

SUMMARY
Gram-negative bacteria are intrinsically resistant to drugs, due to their double-membrane envelope structure that acts as a permeability barrier and as an anchor for efflux pumps. Antibiotics are blocked and expelled from cells, and cannot reach high enough intracellular concentrations to exert a therapeutic effect. Efforts to target one membrane protein at a time have been ineffective. Here, we show that m1G37-tRNA methylation determines the synthesis of a multitude of membrane proteins via its control of translation at proline codons near the start of open-reading frames. Decreases in m1G37 levels in Escherichia coli and Salmonella impair membrane structure and sensitize these bacteria to multiple classes of antibiotics, rendering them unable to develop resistance or persistence. Codon engineering of membrane-associated genes reduces their translational dependence on m1G37 and confers resistance. These findings highlight the potential of tRNA methylation in codon-specific translation to control the development of multi-drug resistance in Gram-negative bacteria.
Keywords: membrane barrier, drug efflux, m1G37-tRNA, TrmD, proline codons, resistance, persistence, tRNAPro
Blurb
Bacterial multi-drug resistance is driven by the membrane barrier and efflux activity that bars and expels antibiotics. The TrmD-synthesized m1G37 methylation of tRNA is a global determinant of biosynthesis of membrane proteins in bacteria. Depletion of TrmD, and thus m1G37-tRNA, impairs the membrane barrier and efflux activity, sensitizes bacteria to antibiotic killing, and suppresses their development of resistance and persistence upon antibiotic exposure.
Graphical Abstract
Multi-drug resistance of Gram-negative bacteria is a critical and expanding medical challenge. In many cases, antibiotics are blocked from entry and expelled from cells, and hence cannot reach high enough intracellular concentrations to exert a therapeutic effect. This problem is due in large part to the double-membrane structure of the cell envelope of Gram-negative bacteria, which acts both as a permeability barrier and as a platform for efflux machineries that export drugs (Payne et al., 2007; Silver, 2011). In previous efforts focusing on targeting one membrane protein or one efflux pump at a time, resistance mutations were quick to develop (Murakami et al., 2006). Such mutations are selected upon the antibiotic challenge during therapy, giving rise to a resistant population (Silver, 2011, 2012). Inhibition of a process that simultaneously controls the expression of multiple membrane-associated genes would be a more powerful strategy for enhancing antibiotic efficacy. Such a global mechanism, which has not yet been identified, could provide a new anti-bacterial strategy to enable multiple drugs to take action, render resistance less likely, and accelerate bactericidal action.
The cell envelope of Gram-negative bacteria consists of a plasma inner membrane (IM), a cell wall, and an outer membrane (OM). The IM is a fluid lipid bilayer, while the cell wall is a rigid and cross-linked matrix of peptidoglycan that endows the cell with mechanical strength (Holtje, 1998). The OM is made up of phospholipids in the inner leaflet and lipopolysaccharides in the outer leaflet, forming an asymmetric bilayer that prevents compounds from diffusing into the periplasm or cytosol, and also expels compounds to the external medium through membrane-bound efflux transporters (Nikaido, 1998). We recently showed that, in addition to its barrier function, the OM of Escherichia coli confers mechanical stiffness to the cell on par with the cell wall (Rojas et al., 2018), indicating that robust OM biogenesis is important for cellular mechanical integrity. The biogenesis of both IM and OM requires extensive integration with protein components, which also regulate cell-wall synthesis (Typas et al., 2011). Thus, the production of membrane proteins determines the quality of the entire Gram-negative cell envelope; it is essential for establishing a permeability barrier and efflux activity against drugs and for defining cell shape and stability during cell growth.
One mechanism for global coordination of protein biosynthesis is via codon-specific translation, which directly impacts the speed and quality of translation at specific codons and has the ability to reprogram gene expression for disease development and drug resistance (Rapino et al., 2018). This regulation is distinct from transcriptional regulation via promoters or translational regulation via ribosome-binding sites. Mechanistically, codon-specific translation is mediated by post-transcriptional modifications of the tRNA anticodon or adjacent nucleotides. For membrane-associated genes, the translation of proline (Pro) codons (CCN) is critical, because Pro is the unique amino acid that is required for the creation of kinks in polypeptides and for the structure and activity of trans-membrane domains (Schmidt et al., 2016). We previously showed that the translation of Pro codons, particularly CC[C/U] codons, requires the conserved N1 - methylation of G37 on the 3’-side of the tRNA anticodon (Gamper et al., 2015a, b). Without m1G37, tRNA is highly prone to stalling and +1 frameshifting (Gamper et al., 2015a, b), which are errors that disrupt the reading frame and prematurely terminate protein synthesis. The synthesis of m1G37 in bacteria is by the conserved tRNA methyl transferase TrmD, using S-adenosyl-methionine as the methyl donor (Hou et al., 2017) (Figure 1A, B). Depletion of TrmD, and consequently m1G37-tRNA, accumulates ribosomal frameshifts and leads to cell death (Gamper et al., 2015a). We found that CC[C/U] codons are prevalent in Gram-negative membrane-associated genes (Figure 1C), raising the possibility that the m1G37 methylation of tRNA by TrmD can provide a general mechanism to control the biosynthesis of membrane proteins.
Figure 1: m1G37-tRNA is important for expression of membrane-associated genes.

A) TrmD (PDB: 1UAK) synthesizes m1G37-tRNA.
B) Translation of CCC codon requires m1G37-tRNAPro to suppress +1 frameshifts at the P-site.
C) Gram-negative genes for membrane-associated proteins often contain CC[C/U] codons (red) near the start of the ORF. Five examples from E. coli (Ec) and Salmonella enterica (Se) are shown.
D) Western blots of trmD-KD cells showed that human Trm5 is unstable upon removing the inducer Ara. Overnight cultures with 0.2% Ara were diluted 1:100 into fresh LB in Ara+/− conditions. Cells were sampled over time and levels of Trm5 and CysRS were determined using antibodies.
E) Expression of trm5 is required for viability of trmD-KD cells. Overnight cultures in LB with 0.2% Ara were maintained in a viable state by expression of the plasmid-borne PBAD- controlled human trm5. Cells were serially diluted and spotted on LB plates with or without 0.2% Ara. Growth was assayed after overnight incubation at 37 °C.
F) Primer extension analysis of of m1G37 in tRNAPro/UGG. Cells were prepared as in (D), diluted after 5 h to OD600 = 0.1 in fresh LB (Ara+/−), incubated for another 2 h at 37 °C, and total small RNA was purified. (Top) Primer extension was blocked at m1G37 in cells grown with Ara+ (+), whereas the primer read through to nucleotide C1 in cells grown without Ara (Ara−). (Bottom) m1G37 levels are shown as mean ± standard error of the mean (SEM), n = 3. Welch’s t-test: **p < 0.05, ***p < 0.01.
G) Mass spectrometry analysis of m1G37 levels in tRNAPro/UGG. Cells were prepared as in (D) and the tRNA was isolated by affinity purification. m1G37 levels are shown as mean ± SEM, n = 3. The fraction of m1G among total Gs in the tRNA was 0.055, representing ~100% methylation as compared to the theoretical value (one m1G among 25 Gs = 0.04, Figure S3B). Welch’s t-test: **p < 0.05, ***p < 0.01.
See also Figures S1 and S2.
Here, we demonstrate that TrmD is a global determinant of membrane biosynthesis in E. coli and Salmonella enterica serovar Typhimurium (hereafter Salmonella), two major Gram-negative pathogens. We show that m1G37 deficiency caused by TrmD depletion disrupts the OM structure and rigidity, sensitizes E. coli and Salmonella to various classes of antibiotics, and suppresses their development of resistance or persistence upon antibiotic exposure. Engineering of the CC[C/U] codon to the less vulnerable CCG codon in membrane-associated genes reduces the translational dependence on m1G37 and confers drug resistance to bacteria. We also show that the conservation of m1G37 is required for codon-specific translation of CC[C/U], and that the methylation cannot be substituted by any other nucleotides. These results demonstrate that by simultaneously affecting codon-specific translation of Pro in entire classes of genes encoding membrane-associated proteins, TrmD-mediated methylation of tRNA is a major determinant of multi-drug resistance in Gram-negative bacteria.
RESULTS
m1G37-deficient E. coli and Salmonella have lower levels of membrane proteins
We previously showed that m1G37 has the strongest effect on codon-specific translation of CC[C/U] at the 2nd codon position of an open reading frame, and that this effect gradually decreases over the next 15 codons (Gamper et al., 2015a). In an analysis of the E. coli MG1655 genome, we found that the occurrence of CC[C/U] at the 2nd codon position is 2-fold higher for genes encoding membrane-associated proteins relative to non-membrane-associated proteins (1.8% vs. 0.8%, n = 4,289, p < 0.05, Fisher’s exact test with Bonferroni correction) (Hou et al., 2017). This enrichment was also observed when considering both the 2nd and 3rd codon positions (3.7% vs. 1.5%, n = 4,289, p < 0.0005, Fisher’s exact test with Bonferroni correction). The over-representation of CC[C/U] is also evident in the genome of Salmonella LT2 (Hou et al., 2017). Among genes with CC[C/U] at the 2nd codon position, 31% and 26% encode membrane-associated proteins in E. coli and Salmonella, respectively (Figures 1C and S1). The high prevalence of Pro near the N-terminus of membrane proteins is consistent with its role in creating turns of transmembrane domains that cross a lipid bilayer (Yohannan et al., 2004).
To determine how m1G37 controls codon-specific translation of membrane-associated genes, we created trmD-KD (knockdown) strains of E. coli and Salmonella. Since trmD is essential for cell viability (Gamper et al., 2015a) and cannot be deleted, we created each trmD-KD strain by deleting the chromosomal trmD (Figure S2A–B) while expressing the human counterpart trm5 from a plasmid with an arabinose (Ara)-inducible promoter. We previously showed that Trm5 is capable of supplying m1G37-tRNA to support bacterial viability (Christian et al., 2004), but that it is unstable in bacteria and can be removed rapidly (Christian et al., 2013). In the E. coli and Salmonella trmD-KD strains, the level of human Trm5 upon Ara induction increased with time and reached a steady state in 1–2 h, but decreased rapidly within 30 min upon Ara removal (Figure 1D). Cells with Trm5-produced m1G37 formed colonies up to a 104-fold dilution, whereas m1G37-deficient cells were not viable even without dilution (Figure 1E). To determine intracellular m1G37 levels, cells were grown with 0.2% Ara to saturation and diluted 1:100 into fresh Luria broth (LB) with or without Ara for 4 h, followed by another dilution to OD600 = 0.1 in fresh LB with or without Ara and grown for 3 h. These serial passages were necessary to deplete cells of pre-existing m1G37-tRNA (Figure S3A). Primer extension analysis validated that the UGG isoacceptor of tRNAPro in trmD-KD cells contained m1G37 at 70% and 12% in cultures with and without Ara (Figure 1F). This pattern was preserved for the GGG isoacceptor (Figure S3B,C) and was consistent with quantitative mass spectrometry analyses of the UGG isoacceptor (Figure 1G).
To determine the effect of m1G37 deficiency on the biosynthesis of membrane proteins, we used quantitative proteomics to measure protein levels in the membrane fraction of E. coli trmD-KD cells grown with or without Ara. A total of 226 membrane proteins, 47 of which were associated with the OM, were analyzed by label-free quantification to determine fold-changes between Ara+ and Ara− conditions. While non-OM proteins were on average up-regulated in the absence of Ara by 16% (median increase of 20.22 = 1.16), OM proteins were on average down-regulated by 21% (median decrease of 2−0.33 = 0.79) (Figure 2A). Of interest were LolB and OmpA, responsible for stable anchoring of drug-efflux pumps to the OM (Hayashi et al., 2014; Tsukahara et al., 2009) and for anchoring the OM to the peptidoglycan cell wall, respectively. lolB and ompA are enriched with Pro codons relative to the average codon usage in E. coli protein-coding genes (Figure 2B, lolB: CCN (6.7% vs. 4.3%) and CC[C/U] (2.4 vs. 1.1%) and ompA CCN (5.5 vs. 4.3%)). This enrichment is specific, because their usage of Leu codons (CUN), which also require m1G37 for translation, is typical (Figure S3D). The enrichment of Pro codons in lolB and ompA supports the notion that their decrease in protein levels is correlated with the poor translation of Pro codons in m1G37-deficient cells. Western blot analysis showed that the amount of LolB relative to the cytosolic cysteinyl-tRNA synthetase CysRS (Hou et al., 1991; Lipman and Hou, 1998) in m1G37-deficient cells decreased to 26% in E. coli and to 56% in Salmonella (Figures 2C and S3E), while relative mRNA levels were unaffected (Figure S3F), indicating that the reduction in protein levels was due to reduced translation. These data are consistent with the notion that translation of lolB involves a TrmD-dependent codon at the 2nd and 4th positions of the E. coli gene and at the 4th position of the Salmonella gene (Figure 1A), whereas translation of cysS (for CysRS) involves no such codons in the first 16 positions. Western blot analysis also showed that the amount of OmpA relative to CysRS decreased to 72% in m1G37-deficient E. coli cells (Figure 2D), providing additional support for the notion that translation of membrane-associated genes that are enriched with Pro codons is sensitive to loss of m1G37.
Figure 2: m1G37 deficiency in E. coli and Salmonella affects cell viability.

A) Quantitative mass spectrometry analysis of membrane proteins in E. coli trmD-KD cells isolated from Ara− and Ara+ conditions. The label-free quantification intensity is compared to the signal of log2 (fold-change) (Ara−/Ara+). OM proteins are plotted in black with a vertical line indicating the median of −0.33 (equivalent to a decrease of 21%), while non-OM proteins are plotted in blue with a vertical line showing the median of 0.22 (equivalent to an increase of 16%). p < 0.001 by a Kolmogorov-Smirnov analysis.
B) Frequency of Pro codons CCN (top) and CC[C/U] (bottom) in genes whose OM proteins are reduced in Ara− vs. Ara+ in (A). Each frequency is compared to the average frequency of respective Pro codons in E. coli protein-coding genes.
C,D) m1G37 deficiency decreased LolB levels (C) to 26% in E. coli and to 56% in Salmonella, and decreased OmpA levels in E. coli to 72% (D) in Western blots (top). Overnight cultures of trmD-KD cells were diluted 1:100 into fresh LB with or without 0.2% Ara and grown for 4 h at 37 °C. Cells were inoculat ed into fresh LB in Ara+/− conditions for another 3 h. Data and error bars represent mean ± SD, n = 4.
See also Figure S3.
m1G37 deficiency causes membrane damage and reduces OM stiffness
We hypothesized that the reduced biosynthesis of membrane proteins in m1G37-deficient cells would damage membrane structural integrity. We observed increased intracellular accumulation in m1G37-deficient bacteria of both the redox sensor AlamarBlue, which becomes fluorescent inside cells, and the DNA fluorescent stain Hoechst 33342, indicating increased membrane permeability (Figures 3A, B, S4A). The accumulation of each dye was measured during exponential growth, and dye exposure was initiated in the presence of carbonyl cyanide m-chlorophenyl hydrazine (CCCP) to inactivate membrane efflux. To validate that AlamarBlue fluorescence reflected the permeability of the OM, we treated E. coli and Salmonella m1G37+ cells with sublethal doses of polymyxin B, which binds to lipopolysaccharide in the OM and permeabilizes the double-membrane envelope. We showed that intracellular AlamarBlue fluorescence increased as a function of polymyxin B dose (Figure S4B), and that the maximum increase (4- to 5-fold) at a lethal dose of polymyxin B was in the same range as the observed increases in m1G37-deficient cells relative to m1G37+ cells (2- to 3-fold, Figure 3A, B). We further showed that the intracellular AlamarBlue increase due to m1G37 deficiency was similar to the increase in E. coli cells expressing a defective OM pore protein relative to the control (Figure S4C). This defective pore protein was created by mutations in the siderophore transporter protein FhuA to enlarge the pore size, rendering the OM hyperpermeable to a wide range of compounds without affecting efflux (Krishnamoorthy et al., 2016).
Figure 3: m1G37 deficiency weakens the cell envelope.

A,B) E. coli (A) and Salmonella (B) trmD-KD cells in m1G37 deficiency (m1G37−) show increased membrane permeability relative to m1G37+ cells. Cells were grown as in Figure 2C and the intracellular accumulation of AlamarBlue in m1G37+ (Ara+, blue) and m1G37-deficient (Ara−, red) conditions was monitored in the presence of CCCP. Levels of intracellular dye accumulation were normalized by OD600. Data and error bars are mean ± SD, n = 3.
C-E) E. coli (C) and Salmonella (D) trmD-KD cells showed reduced Nile Red efflux in m1G37− vs. m1G37+ conditions. Cells pre-loaded with Nile Red were de-energized with CCCP for 100 s, followed by addition of 50 mM glucose (Glc) to activate efflux, and the time course of Nile Red efflux was monitored for 200 s in cells grown in Ara+/− conditions. The time required to efflux 50% pre-loaded Nile Red (tefflux 50%) was longer for m1G37− relative to m1G37+ cells (E). The lack of efflux in m1G37+ cells in the presence of CCCP were negative controls. Data and error bars are mean ± SD, n > 3.
F) Membrane potential was reduced in m1G37− vs. m1G37+ cells as measured by ThT fluorescence. E. coli and Salmonella trmD-KD cells were inoculated in LB from a 1:100 dilution of an overnight culture without or with 0.2% Ara and grown for 4 h at 37 °C, followed by dilution in LB in Ara+/− conditions to OD600 of 0.1 and grown for 3 h at 37 °C. ThT fluorescence was normalized by OD600. Data and error bars are mean ± SD, n > 3.
G) The population-averaged length of the cell envelope during 100-mM oscillatory osmotic shocks was shorter in Ara− (red) than Ara+ (blue) E. coli cells. Data are mean ± SD, n = 3. Inset: Phase-contrast microscopy showed that E. coli trmD-KD cells were smaller in m1G37− (red) relative to m1G37+ (blue) conditions. Scale bars: 2 μm.
H) The fractional extension of the cell envelope was larger in m1G37− relative to m1G37+ cells. The extension was calculated as (l - lav)/l, where l is the effective population-averaged envelope length and lav is the time-averaged value of l using the period of the oscillatory cycles as an averaging window. Data are mean ± SD, n = 3.
I) The amplitude of length oscillations in (H) averaged over oscillatory cycles was larger in m1G37− relative to m1G37+ cells, averaged over oscillatory cycles. Data and error bars are mean ± SD from n > 67 cells. ***: p < 0.0001 by Student’s t-test. In a replicate experiment, the ratio of the amplitude of length oscillations between m1G37+ and m1G37− cells measured after sufficient m1G37 depletion to reduce growth rate to < 0.2 h−1 was 1.43 (n > 609 cells).
See also Figures S4 and S5.
To further validate the significance of the AlamarBlue increase due to m1G37 deficiency, we created proS-KD and cysS-KD strains, in which the essential genes responsible for amino-acid charging of tRNAPro (proS) and tRNACys (cysS), respectively, were deleted from the chromosome and cell viability was maintained by Ara-dependent, plasmid-borne expression of each native gene. The proS-KD strain was a positive control to determine whether the deficiency of Pro-tRNAPro affected translation of Pro codons in a manner similar to the deficiency of m1G37, while the cysS-KD strain was a negative control for how depletion of an essential protein that is unlikely to be involved in OM protein biogenesis would affect membrane permeability. The relative AlamarBlue increase due to proS depletion (2- to 3-fold) was comparable to that due to m1G37 deficiency, whereas the relative change due to cysS depletion was not significant (<1.3-fold, Figure S4C). Together, these data show that m1G37 deficiency increases membrane permeability to the same extent as the deficiency caused by a hyperpermeable pore or by reduced levels of charged tRNA for translation of Pro codons.
m1G37 deficiency also reduced membrane efflux, as indicated by the increased time required to pump out 50% of pre-loaded Nile Red dye (from 36 ± 1 to 66 ± 2 s for E. coli and 32 ± 3 to 45 ± 3 s for Salmonella in m1G37-deficient relative to m1G37+ cells, Figure 3C–E). The extensions of efflux time (1.8- and 1.4-fold for E. coli and Salmonella, respectively) were smaller than that due to deletion of acrB relative to wildtype (> 4-fold) (Figure S5A, B); this smaller effect is expected, because m1G37 deficiency reduces but does not eliminate levels of efflux pumps, whereas acrB deletion (ΔacrB) eliminates a component of the AcrAB-TolC complex, which is the major efflux pump responsible for expelling most antibiotics. The reduction in efflux due to m1G37 deficiency was also observed by monitoring ethidium bromide (Figure S5C, D), which showed an increase in the efflux time as a function of polymyxin B dose (Figure S5E, F). As expected, the extension time required for expelling ethidium bromide was smaller compared with the effect of ΔtolC on the AcrAB-TolC complex (Figure S5C, D). We also used Thioflavin T (ThT) to probe the membrane potential (Prindle et al., 2015) and confirmed that m1G37 deficiency reduced the fluorescence of ThT in E. coli and Salmonella (Figure 3F), further supporting our conclusion that the OM was impaired.
To determine how m1G37 deficiency affected the cell envelope structure, we measured cellular mechanical stiffness using an assay that we recently developed and utilized to demonstrate that the OM makes a surprisingly large contribution to the overall stiffness of the E. coli cell envelope (Rojas et al., 2018). Perturbation of the OM by chemical agents or genetic mutations caused large reductions in stiffness and rendered cells susceptible to lysis under oscillatory osmotic shocks (Rojas et al., 2018). We previously showed that deletion of ompA and lpp and introduction of a mutant allele of lptD each decreased OM stiffness (Rojas et al., 2018). While ompA and lpp encode abundant OM proteins, the mutant lptD allele encodes a variant of the lipopolysaccharide assembly machinery that is known to increase the OM permeability to antibiotics (Ruiz et al., 2005). We thus hypothesized that the altered OM composition during m1G37 deficiency would decrease the stiffness of the cell envelope.
Our assay involves application of force to the cell envelope by subjecting cells to oscillatory osmotic shocks using a microfluidic device and measurement of the resulting deformations of the cell envelope (Rojas et al., 2014; Rojas et al., 2018). For small shock magnitudes (100 mM sorbitol), the plasma membrane essentially remains in contact with the cell envelope (Rojas et al., 2014), so that the boundary of the cytoplasm detected from phase-contrast images can be used to track the envelope contour. The degree to which the envelope deforms, as defined by the amplitude of the cell-length oscillations in response to oscillatory osmotic shocks, is inversely correlated with envelope stiffness (Rojas et al., 2018). During m1G37 deficiency due to growth without Ara for ~4 h, cells grew more slowly and were smaller than cells grown in the presence of Ara (Figure 3G). The amplitude of response to 100 mM oscillatory osmotic shocks increased substantially in Ara− cells relative to Ara+ cells (n = 2 experiments with 67–713 cells; Figure 3G–I), indicating a decrease in envelope stiffness. This increase in amplitude runs counter to the expectation based on the reduction in cell size alone, whereby the mechanical expansion of a thin shell under load is predicted to be larger for a cell with a larger radius than for a shell of the same material and thickness with a smaller radius. In sum, these data suggest that m1G37 deficiency changes the composition of the cell envelope, resulting in lower load-bearing capacity and higher permeability.
m1G37 deficiency sensitizes Gram-negative bacteria to multiple antibiotics
We hypothesized that m1G37 deficiency would sensitize Gram-negative cells to antibiotics due to compromised permeability and mechanics of the cell envelope. We assessed antibiotics with various mechanisms of action (Silver, 2011), including: the β-lactams ampicillin and carbenicillin, which target cell-wall biosynthesis; the aminoglycosides kanamycin and gentamicin, which inhibit protein synthesis; paromomycin, which reduces fidelity of the 30S ribosomal subunit; the ansamycin polyketide rifampicin, which targets RNA polymerase; and the quinolone ciprofloxacin, which targets DNA gyrase. This diverse collection of antibiotics accesses different mechanisms of membrane permeability and efflux pumps, allowing us to determine the general impact of m1G37 deficiency. We inoculated E. coli and Salmonella at 106 colony-forming units (CFUs)/mL and grew these cells with each antibiotic for 18 h. Defining growth as an increase in cell density above OD600 of 0.15 for the purpose of determining the minimum inhibitory concentration (MIC), we found that m1G37-deficient E. coli and Salmonella showed at least 2-fold lower MICs relative to controls for all antibiotics (Figures 4A, B, S6A, B). In most cases, these reductions were in the same range as those reported previously for ΔtolC cells (Krishnamoorthy et al., 2016), and also in the same range as the reductions in cells treated with a sublethal dosage of polymyxin B (Figure 4A, B). For example, the fold-changes in the MICs of ampicillin and carbenicillin between m1G37+ and m1G37-deficient cells of E. coli (2.0- and 2.7-fold) and Salmonella (2.7- and 3.0-fold) were similar to those between untreated and polymyxin-treated m1G37+ cells (1.5- and 2.0-fold and 0.8- and 1.0-fold, respectively). This similarity held generally for all tested antibiotics, indicating that m1G37 deficiency has similar effects as polymyxin on membrane permeability to antibiotics. To further validate the magnitude of m1G37 effects on antibiotic sensitivity, we showed that the fold-change in MIC of antibiotics during m1G37 deficiency was generally larger than the effect of Δefp (Figure S6C), the gene encoding protein-synthesis elongation factor P, which has a role in antibiotic susceptibility (Navarre et al., 2010). The broad spectrum of antibiotics exhibiting a reduction in MIC in m1G37-deficient cells indicates that multiple membrane proteins were affected, resulting in a generally compromised membrane similar to the damage caused by polymyxin B.
Figure 4: m1G37 deficiency sensitizes trmD-KD cells to multiple antibiotic classes.

A,B) m1G37− cells had at least 2-fold lower MICs than m1G37+ cells. The fold-decrease in MIC of each antibiotic was calculated for E. coli (A) and Salmonella (B) trmD-KD cells as the ratio of the MIC in m1G37+ and m1G37− cells (red) and was compared with the relative decrease of m1G37+ cells upon treatment with polymyxin B (PMB) at 0.25X MIC (blue). Overnight cultures were inoculated into fresh LB at 106 CFUs/mL and incubated with an antibiotic in serial dilutions. After 18 h of incubation at 37 °C, cell densities lower than OD600 = 0.15 were scored as no growth. Fold-changes are taken from Figure S6A, where data and errors are mean ± SD, n > 4. Amp, ampicillin; Cbc, carbenicillin; Rif, rifampicin; Kan, kanamycin; Gen, gentamicin; Par, paromomycin; Cip, ciprofloxacin; Van, vancomycin.
C,D) Time-kill analyses of E. coli (C) and Salmonella (D) trmD-KD cells indicate that m1G37+ cells (blue) recovered from antibiotic exposure, but that m1G37− (red) cells did not. Overnight cultures (106 CFUs/mL) were inoculated into fresh LB with an antibiotic at the indicated concentration and grown at 37 °C. Dat a and error bars show mean ± SD, n > 3.
E,F) Percent survival of m1G37− E. coli trmD-KD cells upon exposure to 25 μg/mL carbenicillin (E) or 256 μg/mL vancomycin (F), showing a decrease in survival comparable to proS-KD cells but faster and to a greater extent compared with cysS-KD cells. Data and error bars show mean ± SD, n > 3.
See also Figure S6.
As an additional probe of membrane structure, we tested vancomycin, a linear hepta-peptide that inhibits cell-wall synthesis (Ruiz et al., 2005). Vancomycin is typically only active against Gram-positive bacteria, although disruption of the OM in Gram-negative bacteria permits its passage and action (Shlaes et al., 1989; Young and Silver, 1991). We observed a 2- to 4-fold reduction in the MIC of vancomycin in m1G37-deficient cells (Figure 4A, B), a 4- to 5-fold reduction in polymyxin-treated m1G37+ cells (Figure 4A, B), and a 2-fold reduction in Δefp cells (Figure S6C). These effects further highlight the damage to the OM in m1G37-deficient cells.
While we could not quantify the full extent of the effect of m1G37 deficiency on antibiotic sensitivity, due to the essentiality of TrmD, we were interested in determining whether the OM damage in m1G37-deficient cells increased intracellular drug concentrations sufficiently to accelerate bactericidal action. By incubating 106 CFUs of cells with increasing concentrations of each antibiotic and measuring CFUs/mL over time within the first 24 h of treatment, we demonstrated that m1G37-deficient cells were killed faster relative to controls. The concentration of each drug that displayed the strongest effect due to m1G37 deficiency was selected for in-depth analysis (Figure 4C, D). The time-kill kinetics of carbenicillin and ampicillin showed that the viability of both m1G37+ and m1G37-deficient cells remained relatively stable within 5–7 h of exposure, after which the viability of m1G37-deficient cells declined while m1G37+ cells regrew. By contrast, the time-kill kinetics of gentamicin and kanamycin showed a 103- to 104-fold decrease in viability immediately upon exposure, after which m1G37-deficient cells remained low in viability up to 24 h while m1G37+ cells recovered. The more robust regrowth of aminoglycoside-treated cells relative to carbenicillin- or ampicillin-treated cells is likely driven by the development of adaptive resistance through aminoglycoside-induced down-regulation of drug uptake and up-regulation of efflux (Mohamed et al., 2012). The presence of m1G37 may confer adaptive resistance by promoting biosynthesis of high-quality pumps. In the time-kill kinetics of vancomycin, m1G37 deficiency immediately decreased cell viability upon exposure, while m1G37+ cells simply increased in number over time.
Our cysS-KD and proS-KD uptake data (Figure S4C) suggest that the reduced viability of m1G37-deficient cells was due to translational defects at Pro codons, and not to the nonspecific loss of an essential gene. Further supporting this conclusion, time-kill kinetics with carbenicillin and vancomycin revealed that m1G37-deficient proS-KD cells were killed faster and to a greater extent than cysS-KD cells (Figure 4E, F). To query whether the reduced cell viability during m1G37 deficiency was due to an unrelated stress response, we determined that m1G37+ and m1G37-deficient cells had virtually identical time-kill kinetics when incubated with 2 mM H2O2 (Figure S6E, F), indicating that the expression of genes in response to oxidative stress, unlike those for biosynthesis of the cell envelope, is not affected by m1G37 deficiency. Thus, m1G37 deficiency has a specific effect on bacterial survival in antibiotic exposure, likely due to the reduced synthesis of membrane proteins.
m1G37-deficient cells exhibit reduced resistance and persistence to antibiotics
We hypothesized that the faster antibiotic killing of m1G37-deficient cells would preempt their ability to develop mutations that confer resistance. We chose a concentration for each drug near 1X MIC for m1G37+ cells and determined the relative frequency of resistance in m1G37-deficient cells. Log-phase cells were grown on plates containing each antibiotic and the frequency of resistance was determined by the number of colonies that appeared after three days of incubation. Consistently across E. coli and Salmonella, analysis of a broad spectrum of antibiotics showed that m1G37-deficient cells produced significantly fewer resistant colonies than m1G37+ cells from an inoculum of 105 CFUs (Figure 5A, B). We confirmed that selected resistant colonies indeed exhibited an increase in MIC (by 3- to 6-fold) to the tested drug (Figure 5C). When we tested each drug at 1X MIC for m1G37+ and m1G37-deficient cells, respectively, m1G37-deficient cells remained compromised in the frequency of resistance relative to m1G37+ cells (Figure S6G, H).
Figure 5: m1G37 deficiency decreases resistance and persistence to antibiotic treatment.

A-C) Resistance arises less frequently in m1G37− (red) E. coli (A) and Salmonella (B) trmDKD cells than in m1G37+ (blue) cells. An overnight culture of cells at 105 CFUs was plated onto an LB agar plate containing the indicated concentration of gentamicin (Gen), kanamycin (Kan), ampicillin (Amp), or vancomycin (Van). Each concentration was near 1X MIC for m1G37+ cells. Resistant colonies were counted after incubation at 37 °C for 3 days. Mutants were verified to have an increase in MIC to the respective antibiotic (C). Data and error bars are mean ± SD, n = 3. Welch’s t-test: *p < 0.1, **p < 0.05, ***p < 0.01.
D-F) Persistence of Salmonella trmD-KD cells, showing CFUs/mL over time (left) and the average CFUs/mL at 6 h post-treatment in m1G37+ and m1G37− conditions (right). Untreated Salmonella trmD-KD cells had similar CFUs/mL in the two conditions (D), while persistence arose more frequently in m1G37+ than m1G37− cells treated with 20 μg/mL Gen (3.7X and 8.5X MIC for m1G37+ and m1G37− conditions) (E) and with 100 μg/mL paromomycin (Par; 2.7X and 10.7X MIC for m1G37+ and m1G37− conditions) (F). An overnight culture in LB with 0.2% Ara was diluted 1:100 into fresh LB with or without 0.2% Ara and incubated at 37 °C for 3 h. Cells were treated with water (no drug), Gen or Par for 0, 1, 2, 4, and 6 h, collected, washed, and plated on LB with Ara. Horizontal lines on the right represent the median, n = 5. Mann-Whitney U test: **p < 0.05, ***p < 0.01.
Unlike resistance that arises from genetic mutations upon drug treatment, persistence arises from noise in gene expression that gives rise to drug tolerance in a subpopulation of isogenic cells (Brauner et al., 2016). This subpopulation of persisters typically survives for some time, contributing to the recurrence of chronic infections. Although the mechanisms underlying persistence are complex, one major pathway is to enhance efflux to pump out the drug (Pu et al., 2016) while shutting down all other biological processes. We hypothesized that by reducing protein synthesis of efflux pumps and OM proteins (Figure 2C, D), m1G37 deficiency would reduce the frequency of persistence under antibiotic treatment.
We studied persistence using Salmonella, which showed a greater response in uptake due to m1G37 deficiency than E. coli (Figure 3A, B) and hence was predicted to manifest a larger effect on persistence. Salmonella cells were treated with a lethal dosage (2–3X MIC) of gentamicin or paromomycin, and viability was measured over time after the start of treatment. While untreated cells maintained viability, drug-treated cells displayed bi-phasic time-kill curves (Figure 5D–F) that signify a heterogeneous response of persistent and non-persistent sub-populations (Balaban et al., 2004). The faster phase of the bi-phasic curve represented killing of the susceptible population, while the slower phase reflected killing of the persistent population. The greater extent of killing in the faster phase was consistent with the susceptible population being the larger fraction. After 6 h of treatment, m1G37-deficient cells exhibited a >10-fold reduction in the frequency of persistence relative to m1G37+ controls, indicating that m1G37 deficiency compromised Salmonella’s ability to tolerate high drug concentrations. Together, these data support the notion that, when the cell envelope was disrupted by m1G37 deficiency, more antibiotics penetrated into and accumulated inside cells to accelerate bactericidal action before resistance or persistence can develop.
Codon composition determines the effect of m1G37 methylation
We tested the hypothesis that the reduced synthesis of membrane proteins in m1G37-deficient cells was due to the poor translation of Pro codons by the unmethylated tRNAPro. We examined the translation of E. coli lolB, which has a CCC-C sequence at the 2nd codon and a CCC-G sequence at the 4th codon (Figure 1C). To maintain the natural gene dosage, we changed the m1G37-dependent CCC at both positions on the chromosome to the less-dependent CCG codon. We used λ-Red recombination for codon engineering, which left a scar in the genome.
Western blot analysis of lysates of cells with the scar showed that, while m1G37 deficiency reduced the translation of the unedited lolB to 89%, it had the opposite effect on the translation of the edited gene by increasing it to 131% (Figure 6A). Each measurement of lolB translation was normalized to that of cysS. The increase in lolB translation by single-nucleotide synonymous changes illustrates the effect of m1G37 on codon-specific translation.
Figure 6: m1G37-tRNA is required for translation of CC[C/U] codons.

A) Western blot analysis showed that m1G37− (red) E. coli trmD-KD cells had lower lolB expression relative to cysS from the native gene than m1G37+ (blue) cells, but higher expression from the codon-engineered gene. Data and error bars are mean ± SD, n = 6. Welch’s t-test: **p < 0.05.
B) m1G37− Salmonella trmD-KD cells survived better in novobiocin treatment when expressing the engineered CCG codon at the 6th position of tolC than when expressing the natural CCC codon. Cells were grown in the presence of 12.5 μg/mL novobiocin for 24 h and the fold-change in CFUs relative to t = 0 was compared. One sample t-test: *p < 0.1, n = 5.
C) E. coli cells expressing C37-tRNAPro/UGG (red) grew poorly compared to cells expressing the G37 version (blue). Data and error bars are mean ± SEM, n = 3.
D) Cells expressing C37-tRNAPro/UGG (red) accumulated more Hoechst 33342 dye than cells expressing G37-tRNAPro/UGG (blue). Data and error bars are mean ± SEM, n = 3.
E) Cells expressing C37-tRNAPro/UGG (red) showed lower MICs than cells expressing G37- tRNAPro/UGG (blue). Data and error bars are mean ± SEM, n = 3. Welch’s t-test: **p < 0.05, ***p < 0.01.
F) Cells expressing C37-tRNAPro/UGG (red) died faster than cells expressing G37-tRNAPro/UGG (blue) after exposure to gentamicin or vancomycin. Data and error bars are mean ± SEM, n = 3.
As a second test, we changed the CCC codon at the 6th position of tolC in Salmonella (Figure 1) to CCG. This single-nucleotide synonymous change would lessen the translational dependence on m1G37 relative to the unedited gene, thereby increasing tolC translation and reducing susceptibility to antibiotics in m1G37 deficiency. We focused on novobiocin, which is cell-permeable but subject to TolC-mediated efflux (Kodali et al., 2005). Survival of m1G37-deficient cells under novobiocin treatment was 2.7-fold higher when expressing the edited tolC relative to cells expressing the unedited gene (Figure 6B), supporting the codon-specific effect of m1G37. The Pro at the 6th position of TolC is conserved among Gram-negative bacteria, and substitution of Pro with Ala by mutating the CCC codon to GCG reduced the protein to undetectable levels (data not shown), probably due to membrane mistargeting and destabilization (Masi et al., 2009). These data suggest that the conservation of Pro at the 6th position is critical for TolC structure and function, and that its incorporation into the protein is regulated at the codon level by m1G37.
The importance of m1G37 in the UGG isoacceptor of tRNAPro
E. coli and Salmonella both express three isoacceptors of tRNAPro (http://trna.bioinf.uni-leipzig.de/), all of which contain m1G37. Of the three, the UGG isoacceptor is the most sensitive to loss of m1G37 (Gamper et al., 2015a). This isoacceptor is capable of reading all Pro codons via an additional cmo5U34 modification at the wobble position (Nasvall et al., 2004), and it is also the only one that is required for cell growth and survival. We tested whether an alternative nucleotide could substitute for m1G37 in the UGG tRNA to eliminate the need for trmD. We created a derivative of E. coli MG1655 that lacked the tRNA gene on the chromosome and expressed the isoacceptor from a plasmid to maintain viability. This strain also lacked the gene for the GGG isoacceptor on the chromosome, so that the translation of CC[C/U] was completely dependent on the plasmid-borne UGG tRNA. While we designed strains with all three non-G substitutions on the plasmid-borne tRNA, we only recovered the C37 variant (data not shown), suggesting that the A37 and U37 variants were lethal. We previously showed that the C37 variant is not methylated by TrmD (Christian et al., 2004).
The strain expressing the C37 variant of the UGG tRNA was severely defective in growth relative to the G37 version (Figure 6C), even though trmD was intact. Cells expressing the C37-tRNA accumulated more Hoechst dye (Figure 6D), indicating the disruption of the membrane barrier. Cells expressing the C37-tRNA were also more sensitive to antibiotic killing than cells expressing the G37 version, with MIC decreases of 8.2-fold for gentamicin and 4.0-fold for vancomycin (Figure 6E). These decreases for two unrelated antibiotics suggest that the envelope structure is disrupted in cells expressing the C37-tRNA. Expression of the C37-tRNA led to more rapid killing upon exposure to gentamicin or vancomycin (Figure 6F). Collectively, these data indicate that C37-tRNA is unable to support the biosynthesis of membrane proteins at the levels of m1G37-tRNA, and that the single G37C substitution is sufficient to cause general damage to the cell envelope, leading to faster antibiotic killing. Thus, m1G37 methylation by TrmD is necessary for the function of UGG tRNA and cannot be replaced.
DISCUSSION
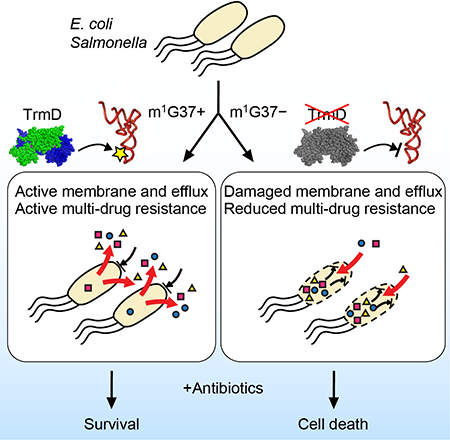
Multi-drug resistance among Gram-negative bacteria is a major human health problem. We report here the discovery of m1G37 methylation of tRNA as a global determinant of multi-drug resistance in E. coli and Salmonella. The mechanism of this methylation is at the codon level during the elongation phase of protein synthesis, rather than at the initiation of transcription or translation. Because protein synthesis is the last step of gene expression in a highly energy-demanding process, the control of its speed and quality at individual codons provides enormous capacity to influence the proteome of a cell. The m1G37 methylation is present in all isoacceptors of Pro, two isoacceptors (GAG and CAG) of Leu, and one isoacceptor (CCG) of Arg. The complete association of m1G37 with tRNAPro species emphasizes its ability to regulate translation of genes enriched with Pro codons (particularly the CC[C/U] codons), which include many Gram-negative genes encoding OM proteins. With few exceptions, most of these genes are not operon-organized and cannot be simultaneously regulated by transcription or translation initiation. Instead, their dependence on translation of Pro codons to generate transmembrane domains provides a common thread that unites them under the control of m1G37 methylation. Our data support a model in which m1G37 ensures robust biosynthesis of Gram-negative OM membrane proteins to produce an effective envelope barrier and efflux activity, which confers multi-drug resistance, whereas m1G37 deficiency reduces the levels of OM proteins, thereby permeabilizing the OM structure and sensitizing cells to antibiotic killing (Figure 7A). While m1G37 deficiency does not act on all genes for membrane proteins, the effects are sufficiently widespread (e.g. lolB, ompA, and tolC) and impactful to accelerate bactericidal action of antibiotics and to halt resistance or persistence upon antibiotic exposure. Our data are generally consistent across E. coli and Salmonella, and are likely applicable to a broad spectrum of Gram-negative pathogens, including Pseudomonas aeruginosa, Yersinia pestis, Serratia marcescens, and Shigella dysenteriae, in which CC[C/U] codons are widely present near the start of membrane-associated genes (Figure S7). Strikingly, the CC[C/U] codon at the 6th position of tolC is conserved among g-proteobacteria (Figure 7B), indicating that the efflux activity of the gene and multi-drug resistance of these Gram-negative bacteria is determined by m1G37.
Figure 7: m1G37-dependent regulation of bacterial multi-drug resistance.

A) Gram-negative membrane-associated genes are enriched with CC[C/U] codons, which depend on TrmD synthesis of m1G37-tRNA for translation. In the m1G37+ condition (top), translation of CC[C/U] is active to establish a robust envelope barrier and efflux activity that confers multi-drug resistance. In the m1G37-deficient condition (m1G37−, bottom), translation of CC[C/U] is impaired, decreasing the barrier and efflux activity, permitting intracellular accumulation of multiple drugs, accelerating bactericidal action, and inhibiting the development of resistance and persistence.
B) The tolC gene is conserved with the CC[C/U] codon at the 6th position among many Gram-negative g-proteobacterial pathogens.
See also Figure S7.
m1G37 is distinct from the >100 post-transcriptional modifications that have been associated with tRNA to date (http://modomics.genesilico.pl/). Crucially, m1G37 is both essential and is conserved across all three kingdoms of life (Bjork et al., 2001). In bacteria, where m1G37 is synthesized by TrmD, its level is stable across various growth phases (Gamper et al., 2015a). Even when E. coli cells are deep in stationary phase, when glucose and all other nutrients are depleted, m1G37 levels remain at ~100% (Gamper et al., 2015a). By contrast, levels of most tRNA post-transcriptional modifications are variable depending on cellular conditions. The synthesis of m1A58, required for tRNA translation in eukaryotes, is subject to demethylation during glucose deprivation (Liu et al., 2016). The formation of s4U8 in bacteria is induced by near-UV radiation (Favre et al., 1971) and that of cmo5U34 is activated by hypoxia (Chionh et al., 2016). The formation of m5C34 in yeast is induced by oxidative stress (Chan et al., 2012) and that of mcm5U34 and mcm5s2U34 is by alkylation damage (Begley et al., 2007). The stability of m1G37 levels emphasizes the potential of targeting TrmD for antibacterial therapies.
TrmD is a high-priority antibacterial target (White and Kell, 2004). Besides its essentiality for bacterial growth and survival (Gamper et al., 2015a), TrmD is broadly conserved among bacterial species, has a methyl-donor binding site for drug targeting, and is fundamentally distinct from its human counterpart Trm5 in structure and mechanism (Christian et al., 2004; Christian and Hou, 2007; Christian et al., 2010; Christian et al., 2016; Lahoud et al., 2011; Sakaguchi et al., 2012; Sakaguchi et al., 2014), enabling the development of bacteria-selective compounds. However, while pharmaceutical companies have attempted to target TrmD, progress has stalled, because the isolated inhibitors have failed to overcome the OM barrier and efflux activity (Hill et al., 2013). This obstacle resonates with the major challenge that confronts current antibacterial discovery – the inability to make compounds that penetrate bacteria, especially Gram-negative species (Tommasi et al., 2015). Our finding that TrmD is a global determinant of the biosynthesis of Gram-negative membrane proteins provides new insight into how to address this problem.
To target TrmD, we suggest exploiting its ability to control the translation of CC[C/U] in membrane-associated genes. The CCC codon at the 6th position of tolC is an example, which is conserved among γ-proteobacterial pathogens and is required for protein stability, acting as an Achilles heel that is required for efflux activity of tolC but is also subject to regulation by TrmD for translation. While the AcrAB-TolC pump exports a wide range of antibiotics (Li et al., 1995; Okusu et al., 1996), it does not act on gentamicin-like aminoglycosides (Edgar and Bibi, 1997). Thus, primary inhibitors of TrmD should be gentamicin-like molecules, capable of entering cells without being expelled by AcrAB-TolC. Once inside cells, these inhibitors can target TrmD and reduce the synthesis of TolC, as well as many other membrane proteins and efflux pumps that depend on TrmD for translation. By targeting TrmD while exerting collateral damage on the cell envelope, primary inhibitors can destabilize the membrane barrier to allow secondary inhibitors with distinct mechanisms of action to enter cells and function. In this two-tiered strategy, accelerated bactericidal action should reduce the likelihood of resistance and persistence and improve the efficiency of antibacterial treatments, yielding a general strategy for mitigating bacterial multi-drug resistance. This study demonstrates that tRNA methylation events such as m1G37 have broad effects on cellular physiology and membrane biology, which can be exploited for novel drug discovery.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
Methods Details
Strain constructions
The Escherichia coli MG1655 trmD-KD (E. coli trmD-KD) strain was made via P1 transduction of E. coli K-12 MG1655, using phage lysate prepared from an E. coli BL21(DE3) trmD-KD strain (Gamper et al., 2015a, b). The Salmonella enterica serovar Typhymurium LT2 trmD-KD (Salmonella trmD-KD) strain was made using the λ-Red recombinase system (Datsenko and Wanner, 2000). A kanamycin resistance marker (kanR) was amplified from pKD4 using primers in Table S1 and purified using a PCR clean-up kit (Macherey-nagel). Salmonella LT2 cells were transformed with the λ-Red recombinase plasmid pKD46 and also with a pACYC-araC-PC-PBAD-human trm5 that encodes human trm5 under the arabinose (Ara)-controlled PBAD promoter and the repressor araC under the PC promoter. Salmonella cells were grown with expression of λ-Red recombinase, harvested in mid-log phase, and made electro-competent after two washes with cold 10% glycerol. Cells were electroporated with the indicated PCR-amplified and purified fragment using MicroPulser Electroporator (BIO-RAD), and cells exhibiting kanR were analyzed for marker insertion to the chromosomal trmD locus by PCR using primers in Table S1. Insertion was confirmed via sequencing (data not shown). After overnight growth at 43 °C to remove pKD46, cells were transformed with the FLP-recombinase plasmid pCP20 at 30 °C and removal of the kanR marker and the remaining ~100 bp scar sequence was confirmed via PCR and subsequent sequencing (data not shown). Finally, pCP20 was cured from cells by incubating them at 43 °C overnight. After confirmation of the trmD-KD genotypes via PCR using primers at flanking regions of the trmD locus (Figure S2A), cells were grown in Luria broth (LB) supplemented with 0.2% Ara overnight at 37 °C. Cell s were inoculated at a 1:100 dilution into fresh LB without Ara but with 0.2% D-glucose (Glc) and grown for 3 h at 37 °C to deplete pre-existing Trm5 and methylated tRNAs. A 10-fold serial dilution of cells was spotted onto LB plates with 0.2% Ara or 0.2% Glc for m1G37+ and m1G37− conditions, respectively, and growth was examined after overnight incubation at 37 °C.
To create E. coli cysS-KD and proS-KD strains, we first created the maintenance plasmids that expressed E. coli cysS and proS respectively with a C-terminal degron tag for rapid depletion. The ORFs were each amplified from extracted genomic DNA of E. coli MG1655 using primers in Table S1. These PCR products encoded a C-terminal 6x His tag followed by a GGS linker and a degron tag YALAA. The plasmid backbone sequence was amplified from the pACYC-araC-PC-PBAD maintenance plasmid that already encoded a GGS linker and a degron tag using primers in Table S1. Each PCR product was ligated to the linearized plasmid using a Gibson cloning kit (New England Biolabs) and the correct clone was confirmed by sequencing analysis. E. coli MG1655 harboring pKD46 λ-Red recombinase plasmid and a cysS or proS maintenance plasmid was prepared as electrocompetent cells. A kanamycin marker targeting the chromosomal cysS or proS was amplified from pKD4 using primers in Table S1 and electroporated into competent cells of MG1655 for recombination and gene deletion. Cells were screened for kanamycin resistance and the chromosomal locus was confirmed by PCR using primers in Table S1. The kanamycin marker was then removed by FLP recombination using pCP20 and removal was confirmed by sequencing.
The E. coli MG1655 strain expressing the variant C37-tRNAPro/UGG was constructed using the λ-Red system to remove the native tRNA gene (proM) from the chromosome. A kanR marker was amplified via PCR with homologous extensions to the flanking regions of the proM locus using primers in Table S1. E. coli MG1655 was transformed with pKD46 and the maintenance plasmid pKK223–3 E. coli G37-UGG tRNA expressing E. coli proM at the EcoRI and PstI sites. Cells were electroporated with the PCR-amplified kanR to introduce the marker into the chromosomal proM locus. Non-G37 (namely, A37, C37, and U37) variants of E. coli tRNAPro/UGG were created using Quikchange mutagenesis (Agilent) of pKK223–3 E. coli G37-UGG tRNA. Transformation of E. coli MG1655 with these variants, followed by P1 transduction of the proM-KD locus, recovered only the C37 variant. For cells expressing the G37 or C37 version of the UGG tRNA from the maintenance plasmid, the GGG tRNA gene (proL), which reads CCC and CCU codons but is not essential for growth, was removed via λ-Red recombination in E. coli MG1655, followed by P1 transduction into the respective strain (see Table S1 for primers). After selection for kanR, the marker was removed with FLP recombinase from pCP20 and purified. Each purified G37 and C37 clone was grown overnight and inoculated into fresh LB to OD600 = 0.05 with 100 μg/mL ampicillin and growth in a 40-mL culture was monitored by OD600 for 13 h at 37 °C.
MS analysis of membrane proteomes
An overnight E. coli trmD-KD culture was inoculated at a 1:100 dilution into fresh LB with or without 0.2% Ara and grown for 5 h at 37 °C. Cells were then diluted to OD600 = 0.1 in fresh LB with or without 0.2% Ara and grown for another 2 h. Cells were harvested and a membrane fraction was prepared by method 4 in (Thein et al., 2010). Extracted membrane proteins (30–40 μg) were boiled in 4% SDS in 100 mM Tris pH 6.8, separated into three technical replicates, and run on a 10% SDS-PAGE gel. Proteins were visualized, digested with trypsin, and analyzed on an Impact II QTOF mass spectrometer (Bruker Daltonics) (Gibbs et al., 2017). Mass spectrometry data were analyzed with MaxQuant v. 1.5.3.30 (Tyanova et al., 2016) against the UNIPROT Escherichia coli K12 protein sequence database (downloaded on May 12, 2015; 4,481 entries) plus common contaminants (245 entries) with variable modifications of methionine oxidation, N-acetylation of proteins, and fixed modification of cysteine carbamidomethylation. The false discovery rate was set to 1% for both proteins and peptides. Technical replicates of the two treatments were searched together using MaxQuant’s “match between run” and label-free quantification options.
Western blotting
E. coli trmD-KD and Salmonella trmD-KD cells were grown in LB supplemented with 0.2% Ara overnight at 37 °C. Cells were inoculated at a 1:10 0 dilution into fresh LB with 0.2% Ara or 0.2% Glc and grown at 37 °C. To monitor the depletion of Trm5 (the maintenance protein), cells were sampled over 3 h and whole-cell lysates were prepared via repeated heating at 95 °C and vortexing. Cell lysates containing 15–20 μg proteins were separated via 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to an Immobilon-P PVDF membrane (Millipore). The membrane was incubated with primary rabbit antibodies against human Trm5 (Sigma-Aldrich) at a 1:1,000 dilution or against E. coli CysRS at a 1:10,000 dilution and secondary goat antibody against rabbit IgG (Sigma-Aldrich), followed by incubation with SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) and imaging with Chemi-Doc XRS+ (BIO-RAD). The absence of chromosomally expressed TrmD was confirmed by Western blotting using rabbit antibodies against E. coli TrmD (a gift from Dr. Glenn Bjork). For LolB and OmpA quantification, after inoculation into fresh LB, cells were grown for 4 h at 37 °C, diluted to OD 600 = 0.1 into fresh LB with or without 0.2% Ara, and grown for another 3 h. Cells were harvested, precipitated with 10% (w/v) trichloroacetic acid (TCA), washed with ice-cold acetone, and sonicated using a Bransonic 1210 Ultrasonic Cleaner (Branson) until the pellet was dissolved. Proteins were pelleted by centrifuge at 16,000g for 10 min at 4 °C, dried and resuspended in water, then b oiled in 1x SDS buffer at 95 °C for 5 min; total protein content was analyzed via 12% SDS-PAGE. Rabbit polyclonal antibodies against LolB and OmpA were kind gifts from Dr. Hajime Tokuda (Morioka University). Levels of LolB and OmpA relative to CysRS and total membrane proteins, respectively, were quantified using Image Lab v. 6.0 (BIO-RAD).
Primer-extension analysis of m1G37
E. coli trmD-KD and Salmonella trmD-KD cells were grown in LB overnight as for Western blotting. Cells were diluted 1:100 into fresh LB with or without 0.2% Ara and grown for 4 h at 37 °C. Cells were then diluted to OD600 = 0.1 in fresh LB with or without 0.2% Ara and grown for another 3 h. To analyze the initial pre-depletion of methylated tRNA (Figure S3A), the overnight culture was diluted 1:100 into fresh LB without Ara and cells were collected every hour up to t = 3 h. Cells were harvested via centrifugation at 4,000g for 10 min at 4 °C and pellets were stored at −20 °C until use. Total small RNA was extracted from cell pellets as described previously (Frenkel-Morgenstern et al., 2012). Briefly, cell pellets were suspended in buffer A (1 mM Tris-HCl [pH 7.5] and 10 mM MgCl2), mixed with an equal volume of water-saturated phenol, and vortexed three times each for 45 s. After centrifugation at 12,000g for 5 min, the aqueous phase was collected and the phenol phase was extracted three times with an equal volume of buffer A. Total small RNAs in the pooled aqueous phase were pelleted via ethanol precipitation and centrifugation. RNA pellets were dissolved in TE buffer (10 mM Tris-HCl [pH 8.0] and 1 mM ethylenediaminetetraacetic acid) and stored at −20 °C. The level of m 1G37 in tRNAPro/UGG was quantified via primer extension on 2 μg of total small RNA with Superscript III reverse transcriptase (Invitrogen) at 50 °C for 40 min as d escribed previously (Christian et al., 2004). The primer (Table S1) was designed to hybridize to the tRNA to enable a 2-nucleotide extension to m1G37 and was labeled at the 5′-end with [γ−32P]-ATP (PerkinElmer) using T4 polynucleotide kinase (New England Biolabs). The same primer was used for analysis of tRNAPro/UGG in E. coli and Salmonella, which share an identical sequence. Primer extension was stopped via heating at 65 °C for 5 min and separation was achieved on 1 2% polyacrylamide/7 M urea gels. Gels were imaged via phosphorimaging using a Typhoon IP Imaging system (GE Healthcare) and analyzed with ImageJ v. 1.51 (NIH). A similar analysis was performed for tRNAPro/GGG (Table S1, Figure S3B, S3C). The amount of m1G37 was calculated as the percentage by the band intensity of the primer stop at position 37 over the sum of stops and read-through to nucleotide 1.
LC-MS/MS analysis of m1G37
E. coli trmD-KD and Salmonella trmD-KD cells were grown in Ara+ and Ara− conditions and total small RNA was prepared as in primer-extension analysis. The tRNA fraction was enriched using NucleoBond AX 2000 (Macherey-nagel). Briefly, the column was first equilibrated with a buffer containing 100 mM Tris-acetate [pH 6.3], 15% EtOH, and 200 mM KCl. The RNA sample was loaded and washed with a buffer containing 100 mM Tris-acetate [pH 6.3], 15% EtOH, and 400 mM KCl. The enriched tRNA fraction was then eluted from the column with a buffer containing 100 mM Tris-acetate [pH 6.3], 15% EtOH, and 750 mM KCl, and tRNAPro/UGG isoacceptor was affinity-purified (Masuda et al., 2018) and the salt adducts were removed by repeated ethanol-precipitation in the presence of a high concentration of NH4OAc. Approximately 200–300 ng tRNA was digested with nuclease P1 (1 U, Sigma-Aldrich) in 20 μL reaction buffer containing 10 mM of NH4OAc [pH 5.3] at 42 °C for 2 h. With the addition of 2.5 μL NH4HCO3 (1M, freshly prepared in water), 1 U of alkaline phosphatase (Sigma-Aldrich) was added and the sample was incubated at 37 °C for 2 h. After the incubation, the sample was diluted with an additional 40 μL water and filtered with 0.22-μm filters (4 mm diameter, Millipore) and 8 μL of the entire solution was injected into an LC-MS/MS. Nucleosides were separated by reverse phase ultra-performance liquid chromatography on a C1 column with on-line mass spectrometry detection by an Agilent 6410 QQQ triple-quadruple LC mass spectrometer in positive electrospray ionization mode. The nucleosides were quantified with retention time and the nucleoside-to-base ion mass transition of 284–152 (G), 268–136 (A), and 298.1–166.1 (m1G). Quantification was performed in comparison with a standard curve, obtained from pure nucleoside standards running with the same batch of samples. The m1G level was calculated as the ratio of m1G to G based on calibrated concentration curves.
Quantification of lolB expression with a YFP reporter
The native promoter of lolB was PCR-amplified from E. coli MG1655 genomic DNA and inserted into the pZS2R plasmid, a vector of 4.3 kb in length that carries a KanR marker, is amplified from the pSC101 replication origin, and contains YFP under the control of the strong and constitutive λ phage promoter R (a kind gift from Dr. Roy Kishony) (Kelsic et al., 2015)). The insertion replaced the original promoter with the PlolB promoter to generate pZS2R-PlolB-YFP for transcriptional analysis of YFP. E. coli trmD-KD cells harboring this plasmid were grown in LB, diluted 1:100 into fresh LB, and grown for 4 h at 37 °C with or without 0.2% Ara. Cells were diluted to OD600 = 0.1 in fresh LB with or without 0.2% Ara and grown for another 3 h. Cells were then harvested by centrifugation at 7,000g for 1 min and suspended in M9, and the YFP intensity from the suspension was measured in an Infinite M200 PRO (Tecan) plate reader at excitation and emission wavelengths of 500 nm and 540 nm, respectively. After normalization based on OD600, the signal intensity was calculated for the m1G37-deficient condition relative to the m1G37+ condition (Figure S3F).
AlamarBlue accumulation assay
E. coli trmD-KD and Salmonella trmD-KD cells were grown in Ara+ and Ara− conditions as in RNA analyses above. Cells were washed with and resuspended in 150 μL of 20 mM potassium phosphate buffer pH 7.0 containing 1 mM MgCl2 (PPB) at 4 × 108 CFU/mL in a 96-well plate. At t = 0, a 1/10 volume of AlamarBlue (Invitrogen) in the stock concentration was added and fluorescence signal at Ex565nm/Em590nm was monitored over 30 min as the uptake of AlamarBlue. The signal was normalized by OD600 and plotted over time. E. coli cysS-KD and E. coli proS-KD cells were grown in the same way and used for the assay. As a control, an E. coli hyper-permeable strain (Krishnamoorthy et al., 2016) (a gift from Dr. Helen Zgurskaya) was used. This strain has a highly permeable outer membrane, due to a mutant form of the outer membrane protein FhuA that is driven from an arabinose promoter. The hyper-permeable strain was grown for 5 h in the presence or absence of arabinose and AlamarBlue uptake was monitored as described above. Another control was the use of polymyxin B (PMB), which disrupts and permeabilizes the outer membrane by binding to lipids. E. coli trmD-KD and Salmonella trmD-KD cells grown with Ara were used for the assay, and the AlamarBlue uptake was monitored for 10 min without PMB, followed by an additional 30 min of incubation in the presence of varying concentrations of PMB (1 to 20 μg/mL).
Hoechst accumulation assay
Hoechst H33342 dye is an intercalating agent that fluoresces when bound to DNA (van den Berg van Saparoea et al., 2005), and hence is commonly used for measuring outer-membrane permeability. The fluorescence intensity of the dye accumulated in the cell serves as a proxy for cellular uptake and efflux. Accumulation of H33342 was monitored in the presence of cyanide 3-chlorophenylhydrazone (CCCP) to inhibit energy-dependent efflux. In accordance with a protocol adapted from a previous assay (Murata et al., 2007), cells were grown to saturation overnight with shaking at 37 °C in LB with chloramp henicol (34 μg/mL) and Ara (0.2% w/v), diluted 1:100 into fresh LB with chloramphenicol in the presence of Ara or Glc (0.2% w/v), and grown for 3 h to OD600~0.8 at 37 °C until cells reached exponential phase. Cells were then diluted 1:10 into fresh LB with chloramphenicol in the presence of the same carbon source (Ara or Glc) as in the first round of culturing and grown for 5 h. Cells were harvested via centrifugation (5,400 g for 5 min) at room temperature, washed with 1X phosphate-buffered saline (PBS) [pH 7.4], suspended in PBS, and adjusted to OD600~0.6. Each cell suspension (100 μL) was transferred to a well in a black opaque 96-well microplate (Greiner Bio-One) and mixed with 100 μL of 3 μM H33342 dye (Sigma-Aldrich) in 1X PBS [pH 7.4] to a final concentration of 1.5 μM. The plate was covered with a transparent sealing film (Excel Scientific) and fluorescence intensity was monitored every minute for 30 min with shaking in a Synergy H1 Hybrid Multi-Mode Reader (Biotek) or Infinite M200 PRO (Tecan) plate reader at 37 °C. Fluorescence was recorded with excitation and emission wavelengths of 355 and 460 nm, respectively. In experiments in which ATP-dependent efflux was abolished, freshly prepared CCCP (Sigma-Aldrich) was added to a final concentration of 50 μM (from a 50 mM stock solution prepared in dimethyl sulfoxide (DMSO)) together with 1.5 μM H33342. Each experiment was repeated at least three times. For E. coli C37-tRNAPro/UGG cells, a mid-log culture was used for the assay.
Nile Red efflux assay
The Nile Red efflux assay was modified from a previous protocol (Bohnert et al., 2010). Cells were grown as for Hoechst assays for 5 h and then shifted to room temperature and prepared as follows: each culture (6 mL) was centrifuged for 10 min at 3,829g and the pellet was suspended in PPB. After another round of centrifugation and resuspension, cells were adjusted to OD600~0.9–1.0 in PPB (potassium phosphate buffer: 20 mM potassium phosphate buffer pH 7.0 containing 1 mM MgCl2) and mixed with CCCP (1 mM stock solution in 50% DMSO) to a final concentration of 5 μM. After incubation at room temperature for ~20 min, cells were transferred to 10 mL glass tubes. Nile Red (Acros Organics; 1 mM stock in anhydrous DMSO) was added to a final concentration of 5 μM, and the tubes were incubated at 37 °C and shaken at 140 rpm for 3 h. Cells were shifted to room temperature for 1 h without shaking and then centrifuged for 5 min at 3,829g. The supernatant was discarded, any droplets left clinging to the tube walls were removed with Kimwipes, and cells were suspended in PPB at OD600~0.9–1.0. Cell suspensions (0.2 mL) were quickly transferred to a quartz cuvette (Starna Cells Inc.) containing 1.8 mL PPB. Fluorescence emission was recorded with a QuantaMaster 220 spectrofluorometer (Photon Technology International) using the PTI Felix32 software. Cell suspensions were continuously stirred with a magnetic stirrer inside the cuvette. The slit width was set to 10 nm and the excitation and emission wavelengths were set to 552 nm and 636 nm, respectively. The fluorescence of each cell suspension was followed over 100 s, and Nile Red efflux was triggered via rapid energization with the addition of 100 μL of 1 M glucose.
Fluorescence was monitored for another 200 s. Trials displaying no pre-energization efflux were included in the analysis, whereas trials that showed substantial pre-energization efflux were discarded. The time required for 50% Nile Red efflux (tefflux 50%) was calculated for at least three independent measurements per sample as described previously (Bohnert et al., 2010). A ΔacrB strain from the Coli Genetic Stock Center (CGSC) at Yale University was tested as a control.
Ethidium bromide efflux assay
E. coli trmD-KD and Salmonella trmD-KD cells were grown in Ara+ and Ara− conditions as in RNA analyses above. Cells were adjusted to OD600~0.9–1.0 in PPB and incubated with 20 μM CCCP and 10 μg/mL ethidium bromide (EtBr) for 2 h at 30 °C. Cells were spun, washed and resuspended in a fresh PPB at 5×108 CFU/mL in a 96-well plate. The fluorescence signal of EtBr at Ex530nm/Em600nm was monitored for the first 3 min, then efflux was activated by addition of 50 mM Glc, and the signal was monitored for 30 min. The EtBr signal was normalized by OD600 and plotted over time. An E. coli tolC-KO strain was purchased from CGSC, and after the kanamycin marker was removed by a pCP20 plasmid transformation, it was used for the assay as a control. Efflux was also assayed for E. coli trmD-KD and Salmonella trmD-KD cells with various concentrations of polymyxin B added to the cell resuspension 5 min prior to Glc addition.
Thioflavin T fluorescence assay
Overnight cultures of E. coli and Salmonella trmD-KD cells were inoculated in LB at 1:100 dilution without or with 0.2% Ara and grown for 4 h at 37 °C. Cells were diluted in LB in Ara+/− conditions to OD600 = 0.1 and grown for another 3 h at 37 °C. Cells we re then washed with M9 medium and incubated in M9 containing 20 μM Thioflavin T (ThT) for 2 h at 37 °C with Ara or Glc for m1G37+ and m1G37-deficient conditions, respectively, and the ThT fluorescence was measured at Ex446nm/Em482nm and normalized by OD600 (Prindle et al., 2015).
Imaging in microfluidic devices
Overnight E. coli trmD-KD cultures were grown in LB + 0.2% Ara and 30 ug/mL chloramphenicol. These cultures were diluted 1:100 into 1 mL fresh LB with 0.2% Ara or 0.2% Glc (to deplete the pre-existing Trm5 and m1G37 tRNA) and grown with shaking at 37 °C for 3.5 h. Cells were then transferred to B04A microfluidic perfusion plates (CellASIC Corp.) that had been loaded with medium and pre-warmed to 37 °C, and cells were incu bated at 37 °C for >1 h before imaging. The medium was exchanged using the ONIX microfluidic platform (CellASIC Corp.). The osmolarity of the growth medium or phosphate-buffered saline (PBS) was modulated with sorbitol (Sigma). For oscillatory osmotic shocks, cells were allowed to grow for 5 min in medium in the imaging chamber before being subjected to 100-mM oscillatory osmotic shocks by switching between LB and LB + 100 mM sorbitol.
Cells were imaged on a Nikon Eclipse Ti-E inverted fluorescence microscope with a 100X (NA 1.40) oil-immersion objective. Images were collected on a DU885 electron multiplying charged couple device camera (Andor) using μManager v. 1.4 (Edelstein et al., 2014). Cells were maintained at 37 °C during imaging with an active-control environmental chamber (HaisonTech).
Cell tracking and analysis
To calculate the amplitude of length oscillations during oscillatory osmotic shocks, cells were tracked over time using custom MATLAB algorithms, similar to previous studies (Rojas et al., 2014). First, cell-wall lengths (l) were automatically identified. The effective population-averaged length leff at time t1 was calculated by integrating the population-averaged elongation rate over time (Rojas et al., 2014):
where l0 is the mean initial cell length at time t0, and ė is the instantaneous growth rate. The effective population-averaged length was then smoothed with a mean filter with window size equal to the period of oscillation, and subtracted from the unsmoothed trace to obtain the deviation of the length oscillations around the smoothed trace. The peak-to-peak amplitude was calculated for each cycle. The mean amplitude was calculated by averaging the peak-to-peak amplitude over cycles. Uncertainty was estimated as the standard deviation of the mean amplitude over cycles.
Minimal inhibitory concentration (MIC) measurements
Overnight cultures of trmD-KD cells with 0.2% Ara were diluted 1:100 into LB without Ara and grown at 37 °C for 1 and 3 h for Salmonella and E. coli, respectively, to pre-deplete Trm5 and methylated tRNAs (Figure S3). This short pre-depletion was appropriate for MIC analysis, because longer pre-depletion made cells in m1G37-deficient conditions too weak to distinguish death by antibiotic killing from death by lack of m1G37 (data not shown). After pre-depletion, cells for all but ciprofloxacin analysis were diluted to 106 CFU/mL and grown in 96-well plates with 0.2% Ara or Glc in the presence of an antibiotic across a 2-fold serial dilution. After 18 h of incubation at 37 °C, OD600 was measured and the MIC was determined based on a threshold for growth of OD600 = 0.15 (Kim et al., 2010). For analysis of MIC of ciprofloxacin, 105 CFU/mL cells were inoculated and grown for 24 h at 37 °C. The MI C of polymyxin B for E. coli trmD-KD and Salmonella trmD-KD cells grown in the Ara+ condition was determined as 0.5 and 1.0 μg/mL, respectively. To test the effect of polymyxin B on the permeability of each antibiotic, we used polymyxin B at 0.25X MIC for E. coli trmD-KD and Salmonella trmD-KD cells. For E. coli C37-tRNAPro/UGG cells, overnight cultures were inoculated into fresh LB at 106 CFU/mL and the MICs were determined for gentamicin and vancomycin. A Δefp strain was purchased from CGSC and the kanamycin marker was removed by transformation of the pCP20 plasmid. The MICs were determined for ampicillin, gentamicin and vancomycin.
Time-kill analyses
Cells depleted of Trm5 and m1G37-tRNA were prepared as for MIC analyses and were inoculated into fresh LB at 106 CFU/mL with 0.2% Ara or 0.2% Glc in the presence of an antibiotic. Several concentrations were tested for each drug, ranging from 0.6X to 6.4X MIC (Figure S6D); the concentration that yielded the largest difference between m1G37+ and m1G37-deficient conditions was selected (12.5 μg/mL carbenicillin, 3.125 μg/mL ampicillin, 6.25 μg/mL gentamicin, 12.5 μg/mL kanamycin, and 256 μg/mL vancomycin). In the presence of the chosen concentration of each drug, cells were grown at 37 °C and sampled up to 18–24 h. At each time point, 10-fold serial dilutions of cells were spotted onto LB plates with 0.2% Ara and grown overnight. The number of viable colonies was counted and converted to CFU/mL. For analysis of E. coli cysS-KD and proS-KD cells, cells depleted of the protein product of each gene were prepared in a similar manner as for trmD-KD cells and the time-kill curve was determined for 25 μg/mL carbenicillin and 256 μg/mL vancomycin in the absence of Ara. After counting viable colonies from an LB plate, the survival rate was calculated relative to t = 0. The same CFU counting method was used for a control experiment with 2 mM H2O2 (Fig. S6); the time course was followed up to 6 h. For analysis of E. coli C37-tRNAPro/UGG cells, overnight cultures were inoculated into fresh LB at 106 CFU/mL with 3.125 μg/mL gentamicin or 512 μg/mL vancomycin and analyzed as above.
Resistance analyses
Cells depleted of Trm5 and m1G37-tRNA were cultured as for MIC and time-kill assays. Cells were diluted to 105 CFU based on the calibration that OD600 = 1 corresponds to 8 × 108 CFU/mL, and plated on LB with 0.2% Ara or 0.2% Glc in the presence of an antibiotic at a concentration near 1X MIC of m1G37+ cells or at a concentration of 1X MIC for each specific type of cells: gentamicin at 2.7 μg/mL (m1G37+) and 0.88 μg/mL (m1G37-deficient) for E. coli and 5.5 μg/mL (m1G37+) and 2.3 μg/mL (m1G37-deficient) for Salmonella; ampicillin at 9.4 μg/mL (m1G37+) and 4.7 μg/mL (m1G37-deficient) for E. coli and 2.4 μg/mL (m1G37+) and 0.78 μg/mL (m1G37-deficient) for Salmonella; vancomycin at 341 μg/mL (m1G37+) and 128 μg/mL (m1G37-deficient) for E. coli and 512 μg/mL (m1G37+) and 192 μg/mL (m1G37-deficient) for Salmonella. After incubation at 37 °C for 3 days, CFUs were counted. A representative gentamicin-resistant clone was purified and an increase in MIC was confirmed (Fig. 5C).
Persistence analyses
Salmonella trmD-KD cells were grown in LB with 0.2% Ara overnight, diluted 1:100 into fresh LB with 0.2% Ara or 0.2% Glc, grown at 37 °C for 3 h, and challenged with a specific antibiotic for 6 h. At each time point, cells were washed three times with saline (0.9% NaCl) and 10-fold dilutions were spotted on LB plates with 0.2% Ara at 37 °C. CFUs were counted the next day.
Codon engineering
Codon engineering of E. coli lolB on the chromosome was performed with the λ-Red recombinase system. The 5′ end of lolB with the flanking sequence was amplified via PCR using primers with mutations to change the second and fourth codons in the MG1655 genome AUG-CCC-CUG-CCC-GAU to AUG-CCG-CUG-CCG-GAU. The PCR product was connected with the kanR sequence of pKD4 via a second PCR, followed by a third PCR to expand coverage to the entire lolB sequence for homologous recombination. The resultant PCR product was introduced into E. coli MG1655 cells expressing λ-Red recombinase from pKD46. After selection for the kanR marker and confirmation via sequencing (data not shown), the mutated locus was moved into E. coli trmD-KD with the maintenance plasmid expressing human trm5. The desired clone was selected with the kanR marker, which was subsequently removed via pCP20-mediated FLP recombination to leave a scar. The scar-carrying mutant with the engineered codon was purified from single colonies. An isogenic strain carrying the wild-type sequence was also created from trmD-KD cells with the scar sequence. LolB protein levels were determined through Western blotting.
Codon engineering of Salmonella tolC on the chromosome was accomplished with λ-Red recombination. The 5′ end of tolC with the flanking sequence was amplified by PCR with primers containing a mutation to change the sixth codon from AUG-AAG-AAA-UUG-CUC-CCC-AUC to AUG-AAG-AAA-UUG-CUC-CCG-AUC. After the second and third PCRs, recombination was performed in Salmonella LT2 cells expressing λ-Red recombinase from pKD46. The mutation was confirmed via sequencing (data not shown). A clone containing the mutation but without phage contamination was isolated using a green plate (Chan et al., 1972) and the mutated locus was transferred to Salmonella trmD-KD with the maintenance plasmid expressing human trm5. The desired clone was selected with the kanR marker, which was removed via pCP20-mediated FLP recombination to leave a scar. An isogenic strain carrying the wild-type sequence was isolated from trmD-KD cells and grown in LB at 37 °C along with the muta nt clone with 0.2% Ara. Cells were inoculated into fresh Ara-free LB at 106 CFU/mL, supplemented with 12.5 μg/mL novobiocin, and grown without pre-depletion at 37 °C. After 24 h of growth, 10-fold dilutions were spotted onto LB plates with 0.2% Ara for CFU analysis. The fold-increase of CFUs after 24 h at 37 °C relative to t = 0 was calculated and normalized to growth of the wild-type clone.
Quantification and statistical analyses
All experiments were repeated at least three times with biological replicates; mean or median values are shown. Statistical significance was determined using an unpaired, two-tailed Welch’s t test, a one-sample Student’s t test, a Wilcoxon rank-sum test, or Fisher’s exact test. Statistics were computed with R v. 3.1.3 (R Core Team, Vienna, Austria) or Microsoft Excel. Statistical significance was defined as p < 0.05.
Supplementary Material
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-LolB antibodies | (Matsuyama et al., 1997) | N/A |
| Rabbit polyclonal anti-CysRS antibodies | This paper | N/A |
| Rabbit polyclonal anti-TrmD antibodies | (Li and Bjork, 1999) | N/A |
| Rabbit polyclonal anti-hTrm5 antibodies | Sigma-Aldrich | Cat.#SAB2102581; QC18187 |
| Rabbit polyclonal anti-OmpA antibodies | (Tani et al., 1990) | N/A |
| Goat polyclonal anti-rabbit IgG antibodies peroxidase conjugate | Sigma-Aldrich | Cat. #A0545 |
| Bacterial and Virus Strains | ||
| Escherichia coli strain K-12 substrain MG1655 | ATCC | 700926 |
| E. coli strain BW25113 | The Coli Genetic Stock Center (CGSC) | CGSC#: 7636 |
| E. coli ΔacrB | CGSC | JW0451–2 |
| E. coli Δefp | CGSC | JW4107–1 |
| E. coli ΔtolC | CGSC | JW5503–1 |
| Salmonella enterica serovar Typhimurium strain LT2 | ATCC | 700720 |
| Bacteriophage P1vir | Goulian lab collection | N/A |
| Bacteriophage P22 | ATCC | 97540 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| L-(+)-arabinose | Acros Organics | Cat. #365181000 |
| D-(+)-glucose | MG Scientific | Cat. #MAL4912 |
| EcoRI | New England BioLabs | Cat. #R0101S |
| Pstl | New England BioLabs | Cat. #R0140S |
| PfuUltrall fusion HS DNA polymerase | Agilent Technologies | Cat. #600670 |
| Nuclease P1 | Sigma-Aldrich | Cat. #N8630 |
| Alkaline phosphatase | Sigma-Aldrich | Cat. #P5931 |
| 1-methylguanosine (QQQ standard) | Boc Sciences | Cat. #2140–65–0 |
| Guanosine (QQQ standard) | Sigma-Aldrich | Cat. #G6752 |
| [γ-32P]-ATP | PerkinElmer | Cat. #NEG002A |
| T4 polynucleotide kinase | New England Biolabs | Cat. #M0201 |
| AlamarBlue Dye | Invitrogen | Cat. #DAL1025 |
| Hoechst 33342 (H33342) | Sigma-Aldrich | Cat. #B2261 |
| Carbonyl cyanide m-chlorophenyl hydrazine (CCCP) | Sigma-Aldrich | Cat. #C2759 |
| Ethidium bromide | Sigma-Aldrich | Cat. #E7637 |
| Nile Red | Acros Organics | Cat. #415711000 |
| Thioflavin T | Sigma-Aldrich | Cat. #T3516 |
| Sorbitol | Sigma-Aldrich | Cat. #S1876 |
| Ampicillin | Fisher Scientific | Cat. #BP1760 |
| Carbenicillin | Fisher Scientific | Cat. #BP2648 |
| Chloramphenicol | Gold Biotechnology | Cat. #G-105 |
| Ciprofloxacin | Sigma-Aldrich | Cat. #17850 |
| Gentamicin | Gold Biotechnology | Cat. #G-400 |
| Kanamycin | Gemini Bio-products | Cat. #400–114P |
| Novobiocin | Sigma-Aldrich | Cat. #N 1628 |
| Paromomycin | Sigma-Aldrich | Cat. #P5057 |
| Polymyxin B | Sigma-Aldrich | Cat. #P4932 |
| Rifampicin | Sigma-Aldrich | Cat. #R3501 |
| Vancomycin | Sigma-Aldrich | Cat. #SBR00001 |
| Critical Commercial Assays | ||
| SuperSignal West Pico Chemiluminescent Substrate | Thermo Fisher Scientific | Cat. #34080 |
| Experimental Models: Organisms/Strains | ||
| E. coli BL21(DE3) trmD-KD | (Gamper et al., 2015a) | N/A |
| E. coli MG1655 trmD-KD | This paper | N/A |
| Salmonella enterica serovar Typhymurium LT2 trmD-KD | This paper | N/A |
| E. coli MG1655 cysS-KD | This paper | N/A |
| E. coli MG 1655 proS-KD | This paper | N/A |
| E. coli proM-KD C37-UGG tRNA | This paper | N/A |
| E. coli trmD-KD codon-engineered lolB | This paper | N/A |
| Salmonella trmD-KD codon-engineered tolC | This paper | N/A |
| Oligonucleotides | ||
| Oligo DNA primers for strain construction, plasmid construction, codon engineering, primer extension and affinity tRNA purification | Table S1 | N/A |
| Recombinant DNA | ||
| pKD4 | CGSC | CGSC #7632 |
| pKD46 | CGSC | CGSC #7634 |
| PCP20 | CGSC | CGSC #7629 |
| pZS2R | (Kelsic et al., 2015) | N/A |
| pACYC-araC-PC-PBAD-human trm5 | (Gamper et al., 2015a) | N/A |
| pACYC-araC-PC-PBAD-Ec cysS-His-deg | This paper | N/A |
| pACYC-araC-Pc-PBAD-Ec proS-His-deg | This paper | N/A |
| pKK223-3 E. coli G37-UGG tRNA | This paper | N/A |
| pKK223-3 E. coli C37-UGG tRNA | This paper | N/A |
| pZS2R-PlolB-YFP | This paper | N/A |
| Software and Algorithms | ||
| MaxQuant v. 1.5.3.30 | (Tyanova et al., 2016) | http://www.biochem.rnpg.de/5111795/maxquant |
| tRNA MS analysis software v.B.07.00 | MassHunter Workstation, qualitative analysis | N/A |
| Image Lab v. 6.0 | BIO-RAD | http://www.bio-rad.com/en-us/product/image-lab-software |
| ImageJ v. 1.51 | NIH | https://imagej.nih.gov |
| Felix32 | Photon Technology International | N/A |
| μManager v. 1.4 | (Edelstein et al.,2014) | N/A |
| MATLAB 2016b | MathWorks | https://www.mathworks.com/products/matlab.html |
| Other | ||
| NucleoSpin Gel and PCR Clean-up | Macherey-Nagel | Cat. #740609 |
| Gibson Assembly Master Mix | New England BioLabs | Cat. #E2611L |
| MicroPulser Electroporator | BIO-RAD | Cat. #1652100 |
| NucleoBond AX 2000 | Macherey-Nagel | Cat. #740525 |
| Impact II Qtof Mass Spectrometer | Bruker Daltonics | N/A |
| 0.22-μm filter | Millipore | SLGV004SL |
| 6410 QQQ triple-quadrupole LC mass spectrometer | Agilent | N/A |
| Immobilon-P PVDF Membrane | Millipore | IPVH00010 |
| Bransonic 1210 Ultrasonic Cleaner | Branson | N/A |
| Chemi-Doc XRS+ System | BIO-RAD | Cat. #1708265 |
| Typhoon IP Imaging system | GE Healthcare | N/A |
| Infinite M200 PRO plate reader | Tecan | N/A |
| Black opaque 96-well microplate | Greiner Bio-One | Cat. #655077 |
| Transparent sealing film | Excel Scientific | Cat. #STR-SEAL-PLT |
| Synergy H1 Hybrid Multi-Mode Reader | BioTek | N/A |
| Quartz cuvette | Starna Cells | Cat. #3-Q-10 |
| QuantaMaster 220 spectrofluorometer | Photon Technology International | N/A |
| Microfluidic perfusion plates | CellASIC | Cat. #B04a |
| ONIX microfluidic platform | CellASIC | N/A |
| Nikon Eclipse Ti-E inverted fluorescence microscope | Nikon | N/A |
| DU885 electron multiplying charged coupled device camera | Andor | N/A |
| Active-control environmental chamber | Haison Technology | N/A |
Highlights.
Bacterial multi-drug resistance is driven by the membrane barrier/efflux activity
Synthesis of membrane proteins requires m1G37-tRNAs translation of CC[C/U] codons
m1G37-deficient bacteria are membrane-impaired and sensitive to antibiotic killing
Deficiency of m1G37-tRNAs reduces bacterial multi-drug resistance
ACKNOWLEDGMENTS
We acknowledge support by grants from the US National Institutes of Health (GM108972 and GM114343 to YMH; GM080279 to MG) and the National Science Foundation (DMR-1120901 to MG; MCB-1149328 to KCH), the Allen Discovery Center at Stanford on Systems Modeling of Infection (to KCH), the Canadian Institutes of Health Research (CIHR MOP-77688 to LF), and a JSPS postdoctoral fellowship (to IM). KCH is a Chan Zuckerberg Biohub Investigator. We thank Chuan He, Mark Brynildsen, Hiroshi Nikaido, and Sean Moore for discussions, Hajime Tokuda and Glenn Björk for antibodies against LolB, OmpA, and TrmD, Roy Kishony for the YFP plasmid, Harvey Rubin, Trevor Selwood, Lynn Silver, and Hong-Suk Kim for discussion, Sunita Maharjan for help with experiments, Helen Zgurskaya for providing the E. coli hyper-permeable strain, and Laszlo Csonka for providing the Salmonella LT2 strain.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental information includes 7 figures, which can be found with this article online at….
Table S1: Primers used in this study. (Related to STAR Methods)
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Balaban NQ, Merrin J, Chait R, Kowalik L, and Leibler S (2004). Bacterial persistence as a phenotypic switch. Science 305, 1622–1625. [DOI] [PubMed] [Google Scholar]
- Begley U, Dyavaiah M, Patil A, Rooney JP, DiRenzo D, Young CM, Conklin DS, Zitomer RS, and Begley TJ (2007). Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Mol Cell 28, 860–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjork GR, Jacobsson K, Nilsson K, Johansson MJ, Bystrom AS, and Persson OP (2001). A primordial tRNA modification required for the evolution of life? EMBO J 20, 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnert JA, Karamian B, and Nikaido H (2010). Optimized Nile Red efflux assay of AcrAB-TolC multidrug efflux system shows competition between substrates. Antimicrob Agents Chemother 54, 3770–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauner A, Fridman O, Gefen O, and Balaban NQ (2016). Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat Rev Microbiol 14, 320–330. [DOI] [PubMed] [Google Scholar]
- Chan CT, Pang YL, Deng W, Babu IR, Dyavaiah M, Begley TJ, and Dedon PC (2012). Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat Commun 3, 937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RK, Botstein D, Watanabe T, and Ogata Y (1972). Specialized transduction of tetracycline resistance by phage P22 in Salmonella typhimurium. II. Properties of a high-frequency-transducing lysate. Virology 50, 883–898. [DOI] [PubMed] [Google Scholar]
- Chionh YH, McBee M, Babu IR, Hia F, Lin W, Zhao W, Cao J, Dziergowska A, Malkiewicz A, Begley TJ, et al. (2016). tRNA-mediated codon-biased translation in mycobacterial hypoxic persistence. Nat Commun 7, 13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Evilia C, Williams S, and Hou YM (2004). Distinct origins of tRNA(m1G37) methyltransferase. J Mol Biol 339, 707–719. [DOI] [PubMed] [Google Scholar]
- Christian T, Gamper H, and Hou YM (2013). Conservation of structure and mechanism by Trm5 enzymes. RNA 19, 1192–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, and Hou YM (2007). Distinct determinants of tRNA recognition by the TrmD and Trm5 methyl transferases. J Mol Biol 373, 623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Lahoud G, Liu C, and Hou YM (2010). Control of catalytic cycle by a pair of analogous tRNA modification enzymes. J Mol Biol 400, 204–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Sakaguchi R, Perlinska AP, Lahoud G, Ito T, Taylor EA, Yokoyama S, Sulkowska JI, and Hou YM (2016). Methyl transfer by substrate signaling from a knotted protein fold. Nat Struct Mol Biol 23, 941–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, and Wanner BL (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein A, Amodaj N, Hoover K, Vale R, and Stuurman N (2014). Computer control of microscopes using uManager. Journal of Biological Methods 1, e10.25606571 [Google Scholar]
- Edgar R, and Bibi E (1997). MdfA, an Escherichia coli multidrug resistance protein with an extraordinarily broad spectrum of drug recognition. J Bacteriol 179, 2274–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre A, Michelson AM, and Yaniv M (1971). Photochemistry of 4-thiouridine in Escherichia coli transfer RNA1Val. J Mol Biol 58, 367–379. [DOI] [PubMed] [Google Scholar]
- Frenkel-Morgenstern M, Danon T, Christian T, Igarashi T, Cohen L, Hou YM, and Jensen LJ (2012). Genes adopt non-optimal codon usage to generate cell cycle-dependent oscillations in protein levels. Mol Syst Biol 8, 572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper HB, Masuda I, Frenkel-Morgenstern M, and Hou YM (2015a). Maintenance of protein synthesis reading frame by EF-P and m(1)G37-tRNA. Nat Commun 6, 7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper HB, Masuda I, Frenkel-Morgenstern M, and Hou YM (2015b). The UGG Isoacceptor of tRNAPro Is Naturally Prone to Frameshifts. Int J Mol Sci 16, 14866–14883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs MR, Moon KM, Chen M, Balakrishnan R, Foster LJ, and Fredrick K (2017). Conserved GTPase LepA (Elongation Factor 4) functions in biogenesis of the 30S subunit of the 70S ribosome. Proc Natl Acad Sci U S A 114, 980–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Tsurumizu R, Tsukahara J, Takeda K, Narita S, Mori M, Miki K, and Tokuda H (2014). Roles of the protruding loop of factor B essential for the localization of lipoproteins (LolB) in the anchoring of bacterial triacylated proteins to the outer membrane. J Biol Chem 289, 10530–10539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill PJ, Abibi A, Albert R, Andrews B, Gagnon MM, Gao N, Grebe T, Hajec LI, Huang J, Livchak S, et al. (2013). Selective inhibitors of bacterial t-RNA-(N(1)G37) methyltransferase (TrmD) that demonstrate novel ordering of the lid domain. J Med Chem 56, 7278–7288. [DOI] [PubMed] [Google Scholar]
- Holtje JV (1998). Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol Mol Biol Rev 62, 181–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou YM, Matsubara R, Takase R, Masuda I, and Sulkowska JI (2017). TrmD: A methyl transferase for tRNA methylation with m1G37 In The Enzymes (Academic Press, Elsevier; ), pp. 89– 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou YM, Shiba K, Mottes C, and Schimmel P (1991). Sequence determination and modeling of structural motifs for the smallest monomeric aminoacyl-tRNA synthetase. Proc Natl Acad Sci U S A 88, 976–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsic ED, Zhao J, Vetsigian K, and Kishony R (2015). Counteraction of antibiotic production and degradation stabilizes microbial communities. Nature 521, 516–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Nagore D, and Nikaido H (2010). Multidrug efflux pump MdtBC of Escherichia coli is active only as a B2C heterotrimer. J Bacteriol 192, 1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodali S, Galgoci A, Young K, Painter R, Silver LL, Herath KB, Singh SB, Cully D, Barrett JF, Schmatz D, et al. (2005). Determination of selectivity and efficacy of fatty acid synthesis inhibitors. J Biol Chem 280, 1669–1677. [DOI] [PubMed] [Google Scholar]
- Krishnamoorthy G, Wolloscheck D, Weeks JW, Croft C, Rybenkov VV, and Zgurskaya HI (2016). Breaking the Permeability Barrier of Escherichia coli by Controlled Hyperporination of the Outer Membrane. Antimicrob Agents Chemother 60, 7372–7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahoud G, Goto-Ito S, Yoshida K, Ito T, Yokoyama S, and Hou YM (2011). Differentiating analogous tRNA methyltransferases by fragments of the methyl donor. RNA 17, 1236–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JN, and Bjork GR (1999). Structural alterations of the tRNA(m1G37)methyltransferase from Salmonella typhimurium affect tRNA substrate specificity. RNA 5, 395–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XZ, Nikaido H, and Poole K (1995). Role of mexA-mexB-oprM in antibiotic efflux in Pseudomonas aeruginosa. Antimicrob Agents Chemother 39, 1948–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipman RS, and Hou YM (1998). Aminoacylation of tRNA in the evolution of an aminoacyltRNA synthetase. Proc Natl Acad Sci U S A 95, 13495–13500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Clark W, Luo G, Wang X, Fu Y, Wei J, Wang X, Hao Z, Dai Q, Zheng G, et al. (2016). ALKBH1-Mediated tRNA Demethylation Regulates Translation. Cell 167, 816–828 e816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masi M, Duret G, Delcour AH, and Misra R (2009). Folding and trimerization of signal sequence-less mature TolC in the cytoplasm of Escherichia coli. Microbiology 155, 1847–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda I, Takase R, Matsubara R, Paulines MJ, Gamper H, Limbach PA, and Hou YM (2018). Selective terminal methylation of a tRNA wobble base. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S, Yokota N, and Tokuda H (1997). A novel outer membrane lipoprotein, LolB (HemM), involved in the LolA (p20)-dependent localization of lipoproteins to the outer membrane of Escherichia coli. EMBO J 16, 6947–6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed AF, Nielsen EI, Cars O, and Friberg LE (2012). Pharmacokinetic-pharmacodynamic model for gentamicin and its adaptive resistance with predictions of dosing schedules in newborn infants. Antimicrob Agents Chemother 56, 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Nakashima R, Yamashita E, Matsumoto T, and Yamaguchi A (2006). Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 443, 173–179. [DOI] [PubMed] [Google Scholar]
- Murata T, Tseng W, Guina T, Miller SI, and Nikaido H (2007). PhoPQ-mediated regulation produces a more robust permeability barrier in the outer membrane of Salmonella enterica serovar typhimurium. J Bacteriol 189, 7213–7222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasvall SJ, Chen P, and Bjork GR (2004). The modified wobble nucleoside uridine-5-oxyacetic acid in tRNAPro(cmo5UGG) promotes reading of all four proline codons in vivo. RNA 10, 1662–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarre WW, Zou SB, Roy H, Xie JL, Savchenko A, Singer A, Edvokimova E, Prost LR, Kumar R, Ibba M, et al. (2010). PoxA, yjeK, and elongation factor P coordinately modulate virulence and drug resistance in Salmonella enterica. Mol Cell 39, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H (1998). Antibiotic resistance caused by gram-negative multidrug efflux pumps. Clin Infect Dis 27 Suppl 1, S32–41. [DOI] [PubMed] [Google Scholar]
- Okusu H, Ma D, and Nikaido H (1996). AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J Bacteriol 178, 306–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne DJ, Gwynn MN, Holmes DJ, and Pompliano DL (2007). Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 6, 29–40. [DOI] [PubMed] [Google Scholar]
- Prindle A, Liu J, Asally M, Ly S, Garcia-Ojalvo J, and Suel GM (2015). Ion channels enable electrical communication in bacterial communities. Nature 527, 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu Y, Zhao Z, Li Y, Zou J, Ma Q, Zhao Y, Ke Y, Zhu Y, Chen H, Baker MAB, et al. (2016). Enhanced Efflux Activity Facilitates Drug Tolerance in Dormant Bacterial Cells. Mol Cell 62, 284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapino F, Delaunay S, Rambow F, Zhou Z, Tharun L, De Tullio P, Sin O, Shostak K, Schmitz S, Piepers J, et al. (2018). Codon-specific translation reprogramming promotes resistance to targeted therapy. Nature 558, 605–609. [DOI] [PubMed] [Google Scholar]
- Rojas E, Theriot JA, and Huang KC (2014). Response of Escherichia coli growth rate to osmotic shock. Proc Natl Acad Sci U S A 111, 7807–7812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas ER, Billings G, Odermatt PD, Auer GK, Zhu L, Miguel A, Chang F, Weibel DB, Theriot JA, and Huang KC (2018). The outer membrane is an essential load-bearing element in Gram-negative bacteria. Nature 559, 617–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz N, Falcone B, Kahne D, and Silhavy TJ (2005). Chemical conditionality: a genetic strategy to probe organelle assembly. Cell 121, 307–317. [DOI] [PubMed] [Google Scholar]
- Sakaguchi R, Giessing A, Dai Q, Lahoud G, Liutkeviciute Z, Klimasauskas S, Piccirilli J, Kirpekar F, and Hou YM (2012). Recognition of guanosine by dissimilar tRNA methyltransferases. RNA 18, 1687–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi R, Lahoud G, Christian T, Gamper H, and Hou YM (2014). A divalent metal ion-dependent N(1)-methyl transfer to G37-tRNA. Chem Biol 21, 1351–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T, Situ AJ, and Ulmer TS (2016). Structural and thermodynamic basis of proline-induced transmembrane complex stabilization. Sci Rep 6, 29809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlaes DM, Shlaes JH, Davies J, and Williamson R (1989). Escherichia coli susceptible to glycopeptide antibiotics. Antimicrob Agents Chemother 33, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver LL (2011). Challenges of antibacterial discovery. Clin Microbiol Rev 24, 71–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver LL (2012). Rational approaches to antibacterial discovery: Pre-genomic directed and phenotypic screening In Antibiotic discovery and development, Dougherty TJ, and Pucci MJ, eds. (Springer Science + Business Media; ), pp. 33–75. [Google Scholar]
- Tani K, Tokuda H, and Mizushima S (1990). Translocation of ProOmpA possessing an intramolecular disulfide bridge into membrane vesicles of Escherichia coli. Effect of membrane energization. J Biol Chem 265, 17341–17347. [PubMed] [Google Scholar]
- Thein M, Sauer G, Paramasivam N, Grin I, and Linke D (2010). Efficient subfractionation of gram-negative bacteria for proteomics studies. J Proteome Res 9, 6135–6147. [DOI] [PubMed] [Google Scholar]
- Tommasi R, Brown DG, Walkup GK, Manchester JI, and Miller AA (2015). ESKAPEing the labyrinth of antibacterial discovery. Nat Rev Drug Discov 14, 529–542. [DOI] [PubMed] [Google Scholar]
- Tsukahara J, Mukaiyama K, Okuda S, Narita S, and Tokuda H (2009). Dissection of LolB function--lipoprotein binding, membrane targeting and incorporation of lipoproteins into lipid bilayers. FEBS J 276, 4496–4504. [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, and Cox J (2016). The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc 11, 2301–2319. [DOI] [PubMed] [Google Scholar]
- Typas A, Banzhaf M, Gross CA, and Vollmer W (2011). From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol 10, 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg van Saparoea, Lubelski HB, van Merkerk J, Mazurkiewicz R, S. P, and Driessen AJ (2005). Proton motive force-dependent Hoechst 33342 transport by the ABC transporter LmrA of Lactococcus lactis. Biochemistry 44, 16931–16938. [DOI] [PubMed] [Google Scholar]
- White TA, and Kell DB (2004). Comparative genomic assessment of novel broad-spectrum targets for antibacterial drugs. Comp Funct Genomics 5, 304–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yohannan S, Yang D, Faham S, Boulting G, Whitelegge J, and Bowie JU (2004). Proline substitutions are not easily accommodated in a membrane protein. J Mol Biol 341, 1–6. [DOI] [PubMed] [Google Scholar]
- Young K, and Silver LL (1991). Leakage of periplasmic enzymes from envA1 strains of Escherichia coli. J Bacteriol 173, 3609–3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.