

Abstract
Stem cells are emerging as a therapeutic option for incurable diseases, such as Amyotrophic Lateral Sclerosis (ALS). However, critical issues are related to their origin as well as to the need to deepen our knowledge of the therapeutic actions exerted by these cells. Here, we investigate the therapeutic potential of clinical-grade human neural stem cells (hNSCs) that have been successfully used in a recently concluded phase I clinical trial for ALS patients (NCT01640067). The hNSCs were transplanted bilaterally into the anterior horns of the lumbar spinal cord (four grafts each, segments L3–L4) of superoxide dismutase 1 G93A transgenic rats (SOD1 rats) at the symptomatic stage. Controls included untreated SOD1 rats (CTRL) and those treated with HBSS (HBSS). Motor symptoms and histological hallmarks of the disease were evaluated at three progressive time points: 15 and 40 days after transplant (DAT), and end stage. Animals were treated by transient immunosuppression (for 15 days, starting at time of transplantation). Under these conditions, hNSCs integrated extensively within the cord, differentiated into neural phenotypes and migrated rostro-caudally, up to 3.77 ± 0.63 cm from the injection site. The transplanted cells delayed decreases in body weight and deterioration of motor performance in the SOD1 rats. At 40DAT, the anterior horns at L3–L4 revealed a higher density of motoneurons and fewer activated astroglial and microglial cells. Accordingly, the overall survival of transplanted rats was significantly enhanced with no rejection of hNSCs observed. We demonstrated that the beneficial effects observed after stem cell transplantation arises from multiple events that counteract several aspects of the disease, a crucial feature for multifactorial diseases, such as ALS. The combination of therapeutic approaches that target different pathogenic mechanisms of the disorder, including pharmacology, molecular therapy and cell transplantation, will increase the chances of a clinically successful therapy for ALS.
Subject terms: Amyotrophic lateral sclerosis, Neural stem cells
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive disease that targets primarily motor neurons (MNs) and leads to fatal paralysis1,2. The molecular mechanisms that initiate and drive the inherent neurodegenerative process are largely unknown. Recent reports strongly implicate neuroinflammatory processes, such as astrogliosis, microgliosis and the infiltration of T lymphocytes, in the progressive degeneration of MNs:3–5 These non-cell-autonomous mechanisms are promising therapeutic targets.
ALS is a multifactorial disorder that involves genetic as well as environmental factors. Most (90–95%) of ALS forms are sporadic (sALS), while about 10% of cases are familial (fALS) and are associated with mutations in specific genes, such as superoxide dismutase 1 (SOD1, 20% of fALS), TARDBP and FUS (5% of fALS) and C9ORF72 (40% of fALS)6–8. SOD1 was the first mutated protein that was correlated with the development of ALS9, and it has been leveraged to generate animal models of ALS—these include the SOD1 rats used here10, which reproduce many of pathological and symptomatic features of the human disorder and have been used for developing therapeutic strategies, such as stem-cell transplantation.
Preclinical studies show that intraspinally transplanted human neural stem cells (hNSCs) provide trophic support to damaged cells, and also modulate the immune cell environment, thus acting on disease mechanisms at multiple levels;11–20 based on these results, the approach was translated into the clinic, and two phase I21–23 and phase II24,25 studies with use of hNSCs have been successfully completed. The exact mechanisms through which these cells exert their beneficial effects have not been completely identified. Moreover, the use of hNSCs derived from different CNS sources, using a variety of methods, further confounds the direct comparisons of findings from different labs. For clinical applications, a standardised protocol that guarantees the reproducibility, safety and efficacy of hNSCs is of utmost importance. Our group has established a Cell Factory and Biobank at the Hospital Santa Maria in Terni that is currently producing hNSC lines from the foetal brain, using methods26 that are fully compliant with current Good Manufacturing Practice (cGMP) guidelines, and are approved for clinical applications by the Italian Medicine Agency (AIFA, aM 154/2018). The cell lines are characterised by a consolidated paradigm to assess their stemness and safety. Consistent with this rigorous approach, the hNSCs have been successfully used in the phase I trial for ALS patients23, EudraCT 2009–014484–39 NCT01640067), and are also currently being evaluated in a phase I study for the treatment of Secondary Progressive Multiple Sclerosis (EudraCT 2015–004855–37 NCT03282760).
As a complement to the phase I trial, and preliminary to phase II, we evaluate here the therapeutic potential of using a GMP-grade hNSC line in the SOD1 rat model of ALS. hNSCs were delivered by intraspinal cord transplantation, using the same strategy as for ALS patients23,24. Because we intended to unveil the role played by hNSCs in delaying neural degeneration, e.g., by modulating neuroinflammation11, we also evaluated the symptomatic hallmarks of ALS, together with astrogliosis and microgliosis, at different stages of disease progression
Results
Hallmarks of symptomatic progression in SOD1 rats
We evaluated disease progression in SOD1 rats by monitoring the gradual deterioration of the motor system as reflected by rotarod performance, motor score and weight assessment, in n = 22 SOD1 rats (CTRL) compared with n = 9 wild-type (WT) animals27–29. This analysis led us to identify the following four subsequent clinical stages (Fig. 1a), based on the worsening of symptoms:
-
(i)
Early-symptomatic stage (ESS): this phase started with subtle decrease in limb control (resulting in a 5% reduction in the motor score) and/or a two consecutive failures of the rotarod test in the same week. These changes occurred variably between P92 and P115 (Fig. 1).
-
(ii)
Mid-symptomatic stage (MSS): the symptoms became gradually more evident during the following month (~27 days after ESS; Fig. 1a), at the end of which a decline in the weight of SOD1 rats, due to denervation-induced muscle atrophy, started to become evident, (peak body weight at 21 days after ESS, orange arrow in Supplementary Fig. 1A). Concomitantly, motor impairments were now overt and stable, with a significant decrease in the motor and rotarod performance scores relative to the scores recorded for ESS (18%, p < 0.05 vs. WT rats, orange arrow in Supplementary Fig. 1B; and 25%, p < 0.01 vs. WT, orange arrow in Supplementary Fig. 1C, respectively).
-
(iii)
Late-symptomatic stage (LSS): transition from MSS to LSS was characterised by a rapid decline of weight and motor functions (Fig. 1a). At 40 days after ESS, the body mass was reduced by 10% (red arrows in Supplementary Fig. 1A) and the performance on both motor tests was decreased by 50% (red arrows in Supplementary Fig. 1B, C). At about 60 days after ESS, SOD1 rats displayed minimal motility and were unable to perform the rotarod challenge (Supplementary Fig. 1A–C).
-
(iv)
End stage (E-ST): the day of E-ST was established by the loss of the righting reflex (∼70 days after ESS, Fig. 1a).
Fig. 1. Experimental plan and migration of transplanted hNSCs.

a Experimental plan: rats were transplanted with hNSCs or HBSS when showed the initial symptoms of the disease (ESS), age-matched not-transplated controls were also analysed. Behavioural test was performed for all the durations of animals life. Histological analysis of the spinal cord was performed at 15DAT, 40DAT and at E-ST, that approximately correspond to MSS, LSS and E-ST. b Schematic showing the transplant strategy and migration of hNSCs (green dots). c Schematic representation of the spinal cord organisation. hNSC or HBSS were transplanted into the anterior horns (asterisk) and were able to radially migrate into the anterior funiculus (red) and the lateral funiculus (blue). d Injection site with an evident cluster of grafted hNSCs (huN +) in the anterior horn of an animal at 40DAT. e At E-ST hNSCs (huN +) reached a higher grade of dispersion into the parenchyma. f, g Charts showing the differences in hNSCs migration (f) and survival (g) at 15, 40DAT or E-ST. ESS early-symptomatic stage, MSS mid-symptomatic stage, E-ST end stage, DAT days after transplantation, a.f. anterior funiculus, a.h. anterior horn. Scale bars: 100 μm
The day of transplantation was established as that day when early motor symptoms first appear (referred to as day 0, occurring within the ESS, at ~P105-P115; Fig. 1a and yellow arrows in Supplementary Fig. 1A–C). SOD1 rats received four bilateral injections of hNSCs (hNSC group, a total of 400,000 cells/animal, n = 15), or of only HBSS (HBSS group, n = 15) into the anterior horn of the lumbar spinal cord (L3–L4; Fig. 1b, c). Another control group of SOD1 rats did not receive any surgical procedure (CTRL group, n = 22).
Histological analysis of the spinal cord was performed at three different time points (Fig. 1a): 15 days after transplantation (15DAT) during the MSS, 40DAT during the LSS and at E-ST.
Transplanted hNSCs survive, migrate and differentiate in the spinal cord of SOD1 rats
Viable hNSCs grafts (huN +, Fig. 1d–e) were found in all animals. At 15 and 40DAT, most of hNSCs were integrated into laminae VIII–IX of the anterior horns (Fig. 1c, d, AH) and few had migrated into the lateral and anterior funiculi (Fig. 1d, AF). At E-ST, huN + cells were dispersed within the spinal cord and occurred individually (not clustered, Fig. 1e); the cells had migrated extensively along the rostro-caudal axis of the cord, from the thoracic and sacral cord segments (1.69 ± 0.47 cm at 15DAT, 1.02 ± 0.39 cm at 40DAT and 3.77 ± 0.63 cm at E-ST, Fig. 1f).
A high percent of the transplanted cells had survived, and were detected at all three time points of analysis (53.29 ± 32.34% at 15DAT; 77.81 ± 12.89% at 40DAT; and 53.65 ± 19.41% at E-ST—Fig. 1g, n = 3/time point). At each time point, we also detected a similar fraction of transplanted cells that were actively proliferating (Ki67 + cells: 9.11 ± 2.5% at 15DAT, 14.1 ± 4.3% at 40DAT and 15.2 ± 2.1% at E-ST; Fig. 2a, b), suggesting that new progenitors were generated in vivo. Transplanted hNSCs (Fig. 2a, b) expressed Nestin (26.9 ± 7.3% at 15DAT; 21.7 ± 7.7% at 40DAT; and sporadic at E-ST) and differentiated into astroglia (GFAP +: 25 ± 4.7% at 15DAT, 20.5 ± 7.7% at 40DAT and 34.4 ± 3% at E-ST), neurons (β-Tub III +, 16.1 ± 2.2% at 15DAT, 10.7 ± 5.4% at 40DAT and 4.5 ± 2% at E-ST) and oligodendrocyte precursors (PDGFRa +: 9.6 ± 3.7% at 15DAT; 17.6 ± 2.9% at 40DAT; and 9.4 ± 1.6% at E-ST). These data indicate that hNSCs efficiently survive, transiently proliferate and differentiate in the spinal cord of SOD1 rats.
Fig. 2. Differentiation of transplanted hNSCs within the SOD1 rat spinal cord.
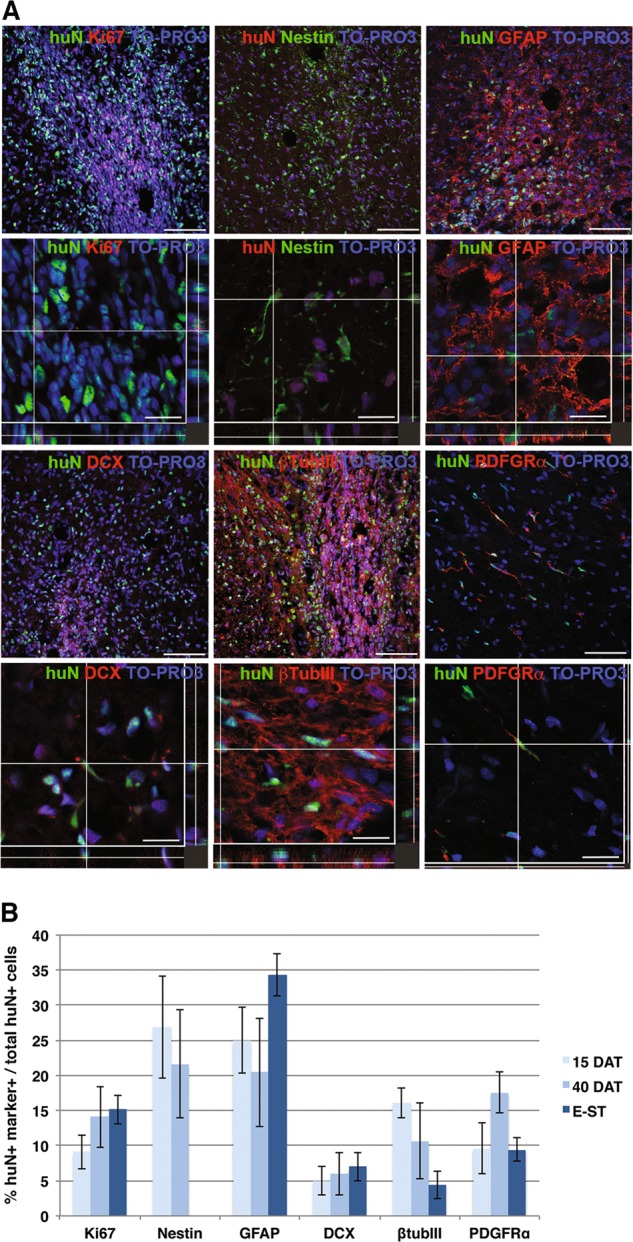
a Representative confocal images showing the colocalization of the human nuclear antigen (huN) with proliferating cells (Ki67) and neural phenotype markers (Nestin, GFAP, DCX, btubIII and PDGFR-a) in transplanted hNSCs at 40DAT. b Quantification of each markers. Scale bars: 75 μm for the maximum projection images and 15–25 μm for the colocalization images
Transplanted hNSCs prolong survival and delay the progression of motor symptoms
The median survival time for hNSCs-transplanted rats was 83.5 days, ~23 days longer than the control groups, which had median survival times of 60.5 days (CTRL animals, HR: 0.08; 95% CI = 0.01–0.65; p = 0.018) and 57.0 days (HBSS group, HR: 0.08; 95%CI = 0.01–0.72; p = 0.024, see Fig. 3a). The curves for the two control groups were overlapping (HR: 0.94; 95% CI = 0.31–2.79; p = 0.905).
Fig. 3. Extention of lifespan and slow-down of behavioral deterioration in hNSCs-treated rats.

a Kaplan–Meier survival probability analysis, n = 14 for untreated SOD1 (CTRL, green) and n = 5 for HBSS-treated SOD1 (HBSS, red) and hNSCs-treated SOD1 (hNSCs, blue). b–d Trend of weight, motor score and rotarod performance decline in CTRL (green, n = 22), HBSS (red, n = 15) and hNSCs (blue, n = 15) rats. The curves were obtained by appliyng statistical modelling of the progressive reduction of the mean weight value (b) or mean motor score value (c) or the mean time spent on the rotarod (d). Where the hNSCs-treated animals showed a statistically significant improved value respect to CTRL is indicated with asterisks above the green line, respect to HBSS animals with asterisks above the red line. On the x axis, the days are shown after ESS and coloured bars indicate the stage of the disease. *p ≤ 0,05; **p ≤ 0,01; ***p ≤ 0,001. Data are reported as mean ± SEM
Disease progression was significantly delayed in hNSCs-transplanted animals, relative to CTRL and HBSS-treated animals (Fig. 3b–d), as evaluated by weight decline (p < 0.0001 versus CTRL and p = 0.023 versus HBSS), motor score (p < 0.0001 versus CTRL and p = 0.001 versus HBSS) as well as rotarod performance (p < 0.0001 versus CTRL and p = 0.011 versus HBSS). The transplantation surgery led to a worsening of motor performance for about 10 days, during which time the treated rats were not always able to perform on the behavioural tests. In fact, during the MSS, the performance of both treated groups (hNSC and HBSS) was similar (p > 0.5), and was significantly worse than that of the CTRL group (p < 0.03).
At the LSS stage, the performance of the hNSCs-treated rats appeared to be stabilised, and was reliably better than that of the HBSS-treated animals. The motor score was 2.02 ± 0.3 for hNSC and 1.17 ± 0.31 for HBSS at 39DAT (p < 0.05, Fig. 3c); the rotarod score was 103.37 ± 22.84 s for hNSC and 30.47 ± 25.99 s for HBSS at 48DAT (p < 0.04, Fig. 3d); and the weight was 323.51 ± 12.56 g for hNSC rats and 280.12 ± 15.86 g for HBSS rats at 57DAT (p < 0.04, Fig. 3b). Of note, disease progression was ameliorated in hNSCs-transplanted animals, even when compared with CTRL animals, although this difference was evident later than relative to the HBSS groups. In particular, the motor scores were 2.02 ± 0.31 for hNSC rats and 1.23 ± 0.25 for CTRL at 51DAT (p < 0.05, Fig. 3c); the rotarod score was 96.62 ± 24.13 s for hNSC rats and 23.65 ± 20.94 s for the CTRL group at 57DAT (p < 0.03, Fig. 3d); and weights were 313.89 ± 13.04 g for hNSC rats versus 269.96 ± 12.59 g for the CTRL group at 63DAT (p < 0.02, Fig. 3b).
At E-ST (60DAT), while animals in both control groups had almost completely lost their motor ability (rotarod and motor score values = 0), motor function was still partially preserved in the hNSCs-transplanted animals (50% of motor score, 1.93 ± 0.34, Fig. 3c), as was their ability to run on the rod (30% of rotarod performance, 93.47 ± 24.54 s, Fig. 3d).
hNSCs induce a significant decrease in the degeneration of MNs
The functional data were validated by histopathological findings, which mirrored a significant delay in the degeneration process of MNs at 40DAT, as demonstrated by the results of stereological analysis of MN cell density in the treated region of the spinal cord (Fig. 4). Preservation of MNs was maximal around the cell injection site, namely within segments L3–L4: hNSCs-treated SOD1 rats displayed a significantly higher density of MNs (336.65 ± 28.73 MNs/mm3) relative to control groups (204.25 ± 19.47 for CTRL p ≤ 0.01 and 231.60 ± 24.05 for HBSS p ≤ 0.05; Fig. 4a, b). To corroborate the beneficial effect of hNSCs, we evaluated the presence of misfolded SOD1 deposits by using the SEDI antibody30,31. Of note, in the anterior horns of hNSC animals, at 40DAT, SOD1 deposits appeared reduced respect to both control groups (Fig. 4c), in which SOD1 was apparent and predominantly accumulated within MNs (Fig. 4c, arrow).
Fig. 4. Reduction of motor neurons degeneration and accumulation of misfolded SOD1 in hNSCs-treated SOD1 rats.

a Chart showing the motor neurons density (MNs/cm3) in the L1-L4 tract, n = 5 for each experimental group. *p ≤ 0.05; **p ≤ 0.01. Data are reported as mean ± SEM. b Representative images of the MNs in the anterior horn (AH) of the spinal cord (Nissle staining). Scale bars: 100 μm. c Representative confocal images of misfolded SOD1 deposits in the AH of CTRL, HBSS or hNSCs rats (n = 3/group) at 40DAT. Scale bars: 20 μm
hNSCs immunomodulate reactive astrogliosis and microgliosis
To assess whether hNSCs can influence resident inflammatory cells, we evaluated the morphology and quantity of GFAP+ cells (for astrogliosis, Fig. 5) and of Iba1+ and CD68+ cells (for microgliosis, Figs. 6, 7), in the L3–L4 segments of the spinal cord around the grafted area, and looked for how the activation correlated to disease progression (at 15, 40DAT and E-ST). To determine the magnitude of the effect that hNSCs have on the different components of the motor system, we separately evaluated the anterior horns (which contain the somata of α-MNs) and the white matter, specifically the anterior and lateral funiculi (which comprise the descending fibres of the pyramidal and extrapyramidal tracts, respectively). These areas, colonised by transplanted hNSCs (Fig. 1c–e), are directly involved in the neuronal circuitry for motor control and majorly affected by the inherent degeneration process in this animal model of ALS12.
Fig. 5. Reduction of astrogliosis in hNSCs-treated rats.

a Representative confocal images of activated astroglial cells (GFAP+) in the anterior horn (AH) of CTRL, HBSS or hNSCs rats. b Chart showing the quantification analysis of the number of GFAP+ cells in the anterior horn, anterior funiculus or whole (anterior horn, anterior funiculus and lateral funiculus). Scale bars: 75 μm
Fig. 6. Reduction of microgliosis in hNSCs-treated rats.

a Representative confocal images of activated microglial cells (Iba1+) in the anterior horn (AH) of CTRL, HBSS or hNSCs rats. b Chart showing the quantification analysis of the number of Iba1+ cells in the anterior horn, anterior funiculus or whole (anterior horn, anterior funiculus and lateral funiculus). Scale bars: 75 μm
Fig. 7. Reduction of activated CD68+ microglial cells in hNSCs-treated rats.

a Representative confocal images of activated microglial cells (Iba1 + CD68+) in the anterior horn (AH) of CTRL, HBSS or hNSCs rats. b Chart showing the quantification analysis of the number of CD68+ cells in the anterior horn, anterior funiculus or whole (anterior horn, anterior funiculus and lateral funiculus). Scale bars: 75 μm
Astrogliosis
At 15DAT, in both control groups, almost all the astrocytes in the anterior horns were in a reactive state, displaying hypertrophic and thick processes strongly immunopositive for GFAP (Fig. 5a, arrows). At 40DAT and at E-ST, astrogliosis was further enhanced (Fig. 5a, arrowheads). In contrast, in hNSC-transplanted SOD1 rats, the majority of astrocytes displayed thinner processes and a smaller soma. These morphological differences between animal groups were more evident at 40DAT (Fig. 5a; Supplementary Fig. 2A). A similar immunomodulatory effect of transplanted hNSC was also observed in white matter regions (Supplementary Fig. 2B). The quantitative analysis mirrored the morphological observations (Fig. 5b) by demonstrating, as early as 15DAT, a statistically relevant reduction of the number of astrocytes in hNSCs-transplanted SOD1 rats relative to the HBSS-treated group, both in the anterior horns and funiculi; this difference became more striking at 40DAT (p < 0.01 versus untreated SOD1 and p < 0.05 versus HBSS-treated SOD1). Finally, at E-ST, the hNSC-transplanted rats still showed a lower level of astrogliosis in the white matter tracts (Fig. 5b, anterior funiculi and whole, p < 0.05 versus the same in untreated SOD1 rats). To note, hNSC-treated animals reached E-ST at a significantly later age (mean age, P204) compared with control animals (mean age P170).
Astrocyte fractions were comparable at the mid-to-late stage transition (15 to 40DAT) in most animals that belonged to the same treatment group, but had decreased at E-ST, likely due to generalised cell damage that also involved astrocytes32–34.
Microgliosis
The immunomodulatory effect of hNSCs on the microglial component was mostly marked at 40DAT. In fact, we observed that at 15DAT, Iba1 + microglial cells displayed thick, rough prolongations (Fig. 6a, arrows) and were comparable among all groups. This inflammatory process was consistently enhanced within the 15 to 40DAT transition in the spinal cord of control groups. At these time points, Iba1+ cells were hypertrophic, with short processes and an “amoeboid” (activated) morphology (Fig. 6a, arrowheads; Supplementary Fig. 3A). In contrast, at 40DAT, in the anterior horns of hNSCs treated SOD1 rats, the switch of microglial cells to an enhanced activated state, appeared arrested (Fig. 6a; Supplementary Fig. 3A). No difference was observed between the groups at the E-ST (Fig. 6a). A similar pattern was also observed in white matter regions (Supplementary Fig. 3B). The quantification analysis correlated with the morphological observations, showing the reduced numbers of Iba1+ cells in hNSC-transplanted SOD1 rats at 40DAT (from p ≤ 0.05 to p ≤ 0.01 versus controls, Fig. 6b).
To evaluate the contribute of activated microglial/macrophages cells, we analysed the expression of CD68. Consistent with the above findings, CD68+ microglial cells displayed a morphologically activated state in the spinal cord of each experimental group (Fig. 7a; Supplementary Fig. 4A), and the CD68 protein co-localised with the pan-microglial antigen Iba1 (Supplementary Fig. 4B). The quantitative analysis confirmed that, at 40DAT, the number of activated CD68+ cells was reduced in hNSC-treated animals (p < 0.05 in the anterior horn, Fig. 7b).
Discussion
ALS is a multifactorial disease with no available effective cure1,2. We present compelling evidence that intraspinal delivery of hNSCs ameliorates the course of the disease, delays the deterioration of motor functions and extends overall survival in a transgenic rat model of ALS. These favourable symptomatic outcomes (that peaked between 40–60DAT) are accompanied by a delay in the accumulation of histopathological markers of ALS, referable to a hNSCs pleiotropic mode of action35–38. In particular, anterior horn MNs are preserved and astrogliosis and microgliosis are reduced in the spinal cord tract of hNSC-treated SOD1 rats.
Relevance to clinical studies
The positive effects of hNSCs transplantation on disease progression in models of ALS have been widely documented11–15,22,39,40, but the variability in the methodologies used to derive and expand hNSCs has often confounded interpretations. Our data are remarkably relevant because we used hNSCs derived with the AIFA-authorised protocol (aM 154/2018) that has been used for the ALS Phase I trial (EudraCT:2009–014484–39 trial)23,26,41. The rigorous experimental conditions that we describe in this study mimic the clinical setting with the goal of having our results be relevant to future phase II studies in patients.
In preclinical studies, often stem cells are delivered at the pre-symptomatic stage of the disease, in order to maximise the window during which the cells can exert their therapeutic actions12,15,40. But while this approach is optimal for obtaining positive results in a basic research setting, translating this therapeutic sequence—which involves making transplantations prior to the appearance of symptoms—to the bedside is clearly not realistic, because ALS diagnosis only occurs at the symptomatic stage. We thus chose to treat the animals when the initial motor symptoms of the pathology are recognisable. But despite the treatment being administered after disease onset and a corresponding reduction of the therapeutic window, hNSC-treated rats showed better motor performances compared with controls.
Because intraspinal cord injection partially disrupts the parenchymal structure and triggers the inflammatory reaction, worsening the animal’s motor functions42,43, we also compared the symptomatic progression of SOD1 rats treated with hNSCs to untreated SOD1 rats (CRTL)—the goal here was to control for possible detrimental effects of the injection procedure. As expected, the motor performances of treated animals were reduced compared with CTRL, in the immediate post-injection period. Notwithstanding, as early as 50DAT, hNSC-transplanted rats completely overcame the surgery-induced impairment, displaying a significantly improved motor performance relative to both control groups. Under our stringent experimental conditions, a symptomatic improvement relative to untreated animals has never been documented, further strengthening the clinical relevance of our approach and supporting our surgical procedure.
Symptomatic improvement
Intra-spinal delivery is aimed at preventing the degeneration of secondary MNs that leads to paralysis, and ultimately to respiratory failure. The major limitation of this strategy is that therapeutic hNSCs are supplied to a restricted area of the motor control system, while ALS affects the entire motor neuraxis, including upper MNs in the cortex. Notwithstanding this, the survival of hNSC-treated rats was extended by ~30 days relative to control animals. Similar studies have reported a smaller increase in lifespan (between 11 and 17 days14,15 or no effect on survival12).
Extensive migration and integration and no rejection
hNSCs migrated radially, from the anterior horn into the lateral and anterior funiculi of the white matter (comprising descending extrapyramidal and pyramidal fibres) as well as along the rostro-caudal axis, reaching the thoracic and sacral segments. The observation that different hNSC lines display an intrinsic variability in migration (unpublished data), suggests the necessity of an accurate evaluation of this parameter in preclinical phases, in order to improve the therapeutic efficacy of these stem cells in phase II trials.
Moreover, under conditions of transient immunosuppression, hNSCs are able to survive in SOD1 rats, up to the ESS, with no rejection and no adverse immunological effects due to cross-species differences, consistently with our previous studies44. Our findings validate the wide therapeutic potential of hNSC in that they show virtually zero immunogenicity. This aspect, of course, must be further verified with regard to the absence of rejection after the cells have been transplanted.
The slow-down of symptomatic worsening corresponds to an amelioration of the pathogenic environment including neuroinflammation blunting by hNSCs
Here we present, for the first time, a time course analysis to evaluate the progression of astrogliosis and microgliosis in order to reinforce the notion that the inflammatory cascade is an efficacious therapeutic target for the transplanted hNSCs44–46. Our analysis points to a correspondence between the delay in worsening of symptoms observed in the hNSCs-treated SOD1 rats and the reduced inflammatory response in the anterior horns. In fact, at 15DAT, the three experimental groups showed minimal differences, concerning both neuroinflammation and behavioural symptoms. While at 40DAT, activated astroglial and microglial cells were significantly reduced in hNSCs-treated rats, at the morphological level as well as by quantitative evaluation. These differences were accompanied by evidence of the first symptomatic benefits (motor score at 40DAT), which continued through the subsequent evaluation period (40–60DAT).
Within the same time frame (40DAT), the misfolded SOD1 deposits were less present in the anterior horns of hNSCs-transplanted rats respect to both control groups. Although preliminary, this observation is consistent with the hypothesis that hNSCs ameliorate the pathogenic surroundings of MNs, in fact it is now well established that misfolded SOD1 accumulation is a key element of SOD1-mediated ALS, and that the reduction of its level determines significant beneficial effects in preclinical model47,48.
Hefferan et al.12 have demonstrated that axons of descending primary MNs in the SOD1 rat model are impaired. Notably, these degenerative processes are more evident in the lateral (mostly rubrospinal tract) and anterior (pyramidal pathway) white matter. In addition, when the corticospinal tract is damaged, lateral columns provide de novo innervating cells to MNs in the anterior horns49,50, suggesting that lateral columns contribute to recovery mechanisms following degenerative insults to the motor system. Considering the extensive migration of hNSCs from the anterior horns to the aforementioned areas (lateral and anterior white matter), we evaluated and quantified a significant reduction of neuroinflammation in all these areas that were maintained until the late stages of the disease, with an obvious amelioration of the local pathogenic environment. Given the reports of axon dieback (i.e., axonal degeneration might occur prior to the onset of symptoms) in the pathogenesis of ALS51–53, the amelioration of parenchymal milieu surrounding distal axons—such as the reduction of neuroinflammation—could retrogradely affect the MN soma located in the motor cortex. Thus, it probably contributes to the generalised beneficial effect of hNSCs, in spite of their local delivery.
In conclusion, our study points to the potential therapeutic efficacy of using clinical-grade hNSCs that are already in use in a clinical trial on ALS patients. Our findings validate that extensive and non-invasive intraparenchymal transplantation of hNSC lines, which have been cultured under standardised conditions, can work as a therapy for ALS.
Materials and methods
Animal model—Ethics Statement and Institutional Animal Care and Use Committee (IACUC) approvals
SOD1 transgenic male rats (Taconic, USA) were bred with Sprague Dawley female rats (Harlan Laboratories, USA) to obtain and maintain hemizygous transgenic progeny—the transgenic offspring were identified by polymerase chain reaction (PCR), in which DNA extracted from the tails at P22 was amplified and used to verify the presence of the exogenous human SOD1 gene28. All animal care and experimental procedures were carried out according to the current national and international animal ethics guidelines, and were approved by the Italian Ministry of Health (authorisation number # 286/2013 -B).
Preparing hNSCs for implantation
The hNSCs used in the study have been produced and characterised in the Cell Factory and Biobank of Santa Maria Hospital (Terni, Italy), authorised by the Italian Medicine Agency (AIFA) for the production of hNSCs to be used for clinical trials (aM 54/2018). The methodology applied to isolate, expand, characterise and cryopreserve the lines is based on the Neurosphere Assay26,41,54, and has been used for the production of the cells utilised in phase I trials for Amyotrophic Lateral Sclerosis patients (NCT0164006723) and for Secondary Progressive Multiple Sclerosis patients (NCT03282760, ongoing).
The entire production process, starting from tissue procurement to cryopreservation is compliant to cGMP guidelines and approved by AIFA. The hNSCs are obtained from foetal brain tissue derived from fetuses that underwent miscarriage or natural in utero death upon receiving the signed informed consent from the mother. Forty-eight hours prior to implantation, hNSCs were plated in the growth medium at a concentration of 10,000 cells/cm2. On the day of surgery, hNSCs were collected by centrifugation, viable cells were counted by Trypan blue exclusion criteria, and the correct number of cells were re-suspended in HBSS for the transplant.
Experimental groups
SOD1 transgenic male rats were randomly divided into three experimental groups: (i) transplanted with hNSCs (hNSC rats, n = 15); (ii) treated with HBSS (HBSS rats, n = 15) and (iii) untreated (CTRL rats, n = 22). An additional group of non-transgenic littermates (wild-type, WT, n = 9) were used as controls for symptomatic evaluation of the colony. Tacrolimus (FK506) and cyclosporine (cyclosporin A) are the principal immunosuppressive drugs that have been applied for solid organ transplantation55,56 and have been translated to stem cell treatments for PD57 and ALS22. In animal models, despite differences in potency, both drugs showed excellent survival rates for grafts across many comparative studies58,59. Our previous results44,45 showed that hNSCs can survive—without signs of rejection—in the rat brain up to 6 months under transient immunosuppression treatment, with cyclosporin A. On the bases of these results, we applied the same immunosuppressive treatment with administration of cyclosporine A (15 mg/kg/day subcutaneous; Sandimmne, Novartis) that was initiated on the day of transplantation and continued for 15 days after surgery (for all animal groups).
Implanting hNSCs into spinal cord
Cells were transplanted at the early-symptomatic stage (ESS) of the disease, i.e., when the SOD1 rats presented with initial signs of a decrease in limb control and/or with initial failure on the rotarod. Animals were anaesthetised with isofluorane (1.5–2% maintenance; in room air), placed in a spinal clamp apparatus (David Kopf Instruments, Tujunga, CA), and a partial L1–L2 laminectomy was performed. The cells (or HBSS) were injected bilaterally with use of a Hamilton syringe (33-gauge needle) into the ventral horns of the L3–L4 segments of the cord. Injections were made on both sides, at four sites, set 1 mm apart. At each site, we delivered 1 μl of HBSS containing the hNSCs (100,000 cells/μl), or 1 μl of HBSS only (in control group), applied over 60 s; the needle was left in place for an additional 180 s, then withdrawn. After all injections were completed, the incision was sutured and animals were placed back into their cage with ad libitum access to food and water.
Assessment of neurological function and disease progression
Motor functions were evaluated using three different tests: rotarod, motor score and body weight27,28— these tests allowed us to define the ESS, the MSS, the LSS and the E-ST of the disease (see details in the Results section). All behavioural measures were recorded by individuals blinded to the treatment group.
Rotarod test
The rotarod test was used to evaluate general motor coordination, and the strength and balance of the animal. Starting on P70, animals were trained on the rotating rod, increasing the sped from 4 to 22 rotations per min within 300 s; the rats were trained three times a week, and then tested twice a week27). Each test consisted of three runs on the rotarod (lasting a maximum of 300 s) and performance was quantified by calculating the mean time (seconds) spent on the rod.
Motor score
Rats were scored twice a week, starting on P70. Hind-limb splay defects were scored on a scale of 0–4, with 4 = normal; 3.5 = onset of hind-limb splay defect; 3 = abnormal gait; 2 = partial hind-limb paralysis (first signs of dragging); 1 = hind-limb paralysis plus forelimb weakness; 0 = significant fore- as well as hind- limb paralysis.
Body weight evaluation
Rats were weighed twice a week, starting on p70.
Statistical evaluation of symptomatic analysis
Temporal evolution of rotarod performance, motor score and body weight were assessed using a longitudinal linear model, accounting for the repeated, unequally spaced, measurements design using a spatial power covariance matrix. Nonlinear evolutions were modelled by adding both linear and time quadratic terms into the model. Pairwise group comparisons (CTRL vs. HBSS vs. hNSCs) of overall and specific time points were assessed with use of suitable statistical contrasts. Estimated means, and their temporal evolution, were graphically represented. Time to death analysis was conducted with use of a proportional hazard Cox regression model, and risks were reported as hazard ratios (HR), along with their 95% confidence intervals (95% CI). The time variable was defined as the time between the surgery (or, for CTRL rats, the time of initial deterioration of symptoms) and the date of death. Kaplan–Meier curves were also displayed. A p-value of < 0.05 was considered to be statistically significant. All analyses were performed using SAS Release 9.3 (SAS Institute, Cary, NC). Note that we performed a sensitivity analysis by choosing a CTRL group that presented, on day 0, a rotarod and motor score value that was better than that for the hNSC and HBSS animals (p < 0.05).
Immunohistochemistry
Histological analyses were performed at three time points: 15DAT, 40DAT and END STAGE. To the scope, we killed n = 5 animals/time point out of the previously described experimental group (transplanted with hNSCs, treated with HBSS and untreated). To retrieve the spinal cord, animals were euthanized by overdose of anaesthesia and perfused/fixed with paraformaldehyde solution (4%). Immunohistochemistry was performed on cryopreserved coronal sections of the spinal cord fixed in 4% PFA made up in PBS. Cords were removed, immersed overnight in 4% PFA at 4 °C, cryoprotected in sucrose gradients (10, 20 and 30%) and sectioned at 30 μm on a cryostat. Parallel series were collected on glass slides. For immunohistochemistry, sections were incubated for 90 min with blocking solution (PBS-1X with 10% normal goat serum (NGS) and TRITON X-100 at 0.1%) at room temperature, then immersed overnight in the following primary antibodies, diluted in the blocking solution, at 4 °C: human nuclei (huN mAb, Millipore), glial fibrillary acidic protein (GFAP pAb, Dako), Ki67 antigen nuclear (Ki67 pAb, Monosan), Ionised calcium-binding adapter molecule 1 (Iba1 pAb, Wako), cluster of differentiation 68 (CD68, mAb, Biorad), doublecortin (DCX, pAb, Santa Crouz), platlet-derived growth factor receptor-α (PDGFR-α, pAb, Euroclone), human Nestin (hNestin, mAb, R&D system), β-tubulin III (β-tubIII, mAb, Covance), anti SOD1 exposed dimer interface (SEDI, pAb StressMarq Bioscineces Inc., SPC-206P, generously provided by Corona C. and Crociara P.). Tissues were rinsed with PBS. Samples were then incubated for 90 min at room temperature, in the following secondary antibodies: Alexa Fluor 594- or 488-labelled anti-mouse and/or anti-rabbit (Molecular Probes). After washing with PBS, the cell nuclei were labelled with DAPI for 20 min at room temperature. Finally, the slides were rinsed in PBS, coverslipped, and left to dry at room temperature in the dark. Microphotographs were acquired using a Zeiss Axiovert 200 direct epifluorescence microscope (Axioplan 2, Carl Zeiss, Jena, Germany) or by confocal microscopy (Leica DM IRE2 or Nikon).
Stereological counts of MNs
One set of serial sections (every 1050 μm) from each animal was Nissl-stained for stereological counting. Nucleoli of MNs in the L1–L4 spinal cord (where cells were delivered) were identified and counted at 40x in (hNSC rats, n = 5; HBSS rats, n = 5; CTRL rats, n = 5): only neurons ≥ 200 μm2 in area (attributable to alpha motor neurons) and located in a proper position (http://store.elsevier.com/The-Spinal-Cord/isbn-9780123742476) were included. A 3.5 -µm guard zone, a 150 -µm × 150 -µm counting frame size and a 200 -µm × 200 -µm scan grid size were set. The density of MNs (cells/mm3) was calculated by counting cells with the Optical Fractionator stereological technique60, on a computer-assisted a Nikon Eclipse E600 microscope equipped with the StereoInvestigator software (MicroBrightField, Williston, VT, USA). Cells were counted on the computer screen, with the help of an Optronics MicroFire digital camera.
Quantitative analysis of immunohistochemistry
Surviving transplanted hNSCs were determined by counting all huN + cells in serial sections through the spinal cord sections that spanned the graft area. The total number of hNSCs was calculated for the entire graft region, using the Abercombie formula. Data were presented as the percentage of surviving cells over the total number of transplanted cells. The full extent of anteroposterior migration was calculated by evaluating the distance between the most proximal and most caudal sections containing huN + cells. Differentiation of hNSCs was assessed by co-localisation of huN reactivity with markers depicting differentiation of the neural lineage: Ki67, Nestin, GFAP, DCX, β-Tub III, PDGFR-a. Analysis was performed on at least three sections within the transplant area, using three fields (×40 magnification) for each section.
To determine the numbers of active astroglia and microglia, we counted cells positive for GFAP, Iba1 or CD68 and divided by the total number of DAPI-stained nuclei. Data were quantified on bilateral epifluorescence images of the anterior horns, anterior funiculi and lateral funiculi acquired on the camera from at least three sections within the transplanted area (L3–L4 region).
Statistical analysis was performed by one-way ANOVA. Data were reported as means ± SEM. Each value represented the average of n = 5, animals, unless stated otherwise. Data were not considered statistically significant unless indicated in the figures (*p < 0.05, **p < 0.01, ***p < 0.001).
Supplementary information
Acknowledgements
This work was supported by Italian Ministry of Health, Ricerca Corrente 2014–2017 to ALV; Association Revert Onlus, Fondazione Cellule Staminali, Fondazione Stefano Borgonovo Onlus, Assicurazioni Generali SpA, Associazione Pro Roberto, Fondazione Milan, Fondazione Cassa di RIsparmio di Terni e Narni. We would like to thank for precious technical help: Andrea Raspa (neuro surgery), Gianmarco Muzi, Claudia Ricciolini, Massimo Projetti-Pensi, Giada Sgaravizzi, (clinical-grade hNSCs production) Alessio Giavazzi, Salvatore Spano, Marta De Luca (histology).
Competing interests
The authors declare that they have no conflict of interest.
Footnotes
Edited by B. Joseph
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Cristina Zalfa, Laura Rota Nodari
Contributor Information
Angelo Luigi Vescovi, Email: vescovia@gmail.com.
Daniela Ferrari, Email: daniela.ferrari@unimib.it.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41419-019-1582-5).
References
- 1.Brown RH, Jr., Al-Chalabi A. N. Engl. J. Med. 2017;377:1602. doi: 10.1056/NEJMra1603471. [DOI] [PubMed] [Google Scholar]
- 2.Ludolph AC, Brettschneider J, Weishaupt JH. Amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2012;25:530–535. doi: 10.1097/WCO.0b013e328356d328. [DOI] [PubMed] [Google Scholar]
- 3.Yamanaka K, et al. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc. Natl. Acad. Sci. U S A. 2008;105:7594–7599. doi: 10.1073/pnas.0802556105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011;7:616–630. doi: 10.1038/nrneurol.2011.152. [DOI] [PubMed] [Google Scholar]
- 5.Serio A, Patani R. Concise review: the cellular conspiracy of amyotrophic lateral sclerosis. Stem Cells. 2018;36:293–303. doi: 10.1002/stem.2758. [DOI] [PubMed] [Google Scholar]
- 6.Alsultan AA, Waller R, Heath PR, Kirby J. The genetics of amyotrophic lateral sclerosis: current insights. Degener. Neurol. Neuromuscul. Dis. 2016;6:49–64. doi: 10.2147/DNND.S84956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chio A, et al. The multistep hypothesis of ALS revisited: the role of genetic mutations. Neurology. 2018;91:e635–e642. doi: 10.1212/WNL.0000000000005996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keller MF, et al. Genome-wide analysis of the heritability of amyotrophic lateral sclerosis. JAMA Neurol. 2014;71:1123–1134. doi: 10.1001/jamaneurol.2014.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 10.Nagai M, et al. Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J. Neurosci. 2001;21:9246–9254. doi: 10.1523/JNEUROSCI.21-23-09246.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teng YD, et al. Multimodal actions of neural stem cells in a mouse model of ALS: a meta-analysis. Sci. Transl. Med. 2012;4:165ra164. doi: 10.1126/scitranslmed.3004579. [DOI] [PubMed] [Google Scholar]
- 12.Hefferan MP, et al. Human neural stem cell replacement therapy for amyotrophic lateral sclerosis by spinal transplantation. PLoS One. 2012;7:e42614. doi: 10.1371/journal.pone.0042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corti S, et al. Neural stem cells LewisX+CXCR4+modify disease progression in an amyotrophic lateral sclerosis model. Brain. 2007;130:1289–1305. doi: 10.1093/brain/awm043. [DOI] [PubMed] [Google Scholar]
- 14.Xu L, Shen P, Hazel T, Johe K, Koliatsos VE. Dual transplantation of human neural stem cells into cervical and lumbar cord ameliorates motor neuron disease in SOD1 transgenic rats. Neurosci. Lett. 2011;494:222–226. doi: 10.1016/j.neulet.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu L, et al. Human neural stem cell grafts ameliorate motor neuron disease in SOD-1 transgenic rats. Transplantation. 2006;82:865–875. doi: 10.1097/01.tp.0000235532.00920.7a. [DOI] [PubMed] [Google Scholar]
- 16.Knippenberg Sarah, Rath Klaus Jan, Böselt Sebastian, Thau-Habermann Nadine, Schwarz Sigrid C., Dengler Reinhard, Wegner Florian, Petri Susanne. Intraspinal administration of human spinal cord-derived neural progenitor cells in the G93A-SOD1 mouse model of ALS delays symptom progression, prolongs survival and increases expression of endogenous neurotrophic factors. Journal of Tissue Engineering and Regenerative Medicine. 2015;11(3):751–764. doi: 10.1002/term.1972. [DOI] [PubMed] [Google Scholar]
- 17.Suzuki M, et al. GDNF secreting human neural progenitor cells protect dying motor neurons, but not their projection to muscle, in a rat model of familial ALS. PLoS ONE. 2007;2:e689. doi: 10.1371/journal.pone.0000689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fricker RA, et al. Site-specific migration and neuronal differentiation of human neural progenitor cells after transplantation in the adult rat brain. J. Neurosci. 1999;19:5990–6005. doi: 10.1523/JNEUROSCI.19-14-05990.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan J, et al. Combined immunosuppressive agents or CD4 antibodies prolong survival of human neural stem cell grafts and improve disease outcomes in amyotrophic lateral sclerosis transgenic mice. Stem Cells. 2006;24:1976–1985. doi: 10.1634/stemcells.2005-0518. [DOI] [PubMed] [Google Scholar]
- 20.Ostenfeld T, et al. Human neural precursor cells express low levels of telomerase in vitro and show diminishing cell proliferation with extensive axonal outgrowth following transplantation. Exp. Neurol. 2000;164:215–226. doi: 10.1006/exnr.2000.7427. [DOI] [PubMed] [Google Scholar]
- 21.Feldman EL, et al. Intraspinal neural stem cell transplantation in amyotrophic lateral sclerosis: phase 1 trial outcomes. Ann. Neurol. 2014;75:363–373. doi: 10.1002/ana.24113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mazzini L, et al. Advances in stem cell therapy for amyotrophic lateral sclerosis. Expert. Opin. Biol. Ther. 2018;18:865–881. doi: 10.1080/14712598.2018.1503248. [DOI] [PubMed] [Google Scholar]
- 23.Mazzini L, et al. Human neural stem cell transplantation in ALS: initial results from a phase I trial. J. Transl. Med. 2015;13:17. doi: 10.1186/s12967-014-0371-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goutman SA, et al. Long-term phase 1/2 intraspinal stem cell transplantation outcomes in ALS. Ann. Clin. Transl. Neurol. 2018;5:730–740. doi: 10.1002/acn3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glass Jonathan D., Hertzberg Vicki S., Boulis Nicholas M., Riley Jonathan, Federici Thais, Polak Meraida, Bordeau Jane, Fournier Christina, Johe Karl, Hazel Tom, Cudkowicz Merit, Atassi Nazem, Borges Lawrence F., Rutkove Seward B., Duell Jayna, Patil Parag G., Goutman Stephen A., Feldman Eva L. Transplantation of spinal cord–derived neural stem cells for ALS. Neurology. 2016;87(4):392–400. doi: 10.1212/WNL.0000000000002889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vescovi AL, et al. Isolation and cloning of multipotential stem cells from the embryonic human CNS and establishment of transplantable human neural stem cell lines by epigenetic stimulation. Exp. Neurol. 1999;156:71–83. doi: 10.1006/exnr.1998.6998. [DOI] [PubMed] [Google Scholar]
- 27.Mancuso R, et al. Effect of genetic background on onset and disease progression in the SOD1-G93A model of amyotrophic lateral sclerosis. Amyotroph. Lateral. Scler. 2012;13:302–310. doi: 10.3109/17482968.2012.662688. [DOI] [PubMed] [Google Scholar]
- 28.Matsumoto A, et al. Disease progression of human SOD1 (G93A) transgenic ALS model rats. J. Neurosci. Res. 2006;83:119–133. doi: 10.1002/jnr.20708. [DOI] [PubMed] [Google Scholar]
- 29.Miana-Mena FJ, et al. Optimal methods to characterize the G93A mouse model of ALS. Amyotroph. Lateral. Scler. Other. Motor. Neuron. Disord. 2005;6:55–62. doi: 10.1080/14660820510026162. [DOI] [PubMed] [Google Scholar]
- 30.Pickles S, et al. ALS-linked misfolded SOD1 species have divergent impacts on mitochondria. Acta Neuropathol. Commun. 2016;4:43. doi: 10.1186/s40478-016-0313-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crociara P, et al. Motor neuron degeneration, severe myopathy and TDP-43 increase in a transgenic pig model of SOD1-linked familiar ALS. Neurobiol. Dis. 2019;124:263–275. doi: 10.1016/j.nbd.2018.11.021. [DOI] [PubMed] [Google Scholar]
- 32.Bruijn LI, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/S0896-6273(00)80272-X. [DOI] [PubMed] [Google Scholar]
- 33.Lasiene J, Yamanaka K. Glial cells in amyotrophic lateral sclerosis. Neurol. Res. Int. 2011;2011:718987. doi: 10.1155/2011/718987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vargas MR, Johnson JA. Astrogliosis in amyotrophic lateral sclerosis: role and therapeutic potential of astrocytes. Neurotherapeutics. 2010;7:471–481. doi: 10.1016/j.nurt.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cossetti C, Alfaro-Cervello C, Donegà M, Tyzack G, Pluchino S. New perspectives of tissue remodelling with neural stem and progenitor cell-based therapies. Cell Tissue Res. 2012;349:321–329. doi: 10.1007/s00441-012-1341-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drago D, et al. The stem cell secretome and its role in brain repair. Biochimie. 2013;95:2271–2285. doi: 10.1016/j.biochi.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giusto E, Donegà M, Cossetti C, Pluchino S. Neuro-immune interactions of neural stem cell transplants: from animal disease models to human trials. Exp. Neurol. 2014;260:19–32. doi: 10.1016/j.expneurol.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pluchino S, Cossetti C. How stem cells speak with host immune cells in inflammatory brain diseases. Glia. 2013;61:1379–1401. doi: 10.1002/glia.22500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mitrecić D, Nicaise C, Gajović S, Pochet R. Distribution, differentiation, and survival of intravenously administered neural stem cells in a rat model of amyotrophic lateral sclerosis. Cell Transplant. 2010;19:537–548. doi: 10.3727/096368910X498269. [DOI] [PubMed] [Google Scholar]
- 40.Lee HJ, et al. Human motor neurons generated from neural stem cells delay clinical onset and prolong life in ALS mouse model. PLoS ONE. 2014;9:e97518. doi: 10.1371/journal.pone.0097518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gelati M, et al. Culturing and expansion of “clinical grade” precursors cells from the fetal human central nervous system. Methods Mol. Biol. 2013;1059:65–77. doi: 10.1007/978-1-62703-574-3_6. [DOI] [PubMed] [Google Scholar]
- 42.Federici, T. et al. Surgical technique for spinal cord delivery of therapies: demonstration of procedure in gottingen minipigs. J. Vis. Exp., e4371, 10.3791/4371 (2012). [DOI] [PMC free article] [PubMed]
- 43.Gutierrez J, et al. Preclinical validation of multilevel intraparenchymal stem cell therapy in the porcine spinal cord. Neurosurgery. 2015;77:604–612. doi: 10.1227/NEU.0000000000000882. [DOI] [PubMed] [Google Scholar]
- 44.Rota Nodari L, et al. Long-term survival of human neural stem cells in the ischemic rat brain upon transient immunosuppression. PLoS ONE. 2010;5:e14035. doi: 10.1371/journal.pone.0014035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferrari D, et al. Differential pathotropism of non-immortalized and immortalized human neural stem cell lines in a focal demyelination model. Cell. Mol. Life Sci. 2012;69:1193–1210. doi: 10.1007/s00018-011-0873-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pluchino S, et al. Human neural stem cells ameliorate autoimmune encephalomyelitis in non-human primates. Ann. Neurol. 2009;66:343–354. doi: 10.1002/ana.21745. [DOI] [PubMed] [Google Scholar]
- 47.Gros-Louis F, Soucy G, Lariviere R, Julien JP. Intracerebroventricular infusion of monoclonal antibody or its derived Fab fragment against misfolded forms of SOD1 mutant delays mortality in a mouse model of ALS. J. Neurochem. 2010;113:1188–1199. doi: 10.1111/j.1471-4159.2010.06683.x. [DOI] [PubMed] [Google Scholar]
- 48.Liu HN, et al. Targeting of monomer/misfolded SOD1 as a therapeutic strategy for amyotrophic lateral sclerosis. J. Neurosci. 2012;32:8791–8799. doi: 10.1523/JNEUROSCI.5053-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Castro AJ. Motor performance in rats. The Effects of Pyramidal Tract Section. Brain Res. 1972;44:313–323. doi: 10.1016/0006-8993(72)90305-8. [DOI] [PubMed] [Google Scholar]
- 50.Raineteau O, Fouad K, Bareyre FM, Schwab ME. Reorganization of descending motor tracts in the rat spinal cord. Eur. J. Neurosci. 2002;16:1761–1771. doi: 10.1046/j.1460-9568.2002.02243.x. [DOI] [PubMed] [Google Scholar]
- 51.Dadon-Nachum M, Melamed E, Offen D. The “dying-back” phenomenon of motor neurons in ALS. J. Mol. Neurosci. 2011;43:470–477. doi: 10.1007/s12031-010-9467-1. [DOI] [PubMed] [Google Scholar]
- 52.Fischer LR, Glass JD. Axonal degeneration in motor neuron disease. Neurodegener. Dis. 2007;4:431–442. doi: 10.1159/000107704. [DOI] [PubMed] [Google Scholar]
- 53.Clark JA, Southam KA, Blizzard CA, King AE, Dickson TC. Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J. Chem. Neuroanat. 2016;76:35–47. doi: 10.1016/j.jchemneu.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 54.Gritti, A., Galli, R. & Vescovi, A. Cultures of Stem Cells of the Central Nervous System. Protocols for Neural Cell Culture. (ed Fedoroff, S.), 173–197, Humana Press: Totowa, NJ, USA (2000).
- 55.Kapturczak MH, Meier-Kriesche HU, Kaplan B. Pharmacology of calcineurin antagonists. Transplant. Proc. 2004;36:25s–32s. doi: 10.1016/j.transproceed.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 56.Williams D, Haragsim L. Calcineurin nephrotoxicity. Adv. Chronic. Kidney. Dis. 2006;13:47–55. doi: 10.1053/j.ackd.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 57.Kirkeby A, Parmar M, Barker RA. Strategies for bringing stem cell-derived dopamine neurons to the clinic: a European approach (STEM-PD) Prog. Brain. Res. 2017;230:165–190. doi: 10.1016/bs.pbr.2016.11.011. [DOI] [PubMed] [Google Scholar]
- 58.Maes BD, Vanrenterghem YF. Cyclosporine: advantages versus disadvantages vis-a-vis tacrolimus. Transplant. Proc. 2004;36:40s–49s. doi: 10.1016/j.transproceed.2004.01.038. [DOI] [PubMed] [Google Scholar]
- 59.Diehl R, et al. Immunosuppression for in vivo research: state-of-the-art protocols and experimental approaches. Cell. Mol. Immunol. 2017;14:146–179. doi: 10.1038/cmi.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.