

Abstract
Background
Very small genomes have evolved repeatedly in eubacterial lineages that have adopted obligate associations with eukaryotic hosts. Complete genome sequences have revealed that small genomes retain very different gene sets, raising the question of how final genome content is determined. To examine the process of genome reduction, the tiny genome of the endosymbiont Buchnera aphidicola was compared to the larger ancestral genome, reconstructed on the basis of the phylogenetic distribution of gene orthologs among fully sequenced relatives of Escherichia coli and Buchnera.
Results
The reconstructed ancestral genome contained 2,425 open reading frames (ORFs). The Buchnera genome, containing 564 ORFs, consists of 153 fragments of 1-34 genes that are syntenic with reconstructed ancestral regions. On the basis of this reconstruction, 503 genes were eliminated within syntenic fragments, and 1,403 genes were lost from the gaps between syntenic fragments, probably in connection with genome rearrangements. Lost regions are sometimes large, and often span functionally unrelated genes. In addition, individual genes and regulatory regions have been lost or eroded. For the categories of DNA repair genes and rRNA genes, most lost loci fall in regions between syntenic fragments. This history of gene loss is reflected in the sequences of intergenic spacers at positions where genes were once present.
Conclusions
The most plausible interpretation of this reconstruction is that Buchnera lost many genes through the fixation of large deletions soon after the acquisition of an obligate endosymbiotic lifestyle. An implication is that final genome composition may be partly the chance outcome of initial deletions and that neighboring genes influence the likelihood of loss of particular genes and pathways.
Background
Genome sizes in the eubacteria and archaebacteria range from 0.58 megabases (Mb) to around 10 Mb [1]. The smallest of these genomes do not represent ancestral states, as was once believed, but are derived from larger genomes through massive loss of genes. The conclusion that small genome size is evolutionarily derived is based on a combination of molecular phylogenetic results, genome size determinations and full genome sequences [2,3,4,5,6,7,8,9]. Extreme genome reduction, producing bacterial genome sizes in the range of 1 Mb or less, is closely correlated with symbiotic or pathogenic lifestyles involving obligate associations with eukaryotic hosts. Thus, when lineages make the transition from potentially free-living lifestyles to obligately host-associated ones, genome reduction ensues.
An immediate question is whether this reduction occurs in large steps, involving relatively few large losses, or whether it is entirely gradual, consisting of loss of individual genes one-by-one. Reconstruction of steps leading to genome reduction requires comparative analysis of related small and large genomes. Public databases now contain complete sequences for many bacterial genomes of varying sizes, but most of the fully sequenced genomes under 1.5 Mb are very distantly related to genomes of larger size. For example, the smallest genome, from Mycoplasma genitalium (0.58 Mb) belongs to the Mollicutes, a large and ancient clade that consists entirely of bacteria with reduced genomes [2,10,11]. Likewise, Rickettsia prowazekii (1.1 Mb) is embedded in a large clade within the alpha-Proteobacteria that contains only small-genome, intracellular inhabitants such as Ehrlichia, Wolbachia pipientis and mitochondria [12]. Similarly, the Chlamydiae, including several fully sequenced organisms, are an ancient clade, all characterized by small genomes (1.0-1.2 Mb) [13]. This phylogenetic distribution hinders reconstruction of the large-genome ancestors of pathogenic lineages and of the events leading from a large genome to a very small one.
Among fully sequenced published genomes, the only instance of a highly reduced genome that shows a close relationship to large genomes is Buchnera aphidicola, the obligate endosymbiont of aphids (Insecta). On the basis of gene content and similarity of orthologous genes, Buchnera is closely related to enteric bacteria, including Escherichia coli (gamma-3 Proteobacteria). The Buchnera endosymbiont of the aphid Acyrthosiphon pisum has a genome of 643 kb [8], only one-seventh the size of the genome of E. coli MG1655 (4.6 Mb) [14]. The E. coli size is similar to that of other enterics such as Salmonella, Klebsiella and Yersinia [1]. The Buchnera gene inventory, consisting of only 564 ORFs, 32 tRNAs and a single copy of each rRNA gene, is essentially a subset of that of E. coli [8]. (The four annotated genes lacking obvious homology with E. coli genes seem to be either recently lost in E. coli or fast-evolving genes or pseudogenes for which orthology would be difficult to detect (I. Tamas et al.; unpublished results)). Orthologous pairs show an average amino-acid identity of 62% and 16S rDNA identity of 89% between E. coli and Buchnera.
Buchnera is a mutualistic endosymbiont of its host, but the pattern of reductions of numbers of genes among functional categories is similar between Buchnera and other fully sequenced small-genome bacteria, all obligate pathogens [8,12,15,16]. The major exception is that the Buchnera genome contains 55 loci (10% of the genome) that specify the biosynthesis of amino acids needed by its host [8]. In contrast, pathogenic species with small genomes have lost these loci and acquire amino acids from host cells. Both Buchnera and small-genome pathogens show reduction in numbers of genes in many functional categories, including biosynthesis of metabolic intermediates, basic cellular processes (transcription, translation, replication, cell division), biosynthesis of phospholipids, and repair and recombination. Small-genome bacteria show convergent features, including accelerated sequence evolution and genome-wide base compositional bias favoring A and T [5,17].
In attempting to characterize the evolutionary forces underlying genome reduction, a critical question concerns the size and content of the deletions. If genes or operons have been eliminated individually in separate events, their loss may be governed by the functional roles and independent fitness effects of individual loci. But, if a substantial portion of the reduction has occurred as large deletion events spanning many kilobases and functionally unrelated genes, the set of genes lost is more reasonably interpreted as the result of selection acting on the composite fitness effects of the set of loci deleted. The content of early deletions is expected to affect the strength of selection for retention of other genes. Thus, the final gene inventory might depend in part on chance combinations of gene order and deletions occurring early in the process of genome reduction. If large deletions have a role in the early stages of genome reduction, then large contiguous regions of the ancestral genome will be found to be missing from the reduced genome.
Here we examine the process of genome reduction in the lineage leading to Buchnera through comparison of its genome with that of a hypothetical ancestor reconstructed on the basis of the genomes of E. coli MG1655 and several other sequenced genomes of enteric bacteria.
Results
Gene inventory of the reconstructed ancestor relative to that of Buchnera
A total of 2,425 protein-coding genes were retained in the ancestor, out of a total of 4,351 genes in E. coli. Under the procedures for reconstructing the ancestral genome (A in Figure 1), a requirement for inclusion in the ancestor was presence in E. coli MG1655. As a result, the reconstructed genome is expected to be smaller than that of the real ancestor, because it does not include genes that were present in the ancestor but lost independently from both E. coli and Buchnera lineages. Despite the removal of 44% of the E. coli genes, over 99% of genes present in Buchnera were also retained in the reconstructed ancestor. This outcome strongly supports this parsimony approach to reconstructing the gene inventory of the shared ancestor of E. coli and Buchnera, and also of the hypothesis that Buchnera evolved from such an ancestor through gene loss. Only two of the 560 protein-coding genes that show clear orthology between E. coli and Buchnera would have been removed (due to absence from both Vibrio cholerae and Yersinia pestis). The two genes (dnaC and dnaT) are linked in both Buchnera and E. coli and were retained in the ancestor.
Figure 1.
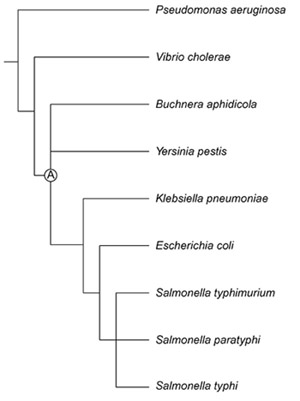
Phylogenetic relationships of Buchnera, E. coli and related taxa used in reconstructing the genome of the free-living ancestor (A) of Buchnera.
Rearrangements in the Buchnera lineage
Fragments for which the order and orientation of genes are the same in Buchnera and E. coli were assumed to be present in the common ancestor. A syntenic fragment was recognized if E. coli and Buchnera showed the same order and orientation apart from missing genes in Buchnera, and if these missing genes could not be located elsewhere in the Buchnera genome (example in Figure 2). Syntenic regions always terminated with loci that were present in both species.
Figure 2.
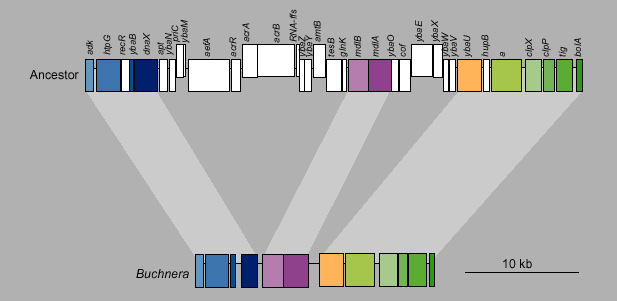
Part of a syntenic fragment from Buchnera and the ancestor (same as E. coli for this region). Deleted loci are white in the ancestor; orthologous genes are color-coded. Genes shifted up in the figure are oriented forward in the genome; genes shifted down are oriented backwards.
This criterion resulted in 91 fragments in the ancestor that contained at least two genes and that corresponded to regions of synteny between Buchnera and E. coli (Figures 3,4). In addition, there were 62 single genes. The longest syntenic fragment, containing the megaoperon of ribosomal proteins that is widely conserved among bacteria, consisted of 89 kb spanning 77 genes in the ancestor and 45 genes in Buchnera. In addition, there were 143 ancestral regions between these retained fragments, containing genes inferred to be lost in Buchnera. (This number is slightly less than the number of retained fragments (153) because, whereas most of the retained fragments were flanked by lost regions, a few directly bordered other retained fragments.) The ancestral genome was therefore divided into a total of 306 fragments, just over half of which were retained in the Buchnera genome, and there are 306 junctions between these fragments on the circular chromosome (Figure 3).
Figure 3.
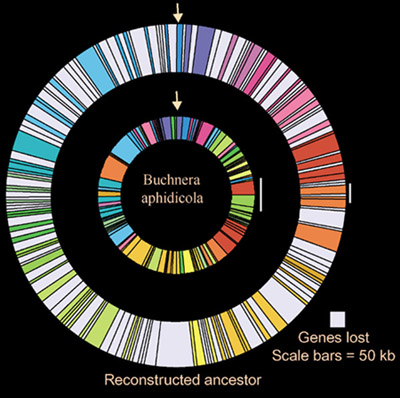
Graphic depiction of syntenic fragments and lost regions in the genome of the reconstructed ancestor and in Buchnera. Syntenic fragments are color-coded based on position in the ancestor. Lost regions occurring between syntenic fragments are gray.
Figure 4.
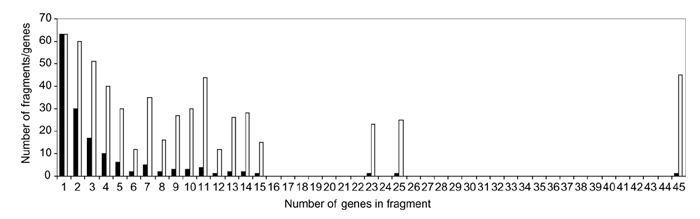
Sizes of Buchnera regions that are syntenic with regions in the reconstructed ancestor. Solid bars, number of fragments; open bars, number of genes.
The evolution of Buchnera was clearly accompanied by many chromosome rearrangements and massive changes in genome size (Figures 3,5). Remarkably, a comparison of ortholog positions between Buchnera and E. coli indicates that a pattern of approximate symmetry of gene distance from the replication origin and terminus was maintained (Figure 6). This is indicated by an X-pattern when the positions in the two genomes are plotted against one another with the origins of replication as endpoints (the replication origin in Buchnera was designated by Shigenobu et al. [8] on the basis of the position of the only DnaA box present on the chromosome and on a shift in the GC-skew value at third codon positions around that region). This X-pattern has recently been found to be typical for comparisons of orthologous regions between pairs of closely related bacteria [18,19,20]. It is most readily explained as the result of successive inversions around the replication terminus or origin.
Figure 5.
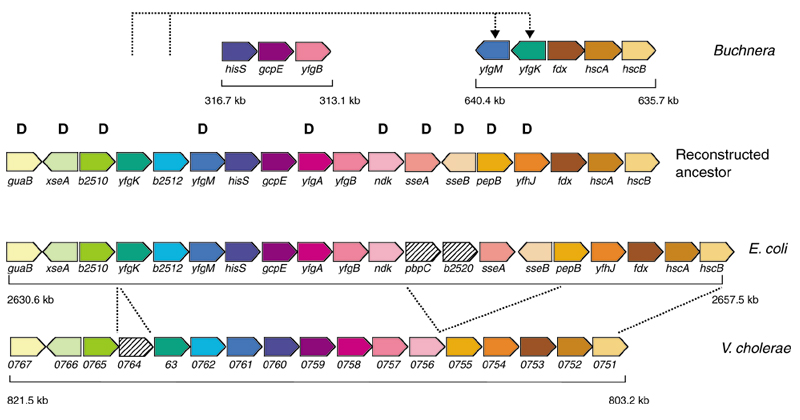
Region rearranged in the Buchnera lineage, based on the order in Vibrio cholerae and E. coli. Orthologous genes are in matching colors. In Buchnera, yfgK and yfgM have been translocated and inverted. Numbers under the genes denote genomic position and size. Genes marked with 'D' were eliminated in the evolution of Buchnera.
Figure 6.
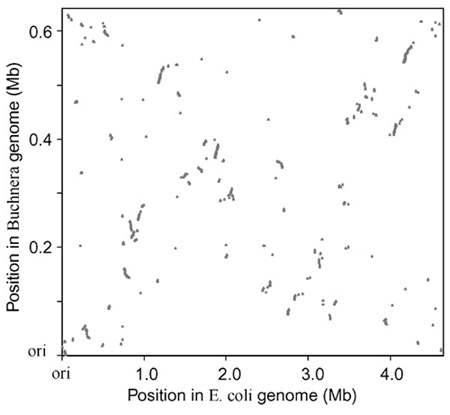
Relative positions of orthologs in the Buchnera and the E. coli genomes, with the origin of replication for each genome at the origin. Each gene is positioned by its starting base within each genome. Despite the difference in genome size, an X-pattern is evident, indicating preservation of the relative absolute distance from the origin of replication.
Genes lost as deletions within and between regions of synteny
On the basis of the reconstructed ancestor, a total of 503 genes in 156 locations were lost in deletions within regions of synteny (Figure 7, top). These deleted regions are sometimes large, with 11 regions spanning 10 or more genes (Figure 7, top). A much larger proportion of the reduction can be attributed to regions lost between syntenic fragments; 1,449 genes were lost in 143 such gaps (Figure 7, bottom).
Figure 7.
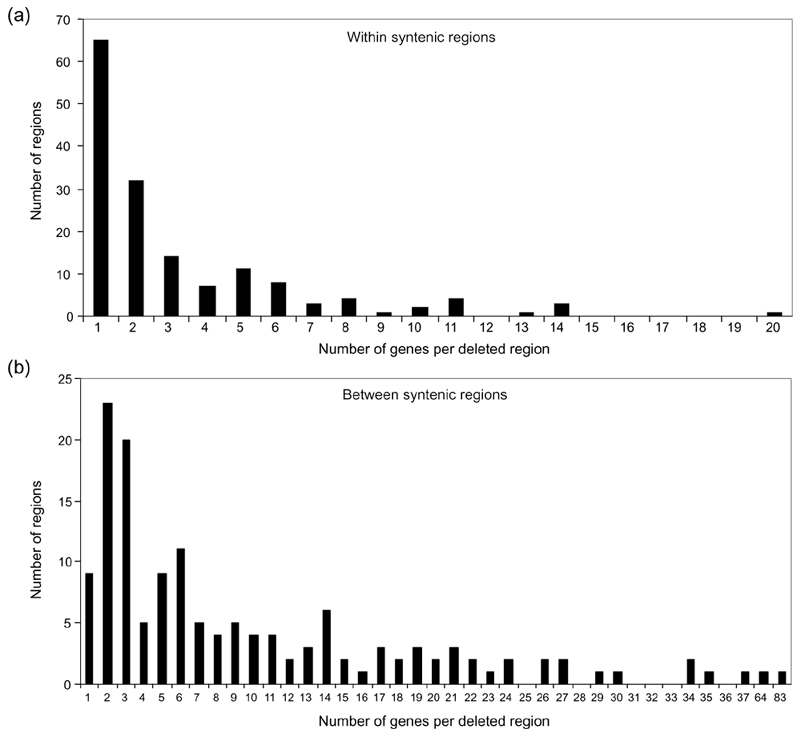
Numbers of genes in deleted regions. (a) Numbers of genes in deleted regions occurring within fragments syntenic between Buchnera and its ancestor. (b) Numbers of genes in deleted regions occurring between fragments syntenic between Buchnera and its ancestor.
For losses occurring both within and between syntenic fragments, a single lost region frequently contains multiple genes, often involved in seemingly unrelated functions (see, for example, Figures 2 and 5).
Gene erosion and spacer lengths
Some genes deleted within regions of synteny persist as partially degraded sequences that range from pseudogenes having clear homology with the E. coli ortholog [8], to much shortened sequences with no recognizable sequence homology. These sequences indicate that function has sometimes been eliminated, initially through a small deletion or substitution, with the remaining DNA sequence subsequently eroded by multiple mutations that are biased in favor of deletion over insertion, as documented for Rickettsia [7,9] and other bacteria including Buchnera [21]. An analysis of Buchnera pseudogenes showed that deletions outnumber insertions (31 versus 2) and that the number of nucleotides deleted is over 100-fold greater than those inserted [21].
An expected result of this gradual gene degradation is that intergenic spacers that contain gene remnants will be longer than spacers where no gene loss has occurred. Buchnera spacers can be categorized into three groups: those flanked by the same genes in Buchnera and the ancestor, those that occur within regions of synteny at positions where gene(s) are missing in Buchnera, and those that occur between syntenic fragments. Spacers in the first category, which can be considered to be descended from ancestral spacers, have an average length of 55 bp (n = 272). These ancient spacers are much shorter than spacers occurring where ancestral genes have been deleted within syntenic fragments (188 bp, n = 165) or than spacers occurring between syntenic fragments (also 188 bp, n = 162). Considering only spacers within syntenic fragments, spacer length does not increase further when the number of genes lost is greater than one (Figure 8).
Figure 8.
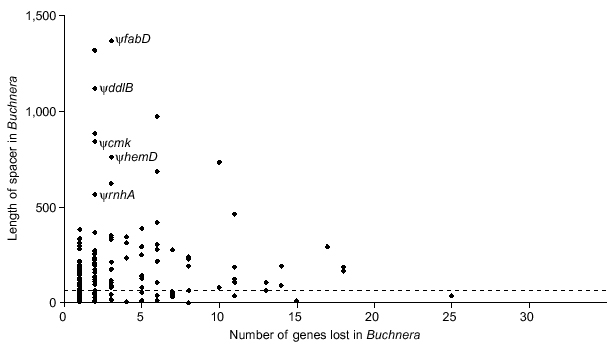
Lengths of intergenic spacers in Buchnera relative to number of genes deleted from the corresponding position within syntenic fragments. Five spacers that included remnants of recognizable pseudogenes (ψ) are indicated. The dotted line indicates the average length of spacers at sites where no genes have been eliminated (mean = 55 nucleotides, N = 272).
Promoter loss and fusion of transcription units
Small bacterial genomes typically contain a smaller proportion of regulatory elements than do larger genomes [22,23]. Consistent with this trend, the genome of Buchnera has been noted to lack virtually all regulatory proteins [8]. We examined promoters within intergenic spacers flanked by the same genes in both Buchnera and E. coli under the assumption that these evolved from a common ancestral sequence and thus represent orthologous spacers. In 44 of the orthologous spacers, a σ-70 promoter is annotated in E. coli on the basis of experimental evidence and/or presence of a promoter consensus sequence [14]. The consensus sequence for this promoter is TTGACA-(l7 nucleotides)-TATAAT, from which the -35 and -10 regions are each defined primarily by three base pairs:TTG--- for the -35 region and TA---T for the -10 region [24,25]. We applied a conservative criterion for presence of a promoter in Buchnera, by requiring retention of as few as four of these six nucleotides with a separation of 16-18 nucleotides. In 16 of the 33 spacers in which flanking genes are encoded on the same strand and in which E. coli has an annotated promoter, Buchnera lacks any recognizable promoter. All such apparent losses occurred when the flanking genes were encoded on the same strand, suggesting that promoter loss is frequent when a group of newly contiguous genes can be translated as a single transcriptional unit. In some cases, fusion of genes into the same polycistron has occurred following the loss of intervening genes oriented in the opposite direction (Figure 9). In contrast, in none of the 11 cases in which orthologous spacers were located between genes oriented in opposite directions did Buchnera lose promoters.
Figure 9.
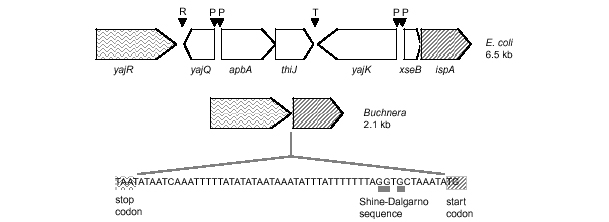
Example of a syntenic fragment in which Buchnera has lost genes of opposite orientation to flanking genes and has fused the new neighbors into a polycistron, through loss of intervening promoters. R = repetitive sequence; P = promoter sequence; T= termination sequence.
On the basis of sequence criteria, Buchnera also shows degeneration of the Shine-Dalgarno (SD) sequences that promote initiation of translation by binding with the anti-SD complement in rRNA (which is widely conserved and identical for Buchnera and E. coli). Changes in the SD sequence can impede initiation of translation [26,27]. In comparisons of spacers orthologous between E. coli and Buchnera, Buchnera showed a lower number of matches to the core eight nucleotides of the SD sequence in 37 of 51 cases (73%). Also, a total of 20 protein-binding sites were annotated within E. coli spacers orthologous to Buchnera spacers (that is, with the same flanking genes in the same orientation). In Buchnera, only one was preserved, four had half of the sequence maintained, and the remaining 15 were not detectable.
These hypothetical losses of regulatory regions from the reduced genome of Buchnera are based on levels of sequence similarity with the known consensus sequence in modern E. coli. There is no experimental evidence to eliminate the possibility that the degraded promoters in Buchnera are still functional, or that the symbiont uses different promoters from those identified in E. coli. In addition, inaccuracies in the annotated positions of Buchnera genes, in which positions of start codons are not verified experimentally, could bias analyses of SD sequences.
Discussion
Genetic drift as a basis for genome reduction
Endosymbiosis or chronic pathogenesis involves a metabolite-rich environment within host tissues, low growth rates and isolation of strains within host individuals. This combination of factors results in relaxed selection at many loci and higher levels of genetic drift affecting the entire genome. The reduced effectiveness of selection is reflected in patterns of sequence evolution in Buchnera and other endosymbionts [17,28,29]. It is also expected to affect genome evolution, accelerating the loss of genes that are entirely superfluous as well as those that are beneficial but not essential.
Small genome size itself is likely to reflect lack of selection for gene retention rather than direct selection for a compact genome [21]. Especially in the case of Buchnera, which possesses 50-200 chromosomes per cell [30] and in which nonfunctional pseudogenes can persist for long periods [31], selection for reduced DNA content seems an untenable explanation for its small genome. The polyploidy itself may be a consequence of the loss, through genetic drift, of one or more loci involved in regulating and resolving chromosome replication during the cell cycle.
One pattern that has emerged from comparative analyses of fully sequenced genomes is that, although different pathogen and symbiont lineages approach the same minimal genome size, the inventories of genes retained by these organisms are extremely different [3,32,33,34,35,36]. Relatively few genes are universally distributed, and universal cellular processes depend in part on nonorthologous genes. The divergence in gene inventories among small-genome bacteria implies redundancy in ancestral genomes underlying central cell processes, including DNA processing, transcription and translation. For the most part, this redundancy does not arise from the presence of paralogous genes, which are relatively few in bacteria. An examination of the process of genome reduction, as exemplified by Buchnera, could yield insight into why gene inventories differ among small genomes.
Deletion sizes in the evolution of Buchnera
The comparison of the Buchnera genome to the reconstructed ancestral genome suggests that a considerable part of genome reduction occurred through large deletions accompanying chromosome rearrangements. One indicator that supports gene loss partly through large deletions spanning multiple genes is the distribution of sizes of these regions in the ancestor (Figure 7, bottom). If genes were lost one by one, the expectation is that retained genes would be randomly mixed with lost genes within the ancestral genome. In the observed distribution, lost genes are aggregated. Although part of this clustering might be attributed to the linkage of functionally related genes, this would not account for the larger segments. Another indicator of gene loss through large deletions is the lack of positive relationship between spacer length and number of genes lost for gene deletions that occurred within syntenic fragments (Figure 8). Thus, the most plausible explanation for much of the gene loss occurring in the evolution of Buchnera is the fixation of single large deletions spanning many genes, although it is also clear that some genes were eliminated through smaller deletions and gradual erosion. Large deletions spanning multiple genes are possible only early in the reductive process, when many genes are nonessential. The content of initial large deletions will determine the degree of selection on remaining loci and thus will govern the ultimate composition of the reduced genome.
Loss of RNA genes
One distinctive aspect of the Buchnera genome is the presence of only one copy each of the genes encoding 16S, 23S and 5S rRNA, and the separation of these genes into two transcriptional units with the 16S rRNA gene (rrs) apart from the others (rrf and rrl) [37]. Typically, bacteria possess multiple rRNA operons, each containing all three genes; the Buchnera ancestor is inferred to have possessed at least five operons and modern E. coli has seven. The remaining rRNA genes descend from two different ancestral operons, with other rRNA operons lost entirely. From flanking genes, it is evident that the two retained transcription units correspond to rrsH and rrfD-rrlD of E. coli. The missing parts of these operons (rrfH, rrlH, and rrsD) were lost in deletions within syntenic fragments. All other rRNA operons were lost in entirety as part of regions occurring between syntenic fragments. Thus, the modern number and arrangement of rRNA genes is the result of the loss of large regions, possibly in the course of rearrangements, as well as the elimination of individual genes within operons.
E. coli has 86 tRNAs, and most of these appear to be present in the ancestor (the parsimony criterion for presence of a tRNA in the ancestor, based on distribution in sequenced genomes, is somewhat unreliable because of sequence homology among different tRNA genes). Buchnera has only 32 tRNAs, with most amino acids having only one. Assuming that the ancestor had the same complement of tRNAs as E. coli, 15 were lost from within syntenic fragments and 39 were lost in regions between syntenic fragments. Selection enforces the retention of at least one tRNA for each amino acid, as observed in modern Buchnera, so the early fixation of large deletions containing some tRNAs would have created a selective requirement for the retention of others.
Loss of DNA repair pathways
One example of a functional category that is routinely reduced in small-genome bacteria is that consisting of genes for DNA repair and recombination. As is typical for small genome bacteria, Buchnera has lost a large number of repair genes (Table 1). In this functional category, as in others, the set of retained genes differs among reduced genomes [15]. For example, Buchnera is unique among sequenced genomes in having lost recA, which functions in homologous recombination and repair. In contrast, Buchnera retains recBCD, which has been lost by several other small genomes [8,12,15]. The reconstructed events underlying the loss of individual repair genes include many large deletions, encompassing many genes (Table 1). For example, under the reconstruction, recA is part of a contiguous deleted region of about 10 kb containing ten genes. Also unusual in Buchnera is the lack of uvrA, uvrB and uvrC (encoding an excision nuclease involved in repair of UV damage to DNA). The uvrB and uvrC genes fall in separate large deleted regions between syntenic fragments, whereas uvrA is one of six genes deleted within a syntenic region, with the corresponding position in Buchnera occupied by a 618 bp spacer (Table 1).
Table 1.
Characteristics of deleted regions containing DNA repair loci that have been lost during the evolution of Buchnera
| Deleted gene | Functional role | Within/between syntenic fragments | Size of deleted region (nucleotides) | Number of genes in deleted region |
| ada | Direct damage reversal | Between | 25,426 | 27 |
| ogt | Direct damage reversal | Between | 88,327 | 83 |
| tag | Base excision repair | Between | 33,065 | 30 |
| mutM | Base excision repair | Between | 809 | 1 |
| mutH | Mismatch repair | Within | 9,601 | 8 |
| recJ | Mismatch repair | Within | 4,533 | 6 |
| uvrD | Mismatch repair | Between | 11,325 | 12 |
| recA | Recombinase pathway | Between | 9,884 | 10 |
| recF | Recombinase pathway | Within | 1,073 | 1 |
| recN | Recombinase pathway | Within | 1,661 | 1 |
| uvrA | UV excision repair | Within | 6,579 | 6 |
| uvrB | UV excision repair | Between | 60,515 | 64 |
| uvrC | UV excision repair | Between | 27,558 | 34 |
The most plausible interpretation of this pattern is that some of these repair functions were initially lost as the result of large deletions, sometimes occurring in combination with chromosomal rearrangements. These deletions were followed by gradual loss of other genes in the same pathways. The process of fixation of these large deletions reflects not only selection on the repair functions but also selection on other genes on the deleted fragments. A gene flanked by required loci is less likely to be lost in an early large deletion than a gene flanked by nonessential loci. For example, if the 10 kb segment containing recA was lost as a single deletion, then the loss of recA was dependent on the fact that the neighboring genes included in the deletion were not essential for survival. The retention of recBCD might have been promoted by Buchnera's requirement for the flanking gene, argA, which encodes an enzyme for biosynthesis of arginine. Other small-genome bacteria lack argA and other genes underlying amino-acid biosynthesis, whereas Buchnera retains all genes required for biosynthesis of essential amino acids, which are needed by the host.
Many genes were lost singly, implying insufficient selection for conservation of individual genes. A consequence is the correlation in presence/absence among genes in the same pathway, even if they occupy separate locations on the chromosome: if pathway function is lost, all genes in the pathway are invariably lost or degraded. An example in the case of the recombinase genes is the loss of recF, which is required for some recA functions [38] and which was deleted individually with both flanking genes retained (Table 1). A plausible scenario for the loss of this pathway is that, once recA was eliminated in the context of a large deletion, there was no selection for recF retention and it was subject to successive small deletions and substitutions. In both Buchnera of A. pisum and Buchnera of Schizaphis graminum, recF is replaced by 127-138 nucleotides, with base composition 87-89% A+T [8,39]. The process of shrinkage and A+T accumulation could result strictly from mutational patterns, which are biased towards deletions [21] and nucleotide substitutions ending in A or T [31].
Large deletions in other bacterial genomes
Genome comparisons across other groups of related bacteria suggest that deletion events encompassing multiple loci are frequent in bacterial evolution. For example, deletions of over 20 kb and 20 loci have occurred in natural isolates of E. coli that are very closely related on the basis of sequence homology [40]. On the basis of comparison with its close relative Mycobacterium tuberculosis, M. leprae shows a pattern of genome reduction that includes both loss of large fragments and also loss of gene function through slight changes in sequence [41]. Different strains of M. tuberculosis have been found to contain at least 25 long deletions, with one event removing as many as 16 ORFs [42].
Consequences of genome reduction for gene regulation
The changes in sequences underlying initiation of both transcription and translation, as well as the loss of regulatory proteins [8], give an impression of genome-wide degeneration of regulatory functions in Buchnera. The hypothesized elimination of promoters and genes seems to have produced newly formed polycistronic regions (Figure 9). The fusion of genes into single transcriptional units has been interpreted as the result of selection favoring small genome size [43] or favoring efficiency in transcription or translation [44]. In Buchnera, however, the hypothesized loss of promoters suggests a general genomic decay influencing transcriptional regulation; this is supported by the observation that SD sequences are also degenerate, and by the finding that some of the most frequently used codons in the genome do not have corresponding tRNAs because they have been deleted throughout Buchnera's evolution. Experiments on gene expression patterns are needed to test the potential deterioration of regulatory capabilities.
Conclusions
Buchnera provides the first case of a fully sequenced, highly reduced genome for which closely related large genomes exist and have been sequenced, allowing reconstruction of the steps in genome reduction. The most plausible interpretation of the deletion of large contiguous regions during the evolution of Buchnera is that the transition to the endosymbiotic lifestyle was accompanied, or soon followed, by large deletions. The location and content of early deletions might well have shaped the ultimate gene inventory of the fully reduced genome found in modern Buchnera.
The similarity in genome size across Buchnera species [45] suggests that most genome reduction occurred in the shared ancestral lineage and that the ancestor of modern Buchnera already had reached a near-minimum size. The extremely stable genome content of modern Buchnera is also indicated by comparison of the full genome sequences of Buchnera of A. pisum and Buchnera of Schizaphis graminum (I. Tamas et al. unpublished results).
The ability to examine further why some genes are retained and some are lost will be improved as additional closely related genomes of different sizes are sequenced and annotated. The gamma-3 Proteobacteria contain free-living bacteria with large genomes and also numerous symbiotic lineages showing varying degrees of reduction in genome size [46,47]. With improved ability to reconstruct ancestral genomes, this group will provide an excellent opportunity to determine the role of chance and selection in the determination of genome content and in the evolution of gene regulatory systems.
Materials and methods
Reconstruction of the ancestor
Phylogenetic studies based on 16S rDNA sequences indicate that Buchnera belongs to the gamma-3 Proteobacteria and is closely related to E. coli (Figure 1). The precise phylogenetic position of Buchnera has varied depending on the method of analysis and the taxa included in the analysis. This instability is partially the result of the accelerated evolution and AT bias that affect all Buchnera genes including the 16S rDNA [17]. Most studies place Buchnera either within or just outside the Enterobacteriaceae, suggesting that it diverged near the time of the common ancestor of this clade [48,49,50,51]. Thus, the reconstructed ancestor of the clade corresponding to Enterobacteriaceae (A in Figure 1) was considered to constitute a close approximation of the free-living ancestor that gave rise to Buchnera.
The genes absent from Buchnera and present in E. coli include many that are present in other related bacteria such as Haemophilus influenzae and V. cholerae. This supports the view that Buchnera was derived through loss of genes from an ancestor similar to E. coli, a conclusion also reached by Shigenobu et al. [8]. However, some genes were acquired recently by E. coli, after divergence from the Buchnera lineage [52]. The ancestral genome reconstruction was based on the E. coli MCl655 genome [14], following the removal of genes that seemed to have been acquired after the E. coli-Buchnera divergence. The rules for removing a gene were based on phylogenetic distribution of orthologs among the unannotated genomes of three serovars of Salmonella enterica (sv. Typhi, sv. Paratyphi and sv. Typhimurium), Klebsiella pneumoniae and Yersinia pestis and the annotated genome of V. cholerae (GenBank accession number NC 002505) [53]. The unpublished sequence data were produced by the Salmonella, Klebsiella and Yersinia Sequencing Groups at the Sanger Centre (for S. enterica sv. Typhi, K. pneumoniae and Y. pestis) [54] and the Washington University Genome Sequencing Center (for S. enterica sv. Typhimurium and S. enterica sv. Paratyphi) [55]. Determination of phylogenetic distribution was based on the 'Pip-maker' analyses [56] for viewing orthology among enteric and related bacteria as produced by McClelland et al. [57,58]. To qualify as an ortholog to an E. coli gene, a sequence had to show at least 40% amino-acid identity for at least half of the E. coli gene.
Inclusion of a gene in the ancestor was not dependent on its presence in all the descendant taxa, as a gene could be ancestral but lost from some lineages or acquired after the ancestor but before divergence of some descendant taxa. Therefore, to be retained in the ancestor, an ortholog had to be present in at least one of the close relatives of E. coli (S. enterica sv. Paratyphi, Typhi or Typhimurium, or K. pneumoniae) and also in at least one of the more distant relatives, either Y. pestis (representing a more basal branch of the Enterobacteriaceae) or V. cholerae (branching just outside the Enterobacteriaceae). These requirements effectively impose a parsimony criterion, minimizing the number of gains and losses of genes. This criterion is likely to be valid for most genes across this relatively shallow phylogeny. Sequences most likely to be misrepresented in the ancestor by this reconstruction are phage genes or insertion elements, which may move in and out of genomes too frequently for ancestral states to be reconstructed using parsimony. Buchnera lacks phage or insertion sequences [8].
The ancestor was assigned the gene order of modern E. coli. Although this order is almost certainly not entirely correct, comparison with other bacteria indicates that it is true for a majority of the borders between the 306 fragments (including those lost and those retained). For example, the same linkages occur in E. coli and V. cholerae and/or E. coli and Y. pestis for 219 of the junctions between fragments in Buchnera (example in Figure 5). This implies that most of the E. coli arrangements were present in the ancestor of Buchnera and E. coli and that the ends of syntenic fragments mostly represent sites at which rearrangements occurred in the evolution of Buchnera. A concentration of rearrangements in the Buchnera lineage is also suggested by the much longer average length of spacers that occur between syntenic fragments as compared to spacers that have the same flanking genes in the ancestor and in E. coli (see above). This longer spacer length can be interpreted as arising from remnants of genes that were rendered functionless when rearrangements occurred. When the relevant genomes currently in progress are complete and annotated, a more accurate reconstruction of ancestral gene order maybe possible.
Acknowledgments
Acknowledgements
We thank Howard Ochman for comments on the manuscript. Financial support was from a US National Science Foundation grant (DEB-9978518) to N.M.
References
- Casjens S. The diverse and dynamic structure of bacterial genomes. Annu Rev Genet. 1998;32:339–377. doi: 10.1146/annurev.genet.32.1.339. [DOI] [PubMed] [Google Scholar]
- Woese CR. Bacterial evolution. Microbiol Rev. 1987;51:221–271. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniloff J. The minimal cell genome: "On being the right size". Proc Natl Acad Sci USA. 1996;93:10004–10006. doi: 10.1073/pnas.93.19.10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmelreich R, Plagens H, Hilbert H, Reiner B, Herrmann R. Comparative analysis of the genomes of the bacteria Mycoplasma pneumoniae and Mycoplasma genitalium. Nucleic Acids Res. 1997;25:701–712. doi: 10.1093/nar/25.4.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson SGE, Kurland CG. Reductive evolution of resident genomes. Trends Microbiol. 1998;6:263–268. doi: 10.1016/s0966-842x(98)01312-2. [DOI] [PubMed] [Google Scholar]
- Andersson JO, Andersson SGE. Insights into the evolutionary process of genome degradation. Curr Opin Genet Dev. 1999;9:664–671. doi: 10.1016/s0959-437x(99)00024-6. [DOI] [PubMed] [Google Scholar]
- Andersson JO, Andersson SGE. Genome degradation is an ongoing process in Rickettsia. Mol Biol Evol. 1999;16:1178–1191. doi: 10.1093/oxfordjournals.molbev.a026208. [DOI] [PubMed] [Google Scholar]
- Shigenobu S, Watanabe H, Hattori M, Sakaki Y, Ishikawa H. Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature. 2000;407:81–86. doi: 10.1038/35024074. [DOI] [PubMed] [Google Scholar]
- Andersson JO, Andersson SGE. Pseudogenes, junk DNA, and the dynamics of Rickettsia genomes. Mol Biol Evol. 2001;18:829–839. doi: 10.1093/oxfordjournals.molbev.a003864. [DOI] [PubMed] [Google Scholar]
- Fraser CM, Gocayne JD, White O, Adams MD, Clayton RA, Fleischmann RD, Bult CJ, Kerlavage AR, Sutton GG, Kelley JM. The minimal gene complement of Mycoplasma genitalium. Science. 1995;270:397–403. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- Glass JI, Lefkowitz EJ, Glass JS, Heiner CR, Chen EY, Cassell GH. The complete sequence of the mucosal pathogen Ureaplasma urealyticum. Nature. 2000;407:757–762. doi: 10.1038/35037619. [DOI] [PubMed] [Google Scholar]
- Andersson SGE, Zomorodipour A, Andersson JO, Sicheritz-Ponten T, Alsmark UC, Podowski RM, Naslund AK, Eriksson AS, Winkler HH, Kurland CG. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396:133–140. doi: 10.1038/24094. [DOI] [PubMed] [Google Scholar]
- Read TD, Brunham R, Shen C, Gill SR, Heidelberg JF, White O, Hickey EK, Peterson J, Umayam LA, Utterback T, et al. Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res. 2000;28:1397–1406. doi: 10.1093/nar/28.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, ColladoVides J, Glasner JD, Rode CK, Mayhew GF, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- Moran NA, Wernegreen JJ. Are mutualism and parasitism irreversible evolutionary alternatives for endosymbiotic bacteria? Insights from molecular phylogenetics and genomics. Trends Ecol Evol. 2000;15:321–326. doi: 10.1016/s0169-5347(00)01902-9. [DOI] [PubMed] [Google Scholar]
- Tamas I, Klasson LM, Sandström JP, Andersson SGE. Mutualists and parasites: how to paint yourself into a (metabolic) corner. FEBS Lett. 2001;498:135–139. doi: 10.1016/s0014-5793(01)02459-0. [DOI] [PubMed] [Google Scholar]
- Moran NA. Accelerated evolution and Muller's ratchet in endosymbiotic bacteria. Proc Natl Acad Sci USA. 1996;93:2873–2878. doi: 10.1073/pnas.93.7.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama M, Bork P. Evolution of prokaryotic gene order: genome rearrangements in closely related species. Trends Genet. 2001;17:10–13. doi: 10.1016/s0168-9525(00)02159-4. [DOI] [PubMed] [Google Scholar]
- Tillier ERM, Collins RA. Genome rearrangement by replication-directed translocation. Nat Genet. 2000;26:195–197. doi: 10.1038/79918. [DOI] [PubMed] [Google Scholar]
- Eisen JA, Heidelberg JF, White O, Salzberg SL. Evidence for symmetric chromosomal inversions around the replication origin in bacteria. Genome Biol. 2000;1:research0011.1–0011.9. doi: 10.1186/gb-2000-1-6-research0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira A, Ochman H, Moran NA. Deletional bias and the evolution of bacterial genomes. Trends Genet. 2001;17:589–596. doi: 10.1016/s0168-9525(01)02447-7. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Mushegian AR, Rudd KE. Sequencing and analysis of bacterial genomes. Curr Biol. 1996;6:404–416. doi: 10.1016/s0960-9822(02)00508-0. [DOI] [PubMed] [Google Scholar]
- Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FSL, Hufnagle WO, Kowalik DJ, Lagrou M, et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- Hawley DK, McClure WR. Compilation and analysis of Escherichia coli promoter DNA sequences. Nucleic Acids Res. 1983;11:2237–2255. doi: 10.1093/nar/11.8.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wosten MMSM. Eubacterial sigma-factors. FEMS Microbiol Rev. 1998;22:127–150. doi: 10.1111/j.1574-6976.1998.tb00364.x. [DOI] [PubMed] [Google Scholar]
- Dunn JJ, Buzash-Pollert E, Studier FW. Mutations of bacteriophage T7 that affect the initiation of synthesis of gene 0.3 protein. Proc Natl Acad Sci USA. 1978;75:2741–2745. doi: 10.1073/pnas.75.6.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999;234:187–208. doi: 10.1016/s0378-1119(99)00210-3. [DOI] [PubMed] [Google Scholar]
- Wernegreen JJ, Moran NA. Evidence for genetic drift in endosymbionts: analyses of protein-coding genes. Mol Biol Evol. 1999;16:83–97. doi: 10.1093/oxfordjournals.molbev.a026040. [DOI] [PubMed] [Google Scholar]
- Spaulding AW, von Dohlen CD. Psyllid endosymbionts exhibit patterns of co-speciation with hosts and destabilizing substitutions in ribosomal RNA. Insect Mol Biol. 2001;10:57–67. doi: 10.1046/j.1365-2583.2001.00231.x. [DOI] [PubMed] [Google Scholar]
- Komaki K, Ishikawa H. Intracellular symbionts of aphids possess many genome copies per bacterium. J Mol Evol. 1999;48:717–722. doi: 10.1007/pl00006516. [DOI] [PubMed] [Google Scholar]
- Wernegreen JJ, Moran NA. Decay of mutualistic potential in aphid endosymbionts through silencing of biosynthetic loci: Buchnera of Diuraphis. Proc R Soc Lond B. 2000;267:1423–1431. doi: 10.1098/rspb.2000.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mushegian AR, Koonin E. A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc Natl Acad Sci USA. 1996;93:10268–10273. doi: 10.1073/pnas.93.19.10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mushegian AR. The minimal genome concept. Curr Opin Genet Dev. 1999;9:709–714. doi: 10.1016/s0959-437x(99)00023-4. [DOI] [PubMed] [Google Scholar]
- Hutchison CN, Peterson SN, Gill SR, Cline RT, White O, Fraser CM, Smith HO, Venter JC. Global transposon mutagenesis and a minimal Mycoplasma genome. Science. 1999;286:2165–2169. doi: 10.1126/science.286.5447.2165. [DOI] [PubMed] [Google Scholar]
- Koonin EV. How many genes can make a cell: the minimal-gene-set concept. Annu Rev Genom Human Genet. 2000;1:99–116. doi: 10.1146/annurev.genom.1.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynen M, Bork P. Measuring genome evolution. Proc Natl Acad Sci USA. 1998;95:5849–5856. doi: 10.1073/pnas.95.11.5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterman BM, Baumann P, McLean DL. Pea aphid symbiont relationships established by analysis of 16S rRNAs. J Bacteriol. 1989;171:2970–2974. doi: 10.1128/jb.171.6.2970-2974.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasior SL, Olivares H, Ear U, Hari DM, Weichselbaum R, Bishop DK. Assembly of RecA-like recombinases: Distinct roles for mediator proteins in mitosis and meiosis. Proc Natl Acad Sci USA. 2001;98:8411–8418. doi: 10.1073/pnas.121046198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MA, Baumann L, Baumann P. Sequence analysis of a 34.7-kb DNA segment from the genome of Buchnera aphidicola (endosymbiont of aphids) containing groEL, dnaA, the atp operon, gidA, and rho. Curr Microbiol. 1998;36:158–163. doi: 10.1007/pl00006760. [DOI] [PubMed] [Google Scholar]
- Ochman H, Jones IB. Evolutionary dynamics of full genome content in Escherichia coli. EMBO J. 2000;19:6637–6643. doi: 10.1093/emboj/19.24.6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole ST, Eiglmeier K, Parkhill J, James KD, Thomson NR, Wheeler PR, Honore N, Garnier T, Churcher C, Harris D. Massive gene decay in the leprosy bacillus. Nature. 2001;409:1007–1011. doi: 10.1038/35059006. [DOI] [PubMed] [Google Scholar]
- Kato-Maeda M, Rhee JT, Gingeras TR, Salamon H, Drenkow J, Smittipat N, Small PM. Comparing genomes within the species Mycobacterium tuberculosis. Genome Res. 2001;11:547–554. doi: 10.1101/gr166401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selosse M-A, Albert B, Godelle B. Reducing the genome size of organelles favors gene transfer to the nucleus. Trends Ecol Evol. 2001;16:135–141. doi: 10.1016/s0169-5347(00)02084-x. [DOI] [PubMed] [Google Scholar]
- Scherbakov DV, Garber MB. Overlapping genes in bacterial and phage genomes. Mol Biol. 2000;34:485–495. [PubMed] [Google Scholar]
- Wernegreen JJ, Ochman H, Jones I, Moran NA. The decoupling of genome size and sequence divergence in a symbiotic bacterium. J Bacteriol. 2000;182:3867–3869. doi: 10.1128/jb.182.13.3867-3869.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akman L, Aksoy S. A novel application of gene arrays: Escherichia coli array provides insight into the biology of the obligate endosymbiont of tsetse flies. Proc Natl Acad Sci USA. 2001;98:7546–7551. doi: 10.1073/pnas.131057498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akman L, Rio RVM, Beard CB, Aksoy S. Genome size determination and coding capacity of Sodalis glossinidius, an enteric symbiont of tsetse flies, as revealed by hybridization to Escherichia coli gene arrays. J Bacteriol. 2001;183:4517–4525. doi: 10.1128/JB.183.15.4517-4525.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA, Munson MA, Baumann P, Ishikawa H. A molecular clock in endosymbiotic bacteria is calibrated using the insect hosts. Proc R Soc Lond B. 1993;253:167–171. [Google Scholar]
- van Ham RCHJ, Moya A, Latorre A. Putative evolutionary origin of plasmids carrying the genes involved in leucine biosynthesis in Buchnera aphidicola (endosymbiont of aphids). J Bacteriol. 1997;179:4768–4777. doi: 10.1128/jb.179.15.4768-4777.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaulding AW, von Dohlen CD. Phylogenetic characterization and molecular evolution of bacterial endosymbionts in psyllids (Hemiptera: Sternorrhyncha). Mol Biol Evol. 1998;15:1506–1513. doi: 10.1093/oxfordjournals.molbev.a025878. [DOI] [PubMed] [Google Scholar]
- Sauer C, Stackebrandt E, Gadau J, Holldobler B, Gross R. Systematic relationships and cospeciation of bacterial endosymbionts and their carpenter ant host species: proposal of the new taxon Candidatus Blochmannia gen. nov. Int J Syst Evol Microbiol. 2000;50:1877–1886. doi: 10.1099/00207713-50-5-1877. [DOI] [PubMed] [Google Scholar]
- Lawrence JG, Ochman H. Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci USA. 1998;95:9413–9417. doi: 10.1073/pnas.95.16.9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Umayam L, et al. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature. 2000;406:477–483. doi: 10.1038/35020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger Centre: microbial genomes http://www.sanger.ac.uk/Projects/Microbes/
- Washington University Genome Sequencing Center: bacterial genomes http://genome.wustl.edu/gsc/Projects/bacteria.shtml
- Schwartz S, Zhang Z, Frazer KA, Smit A, Riemer C, Bouck J, Gibbs R, Hardison R, Miller W. PipMaker: A web server for aligning two genomic DNA sequences. Genome Res. 2000;10:577–586. doi: 10.1101/gr.10.4.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland M, Florea L, Sanderson K, Clifton SW, Parkhill J, Churcher C, Dougan G, Wilson RK, Miller W. Comparison of the Escherichia coli K-12 genome with sampled genomes of a Klebsiella pneumoniae and three Salmonella enterica serovars, Typhimurium, Typhi and Paratyphi. Nucleic Acids Res. 2000;28:4974–4986. doi: 10.1093/nar/28.24.4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Enteric server http://galapagos.cse.psu.edu/enterix/enteric/enteric.html