

Abstract
The bottom-up design and construction of functional metalloproteins remains a formidable task in biomolecular design. While numerous strategies have been used to create new metalloproteins, preexisting knowledge of the tertiary and quaternary protein structure is often required to generate suitable platforms for robust metal coordination and activity. Here we report an alternative and easily implemented approach (Metal Active Sites by Covalent Tethering or MASCoT) whereby folded protein building blocks are linked by a single disulfide bond to create diverse metal coordination environments within evolutionarily naïve protein-protein interfaces. Metalloproteins generated with this strategy uniformly bind a wide array of first-row transition metal ions (MnII, FeII, CoII, NiII, CuII, ZnII and vanadyl) with physiologically relevant thermodynamic affinities (dissociation constants ranging from 700 nM for MnII to 50 fM for CuII). MASCoT readily affords coordinatively unsaturated metal centers, including a five-His coordinated non-heme Fe site, and well-defined binding pockets that can accommodate modifications and enable coordination of exogenous ligands like nitric oxide to the interfacial metal center.
Graphical Abstract
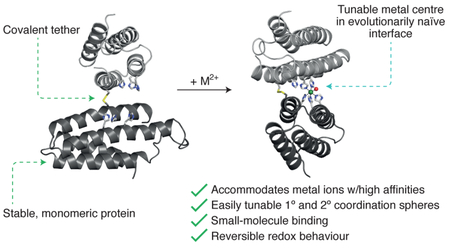
Introduction
Metalloproteins are amongst the most critical components of a living organism. All natural metalloproteins derive their particular function or competitive advantage via robust coordination of a particular metal ion or metallocofactor. Proteins that feature transition metal ions (e.g., MnII, FeII or CuII) often constitute a significant fraction (up to ~60%) of an organism’s proteome.1 Metalloproteins are key pharmaceutical targets2 and progenitors of oncogenesis,3 involved in immune response,4,5 and play central roles in bioenergetics and metabolism.6,7 The proteins involved in these processes have independently evolved unique metal coordination motifs that feature combinations of a small number of weak-field, amino-acid derived ligands, such as imidazole or carboxylates. How metalloproteins coordinate their respective metal ions with high affinities/selectivities and perform remarkably diverse functions despite a limited ligand repertoire remains a topic of great interest for biochemistry, inorganic chemistry and protein design.8-10
Efforts to emulate these features with designed metalloproteins provide a potential avenue towards understanding critical relationships between protein structure and metal coordination. The design of artificial metalloproteins is not unlike the development of human-made catalysts, pharmaceuticals or materials in that they involve the exploration of disparate synthetic approaches.11-13 Some prominent strategies (Fig. 1a-d) include the incorporation of unnatural cofactors or ligands onto preexisting host scaffolds,14-21 redesign of existing scaffolds to enable metal coordination,22-23 and de novo design using consensus sequence/structure motifs and/or computation.24-29 In addition to these design efforts, directed evolution of natural metalloenzymes has also figured prominently in obtaining novel or improved reactivities.30-31 In general, a requirement in most of these approaches is that the generation of a functional metal center relies on a preexisting site in the interior of a tertiary or quaternary structure donated by the host scaffold or derived from sequence/structural similarity to natural scaffolds. The significance of this constraint is nontrivial not only in terms of protein design but also in the context of natural evolution: how did the first functional metalloproteins come into existence without access to the large library of contemporary protein folds?32 This evolutionary question is directly related to a synthetic one: can we construct and diversify functional metalloproteins without the internal constraints of a tertiary/quaternary structure (corresponding to evolutionary innovation) and with minimal engineering steps or mutations (corresponding to evolutionary efficiency)? Along these lines, we previously developed an approach, termed Metal Templated Interface Redesign (MeTIR) (Fig. 1e), in which a small protein building block (cyt cb562) could be engineered on its surface to self-assemble via metal coordination into discrete oligomeric architectures.33 The resultant interfaces in these assemblies could subsequently be tailored with computationally prescribed non-covalent interactions and reinforced with disulfide linkages to generate stable protein complexes that displayed functional properties such as allostery and in vivo catalysis.34-37 While MeTIR represents a streamlined protein design process, it has still involved the incorporation of 10 to 15 surface mutations onto the target protein building block, thereby incurring a non-negligible design/genetic burden.
Fig. 1 ∣. Design strategies for the construction of functional metalloproteins.

a-e, Schematic representations of previously reported metalloprotein design and engineering strategies. f, The MASCoT strategy described herein. MASCoT utilizes intermolecular disulfide linkages to create flexible protein-protein interfaces that serve as an evolutionarily-naïve surface on which to forge diverse metal-binding sites. This strategy does not require unnatural amino acids or cofactors, benefits from an expedient workflow and is potentially generalizable to a wide range of protein monomers.
Cysteine-derived disulfide bonds have been widely exploited to stabilize pre-existing protein architectures (as in the case of MeTIR).34-39 Dutton and colleagues pioneered the use of disulfide bonds for linking together de novo designed coiled-coil peptides to form four-helix bundle “maquettes” that selectively and stably bound various macrocyclic metallocofactors.26,40-42 We wondered whether such covalent linkages could also prove useful in the construction of metal active sites between two arbitrary proteins in the absence of additional stabilizing interactions. We surmised that the formation of a single disulfide linkage between two well-folded proteins would give rise to a malleable protein-protein interface that can be conveniently engineered for the construction of metal coordination sites, with the advantages that a) the protein building blocks are stable and therefore amenable to extensive modifications (in contrast to peptidic building blocks), and b) their well-defined surface features in combination with the restrictions imposed by the disulfide bond would yield robust active site environments (Fig. 1f). We term this strategy MASCoT (Metal Active Sites through Covalent Tethering). As we describe below, this strategy has allowed the simultaneous attainment of several functional features while requiring minimal design and engineering steps: 1) a singular metal coordination motif that can accommodate the entire mid-to-late first-row transition metal ion series with high affinities as well as a metal ion (Fe) in different oxidation states, 2) facile access to coordinative unsaturation and tolerance to large changes in both the primary and secondary coordination spheres, 3) a naturally rare penta-histidine Fe coordination center that can reversibly bind small gaseous molecules, and 4) homo-oligomeric protein assemblies that display both local and global asymmetry.
Results and Discussion
Implementation of MASCoT to construct the dimeric metal-binding complex CH3
As a model building block for MASCoT, we employed cytochrome cb562, a four-helix bundle heme protein, whose high stability, solubility and uniform α-helical composition proved instrumental in our earlier design efforts. To remove any ‘structural memory’ imposed during these efforts, our work herein has focused on the engineering of the original, unadultered cb562 scaffold.43 With the idea that the largest interface between two copies of cyt cb562 would be obtained if the disulfide tether were located centrally on the protein surface, we opted for Cys substitution at position 96 which lies in the middle of Helix 4, the longest of the four α-helices. In order to obtain a stable metal binding site with a high coordination number, we placed a set of three His residues at positions 67 and 71 on Helix 3 and position 97 on Helix 4 that flank position 96. We predicted by inspection of the cyt cb562 crystal structure that this particular placement of three His residues would afford a stable coordination motif on the surface of each protein monomer, and that their combination via a Cys96-Cys96 disulfide tether would generate a clamshell-like metal-chelating motif in the nascent protein interface. Furthermore, we envisioned that the placement of the 3His motif immediately next to the disulfide bond would increase their net local concentration for efficient interfacial metal chelation and could enable the formation of strained coordination geometries. In essence, this strategy is quite analogous to the synthesis of a multi-dentate ligand scaffold, wherein the specific covalent connectivity of the donor atoms determines the flexibility and bite angle of the chelate, ultimately dictating the nuclearity and coordination geometry of the metal center.44
We first investigated the ability of the 3His motif to act as a standalone surface coordination site. Towards this end, we generated the H67/H71/H97cyt cb562 variant (denoted H3), overexpressed it in E. coli and purified it as a soluble, monomeric protein. H3 coordinates CoII, NiII, and ZnII ions with μM dissociation constants (Kd) (Table 1) in a 1:1 stoichiometry, as determined by analytical ultracentrifugation and metal-binding competition experiments (Supplementary Figs. 1 and 2). We obtained single crystals of CoII-bound H3 (Co-H3) and determined its structure at 2.0-Å resolution (Fig. 2a). As planned, the engineered His residues ligate an octahedral CoII center via their Nε atoms in a facial arrangement reminiscent of the non-heme iron enzyme, EgtB, and other analogs from the DinB superfamily;45 three aquo ligands are also well resolved. The backbone alignment of Co-H3 with the parent cyt cb562 shows that the two proteins are essentially identical (RMSD = 0.34 Å), indicating that the core four-helix bundle structure remains unperturbed by surface mutations and metal coordination.
Table 1 ∣.
Dissociation constants (Kd) (in M) determined for the metal complexes of various protein constructs.
| H3 | CH3 | CH3Y | CH2E | CH2EY | CH3Y*d | |
|---|---|---|---|---|---|---|
| Mn∥ | n.d.b | > 2 × 10−5 | n.d.b | 1.6(4) × 10−5 | 4.7(8) × 10−6 | n.d.b |
| aMn∥pH 8.5 | n.d.b | n.d.b | n.d.b | 4.8(5) × 10−6 | 7(3) × 10−7 | n.d.b |
| Fe∥c | n.d.b | n.d.b | n.d.b | n.d.b | n.d.b | 3.7(3) × 10−8 |
| Co∥ | 7(2) × 10−6 | 7(3) × 10−9 | 8(1) × 10−9 | 3.4(4) × 10−8 | n.d.b | 1.9(3) × 10−9 |
| Ni∥ | 6(4) × 10−7 | 9(2) × 10−10 | 1.5(5) × 10−9 | 7(1) × 10−9 | n.d.b | n.d.b |
| Cu∥ | n.d.a | 4(1) × 10−14 | 2.1(8) × 10−13 | 1.4(2) × 10−13 | n.d.b | 5(2) × 10−14 |
| Zn∥ | 8(2) × 10−6 | 1.9(5) × 10−8 | 1.6(3) × 10−9 | 1.4(3) × 10−8 | n.d.b | 8(4) × 10−9 |
All titrations were performed in a metal-free buffer solution (15 mM MOPS) at pH 6.5 with 150 mM NaCl unless stated otherwise.
Titration was performed in a metal-free buffer solution (15 mM Tris-HCl) at pH 8.5 with 150 mM NaCl.
Not determined.
Titration was performed under anaerobic conditions.
CH3Y* refers to a variant of CH3Y wherein the heme cofactor has been removed via steric occlusion.
Fig. 2 ∣. Implementation of MASCoT.

a, Cartoon representation of M-H3 and (bottom) closeup of the coordination environment of Co-H3. b, Cartoon representation of apo-CH3. c, Cartoon and surface representations of M-CH3. d, Closeup of the coordination environment of Co-CH3. e, Salient interfacial interactions that characterize the asymmetric quaternary structure. Water ligands are shown as red spheres. In a and e, the 2Fo-Fc electron density maps are shown in gray and contoured at 2.0 σ and the anomalous difference maps are shown in magenta and contoured at 8.0 σ. In a-c, heme cofactors are not shown for clarity.
Next, we incorporated the T96C mutation into H3 to generate the clamshell CH3 variant. As shown by SDS PAGE analysis, purification of CH3 under oxidizing conditions exclusively afforded a dimeric species (Supplementary Fig. 3), which was subsequently crystallized both in the absence and presence of various divalent metal ions (FeII, CoII, CuII). The resulting crystal structures, with resolutions ranging from 2.75 Å to 1.33 Å (Supplementary Table 3), reveal marked topological differences between apo-CH3 and the metal-bound forms (M-CH3) (Fig. 2b and c) arising from the flexibility of the disulfide linkage. Apo-CH3 displays a roughly perpendicular arrangement of two protein monomers with an interprotein dihedral angle (θip) of 112°. There is minimal interfacial contact between the two monomers (buried surface area = 174 Å2),46 suggesting that the apo-CH3 likely has a fluxional structure in solution and that the observed conformation is stabilized by crystal packing interactions. In contrast, all three M-CH3 complexes possess a singular, compact conformation (RMSD = 0.261 - 0.399 Å) with an antiparallel arrangement of the protein monomers (θip = 163°) and a single, mononuclear metal center adjacent to the disulfide linkage. The dimer topology deviates considerably from C2 symmetry as highlighted by the structure of the Co-CH3 complex (Fig. 2c), which features close protein-protein contacts across only one half of the interface (Fig. 2e).
The topological asymmetry is also projected onto the metal center that possesses an unusual penta-His coordination sphere completed by five of the six designed His residues, whereby a single H97 side chain remains unbound (Fig. 2d). In Fe- and Co-CH3 complexes, the metal coordination is completed by a single aquo ligand, whereas in Cu-CH3, a sixth ligand is not observed as expected from the d9 electronic configuration of the CuII center (Fig. 3a). In each of the three complexes, we observe an ideal square pyramidal arrangement (angular structural parameter τ5 = 0.03 - 0.08)47 of the His ligands with nearly identical MII-N coordination distances (2.0-2.2 Å), including the axial His-Cu bond in Cu-CH3 (2.2 Å). We also prepared a vanadyl adduct of CH3 by reacting the protein with a stoichiometric amount of [VIV≡O][SO4] and characterized it by electron paramagnetic resonance (EPR) spectroscopy in solution (vide infra). The 51V hyperfine parameters of frozen solutions of (VIV≡O)-CH3 are consistent with a monomeric vanadyl species bound to five His ligands.48 The square-pyramidal, 5-His coordination motif is exceedingly rare in natural proteins, with only a handful of representatives in the Protein Data Bank, including the urease maturation protein, UreE, and integral membrane hydroxylases.49, 50 In all M-CH3 crystal structures, the primary and secondary coordination spheres display local B-factors that are 131 - 154% smaller than the mean protein B-factors, consistent with a high degree of structural rigidity found within the protein-protein interface (Supplementary Fig. 4). From a design perspective, our observations indicate that a properly placed disulfide linkage in a sterically encumbered protein-protein interface can impose a strong structural restraint, engendering a robust, coordinatively unsaturated metal coordination geometry that is normally achieved only within the well-defined interior of a folded protein. Importantly, they show that it is possible with MASCoT to create substantial local and global structural asymmetry in a homo-oligomeric system.8,51
Fig. 3 ∣. Structural, spectroscopic and analytical data on Cu-, Fe-, and VIVO bound metalloproteins.

Closeup views of Cu binding sites in a, Cu-CH3 and b, Cu-CH3Y. The 2Fo-Fc maps are shown in gray and contoured at 2.0 σ. c, EPR spectra of Cu-bound metalloproteins and simulations (gray-dotted lines). Conditions: 400 μM Cu, pH 6.5 MOPS, 150 mM NaCl, X-Band, 40 K, 20 mW. d, Cu-binding isotherm for Fura-2-CH3 competition experiments. Protein samples (20 μM/monomer) were combined with 10 μM Fura-2 (pH 6.5, 20 mM MOPS, 150 mM NaCl) and Cu was added sequentially. The gray line represents a simulated isotherm in which CH3 does not compete with Fura-2 for Cu. Closeup views of metal binding sites e, in Fe-CH3Y and f, in VIVO-CH3Y. The 2Fo-Fc maps are shown in gray and contoured at 1.4 σ. g, EPR spectra of VIVO-bound metalloproteins and simulations (gray-dotted lines). Conditions: 400 μM VIVO, pH 6.5 MOPS, 150 mM NaCl, X-Band, 10K, 100 μW. h, Fe-binding isotherm for Fura-2-CH3Y* competition experiments. Protein samples (20 μM/monomer) were combined with 10 uM Fura-2 (pH 6.5, 20 mM MOPS, 150 mM NaCl) and Fe was added sequentially under anaerobic conditions. The gray line represents a simulated isotherm in which CH3Y* does not compete with Fura-2 for Fe.
We next sought to gauge the affinity of the penta-His motif for divalent transition metal ions. Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) analyses of M-CH3 samples that were extensively buffer exchanged confirmed a 1:1 M2+:CH3 stoichiometry (Supplementary Fig. 5). Metal competition titrations of CH3 using the fluorescent metal chelator, Fura-2,52 revealed Kd’s ranging from low μM for Mn and low-to-sub nM for Co, Ni, Zn to low fM for Cu, in accord with the expected trend of the Irving-Williams series (Table 1) (for discussion on Fe, see below). Affinities determined for Co-H3 and Co-CH3 differ by 3 orders of magnitude (corresponding to ca. 4 kcal/mol), providing a quantitative measure on the enhancement of metal binding thermodynamics due to increased denticity/chelate effect afforded by disulfide crosslinking. The values found for CH3 represent some of the highest metal affinities reported for His-rich designed metalloproteins,53-55 consistent with the high denticity of the CH3 scaffold.
Modulation of the primary and secondary coordination spheres in CH3
Given the robustness of the metal coordination environment in CH3 and the fact it is built in a minimally engineered interface formed by the outer surfaces of protein building blocks, we reasoned that it should readily accommodate changes in its primary and secondary coordination spheres. We first targeted positions 70 (natively Gly residues) which lie just above the equatorial M(His)4 plane (Fig. 2d) and could, upon substitution with an appropriate residue, enable the formation of an active-site pocket above the metal center. We opted for replacement of G70 with a Tyr residue (to create variant CH3Y) because of its large but polar side chain. The metal-binding capacity and thermodynamics of CH3Y closely match those of CH3 (Table 1, Supplementary Fig. 5), suggesting that the G70Y mutation must not significantly perturb the quaternary protein structure or the coordination environment. Furthermore, EPR spectra collected on frozen solutions of (VIV≡O)- or Cu2+-bound CH3 and CH3Y variants and pertinent Hamiltonian parameters are nearly identical for each metal ion (Fig. 3, Supplementary Tables 5-6). These findings are corroborated by the crystal structures of several M-CH3Y complexes (M = Fe, Cu, VIV≡O) (resolutions of 1.1 to 1.8 Å) which are isomorphous with their G70 counterparts (RMSDCα = 0.418 Å) (Fig. 3). The exceptions are the well-resolved Y70 side chains that span the dimeric interface above the metal ion and displace numerous ordered water molecules that are observed in M-CH3 (Fig. 3a), effectively creating a small hydrophobic pocket in the interfacial crevice.
We subsequently targeted the primary coordination sphere with the particular aim of creating a stable Mn binding site. Mn lies at the bottom of the Irving-Williams series due to its relatively low Lewis acidity and lack of crystal-field stabilization energy, rendering the engineering of high-affinity Mn coordination sites inherently challenging.56 In light of the predominance of carboxylate-rich coordination motif in natural Mn-proteins,6-7,56-58 we hypothesized that the replacement of one or more of the designed His residues in CH3 and CH3Y with Glu would increase the Mn affinity of these constructs. Two variants, CH2E and CH2EY, were thus created through the H97E mutation. CH2E and CH2EY were found to retain significant fractions of Mn (0.56 and 0.72 equiv per protein dimer, respectively) following incubation with 1 equiv of Mn and subsequent buffer exchange (20 mM MOPS, pH 6.5, 150 mM Tris-HCl), a trait not observed for the CH3 and CH3Y congeners (<0.03 equiv per protein dimer) (Supplementary Fig. 5). Accordingly, these variants were able to compete with the chelating indicator Mag-Fura-257 for Mn binding, allowing us to determine Kd’s of 16 μM (pH 7) or 5 μM (pH 8.5) for Mn-CH2E and 5 μM (pH 7) or 700 nM (pH 8.5) for Mn-CH2EY (Fig. 4a) (Table 1). These values approximate those of natural Mn transcription factors and metalloenzymes.57-58
Fig. 4 ∣. Mn binding by CH2E and CH2EY.

a, Mn-binding isotherm for Mag-Fura-2-CH2EY competition experiments. Protein samples (20 μM/monomer) were combined with 10 μM Mag-Fura-2 (pH 8.5, 20 mM Tris, 150 mM NaCl) and Mn2+ was added sequentially. The gray line represents a simulated isotherm in which CH2EY does not compete with Mag-Fura-2 for Mn2+. b, Comparison of the quaternary structures of Mn-CH2E and Co-CH3 highlighting the difference in inter-protein dihedral angles and emphasizing the tolerance of the MASCoT approach to gross structural perturbations. c, Closeup view of the Mn coordination sphere present in Mn-CH2E. d, Closeup view of the Mn coordination sphere present in Mn-CH2EY. 2Fo-Fc maps are shown in gray and contoured at 2.0 σ. Panels c and d illustrate that secondary sphere point mutations can diversify metal-binding coordination spheres.
Both the structurally unique coordination spheres and secondary-sphere H-bonding networks help rationalize the high-affinity Mn coordination observed for CH2E and CH2EY. The overall topologies of Mn-CH2E and Mn-CH2EY (Fig. 4b) diverge markedly from those of metal-bound CH3 or CH3Y structures (RMSD = 3.7 - 3.9 Å), with the major structural differences arising from a near-perfect antiparallel arrangement of the individual protein monomers (θip = 176° and 174° for Mn-CH2E and Mn-CH2EY, respectively). The Mn coordination sphere in Mn-CH2E (Fig. 4c) includes three meridional His side chains, and a single κ1-bound Glu that completes a square planar ligand arrangement around a trans-(OH2)2MnII unit. These aquo ligands are in turn engaged in strong H-bonding interactions with the two other engineered His and Glu side chains. In contrast, the MnII coordination sphere determined for Mn-CH2EY (Fig. 4d) includes all four designed histidine residues and a κ2-bound glutamate, collectively reminiscent of the non-heme Fe site found in the photosynthetic reaction center of R. sphaeroides.59 The remaining E97 residue is H-bonded to the non-coordinating Nδ of H67A. Apparently, the steric pressure exerted by the adjacent Y70 residues effectively prevents alternative rotameric configurations of H67B, guiding its coordination to the Mn ion in Mn-CH2EY.
Unlike the other divalent metal ions examined in this work, solution studies on FeII have been complicated by adventitious redox reactions involving the ferric heme cofactor that, for example, have prevented the quantitative determination of the Kd’s for the Fe complexes of all variants discussed thus far. Hence, we engineered heme-free variants of CH3Y and CH2EY, denoted CH3Y* and CH2EY*, respectively, in which the heme binding pocket has been engineered with several hydrophobic and largely bulky residues (M7W, C98R, C101A, H102I, R106L)60 that occlude the cofactor. Crystal structures of Fe- and (VIV≡O)-CH3Y* complexes reveal the expected penta-His primary coordination environment (Fig. 5, Supplementary Fig. 7) and affords CoII, CuII and ZnII affinities that are comparable to those for CH3Y (Table 1). Most notably, the Kd for the Fe-CH3Y* complex is 37(3) nM, which compares well with natural and designed non-heme FeII metalloproteins, such as α-ketoglutarate dioxygenase enzymes (Kd = 7.5 μM), cytoplasmic FeII-sensors (Kd = 1.2 μM), and the de novo designed protein DF2 (Kd = 17.8 μM).58,61 The removal of the redox-active heme group also enables unobstructed analyses of the redox properties of the bound non-heme Fe center. In this regard, preliminary electrochemical measurements of the Fe-CH2EY* system reveal a quasireversible FeIII/II redox couple centered at 0.49 V (vs NHE) at pH 6.0 (Supplementary Figure 8), indicating that the dimeric protein scaffold can accommodate multiple oxidation states of a metal center.
Fig. 5 ∣. Nitric oxide binding properties of Fe-CH3Y*.

a, Closeup view of FeNO-CH3Y* with superimposed 2Fo-Fc (gray, 1.6 σ) and NO Fo-Fc omit maps (magenta, 10.0 σ) that illustrate well-ordered electron density arising from an Fe-bound NO ligand. b, FeNO-CH3Y* coordination environment with salient metrical parameters of a protein-bound {FeNO}7. c, UV-visible spectra of Fe-CH3Y* (black) and FeNO-CH3Y* (orange). (inset) X-band EPR spectra of FeNO-CH3Y*. d, 57Fe Mössbauer spectra of FeNO-CH3Y* at 4.2 K in the presence of 50-mT magnetic field. The absence of 57Fe-containing contaminants indicates that NO addition to Fe-CH3Y* in solution yields a single, homogenous {FeNO}7 subunit.
Nitric oxide-binding properties of the Fe-CH3Y* complex
One of the unique functions of metalloproteins is the binding of small diatomic molecules such as O2, CO and NO, which is essential for their storage, transport and chemical activation in living systems. Despite their prevalence in biology, there are few de novo designed metalloproteins with a demonstrated ability to bind small gaseous molecules and such systems rely almost exclusively on the use of privileged cofactors like the heme group.8,26,62-64 There is only one previous report demonstrating NO coordination to an engineered non-heme iron protein.64 Having established the formation of a robust FeII coordination site and a nascent binding pocket in Fe-CH3Y*, we explored its NO binding properties through a battery of spectroscopic and structural investigations. Addition of a suitable NO donor (diethylammonium NONOate) to anaerobic solutions of Fe-CH3Y* resulted in the development of an intense amber hue and visible charge transfer bands characteristic of a mononuclear, intermediate-spin {FeNO}7 unit (Fig. 5c).65 In addition, sharp features emerged at g ~ 4.0 in the X-band EPR spectra of similarly prepared solutions that confirmed the presence of an Fe center with a Stot = 3/2 ground state (Fig. 5c-inset).66-67 Mössbauer spectra collected on frozen solutions of 57Fe-enriched Fe-CH3Y* revealed a single quadrupole doublet whose parameters diverge from those of 57FeSO4 collected in an identical buffer system (Supplementary Fig. 9). Addition of NONOate to these solutions gave rise to magnetic field-dependent multiline absorption features arising from unquenched magnetic interactions with the electronic spin manifold (Fig. 5d).67-71 These features could be well simulated with a Stot = 3/2 spin-Hamiltonian, a positive zero field splitting (>10 cm−1) and low rhombicity (E/D ~ 0), which are similar to those found in synthetic non-heme {FeNO}7 complexes.67,69-71 In contrast, addition of NONOate to solutions of 57FeSO4 in the absence of CH3Y* gives rise to complex spectra suggestive of multiple Fe-containing species (Supplementary Fig. 10).
To gain complementary structural insight into this protein-bound Fe-nitrosyl complex, we soaked pre-formed crystals of Fe-CH3Y* with diethylammonium NONOate and observed the development of an amber hue within each individual crystal (Supplementary Fig. 11). The 2.0-Å resolution X-ray diffraction data obtained from these crystals show that (FeNO)-CH3Y* is isostructural with Fe-CH3Y* (RMSD = 0.11 Å) and contains a distinct electron density above the Fe center which is consistent with a bound NO ligand (Fig. 5a and b). Structural modeling and refinement of the Fe-NO moiety revealed a Fe-N distance of 1.8 Å and a rather acute Fe-N-O angle of 135°, which is unusual among synthetic non-heme {FeNO}7 complexes that have been crystallographically characterized (typical angles range from 147 to 179°).67, 69-71 We cannot rule out the possibility of X-ray induced reduction of the Fe center to generate an {FeNO}8 species that would be anticipated to display such an acute Fe-N-O angle.70,71 Alternatively, it is noteworthy that the NO ligand occupies a rather confined pocket defined by the Tyr70 side chains which may impose steric constraints on the ligand geometry.
Although NO readily coordinates the ferrous center of Fe-CH3Y*, extended exposure of this complex to a O2-rich atmosphere does not induce similar coordination of O2. Monitoring these oxygenation reactions with 57Fe Mossbauer and EPR methods reveals that only 18% of the Fe content in Fe-CH3Y* is slowly oxidized over 24 hours to a single high-spin FeIII species with spectroscopic parameters inconsistent with a ferric superoxide (Supplementary Fig. 12).72 We surmise that the neutral-, nitrogen-rich coordination environment provided by CH3Y* serves to elevate the FeIII/II reduction potential into a regime that is incompatible with effective metal-to-O2 charge transfer that is thought to be essential for O2 coordination in Fe metalloenzymes.1, 72 The high reduction potential measured for the related Fe-CH2EY* species (0.49 V) is suggestive of a thermodynamically unfavorable Fe-O2 interaction. The hexa-histidine site of calprotectin similarly affords a coordination environment that selectively stabilizes the ferrous redox state.73 Given our ability to introduce H-bonding interactions in related constructs (Fig. 4), ongoing efforts are directed at increasing the donor strength of the ligating His residues via secondary sphere tuning to facilitate robust O2 coordination and subsequent activation. Nonetheless, the demonstrated competence of Fe-CH3Y* to coordinate a diatomic ligand (NO) in a structurally well-defined manner is a testament to the functional promise of this scaffold.
Conclusions
The construction of functional proteins in the laboratory is a multi-step process much like the synthesis of a complex natural product from simpler molecules. Both processes involve a careful choice of the building blocks and of the design/synthetic strategy to assemble them, as well as a considerable amount of optimization of the design/synthetic steps to maximize the yield and functional properties of the target structure. While the elegance and heroism of >30-step syntheses are undisputed in organic chemistry, it has been also recognized that a large number of steps is detrimental to the feasibility and practicality of a synthetic route. Thus, under the principle of “step economy”, the focus has considerably shifted to the invention of new types of reactions which minimize the number of steps to reach the desired target.13 It is also preferable if new synthetic strategies also allow for increased chemical diversification at each step, which translates into a wider array of functional molecules that can be obtained with minimal additional effort. This synthetic principle has obvious parallels to the natural evolution of proteins as well: if an evolutionary route involves fewer genetic perturbations to produce a protein with diversifiable functions, it will likely be more efficient than one that requires more perturbations to obtain a protein with a non-diversifiable function.
In the spirit of step economy, we have introduced MASCoT as a readily accessible strategy for the design of oligomeric metalloproteins. A key rate-limiting step in the design of functional proteins is the generation of a stable, yet sufficiently malleable protein architecture through the implementation of numerous non-covalent interactions. This step was streamlined in MASCoT through the tethering of two arbitrary but well-folded protein building blocks via a single disulfide bond, thereby creating a new structural context between two proteins through only one mutation. The elaboration of the resulting interface led to the generation of unusual tetra- and penta-dentate metal coordination motifs that uniformly accommodate a wide variety of coordinatively saturated or unsaturated metal ions, enable substantial alterations in their primary and secondary coordination spheres and bind small gaseous molecules. The fact that each of these functional features are difficult to design on their own but are simultaneously achieved through MASCoT through minimal engineering attests to the expediency of the covalent tethering strategy to build new functional sites in protein interfaces. As this strategy is predicated upon the use of well-folded protein building blocks and natural amino acids, its application in the laboratory evolution of enzymatically-active metalloproteins operative in oxidative or hydrolytic processes can be readily envisioned.35
Supplementary Material
Acknowledgments
This work was supported by the National Science Foundation (Award CHE1607145 to F.A.T.). J.R. was supported by a postdoctoral fellowship from the National Institute of General Medical Sciences of the National Institute of Health (Grant F32GM120981). Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource at the Stanford Linear Accelerator Center and the Advanced Light Source at the Lawrence Berkeley National Laboratory, which are supported by the DOE, Office of Science, Office of Basic Energy Sciences under contracts DE-AC02-76SF00515 and DE-AC02-05CH11231, respectively. We thank C. Moore and M. Gembicky for assistance with XRD experiments.
Footnotes
Competing Financial Interests
The authors declare no competing interests.
Methods and Data Availability
The principal data supporting the findings of this work are available within the figures and the Supplementary Information file. Coordinates and structure factor files for Co-H3 (6DYI), apo-CH3 (6DYB), Co-CH3 (6DYC), Fe-CH3 (6DYE), Cu-CH3 (6DYD), Fe-CH3Y (6DYG), Cu-CH3Y (6DYF), VIVO-CH3Y (6DYH), Mn-CH2E (6DY6), Fe-CH2E (6DY4), Mn-CH2EY (6DY8), Fe-CH3Y* (6DYJ), FeNO-CH3Y* (6DYK), and VIVO-CH3Y* (6DYL) have been deposited to the Protein Data Bank with the corresponding PDB ID codes. Additional data that support the findings of this study are available from the corresponding author on request.
References
- (1).Gray HB, Stiefel EI, Valentine JS & Bertini I Biological Inorganic Chemistry: Structure and Reactivity (University Science Books, USA, 2007). [Google Scholar]
- (2).Chen AY, Adamek RN, Dick BL, Credille CV, Morrison CN & Cohen SM Targetting Metalloenzymes for Therapeutic Intervention. Chem. Rev. 118 ASAP (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Matés JM, Segura JA, Alonso FJ & Márques J Intracellular redox status and oxidative stress: implications for cell proliferation, apoptosis, and carcinogenesis. Arch. Toxicol. 82, 273–299 (2008). [DOI] [PubMed] [Google Scholar]
- (4).Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, et al. Metal Chelation and Inhibition of Bacterial Growth in Tissue Abscesses. Science 319, 962–965 (2008). [DOI] [PubMed] [Google Scholar]
- (5).Hayden JA, Brophy MB, Cunden LS & Nolan EM High-Affinity Manganese Coordination by Human Calprotectin is Calcium-Dependent and Requires a Histidine-Rich Site Formed at the Dimer Interface. J. Am. Chem. Soc. 135, 775–787 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Umena Y, Kawakami K, Shen JR & Kamiya N Crystal Structure of Oxygen-Evolving Photosystem II at a Resolution of 1.9 Angstrom. Nature 473, 55–65 (2011). [DOI] [PubMed] [Google Scholar]
- (7).de Montellano PRO, Cytochrome P450: Structure, Mechanism, and Biochemistry (Kluwer Academic/Plenum, New York, ed. 3, 2005). [Google Scholar]
- (8).Yu F, Cangelosi VM, Zastrow ML, Tegoni M, Plegaria JS, et al. Protein Design: Toward Functional Metalloenzymes. Chem. Rev. 114, 3495–3578 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Schwizer F, Okamoto Y, Heinisch T, Gu Y, Pellizzoni MM, et al. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 118 142–231 (2018). [DOI] [PubMed] [Google Scholar]
- (10).Lu Y, Yeung N, Sieracki N & Marshall NM Design of Functional Metalloproteins. Nature. 460, 855–562 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Soler-Illia GJAA, Sanchez C, Lebeau B & Patarin J Chemical Strategies to Design Textured Materials: from Microporous and Mesoporous Oxides to Nanonetworks and Hierarchical Structures. Chem. Rev. 102, 4093–4138 (2002). [DOI] [PubMed] [Google Scholar]
- (12).Fürstner A Olefin Metathesis and Beyond. Angew. Chem., Int. Ed. 39, 3012–3043 (2000). [PubMed] [Google Scholar]
- (13).Wender PA, Verma VA, Paxton TJ & Pillow TH Function-Oriented Synthesis, Step Economy, and Drug Design. Acc. Chem. Res. 41, 40–49 (2008). [DOI] [PubMed] [Google Scholar]
- (14).Hyster TK, Knorr L, Ward TR & Rovis T Biotinylated Rh(III) Complexes in Engineered Streptavidin for Accelerated Asymmetric C-H Activation. Science 338, 500–503 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Key HM, Dydio P, Clark DS & Hartwig JF Abiological Catalysis by Artificial Haem Proteins Containing Nobel Metals in Place of Iron. Nature 534, 534–537 (2016). [DOI] [PubMed] [Google Scholar]
- (16).Bos J, Fusetti F, Diressen AJM & Roelfes G Enantioselective Artificial Metalloenzymes by Creation of a Novel Active Site at the Protein Dimer Interface. Angew. Chem., Int. Ed. 51, 7472–7475 (2012). [DOI] [PubMed] [Google Scholar]
- (17).Cavazza C, Bochot C, Rousselot-Pailley P, Carpentier P, Cherrier MV, et al. Crystallographic snapshots of the reaction of aromatic C-H with O2 catalyzed by a protein-bound iron complex. Nat. Chem. 2, 1069–1076 (2010). [DOI] [PubMed] [Google Scholar]
- (18).Hayashi T, Hilvert D & Green AP Engineered Metalloenzymes with Non-Canonical Coordination Environments. Chem. Eur. J. 24, 1–11 (2018). [DOI] [PubMed] [Google Scholar]
- (19).Jeschek M, Reuter R, Heinisch T, Trindler C, Klehr J, et al. Directed Evolution of Artificial Metalloenzymes for in vivo Metathesis. Nature 537, 661–665 (2016). [DOI] [PubMed] [Google Scholar]
- (20).Yang H, Swartz AM, Park HJ, Srivastava P, Ellis-Guardiola K, et al. Evolving Artificial Metalloenzymes via Random Mutagenesis. Nat. Chem. 10, 318–324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Mann SI, Heinisch T, Ward TR & Borovik AS Peroxide Activation Regulated by Hydrogen Bonds within Artificial Cu Proteins. J. Am. Chem. Soc 139, 17289–17292 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Yeung N, Lin Y-W, Gao Y-G, Zhao X, Russel BS, et al. Rational Design of a Structural and Functional Nitric Oxide Reductase. Nature 462, 1079–1082 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Toscano MD, Woycechowsky KJ & Hilvert D Minimalist Active-site Redesign: Teaching Old Enzymes New Tricks. Angew. Chem., Int. Ed. 46, 3212–3236 (2007). [DOI] [PubMed] [Google Scholar]
- (24).Lombardi A, Summa CM, Geremia S, Randaccio L, Pavone V & DeGrado WF Retrostructural Analysis of Metalloproteins: Application to the Design of a Minimal Model for Diiron Proteins. Proc. Natl. Acad. Sci. USA 97, 6298–6305 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Reig AJ, Pires MM, Snyder RA, Wu Y, Jo H, et al. Alteration of the Oxygen-Dependent Reactivity of de novo Due Ferri Proteins. Nat. Chem. 4, 900–906 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Koder RL, Anderson JLR, Solomon LE, Reddy KS, Moser CC & Dutton PL Design and Engineering of an O2 Transport Protein. Nature. 458, 305–310 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lombardi A, Marsco D, Maglio O, Costanzo LD, Nastri F & Pavone V Miniaturized Metalloproteins: Applications to Iron-Sulfur Proteins. Proc. Natl. Acad. Sci. USA 97, 11922–11927 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zastrow ML, Peacock AFA, Stuckey JA & Pecoraro VL Hydrolytic Catalysis and Structural Stabilization in a Designed Metalloprotein. Nat. Chem. 4, 118–123 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Der BS, Edwards DR & Kuhlman B Catalysis by a De Novo Zinc-Mediated Protein Interface: Implications for Natural Enzyme Evolution and Rational Enzyme Engineering. Biochemistry 51, 3933–3940 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Arnold FH Directed Evolution: Bringing New Chemistry to Life. Angew. Chem., Int. Ed 57, 4143–4148 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Reetz MT Combinatorial and Evolution-Based Methods in the Creation of Enantioselective Catalysts. Angew. Chem., Int. Ed. 40, 284–310 (2001). [PubMed] [Google Scholar]
- (32).Rufo CM, Moroz YS, Moroz OV, Stohr J, Smith TA, et al. Short peptides self-assemble to produce catalytic amyloids. Nat. Chem 6, 303–309 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Salgado EN, Radford RJ & Tezcan FA Metal-Directed Protein Self-Assembly. Acc. Chem. Res. 43, 661–672 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Churchfield LA, Medina-Morales A, Brodin JD, Perez A & Tezcan FA De Novo Design of an Allosteric Metalloprotein Assembly with Strained Disulfide Bonds. J. Am. Chem. Soc. 138, 13163–13166 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Churchfield LA, Alberstein RG, Williamson LM & Tezcan FA Determining the Structural and Energetic Basis of Allostery in a De Novo Designed Metalloprotein Assembly. J. Am. Chem. Soc 140, 10043–10053 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Song WJ & Tezcan FA A designed supramolecular protein assembly with in vivo enzymatic activity. Science 346, 1525–1528 (2014). [DOI] [PubMed] [Google Scholar]
- (37).Song WJ, Yu J & Tezcan FA Importance of Scaffold Flexibility/Rigidity in the Design and Directed Evolution of Artificial Metallo-β-lactamases. J. Am. Chem. Soc. 139, 16772–16779 (2017). [DOI] [PubMed] [Google Scholar]
- (38).Eijsink VGH, Bjørk A, Gåseidnes S, Sirevåg R, Synstad B, van den Burg B & Vriend G Rational engineering of enzyme stability. J. Biotech 113, 105–120 (2004). [DOI] [PubMed] [Google Scholar]
- (39).Dombkowski AA, Sultana KZ & Craig DB Protein disulfide engineering. FEBS Letters 588, 206–212 (2014). [DOI] [PubMed] [Google Scholar]
- (40).Robertson DE, Farid RS, Moser CC, Urbauer JL, Mulholland SE, et al. Design and synthesis of multi-haem proteins. Nature 368, 425–432 (1994). [DOI] [PubMed] [Google Scholar]
- (41).Lichtenstein BR, Farid TA, Kodali G, Solomon LA, Ross JL, et al. Engineering Oxidoreductases: maquette proteins designed from scratch. Biochem. Soc. Trans 40, 561–566 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Gibney BR, Isogai Y, Rabanal F, Reddy KS, Grosset AM, Moser CC & Dutton PL Self-Assembly of Heme A and Heme B in a Designed Four-Helix Bundle: Implications for a Cytochrome c Oxidase Maquette. Biochemistry 39, 11041–11049 (2000). [DOI] [PubMed] [Google Scholar]
- (43).Faraone-Mennella J, Tezcan FA, Gray HB & Winkler JR Stability and Folding Kinetics of Structural Characterized Cytochrome c-b562. Biochemistry 45, 10504–10511 (2006). [DOI] [PubMed] [Google Scholar]
- (44).Black DSC & Hartshorn AJ Ligand Design and Synthesis. Coord. Chem. Rev. 9, 219–274 (1972). [Google Scholar]
- (45).Goncharenko KV, Vit A, Blankenfeldt W & Seebeck FP Structure of the Sulfoxide Synthase EgtB from the Ergothioneine Biosynthetic Pathway. Ang. Chem., Int. Ed. 54, 2821–2824 (2015). [DOI] [PubMed] [Google Scholar]
- (46).Krissinel E & Henrick K Interface of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007). [DOI] [PubMed] [Google Scholar]
- (47).Addison AW, Rao TN, Reedijk J, van Rijn J & Verschoor GC Synthesis, Structure, and Spectroscopic Properties of Copper(II) Compounds Containing Nitrogen-Sulphur Donor Ligands; the Crystal and Molecular Structure of aqua[1,7-bis(N-methylbenzimidazol-2’-yl)-2,6-dithiaheptane]copper(II) Perchlorate. J. Chem. Soc., Dalton. Trans 1984, 1349–1356 (1984). [Google Scholar]
- (48).Smith TS, LoBrutto R & Pecoraro VL Paramagnetic Spectroscopy of Vanadyl Complexes and its Applications to Biological Systems. Coord. Chem. Rev. 228, 1–18 (2002). [Google Scholar]
- (49).Shi R, Munger C, Asinas A, Benoit SL, Miller E, et al. Crystal Structures of Apo and Metal-Bound Forms of the UreE Protein from Helicobacter pylori: Role of Multiple Metal Binding Sites. Biochemistry 49, 7080–7088 (2010). [DOI] [PubMed] [Google Scholar]
- (50).Zhu G, Koszelak-Rosenblum M, Connelly SM, Dumont ME, Malkowski MG The Crystal Structure of an Integral Membrane Fatty Acid α-Hydroxylase. J. Biol. Chem. 290, 29820–29833 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Parmar AS, Pike D & Nanda V Chapter 12: Computational Design of Metalloproteins in Protein Design Methods and Applications (Humana Press, New York, 2014). [DOI] [PubMed] [Google Scholar]
- (52).Grynkiewicz G, Poenie M & Tsien RY A New Generation of Ca2+ Indicators with Greatly Improved Fluorescence Properties. J. Biol. Chem. 260, 3440–3450 (1985). [PubMed] [Google Scholar]
- (53).Zastrow ML & Pecoraro VL Designing Hydrolytic Zinc Metalloenzymes. Biochemistry 53, 957–978 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Vita C, Roumestand C, Toma F & Ménez A Scorpion Toxins as Natural Scaffolds for Protein Engineering. Proc. Natl. Acad. Sci. USA 92, 6404–6408 (1995) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Brodin JD, Medina-Morales A, Ni T, Salgado EN, Ambroggio XI & Tezcan FA Evolution of Metal Selectivity in Templated Protein Interfaces. J. Am. Chem. Soc 132, 8610–8617 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Hosseinzadeh P, Mirt EN, Pfister TD, Gao Y-G, Mayne C, et al. Enhancing Mn(II)-Binding and Manganese Peroxidase Activity in a Designed Cytochrome c Peroxidase through Fine-Tuning Secondary-Sphere Interactions. Biochemistry 55, 1494–1502 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Golynskiy MV, Gunderson WA, Hendrich MP & Cohen SM Metal Binding Studies and EPR Spectroscopy of the Manganese Transport Regulator MntR. Biochemistry 45, 15359–15372 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Cotruvo JA & Stubbe J Metallation and Mismetallation of Iron and Manganese Proteins in vitro and in vivo: the Class I Ribonucleotide Reductases as a Case Study. Metallomics 4, 1020–1036 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Koepke J, Krammer EM, Klingen AR, Sebban P, Ullman GM & Fritzsch G pH Modulates the Quinone Position in the Photosynthetic Reaction Center from Rhodobacter Sphaeroides in the Neutral and Charge Separated States. J. Mol. Biol 371, 396–409 (2007). [DOI] [PubMed] [Google Scholar]
- (60).Chu R, Takei J, Knowlton JR, Andrykovitch M, Pei W, et al. Redesign of a Four-helix Bundle Protein by Phage Display Coupled with Proteolysis and Structural Characterization by NMR and X-ray Crystallography. J. Mol. Biol 323, 253–262 (2002). [DOI] [PubMed] [Google Scholar]
- (61).Pasternak A, Kaplan J, Lear JD & DeGrado WF Proton and Metal Ion-dependent Assembly of a Model Diiron Protein. Protein Sci. 10, 958–969 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Zhuang J, Amoroso JH, Kinloch R, Dawson JH, Baldwin MJ & Gibney BR Design of a Five-Coordinate Heme Protein Maquette: A Spectroscopic Model of Deoxymyoglobin. Inorg. Chem. 43, 8218–8220 (2004). [DOI] [PubMed] [Google Scholar]
- (63).McLaughlin MP, Retegan M, Bill E, Payne TM, Shafaat HS, et al. Azurin as a protein scaffold for a low-coordinate non-heme iron site with a small-molecule binding pocket. J. Am. Chem. Soc 134, 19746–19757 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Chakraborty S, Reed J, Ross M, Nilges MJ, Petrik ID, Ghosh S, et al. Spectroscopic and Computational Study of a Nonheme Iron nitrosyl Center in a Biosynthetic Model of Nitric Oxide Reductase. Angew. Chem., Int. Ed 126, 2449–2453 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Orville AM & Lipscomb JD Simultaneous Binding of Nitric Oxide and Isotopically-Labeled Substrates of Inhibitors by Reduced Protocatechuate 3,4-Dioxygenase. J. Biol. Chem. 268, 8596–8607 (1993). [PubMed] [Google Scholar]
- (66).Martini RJ, Livada J, Chang W, Green MT, Krebs C, et al. Experimental Correlation of Substrate Position with Reaction Outcome in the Aliphatic Halogenase, SyrB2. J. Am. Chem. Soc. 137, 6912–6919 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Li M, Bonnet D, Bill E, Neese F, Weyhermüller T, Blum N, et al. Tuning the Electronic Structure of Octahedral Iron Complexes [FeL(X)] (L = 1-alkyl-4,7-bis(4-tert-butyl-2-mercaptobenzyl)-1,4,7-triazacyclononane, X = Cl, CH3O, CN, NO). The S = 1/2, S = 3/2 Spin Equilibrium of [FeLPr(NO)]. Inorg. Chem. 41, 3444–3456 (2002). [DOI] [PubMed] [Google Scholar]
- (68).Münck E Chapter 6. Aspects of 57Fe Mössbauer Spectroscopy in Physical Methods in Bioinorganic Chemistry (University Science Books, California, 2000). [Google Scholar]
- (69).Ray M, Golombek AP, Hendrich MP, Yap GPA, Liable-Sands LM, et al. Structure and Magnetic Properties of Trigonal Bipyramidal Iron Nitrosyl Complexes. Inorg. Chem. 38, 3110–3115 (1999). [Google Scholar]
- (70).Speelman AL, White CJ, Zhang B, Alp EE, Zhao J, et al. Non-heme High-Spin {FeNO}6-8 Complexes: One Ligand Platform Can Do It All. J Am. Chem. Soc. 140, 11341–11359 (2018). [DOI] [PubMed] [Google Scholar]
- (71).Serres RG, Grapperhaus CA, Bothe E, Bill E, Weyhermüller T, Neese F & Wieghardt K Structural, Spectroscopic, and Computational Study of an Octahedral, Non-Heme {Fe-NO}6-8 Series: [Fe(NO)(cyclam-ac)]2+/+/0. J. Am. Chem. Soc. 126, 5138–5153 (2004). [DOI] [PubMed] [Google Scholar]
- (72).Mbughuni MM, Chakrabarti M, Hayden JA, Bominaar EL, Hendrich MP, Munck E & Lipscomb JD Trapping and Spectroscopic Characterization of an FeIII-superoxo Intermediate from a Nonheme Mononuclear Iron-containing Enzyme. Proc. Natl. Acad. Sci. USA 107, 16788–16793 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Nakashige TG & Nolan EM Human Calprotectin Affects the Redox Speciation of Iron. Metallomics 9, 1086–1095 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.