

Abstract
Objective:
CD4 germinal center (GC)-T follicular helper (Tfh) cells are important in the pathogenesis of autoimmune arthritis. Previous studies have shown that adenosine 2a receptor (A2aR, Adora2a) signaling can divert CD4 T cells away from the GC-Tfh cell lineage during the primary response to foreign antigens. Therefore, we examined the effects of A2aR signaling on CD4 T cells during the recognition of a self-antigen using a mouse model of autoimmune arthritis.
Methods:
Wild type and Adora2a-deficient KRN TCR-transgenic CD4 T cells specific for glucose-6-phosphate isomerase (GPI)/I-Ag7 were transferred into immunodeficient Tcra–/– I-Ag7-expressing mice to induce arthritis. Recipients were then treated with either the selective A2aR agonist CGS-21680 (CGS) or PBS vehicle alone. Severity of disease, autoantibody titers, KRN T cell numbers and phenotype, and GPI-specific isotype class-switched plasmablasts were tracked.
Results:
CGS treatment inhibited arthritis development and KRN GC-Tfh cell differentiation, blocked the appearance of high affinity GPI-specific and IgG1 isotype class-switched polyclonal plasmablasts, and led to a reduction in anti-GPI IgG1 titers. Additionally, therapeutic administration of CGS after the onset of arthritis blocked further disease progression in association with reduced KRN GC-Tfh cell numbers and anti-GPI IgG1 titers.
Conclusion:
Strong A2aR signaling diverts autoreactive CD4 T cell differentiation away from the GC-Tfh lineage, thus reducing help for the differentiation of dangerous autoreactive B cells that promote arthritis. Therefore, our data suggest that the A2aR and its downstream signaling pathways in CD4 T cells may be promising therapeutic targets to interfere with autoreactive GC-Tfh cell differentiation.
Extracellular adenosine is an immunosuppressive purine nucleoside that reinforces immunological tolerance (1). Numerous investigations in animal models have suggested that activation of adenosine receptors, or biochemical pathways downstream of adenosine receptor signaling (e.g., cyclic adenosine monophosphate (cAMP) accumulation, protein kinase A (PKA) activation), can have ameliorative effects on autoimmune arthritis (2). Nevertheless, the translation of this information into successful therapies for human autoimmune diseases has been hampered by insufficient knowledge regarding the adenosine receptors utilized for this immunosuppressive effect, and the consequences of adenosine receptor engagement on immune cell differentiation and function.
Adenosine 2a receptors (A2aR) have been observed to act as a barrier to autoimmunity by limiting inflammation and negatively regulating adaptive immune responses (3, 4). A2aR signaling can maintain immune homeostasis by promoting the induction of CD4 T cell anergy and the differentiation of Foxp3+ regulatory T (Treg) cells, as well as by blocking the function of effector/memory CD4 T cells (3, 5). A2aRs are expressed at high levels on human T cells during the course of autoimmune disease (6), and the immunosuppressive agents methotrexate and sulfasalazine demonstrate efficacy in diseases such as rheumatoid arthritis (RA) through their abilities to increase A2aR expression and activation by extracellular adenosine (7). Consistent with this, the phosphodiesterase-4 inhibitor apremilast controls psoriatic arthritis (PsA) disease activity as a result of its ability to slow the turnover of the A2aR second-messenger cAMP (8).
Our laboratory and others have recently determined that A2aR signaling during the primary response to a foreign antigen in strong adjuvant has the ability to divert CD4 T cell differentiation away from the T follicular helper (Tfh) and germinal center (GC)-Tfh cell lineages, and consequently reduce help for the differentiation of antigen-specific GC B cells and isotype class-switched plasmablasts (9, 10). In RA, autoreactive B cells are thought to be induced by Tfh cells to undergo clonal expansion, isotype class-switch recombination, and somatic hypermutation within the germinal centers (GC), and this ultimately leads to the differentiation of anti-citrullinated protein antibody (ACPA)-secreting plasma cells (11–13). Additionally, the frequency of circulating Tfh cells is increased in the blood of patients with antibody-mediated autoimmune disorders (14–16), although this has not been universally observed (17). The nuclear factor Bcl6 is responsible for the differentiation of CD4 T cells to the Tfh and GC-Tfh lineage fates, in part, by repressing other effector lineage-specific transcription factors such as Tbet and RORγt (18, 19). Pre-clinical studies using a mouse model of autoimmune arthritis have indicated that Tfh and GC-Tfh cells contribute to the formation of germinal centers, high affinity autoantibody production, and arthritis disease manifestations (20). Consistent with this, T cell-specific deficiencies of Tfh molecules such as the chemokine (C-X-C motif) receptor 5 (CXCR5), signaling lymphocytic activation molecule–associated protein (SAP), and IL-21 lead to protection from the development of severe arthritis (21).
To determine whether strong A2aR signaling can preserve or restore immune homeostasis in autoreactive GC-Tfh cells during the recognition of self-antigens, we utilized a mouse model of autoimmune arthritis wherein KRN TCR-transgenic CD4 T cells specific for the self-antigen glucose-6-phosphate isomerase (GPI)/I-Ag7 differentiate into Tfh and GC-Tfh during the course of arthritis induction (22). Our data now show that the activation of A2aRs on the KRN T cells using the selective agonist CGS-21680 (CGS) blocks the initial development of arthritis as well as arrests disease progression. This therapeutic effect of A2aR signaling occurred in association with reductions in KRN Tfh/GC-Tfh cell differentiation and secondary anti-GPI humoral immunity. Therefore, this study suggests that strong A2aR downstream signals within autoreactive CD4 T cells raise the threshold for the differentiation of dangerous GC-Tfh cell effectors and can ultimately reduce the B cell-dependent immune responses that contribute to autoimmune arthritis.
MATERIAL AND METHODS
Mice
B6 mice were purchased from Charles River Breeding Laboratories under a contract from the National Cancer Institute (Frederick, MD). B6.g7 (H-2g7 congenic) mice and B6 strain KRN mice that express a TCR-transgene specific for GPI/I-Ag7 were gifts from Drs. Diane Mathis and Christophe Benoist (Harvard Medical School, Boston, MA) and the Institut de Génétique et de Biologie Moléculaire et Cellulaire (Strasbourg, France) (23). Wild type and Tcra–/– (B6 x B6.g7) F1 (F1) mice were bred as previously outlined (22). Adora2af/f mice containing loxP sites flanking exon 2 of the Adora2a gene (a gift from Joel Linden, La Jolla Institute for Allergy and Immunology, La Jolla, CA) (24) were crossed with CD4-Cre mice (gift from Michael Farrar, University of Minnesota, Minneapolis, MN) to generate conditional A2aR T cell knockout (KO) mice. The breeding of healthy normal CD45.1+ KRN (WT), CD4-Cre Adora2af/f CD45.1+ KRN (KO), wild type F1, and Tcra–/– F1 mice was carried out in our own colonies. All experimental protocols were performed in accordance with guidelines of the University of Minnesota Institutional Animal Care and Use Committee and the National Institutes of Health.
Adoptive transfer and CGS-21680 treatment
Wild type and Tcra–/– F1 hosts were depleted of NK cells by the administration of anti-asialo GM1 Ab (Wako Chemicals USA, Richmond, VA) as previously described (22). Donor WT and Adora2a-KO KRN spleen and lymph node cells were enriched for naïve CD4 T cells using a Mouse CD4 T Cell Negative Isolation Kit (Stem Cell) per the manufacturer’s instructions. Purified naïve KRN T cells were then adoptively transferred (approximately 10,000 per recipient) via tail vein injection into either the wild type or Tcra–/– F1 host mice to initiate the recognition of the GPI/I-Ag7 self-antigen (Fig. 1). Beginning 1 day after adoptive transfer, host mice were given twice daily i.p. injections with the selective A2aR agonist CGS-21680 (CGS) 2.5 mg/kg (Tocris) or with vehicle alone (PBS) as previously described (3, 9).
Figure 1. T cell adoptive transfer protocols for the assessment of autoimmune arthritis.
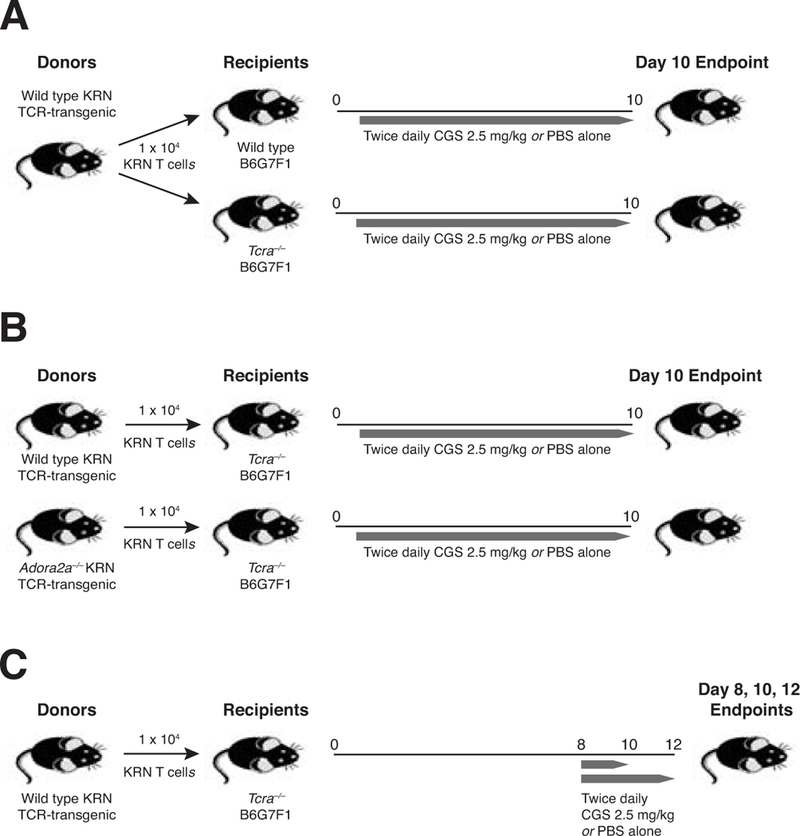
(A) Naïve wild type CD45.1+ GPI/I-Ag7–specific KRN CD4 T cells (104) were adoptively transferred into CD45.2+ wild type and Tcra–/– B6G7F1 (F1) hosts. After 24 hours, mice were injected i.p. twice daily with CGS 2.5 mg/kg or with the vehicle alone (PBS) until the endpoint at day 10. (B) Naïve CD45.1+ KRN CD4 T cells were isolated from both CD4-Cre Adora2af/f conditional knock-out (Adora2a–/–) as well as wild type donor KRN mice, and then the T cells adoptively transferred (104) into CD45.2+ Tcra–/– F1 hosts to induce autoimmune arthritis. Mice were subsequently treated twice daily with either CGS or PBS alone for the duration of the experiment (10 d). (C) Naïve wild type CD45.1+ KRN CD4 T cells were adoptively transferred into CD45.2+ Tcra–/– F1 hosts to induce arthritis. Beginning on day 8, mice were injected i.p. twice daily with CGS 2.5 mg/kg in PBS or else PBS alone as a control for the remainder of the experiment (with endpoints on days 8, 10, and 12 as indicated).
Cell enrichment and flow cytometry
At the completion of all experiments, KRN CD4 T cells were analyzed by pooling the spleen and all LN cells and staining with PE-conjugated Ab to CD45.1 (A20; eBioscience). KRN T cells were enriched using a PE positive selection kit (STEM cell) per the manufacturer’s instructions. Anergy in KRN T cells was assessed based on the ability of T cells to be stimulated for intracellular cytokine accumulation as previously described (9, 22, 25). KRN T cell differentiation was also assessed by flow cytometry for Bcl6, Tbet, RORγt, and Foxp3 using a previously described protocol (9). KRN T cells were incubated in vitro for 3 h at 37°C in RPMI medium 1640 + 10% FCS in the presence of 50 ng/ml PMA (Sigma-Aldrich, St. Louis, MO) and 1 μM ionomycin (EMD Chemicals, Gibbstown, NJ), in the final 2 hours KRN cells were incubated with 10 mg/ml brefeldin A (Sigma-Aldrich). After incubation of the cells, cell surface and intracellular staining was performed as previously described (22, 25). To assess GPI-specific IgG1 plasmablasts, bulk polyclonal lymphocytes were stained with antibodies to B220 (RA3–6B2), GL7 (GL-7), CD38 (90), IgM (RMM-1), and IgD (11–26c.2a), as well as with the irrelevant cell exclusion antibodies CD11c (N418), CD4 (GK1.5), CD8 (53–6.7), and F4/80(BM8), treated with a fixation/permeabilization kit (eBioscience), and then intracellular stained with goat anti-mouse Ig (H+L) (A11068), biotin-conjugated recombinant mouse GPI (kindly provided by Dr. Haochu Huang, University of Chicago, Chicago, IL), and anti-IgG1 (RMG1–1) (31). All cells were analyzed using a BD LSR II flow cytometer (BD Biosciences). Data were analyzed with FlowJo software (Tree Star, Ashland, OR). See Supplemental figure 1 for examples of the flow cytometry gating strategy.
Arthritis scoring
Ankle swelling was measured using a Quick-Mini Series 700 comparator (Mitutoyo, Aurora, IL). Changes were reported as the percent change in ankle thickness from day 0 or day 8, as indicated in the experiment. The arthritis disease activity index was also calculated by assigning a score from 0–3 for each paw based on the erythema/swelling and then summing the scores, as previously described (22).
Anti-GPI isotype-specific IgG antibody measurements
Serum was isolated from recipient mice on the specified days and measured for anti-GPI IgG1, IgG2b, IgG2c, IgG3, and total IgG Abs by ELISA using recombinant mouse GPI together with isotype-specific anti-mouse Ig reagents as previously described (22).
Serum transfer arthritis
Pooled serum from adult arthritic K/BxN mice was a kind gift from Dr. Bryce Binstadt (University of Minnesota). Age-matched wild type F1 mice were injected i.p. twice with 200 μL pooled serum on days 0 and 2. Clinical disease activity and ankle thickness were measured daily. CGS or PBS vehicle-alone treatments were initiated beginning at day 6 (Supplemental Fig. 1).
Joint histology
For histological analysis, ankles and feet were dissected, frozen in O. C. T. medium (Sakura Finetek U.S.A., Inc., Torrance, CA), cryo-sectioned to a thickness of 10μm and stained with hematoxylin and eosin.
Statistical analysis
Statistical tests were performed using Prism (GraphPad) software, and p-values were obtained using an unpaired one-tailed Student’s t-test with a 95% confidence interval. Mean arthritis clinical arthritis disease activity scores were compared using the Mann–Whitney U test.
RESULTS
Adenosine 2a receptor signaling with CGS treatment reinforces immune self tolerance and prevents autoimmune arthritis development
To initially assess whether A2aR signals can act to maintain immune cell homeostasis during self antigen-recognition, we made use of an adoptive transfer of GPI-specific KRN TCR-transgenic CD4 T cells into mice that naturally express GPI/I-Ag7 complexes (see protocol Fig. 1a) (22). As expected, PBS-treated Tcra–/– F1 hosts that lack a population of Foxp3+ Treg cells developed joint swelling and signs of severe arthritis by 8 days after the transfer of the KRN T cells (Fig. 2a, b). Arthritis signs in these paws were accompanied by degradation of the articular cartilage, intense periarticular immune cell infiltration, and erosions of marginal cortical bone tissue (Supplemental Fig. 2). In contrast, the adoptive transfer of KRN T cells into wild type F1 hosts did not interfere with normal peripheral immune self-tolerance mechanisms, and no arthritis was observed. Importantly, lymphopenic Tcra–/– F1 hosts given KRN T cells and then immediately treated with the selective A2aR agonist CGS demonstrated essentially no ankle swelling and significantly reduced clinical disease activity scores as compared to PBS-treated Tcra–/– F1 mice. Joint infiltration and damage were also reduced with this CGS treatment regimen.
Figure 2. Treatment of mice with the selective A2aR agonist CGS-21680 blocks KRN T cell-dependent autoimmune arthritis development.
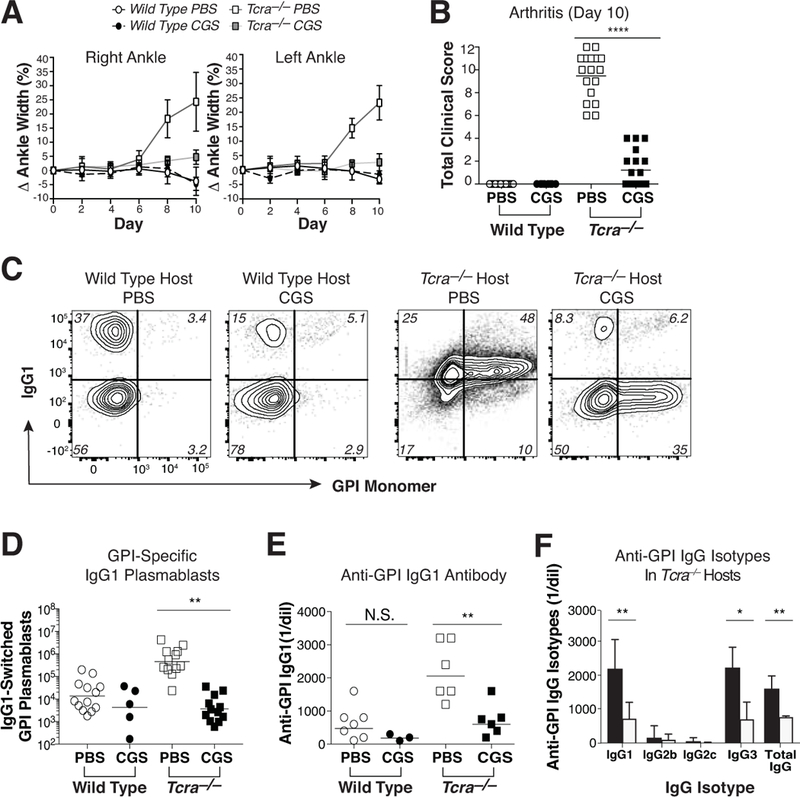
KRN CD4 T cells were adoptively transferred into wild type and Tcra–/– F1 hosts to initiate self-antigen recognition. (A) Comparison of a treatment regimen with twice daily CGS vs. vehicle alone (PBS) on the mean (±SEM) percent change in ankle swelling/size over the duration of the 10 d experiment. (B) Aggregate clinical disease activity scores observed at day 10. (C, D) Frequency (C) and number (D) of host polyclonal B220dim intracellular Ig (H+L)hi GL7– IgG1+ GPI-specific plasmablasts isolated from the spleen and pooled LNs on day 10. Note that weak IgG1 staining in the PBS-treated Tcra–/– group is typical for samples containing high numbers of class-switched IgG1-expressing plasmablasts. (E) Serum anti-GPI IgG1 titers measured by ELISA at day 10. (F) Individual anti-GPI serum IgG isotype titers in Tcra–/– hosts by isotype specific ELISA at day 10. Data are representative of 2–5 independent experiments (n = 3–17). ***P < 0.001 by the Mann-Whitney U test. **P < 0.01 and *P <0.05 by the Students t-test.
GPI-specific autoantibody production is important to the pathogenesis of KRN T cell-dependent arthritis. IgG1 is the dominant autoantibody isotype found in these arthritic animals, and this is associated with the differentiation of GPI-specific IgG1 isotype class-switched polyclonal plasmablasts in the spleen and lymph nodes (LNs) (20, 22). Consistent with these previous reports, polyclonal IgG1 isotype class-switched GPI-specific plasmablasts were found at increased frequencies (averaging 0.51 ± 0.54% of total B cells) in the spleen and LNs of arthritic PBS-treated Tcra–/– F1 hosts as compared to healthy wild type F1 mice (a mean fold-increase of 45.8 ± 10.9). However, CGS treatment significantly reduced the number of GPI-specific IgG1+ plasmablasts present in the Tcra–/– F1 hosts (Fig. 2c, d). In contrast to IgG1+ plasmablasts, GPI-specific plasmablasts negative for IgG1 expression were only infrequently observed in Tcra–/– hosts (averaging 0.08 ± 0.09% of total B cells), but these appeared resistant to the effects of the A2aR agonist (mean fold-decrease of 1.1 ± 0.1) and represented the majority of GPI-binding plasmablasts remaining after the CGS treatment (Fig. 2c, and data not shown). Anti-GPI IgG1, IgG3, and total IgG serum antibody levels mirrored these differences in GPI-specific IgG1+ plasmablast differentiation (Fig. 2e, f). In contrast to lymphopenic Tcra–/– F1 recipients, wild type F1 hosts remained protected from a KRN T cell-mediated breakdown of immune tolerance in the polyclonal B cell compartment regardless of treatment group, and demonstrated persistently low levels of autoreactive plasmablasts and autoantibody (Fig. 2c-e, and data not shown). Thus, A2aR-mediated suppression of autoimmune arthritis occurs in association with the inhibition of the pathogenic anti-GPI IgG1 isotype class-switch.
A2aR signals do not promote KRN CD4 T cell anergy or Treg induction
A2aR signals are thought to inhibit CD4 T cell reactivity in part through the induction of anergy and the differentiation of Foxp3+ Treg cells (3). However, KRN CD4 T cells recovered from day 10 Tcra–/– F1 adoptive transfer recipients demonstrated a robust clonal expansion relative to healthy wild type F1 hosts regardless of whether they were treated with CGS (Fig. 3a). In fact, proliferation by KRN T cells in Tcra–/– F1 hosts appeared enhanced by CGS, based on the expression of the Ki67 proliferative marker (Fig. 3b). To formally test the effects of CGS on the functional responsiveness of the KRN T cells, day 10 cells were stimulated in vitro with phorbol myristate acetate (PMA) plus ionomycin and intracellular cytokine accumulation was then measured. The synthesis of TNFα and IL-2 was similarly induced by stimulation of KRN T cells isolated from both CGS- and PBS-treated Tcra–/– F1 hosts; therefore, anergy induction in the presence of CGS appeared unlikely (Fig. 3c, d). Nonetheless, the treatment of Tcra–/– F1 mice with CGS did interfere with the differentiation of KRN T effector/memory cells capable of secreting the helper cytokine IL-21 (Fig. 3e). This was an isolated defect, as KRN T cells treated with CGS continued to differentiate normally into IFNγ– and IL-17a–producing effector T cells in the Tcra–/– F1 mice (Fig. 3f, g). Given that differentiation of GPI-specific GC B cells and isotype class switch to both IgG1 and IgG3 depend on T cell production of IL-21 (26, 27), this reduced synthesis of IL-21 by KRN T cells in the setting of CGS treatment may have contributed to the reductions in GPI-specific IgG1+ plasmablasts and anti-GPI IgG1 antibody seen here.
Figure 3. A2aR activation does not promote KRN T cell anergy or Foxp3+ Treg cell differentiation in either normal or T cell-lymphopenic hosts.
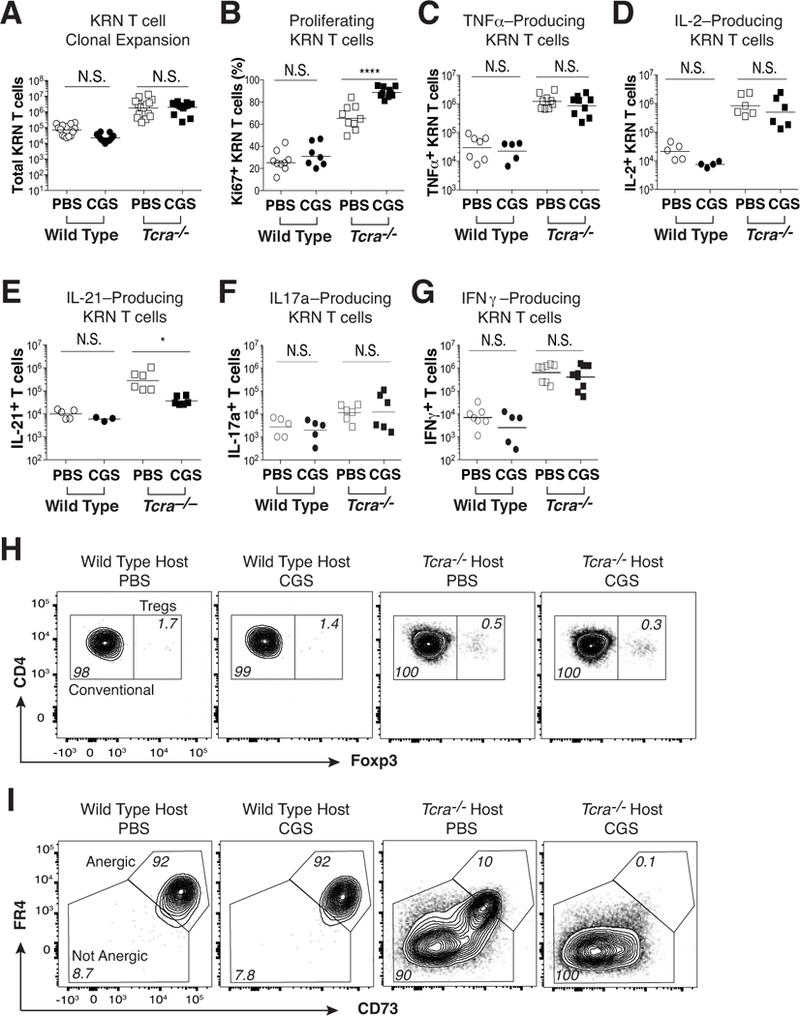
Naïve KRN CD4 T cells were adoptively transferred into wild type and Tcra–/– F1 hosts, and then mice were injected twice daily with CGS or PBS for the duration of the experiment (10 d). (A) Total numbers of KRN T cells. (B) Percent Ki67-expressing conventional Foxp3– KRN T cells. (C-G) Production of TNFα (C), IL-2 (D), IL-21 (E), IL-17a (F), and IFNγ (G) by KRN T cells following 3 h of in vitro ionomycin/PMA stimulation. (H) Frequency of KRN Foxp3+ T regulatory cells. (I) Frequency of conventional Foxp3– FR4+ CD73+ anergic phenotype KRN T cells. Flow cytometry data are representative of at least 3 independent experiments (n = 5–10 mice per group). *P < 0.05 and ***P < 0.001 by the Students t-test. N.S. = not significant.
Foxp3+ Treg cells are indispensable for peripheral immune tolerance (4). We previously reported that a reconstitution of the Foxp3+ Treg cell compartment in Tcra–/– F1 mice restores immune homeostasis and suppresses KRN T cell-mediated adoptive transfer arthritis (22). To determine whether CGS treatment inhibits B cell-dependent arthritis development by promoting KRN Foxp3+ Treg cell differentiation, we looked for expression of Foxp3 within the expanded KRN CD4 T cell population. As shown, CGS treatment was not associated with the enhanced differentiation of KRN Foxp3+ Treg cells in either wild type or Tcra–/– F1 mice (Fig. 3h). In Tcra–/– F1 hosts, the expression of FR4 and CD73 on KRN T cells is typically only intermediate to low, consistent with the avoidance of anergy and successful T effector/memory cell differentiation (22, 25). This is in contrast to wild type F1 hosts where anergic KRN T cells express high levels of FR4 and CD73. Perhaps surprisingly, treatment of the Tcra–/– F1 hosts with CGS failed to induce anergy in the KRN T cells, and instead led to a loss of the subpopulation of T effector/memory cells that expresses intermediate levels of FR4 and CD73 (Fig. 3i). Therefore, A2aR signals promoted neither anergy induction nor Treg differentiation in this arthritis model system, and instead led to alterations in the capacity of the KRN T effector/memory cell population to produce IL-21 and express FR4 and CD73 at intermediate levels.
A2aR signals divert KRN CD4 T effector/memory cells away from the Tfh and GC-Tfh lineage fates
CD4 GC-Tfh cells expressing the highest levels of Bcl6 and CXCR5 are thought to provide cognate help in the form of CD40L and IL-21 to antigen-specific B cells within germinal centers (18, 28, 29). Additionally, CXCR5+ Tfh cells have been reported to express FR4 and CD73 (30). Therefore, we reasoned that the ability of CGS to block both KRN T cell IL-21 production and the differentiation of GPI-specific IgG1 class-switched plasmablasts might stem from its capacity to inhibit Tfh and GC-Tfh cell differentiation. In arthritic Tcra–/– F1 mice, we observed increased levels of CXCR5 on the proportion of KRN T effector/memory cells that co-expressed low but significant amounts of FR4 and CD73 (Fig. 4a, and data not shown). In contrast to PBS-treated mice, KRN T cells in CGS-treated Tcra–/– F1 hosts lacked this subpopulation altogether. Given that strong A2aR signaling has been shown to directly antagonize the differentiation of Tfh and GC-Tfh cells during immunization with a foreign antigen in adjuvant (9), this result suggested the possibility of a similar loss of KRN Tfh and GC-Tfh cell differentiation during the recognition of self-antigen in the presence of CGS. Consistent with this, PBS-treated Tcra–/– F1 adoptive transfer hosts developed greatly expanded KRN populations of Bcl6lo CXCR5lo Tfh and Bcl6hi CXCR5hi GC-Tfh cells, whereas Tfh and GC-Tfh cell differentiation in CGS-treated Tcra–/– F1 mice were significantly reduced (Fig. 4b-d). KRN T effector/memory cells in CGS-treated Tcra–/– F1 mice were instead more likely to display a non-Tfh cell (Bcl6– CXCR5–) phenotype; however, this difference was small and was not associated with any significant change in the numbers of Th1 or Th17 lineage cells (Fig. 4b, e-g). Thus, the data suggested that strong A2aR signaling specifically interferes with the differentiation of KRN CD4 Tfh and GC-Tfh cells during self-antigen recognition.
Figure 4. A2aR agonist treatment blocks Tfh and GC-Tfh cell differentiation during self-antigen recognition.
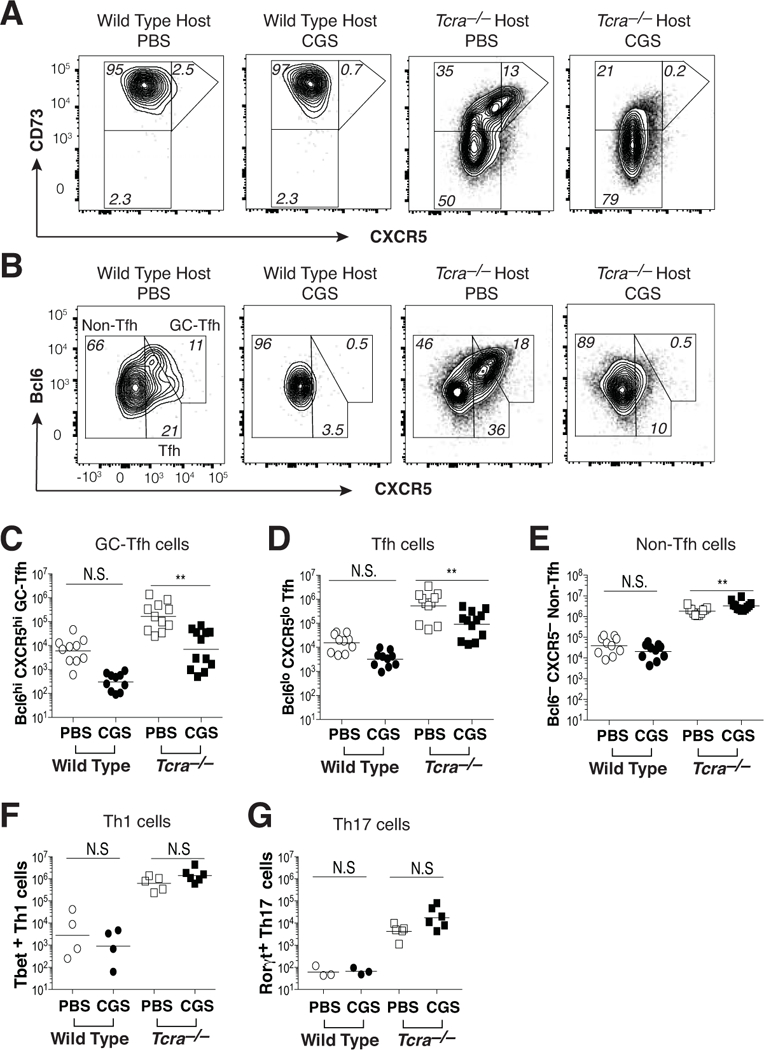
KRN CD4 T cells were transferred into wild type and Tcra–/– F1 hosts, and then mice were treated twice daily for 10 days with either CGS or PBS. (A) Expression of CD73 and CXCR5 on conventional Foxp3– KRN T cells. (B) Bcl6 and CXCR5 expression by conventional Foxp3– KRN T cells. (C-E) Aggregate numbers of Bcl6hi CXCR5hi (GC-Tfh) (C), Bcl6lo CXCR5lo (Tfh) (D), and Bcl6– CXCR5– (non-Tfh) (E) conventional Foxp3– KRN T cells. (F, G) Th1 (Tbet+) (F) and Th17 (RORγt+) (G) lineage T cells within the non-Tfh (Bcl6– CXCR5–) fraction of conventional Foxp3– KRN T cells. Data are representative of 3–4 independent experiments (n = 4–12 mice). **P < 0.01 by the Students t-test. N.S. = not significant.
A2aR-mediated protection from autoimmune arthritis is CD4 T cell-intrinsic
A2aRs are expressed on CD4 T cells following TCR ligation; however, A2aRs can also be expressed on other cells of hematopoietic origin (6, 24, 31). Therefore, it was important to determine whether the effects of CGS on GC-Tfh cell differentiation and arthritis susceptibility were the result of direct A2aR engagement on KRN T cells or instead an indirect effect of A2aR activation on some other cell type. To test this, we generated KRN transgenic CD4-Cre Adora2af/f conditional knock-out (KO) mice lacking A2aRs only on their T cells. Adora2a-KO and wild type KRN T cells were then transferred into Tcra–/– F1 recipients that lacked their own T cells, but appropriately expressed A2aRs on other cell types (see protocol Fig. 1b). In the absence of KRN T cell-specific A2aRs, CGS failed to inhibit T cell Bcl6 and CXCR5 expression, and the frequency of Tfh and GC-Tfh cells in Tcra–/– F1 mice was unperturbed (Fig. 5a, b). Likewise, CGS treatment failed to promote the expansion of non-Tfh cells in Tcra–/– hosts when responder KRN T cells were A2aR-deficient. A2aR-mediated inhibitions of autoreactive IgG1 isotype class-switched plasmablast differentiation and anti-GPI IgG1 autoantibody production in Tcra–/– F1 mice were also lost when A2aRs were absent from the responding KRN T cells (Fig. 5c, d and data not shown). Consistent with all of this, CGS treatment failed to protect Tcra–/– F1 recipients of Adora2a-KO KRN T cells from severe autoimmune arthritis development (Fig. 5e).
Figure 5. The CGS-mediated block in GC-Tfh differentiation and protection against arthritis are T cell-intrinsic.
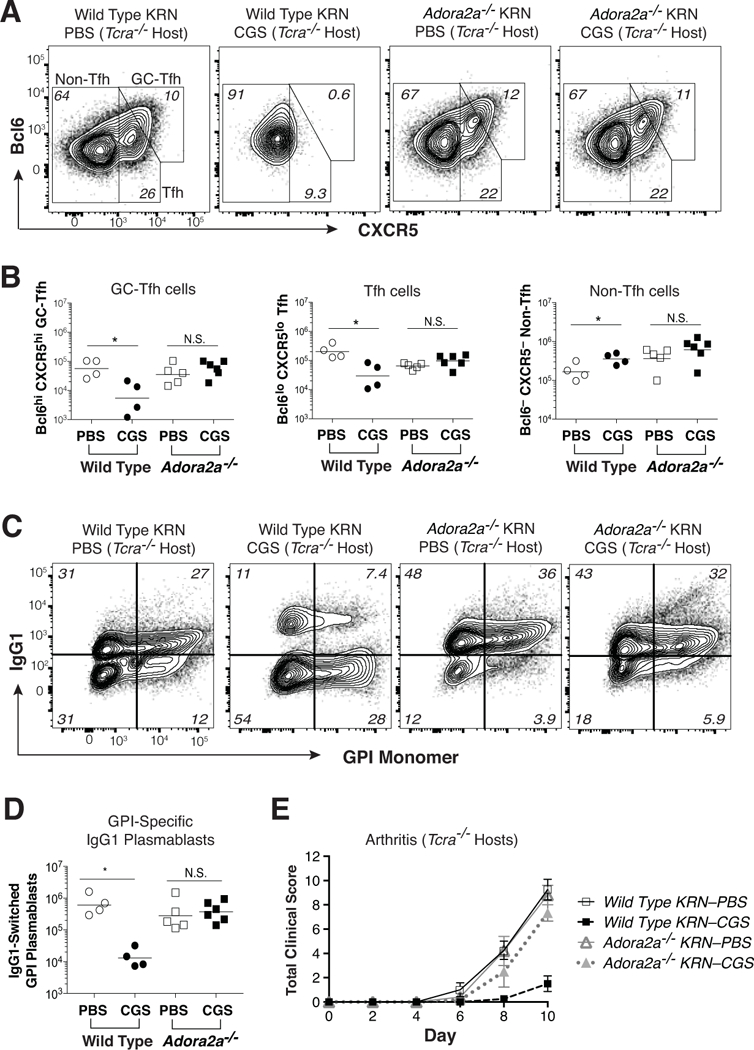
KRN CD4 T cells (either wild type or Adora2a–/–) were adoptively transferred into Tcra–/– F1 hosts, and then mice were injected twice daily with CGS or PBS for the duration of the experiment (10 d). (A) Representative Bcl6 and CXCR5 expression patterns for conventional Foxp3– KRN T cells. (B) Absolute numbers of GC-Tfh (Bcl6hi CXCR5hi), Tfh (Bcl6lo CXCR5lo), and non-Tfh (Bcl-6– CXCR5–) conventional Foxp3– KRN T cells. (C, D) Frequency (C) and number (D) of host polyclonal B220dim intracellular Ig (H+L)hi GL7– IgG1+ GPI-specific plasmablasts in the same secondary lymphoid organs. (E) Mean total clinical disease activity scores (±SEM) over time. Data are representative of 3–4 independent experiments (n = 4–12 mice). *P < 0.05 by Students t-test. N.S. = not significant.
CGS therapy blocks ongoing CD4 T cell-mediated autoimmune arthritis disease progression
The abrogation of dangerous CD4 GC-Tfh cell differentiation using T cell-intrinsic strong A2aR signaling has potential therapeutic application. Nevertheless, the most efficacious strategies for the treatment of autoimmune arthritis will require an ability to arrest clinical activity even after disease onset. Therefore, CGS was examined for its ability to interrupt KRN GC-Tfh differentiation and survival as well as inhibit disease progression when administered only after the onset of arthritis. Wild type naïve KRN CD4 T cells were adoptively transferred into Tcra–/– F1 hosts to induce arthritis, and 8 days later CGS (or PBS alone) administrations were initiated (see protocol Fig. 1c). Mice were again monitored for signs of clinical disease activity, KRN T effector/memory cell differentiation, and polyclonal class-switched IgG1+ GPI-specific plasmablast differentiation within the spleen and LN during the period between days 8 and 12. In PBS-treated control mice, clinical signs of arthritis worsened over time as expected (Fig. 6a, b, and Supplemental Fig. 3). However, activation of A2aRs significantly reduced the clinical disease activity score and joint swelling after just 4 twice-daily CGS treatments (day 10) and these remained low after 8 treatments (day 12). Consistent with these arthritis results, activation of A2aRs beginning on day 8 after KRN T cell adoptive transfer led to a significant reduction in the frequency and number of Bcl6hi CXCR5hi GC-Tfh cells by day 10, as compared to PBS-treated control animals (Fig. 6c, d). GC-Tfh cell numbers remained low following CGS treatment through day 12. On the other hand, Bcl6lo CXCR5lo Tfh cell percentages and numbers at these late time points were unchanged by CGS exposure (Fig. 6c, e). In contrast to the GC-Tfh and Tfh cells, CGS treatment resulted in a small yet significant increase in the KRN non-Tfh cell percentages and numbers (Fig. 6c, f). As before, no alterations in KRN Foxp3+ Treg, Th1, or Th17 cell lineage differentiation events were observed in the presence of CGS (data not shown). Furthermore, the delay of CGS administration until day 8 resulted in a loss of its capacity to interfere with IgG1+ GPI-specific plasmablast differentiation (Fig. 6G). Nevertheless, this late CGS treatment regimen did blunt the rise in anti-GPI IgG1 antibody titer measured at day 12 (Fig. 6H). Note that unlike this KRN CD4 T cell-mediated autoimmune arthritis model described here, K/BxN serum transfer arthritis with its anti-GPI IgG1 antibody-mediated inflammation proved to be completely resistant to treatment with CGS (23, 32, 33) (Supplemental Fig. 4).
Figure 6. Treatment with CGS arrests GC-Tfh differentiation and blocks the progression of autoimmune arthritis.
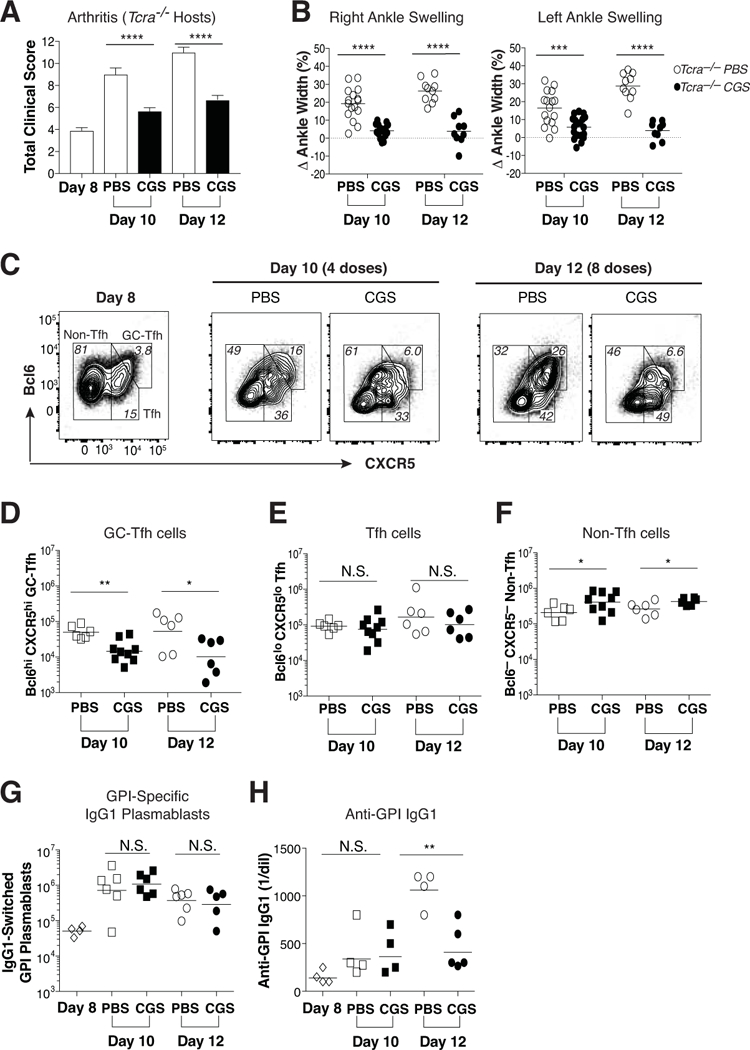
Wild type KRN CD4 T cells were adoptively transferred into Tcra–/– F1 hosts. Beginning on day 8, arthritic mice were injected twice daily with CGS or PBS alone. (A) Mean total clinical disease activity scores (±SEM) observed on days 8, 10, and 12. (B) Percent change in right and left ankle swelling/size from the day 8 measurements. Data are representative of at least three independent experiments (n = 9–16). ***P < 0.001 by Mann Whitney U test and Students t-test, respectively. (C) Representative Bcl6 and CXCR5 expression patterns for conventional Foxp3– KRN T cells at days 8, 10, and 12. (D-F) Absolute numbers of GC-Tfh (Bcl6hi CXCR5hi) (D), Tfh (Bcl6lo CXCR5lo) (E), and non-Tfh (Bcl6– CXCR5–) (F) phenotypes among the conventional Foxp3– KRN T cells observed on days 10 and 12. (G, H) Host polyclonal B220dim intracellular Ig (H+L)hi GL7– IgG1+ GPI-specific plasmablasts (G) and serum anti-GPI IgG1 antibody titers (H) measured at days 8, 10, and 12. These data are representative of at least three independent experiments (n = 4–9). *P < 0.05 and **P < 0.01 by Students t-test. N.S. = not significant.
DISCUSSION
Extracellular adenosine and its receptors have previously demonstrated an ability to suppress animal models of inflammatory arthritis (2). Nevertheless, the mechanisms responsible for their ameliorating effects have remained obscure. A2aR activation can in some circumstances limit the secretion of pro-inflammatory cytokines and chemokines, including the production of IL-2 by T cells (5, 6). A2aR agonists have also been reported to promote anergy induction and divert T effector cell differentiation toward a Foxp3+ Treg cell lineage (3). In this study, we have confirmed that the A2aR agonist CGS blocks the development of autoimmune arthritis, but only when the GPI-specific CD4 T cells express their own A2aRs. While this requirement for A2aRs on the KRN T cells does not exclude the possibility of important CGS actions on additional immune cell types, the result is consistent with the key role that autoreactive CD4 T cells play in arthritis development and suggests that therapies designed to increase A2aR downstream signaling specifically within CD4 T cells can be effective.
Following the adoptive transfer of normal naïve KRN T cells to Tcra–/– F1 hosts, arthritis development proved tightly associated with the expansion and differentiation of IL-21–producing GPI-specific Tfh and GC-Tfh cells, as previously reported (26). Consistent with a role for IL-21–producing KRN Tfh and GC-Tfh cells in GPI/I-Ag7–directed T-B cell collaboration in this model of autoimmune arthritis, polyclonal GPI-specific IgG1 isotype class-switched plasmablasts as well as serum anti-GPI IgG1 (and IgG3) were increased over the course of the arthritis response. Importantly, strong activation of A2aRs on the KRN CD4 T cells using CGS resulted in a selective block of both the Tfh and GC-Tfh cell differentiation and the accompanying T-dependent B cell responses. This inhibition of CXCR5- and Bcl6-expressing Tfh and GC-Tfh cells led to fewer KRN T cells capable of producing IL-21, although the secretion of IL-2, TNFα, IFN-γ, and IL-17a, and the differentiation of Foxp3+ Treg cells were unchanged in the population. Therefore, blunted expression of the Bcl6 gene in KRN T cells in the setting of strong A2aR downstream activation and resultant reductions in CXCR5 and Il21 gene expression represent a plausible mechanism for the inhibition of autoimmune arthritis by CGS. Note that A2aR activation did not appear to influence the strength of the CD4 T cell clonal expansion or the general survival of KRN CD4 T cells, as the subpopulations of KRN non-Tfh cells remained stable or increased.
We noted that neither the blocking of A2aR signaling with a selective A2aR antagonist, nor the use of Adora2a-deficient KRN CD4 T cells for adoptive transfer reliably enhanced Tfh or GC-Tfh cell differentiation in our experimental model system or provoked the development of arthritis in wild type F1 hosts, even though both were sufficient to interfere with the effects of CGS (data not shown). It is possible that compensatory mechanisms within autoreactive CD4 T cells make up for the loss of A2aR signaling, as adenosine 2b receptors can be expressed on some lymphocytes, are homologous to A2aRs, and share the same cAMP/PKA signaling pathways (6, 31, 34, 35). Therefore, additional studies will be necessary to examine the relationship between endogenous extracellular adenosine and A2aRs to reveal their physiological roles in the regulation of autoreactive Tfh/GC-Tfh cell differentiation and arthritis development.
The therapeutic benefits of increased adenosine receptor signaling for autoimmune disorders have previously been suggested in patients treated with methotrexate and sulfasalazine, two important disease-modifying anti-rheumatic drugs commonly prescribed to individuals suffering from systemic rheumatologic diseases like RA (7, 36). The anti-inflammatory effects of methotrexate and sulfasalazine have been attributed to augmented levels of extracellular adenosine signaling (7, 36). A recent study found that a combination therapy consisting of low-dose methotrexate and astilbin, a flavonoid compound that upregulates A2aR expression, significantly alleviates collagen-induced arthritis (36). Simultaneous treatment with the A2aR-specific antagonist, ZM241385, blocked the therapeutic benefits of this combination suggesting that their synergistic efficacy was due to A2aR signaling (36). Therefore, identifying those molecular events downstream of A2aR signaling in CD4 T cells that interrupt Bcl6 gene expression and GC-Tfh cell differentiation may offer a unique opportunity to develop more targeted and effective therapies for individuals suffering from autoimmune disorders.
Supplementary Material
Acknowledgments
Funding: National Institutes of Health (NIH) Grant P01AI035296 (to D.L.M); NIH Institutional Pre-Doctoral Training Grant T32AI00731 (to S.E.S.); Lupus Link of Minnesota (to D.L.M.)
Footnotes
Competing Interests: The authors have declared that no conflict of interest exists.
REFERENCES
- 1.Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7(9):759–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol. 2016;16(3):177–92. [DOI] [PubMed] [Google Scholar]
- 3.Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J, et al. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood. 2008;111(1):251–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zarek PE, Powell JD. Adenosine and anergy. Autoimmunity. 2007;40(6):425–32. [DOI] [PubMed] [Google Scholar]
- 5.Naganuma M, Wiznerowicz EB, Lappas CM, Linden J, Worthington MT, Ernst PB. Cutting edge: Critical role for A2A adenosine receptors in the T cell-mediated regulation of colitis. J Immunol. 2006;177(5):2765–9. [DOI] [PubMed] [Google Scholar]
- 6.Streitova D, Sefc L, Savvulidi F, Pospisil M, Hola J, Hofer M. Adenosine A(1), A(2a), A(2b), and A(3) receptors in hematopoiesis. 1. Expression of receptor mRNA in four mouse hematopoietic precursor cells. Physiol Res. 2010;59(1):133–7. [DOI] [PubMed] [Google Scholar]
- 7.Morabito L, Montesinos MC, Schreibman DM, Balter L, Thompson LF, Resta R, et al. Methotrexate and sulfasalazine promote adenosine release by a mechanism that requires ecto-5’-nucleotidase-mediated conversion of adenine nucleotides. J Clin Invest. 1998;101(2):295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kavanaugh A, Mease PJ, Gomez-Reino JJ, Adebajo AO, Wollenhaupt J, Gladman DD, et al. Treatment of psoriatic arthritis in a phase 3 randomised, placebo-controlled trial with apremilast, an oral phosphodiesterase 4 inhibitor. Ann Rheum Dis. 2014;73(6):1020–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmiel SE, Yang JA, Jenkins MK, Mueller DL. Cutting Edge: Adenosine A2a Receptor Signals Inhibit Germinal Center T Follicular Helper Cell Differentiation during the Primary Response to Vaccination. J Immunol. 2017;198(2):623–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abbott RK, Silva M, Labuda J, Thayer M, Cain DW, Philbrook P, et al. The GS Protein-coupled A2a Adenosine Receptor Controls T Cell Help in the Germinal Center. J Biol Chem. 2017;292(4):1211–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schroder AE, Greiner A, Seyfert C, Berek C. Differentiation of B cells in the nonlymphoid tissue of the synovial membrane of patients with rheumatoid arthritis. Proc Natl Acad Sci U S A. 1996;93(1):221–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim HJ, Krenn V, Steinhauser G, Berek C. Plasma cell development in synovial germinal centers in patients with rheumatoid and reactive arthritis. J Immunol. 1999;162(5):3053–62. [PubMed] [Google Scholar]
- 13.Amara K, Steen J, Murray F, Morbach H, Fernandez-Rodriguez BM, Joshua V, et al. Monoclonal IgG antibodies generated from joint-derived B cells of RA patients have a strong bias toward citrullinated autoantigen recognition. J Exp Med. 2013;210(3):445–55. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Ma J, Zhu C, Ma B, Tian J, Baidoo SE, Mao C, et al. Increased frequency of circulating follicular helper T cells in patients with rheumatoid arthritis. Clin Dev Immunol. 2012;2012:827480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Shan Y, Jiang Z, Feng J, Li C, Ma L, et al. High frequencies of activated B cells and T follicular helper cells are correlated with disease activity in patients with new-onset rheumatoid arthritis. Clin Exp Immunol. 2013;174(2):212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simpson N, Gatenby PA, Wilson A, Malik S, Fulcher DA, Tangye SG, et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum. 2010;62(1):234–44. [DOI] [PubMed] [Google Scholar]
- 17.Chakera A, Bennett SC, Morteau O, Bowness P, Luqmani RA, Cornall RJ. The phenotype of circulating follicular-helper T cells in patients with rheumatoid arthritis defines CD200 as a potential therapeutic target. Clin Dev Immunol. 2012;2012:948218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X, Nurieva RI, Dong C. Transcriptional regulation of follicular T-helper (Tfh) cells. Immunol Rev. 2013;252(1):139–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hatzi K, Nance JP, Kroenke MA, Bothwell M, Haddad EK, Melnick A, et al. BCL6 orchestrates Tfh cell differentiation via multiple distinct mechanisms. J Exp Med. 2015;212(4):539–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ditzel HJ. The K/BxN mouse: a model of human inflammatory arthritis. Trends Mol Med. 2004;10(1):40–5. [DOI] [PubMed] [Google Scholar]
- 21.Chevalier N, Macia L, Tan JK, Mason LJ, Robert R, Thorburn AN, et al. The Role of Follicular Helper T Cell Molecules and Environmental Influences in Autoantibody Production and Progression to Inflammatory Arthritis in Mice. Arthritis Rheumatol. 2016;68(4):1026–38. [DOI] [PubMed] [Google Scholar]
- 22.Martinez RJ, Zhang N, Thomas SR, Nandiwada SL, Jenkins MK, Binstadt BA, et al. Arthritogenic self-reactive CD4+ T cells acquire an FR4hiCD73hi anergic state in the presence of Foxp3+ regulatory T cells. J Immunol. 2012;188(1):170–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsumoto I, Staub A, Benoist C, Mathis D. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science. 1999;286(5445):1732–5. [DOI] [PubMed] [Google Scholar]
- 24.Cekic C, Sag D, Day YJ, Linden J. Extracellular adenosine regulates naive T cell development and peripheral maintenance. J Exp Med. 2013;210(12):2693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalekar LA, Schmiel SE, Nandiwada SL, Lam WY, Barsness LO, Zhang N, et al. CD4(+) T cell anergy prevents autoimmunity and generates regulatory T cell precursors. Nat Immunol. 2016;17(3):304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Block KE, Huang H. The cellular source and target of IL-21 in K/BxN autoimmune arthritis. J Immunol. 2013;191(6):2948–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ozaki K, Spolski R, Feng CG, Qi CF, Cheng J, Sher A, et al. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298(5598):1630–4. [DOI] [PubMed] [Google Scholar]
- 28.Ise W, Inoue T, McLachlan JB, Kometani K, Kubo M, Okada T, et al. Memory B cells contribute to rapid Bcl6 expression by memory follicular helper T cells. Proc Natl Acad Sci U S A. 2014;111(32):11792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kitano M, Moriyama S, Ando Y, Hikida M, Mori Y, Kurosaki T, et al. Bcl6 protein expression shapes pre-germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity. 2011;34(6):961–72. [DOI] [PubMed] [Google Scholar]
- 30.Iyer SS, Latner DR, Zilliox MJ, McCausland M, Akondy RS, Penaloza-Macmaster P, et al. Identification of novel markers for mouse CD4(+) T follicular helper cells. Eur J Immunol. 2013;43(12):3219–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koshiba M, Rosin DL, Hayashi N, Linden J, Sitkovsky MV. Patterns of A2A extracellular adenosine receptor expression in different functional subsets of human peripheral T cells. Flow cytometry studies with anti-A2A receptor monoclonal antibodies. Mol Pharmacol. 1999;55(3):614–24. [PubMed] [Google Scholar]
- 32.Craft JE. Follicular helper T cells in immunity and systemic autoimmunity. Nat Rev Rheumatol. 2012;8(6):337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mak A, Kow NY. The pathology of T cells in systemic lupus erythematosus. J Immunol Res. 2014;2014:419029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2(8):599–609. [DOI] [PubMed] [Google Scholar]
- 35.Wen AY, Sakamoto KM, Miller LS. The role of the transcription factor CREB in immune function. J Immunol. 2010;185(11):6413–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma Y, Gao Z, Xu F, Liu L, Luo Q, Shen Y, et al. A novel combination of astilbin and low-dose methotrexate respectively targeting A2AAR and its ligand adenosine for the treatment of collagen-induced arthritis. Biochem Pharmacol. 2018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.