. The time may be partly determined by women's decisions about when to attend hospital, which may depend on a broad range of physiological, psychological, social and practical factors. For example, a woman experiencing unpleasant side effects, living at a distance from emergency facilities may seek early admission; under these circumstances the outcome does not serve as a good proxy for progress in labour. Criteria for admission to hospital in the trials were frequently not specified and included active labour (variously defined), ruptured membranes and a range of other indications. Further, a short interval to admission is not necessarily a good thing; a very short interval means that sending women home may not be worthwhile, a long interval may not be harmful provided women are reasonably comfortable and there is no urgent need for birth. A short interval to admission is also meaningless if it is offset by prolonged and painful labour. Reporting these two outcomes separately may not, therefore, be helpful.
Measures of cervical change (Bishop score) may also be problematic, for example, mean increases in Bishop scores on hospital admission, or Bishop scores reaching a certain level at given time points, are not straightforward to interpret. Such outcomes may not give any clear idea of when birth will occur, whether more rapid cervical dilatation is predictive of a more rapid labour, or whether the birth will be more or less likely to be normal.
Cost
Health service providers may also assume that transferring care to community or outpatient settings may reduce the total costs of care; we have no evidence to support this assumption. In the absence of formal economic evaluation, descriptive information on the total length of hospital stay for mothers and babies receiving active or placebo interventions may have been helpful in understanding the impact of outpatient procedures on health service utilisation. Such information was generally not provided. Instead, studies tended to focus on proxy measures for progress in labour, but we would advise caution in the way such information is collected and interpreted.
It is possible that different induction agents perform quite differently at different stages of cervical dilatation or at different gestational ages. Most included studies recruited women requiring induction for 'postdates pregnancy'. In different studies 'postdates' was defined differently, and may have been any gestational age between approximately 39 to 44 weeks; in some studies women were recruited from 37 weeks. The cervical status at recruitment also varied considerably with Bishop scores at recruitment being any value less than nine. One of the included studies recruited women with diabetes; there is insufficient evidence to know whether outpatient induction is safe and acceptable for women in high‐risk groups.
With one or two exceptions, information on costs to women was generally not reported in the included trials. In the absence of such data the assumption must be that women were not asked for their views on care, or about costs or inconvenience associated with hospital or outpatient care. The potential importance of such outcomes (patient‐related outcome measures) is increasingly being recognised by commissioners of healthcare services.
Quality of the evidence
Most included studies were assessed as being at relatively low risk of bias; most of the trials were placebo controlled with adequate methods of randomisation and low levels of attrition. There was no blinding in eight trials where interventions were compared with no intervention or routine care. Lack of blinding may be a particular problem in these studies, as many of the outcomes reported may have depended on clinical judgements by staff (e.g. need for hospital admission, prescription of additional drugs to induce or augment labour, and other interventions in labour). In other words, clinical decisions may have been affected by knowledge of treatment allocation. Summaries of 'Risk of bias' assessments are presented in Figure 3 and Figure 2.
For the comparison of vaginal PGE₂ versus placebo or expectant management for the induction of labour in outpatient settings, we graded evidence for uterine hyperstimulation (fetal heart rate (FHR) changes unclear), caesarean section, and neonatal intensive care unit (NICU) admission as low quality (Table 1). There was no evidence for vaginal birth not achieved within 24 hours, vaginal birth not achieved in 48 to 72 hours, serious neonatal morbidity or death, or serious maternal morbidity or death.
For the comparison of intracervical PGE₂ versus placebo, we graded the evidence for vaginal birth not achieved in 48 to 72 hours, uterine hyperstimulation (with FHR changes), and NICU admission as low quality, and evidence for caesarean section as moderate quality (Table 2). Vaginal birth not achieved within 24 hours, serious neonatal morbidity or death, and serious maternal morbidity or death were not reported.
For the comparison of vaginal misoprostol versus placebo, we graded the evidence as low for uterine hyperstimulation (with FHR changes), caesarean section, serious neonatal morbidity or death, and NICU admission (Table 3). Vaginal birth not achieved in 24 hours, vaginal birth not achieved within 48 and 72 hours, and serious maternal morbidity or death were not reported.
Evidence across the three comparisons was downgraded for imprecision of effect estimates, few events, and small sample sizes.
Potential biases in the review process
We acknowledge that there was a possibility of introducing bias at every stage of the review process. We attempted to minimise bias in a number of ways; two review authors assessed eligibility for inclusion, carried out data extraction and assessed risk of bias. Each worked independently. Nevertheless, the process of assessing risk of bias, for example, is not an exact science and includes many personal judgements. Further, the process of reviewing research studies is known to be affected by prior beliefs and attitudes. It is difficult to control for this type of bias.
While we attempted to be as inclusive as possible in the search strategy, the literature identified was predominantly written in English and published in North American and European journals. We did not attempt to formally assess reporting bias, constraints of time meant that assessment of risk of bias largely relied on information available in the published trial reports and thus, reporting bias was not usually apparent. Too few studies were included in each comparison in the review to allow us to explore possible publication bias.
Agreements and disagreements with other studies or reviews
A number of related Cochrane Reviews have examined the same methods of induction of labour considered in this review, namely: vaginal PGE₂ (Thomas 2014), intracervical PGE₂ (Boulvain 2008), vaginal misoprostol (Hofmeyr 2010), oral misoprostol (Alfirevic 2014), mifepristone (Hapangama 2009), oestrogens (Thomas 2001), nitric oxide donors (Ghosh 2016) and acupuncture (Smith 2013). Compared with these other reviews (which included both hospital (inpatient) and home (outpatient) inductions), the current review contains relatively few studies, and therefore, has insufficient statistical power to demonstrate differences between groups. This is particularly the case for relatively rare outcomes such as uterine rupture, but is also true for more common complications such as uterine hyperstimulation.
Evidence from the related Cochrane Reviews is mainly in agreement with the findings of this review. Findings from these reviews indicate that compared with placebo, PGE₂ (vaginal and intracervical) and vaginal and oral misoprostol are effective induction agents in that vaginal birth within 24 hours was more likely for women receiving these agents. There is less evidence regarding the effectiveness of mifepristone, oestrogens, nitric oxide donors (including IMN) and acupuncture. Findings regarding safety suggest that some methods of induction (PGE₂ and vaginal misoprostol) may be associated with an increased risk of uterine hyperstimulation. However, despite the relatively large number of studies included in some of these reviews, even pooled results from studies do not provide strong evidence regarding serious maternal and neonatal morbidity and death; as we have discussed above, with such rare outcomes very large trials are needed to exclude excess risk, or risk must be imputed by examining surrogate outcomes. None of these reviews specifically considered the issue of outpatient induction and we must remain cautious about assuming that methods that appear safe in hospital will achieve the same levels of safety (and indeed effectiveness) in outpatient settings. As we have indicated in this review, related reviews also illustrate that very little attention has been paid to consumer views or the costs of care.
Most of the related Cochrane Reviews examined the effectiveness of induction agents compared with placebo. Relatively few studies have examined different methods of induction directly. Where different agents have been compared (e.g. IMN with vaginal PGE₂ (Osman 2006)) some agents may have advantages over others, and the safety profile of different agents (and doses) may differ. This may mean that they are more or less suitable for outpatient use.
Another Cochrane Review compared the same method of induction in home and hospital settings directly, but this review contained only four trials and was unable to shed much light on issues of either the relative effectiveness, safety or costs associated with outpatient induction (Kelly 2013).
Authors' conclusions
Implications for practice.
Induction of labour in outpatient settings appears feasible. We do not have sufficient evidence to determine which methods are most safe or effective in outpatient settings.
Implications for research.
There have been very few direct comparisons between different methods of labour induction in outpatient settings. Although it is likely that impact on cervix and uterine contractility will be similar in both inpatient and outpatient settings, it would be unwise to extrapolate the clinical outcomes from inpatient to outpatient settings. For this reason, it would be important to carry out further studies where various women‐friendly outpatient protocols are compared head‐to‐head. As part of such work it is important to ask women what sort of management they would prefer. There needs to be more careful consideration of outcomes purporting to measure progress in labour and more consistency in what is measured in trials. Little is known regarding women's preferences, and what combinations of treatment and setting would be most preferred.
It would be particularly helpful to carry out formal cost‐effectiveness analysis which includes the use of emergency services. Data on the utilisation of out of hours community health services and emergency ambulance services might enable those providing health services to decide the best types of induction agents to use, to set out criteria for selecting women for outpatient induction, and would enable women to make more informed choices about their care.
What's new
| Date | Event | Description |
|---|---|---|
| 30 November 2016 | New citation required but conclusions have not changed | Six new studies have been included (Agarwal 2012; Attanayake 2014; Gaffaney 2009; Ghanaie 2013; Rijnders 2011; Schmitz 2014). Conclusions have not changed. |
| 30 November 2016 | New search has been performed | Search updated and 10 new reports identified. Three GRADE 'Summary of findings' tables have been added. |
Acknowledgements
As part of the pre‐publication editorial process, this review has been commented on by three peers (an editor and two referees who are external to the editorial team), a member of the Pregnancy and Childbirth Group's international panel of consumers and the Group's Statistical Adviser.
We thank Dr Elham Shakibazadeh for her assistance with translation and data extraction for one paper. We thank Ms Nancy Medley for her assistance with GRADE tables. We thank Therese Dowswell for her contribution as an author on previous versions of this review.
The World Health Organization, Zarko Alfirevic, Alfred Osoti, Anthony Kelly, Stephania Livio and Jane Norman retain copyright and all other rights in their respective contributions to the manuscript of this Review as submitted for publication.
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Pregnancy and Childbirth Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Data and analyses
Comparison 1. Intravaginal PGE₂ gel versus placebo or expectant management.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Additional induction agents required | 2 | 150 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.52 [0.27, 0.99] |
| 2 Epidural | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.62, 1.12] |
| 3 Uterine hyperstimulation (FHR changes unclear) | 4 | 244 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.76 [0.64, 22.24] |
| 4 Caesarean section | 4 | 288 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.49, 1.31] |
| 5 Apgar score < 7 at 5 minutes | 2 | 180 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.45 [0.07, 2.93] |
| 6 NICU admission | 3 | 230 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.10, 1.03] |
| 7 Chorioamnionitis | 2 | 180 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.37 [0.15, 0.90] |
| 8 Indicator of 'progress' in labour ‐ Cervix unchanged at follow up (not pre‐specified) | 1 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.13 [0.03, 0.47] |
| 9 'Spontaneous labour' within 48 hours | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.43 [2.12, 19.48] |
| 10 Indicator of 'progress' in labour ‐ Admitted to hospital for labour | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.7 [1.47, 4.97] |
| 11 Time to birth ‐ Gestational age at birth (weeks) | 1 | 100 | Mean Difference (IV, Fixed, 95% CI) | ‐0.60 [‐0.99, ‐0.21] |
| 12 Time to birth ‐ Gestational age on admission (days) | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | ‐2.0 [‐4.17, 0.17] |
Comparison 2. Intracervical PGE₂ versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Additional induction agent required (induction with oxytocin or other means) | 3 | 445 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.74, 1.32] |
| 2 Additional induction agents required (further prostaglandin required) | 1 | 90 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.61 [0.22, 1.67] |
| 3 Uterine rupture | 1 | 294 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4 Birth not achieved in 48 to 72 hours | 1 | 43 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.68, 1.02] |
| 5 Oxytocin augmentation | 1 | 84 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.40, 1.12] |
| 6 Uterine hyperstimulation (with FHR changes) | 4 | 488 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.66 [0.63, 11.25] |
| 7 Assisted (instrumental) vaginal birth | 4 | 538 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.29 [0.85, 1.96] |
| 8 Caesarean section | 7 | 674 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.72, 1.12] |
| 9 Apgar score < 7 at 5 minutes | 4 | 515 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.42, 1.60] |
| 10 NICU admission | 3 | 215 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.61 [0.43, 6.05] |
| 11 Postpartum haemorrhage (> 500 mL) | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.10 [0.13, 73.16] |
| 12 Chorioamnionitis | 3 | 468 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.03 [0.66, 6.18] |
| 13 Endometritis | 2 | 174 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.60 [0.27, 9.37] |
| 14 Side effects ‐ Maternal side effects | 2 | 384 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.13, 2.77] |
| 15 Time to birth ‐ Interval from intervention to birth (days) | 2 | 133 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.20 [‐0.55, 0.14] |
| 16 Time to birth ‐ Gestational age at birth (weeks) | 2 | 156 | Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐0.35, 0.23] |
| 17 Indicator of 'progress' in labour ‐ Induction for gestational age > 42 weeks | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 18 Time to birth ‐ Birth within 48 hours of treatment (all births) | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.1 [1.29, 7.47] |
Comparison 3. Vaginal misoprostol versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Serious neonatal morbidity or death | 1 | 77 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.01, 8.14] |
| 2 Epidural | 1 | 50 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.77, 1.26] |
| 3 Uterine hyperstimulation (with FHR changes) | 3 | 265 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.97 [0.43, 9.00] |
| 4 Uterine hyperstimulation (without FHR changes) | 2 | 137 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.64 [0.15, 85.97] |
| 5 Assisted (instrumental) vaginal birth | 2 | 145 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.50, 1.67] |
| 6 Caesarean section | 4 | 325 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.61, 1.46] |
| 7 Apgar score < 7 at 5 minutes | 3 | 248 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.21 [0.01, 4.25] |
| 8 NICU admission | 4 | 325 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.54, 1.47] |
| 9 Perinatal death | 1 | 77 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.01, 8.14] |
| 10 Neonatal infection | 1 | 68 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.30 [0.07, 1.36] |
| 11 Indicator of 'progress' in labour ‐ Oxytocin dose used (mU) | 1 | 72 | Mean Difference (IV, Fixed, 95% CI) | 1508.70 [‐2357.55, 5374.95] |
| 12 Indicator of 'progress' in labour ‐ Number of medication dose | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.44 [‐0.49, ‐0.39] |
| 13 Indicator of 'progress' in labour ‐ Number of women requiring dosing on day 2 | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.61 [0.43, 0.87] |
| 14 Indicator of 'progress' in labour ‐ Number of women requiring induction on day 3 | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.13 [0.04, 0.38] |
| 15 Indicator of 'progress' in labour ‐ Days to admission (all) (days) | 1 | 77 | Mean Difference (IV, Fixed, 95% CI) | ‐2.90 [‐4.99, ‐0.81] |
| 16 Indicator of 'progress' in labour ‐ Days to admission (subgroups by parity) (days) | 1 | 77 | Mean Difference (IV, Fixed, 95% CI) | ‐3.15 [‐5.40, ‐0.89] |
| 16.1 Nulliparous women | 1 | 41 | Mean Difference (IV, Fixed, 95% CI) | ‐3.20 [‐6.44, 0.04] |
| 16.2 Parous women | 1 | 36 | Mean Difference (IV, Fixed, 95% CI) | ‐3.10 [‐6.24, 0.04] |
| 17 Indicator of 'progress' in labour ‐ Gestational age at labour (weeks) | 1 | 77 | Mean Difference (IV, Fixed, 95% CI) | ‐0.80 [‐1.05, ‐0.55] |
| 18 Indicator of 'progress' in labour ‐ Days to admission (parous) (weeks) | 1 | 36 | Mean Difference (IV, Fixed, 95% CI) | ‐3.10 [‐6.24, 0.04] |
| 19 Indicator of 'progress' in labour ‐ Days to PROM (days) | 1 | 77 | Mean Difference (IV, Fixed, 95% CI) | ‐2.5 [‐4.14, ‐0.86] |
| 20 Time to birth ‐ Interval from intervention to vaginal birth (days) | 1 | 50 | Mean Difference (IV, Fixed, 95% CI) | ‐1.4 [‐3.51, 0.71] |
| 21 Time to birth ‐ Days to birth (all) (days) | 1 | 68 | Mean Difference (IV, Fixed, 95% CI) | ‐1.90 [‐3.74, ‐0.06] |
| 22 Time to birth ‐ Days to birth (subgroups by parity) (days) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 22.1 Nulliparous women | 1 | 39 | Mean Difference (IV, Fixed, 95% CI) | ‐3.0 [‐5.42, ‐0.58] |
| 22.2 Parous women | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | ‐0.60 [‐3.51, 2.31] |
3.9. Analysis.
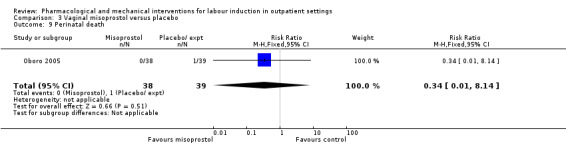
Comparison 3 Vaginal misoprostol versus placebo, Outcome 9 Perinatal death.
3.18. Analysis.
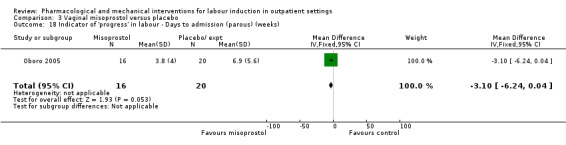
Comparison 3 Vaginal misoprostol versus placebo, Outcome 18 Indicator of 'progress' in labour ‐ Days to admission (parous) (weeks).
Comparison 4. Vaginal misoprostol 25 µg versus 50 µg.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Additional induction agents required (oxytocin) | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.26 [0.22, 23.33] |
| 2 Uterine hyperstimulation | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Caesarean section | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.33, 2.68] |
| 4 NICU admission | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.57 [0.05, 5.83] |
| 5 Interval from treatment to birth (in days, all births) | 1 | 49 | Mean Difference (IV, Fixed, 95% CI) | 1.5 [1.19, 1.81] |
Comparison 5. Intracervical PGE₂ versus vaginal misoprostol.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Uterine hyperstimulation (with or without FHR changes) | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.26 [0.03, 2.73] |
| 2 Caesarean section | 1 | 84 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.38, 2.08] |
| 3 Apgar score < 7 after 5 minutes | 1 | 84 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 7.96] |
| 4 Admission to NICU | 1 | 84 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.36, 4.33] |
| 5 Indicator of 'progress' in labour ‐ Interval from administration to admission (hours) | 1 | 75 | Mean Difference (IV, Fixed, 95% CI) | 2.5 [2.22, 2.78] |
| 6 Indicator of 'progress' in labour ‐ Labour or SROM during ripening | 1 | 83 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.31 [0.14, 0.69] |
| 7 Time to birth ‐ Birth within 24 hours | 1 | 83 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.75, 1.07] |
| 8 Time to birth ‐ Birth within 48 hours (cumulative) | 1 | 83 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.81, 1.06] |
Comparison 6. Oral misoprostol versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Vaginal delivery not achieved within 24 hours | 1 | 87 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.48, 0.86] |
| 2 Additional induction agents required | 2 | 127 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.60 [0.37, 0.97] |
| 3 Oxytocin augmentation | 1 | 87 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.61, 1.08] |
| 4 Uterine hyperstimulation (with FHR changes) | 1 | 87 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.53 [0.47, 5.06] |
| 5 Uterine hyperstimulation (FHR changes unclear) | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.61 [0.06, 6.21] |
| 6 Instrumental vaginal birth | 1 | 87 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.17, 1.57] |
| 7 Caesarean section | 1 | 86 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.28, 1.33] |
| 8 Apgar score < 7 at 5 minutes | 1 | 87 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9 Neonatal intensive care unit admission | 1 | 87 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.07, 15.84] |
| 10 Postpartum haemorrhage | 1 | 87 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.11 [0.25, 103.51] |
| 11 Chorioamnionitis | 2 | 124 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.52, 2.17] |
| 12 Endometritis | 1 | 87 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.05, 5.44] |
| 13 Indicator of 'progress' in labour ‐ Time from first dose to active labor (hours) | 2 | 127 | Mean Difference (IV, Fixed, 95% CI) | ‐37.08 [‐52.44, ‐21.72] |
| 14 Time to birth ‐ First dose to birth (hours) | 1 | 87 | Mean Difference (IV, Fixed, 95% CI) | ‐37.94 [‐57.97, ‐17.91] |
| 15 Indicator of 'progress' in labour ‐ Total doses of medication | 1 | 40 | Mean Difference (IV, Fixed, 95% CI) | ‐0.51 [‐0.92, ‐0.10] |
Comparison 7. Mifepristone versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Additional induction agents required | 4 | 311 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.37, 0.95] |
| 2 Serious neonatal morbidity or death | 1 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.07, 35.67] |
| 3 Oxytocin augmentation | 2 | 116 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.63, 1.26] |
| 4 Epidural | 1 | 112 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.73, 1.03] |
| 5 Assisted (instrumental) vaginal birth | 5 | 343 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.35 [0.93, 1.97] |
| 6 Caesarean section | 5 | 343 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.62, 1.25] |
| 7 Apgar score < 7 at 5 minutes | 2 | 119 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.07, 35.67] |
| 8 NICU admission | 2 | 163 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.31, 2.79] |
| 9 Perinatal death | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 10 Uterine scar separation | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.07, 14.64] |
| 11 Chorioamnionitis | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.20, 19.91] |
| 12 Indicator of 'progress' in labour ‐ Labour or ripe cervix in 48 hours | 1 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [1.00, 4.00] |
| 13 Indicator of 'progress' in labour ‐ Cervix unchanged after 24/48 hours | 2 | 119 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.20, 0.63] |
| 14 Indicator of 'progress' in labour ‐ Spontaneous labour within 72 hours | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.46 [0.68, 3.10] |
| 15 Indicator of 'progress' in labour ‐ Spontaneous labour within 48 hours | 1 | 83 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.05 [1.27, 3.30] |
| 16 Indicator of 'progress' in labour ‐ Oxytocin requirements (IU) | 1 | 32 | Mean Difference (IV, Fixed, 95% CI) | ‐2.56 [‐4.01, ‐1.11] |
| 17 Indicator of 'progress' in labour ‐ Interval between day 1 and start of labour (hours) | 1 | 32 | Mean Difference (IV, Fixed, 95% CI) | ‐22.15 [‐35.96, ‐8.34] |
Comparison 8. Oestrogens versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Oxytocin augmentation | 1 | 87 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.61, 1.43] |
| 2 Assisted (instrumental) vaginal birth | 1 | 87 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.44, 1.60] |
| 3 Caesarean section | 1 | 87 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.27 [0.63, 2.58] |
| 4 NICU admission | 1 | 87 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.06, 15.13] |
| 5 Chorioamnionitis | 1 | 87 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.95 [0.38, 10.12] |
| 6 Endometritis | 1 | 87 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.93 [0.32, 27.10] |
Comparison 9. Vaginal isosorbide mononitrate (IMN) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Vaginal birth not achieved in 24/48 hours | 1 | 238 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.83, 1.15] |
| 2 Additional induction agents required | 4 | 1921 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.75, 1.00] |
| 3 Maternal satisfaction | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 How do you think your labour went? (easy/very difficult) | 1 | 227 | Mean Difference (IV, Fixed, 95% CI) | ‐0.34 [‐0.94, 0.26] |
| 3.2 What do you think about home treatment? (extremely good/not at all good) | 1 | 227 | Mean Difference (IV, Fixed, 95% CI) | 0.61 [0.03, 1.19] |
| 3.3 How painful was the treatment at home? (not at all/very) | 1 | 227 | Mean Difference (IV, Fixed, 95% CI) | 0.58 [‐0.00, 1.16] |
| 3.4 How anxious were you being at home taking the treatment? (not at all/very) | 1 | 227 | Mean Difference (IV, Fixed, 95% CI) | 0.11 [‐0.39, 0.61] |
| 3.5 Would you have the same treatment at home again? (definitely/definitely not) | 1 | 227 | Mean Difference (IV, Fixed, 95% CI) | 0.62 [‐0.02, 1.26] |
| 3.6 Would you advise a friend to have the same treatment at home? (definitely/definitely not) | 1 | 227 | Mean Difference (IV, Fixed, 95% CI) | 0.41 [‐0.17, 0.99] |
| 4 Maternal satisfaction | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 Felt satisfied (very or extremely) | 1 | 1049 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.67, 0.94] |
| 4.2 Would recommend the same treatment | 1 | 1049 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.77, 0.90] |
| 4.3 Would recommend procedure | 1 | 193 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.94, 1.08] |
| 5 Perinatal death | 2 | 1712 | Risk Ratio (M‐H, Random, 95% CI) | 1.61 [0.08, 33.26] |
| 6 Neonatal trauma (long bone fracture, collarbone fracture, basal skull fracture, brachial plexus palsy, facial nerve palsy, phrenic nerve palsy, or subdural haemorrhage) | 1 | 1362 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.19, 2.37] |
| 7 Neonatal convulsions in the first 24 hours | 1 | 1362 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8 Tracheal ventilation > 24 hours | 1 | 1362 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.14, 7.14] |
| 9 Neonatal ICU admission for 5 or more days | 1 | 1362 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.19, 2.37] |
| 10 Neonatal transfer | 1 | 1362 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.67, 1.70] |
| 11 Maternal death | 1 | 1362 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 12 Severe postpartum haemorrhage | 1 | 1362 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.55 [0.78, 3.09] |
| 13 Deep vein thrombosis | 1 | 1362 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.03 [0.12, 74.16] |
| 14 Oxytocin augmentation | 3 | 1816 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.78, 1.14] |
| 15 Epidural | 1 | 350 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.82, 1.09] |
| 16 Uterine hyperstimulation (with FHR changes) | 1 | 102 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.2 [0.01, 4.07] |
| 17 Uterine hyperstimulation (FHR changes unclear) | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.09 [0.01, 1.62] |
| 18 Assisted (instrumental) vaginal birth | 2 | 1712 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.61, 1.07] |
| 19 Caesarean section | 6 | 2286 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.87, 1.14] |
| 20 Apgar score < 7 at 5 minutes | 5 | 2214 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.44, 1.76] |
| 21 NICU (or SCBU) admission | 6 | 1068 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.59, 1.36] |
| 22 Postpartum haemorrhage (> 500 mL) | 5 | 2214 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.95, 1.36] |
| 23 Neonatal infection | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.26, 3.89] |
| 24 Side effects ‐ Maternal side effect ‐ nausea | 4 | 1926 | Risk Ratio (M‐H, Random, 95% CI) | 2.39 [1.54, 3.70] |
| 25 Side effects ‐ Maternal side effect ‐ headache | 7 | 2300 | Risk Ratio (M‐H, Random, 95% CI) | 5.45 [3.38, 8.81] |
| 26 Side effects ‐ Maternal side effects ‐ severe headache | 1 | 220 | Risk Ratio (M‐H, Fixed, 95% CI) | 21.21 [2.91, 154.65] |
| 27 Indicator of 'progress' in labour ‐ Admitted in established labour within 24 hours | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.75 [1.29, 5.88] |
| 28 Indicator of 'progress' in labour ‐ Bishop score > 6 or active labour at 36 hours | 1 | 102 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.8 [1.54, 9.40] |
| 29 Time to birth ‐ Time in hours from admission to birth (all women) | 3 | 374 | Mean Difference (IV, Random, 95% CI) | ‐4.70 [‐6.08, ‐3.31] |
| 30 Indicator of 'progress' in labour ‐ Bishop score on admission after treatment | 1 | 200 | Mean Difference (IV, Fixed, 95% CI) | 2.73 [2.17, 3.29] |
| 31 Indicator of 'progress' in labour ‐ Change in Bishop score | 2 | 272 | Mean Difference (IV, Fixed, 95% CI) | 2.76 [2.48, 3.03] |
| 32 Time to birth ‐ Interval from onset of labour to birth (hours) | 1 | 200 | Mean Difference (IV, Fixed, 95% CI) | ‐1.24 [‐1.82, ‐0.66] |
| 33 Indicator of 'progress' in labour ‐ Cervix unchanged after 48 hours | 1 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.70, 0.97] |
| 34 Time to birth ‐ Interval from admission to vaginal birth (hours) | 1 | 128 | Mean Difference (IV, Fixed, 95% CI) | ‐0.70 [‐6.11, 4.71] |
| 35 Total cost of care package (GBP) | 1 | 350 | Mean Difference (IV, Fixed, 95% CI) | 11.98 [‐105.34, 129.30] |
Comparison 10. Acupuncture versus routine care.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Additional induction agents required | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.6 [0.31, 1.17] |
| 2 Caesarean section | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.17, 1.11] |
Comparison 11. Outpatient amniotomy for induction versus routine care.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Maternal satisfaction ‐ I look back positively on the treatment I received | 1 | 404 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.97, 1.10] |
| 2 Maternal satisfaction ‐ In retrospect, I would have preferred another treatment than received | 1 | 472 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.36, 0.72] |
| 3 Augmentation, induction or both | 1 | 521 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.71, 0.97] |
| 4 Epidural, opioids or both for pain relief | 1 | 521 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.76, 1.30] |
| 5 Instrumental vaginal birth | 1 | 521 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.70 [0.46, 1.08] |
| 6 Caesarean section | 1 | 521 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.20 [0.78, 1.86] |
| 7 Apgar < 7 at 5 minutes | 1 | 521 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.86 [0.34, 10.06] |
| 8 Neonatal intensive care unit admission | 1 | 521 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.64, 1.85] |
| 9 Duration of birth (hours) | 1 | 521 | Mean Difference (IV, Fixed, 95% CI) | 0.40 [‐0.72, 1.52] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Agarwal 2012.
| Methods | Parallel randomised, single‐blind, placebo‐controlled trial. | |
| Participants | Setting: Safdarjung Hospital, New Delhi, India. 200 women randomised. Inclusion criteria: singleton pregnancy, > 40 completed weeks, unfavourable cervix (Bishop score < 6), absence of uterine contractions, intact membranes. Exclusion criteria: fetal malpresentation, pre‐partum haemorrhage, previous uterine incision, ruptured membranes, high‐risk factors such as pre‐eclampsia, oligohydramnios, intrauterine growth restriction, diabetes mellitus, heart disease, and hypertension, or any contraindication to receive IMN or prostaglandins such as a known allergy to the drugs, bronchial asthma, hypotension, and palpitations. |
|
| Interventions | Intervention group: 2 x 40 mg tablets of IMN self‐administered at home, vaginally, 1 of the tablets at 9 AM and the other at 9 PM the same day and to report to the hospital the next day at 9 AM for admission. Control group: 2 x 40 mg tablets of pyridoxine as placebo IMN self‐administered at home, vaginally, 1 of the tablets at 9 AM and the other at 9 PM the same day and to report to the hospital the next day at 9 AM for admission. Both arms received labour induction protocol on return to hospital. |
|
| Outcomes | Primary outcomes
Secondary outcomes
|
|
| Notes | Added for 2017 update. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Paper states the randomisation codes were generated using a random allocation sequence, and that the random number table was generated by the statistician using a computerised random number table. |
| Allocation concealment (selection bias) | Unclear risk | The participants were enrolled by the first author and assignment to the study or control group was done in accordance with the list of codes, which was generated by the second author. However, did not state whether random sequence was concealed or not. |
| Blinding (performance bias and detection bias) Women | Low risk | It was a single‐blind trial as the participants did not know whether they were given IMN. However, it seems that personnel were unblinded. |
| Blinding (performance bias and detection bias) Clinical staff | High risk | It was a single‐blind trial as the participants did not know whether they were given IMN. However, it seems that personnel were unblinded. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Not specifically stated, however based on above it is possible outcome assessors were unblinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Outcome data complete for all participants. |
| Selective reporting (reporting bias) | Low risk | No evidence of selective outcome reporting. |
| Other bias | Low risk | None identified. |
Attanayake 2014.
| Methods | Double blind RCT. | |
| Participants | Setting: Academic Obstetric Unit of the Teaching Hospital Mahamodara, Galle, Sri Lanka. Inclusion criteria: uncomplicated pregnancy at 39 weeks' gestational age (GA) with a singleton fetus having a cephalic presentation and a modified Bishop score < 5 out of 10, and consenting to self‐administer the vaginal tablets every other day for 5 days. Exclusion criteria: any pregnancy complications, e.g. hypertension or hyperglycaemia in pregnancy, multiple pregnancies, planned caesarean birth, fetal growth restriction and history of hypersensitivity or idiosyncratic reaction to nitrates. |
|
| Interventions | Intervention: self‐administer vaginally at home every other day, 5 doses of 60 mg of the sustained release form of isosorbide mononitrate (ISMN) from 273 days to 282 days. Control: pyroxidine 10 mg, using same regimen. In both arms, participants were instructed to self‐administer the tablets vaginally at home at GAs of 39 weeks, 39 weeks + 2 days, 39 weeks + 4 days, 39 weeks + 6 days and 40 weeks + 1 day, unless spontaneous onset of labour (SOL) was established and she needed admission to hospital. If SOL was not established by 40 weeks + 2 days, all participants were admitted to hospital, the MBS was re‐assessed and artificial separation of membranes was carried out if feasible, and if not feasible, a cervical massage were carried out. Thereafter the routine management guideline for cervical ripening and IOL of the unit were followed using artificial separation of membranes, prostaglandin (PGE₂ 3 mg tablets) vaginally or intra cervical Foley catheter, followed by amniotomy and intravenous oxytocin infusion, if SOL was not established by 41 weeks. On admission to hospital either with SOL or at 40 weeks + 2 days, compliance to the interventions was assessed by checking the cards which the participants had been requested to maintain, indicating when they self administered the study medication. |
|
| Outcomes | Outcomes stratified:
Satisfaction and acceptability For pregnancies reaching 41 weeks:
|
|
| Notes | This study was reported in a brief abstract. We attempted to contact authors for further information (1 September 2016), who provided an unpublished version of the manuscript (accepted for publication). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Article states "Using computer generated random numbers participants were allocated into the study and control groups by stratified (Primips / Multips) block randomization". |
| Allocation concealment (selection bias) | Low risk | Article states "Two sets of sequentially numbered opaque envelopes (one for Primips and one for Multips) were packed with five tablets of either ISMN–SR 60mgs (Angifree – SR, Microlabs, Bangalore, India) or five tablets of Pyridoxine 10mgs (HealthAid Vitamins, Harrow, Middlesex, United Kingdom ) according to the random allocation sequence in blocks of four, by the second author". |
| Blinding (performance bias and detection bias) Women | Unclear risk | Study title describes as double blind, no further detail provided. |
| Blinding (performance bias and detection bias) Clinical staff | Unclear risk | Study title describes as double blind, no further detail provided. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Study title describes as double blind, no further detail provided. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Outcome data reported for all participants, except for 1 drop out from intervention arm (ISMN group) who discontinued due to anxiety. |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting. |
| Other bias | Low risk | No other forms of bias identified. |
Bollapragada 2006a.
| Methods | RCT. | |
| Participants | Setting: large teaching hospital in Glasgow, Scotland, UK. 350 women randomised. Inclusion criteria: primiparous women at term (gestational age > 37 weeks) with singleton pregnancy and Bishop score < 7. Women were scheduled for induction (97% for prolonged pregnancy: 40 weeks + 10 days gestation). Women recruited were willing to self‐administer vaginal tablets. Exclusion criteria: women with ruptured membranes, aged < 16 years age, who needed birth within the next 48 h, or with fetal compromise requiring daily fetal monitoring. |
|
| Interventions | Intervention group: self‐administered vaginal IMN 40 mg every 16 h to maximum of 3 doses (48 h, 32 h and 16 h prescheduled admission for induction). Comparison group: self‐administered placebo, same regimen as intervention group. |
|
| Outcomes | Time from hospital admission to birth, women's views on induction process, pain, mode of birth, cost to NHS, neonatal outcomes. | |
| Notes | A review author, Jane Norman (JN), was an investigator on this trial. JN was not involved in assessing the eligibility of the study for inclusion, data extraction or assessment of risk of bias. See Eddama 2009 for associated paper on cost outcomes. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated sequence. |
| Allocation concealment (selection bias) | Low risk | Central randomisation with automated telephone service. Women were given information and consented after the decision to induce labour had been made. Randomisation in the antenatal clinic up to 9 days before treatment commenced. |
| Blinding (performance bias and detection bias) Women | Low risk | Treatment packs for intervention and control groups were described as identical, prepared by pharmacy. |
| Blinding (performance bias and detection bias) Clinical staff | Low risk | Treatment packs for intervention and control groups were described as identical, prepared by pharmacy. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Blinding of outcomes assessors not explicitly stated. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 350 randomised. 80 women did not initiate treatment as they went in to labour before the scheduled time for taking medication, a further 11 women withdrew (including 2 with breech presentation). All women randomised were included in an ITT analysis for primary outcomes (but not in secondary analysis). |
| Selective reporting (reporting bias) | Low risk | We examined the protocol for this study and there is no evidence of reporting bias. |
| Other bias | Low risk | No baseline imbalance apparent. |
Bullarbo 2007.
| Methods | RCT. | |
| Participants | Setting: 2 hospitals in Gothenburg, Sweden. 200 women randomised. Inclusion criteria: women with uncomplicated pregnancies, singleton, cephalic presentation, intact membranes, > 42 weeks' gestation (confirmed by ultrasound before 20 weeks) normal AFI, reactive NST. Exclusion criteria: serious medical or obstetric complication (daily use of medication), history of headache, regular contractions, alcohol abuse, intolerance of IMN. |
|
| Interventions | Intervention group: 40 mg IMN intravaginal. Comparison: placebo. Review arranged for the next day, if labour had not started then IOL was carried out according to local protocol. |
|
| Outcomes | Additional induction agents required, maternal satisfaction, CS PPH. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number tables. |
| Allocation concealment (selection bias) | Low risk | Sealed sequentially numbered envelopes. |
| Blinding (performance bias and detection bias) Women | Low risk | Described as double blind. Women unaware of assignment. |
| Blinding (performance bias and detection bias) Clinical staff | Low risk | Staff unaware of treatment assignment; placebo and treatment identical. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Blnding of outcome assessors not described. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Report that all women completed the study. |
| Selective reporting (reporting bias) | Low risk | Stated outcomes are reported. |
| Other bias | Low risk | Baseline characteristics comparable. |
Buttino 1990.
| Methods | RCT, 2 arm trial. | |
| Participants | Setting: 43 women attending antenatal clinics in California, USA. Inclusion criteria: women with "post‐dates" pregnancies (gestational age > 41 weeks and 6 days based on reliable menstrual history and early ultrasound confirmation) with reactive NST. Exclusion criteria: contraindications to prostaglandins. |
|
| Interventions | Intervention group: intracervical PGE₂ 0.5 mg. Comparison group: visually identical placebo gel. Women in both groups were observed for 1 h with external fetal monitoring and then discharged home. |
|
| Outcomes | Bishop score on admission, mode of birth, interval to birth, length of labour, infant birthweight and Apgar score. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | External sequence generation by hospital pharmacy. |
| Allocation concealment (selection bias) | Low risk | Coded syringes of identical appearance were dispensed from pharmacy. |
| Blinding (performance bias and detection bias) Women | Low risk | Placebo controlled trial. Women and physicians unaware of group assignment. Identical treatment and placebo. |
| Blinding (performance bias and detection bias) Clinical staff | Low risk | Placebo controlled trial. Women and physicians unaware of group assignment. Identical treatment and placebo. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Not clear when code was revealed, but investigators were not involved in the inpatient care of women. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All women randomised appeared to be included in the analyses. |
| Selective reporting (reporting bias) | Low risk | Stated outcomes are reported. |
| Other bias | Low risk | No other bias apparent. |
Elliott 1998.
| Methods | RCT. 4 arm trial. | |
| Participants | Setting: Edinburgh, UK. 80 women recruited with IOL scheduled 72 h after recruitment. Inclusion criteria: primiparous women aged 18 to 40 years, normal viable fetus, 37 to 41 weeks (confirmed by first trimester ultrasound scan), cephalic presentation, Bishop score < 5. Exclusion criteria: women who showed signs of labour onset, placental insufficiency or contraindication to mifepristone, |
|
| Interventions | Intervention: group 1: (25 women) oral mifepristone 50 mg. Group 2: (25 women) oral mifepristone 200 mg. (In this review we combined both groups in the analysis although it was not clear how randomisation was achieved in the higher dose study.) Comparison groups: placebo (2 groups of women 25 compared with the lower dose and 5 with the higher dose. We have combined placebo groups in the analysis in this review as data were reported together in the results in the study reports; group size was very unbalanced for the second part of the study). |
|
| Outcomes | Additional induction agents required, labour within 72 h, CS, oxytocin augmentation. NICU admission. | |
| Notes | It was not clear why the placebo group for the higher dose comparison was so small (5 women) or how randomisation was performed to achieve the unbalanced intervention and control groups. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Pre‐determined randomisation code." |
| Allocation concealment (selection bias) | Unclear risk | "Treatment in predetermined numeric order." It was not clear why the group allocation in the placebo arms of the trial were very unbalanced. |
| Blinding (performance bias and detection bias) Women | Low risk | "Neither the patient nor the physician had knowledge of whether a simple oral dose of mifepristone or placebo was given." |
| Blinding (performance bias and detection bias) Clinical staff | Low risk | "Neither the patient nor the physician had knowledge of whether a simple oral dose of mifepristone or placebo was given." |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Blinding of outcome assessors not described. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | All women randomised seemed to be accounted for in the analysis, although there was serious imbalance in group size. |
| Selective reporting (reporting bias) | Low risk | Stated outcomes are reported. |
| Other bias | Unclear risk | In the second part of the study (higher dose) the treatment to placebo ratio was 1:5. It was not clear how randomisation was performed, or why the control group was so small. |
Frydman 1992.
| Methods | RCT 2 arm parallel group design. | |
| Participants | 120 women attending an antenatal clinic in a hospital in France, 1990 to 1991. Inclusion criteria: term pregnancy scheduled for induction (range of indications), Bishop score < 4. Exclusion criteria: malpresentation, ruptured membranes, multiple pregnancy, > 1 previous CS or known medical condition. |
|
| Interventions | Intervention group: active tablets mifepristone 200 mg. All women received a box with 2 tablets, the first to be taken on the morning of day 1 and the second on the morning of day 2. Comparison group: placebo tablets. Same regimen as intervention group. IOL scheduled for 4 days after intervention, women reported to the hospital each day over the 4 day study period and were asked to report drug reactions, pain, bleeding or contractions. |
|
| Outcomes | Labour within 4‐day study period, other induction agents required, duration of labour, mode of birth, Apgar score < 7 at 5 min. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Tablets were supplied by pharmacy according to a "balanced randomisation list". Block size 4. |
| Allocation concealment (selection bias) | Low risk | Small block size might mean that allocation order could potentially be anticipated in advance but the drug packs were described as being of similar appearance. |
| Blinding (performance bias and detection bias) Women | Low risk | Described as a double‐blind study. |
| Blinding (performance bias and detection bias) Clinical staff | Low risk | Placebo described as being of similar appearance. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Blinding of outcome assessors not described. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 120 women were randomised but 8 were excluded from the results because of a deterioration in their condition within 12 h of the first pill (3 in the mifepristone group and 5 in the placebo group). |
| Selective reporting (reporting bias) | Unclear risk | Efficacy and safety outcomes not specified in methods text but many labour and infant outcomes reported. |
| Other bias | Unclear risk | Additional induction agents were used for some women so labour and other outcomes may be affected by co‐interventions. |
Gaffaney 2009.
| Methods | Double‐blind, placebo‐controlled RCT (pilot study). | |
| Participants | Setting: Women’s Pavilion at Miller Children’s Hospital, Long Beach Memorial Medical Center, Long Beach, California. Inclusion criteria: at hospital for prolonged pregnancy surveillance, women at gestational age of 40 to 42 weeks, singleton gestation, Bishop score < 6/ unfavourable cervix, vertex presentation, intact membranes, reactive NST, AFI of more than or equal to 5, willing to forgo induction for 72 h. Exclusion criteria: none specified. |
|
| Interventions | Intervention: cervical ripening regimen (N = 43) Subjects were treated daily for up to 3 days with oral capsule containing 100 mg of misoprostol. Electronic fetal monitoring for 2 h after administration. Women were asked to return in 24 h to be evaluated for a repeat dosage. During the 3 days of study observation, labour induction was not allowed. If adequate cervical ripening was achieved on days 1 or 2, the next doses of study drug were withheld. If the Bishop score was 6 or greater or if the patient went into active labour, she was removed from the study protocol and managed according to standard hospital protocol. After 3 days, all women removed and management was according to routine care. In hospital, maternal FHR monitoring for 2 h. Control: placebo (unspecified content) daily for 3 days, according to same regimen for women in the intervention arm (N = 44). |
|
| Outcomes | Primary outcome: time from study drug administration to birth Secondary outcome
Adverse events:
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Article states "Randomization of subjects was completed using a computerized random number generator (True Epistat). Randomization was coordinated by the Labor and Delivery pharmacist, who was apprised of each candidate’s eligibility and assigned the treatments in sequence based on the computer‐generated randomization scheme". |
| Allocation concealment (selection bias) | Low risk | Article states "All study drugs were prepared by the research pharmacy staff and packaged to maintain the blinded assignment". |
| Blinding (performance bias and detection bias) Women | Low risk | Article states "All study drugs were prepared by the research pharmacy staff and packaged to maintain the blinded assignment". |
| Blinding (performance bias and detection bias) Clinical staff | Low risk | Article states "All study drugs were prepared by the research pharmacy staff and packaged to maintain the blinded assignment". |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Not specifically stated. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 9 women were excluded from analysis post‐randomisation:
Data on these 9 women not reported. |
| Selective reporting (reporting bias) | Low risk | Pre‐specified outcome of being undelivered by 72 h not reported, but results imply all women still undelivered at that time. All other outcomes reported. |
| Other bias | Low risk | None identified. |
Ghanaie 2013.
| Methods | A double‐blind, placebo‐controlled RCT. | |
| Participants | Setting: Alzahra educational hospital in Rasht city, Iran. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Intervention (N = 36): 20 mg isosorbide‐5‐mononitrate tablets vaginally twice each 12 h prior to admission for IOL. Women asked to come back urgently to the hospital in case they had leakage, contractions or bleeding. If they had no symptoms they should come back after 12 h. In the next visit, women were asked about the side effects of the tablets including headache and palpitations. If the contraction had not started, another 20 mg of IMN was administered and the patients were asked to come back after 12 h. Immediately after hospitalisation, the Bishop score was assessed and induction with oxytocin was commenced. Control (N = 36): 2 placebo tablets of similar design inserted vaginally twice each 12 h, prior to admission for IOL (according to regimen described above). |
|
| Outcomes | Change in Bishop score Mean time to active phase of labour Admission to birth interval Type of birth CS indications (meconium, failure to progress, fetal distress) Min Apgar Headache Palpitation Need for NICU Fetal complications Need for blood transfusion |
|
| Notes | Article abstract in English, full article in Iranian. An Iranian‐speaking colleague (E Shakibazadeh) kindly completed data extraction for risk of bias assessment and additional data. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomly allocated to 2 intervention and control groups using random blocks. |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding (performance bias and detection bias) Women | Unclear risk | The authors have suggested that their study is a double blind study. However, there is no further information provided. |
| Blinding (performance bias and detection bias) Clinical staff | Unclear risk | The authors have suggested that their study is a double blind study. However, there is no further information provided. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | The authors have suggested that their study is a double blind study. However, there is no further information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Full translation required. |
| Selective reporting (reporting bias) | Unclear risk | all specified outcomes were reported. |
| Other bias | Unclear risk | Full translation required. |
Giacalone 1998.
| Methods | RCT, 2 arm trial. | |
| Participants | Setting: study carried out in 2 hospitals in France, 1991 to 1992. 84 women randomised. Inclusion criteria: women with gestational age 41 weeks and 3 days or more and scheduled for induction for "post‐dates" pregnancy, Bishop score < 6, induction could be postponed for 48 h. Exclusion criteria: women with multiple pregnancies, ruptured membranes, contraindication to vaginal birth, no uterine scarring, parity < 4, no FHR abnormalities, serious medical disease or obstetric complication. |
|
| Interventions | Intervention group: mifepristone 400 mg, single oral dose. Comparison group: placebo tablets of identical appearance. Women in both groups returned after 1 day for review. If Bishop score > 6 then women had labour induction or returned the next day for labour induction. |
|
| Outcomes | Change in Bishop score after 48 h, treatment to birth interval, mode of birth, oxytocin augmentation, neonatal condition at birth. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Balanced randomisation list in permuted blocks (block size not stated). |
| Allocation concealment (selection bias) | Low risk | Coded drug bottles. The "code for each subject was to be kept sealed in an opaque envelope to be opened in case of an emergency". |
| Blinding (performance bias and detection bias) Women | Low risk | Described as double blind study. Placebo described as being of identical appearance. |
| Blinding (performance bias and detection bias) Clinical staff | Low risk | Obstetricians were blinded to group assignment. |
| Blinding (performance bias and detection bias) Outcome assessor | Low risk | Outcome assessors were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 84 women were recruited, 1 woman (from the mifepristone group) was lost to follow up. |
| Selective reporting (reporting bias) | Low risk | Specified outcomes are reported. |
| Other bias | Low risk | Baselind characteristics of groups are comparable. |
Gittens 1996.
| Methods | RCT (little information on study methods). | |
| Participants | 32 women. Setting: New Jersey, USA Inclusion criteria: women with previous CS, gestational age 39 weeks with Bishop score < 6. |
|
| Interventions | Intervention group: intracervical PGE₂ repeated weekly. Comparison group: expectant management. |
|
| Outcomes | CS. | |
| Notes | Brief abstract, little information provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information. |
| Allocation concealment (selection bias) | Unclear risk | "prospectively randomised." |
| Blinding (performance bias and detection bias) Women | High risk | Not feasible. |
| Blinding (performance bias and detection bias) Clinical staff | High risk | No information. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | No information. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Trial reported as abstract only; numbers unclear. |
| Selective reporting (reporting bias) | Unclear risk | Not enough information to assess. |
| Other bias | Unclear risk | Not enough information to assess. |
Habib 2008.
| Methods | RCT. | |
| Participants | Setting: 102 women in a Cairo hospital, Egypt. Inclusion criteria: women at term (> 37 weeks' gestation) scheduled for induction, singleton viable fetus, intact membranes, no uterine contractions. Exclusion criteria: malpresentation, placenta previa, previous uterine surgery, contraindications to induction. |
|
| Interventions | Intervention group: self‐administered IMN, 40 mg, 3 doses 12 h apart (scheduled for 36 h, 24 h and 12 h before induction. Comparison group: placebo same regiment as intervention group. |
|
| Outcomes | CS, further induction agents required, PPH, Apgar score > 7 at 5 minutes, NICU admission, side effects. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated sequence. |
| Allocation concealment (selection bias) | Low risk | Coded treatment packs prepared by pharmacy. |
| Blinding (performance bias and detection bias) Women | Low risk | Placebo controlled trial. |
| Blinding (performance bias and detection bias) Clinical staff | Low risk | Tablets for intervention and placebo not described as though physicians would not know the difference between them. Staff and women would be unaware of assignment. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Blinding of outcome assessors not described specifically, though obstetric staff were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All women randomised appear to be accounted for in the analysis. |
| Selective reporting (reporting bias) | Low risk | Stated outcomes are reported. |
| Other bias | Low risk | Demographic characteristics similar. No other bias noted. |
Hage 1993.
| Methods | RCT, placebo controlled trial. | |
| Participants | Setting: not clear but probably USA. 36 women. Inclusion criteria: healthy, nulliparous women, 41 weeks' gestation and Bishop score < 9. |
|
| Interventions | Intervention group: 2.5 mg intravaginal PGE₂, with second dose if labour not established 24 h later. Comparison group: placebo gel, with second dose after 24 h if labour was not established. |
|
| Outcomes | Change in cervix after 48 h. | |
| Notes | Information from brief abstract. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as "randomized". |
| Allocation concealment (selection bias) | Unclear risk | No information. |
| Blinding (performance bias and detection bias) Women | Low risk | Described as double‐blind trial with placebo gel. |
| Blinding (performance bias and detection bias) Clinical staff | Unclear risk | Not described specifically but treatment and placebo both described as gel. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | No information. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Little information on methods. It appeared that all women were available at follow up. |
| Selective reporting (reporting bias) | Unclear risk | Abstract only with limited outcome data available. |
| Other bias | Unclear risk | Trial reported as abstract only so not able to assess for other bias. |
Harper 2006.
| Methods | RCT with block randomisation. | |
| Participants | Setting: outpatient clinic in North Carolina, USA. 56 women randomised. Inclusion criteria: primiparous women at term (39 weeks and 4 days to 41 weeks) with singleton, cephalic, pregnancy and Bishop score < 7. Exclusion criteria: cannot tolerate acupuncture, uncertain dates, breech presentation, placenta praevia, contra‐indication to vaginal birth. |
|
| Interventions | Cervical examination and ultrasound at recruitment. Intervention group: acupuncture + routine care on 3 of 4 consecutive days, visits also included fetal monitoring, treatment by trained acupuncturist to hands, legs and lower back and low voltage stimulation. Comparison group: routine care with follow up after 3 or 4 days. |
|
| Outcomes | Vaginal birth not achieved in 24 h, additional induction agents required. CS, mean time to birth. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated sequence in balanced blocks of 2 or 4. |
| Allocation concealment (selection bias) | Low risk | Sealed, opaque, sequentially numbered envelopes. |
| Blinding (performance bias and detection bias) Women | High risk | Blinding not feasible. |
| Blinding (performance bias and detection bias) Clinical staff | High risk | Women and staff would have been aware of treatment. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Not described. Birth outcomes would have been assessed separately from the intervention and control (outpatient acupuncture or no treatment), but report states that staff were not blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Data were available for all women randomised but denominators were not clear for some outcomes. |
| Selective reporting (reporting bias) | Low risk | Stated primary and secondary outcomes are reported with neonatal outcomes. |
| Other bias | Unclear risk | Women receiving acupuncture attended for 3 additional visits where other interventions occurred as well as acupuncture that may have affected outcomes. |
Incerpi 2001.
| Methods | RCT, 2 arm trial. | |
| Participants | Setting: Los Angeles hospital, USA, 1996 to 2000. 120 women with diabetes. Inclusion criteria: women with insulin dependent or other diabetes, gestational age 38 weeks (confirmed by ultrasound), not in labour, normal AFI (> 5 cm), normal FHR. Women compliant with hospital appointments and home glucose monitoring. Exclusion criteria: women with multiple pregnancies, ruptured membranes, vaginal bleeding, prior uterine surgery, active genital herpes, glaucoma, serious medical disease, parity > 5, fetal weight > 4500 g or < 2000 g. |
|
| Interventions | Study over 7 days. Intervention group: single dose of vaginal misoprostol 25 µg. Comparison group: placebo of similar appearance. Both groups were observed for 4 h and if there were no signs of fetal distress of painful contractions women were sent home. Reviewed after 3 to 4 days. If labour had not started then intervention/placebo was repeated. At 7 days women not in labour were induced. |
|
| Outcomes | Additional induction agents required (oxytocin), mode of birth, uterine hyperstimulation, neonatal condition at birth. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated sequence. |
| Allocation concealment (selection bias) | Low risk | Coded drug boxes. Pharmacy prepared and distributed medication according to the randomisation schedule. |
| Blinding (performance bias and detection bias) Women | Low risk | Placebo controlled trial. |
| Blinding (performance bias and detection bias) Clinical staff | Low risk | Placebo and intervention tablets were similar in appearance. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Not clear when code revealed. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 120 women randomised and no loss to follow up was apparent but denominators in the data tables were not always clear. |
| Selective reporting (reporting bias) | Unclear risk | Apart from outcome used for sample size, outcomes not specified in the methods text. |
| Other bias | Low risk | Baseline characteristics comparable. |
Kipikasa 2005.
| Methods | RCT 2‐arm parallel group design (dose comparison study). | |
| Participants | 52 women attending a large teaching hospital and scheduled for IOL. Inclusion criteria: singleton, cephalic presentation, not in active labour, gestational age > 40 weeks (confirmed by menstrual dates and ultrasound before 20 weeks). Exclusion criteria: previous CS, FHR abnormalities, contraindication to prostaglandin or vaginal birth. |
|
| Interventions | Intervention group: 50 µg oral misoprostol. Comparison group: 25 µg misoprostol. Prior to randomisation women received an ultrasound to assess fetal growth and AFV and a fetal NST was carried out. In both groups medication was administered by a nurse and in the absence of labour or contraindications the dose was repeated after 3 days to a maximum of 3 doses over 9 days. Women returned to hospital every 3 days unless labour started or there was any reduction in fetal kicks. |
|
| Outcomes | Days to birth, uterine hyperstimulation, further induction agents required, CS, Apgar score < 6 at 5 min, NICU admission, meconium staining, perinatal death. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated sequence. |
| Allocation concealment (selection bias) | Low risk | Coded drug boxes. |
| Blinding (performance bias and detection bias) Women | Unclear risk | Intervention and placebo tablets were cut from larger tablets (1/4 or 1/8) and described as appearing the same. |
| Blinding (performance bias and detection bias) Clinical staff | Unclear risk | Staff were said to be blinded because placebo and intervention tablets indistinguishable. We were unsure if they were indistinguishable to knowledgeable staff because they were cut from larger tablets (1/4 or 1/8). |
| Blinding (performance bias and detection bias) Outcome assessor | Low risk | Described as blind. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There were some inconsistencies in the figures; while 49 women seem to have been randomised there were 52 in the results tables. |
| Selective reporting (reporting bias) | Unclear risk | This was a pilot study and secondary measures for women and infants not specified in the methods text. |
| Other bias | Unclear risk | This was a pilot study with limited sample size. Authors state secondary outcomes analysed without stratification, but not what characteristic on which the sample would be stratified. The authors state the possibility of type II error due to inadequate sample size to evaluate neonatal outcomes. |
Larmon 2002.
| Methods | RCT, 3 arm trial. | |
| Participants | Setting: Mississippi, USA (outpatient setting). 136 women randomised. Inclusion criteria: women at term (37 weeks' gestation), Bishop score < 6, candidates for vaginal birth with uncomplicated pregnancy. Exclusion criteria: women with diabetes or serious pregnancy complications including hypertension, or chronic medical conditions. |
|
| Interventions | Intervention group (1): PGE₂ 0.5 mg intracervical. Intervention group (2): vaginal oestrogen cream (estradiol) 4 mg. Comparison group: inert lubricant vaginal jelly. Women were assessed weekly until an indication for birth arose. Medication was repeated weekly. |
|
| Outcomes | Mode of birth, use of oxytocin, condition of newborn. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number tables. |
| Allocation concealment (selection bias) | Low risk | Opaque, sequentially numbered envelopes. |
| Blinding (performance bias and detection bias) Women | Low risk | Placebo controlled. |
| Blinding (performance bias and detection bias) Clinical staff | Unclear risk | Interventions not identical; placebo jelly distinguishable from estradiol cream for staff. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Assessment of outcomes remote from intervention and placebo administration, but unclear if staff would have been aware of group assignment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 136 women were randomised, 8 were excluded after randomisation. |
| Selective reporting (reporting bias) | Unclear risk | Specific outcomes not stated in methods text, apart from sample size calculation. |
| Other bias | Unclear risk | Baseline group characteristics are similar. |
Lelaidier 1994.
| Methods | RCT. | |
| Participants | Setting: not clear. 32 women. Inclusion criteria: women who had a previous CS with gestational age > 38 and < 42 weeks confirmed by ultrasound. All women were scheduled for induction (21 for "post‐dates", 7 for hypertension and 4 for FGR); Bishop score < 4. |
|
| Interventions | The study was carried out over a 4 day observation period, induction was planned for the fourth day (PGE₂ and amniotomy or oxytocin induction if Bishop score > 3). Women attended the outpatient's department for NST daily. Intervention group: 200 mg oral mifepristone on days 1 and 2. Comparison group: placebo, same regime as intervention group. |
|
| Outcomes | CS, assisted birth, uterine scar separation, fetal distress. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as "randomisation list" using block design (block size 4). |
| Allocation concealment (selection bias) | Low risk | Coded drug boxes. |
| Blinding (performance bias and detection bias) Women | Low risk | Described as double‐blind placebo controlled study. "External appearance of the tablets was similar." |
| Blinding (performance bias and detection bias) Clinical staff | Low risk | Intervention and placebo tablet described as similar. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Blinding of outcome assessors not described. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All women appeared to be accounted for in the analysis. |
| Selective reporting (reporting bias) | Unclear risk | Outcomes not stated in methods text |
| Other bias | Unclear risk | Baseline characteristics similar but no formal test (P value) reported. |
Lien 1998.
| Methods | RCT 2 arm parallel group design. | |
| Participants | 90 women attending 4 USA hospitals. Inclusion criteria: women with post‐dates pregnancy (gestational age > 40 weeks + 3 days) attending for FHR testing. Gestation confirmed by ultrasound before 24 weeks, AFI > 5 cm, reactive NST. Exclusion criteria: malpresentation, multiple pregnancy, previous CS, evidence of hyperstimulation or suspicious FHR patterns, grand multiparity (> 4 previous deliveries), placenta praevia or other contraindications to vaginal birth. |
|
| Interventions | Intervention group: intracervical PGE₂ gel (Prepidil) 0.5 mg. Comparison group: placebo gel. Gel was inserted by doctor or midwife in an antenatal testing centre or in the labour unit within rapid transport distance of birth facilities. After insertion there was 40 min of continuous monitoring. Women returned to hospital after 3 to 4 days for fetal testing and further gel up to a maximum of 4 doses. |
|
| Outcomes | Further induction agents required, CS rates, uterine hyperstimulation, FHR changes and side effects. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated sequence (permuted block design, but block size not stated). |
| Allocation concealment (selection bias) | Low risk | Central randomisation with coded drug boxes. |
| Blinding (performance bias and detection bias) Women | Low risk | Treatment and placebo gels were identical and produced by manufacturer. |
| Blinding (performance bias and detection bias) Clinical staff | Low risk | Treatment and placebo gels were identical and produced by manufacturer. |
| Blinding (performance bias and detection bias) Outcome assessor | Low risk | Unblinding was reported to occur only after completion of all the data collection. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 2 women that were randomised were not included in the analysis as they did not meet the inclusion criteria (the study was described as ITT). |
| Selective reporting (reporting bias) | Unclear risk | Specific outcomes not mentioned in results section apart from sample size calculation. |
| Other bias | Unclear risk | This is a pilot study. Women in the prostaglandin group were further over due than women in the control group, but other baseline characteristics similar. |
Lyons 2001.
| Methods | RCT, 2 arm trial. | |
| Participants | Setting: 40 women, setting not clear. Inclusion criteria: women with gestational age 40 to 42 weeks, singleton, cephalic presentation, intact membranes, Bishop score < 6, reassuring FHR and < 3 contractions in 10 minutes. |
|
| Interventions | Intervention group: 100 mg oral misoprostol, dose repeated every 24 h with maximum of 3 doses. 2 h continual fetal monitoring after each dose. Comparison group: placebo, with same regime and monitoring as the intervention group. |
|
| Outcomes | Chorioamnionitis, meconium aspiration, uterine hyperstimulation, mean time to active labour. | |
| Notes | Study reported in brief abstract. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as "randomized". |
| Allocation concealment (selection bias) | Unclear risk | Placebo controlled, no information on randomisation procedure. |
| Blinding (performance bias and detection bias) Women | Low risk | Described as double‐blind, placebo controlled study. |
| Blinding (performance bias and detection bias) Clinical staff | Low risk | Described as double‐blind, placebo controlled study. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Not described. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | All women appeared to have been followed up, but little information. |
| Selective reporting (reporting bias) | Unclear risk | Study reported in brief abstract; unable to assess this bias domain. |
| Other bias | Unclear risk | Study reported in brief abstract; unable to assess this bias domain. |
Magann 1998.
| Methods | RCT. 3 arm trial. | |
| Participants | Setting: California, USA, women attending a naval medical centre. 70 women included in the analysis (2 of 3 treatment arms included, total recruited 105 women). Inclusion criteria: women with "post dates" pregnancy ‐ gestational age 41 weeks confirmed by menstrual dates and pre‐20 weeks ultrasound. Uncomplicated pregnancy. Bishop score < 5. Exclusion criteria: women with any contraindication to vaginal birth. |
|
| Interventions | (1 intervention group had daily membrane stripping; this group has not been included in the analysis in this review.) Intervention group: intracervical PGE₂ 0.5 mg, daily for 3 days. Comparison group: gentle cervical examination, daily for 3 days. Women were instructed to return to hospital is they had bleeding, membrane rupture, regular contractions of reduction in fetal movements. Once Bishop score = 8 or women reached 42 weeks they were admitted to hospital for induction. |
|
| Outcomes | Induced at 42 weeks, CS, instrumental birth. Apgar score < 7 at 5 min, admission to NICU. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number tables. |
| Allocation concealment (selection bias) | Low risk | Sealed, opaque, sequentially numbered envelopes. |
| Blinding (performance bias and detection bias) Women | High risk | Not feasible. |
| Blinding (performance bias and detection bias) Clinical staff | High risk | Not feasible. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Outcome assessment of cervical changes were reported to be blind; blinding not described for other outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent loss to follow up. |
| Selective reporting (reporting bias) | Unclear risk | Outcomes not stated in methods text. |
| Other bias | Low risk | Baseline demographics similar. |
McKenna 1999.
| Methods | RCT. | |
| Participants | Setting: Ohio hospital USA (65 women). Inclusion criteria: women at term (gestational age > 37 weeks), age > 17 years, Bishop score < 7. "Well dated pregnancy" with no indication for immediate induction. Exclusion criteria: multiple pregnancy, insulin dependent diabetes, ruptured membranes, non‐reassuring NST, contraindications to a trial of labour, chronic hypertension. |
|
| Interventions | Intervention group: intracervical PGE₂ 0.5 mg. Comparison group: placebo. Both groups had continuous monitoring for 1 h, if labour started women were admitted to hospital, otherwise they were discharged home. |
|
| Outcomes | Uterine hyperstimulation, further induction agents required,uterine hyperstimulation, CS. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number tables. |
| Allocation concealment (selection bias) | Low risk | Placebo controlled trial. |
| Blinding (performance bias and detection bias) Women | Low risk | Placebo was described as identical to active PGE₂. |
| Blinding (performance bias and detection bias) Clinical staff | Unclear risk | Investigators who administered the gel were blinded; however, prenatal care sometimes delivered by other staff who were aware of study participation but not treatment. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Unclear if obstetric care staff were aware of study participation and/or group assignment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 65 women were randomised, there were 4 post randomisation exclusions. |
| Selective reporting (reporting bias) | Unclear risk | Trial outcomes not specified in methods text. |
| Other bias | Low risk | No baseline imbalance apparent. |
McKenna 2004.
| Methods | RCT, 2 arm trial. | |
| Participants | Setting: not clear. 68 women included. Inclusion criteria: women with "well‐dated" pregnancies with gestational age > 40 weeks and Bishop score < 9. Exclusion criteria: current indication for IOL, malpresentation, multiple pregnancy, previous CS, oligohydramnios (AFI < 5 cm). any contraindication to a trial of labour, current regular contractions. |
|
| Interventions | All women were assessed prior to randomisation. Intervention group: vaginal misoprostol 25 µg. Comparison group: placebo gel. Fetal and uterine monitoring for 1 h after treatment then women were discharged home. Labour was induced if BIshop score > 8 after 41 weeks or all women after 42 weeks. |
|
| Outcomes | Uterine hyperstimulation, mode of birth, epidural, Apgar score, NICU admission. (Women with PROM were given oxytocin to "stimulate labour" but were not included as inductions in the analyses.) | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated sequence performed in hospital pharmacy. |
| Allocation concealment (selection bias) | Low risk | Placebo controlled trial. |
| Blinding (performance bias and detection bias) Women | Low risk | Placebo of similar appearance. |
| Blinding (performance bias and detection bias) Clinical staff | Unclear risk | Investigators blinded but other prenatal care providers aware of study participation. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Blinding of outcomes assessors not described. Obstetric staff aware of study participation. Birth and obstetric data taken from computerised records. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 68 women were randomised, 4 were excluded after randomisation and did not receive the study medication, but were included in an ITT analysis. |
| Selective reporting (reporting bias) | Unclear risk | Outcomes (apart from sample size calculation) not mentioned in methods text. |
| Other bias | Unclear risk | No baseline imbalance apparent. |
Meyer 2005.
| Methods | RCT 2‐arm parallel group design. | |
| Participants | 84 women attending a USA hospital between 1999 to 2001. Inclusion criteria: singleton, cephalic presentation, intact membranes, Bishop score of 6 or less, reactive NST. Exclusion criteria: ruptured membranes, Bishop score > 6, contraindication to induction, > 3 contractions in 10 min, uterine scar. |
|
| Interventions | Intervention group: vaginal misoprostol 25 µg. Comparison group: intracervical PGE₂ gel (dinoprostone) 0.5 mg. Women in both groups were randomised after a reactive NST. After drug administration women had continuous FHR monitoring for 3 h with discharge home if clinically stable. Women were asked to return the next day (after 18 h) for oxytocin induction if labour was not established. |
|
| Outcomes | Vaginal birth within 24 or 48 h, uterine hyperstimulation, CS, oxytocin required, Apgar score < 7 at 5 min, NICU admission. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number table. |
| Allocation concealment (selection bias) | Low risk | Opaque sequentially numbered envelopes (not stated whether sealed). |
| Blinding (performance bias and detection bias) Women | High risk | Blinding women would be feasible but the study was not blinded. |
| Blinding (performance bias and detection bias) Clinical staff | High risk | Study not blinded. |
| Blinding (performance bias and detection bias) Outcome assessor | High risk | Study not blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 84 women were randomised (42 in each group), 2 women were lost to follow up in the misoprostol group but were included in the denominators. |
| Selective reporting (reporting bias) | Low risk | Primary and secondary outcomes stated in methods text and reported. |
| Other bias | Low risk | None apparent. |
Newman 1997.
| Methods | RCT, 2 arm trial. | |
| Participants | 58 women. South Carolina, USA. Inclusion criteria: women with diabetes at term or women with prolonged pregnancy (> 42 weeks) requiring induction, Bishop score < 7. |
|
| Interventions | Intervention group: 2 mg intravaginal PGE₂ after reassuring NST, then continuous fetal monitoring for 3 h. Women were admitted if labour started or cervix favourable. Treatment repeated after 24 h and 48 h and admitted after third dose. Comparison group: expectant management with weekly assessment of AFI and NST. Admission in labour or if signs of fetal distress. IOL at 44 weeks. |
|
| Outcomes | Spontaneous labour within 48 h, uterine hyperstimulation, CS. | |
| Notes | Results reported in brief abstract. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as "prospectively randomised". |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding (performance bias and detection bias) Women | High risk | Not feasible. |
| Blinding (performance bias and detection bias) Clinical staff | High risk | Not feasible. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Outcomes assessors not mentioned in brief abstract. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Little information. No loss to follow up apparent. |
| Selective reporting (reporting bias) | Unclear risk | Trial reported only in brief abstract so unable to assess this bias domain. |
| Other bias | Unclear risk | Trial reported only in brief abstract so unable to assess this bias domain. |
O'Brien 1995.
| Methods | Placebo controlled RCT. | |
| Participants | Setting: outpatient clinic in Memphis, USA. 100 women recruited. Inclusion criteria: gestation 38 to 40 weeks with Bishop score < 7. Exclusion criteria: non‐reactive NST, oligohydramnios (AFI < 5.0 cm) macrosomia (> 4000 g or 10th centile), medical indication for birth, > 1 previous CS. |
|
| Interventions | All women underwent NST, AFV and ultrasound assessment. Intervention group: 2 mg intravaginal PGE₂ for 5 consecutive days. Comparison group: identical placebo for 5 consecutive days. After each dose women were monitored for 30 min to rule out labour or fetal distress. Women were reviewed twice weekly (NST and AFV). |
|
| Outcomes | Other induction agents required, uterine hyperstimulation, CS, epidural, chorioamnionitis, Apgar score, NICU admission, gestational age at birth. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number table. Permuted blocks with variable block size. The randomisation schedule was kept in pharmacy. |
| Allocation concealment (selection bias) | Low risk | Coded drug boxes prepared by pharmacy. |
| Blinding (performance bias and detection bias) Women | Low risk | Placebo controlled trial. |
| Blinding (performance bias and detection bias) Clinical staff | Low risk | Investigators blind. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Unclear if obstetric staff other than investigators blind to study participation. Outcome assessment not mentioned. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | State that "no post randomisation exclusions were allowed". All women included in the analysis. |
| Selective reporting (reporting bias) | Unclear risk | Specific outcomes not specified in methods text, though categories were such as 'neonatal outcomes'. |
| Other bias | Low risk | No baseline imbalance apparent. |
Oboro 2005.
| Methods | RCT, 2 arm trial. | |
| Participants | Setting: district hospital in southern Nigeria, 2000 to 2001. 77 women randomised. Inclusion criteria: women with gestational age > 40 weeks, Bishop score < 9, uncomplicated pregnancy, candidates for vaginal birth (lack of current indication for induction), singleton gestation in cephalic presentation. Exclusion criteria: women with previous CS, vaginal bleeding, ruptured membranes of indication for immediate IOL, uncertain dates, non reactive stress test or estimated fetal weight > 4500 g. |
|
| Interventions | Intervention group: vaginal misoprostol 25 µg (quarter of 100 µg tablet). Comparison group: expectant management with gentle vaginal examinations only. Women were monitored for 1 h after treatment. If regular contractions started women were admitted otherwise they were discharged home. |
|
| Outcomes | Time to birth, GA at birth, proportion of women requiring induction for post‐term birth, length of labour, incidence and severity of side effects, perinatal mortality, Apgar score, NICU admission. | |
| Notes | Unbalanced randomisation 24 in intervention group versus 12 in control group. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated. |
| Allocation concealment (selection bias) | Low risk | Sealed, sequentially numbered envelopes (not stated that envelopes opaque). |
| Blinding (performance bias and detection bias) Women | High risk | Described as an "open" RCT. |
| Blinding (performance bias and detection bias) Clinical staff | High risk | Blinding not mentioned; trial described as open. |
| Blinding (performance bias and detection bias) Outcome assessor | High risk | Blinding not mentioned; trial described as open. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Outcome data were available for all women randomised. |
| Selective reporting (reporting bias) | Unclear risk | Outcomes mentioned in the methods text are reported, but so are many other outcomes. Side effects are mentioned in the methods text and abstract but not defined specifically, so unable to say if these are reported. |
| Other bias | High risk | Nulliparous women was different in either arm (58% in misoprostol arm versus 49% in control arm). Groups otherwise similar at baseline. |
Rayburn 1999.
| Methods | RCT, 2 arm trial. | |
| Participants | Setting: USA. FHR tracings and uterine activity monitored for 20 minutes before randomisation. Inclusion criteria: 294 women who had 1 previous CS and were candidates for vaginal birth with accurate gestational age dating (39 to 41 weeks) by ultrasound before 20 weeks, with no signs of labour, no fetal growth abnormalities and reassuring FHR tracing. Bishop score < 6. Exclusion criteria: malpresentation, multiple pregnancies, diabetes, hypertension, vaginal bleeding, ruptured membranes, cephalopelvic disproportion, contraindication to oxytocic drugs or hypersensitivity to PGE₂, > 1 previous CS. |
|
| Interventions | Intervention group: intracervical PGE₂ 0.5 mg. Women were monitored for 2 h after insertion. Comparison group: expectant management. Women in both groups were reviewed at 40 and 41 weeks for routine assessments. |
|
| Outcomes | Further induction agents required, uterine hyperstimulation, CS, instrumental vaginal birth, maternal infection, Apgar score at 5 min, side effects, birthweight. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated sequence provided by pharmaceutical company. |
| Allocation concealment (selection bias) | Unclear risk | "Blocks of the list were sent with the drugs to the study centres where new subjects were assigned to the next number on the list to determine treatment group." |
| Blinding (performance bias and detection bias) Women | High risk | Study described as "open‐label"; women in the intervention group would have been aware of having to go for additional appointments to receive a gel. |
| Blinding (performance bias and detection bias) Clinical staff | High risk | Not feasible. Study says investigators masked to assignment but unclear if study investigators were in charge of prenatal and obstetric care of all participant women. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Outcome assessors not mentioned. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 300 were enrolled but 6 were not included in analysis "because of improper entry or non compliance with clinic visits". |
| Selective reporting (reporting bias) | Unclear risk | Several categories of possible outcomes mentioned in methods text. |
| Other bias | Unclear risk | Groups appeared similar at baseline. Research was supported by the manufacturers of the study intervention (Prepidil). |
Rijnders 2011.
| Methods | Unblinded, pragmatic, parallel multicenter RCT. | |
| Participants | Setting: multicentre, midwifery practices in Netherlands. The study began in 4 midwifery practices, but by the end of the study period recruitment had been rolled out to 46 midwifery practices in the Netherlands. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Intervention group (N = 270): amniotomy in an outpatient setting (at home) for induction between 292 and 294 days gestation. Control group (N = 251): routine care following the Dutch guideline for management of post term pregnancy. The Guideline prescribed referral to an obstetrician for fetal assessment on the morning of day 294. |
|
| Outcomes | Primary outcomes
Secondary outcomes
Intervention group only
|
|
| Notes | PhD thesis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computerised randomisation service was carried out by an independent Medical Call Centre available for telephone contact 24 h per day, 7 days a week. While random sequence generation was adequate, there were 2 problems:
Above issues do not appear to have affected the random sequence itself. Hence, low risk of bias. |
| Allocation concealment (selection bias) | Low risk | Telephone assignment. |
| Blinding (performance bias and detection bias) Women | High risk | It was not possible to blind participants. |
| Blinding (performance bias and detection bias) Clinical staff | High risk | It was not possible to blind participants, midwives, or other caregivers. |
| Blinding (performance bias and detection bias) Outcome assessor | High risk | Outcome assessors stated as not blind. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Intervention
Control
Responses to the satisfaction survey were not balanced (221 and 183), the response rate was likely affected by the intervention (women in intervention arm were in the home, so response rate was higher). |
| Selective reporting (reporting bias) | Low risk | Stated outcomes are reported. |
| Other bias | Low risk | Baseline demographics similar. |
Sawai 1991.
| Methods | RCT. | |
| Participants | Setting: post‐dates clinic in Florida hospital USA. 50 women with prolonged pregnancy (> 41 weeks, 287 days). Inclusion criteria: reactive NST and normal ultrasound, EDD confirmed by menstrual dates, clinical exam and early ultrasound. Bishop score < 9. Exclusion criteria: malpresentations, multiple pregnancy, diabetes, hypertension, vaginal bleeding, abnormal FHR, established contractions, macrosomia (> 4500 g), FGR, fetal abnormalities or oligohydramnios. |
|
| Interventions | Intervention group: Intravaginal PGE₂ gel 2 mg. Repeated twice weekly. Comparison group: placebo gel. Repeated twice weekly. Uterine activity and FHR was monitored for 1 to 2 h after gel insertion, if no regular contractions or side effects, women were discharged home returning for weekly sonograms and AFV assessment, and returning twice weekly for NST and repeat interventions. |
|
| Outcomes | Further induction agents required, uterine hyperstimulation, CS, NICU admission. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly generated assignments." |
| Allocation concealment (selection bias) | Unclear risk | "drawing of envelopes." |
| Blinding (performance bias and detection bias) Women | Low risk | Placebo controlled study. |
| Blinding (performance bias and detection bias) Clinical staff | Low risk | Placebo controlled study. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All women randomised accounted for in the analysis. |
| Selective reporting (reporting bias) | Low risk | Stated outcomes are reported. |
| Other bias | Low risk | Baseline demographics similar between groups. |
Sawai 1994.
| Methods | RCT. | |
| Participants | Setting: 91 women with prolonged pregnancy (gestational age > 41 weeks) attending a Florida, USA hospital. Inclusion criteria: uncomplicated pregnancy, reliable dating, Bishop score < 9, reactive NST and ultrasound. Exclusion criteria: vaginal bleeding, ruptured membranes, macrosomia (estimated fetal weight > 4500 g) previous uterine surgery or stillbirth, abnormal FHR or ultrasound, regular contractions. |
|
| Interventions | Intervention group: daily self‐administered vaginal PGE₂ 2 mg before bed (women were given instructions re placement and storage of suppositories). Comparison group: self‐administered placebo. Telephone contact available 24 h for both groups. Twice weekly clinic attendance for post‐dates surveillance (NST and AFV); induction if indicated or at 44 weeks. |
|
| Outcomes | CS rates, chorioamnionitis, Apgar score at 5 min, NICU admission. | |
| Notes | Costs data reported. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated. |
| Allocation concealment (selection bias) | Low risk | Coded drug boxes. |
| Blinding (performance bias and detection bias) Women | Low risk | Described as "double blind" placebo controlled. |
| Blinding (performance bias and detection bias) Clinical staff | Low risk | Described as "double blind" placebo controlled. |
| Blinding (performance bias and detection bias) Outcome assessor | Unclear risk | Outcomes assessors not mentioned. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 91 were enrolled but 11 were lost to follow up (3 were excluded as they were non compliant). |
| Selective reporting (reporting bias) | Low risk | Outcomes stated in methods text are reported. |
| Other bias | Low risk | baseline demographics similar between groups. |
Schmitz 2014.
| Methods | Randomised, multicentre, double‐blind, placebo‐controlled RCT. | |
| Participants | Setting: 11 French university hospital referral maternity units that collaborate in the “Groupe de Recherche en Obstétrique et Gynécologie” (Obstetrics and Gynecology Research Group). Inclusion criteria
Exclusion criteria
Contra‐indications to IMN treatment (known hypersensitivity to it, cardiovascular collapse, aortic stenosis, mitral stenosis, obstructive myocardial hypertrophy, systolic blood pressure < 95 mm Hg). |
|
| Interventions | Experimental intervention (N = 684) Cervical ripening: 2 tablets of 20 mg isosorbide‐5‐mononitrate were taken from identical blister packs and inserted by midwives into the posterior vaginal fornix Intervention protocol: administered at 41 + 0, 41 + 2 and 41 + 4 weeks, or until cervix favourable, or fetal status abnormal, where labour was induced If 41 + 5 weeks was reached, labour was induced (in hospital) Control (N = 689) 2 placebo tablets of similar design were taken from identical blister packs and inserted by midwives into the posterior vaginal fornix |
|
| Outcomes | Primary outcome
Secondary outcomes
|
|
| Notes | Schmitz 2014 is a brief abstract published on the same trial. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Eligible women were randomly assigned by obstetricians or midwives using a web‐based application in a 1‐to‐1 ratio to the IMN or placebo groups; the application was based on a computer‐generated list with permuted blocks of 4 stratified by maternity units. |
| Allocation concealment (selection bias) | Low risk | The allocation sequence was not available to any member of the research team until the database was completed and locked. Patients, study staff, and data analysts were masked to assignment. |
| Blinding (performance bias and detection bias) Women | Low risk | Article states “Patients, study staff, and data analysts were masked to assignment". |
| Blinding (performance bias and detection bias) Clinical staff | Low risk | Article states “Patients, study staff, and data analysts were masked to assignment". |
| Blinding (performance bias and detection bias) Outcome assessor | Low risk | Trained research nurses recorded outcomes from hospital notes and entered data into a web‐based data‐capture system. Article states “Patients, study staff, and data analysts were masked to assignment". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Only 1 woman lost to follow up. Missing values accounted for < 1% of all results, except for arterial cord blood pH (18%) and maternal satisfaction criteria (23%). Article states a post‐randomisation exclusion, that were not included in analysis: “Ten women (0.7%), five in each group, were secondarily excluded from the analysis because they did not meet the inclusion criteria”. While balanced, this exclusion may cause bias. |
| Selective reporting (reporting bias) | High risk | Maternal and neonatal ICU admission rates not reported, although were pre‐specified outcomes. |
| Other bias | Low risk | Baseline characteristics similar apart from maternal age; women were slightly older in the treatment group. |
Stenlund 1999.
| Methods | RCT, 2 arm trial. | |
| Participants | Setting: 36 women attending hospital in Stockholm, Sweden. Inclusion criteria: maternal or fetal indications for labour induction, women in whom labour induction could be deferred for 48 h, Bishop score < 6, single pregnancy, head presentation and intact membranes. All women were 14 days post‐term and scheduled for induction, but where IOL could be postponed for 48 h. Exclusion criteria: parity > 4, contra‐indications to vaginal birth, oligohydramnios, prior uterine surgery, obstetric or medical complications. |
|
| Interventions | Intervention group: 400 mg mifepristone. Comparison group: placebo. Women returned for review after 24 h and 48 h if labour did not start. If Bishop score > 6 then ARM and oxytocin induction, if < 6 then PGE₂ 0.5 mg intracervical up to 2 treatments. |
|
| Outcomes | Labour within 48 h, Mode of onset of labour, ripe cervix within 48 h, birth within 48 h, need for PGE₂ for cervical ripening, change in Bishop score, duration of labour, interval from treatment to admission in labour, Apgar score, umbilical pH, maternal and neonatal serum concentrations of mifepristone at birth. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number tables. |
| Allocation concealment (selection bias) | Low risk | Coded drug boxes, "sealed pre‐numbered boxes containing either mifepristone or placebo tablets". |
| Blinding (performance bias and detection bias) Women | Low risk | "...the type of treatment the women were given was not known until the entire study was finished". |
| Blinding (performance bias and detection bias) Clinical staff | Low risk | Study described as blinded. |
| Blinding (performance bias and detection bias) Outcome assessor | Low risk | Study described as blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses to follow up apparent. |
| Selective reporting (reporting bias) | Unclear risk | Many more outcomes reported than mentioned in methods text. FHR and uterine contractility not mentioned specifically in results text. |
| Other bias | Unclear risk | Some baseline imbalance, intervention group 79% primiparous versus 58% in the control group. |
Stitely 2000.
| Methods | RCT. | |
| Participants | Setting: USA, naval medical centre. 50 women. Inclusion criteria: women with prolonged pregnancy (41 to 42 weeks' gestation) confirmed by ultrasound, clinical examination and menstrual dates. Singleton, cephalic presentation, intact membranes, Bishop score < 5, < 8 contractions per h, AFI > 5 cm, reactive NST, maternal age > 18, < 50 years. Exclusion criteria: malpresentations, multiple pregnancy, previous CS, vaginal bleeding, ruptured membranes, non reactive NST, estimated fetal weight > 4500 g or < 2000 g, placenta previa, active herpes, hypersensitivity to prostaglandin, signs of infection, asthma or serious disease. |
|
| Interventions | Intervention group: vaginal misoprostol 25 µg (with second dose after 24 h). Comparison group: placebo, packaged and labelled to appear indistinguishable. Both groups were observed for 4 h with FHR and uterine activity monitoring. If women showed no sign of labour of fetal distress they were discharged and asked to return after 24 h for a second dose, then review after a further 24 h for inpatient management. |
|
| Outcomes | Uterine hyperstimulation, CS, Apgar score < 7 at 5 min, meconium staining. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated sequence by pharmacy (permuted block design). |
| Allocation concealment (selection bias) | Low risk | The list was maintained by inpatient pharmacy and drugs were dispensed to appear identical. |
| Blinding (performance bias and detection bias) Women | Low risk | Women would not have been aware of assignment; treatment and placebo identical. |
| Blinding (performance bias and detection bias) Clinical staff | Low risk | Investigators and other obstetric staff blind to group assignment. |
| Blinding (performance bias and detection bias) Outcome assessor | Low risk | Outcome assessors not described, but all staff described as blind until analysis completed. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent loss to follow up. |
| Selective reporting (reporting bias) | Unclear risk | Only primary outcome stated in methods text; fetal outcomes not specified. |
| Other bias | Low risk | Baseline demographics comparable. |
AFI: amniotic fluid index AFV: amniotic fluid volume ARM: artificial rupture of membranes CPD: cephalo‐pelvic disproportion CS: caesarean section EDD: expected date of delivery EFW: estimated fetal weight FGR: fetal growth retardation FHR: fetal heart rate GA: gestational age h: hour/s IMN: isosorbide mononitrate ISMN:isosorbide mononitrate IOL: induction of labour ITT: intention‐to‐treat NHS: National Health Service (UK) NICU: neonatal intensive care unit NST: non‐stress test PGE: progesterone PPH: postpartum haemorrhage PROM: premature rupture of the membranes RCT: randomised controlled trial SOL: spontaneous onset of labour
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Adewole 1993 | This study examined breast stimulation and used a cross‐over design. Women were allocated to either breast stimulation versus no stimulation; after 3 days, if labour had not started women crossed over into the other study group. |
| Damania 1988 | Very little information was provided on study methods. It was not clear this was a RCT. |
| Damania 1992 | In this study breast stimulation was compared with an oxytocin infusion. It was not clear that women in the oxytocin group were discharged home. |
| Di Lieto 1989 | This study used a cross‐over design. |
| Doany 1997 | In this study intravaginal PGE₂ with or without membrane sweeping was compared with placebo with or without membrane sweeping. Complex interventions or interventions involving membrane sweeping are not included in this review. |
| Dorfman 1987 | In this study women received a range of homeopathic herbal preparations versus placebo. The intervention was to prepare women for childbirth generally rather than to induce labour. |
| Elliott 1984 | This study focused on breast stimulation and used a cross‐over design. |
| Evans 1983 | It was not clear that this was a RCT: "the assignment [of medication] to patients was by consecutive entry into either of the studies". The paper described findings for 2 separate studies both examining the use of intracervical porcine ovarian relaxin. The first study appeared to be conducted in hospital and women receiving medication were compared with a control group. In the "outpatient study" there was no control group; women received either 2 mg or 4 mg of relaxin 5 to 7 days before scheduled induction; no outcomes were reported relevant for inclusion in the review. |
| Garry 2000 | This study compared castor oil with no treatment, women were alternately allocated to groups; otherwise there was little information on methods. |
| Griffin 2003 | This study was reported in a brief abstract and insufficient information was available on methods and results to include the study. We contacted the study author and further data are not available. |
| Herabutya 1992 | This study examined intracervical prostaglandin. Little information was provided on study methods. Women "randomized" to the intervention group received intracervical PGE₂ and then monitored for 4 h to 6 h, some had a repeat dose after 6 h, some had a repeat dose the next day and if labour did not start on the third day these women were admitted to hospital for amniotomy and oxytocin infusion. It was not clear what happened to women in the control group other than that they had weekly fetal monitoring; these women were not admitted unless there were signs of abnormality or until they reached 44 weeks' gestation. The management of women in the 2 groups was so different that results are difficult to interpret. |
| Kadar 1990 | This study focused on nipple stimulation. Group allocation was by a quasi‐randomised method; there were serious protocol violations and analysis was not by randomisation group making results very difficult to interpret. |
| Kaul 2004 | This study focused on membrane sweeping. This intervention is not included in this review. |
| Krammer 1995 | This study was reported in a very brief abstract. No original data were presented in the results. |
| Magann 1999 | This study compared PGE₂ and membrane sweeping. Membrane sweeping is not included in this review. |
| Manidakis 1999 | This study was reported in a brief abstract. It was not clear that it was a RCT. We were unable to find contact details for the author to obtain further information. |
| Moghtadaei 2007 | This study focused on extra‐amniotic saline infusion, an intervention rarely used nowadays. It was not clear that this intervention was carried out in an outpatient setting. |
| Ohel 1996 | This quasi randomised trial compared women receiving vaginal PGE₂ with expectant management. Analysis was not by randomisation group. Of 96 cases randomised to PGE₂ 26 preferred expectant management and were therefore omitted from the analysis. As there was no intention‐to‐treat analysis results of this study were very difficult to interpret. |
| Rayburn 1988 | In this study some of the women included in the study were admitted to hospital rather that being treated as outpatients. No separate results were available for women in the outpatient group. |
| Rezk 2014 | This RCT was not conducted in outpatient setting. |
| Salamalekis 2000 | In this study membrane sweeping was compared with oxytocin for labour induction. It was not clear that women were discharged home after interventions and membrane sweeping is not included in this review. |
| Salmon 1986 | This study focused on breast stimulation and used a cross‐over design. Women were allocated to either breast stimulation versus no stimulation; after 3 days, if labour had not started women crossed over into the other study group. |
| Spallicci 2007 | The intervention in this trial was an intracervical injection of hyaluronidase. This intervention is no longer used in clinical practice. |
| Voss 1996 | It was not clear that this intervention was carried out in an outpatient setting or that women were discharged home after treatment. |
| Ziaei 2003 | This study compared dexamethasone with oxytocin. it was not clear that the intervention was carried out in an outpatient setting. |
h: hour/s RCT: randomised controlled trial
Characteristics of studies awaiting assessment [ordered by study ID]
Ascher‐Walsh 2000.
| Methods | Double blind RCT. |
| Participants | 30 women at term (40 to 41 weeks) with a Bishop score < 7. |
| Interventions | Intervention: (2 groups) 200 µg or 100 µg of oral misoprostol. Comparison group: placebo. FHR and uterine monitoring for 2 h after medication. Procedure was repeated after 3 days if labour did not start until 42 weeks. |
| Outcomes | Interval to birth, CS, Induction at 42 weeks, hyperstimulation, Apgar scores. |
| Notes | This study was reported in a brief abstract and the data were described as "preliminary". We attempted to contact authors for further information (8 September 2009). A repeat attempt was made to contact authors as part of the update of this review (1 September 2016). |
Mostaghel 2009.
| Methods | Open RCT. |
| Participants | Setting: Mahdieh hospital, Tehran, Iran. Eligibility criteria not specified. |
| Interventions | Randomised at 40 weeks' gestation to receive 25 µg vaginal misoprostol (N = 22) or placebo (N = 22) on an outpatient basis. Women allowed to go into spontaneous labour, unless an indication for induction developed. |
| Outcomes | Incidence of post term birth, misoprostol side‐effects and neonatal outcomes. |
| Notes | This study was reported in a brief abstract, and available data did not align with primary outcomes of the review. We attempted to contact authors for further information (1 September 2016). |
Thakur 2005.
| Methods | Double blind RCT. |
| Participants | 50 primiparous women with unfavourable cervix with gestational age > 41 weeks. |
| Interventions | Intervention group: 2 tablets (400 mg) mifepristone 48 h before scheduled induction of labour. Comparison group: 2 tablets placebo. |
| Outcomes | Interval to birth, CS, onset of spontaneous labour. |
| Notes | This study was reported in a brief abstract. The setting was not clear. We attempted to contact the authors for further information (11 September 2009). A repeat attempt was made to contact authors as part of the update of this review (1 September 2016). |
CS: caesarean section FHR: fetal heart rate h: hour/s RCT: randomised controlled trial
Differences between protocol and review
We have added a number of additional (non‐prespecified) outcomes focusing on proxy measures of progress towards birth, and potential adverse effects. These were added to capture additional outcome data that relate to the effectiveness and potential harms of the treatments in outpatient settings.
Maternal and caregiver satisfaction were previously defined as "mother (or caregiver) not satisfied", however these were revised during this update.
Three GRADE 'Summary of findings' tables have been added for this update (2017).
The title has changed from the 2010 version of the review from, Different methods for the induction of labour in outpatient settings, to Pharmacological and mechanical interventions for labour induction in outpatient settings, in this 2017 update.
The objectives have been amended in this update (2017) to:
To examine pharmacological and mechanical interventions to induce labour or ripen the cervix in outpatient settings in terms of effectiveness, maternal satisfaction, healthcare costs and, where information is available, safety.
In the previous version of this review,(2010), the objectives were:
To examine pharmacological and mechanical interventions to induce labour in outpatient settings in terms of feasibility, effectiveness, maternal satisfaction, healthcare costs and, where information is available safety. The review complements existing reviews on labour induction examining effectiveness and safety.
Contributions of authors
For the trials added to this review update, Joshua Vogel and Alfred Osoti assessed study eligibility, carried out data extraction and data entry, analysed results and drafted text of the review. All authors reviewed and agreed on the final text of this review. Zarko Alfirevic is guarantor for the review.
Sources of support
Internal sources
The University of Liverpool, UK.
External sources
No sources of support supplied
Declarations of interest
Joshua P Vogel: none known.
Alfred O Osoti: none known.
Anthony J Kelly: none known.
Stefania Livio: none known.
Jane E Norman: Jane Norman was an investigator on two trials included in this review (Bollapragada 2006b; Osman 2006); the reports from these trials were independently assessed by two other review authors. Jane Norman has received a grant of GBP 11,000 (paid to her institution) from the Chief Scientist's Office, Scottish Executive, for an epidemiological study entitled: "Ferguson EF, Norman JE, Chalmers J, Shanks E, Finlayson A. Investigation of the beneficial and adverse effects of induction of labour." Jane Norman has received a number of research grants (paid to her institution) to support research into improving perinatal outcome ‐ none specifically related to immediate versus deferred delivery. Jane has also received small amounts of money for speaking at meetings about prematurity but not immediate versus deferred delivery.
Zarko Alfirevic: none known.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Agarwal 2012 {published data only}
- Agarwal K, Batra A, Dabral A, Aggarwal A. Evaluation of isosorbide mononitrate for cervical ripening prior to induction of labor for postdated pregnancy in an outpatient setting. International Journal of Gynecology and Obstetrics 2012;118(3):205‐9. [DOI] [PubMed] [Google Scholar]
Attanayake 2014 {published data only}
- Attanayake K, Goonewardene M. Outpatient cervical ripening with vaginal isosorbide mononitrate in uncomplicated singleton pregnancies at 39 weeks gestation: A double blind randomized controlled trial. Sri Lanka Journal of Obstetrics and Gynaecology 2014;36(Suppl 1):28, Abstract no: FC 10.5. [DOI] [PubMed] [Google Scholar]
Bollapragada 2006a {published data only}
- Bollapragada S, Mackenzie F, Norrie J, Petrou S, Reid M, Greer I, et al. IMOP: randomised placebo controlled trial of outpatient cervical ripening with isosorbide mononitrate (IMN) prior to induction of labour ‐ clinical trial with analyses of efficacy, cost effectiveness and acceptability. BMC Pregnancy and Childbirth 2006;6:25. [DOI: 10.1186/1471-2393-6-25] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollapragada SS, MacKenzie F, Norrie J, Petrou S, Reid M, Greer IA, et al. Randomized placebo controlled trial of outpatient cervical ripening with isosorbide mononitrate (IMN) prior to induction of labour ‐ clinical trial with analyses of efficacy, cost effectiveness and acceptability. The IMOP study [abstract]. Journal of Obstetrics and Gynaecology 2007;27(Suppl 1):S22. [DOI] [PubMed] [Google Scholar]
- Bollapragada SS, MacKenzie F, Norrie JD, Eddama O, Petrou S, Reid M, et al. Randomised placebo‐controlled trial of outpatient (at home) cervical ripening with isosorbide mononitrate (IMN) prior to induction of labour‐clinical trial with analyses of efficacy and acceptability. The IMOP study. BJOG: an international journal of obstetrics and gynaecology 2009;116(9):1185‐95. [DOI] [PubMed] [Google Scholar]
- Eddama O, Petrou S, Schroeder L, Bollapragada SS, Mackenzie F, Norrie J, et al. The cost‐effectiveness of outpatient (at home) cervical ripening with isosorbide mononitrate prior to induction of labour. BJOG: an international journal of obstetrics and gynaecology 2009;116(9):1196‐203. [DOI] [PubMed] [Google Scholar]
- Reid M, Lorimer K, Norman JE, Bollapragada SS, Norrie J. The home as an appropriate setting for women undertaking cervical ripening before the induction of labour. Midwifery 2011;27(1):30‐5. [DOI] [PubMed] [Google Scholar]
Bullarbo 2007 {published data only}
- Bullarbo M, Orrskog ME, Andersch B, Granström L, Norström A, Ekerhovd E. Outpatient vaginal administration of the nitric oxide donor isosorbide mononitrate for cervical ripening and labor induction postterm: a randomized controlled study. American Journal of Obstetrics and Gynecology 2007;196(1):50.e1‐5. [DOI] [PubMed] [Google Scholar]
Buttino 1990 {published data only}
- Buttino LT Jr, Garite TJ. Intracervical prostaglandin in postdate pregnancy. A randomized trial. Journal of Reproductive Medicine 1990;35(2):155‐8. [PubMed] [Google Scholar]
Elliott 1998 {published data only}
- Elliot CL, Brennand JE, Calder AA. The effect of mifepristone (RU486) on cervical ripening and induction of labour in human pregnancy. 27th British Congress of Obstetrics and Gynaecology; 1995 July 4‐7; Dublin, Ireland. 1995:207.
- Elliott CL, Brennand JE, Calder AA. The effects of mifepristone on cervical ripening and labor induction in primigravidae. Obstetrics and Gynecology 1998;92(5):804‐9. [DOI] [PubMed] [Google Scholar]
Frydman 1992 {published data only}
- Frydman R, Baton C, Lelaidier C, Vial M, Bourget P, Fernandez H. Mifepristone for induction of labour. Lancet 1991;337(8739):488‐9. [DOI] [PubMed] [Google Scholar]
- Frydman R, Lelaidier C, Baton‐Saint‐Mleux C, Fernandez H, Vial M, Bourget P. Labor induction in women at term with mifepristone (RU 486): a double blind, randomized, placebo‐controlled study. Obstetrics and Gynecology 1992;80(6):972‐5. [PubMed] [Google Scholar]
- Frydman R, Lelaidier C, Baton‐Saint‐Mleux C, Fernandez H, Vial M, Bourget P. Labor induction in women at term with mifepristone (RU 486): a double‐blind, randomized, placebo‐controlled study. International Journal of Gynecology and Obstetrics 1993;42(2):220. [PubMed] [Google Scholar]
- Frydman R, Taylor S, Paoli C, Pourade A. RU 486 (mifepristone): A new tool for labour induction in term women with fetus alive [Le RU 486 (mifepristone) un novel outil pour le declenchment du travail a terme]. Contraception, Fertilite, Sexualite 1992;20(12):1133‐6. [PubMed] [Google Scholar]
- Lelaidier C, Benifla JL, Fernandez H, Baton C, Bourget P, Bourrier MC, et al. RU 486 (mifepristone) in medical indications for labour induction in pregnancies at term: results of a randomized, double‐blind study of RU 486 vs placebo. Journal de Gynecologie, Obstetrique et Biologie de la Reproduction 1993;22:92‐100. [PubMed] [Google Scholar]
Gaffaney 2009 {published data only}
- Gaffaney CA, Saul LL, Rumney PJ, Morrison EH, Thomas S, Nageotte MP, et al. Outpatient oral misoprostol for prolonged pregnancies: a pilot investigation. American Journal of Perinatology 2009;26(9):673‐7. [DOI] [PubMed] [Google Scholar]
Ghanaie 2013 {published data only}
- Ghanaie MM, Mirblouk F, Godarzi R, Shakiba M. Effect of outpatient isorbide mononitrate on success of labor induction. Journal of Babol University of Medical Sciences 2013;15(2):12‐7. [Google Scholar]
Giacalone 1998 {published data only}
- Giacalone PL, Targosz V, Laffargue F, Boog G, Faure JM. Cervical ripening with mifepristone before labor induction: a randomized study. Obstetrics and Gynecology 1998;92(4 Pt 1):487‐92. [DOI] [PubMed] [Google Scholar]
Gittens 1996 {published data only}
- Gittens L, Schenkel C, Strassberg S, Apuzzio J. Vaginal birth after cesarean section: comparison of outpatient use of prostaglandin gel to expectant management. American Journal of Obstetrics and Gynecology 1996;174(1 Pt 2):354. [Google Scholar]
Habib 2008 {published data only}
- Habib SM, Emam SS, Saber AS. Outpatient cervical ripening with nitric oxide donor isosorbide mononitrate prior to induction of labor. International Journal of Gynaecology and Obstetrics 2008;101(1):57‐61. [DOI] [PubMed] [Google Scholar]
Hage 1993 {published data only}
- Hage P, Shaw J, Zarou D, Fleisher J, Wehbeh H. Double blind randomized trial to evaluate the role of outpatient use of PGE2 in cervical ripening. American Journal of Obstetrics and Gynecology 1993;168(1 Part 2):430. [Google Scholar]
Harper 2006 {published data only}
- Harper TC, Coeytaux RR, Chen W, Campbell K, Kaufman JS, Moise KJ, et al. A randomized controlled trial of acupuncture for initiation of labor in nulliparous women. Journal of Maternal‐Fetal and Neonatal Medicine 2006;19(8):465‐70. [DOI] [PubMed] [Google Scholar]
Incerpi 2001 {published data only}
- Incerpi M, Fassett M, Kjos S, Tran S, Wing D. Vaginally administered misoprostol for outpatient labor induction in pregnancies with diabetes mellitus [abstract]. American Journal of Obstetrics and Gynecology 2001;184(1):S120. [DOI] [PubMed] [Google Scholar]
- Incerpi MH, Fassett MJ, Kjos SL, Tran SH, Wing DA. Vaginally administered misoprostol for outpatient cervical ripening in pregnancies complicated by diabetes mellitus. American Journal of Obstetrics and Gynecology 2001;185(4):916‐9. [DOI] [PubMed] [Google Scholar]
Kipikasa 2005 {published data only}
- Kipikasa JH, Adair CD, Williamson J, Breen JM, Medford LK, Sanchez‐Ramos L. Use of misoprostol on an outpatient basis for postdate pregnancy. International Journal of Gynaecology and Obstetrics 2005;88(2):108‐11. [DOI] [PubMed] [Google Scholar]
Larmon 2002 {published data only}
- Larmon JE, Magann EF, Dickerson GA, Morrison JC. Outpatient cervical ripening with prostaglandin E2 and estradiol. Journal of Maternal‐Fetal and Neonatal Medicine 2002;11(2):113‐7. [DOI] [PubMed] [Google Scholar]
Lelaidier 1994 {published data only}
- Lelaidier C, Baton C, Benifla JL, Fernandez H, Bourget P, Frydman R. Mifepristone for labour induction after previous caesarean section. British Journal of Obstetrics and Gynaecology 1994;101(6):501‐3. [DOI] [PubMed] [Google Scholar]
Lien 1998 {published data only}
- Lien JM, Morgan MA, Garite TJ, Kennedy KA, Sassoon DA, Freeman RK. Antepartum cervical ripening: applying prostaglandin e2 gel in conjunction with scheduled nonstress tests in postdate pregnancies. American Journal of Obstetrics and Gynecology 1998;179(2):453‐8. [DOI] [PubMed] [Google Scholar]
Lyons 2001 {published data only}
- Lyons C, Rumney P, Huang W, Morrison E, Thomas S, Nageotte M, et al. Outpatient cervical ripening with oral misoprostol post‐term: induction rates decreased. American Journal of Obstetrics and Gynecology 2001;184(1):S116. [Google Scholar]
Magann 1998 {published data only}
- Magann E, Chauhan SP, Nevils BG, McNamara MF, Kinsella MJ, Morrison JC. Management of pregnancies beyond forty‐one weeks' gestation with an unfavorable cervix. American Journal of Obstetrics and Gynecology 1998;178(6):1279‐87. [DOI] [PubMed] [Google Scholar]
McKenna 1999 {published data only}
- McKenna DS, Costa SW, Samuels P. Prostaglandin E2 cervical ripening without subsequent induction of labor. Obstetrics and Gynecology 1999;94(1):11‐4. [DOI] [PubMed] [Google Scholar]
McKenna 2004 {published data only}
- McKenna DS, Ester JB, Proffitt M, Waddell KR. Misoprostol outpatient cervical ripening without subsequent induction of labor: a randomized trial. Obstetrics and Gynecology 2004;104(3):579‐84. [DOI] [PubMed] [Google Scholar]
Meyer 2005 {published data only}
- Meyer M, Pflum J. Outpatient administration of misoprostol decreases induction time. American Journal of Obstetrics and Gynecology 2002;187(6 Pt 2):S167. [Google Scholar]
- Meyer M, Pflum J, Howard D. Outpatient misoprostol compared with dinoprostone gel for preinduction cervical ripening: a randomized controlled trial. Obstetrics and Gynecology 2005;105(3):466‐72. [DOI] [PubMed] [Google Scholar]
Newman 1997 {published data only}
- Newman M, Newman R. Multiple‐dose PGE2 cervical ripening on an outpatient basis: safety and efficacy. American Journal of Obstetrics and Gynecology 1997;176(1 Pt 2):S112. [Google Scholar]
O'Brien 1995 {published data only}
- O'Brien JM, Mercer B, Cleary N, Sibai BM. Efficacy of outpatient induction with low dose intravaginal prostaglandin E2: A randomized, double‐blind, placebo‐controlled trial. American Journal of Obstetrics and Gynecology 1995;172:424. [DOI] [PubMed] [Google Scholar]
- O'Brien JM, Mercer BM, Cleary NT, Sibai BM. Efficacy of outpatient induction with low‐dose intravaginal prostaglandin E2: A randomized, double blind, placebo‐controlled trial. American Journal of Obstetrics and Gynecology 1995;173(6):1855‐9. [DOI] [PubMed] [Google Scholar]
Oboro 2005 {published data only}
- Oboro VO, Tabowei TO. Outpatient misoprostol cervical ripening without subsequent induction of labor to prevent post‐term pregnancy. Acta Obstetricia et Gynecologica Scandinavica 2005;84(7):628‐31. [DOI] [PubMed] [Google Scholar]
Rayburn 1999 {published data only}
- Rayburn W, Lucas M, Gittens L, Goodwin TM, Baxi L, Gall S, et al. Attempted vaginal birth after cesarean section: a multicenter comparison of outpatient prostaglandin E2 gel with expectant management. Primary Care Update for Ob/Gyns 1998;5(4):182‐3. [DOI] [PubMed] [Google Scholar]
- Rayburn WF, Gittens LN, Lucas MJ, Gall SA, Martin ME. Weekly administration of prostaglandin e2 gel compared with expectant management in women with previous cesareans Prepidil gel study group. Obstetrics and Gynecology 1999;94(2):250‐4. [DOI] [PubMed] [Google Scholar]
Rijnders 2011 {published data only}
- ISRCTN47736435. Costs and effects of amniotomy at home for induction of post term pregnancy. isrctn.com/ISRCTN47736435 (first received 9 January 2006).
- Rijnders MEB. A randomised controlled trial of amniotomy at home for induction between 292 and 294 days gestation. Interventions in Midwife Led Care in the Netherlands to Achieve Optimal Birth Outcomes: Effects and Women's Experiences. Amsterdam: University of Amsterdam, 2011. [Google Scholar]
Sawai 1991 {published data only}
- Sawai SK, Williams MC, O'Brien WF, Angel JL, Mastrogiannis DS, Johnson L. Sequential outpatient application of intravaginal prostaglandin E2 gel in the management of postdates pregnancies. Obstetrics and Gynecology 1991;78(1):19‐23. [PubMed] [Google Scholar]
- Williams MG, O'Brien WF, Sawai SK, Knuppel RA. Outpatient cervical ripening in the postdates pregnancy. Proceedings of 10th Annual Meeting of Society of Perinatal Obstetricians; 1990 Jan 23‐27; Houston, Texas, USA. 1990:533.
Sawai 1994 {published data only}
- Sawai SK, O'Brien WF, Mastrogiannis DS, Krammer J, Mastry MG, Porter GW. Patient‐administered outpatient intravaginal prostaglandin E2 suppositories in post‐date pregnancies: a double‐blind, randomized, placebo‐controlled study. Obstetrics and Gynecology 1994;84(5):807‐10. [PubMed] [Google Scholar]
- Sawai SK, O'Brien WF, Mastrogiannis MS, Mastry MG, Porter GW, Johnson L. Outpatient prostaglandin E2 suppositories in postdates pregnancies. American Journal of Obstetrics and Gynecology 1992;166(1 Pt 2):400. [Google Scholar]
Schmitz 2014 {published data only}
- Schmitz T, Closset E, Fuchs F, Maillard F, Rozenberg P, Anselem O, et al. Outpatient cervical ripening with nitric oxide (NO) donors for prolonged pregnancy in nullipara: the NOCETER randomized, multicentre, double‐blind, placebo‐controlled trial. American Journal of Obstetrics and Gynecology 2014;210(1 Suppl):S19. [Google Scholar]
- Schmitz T, Fuchs F, Closset E, Rozenberg P, Winer N, Perrotin F, et al. Outpatient cervical ripening by nitric oxide donors for prolonged pregnancy: a randomized controlled trial. Obstetrics and Gynecology 2014;124(6):1089‐97. [DOI] [PubMed] [Google Scholar]
Stenlund 1999 {published data only}
- Stenlund PM, Bygdeman M, Ekman G. Induction of labor with mifepristone (RU 486). A randomized double‐blind study in post‐term pregnant women with unripe cervices. Acta Obstetricia et Gynecologica Scandinavica Supplement 1994;73(161):Abstract FP50. [Google Scholar]
- Stenlund PM, Ekman G, Aedo AR, Bygdeman M. Induction of labour with mifepristone ‐ a randomized double‐blind study versus placebo. Acta Obstetricia et Gynecologica Scandinavica 1999;78:793‐8. [PubMed] [Google Scholar]
Stitely 2000 {published data only}
- Stitely ML, Browning J, Fowler M, Gendron RT, Gherman RB. Outpatient cervical ripening with intravaginal misoprostol. Obstetrics and Gynecology 2000;96(5 Pt 1):684‐8. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Adewole 1993 {published data only}
- Adewole IF, Franklin O, Matiluko AA. Cervical ripening and induction of labour by breast stimulation. African Journal of Medicine and Medical Sciences 1993;22(4):81‐6. [PubMed] [Google Scholar]
Damania 1988 {published data only}
- Damania KR, Nanavati MS, Dastur NA, Daftary SN. Breast stimulation for cervical ripening. Journal of Obstetrics and Gynaecology of India 1988;58:663‐5. [Google Scholar]
Damania 1992 {published data only}
- Damania KK, Natu U, Mhatre PN, Mataliya M, Mehta AC, Daftary SN. Evaluation of two methods employed for cervical ripening. Journal of Postgraduate Medicine 1992;38(2):58‐9. [PubMed] [Google Scholar]
Di Lieto 1989 {published data only}
- Lieto A, Miranda L, Ardito P, Favale P, Albano G. Changes in the bishop score induced by manual nipple stimulation. A cross‐over randomized study. Clinical and Experimental Obstetrics and Gynecology 1989;16(1):26‐9. [PubMed] [Google Scholar]
Doany 1997 {published data only}
- Doany W. Outpatient management of postdate pregnancy with intravaginal prostaglandin E2 and membrane stripping. American Journal of Obstetrics and Gynecology 1996;174(1Pt 2):351. [Google Scholar]
- Doany W, McCarty J. Outpatient management of the uncomplicated postdate pregnancy with intravaginal prostaglandin E2 gel and membrane stripping. Journal of Maternal‐Fetal Medicine 1997;6(2):71‐8. [DOI] [PubMed] [Google Scholar]
Dorfman 1987 {published data only}
- Dorfman P, Lasserre MN, Tetau M. Preparation for childbirth by homeopathy [Preparation a l'accouchement par homeopathie: experimentation en double insu versus placebo]. Cahiers de Biothérapie 1987;94:77‐81. [Google Scholar]
Elliott 1984 {published data only}
- Elliott JP, Flaherty JF. The use of breast stimulation to prevent postdate pregnancy. American Journal of Obstetrics and Gynecology 1984;149(6):628‐32. [DOI] [PubMed] [Google Scholar]
- Elliott JP, Flaherty JF. The use of breast stimulation to ripen the cervix in term pregnancies. American Journal of Obstetrics and Gynecology 1983;145(5):553‐6. [DOI] [PubMed] [Google Scholar]
Evans 1983 {published data only}
- Evans MI, Dougan MB, Moawad AH, Evans WJ, Bryant‐Greenwood GD, Greenwood FC. Ripening of the human cervix with porcine ovarian relaxin. American Journal of Obstetrics and Gynecology 1983;147(4):410‐4. [DOI] [PubMed] [Google Scholar]
Garry 2000 {published data only}
- Garry D, Figueroa R, Guillaume J, Cucco V. Use of castor oil in pregnancies at term. Alternative Therapies in Health and Medicine 2000;6(1):77‐9. [PubMed] [Google Scholar]
Griffin 2003 {published data only}
- Griffin C. Outpatient cervical ripening using sequential oestrogen ‐ a randomised controlled pilot study. Australian and New Zealand Journal of Obstetrics and Gynaecology 2003;43:183. [Google Scholar]
Herabutya 1992 {published data only}
- Herabutya Y, Prasertsawat PO, Tongyai T, Isarangura N, Ayudthya N. Prolonged pregnancy: the management dilemma. International Journal of Gynaecology and Obstetrics 1992;37(4):253‐8. [DOI] [PubMed] [Google Scholar]
Kadar 1990 {published data only}
- Kadar N, Tapp A, Wong A. The influence of nipple stimulation at term on the duration of pregnancy. Journal of Perinatology 1990;10(2):164‐6. [PubMed] [Google Scholar]
Kaul 2004 {published data only}
- Kaul V, Aggarwal N, Ray P. Membrane stripping versus single dose intracervical prostaglandin gel administration for cervical ripening. International Journal of Gynaecology and Obstetrics 2004;86(3):388‐9. [DOI] [PubMed] [Google Scholar]
Krammer 1995 {published data only}
- Krammer J, O'Brien W, Williams M. Outpatient cervical ripening does not affect gestational age at delivery. American Journal of Obstetrics and Gynecology 1995;172:425. [Google Scholar]
Magann 1999 {published data only}
- Magann EF, Chauhan SP, McNamara MF, Bass JD, Estes CM, Morrison JC. Membrane stripping vs dinoprostone vaginal insert in the management of pregnancies beyond 41 weeks with unfavorable cervix. American Journal of Obstetrics and Gynecology 1998;178(1 Pt 2):S30. [Google Scholar]
- Magann EF, Chauhan SP, McNamara MF, Bass JD, Estes CM, Morrison JC. Membrane sweeping versus dinoprostone vaginal insert in the management of pregnancies beyond 41 weeks with an unfavorable cervix. Journal of Perinatology 1999;19(2):88‐91. [DOI] [PubMed] [Google Scholar]
Manidakis 1999 {published data only}
- Manidakis G, Sifakis S, Orfanoudaki E, Mikelakis G, Prokopakis P, Magou M, et al. Prostaglandin versus stripping of membranes in management of pregnancy beyond 40‐41 weeks [abstract]. European Journal of Obstetrics, Gynecology, and Reproductive Biology 1999;86:S79‐S80. [Google Scholar]
Moghtadaei 2007 {published data only}
- Moghtadaei P. A randomized trial comparing outpatient vaginal isosorbide‐mononitrate versus extra‐amnion saline infusion with concurrent oxytocin for cervical ripening and labor induction in nulliparous women. American Journal of Obstetrics and Gynecology 2007;197(6 Suppl 1):S103. [Google Scholar]
Ohel 1996 {published data only}
- Ohel G, Rahav D, Rothbart H, Ruach M. Randomised trial of outpatient induction of labor with vaginal PGE2 at 40‐41 weeks of gestation versus expectant management. Archives of Gynecology and Obstetrics 1996;258(3):109‐12. [DOI] [PubMed] [Google Scholar]
Rayburn 1988 {published data only}
- Rayburn W, Gosen R, Ramadei C, Woods R, Scott J Jr. Outpatient cervical ripening with prostaglandin E2 gel in uncomplicated postdate pregnancies. American Journal of Obstetrics and Gynecology 1988;158(6 Pt 1):1417‐23. [DOI] [PubMed] [Google Scholar]
Rezk 2014 {published data only}
- Rezk M, Sanad Z, Dawood R, Masood A, Emarh M, Halaby AA. Intracervical foley catheter versus vaginal isosorbid mononitrate for induction of labor in women with previous one cesarean section. Journal of Clinical Gynecology and Obstetrics 2014;3(2):55‐61. [Google Scholar]
Salamalekis 2000 {published data only}
- Salamalekis E, Vitoratos N, Kassanos D, Loghis C, Batalias L, Panayotopoulos N, et al. Sweeping of the membranes versus uterine stimulation by oxytocin in nulliparous women: a randomized controlled trial. Gynecologic and Obstetric Investigation 2000;49(4):240‐3. [DOI] [PubMed] [Google Scholar]
Salmon 1986 {published data only}
- Salmon YM, Kee WH, Tan SL, Jen SW. Cervical ripening by breast stimulation. Obstetrics and Gynecology 1986;67(1):21‐4. [PubMed] [Google Scholar]
Spallicci 2007 {published data only}
- Spallicci MD, Chiea MA, Singer JM, Albuquerque PB, Bittar RE, Zugaib M. Use of hyaluronidase for cervical ripening: a randomized trial. European Journal of Obstetrics, Gynecology, and Reproductive Biology 2007;130(1):46‐50. [DOI] [PubMed] [Google Scholar]
- Spallicci MDB, Bittar RE. Randomized double blind study of ripening the cervix with hyaluronidase in term gestations [Estudo clinico aleatorizado com grupo controle e mascaramento duplo da maturacao do colo uterino pela hialuronidase em gestacoes a termo]. Revista Brasileira de Ginecologia e Obstetricia 2003;25(1):67. [Google Scholar]
Voss 1996 {published data only}
- Voss DH, Cumminsky KC, Cook VD, Nethers MS, Spinnato JA, Gall SA. Effect of three concentrations of intracervical prostaglandin E2 gel for cervical ripening. Journal of Maternal‐Fetal Medicine 1996;5(4):186‐93. [DOI] [PubMed] [Google Scholar]
Ziaei 2003 {published data only}
- Ziaei S, Rosebehani N, Kazeminejad A, Zafarghandi S. The effects of intramuscular administration of corticosteroids on the induction of parturition. Journal of Perinatal Medicine 2003;31(2):134‐9. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Ascher‐Walsh 2000 {published data only}
- Ascher‐Walsh C, Burke B, Baxi L. Outpatient management of prolonged pregnancy with misoprostol (mp): a randomized double‐blind placebo controlled study, prelim data. American Journal of Obstetrics and Gynecology 2000;182(1 Pt 2):S20. [Google Scholar]
Mostaghel 2009 {published data only}
- Mostaghel N, Nakhai F. Outpatient vaginal misoprostol and its effect on post‐term pregnancy. International Journal of Gynaecology and Obstetrics 2009;107(Suppl 2):S586. [Google Scholar]
Thakur 2005 {published data only}
- Thakur V, Dorman E, Sanu L, Harrington K. Mifepristone is an effective ripening agent in postdates primips with cervical length ≥ 2.5cm, but mode of delivery correlates with birthweight: a randomised, placebo controlled double blind study. Ultrasound in Obstetrics & Gynecology 2005;26:452. [Google Scholar]
Additional references
Alfirevic 2014
- Alfirevic Z, Aflaifel N, Weeks A. Oral misoprostol for induction of labour. Cochrane Database of Systematic Reviews 2014, Issue 6. [DOI: 10.1002/14651858.CD001338.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bollapragada 2006b
- Bollapragada S, Mackenzie F, Norrie J, Petrou S, Reid M, Greer I, et al. IMOP: randomised placebo controlled trial of outpatient cervical ripening with isosorbide mononitrate (IMN) prior to induction of labour ‐ clinical trial with analyses of efficacy, cost effectiveness and acceptability. BMC Pregnancy and Childbirth 2006;6:25. [DOI: 10.1186/1471-2393-6-25] [DOI] [PMC free article] [PubMed] [Google Scholar]
Boulvain 2008
- Boulvain M, Kelly AJ, Irion O. Intracervical prostaglandins for induction of labour. Cochrane Database of Systematic Reviews 2008, Issue 1. [DOI: 10.1002/14651858.CD006971] [DOI] [PubMed] [Google Scholar]
Curtis 1987
- Curtis P, Evans S, Resnick J. Uterine hyperstimulation. The need for standard terminology. Journal of Reproductive Medicine 1987;32(2):91‐5. [PubMed] [Google Scholar]
Eddama 2009
- Eddama O, Petrou S, Schroeder L, Bollapragada S, Mackenzie F, Norrie J, et al. The cost‐effectiveness of outpatient (at home) cervical ripening with isosorbide mononitrate prior to induction of labour. BJOG: an international journal of obstetrics and gynaecology 2009;116:1196‐1203. [DOI] [PubMed] [Google Scholar]
Elliott 1992
- Elliott JP, Clewell WH, Radin TG. Intracervical prostaglandin E2 gel. Safety for outpatient cervical ripening before induction of labor. Journal of Reproductive Medicine 1992;37(8):713‐6. [PubMed] [Google Scholar]
Ghosh 2016
- Ghosh A, Lattey KR, Kelly AJ. Nitric oxide donors for cervical ripening and induction of labour. Cochrane Database of Systematic Reviews 2016, Issue 12. [DOI: 10.1002/14651858.CD006901.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Glantz 2003
- Glantz JC. Labor induction rate variation in upstate New York: what is the difference?. Birth 2003;30(3):168‐74. [DOI] [PubMed] [Google Scholar]
Guerra 2009
- Guerra G, Cecatti J, Souza J, Faundes A, Morais SS, Gulmezoglu AM, et al. Factors and outcomes associated with the induction of labour in Latin America. BJOG: an international journal of obstetrics and gynaecology 2009;116(113):1762‐72. [DOI] [PubMed] [Google Scholar]
Gülmezoglu 2012
- Gülmezoglu AM, Crowther CA, Middleton P, Heatley E. Induction of labour for improving birth outcomes for women at or beyond term. Cochrane Database of Systematic Reviews 2012, Issue 6. [DOI: 10.1002/14651858.CD004945.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hapangama 2009
- Hapangama D, Neilson JP. Mifepristone for induction of labour. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD002865.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hofmeyr 2009
- Hofmeyr GJ, Alfirevic Z, Kelly AJ, Kavanagh J, Thomas J, Neilson JP, et al. Methods for cervical ripening and labour induction in late pregnancy: generic protocol. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD002074] [DOI] [Google Scholar]
Hofmeyr 2010
- Hofmeyr GJ, Gülmezoglu AM, Pileggi C. Vaginal misoprostol for cervical ripening and induction of labour. Cochrane Database of Systematic Reviews 2010, Issue 10. [DOI: 10.1002/14651858.CD000941.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
ISRCTN47736435
- ISRCTN47736435. Costs and effects of amniotomy at home for induction of post term pregnancy. isrctn.com/ISRCTN47736435 (first received 9 January 2006).
Kelly 2013
- Kelly AJ, Alfirevic Z, Ghosh A. Outpatient versus inpatient induction of labour for improving birth outcomes. Cochrane Database of Systematic Reviews 2013, Issue 11. [DOI: 10.1002/14651858.CD007372.pub3] [DOI] [PubMed] [Google Scholar]
Kirby 2004
- Kirby RS. Trends in labor induction in the United States: is it true that what goes up must come down?. Birth 2004;31(2):148‐51. [DOI] [PubMed] [Google Scholar]
McGill 2007
- McGill J, Shetty A. Mifepristone and misoprostol in the induction of labor at term. International Journal of Gynaecology and Obstetrics 2007;96(2):80‐4. [DOI] [PubMed] [Google Scholar]
Neale 2002
- Neale E, Pachulski A, Whiterod S, McGuinness E, Gallagher N, Wallace R. Outpatient cervical ripening prior to induction of labour. Journal of Obstetrics and Gynaecology 2002;22(6):634‐5. [DOI] [PubMed] [Google Scholar]
NHS 2014‐15
- Hospital Episode Statistics Analysis, Health and Social Care Information Centre. Hospital Episode Statistics ‐ NHS Maternity Statistics – England, 2014‐15. http://content.digital.nhs.uk/catalogue/PUB19127/nhs‐mate‐eng‐2014‐15‐summ‐repo‐rep.pdf. London: National Statistics, The Information Centre, 25 November 2015.
Osman 2006
- Osman I, MacKenzie F, Norrie J, Murray HM, Greer IA, Norman JE. The "PRIM" study: a randomized comparison of prostaglandin E2 gel with the nitric oxide donor isosorbide mononitrate for cervical ripening before the induction of labor at term. American Journal of Obstetrics and Gynecology 2006;194(4):1012‐21. [DOI] [PubMed] [Google Scholar]
Ramsey 2005
- Ramsey PS, Meyer L, Walkes BA, Harris D, Ogburn PL Jr, Heise RH, et al. Cardiotocographic abnormalities associated with dinoprostone and misoprostol cervical ripening. Obstetrics and Gynecology 2005;105(1):85‐90. [DOI] [PubMed] [Google Scholar]
Rayburn 2002
- Rayburn WF, Zhang J. Rising rates of labor induction: present concerns and future strategies. Obstetrics and Gynecology 2002;100(1):164‐7. [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Salvador 2009
- Salvador SC, Simpson ML, Cundiff GW. Dinoprostone vaginal insert for labour induction: a comparison of outpatient and inpatient settings. Journal of Obstetrics and Gynaecology Canada 2009;31(11):1028‐34. [DOI] [PubMed] [Google Scholar]
Sawai 1995
- Sawai SK, O'Brien WF. Outpatient cervical ripening. Clinical Obstetrics and Gynecology 1995;38(2):301‐9. [DOI] [PubMed] [Google Scholar]
Shetty 2005
- Shetty A, Burt R, Rice P, Templeton A. Women's perceptions, expectations and satisfaction with induced labour ‐ a questionnaire‐based study. European Journal of Obstetrics, Gynecology, and Reproductive Biology 2005;123(1):56‐61. [DOI] [PubMed] [Google Scholar]
Smith 2013
- Smith CA, Crowther CA, Grant SJ. Acupuncture for induction of labour. Cochrane Database of Systematic Reviews 2013, Issue 8. [DOI: 10.1002/14651858.CD002962.pub3] [DOI] [PubMed] [Google Scholar]
Thomas 2001
- Thomas J, Kelly AJ, Kavanagh J. Oestrogens alone or with amniotomy for cervical ripening or induction of labour. Cochrane Database of Systematic Reviews 2001, Issue 4. [DOI: 10.1002/14651858.CD003393] [DOI] [PMC free article] [PubMed] [Google Scholar]
Thomas 2014
- Thomas J, Fairclough A, Kavanagh J, Kelly AJ. Vaginal prostaglandin (PGE2 and PGF2a) for induction of labour at term. Cochrane Database of Systematic Reviews 2014, Issue 6. [DOI: 10.1002/14651858.CD003101.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Vogel 2013
- Vogel J, Souza JP, Gülmezoglu AM. Patterns and outcomes of induction of labour in Africa and Asia: a secondary analysis of the WHO Global Survey on Maternal and Neonatal Health. PLoS One 2013;8(6):e65612. [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2011
- World Health Organization. WHO recommendations for Induction of labour. apps.who.int/iris/bitstream/10665/44531/1/9789241501156_eng.pdf. Geneva: World Health Organization, (accessed prior to 7 August 2017).
References to other published versions of this review
Dowswell 2010
- Dowswell T, Kelly AJ, Livio S, Norman JE, Alfirevic Z. Different methods for the induction of labour in outpatient settings. Cochrane Database of Systematic Reviews 2010, Issue 8. [DOI: 10.1002/14651858.CD007701.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kelly 2009
- Kelly AJ, Alfirevic Z, Norman JE, Dowswell T. Different methods for the induction of labour in outpatient settings. Cochrane Database of Systematic Reviews 2009, Issue 2. [DOI: 10.1002/14651858.CD007701] [DOI] [PMC free article] [PubMed] [Google Scholar]